
m hore uvedených výnimiek) alebo život ohrozujúcu toxicitu alebo toxicitu spôsobujúcu trvalé následky (toxicita 4. stupňa podľa US NCI CTC), musí sa liečba klofarabínom ukončiť (pozri časť 4.4).
Osobitné skupinypacientov
Porucha funkcie obličiek
Obmedzené údaje, ktoré máme k dispozícii, svedčia o možnosti hromadenia klofarabínu u pacientov so zníženým klírensom kreatinínu (pozri časti 4.4 a 5.2). Kontraindikované je použitie klofarabínu
u pacientov so závažnou insuficienciou obličiek (pozri časť 4.3), a u pacientov so slabou až miernou insuficienciou je potrebné postupovať opatrne (pozri časť 4.4).
Pacienti so stredne závažnou poruchou funkcie obličiek (klírens kreatinínu 30 – < 60 ml/min)
vyžadujú zníženie dávky na polovicu (pozri časť 5.2).
Pacienti s poruchou funkciou pečene
Neexistujú žiadne skúsenosti s pacientmi s poruchou funkciou pečene (bilirubín v sére ≥ 1,5 x ULN plus AST a ALT > 5 x ULN), pritom je pečeň potenciálny cieľový orgán z hľadiska toxicity. Z tohto dôvodu je klofarabín kontraindikovaný u pacientov so závažnou poruchou funkcie pečene (pozri
časť 4.3) a u pacientov so slabo až mierne narušenou funkciou pečene je potrebná opatrnosť (pozri časť 4.4).
Spôsob podávania
Odporúčaná dávka sa podáva intravenóznou infúziou, hoci v prebiehajúcich klinických skúškach bola
podávaná centrálnym venóznym katétrom. Evoltra sa nesmie zmiešať s inými liekmi ani súbežne podávať s inými liekmi použitím tej istej intravenóznej hadičky (pozri časť 6.2). Pokyny na nariedenie lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Použitie u pacientov s ťažkou insuficienciou obličiek alebo závažným narušením funkcie pečene. Dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Evoltra je veľmi účinná antineoplastická látka s potenciálne významnými hematologickými a nehematologickými vedľajšími účinkami (pozri časť 4.8).
U pacientov liečených klofarabínom potrebné starostlivé monitorovanie nasledovných parametrov:
· V pravidelných intervaloch sa musí sledovať komplexný krvný obraz a počty krvných doštičiek, častejšie u pacientov, ktorí trpia cytopéniou.
· Funkcie obličiek a pečene pred začatím liečby, počas aktívnej liečby a po liečbe. Liečba klofarabínom sa musí okamžite ukončiť po zistení významného zvýšenia kreatinínu, pečeňových enzýmov a/alebo bilirubínu.
· Stav dýchacieho traktu, krvný tlak, rovnováha tekutín a hmotnosť po celú dobu a okamžite po päťdennom období podávania klofarabínu.
Treba rátať s potlačením funkcie kostnej drene. Spravidla býva reverzibilné a zdá sa, že závisí od dávky. U pacientov liečených klofarabínom bola zaznamenaná závažná supresia kostnej drene vrátane neutropénie, anémie a trombocytopénie. Bolo hlásené krvácanie, vrátane mozgového, gastrointestinálneho a pľúcneho, ktoré môže byť fatálne. Väčšina prípadov súvisela s trombocytopéniou (pozri časť 4.8). Okrem toho má väčšina pacientov na začiatku liečby pri
klinických štúdiách oslabenú hematológiu ako prejav leukémie. S ohľadom na predchádzajúcu zníženú imunitu týchto pacientov a dlhdobú neutropéniu, ktorá môže byť spôsobovaná klofarabínom, pacientom hrozí zvýšené riziko závažných oportúnnych infekcií vrátane sepsy s potenciálne letálnym priebehom. Pacientov je potrebné monitorovať na príznaky a symptómy infekcie a bezodkladne ich liečiť.
Popri liečbe klofarabínom bol hlásený výskyt enterokolitídy vrátane neutropenickej kolitídy, zápalu slepého čreva a kolitídy C. difficile. Táto sa vyskytovala častejšie do 30 dní od liečby a v podmienkach kombinovanej chemoterapie. Enterokolitída môže viesť ku komplikáciám ako nekróza, perforácia alebo sepsa a môže byť spojená s fatálnym následkom (pozri časť 4.8). Pacienti majú byť kvôli znakom a príznakom enterokolitídy monitorovaní.
Hlásený bol tiež Stevensov-Johnsonov syndróm (SJS) a toxická epidermálna nekrolýza (TEN), vrátane fatálnych prípadov (pozri časť 4.8). V prípade exfoliatívnej alebo pľuzgierovitej vyrážky, alebo v prípade podozrenia na SJS alebo TEN musí byť podávanie klofarabínu prerušené.
Podávanie klofarabínu vedie k rýchlemu zníženiu počtu periférnych leukemických buniek. Počas liečby klofarabínom sa pacienti musia sledovať a vyhodnocovať z hľadiska príznakov a symptómov syndrómu lýzy nádoru a uvoľňovania cytokínu (t.j. tachypnoe, tachykardia, hypotenzia, pľúcny edém), ktoré by sa mohli rozvinúť na syndróm systémovej zápalovej odpovede organizmu (SIRS), syndrómu presakovania vlásočníc a/alebo orgánovú dysfunkciu (pozri časť 4.8).
· Treba zvážiť profylaktické podávanie allopurinolu, ak sa očakáva hyperurikémia (lýza nádoru).
· Pacienti majú dostávať intravenózne tekutiny po celé 5. denné obdobie podávania klofarabínu na zníženie účinkov lýzy nádoru a iných príhod.
· Používanie profylaktických steroidov (napr. 100 mg/m2 hydrokortizónu v prvý až tretí deň) môže byť prínosom pri prevencii symptómov alebo príznakov SIRS alebo presakovania vlásočníc.
Ak pacient vykazuje prvé príznaky alebo symptómy SIRS/syndrómu presakovania vlásočníc alebo závažnú orgánovú dysfunkciu, liečba klofarabínom sa musí okamžite prerušiť a musia byť zavedené vhodné podporné opatrenia. Liečbu klofarabínom treba prerušiť aj v prípade, ak sa u pacienta vyvinie hypotenzia z akéhokoľvek dôvodu počas 5 dní podávania. Ďalšiu liečbu klofarabínom, vo
všeobecnosti so zníženou dávkou, možno zvážiť potom, ako sa pacienti stabilizujú a orgánová funkcia sa vráti do východiskového stavu.
Väčšina pacientov, ktorí reagujú na klofarabín, dosiahnu reakciu organizmu po 1 alebo 2 liečebných cykloch (pozri časť 5.1). Ošetrujúci lekár by preto mal zhodnotiť potenciálne prínosy a riziká spojené s pokračovaním liečby u pacientov, ktorí po 2 liečebných cykloch nevykazujú hematologické ani klinické zlepšenie.
Pacienti so srdcovým ochorením a pacienti užívajúci lieky, ktoré ovplyvňujú krvný tlak alebo funkciu srdca, musia byť počas liečby klofarabínom starostlivo monitorovaní (pozri časti 4.5 a 4.8).
