
e potrebná žiadna úprava dávky vzhľadom na vek. Dávka sa má upraviť podľa funkcie obličiek pacienta (pozri odporúčané dávkovanie pri poruche funkcie obličiek a časť 5.2).
Pohlavie a rasa: nie je potrebná žiadna úprava dávky vzhľadom na pohlavie alebo rasu.
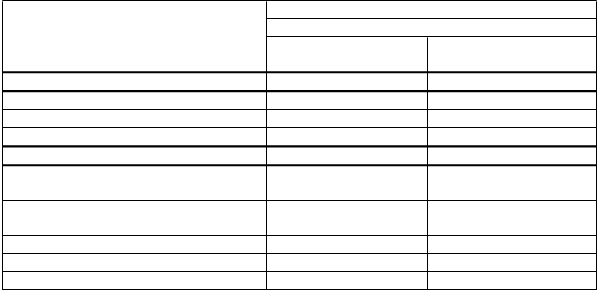
Porucha funkcie obličiek: klírens entekaviru sa znižuje so znižujúcim sa klírensom kreatinínu (pozri časť 5.2). U pacientov s klírensom kreatinínu < 50 ml/min, vrátane pacientov na hemodialýze alebo kontinuálnej ambulantnej peritoneálnej dialýze (CAPD), sa odporúča úprava dávky. Odporúča sa redukcia dennej dávky perorálneho roztoku entekaviru, ako je uvedené v tabuľke. Alternatívne, ak nie je perorálny roztok dostupný, môže sa dávka prispôsobiť zvýšením dávkovacieho intervalu tak, ako je uvedené v tabuľke. Navrhovaná úprava dávky je založená na extrapolácii limitovaných údajov a ich bezpečnosť a účinnosť nebola klinicky hodnotená. Preto sa má virologická odpoveď starostlivo monitorovať.
Dávkovanie Entecavir Accord*
K
l
í
rens kreatinínu
(
m
l
/
mi
n)
Pacienti bez predchádzajúcej
liečby nukleozidmi
Pacienti refraktérni na lamivudín alebo
s dekompenzovaným ochorením pečene
≥ 50 0,5 mg jedenkrát denne 1 mg jedenkrát denne
30 – 49 0,25 mg jedenkrát denne*

alebo
0,5 mg každých 48 hodín
10 – 29 0,15 mg jedenkrát denne*
alebo
0,5 mg každých 72 hodín
0,5 mg jedenkrát denne
0,3 mg jedenkrát denne*
alebo
0,5 mg každých 48 hodín
< 10
Hemodialýza alebo
CAPD**
0,05 mg jedenkrát denne*
alebo
0,5 mg každých 5-7 dní
0,1 mg jedenkrát denne*
alebo
0,5 mg každých 72 hodín
* pre dávky < 0,5 mg sa odporúča perorálny roztok entekaviru.
** v dňoch hemodialýzy podajte entekavir po hemodialýze.
Porucha funkcie pečene: u pacientov s poruchou funkcie pečene nie je potrebná žiadna úprava dávky.
Spôsob podávania
Entecavir Accord sa má užívať perorálne.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Porucha funkcie obličiek: pre pacientov s poruchou funkcie obličiek sa odporúča úprava dávkovania (pozri časť 4.2). Navrhovaná úprava dávky je založená na extrapolácii limitovaných údajov a ich bezpečnosť a účinnosť nebola klinicky hodnotená. Preto sa má virologická odpoveď starostlivo monitorovať.
Exacerbácie hepatitídy: spontánne exacerbácie chronickej hepatitídy B sú relatívne časté a sú charakterizované prechodnými zvýšeniami sérovej ALT. Po začatí antivírusovej liečby sa môže u niektorých pacientov sérová ALT zvýšiť, zatiaľ čo sérové hladiny HBV DNA klesajú (pozri
časť 4.8). U pacientov liečených entekavirom bol stredný čas do nástupu exacerbácií počas liečby
4 – 5 týždňov. U pacientov s kompenzovaným ochorením pečene tieto zvýšenia sérovej ALT zvyčajne nie sú sprevádzané zvýšením koncentrácií bilirubínu v sére alebo dekompenzáciou pečene. Pacienti
s pokročilým ochorením pečene alebo s cirhózou môžu byť vystavení vyššiemu riziku dekompenzácie
pečene po exacerbácii hepatitídy a preto musia byť počas liečby pozorne sledovaní.
Akútna exacerbácia hepatitídy bola hlásená aj u pacientov, ktorí prerušili liečbu hepatitídy B (pozri časť 4.2). Exacerbácie po liečbe sú zvyčajne spojené s vzostupom HBV DNA a zdá sa, že väčšina z nich spontánne vymizne. Boli však hlásené ťažké exacerbácie, zahŕňajúce fatálne prípady.
U pacientov bez predošlej liečby nukleozidmi, ktorí boli liečení entekavirom, bol stredný čas do nástupu exacerbácií po liečbe 23 - 24 týždňov a väčšinou boli hlásené u HBeAg negatívnych pacientov (pozri časť 4.8). Minimálne po dobu 6 mesiacov po prerušení liečby hepatitídy B má byť funkcia pečene sledovaná v pravidelných intervaloch pomocou klinických a laboratórnych vyšetrení.
V prípade potreby sa môže zvážiť opätovné začatie liečby hepatitídy B.
Pacienti s dekompenzovaným ochorením pečene: u pacientov s dekompenzovaným ochorením pečene, najmä u tých s ochorením podľa Child-Pugh-Turcotte (CPT) stupňa C, bol v porovnaní s pacientmi s kompenzovanou funkciou pečene pozorovaný vyšší výskyt závažných hepatálnych nežiaducich účinkov (bez ohľadu na kauzalitu). Taktiež pacienti s dekompenzovaným ochorením pečene môžu byť vo vyššom riziku pre laktátovú acidózu a pre špecifické renálne nežiaduce účinky ako je
hepatorenálny syndróm. V tejto populácii pacientov sa preto majú starostlivo sledovať klinické
a laboratórne parametre (pozri tiež časť 4.8 a 5.1).
Laktátová acidóza a ťažká hepatomegália so steatózou: pri použití nukleozidových analógov boli
hlásené prípady laktátovej acidózy (pri absencii hypoxémie), niekedy fatálnej, zvyčajne súvisiacej
s ťažkou hepatomegáliou a steatózou pečene. Keďže entekavir je nukleozidový analóg, toto riziko nie je možné vylúčiť. Liečba nukleozidovými analógmi musí byť prerušená, keď sa objavia rýchlo sa
zvyšujúce hladiny aminotransferáz, progresívna hepatomegália alebo metabolická/laktátová acidóza
neznámej etiológie. Benígne digestívne symptómy, ako sú nauzea, vracanie a bolesť brucha, môžu naznačovať rozvoj laktátovej acidózy. Ťažké prípady, niekedy s fatálnym koncom, boli spojené
s pankreatitídou, zlyhaním pečene/steatózou pečene, zlyhaním obličiek a vyššími hladinami sérového
laktátu. Opatrnosť je potrebná pri predpisovaní nukleozidových analógov akémukoľvek pacientovi (obzvlášť obéznym ženám) s hepatomegáliou, hepatitídou alebo inými známymi rizikovými faktormi pre ochorenie pečene. Títo pacienti musia byť starostlivo sledovaní.
Lekári sa musia ubezpečiť, že zmeny ALT sú spojené so zlepšením ostatných laboratórnych ukazovateľov chronickej hepatitídy B, aby rozlíšili zvýšenie aminotransferáz v dôsledku odpovede na liečbu od zvýšenia potenciálne súvisiaceho s laktátovou acidózou.
R
ezistencia a špeciálne opatrenia pre pacientov refraktérnych na lamivudín: mutácie HBV polymerázy, ktorá kóduje lamivudín rezistentné substitúcie, môžu vyústiť do následného objavenia sa sekundárnych substitúcií, vrátane tých, ktoré sú spojené s rezistenciou na entekavir (ETVr). U malého percenta pacientov refraktérnych na lamivudín boli ETVr substitúcie na rezíduách rtT184, rtS202 alebo rtM250. Pacienti s HBV rezistentnou na lamivudín majú vyššie riziko rozvoja následnej rezistencie na entekavir ako pacienti bez rezistencie na lamivudín. Kumulatívna pravdepodobnosť rozvinutej genotypovej rezistencie po 1. 2. 3. 4. a 5. roku liečby v štúdiách refraktérnych na lamivudín bola 6 %, 15 %, 36 %, 47 % a 51 %. V populácii refraktérnej na lamivudín sa virologická odpoveď musí často sledovať a majú sa vykonávať odpovedajúce testy na rezistenciu. U pacientov so suboptimálnou virologickou odpoveďou po 24 týždňoch liečby entekavirom sa má zvážiť modifikovaná liečba (pozri časti 4.5 a 5.1). Pri začatí liečby pacientov s potvrdenou HBV rezistentnou na lamivudín v anamnéze sa má prednostne zvážiť použitie kombinácie entekaviru s druhou antivírusovou látkou (ktorá sa nepodieľa na skríženej rezistencii s lamivudínom ani s entekavirom) pred monoterapiou entekavirom.
