
roflumilastu je kombinovaný účinok obidvoch zložiek, roflumilastu aj roflumilast N-oxidu. Štúdie
interakcií s inhibítorom CYP1A2/3A4 enoxacínom a inhibítormi CYP1A2/2C19/3A4 fluvoxamínom a
cimetidínom preukázali zvýšenú celkovú PDE4 inhibičnú aktivitu o 25%, 47% a 59%. Testovaná dávka fluvoxamínu bola 50 mg. Kombinácia roflumilastu s týmito liečivami môže viesť k zvýšenej expozícii a pretrvávajúcej intolerancii. V takomto prípade sa má liečba roflumilastom prehodnotiť (pozri časť 4.4).
Pri aplikácii rifampicínu, ktorý indukuje cytochrómový enzým P450, sa znížila celková PDE4 inhibičná aktivita približne o 60%. Použitie silných induktorov cytochrómu P450 (napr. fenobarbital, karbamazepín, fenytoín) môže preto znižovať terapeutickú účinnosť roflumilastu. Z tohto dôvodu sa pacientom užívajúcim silné inhibítory cytochrómu P450 liečba roflumilastom neodporúča.
Klinické interakčné štúdie s inhibítormi CYP3A4 erytromycínom a ketokonazolom preukázali zvýšenie celkovej inhibičnej aktivity PDE4 o 9%. Súbežná aplikácia s teofylínom spôsobila zvýšenie
celkovej inhibičnej aktivity PDE4 o 8% (pozri časť 4.4). V štúdii interakcie s perorálnymi kontraceptívami, ktoré obsahovali gestodén a etinylestradiol, sa celková PDE4 inhibičná aktivita zvýšila o 17%. Z tohto dôvodu, u pacientov, ktorí užívajú uvedené liečivá, nie je nutná úprava dávkovania.
Nezistili sa žiadne klinicky významné interakcie s inhalovaným salbutamolom, formoterolom, budezonidom a perorálne aplikovaným montelukastom, digoxínom, warfarínom, sildenafilom a midazolamom.
Súbežná aplikácia s antacidom (kombinácia hydroxidu hlinitého a magnézium hydroxidu) nezmenila absorpciu ani farmakokinetiku roflumilastu alebo jeho N-oxidu.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby. Roflumilast sa neodporúča u žien vo fertilnom veku, ktoré nepoužívajú antikoncepciu.
Gravidita
Údaje o podávaní roflumilastu gravidným ženám sú obmedzené.
Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Roflumilast sa neodporúča počas gravidity.
U gravidných samíc potkanov sa preukázalo, že roflumilast prechádza placentou. Dojčenie
Dostupné farmakokinetické údaje na zvieratách preukázali vylučovanie roflumilastu alebo jeho
metabolitov do materského mlieka. Nie je možné vylúčiť riziko pre dojčené dieťa. Roflumilast sa nemá používať počas dojčenia.
Fertilita
V štúdii spermatogenézy u ľudí nemala dávka 500 mikrogramov roflumilastu žiaden účinok na parametre semena alebo reprodukčné hormóny počas 3-mesačnej liečby ani nasledujúce 3 mesiace po jej ukončení.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Daxas nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
V klinických štúdiách CHOCHP sa u približne 16% pacientov prejavili nežiaduce reakcie roflumilastu (v porovnaní s 5% výskytom u placeba). Najčastejšie hlásenými nežiaducimi reakciami boli hnačka (5,9%), pokles hmotnosti (3,4%), nauzea (2,9%), bolesť brucha (1,9%) a bolesť hlavy (1,7%). Väčšina nežiaducich reakcií bola mierna až stredná. Nežiaduce reakcie sa vyskytli hlavne v prvých týždňoch liečby a pokračovaním liečby väčšinou vymizli.
Tabuľkovýprehľadnežiaducichreakcií
Frekvencia výskytu nežiaducich reakcií v nasledujúcej tabuľke je uvedená podľa klasifikácie
MedDRA:
veľmi časté (³1/10); časté (³1/100 až <1/10); menej časté (³1/1 000 až <1/100); zriedkavé (³1/10 000
až <1/1 000); veľmi zriedkavé (<1/10 000), neznáme (z dostupných údajov).
V každej skupine frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
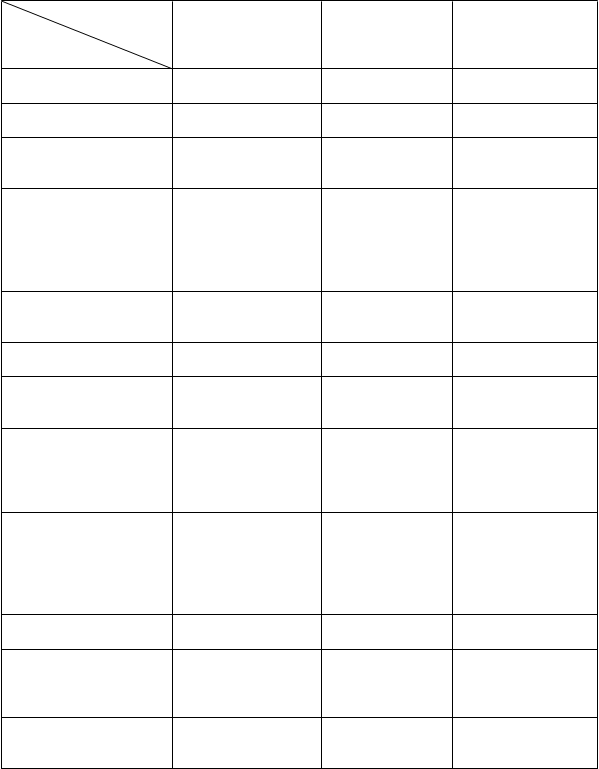
Tabuľka 1. Nežiaduce reakcie roflumilastu v klinických štúdiách CHOCHP a postmarketingové údaje
Frekvencia
Čast
é Menej časté Zriedkavé
Trieda
orgánovýc
h systémov Poruchy imunitného systému
Poruchy endokrinného systému
Poruchy metabolizmu a výživy
Zníženie hmotnosti Zníženie chuti do jedla
Precitlivenosť Angioedém
Gynekomastia
Psychické poruchy Nespavosť Úzkosť Samovražedné myšlienky
a správanie* Depresia Nervozita
Záchvat paniky
Poruchy nervového systému
Poruchy srdca a srdcovej činnosti
Poruchy dýchacej sústavy, hrudníka a mediastína
Bolesť hlavy Tras Vertigo Závraty Palpitácie
Poruchy chuti
Infekcie dýchacej sústavy (mimo zápalu pľúc)
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
Hnačka
Nauzea
Bolesť brucha
Gastritída Vracanie Gastroezofageálna refluxná choroba Dyspepsia
Krv v stolici
Obstipácia
Zvýšenie
gamma-glutamyl transpeptidázy Zvýšenie aspartátaminotransfe- rázy (AST)
Poruchy kože a podkožného tkaniva
Vyrážka Žihľavka
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Celkové poruchy a reakcie v mieste podania
Svalové kŕče a slabosť Bolesť svalov Bolesť chrbta Malátnosť Slabosť
Únava
Zvýšenie hladiny kreatínfosfokinázy (CPK) v krvi
Popis
zvolených
nežiaducich
reakcií
* V klinických štúdiách a po zavedení lieku na trh boli pozorované zriedkavé prípady samovražedných myšlienok a správania, vrátane samovraždy. Pacientov a opatrovateľov treba poučiť, aby oznámili predpisujúcemu lekárovi akékoľvek samovražedné myšlienky (pozri časť 4.4).
