
b>systémov
V
eľmi časté Časté Menej časté
P
oruchy ucha a labyrintu závrat
P
oruchy gastrointestinálneho traktu Poruchy kože
a podkožného tkaniva
C
elkové poruchy a reakcie v mieste podania

bolesť v mieste podania injekcie, reakcie v mieste podania injekciea
zápcha
pruritus urtikária
a Najčastejšie hlásené (≥ 1 %) boli: reakcia v mieste podania injekcie, erytém v mieste podania, pruritus v mieste podania, podliatina v mieste podania, opuch v mieste podania.
O
pis vybraných nežiaducich reakcií
B
olesť alebo reakcie v mieste podania injekcie
Väčšina prípadov súvisiacich s miestom podania injekcie bola mierna až stredne závažná a menej ako
0,5 % pacientov vystavených pôsobeniu galkanezumabu v priebehu štúdií fázy 3 prerušilo liečbu
v dôsledku reakcie v mieste podania injekcie. Väčšina reakcií v mieste podania bolo hlásených do 1 dňa a vymizli v priemere počas 5 dní. U 86 % pacientov, ktorí hlásili bolesť v mieste podania injekcie, sa táto udalosť vyskytla do 1 hodiny po podaní injekcie a vymizla v priemere za 1 deň. U jedného percenta pacientov vystavených pôsobeniu galkanezumabu v priebehu štúdií fázy 3 sa objavila silná bolesť v mieste podania injekcie.
Urtikária
Zatiaľ čo urtikária je menej častá, v klinických štúdiách s galkanezumabom boli hlásené závažné prípady urtikárie.
Imunogenita
V klinických štúdiách v priebehu dvojito zaslepenej fázy bola incidencia výskytu protilátok proti lieku u 4,8 % pacientov užívajúcich galkanezumab jedenkrát mesačne (všetci okrem jedného mali
neutralizačnú aktivitu in vitro). Počas 12-mesačnej liečby sa až u 12,5 % pacientov liečených
galkanezumabom vytvorili protilátky proti lieku, väčšina ktorých mala nízky titer a pozitívny test na
neutralizačnú aktivitu in vitro. Prítomnosť protilátok proti lieku však nemala vplyv na
farmakokinetiku, účinnosť ani bezpečnosť galkanezumabu.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.*
4.9 Predávkovanie
Dávky až do 600 mg boli podávané subkutánne ľuďom bez toxicity obmedzujúcej dávku. V prípade predávkovania sa odporúča u pacienta sledovať akékoľvek prejavy alebo príznaky nežiaducich reakcií a aby sa okamžite začalo s vhodnou symptomatickou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: analgetiká, iné antimigrenotiká, ATC kód: N02CX08
Mechanizmus účinku
Galkanezumab je humanizovaná monoklonálna protilátka typu IgG4, ktorá sa viaže na
kalcitonínovému génu príbuzný peptid (calcitonin gene-related peptide, CGRP), čím zabraňuje jeho biologickej aktivite. Zvýšené koncentrácie CGRP v krvi súvisia so záchvatmi migrény. Galkanezumab
sa s vysokou afinitou (KD = 31 pM) a vysokou špecifickosťou (> 10 000-násobne vyššou oproti príbuzným peptidom adrenomedulínu, amylínu, kalcitonínu a intermedínu) viaže na CGRP.
Klinická účinnosť abezpečnosť
Účinnosť a bezpečnosť galkanezumabu bola skúmaná v 3 randomizovaných, placebom
kontrolovaných, dvojito zaslepených štúdiách fázy 3 u dospelých pacientov (N = 2886). Do 2 štúdií epizodickej migrény (EVOLVE-1 a EVOLVE-2) boli zaradení pacienti, ktorí spĺňali kritériá Medzinárodnej klasifikácie bolestí hlavy (International Classification of Headache Disorders - ICHD) pre diagnózu migrény s aurou alebo bez aury a s 4-14 dňami migrény za mesiac. Do štúdie chronickej migrény (REGAIN) boli zaradení pacienti, ktorí spĺňali kritériá ICHD pre chronickú migrénu
s ≥ 15 dňami s bolesťou hlavy za mesiac, z ktorých najmenej 8 malo znaky migrény. Pacienti
s nedávnymi akútnymi kardiovaskulárnymi príhodami (vrátane infarktu myokardu, nestabilnej angíny pectoris, bajpasom koronárnych tepien, mozgovej mŕtvice a hlbokej žilovej trombózy) a/alebo tí,
u ktorých sa predpokladalo závažné kardiovaskulárne riziko, boli z klinických skúšaní s galkanezumabom vylúčení. Pacienti vo veku > 65 rokov boli tiež vylúčení.
Pacientom bolo podávané placebo, galkanezumab 120 mg/mesiac (s počiatočnou nasycovacou dávkou
240 mg v prvom mesiaci) alebo galkanezumab 240 mg/mesiac a mali povolené užívať lieky na liečbu
akútnej migrény. Vo všetkých 3 štúdiách boli pacientmi prevažne ženy (> 83 %) s priemerným vekom
41 rokov a s priemernou anamnézou migrény v dĺžke 20 až 21 rokov. U približne jednej tretiny pacientov vo všetkých štúdiách sa vyskytlo z dôvodov účinnosti najmenej 1 predchádzajúce zlyhanie
profylaktickej liečby migrény a približne u 16 % pacientov vo všetkých štúdiách sa vyskytli
z dôvodov účinnosti najmenej 2 predchádzajúce zlyhania profylaktickej liečby.
Vo všetkých 3 štúdiách bola primárnym ukazovateľom účinnosti celková priemerná zmena počtu dní s migrénou (Migraine Headache Day, MHD) za mesiac od začiatku liečby. Mierou odpovede je priemerný percentuálny podiel pacientov spĺňajúcich definovanú hranicu zníženého počtu MHD za mesiac (≥ 50 %, ≥ 75 % a 100 %) v priebehu dvojito zaslepeného obdobia liečby. Vplyv migrény na funkčnosť sa hodnotil pomocou položky Role Function-Restrictive v Dotazníku kvality života
s migrénou (Migraine-Specific Quality of Life Questionnaire - MSQ) verzia 2.1 a Dotazníka hodnotiaceho zdravotné postihnutie pri migréne (Migraine Disability Assessment - MIDAS). Dotazník
MSQ meria vplyv migrény na prácu a každodenné aktivity, vzťahy s rodinou a priateľmi, voľný čas,
produktivitu, sústredenie, vitalitu a únavu. Skóre sa pohybuje od 0 do 100, pričom vyššie skóre označuje menšie zdravotné postihnutie, čiže u pacientov sa vyskytuje menej obmedzení pri
vykonávaní každodenných aktivít. V dotazníku MIDAS vyššie skóre označuje vyššie zdravotné
postihnutie. Skóre dotazníka MIDAS na začiatku liečby vyjadrovalo závažné zdravotné postihnutie pacientov súvisiace s migrénou v štúdiách EVOLVE-1 a EVOLVE-2 (priemer 33,1) a veľmi závažné zdravotné postihnutie u pacientov (priemer 67,2) v štúdii REGAIN.
Epizodická migréna
Štúdie EVOLVE-1 a EVOLVE-2 mali 6-mesačnú, dvojito zaslepenú, placebom kontrolovanú fázu liečby. Miera ukončenia účasti v dvojito zaslepenej fáze liečby u pacientov, ktorým bol podávaný galkanezumab, sa pohybovala od 82,8 % do 87,7 %.
Obe galkanezumabom 120 mg a 240 mg liečené skupiny preukázali štatisticky významné a klinicky významné zlepšenie od začiatku liečby v porovnaní s placebom v priemernej zmene v MHD (pozri tabuľku č. 2). U pacientov liečených galkanezumabom sa vyskytla vyššia miera odpovede
a výraznejšie zníženie počtu MHD za mesiac, kedy užívali akútnu úľavovú liečbu, v porovnaní s pacientmi liečenými placebom. Pacienti liečení galkanezumabom mali počnúc 1. mesiacom
výraznejšie zlepšenie funkčnosti (podľa merania položky Role Function-Restrictive dotazníka MSQ)
v porovnaní s pacientmi liečenými placebom. Viac pacientov liečených galkanezumabom dosiahlo klinicky významnú úroveň zlepšenia funkčnosti (miera odpovede v položke Role Function Restrictive
dotazníka MSQ) v porovnaní s pacientmi liečenými placebom. Galkanezumab sa spájal so štatisticky
významným znížením zdravotného postihnutia oproti placebu.