Existujúca HBV rezistentná na lamivudín sa spája so zvyšujúcim rizikom následnej rezistencie na entekavir bez ohľadu na stupeň ochorenia pečene; u pacientov s dekompenzovaným ochorením pečene môže byť virologický prienik spojený so závažnými klinickými komplikáciami základného ochorenia pečene. Preto, u pacientov s oboma ochoreniami, dekompenzovaným ochorením pečene aj s HBV rezistentným na lamivudín, sa má prednostne zvážiť kombinácia entekaviru s inou antivírusovou
látkou (bez skríženej rezistencie buď na lamivudín alebo na entekavir) pred monoterapiou
entekavirom.
Pediatrická populácia: U pediatrických pacientov s východiskovými hodnotami HBV
DNA ≥ 8,0 log10 IU/ml sa pozorovala nižšia miera virologickej odpovede (HBV DNA < 50 IU/ml) (pozri časť 5.1). Entekavir sa má u týchto pacientov používať iba, ak potenciálny prínos zdôvodní možné riziko u dieťaťa (napr. rezistencie). Keďže u niektorých pediatrických pacientov môže byť potrebná dlhodobá či dokonca celoživotná liečba chronickej aktívnej hepatitídy B, má sa zohľadniť vplyv entekaviru na budúce možnosti liečby.
Príjemcovia transplantátu pečene: u príjemcov transplantátu pečene užívajúcich cyklosporín alebo takrolimus má byť pred a počas liečby entekavirom pozorne zhodnotená funkcia obličiek (pozri časť 5.2).
Súbežná infekcia vírusom hepatitídy C alebo D: neexistujú údaje o účinnosti entekaviru u pacientov súbežne infikovaných vírusom hepatitídy C alebo D.
Pacienti súbežne infikovaní vírusom humánnej imunodeficiencie (HIV)/HBV neužívajúci súbežnú antiretrovírusovú terapiu: entekavir nebol hodnotený u pacientov súbežne infikovaných HIV/HBV a nesúbežne užívajúcich účinnú HIV liečbu. Výskyt HIV rezistencie sa pozoroval, keď sa entekavir používal na liečbu chronickej hepatitídy B u pacientov s HIV infekciou neužívajúcich vysoko aktívnu antiretrovírusovú terapiu (HAART) (pozri časť 5.1). Preto sa liečba entekavirom nemá používať u pacientov súbežne infikovaných HIV/HBV, ktorí neužívajú HAART. Entekavir nebol skúmaný ako terapia pre HIV infekciu a preto sa neodporúča pre toto použitie.
Pacienti súbežne infikovaní HIV/HBV užívajúci súbežnú antiretrovírusovú terapiu: entekavir sa skúmal u 68 dospelých súbežne infikovaných HIV/HBV užívajúcich lamivudín zahrnutý do HAART režimu (pozri časť 5.1). Nie sú dostupné údaje o účinnosti entekaviru u HBeAg negatívnych pacientov súbežne infikovaných vírusom HIV. K dispozícii sú len obmedzené údaje o pacientoch súbežne infikovaných vírusom HIV, ktorí majú nízky počet CD4 buniek (< 200 buniek/mm3).
Všeobecne: pacienti majú byť oboznámení s tým, že nebolo dokázané, že liečba entekavirom znižuje
riziko prenosu HBV, a preto je potrebné naďalej dodržiavať príslušné preventívne opatrenia.
Sójové polysacharidy: Tento liek obsahuje sójové polysacharidy. Ak ste alergický na sóju, nepoužívajte tento liek.
4.5 Liekové a iné interakcie
Vzhľadom na to, že entekavir sa vylučuje prevažne obličkami (pozri časť 5.2), súbežné podávanie s liekmi, ktoré znižujú funkciu obličiek alebo súperia o aktívnu tubulárnu sekréciu, môže zvýšiť sérové koncentrácie niektorého lieku. S výnimkou lamivudínu, adefovir dipivoxilu a tenofovir- dizoproxilfumarátu nebol hodnotený vplyv súbežného podávania entekaviru s liekmi, ktoré sú vylučované obličkami, alebo ktoré ovplyvňujú funkciu obličiek. Pri súbežnom podávaní entekaviru s takýmito liekmi majú byť pacienti pozorne sledovaní kvôli nežiaducim reakciám.
Medzi entekavirom a lamivudínom, adefovirom alebo tenofovirom neboli pozorované žiadne farmakokinetické interakcie.
Entekavir nie je substrátom, induktorom ani inhibítorom enzýmov cytochrómu P450 (CYP450) (pozri časť 5.2). Preto, výskyt liekových interakcií entekaviru sprostredkovaný CYP450 nie je pravdepodobný.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku: vzhľadom na to, že nie sú známe potenciálne riziká pre vyvíjajúci sa plod,
ženy vo fertilnom veku majú používať účinnú antikoncepciu.
Gravidita: nie sú k dispozícii dostatočné údaje o použití entekaviru u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu pri vysokých dávkach (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí. Entecavir Accord má byť užívaný počas gravidity iba v nevyhnutných prípadoch. Neexistujú údaje o vplyve entekaviru na prenos HBV z matky na novorodenca. Z tohto dôvodu sa majú použiť príslušné intervencie na prevenciu neonatálnej infekcie HBV.
Dojčenie: nie je známe, či sa entekavir vylučuje do ľudského mlieka. Dostupné toxikologické údaje u zvierat preukázali vylučovanie entekaviru do mlieka (pre podrobné informácie pozri časť 5.3). Riziko pre dojčatá nie je možné vylúčiť. Dojčenie sa má počas liečby s Entecavir Accord ukončiť.
Fertilita: toxikologické štúdie na zvieratách, ktorým bol podávaný entekavir, nepreukázali žiadny
dôkaz poruchy fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Neuskutočnili sa žiadne štúdie o účinkoch na schopnosť viesť vozidlá a obsluhovať stroje. Závrat,
únava a somnolencia sú časté vedľajšie účinky, ktoré môžu zhoršiť schopnosť viesť vozidlá
a obsluhovať stroje.
4.8 Nežiaduce účinky
a. Prehľad bezpečnostného profilu
V klinických štúdiách u pacientov s kompenzovaným ochorením pečene, najčastejšími nežiaducimi reakciami akejkoľvek závažnosti s prinajmenšom možným vzťahom k entekaviru boli bolesť hlavy
(9 %), únava (6 %), závrat (4 %) a nauzea (3 %). Tiež bola hlásená exacerbácia hepatitídy počas a po
ukončení liečby entekavirom (pozri časť 4.4 a c. Opis vybraných nežiaducich reakcií).
b. Tabuľkový zoznam nežiaducich reakcií
Hodnotenie nežiaducich reakcií sa zakladá na skúsenostiach z postmarketingového sledovania a štyroch klinických štúdiách, v ktorých 1720 pacientov s chronickou infekciou hepatitídy B
a kompenzovaným ochorením pečene dostávalo dvojito zaslepenú liečbu entekavirom (n = 862) alebo
lamivudínom (n = 858) až po dobu 107 týždňov (pozri časť 5.1). V týchto štúdiách boli profily
bezpečnosti, vrátane laboratórnych abnormalít, porovnateľné pre entekavir 0,5 mg denne (679 HBeAg
pozitívnych alebo negatívnych pacientov bez predošlej liečby nukleozidmi liečených v mediáne
53 týždňov), entekavir 1 mg denne (183 pacientov refraktérnych na lamivudín liečených v mediáne
69 týždňov) a lamivudín.
Nežiaduce reakcie pokladané za prinajmenšom možno súvisiace s liečbou entekavirom sú uvedené podľa tried orgánových systémov. Frekvencia je definovaná ako veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až 1/100); zriedkavé (≥ 1/10 000 až 1/1 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Poruchy imunitného systému
zriedkavé : anafylaktická reakcia
Psychické poruchy
časté : nespavosť
Poruchy nervového systému:
časté : bolesť hlavy, závrat, somnolencia
Poruchy gastrointestinálneho traktu
časté : vracanie, hnačka, nauzea, dyspepsia
Poruchy pečene a žlčových ciest
časté : zvýšenie hladín transamináz
Poruchy kože a podkožného tkaniva
menej časté : vyrážka, alopécia
Celkové poruchy a reakcie v mieste podania
časté : únava
Boli hlásené prípady laktátovej acidózy, často spojené s dekompenzáciou pečene, inými vážnymi
medicínskymi okolnosťami alebo liekovými expozíciami (pozri časť 4.4).
Liečba po 48 týždňoch: pokračujúca liečba entekavirom po strednú dĺžku trvania 96 týždňov neodhalila žiadne nové bezpečnostné signály.
c. Opis vybraných nežiaducich reakcií
Abnormality laboratórnych testov: V klinických štúdiách u pacientov bez predošlej liečby nukleozidmi, 5 % mali zvýšenia ALT > 3-násobok bazálnych hodnôt a < 1 % malo zvýšenia ALT > 2-násobok bazálnych hodnôt spolu s celkovým bilirubínom > 2-násobok hornej hranice normálu (Upper Limit of Normal, ULN) a > 2-násobok bazálnych hodnôt. Hladiny albumínu
< 2,5 g/dl sa vyskytli u < 1 % pacientov, hladiny amylázy > 3-násobok bazálnych hodnôt u 2 %, hladiny lipázy > 3-násobok bazálnych hodnôt u 11 % a krvné doštičky < 50 000/mm3 u < 1 %.
V klinických štúdiách u pacientov refraktérnych na lamivudín, 4 % mali zvýšenia ALT > 3-násobok bazálnych hodnôt a < 1 % malo zvýšenia ALT > 2-násobok bazálnych hodnôt spolu s celkovým bilirubínom > 2-násobok ULN a > 2-násobok bazálnych hodnôt. Hladiny amylázy > 3-násobok bazálnych hodnôt sa vyskytli u 2 % pacientov, hladiny lipázy > 3-násobok bazálnych hodnôt u 18 % a krvné doštičky < 50 000/mm3 u < 1 %.