Inéosobitnépopulácie
Vyšší výskyt porúch spánku (hlavne nespavosti) u pacientov vo veku ≥ 75 rokov alebo starších sa pozoroval v štúdii RO-2455-404-RD u pacientov liečených roflumilastom v porovnaní s pacientmi
užívajúcimi placebo (3,9% oproti 2,3%). Pozorovaný výskyt bol tiež vyšší u pacientov mladších ako
75 rokov, ktorí boli liečení roflumilastom, v porovnaní s pacientmi užívajúcimi placebo (3,1% oproti
2,0%).
Vyšší výskyt porúch spánku (hlavne nespavosti) u pacientov so základnou telesnou hmotnosťou
< 60 kg sa pozoroval v štúdii RO-2455-404-RD u pacientov liečených roflumilastom v porovnaní
s pacientmi užívajúcimi placebo (6,0% oproti 1,7%). U pacientov so základnou telesnou hmotnosťou
≥ 60 kg liečených roflumilastom bol výskyt 2,5% oproti 2,2% pacientov užívajúcich placebo.
Súbežnáliečbas dlhodobopôsobiacimi antagonistamimuskarínovýchreceptorov(LAMA)Vyšší výskyt poklesu telesnej hmotnosti, zníženej chuti do jedla, bolesti hlavy a depresie sa pozoroval
počas štúdie RO-2455-404-RD u pacientov súbežne užívajúcich roflumilast a dlhodobo pôsobiace antagonisty muskarínových receptorov (LAMA) spolu so súbežne inhalovanými kortikosteroidmi (ICS) a dlhodobo pôsobiacimi agonistami B2 receptorov (LABA) v porovnaní s pacientmi, ktorí sa súbežne liečili len roflumilastom, ICS a LABA. Rozdiel výskytu medzi roflumilastom a placebom bol kvantitatívne vyšší so súbežne užívaným LAMA pre pokles hmotnosti (7,2% oproti 4,2%), zníženú chuť do jedla (3,7% oproti 2,0%), bolesť hlavy (2,4% oproti 1,1%) a depresiu (1,4% oproti -0,3%).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePríznakyV štúdiách vo fáze I sa po jednorazových perorálnych dávkach 2 500 mikrogramov a jednej dávke
5 000 mikrogramov (desaťnásobok odporučenej dávky) pozoroval zvýšený výskyt nasledujúcich
príznakov: bolesť hlavy, gastrointestinálne poruchy, závraty, palpitácie, mierne vertigo, zvýšené potenie a arteriálna hypotenzia.
LiečbaPokiaľ dôjde k predávkovaniu, odporúča sa poskytnúť vhodnú podpornú medicínsku starostlivosť. Vzhľadom na to, že sa roflumilast výrazne viaže na bielkoviny, hemodialýza pravdepodobne nie je účinný spôsob na odstránenie liečiva. Nie je známe, či je roflumilast dialyzovateľný peritoneálnou dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Lieky na obštrukčné ochorenia dýchacích ciest, iné systémové liečivá ochorení spojených s obštrukciou dýchacích ciest, ATC kód: R03DX07
MechanizmusúčinkuRoflumilast je inhibítor PDE4, nesteroidové protizápalové liečivo s cieleným účinkom na systémové a pľúcne zápaly spojené s CHOCHP. Mechanizmom účinku je inhibícia PDE 4, hlavného enzýmu, ktorý metabolizuje cyklický adenozín monofosfát (cAMP) a zistilo sa, že je dôležitý pre patogenézu CHOCHP v štrukturálnych a zápalových bunkách. Roflumilast pôsobí na spojené varianty reťazca PDE4A, 4B a 4D s podobnou potenciou na nanomolárnej úrovni. Afinita k spojenému variantu PDE4C je 5 až 10 násobne nižšia. Tento mechanizmus účinku a selektívnosť platí aj pre roflumilast N-oxid, čo je hlavný aktívny metabolit roflumilastu.
FarmakodynamickéúčinkyInhibícia PDE4 vedie k zvýšeniu hladiny vnútrobunkového cAMP a v experimentálnych modeloch zmierňuje porušené funkcie leukocytov, hladkých svalov buniek dýchacích ciest a v pľúcnych cievach,
endoteliálnych bunkách a epiteliálnych bunkách dýchacích ciest a fibroblastov, ktoré boli porušené dôsledkom CHOCHP. Po in vitro stimulácii ľudských neutrofilov, monocytov, makrofágov alebo lymfocytov roflumilast a roflumilast N-oxid potláčal uvoľňovanie mediátorov zápalu, napr. leukotriénu B4, reaktívnych foriem kyslíka, tumor nekrotizujúceho faktoru α, interferónu γ
a granzymu B.
U pacientov s CHOCHP roflumilast znižoval počet neutrofilov v spúte. Okrem toho roflumilast zoslaboval prienik neutrofilov a eozinofilov do dýchacích ciest u zdravých dobrovoľníkov, ktorým sa aplikoval endotoxín.
Klinickáúčinnosťabezpečnosť
V dvoch konfirmačných opakovaných jednoročných štúdiách (M-124 a M-125) a v dvoch doplňujúcich šesťmesačných štúdiách (M2-127 a M2-128) bolo randomizovaných celkovo 4 768 pacientov, z toho bolo 2 374 liečených roflumilastom. Štúdie mali paralelné skupiny, boli dvojito zaslepené a kontrolované placebom.
V jednoročných štúdiách sledovali pacientov s anamnézou závažnej alebo veľmi závažnej CHOCHP
[FEV1 (úsilný expiračný objem za sekundu) ≤ 50% predpokladanej hodnoty] spojenej s chronickou bronchitídou, aspoň s jednou dokumentovanou exacerbáciou v predchádzajúcom roku a s príznakmi
určenými vstupnými údajmi podľa skóre kašľa a spúta. Používanie dlhodobo účinkujúcich
beta-agonistov (LABA) bolo počas štúdie povolené, použilo ich približne 50% pacientov. Krátko
účinkujúce anticholinergiká (SAMA) boli povolené pre pacientov, ktorí neužívali LABA. Záchranná medikácia (salbutamol alebo albuterol) bola povolená podľa potreby. Používanie inhalačných kortikosteroidov a teofylínu bolo počas štúdie zakázané. Pacienti bez exacerbácií v anamnéze boli vylúčení.
V súhrnnej analýze jednoročných štúdií M2-124 a M2-125 roflumilast v dávke 500 mikrogramov jeden krát denne signifikantne zlepšoval funkciu pľúc v porovnaní s placebom, priemerne o 48 ml (FEV1 pred podaním bronchodilatátora, primárny cieľ, p<0,0001) a o 55 ml (FEV1 po podaní bronchodilatátora, p<0,0001). Zlepšenie funkcie pľúc bolo zreteľné pri prvej návšteve po 4 týždňoch a pretrvávalo 1 rok (ukončenie liečby). Pomer (pacient/rok) stredných exacerbácií (vyžadujúcich podávanie systémových glukokortikosteroidov) alebo závažných exacerbácií (vyžadujúcich hospitalizáciu a/alebo vedúcich k úmrtiu) po jednom roku bol 1,142 u roflumilastu a 1,374 u placeba, čo zodpovedá zníženiu relatívneho rizika o 16,9% (95% IS: 8,2% až 24,8%) (primárny cieľ, p=0,0003). Účinky boli podobné, nezávislé od predchádzajúcej liečby inhalačnými kortikosteroidmi
alebo liečbou LABA. V podskupine pacientov s anamnézou častých exacerbácií (aspoň 2 exacerbácie počas posledného roka), bol pomer exacerbácií 1,526 u roflumilastu a 1,941 u placeba, čo zodpovedá zníženiu relatívneho rizika o 21,3% (95% IS: 7,5% až 33,1%). Roflumilast signifikantne neznižoval výskyt exacerbácií v porovnaní s placebom v podskupine pacientov so strednou CHOCHP.