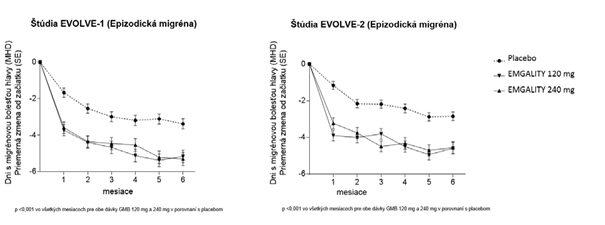
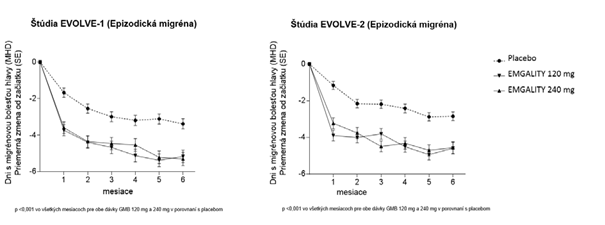
V porovnaní s pacientmi liečenými placebom sa u pacientov liečených galkanezumabom 120 mg alebo 240 mg v 1. mesiaci a všetkých nasledujúcich mesiacoch až do 6. mesiaca vyskytlo významne výraznejšie priemerné zníženie počtu MHD za mesiac od začiatku liečby (pozri obrázok č. 1). Okrem toho, v 1. mesiaci pacienti liečení galkanezumabom (nasycujúca dávka 240 mg) preukázali významne menej MHD za týždeň v 1. týždni a každom nasledujúcom týždni v porovnaní s pacientmi liečenými placebom.
O
brázok č. 1: Zníženie počtu dní s migrénou za mesiac, v priebehu štúdií EVOLVE-1
a EVOLVE-2
 T
abuľka č. 2: Účinnosť a pacientmi hlásené výsledky
E
VOLVE 1 – Epizodická migréna EVOLVE 2 - Epizodická migréna
T
abuľka č. 2: Účinnosť a pacientmi hlásené výsledky
E
VOLVE 1 – Epizodická migréna EVOLVE 2 - Epizodická migréna
Emgality

Placebo
Emgality
Placebo
Výsledná
účinnosť
a
MHD
120 mg 240 mg 120 mg 240 mg
N = 210 N = 208 N = 425 N = 226 N = 220 N = 450

Začiatočná hodnota 9,21 9.14 9,08 9,07 9,06 9,19
Priemerná zmena -4,73 -4,57 -2,81 -4,29 -4,18 -2,28
Rozdiel v liečbe -1,92 -1,76 -2,02 -1,90
CI95% (-2,48; -1,37) (-2,31; -1,20) (-2,55; -1,48) (-2,44; -1,36) P-hodnota <0,001d <0,001d <0,001d <0,001d
≥ 50% MHD respondériPercento, % 62,3 60,9 38,6 59,3 56,5 36,0
P-hodnota <0,001d <0,001d <0,001d <0,001d
≥ 75% MHD respondériPercento, % 38,8 38,5 19,3 33,5 34,3 17,8
P-hodnota <0,001d <0,001d <0,001d <0,001d
100 % respondériPercento, % 15,6 14,6 6,2 11,5 13,8 5,7
P-hodnota <0,001d <0,001d <0,001d <0,001d
MHD s použitím liečby naakútnu migrénuZačiatočná hodnota 7,42 7,34 7,38 7,47 7,47 7,62
Priemerná zmena -3,96 -3,76 -2,15 -3,67 -3,63 -1,85
Rozdiel v liečbe -1,81 -1,61 -1,82 -1,78
CI95% (-2,28; -1,33) (-2,09; -1,14) (-2,29; -1,36) (-2,25; -1,31) P-hodnota <0,001d <0,001d <0,001d <0,001d
Pacientmi hlásené výsledkyMSQ položka RoleFunction-Restrictive b
N 189 184 377 213 210 396
Začiatočná hodnota 51,39 48,76 52,92 52,47 51,71 51,35
Priemerná zmena 32,43 32,09 24,69 28,47 27,04 19,65
Rozdiel v liečbe 7,74 7,40 8,82 7,39
CI95% (5,20; 10,28) (4,83; 9,97) (6,33; 11,31) (4,88; 9,90) P-hodnota <0,001d <0,001d <0,001d <0,001d
E
VOLVE 1 – Epizodická migréna EVOLVE 2 - Epizodická migréna
Emgality

Placebo
Emgality
Placebo
Respondéri MSQ položky
Role Function-Restrictivec
120 mg 240 mg 120 mg 240 mg
N = 210 N = 208 N = 425 N = 226 N = 220 N = 450

N 189 184 377 213 210 396
Percento, % 63,5 69,6 47,2 58,2 60,0 43,4
P-hodnota <0,001f <0,001f <0,001f <0,001f
MIDAS Celkové skóre e
N 177 170 345 202 194 374
Začiatočná hodnota 32,93 36,09 31,84 30,87 32,75 34,25
Priemerná zmena -21,16 -20,06 -14,87 -21,17 -20,24 -12,02
Rozdiel v liečbe -6,29 -5,19 -9,15 -8,22
CI95% (-9,45; -3,13) (-8,39; -1,98) (-12,61; -5,69) (-11,71; -4,72) P-hodnota <0,001f 0,002f <0,001f <0,001f
N = počet pacientov; CI95% = 95 % interval spoľahlivosti. aVýsledky účinnosti boli hodnotené v priebehu 1. - 6. mesiaca. bHodnotené v priebehu 4. - 6. mesiaca.
cDefinovaní ako pacienti so zlepšením o ≥ 25 bodov u epizodickej migrény v priemere 4. - 6. mesiaca.
dŠtatisticky významné po úprave na viacnásobné porovnanie.
eHodnotené v 6. mesiaci.
fNeupravené na viacnásobné porovnanie.
V súhrnných údajoch zo štúdií EVOLVE-1 a EVOLVE-2, u pacientov, u ktorých z dôvodov účinnosti
zlyhali jedna alebo viaceré profylaktické liečby, bol pozorovaný rozdiel v liečbe zameranej na zníženie počtu priemerných MHD za mesiac medzi 120 mg galkanezumabu a placebom -2,69 dňa
(p < 0,001) a medzi 240 mg galkanezumabu a placebom -2,78 dňa (p < 0,001). U pacientov, u ktorých zlyhali dve alebo viaceré profylaktické liečby, bol rozdiel v liečbe -2,64 dňa (p < 0,001) medzi 120 mg a placebom a -3,04 dňa (p < 0,001) medzi 240 mg a placebom.
Chronická migrénaŠtúdia REGAIN mala 3-mesačnú, dvojito zaslepenú, placebom kontrolovanú fázu liečby, po ktorej nasledovala 9-mesačná otvorená pokračovacia fáza. Približne 15 % pacientov pokračovalo v súbežnej liečbe topiramátom alebo propranololom tak, ako to umožňoval protokol pre profylaxiu migrény. Miera dokončenia fázy dvojito zaslepenej liečby u pacientov, ktorým bol podávaný galkanezumab, bola 95,3 %.