Exacerbáciepočasliečby: v štúdiách u pacientov bez predošlej liečby nukleozidmi sa zvýšenia ALT
> 10-násobok ULN a > 2-násobok bazálnych hodnôt vyskytli počas liečby u 2 % pacientov liečených entekavirom oproti 4 % pacientov liečených lamivudínom. V štúdiách u pacientov refraktérnych na lamivudín sa zvýšenia ALT > 10-násobok ULN a > 2-násobok bazálnych hodnôt vyskytli počas liečby u 2 % pacientov liečených entekavirom oproti 11 % pacientov liečených lamivudínom. U pacientov liečených entekavirom bol stredný čas do nástupu zvýšení ALT počas liečby 4 – 5 týždňov, zvyčajne ustúpili s pokračujúcou liečbou a vo väčšine prípadov boli spojené s ≥ 2 log10/ml znížením vo
vírusovej záťaži, ktoré predchádzalo alebo sa vyskytovalo súbežne so zvýšením ALT. Počas liečby sa
odporúča pravidelné sledovanie funkcie pečene.
E
xacerbácie
p
o
prerušení
liečby: u pacientov, ktorí prerušili liečbu hepatitídy B, vrátane liečby entekavirom, boli hlásené akútne exacerbácie hepatitídy (pozri časť 4.4). V štúdiách u pacientov bez predošlej liečby nukleozidmi sa počas sledovania po liečbe vyskytli zvýšenia ALT (> 10-násobok ULN a > 2-násobok referenčnej hodnoty [minimálna bazálna hodnota alebo posledné meranie na konci dávkovacieho intervalu]) u 6 % pacientov liečených entekavirom a u 10 % pacientov liečených
lamivudínom. U pacientov liečených entekavirom bez predošlej liečby nukleozidmi bol stredný čas do nástupu zvýšení ALT 23 - 24 týždňov a 86 % (24/28) zvýšení ALT sa vyskytlo u HBeAg negatívnych pacientov. V štúdiách u pacientov refraktérnych na lamivudín, v ktorých boli sledované len
obmedzené počty pacientov, sa počas sledovania po liečbe vyskytli zvýšenia ALT u 11 % pacientov
liečených entekavirom a nevyskytli sa u pacientov liečených lamivudínom.
V klinických štúdiách bola liečba entekavirom prerušená, keď pacienti dosiahli vopred špecifikovanú odpoveď. Ak sa liečba preruší bez ohľadu na odpoveď na liečbu, výskyt prudkých zvýšení ALT po liečbe môže byť vyšší.
d. Pediatrická populácia
Bezpečnosť entekaviru u pediatrických pacientov vo veku od 2 do < 18 rokov sa zakladá na dvoch prebiehajúcich klinických skúšaniach s jedincami s infekciou chronickej HBV; jedno farmakokinetické skúšanie vo fáze 2 (štúdia 028) a jedno skúšanie vo fáze 3 (štúdia 189). Tieto klinické skúšania poskytujú skúsenosti so 195 HBeAg pozitívnymi jedincami bez predchádzajúcej liečby nukleozidmi liečenými entekavirom s mediánom dĺžky liečby 99 týždňov. Nežiaduce reakcie pozorované u pediatrických jedincov, ktorí dostávali liečbu entekavirom, boli zhodné s tými, ktoré sa pozorovali v klinických skúšaniach s entekavirom s dospelými (pozri a. Prehľad bezpečnostného profilu a časť 5.1).
e. Ostatné osobitné skupiny pacientov
Skúsenosti u pacientov s dekompenzovanýmochorenímpečene: profil bezpečnosti entekaviru
u pacientov s dekompenzovaným ochorením pečene bol hodnotený v randomizovanej otvorenej porovnávacej štúdii v ktorej boli pacienti liečený entekavirom 1 mg/deň (n = 102) alebo adefovir
dipivoxilom 10 mg/deň (n = 89) (štúdia 048). V porovnaní s nežiaducimi reakciami uvedenými v časti
b. Zoznam nežiaducich reakcií zostavených do tabuľky, bola v 48. týždni u pacientov liečených entekavirom hlásená ďalšia nežiaduca reakcia [pokles hydrogenuhličitanu v krvi (2 %)]. Kumulatívna miera úmrtí v štúdií bola 23 % (23/102) a príčiny úmrtí vo všeobecnosti súviseli s pečeňou tak, ako sa u tejto populácie očakávalo. Kumulatívna miera hepatocelulárneho karcinómu v štúdii (hepatocellular carcinoma, HCC) bola 12 % (12/102). Závažné nežiaduce účinky v štúdii, s kumulatívnou frekvenciou
69 %, vo všeobecnosti súviseli s pečeňou. U pacientov s vysokou bazálnou hodnotou CPT skóre bolo vyššie riziko rozvoja závažných nežiaducich účinkov (pozri časť 4.4).
Abnormality laboratórnych testov: V 48. týždni nemal žiaden z pacientov s dekompenzovaným ochorením pečene, liečený entekavirom zvýšenia ALT > 10-násobok ULN a zároveň > 2-násobok bazálnych hodnôt, a 1 % pacientov malo zvýšenia ALT > 2-násobok bazálnych hodnôt spolu
s celkovým bilirubínom > 2-násobok ULN a > 2-násobok bazálnych hodnôt. Hladiny albumínu
< 2,5 g/dl sa vyskytli u < 30 % pacientov, hladiny lipázy > 3-násobok bazálnych hodnôt u 10 %
a krvné doštičky < 50 000/mm3 u < 20 %.
Skúsenosti u pacientov súbežne infikovaných HIV: profil bezpečnosti entekaviru u obmedzeného počtu pacientov súbežne infikovaných HIV/HBV, ktorí dostávali režimy HAART (vysoko účinnej antiretrovírusovej liečby) obsahujúce lamivudín, bol podobný profilu bezpečnosti u HBV monoinfikovaných pacientov (pozri časť 4.4).
Pohlavie/vek: nebol žiadny zjavný rozdiel v profile bezpečnosti entekaviru s ohľadom na pohlavie
(≈ 25 % žien v klinických štúdiách) alebo vek (≈ 5 % pacientov > 65 rokov). Hlásenie podozrení na nežiaduce reakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieSkúsenosti s predávkovaním entekavirom hláseným od pacientov sú limitované. Zdraví jedinci, ktorí užívali až 20 mg/deň po dobu 14 dní a jednorazové dávky až 40 mg, nemali žiadne neočakávané nežiaduce reakcie. Ak dôjde k predávkovaniu, pacient musí byť sledovaný kvôli príznakom toxicity
a v prípade potreby dostať štandardnú podpornú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antivirotiká na systémové použitie, nukleozidové a nukleotidové inhibítory reverznej transkriptázy
ATC kód: J05AF10
Mechanizmus účinku: entekavir, nukleozidový analóg guanozínu s aktivitou proti HBV polymeráze, je účinne fosforylovaný na aktívnu trifosfátovú (TP) formu, ktorá má intracelulárny polčas 15 hodín. Prostredníctvom kompetície s prirodzeným substrátom deoxyguanozínom TP, entekavir-TP funkčne inhibuje 3 aktivity vírusovej polymerázy: (1) pripojenie (“priming”) HBV polymerázy, (2) reverznú transkripciu negatívneho reťazca DNA z pregenómovej mediátorovej RNA a (3) syntézu pozitívneho reťazca HBV DNA. Hodnota Ki (inhibičnej konštanty) entekaviru-TP pre HBV DNA polymerázu je
0,0012 μM. Entekavir-TP je slabým inhibítorom celulárnych DNA polymeráz α, β a δ s hodnotami Ki
18 až 40 μM. Vysoké expozície entekaviru okrem toho nemali žiadne významné nežiaduce účinky na
polymerázu γ alebo syntézu mitochondriálnej DNA v HepG2 bunkách (Ki > 160 μM).
Antivírusová aktivita: entekavir inhiboval syntézu HBV DNA (50 % zníženie, EC50) pri koncentrácii
0,004 μmol/l v ľudských HepG2 bunkách transfekovaných divokým typom HBV. Stredná hodnota
EC50 entekaviru proti HBV rezistentnému na LVD (rtL180M a rtM204V) bola 0,026 μmol/l (rozsah
0,010 – 0,059 μmol/l). Rekombinantné vírusy kódujúce substitúcie spôsobujúce rezistenciu na
adefovir buď na rtN236T, alebo rtA181V, zostali úplne citlivé na entekavir.
Analýza inhibičnej aktivity entekaviru voči skupine laboratórnych a klinických HIV-1 izolátov, využijúc typy buniek a podmienky testu, poskytla hodnoty EC50 v rozmedzí od 0,026 do > 10 µmol/l; nižšia hodnota EC50 sa zaznamenala, keď sa v teste použili znížené hladiny vírusu. V bunkovej kultúre entekavir selektovaný pre M184I substitúciu v mikromolárnych koncentráciách potvrdil inhibičný tlak vo vysokých koncentráciách entekaviru. HIV varianty obsahujúce M184V substitúciu ukázali stratu citlivosti na entekavir (pozri časť 4.4).