Počet stredných alebo závažných exacerbácií u pacientov s roflumilastom a LABA v porovnaní
s placebom a LABA sa znížil priemerne 21% (p=0,0011). Pokles exacerbácií u pacientov bez súbežnej liečby s LABA bol v priemere 15% (p=0,0387). Počet pacientov, ktorí zomreli z akýchkoľvek dôvodov, bol rovnaký v skupine pacientov, ktorí boli liečení roflumilastom alebo dostávali placebo
(42 úmrtí v každej skupine; 2,7% v každej skupine; súhrnná analýza).
Do dvoch podporných randomizovaných 1 ročných štúdií (M2-111 a M2-112) bolo zaradených a randomizovaných celkovo 2 690 pacientov. Na rozdiel od dvoch konfirmatívnych štúdií sa na zaradenie do štúdie nevyžadovala anamnéza chronickej bronchitídy a CHOCHP exacerbácií. Inhalačné kortikosteroidy použilo 809 pacientov (61%), ktorí boli liečení roflumilastom, zatiaľ čo používanie LABA a teofylínu bolo zakázané. Roflumilast 500 mikrogramov jedenkrát denne signifikantne zlepšil funkciu pľúc v porovnaní s placebom, v priemere o 51 ml (FEV1 pred podaním bronchodilatátora, p<0,0001) a o 53 ml (FEV1 po podaní bronchodilatátora, p <0,0001). Výskyt exacerbácií (tak ako boli definované v protokoloch) v jednotlivých štúdiách roflumilast signifikantne neznižoval (zníženie relatívneho rizika: 13,5% v štúdii M2-111 a 6,6% v štúdii M2-112; p=nesignifikantné). Výskyt nežiaducich účinkov nezávisel od súbežnej liečby inhalačnými kortikosteroidmi.
Do dvoch podporných klinických štúdií (M2-127 a M2-128) boli zaradení pacienti s anamnézou CHOCHP diagnostikovanou najmenej 12 mesiacov pred zaradením do štúdie. Do obidvoch štúdií sa zaradili pacienti so strednou až ťažkou nereverzibilnou obštrukciou dýchacích ciest a FEV1 40% až
70% predpokladanej hodnoty. Roflumilast alebo placebo sa pridali k pokračujúcej liečbe dlhodobo účinkujúcim bronchodilatátorom, hlavne k salmeterolu v štúdii M2-127 alebo k tiotropiu v štúdii M2-128. V dvoch 6-mesačných štúdiách sa FEV1 pred podaním bronchodilatátora signifikantne
zlepšilo o 49 ml (primárny cieľ, p<0,0001) v súbežnej liečbe salmeterolom v štúdii M2-127 a o 80 ml
(primárny cieľ, p<0,0001) v súbežnej liečbe tiotropiom v štúdii M2-128.

Štúdia RO-2455-404-RD bola jednoročná štúdia u pacientov s CHOCHP s počiatočnou hodnotou (pred podaním bronchodilatátora) FEV1 < 50% predpokladaných normálnych a minulých častých exacerbácií. Štúdia vyhodnocovala účinok roflumilastu na mieru exacerbácií CHOCHP u pacientov liečených pevnou kombináciou LABA a inhalovaných kortikosteroidov, v porovnaní s placebom. Celkom 1 935 pacientov bolo randomizovaných do dvojito zaslepenej liečby a približne 70% tiež používalo v priebehu štúdie dlhodobo pôsobiace antagonisty muskarínových receptorov (LAMA). Primárnym cieľom bolo zníženie miery stredných alebo závažných exacerbácií CHOCHP na pacienta na rok. Miera závažných exacerbácií CHOCHP a zmeny hodnôt FEV1 sa vyhodnocovali ako kľúčové sekundárne ciele.
Tabuľka 2. Súhrn cieľov týkajúcich sa exacerbácií CHOCHP v štúdii RO-2455-404-RD
Roflumilast
(N=969)
Placebo
(N=966)
P
o
me
r Roflumilast/Placebo
2-
stranná
Kateg
ória
exacerbácie
Stredná alebo
ModelanalýzyPoissono
Miera(n)0,805
Miera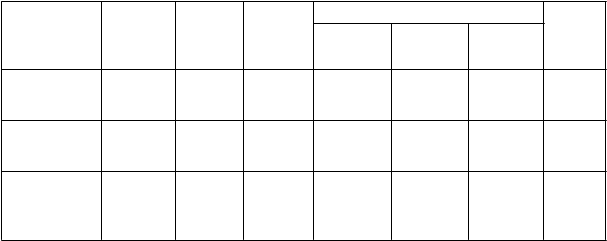 (n)
(n)0,927
PomermierZmena(%) 95% IShodnot a p
závažná
vá regresia
(380)
(432) 0,868 -13,2 0,753;
1,002
0,0529
Stredná Poissono vá
regresia
Závažná negatívna binominá
0,574 (287)
0,239 (151)
0,627
(333) 0,914 -8,6 0,775;
1,078
0,315
(192) 0,757 -24,3 0,601;
0,2875
lna regresia
0,952 0,0175
Vyskytol sa trend smerom k zníženiu stredných a závažných exacerbácií u pacientov liečených
roflumilastom v porovnaní s placebom po dobu 52 týždňov, ktorý však nedosiahol štatistický význam
(tabuľka 2). Predurčená analýza citlivosti s použitím modelu binominálnej regresie preukázala štatistický významný rozdiel -14,2% (pomer mier: 0,86; 95% IS: 0,74 až 0,99).
Pomery mier analýzy s Poissonovou regresiou podľa protokolu a analýzy pôvodného zámeru liečby vylučujúcej nevýznamnú citlivosť s Poissonovou regresiou boli 0,81 (95% IS: 0,69 až 0,94) a 0,89 (95% IS: 0,77 až 1,02), v uvedenom poradí.
Zníženia sa dosiahli v podskupine pacientov súbežne liečených LAMA (pomer mier: 0,88; 95% IS:
0,75 až 1,04) a v podskupine súbežne neliečenej LAMA (pomer mier: 0,83; 95% IS: 0,62 až 1,12).
Miera závažných exacerbácií sa znížila v celkovej skupine pacientov (pomer mier: 0,76; 95% IS: 0,60 až 0,95) s mierou 0,24 na pacienta/rok v porovnaní s mierou 0,32 na pacienta/rok u pacientov užívajúcich placebo. Podobné zníženie sa dosiahlo v podskupine pacientov súbežne liečenej LAMA (pomer mier: 0,77; 95% IS: 0,60 až 0,99) a v podskupine súbežne neliečenej LAMA (pomer mier:
0,71; 95% IS: 0,42 až 1,20).
Roflumilast zlepšil funkciu pľúc po 4 týždňoch (ktorá sa zachovala po dobu 52 týždňov). Hodnoty
FEV1 po podaní bronchodilatátora sa zvýšili v skupine s roflumilastom o 52 ml (95% IS: 40, 65 ml)
a znížili v skupine s placebom o 4 ml (95% IS: -16, 9 ml). Hodnoty FEV1 po podaní bronchodilatátora ukázali klinicky významné zlepšenie v prospech roflumilastu o 56 ml nad placebom (95% IS: 38,
73 ml).
Sedemnásť (1,8%) pacientov v skupine s roflumilastom a 18 (1,9%) pacientov v skupine s placebom zomrelo počas dvojito zaslepenej fázy z akýchkoľvek dôvodov a 7 (0,7%) pacientov v každej skupine zomrelo z dôvodu exacerbácie CHOCHP. Pomer pacientov, u ktorých sa vyskytol aspoň 1 nežiaduci účinok počas dvojito zaslepenej fázy bol 648 (66,9%) pacientov a 572 (59,2%) pacientov v skupinách
s roflumilastom a placebom, v uvedenom poradí. Pozorované nežiaduce účinky pre roflumilast v štúdii
RO-2455-404-RD boli v súlade s nežiaducimi účinkami už zahrnutými v časti 4.8.
