
denia mozgu, nedávnej mozgovej príhody (do jedného roka), primárnych mozgových nádorov alebo mozgových metastáz. Ak sa počas liečby Erleadou vyskytne záchvat, liečba sa má natrvalo prerušiť. Riziko záchvatu môže byť zvýšené u pacientov užívajúcich súbežne lieky, ktoré znižujú prahovú hodnotu záchvatov.
Záchvat sa vyskytol u 0,2 % pacientov, ktorí dostávali Erleadu v klinických štúdiách. Z účasti na týchto štúdiách boli vylúčení pacienti s anamnézou záchvatov alebo s predispozičnými faktormi pre záchvaty.
Nie sú žiadne klinické skúsenosti s opätovným podávaním Erleady pacientom, u ktorých sa vyskytol záchvat.
Pádyazlomeniny
U pacientov užívajúcich Erleadu sa vyskytli pády a zlomeniny (pozri časť 4.8). Pacienti majú byť posúdení z hľadiska rizika zlomenín a pádov pred začatím liečby Erleadou a majú byť naďalej
sledovaní, zlomeniny sa majú liečiť podľa stanovených liečebných postupov a má sa zvážiť použitie
liekov cielených na zachovanie kostí.
Súbežnéužívaniesinýmiliekmi
Apalutamid je silný induktor enzýmov a môže viesť k strate účinnosti mnohých bežne používaných liekov (pozri časť 4.5). Pri začatí liečby apalutamidom sa má preto vykonať kontrola súbežne
podávaných liekov. Súbežnému používaniu apalutamidu a liekov, ktoré sú citlivými substrátmi mnohých metabolizujúcich enzýmov alebo transportérov (pozri časť 4.5), sa má vo všeobecnosti vyhnúť, ak má ich terapeutický účinok veľký význam pre pacienta a ak nie je možné ľahko vykonať úpravy dávky na základe monitorovania účinnosti alebo plazmatických koncentrácií.
Je potrebné vyhnúť sa súbežnému podávaniu s warfarínom a antikoagulanciami kumarínového typu. Ak sa Erleada podáva súbežne s antikoagulanciami metabolizovanými CYP2C9 (ako je warfarín alebo acenokumarol), má sa vykonať dodatočné monitorovanie medzinárodného normalizovaného pomeru (INR) (pozri časť 4.5).
Nedávnekardiovaskulárneochorenie
Pacienti s klinicky významným kardiovaskulárnym ochorením za posledných 6 mesiacov vrátane závažnej/nestabilnej angíny pektoris, infarktu myokardu, symptomatického kongestívneho zlyhania
srdca, arteriálnych alebo venóznych tromboembolických príhod (napr. pľúcna embólia, cerebrovaskulárna príhoda vrátane prechodných ischemických záchvatov) alebo klinicky významných
ventrikulárnych arytmií boli vylúčení z klinických štúdií. Z toho dôvodu nebola bezpečnosť apalutamidu u týchto pacientov preukázaná. Ak je predpísaná Erleada, pacienti s klinicky významným kardiovaskulárnym ochorením majú byť sledovaní z hľadiska rizikových faktorov, ako je
hypercholesterolémia, hypertriglyceridémia alebo iné kardiometabolické poruchy (pozri časť 4.8). Pokiaľ je to potrebné, po začatí liečby Erleadou majú byť pacienti liečení podľa stanovených
liečebných postupov.
AndrogénnadeprivačnáterapiamôžepredlžovaťQTinterval
U pacientov s anamnézou alebo s rizikovými faktormi predĺženia QT a u pacientov užívajúcich súbežne lieky, ktoré môžu predĺžiť QT interval (pozri časť 4.5), majú lekári zvážiť pomer prínosu a rizika vrátane potenciálneho rizika Torsade de pointes pred začatím liečby Erleadou.
4.5 Liekové a iné interakcie
Eliminácia apalutamidu a tvorba jeho aktívneho metabolitu, N-demetyl apalutamidu, je sprostredkovaná tak CYP2C8, ako aj CYP3A4 v podobnom rozsahu v rovnovážnom stave. Neočakávajú sa klinicky významné zmeny ich celkovej expozície v dôsledku liekovej interakcie s inhibítormi alebo induktormi CYP2C8 alebo CYP3A4. Apalutamid je induktorom enzýmov a transportérov a môže viesť k zvýšeniu eliminácie mnohých bežne používaných liekov.
Možný účinokinýchliekovnaexpozíciuapalutamidu
Lieky, ktoré inhibujú CYP2C8
CYP2C8 zohráva úlohu pri eliminácii apalutamidu a pri tvorbe jeho aktívneho metabolitu. V štúdii liekových interakcií sa hodnota Cmax apalutamidu znížila o 21 %, zatiaľ čo hodnota AUC sa zvýšila
o 68 % po súbežnom podaní jednorazovej dávky apalutamidu 240 mg a gemfibrozilu (silný inhibítor
CYP2C8). Pre aktívne zložky (súčet apalutamidu a aktívneho metabolitu s upravenou silou) hodnota Cmax klesla o 21 %, zatiaľ čo AUC sa zvýšila o 45 %. Pri súbežnom podávaní Erleady so silným inhibítorom CYP2C8 (napr. gemfibrozil, klopidogrel) nie je potrebná úprava úvodnej dávky, má sa však zvážiť zníženie dávky Erleady na základe znášanlivosti (pozri časť 4.2). Neočakáva sa, že mierne alebo stredne silné inhibítory CYP2C8 ovplyvnia expozíciu apalutamidu.