V kombinovaných testoch s HBV v bunkovej kultúre, abakavir, didanozín, lamivudín, stavudín, tenofovir alebo zidovudín nepôsobili antagonisticky voči anti-HBV aktivite entekaviru v širokom rozsahu koncentrácií. V antivírusových testoch HIV entekavir v mikromolárnych koncentráciách nepôsobil antagonisticky voči anti HIV aktivite v bunkovej kultúre týchto šiestich NRTIs alebo emtricitabínu.
Rezistencia v bunkovej kultúre: v porovnaní s divokým typom HBV, vírusy rezistentné na LVD obsahujúce substitúcie rtM204V a rtL180M v rámci reverznej transkriptázy vykazujú 8-násobne zníženú citlivosť na entekavir. Včlenenie ďalších ETVr aminokyselín spôsobujúcich zmeny rtT184, rtS202 alebo rtM250 znižuje citlivosť na entekavir v bunkovej kultúre. Substitúcie zistené na klinických izolátoch (rtT184A, C, F, G, I, L, M alebo S; rtS202 C, G alebo I a/alebo rtM250I, L alebo V) viedli k 16- až 741-násobnému zníženiu citlivosti na entekavir v porovnaní s divokým typom vírusu. ETVr substitúcie na rezíduách rtT184, rtS202 a rtM250 samotné majú iba mierny účinok na citlivosť na entekavir a nepozorovali sa za neprítomnosti LVDr substitúcií u viac než
1000 pacientskych sekvenčných vzoriek. Rezistencia je sprostredkovaná redukovanou väzbou inhibítora na zmenenú HBV reverznú transkriptázu a rezistentná HBV sa prejavuje redukovanou replikačnou kapacitou v bunkovej kultúre.
Klinické skúsenosti: preukázanie prínosu sa zakladá na histologických, virologických, biochemických a serologických odpovediach po 48 týždňoch liečby v klinických štúdiách kontrolovaných aktívnou látkou u 1 633 dospelých s chronickou infekciou vírusom hepatitídy B, s dôkazom o vírusovej replikácii a kompenzovaným ochorením pečene. Bezpečnosť a účinnosť entekaviru boli tiež hodnotené v klinickej štúdii kontrolovanej aktívnou látkou u 191 pacientov infikovaných HBV, s dekompenzovaným ochorením pečene a v klinickej štúdii u 68 pacientov súčasne infikovaných HBV aj HIV.
V štúdiách u pacientov s kompenzovaných ochorením pečene, bolo histologické zlepšenie definované ako ≥ 2-bodové zníženie od bazálnej hodnoty v Knodellovom skóre nekrotického zápalu bez zhoršenia Knodellovho skóre fibrózy. Odpovede u pacientov, ktorí mali v Knodellovom skóre fibrózy bazálnu hodnotu 4 (cirhóza), boli porovnateľné s celkovými odpoveďami vo všetkých meraniach výsledku účinnosti (všetci pacienti mali kompenzované ochorenie pečene). Vysoké skóre Knodellovho indexu nekrotického zápalu v bazálnej hodnote (> 10) bolo spojené s väčším histologickým zlepšením
u pacientov bez predošlej liečby nukleozidmi. Bazálne hladiny ALT ≥ 2-násobok ULN aj bazálne hodnoty HBV DNA ≤ 9,0 log10 kópií/ml boli spájané s vyššou virologickou odpoveďou (48. týždeň, HBV DNA < 400 kópií/ml) u HBeAg pozitívnych pacientov bez predošlej liečby nukleozidmi. Bez ohľadu na východiskové charakteristiky väčšina pacientov preukázala histologickú a virologickú odpoveď na liečbu.
Skúsenostiupacientovskompenzovanýmochorenímpečenebezpredošlejliečbynukleozidmi:Výsledky v 48. týždni randomizovaných, dvojito zaslepených štúdií porovnávajúcich entekavir (ETV) s lamivudínom (LVD) u HBeAg pozitívnych (022) a HBeAg negatívnych (027) pacientov sú uvedené v tabuľke.
Pacienti bez predošlej liečby nukleozidmi
HBeAg pozitívni
(štúdia 022)
HBeAg negatívni
(štúdia 027)

ETV 0,5 mg jedenkrát denne
LVD
100 mg jedenkrát denne
ETV 0,5 mg jedenkrát denne
LVD
100 mg jedenkrát denne
N 314a 314a 296a 287a Histologické zlepšenieb 72 %* 62 % 70 %* 61 % Zlepšenie v Ishakovom skóre fibrózy 39 % 35 % 36 % 38 % Zhoršenie v Ishakovom skóre fibrózy 8 % 10 % 12 % 15 % N 354 355 325 313
Zníženie vírusovej záťaže (log10 kópií/ml)c Nedetegovateľná HBV DNA
(< 300 kópií/ml metódou PCR)c
-6,86* -5,39 -5,04* -4,53
67 %* 36 % 90 %* 72 %
Normalizácia ALT (≤ 1-násobok ULN) 68 %* 60 % 78 %* 71 %
HBeAg Sérokonverzia 21 % 18 %
*p hodnota oproti lamivudínu < 0,05
a pacienti s hodnotiteľnou bazálnou histológiou (bazálne Knodellovo skóre nekrotického zápalu ≥ 2)
b primárny koncový ukazovateľ
c Roche Cobas Amplicor PCR assay (LLOQ = 300 kópií/ml)
Skúsenosti u pacientov refraktérnych na lamivudín s kompenzovanýchochorenímpečene:
V randomizovanej, dvojito zaslepenej štúdii u HBeAg pozitívnych pacientov refraktérnych na lamivudín (026), s 85 % pacientov, u ktorých boli na začiatku liečby prítomné LVDr mutácie, pacienti užívajúci lamivudín po zaradení do štúdie prešli buď na entekavir 1 mg jedenkrát denne, bez obdobia
bez liečby (“washout”) aj bez obdobia súbežnej liečby (“overlap”) (n = 141), alebo pokračovali na
lamivudíne 100 mg jedenkrát denne (n = 145). Výsledky v 48. týždni sú uvedené v tabuľke.
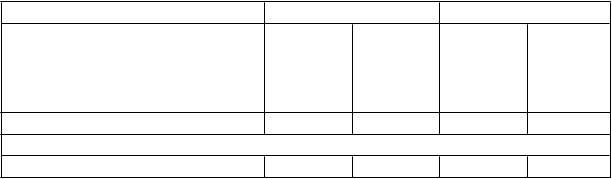
Pacienti refraktérni na lamivudín
HBeAg pozitívni (štúdia 026)
ETV 1,0 mg jedenkrát denne
LVD 100 mg jedenkrát denne
N 124a 116a Histologické zlepšenieb 55 %* 28 % Zlepšenie v Ishakovom skóre fibrózy 34 %* 16 % Zhoršenie v Ishakovom skóre fibrózy 11 % 26 % N 141 145
Zníženie vírusovej záťaže (log10 kópií/ml)c Nedetegovateľná HBV DNA
(< 300 kópií/ml metódou PCR)c
-5,11* -0,48
19 %* 1 %
Normalizácia ALT(≤ 1-násobok ULN) 61 %* 15 %
HBeAg Sérokonverzia 8 % 3 %
*p hodnota oproti lamivudínu < 0,05
a pacienti s hodnotiteľnou bazálnou histológiou (bazálne Knodellovo skóre nekrotického zápalu ≥ 2)
b primárny koncový ukazovateľ.
c Roche Cobas Amplicor PCR assay (LLOQ = 300 kópií/ml)
Výsledky po 48 týždňochliečby:
Liečba bola prerušená vtedy, keď boli splnené vopred špecifikované kritériá pre odpoveď buď
v 48. týždni, alebo počas druhého roku liečby. Kritériá pre odpoveď boli virologická supresia HBV
(HBV DNA < 0,7 MEq/ml analýzou bDNA) a strata HBeAg (u HBeAg pozitívnych pacientov) alebo ALT < 1,25-násobok ULN (u HBeAg negatívnych pacientov). Pacienti s odpoveďou boli sledovaní ďalších 24 týždňov po ukončení liečby. Pacienti, ktorí splnili kritériá pre virologickú ale nie pre serologickú alebo biochemickú odpoveď, pokračovali zaslepenou liečbou. Pacientom, ktorí nemali virologickú odpoveď, bola ponúknutá alternatívna liečba.
Pacienti bez predošlej liečby nukleozidmi:
HBeAg pozitívni pacienti (štúdia 022): liečba entekavirom po dobu až 96 týždňov (n = 354) viedla ku kumulatívnej miere odpovedí v hodnote 80 % pre HBV DNA < 300 kópií/ml metódou PCR, 87 % pre normalizáciu ALT, 31 % pre HBeAg sérokonverziu a 2 % pre HBsAg sérokonverziu (5 % pre stratu HBsAg). Kumulatívna miera odpovedí pre lamivudín (n = 355) bola v hodnote 39 % pre HBV DNA
< 300 kópií/ml metódou PCR, 79 % pre normalizáciu ALT, 26% pre HBeAg sérokonverziu a 2 % pre
HBsAg sérokonverziu (3 % pre stratu HBsAg).
Na konci dávkovacieho intervalu malo spomedzi pacientov, ktorí pokračovali v liečbe po 52 týždňoch
(stredná doba 96 týždňov) 81 % z 243 pacientov liečených entekavirom a 39 % zo 164 pacientov
liečených lamivudínom HBV DNA < 300 kópií/ml metódou PCR, kým k normalizácii
ALT (≤ 1-násobok ULN) došlo u 79 % pacientov liečených entekavirom a u 68 % pacientov liečených
lamivudínom.