Viac pacientov v skupine s roflumilastom (27,6%) v porovnaní s placebom (19,8%) ukončilo užívanie skúmaného lieku z akéhokoľvek dôvodu (pomer rizika: 1,40; 95% IS: 1,19 až 1,65). Najhlavnejšími dôvodmi ukončenia účasti na štúdii boli stiahnutie súhlasu a hlásenie nežiaducich účinkov.
Skúšanie titrácie úvodnejdávky
Znášanlivosť roflumilastu bola hodnotená v 12-týždňovom randomizovanom, dvojito zaslepenom
skúšaní s paralelnými skupinami (RO-2455-302-RD) u pacientov so závažnou CHOCHP súvisiacou s chronickou bronchitídou. Počas skríningu museli mať pacienti aspoň jednu exacerbáciu
v predchádzajúcom roku a museli dostávať štandardnú udržiavaciu liečbu CHOCHP počas minimálne
12 týždňov. Celkovo 1 323 pacientov bolo randomizovaných, aby dostávali 500 mikrogramov roflumilastu raz denne počas 12 týždňov (n=443), 500 mikrogramov roflumilastu každý druhý deň počas 4 týždňov a potom 500 mikrogramov roflumilastu raz denne počas 8 týždňov (n=439) alebo
250 mikrogramov roflumilastu raz denne počas 4 týždňov a potom 500 mikrogramov roflumilastu raz denne počas 8 týždňov (n=441).
Počas celého obdobia štúdie 12 týždňov bolo percento pacientov, ktorí vysadili liečbu z akéhokoľvek dôvodu, štatisticky významne nižšie u pacientov, ktorí na začiatku užívali 250 mikrogramov roflumilastu raz denne počas 4 týždňov a potom 500 mikrogramov roflumilastu raz denne počas 8 týždňov (18,4%) v porovnaní s pacientmi, ktorí dostávali 500 mikrogramov roflumilastu raz denne počas 12 týždňov (24,6%; pomer pravdepodobnosti 0,66, 95% IS [0,47; 0,93], p=0,017). Miera vysadenia u pacientov, ktorí užívali 500 mikrogramov každý druhý deň počas 4 týždňov a potom
500 mikrogramov raz denne počas 8 týždňov, nebola štatisticky významne odlišná ako u pacientov,
ktorí užívali 500 mikrogramov raz denne počas 12 týždňov. Percento pacientov, u ktorých sa vyskytovali sledované nežiaduce udalosti súvisiace s liečbou (Treatment Emergent Adverse Event, TEAE), definované ako hnačka, nauzea, bolesť hlavy, znížená chuť do jedla, nespavosť a bolesť brucha (sekundárny cieľ), bolo štatisticky významne nižšie u pacientov, ktorí na začiatku užívali
250 mikrogramov roflumilastu raz denne počas 4 týždňov a potom 500 mikrogramov roflumilastu raz denne počas 8 týždňov (45,4%) v porovnaní s pacientmi, ktorí užívali 500 mikrogramov roflumilastu raz denne počas 12 týždňov (54,2%, pomer pravdepodobnosti 0,63, 95% IS [0,47; 0,83], p=0,001). Miera výskytu sledovaných TEAE u pacientov užívajúcich 500 mikrogramov každý druhý deň počas
4 týždňov a potom 500 mikrogramov raz denne počas 8 týždňov nebola štatisticky významne odlišná ako u pacientov užívajúcich 500 mikrogramov raz denne počas 12 týždňov.
Pacienti užívajúci 500 mikrogramov raz denne mali medián inhibičnej aktivity PDE4 1,2 (0,35; 2,03)
a pacienti užívajúci 250 mikrogramov raz denne mali medián inhibičnej aktivity PDE4 0,6 (0,20;
1,24). Dlhodobé podávanie v dávke 250 mikrogramov nemusí indukovať dostatočnú inhibíciu PDE4
potrebnú na vyvolanie klinickej účinnosti. 250 mikrogramov raz denne je subterapeutická dávka a má sa používať len ako úvodná dávka počas prvých 28 dní (pozri časti 4.2 a 5.2).
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s roflumilastom vo všetkých podskupinách pediatrickej populácie s chronickým obštrukčným ochorením pľúc (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Roflumilast sa u ľudí extenzívne metabolizuje a tvorí sa hlavný farmakodynamicky aktívny metabolit roflumilast N-oxid. Vzhľadom na to, že roflumilast aj roflumilast N-oxid sa podieľajú na PDE4 inhibičnej aktivite in vivo, farmakokinetické pozorovania sú založené na celkovej PDE4 inhibičnej aktivite (teda celkovej expozícii roflumilastu aj roflumilast N-oxidu).
Absorpcia
Absolútna biologická dostupnosť roflumilastu po perorálnej dávke 500 mikrogramov je približne 80%. Maximálne plazmatické koncentrácie roflumilastu sa dosahujú približne 1 hodinu po užití dávky (od
0,5 h po 2 h) nalačno. Maximálne koncentrácie metabolitu N-oxidu sa dosiahnu približne po 8 hodinách (od 4 h po 13 h). Príjem potravy neovplyvňuje PDE4 inhibičnú aktivitu, ale spomaľuje čas na dosiahnutie maximálnej koncentrácie (tmax) roflumilastu o 1 hodinu a znižuje Cmax približne o 40%. Cmax a tmax roflumilast N-oxidu však nie sú ovplyvnené.
Distribúcia
Na bielkoviny sa viaže približne 99% roflumilastu a 97% roflumilast N-oxidu. Distribučný objem pre jednorazovú dávku 500 mikrogramov roflumilastu je okolo 2,9 l/kg. Vzhľadom na fyzikálno-chemické vlastnosti sa roflumilast rýchlo distribuuje do orgánov a tkanív včítane tukových tkanív myší,
škrečkov a potkanov. Po skorej distribučnej fáze so zjavnou penetráciou do tkanív dochádza k zjavnej eliminačnej fáze z tukových tkanív, najpravdepodobnejšie ako dôsledok rozkladu pôvodnej zlúčeniny na roflumilast N-oxid. Tieto štúdie na potkanoch s rádioaktívne značeným roflumilastom indikujú aj nízku penetráciu cez krvno-mozgovú bariéru. Nie je žiaden dôkaz špecifickej akumulácie alebo zadržiavania roflumilastu alebo jeho metabolitov v orgánoch a tukových tkanivách.
Biotransformácia
Roflumilast sa extenzívne metabolizuje reakciami fázy I (cytochróm P450) a fázy II (konjugácia).
N-oxid je hlavným metabolitom zisteným v ľudskej plazme. Plazmatické AUC metabolitu N-oxidu je priemerne 10-násobne vyššie ako plazmatické AUC roflumilastu. Preto sa predpokladá, že N-oxid sa významne podieľa na celkovej PDE4 inhibičnej aktivite in vivo.
Štúdie in vitro a štúdie klinických interakcií naznačujú, že k metabolizovaniu roflumilastu na jeho N-oxid dochádza prostredníctvom CYP1A2 a 3A4. Z ďalších výsledkov in vitro v mikrozómoch ľudskej pečene vyplýva, že terapeutické plazmatické koncentrácie roflumilastu a roflumilast N-oxidu neinhibujú CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5 alebo 4A9/11. Z toho vyplýva, že je malá pravdepodobnosť relevantných interakcií so zlúčeninami metabolizovanými týmito P450 enzýmami. Okrem toho sa v in vitro štúdiách nepreukázala žiadna indukcia CYP1A2, 2A6, 2a9, 2C19 alebo 3A4/5 a len slabá indukcia CYP2B6 roflumilastom.
Eliminácia
Plazmatický klírens po krátkodobej intravenóznej infúzii roflumilastu je približne 9,6 l/h. Po perorálnej dávke je stredný účinný plazmatický polčas roflumilastu a jeho metabolitu N-oxidu
približne 17 a 30 hodín. Rovnovážne plazmatické koncentrácie roflumilastu a jeho metabolitu N-oxidu sa dosiahnu približne po 4 dňoch u roflumilastu a po 6 dňoch u jeho metabolitu N-oxidu po podaní jednej dávky denne. Po intravenóznom alebo perorálnom podaní rádioaktívne značeného roflumilastu
sa vylúči stolicou okolo 20% rádioaktivity a 70% močom ako neaktívne metabolity.
