
b>n alebo ml/min/1,73 m
2
)*
Ú
vodná dávka
U
držiavacia dávka
Schéma podávania udržiavacej dávky
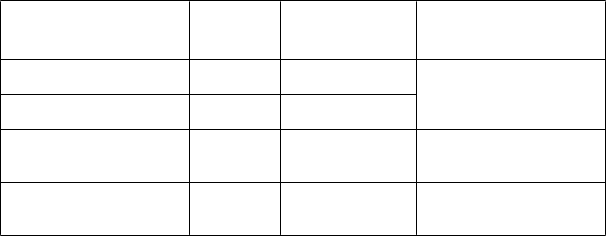
400 mg dvakrát
denne Začnite podávať udržiavaciu
dávku po 12 hodinách
30 až < 50 600 mg 250 mg dvakrát denne
15 až < 30 600 mg 150 mg dvakrát denne
< 15 600 mg 60 mg dvakrát denne
od úvodnej dávky
Začnite podávať udržiavaciu
dávku po 24 hodinách od úvodnej dávky
Začnite podávať udržiavaciu
dávku po 48 hodinách od úvodnej dávky
*Jednotky CLcr alebo CLCRRT v ml/min pre dospievajúcich vo veku 13 rokov až menej ako 18 rokov, alebo v ml/min/1,73 m2 pre deti vo veku 6 rokov až menej ako 13 rokov.
Deti a dospievajúci (vo veku 6 rokov až menej ako 18 rokov s telesnou hmotnosťou nižšou ako 50 kg)
a dojčatá a deti (vo veku 6 mesiacov až menej ako 6 rokov) s klírensom kreatinínu (CLcr) alebo
s klírensom pri kontinuálnej liečbe nahrádzajúcej funkciu obličky (CLCRRT) < 80 ml/min majú dostať úvodnú dávku, po ktorej nasleduje podávanie vhodnej udržiavacej dávky dvakrát denne, ako je uvedené v tabuľkách 3, 4 a 5.
T
abuľka 3: Úvodná dávka a schéma podávania udržiavacej dávky pre deti a dospievajúcich (vo veku 6 rokov až menej ako 18 rokov, s telesnou hmotnosťou nižšou ako 50 kg) s poruchou funkcie obličiek
CL
cr alebo CL
CRRT
(
m
l
/
mi
n alebo ml/min/1,73 m
2
)*
Ú
vodná dávka
U
držiavacia dávka
Schéma podávania udržiavacej dávky
50 až < 80 12 mg/kg 8 mg/kg dvakrát denne
5 mg/kg dvakrát
Začnite podávať udržiavaciu
dávku dvakrát denne
po 12 hodinách od úvodnej
30 až < 50 12 mg/kg
denne
dávky
Začnite podávať udržiavaciu
15 až < 30 12 mg/kg 3 mg/kg dvakrát denne
< 15 12 mg/kg 1,2 mg/kg dvakrát denne
dávku dvakrát denne
po 24 hodinách od úvodnej dávky
Začnite podávať udržiavaciu
dávku dvakrát denne
po 48 hodinách od úvodnej dávky
*Jednotky CLcr alebo CLCRRT v ml/min pre dospievajúcich vo veku 13 rokov až menej ako 18 rokov, alebo v ml/min/1,73 m2 pre deti vo veku 6 rokov až menej ako 13 rokov.