Neexistujú skúsenosti vo forme klinických štúdií u pediatrických pacientov s insuficienciou obličiek (definovanou v klinických štúdiách ako kreatinín v sére ≥ 2 x ULN pre daný vek) a klofarabín sa vylučuje prevažne cez obličky. Z tohto dôvodu sa klofarabín musí používať so zvýšenou obozretnosťou u pacientov so slabou až miernou insuficienciou obličiek (pozri časti 4.2 a 4.3). V súčasnosti nie sú k dispozícii dostatočné údaje o farmakokinetike klofarabínu u pediatrických pacientov so zníženým klírensom kreatinínu. Obmedzené údaje, ktoré máme k dispozícii, však nasvedčujú na možnosť hromadenia klofarabínu u pacientov so zníženým klírensom kreatinínu (pozri časti 4.2 a 5.2). Predovšetkým počas päťdňového obdobia podávania klofarabínu sa treba vystríhať súbežného používania liekov, s ktorými sa spája obličková toxicita a liekov, ktoré sa vylučujú sekréciou v tubuloch, ako sú NSAID, amfotericín B, mototrexát, aminozidy, organoplatiny, foscarnet, pentamidiín, cyklosporín, tacrolimus, acyklovír a valnganciklovír; prednosť treba dávať liekom, ktoré nie sú nefrotoxické (pozri časti 4.5 a 4.8). Ako dôsledok infekcií, sepsy a syndrómu lýzy tumoru (pozri časť 4.8) bolo pozorované zlyhanie obličiek alebo akútne zlyhanie obličiek. Pacienti majú byť sledovaní kvôli toxicite obličiek a, ak je to potrebné, klofarabín sa má vysadiť.
Pri užívaní klofarabínu v kombinácii bola zaznamenaná zvýšená frekvencia a závažnosť nežiaducich reakcií, najmä infekcie, myelosupresie (neutropénia) a hepatotoxicity. V tejto súvislosti, keď sa klofarabín používa v kombinovaných režimoch, majú byť pacienti starostlivo monitorovaní.
Pacienti dostávajúci klofarabín môžu trpieť vracaním a hnačkami; preto by mali byť poučení o vhodných opatreniach proti dehydratácii. Pacienti by mali byť poučení, že majú vyhľadať pomoc lekára ak pocítia symptómy závratu, záchvaty mdloby alebo zníženie výdaja moču.
Treba zvážiť profylaktické použitie antiemetických liekov.
Neexistujú žiadne skúsenosti s pacientmi s narušenou funkciou pečene (bilirubín v sére ≥ 1,5 x ULN plus AST a ALT > 5 ULN), pritom je pečeň potenciálny cieľový orgán z hľadiska toxicity. Z tohto dôvodu sa klofarabín musí používať so zvýšenou obozretnosťou u pacientov s miernou až stredne ťažkou poruchou funkcie pečene (pozri časti 4.2 a 4.3). Vždy ak to je možné sa treba vystríhať súbežného používania liekov, s ktorými sa spája hepatická toxicita (pozri časti 4.5 a 4.8). Ak pacient trpí hematologickou toxicitou neutropénie 4. stupňa (ANC < 0,5 x 109/l) trvajúcou ≥ 4 týždne, dávka v nasledujúcom cykle má byť znížená o 25 %.
U každého pacienta, ktorý pociťuje závažnú nehematologickú toxicitu (toxicita 3. stupňa podľa US NCI CTC) pri treťom pokuse, život ohrozujúcu toxicitu, ktorá neustúpi do 14 dní (okrem nauzey alebo vracania) alebo život ohrozujúcu neinfekčnú nehematologickú toxicitu alebo toxicitu spôsobujúcu trvalé následky (toxicita 4. stupňa podľa US NCI CTC), musí byť liečba klofarabínom ukončená (pozri časť 4.2).
Pacientom, ktorí predtým dostali transplantáciu kmeňových buniek krvotvorby (HSCT), môže hroziť zvýšené riziko hepatotoxicity pripomínajúcej veno-okluzívne ochorenie (VOD) po liečbe klofarabínom (40 mg/m2), ak sa používa v kombinácii s etopozidom (100 mg/m2) a cyklofosfamidom (440 mg/m2). V post-marketingovom období po liečbe klofarabínom boli závažné hepatotoxické nežiaduce reakcie VOD u pediatrických aj dospelých pacientov spojené s fatálnymi následkami. Pri liečbe klofarabínom boli hlásené prípady hepatitídy a zlyhania pečene, vrátane fatálnych následkov (pozri časť 4.8).
Väčšina pacientov prijala udržiavacie liečby, ktoré zahŕňali busulfan, melfalan, a/alebo kombináciu cyklofosfamidu a celkové ožiarenie tela.V kombinovanej štúdii klofarabínu fázy 1/2 boli u pediatrických pacientov s relapsujúcou alebo refraktórnou akútnou leukémiou zaznamenané závažné hepatotoxické príhody.
V súčasnosti sú k dispozícii obmedzené údaje o bezpečnosti a účinnosti klofarabínu, ak sa podáva dlhšie ako počas 3 liečebných cyklov.
Každá injekčná liekovka Evoltry obsahuje 180 mg chloridu sodného. Je to ekvivalent 3,08 mmol
(alebo 70,77 mg) sodíka a musí sa zohľadňovať u pacientov, ktorí majú kontrolovanú sodíkovú diétu.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Neexistujú žiadne známe klinicky významné interakcie s inými liekmi alebo laboratórnymi testami.
Klofarabín nie je detekovateľne metabolizovaný enzýmovým systémom cytochrómu P450 (CYP). Z tohto dôvodu nie je pravdepodobné, že bude interagovať s liečivami, ktoré inhibujú alebo indukujú enzýmy cytochrómu P450. Okrem toho nie je pravdepodobné, aby klofarabín inhiboval ktorékoľvek
z 5 najvýznamnejších ľudských izoforiem CYP (1A2, 2C9, 2C19, 2D6 a 3A4), alebo aby indukoval 2
z týchto izoforiem (1A2 a 3A4) pri koncentráciách v plazme dosahovaných po intravenóznej infúzii
52 mg/m2/deň. V súvislosti s tým sa neočakáva, že by ovplyvňoval metabolizmus liečiv, ktoré sú známymi substrátmi pre tieto enzýmy.
Klofarabín sa vylučuje hlavne cez obličky. To znamená, že predovšetkým počas päťdňového obdobia podávania klofarabínu sa treba vystríhať súbežného používania liekov, s ktorými sa spája obličková toxicita a liekov, ktoré sa vylučujú tubulárnou sekréciou, ako sú NSAIDs, amfotericín B, metotrexát,
aminozidy, organoplatiny, foskarnet, pentamidín, cyklosporín, takrolimus, acyklovír a valganciklovír
(pozri časti 4.5, 4.8 a 5.2).
Pečeň je potenciálny cieľový orgán pre toxicitu. Preto vždy, ak to je možné, sa treba vystríhať súbežného používania liekov, s ktorými sa spája hepatická toxicita (pozri časti 4.4 a 4.8).
Pacienti užívajúci lieky, ktoré ovplyvňujú krvný tlak alebo funkciu srdca, musia byť počas liečby klofarabínom starostlivo monitorovaní (pozri časti 4.4 a 4.8).
4.6 Fertilita, gravidita a laktácia
Antikoncepcia umužovažien
Ženy vo fertilnom veku a pohlavne aktívni muži musia počas liečby používať účinnú antikoncepciu.
Gravidita
Nie sú k dispozícii žiadne údaje o použití klofarabínu u gravidných žien. Štúdie na zvieratách
preukázali reprodukčnú toxicitu vrátane teratogenicity (pozri časť 5.3). Klofarabín môže spôsobiť závažné vrodené chyby, keď je podávaný počas gravidity. Z tohto dôvodu sa Evoltra má užívať počas gravidity, predovšetkým počas prvého trimestra, iba v nevyhnutných prípadoch (t.j. iba ak potenciálne prínosy pre matku prevažujú nad rizikom pre plod). Ak pacientka otehotnie počas liečby
klofarabínom, musí byť oboznámená s možnými nebezpečenstvami pre plod.
Dojčenie
Nie je známe, či sa klofarabín alebo jeho metabolity vylučujú do ľudského materského mlieka.
Vylučovanie klofarabínu do mlieka sa neštudovalo na zvieratách. S ohľadom na potenciálne vážne nežiaduce reakcie u dojčiat však dojčenie treba prerušiť pred začatím liečby Evoltrou, počas liečby a po jej ukončení (pozri časť 4.3).