Obidve liečebné skupiny s galkanezumabom v dávke 120 mg aj 240 mg preukázali štatisticky signifikantné a klinicky významné zlepšenie od začiatku liečby v porovnaní s placebom v priemernej zmene v MHD (pozri tabuľku č. 3). Pacienti liečení galkanezumabom mali vyššiu mieru odpovede
a výraznejšie zníženie počtu MHD za mesiac, kedy užívali akútnu úľavovú liečbu, v porovnaní s
pacientmi liečenými placebom. Pacienti liečení galkanezumabom mali počnúc 1. mesiacom výraznejšie zlepšenie vo funkčnosti (podľa merania položky Role Function-Restrictive dotazníka
MSQ) v porovnaní s pacientmi liečenými placebom. Viac pacientov liečených galkanezumabom
dosiahlo klinicky významnú úroveň zlepšenia vo fungovaní (miera odpovede na základe položky Role
Function Restrictive dotazníka MSQ) v porovnaní s pacientmi liečenými placebom. Dávka 120 mg sa spájala so štatisticky významným znížením zdravotného postihnutia oproti placebu.
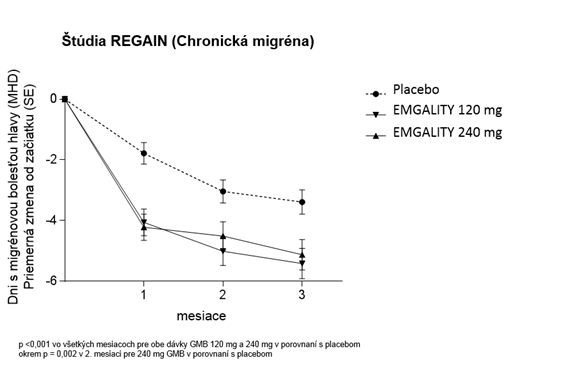
V porovnaní s pacientmi liečenými placebom mali pacienti liečení 120 mg alebo 240 mg galkanezumabu v prvom mesiaci a všetkých nasledujúcich mesiacoch až do 3. mesiaca významne výraznejšie priemerné zníženie počtu MHD za mesiac od začiatku liečby (pozri obrázok č. 2). Okrem toho, v 1. mesiaci pacienti liečení galkanezumabom (nasycovacia dávka 240 mg) preukázali významne menej MHD za týždeň v porovnaní s pacientmi liečenými placebom v 1. týždni a v každom nasledujúcom týždni.
O
brázok č. 2: Zníženie počtu dní s migrénou za mesiac v priebehu štúdie REGAIN

T
abuľka č. 3. Účinnosť a pacientmi hlásené výsledky
 REGAIN – Chronická migréna
REGAIN – Chronická migréna
Emgality
Placebo
Výsledná
účinnosť
a
MHD
120 mg 240 mg
N = 273 N = 274 N = 538
Začiatočná hodnota 19,36 19,17 19,55
Priemerná zmena -4,83 -4,62 -2,74
Rozdiel v liečbe -2,09 -1,88
CI95% (-2,92; -1,26) (-2,71; -1,05) P-hodnota < 0,001c < 0,001c
≥ 50% MHD respondériPercento, % 27,6 27,5 15,4
P-hodnota < 0,001c < 0,001c
≥ 75% MHD respondériPercento, % 7,0 8,8 4,5
P-hodnota 0,031d < 0,001c
100 % respondériPercento, % 0,7 1,3 0,5
P-hodnota > 0,05d > 0,05d
MHD s použitím liečby akútnej migrényZačiatočná hodnota 15,12 14,49 15,51
Priemerná zmena -4,74 -4,25 -2,23

Rozdiel v liečbe -2,51 -2,01
CI95% (-3,27; -1,76) (-2,77; -1,26) P-hodnota < 0,001d < 0,001 c
Pacientmi hlásené výsledkyb
MSQ položka Role Function-RestrictiveN 252 253 494
Začiatočná hodnota 39,29 38,93 38,37
Priemerná zmena 21,81 23,05 16,76
Rozdiel v liečbe 5,06 6,29
CI95% (2,12; 7,99) (3,03; 9,55) P-hodnota < 0,001d < 0,001c
Respondéri MSQ položky Role Function- Restrictivec
N 252 253 494
Percento, % 64,3 64,8 54,1
P-hodnota 0,003e 0,002e
MIDAS Celkové skóreN 254 258 504
Začiatočná hodnota 62,46 69,17 68,66
Priemerná zmena -20,27 -17,02 -11,53
Rozdiel v liečbe -8,74 -5,49
CI95% (-16,39; -1,08) (-13,10; 2,12) P-hodnota 0,025e > 0,05e
N = počet pacientov; CI95% = 95% interval spoľahlivosti.
aVýsledky účinnosti boli hodnotené v priebehu 1. - 3. mesiaca.
bVýsledky hlásené pacientmi boli hodnotené v 3. mesiaci. Respondenti položky Role function restrictive dotazníka
MSQ boli definovaní ako pacienti so zlepšením o ≥ 17,14 bodov u chronickej migrény v 3. mesiaci.
cŠtatisticky významné po úprave na viacnásobné porovnanie. dŠtatisticky nevýznamné po úprave na viacnásobné porovnanie. eNeupravené na viacnásobné porovnanie.
U pacientov, u ktorých zlyhala z dôvodu účinnosti jedna alebo viaceré profylaktické liečby, bol pozorovaný rozdiel v liečbe v znížení priemerného počtu MHD za mesiac medzi 120 mg galkanezumabu a placebom -3,54 dňa (p < 0,001) a medzi 240 mg galkanezumabu a placebom
- 1,37 dňa (p < 0,05). U pacientov, u ktorých zlyhali dve alebo viac profylaktických liečob, bol rozdiel
v liečbe - 4,48 dňa (p < 0,001) medzi 120 mg a placebom a -1,86 dňa (p < 0,01) medzi 240 mg a placebom.
U šesťdesiatich štyroch percent pacientov sa vyskytlo nadmerné užívanie liekov na akútnu bolesť
hlavy na začiatku liečby. U týchto pacientov bol pozorovaný rozdiel v liečbe na zníženie MHD medzi
120 mg galkanezumabu a placebom, a medzi 240 mg galkanezumabu a placebom, -2,53 dňa
(p < 0,001) a -2,26 dňa (p < 0,001) v uvedenom poradí.
Dlhodobá účinnosť
Účinnosť sa udržala až 1 rok v otvorenej štúdii, v ktorej bol pacientom s epizodickou alebo chronickou migrénou (s priemernou vstupnou hodnotou 10,6 MHD za mesiac) podávaný galkanezumab
120 mg/mesiac (s počiatočnou nasycovacou dávkou 240 mg v prvom mesiaci) alebo galkanezumab
240 mg/mesiac. 77,8 % pacientov dokončilo celé obdobie liečby. Celkové priemerné zníženie od začiatku liečby v počte MHD za mesiac spriemerované za celú liečebnú fázu bolo 5,6 dňa u skupiny s dávkou 120 mg a 6,5 dňa u skupiny s dávkou 240 mg. Viac ako 72 % pacientov, ktorí dokončili celú štúdiu, hlásilo 50 % zníženie MHD v 12. mesiaci. V združených údajoch zo štúdií EVOLVE-1 a EVOLVE-2 si viac ako 19 % pacientov liečených galkanezumabom udržalo ≥ 50 % odpoveď
od 1. mesiaca do 6. mesiaca oproti 8 % pacientom na placebe (p < 0,001).
Pediatrická populácia
Európska lieková agentúra udelila odklad z povinnosti predložiť výsledky štúdií s galkanezumabom
v jednej alebo vo viacerých podskupinách pediatrickej populácie v profylaxii migrénových bolestí hlavy (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Na základe populačnej analýzy farmakokinetiky (PK), bola po nasycovacej dávke 240 mg najvyššia
sérová koncentrácia (Cmax) galkanezumabu približne 30 μg/ml (27 % koeficient variácie (CV)) a čas
do Cmax bol 5 dní od podania dávky.
Mesačné dávky 120 mg alebo 240 mg dosiahli ustálený stav Cmax (Cmax, ss) s priemernou hodnotou
28 μg/ml (35 % CV) alebo 54 μg/ml (31 % CV), v uvedenom poradí. Cmax, ss galkanezumabu pri mesačných dávkach 120 mg sa dosiahne po nasycovacej dávke 240 mg.
Miesto podania injekcie (brucho, stehno, zadok a rameno) nemalo významný vplyv na absorpciu galkanezumabu.