Tabuľka 4: Úvodná dávka a schéma podávania udržiavacej dávky pre dojčatá a detí (vo veku
6 mesiacov až menej ako 6 rokov, s telesnou hmotnosťou 42,8 kg alebo vyššou) s poruchou
funkcie obličiek
CL
cr alebo CL
CRRT
(
m
l
/
mi
n
/
1,73 m
2
)
Ú
vodná
dávka
U
držiavacia dávka
Schéma podávania
udržiavacej dávky
50 až < 80 600 mg 400 mg dvakrát denne
250 mg dvakrát
Začnite podávať udržiavaciu
dávku dvakrát denne
po 12 hodinách od úvodnej
30 až < 50 600 mg
denne
dávky
Začnite podávať udržiavaciu
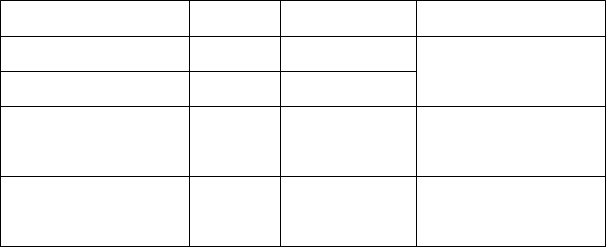
 15 až < 30
15 až < 30 600 mg 150 mg dvakrát denne
< 15 600 mg 60 mg dvakrát denne
dávku dvakrát denne
po 24 hodinách od úvodnej dávky
Začnite podávať udržiavaciu
dávku dvakrát denne
po 48 hodinách od úvodnej dávky
T
abuľka 5: Úvodná dávka a schéma podávania udržiavacej dávky pre dojčatá a deti (vo veku
6 mesiacov až menej ako 6 rokov, s telesnou hmotnosťou nižšou ako 42,8 kg) s poruchou funkcie
obličiek
CL
cr alebo CL
CRRT
(
m
l
/
mi
n
/
1,73 m
2
)
Ú
vodná
dávka
U
držiavacia dávka
Schéma podávania
udržiavacej dávky
50 až < 80 14 mg/kg 9,3 mg/kg dvakrát denne
5,8 mg/kg dvakrát
Začnite podávať udržiavaciu
dávku dvakrát denne
po 12 hodinách od úvodnej
30 až < 50 14 mg/kg

denne
dávky
Začnite podávať udržiavaciu
15 až < 30 14 mg/kg 3,5 mg/kg dvakrát denne
< 15 14 mg/kg 1,4 mg/kg dvakrát denne
dávku dvakrát denne
po 24 hodinách od úvodnej dávky
Začnite podávať udržiavaciu
dávku dvakrát denne
po 48 hodinách od úvodnej dávky
Pacientom, ktorí dostávajú intermitentnú hemodialýzu alebo intermitentnú peritoneálnu dialýzu, sa má
dávka podať po skončení dialýzy.
U pacientov, ktorí dostávajú kontinuálnu liečbu nahrádzajúcu funkciu obličky, sa má dávka zvoliť
pomocou vhodného CRRT klírensu (CLCRRT v ml/min).
Porucha funkcie pečene
Nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Spôsob podávania
Intravenózne použitie
Dectova sa podáva intravenóznou infúziou trvajúcou 30 minút.
Pokyny na riedenie lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Porucha funkcie obličiek
Zanamivir sa eliminuje vylučovaním obličkami, preto sa dávka Dectovy podávaná intravenózne musí
znížiť u pacientov s poruchou funkcie obličiek (pozri časť 4.2). Funkcia obličiek sa u všetkých
pacientov musí zhodnotiť pred začiatkom liečby a v pravidelných intervaloch počas liečby.
Závažné reakcie z precitlivenosti
Počas liečby zanamivirom boli hlásené anafylaktické reakcie a závažné kožné reakcie (zahŕňajúce
multiformný erytém, toxickú epidermálnu nekrolýzu a Stevensov-Johnsonov syndróm) (pozri
časť 4.8). Ak sa počas podávania infúzie Dectovy vyskytne akákoľvek reakcia z precitlivenosti, podávanie infúzie sa musí ihneď zastaviť a má sa začať vhodná liečba.
N
europsychické nežiaduce udalosti
Chrípka môže súvisieť s rôznymi neurologickými a behaviorálnymi príznakmi. Počas podávania
zanamiviru pacientom s chrípkou boli hlásené neuropsychické nežiaduce udalosti zahŕňajúce záchvaty kŕčov, delírium, halucinácie a nezvyčajné správanie, najmä u detí a dospievajúcich. Preto sa
u pacientov majú prísne sledovať zmeny v správaní a u každého pacienta sa majú starostlivo zhodnotiť prínosy a riziká pokračujúcej liečby (pozri časť 4.8).
Rezistencia u pacientov s oslabeným imunitným systémom
Rezistencia vznikajúca počas liečby je pri zanamivire zriedkavá (pozri časť 5.1). U pacientov
s oslabeným imunitným systémom je vyššia pravdepodobnosť selekcie rezistentných vírusov chrípky
po liečbe antivírusovými liekmi vrátane liečby Dectovou; preto je u nich dôležité sledovať rezistenciu
a zvážiť prechod na alternatívnu liečbu, ak je to vhodné.
Obmedzenia klinických údajov
Účinnosť Dectovy v liečbe komplikovanej infekcie spôsobenej vírusom chrípky typu A alebo B
u dospelých a detí vo veku od 6 mesiacov bola vyvodená z:
• aktivity zanamiviru v pomienkach in vitro;
• klinickej a virologickej aktivity zanamiviru v porovnaní s placebom v štúdii využívajúcej
infikovanie dobrovoľníkov („challenge study“) vírusom chrípky;
• hladín zanamiviru v tekutine pokrývajúcej epitel (epithelial lining fluid, ELF) priedušiek a z hladín zanamiviru v sére zistených v štúdii využívajúcej bronchoalveolárnu laváž;
• hladín zanamiviru v sére zistených u pacientov s komplikovanou chrípkou (pozri časť 5.1).