Fertilita
Toxicita v závislosti od dávky pre mužské reprodukčné orgány sa pozorovala u myší, potkanov a psov
a toxicita na ženské reprodukčné orgány sa pozorovala u myší (pozri časť 5.3). Pretože účinok liečby klofarabínom na ľudskú plodnosť je neznámy, podľa potreby sa treba s pacientmi porozprávať
o plánovaní rodičovstva.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch klofarabínu na schopnosť viesť vozidlá a obsluhovať stroje. Pacientov treba poučiť, že počas liečby môžu pociťovať nežiaduce účinky, ako je točenie hlavy,
závrat alebo epizóda mdloby a že za takýchto okolností nemajú viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Takmer všetci pacienti (98 %) pociťovali najmenej jednu nežiaducu reakciu, ktorú realizátor štúdie
považoval za súvisiacu s klofarabínom. Medzi najčastejšie zaznamenané patria nauzea (61 % pacientov), vracanie (59 %), febrilná neutropénia (35 %), bolesti hlavy (24 %), vyrážka (21 %), hnačka (20 %), svrbenie (20 %), pyrexia (19 %), syndróm palmárno-plantárnej erytrodyzestézie
(15 %), únava (14 %), úzkosť (12 %), zápal sliznice (11 %) a návaly tepla (11 %). Šesťdesiat osem pacientov (59 %) pocítilo najmenej jednu závažnú nežiaducu príhodu súvisiacu s klofarabínom. Jeden pacient prerušil liečbu preto, že pocítil hyperbilirubinémiu 4. stupňa, považovanú za súvisiacu s klofarabínom po podaní 52 mg/m2 klofarabínu. Traja pacienti zomreli na nežiaduce účinky
považované realizátorom štúdie za súvisiace s liečbou klofarabínom: jeden na syndróm dýchacích ťažkostí, hepatocelulárne poškodenie a syndróm presakovania vlásočníc; jeden pacient na sepsu VRE a multiorgánové zlyhanie; a jeden pacient na septický šok a multiorgánové zlyhanie.
Zoznam nežiaducich reakciívtabuľke
Poskytované informácie sú založené na údajoch získaných z klinických skúšok, pri ktorých 115
pacientov (vo veku > 1 a < 21 rokov), ktorí mali ALL alebo akútnu myeloblastovú leukémiu (AML), dostalo najmenej jednu dávku klofarabínu s odporúčaným dávkovaním 52 mg/m2 za deň x 5. Nežiaduce reakcie sú zoradené podľa tried orgánových systémov a frekvencie (veľmi časté (³1/10); časté (³ 1/100 až < 1/10), menej časté (³ 1/1 000 až < 1/100); zriedkavé (³1/10 000 až <1/1 000) a veľmi zriedkavé (< 1/10 000)) v tabuľke uvedenej ďalej. Nežiaduce reakcie hlásené po uvedení lieku na trh sú tiež uvedené v nasledujúcej tabuľke vo frekvenčnej kategórii „neznáme“ (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Pacienti v pokročilých štádiách ALL alebo AML mohli byť v pomerne nejasnom zdravotnom stave, ktorý komplikoval hodnotenie kauzality nežiaducich účinkov s ohľadom na rôzne symptómy súvisiace so základným ochorením, jeho postupom a súčasným podávaním veľkého počtu liekov.
Nežiaduce reakcie považované za súvisiace s klofarabínom zaznamenané s frekvenciou ≥ 1/1000
(t.j. u > 1/115 pacientov) pri klinických skúškach a po uvedení lieku na trh
Infekcie a nákazy Časté: septický šok*, sepsa, bakterémia, pneumónia, herpes zoster, herpes simplex, orálna kandidóza Neznáme: kolitída spôsobená C. difficile
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
Časté: syndróm rozpadu nádoru*
Poruchy krvi a lymfatického systému Veľmi časté: febrilná neutropénia
Časté: neutropénia
Poruchy imunitného systému Časté: precitlivenosť
Poruchy metabolizmu a výživy Časté: anorexia, znížená chuť do jedla. Dehydratácia
Neznáma frekvencia: hyponatrémia
Psychické poruchy Veľmi časté: pocit úzkosti
Časté: vzrušenie, nepokoj, zmeny duševného stavu
Poruchy nervového systému Veľmi časté: bolesť hlavy
Časté: chorobná spavosť, periférna neuropatia, parestézia, závraty, tras
Poruchy ucha a labyrintu Časté: hypoakúzia
Poruchy srdca a srdcovej činnosti Časté: perikardiálna efúzia*, tachykardia* Poruchy ciev Veľmi časté: návaly horúčavy*
Časté: hypotenzia*, syndróm presakovania vlásočníc,
hematóm
Poruchy dýchacej sústavy, hrudníka a mediastína
Časté: respiračný distres, epistaxia, dyspnoe, tachypnoe, kašeľ

Poruchy gastrointestinálneho traktu
Veľmi časté: vracanie, nauzea, hnačka
Časté: krvácanie z úst, krvácanie ďasien, hemateméza, bolesti brucha, stomatitída, bolesti v hornej časti brušnej dutiny, proktalgia, ulcerácia úst
Neznáme: Pankreatitída, zvýšená sérová amyláza a
lipáza, enterokolitída, neutropenická kolitída, zápal slepého čreva
Poruchy pečene a žlčových ciest
Časté: hyperbilirubinémia, žltačka, veno-okluzívna
Celkové poruchy a reakcie v mieste podania
choroba, zvýšenie alanín- (ALT)* a aspartát- (AST)*
aminotransferázy, zlyhanie pečene
Menej časté: Hepatitída
Veľmi časté: únava, pyrexia, zápal sliznice,
Časté: multiorgánová porucha, syndróm systémovej zápalovej odpovede*, bolesť, triaška, dráždivosť, edém, periférny edém, pocit tepla, abnormálny pocit
Poruchy kože a podkožného tkaniva Veľmi časté: syndróm palmárno-plantárnej erytrodysestézie, pruritus
Časté: makulopapulárny exantém, petechia, erytém, pruritický exantém, exfoliácia kože, generalizovaný exantém, alopécia, hyperpigmentácia kože, generalizovaný erytém, erytematózny exantém, suchá pokožka, hyperhidróza
Neznáme: Stevensov-Johnsonov syndróm (SJS), toxická epidermálna nekrolýza (TEN)
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté: bolesti končatín, myalgia, bolesti kostí, bolesti prsnej steny, artralgia, bolestí krku a chrbta
Poruchy obličiek a močových ciest Časté: hematúria*
Časté: Zlyhanie obličiek, akútne zlyhanie obličiek
Laboratórne a funkčné vyšetrenia Časté: zníženie telesnej hmotnosti
Úrazy, otravy a komplikácie liečebného postupu
* = pozri dolu
Časté: pomliaždenina
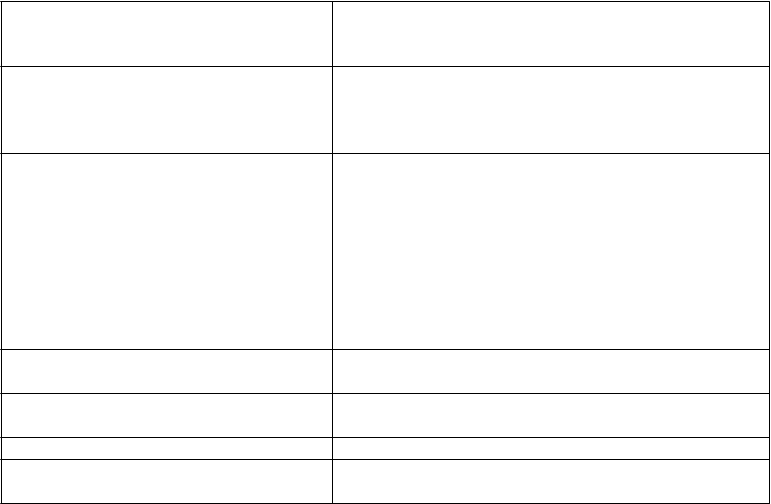
**V tejto tabuľke sú uvedené všetky nežiaduce príznaky, ktoré sa vyskytli najmenej dva razy (t.j. 2
alebo viac reakcií (1,7 %))
PopisvybranýchnežiaducichreakciíPoruchykrvialymfatickéhosystémuNajčastejšie hematologické laboratórne abnormality pozorované u pacientov liečených klofarabínom boli anémia (83,3 %; 95/114); leukopénia (87,7 %; 100/114); lymfopénia (82,3 %; 93/113), neutropénia (63,7 %; 72/113), a trombocytopénia (80,7 %; 92/114). Väčšina týchto príhod boli klasifikované do kategórie ³ 3.