HBeAg negatívni pacienti (štúdia 027): liečba entekavirom po dobu až 96 týždňov (n = 325) viedla ku kumulatívnej miere odpovedí v hodnote 94 % pre HBV DNA < 300 kópií/ml metódou PCR a 89 %
pre normalizáciu ALT oproti 77 % pre HBV DNA < 300 kópií/ml metódou PCR a 84 % pre
normalizáciu ALT u pacientov liečených lamivudínom (n = 313).
V skupine 26 pacientov liečených entekavirom a 28 pacientov liečených lamivudínom, ktorí pokračovali v liečbe po 52 týždňoch (stredná doba 96 týždňov), malo na konci dávkovacieho intervalu
HBV DNA < 300 kópií/ml metódou PCR 96 % pacientov liečených entekavirom a 64 % pacientov
liečených lamivudínom. K normalizácii ALT (≤ 1-násobok ULN) na konci dávkovacieho intervalu došlo u 27 % pacientov liečených entekavirom a 21 % pacientov liečených lamivudínom.
U pacientov, ktorí splnili protokolom definované kritériá pre odpoveď, trvala odpoveď počas celého
24-týždňového sledovania po liečbe u 75 % (83/111) pacientov odpovedajúcich na entekavir oproti
73 % (68/93) pacientov odpovedajúcich na lamivudín v štúdii 022 a u 46 % (131/286) pacientov odpovedajúcich na entekavir oproti 31 % (79/253) pacientov odpovedajúcich na lamivudín v štúdii
027. V priebehu 48 týždňov sledovania po liečbe došlo u veľkého množstva HBeAg negatívnych
pacientov k strate odpovede.
Výsledky biopsie pečene: U 57 pacientov z pilotnej štúdie bez predchádzajúcej liečby nukleozidmi
022 (HBeAg pozitívni) a 027 (HBeAg negatívni), ktorí sa zúčastnili dlhodobej prechodovej štúdie,
boli vyhodnocované dlhodobé výsledky histológie pečene. V pilotných štúdiách bola dávka entecaviru
0,5 mg denne (priemerná expozícia 85 týždňov) a 1 mg denne v prechodovej štúdii (priemerná expozícia 177 týždňov) a 51 pacientov v prechodovej štúdii dostávalo aj lamivudín (priemerne po dobu 29 týždňov). 55/57 (96 %) pacientov zaznamenalo histologické zlepšenie ako už bolo spomenuté (vyššie) a 50/57 (88 %) zaznamenalo zníženie o ≥ 1- podľa Ishakovho skóre. Pacientom s počiatočnou hodnotou Ishakovho skóre ≥ 2, 25/43 (58 %) kleslo skóre o ≥ 2- body. Všetci pacienti (10/10)
s pokročilou fibrózou alebo cirhózou zaznamenali na začiatku (Ishakovo skóre 4,5 alebo 6) pokles o ≥ 1 bod (priemerný pokles východiskovej hodnoty bol 1,5 bodu). V čase dlhodobej biopsie mali všetci pacienti HBV DNA < 300 kópií/ml a 49/57 (86 %) mali sérum ALT ≤ 1-násobok ULN. Všetci
57 pacienti zotrvali HBsAg pozitívny.
Pacienti refraktérni na lamivudín:
HBeAg pozitívni pacienti (štúdia 026): liečba entekavirom po dobu až 96 týždňov (n = 141) viedla ku kumulatívnej miere odpovedí v hodnote 30 % pre HBV DNA < 300 kópií/ml metódou PCR a 85 %
pre normalizáciu ALT a 17 % pre HBeAg sérokonverziu.
V skupine 77 pacientov, ktorí pokračovali v liečbe entekavirom po 52 týždňoch (stredná doba
96 týždňov), malo na konci dávkovacieho intervalu HBV DNA < 300 kópií/ml metódou PCR 40 %
pacientov a k normalizácii ALT (≤ 1-násobok ULN) došlo u 81 %.
Vek/pohlavie:
Nepozoroval sa viditeľný rozdiel v účinnosti entekaviru v závislosti na pohlaví (≈ 25 % žien v klinických štúdiách) alebo veku (≈ 5 % pacientov starších ako 65 rokov).
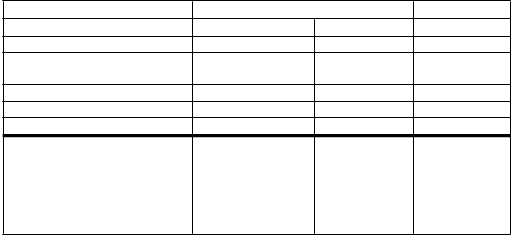
Osobitné skupiny pacientov
Pacienti s dekompenzovaným ochorením pečene: v štúdii 048, 191 pacientov s HBeAg pozitívnou alebo negatívnou chronickou infekciou HBV a dôkazom hepatálnej dekompenzácie definovanej ako
CPT skóre 7 alebo vyššie, dostávalo entekavir 1 mg jedenkrát denne alebo adefovir dipivoxil 10 mg
jedenkrát denne. Pacienti boli buď predliečení na HBV alebo bez predchádzajúcej liečby HBV (okrem predchádzajúcej liečby entekavirom, adefovir dipivoxilom alebo tenofovir disoproxil fumarátom). Na
začiatku liečby mali pacienti priemerné CPT skóre 8,59 a 26 % pacientov malo CPT stupňa C.
Priemerná bazálna hodnota skóre modelu pre konečné štádium ochorenia pečene (Model for End Stage Liver Disease, MELD) bola 16,23. Priemerná hodnota HBV DNA v sére bola 7,83 log10 kópií/ml metódou PCR a priemerná sérová ALT bola 100 U/l, 54 % pacientov bolo HBeAg pozitívnych a 35 % pacientov malo na začiatku liečby LVDr substitúcie. Entekavir bol významne účinnejší ako adefovir dipivoxil v primárnom koncovom ukazovateli priemernej zmeny od bazálnej hodnoty sérovej HBV DNA metódou PCR v 24. týždni. Výsledky koncových ukazovateľov jednotlivých štúdii v 24.
a 48. týždni sú uvedené v tabuľke.
Týždeň 24 Týždeň 48
ETV
1 mg jedenkrát
denne
Adefovir
Dipivoxil
10 mg jedenkrát denne
ETV
1 mg jedenkrát
denne
Adefovir
Dipivoxil
10 mg jedenkrát denne
N 100 91 100 91
HBV DNAa
Nedetegovateľný podiel (<300 kópií/ml)b 49 %* 16 % 57 %* 20 %
Týždeň 24 Týždeň 48
ETV
1 mg jedenkrát

denne
Adefovir
Dipivoxil
10 mg jedenkrát denne
ETV
1 mg jedenkrát
denne
Adefovir
Dipivoxil
10 mg jedenkrát denne
N 100 91 100 91
Priemerná zmena bazálnej hodnoty
(log10 kópií/ml)c
-4,48* -3,40 -4,66 -3,90
Stabilné alebo zlepšené CTP skóreb,d 66 % 71 % 61 % 67 % MELD skóre
Priemerná zmena bazálnej hodnotyc,e -2,0 -0,9 -2,6 -1,7
Strata HBsAgb 1 % 0 5 % 0
Normalizácia:f
ALT (≤1 X ULN)b 46/78 (59 %)*
28/71 (39 %) 49/78 (63 %)*
33/71 (46 %)
Albumín (≥1 X LLN)b 20/82 (24 %) 14/69 (20 %) 32/82 (39 %) 20/69 (29 %) Bilirubín (≤1 X ULN)b 12/75 (16 %) 10/65 (15 %) 15/75 (20 %) 18/65 (28 %) Protrombínový čas (≤1 X ULN)b 9/95 (9 %) 6/82 (7 %) 8/95 (8 %) 7/82 (9 %)
a Roche COBAS Amplicor PCR assay (LLOQ = 300 kópií/ml).
b NC = F (nedokončil = zlyhanie), znamená ukončenia liečby pred týždňom kedy mala byť urobená analýza vrátane dôvodov ako sú úmrtie, nedostatočná účinnosť, nežiaduca udalosť, noncompliance/ vynechanie následnej kontroly, ktoré sú považované za zlyhania (napr. HBV DNA ≥ 300 kópii/ml).
c NC = M (nedokončili = chýba)
dDefinovaný ako nárast alebo bez zmeny bazálnej hodnoty CPT skóre
e Priemerná bazálna hodnota MELD skóre bola 17,1 pre ETV a 15,3 pre adefovir dipivoxil.
f Menovateľ je pacient s abnormálnymi hodnotami na začiatku liečby.
*p<0,05
ULN = horná hranica normálu, LLN = spodná hranica normálu.
Čas do nástupu HCC alebo úmrtia (podľa toho čo nastalo skôr) bol porovnateľný v dvoch liečených skupinách; kumulatívne miery úmrtí v štúdií boli 23 % (23/102) u pacientov liečených entekavirom a 33 % (29/89) u pacientov liečených adefovir dipivoxilom a kumulatívne miery HCC v štúdií boli
12 % (12/102) u pacientov liečených entekavirom a 20% (18/89) u pacientov liečených adefovir
dipivoxilom.
U pacientov s LVDr substitúciami na začiatku liečby bolo percento pacientov s HBV
DNA <300 kópií/ml 44 % pre entecavir a 20 % pre adefovir v 24 týždni a 50 % pre entekavir a 17 %
pre adefovir v 48. týždni.