Distribúcia
Na základe populačnej PK analýzy bol zdanlivý distribučný objem galkanezumabu 7,3 l.
Biotransformácia
Predpokladá sa, že galkanezumab ako humanizovaná IgG4 monoklonálna protilátka sa bude rozkladať
na malé peptidy a aminokyseliny prostredníctvom katabolických dráh rovnakým spôsobom ako endogénny IgG.
E
li
m
i
nácia
Na základe populačnej PK analýzy bol zdanlivý klírens galkanezumabu približne 0,008 l/hod. a polčas
galkanezumabu bol 27 dní.
Linearita/nelinearita
Expozícia galkanezumabu sa zvyšuje úmerne s dávkou.
Na základe populačnej PK analýzy, ktorá zahŕňala dávky v rozpätí 5 – 300 mg, rýchlosť absorpcie,
zdanlivý klírens a zdanlivý distribučný objem boli od dávky nezávislé.
Vek, pohlavie,hmotnosť,rasa,etnickápríslušnosť
Úprava dávky podľa veku (18 - 65 rokov), pohlavia, hmotnosti, rasy ani etnickej príslušnosti nie je
potrebná, pretože nebol žiadny klinicky významný účinok týchto faktorov na zdanlivý klírens ani na zdanlivý distribučný objem galkanezumabu.
Porucha funkcie obličiek a pečene
Neuskutočnili sa žiadne špecifické klinické farmakologické štúdie na vyhodnotenie účinkov poruchy
funkcie obličiek a pečene na PK galkanezumabu. Renálna eliminácia IgG monoklonálnej protilátky je
nízka. Podobne sa aj IgG monoklonálne protilátky eliminujú prostredníctvom intracelulárneho katabolizmu a neočakáva sa, že porucha funkcie pečene bude mať vplyv na klírens galkanezumabu.
Na základe populačnej PK analýzy nemala koncentrácia bilirubínu ani Cockcroft-Gaultov klírens
kreatinínu (rozsah: 24 - 308 ml/min) významný vplyv na zdanlivý klírens galkanezumabu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje nepreukázali žiadne špecifické riziko pre ľudí na základe štúdií toxicity
po opakovanom podávaní vykonaných na potkanoch a makakoch dlhochvostých a vyhodnotení farmakologickej bezpečnosti uskutočnenej na makakoch dlhochvostých s expozíciami približne 10 až
80-krát vyššími ako sú klinické expozície u pacientov užívajúcich dávku 240 mg.
Neuskutočnili sa žiadne predklinické štúdie na vyhodnotenie karcinogénneho ani mutagénneho potenciálu galkanezumabu. Neexistujú žiadne dôkazy, ktoré by naznačovali, že by chronická liečba galkanezumabom zvyšovala riziko karcinogenézy a ktoré by vyplývali z údajov farmakológie
a chronických toxikologických štúdií s galkanezumabom, ani z vyhodnotenia literatúry týkajúcej sa
CGRP.
Na potkanoch, ktorým bol podávaný galkanezumab (expozície približne 4 - 20-krát vyššie ako expozícia 240 mg dávke u ľudí) nebol pozorovaný žiaden vplyv na parametre fertility ako sú estrálny cyklus, analýza spermií alebo výkonnosť pri párení a reprodukcii. V štúdii fertility samčekov sa hmotnosť pravých semenníkov výrazne znížila pri expozíciách až 4-násobku ľudskej expozície pri 240 mg.
V 20. deň gestačného vývoja v štúdii toxicity embryo-fetálneho vývoja na potkanoch došlo k nárastu počtu plodov a vrhov s krátkymi rebrami a zníženiu počtu osifikovaných kaudálnych stavcov, pri expozícii približne 20-násobne vyššej ako expozícia 240 mg u ľudí. Tieto zistenia neboli zaznamenané pri materskej toxicite a boli považované za súvisiace s galkanezumabom, ale nie nežiaduce.
V 29. deň gestačného vývoja v štúdii toxicity embryo-fetálneho vývoja na králikoch sa objavili anomálie na lebke u jedného mužského plodu matky liečenej galkanezumabom, pri expozícii približne
33-násobne vyššej ako expozícia 240 mg u ľudí.
V juvenilnej toxikologickej štúdii, v ktorej bol potkanom podávaný galkanezumab dvakrát týždenne od 21. do 90. dňa po narodení, boli systémové účinky obmedzené na reverzibilné, minimálne,
nevýrazné zníženia celkového obsahu minerálov v kostiach a kostnej minerálnej hustoty pri expozíciách až 50-krát vyšších ako sú expozície u ľudí pri dávke 240 mg.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
L-histidíniumchlorid, monohydrát polysorbát 80
chlorid sodný
voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 ºC - 8 ºC).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Emgality sa môže uchovávať mimo chladničky po dobu najviac 7 dní, ak sa uchováva pri teplotách nižších ako 30 °C. Ak sa tieto podmienky prekročia, naplnené pero sa musí zlikvidovať.
6.5 Druh obalu a obsah balenia
1 ml roztoku v injekčnej striekačke z číreho skla typu I. Striekačka je zabudovaná v jednorazovom pere s jednou dávkou. Balenie obsahuje 1, 2 alebo 3 naplnené perá. Na trh nemusia byť uvedené všetky veľkosti balenia.
Ihla, ktorá je súčasťou balenia, je vhodná len na subkutánnu injekciu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Návod na používanie
Návod na používanie pera, ktorý je súčasťou písomnej informácie pre pacienta, sa musí starostlivo
dodržiavať. Musí sa podať celý obsah naplneného pera.
Naplnené pero sa má pred použitím vizuálne skontrolovať. Emgality sa nemá používať vtedy, ak je roztok zakalený, zmenil farbu, obsahuje častice alebo ak sa zdá, že niektorá časť pomôcky je poškodená.
Nepretrepávajte.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528BJ Utrecht, Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/18/1330/001
EU/1/18/1330/002
EU/1/18/1330/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 14. novembra 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
http://www.ema.europa.eu.

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUEmgality 120 mg injekčný roztok v naplnenej injekčnej striekačke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá naplnená injekčná striekačka obsahuje 120 mg galkanezumabu v 1 ml.
Galkanezumab je rekombinantná humanizovaná monoklonálna protilátka produkovaná ovariálnymi bunkami čínskeho škrečka (Chinese Hamster Ovary, CHO).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInjekčný roztok (injekcia).
Roztok je číry a bezfarebný až žltkastý.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieEmgality je indikovaná na profylaxiu migrény u dospelých pacientov, ktorí majú migrénu minimálne
4 dni mesačne.
4.2 Dávkovanie a spôsob podávaniaLiečbu majú iniciovať lekári, ktorí majú skúsenosti s diagnostikovaním a liečbou migrény.
DávkovanieOdporúčaná dávka je 120 mg galkanezumabu podávaná subkutánne jedenkrát mesačne, s počiatočnou
nasycovacou dávkou 240 mg.
Pacienti majú byť poučení, aby si vynechanú dávku injekčne podali čo najskôr a potom pokračovali
v podávaní dávok každý mesiac.
Prínos liečby sa má vyhodnotiť do 3 mesiacov od začiatku liečby. Akékoľvek ďalšie rozhodnutie
o pokračovaní v liečbe sa má prijať u každého pacienta individuálne. Potom sa odporúča pravidelné
posudzovanie potreby pokračovať v liečbe.
Starší pacienti (> 65 rokov)Galkanezumab nebol skúmaný u starších pacientov. Úprava dávky sa nevyžaduje, pretože vek nemá vplyv na farmakokinetiku galkanezumabu.
P
orucha funkcie obličiek/porucha funkcie pečene
Úprava dávky sa nevyžaduje u pacientov s miernou až stredne závažnou poruchou funkcie obličiek,
ani s poruchou funkcie pečene (pozri časť 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť galkanezumabu u detí vo veku 6 až 18 rokov nebola doteraz stanovená. K dispozícii nie sú žiadne údaje.
Použitie galkanezumabu na prevenciu migrény u detí vo veku do 6 rokov nie je relevantné. Spôsob podávania
Subkutánne použitie.
