
jekčne podávaných očkovacích látkach, vždy musí byť ihneď k dispozícii primeraná liečba a lekársky dohľad pre prípad anafylaktickej udalosti po podaní
očkovacej látky.
Trvanie ochrany
Očkovanie Ervebom nemusí mať za následok ochranu všetkých očkovaných osôb. Účinnosť
očkovacej látky bola preukázaná v období ≥ 10 až ≤ 31 dní po očkovaní, trvanie ochrany však nie je známe (pozri časť 5.1). Používanie iných opatrení na kontrolu eboly sa preto nemá prerušiť.
Očkovanie osôb, ktoré boli v kontakte s prípadmi eboly sa má vykonať ihneď ako je to možné (pozri
časť 5.1).
Štandardnéopatreniapristarostlivostiopacientovsoznámymochorenímebolaalebospodozrenímnatotoochorenie
Očkovanie Ervebom nevylučuje potrebu použitia štandardných opatrení pri starostlivosti o pacientov
so známym ochorením ebola alebo s podozrením na toto ochorenie. Všetci zdravotnícki pracovníci
a iní pomocní poskytovatelia, ktorí boli zaočkovaní, nemajú po očkovaní meniť svoje postupy týkajúce sa bezpečného podávania očkovacej látky, hygieny a používania osobných ochranných prostriedkov (OOP).
Zdravotnícki pracovníci, ktorí sa starajú o pacientov s podozrením na vírus eboly alebo s potvrdeným vírusom eboly, majú uplatňovať ďalšie opatrenia na kontrolu infekcie, aby zabránili kontaktu s krvou a telesnými tekutinami pacienta a s kontaminovanými povrchmi alebo materiálmi, ako sú oblečenie
a posteľná bielizeň. So vzorkami od ľudí a zvierat určenými na vyšetrenie infekcie vírusom eboly má zaobchádzať vyškolený personál a vzorky majú byť spracovávané len vo vhodne vybavených
laboratóriách.
Osoby podávajúce očkovaciu látku majú poučiť očkované osoby, aby sa naďalej chránili pomocou vhodných opatrení.
Osoby soslabenýmimunitnýmsystémom
Bezpečnosť a účinnosť Erveba neboli hodnotené u osôb s oslabeným imunitným systémom. Osoby
s oslabeným imunitným systémom nemusia na Ervebo odpovedať rovnako ako osoby s normálnou funkciou imunitného systému. Ako preventívne opatrenie sa odporúča vyhnúť sa použitiu Erveba
u osôb so známymi stavmi spojenými s oslabením imunitného systému alebo ktoré dostávajú
imunosupresívnu liečbu, vrátane nasledujúcich stavov:
· Závažná humorálna alebo bunková (primárna alebo získaná) imunodeficiencia, napr. závažná kombinovaná imunodeficiencia, agamaglobulinémia a AIDS alebo symptomatická infekcia HIV. Hraničná hodnota počtu CD4+ T-lymfocytov pre použitie u asymptomatických HIV- pozitívnych osôb nebola stanovená.
· Prebiehajúca liečba imunosupresívami vrátane vysokých dávok kortikosteroidu. Netýka sa to osôb, ktoré dostávajú lokálne kortikosteroidy, inhalačné kortikosteroidy alebo nízku dávku parenterálnych kortikosteroidov (napr. na profylaxiu astmy alebo substitučnú liečbu).
· Ochorenia krvi ako sú leukémia, lymfómy akéhokoľvek typu alebo iné malígne novotvary postihujúce hematopoetický a lymfatický systém.
· Vrodená alebo dedičná imunodeficiencia v rodinnej anamnéze, pokiaľ sa nepreukáže normálna funkcia imunitného systému potenciálneho príjemcu očkovacej látky.
Gravidnéa dojčiace ženy
Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Erveba počas gravidity. Pozri časť 4.6.
Prenos
Vírus očkovacej látky môže byť prítomný v biologických tekutinách ako sú krv, moč, sliny, sperma,
vaginálne tekutiny, komorový mok, materské mlieko, stolica, pot, plodová voda a placenta. U väčšiny dospelých osôb bola v plazme pomocou PCR zistená RNA vírusu očkovacej látky. RNA vírusu očkovacej látky bola detegovaná predovšetkým v 1. až 7. dni. U 19 z 299 dospelých osôb bolo pomocou PCR zistené vylučovanie vírusu očkovacej látky močom alebo slinami a u 4 z 10 dospelých osôb kožnými vezikulami. U jednej osoby zo štyroch bola v 12. deň po podaní očkovacej látky zistená RNA vírusu očkovacej látky v kožných vezikulách.
Vylučovanie vírusu sa pozorovalo častejšie u detí a dospievajúcich (28/39) v porovnaní s dospelými. Prenos vírusu očkovacej látky pri blízkom osobnom kontakte sa považuje za teoreticky možný.