Riziko vzniku bakteriálnych infekcií
Nepreukázalo sa, že Dectova znižuje riziko vzniku bakteriálnych komplikácií súvisiacich s chrípkovou
infekciou.
Pomocné látky
Tento liek obsahuje 70,8 mg sodíka na injekčnú liekovku, čo zodpovedá 3,54 % WHO odporúčaného
maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
4.5 Liekové a iné interakcie
Potenciál vzniku interakcií s inými liekmi je nízky, vychádzajúc zo známej cesty eliminácie zanamiviru.
Zanamivir v klinicky významných koncentráciách nie je substrátom, inhibítorom ani induktorom izoenzýmov cytochrómu P450, ani nie je substrátom alebo inhibítorom renálnych a hepatálnych transportérov (pozri časť 5.2).
V klinickej štúdii sa nepreukázala interakcia s perorálne podávaným oseltamivirom.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii sú obmedzené údaje o použití zanamiviru u gravidných žien. Štúdie na zvieratách
nepreukázali priame alebo nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Reprodukčné štúdie vykonané na potkanoch a králikoch preukázali, že dochádza k prechodu zanamiviru placentou a nezistili žiadne známky teratogenity. Výsledky štúdie perinatálneho
a postnatálneho vývinu vykonanej na potkanoch neukázali klinicky významné poruchy vývinu
potomkov. K dispozícii však nie sú žiadne údaje o prechode placentou u ľudí.
Vzhľadom na obmedzené skúsenosti sa má použitie Dectovy v období gravidity zvážiť, iba ak možný prínos pre pacientku prevyšuje akékoľvek možné riziko pre plod.
Dojčenie
Nie je známe, či sa zanamivir vylučuje do ľudského mlieka. U potkanov sa preukázalo, že zanamivir
sa v malom množstve vylučuje do mlieka.
Vzhľadom na obmedzené skúsenosti sa má použitie zanamiviru u dojčiacich matiek zvážiť, iba ak možný prínos pre matku prevyšuje akékoľvek možné riziko pre dieťa.
Fertilita
Štúdie na zvieratách nepreukázali žiadne klinicky významné účinky zanamiviru na samčiu alebo
samičiu fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Dectova nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Bezpečnostný profil Dectovy je založený hlavne na údajoch z jednej štúdie fázy II a z jednej štúdie
fázy III, s podporou údajov zo štúdií fázy I, z programu „compassionate use“ (program na použitie neregistrovaného lieku v nevyhnutých prípadoch) a údajov o nežiaducich reakciách na liek hlásených pri inhalačne podávanom zanamivire. Frekvencia nežiaducich reakcií je založená na počte hlásení
v populácii dospelých, ktorým bol zanamivir podávaný intravenózne v dávke 600 mg dvakrát denne v štúdii fázy II a v štúdii fázy III. Nežiaduce reakcie sú uvedené podľa triedy orgánových systémov
MedDRA.
Najčastejšie hlásenými nežiaducimi reakciami, o ktorých sa usúdilo, že možno alebo pravdepodobne súvisia s liečbou Dectovou, sú zvýšená hladina alanínaminotransferázy (2 %), zvýšená hladina aspartátaminotransferázy (1 %), hepatocelulárne poškodenie (1 %), hnačka (1 %) a vyrážka (1 %). Najzávažnejšou nežiaducou reakciou bolo hepatocelulárne poškodenie, ktoré bolo pozorované u dvoch pacientov (1 %).