V období po uvedení lieku na trh bola hlásená dlhodobá cytopénia (trombocytopénia, anémia, neutropénia a leukopénia) a zlyhanie kostnej drene. V prípade trombocytopénie boli pozorované krvácavé udalosti. Bolo hlásené krvácanie, vrátane mozgového, gastrointestinálneho a pľúcneho. Môže byť spojené s fatálnym následkom (pozri časť 4.4).
PoruchycievŠesťdesiatštyri zo 115 pacientov (55,7 %) malo najmenej jednu nežiaducu udalosť vaskulárnej poruchy. Dvadsaťtri zo 115 pacientov trpelo vaskulárnou poruchou považovanou za súvisiacu s klofarabínom, pričom najčastejšie boli hlásené návaly tepla (13 príhod, nie závažných) a hypotenzia
(5 príhod, z ktorých boli všetky považované za závažné, pozri časť 4.4). Väčšina týchto hypotenzných príhod bola hlásená u pacientov, ktorí súčasne trpeli aj závažnými infekciami.
PoruchysrdcaasrdcovejčinnostiPäťdesiat percent pacientov malo najmenej jednu nežiaducu udalosť poruchy srdca. Jedenásť príhod u
115 pacientov bolo považovaných za súvisiace s klofarabínom, žiadna z nich nebola ťažká a najčastejšie bola hlásená tachykardia (35 %) (pozri časť 4.4). 6,1 % (7/115) týchto príhod tachykardie u pacientov bola považovaná za súvisiacu s klofarabínom. Väčšina nežiaducich účinkov z triedy porúch srdca bola hlásená v prvých dvoch cykloch.
Perikardiálna efúzia a perikarditída sa zaznamenali ako nežiaduce účinky u 9 % (10/115) pacientov. Tri z týchto udalostí boli následne vyhodnotené ako súvisiace s klofarabínom: perikardiálna efúzia
(2 príhody; z ktorých 1 bola ťažká) a perikarditída (1 príhoda; nie ťažká). U väčšiny pacientov (8/10) bola perikardiálna efúzia a perikarditída považovaná za asymptomatickú a s nízkou alebo žiadnou klinickou významnosťou podľa echokardiografického hodnotenia. Perikardiálna efúzia však bola klinicky významná u 2 pacientov s určitým súvisiacim zhoršením hemodynamiky.
Infekcieanákazy
Štyridsať osem percent pacientov malo pred podaním liečby klofarabínom jednu alebo niekoľko pokračujúcich infekcií. Celkovo 83 % pacientov malo najmenej jednu infekciu po liečbe klofarabínom, vrátane hubových, vírusových a bakterialných infekcií (pozri časť 4.4). Dvadsať jeden (18,3 %) príhod sa považovalo za súvisiace s klofarabínom, z toho infekcia súvisiaca s katétrom (1 príhoda), sepsa (2 príhody) a septický šok (2 príhody; 1 pacient zomrel – pozri vyššie) boli považované za ťažké.
V období po uvedení lieku na trh boli hlásené prípady bakteriálnych, fungálnych a vírusových infekcií, ktoré môžu byť fatálne. Tieto infekcie môžu viesť k septickému šoku, zlyhaniu dýchania, zlyhaniu funkcie obličiek a/alebo zlyhaniu viacerých orgánov.
Poruchyobličiekamočovýchciest
štyridsať jeden zo 115 pacientov (35,7 %) zaznamenalo najmenej jednu nežiaducu udalosť z kategórie poruchy obličiek a močových ciest. Najrozšírenejšou renálnou toxicitou u pediatrických pacientov bol zvýšený kreatinín. Zvýšenie kreatinínu 3. alebo 4. stupňa sa vyskytlo u 8 % pacientov. K renálnej toxicite môžu prispievať nefrotoxické lieky, lýza tumoru a lýza tumoru s hyperurikémiou (pozri časti
4.3 a 4.4). Hematúria bola pozorovaná celkom u 13 % pacientov. Štyri nežiaduce reakcie u 115 pacientov bolo považovaných za súvisiace s klofarabínom, žiadna z nich nebola ťažká; hematúria (3 príhody) a akútna porucha funkcie obličiek (1 príhoda) (pozri časti 4.3 a 4.4).
Poruchypečeneažlčovýchciest
Pečeň je potenciálny cieľový orgán pre toxicitu klofarabínu a 25,2 % pacientov malo najmenej jednu príhodu nežiaduceho účinku na pečeň a žlčové cesty (pozri časti 4.3 a 4.4). Šesť príhod bolo považovaných za súvisiace s klofarabínom, z toho boli považované za ťažké akútna cholecystitída (1 príhoda), cholelitiáza (1 príhoda), hepatocelulárne poškodenie (1 príhoda; pacient zomrel – pozri vyššie) a hyperbilirubinémia (1 príhoda; pacient predčasne ukončil liečbu – pozri vyššie). Dve pediatrické hlásenia (1,7 %) veno-okluzívneho ochorenia (VOD) boli považované za súvisiaci so skúmaným liekom.
Prípady VOD hlásené v období po uvedení lieku na trhboli u detí a dospievajúcich a dospelých pacientov spojené s fatálnym následkom(pozri časť 4.4).
Navyše 50/113 pacientov, ktorým sa podával klofarabín, malo aspoň závažne (najmenej 3. triedy podľa US NCI CTC) zvýšené hladiny ALT, 36/100 zvýšené hladiny AST a 15/114 zvýšené hladiny bilirubínu. Väčšina zvýšených hladín ALT a AST sa zistila do 10 dní od podávania klofarabínu a do
15 dní sa vrátila na £ 2. stupeň. Zvýšené hladiny bilirubínu, hoci menej často sa vyskytujúce, sa ukázali ako trvajúce dlhšie. V tých prípadoch, kde sú dispozícii údaje z následného sledovania,
väčšina zvýšených hladín bilirubínu sa vrátili na £ 2. stupeň do 10 dní.
Syndrómsystémovejzápalovejodpovedeorganizmu(SIRS)alebosyndrómpresakovaniavlásočnícSIRS alebo syndróm presakovania vlásočníc (príznaky a symptómy uvoľňovania cytokínu, napr. tachypnoe, tachykardia, hypotenzia, pľúcny edém) sa zaznamenali ako nežiaduce účinky u 5 % (6/115) pediatrických pacientov (5 ALL, 1 AML) (pozri časť 4.4). Bolo zaznamenaných trinásť udalostí syndrómu lýzy tumoru, syndrómu presakovania vlásočníc alebo SIRS; SIRS (2 príhody; obe boli považované za ťažké), syndróm presakovania vlásočníc (4 príhody; z ktorých 3 boli považované za ťažké) a syndróm lýzy nádoru (7 príhod; 6 z nich boli považované za súvisiace a 3 z nich boli ťažké).
Prípady syndrómu presakovania vlásočníc hlásené v období po udelení rozhodnutia o registrácii boli spojené s fatálnym následkom (pozri časť 4.4).