Lieky, ktoré inhibujú CYP3A4
CYP3A4 zohráva úlohu pri eliminácii apalutamidu a pri tvorbe jeho aktívneho metabolitu. V štúdii liekových interakcií sa hodnota Cmax apalutamidu znížila o 22 %, zatiaľ čo hodnota AUC bola podobná po súbežnom podaní jednorazovej dávky Erleady 240 mg a itrakonazolu (silný inhibítor CYP3A4).
Pre aktívne zložky (súčet apalutamidu a aktívneho metabolitu s upravenou silou) hodnota Cmax klesla o 22 %, zatiaľ čo AUC bola opäť podobná. Pri súbežnom podávaní Erleady so silným inhibítorom CYP3A4 (napr. ketokonazol, ritonavir, klaritromycín) nie je potrebná úprava úvodnej dávky, má sa však zvážiť zníženie dávky Erleady na základe znášanlivosti (pozri časť 4.2). Neočakáva sa, že mierne alebo stredne silné inhibítory CYP3A4 ovplyvnia expozíciu apalutamidu.
Lieky, ktoré indukujú CYP3A4 alebo CYP2C8
Účinky induktorov CYP3A4 alebo CYP2C8 na farmakokinetiku apalutamidu neboli hodnotené in vivo. Na základe výsledkov štúdie liekových interakcií so silným inhibítorom CYP3A4 alebo silným inhibítorom CYP2C8 sa nepredpokladá, že induktory CYP3A4 alebo CYP2C8 majú klinicky významné vplyvy na farmakokinetiku apalutamidu a aktívnych zložiek, preto nie je potrebná úprava dávky, keď sa Erleada podáva súbežne s induktormi CYP3A4 alebo CYP2C8.
Možný účinok apalutamidunaexpozíciuinýchliekov
Apalutamid je silný induktor enzýmov a zvyšuje syntézu mnohých enzýmov a transportérov; preto sa
očakáva interakcia s mnohými bežnými liekmi, ktoré sú substrátmi enzýmov alebo transportérov. Zníženie plazmatických koncentrácií môže byť podstatné a môže viesť k strate alebo zníženiu klinického účinku. Existuje aj riziko zvýšenej tvorby aktívnych metabolitov.
Enzýmy metabolizujúce lieky
Štúdie in vitro ukázali, že apalutamid a N-demetyl apalutamid sú stredne silnými až silnými induktormi CYP3A4 a CYP2B6, miernymi inhibítormi CYP2B6 a CYP2C8 a slabými inhibítormi CYP2C9, CYP2C19 a CYP3A4. Apalutamid a N-demetyl apalutamid neovplyvňujú CYP1A2 a CYP2D6 v terapeuticky relevantných koncentráciách. Účinok apalutamidu na substráty CYP2B6 nebol hodnotený in vivo a čistý účinok nie je v súčasnosti známy. Keď sa s Erleadou podávajú substráty CYP2B6 (napr. efavirenz), má sa sledovať nežiaduca reakcia a vyhodnocovať strata účinnosti substrátu, a môže byť potrebná úprava dávky substrátu na udržanie optimálnych plazmatických koncentrácií.
U ľudí je Erleada silným induktorom CYP3A4 a CYP2C19 a slabým induktorom CYP2C9. V štúdii liekových interakcií s použitím koktailového prístupu súbežné podávanie Erleady s jednorazovými perorálnymi dávkami citlivých substrátov CYP viedlo k 92 % zníženiu hodnoty AUC midazolamu (substrát CYP3A4), 85 % zníženiu hodnoty AUC omeprazolu (substrát CYP2C19), a 46 % zníženiu hodnoty AUC S-warfarínu (substrát CYP2C9). Erleada nespôsobila klinicky významné zmeny
v expozícii substrátu CYP2C8. Súbežné užívanie Erleady s liekmi, ktoré sú primárne metabolizované
CYP3A4 (napr. darunavir, felodipín, midazolam, simvastatín), CYP2C19 (napr. diazepam, omeprazol) alebo CYP2C9 (napr. warfarín, fenytoín), môže viesť k nižšej expozícii týmto liekom. Ak je to možné, odporúča sa nahradiť tieto lieky alebo sa má vyhodnotiť strata účinnosti, ak sa v podávaní lieku pokračuje. Ak sa podáva s warfarínom, počas liečby Erleadou sa má monitorovať INR.
Indukcia CYP3A4 apalutamidom naznačuje, že UDP-glukuronozyltransferáza (UGT) môže byť tiež indukovaná aktiváciou nukleárneho pregnanového X receptora (PXR). Súbežné podávanie Erleady
s liekmi, ktoré sú substrátmi UGT (napr. levotyroxín, kyselina valproová), môže viesť k nižšej expozícii týmto liekom. Keď sa s Erleadou podávajú substráty UGT, má sa vyhodnotiť strata účinnosti substrátu a môže byť nutná úprava dávky substrátu na udržanie optimálnych plazmatických
koncentrácií.