Tabuľkový zoznamnežiaducichreakcií
Frekvencia nežiaducich reakcií je definovaná pomocou nasledujúcej konvencie: veľmi časté (≥ 1/10);
časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000);
veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
T
rieda orgánových systémov Nežiaduce reakcie Frekvencia
Poruchy imunitného systému orofaryngálny edém opuch tváre anafylaktické/anafylaktoidné reakcie
neznáme
Psychické poruchy nezvyčajné správanie
halucinácie delírium
neznáme
Poruchy nervového systému kŕče
znížená úroveň vedomia
neznáme
Poruchy gastrointestinálneho
traktu
hnačka časté
Poruchy pečene a žlčových
ciest
zvýšená hladina
alanínaminotransferázy (ALT)
a/alebo zvýšená hladina aspartátaminotransferázy (AST) hepatocelulárne poškodenie
časté
zvýšená hladina alkalickej
fosfatázy
menej časté
Poruchy kože a podkožného
tkaniva
vyrážka časté
urtikária menej časté
multiformný erytém
Stevensov-Johnsonov syndróm toxická epidermálna nekrolýza
neznáme
Pediatrická populácia
Profil nežiaducich reakcií v pediatrickej populácii je založený na 71 pacientoch vo veku ≥ 6 mesiacov
až < 18 rokov zo štúdie fázy II. Bezpečnostný profil u pediatrických pacientov bol celkovo podobný
bezpečnostnému profilu pozorovanému u dospelých v klinických štúdiách.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieExistujú obmedzené skúsenosti s predávkovaním počas podávania Dectovy. K dispozícii nie je
špecifické antidotum na liečbu predávkovania týmto liekom. Liečba predávkovania má pozostávať
zo všeobecných podporných opatrení vrátane monitorovania vitálnych funkcií a sledovania klinického stavu pacienta. Zanamivir sa vylučuje renálnou exkréciou a predpokladá sa, že sa dá odstrániť hemodialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antivirotiká na systémové použitie, inhibítory neuraminidázy
ATC kód: J05AH01
Mechanizmus účinku
Zanamivir je inhibítor neuraminidázy vírusu chrípky, enzýmu, ktorý uvoľňuje vírusové častice
z plazmatickej membrány infikovaných buniek a podporuje šírenie vírusu v dýchacích cestách.
Aktivita v podmienkach in vitro
V podmienkach in vitro došlo k inhibícii neuraminidázy pri veľmi nízkych koncentráciách zanamiviru,
pričom medián inhibičných (IC50) hodnôt bol 0,33 nmol/l až 5,77 nmol/l proti kmeňom vírusu chrípky
typu A a B v uvedenom poradí.
Rezistencia
Selekcia rezistencie počas liečby zanamivirom je zriedkavá. Znížená citlivosť na zanamivir je spájaná
s mutáciami, ktoré spôsobujú zmeny aminokyselín vo vírusovej neuraminidáze alebo vo vírusovom hemaglutiníne alebo v oboch. V ľudských vírusoch a vo vírusoch so zoonotickým potenciálom sa počas liečby zanamivirom objavili substitúcie v neuraminidáze, ktoré spôsobili zníženú citlivosť
na zanamivir: E119D, E119G, I223R, R368G, G370D, N434S (A/H1N1); N294S, T325I (A/H3N2); R150K (B); R292K (A/H7N9). Substitúcia v neuraminidáze Q136K (A/H1N1 a A/H3N2) spôsobuje
vysoko-úrovňovú rezistenciu na zanamivir, ale bola vyselektovaná počas adaptácie na bunkovú
kultúru a nie počas liečby.
Klinický dosah zníženej citlivosti týchto vírusov nie je známy a vplyv špecifických substitúcií na citlivosť vírusu na zanamivir môže závisieť od kmeňov vírusu.
Skrížená rezistencia
V neuraminidázovom inhibičnom teste bola pozorovaná skrížená rezistencia medzi zanamivirom a oseltamivirom alebo peramivirom. Niekoľko substitúcií aminokyselín v neuraminidáze, ktoré vznikajú počas liečby oseltamivirom alebo peramivirom, má za následok zníženú citlivosť
na zanamivir. Klinický dosah substitúcií súvisiacich so zníženou citlivosťou na zanamivir a na iné inhibítory neuraminidázy je rôzny a môže závisieť od kmeňov vírusov.
Súbstitúcia H275Y je najčastejšou substitúciou v neuraminidáze súvisiacou s rezistenciou a spája sa so zníženou citlivosťou na peramivir a oseltamivir. Táto substitúcia nemá žiadny vplyv na zanamivir; preto si vírusy so substitúciou H275Y zachovávajú úplnú citlivosť na zanamivir.