Linearita/nelinearita
Farmakokinetika roflumilastu a jeho metabolitu N-oxidu sú úmerné dávke v rozsahu dávok od
250 mikrogramov po 1 000 mikrogramov.
Osobitnépopulácie
U starších ľudí, žien a osôb inej ako bielej pleti bola celková inhibičná aktivita PDE4 zvýšená. Celková inhibičná aktivita PDE4 bola mierne znížená u fajčiarov. Žiadna z týchto zmien sa nepovažovala za klinicky významnú. U týchto pacientov nie je potrebné upravovať dávku. Kombinácia faktorov ako nefajčiarka čiernej pleti môže viesť k zvýšenej expozícii a pretrvávajúcej netolerovateľnosti. V takomto prípade je potrebné liečbu roflumilastom prehodnotiť (pozri časť 4.4).
V štúdii RO-2455-404-RD, pri porovnaní s celkovou populáciou bola zistená celková inhibičná aktivita PDE4 určená z ex vivo neviazaných frakcií, o 15% vyššia u pacientov vo veku ≥ 75 rokov a o 11% vyššia u pacientov so základnou telesnou hmotnosťou < 60 kg (pozri časť 4.4).
Porucha funkcie obličiek
U pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu 10-30 ml/min) bola celková inhibičná aktivita PDE4 znížená o 9%. Nie je potrebná úprava dávky.
Porucha funkcie pečene
Farmakokinetika roflumilastu v dávke 250 mikrogramov jedenkrát denne sa hodnotila u 16 pacientov
s miernou až stredne ťažkou poruchou funkcie pečene klasifikovanou ako Child-Pugh A a B. U týchto
pacientov sa celková inhibičná aktivita PDE4 zvýšila približne o 20% u pacientov s Child-Pugh
A a približne o 90% u pacientov s Child-Pugh B. Simulácie podporujú proporcionalitu dávok medzi roflumilastom 250 mikrogramov a 500 mikrogramov u pacientov s miernou až stredne ťažkou poruchou funkcie pečene. U pacientov s Child-Pugh A je potrebná opatrnosť (pozri časť 4.2). Pacienti so stredne ťažkou alebo ťažkou poruchou funkcie pečene klasifikovanou ako Child-Pugh B alebo C nemajú užívať roflumilast (pozri časť 4.3).
5.3 Predklinické údaje o bezpečnosti
Nie je žiaden dôkaz pre imunotoxický, kožu senzibilizujúci alebo fototoxický potenciál.
V spojení epididymálnou toxicitou u potkanov bolo popísané mierne zníženie fertility samcov. Epididymálna toxicita alebo zmeny parametrov semena sa nezistili u hlodavcov alebo iných druhov včítane opíc ani pri vysokej expozícii.
V jednej z dvoch štúdií embryofetálneho vývoja u potkanov je popísaná vyššia incidencia nekompletnej osifikácie kostí lebky pri dávkach, ktoré spôsobili toxicitu u matky. V jednej z troch štúdií fertility a embryofetálneho vývoja u potkanov sa pozorovali poimplantačné straty. Poimplantačné straty sa nepozorovali u králikov. Predĺžená gestácia sa pozorovala u myší.
Závažnosť týchto pozorovaní pre človeka nie je známa.
Najzávažnejšie zistenia v štúdiách bezpečnej farmakológie a toxikológie sa pozorovali pri vysokých dávkach a expozícii dlhších ako bola predpokladaná dávka pre klinické použitie. Tieto zistenia sa týkali gastrointestinálnych nálezov (napr. vracanie, zvýšená žalúdočná sekrécia, erózie žalúdka, zápal čriev) a kardiálnych nálezov (napr. fokálnych krvácaní, hemosiderického depozitu a infiltrácie
lymfo-histiocytárnych buniek do pravého atria psov, zníženie krvného tlaku a zvýšenie frekvencie
srdca u potkanov, morčiat a psov).
Toxicita špecifická pre hlodavce sa pozorovala na sliznici nosa v štúdiách toxicity po opakovaných dávkach a v štúdiách karcinogenity. Predpokladá sa, že tento efekt je dôsledkom ADCP
(4-amino-3,5-dichloropyridín) N-oxidu, medziproduktu, ktorý sa tvorí na očnej sliznici hlodavcov a má špecifickú väzbovú afinitu u týchto druhov (napr. myš, potkan a škrečok).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
monohydrát laktózy kukuričný škrob povidón magnéziumstearát
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti4 roky
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaPVC/PVDC hliníkové blistre v balení po 28 tabliet. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLOEU/1/10/636/008
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 5. júl 2010
Dátum posledného predĺženia registrácie: 24. apríl 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.

Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUDaxas 500 mikrogramov filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá tableta obsahuje 500 mikrogramov roflumilastu.
Pomocná látka soznámym účinkom:Každá filmom obalená tableta obsahuje 188,72 mg laktózy vo forme monohydrátu.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAFilmom obalená tableta.
Žltá filmom obalená 9 mm tableta tvaru D s embosovaným “D” na jednej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieDaxas je indikovaný na udržiavaciu liečbu závažnej chronickej obštrukčnej choroby pľúc (CHOCHP) (FEV1 po podaní bronchodilatátora menej ako 50% predpokladanej hodnoty) spojenej s chronickou bronchitídou u dospelých pacientov s častými exacerbáciami v anamnéze ako prídavný liek
k bronchodilatačnej liečbe.
4.2 Dávkovanie a spôsob podávaniaDávkovanieÚvodnádávkaOdporúčaná úvodná dávka je jedna tableta 250 mikrogramov roflumilastu podávaná raz denne počas
28 dní.
Táto úvodná dávka má znížiť výskyt nežiaducich udalostí a vysadenia lieku u pacienta na začiatku liečby, avšak je to subterapeutická dávka. Dávka 250 mikrogramov sa má preto používať len ako úvodná dávka (pozri časti 5.1 a 5.2).
UdržiavaciadávkaPo 28 dňoch liečby úvodnou dávkou 250 mikrogramov sa musí dávka u pacienta titrovať na jednu
tabletu 500 mikrogramov roflumilastu podávanú raz denne.
Maximálny účinok Daxasu 500 mikrogramov sa môže prejaviť až po niekoľkých týždňoch užívania (pozri časti 5.1 a 5.2). Daxas 500 mikrogramov sa hodnotil v klinických štúdiách trvajúcich až 1 rok a je určený na udržiavaciu liečbu.
Osobitnépopulácie
Starší pacienti
Nie je potrebná úprava dávky.
Porucha funkcie obličiek
Nie je potrebná úprava dávky.
Porucha funkcie pečene
Klinické údaje o podávaní Daxasu pacientom s miernou poruchou funkcie pečene klasifikovanou ako Child-Pugh A sú nedostatočné na odporučenie úpravy dávkovania (pozri časť 5.2), preto sa má Daxas u týchto pacientov používať s opatrnosťou.
Pacienti so stredne ťažkou až ťažkou poruchou funkcie pečene klasifikovanou ako Child-Pugh B alebo
C nesmú užívať Daxas (pozri časť 4.3).
Pediatrická populácia
V pediatrickej populácii (mladšej ako 18 rokov) nie je používanie Daxasu v indikácii CHOCHP
relevantné.
Spôsob podávania
Perorálne použitie.
Tableta sa má prehltnúť s vodou a užívať v rovnakom dennom čase. Tableta sa môže užívať s jedlom alebo bez jedla.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Stredne ťažká až ťažká porucha funkcie pečene (Child-Pugh B alebo C).