Pacient si môže sám injekčne podať galkanezumab podľa Návodu na použitie. Galkanezumab sa má podať subkutánne do brucha, stehna, zadnej časti ramena alebo do gluteálnej oblasti. Po zaškolení si pacienti môžu sami injekčne podávať galkanezumab, v prípade, ak lekár rozhodne, že je to vhodné. Podrobné pokyny na podávanie sú uvedené v Písomnej informácii pre používateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby bolo možné zlepšiť sledovateľnosť biologických liekov, má sa zrozumiteľne zaznamenať názov
a číslo šarže podaného lieku.
Kardiovaskulárne riziko
Pacienti s určitými závažnými kardiovaskulárnymi ochoreniami boli z klinických štúdií vylúčení
(pozri časť 5.1). U týchto pacientov nie sú k dispozícii žiadne údaje o bezpečnosti.
Závažná hypersenzitivita
V prípade výskytu závažnej reakcie z precitlivenosti sa má okamžite ukončiť podávanie
galkanezumabu a má sa začať s vhodnou liečbou.
Pomocné látky
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v 120 mg dávke, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Neočakávajú sa žiadne farmakokinetické liekové interakcie
súvisiace s vlastnosťami galkanezumabu.
4.6 Fertilita, gravidita a laktácia
Gravidita
Existujú obmedzené údaje o používaní galkanezumabu u gravidných žien. Štúdie na zvieratách
nenaznačujú priame ani nepriame škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Je
známe, že ľudský imunoglobulín (IgG) prestupuje placentárnou bariérou. Ako preventívne opatrenie je vhodnejšie sa vyhnúť používaniu galkanezumabu počas gravidity.
Dojčenie
Nie je známe, či sa galkanezumab vylučuje do ľudského materského mlieka. Je známe, že ľudský IgG
sa v priebehu prvých dní po pôrode vylučuje do materského mlieka a skoro potom jeho koncentrácie klesajú; preto v priebehu tohto krátkeho obdobia nie je možné toto riziko u dojčených detí vylúčiť.
Následne, iba ak je to klinicky potrebné, môže sa zvážiť použitie galcanezumabu počas dojčenia.
Fertilita
Vplyv galkanezumabu na ľudskú fertilitu nebol doteraz stanovený. Štúdie fertility na zvieratách
neuvádzajú škodlivé účinky na plodnosť samcov a samíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Galkanezumab môže mať mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Po podaní
galkanezumabu sa môžu objaviť závraty (pozri časť 4.8).
4.8 Nežiaduce účinky
Zhrnutie bezpečnostného profilu
Vyše 2500 pacientov bolo v klinických štúdiách zameraných na profylaxiu migrény vystavených
pôsobeniu galkanezumabu. Viac ako 1400 pacientov bolo vystavených pôsobeniu galkanezumabu v priebehu dvojito zaslepenej fázy liečby v placebom kontrolovaných štúdiách fázy 3. 279 pacientov
bolo vystavených jeho pôsobeniu po dobu 12 mesiacov.
Hlásené nežiaduce účinky lieku pre 120 mg a 240 mg boli bolesť v mieste podania injekcie (10,1 % /
11,6 %), reakcie v mieste podania injekcie (9,9 % / 14,5 %), závrat (0,7 % / 1,2 %), zápcha (1,0 % /
1,5 %), pruritus (0,7 % / 1,2 %) a urtikária (0,3 % / 0,1 %). Intenzita väčšiny reakcií bola mierna alebo stredne závažná. Menej ako 2,5 % pacientov v týchto štúdiách prerušilo liečbu v dôsledku nežiaducich účinkov.
Zoznam nežiaducich reakcií v tabuľke
Tabuľka č. 1. Zoznam nežiaducich reakcií v klinických štúdiách
Odhad frekvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 to < 1/10), menej časté (≥ 1/1000 to < 1/100).
T
rieda orgánových
systémov
V
eľmi časté Časté Menej časté
P
oruchy ucha a labyrintu závrat
P
oruchy gastrointestinálneho traktu Poruchy kože
a podkožného tkaniva
C
elkové poruchy a reakcie v mieste podania
bolesť v mieste podania injekcie, reakcie v mieste podania injekciea
zápcha
pruritus urtikária
a Najčastejšie hlásené (≥ 1 %) boli: reakcia v mieste podania injekcie, erytém v mieste podania, pruritus v mieste podania, podliatina v mieste podania, opuch v mieste podania.
O
pis vybraných nežiaducich reakcií
B
olesť alebo reakcie v mieste podania injekcie
Väčšina prípadov súvisiacich s miestom podania injekcie bola mierna až stredne závažná a menej ako
0,5 % pacientov vystavených pôsobeniu galkanezumabu v priebehu štúdií fázy 3 prerušilo liečbu
v dôsledku reakcie v mieste podania injekcie. Väčšina reakcií v mieste podania bolo hlásených do 1 dňa a vymizli v priemere počas 5 dní. U 86 % pacientov, ktorí hlásili bolesť v mieste podania injekcie, sa táto udalosť vyskytla do 1 hodiny po podaní injekcie a vymizla v priemere za 1 deň. U jedného percenta pacientov vystavených pôsobeniu galkanezumabu v priebehu štúdií fázy 3 sa objavila silná bolesť v mieste podania injekcie.
Urtikária
Zatiaľ čo urtikária je menej častá, v klinických štúdiách s galkanezumabom boli hlásené závažné prípady urtikárie.
Imunogenita
V klinických štúdiách v priebehu dvojito zaslepenej fázy bola incidencia výskytu protilátok proti lieku u 4,8 % pacientov užívajúcich galkanezumab jedenkrát mesačne (všetci okrem jedného mali
neutralizačnú aktivitu in vitro). Počas 12-mesačnej liečby sa až u 12,5 % pacientov liečených
galkanezumabom vytvorili protilátky proti lieku, väčšina ktorých mala nízky titer a pozitívny test na
neutralizačnú aktivitu in vitro. Prítomnosť protilátok proti lieku však nemala vplyv na
farmakokinetiku, účinnosť ani bezpečnosť galkanezumabu.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.*
4.9 Predávkovanie
Dávky až do 600 mg boli podávané subkutánne ľuďom bez toxicity obmedzujúcej dávku. V prípade predávkovania sa odporúča u pacienta sledovať akékoľvek prejavy alebo príznaky nežiaducich reakcií a aby sa okamžite začalo s vhodnou symptomatickou liečbou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: analgetiká, iné antimigrenotiká, ATC kód: N02CX08
Mechanizmus účinku
Galkanezumab je humanizovaná monoklonálna protilátka typu IgG4, ktorá sa viaže na
kalcitonínovému génu príbuzný peptid (calcitonin gene-related peptide, CGRP), čím zabraňuje jeho
biologickej aktivite. Zvýšené koncentrácie CGRP v krvi súvisia so záchvatmi migrény. Galkanezumab sa s vysokou afinitou (KD = 31 pM) a vysokou špecifickosťou (> 10 000-násobne vyššou oproti príbuzným peptidom adrenomedulínu, amylínu, kalcitonínu a intermedínu) viaže na CGRP.
Klinická účinnosť abezpečnosť
Účinnosť a bezpečnosť galkanezumabu bola skúmaná v 3 randomizovaných, placebom
kontrolovaných, dvojito zaslepených štúdiách fázy 3 u dospelých pacientov (N = 2886). Do 2 štúdií epizodickej migrény (EVOLVE-1 a EVOLVE-2) boli zaradení pacienti, ktorí spĺňali kritériá Medzinárodnej klasifikácie bolestí hlavy (International Classification of Headache Disorders - ICHD) pre diagnózu migrény s aurou alebo bez aury a s 4-14 dňami migrény za mesiac. Do štúdie chronickej migrény (REGAIN) boli zaradení pacienti, ktorí spĺňali kritériá ICHD pre chronickú migrénu
s ≥ 15 dňami s bolesťou hlavy za mesiac, z ktorých najmenej 8 malo znaky migrény. Pacienti
s nedávnymi akútnymi kardiovaskulárnymi príhodami (vrátane infarktu myokardu, nestabilnej angíny pectoris, bajpasom koronárnych tepien, mozgovej mŕtvice a hlbokej žilovej trombózy) a/alebo tí,
u ktorých sa predpokladalo závažné kardiovaskulárne riziko, boli z klinických skúšaní s galkanezumabom vylúčení. Pacienti vo veku > 65 rokov boli tiež vylúčení.