Pacienti súbežne infikovaní HIV/HBV užívajúci súbežnú HAART: štúdia 038 zahŕňala 67 HBeAg pozitívnych a 1 HBeAg negatívneho pacienta, ktorí boli súbežne infikovaní vírusom HIV (HIV RNA
< 400 kópií/ml) s rekurenciami virémie HBV počas liečby režimom HAART obsahujúcom lamivudín.
HAART režimy nezahŕňajú emtricitabín a tenofovir disoproxil fumarát. Na začiatku liečby mali pacienti liečení entekavirom strednú dĺžku trvania predošlej liečby lamivudínom 4,8 rokov a stredný počet CD4 494 buniek/mm3 (iba s 5 jedincami majúcimi počet CD4 < 200 buniek/mm3). Pacienti pokračovali vo svojich režimoch s lamivudínom a boli priradení buď k entekaviru 1 mg jedenkrát denne (n = 51), alebo k placebu (n = 17) po dobu 24 týždňov, po ktorej nasledovalo ďalších
24 týždňov, počas ktorých všetci užívali entekavir. V 24. týždni bolo zníženie vírusovej záťaže HBV významne väčšie u entekaviru (-3,65 oproti vzostupu o 0,11 log10 kópií/ml). U pacientov, ktorí boli pôvodne priradení k liečbe entekavirom, bolo zníženie HBV DNA v 48. týždni -4,20 log10 kópií/ml,
k normalizácii ALT došlo u 37 % pacientov s abnormálnymi bazálnymi hodnotami ALT a u žiadneho
z pacientov nedošlo k HBeAg sérokonverzii.
P
acienti súbežne infikovaní HIV/HBV neužívajúci súbežnú HAART: entekavir nebol hodnotený
u pacientov súbežne infikovaných HIV/HBV a nesúbežne užívajúcich účinnú HIV terapiu. Redukcia HIV RNA bola zaznamenaná u pacientov súbežne infikovaných HIV/HBV užívajúcich entekavir v monoterapii bez HAART. V niektorých prípadoch sa pozorovala selekcia HIV variantu M184V, ktorá má za dôsledok selekciu HAART režimov, ktoré môže pacienti užívať v budúcnosti. Preto sa
entekavir za týchto podmienok nemá používať vzhľadom na potenciál rozvoja HIV rezistencie (pozri časť 4.4).
Príjemcovia transplantátu pečene: bezpečnosť a účinnosť 1 mg entekaviru jedenkrát denne sa hodnotila v jednoramennej štúdii so 65 pacientmi, ktorí dostali transplantát pečene z dôvodu komplikácií chronickej infekcie HBV a v čase transplantácie mali HBV DNA < 172 IU/ml (približne
1 000 kópií/ml). V študovanej skupine pacientov bolo 82 % mužov, 39 % belochov a 37 % aziatov s priemerným vekom 49 rokov; v čase transplantácie malo 89 % pacientov ochorenie s negatívnym
HBeAg. Zo 61 pacientov, u ktorých sa hodnotila účinnosť (dostávalo entekavir minimálne 1 mesiac),
60 dostávalo aj imunoglobulín hepatitídy B (HBIg) ako súčasť profylaktického režimu po
transplantácii. Z týchto 60 pacientov dostalo 49 liečbu HBIg dlhšie ako 6 mesiacov. V 72. týždni po transplantácii sa u žiadneho z 55 sledovaných prípadov nevyskytla rekurencia vírusu HBV
[definovanú ako HBV DNA ≥ 50 IU/ml (približne 300 kópií/ml)] a nehlásila sa ani žiadna rekurencia
vírusu v čase sledovania zvyšných 6 pacientov. Po transplantácii vymizli HBsAg u všetkých
61 pacientov a 2 z nich sa neskôr stali HBsAg pozitívny i napriek zachovaniu nedetekovateľnej HBV
DNA (< 6 IU/ml). Frekvencia a charakter nežiaducich účinkov v tejto štúdii sa zhodovali s tými, ktoré sa očakávali u pacientov, ktorí dostali transplantát pečene a so známym bezpečnostným profilom entekaviru.
Pediatrická populácia: Štúdia 189 je prebiehajúca štúdia účinnosti a bezpečnosti entekaviru so
skupinou 180 detí a dospievajúcich bez predchádzajúcej liečby nukleozidmi vo veku od
2 do < 18 rokov s HBeAg pozitívnou infekciou chronickej hepatitídy B, kompenzovaným ochorením
pečene a zvýšenými hodnotami ALT. Jedinci boli randomizovaní (2:1) na užívanie zaslepenej liečby entekaviru 0,015 mg/kg až 0,5 mg/deň (N = 120) alebo placebo (N = 60). Randomizácia bola rozvrstvená podľa vekových skupín (2 až 6 rokov; > 6 až 12 rokov a > 12 až < 18 rokov). Východiskové demografické charakteristiky a charakteristika ochorenia HBV boli medzi 2 liečenými skupinami a naprieč vekovým kohortám porovnateľné. Pri vstupe do štúdie bola v študovanej'
populácii priemerná hodnota HBV DNA 8,1 log10 IU/ml a priemerná hodnota ALT bola 103 U/l. Výsledok pre hlavné koncové ukazovatele účinnosti v 48. týždni a 96. týždni je uvedený nižšie.
Entekavir Placebo*
48. týždeň 96. týždeň 48. týždeň
n 120 120 60
HBV DNA < 50 IU/ml a
HBeAg sérokonverziaa
24,2 % 35,8 % 3,3 %
HBV DNA < 50 IU/mla 49,2 % 64,2 % 3,3 % HBeAg sérokonverziaa 24,2 % 36,7 % 10,0 % ALT normalizáciaa 67,5 % 81,7 % 23,3 %
HBV DNA < 50 IU/mla
Východisková HBV DNA < 8 log10 IU/ml Východisková HBV DNA
≥ 8 log10 IU/ml
82,6 % (38/46) 82,6 % (38/46) 6,5 % (2/31)
28,4 % (21/74) 52,7 % (39/74) 0 % (0/29)
a NC = F (neukončili liečbu = liečba zlyhala)
* Pacienti randomizovaní na placebo, ktorí nemali HBe- sérokonverziu do 48. týždni prešli do otvorenej štúdie s entekavirom
počas druhého roku štúdie; z tohto dôvodu sú dostupné randomizované porovnávacie údaje iba do 48. týždňa.
Hodnotenie rezistencie u detí sa zakladá na údajoch od pediatrických pacientov s HBeAg-pozitívnou chronickou infekciou HBV predtým neliečených nukleozidmi z dvoch prebiehajúcich klinických skúšaní (028 a 189). Dve skúšania poskytujú údaje o rezistencii u 183 liečených pacientoch
a sledovaných v 1. roku a 180 liečených pacientoch a sledovaných v 2. roku. Hodnotenia genotypu sa
vykonali pre všetkých pacientov s dostupnými vzorkami, ktorí mali virologický prienik do 96. týždňa alebo HBV DNA ≥ 50 IU/ml v 48. týždni alebo v 96. týždni. Počas 2. roku bola detegovaná genotypová rezistencia na ETV u 2 pacientov (1,1 % kumulatívna pravdepodobnosť rezistencie počas
2. roku).
Klinická rezistencia u dospelých: pacienti najprv liečení entekavirom v dávke 0,5 mg (bez predchádzajúcej liečby nukleozidmi) alebo v dávke 1,0 mg (refraktérni na lamivudín) a s výsledkom PCR liečby HBV DNA v 24. alebo po 24. týždni boli monitorovaní na rezistenciu.
Počas týždňa 240 v štúdiách bez predchádzajúcej liečby nukleozidmi bola genotypová evidencia ETVr
substitúcií na rtT184, rtS202 a rtM250 identifikovaná u 3 pacientov liečených entekavirom, 2 z nich
mali skúsenosť virologického prieniku (pozri tabuľku). Tieto substitúcie sa zistili iba za prítomnosti
LVDr substitúcií (rtM204V a rtL180M).
Rozvinutá genotypová rezistencia na entekavir počas 5. roka, Štúdie bez predchádzajúcej liečby
nukleozidmi
Pacienti liečení a monitorovaní
na rezistenciub
Pacienti počas špecifickéhoroka s:- rozvinutá genotypová
ETVrc
- genotypová ETVrc s virologickým prienik d
Kumulatívnapravdepodobnosť:
- rozvinutá genotypová
ETVrc
- genotypová ETVrc s virologickým prienik d
Rok 1 Rok 2 Rok 3a Rok 4a Rok 5a
663 278 149 121 108
1 1 1 0 0
1 0 1 0 0
0,2 % 0,5 % 1,2 % 1,2 % 1,2 %
0,2 % 0,2 % 0,8 % 0,8 % 0,8 %
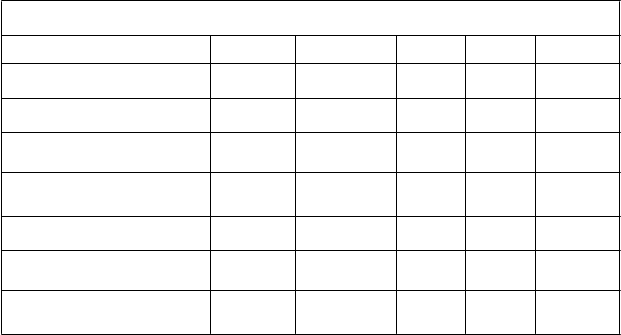
a Výsledky odrážajú použitie 1-mg dávky entekaviru u 147 zo 149 pacientov v Roku 3 a všetkých pacientov v Roku 4 a 5 a
kombinovanej liečby entekavir-lamivudínom (následne po dlhodobej liečbe enetekavirom) s mediánom 20 týždňov u 130 zo
149 pacientov v Roku 3 a 1 týždeň pre 1 zo 121 pacientov v Roku 4 prechádzajúcich z jednej štúdie do druhej (rollover study).
b Zahŕňa pacientov s najmenej jednou terapiou HBV DNA merateľnou prostredníctvom PCR v alebo po 24 týždňoch počas týždňa 58 (Rok 1), po 58 týždňoch počas týždňa 102 (Rok 2) po 102 týždňoch počas týždňa 156 (Rok 3) po 156 týždňoch
počas týždňa 204 (Rok 4) alebo po 204 týždňoch počas týždňa 252 (Rok 5).
c Pacienti, ktorí tiež majú LVDr substitúcie.
d ≥ 1 log10 zvýšenie nad najnižšiu hodnotu u HBV DNA potvrdené postupnými meraniami s PCR alebo na konci časového
rozpätia.