Príjemcovia očkovacej látky sa majú vyhnúť blízkemu kontaktu s vysokorizikovými osobami
a vystaveniu vysokorizikových osôb krvi a telesným tekutinám minimálne 6 týždňov po očkovaní. Medzi vysokorizikové osoby patria:
· osoby s oslabeným imunitným systémom a osoby dostávajúce imunosupresívnu liečbu (pozri časť vyššie),
· gravidné alebo dojčiace ženy (pozri časť 4.6),
· deti vo veku < 1 rok.
Osoby, u ktorých sa po podaní očkovacej látky objaví vezikulárna vyrážka, si majú vezikuly prekryť, kým sa nezahoja, aby sa minimalizovalo riziko možného prenosu vírusu očkovacej látky cez otvorené vezikuly. Kontaminované náplasti zlikvidujte podľa inštitucionálnych usmernení alebo postupu WHO na nakladanie s odpadom zo zdravotnej starostlivosti. Pozri časť 5.3.
Neúmyselný prenos vírusu očkovacej látky na zvieratá a hospodárske zvieratá je tiež teoreticky možný, pozri nižšie.
Osoby, ktorým bolo podané Ervebo, nesmú darovať krv minimálne 6 týždňov po očkovaní. Prenosnazvieratáahospodárskezvieratá
Prenos vírusu očkovacej látky pri blízkom kontakte s hospodárskymi zvieratami sa považuje za
teoreticky možný. Príjemcovia očkovacej látky sa majú pokúsiť predísť vystaveniu hospodárskych zvierat krvi a telesným tekutinám minimálne 6 týždňov po očkovaní. Osoby, u ktorých sa po podaní
očkovacej látky objaví vezikulárna vyrážka, si majú vezikuly prekryť, kým sa nezahoja.
Kontaminované náplasti zlikvidujte podľa inštitucionálnych usmernení alebo postupu WHO na nakladanie s odpadom zo zdravotnej starostlivosti. Pozri časť 5.3.
Súčasné ochorenie
Podanie očkovacej látky sa má odložiť u osôb, ktoré majú stredne závažné alebo závažné febrilné
ochorenie. Prítomnosť miernej infekcie nemá viesť k odloženiu očkovania.
Trombocytopénia akoagulačnéochorenia
Očkovacia látka sa má podávať s opatrnosťou osobám s trombocytopéniou alebo akýmkoľvek
koagulačným ochorením, pretože u týchto osôb môže dôjsť po intramuskulárnom podaní ku krvácaniu alebo tvorbe podliatin.
Ochrana protiochoreniuspôsobenémufilovírusmi
Očkovacia látka nezabráni ochoreniu spôsobenému inými filovírusmi ako je vírus zairskej eboly.
Vplyv nasérologickévyšetrenie
Osoby, ktorým bolo podané Ervebo, môžu mať pozitívny výsledok vyšetrenia na nukleové kyseliny
pre glykoproteín (GP) vírusu eboly, antigény alebo protilátky proti GP eboly, ktoré sú cieľovými látkami pre určité diagnostické vyšetrenia na vírus eboly. Diagnostické vyšetrenie na vírus eboly sa má preto zameriavať na ne-GP časti vírusu eboly.
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Keďže k dispozícii nie sú žiadne údaje o súbežnom podaní Erveba s inými očkovacími látkami, súbežné použitie Erveba s inými očkovacími látkami sa neodporúča.
Súbežne s Ervebom sa nemá podávať imunoglobulín (IG), transfúzia krvi alebo krvnej plazmy. Podanie imunoglobulínov, transfúzia krvi alebo krvnej plazmy podaná 3 mesiace pred alebo až do 1 mesiaca po podaní Erveba môže ovplyvniť očakávanú imunitnú odpoveď.
Nie je známe, či súbežné podanie antivirotík, vrátane interferónov, môže ovplyvniť replikáciu a účinnosť vírusu očkovacej látky.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii je obmedzené množstvo údajov (menej ako 300 ukončených gravidít) o použití Erveba
u gravidných žien, alebo u žien, ktoré otehotneli po podaní očkovacej látky. Bezpečnosť Erveba nebola u gravidných žien stanovená.
Keďže dostupné údaje sú obmedzené, vrátane malého počtu prípadov, pri vyvodzovaní záverov sa má postupovať opatrne. Nedostatok spoľahlivých údajov týkajúcich sa východiskových mier gravidít
a výsledkov gravidít v dotknutých regiónoch tiež spôsobuje, že kontextové hodnotenie údajov je
náročné.
Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Erveba počas gravidity. Vzhľadom na závažnosť EVD sa však očkovanie nemá odoprieť, ak existuje zjavné riziko vystavenia sa infekcii vírusom eboly.
Gravidite sa má predísť 2 mesiace po očkovaní. Ženy v reprodukčnom veku majú používať účinnú metódu antikoncepcie.