Transportéry liekov
Klinicky sa zistilo, že apalutamid je slabým induktorom P-glykoproteínu (P-gp), proteínu rezistencie rakoviny prsníka (BCRP) a polypeptidu transportujúceho organické anióny 1B1 (OATP1B1). V štúdii
liekových interakcií s použitím koktailového prístupu sa ukázalo, že súbežné podávanie Erleady s jednorazovými perorálnymi dávkami citlivých substrátov transportérov viedlo k 30 % zníženiu
hodnoty AUC fexofenadínu (substrát P-gp) a 41 % zníženiu hodnoty AUC rosuvastatínu (substrát BCRP/OATP1B1), ale nemalo žiadny vplyv na hodnotu Cmax. Súbežné užívanie Erleady s liekmi, ktoré sú substrátmi P-gp (napr. kolchicín, dabigatran etexilát, digoxín), BCRP alebo OATP1B1 (napr. lapatinib, metotrexát, rosuvastatín, repaglinid), môže viesť k nižšej expozícii týchto liekov. Keď sa
s Erleadou podávajú substráty Pgp, BCRP alebo OATP1B1, má sa vyhodnotiť strata účinnosti substrátu a môže byť nutná úprava dávky substrátu na udržanie optimálnych plazmatických koncentrácií.
Na základe údajov in vitro sa nedá vylúčiť inhibícia transportéra organických katiónov 2 (OCT2), transportéra organických katiónov 3 (OAT3) a proteínov extrudujúcich viaceré lieky a toxíny (MATE)
apalutamidom a jeho N-demetylovým metabolitom. In vitro neboli pozorované žiadne inhibície transportéra organických aniónov 1 (OAT1).
Lieky,ktorépredlžujúQT interval
Vzhľadom na to, že androgénna deprivačná terapia môže predlžovať QT interval, súbežné podávanie
Erleady s liekmi, o ktorých je známe, že predlžujú QT interval, alebo s liekmi schopnými vyvolať Torsade de pointes, ako sú antiarytmiká triedy IA (napríklad chinidín, disopyramid) alebo triedy III (napríklad amiodarón, sotalol, dofetilid, ibutilid), metadón, moxifloxacín, antipsychotiká (napr. haloperidol) atď., má byť starostlivo zvážené (pozri časť 4.4).
Pediatrickápopulácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Antikoncepciaumužovažien
Nie je známe, či apalutamid alebo jeho metabolity sú prítomné v ejakuláte. Erleada môže byť škodlivá pre vyvíjajúci sa plod. Pacienti, ktorí sexuálne žijú so ženami vo fertilnom veku, majú používať
kondóm spolu s ďalšou vysoko účinnou antikoncepčnou metódou počas liečby a 3 mesiace po poslednej dávke Erleady.
Gravidita
Erleada je kontraindikovaná u žien, ktoré sú tehotné alebo môžu otehotnieť (pozri časť 4.3). Na základe svojho mechanizmu účinku môže Erleada spôsobiť poškodenie plodu, keď sa podáva počas
tehotenstva. Nie sú k dispozícii žiadne údaje o používaní Erleady u tehotných žien. S Erleadou neboli
vykonané reprodukčné štúdie na zvieratách.
Dojčenie
Nie je známe, či sa apalutamid alebo metabolity vylučujú do ľudského mlieka. Riziko pre dojčené dieťa nemožno vylúčiť. Erleada sa nemá užívať počas dojčenia.
Fertilita
Na základe štúdií na zvieratách môže Erleada znížiť plodnosť mužov s reprodukčným potenciálom
(pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Erleada nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U pacientov užívajúcich Erleadu boli však zaznamenané záchvaty. Pacienti majú byť informovaní
o tomto riziku v súvislosti s vedením vozidiel alebo s obsluhou strojov.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšie nežiaduce reakcie sú únava (30 %), kožná vyrážka (24 % akýkoľvek stupeň a 5 % stupeň
3 alebo 4), úbytok hmotnosti (16 %), artralgia (16 %) a pády (16 %). Ďalšie dôležité nežiaduce reakcie zahŕňajú zlomeniny (12 %) a hypotyreózu (8 %).