ETVr substitúcie (okrem LVDr substitúcií rtM204V/I ± rtL180M) sa zistili na základných izolátoch
u 10/187 (5 %) pacientov refraktérnych na lamivudín liečených entekavirom a monitorovaných na rezistenciu naznačujúc tak, že predošlá liečba lamivudínom môže selektovať tieto rezistentné
substitúcie a zistiť ich nízky výskyt pred liečbou entekavirom. Počas 240 týždňa mali 3 z 10 pacientov
spontánny virologický prienik (≥ 1 log10 zvýšenie nad najnižšiu hodnotu). Rozvinutá rezistencia na
entekavir v lamivudín refraktérnych štúdiách počas 240 týždňa je zhrnutá v tabuľke.
Genotypová rezistencia na entekavir počas 5. roka, Lamivudín refraktérne štúdie
Rok 1 Rok 2 Rok 3a Rok 4 a Rok 5 a
Pacienti liečení a monitorovaní
na rezistenciub
Pacienti počas špecifického
roka s:
- rozvinutá genotypová
ETVrc
187 146 80 52 33
11 12 16 6 2
- genotypová ETVrc s virologickým prienik d
Kumulatívnapravdepodobnosť:
e e e e e
14 9 1
- rozvinutá genotypová
ETVrc
- genotypová ETVrc s virologickým prienik d
6,2 % 15 % 36,3 % 46,6 % 51,45 %
1,1 % e 10,7 % e 27 % e 41,3 % e 43,6 % e
a Výsledky odrážajú použitie kombinovanej liečby entekavir-lamivudínom (následne po dlhodobej liečbe
enetekavirom) s mediánom 13 týždňov u 48 z 80 pacientov v Roku 3, mediánom 38 týždňov pre 10 z 52 pacientov v Roku 4 a 16 týždňov pre 1 z 33 pacientov v Roku 5 prechádzajúcich z jednej štúdie do druhej (rollover study).
b Zahŕňa pacientov s najmenej jednou terapiou HBV DNA merateľnou prostredníctvom PCR v alebo po
24 týždňoch počas týždňa 58 (Rok 1), po 58 týždňoch počas týždňa 102 (Rok 2) alebo po 102 týždňoch počas týždňa
156 (Rok 3), po 156 týždňoch počas týždňa 204 (Rok 4) alebo po 204 týždňoch počas týždňa 252 (Rok 5).
c Pacienti, ktorí tiež majú LVDr substitúcie.
d ≥ 1 log10 zvýšenie nad najnižšiu hodnotu u HBV DNA potvrdené postupnými meraniami s PCR alebo na konci
časového rozpätia
e Existujúca ETVr v ktoromkoľvek roku; virologický prienik v špecifickom roku.
U lamivudín refraktérnych pacientov s HBV DNA <107 log10 kópií/ml, 64 % (9/14) sa dosiahla HBV DNA <300 kópií/ml v týždni 48. Títo 14 pacienti mali nižší pomer genotypovej rezistencie na entekavir (kumulatívna pravdepodobnosť 18,8 % počas 5 rokov sledovania) ako celková populácia štúdie (pozri tabuľku). Taktiež pacienti refraktérni na lamivudín, ktorí dosiahli HBV DNA <104 log10 kópií/ml prostredníctvom PCR v týždni 24 mali nižší pomer ako tí, čo ho nedosiahli (5 ročná kumulatívna pravdepodobnosť 17,6 % [n = 50] oproti 60,5 % [n = 135], v uvedenom poradí).
5.2 Farmakokinetické vlastnostiAbsorpcia: entekavir sa rýchlo vstrebáva, pričom maximálne plazmatické koncentrácie sa dosiahnu po
0,5 až 1,5 hodine. Absolútna biologická dostupnosť nebola stanovená. Na základe vylučovania nezmeneného liečiva močom bola biologická dostupnosť odhadnutá na najmenej 70 %. Po
opakovaných dávkach v rozsahu od 0,1 – 1 mg dochádza k vzostupu hodnôt Cmax a AUC úmerných podanej dávke. Rovnovážny stav sa dosiahne po 6 až 10 dňoch po dávke podávanej jedenkrát denne
s ≈ 2-násobnou kumuláciou. V rovnovážnom stave je hodnota Cmax 4,2 ng/ml a Cmin 0,3 ng/ml pre dávku 0,5 mg a hodnota Cmax 8,2 ng/ml a Cmin 0,5 ng/ml pre dávku 1 mg. U zdravých jedincov bola forma tabliet a perorálneho roztoku bioekvivalentná; z tohto dôvodu sa obe formy môžu používať zameniteľne.
Podanie 0,5 mg entekaviru so štandardným jedlom s vysokým obsahom tuku (945 kcal, 54,6 g tuku) alebo s jedlom s nízkym obsahom tuku (379 kcal, 8,2 g tuku) viedlo k minimálnemu predĺženiu absorpcie (1 – 1,5 hodiny po jedle oproti 0,75 hodiny nalačno), k poklesu Cmax o 44 – 46 % a poklesu AUC o 18 – 20 %. Nižšie hodnoty Cmax a AUC po užití s jedlom sa nepovažujú za klinicky významné u pacientov bez predošlej liečby nukleozidmi, ale mohli by ovplyvniť účinnosť u pacientov refraktérnych na lamivudín (pozri časť 4.2).
Distribúcia: odhadovaný distribučný objem entekaviru presahuje celkový objem vody v tele. Väzba na ľudské sérové proteíny
in vitro je ≈ 13 %.
B
i
otransformácia: entekavir nie je substrátom, induktorom ani inhibítorom enzýmového systému CYP450. Po podaní entekaviru značeného 14C neboli pozorované žiadne oxidačné ani acetylované metabolity a boli pozorované malé množstvá metabolitov II. fázy - glukuronidových a sulfátových konjugátov.
Eliminácia: entekavir je vylučovaný hlavne obličkami, pričom v moči sa v rovnovážnom stave zistilo
nezmenené liečivo v množstve približne 75 % dávky. Renálny klírens je nezávislý od dávky a je v rozsahu 360 až 471 ml/min, čo svedčí o tom, že entekavir podlieha glomerulárnej filtrácii aj
tubulárnej sekrécii. Po dosiahnutí maximálnych hladín dochádza k biexponenciálnemu poklesu
plazmatických hladín s konečným eliminačným polčasom ≈ 128 – 149 hodín. Pozorovaný index kumulácie liečiva je ≈ 2-násobný pri dávke podávanej jedenkrát denne, čo svedčí o efektívnom kumulačnom polčase približne 24 hodín.
Porucha funkcie pečene: farmakokinetické parametre u pacientov so stredne ťažkou alebo ťažkou poruchou funkcie pečene boli podobné parametrom u pacientov s normálnou funkciou pečene.
Porucha funkcie obličiek: klírens entekaviru sa znižuje so znižujúcim sa klírensom kreatinínu.

4-hodinová hemodialýza odstránila ≈ 13 % dávky a 0,3 % boli odstránené CAPD. Farmakokinetika entekaviru po jednorazovej 1 mg dávke u pacientov (bez chronickej infekcie vírusom hepatitídy B) je uvedená nižšie v tabuľke:
Bazálny klírens kreatinínu (ml/min)
B
ez poruchy
> 80
Ľahká
porucha
> 50; ≤ 80
Stredne ťažká porucha
30 – 50
Ťažká
porucha
20-< 30
Ťažká porucha liečená hemodialýzou
Ťažká porucha liečená CAPD
(n = 6) (n = 6) (n = 6) (n = 6) (n = 6) (n = 4)

Cmax (ng/ml) (CV%)
AUC(0-T) (ng·h /ml) (CV)
8,1 (30,7)
27,9 (25,6)
10,4 (37,2)
51,5 (22,8)
10,5 (22,7)
69,5 (22,7)
15,3 (33,8)
145,7 (31,5)
15,4 (56,4)
233,9 (28,4)
16,6 (29,7)
221,8 (11,6)
CLR
(ml/min) (SD)
383,2
(101,8)
197,9
(78,1)
135,6
(31,6)
40,3
(10,1)
NA NA
CLT/F
(ml/min) (SD)
588,1
(153,7)
309,2
(62,6)
226,3
(60,1)
100,6
(29,1)
50,6
(16,5)
35,7
(19,6)
 P
o transplantácii pečene:
P
o transplantácii pečene: expozícia entekaviru u príjemcov transplantátu pečene infikovaných HBV
na stabilnej dávke cyklosporínu A alebo takrolimu (n = 9) bola ≈ 2-násobkom expozície u zdravých jedincov s normálnou funkciou pečene. U pacientov po transplantácii pečene zmenená funkcia pečene prispela k zvýšeniu expozície entekaviru (pozri časť 4.4).