Dojčenie
Nie je známe, či sa vírus očkovacej látky vylučuje do ľudského mlieka.
Riziko pre novorodencov/dojčatá sa pri dojčení očkovanými matkami nedá vylúčiť.
Hodnotenie vírusu očkovacej látky v mlieku zvierat nebolo vykonané. Keď sa Ervebo podávalo samiciam potkanov, boli u potomkov zistené protilátky proti vírusu očkovacej látky, pravdepodobne vďaka získaniu materských protilátok placentárnym prenosom počas gravidity a dojčením. Pozri časť
5.3.
Rozhodnutie, či ukončiť dojčenie alebo nepodať Ervebo sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu. Za určitých okolností, ak sú alternatívne spôsoby dojčenia obmedzené, sa majú zohľadniť bezprostredné potreby a zdravotné prínosy pre dojčené dieťa v porovnaní
s matkinou potrebou Erveba. Obe môžu predstavovať nevyhnutné potreby, ktoré sa majú pred očkovaním matky zvážiť.
Fertilita
K dispozícii nie sú žiadne údaje o účinkoch na fertilitu u ľudí.
Štúdie na zvieratách u samíc potkana nenaznačujú škodlivé účinky (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o vplyve Erveba na schopnosť viesť vozidlá a obsluhovať stroje.
Ervebo nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Anafylaxia bola v klinických skúšaniach hlásená veľmi zriedkavo (0,006 %).
Najčastejšie nežiaduce reakcie v mieste podania injekcie boli bolesť v mieste podania injekcie
(70,3 %), opuch (16,7 %) a erytém (13,7 %).
Najčastejšie systémové nežiaduce reakcie hlásené po očkovaní Ervebom boli bolesť hlavy (36,9 %), pyrexia (34,3 %), myalgia (32,5 %), únava (18,5 %), artralgia (17,1 %), nauzea (8,0 %), zimnica
(6,3 %), artritída (3,7 %), vyrážka (3,6 %), hyperhidróza (3,2 %) a abdominálna bolesť (1,4 %). Vo všeobecnosti boli tieto reakcie hlásené do 7 dní po očkovaní, boli miernej až stredne závažnej intenzity
a mali krátke trvanie (menej ako 1 týždeň).
Tabuľkový zoznamnežiaducichreakcií
Frekvencie sú hlásené ako:
veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (nedá sa určiť z dostupných údajov). V rámci každej skupiny frekvencie sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1: Tabuľkový zoznam nežiaducich reakcií považovaných za súvisiace s očkovaním
Trieda orgánových systémov
podľa databázy MedDRA Nežiaduce reakcie Frekvencia
Poruchy imunitného systému: anafylaktická reakcia veľmi zriedkavé
Poruchy nervového systému: bolesť hlavy veľmi časté
Poruchy gastrointestinálneho traktu:
abdominálna bolesť nauzea
časté
Poruchy kože a podkožného tkaniva:
vyrážka§ časté
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva:
artralgia§
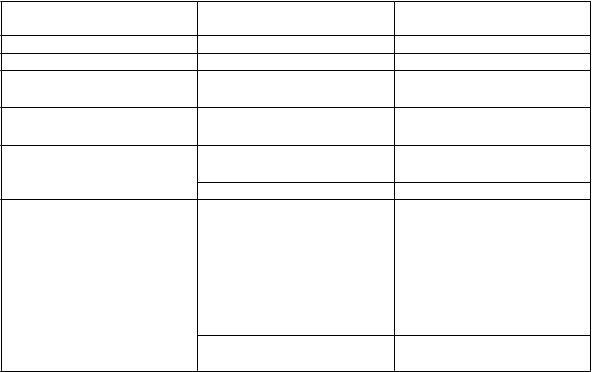
myalgia
veľmi časté
artritída§ časté
Celkové poruchy a reakcie v mieste podania:
pyrexia únava
bolesť v mieste podania
injekcie
erytém v mieste podania injekcie
opuch v mieste podania
injekcie
zimnica
hyperhidróza (potenie)
veľmi časté
časté
§Pozri opis vybraných nežiaducich reakcií.