Tabuľkovýzoznamnežiaducichreakcií
Nežiaduce reakcie pozorované počas klinických štúdií sú uvedené nižšie podľa kategórie frekvencie. Kategórie frekvencie sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej
časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (<1/10 000) a
neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie zistené v klinických štúdiách
Tried
a orgánových systémov Nežiaduca reakcia a frekvencia výskytu
Poruchy endokrinného systému časté: hypotyreóza*
Poruchy metabolizmu a výživy časté: hypercholesterolémia časté: hypertriglyceridémia
Poruchy nervového systému menej časté: záchvat (pozri časť 4.4)
Poruchy srdca a srdcovej činnosti neznáme: predĺženie QT intervalu (pozri časti 4.4 a 4.5)
Poruchy kože a podkožného tkaniva veľmi časté: kožná vyrážka**

časté: svrbenie
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
veľmi časté: zlomenina+
veľmi časté: artralgia
Celkové poruchy a reakcie v mieste podania veľmi časté: únava
Laboratórne a funkčné vyšetrenia veľmi časté: úbytok hmotnosti
Úrazy, otravy a komplikácie liečebného postupu
veľmi časté: pád
* Zahŕňa hypotyreózu, zvýšenie hladiny hormónu stimulujúceho štítnu žľazu v krvi, zníženie hladiny tyroxínu, autoimunitnú tyroiditídu, zníženie hladiny voľného tyroxínu, zníženie trijódtyronínu
** Pozri časť „Kožná vyrážka“ v časti „Opis vybraných nežiaducich reakcií“
+ Zahŕňa zlomeninu rebier, zlomeninu bedrových stavcov, kompresnú zlomeninu chrbtice, zlomeninu chrbtice, zlomeninu chodidla, zlomeninu bedrového kĺbu, zlomeninu humeru, zlomeninu hrudníkových stavcov, zlomeninu
hornej končatiny, zlomeninu krížovej kosti, zlomeninu ruky, zlomeninu lonovej kosti, zlomeninu acetabula,
zlomeninu členku, kompresnú zlomeninu, zlomeninu rebrových chrupaviek, zlomeninu tvárových kostí, zlomeninu dolných končatín, osteoporotickú zlomeninu, zlomeninu zápästia, zlomeninu avulzie, zlomeninu fibuly, zlomeninu
kostrče, zlomeninu panvy, zlomeninu rádia, zlomeninu sterna, únavovú zlomeninu, traumatickú zlomeninu, zlomeninu krčných stavcov, zlomeninu krčka stehnovej kosti, zlomeninu tíbie. Pozri nižšie.
Opisvybranýchnežiaducichreakcií
Kožná vyrážka
Kožná vyrážka spojená s Erleadou bola najčastejšie opísaná ako makulárna alebo makulopapulárna. Kožná vyrážka zahŕňala vyrážku, makulopapulárnu vyrážku, generalizovanú vyrážku, žihľavku, svrbiacu vyrážku, makulárnu vyrážku, konjunktivitídu, multiformný erytém, papulárnu vyrážku, exfoliáciu kože, vyrážku na genitáliách, erytematóznu vyrážku, stomatitídu, liekovú erupciu, ulceráciu v ústach, pustulárnu vyrážku, pľuzgier, papulu, pemfigoid, kožnú eróziu a pľuzgierovitú vyrážku. Nežiaduce reakcie v podobe kožnej vyrážky boli hlásené u 24 % pacientov liečených Erleadou. Pri liečbe Erleadou boli kožné vyrážky 3. stupňa (definované ako pokrytie > 30 % plochy povrchu tela
[BSA]) hlásené u 5,2 % pacientov.
Medián počtu dní do nástupu kožnej vyrážky bol 82 dní v rozmedzí od 1 do 994 dní. U 81 percent pacientov bol problém s vyrážkou vyriešený s mediánom 60 dní do vyriešenia. Používané lieky zahŕňali lokálne kortikosteroidy, systémové kortikosteroidy a perorálne antihistaminiká. U pacientov s kožnou vyrážkou došlo k prerušeniu podávania u 28 % pacientov a k zníženiu dávky u 12 % pacientov (pozri časť 4.2). Kožná vyrážka sa opakovane vyskytla u približne polovice pacientov,
ktorým bol liek opätovne podaný. Kožná vyrážka viedla k ukončeniu liečby Erleadou u 9 % pacientov, u ktorých sa vyskytla kožná vyrážka.
Pády a zlomeniny
V štúdii ARN-509-003 bola zlomenina hlásená u 11,7 % pacientov liečených Erleadou a u 6,5 %
pacientov liečených placebom. Polovica pacientov zaznamenala pád v priebehu 7 dní pred výskytom zlomeniny v obidvoch liečebných skupinách. Pády boli hlásené u 15,6 % pacientov liečených
Erleadou oproti 9,0 % pacientov liečených placebom (pozri časť 4.4).
Hypotyreóza
Hypotyreóza bola hlásená u 8,1 % pacientov liečených Erleadou a u 2,0 % pacientov liečených placebom na základe hodnotenia hormónu stimulujúceho štítnu žľazu (TSH) každé 4 mesiace.
Nevyskytli sa žiadne nežiaduce udalosti 3. alebo 4. stupňa. Hypotyreóza sa vyskytla u 28 % pacientov,
ktorí už dostávali substitučnú terapiu štítnej žľazy v skupine s Erleadou a u 5,9 % pacientov v skupine s placebom. U pacientov, ktorí neužívali substitučnú terapiu štítnej žľazy, sa hypotyreóza vyskytla
u 5,7 % pacientov liečených Erleadou a u 0,8 % pacientov liečených placebom. Keď je substitučná
terapia štítnej žľazy klinicky indikovaná, má sa s ňou začať alebo má byť upravená jej dávka (pozri časť 4.5).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNie je známe žiadne špecifické antidotum na predávkovanie apalutamidom. V prípade predávkovania sa má ukončiť podávanie Erleady a vykonať všeobecné podporné opatrenia, až kým klinická toxicita nebude znížená alebo nepominie. Nežiaduce reakcie v prípade predávkovania neboli doteraz pozorované, očakáva sa však, že takéto reakcie sa budú podobať nežiaducim reakciám uvedeným
v časti 4.8.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Endokrinná liečba, antiandrogény, ATC kód: L02BB05
MechanizmusúčinkuApalutamid je perorálne podávaný selektívny inhibítor androgénneho receptora (AR), ktorý sa viaže priamo na doménu AR viažucu ligand. Apalutamid bráni nukleárnej translokácii AR, inhibuje väzbu
DNA, bráni transkripcii sprostredkovanej AR a chýba mu aktivita agonistu androgénneho receptora.