PoruchygastrointestinálnehotraktuPopri liečbe klofarabínom bol hlásený výskyt enterokolitídy vrátane neutropenickej kolitídy, zápalu slepého čreva a kolitídy
C. difficile. Enterokolitída môže viesť ku komplikáciám ako nekróza, perforácia alebo sepsa a môže byť spojená s fatálnym následkom (pozri časť 4.4).
PoruchykožeapodkožnéhotkanivaStevensov-Johnsonov syndróm (SJS) a toxická epidermálna nekrolýza (TEN), vrátane fatálnych prípadov boli tiež hlásené u pacientov, ktorí užívali, alebo boli v poslednom období liečení klofarabínom. Hlásené boli tiež iné exfoliatívne stavy.
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePríznakyNebol hlásený žiaden prípad predávkovania. Predpokladá sa však, že medzi možné symptómy
predávkovania možno zaradiť nauzeu, vracanie, hnačku a závažnú supresiu kostnej drene. Dosiaľ bola najvyššia denná dávka podaná človeku 70 mg/m2 počas 5 po sebe nasledujúcich dní (2 pediatrickí pacienti s ALL). K toxicite pozorovanej u týchto pacientov patrilo vracanie, hyperbilirubinémia, zvýšené hladiny transaminázy a makulopapulárny exantém.
LiečbaNeexistuje žiadna špecifická antitoxická terapia. Odporúča sa okamžité prerušenie terapie, dôkladné
pozorovanie a nasadenie vhodných podporných opatrení.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatiká, antimetabolity ATC kód: L01BB06.
MechanizmusúčinkuKlofarabín je antimetabolit purínnukleozidu. Predpokladá sa, že jeho protinádorový účinok
vyvolávajú 3 mechanizmy:
· Inhibícia DNA polymerázy a, v dôsledku čoho sa ukončuje predlžovanie reťazca DNA a/alebo syntéza/oprava DNA.
· Inhibícia ribonukleotid reduktázy s redukciou celulárnych akumulácií deoxynukleotid trifosfátu
(dNTP).
· Porušenie celistvosti mitochondriálnej membrány spojenej s uvoľňovaním cytochrómu C a iných proapoptotických faktorov, čo vedie k naprogramovanej smrti bunky dokonca aj v nedeliacich sa lymfocytoch.
Najprv musí dôjsť k difúzii alebo transportu klofarabínu do cieľových buniek, kde sa postupne vnútrobunkovými kinázami fosforyluje na mono- a bifosfát a nakoniec na aktívny konjugát klofarabín
5’-trifosfát. Klofarabín má vysokú afinitu na jeden z aktivačných fosforylačných enzýmov, deoxycytidínkinázu, ktorá prevyšuje afinitu jeho prírodného substrátu – deoxycytidínu.
Okrem toho klofarabín má vyššiu odolnosť proti bunkovej degradácii adenozíndeaminázou a znížený sklon k fosforolytickému štepeniu ako iné liečivá jeho triedy, zatiaľ čo afinita klofarabín trifosfátu na DNA polymerázu α a na ribonukleotidreduktázu je podobná alebo väčšia ako afinita deoxyadenozín trifosfátu.
Farmakodynamické účinky
Štúdie in vitro preukázali, že klofarabín inhibuje rast rýchlo proliferujúcich bunkových radov
hematologických a solídnych nádorov a je voči nim cytotoxický. Tiež prejavoval aktivitu proti inaktívnym lymfocytom a makrofágom. Navyše klofarabín spomaľoval rast nádoru a v niektorých prípadoch spôsobil regresiu nádoru vo vybraných ľudských a myšacích xenoimplantátoch implantovaným myšiam.
Klinická účinnosťabezpečnosť
Klinická účinnosť: Aby bolo možné systematické hodnotenie odpovedí zaznamenaných u pacientov,
stanovil nezaslepený nezávislý panel na hodnotenie odpovedí (IRRP) nasledujúce miery odpovedí na základe definícií vypracovaných Skupinou detskej onkológie (Children’s Oncology Group):
CR = Celková remisia Pacienti, ktorí splnia všetky nasledujúce kritériá:
· Žiadne dôkazy blastov v obehu alebo extramedulárnej choroby
· Kostná dreň M1 (≤ 5% blastov)
· Obnovenie periférnych počtov (krvné doštičky ³ 100
x 109/l a ANC ³ 1,0 x 109/l)
CRp = Celková remisia pri absencii úplnej obnovy krvných doštičiek
· Pacienti, ktorí splnia všetky kritériá pre CR okrem obnovy počtov krvných doštičiek na > 100 x 109/l
PR = Čiastočná remisia Pacienti, ktorí splnia všetky nasledujúce kritériá:
· Úplné zmiznutie blastov v obehu
· Kostná dreň M2 (³ 5 % a ≤ 25 % blastov)
a objavenie sa normálnych progenitorových buniek
· Kostná dreň M1, ktorá nespĺňa požiadavky na CR
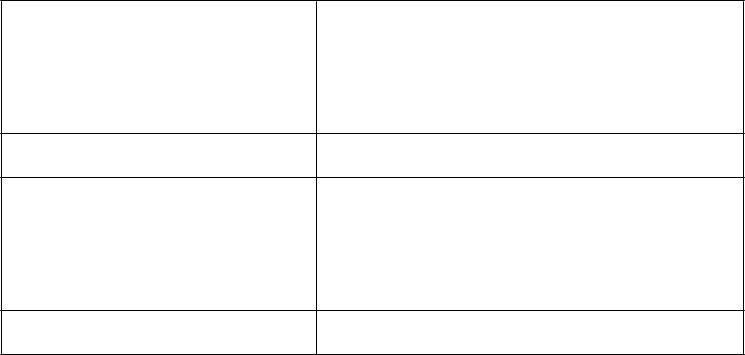
alebo CRp
Miera celkovej remisie (OR) · (Počet pacientov s CR + počet pacientov s CRp) ÷ Počet vhodných pacientov, ktorí dostávali klofarabín
Bezpečnosť a účinnosť klofarabínu sa hodnotila v otvorenej nekomparatívnej štúdii fázy I so
zvyšovaním dávky na 25 pediatrických pacientoch s relapsujúcou alebo refraktórnou leukémiou
(17 ALL; 8 AML), u ktorých štandardná terapia nezabrala, alebo pre ktorých neexistovala žiadna iná terapia. Dávkovanie sa začínalo pri 11,25 mg/m2/deň a postupne sa zvyšovalo na 15, 30, 40, 52
a 70 mg/m2/deň, podávané intravenóznou infúziou počas 5 dní jedenkrát za 2 až 6 týždňov v závislosti od toxicity a od reakcie. Deväť zo 17 pacientov ALL bolo liečených klofarabínom 52 mg/m2 za deň. Zo 17 pacientov ALL 2 dosiahli celkovú remisiu (12 %; CR) a 2 čiastočnú remisiu (12 %; PR) pri rôznych dávkach. Toxicitami obmedzujúcimi dávku boli v tejto štúdii hyperbilirubinémia, zvýšené hladiny transaminázy a makulopapulárny exantém, ktorý bol zaznamenaný pri 70 mg/m2/deň
(2 pacienti ALL; pozri časť 4.9).
Uskutočnila sa otvorená nekomparatívna štúdia klofarabínu fázy II na určenie celkovej miery remisie (OR) u pacientov, ktorí v minulosti absolvovali náročnú liečbu (vo veku ≤ 21 rokov pri počiatočnom stanovení diagnózy) s relapsujúcou alebo refraktórnou ALL definovanou použitím francúzsko- americko-britskej klasifikácie. Maximálna tolerovaná dávka 52 mg/m2/deň klofarabínu, stanovená
vo vyššie popisovanej štúdii fázy I, bola podávaná intravenóznou infúziou počas 5 po sebe
nasledujúcich dňoch každé 2 až 6 týždňov. Nasledujúca tabuľka uvádza zhrnutie najdôležitejších výsledkov tejto štúdie z hľadiska účinnosti.