4.4 Osobitné upozornenia a opatrenia pri používaní
Pred začiatkom liečby Daxasom majú byť všetci pacienti informovaní o rizikách Daxasu a o opatreniach pre jeho bezpečné užívanie a majú dostať kartu pre pacienta.
Záchrannélieky
Daxas nie je indikovaný ako záchranný liek na úľavu akútnych bronchospazmov.
Zníženiehmotnosti
V štúdiách trvajúcich 1 rok (M2-124, M2–125) sa častejšie znížila telesná hmotnosť u pacientov liečených roflumilastom v porovnaní s pacientmi, ktorí užívali placebo. Po prerušení liečby roflumilastom sa u väčšiny pacientov obnovila pôvodná hmotnosť po 3 mesiacoch.
Pacientom so zníženou hmotnosťou sa má telesná hmotnosť kontrolovať pri každej návšteve. Pacientov treba poučiť, aby si svoju hmotnosť pravidelne kontrolovali. V prípade nevysvetleného
a klinicky významného zníženia hmotnosti sa má liečba roflumilastom zastaviť a naďalej sledovať telesná hmotnosť.
Osobitnéklinicképodmienky
Vzhľadom na chýbajúce relevantné skúsenosti sa liečba roflumilastom nemá začínať alebo už začatá liečba roflumilastom sa má zastaviť u pacientov so závažnými imunologickými ochoreniami (napr. infekcia HIV, skleróza multiplex, lupus erythematosus, progresívna multifokálna leukoencefalopatia), so závažnými akútnymi infekčnými ochoreniami, nádorovými ochoreniami (okrem bazálneho bunkového karcinómu) alebo u pacientov liečených imunosupresívnymi liekmi (napr. metotrexát, azatioprin, infliximab, etanercept, alebo dlhodobo liečených perorálnymi kortikosteroidmi; okrem krátko účinkujúcich systémových kortikosteroidov). Skúsenosti s pacientmi s latentnými infekciami ako tuberkulóza, vírusová hepatitída, herpesové vírusové infekcie a herpes zoster sú obmedzené. Pacientom s kongestívnym srdcovým zlyhaním (NYHA stupeň 3 a 4) sa táto liečba neodporúča, pretože neboli sledovaní.
Psychicképoruchy
Roflumilast je spojený so zvýšeným rizikom psychických porúch ako je nespavosť, úzkosť, nervozita a depresie. Zriedkavo boli pozorované prípady samovražedných úvah a správania vrátane samovraždy u pacientov s depresiou v anamnéze alebo bez nej, najčastejšie v prvých týždňoch liečby (pozri časť
4.8). Riziko a prínos začatia alebo pokračovania liečby roflumilastom sa má dôkladne zvážiť
u pacientov, ktorí mali v minulosti alebo majú v súčasnosti psychické príznaky alebo ak sa uvažuje o súbežnej liečbe inými liekmi, ktoré by mohli vyvolať psychické problémy. Roflumilast sa neodporúča u pacientov s depresiou v anamnéze spojenou so samovražednými myšlienkami alebo správaním. Pacientov a ich opatrovateľov treba poučiť, aby oznámili lekárovi, ktorý predpísal liek zmeny v náladách alebo rozmýšľanie o samovražde. Ak pacienti trpia novými alebo zhoršujúcimi sa symptómami, alebo sa vyskytnú myšlienky alebo pokus o samovraždu, odporúča sa liečbu roflumilastom prerušiť.
Pretrvávajúcaintolerancia
Nežiaduce účinky ako hnačka, nauzea, bolesť brucha a bolesť hlavy sa vyskytujú hlavne v prvých
týždňoch liečby, s postupujúcou liečbou väčšinou vymiznú. Liečbu roflumilastom však treba prehodnotiť pri pretrvávajúcej intolerancii. Výskyt je možný u osobitnej populácie s možnou vyššou expozíciou, ako sú ženy nefajčiarky čiernej pleti (pozri časť 5.2) alebo u pacientov súbežne liečených inhibítormi CYP1A2/2C19/3A4 (ako je fluvoxamín alebo cimetidín) alebo inhibítorom CYP1A2/3A4 enoxacínom (pozri časť 4.5).
Telesnáhmotnosť < 60kg
Liečba roflumilastom môže viesť k vyššiemu riziku výskytu porúch spánku (hlavne nespavosti) u pacientov so základnou telesnou hmotnosťou < 60 kg, kvôli celkovej vyššej inhibičnej aktivite PDE4 u týchto pacientov (pozri časť 4.8).
Teofylín
Nie sú žiadne klinické údaje, ktoré podporujú súbežnú liečbu s teofylínom ako udržiavaciu liečbu. Súbežná liečba s teofylínom sa preto neodporúča.
Laktóza
Tablety Daxasu obsahujú monohydrát laktózy. Pacienti so zriedkavou vrodenou intoleranciou
galaktózy, laponskou deficienciou laktázy alebo s poruchou absorpcie glukózy a galaktózy nemajú užívať tento liek.
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Hlavným stupňom metabolizmu roflumilastu je jeho N-oxidácia na roflumilast N-oxid prostredníctvom CYP3A4 a CYP1A2. Roflumilast aj roflumilast N-oxid majú vlastnú inhibičnú aktivitu fosfodiesterázy 4 (PDE4). Predpokladá sa, že celková inhibícia PDE4 po aplikácii roflumilastu je kombinovaný účinok obidvoch zložiek, roflumilastu aj roflumilast N-oxidu. Štúdie
interakcií s inhibítorom CYP1A2/3A4 enoxacínom a inhibítormi CYP1A2/2C19/3A4 fluvoxamínom a cimetidínom preukázali zvýšenú celkovú PDE4 inhibičnú aktivitu o 25%, 47% a 59%. Testovaná dávka fluvoxamínu bola 50 mg. Kombinácia roflumilastu s týmito liečivami môže viesť k zvýšenej expozícii a pretrvávajúcej intolerancii. V takomto prípade sa má liečba roflumilastom prehodnotiť (pozri časť 4.4).
Pri aplikácii rifampicínu, ktorý indukuje cytochrómový enzým P450, sa znížila celková PDE4 inhibičná aktivita približne o 60%. Použitie silných induktorov cytochrómu P450 (napr. fenobarbital, karbamazepín, fenytoín) môže preto znižovať terapeutickú účinnosť roflumilastu. Z tohto dôvodu sa pacientom užívajúcim silné inhibítory cytochrómu P450 liečba roflumilastom neodporúča.
Klinické interakčné štúdie s inhibítormi CYP3A4 erytromycínom a ketokonazolom preukázali zvýšenie celkovej inhibičnej aktivity PDE4 o 9%. Súbežná aplikácia s teofylínom spôsobila zvýšenie celkovej inhibičnej aktivity PDE4 o 8% (pozri časť 4.4). V štúdii interakcie s perorálnymi kontraceptívami, ktoré obsahovali gestodén a etinylestradiol, sa celková PDE4 inhibičná aktivita
zvýšila o 17%. Z tohto dôvodu, u pacientov, ktorí užívajú uvedené liečivá, nie je nutná úprava dávkovania.
Nezistili sa žiadne klinicky významné interakcie s inhalovaným salbutamolom, formoterolom, budezonidom a perorálne aplikovaným montelukastom, digoxínom, warfarínom, sildenafilom a midazolamom.
Súbežná aplikácia s antacidom (kombinácia hydroxidu hlinitého a magnézium hydroxidu) nezmenila absorpciu ani farmakokinetiku roflumilastu alebo jeho N-oxidu.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby. Roflumilast sa neodporúča
u žien vo fertilnom veku, ktoré nepoužívajú antikoncepciu.
Gravidita
Údaje o podávaní roflumilastu gravidným ženám sú obmedzené.
Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Roflumilast sa neodporúča počas gravidity.