Opisvybraných nežiaducichreakcií
Artralgia a artritída
Artralgia bola vo všeobecnosti hlásená počas prvých niekoľkých dní po očkovaní, mala miernu až stredne závažnú intenzitu a odznela do jedného týždňa po výskyte. Artritída (artritída, kĺbový výpotok, opuch kĺbov, osteoartritída, monoartritída alebo polyartritída) bola vo všeobecnosti hlásená počas prvých niekoľkých týždňov po očkovaní. V klinických skúšaniach, v ktorých bola hlásená artritída,
bol medián nástupu medzi 10 až 12 dňami (rozsah od 0 do 25 dní). V klinických skúšaniach hlásili pacienti artritídu s frekvenciou, ktorá sa pohybovala od 0 % v niekoľkých protokoloch až do 23,5 % v jednej štúdii fázy 1. Väčšina reakcií artritídy bola miernej až strednej závažnosti. Medián trvania artritídy v klinických skúšaniach, v ktorých bola artritída hlásená, sa pohyboval od 2 dní do 81,5 dní (vrátane trvania rekurentnej artritídy), maximálne 330 dní. Dôvody rozdielov v hlásení artritídy
v skúšaniach nie sú známe, môžu však byť spôsobené rozdielmi v populáciách štúdie alebo v hlásení výsledkov. V štúdii fázy 1 s najvyššou mierou artritídy malo 6 z 24 pacientov (25 %), ktorí hlásili po očkovaní artritídu, pretrvávajúce príznaky týkajúce sa kĺbov dva roky po očkovaní. Pri malom počte osôb sa vírus očkovacej látky získal zo vzoriek kĺbového výpotku, čo poukazuje na proces vyvolaný vírusom po očkovaní.
VyrážkaVyrážka bola v klinických skúšaniach opísaná viacerými spôsobmi zahŕňajúcimi generalizovanú vyrážku (2,3 %), vezikulárnu vyrážku (0,5 %), dermatitídu (0,3 %) alebo kožnú vaskulitídu (0,01 %).
V iných skúšaniach bola vyrážka hlásená s mediánom nástupu 7,5 až 10,5 dní (rozsah od 0 do 47 dní).
Medián trvania bol medzi 6 a 18 dňami. U 6 z 18 testovaných osôb sa vírus očkovacej látky zistil vo vyrážkach (opísaných ako dermatitída, vezikuly alebo lézie kožnej vaskulitídy), čo poukazuje na proces vyvolaný vírusom po očkovaní.
Prechodné zníženie počtu leukocytovV štúdiách fázy 1/2 sa veľmi často pozorovali prechodné zníženia počtu lymfocytov, neutrofilov
a celkového počtu bielych krviniek počas prvých 3 dní po očkovaní; tieto udalosti sa vo všeobecnosti upravili po prvom týždni po očkovaní. V skúšaniach fázy 1/2 neboli pozorované žiadne nežiaduce
udalosti infekcií.
Pediatrická populáciaV skúšaniach fázy 1 až fázy 3 dostalo 234 detí a dospievajúcich vo veku 6 až 17 rokov dávku Erveba.
Bezpečnostný profil Erveba u detí a dospievajúcich vo veku 6 až 17 rokov bol vo všeobecnosti podobný profilu pozorovanému u dospelých.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeboli hlásené žiadne prípady predávkovania.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Očkovacie látky, vírusové očkovacie látky, ATC kód: J07BX02
Mechanizmus účinkuErvebo pozostáva z vektora obsahujúceho živý, atenuovaný rekombinantný vírus vezikulárnej
stomatitídy, ktorý exprimuje gén glykoproteínového obalu vírusu zairskej eboly
(rVSV∆G-ZEBOV-GP). Imunizácia osôb touto očkovacou látkou vedie k imunitnej odpovedi
a ochrane pred ochorením vyvolaným vírusom zairskej eboly (EVD). Relatívny príspevok prirodzenej, humorálnej a bunkami sprostredkovanej imunity na ochranu pred vírusom zairskej eboly nie je známy.
Klinickáúčinnosť
Program klinického vývoja zahŕňal štyri klinické skúšania fázy 2/3 (protokoly 009 – 012). Všetky
osoby dostali jednu dávku očkovacej látky. Klinická účinnosť Erveba bola hodnotená v Protokole 010.
Protokol 010 (štúdia očkovania okruhu ľudí) bola otvoreným skúšaním očkovania okruhu ľudí (očkovanie osôb, ktoré boli v kontakte s osobami so zdokumentovanou ebolou a očkovanie osôb v kontakte s týmito osobami [contacts and contacts of contacts, CCC]) fázy 3, ktoré hodnotilo
účinnosť a bezpečnosť Erveba v Guinei. V tomto skúšaní bolo 9 096 osôb vo veku ≥ 18 rokov, ktoré boli považované za CCC osoby s osobami so zdokumentovanou laboratórne potvrdenou EVD,
randomizovaných na okamžité (4 539 osôb v 51 skupinách) alebo o 21 dní oneskorené (4 557 osôb v 47 skupinách) očkovanie Ervebom. Z týchto 9 096 osôb dostalo 4 160 Ervebo (2 119 osôb bolo očkovaných okamžite a 2 041 osôb bolo očkovaných oneskorene). Medián veku CCC osôb so
súhlasom bol 35 rokov. Konečná primárna analýza zahŕňala 2 108 osôb (51 skupín) v skupine s okamžitým očkovaním a 1429 osôb (46 skupín) vhodných a so súhlasom v deň 0 v skupine
s oneskoreným očkovaním.