Liečba apalutamidom znižuje proliferáciu nádorových buniek a zvyšuje apoptózu, čo vedie k silnej protinádorovej aktivite. Jeden z hlavných metabolitov, N-demetyl apalutamid, vykazoval jednu tretinu aktivity apalutamidu
in vitro.
KardiálnaelektrofyziológiaÚčinok apalutamidu 240 mg jedenkrát denne na QTc interval bol hodnotený v otvorenej, nekontrolovanej, multicentrickej, jednoramennej klinickej štúdii zameranej na QT u 45 pacientov
s CRPC. V rovnovážnom stave bola maximálna priemerná zmena QTcF oproti východiskovej hodnote
12,4 ms (2-stranná 90 % horná IS 16,0 ms). Analýza vplyvu expozície na QT naznačila nárast QTcF
v závislosti od koncentrácie apalutamidu a jeho aktívneho metabolitu.
KlinickáúčinnosťabezpečnosťCelkovo bolo 1207 subjektov s nmCRPC randomizovaných v pomere 2:1 a následne dostávali buď apalutamid perorálne v dávke 240 mg jedenkrát denne v kombinácii s androgénnou deprivačnou
liečbou (ADT) (medikamentóznou kastráciou alebo predchádzajúcou chirurgickou kastráciou) alebo
placebom v kombinácii s ADT v multicentrickej, dvojito zaslepenej klinickej štúdii (štúdia
ARN-509-003). Pacienti zaradení do štúdie mali čas do zdvojnásobenia prostatického špecifického antigénu (PSADT) ≤ 10 mesiacov, a preto sa považujú za osoby s vysokým rizikom bezprostredného
metastatického ochorenia a úmrtia na rakovinu prostaty. Všetci pacienti, ktorí nepodstúpili chirurgickú kastráciu, dostávali ADT nepretržite počas celej štúdie. Výsledky PSA boli zaslepené a neboli použité
na ukončenie liečby. Pacienti randomizovaní do oboch liečebných skupín mali pokračovať v liečbe až do progresie ochorenia definovanej zaslepeným centrálnym zobrazovacím hodnotením (BICR), do iniciácie novej liečby, neprijateľnej toxicity alebo odstúpenia zo štúdie.
Pacienti boli rozdelení medzi liečebné skupiny rovnomerne podľa nasledujúcich demografických charakteristík a východiskových charakteristík ochorenia. Medián veku bol 74 rokov (rozsah 48 až 97) a 26 % pacientov bolo vo veku 80 rokov alebo viac. Rozdelenie podľa rasy bolo 66 % belosi, 5,6 % černosi, 12 % ázijská rasa a 0,2 % iná. Sedemdesiatsedem percent (77 %) pacientov v oboch liečebných skupinách malo predtým operáciu alebo rádioterapiu prostaty. Väčšina pacientov mala Gleasonovo skóre 7 alebo viac (81 %). Pätnásť percent (15 %) pacientov malo na začiatku štúdie
< 2 cm panvové lymfatické uzliny. Sedemdesiattri percent (73 %) pacientov dostalo predchádzajúcu liečbu antiandrogénom prvej generácie; 69 % pacientov dostalo bikalutamid a 10 % pacientov dostávalo flutamid. Všetci zaradení pacienti boli potvrdení ako nemetastatickí zaslepeným centrálnym zobrazovacím hodnotením a pri zaradení do štúdie mali výkonnostný stav podľa Eastern Cooperative Oncology Group Performance Status (ECOG PS) 0 alebo 1.
Prežitie bez metastáz (MFS) bolo primárnym koncovým ukazovateľom definovaným ako čas od randomizácie až po prvý dôkaz vzdialených metastáz v kostiach alebo mäkkých tkanivách potvrdených BICR alebo úmrtia z akejkoľvek príčiny, podľa toho, čo nastane skôr. Liečba Erleadou výrazne zlepšila MFS. Erleada znížila relatívne riziko vzdialených metastáz alebo úmrtia o 70 %
v porovnaní s placebom (HR = 0,30; 95 % IS: 0,24; 0,36; p < 0,0001). Stredná hodnota MFS pre Erleadu bola 41 mesiacov a pre placebo bola 16 mesiacov (pozri obrázok 1). Konzistentné zlepšenie MFS bolo s Erleadou pozorované vo všetkých vopred definovaných podskupinách, vrátane veku, rasy, pôvodu, nodálneho stavu, predchádzajúceho počtu hormonálnych liečob, východiskovej hodnoty PSA, času zdvojnásobenia PSA, východiskovej hodnoty ECOG a použitia liekov na zachovanie kostí.