Pacientom bolo podávané placebo, galkanezumab 120 mg/mesiac (s počiatočnou nasycovacou dávkou
240 mg v prvom mesiaci) alebo galkanezumab 240 mg/mesiac a mali povolené užívať lieky na liečbu
akútnej migrény. Vo všetkých 3 štúdiách boli pacientmi prevažne ženy (> 83 %) s priemerným vekom
41 rokov a s priemernou anamnézou migrény v dĺžke 20 až 21 rokov. U približne jednej tretiny pacientov vo všetkých štúdiách sa vyskytlo z dôvodov účinnosti najmenej 1 predchádzajúce zlyhanie
profylaktickej liečby migrény a približne u 16 % pacientov vo všetkých štúdiách sa vyskytli
z dôvodov účinnosti najmenej 2 predchádzajúce zlyhania profylaktickej liečby.
Vo všetkých 3 štúdiách bola primárnym ukazovateľom účinnosti celková priemerná zmena počtu dní s migrénou (Migraine Headache Day, MHD) za mesiac od začiatku liečby. Mierou odpovede je priemerný percentuálny podiel pacientov spĺňajúcich definovanú hranicu zníženého počtu MHD za mesiac (≥ 50 %, ≥ 75 % a 100 %) v priebehu dvojito zaslepeného obdobia liečby. Vplyv migrény na funkčnosť sa hodnotil pomocou položky Role Function-Restrictive v Dotazníku kvality života
s migrénou (Migraine-Specific Quality of Life Questionnaire - MSQ) verzia 2.1 a Dotazníka hodnotiaceho zdravotné postihnutie pri migréne (Migraine Disability Assessment - MIDAS). Dotazník
MSQ meria vplyv migrény na prácu a každodenné aktivity, vzťahy s rodinou a priateľmi, voľný čas,
produktivitu, sústredenie, vitalitu a únavu. Skóre sa pohybuje od 0 do 100, pričom vyššie skóre označuje menšie zdravotné postihnutie, čiže u pacientov sa vyskytuje menej obmedzení pri
vykonávaní každodenných aktivít. V dotazníku MIDAS vyššie skóre označuje vyššie zdravotné
postihnutie. Skóre dotazníka MIDAS na začiatku liečby vyjadrovalo závažné zdravotné postihnutie pacientov súvisiace s migrénou v štúdiách EVOLVE-1 a EVOLVE-2 (priemer 33,1) a veľmi závažné zdravotné postihnutie u pacientov (priemer 67,2) v štúdii REGAIN.'
Epizodická migréna
Štúdie EVOLVE-1 a EVOLVE-2 mali 6-mesačnú, dvojito zaslepenú, placebom kontrolovanú fázu liečby. Miera ukončenia účasti v dvojito zaslepenej fáze liečby u pacientov, ktorým bol podávaný galkanezumab, sa pohybovala od 82,8 % do 87,7 %.
Obe galkanezumabom 120 mg a 240 mg liečené skupiny preukázali štatisticky významné a klinicky významné zlepšenie od začiatku liečby v porovnaní s placebom v priemernej zmene v MHD (pozri tabuľku č. 2). U pacientov liečených galkanezumabom sa vyskytla vyššia miera odpovede
a výraznejšie zníženie počtu MHD za mesiac, kedy užívali akútnu úľavovú liečbu, v porovnaní s pacientmi liečenými placebom. Pacienti liečení galkanezumabom mali počnúc 1. mesiacom
výraznejšie zlepšenie funkčnosti (podľa merania položky Role Function-Restrictive dotazníka MSQ)
v porovnaní s pacientmi liečenými placebom. Viac pacientov liečených galkanezumabom dosiahlo
klinicky významnú úroveň zlepšenia funkčnosti (miera odpovede v položke Role Function Restrictive dotazníka MSQ) v porovnaní s pacientmi liečenými placebom. Galkanezumab sa spájal so štatisticky významným znížením zdravotného postihnutia oproti placebu.
V porovnaní s pacientmi liečenými placebom sa u pacientov liečených galkanezumabom 120 mg alebo 240 mg v 1. mesiaci a všetkých nasledujúcich mesiacoch až do 6. mesiaca vyskytlo významne výraznejšie priemerné zníženie počtu MHD za mesiac od začiatku liečby (pozri obrázok č. 1). Okrem toho, v 1. mesiaci pacienti liečení galkanezumabom (nasycujúca dávka 240 mg) preukázali významne menej MHD za týždeň v 1. týždni a každom nasledujúcom týždni v porovnaní s pacientmi liečenými placebom.
O
brázok č. 1: Zníženie počtu dní s migrénou za mesiac, v priebehu štúdií EVOLVE-1
a EVOLVE-2
 T
abuľka č. 2: Účinnosť a pacientmi hlásené výsledky
E
VOLVE 1 – Epizodická migréna EVOLVE 2 - Epizodická migréna
T
abuľka č. 2: Účinnosť a pacientmi hlásené výsledky
E
VOLVE 1 – Epizodická migréna EVOLVE 2 - Epizodická migréna
Emgality
Placebo
Emgality
Placebo
Výsledná
účinnosť
a
MHD
120 mg 240 mg 120 mg 240 mg
N = 210 N = 208 N = 425 N = 226 N = 220 N = 450

Začiatočná hodnota 9,21 9.14 9,08 9,07 9,06 9,19
Priemerná zmena -4,73 -4,57 -2,81 -4,29 -4,18 -2,28
Rozdiel v liečbe -1,92 -1,76 -2,02 -1,90
CI95% (-2,48; -1,37) (-2,31; -1,20) (-2,55; -1,48) (-2,44; -1,36) P-hodnota <0,001d <0,001d <0,001d <0,001d
≥ 50% MHD respondériPercento, % 62,3 60,9 38,6 59,3 56,5 36,0
P-hodnota <0,001d <0,001d <0,001d <0,001d
≥ 75% MHD respondériPercento, % 38,8 38,5 19,3 33,5 34,3 17,8
P-hodnota <0,001d <0,001d <0,001d <0,001d
100 % respondériPercento, % 15,6 14,6 6,2 11,5 13,8 5,7
P-hodnota <0,001d <0,001d <0,001d <0,001d
MHD s použitím liečby naakútnu migrénuZačiatočná hodnota 7,42 7,34 7,38 7,47 7,47 7,62
Priemerná zmena -3,96 -3,76 -2,15 -3,67 -3,63 -1,85
Rozdiel v liečbe -1,81 -1,61 -1,82 -1,78
CI95% (-2,28; -1,33) (-2,09; -1,14) (-2,29; -1,36) (-2,25; -1,31) P-hodnota <0,001d <0,001d <0,001d <0,001d
Pacientmi hlásené výsledkyMSQ položka RoleFunction-Restrictive b
N 189 184 377 213 210 396
Začiatočná hodnota 51,39 48,76 52,92 52,47 51,71 51,35
Priemerná zmena 32,43 32,09 24,69 28,47 27,04 19,65
Rozdiel v liečbe 7,74 7,40 8,82 7,39
CI95% (5,20; 10,28) (4,83; 9,97) (6,33; 11,31) (4,88; 9,90) P-hodnota <0,001d <0,001d <0,001d <0,001d
E
VOLVE 1 – Epizodická migréna EVOLVE 2 - Epizodická migréna
Emgality
Placebo
Emgality
Placebo
Respondéri MSQ položky
Role Function-Restrictivec
120 mg 240 mg 120 mg 240 mg
N = 210 N = 208 N = 425 N = 226 N = 220 N = 450
N 189 184 377 213 210 396
Percento, % 63,5 69,6 47,2 58,2 60,0 43,4
P-hodnota <0,001f <0,001f <0,001f <0,001f
MIDAS Celkové skóre e
N 177 170 345 202 194 374
Začiatočná hodnota 32,93 36,09 31,84 30,87 32,75 34,25
Priemerná zmena -21,16 -20,06 -14,87 -21,17 -20,24 -12,02
Rozdiel v liečbe -6,29 -5,19 -9,15 -8,22
CI95% (-9,45; -3,13) (-8,39; -1,98) (-12,61; -5,69) (-11,71; -4,72) P-hodnota <0,001f 0,002f <0,001f <0,001f
N = počet pacientov; CI95% = 95 % interval spoľahlivosti. aVýsledky účinnosti boli hodnotené v priebehu 1. - 6. mesiaca. bHodnotené v priebehu 4. - 6. mesiaca.
cDefinovaní ako pacienti so zlepšením o ≥ 25 bodov u epizodickej migrény v priemere 4. - 6. mesiaca.
dŠtatisticky významné po úprave na viacnásobné porovnanie.
eHodnotené v 6. mesiaci.
fNeupravené na viacnásobné porovnanie.