Konečná primárna analýza bola zameraná na vyhodnotenie účinnosti proti laboratórne potvrdenej EVD porovnaním výskytu prípadov, ktoré sa vyskytli 10 až 31 dní po randomizácii u osôb, ktoré boli očkované v okruhu s okamžitým očkovaním, oproti výskytu prípadov u osôb, ktoré boli zahrnuté do skúšania v deň 0 do okruhu osôb s oneskoreným očkovaním. Účinnosť očkovacej látky bola 100 % (neprispôsobený 95 % IS: 63,5 % až 100 %; 95 % IS prispôsobený na opakovanie: 14,4 % až 100 %) (0 prípadov v skupine s okamžitým očkovaním; 10 prípadov v 4 okruhoch s oneskoreným očkovaním). Randomizácia sa ukončila po predbežnej analýze s p = 0,0036, ktorá nespĺňala vopred stanovenú hladinu alfa 0,0027. Z 10 prípadov 7 bolo v kontakte a 3 boli v kontakte s osobami, ktoré boli v kontakte s osobami so zdokumentovanou ebolou. Vzhľadom na metodologické obmedzenia
a výnimočné okolnosti, ktoré sa vyskytli počas skúšania, pretrvávajú nejasnosti pokiaľ ide o úroveň, trvanie a typ ochrany.
Klinická imunogenita
Doteraz neboli definované imunologické koreláty ochrany.
Protokol 009 Partnerstvo pre výskum očkovacích látok proti ebole v Libérii (Partnership for Research on Ebola Vaccines in Liberia, PREVAIL) bol randomizovaným, dvojito zaslepeným, placebom kontrolovaným skúšaním fázy 2, ktoré hodnotilo bezpečnosť a imunogenitu potenciálnych očkovacích látok proti ebole vrátane Erveba. Toto skúšanie porovnávalo Ervebo s placebom obsahujúcim fyziologický roztok u 1 000 dospelých vo veku ≥ 18 rokov v Libérii.
Protokol 011 nazvaný Skúšanie v Sierra Leone na zavedenie očkovacej látky proti ebole (Sierra Leone
Trial to Introduce a Vaccine against Ebola, STRIVE) bol randomizovaným, otvoreným skúšaním fázy
2/3, ktoré hodnotilo bezpečnosť a imunogenitu Erveba u dospelých vo veku ≥ 18 rokov pracujúcich v zariadeniach zdravotnej starostlivosti alebo na priamych aktivitách súvisiacich s odpoveďou na
ebolu v Sierra Leone. V tomto skúšaní bolo zahrnutých 8 673 dospelých osôb a 8 651 s platným
súhlasom randomizovaných na okamžité (do 7 dní od zaradenia) alebo oneskorené (18 až 24 týždňov po zaradení) očkovanie Ervebom. Podštúdia imunogenity zahŕňala 508 osôb, ktoré boli očkované
a poskytli vzorky na hodnotenie imunogenity.
Protokol 012 bol randomizovaným, dvojito zaslepeným, placebom kontrolovaným skúšaním fázy 3, ktoré hodnotilo bezpečnosť a imunogenitu troch konzistentných šarží a šarže s vysokou dávkou (približne päťnásobne vyššou ako dávka v konzistentných šaržách a ako dávka použitá v iných skúšaniach fázy 2/3) Erveba v porovnaní s placebom obsahujúcim fyziologický roztok. Celkovo 1 197 zdravých osôb vo veku 18 až 65 rokov bolo zaradených v Spojených štátoch amerických, Kanade
a Španielsku.
Testy imunogenity sa vykonali v protokole 009, protokole 011 a protokole 012 a zahŕňajú hodnotenie väzby imunoglobulínu G (IgG) špecifického pre purifikovaný GP ZEBOV Kikwit pomocou
validovaného testu ELISA (enzyme linked immunosorbent assay) (GP-ELISA), ako aj validovanou neutralizáciou vírusu očkovacej látky pomocou neutralizačného testu zníženia plakov (plaque reduction neutralization test, PRNT).
Ako je uvedené v tabuľkách 2 a 3, priemerné geometrické titre (geometric mean titer, GMT) pre GP- ELISA a PRNT sa pri porovnaní pred očkovaním a po očkovaní zvýšili. Viac ako 93,8 % očkovaných osôb splnilo kritériá sérologickej odpovede definované ako ≥ 2-násobné zvýšenie od východiskového stavu a ≥ 200 EU/ml kedykoľvek po očkovaní na základe GP-ELISA a viac ako 80,4 % osôb splnilo kritériá sérologickej odpovede definované ako ≥ 4-násobné zvýšenie od východiskového stavu kedykoľvek po očkovaní na základe PRNT. Viac ako 80,1 % osôb naďalej spĺňalo kritériá sérologickej odpovede pri GP-ELISA a viac ako 63,5 % očkovaných osôb naďalej spĺňalo kritériá sérologickej odpovede pri PRNT v 12. mesiaci. Klinický význam údajov o imunogenite nie je
v súčasnosti známy.