U gravidných samíc potkanov sa preukázalo, že roflumilast prechádza placentou. Dojčenie
Dostupné farmakokinetické údaje na zvieratách preukázali vylučovanie roflumilastu alebo jeho metabolitov do materského mlieka. Nie je možné vylúčiť riziko pre dojčené dieťa. Roflumilast sa nemá používať počas laktácie.
Fertilita
V štúdii spermatogenézy u ľudí nemala dávka 500 mikrogramov roflumilastu žiaden účinok na
parametre semena alebo reprodukčné hormóny počas 3-mesačnej liečby ani nasledujúce 3 mesiace po jej ukončení.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Daxas nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
V klinických štúdiách CHOCHP sa u približne 16% pacientov prejavili nežiaduce reakcie roflumilastu (v porovnaní s 5% výskytom u placeba). Najčastejšie hlásenými nežiaducimi reakciami boli hnačka (5,9%), pokles hmotnosti (3,4%), nauzea (2,9%), bolesť brucha (1,9%) a bolesť hlavy (1,7%). Väčšina nežiaducich reakcií bola mierna až stredná. Nežiaduce reakcie sa vyskytli hlavne v prvých týždňoch liečby a pokračovaním liečby väčšinou vymizli.
Tabuľkovýprehľadnežiaducichreakcií
Frekvencia výskytu nežiaducich reakcií v nasledujúcej tabuľke je uvedená podľa klasifikácie
MedDRA:
veľmi časté (³1/10); časté (³1/100 až <1/10); menej časté (³1/1 000 až <1/100); zriedkavé (³1/10 000
až <1/1 000); veľmi zriedkavé (<1/10 000), neznáme (z dostupných údajov).
V každej skupine frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 1. Nežiaduce reakcie roflumilastu v klinických štúdiách CHOCHP a postmarketingové údaje
Frekvencia
Čast
é Menej časté Zriedkavé
Trieda
orgánovýc
h systémov Poruchy imunitného systému
Poruchy endokrinného systému
Poruchy metabolizmu a výživy
Zníženie hmotnosti Zníženie chuti do jedla
Precitlivenosť Angioedém
Gynekomastia
Psychické poruchy Nespavosť Úzkosť Samovražedné myšlienky
a správanie*
Depresia Nervozita Záchvat paniky
Poruchy nervového systému
Poruchy srdca a srdcovej činnosti
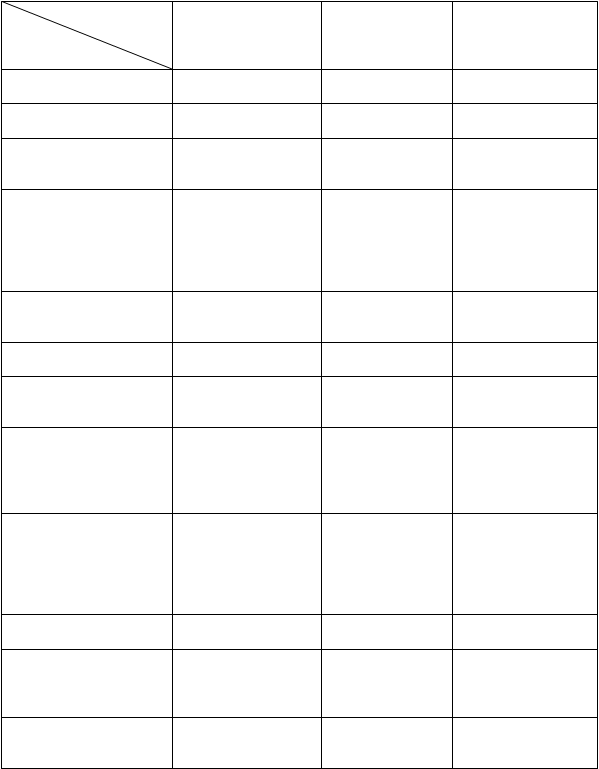
Poruchy dýchacej sústavy, hrudníka a mediastína
Bolesť hlavy Tras Vertigo Závraty Palpitácie
Poruchy chuti
Infekcie dýchacej sústavy (mimo zápalu pľúc)
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
Hnačka
Nauzea
Bolesť brucha
Gastritída Vracanie Gastroezofageálna refluxná choroba Dyspepsia
Krv v stolici
Obstipácia
Zvýšenie
gamma-glutamyl
transpeptidázy Zvýšenie aspartátaminotransfe- rázy (AST)
Poruchy kože a podkožného tkaniva
Vyrážka Žihľavka
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Celkové poruchy a reakcie v mieste podania
Svalové kŕče a slabosť Bolesť svalov Bolesť chrbta Malátnosť Slabosť
Únava
Zvýšenie hladiny kreatinínfosfokinázy (CPK) v krvi
Popi
s
zvolených
nežiaducich
reakcií
* V klinických štúdiách a po zavedení lieku na trh boli pozorované zriedkavé prípady samovražedných myšlienok a správania, vrátane samovraždy. Pacientov a opatrovateľov treba poučiť, aby oznámili predpisujúcemu lekárovi akékoľvek suicidálne myšlienky (pozri časť 4.4).
Inéosobitnépopulácie
Vyšší výskyt porúch spánku (hlavne nespavosti) u pacientov vo veku ≥ 75 rokov alebo starších sa
pozoroval v štúdii RO-2455-404-RD u pacientov liečených roflumilastom v porovnaní s pacientmi užívajúcimi placebo (3,9% oproti 2,3%). Pozorovaný výskyt bol tiež vyšší u pacientov mladších ako
75 rokov, ktorí boli liečení roflumilastom, v porovnaní s pacientmi užívajúcimi placebo (3,1% oproti
2,0%).
Vyšší výskyt porúch spánku (hlavne nespavosti) u pacientov so základnou telesnou hmotnosťou
< 60 kg sa pozoroval v štúdii RO-2455-404-RD u pacientov liečených roflumilastom v porovnaní
s pacientmi užívajúcimi placebo (6,0% oproti 1,7%). U pacientov so základnou telesnou hmotnosťou
≥ 60 kg liečených roflumilastom bol výskyt 2,5% oproti 2,2% pacientov užívajúcich placebo.
Súbežnáliečbas dlhodobopôsobiacimi antagonistamimuskarínovýchreceptorov(LAMA)Vyšší výskyt poklesu telesnej hmotnosti, zníženej chuti do jedla, bolesti hlavy a depresie sa pozoroval počas štúdie RO-2455-404-RD u pacientov súbežne užívajúcich roflumilast a dlhodobo pôsobiace antagonisty muskarínových receptorov (LAMA) spolu so súbežne inhalovanými kortikosteroidmi (ICS) a dlhodobo pôsobiacimi agonistami B2 receptorov (LABA) v porovnaní s pacientmi, ktorí sa súbežne liečili len roflumilastom, ICS a LABA. Rozdiel výskytu medzi roflumilastom a placebom bol kvantitatívne vyšší so súbežne užívaným LAMA pre pokles hmotnosti (7,2% oproti 4,2%), zníženú chuť do jedla (3,7% oproti 2,0%), bolesť hlavy (2,4% oproti 1,1%) a depresiu (1,4% oproti -0,3%).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.'
4.9 PredávkovaniePríznakyV štúdiách vo fáze I sa po jednorazových perorálnych dávkach 2 500 mikrogramov a jednej dávke
5 000 mikrogramov (desaťnásobok odporučenej dávky) pozoroval zvýšený výskyt nasledujúcich príznakov: bolesť hlavy, gastrointestinálne poruchy, závraty, palpitácie, mierne vertigo, zvýšené potenie a arteriálna hypotenzia.