Klinická účinnosť
Štúdia využívajúca infikáciu dobrovoľníkov („challenge study“)
Uskutočnila sa dvojito zaslepená, randomizovaná štúdia skúmajúca profylaktickú protivírusovú aktivitu a účinnosť zanamiviru, podávaného intravenózne v opakovanej 600 mg dávke
každých 12 hodín, v porovnaní s placebom proti infekcii u zdravých dobrovoľníkov mužského pohlavia, ktorí boli naočkovaní vírusom chrípky A/Texas/91 (H1N1). Zanamivir mal významný profylaktický účinok po experimentálnom infikovaní vírusom chrípky A, čo sa preukázalo na základe
nízkeho výskytu infekcie (14 % oproti 100 % v skupine s placebom v zmysle pozitívnych výsledkov sérologického testovania, p < 0,005), izolácie vírusu na vírusových kultúrach (0 % oproti 100 %
v skupine s placebom, p < 0,005), ako aj na základe nižšieho výskytu horúčky (14 % oproti 88 %
v skupine s placebom, p < 0,05), ochorenia horných dýchacích ciest (0 % oproti 100 % v skupine
s placebom, p < 0,005) a celkového skóre príznakov (medián skóre 1 bod oproti 44 bodom v skupine s placebom, p < 0,001).
Štúdia využívajúca bronchoalveolárnu laváž
Uskutočnila sa otvorená štúdia fázy I hodnotiaca farmakokinetiku zanamiviru v sére a v dolných dýchacích cestách po jeho intravenóznom a inhalačnom podávaní zdravým dospelým osobám, a to
s využitím tekutiny získanej bronchoalveolárnou lavážou. Po 600 mg dávke podávanej intravenózne sa koncentrácie zanamiviru v tekutine pokrývajúcej epitel najviac približovali koncentráciám
dosiahnutým po podávaní schválenej 10 mg dávky zanamiviru vo forme inhalačného prášku, pri ktorej sa preukázala účinnosť vo veľkých klinických štúdiách zameraných na liečbu nekomplikovanej chrípky.
Štúdia fázy III u pacientov s komplikovanou chrípkou
Uskutočnila sa dvojito zaslepená štúdia fázy III hodnotiaca účinnosť, protivírusovú aktivitu
a bezpečnosť zanamiviru podávaného intravenózne v dávke 600 mg dvakrát denne v porovnaní
s oseltamivirom podávaným perorálne v dávke 75 mg dvakrát denne a so zanamivirom podávaným intravenózne v dávke 300 mg dvakrát denne u hospitalizovaných pacientov (vo veku > 16 rokov)
s chrípkou. Medián veku pacientov bol 57 rokov a 35 % (218/615) pacientov malo ≥ 65 rokov,
z ktorých 17 % (n = 103) malo 65 až < 75 rokov; 14 % (n = 84) malo 75 až < 85 rokov a 5 % (n = 31) malo ≥ 85 rokov. Pacienti boli pri randomizácii stratifikovaní podľa času, ktorý uplynul od vzniku príznakov do začiatku liečby (≤ 4 dni a 5 až 6 dní). Vhodní pacienti nesmeli podstúpiť predchádzajúcu protivírusovú liečbu trvajúcu > 3 dni. Úvodný 5-dňový cyklus liečby mohol byť predĺžený
až o 5 dodatočných dní, ak si klinické príznaky alebo charakteristiky pacienta vyžadovali ďalšiu
liečbu. Primárnym cieľovým ukazovateľom bol čas do dosiahnutia klinickej odpovede (time to clinical response, TTCR); klinická odpoveď bola definovaná buď ako kombinácia stabilizácie vitálnych
funkcií (teplota, saturácia krvi kyslíkom, stav dýchania, srdcová frekvencia a systolický krvný tlak),
alebo ako prepustenie pacienta z nemocnice. Primárna analýza bola vykonaná u populácie
s pozitívnym nálezom chrípky (Influenza Positive Population, IPP) tvorenej 488 pacientmi. Štúdia
nesplnila svoj vopred špecifikovaný primárny cieľ spočívajúci v preukázaní superiority 600 mg dávky zanamiviru v porovnaní s perorálne podávaným oseltamivirom alebo s 300 mg dávkou zanamiviru
v zmysle TTCR. Nezistili sa žiadne významné rozdiely v TTCR pri porovnaní liečby v celkovej IPP
alebo v dvoch vopred špecifikovaných podskupinách (tabuľka 6).