Údaje o účinnosti sa získali v protokole 010 v Guinei a údaje o imunogenite v protokole 009 v Libérii, protokole 011 v Sierra Leone a protokole 012 v Spojených štátoch, Kanade a Európe. Východisková séropozitivita, ožarovanie vzoriek gama žiarením na zníženie rizika infekcie laboratórnych pracovníkov divokým typom vírusu eboly a ďalšie faktory môžu ovplyvniť imunitnú odpoveď na očkovaciu látku, čo má za následok vyššiu imunitnú odpoveď v protokole 012. Aj keď klinický význam GMT pre GP-ELISA, ako aj sérologickej odpovede nie je v súčasnosti známy, na základe dostupných údajov sa očakáva, že účinnosť očkovacej látky by sa uplatnila na osoby z Guiney,
Libérie, Sierra Leone, Spojených štátov, Kanady, Európy alebo iných častí sveta.
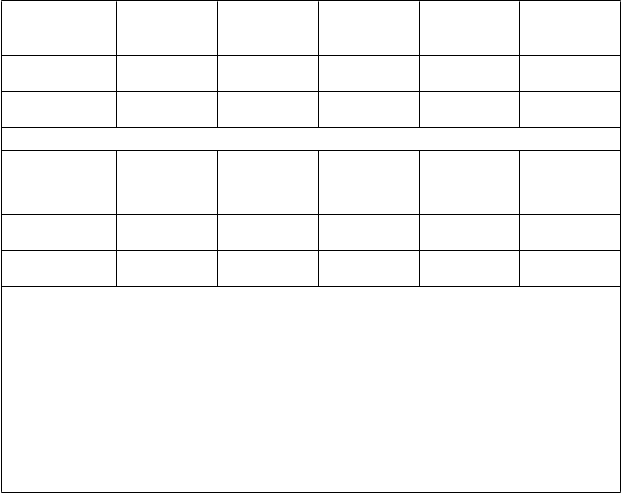
Tabuľka 2: Prehľad priemerných geometrických titrov pre GP-ELISA z protokolov 009, 011 a 012
klinických skúšaní
Skúšanie východiskový
G
M
T (n)
[9
5 % IS]
Protokol 009
§ 117,9 (464)
[107,9; 128,7]
Protokol 011§ 92,7 (503)
[85,3; 100,9]
GMT v 1.
mesiaci (n)
[95 % IS]
994,7 (475)
[915,0; 1 081,3]
964,3 (443)
[878,7; 1 058,3]
GMT v 6.
mesiaci (n)
[95 % IS]
712,2 (477)
[659,4; 769,3]
751,8 (383)
[690,6; 818,4]
GMT v 12.
mesiaci* (n)
[95 % IS]
661,4 (475)
[613,2; 713,4]
760,8 (396)
[697,6; 829,8]
GMT v 24.
mesiaci (n)
[95 % IS]
NA
NA
Protokol 012
Skupina
s kombinovanými konzistentnými šaržami
Skupina s vysokou dávkou
< 36,11 (696)
[<36,11; <36,11]
< 36,11 (219)
[<36,11; <36,11]
1 262,0 (696)
[1 168,9;
1 362,6]
1 291,9 (219)'
[1 126,9;
1 481,2]
1 113,4 (664)
[1 029,5;
1 204,0]
1 189,5 (215)
[1 036,7;
1 364,9]
1 078,4 (327)
[960,6; 1 210,7]
1 135,5 (116)
[934,8; 1 379,3]
920,3 (303)
[820,4; 1 032,3]
1 009,1 (105)
[830,0; 1 226,7]
Placebo skupina < 36,11 (124)
[<36,11; <36,11]
< 36,11 (124)
[<36,11; <36,11]
< 36,11 (123)
[<36,11;
<36;11]
< 36,11 (65)
[<36,11; <36,11]
< 36,11 (65)
[<36,11; <36,11]

Populácia celkovej analyzovanej skupiny bola primárnou populáciou pre analýzy imunogenity v protokoloch 009 a 011 a pozostávala zo všetkých očkovaných osôb so sérologickými údajmi a ktorým bola vzorka séra odobraná v prijateľnom rozsahu dní.
Populácia imunogenity podľa protokolu bola primárnou populáciou pre analýzy imunogenity v protokole 012 a zahŕňa všetky osoby, ktoré boli v súlade s protokolom, očkované, séronegatívne v deň 1 a vzorku séra mali odobranú v jednom alebo viacerých časových bodoch
v prijateľnom rozsahu dní.
n = Počet osôb zahrnutých do analýzy.