Pacienti s ALL nesmeli byť vhodní pre terapiu s vyšším liečivým potenciálom a museli byť v druhom alebo ďalšom relapse a/alebo byť refraktórni, t.j. nedosiahli remisiu po najmenej dvoch predchádzajúcich protokoloch. Pred zaradením do štúdie dostávalo 58 zo 61 pacientov (95 %) 2 až
4 rôzne indukčné protokoly a 18/61 (30 %) týchto pacientov predtým absolvovalo najmenej
1 transplantáciu hematologických kmeňových buniek (HSCT). Priemerný vek liečených pacientov
(37 mužského a 24 ženského pohlavia) bol 12 rokov.
Podávanie klofarabínu viedlo k dramatickému a rýchlemu zníženiu periférnych leukémických buniek u 31 z 33 pacientov (94 %), ktorí mali merateľný absolútny počet blastov na začiatku liečby.
12 pacientov, ktorí dosiahli celkovú remisiu (CR + CRp), dosiahlo ku dňu ukončenia zberu údajov priemernú dobu prežitia 66,6 týždňov. Reakcie sa pozorovali v rôznych imunofenotypoch ALL, vrátane buniek pre-B a T. Hoci podiel transplantácií nepredstavoval konečný bod štúdie, 10/61 pacientov (16 %) pokračovalo transplantáciou HSCT po liečbe klofarabínom (3 po dosiahnutí CR,
2 po CRp, 3 po PR, 1 pacient, ktorého liečbu vyhodnotil IRRP ako neúspešnú a 1 pacient, ktorý podľa IRRP sa považoval za nevhodného na hodnotenie). Doba trvania reakcie je skresľujúca u pacientov, ktorí dostali HSCT.
Výsledná účinnosť z hlavnej štúdie u pacientov (vo veku ≤ 21 rokov pri počiatočnom stanovení diagnózy) s relapsujúcou alebo refraktórnou ALL najmenej po dvoch predchádzajúcich protokoloch
K
ategória reakcie
Celková remisia
(CR + CRp)
Pacienti
ITT* (n = 61)
12 (20 %)
Priemerná dĺžka trvania remisie (v týždňoch) (95 % CI)
32,0
(9,7 až 47,9)
Priemerný čas do progresie (v týždňoch)** (95 % CI)'
38,2 (15,4 až 56,1)
Priemerná doba prežitia (v týždňoch)
(95 % CI)
69,5 (58,6 až -)
CR 7 (12 %)
CRp 5
(8 %) PR 6
(10 %)
CR + CRp + PR 18 (30 %)
47,9 (6,1 až -)
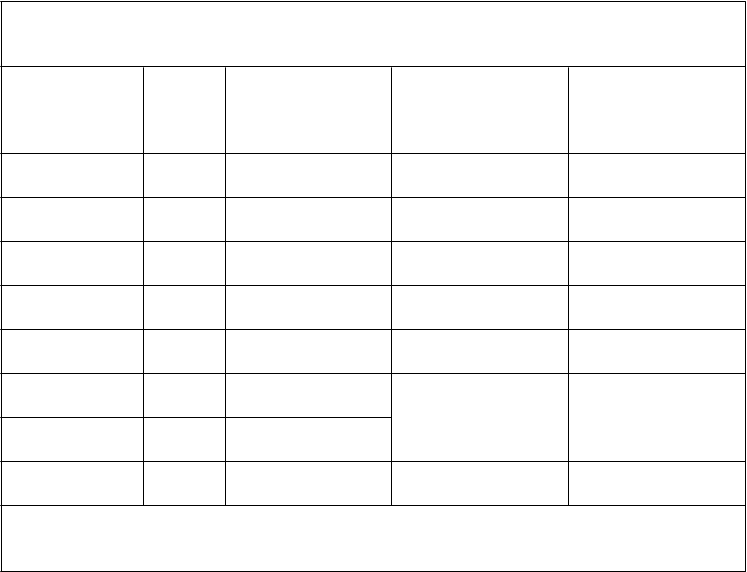
28,6
(4,6 až 38,3)
11,0 (5,0 až -)
21,5
(7,6 až 47,9)
56,1 (13,7 až -)
37,0 (9,1 až 42)
14,4 (7,0 až -)
28,7 (13,7 až 56,1)
72,4 (66,6 až -)
53,7 (9,1 až -)
33,0 (18,1 až -)
66,6 (42,0 až -)
Neúspešná liečba Nevhodný na vyhodnotenie
33 (54 %)
10 (16 %)
N/A N/A
4,0
(3,4 až 5,1)
7,6
(6,7 až 12,6)
Všetci pacienti 61 (100 %)
*ITT = zámer liečiť.
N/A 5,4
(4,0 až 6,1)
12,9
(7,9 až 18,1)
**Živí pacienti živí v štádiu remisie v čase poslednej následnej kontroly boli v tomto čase pre účely analýzy cenzurovaní.
Individuálne údaje o trvaní remisie a o prežívaní pacientov, ktorí dosiahli CR alebo CRp
Č
as do celkovej
N
ajlepšia odpoveď
remisie
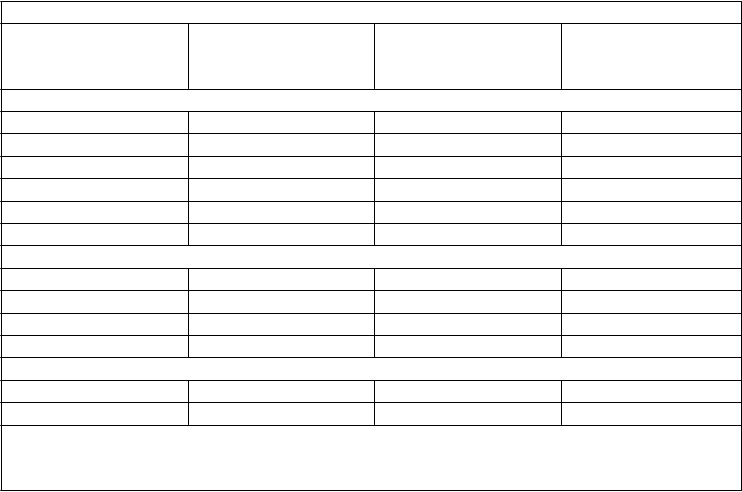 (
v týždňoch)
D
ĺ
ž
ka trvania remisie (v týždňoch)
C
elkové prežívanie
(
v týždňoch)
(
v týždňoch)
D
ĺ
ž
ka trvania remisie (v týždňoch)
C
elkové prežívanie
(
v týždňoch)
P
acienti, ktorí sa podrobili transplantácii
CR 5,7 4,3 66,6
CR 14,3 6,1 58,6
CR 8,3 47,9 66,6
CRp 4,6 4,6 9,1
CR 3,3 58,6 72,4
CRp 3,7 11,7 53,7
Pacienti, ktorí sa podrobili transplantácii počas trvania remisie*
CRp 8,4 11,6+ 145,1+ CR 4,1 9,0+ 111,9+ CRp 3,7 5,6+ 42,0
CR 7,6 3,7+ 96,3+
Pacienti, ktorí sa podrobili transplantácii po alternatívnej terapii alebo relapse*
CRp 4,0 35,4 113,3+** CR 4,0 9,7 89,4***
* Dĺžka trvania remisie cenzorovaná v čase transplantácie
** Pacient sa podrobil transplantácii po alternatívnej terapii
*** Pacient sa podrobil transplantácii po relapse
Tento liek bol registrovaný za takzvaných „mimoriadnych okolností“. To znamená, že v dôsledku
zriedkavosti ochorenia nebolo možné získať kompletné informácie o prínosoch a rizikách tohto lieku. Európska lieková agentúra bude každý rok posudzovať nové informácie o lieku, ktoré budú dostupné a tento súhrn charakteristických vlastností bude podľa potreby aktualizovaný.