V súhrnných údajoch zo štúdií EVOLVE-1 a EVOLVE-2, u pacientov, u ktorých z dôvodov účinnosti
zlyhali jedna alebo viaceré profylaktické liečby, bol pozorovaný rozdiel v liečbe zameranej na zníženie počtu priemerných MHD za mesiac medzi 120 mg galkanezumabu a placebom -2,69 dňa
(p < 0,001) a medzi 240 mg galkanezumabu a placebom -2,78 dňa (p < 0,001). U pacientov, u ktorých zlyhali dve alebo viaceré profylaktické liečby, bol rozdiel v liečbe -2,64 dňa (p < 0,001) medzi 120 mg a placebom a -3,04 dňa (p < 0,001) medzi 240 mg a placebom.
Chronická migrénaŠtúdia REGAIN mala 3-mesačnú, dvojito zaslepenú, placebom kontrolovanú fázu liečby, po ktorej nasledovala 9-mesačná otvorená pokračovacia fáza. Približne 15 % pacientov pokračovalo v súbežnej liečbe topiramátom alebo propranololom tak, ako to umožňoval protokol pre profylaxiu migrény. Miera dokončenia fázy dvojito zaslepenej liečby u pacientov, ktorým bol podávaný galkanezumab, bola 95,3 %.
Obidve liečebné skupiny s galkanezumabom v dávke 120 mg aj 240 mg preukázali štatisticky signifikantné a klinicky významné zlepšenie od začiatku liečby v porovnaní s placebom v priemernej zmene v MHD (pozri tabuľku č. 3). Pacienti liečení galkanezumabom mali vyššiu mieru odpovede
a výraznejšie zníženie počtu MHD za mesiac, kedy užívali akútnu úľavovú liečbu, v porovnaní s
pacientmi liečenými placebom. Pacienti liečení galkanezumabom mali počnúc 1. mesiacom výraznejšie zlepšenie vo funkčnosti (podľa merania položky Role Function-Restrictive dotazníka
MSQ) v porovnaní s pacientmi liečenými placebom. Viac pacientov liečených galkanezumabom
dosiahlo klinicky významnú úroveň zlepšenia vo fungovaní (miera odpovede na základe položky Role
Function Restrictive dotazníka MSQ) v porovnaní s pacientmi liečenými placebom. Dávka 120 mg sa spájala so štatisticky významným znížením zdravotného postihnutia oproti placebu.
V porovnaní s pacientmi liečenými placebom mali pacienti liečení 120 mg alebo 240 mg galkanezumabu v prvom mesiaci a všetkých nasledujúcich mesiacoch až do 3. mesiaca významne výraznejšie priemerné zníženie počtu MHD za mesiac od začiatku liečby (pozri obrázok č. 2). Okrem toho, v 1. mesiaci pacienti liečení galkanezumabom (nasycovacia dávka 240 mg) preukázali významne menej MHD za týždeň v porovnaní s pacientmi liečenými placebom v 1. týždni a v každom nasledujúcom týždni.
O
brázok č. 2: Zníženie počtu dní s migrénou za mesiac v priebehu štúdie REGAIN
T
abuľka č. 3. Účinnosť a pacientmi hlásené výsledky
REGAIN – Chronická migréna
Emgality
Placebo
Výsledná
účinnosť
a
MHD
120 mg 240 mg
N = 273 N = 274 N = 538
Začiatočná hodnota 19,36 19,17 19,55
Priemerná zmena -4,83 -4,62 -2,74
Rozdiel v liečbe -2,09 -1,88
CI95% (-2,92; -1,26) (-2,71; -1,05) P-hodnota < 0,001c < 0,001c
≥ 50% MHD respondériPercento, % 27,6 27,5 15,4
P-hodnota < 0,001c < 0,001c
≥ 75% MHD respondériPercento, % 7,0 8,8 4,5
P-hodnota 0,031d < 0,001c
100 % respondériPercento, % 0,7 1,3 0,5
P-hodnota > 0,05d > 0,05d
MHD s použitím liečby akútnej migrényZačiatočná hodnota 15,12 14,49 15,51
Priemerná zmena -4,74 -4,25 -2,23

Rozdiel v liečbe -2,51 -2,01
CI95% (-3,27; -1,76) (-2,77; -1,26) P-hodnota < 0,001d < 0,001 c
Pacientmi hlásené výsledkyb
MSQ položka Role Function-RestrictiveN 252 253 494
Začiatočná hodnota 39,29 38,93 38,37
Priemerná zmena 21,81 23,05 16,76
Rozdiel v liečbe 5,06 6,29
CI95% (2,12; 7,99) (3,03; 9,55) P-hodnota < 0,001d < 0,001c
Respondéri MSQ položky Role Function- Restrictivec
N 252 253 494
Percento, % 64,3 64,8 54,1
P-hodnota 0,003e 0,002e
MIDAS Celkové skóreN 254 258 504
Začiatočná hodnota 62,46 69,17 68,66
Priemerná zmena -20,27 -17,02 -11,53
Rozdiel v liečbe -8,74 -5,49
CI95% (-16,39; -1,08) (-13,10; 2,12) P-hodnota 0,025e > 0,05e
N = počet pacientov; CI95% = 95% interval spoľahlivosti.
aVýsledky účinnosti boli hodnotené v priebehu 1. - 3. mesiaca.
bVýsledky hlásené pacientmi boli hodnotené v 3. mesiaci. Respondenti položky Role function restrictive dotazníka
MSQ boli definovaní ako pacienti so zlepšením o ≥ 17,14 bodov u chronickej migrény v 3. mesiaci.
cŠtatisticky významné po úprave na viacnásobné porovnanie. dŠtatisticky nevýznamné po úprave na viacnásobné porovnanie. eNeupravené na viacnásobné porovnanie.
U pacientov, u ktorých zlyhala z dôvodu účinnosti jedna alebo viaceré profylaktické liečby, bol pozorovaný rozdiel v liečbe v znížení priemerného počtu MHD za mesiac medzi 120 mg galkanezumabu a placebom -3,54 dňa (p < 0,001) a medzi 240 mg galkanezumabu a placebom
- 1,37 dňa (p < 0,05). U pacientov, u ktorých zlyhali dve alebo viac profylaktických liečob, bol rozdiel
v liečbe - 4,48 dňa (p < 0,001) medzi 120 mg a placebom a -1,86 dňa (p < 0,01) medzi 240 mg a placebom.
U šesťdesiatich štyroch percent pacientov sa vyskytlo nadmerné užívanie liekov na akútnu bolesť
hlavy na začiatku liečby. U týchto pacientov bol pozorovaný rozdiel v liečbe na zníženie MHD medzi
120 mg galkanezumabu a placebom, a medzi 240 mg galkanezumabu a placebom, -2,53 dňa
(p < 0,001) a -2,26 dňa (p < 0,001) v uvedenom poradí.