IS = interval spoľahlivosti; GP-ELISA = Anti-Glycoprotein Human Enzyme-Linked Immunosorbent Assay (EU/ml); GMT = priemerný geometrický titer (Geometric mean titer)
*Protokol 011 z mesiacov 9-12
§V protokoloch 009 a 011 bolo použité ožarovanie vzoriek gama žiarením na zníženie rizika infekcie laboratórnych pracovníkov divokým typom vírusu eboly
Tabuľka 3: Prehľad priemerných geometrických titrov pre PRNT z protokolov 009, 011 a 012
klinických skúšaní
Skúšanie východiskový
G
M
T (n)
[9
5 % IS]
Protokol 009
§ < 35 (428)
[<35; <35]
Protokol 011§ < 35 (438)
[<35; <35]
GMT v 1.
mesiaci (n)
[95 % IS]
116,8 (477)
[106,0; 128,8]
116,0 (437)
[105,7; 127,4]
GMT v 6.
mesiaci (n)
[95 % IS]
76,8 (477)
[69,9; 84,4]
95,3 (382)
[86,3; 105,3]
GMT v 12.
mesiaci* (n)
[95 % IS]
100,4 (476)
[91,4; 110,3]
119,9 (396)
[107,9; 133,2]
GMT v 24.
mesiaci (n)
[95 % IS]
NA
NA
Protokol 012
Skupina
s kombinovanými konzistentnými
šaržami
Skupina s vysokou dávkou
< 35 (696)
[<35; <35]
< 35 (219)
[<35; <35]
202,1 (696)
[187,9; 217,4]
236,1 (219)
[207,4; 268,8]
266,5 (664)
[247,4; 287,0]
302,1 (215)
[265,2; 344,1]
271,4 (327)
[243,4; 302,7]
323,7 (116)
[269,5; 388,8]
267,6 (302)
[239,4; 299,2]
342,5 (105)
[283,4; 414,0]
Placebo skupina < 35 (124)
[<35; <35]
< 35 (123)
[<35; <35]
< 35 (123)
[<35; <35]
< 35 (65)
[<35; <35]
< 35 (65)
[<35; <35]
Populácia celkovej analyzovanej skupiny bola primárnou populáciou pre analýzy imunogenity v protokoloch 009 a 011 a pozostávala zo všetkých očkovaných osôb so sérologickými údajmi a ktorým bola vzorka séra odobraná v prijateľnom rozsahu dní.
Populácia imunogenity podľa protokolu bola primárnou populáciou pre analýzy imunogenity v protokole 012 a zahŕňa všetky osoby, ktoré boli v súlade s protokolom, očkované, séronegatívne v deň 1 a vzorku séra mali odobranú v jednom alebo viacerých časových bodoch
v prijateľnom rozsahu dní.
n = Počet osôb zahrnutých do analýzy.
IS = interval spoľahlivosti; GMT = priemerný geometrický titer (Geometric mean titer); PRNT = neutralizačný test zníženia plakov (plaque reduction neutralization test)
*Protokol 011 z mesiacov 9-12
§V protokoloch 009 a 011 bolo použité ožarovanie vzoriek gama žiarením na zníženie rizika infekcie laboratórnych pracovníkov divokým typom vírusu eboly
PediatrickápopuláciaÚčinnosť sa u detí nehodnotila. V skúšaní fázy 1 u detí vo veku 6 až 17 rokov (medián veku = 10) boli
výsledky nevalidovaného testu ELISA a testu neutralizácie pseudoviriónu (pseudovirion neutralization assay, PsVNA) v 28. dni a 180. dni po očkovaní podobné ako výsledky pozorované u dospelých
v rovnakej štúdii (pozri aj časti 4.4 a 4.8).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Ervebom v jednej alebo vo viacerých podskupinách pediatrickej populácie v prevencii ochorenia eboly (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku.
Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnostiNeaplikovateľné.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní a reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Pri podávaní Erveba samičkám potkana boli u plodov a mláďat detegované protilátky proti vírusu očkovacej látky, pravdepodobne z dôvodu trans-placentárneho prenosu počas gravidity u plodov
a získaním materských protilátok počas dojčenia u mláďat (pozri časť 4.6).
Ervebo podané samičkám potkana nemalo žiadne účinky na reprodukčný výkon, fertilitu alebo embryonálny/fetálny vývin.
Ervebo podané samičkám potkana nemalo žiadne účinky na vývin alebo správanie mláďat. Hodnotenieenvironmentálnehorizika(environmentalriskassessment,ERA)
Vírus očkovacej látky je geneticky modifikovaný organizmus (genetically modified organism, GMO).
ERA bolo vykonané na stanovenie možného vplyvu tejto očkovacej látky na zdravie ľudí a životné prostredie. Pretože je táto očkovacia látka založená na VSV, čo je známy patogén pre hospodárske
zvieratá (napr. kone, dobytok, ošípané), hodnotenie rizika zahŕňalo druhy, ktoré sú relevantné pre
divoký typ (wild type, wt) VSV, ktorý tvorí základ tejto očkovacej látky.