Obrázok 1: Kaplanova-Meierova krivka prežitia bez metastáz (MFS) v štúdii ARN-509-003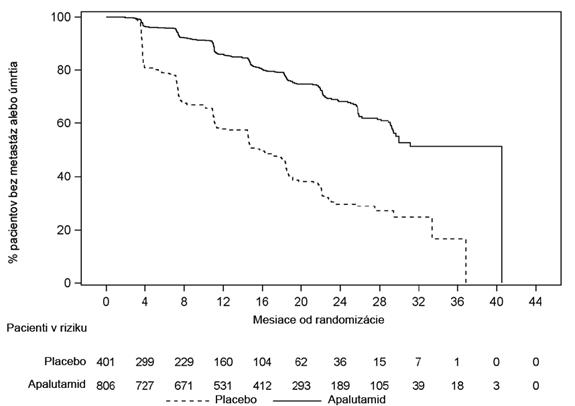
Pacienti liečení Erleadou a ADT vykazovali významné zlepšenie oproti pacientom liečeným samotným ADT pre nasledujúce sekundárne koncové ukazovatele: čas do metastáz (HR = 0,28, 95 % IS: 0,23; 0,34, p < 0,0001), prežitie bez progresie (PFS) (HR = 0,30, 95 % IS: 0,25, 0,36; p < 0,0001); čas do symptomatickej progresie (HR = 0,45, 95 % IS: 0,32, 0,63; p <0,0001) a trend celkového prežitia (OS) (HR = 0,70, 95 % IS: 0,47, 1,04; p = 0,0742).
Čas do symptomatickej progresie bol definovaný ako čas od randomizácie po vznik udalosti súvisiacej so skeletom, bolesť/príznaky vyžadujúce iniciáciu novej systémovej protinádorovej liečby alebo lokálnu/regionálnu nádorovú progresiu vyžadujúcu rádioterapiu/chirurgický zákrok. Napriek tomu, že celkový počet prípadov bol malý, rozdiel medzi dvoma skupinami bol dostatočne veľký na to, aby dosiahol štatistický význam. V skupine s apalutamidom došlo u 64 (7,9 %) pacientov
k symptomatickej progresii v porovnaní so 63 (16 %) v skupine s placebom, s pomerom rizika 0,447 (95 % IS: 0,315; 0,634), čím bola splnená predpísaná hranica účinnosti podľa O’Brien Fleminga pre'
významnosť, p < 0,00008. Medián času do symptomatickej progresie nebol dosiahnutý ani v jednej
z liečebných skupín.
V skupine s apalutamidom zomrelo 62 (7.7 %) pacientov v porovnaní so 42 (10,5 %) pacientmi
v skupine s placebom. Medián prežívania v skupine s apalutamidom nebol dosiahnutý v porovnaní s 39,03 mesiacmi s 95 % IS (39,03; NE) v skupine s placebom. Vo vopred definovanej predbežnej
analýze nebol dosiahnutý štatistický význam v celkovom prežívaní.
Prežitie po progresii (PFS-2, definované ako čas do progresie ochorenia po prvej následnej liečbe alebo do úmrtia) bolo dlhšie u pacientov liečených Erleadou v porovnaní s pacientmi dostávajúcimi placebo (HR = 0,489; 95 % IS: 0,361, 0,662; p < 0,0001).
Neboli pozorované žiadne štatisticky významné rozdiely v zmene oproti východiskovej analýze
Funkčného hodnotenia liečby karcinómu – prostata (FACT-P) pre celkové skóre alebo niektorú
z podskupín medzi pacientmi liečenými Erleadou pridanou k ADT v porovnaní s placebom s ADT.
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Erleadou vo všetkých podskupinách pediatrickej populácie pre pokročilé štádium karcinómu prostaty. Informácie o pediatrickom použití, pozri časť 4.2.
5.2 Farmakokinetické vlastnosti
Po opakovanom dávkovaní jedenkrát denne sa expozícia apalutamidu (Cmax a plocha pod krivkou koncentrácie [AUC]) zvýšila proporcionálne k dávke v rozmedzí dávok od 30 do 480 mg. Po podaní
240 mg jedenkrát denne bol ustálený stav apalutamidu dosiahnutý po 4 týždňoch a priemerný akumulačný pomer bol približne 5-násobný v porovnaní s jednorazovou dávkou. Pri rovnovážnom
stave boli priemerné hodnoty (CV %) Cmax a AUC pre apalutamid 6 μg/ml (28 %) a 100 μg.h/ml
(32 %) v uvedenom poradí. Denné výkyvy v plazmatických koncentráciách apalutamidu boli nízke,
priemerný pomer maximálnej a minimálnej koncentrácie bol 1,63. Zvýšenie zdanlivého klírensu (CL/F) sa pozorovalo pri opakovanom podávaní, pravdepodobne v dôsledku indukcie vlastného metabolizmu apalutamidu.