Pohlavie: hodnota AUC bola o 14 % vyššia u žien ako u mužov, kvôli rozdielom vo funkcii obličiek a telesnej hmotnosti. Po úprave v zmysle rozdielov v klírense kreatinínu a telesnej hmotnosti nebol medzi mužmi a ženami žiadny rozdiel v expozícii.
Staršie osoby: vplyv veku na farmakokinetiku entekaviru bol hodnotený porovnaním starších jedincov vo vekovom rozmedzí 65 – 83 rokov (priemerný vek žien 69 rokov, mužov 74 rokov) s mladými jedincami vo vekovom rozmedzí 20 – 40 rokov (priemerný vek žien 29 rokov, mužov 25 rokov). Hodnota AUC bola o 29 % vyššia u starších jedincov ako u mladých jedincov, hlavne kvôli rozdielom vo funkcii obličiek a telesnej hmotnosti. Po úprave vzhľadom na rozdiel v klírense kreatinínu
a telesnej hmotnosti bola hodnota AUC u starších jedincov o 12,5 % vyššia ako u mladých jedincov.
Populačná farmakokinetická analýza zahŕňajúca pacientov vo vekovom rozmedzí 16 – 75 rokov
neidentifikovala vek ako faktor významne ovplyvňujúci farmakokinetiku entekaviru.
Rasa: populačná farmakokinetická analýza neidentifikovala rasu ako faktor významne ovplyvňujúci farmakokinetiku entekaviru. Závery je však možné urobiť len pre bielu a ázijskú rasu, pretože bolo príliš málo jedincov iných rás.
Pediatrická populácia: farmakokinetika rovnovážneho stavu entekaviru sa hodnotila (štúdia 028) u 24 HBeAg pozitívnych pediatrických jedincov vo veku od 2 do < 18 rokov s kompenzovaným ochorením pečene predtým neliečených nukleozidmi a u 19 liečených lamivudínom. Expozícia entekaviru v skupine jedincov predtým neliečených nukleozidmi, ktorí dostávali jedenkrát denne dávky entekaviru 0,015 mg/kg až maximálnu dávku 0,5 mg bola podobná expozícii, ktorá sa dosiahla u dospelých, ktorí dostávali jedenkrát denne dávky 0,5 mg. Cmax, AUC(0-24) a Cmin u týchto jedincov bola 6,31 ng/ml, 18,33 ng h/ml a 0,28 ng/ml, v uvedenom poradí. Expozícia entekaviru
v skupine jedincov liečených lamivudínom, ktorí dostávali jedenkrát denne dávky entekaviru
0,030 mg/kg až maximálnu dávku 1,0 mg bola podobná expozícii, ktorá sa dosiahla u dospelých, ktorí dostávali jedenkrát denne dávky 1,0 mg. Cmax, AUC(0-24) a Cmin u týchto jedincov bola
14,48 ng/ml, 38,58 ng∙h/ml a 0.47 ng/ml, v uvedenom poradí.
5.3 Predklinické údaje o bezpečnosti
V toxikologických štúdiách s opakovaným podávaním u psov bol pozorovaný reverzibilný perivaskulárny zápal v centrálnom nervovom systéme, pre ktorý neúčinné dávky zodpovedali expozíciám 19- a 10-násobne vyšším ako sú expozície u ľudí (pri dávke 0,5 mg a 1 mg). Toto zistenie nebolo pozorované v štúdiách s opakovaným podávaním u ďalších živočíšnych druhov, vrátane opíc, ktorým bol entekavir podávaný denne po dobu 1 roka pri expozíciách ≥ 100-násobne vyšších ako sú expozície u ľudí.
V reprodukčných toxikologických štúdiách, v ktorých bol zvieratám podávaný entekavir po dobu až
4 týždňov, neboli pozorované žiadne dôkazy o poruche fertility u samcov ani u samíc potkanov pri vysokých expozíciách. Zmeny semenníkov (degenerácia semenných kanálikov) boli zjavné
v toxikologických štúdiách s opakovaným podávaním u hlodavcov a psov pri expozíciách
≥ 26-násobne vyšších ako expozície u ľudí. V 1-ročnej štúdii u opíc neboli zjavné žiadne zmeny semenníkov.
U gravidných potkanov a králikov, ktorým bol podávaný entekavir, neboli pri expozíciách
≥ 21-násobne vyšších ako sú expozície u ľudí pozorované žiadne toxické účinky na embryo ani na
matku. U potkanov boli pozorované toxické účinky na matku, embryonálny a fetálny vývoj (resorpcie), nižšia telesná hmotnosť plodu, malformácie chvostu a chrbtice, znížená osifikácia (stavcov, segmentov hrudnej kosti a článkov prstov) a nadpočetné driekové stavce a rebrá pozorované pri vysokých expozíciách. U králikov boli pri vysokých expozíciách pozorované toxické účinky na vývoj embrya a plodu (resorpcie), znížená osifikácia (jazylky) a zvýšený výskyt 13. rebra. V štúdiách perinatálneho a postnatálneho obdobia u potkanov neboli pozorované žiadne nežiaduce účinky na mláďatá. V osobitnej štúdii, v ktorej bol entekavir podávaný gravidným potkanom počas laktácie
v dávke 10 mg/kg, bola preukázaná fetálna expozícia entekaviru aj vylučovanie entekaviru do mlieka.
U juvenilných potkanov, ktorým sa podával entekavir od 4. do 80. dňa po narodení, sa zaznamenala mierne znížená reakcia na akustické vyplašenie počas obdobia zotavovania sa (110. až 114. deň po narodení), no nie počas obdobia podávania pri hodnotách AUC ≥ 92-násobok tých, ktoré sú u ľudí pri dávke 0,5 mg alebo pri ekvivalentnej pediatrickej dávke. Vzhľadom na rozsah expozície sa tento nález považuje za nález nepravdepodobného klinického významu.
V Amesovom teste mikrobiálnej mutagenity, v teste génových mutácií v bunkách cicavcov a v teste bunkových transformácií v embryonálnych bunkách sýrskeho škrečka neboli pozorované žiadne dôkazy o genotoxicite. Mikronukleový test a DNA reparačný test boli tiež negatívne. Entekavir mal klastogénny účinok v kultúrach ľudských lymfocytov pri koncentráciách značne vyšších ako sú koncentrácie dosiahnuté pri klinickom používaní.
Dvojročné štúdie karcinogenity: u samcov myší bol pozorovaný zvýšený výskyt tumorov pľúc pri expozíciách ≥ 4-násobne (pri 0,5 mg) a ≥ 2-násobne (pri 1 mg) vyšších ako sú expozície u ľudí. Rozvoju tumorov predchádzala proliferácia pneumocytov v pľúcach, ktorá nebola pozorovaná
u potkanov, psov ani opíc, čo naznačuje, že kľúčová udalosť pri rozvoji tumorov pľúc pozorovaná u myší bola pravdepodobne špecifická pre tento živočíšny druh. Zvýšený výskyt iných tumorov zahŕňajúcich gliómy mozgu u samcov a samíc potkanov, karcinómy pečene u samcov myší, benígne vaskulárne tumory u samíc myší a adenómy a karcinómy pečene u samíc potkanov boli pozorované len pri vysokých celoživotných expozíciách. Nebolo však možné presne stanoviť neúčinnú dávku. Predikčná hodnota týchto zistení pre ľudí nie je známa.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety:
Uhličitan vápenatý Škrob, predželatinovaný Sodná soľ karmelózy Sójové polysacharidy
Monohydrát kyseliny citrónovej
Stearylfumarát sodný
Obal tablety:
Entecavir Accord 0,5 mg filmom obalené tablety
Hypromelóza
Oxid titaničitý (E171) Makrogol
Polysorbát 80
Entecavir Accord 1 mg filmom obalené tablety
Hypromelóza
Oxid titaničitý (E171) Makrogol
Červený oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
Po otvorení fľaše použite do 90 dní.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Každá škatuľa obsahuje buď:
§ 30 x 1 filmom obalená tableta; 3 blistre s 10 x 1 filmom obalená tableta, každá z tabliet je v Alu/Alu perforovanom jednodávkovom blistri alebo
§ 90 x 1 filmom obalená tableta; 9 blistrov s 10 x 1 filmom obalená tableta, každá z tabliet je v Alu/Alu perforovanom jednodávkovom blistri
Fľaša z polyetylénu s vysokou hustotou (HDPE), obsahuje nádobku na silikagél, s polypropylénovým uzáverom bezpečným pred deťmi obsahujúca 30 filmom obalených tabliet. Každá škatuľa obsahuje jednu fľašu.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAccord Healthcare Limited
Sage House, 319 Pinner Road
North Harrow, Middlesex, HA1 4HF
Veľká Británia
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)Entecavir Accord 0,5 mg filmom obalené tabletyFľaša: EU/1/17/1211/001
Blister: EU/1/17/1211/002
EU/1/17/1211/003
Entecavir Accord 1 mg filmom obalené tabletyFľaša: EU/1/17/1211/004
Blister: EU/1/17/1211/005
EU/1/17/1211/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.