V štúdii biologickej distribúcie vykonanej na nehumánnych primátoch bola RNA vírusu očkovacej látky zistená v lymfoidných orgánoch až do 112 dní po očkovaní. Infekčný vírus bol však detegovaný na 1. deň a pretrvávajúci infekčný vírus sa v následných sledovaných časových úsekoch (56., 84.
a 112. deň) nezistil.
Na základe obmedzeného vylučovania u dospelých, výsledkov štúdie toxicity u nehumánnych primátov a absencie horizontálneho prenosu u ošípaných, sa celkové riziko Erveba pre zdravie ľudí a životné prostredie považuje za zanedbateľné. Ako preventívne opatrenie sa však očkované osoby majú pokúsiť predísť vystaveniu hospodárskych zvierat krvi a telesným tekutinám minimálne 6 týždňov po očkovaní, aby sa predišlo teoretickému riziku rozšírenia vírusu očkovacej látky. Osoby,
u ktorých sa po podaní očkovacej látky objaví vezikulárna vyrážka, si majú vezikuly prekryť, kým sa nezahoja. Miesto podania očkovacej látky alebo akékoľvek vezikuly prekryte vhodnou náplasťou
(napr. akoukoľvek vankúšikovou náplasťou alebo gázou a leukoplastom), ktorá poskytuje fyzickú
bariéru na ochranu pred priamym kontaktom s tekutinou vo vezikule (pozri časť 4.2). Náplasť sa môže odstrániť, keď nedochádza k viditeľnému úniku tekutiny. Aby sa predišlo neželanej expozícii hospodárskych zvierat, zaistite, aby sa medicínsky odpad a iný čistiaci materiál nedostal do kontaktu
s hospodárskymi zvieratami.
Ďalšie informácie pozri v častiach 4.4 a 6.6.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
rekombinantný ľudský sérový albumín trometamolový tlmivý roztok
voda na injekcie
kyselina chlorovodíková (na úpravu pH)
hydroxid sodný (na úpravu pH)
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte a prepravujte v mraze pri teplote -80 °C až -60 °C.
Po rozmrazení sa má očkovacia látka použiť okamžite; údaje o stabilite počas používania však preukázali, že po rozmrazení sa očkovacia látka môže pred použitím uchovávať až do 14 dní pri teplote 2 °C až 8 °C. Na konci 14. dňa sa má očkovacia látka použiť alebo zlikvidovať. Po vybratí
z mrazničky sa má na očkovacej látke vyznačiť dátum, kedy bola očkovacia látka vybratá z mrazničky a tiež nový dátum likvidácie (namiesto dátumu exspirácie uvedenom na vonkajšom obale). Po
rozmrazení sa očkovacia látka nemôže opätovne zamraziť.
Injekčnú liekovku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Roztok na 1 dávku v injekčnej liekovke (sklo typu I) so zátkou (chlórbutylovou) a vyklápacím plastovým viečkom s hliníkovým tesnením.
Veľkosť balenia: 10 injekčných liekoviek.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
· Očkovacia látka sa uchováva v mraze pri teplote -80 °C až -60 °C a z mrazničky sa má vybrať a rozmrazovať menej ako 4 hodiny, kým nie sú prítomné viditeľné kúsky ľadu. Injekčnú liekovku nerozmrazujte v chladničke, pretože nie je zaručené, že sa liekovka rozmrazí za menej ako 4 hodiny. Rozmrazená injekčná liekovka sa má pred natiahnutím obsahu do injekčnej striekačky niekoľkokrát jemne prevrátiť. Očkovacia látka má byť bezfarebná až slabo hnedo- žltá tekutina bez viditeľných častíc. Ak sú prítomné častice, očkovaciu látku zlikvidujte.
· Natiahnite celý obsah očkovacej látky z injekčnej liekovky pomocou sterilnej ihly a injekčnej striekačky.
Pokiaľ je to možné, tekutina z výplachu očí sa má pred likvidáciou do odtoku zachytiť a dekontaminovať.
Nepoužitú očkovaciu látku alebo odpad vzniknutý z očkovacej látky je potrebné zlikvidovať v súlade s inštitucionálnymi usmerneniami pre geneticky modifikované organizmy alebo biologicky nebezpečný odpad.
Ak dôjde k rozbitiu/rozliatiu, preukázalo sa, že dezinfekčné látky ako napr. aldehydy, alkoholy a detergenty znižujú možnosť vírusovej infekcie po pár minútach.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Merck Sharp & Dohme B.V. Waarderweg 39
2031 BN Haarlem
Holandsko
8. REGISTRAČNÉ ČÍSLO
EU/1/19/1392/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: {DD mesiac RRRR}
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky:
http://www.ema.europa.eu.