5.2 Farmakokinetické vlastnosti
Adsorpcia adistribúcia
Farmakokinetika klofarabínu sa študovala na 40 pacientoch vo veku od 2 do 19 rokov
s relapsujúcou alebo refraktórnou ALL alebo AML. Pacienti boli zaradení do jednofázovej I (n = 12) alebo do dvojfázových II (n = 14 / n = 14) štúdií bezpečnosti a účinnosti a dostávali opakované dávky klofarabínu intravenóznou infúziou (pozri časť 5.1).
F
armakokinetika klofarabínu u pacientoch vo veku od 2 do 19 rokov s relapsujúcou alebo refraktórnou ALL alebo AML po podaní opakovaných dávok klofarabínu intravenóznou infúziou
P
arameter Odhady na základe nekompartmentizačnej
analýzy
(
n = 14 / n = 14)
O
dhady založené na inej analýze
D
i
stribúcia:
Objem distribúcie (stabilizovaný stav)

172 l/m2
Viazanie na proteíny plazmy 47,1 % Albumín v sére 27,0 % Vylučovanie:
b polčas klofarabínu 5,2 hodiny
Polčas trifosfátu klofarabínu > 24 hodín
Celkový klírens 28,8 l/h/m2
Obličkový klírens 10,8 l/h/m2
Dávka vylučovaná cez moč 57 %
Multivariačná analýza ukázala, že farmakokinetika klofarabínu závisí od hmotnosti a hoci bolo
zistené, že na farmakokinetiku klofarabínu má vplyv počet bielych krviniek (WBC), nezdá sa byť dostatočný na určovanie individuálneho protokolu pre pacienta na základe jeho počtu bielych krviniek. Intravenózna infúzia 52 mg/m2 klofarabínu spôsobila ekvivalentnú expozíciu v rámci širokého rozsahu hmotností. Cmax je však nepriamo úmerné hmotnosti pacienta, z tohto dôvodu majú malé deti po skončení infúzie vyššie Cmax ako typické 40 kg dieťa po podaní tej istej dávky klofarabínu na 1 m2. Z uvedeného vyplýva, že u detí s hmotnosťou < 20 kg treba uvažovať s dlhšími časmi infúzie (pozri časť 4.2).
Biotransformácia aeliminácia
Klofarabín sa vylučuje kombináciou renálnej a nerenálnej exkrécie. Po 24 hodinách sa približne 60 %
dávky vylúči bez zmeny v moči. Rýchlosť klírensu klofarabínu sa zdá byť oveľa vyššia ako sú rýchlosti glomerulárnej filtrácie, čo naznačuje, že mechanizmami vylučovania cez obličky sú filtrácia a tubulárna sekrécia. Nakoľko sa však klofarabín detekovateľne nemetabolizuje enzýmovým systémom cytochrómu P450 (CYP), cesty nerenálneho vylučovania zostávajú nateraz neznáme.
Nezistili sa žiadne zjavné rozdiely vo farmakokinetike medzi pacientmi s ALL alebo AML, ani medzi mužmi a ženami.
U tejto populácie nebola stanovená žiadna závislosť medzi expozíciou klofarabínu alebo trifosfátu klofarabínu a účinnosťou alebo toxicitou.
Osobitné skupiny pacientov:
Dospelí(vovekuod21do65rokov)
V súčasnosti nie je k dispozícii dostatok údajov na stanovenie bezpečnosti a účinnosti klofarabínu
u dospelých pacientov. Farmakokinetika klofarabínu u dospelých s relapsujúcou alebo refraktórnou AML po podaní jednej dávka 40 mg/m2 klofarabínu intravenóznou infúziou v priebehu jednej hodiny bola porovnateľná s farmakokinetikou popisovanou vyššie pre pacientov vo veku od 2 do 19 rokov
s relapsujúcou alebo refraktórnou ALL alebo AML po podaní 52 mg/m2 klofarabínu intravenóznou
infúziou v priebehu viac ako 2 hodín po 5 po sebe nasledujúcich dňoch.
Staršieosoby(vovekunad65rokov)
V súčasnosti nie je k dispozícii dostatok údajov na stanovenie bezpečnosti a účinnosti klofarabínu u pacientov vo veku 65 alebo viac rokov.
Pacienti s poruchou funkcieobličiek:
Neexistujú skúsenosti vo forme klinických štúdií u pediatrických pacientov s insuficienciou obličiek
(definovanou v klinických štúdiách ako kreatinín v sére ≥ 2 x ULN pre daný vek) a klofarabín sa vylučuje prevažne cez obličky (pozri časti 4.3 a 4.4). V súčasnosti sú v obmedzenom rozsahu
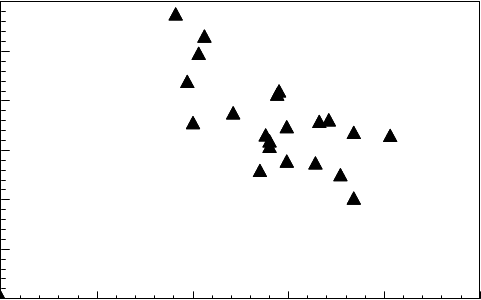
k dispozícii údaje o farmakokinetike klofarabínu u pediatrických pacientov so zníženým klírensom kreatinínu. Tieto údaje však nasvedčujú na možnosť hromadenia klofarabínu u takýchto pacientov (pozri nasledujúci diagram a časti 4.2 a 4.4).
Farmakokinetické údaje pre populácie dospelých a pediatrických pacientov naznačujú, že pacienti so stabilným stredne závažným poškodením funkcie obličiek (klírens kreatinínu 30 – < 60 ml/min), ktorí dostávajú dávku zníženú na polovicu, dosahujú podobnú expozíciu klofarabínu, ako pacienti, ktorí s normálnou funkciou obličiek dostávajú štandardnú dávku. Bezpečnostný profil klofarabínu nebol stanovený u pacientov so závažným poškodením funkcie obličiek alebo pacientov s mimotelovou eliminačnou liečbou.
Závislosť AUC0-24 hodín klofarabínu od odhadovaného počiatočného klírensu kreatinínu
u pacientov vo veku od 2 do 19 rokov s relapsujúcou alebo refraktórnou ALL alebo AML
(n = 11 / n = 12) po podaní viacerých dávok klofarabínu intravenóznou infúziou
(klírens kreatinínu odhadovaný použitím Schwartzovho vzorca)
3000
AUC 0-24 hodín 2500
Klofarabínu
(ng*h/ml)
2000
1500
1000
500
0
 0 50 100 150 200 250
O
d
h
a
d
o
v
a
n
ý k lírens k reatinínu (ml/min)
Pacienti
s
poruchou
funkcie
pečene
0 50 100 150 200 250
O
d
h
a
d
o
v
a
n
ý k lírens k reatinínu (ml/min)
Pacienti
s
poruchou
funkcie
pečene
Neexistujú žiadne skúsenosti s pacientmi s poruchou funkcie pečene (bilirubín v sére
≥ 1,5 x ULN + AST a ALT > 5 x ULN) a pečeň je potenciálny cieľový orgán pre toxicitu (pozri časti
4.3 a 4.4).
5.3 Predklinické údaje o bezpečnostiToxikologické štúdie klofarabínu na myšiach, potkanoch a psoch preukázali, že primárnymi cieľovými orgánmi toxicity boli rýchlo proliferujúce tkanivá.
U potkanov sa pozorovali účinky na srdce, zodpovedajúce kardiomyopatii, ktoré prispeli k príznakom srdcovej slabosti po opakovaných cykloch liečby. Výskyt týchto toxicít bol závislý od podávanej
dávky klofarabínu aj od dĺžky liečby. Zaznamenali sa úrovne expozície (Cmax) približne 7- až 13- násobne vyššie (po 3 alebo viacerých dávkovacích cykloch), resp. 16- až 35-násobne vyššie (po jednom alebo viacerých dávkovacích cykloch) oproti klinickej expozícii. Minimálne účinky pozorované pri nižších dávkach nasvedčujú tomu, že existuje určitá prahové úroveň toxicít na srdce a nelineárna plazmová farmakokinetika u potkanov môže v pozorovaných účinkoch zohrávať určitú úlohu. Potenciálne riziko pre ľudí nie je známe.