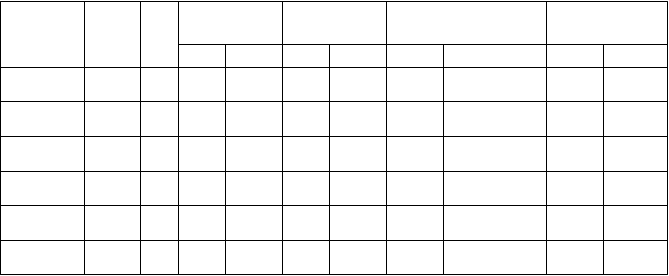
Tabuľka 6: Štatistické porovnania TTCR medzi skupinou so 600 mg dávkou zanamiviru a každou inou skupinou (IPP)
I
n
f
úzny
roztok zanamiviru
 300 mg
I
n
f
úzny
roztok zanamiviru
600 mg
Os
e
lt
am
i
vir
75 mg
300 mg
I
n
f
úzny
roztok zanamiviru
600 mg
Os
e
lt
am
i
vir
75 mg
Populácia s pozitívnym nálezom chrípky, N 163 162 163
Medián TTCR, dni 5,87 5,14 5,63
Medián rozdielu medzi liečbami, dni (95 % IS) -0,73 (-1,79; 0,75) -0,48 (-2,11; 0,97)
p-hodnota z 2-stranného Wilcoxonovho testu
(Wilcoxon rank-sum 2-sided test) Podskupina pacientov na jednotke intenzívnej starostlivosti/vyžadujúcich mechanickú ventiláciu, N
0,25 0,39
68 54 68
Medián TTCR, dni 11,26 12,79 14,58
Medián rozdielu medzi liečbami, dni (95 % IS) 1,53 (-4,29; 8,34) -1,79 (-11,1; 6,92)
p-hodnota z 2-stranného Wilcoxonovho testu 0,87 0,51
Podskupina pacientov, u ktorých od vzniku príznakov
do začiatku liečby uplynuli ≤ 4 dni, N
127 131 121
Medián TTCR, dni 5,63 4,80 4,80
Medián rozdielu medzi liečbami, dni (95 % IS) -0,83 (-1,98; 0,56) 0,00 (-1,05; 0,97)'
p-hodnota z 2-stranného Wilcoxonovho testu 0,09 0,82
Tento liek bol registrovaný za tzv. mimoriadnych okolností. To znamená, že z vedeckých dôvodov
nebolo možné získať všetky informácie o tomto lieku.
Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn
charakteristických vlastností lieku bude podľa potreby aktualizovať.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Dectovou v jednej
alebo vo viacerých podskupinách pediatrickej populácie pri liečbe a prevencii chrípky (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Sérové farmakokinetické parametre zanamiviru podávaného intravenózne sa skúmali u zdravých dobrovoľníkov, ktorí dostávali jednorazové zvyšujúce sa dávky od 1 do 1 200 mg a opakované dávky 600 mg dvakrát denne počas 5 dní. Hospitalizovaní pacienti s chrípkou tiež dostávali 300 mg alebo 600 mg dvakrát denne počas 5 až 10 dní.
Zistilo sa, že hodnoty Cmax a AUC zanamiviru sú úmerné veľkosti dávky a že po opakovaných intravenóznych dávkach až do 600 mg nie je zjavná žiadna kumulácia zanamiviru v sére.
Distribúcia
Väzba zanamiviru na plazmatické bielkoviny je veľmi nízka (menej ako 10 %). Distribučný objem
zanamiviru u dospelých je približne 16 l a je približne rovnaký ako objem extracelulárnej tekutiny.
Po podávaní zanamiviru vo forme infúzneho roztoku dvakrát denne predstavovali jeho koncentrácie v tekutine pokrývajúcej epitel pľúc 60 - 65 % hodnoty koncentrácií v sére po odbere vzoriek
v príslušnom čase, a to po 12 hodinách od podania dávky. Po podávaní zanamiviru vo forme infúzneho roztoku v dávke 600 mg dvakrát denne sa priemerné minimálne („trough“, t. j. namerané
na konci dávkovacieho intervalu tesne pred podaním ďalšej dávky) koncentrácie zanamiviru v tekutine pokrývajúcej epitel pohybovali v rozmedzí od 419 ng/ml do 584 ng/ml a predstavovali
47 - 66 % hodnoty koncentrácií v úvodnej bronchoalveolárnej vzorke po perorálnom podávaní zanamiviru vo forme inhalačného prášku v dávke 10 mg dvakrát denne.
Biotransformácia
K dispozícii nie sú dôkazy o tom, že zanamivir sa metabolizuje.
Eliminácia
Zanamivir sa eliminuje v nezmenenej forme močom prostredníctvom glomerulárnej filtrácie.
U dospelých s normálnou funkciou obličiek je polčas eliminácie približne 2 - 3 hodiny.
Staršie osoby
Farmakokinetika u starších osôb bola podobná farmakokinetike u mladých dospelých osôb.
V populačnej farmakokinetickej analýze nemal vek žiadny významný vplyv na farmakokinetiku zanamiviru.