Dlhodobá účinnosť
Účinnosť sa udržala až 1 rok v otvorenej štúdii, v ktorej bol pacientom s epizodickou alebo chronickou migrénou (s priemernou vstupnou hodnotou 10,6 MHD za mesiac) podávaný galkanezumab
120 mg/mesiac (s počiatočnou nasycovacou dávkou 240 mg v prvom mesiaci) alebo galkanezumab
240 mg/mesiac. 77,8 % pacientov dokončilo celé obdobie liečby. Celkové priemerné zníženie od začiatku liečby v počte MHD za mesiac spriemerované za celú liečebnú fázu bolo 5,6 dňa u skupiny s dávkou 120 mg a 6,5 dňa u skupiny s dávkou 240 mg. Viac ako 72 % pacientov, ktorí dokončili celú štúdiu, hlásilo 50 % zníženie MHD v 12. mesiaci. V združených údajoch zo štúdií EVOLVE-1 a EVOLVE-2 si viac ako 19 % pacientov liečených galkanezumabom udržalo ≥ 50 % odpoveď
od 1. mesiaca do 6. mesiaca oproti 8 % pacientom na placebe (p < 0,001).
Pediatrická populácia
Európska lieková agentúra udelila odklad z povinnosti predložiť výsledky štúdií s galkanezumabom
v jednej alebo vo viacerých podskupinách pediatrickej populácie v profylaxii migrénových bolestí hlavy (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Na základe populačnej analýzy farmakokinetiky (PK), bola po nasycovacej dávke 240 mg najvyššia
sérová koncentrácia (Cmax) galkanezumabu približne 30 μg/ml (27 % koeficient variácie (CV)) a čas
do Cmax bol 5 dní od podania dávky.
Mesačné dávky 120 mg alebo 240 mg dosiahli ustálený stav Cmax (Cmax, ss) s priemernou hodnotou
28 μg/ml (35 % CV) alebo 54 μg/ml (31 % CV), v uvedenom poradí. Cmax, ss galkanezumabu pri mesačných dávkach 120 mg sa dosiahne po nasycovacej dávke 240 mg.
Miesto podania injekcie (brucho, stehno, zadok a rameno) nemalo významný vplyv na absorpciu galkanezumabu.
Distribúcia
Na základe populačnej PK analýzy bol zdanlivý distribučný objem galkanezumabu 7,3 l.
Biotransformácia
Predpokladá sa, že galkanezumab ako humanizovaná IgG4 monoklonálna protilátka sa bude rozkladať
na malé peptidy a aminokyseliny prostredníctvom katabolických dráh rovnakým spôsobom ako endogénny IgG.
E
li
m
i
nácia
Na základe populačnej PK analýzy bol zdanlivý klírens galkanezumabu približne 0,008 l/hod. a polčas
galkanezumabu bol 27 dní.
Linearita/nelinearita
Expozícia galkanezumabu sa zvyšuje úmerne s dávkou.
Na základe populačnej PK analýzy, ktorá zahŕňala dávky v rozpätí 5 – 300 mg, rýchlosť absorpcie,
zdanlivý klírens a zdanlivý distribučný objem boli od dávky nezávislé.
Vek, pohlavie,hmotnosť,rasa,etnickápríslušnosť
Úprava dávky podľa veku (18 - 65 rokov), pohlavia, hmotnosti, rasy ani etnickej príslušnosti nie je
potrebná, pretože nebol žiadny klinicky významný účinok týchto faktorov na zdanlivý klírens ani na zdanlivý distribučný objem galkanezumabu.
Porucha funkcie obličiek a pečene
Neuskutočnili sa žiadne špecifické klinické farmakologické štúdie na vyhodnotenie účinkov poruchy
funkcie obličiek a pečene na PK galkanezumabu. Renálna eliminácia IgG monoklonálnej protilátky je
nízka. Podobne sa aj IgG monoklonálne protilátky eliminujú prostredníctvom intracelulárneho katabolizmu a neočakáva sa, že porucha funkcie pečene bude mať vplyv na klírens galkanezumabu.
Na základe populačnej PK analýzy nemala koncentrácia bilirubínu ani Cockcroft-Gaultov klírens
kreatinínu (rozsah: 24 - 308 ml/min) významný vplyv na zdanlivý klírens galkanezumabu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje nepreukázali žiadne špecifické riziko pre ľudí na základe štúdií toxicity
po opakovanom podávaní vykonaných na potkanoch a makakoch dlhochvostých a vyhodnotení farmakologickej bezpečnosti uskutočnenej na makakoch dlhochvostých s expozíciami približne 10 až
80-krát vyššími ako sú klinické expozície u pacientov užívajúcich dávku 240 mg.
Neuskutočnili sa žiadne predklinické štúdie na vyhodnotenie karcinogénneho ani mutagénneho potenciálu galkanezumabu. Neexistujú žiadne dôkazy, ktoré by naznačovali, že by chronická liečba galkanezumabom zvyšovala riziko karcinogenézy a ktoré by vyplývali z údajov farmakológie
a chronických toxikologických štúdií s galkanezumabom, ani z vyhodnotenia literatúry týkajúcej sa
CGRP.
Na potkanoch, ktorým bol podávaný galkanezumab (expozície približne 4 - 20-krát vyššie ako expozícia 240 mg dávke u ľudí) nebol pozorovaný žiaden vplyv na parametre fertility ako sú estrálny cyklus, analýza spermií alebo výkonnosť pri párení a reprodukcii. V štúdii fertility samčekov sa hmotnosť pravých semenníkov výrazne znížila pri expozíciách až 4-násobku ľudskej expozície pri 240 mg.
V 20. deň gestačného vývoja v štúdii toxicity embryo-fetálneho vývoja na potkanoch došlo k nárastu počtu plodov a vrhov s krátkymi rebrami a zníženiu počtu osifikovaných kaudálnych stavcov, pri expozícii približne 20-násobne vyššej ako expozícia 240 mg u ľudí. Tieto zistenia neboli zaznamenané pri materskej toxicite a boli považované za súvisiace s galkanezumabom, ale nie nežiaduce.
V 29. deň gestačného vývoja v štúdii toxicity embryo-fetálneho vývoja na králikoch sa objavili anomálie na lebke u jedného mužského plodu matky liečenej galkanezumabom, pri expozícii približne
33-násobne vyššej ako expozícia 240 mg u ľudí.
V juvenilnej toxikologickej štúdii, v ktorej bol potkanom podávaný galkanezumab dvakrát týždenne od 21. do 90. dňa po narodení, boli systémové účinky obmedzené na reverzibilné, minimálne,
nevýrazné zníženia celkového obsahu minerálov v kostiach a kostnej minerálnej hustoty pri expozíciách až 50-krát vyšších ako sú expozície u ľudí pri dávke 240 mg.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
L-histidín
L-histidíniumchlorid, monohydrát polysorbát 80
chlorid sodný
voda na injekciu
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 ºC - 8 ºC).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Emgality sa môže uchovávať mimo chladničky po dobu najviac 7 dní, keď sa uchováva pri teplotách nižších ako 30°C. Ak sa tieto podmienky prekročia, naplnená injekčná striekačka sa musí zlikvidovať.
6.5 Druh obalu a obsah balenia
1 ml roztoku v injekčnej striekačke z číreho skla typu I s jednou dávkou. Balenie obsahuje 1, 2 alebo 3
naplnené striekačky. Na trh nemusia byť uvedené všetky veľkosti balenia.
Ihla, ktorá je súčasťou balenia, je vhodná len na subkutánnu injekciu.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Návod na používanie
Návod na používanie naplnenej injekčnej striekačky, ktorý je súčasťou písomnej informácie pre
pacienta, sa musí starostlivo dodržiavať. Musí sa podať celý obsah naplnenej injekčnej striekačky.
Naplnená injekčná striekačka sa má pred použitím vizuálne skontrolovať. Emgality sa nemá používať vtedy, ak je roztok zakalený, zmenil farbu, obsahuje častice alebo ak sa zdá, že niektorá časť pomôcky je poškodená.
Nepretrepávajte.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528BJ Utrecht, Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/18/1330/003
EU/1/18/1330/004
EU/1/18/1330/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 14. novembra 2018
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej liekovej agentúry
http://www.ema.europa.eu.