Glomerulonefropatia bola hlásená u potkanov pri úrovniach expozícii, ktoré sú tri- až päťnásobne vyššie ako klinická plocha pod krivkou (AUC) po 6 dávkovacích cykloch klofarabínu. Prejavovala sa malým zhrubnutím glomerulárnej bazálnej membrány s minimálnym poškodením tubulov a nesúvisela s chemickými zmenami séra.
Účinky na pečeň sa u potkanov pozorovali po chronickom podávaní klofarabínu. Je pravdepodobné, že predstavovali superpozíciu degeneratívnych a regeneratívnych zmien vyvolaných liečebnými cyklami a nesúviseli s chemickými zmenami séra. Histologické dôkazy účinkov na pečeň sa zaznamenali u psov po akútnom podaní vysokých dávok, ani v tomto prípade však neboli sprevádzané chemickými zmenami séra.
Toxicity na mužské pohlavné orgány, závislé od dávky, sa pozorovali u myší, potkanov a psov. Medzi tieto účinky patrila bilaterálna degenerácia semenonosného epitelu so zadržanými spermatidmi
a atrofia intersticiálnych buniek u potkanov pri nadmerných úrovniach expozície (150 mg/m2/deň)
a degenerácia buniek nadsemenníka a degenerácia semenonosného epitelu u psov pri klinicky relevantných úrovniach expozície (> 7,5 mg/m2 klofarabínu za deň).
U samičiek myší sa pozorovala oneskorená atrofia alebo degenerácia vaječníkov a apoptóza sliznice
maternice pri jedinej použitej dávke 225 mg/m2 klofarabínu za deň.
Klofarabín mal teratogénne účinky u potkanov a králikov. U potkanov, ktorí dostávali dávky predstavujúce približne dvoj- až trojnásobok klinickej expozície (54 mg/m2/deň) a u králikov, ktorí dostávali 12 mg/m2 klofarabínu za deň, boli hlásené zvýšené postimplantačné straty, zníženie telesnej hmotnosti plodu a zníženie celkovej hmotnosti vrhu spolu so zvýšením počtu malformácií (veľkých externých malformácií, malformácií mäkkých tkanív) a skeletálnych zmien (vrátane retardovanej osifikácie). (Nie sú k dispozícii žiadne údaje o expozícii u králikov). Za prah vývojovej toxicity bola považovaná dávka 6 mg/m2/deň u potkanov a 1,2 mg/m2/deň u králikov. Úroveň nespozorovateľných účinkov na materskú toxicitu u potkanov bola 18 mg/m2/deň a u králikov viac ako 12 mg/m2/deň. Neuskutočnili sa žiadne štúdie plodnosti.
Štúdie genotoxicity preukázali, že klofarabín nemal mutagénne účinky v analýze reverznej mutácie na baktériách, vyvolal však klastogénne účinky v teste chromozómových aberácií na bunkách vaječníkov čínskeho škrečka (CHO) bez aktivácie a v teste potkanieho mikronukleusu in vivo.
Neuskutočnili sa žiadne štúdie karcinogenity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Chlorid sodný
Voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
3 roky.
Nariedený koncentrát je chemicky a fyzikálne stabilný 3 dni v rozsahu od 2°C do 8°C a pri izbovej teplote (až do 25°C). Z mikrobiologického hľadiska by sa mal použiť okamžite. Ak sa nepoužije okamžite, za čas a podmienky uchovávania pred použitím zodpovedá používateľ. Spravidla sa nesmie uchovávať dlhšie ako 24 hodín pri 2°C - 8°C, ak sa riedenie neuskutočňuje v kontrolovanom
a aseptickom prostredí.
6.4 Špeciálne upozornenia na uchovávanieNeuchovávajte v mrazničke.
Podmienky uchovávania po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah baleniaSklenená injekčná liekovka typu I s bromobutylovou gumovou zátkou, polypropylénovým vyklápacím viečkom a vonkajším hliníkovým tesnením. Injekčné liekovky obsahujú 20 ml infúzneho koncentrátu
a sú zabalené v škatuli.
Každá škatuľa obsahuje 1, 3, 4, 10 alebo 20 liekoviek. Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomŠpeciálne opatrenianapodávanieInfúzny koncentrát Evoltra 1 mg/ml musí byť pred podávaním rozriedený. Musí byť prefiltrovaný cez
sterilný 0,2 mikrometrový filter injekčnej striekačky a potom rozriedený intravenóznou infúziou chloridu sodného 9 mg/ml (0,9 %), aby sa dosiahol celkový objem podľa príkladov uvádzaných v tabuľke nižšie. Konečný nariedený objem sa však môže líšiť v závislosti od klinického stavu pacienta a podľa uváženia lekára. (Ak nie je možné použiť 0,2 mikrometrový filter injekčnej striekačky, koncentrát sa musí najprv prefiltrovať 5 mikrometrovým filtrom, potom sa rozriediť
a nakoniec podávať cez zabudovaný 0,22 mikrometrový filter.)
 Odporúčaná schéma riedenia na základe odporúčaného dávkovania 52 mg klofarabínu na m2a deňPlocha povrchu tela (m2) Koncentrát (ml)* Celkový nariedený objem
Odporúčaná schéma riedenia na základe odporúčaného dávkovania 52 mg klofarabínu na m2a deňPlocha povrchu tela (m2) Koncentrát (ml)* Celkový nariedený objem≤ 1,44 ≤ 74,9 100 ml
1,45 až 2,40 75,4 až 124,8 150 ml
2,41 až 2,50 125,3 až 130,0 200 ml
*Jeden ml koncentrátu obsahuje 1 mg klofarabínu. Každá 20 ml injekčná liekovka obsahuje 20 mg klofarabínu. Z tohto dôvodu pre pacientov s plochou povrchu tela ≤ 0,38 m2 bude potrebná na získanie odporúčanej dennej dávky klofarabínu časť obsahu jednej injekčnej liekovky. Avšak pre pacientov s plochou povrchu tela > 0,38 m2 bude na získanie odporúčanej dennej dávky klofarabínu potrebná 1 až 7 injekčných liekoviek.
Nariedený koncentrát má byť číry, bezfarebný roztok. Roztok sa má pred podávaním vizuálne
skontrolovať na prítomnosť pevných častíc a zmenu sfarbenia.
Návod napoužitieEvoltra je iba na jedno použitie. Všetok nepoužitý liek musí byť zlikvidovaný.
Dodržiavajte postupy na správne zaobchádzanie s antineoplastickými látkami. S cytotoxickými liekmi sa musí zaobchádzať opatrne.
Pri manipulácii s Evoltrou odporúčame používať rukavice na jedno použitie a ochranné odevy. Ak sa liek dostane do kontaktu s očami, kožou alebo sliznicami, okamžite postihnuté miesto vymyte dostatočným množstvom vody.
Tehotné ženy nesmú manipulovať s Evoltrou.
LikvidáciaEvoltra je iba na jednorazové použitie. Nepoužitý liek alebo odpad vzniknutý z lieku má byť
zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGenzyme Europe B.V. Gooimeer 10
1411DD Naarden
Holandsko
Tel: +31 (0) 35 699 12 00
Fax: +31 (0) 35 694 32 14
8. REGISTRAČNÉ ČÍSLAEU/1/06/334/001 3 injekčné liekovky EU/1/06/334/002 4 injekčné liekovky EU/1/06/334/003 10 injekčných liekoviek EU/1/06/334/004 20 injekčných liekoviek EU/1/06/334/005 1 injekčná liekovka
9. DÁTUM PRVEJ REGISTRÁCIE / PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 29. máj 2006
Dátum posledného predĺženia registrácie: 14. január 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
http://www.ema.europa.eu/.