Pri rovnovážnom stave boli priemerné hodnoty (CV %) Cmax a AUC pre hlavný aktívny metabolit, N-demetyl apalutamid, 5,9 μg/ml (18 %) a 124 μg.h/ml (19 %) v uvedenom poradí. N-demetyl- apalutamid je charakterizovaný rovným profilom priebehu koncentrácie v čase v rovnovážnom stave
s priemerným pomerom maximálnej a minimálnej koncentrácie 1,27. Priemerný pomer (CV %) AUC
metabolitu k pôvodnému lieku pre N-demetyl apalutamid po podaní opakovanej dávky bol asi 1,3
(21 %). Na základe systémovej expozície, relatívnej sily a farmakokinetických vlastností N-demetyl- apalutamid pravdepodobne prispieva ku klinickej aktivite apalutamidu.
Absorpcia
Po perorálnom podaní bol medián času do dosiahnutia maximálnej plazmatickej koncentrácie (tmax)
2 hodiny (rozsah: 1 až 5 hodín). Priemerná absolútna perorálna biologická dostupnosť je približne
100 %, čo znamená, že apalutamid sa po perorálnom podaní úplne absorbuje.
Podávanie apalutamidu zdravým jedincom nalačno a s jedlom s vysokým obsahom tuku neviedlo
k žiadnym klinicky významným zmenám Cmax a AUC. Medián času do dosiahnutia tmax bol s jedlom oneskorený približne o 2 hodiny (pozri časť 4.2).
Apalutamid nie je ionizovateľný za príslušných fyziologických podmienok pH, preto sa neočakáva, že látky znižujúce kyselinu (napr. inhibítor protónovej pumpy, antagonista H2 receptorov, antacidum) budú ovplyvňovať rozpustnosť a biologickú dostupnosť apalutamidu.
In vitro apalutamid a jeho metabolit N-demetyl sú substrátmi pre P-gp. Keďže apalutamid sa po perorálnom podaní úplne absorbuje, P-gp neobmedzuje absorpciu apalutamidu, a preto sa neočakáva, že inhibícia alebo indukcia P-gp bude ovplyvňovať biologickú dostupnosť apalutamidu.
Distribúcia
Priemerný zdanlivý distribučný objem pri rovnovážnom stave apalutamidu je asi 276 litrov. Distribučný objem apalutamidu je väčší ako objem celkovej vody v tele, čo poukazuje na rozsiahlu extravaskulárnu distribúciu.
Apalutamid a N-demetyl apalutamid sú na 96 % resp. 95 %, v uvedenom poradí viazané na plazmatické bielkoviny a prevažne sa viažu na sérový albumín bez závislosti od koncentrácie.
Biotransformácia
Po jednorazovom perorálnom podaní apalutamidu 240 mg označeného rádioaktívnym uhlíkom 14C, apalutamid, aktívny metabolit N-demetyl apalutamid a inaktívny metabolit v podobe karboxylovej
kyseliny predstavovali väčšinu rádioaktivity 14C v plazme a predstavovali 45 %, 44 % a 3 % celkovej
hodnoty AUC rádioaktívneho uhlíka 14C v uvedenom poradí.
Metabolizmus je hlavnou cestou eliminácie apalutamidu. Primárne sa metabolizuje prostredníctvom CYP2C8 a CYP3A4 za vzniku N-demetyl apalutamidu. Apalutamid a N-demetyl apalutamid sa ďalej metabolizujú karboxylesterázou za vzniku neaktívneho metabolitu karboxylovej kyseliny. Prispievanie CYP2C8 a CYP3A4 k metabolizmu apalutamidu sa odhaduje na 58 % a 13 % po jednorazovej dávke, ale očakáva sa, že úroveň prispievania sa bude meniť v rovnovážnom stave v dôsledku indukcie CYP3A4 apalutamidom po opakovanej dávke.
Eliminácia
Apalutamid, hlavne vo forme metabolitov, sa eliminuje primárne močom. Po jednorazovom perorálnom podaní rádioaktívne označeného apalutamidu sa 89 % rádioaktivity zistilo do 70 dní po
podaní dávky: 65 % sa zistilo v moči (1,2 % dávky ako nezmenený apalutamid a 2,7 % ako
N-demetyl apalutamid) a 24 % sa zistilo v stolici (1,5 % dávky ako nezmenený apalutamid a 2 % ako
N-demetyl apalutamid).
Hodnota CL/F apalutamidu je po jednorazovom podaní 1,3 l/h a v rovnovážnom stave po dávkovaní jedenkrát denne sa zvyšuje na 2,0 l/h. Priemerný účinný polčas pre apalutamid u pacientov je približne
3 dni pri rovnovážnom stave.
Údaje in vitro naznačujú, že apalutamid a jeho metabolit N-demetyl nie sú substrátmi pre BCRP, OATP1B1 alebo OATP1B3.
Osobitnépopulácie
Účinky poruchy funkcie obličiek, poruchy funkcie pečene, veku, rasy a iných vonkajších faktorov na farmakokinetické vlastnosti apalutamidu sú zhrnuté nižšie.