Pediatrická populácia
Farmakokinetika zanamiviru po intravenóznom podávaní dávky 14 mg/kg dvakrát denne pediatrickým
pacientom vo veku 6 mesiacov až < 6 rokov a 12 mg/kg dvakrát denne pediatrickým pacientom
vo veku 6 rokov až < 18 rokov bola podobná farmakokinetike pozorovanej u dospelých, ktorým bola intravenózne podávaná dávka 600 mg dvakrát denne. Farmakokinetika zanamiviru u osôb vo veku
6 mesiacov až < 18 rokov (ktorým bola podávaná štandardná dávka 12 mg/kg, 14 mg/kg alebo 600 mg
v závislosti od veku a telesnej hmotnosti) a u dospelých osôb (ktorým bola podávaná štandardná dávka
600 mg) bola podobná (tabuľka 7).
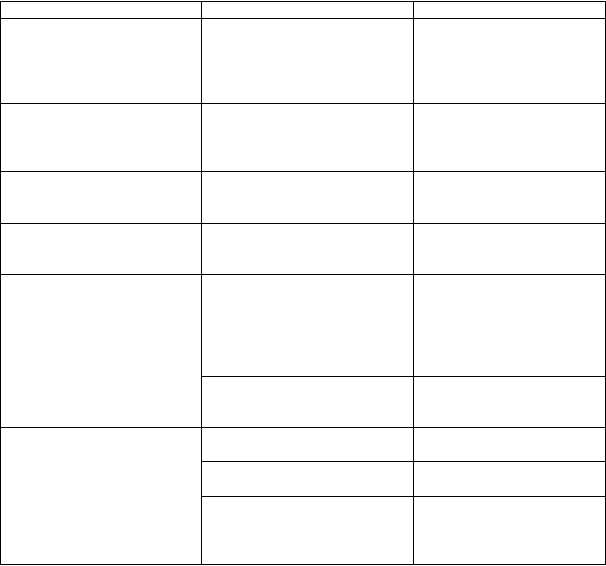
Tabuľka 7: Farmakokinetické parametre u pediatrických a dospelých osôb
V
eková
s
kupina
D
ávka N C
m
a
x
(µg/ml)
AUC(0-∞)
(µg.h/ml)
Cmin
(µg/ml)
T1/2
(h)
6 mesiacov
- < 1 rok
1 - < 2
roky
2 - < 6
rokov
6 - < 13
rokov
13 - < 18
rokov
14
mg/kg
14
mg/kg
14
mg/kg
12
mg/kg
600
mg
GM %CV GM %CV GM Rozmedzie GM %CV
7 36,2 21 75,3 23 NA NA 1,84 19
6 37,8 24 72,4 14 0,305 NA 2,49 118
12 41,5 23 80,3 38 0,277 0,133 - 0,984 1,60 34
16 44,2 47 107 41 0,564 0,111 - 2,31 2,57 55
13 34,5 27 91,1 27 0,211 0,104 - 0,428 2,06 47
> 18 rokov 600
mg
67 32,8 34 82,9 36 0,82 0,1 - 11,4 2,39 31
%CV = variačný koeficient vyjadrený v percentách, GM = geometrický priemer; NA = nie je k dispozícii (Not available)
Porucha funkcie obličiekPolčas zanamiviru v sére sa zvyšuje približne na 12 - 20 hodín u pacientov so závažnou poruchou
funkcie obličiek (klírens kreatinínu < 30 ml/min). Dectova nebola skúmaná u pacientov s ochorením
obličiek v konečnom štádiu.
K dispozícii sú obmedzené údaje o expozícii zanamiviru počas súbežnej kontinuálnej liečby
nahrádzajúcej funkciu obličiek a veľmi obmedzené údaje o jeho expozícii počas dialýzy.
Porucha funkcie pečeneZanamivir sa nemetabolizuje, preto sa neočakáva žiadny vplyv poruchy funkcie pečene.
RasaFarmakokinetické štúdie u zdravých osôb thajského, čínskeho a japonského pôvodu nezistili žiadne
klinicky významné rozdiely vo farmakokinetike zanamiviru v týchto populáciách v porovnaní s osobami belošského (kaukazského) pôvodu.
L
i
ekové interakcie
I
n vitro štúdie preukazujú, že zanamivir nie je inhibítorom ani substrátom proteínu zodpovedného
za rezistenciu pri rakovine prsníka (breast cancer resistance protein, BCRP), P-glykoproteínu, proteínu extrúzie viacerých liekov a toxínov MATE1 (multidrug and toxin extrusion protein 1), MATE2-K, transportéra organických aniónov (Organic Anion Transporter, OAT)1, OAT3, transportného polypeptidu organických aniónov (Organic Anion Transporting Polypeptide, OATP)1B1, OATP1B3 a transportéra organických katiónov (Organic Cation Transporter, OCT)2, ani nie je inhibítorom enzýmov cytochrómu P450 (CYP)1A2, 2B6, 2C8, 2C9, 2C19, 2D6 a 3A4.