Poruchafunkcieobličiek
Štúdia zameraná na poruchu funkcie obličiek pri liečbe apalutamidom sa neuskutočnila. Na základe populačnej farmakokinetickej analýzy s použitím údajov z klinických štúdií u pacientov s karcinómom prostaty rezistentným na kastráciu (CRPC) a zdravých jedincov nebol pozorovaný žiadny významný rozdiel v systémovej expozícii u pacientov s existujúcou miernou až stredne ťažkou poruchou funkcie obličiek (odhadovaná rýchlosť glomerulárnej filtrácie [eGFR] medzi 30 až 89 ml/min/1,73 m2,
N = 585) v porovnaní s jedincami s východiskovou normálnou funkciou obličiek
(eGFR ≥ 90 ml/min/1,73 m2, N = 372). Potenciálny účinok ťažkej poruchy funkcie obličiek alebo ochorenia obličiek v terminálnom štádiu (eGFR ≤ 29 ml/min/1,73 m2) nebol stanovený z dôvodu
nedostatočných údajov.
Poruchafunkciepečene
Štúdia zameraná na poruchu funkcie pečene porovnávala systémovú expozíciu apalutamidu a N- demetyl apalutamidu u pacientov s východiskovou miernou poruchou funkcie pečene (N = 8, trieda A
podľa Childa-Pugha, priemerné skóre = 5,3) alebo stredne ťažkou poruchou funkcie pečene (N = 8,
trieda B podľa Childa-Pugha, priemerné skóre = 7,6) v porovnaní s kontrolnými zdravými jedincami s normálnou funkciou pečene (N = 8). Po jednorazovej perorálnej dávke 240 mg apalutamidu bol priemerný geometrický pomer (GMR) pre AUC a Cmax pre apalutamid u pacientov s miernou
poruchou funkcie pečene 95 % resp. 102 % a GMR pre AUC a Cmax apalutamidu u pacientov so stredne ťažkou poruchou funkcie pečene bol 113 % resp. 104 % v porovnaní so zdravými kontrolnými jedincami. Klinické a farmakokinetické údaje nie sú k dispozícii pre pacientov s ťažkou poruchou funkcie pečene (trieda C podľa Childa-Pugha).
Etnickýpôvodarasa
Na základe populačnej farmakokinetickej analýzy neboli žiadne klinicky významné rozdiely vo farmakokinetických vlastnostiach apalutamidu medzi belochmi (belosi alebo hispánci alebo
latinoameričania, N = 761), černochmi (s africkým pôvodom alebo afro-americkým, N = 71), aziatmi
(okrem Japonska, N = 58 ) a Japoncami (N = 58).
Vek
Populačná farmakokinetická analýza ukázala, že vek (rozsah: 18 až 94 rokov) nemá klinicky významný vplyv na farmakokinetické vlastnosti apalutamidu.
5.3 Predklinické údaje o bezpečnosti
Apalutamid bol negatívny z hľadiska genotoxicity v štandardnej sérii testov in vitro a in vivo. Neuskutočnili sa dlhodobé štúdie na zvieratách na hodnotenie karcinogénneho potenciálu apalutamidu.
Mužská fertilita je pravdepodobne zhoršená liečbou apalutamidom na základe zistení v štúdiách toxicity po opakovanom podávaní, ktoré sú v súlade s farmakologickou aktivitou apalutamidu. V štúdiách toxicity sa po opakovanom podávaní u samcov potkanov a psov pozorovali atrofia,
aspermia/hypospermia, degenerácia a/alebo hyperplázia alebo hypertrofia v reprodukčnom systéme v dávkach zodpovedajúcich expozíciám približne rovnakým ako expozícia u ľudí na základe AUC.
V štúdii fertility u samcov potkanov sa po 4 týždňoch dávkovania v dávkach zodpovedajúcich expozíciám približne rovnakým ako expozícia u ľudí na základe AUC pozorovalo zníženie koncentrácie a pohyblivosti spermií, miera kopulácie a fertility (po párení s neliečenými samicami) spolu so zníženou hmotnosťou sekundárnych pohlavných žliaz a nadsemenníkov. Účinky na samcov potkanov boli reverzibilné po 8 týždňoch od posledného podania apalutamidu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety
Koloidný bezvodý oxid kremičitý
Sodná soľ kroskarmelózy Acetátosukcinát hypromelózy Stearan horečnatý Mikrokryštalická celulóza
Mikrokryštalická celulóza (silicifikovaná)
Filmovýobal
Čierny oxid železitý (E172) Žltý oxid železitý (E172) Makrogol
Polyvinylalkohol (čiastočne hydrolyzovaný) Mastenec
Oxid titaničitý (E171)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanieUchovávajte v pôvodnom obale na ochranu pred vlhkosťou. Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaBiela nepriehľadná fľaša z polyetylénu s vysokou hustotou (HDPE) s polypropylénovým (PP) detským bezpečnostným uzáverom. Jedna fľaša obsahuje 120 filmom obalených tabliet a celkovo 6 g silikagélového vysúšadla.
Fóliový blister PVC-PCTFE prekrytý hliníkovou pretláčacou fóliou uzavretý vo vnútri puzdra.
· Jedna škatuľa na 28 dní obsahuje 112 filmom obalených tabliet v 4 kartónových puzdrách po 28
filmom obalených tabliet.
· Jedna škatuľa na 30 dní obsahuje 120 filmom obalených tabliet v 5 kartónových puzdrách po 24
filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIJanssen-Cilag International NV Turnhoutseweg 30
B-2340 Beerse
Belgicko
8. REGISTRAČNÉ ČÍSLOEU/1/18/1342/001
EU/1/18/1342/002
EU/1/18/1342/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.