Zanamivir nie je induktorom CYP1A2 a 2B6 a i keď sa v in vitro podmienkach pozorovala indukcia CYP3A4 pri koncentrácii 50-násobne vyššej ako sú klinicky významné koncentrácie, na základe fyziologicky založeného farmakokinetického modelovania (physiologically based pharmacokinetic modelling, PBPK) sa nepredpokladá žiadna interakcia so substrátmi CYP3A4.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity
po opakovanom podávaní, genotoxicity, karcinogénneho potenciálu, reprodukčnej toxicity a vývinu okrem štúdie embryo-fetálneho vývinu na potkanoch (subkutánne podávanie) neodhalili žiadne osobitné riziko pre ľudí. V štúdii embryo-fetálneho vývinu na potkanoch sa zistilo zvýšenie výskytu rôznych drobných zmien skeletu a vnútorných orgánov, pričom väčšina z nich sa vyskytovala
v rozmedzí prirodzenej incidencie historicky sa vyskytujúcej u skúmaného kmeňa.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Chlorid sodný
Voda na injekcie
6.2 Inkompatibility
Dectova sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
Dectova sa nemá podávať súčasne s inými intravenózne podávanými liekmi ani pripravovať
v roztokoch obsahujúcich glukózu alebo iné elektrolyty (pozri časť 6.6).
6.3 Čas použiteľnosti
Neotvorené injekčné liekovky
5 rokov.
Po riedení
Z mikrobiologického hľadiska sa má liek použiť ihneď. Ak sa nepoužije ihneď, za čas a podmienky
uchovávania zriedeného lieku pred použitím je zodpovedný používateľ a nemá sa uchovávať dlhšie
ako 24 hodín pri teplote 2 °C až 8 °C, pokiaľ sa riedenie uskutočnilo v kontrolovaných a overených aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Injekčná liekovka z číreho skla (typu I) s objemom 26 ml, so zátkou (potiahnutý chlórbutylkaučuk),
obrubou (hliník) a plastovým vyklápacím („flip-off“) viečkom. Veľkosť balenia: 1 injekčná liekovka.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
Príprava Dectovy
• Objem Dectovy a celkový objem podávaný infúziou budú závisieť od veku, telesnej hmotnosti
a funkcie obličiek pacienta (pozri časť 4.2).
• Dávka sa môže podávať infúziou vo forme dodaného roztoku alebo roztoku zriedeného injekčným roztokom chloridu sodného s koncentráciou 9 mg/ml (0,9 %) na akúkoľvek koncentráciu vyššiu alebo rovnú 0,2 mg/ml.
• Každá injekčná liekovka je určená len na jednorazové použitie; po narušení uzáveru sa zvyšný
objem musí zlikvidovať.
Ako pripraviťinfúziunaintravenóznepodanie:
• Počas celej prípravy dávky používajte aseptické postupy.
• Vypočítajte požadovanú dávku a objem Dectovy.
• Rozhodnite, aký objem injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %)
sa má použiť na infúziu.
• Pomocou sterilnej ihly a injekčnej striekačky odoberte a zlikvidujte objem injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %) (rovnajúci sa objemu Dectovy) z infúzneho vaku.
• Infúzne vaky môžu obsahovať ďalší prebytok injekčného roztoku chloridu sodného
s koncentráciou 9 mg/ml (0,9 %) - tento prebytok sa tiež môže odstrániť, ak sa to považuje
za potrebné.
• Pomocou sterilnej ihly a injekčnej striekačky odoberte objem Dectovy z injekčnej (injekčných)
liekovky (liekoviek) a pridajte do infúzneho vaku.
• Zlikvidujte všetok nepoužitý roztok, ktorý zostal v injekčnej liekovke.
• Infúznym vakom sa má ručne jemne pohýbať, aby sa zaistilo dôkladné premiešanie jeho obsahu.
• Ak sa infúzny vak uchovával v chladničke, má sa z nej vybrať a pred použitím má dosiahnuť
izbovú teplotu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
GlaxoSmithKline Trading Services Limited
12 Riverwalk
Citywest Business Campus
Dublin 24
Írsko
8. REGISTRAČNÉ ČÍSLO
EU/1/18/1349/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 26. apríla 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.