LiečbaPokiaľ dôjde k predávkovaniu, odporúča sa poskytnúť vhodnú podpornú medicínsku starostlivosť. Vzhľadom na to, že sa roflumilast výrazne viaže na bielkoviny, hemodialýza pravdepodobne nie je účinný spôsob na odstránenie liečiva. Nie je známe, či je roflumilast dialyzovateľný peritoneálnou dialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Lieky na obštrukčné ochorenia dýchacích ciest, iné systémové liečivá ochorení spojených s obštrukciou dýchacích ciest, ATC kód: R03DX07
MechanizmusúčinkuRoflumilast je inhibítor PDE4, nesteroidové protizápalové liečivo s cieleným účinkom na systémové a pľúcne zápaly spojené s CHOCHP. Mechanizmom účinku je inhibícia PDE 4, hlavného enzýmu, ktorý metabolizuje cyklický adenozín monofosfát (cAMP) a zistilo sa, že je dôležitý pre patogenézu CHOCHP v štrukturálnych a zápalových bunkách. Roflumilast pôsobí na spojené varianty reťazca PDE4A, 4B a 4D s podobnou potenciou na nanomolárnej úrovni. Afinita k spojenému variantu PDE4C je 5 až 10 násobne nižšia. Tento mechanizmus účinku a selektívnosť platí aj pre roflumilast N-oxid, čo je hlavný aktívny metabolit roflumilastu.
FarmakodynamickéúčinkyInhibícia PDE4 vedie k zvýšeniu hladiny vnútrobunkového cAMP a v experimentálnych modeloch
zmierňuje porušené funkcie leukocytov, hladkých svalov buniek dýchacích ciest a v pľúcnych cievach, endoteliálnych bunkách a epiteliálnych bunkách dýchacích ciest a fibroblastov, ktoré boli porušené
dôsledkom CHOCHP. Po in vitro stimulácii ľudských neutrofilov, monocytov, makrofágov alebo lymfocytov roflumilast a roflumilast N-oxid potláčal uvoľňovanie mediátorov zápalu, napr. leukotriénu B4, reaktívnych foriem kyslíka, tumor nekrotizujúceho faktoru α, interferónu γ
a granzymu B.
U pacientov s CHOCHP roflumilast znižoval počet neutrofilov v spúte. Okrem toho roflumilast zoslaboval prienik neutrofilov a eozinofilov do dýchacích ciest u zdravých dobrovoľníkov, ktorým sa aplikoval endotoxín.
Klinickáúčinnosť abezpečnosť
V dvoch konfirmačných opakovaných jednoročných štúdiách (M-124 a M-125) a v dvoch
doplňujúcich šesťmesačných štúdiách (M2-127 a M2-128) bolo randomizovaných celkovo 4 768 pacientov, z toho bolo 2 374 liečených roflumilastom. Štúdie mali paralelné skupiny, boli dvojito zaslepené a kontrolované placebom.
V jednoročných štúdiách sledovali pacientov s anamnézou závažnej alebo veľmi závažnej CHOCHP
[FEV1 (úsilný expiračný objem za sekundu) ≤ 50% predpokladanej hodnoty] spojenej s chronickou bronchitídou, aspoň s jednou dokumentovanou exacerbáciou v predchádzajúcom roku a s príznakmi určenými vstupnými údajmi podľa skóre kašľa a spúta. Používanie dlhodobo účinkujúcich
beta-agonistov (LABA) bolo počas štúdie povolené, použilo ich približne 50% pacientov. Krátko účinkujúce anticholinergiká (SAMA) boli povolené pre pacientov, ktorí neužívali LABA. Záchranná medikácia (salbutamol alebo albuterol) bola povolená podľa potreby. Používanie inhalačných kortikosteroidov a teofylínu bolo počas štúdie zakázané. Pacienti bez exacerbácií v anamnéze boli vylúčení.
V súhrnnej analýze jednoročných štúdií M2-124 a M2-125 roflumilast v dávke 500 mikrogramov jeden krát denne signifikantne zlepšoval funkciu pľúc v porovnaní s placebom, priemerne o 48 ml (FEV1 pred podaním bronchodilatátora, primárny cieľ, p<0,0001) a o 55 ml (FEV1 po podaní bronchodilatátora, p<0,0001). Zlepšenie funkcie pľúc bolo zreteľné pri prvej návšteve po 4 týždňoch a pretrvávalo 1 rok (ukončenie liečby). Pomer (pacient/rok) stredných exacerbácií (vyžadujúcich podávanie systémových glukokortikosteroidov) alebo závažných exacerbácií (vyžadujúcich hospitalizáciu a/alebo vedúcich k úmrtiu) po jednom roku bol 1,142 u roflumilastu a 1,374 u placeba, čo zodpovedá zníženiu relatívneho rizika o 16,9% (95% IS: 8,2% až 24,8%) (primárny cieľ, p=0,0003). Účinky boli podobné, nezávislé od predchádzajúcej liečby inhalačnými kortikosteroidmi
alebo liečbou LABA. V podskupine pacientov s anamnézou častých exacerbácií (aspoň 2 exacerbácie počas posledného roka), bol pomer exacerbácií 1,526 u roflumilastu a 1,941 u placeba, čo zodpovedá zníženiu relatívneho rizika o 21,3% (95% IS: 7,5% až 33,1%). Roflumilast signifikantne neznižoval výskyt exacerbácií v porovnaní s placebom v podskupine pacientov so strednou CHOCHP.
Počet stredných alebo závažných exacerbácií u pacientov s roflumilastom a LABA v porovnaní
s placebom a LABA sa znížil priemerne 21% (p=0,0011). Pokles exacerbácií u pacientov bez súbežnej liečby s LABA bol v priemere 15% (p=0,0387). Počet pacientov, ktorí zomreli z akýchkoľvek dôvodov, bol rovnaký v skupine pacientov, ktorí boli liečení roflumilastom alebo dostávali placebo
(42 úmrtí v každej skupine; 2,7% v každej skupine; súhrnná analýza).
Do dvoch podporných randomizovaných 1 ročných štúdií (M2-111 a M2-112) bolo zaradených a randomizovaných celkovo 2 690 pacientov. Na rozdiel od dvoch konfirmatívnych štúdií sa na zaradenie do štúdie nevyžadovala anamnéza chronickej bronchitídy a CHOCHP exacerbácií. Inhalačné kortikosteroidy použilo 809 pacientov (61%), ktorí boli liečení roflumilastom, zatiaľ čo používanie LABA a teofylínu bolo zakázané. Roflumilast 500 mikrogramov jedenkrát denne signifikantne zlepšil funkciu pľúc v porovnaní s placebom, v priemere o 51 ml (FEV1 pred podaním bronchodilatátora, p<0,0001) a o 53 ml (FEV1 po podaní bronchodilatátora, p <0,0001). Výskyt exacerbácií (tak ako boli definované v protokoloch) v jednotlivých štúdiách roflumilast signifikantne neznižoval (zníženie relatívneho rizika: 13,5% v štúdii M2-111 a 6,6% v štúdii M2-112; p=nesignifikantné). Výskyt nežiaducich účinkov nezávisel od súbežnej liečby inhalačnými kortikosteroidmi.
Do dvoch podporných klinických štúdií (M2-127 a M2-128) boli zaradení pacienti s anamnézou CHOCHP diagnostikovanou najmenej 12 mesiacov pred zaradením do štúdie. Do obidvoch štúdií sa zaradili pacienti so strednou až ťažkou nereverzibilnou obštrukciou dýchacích ciest a FEV1 40% až
70% predpokladanej hodnoty. Roflumilast alebo placebo sa pridali k pokračujúcej liečbe dlhodobo
účinkujúcim bronchodilatátorom, hlavne k salmeterolu v štúdii M2-127 alebo k tiotropiu v štúdii M2-128. V dvoch 6-mesačných štúdiách sa FEV1 pred podaním bronchodilatátora signifikantne zlepšilo o 49 ml (primárny cieľ, p<0,0001) v súbežnej liečbe salmeterolom v štúdii M2-127 a o 80 ml (primárny cieľ, p<0,0001) v súbežnej liečbe tiotropiom v štúdii M2-128.
Štúdia RO-2455-404-RD bola jednoročná štúdia u pacientov s CHOCHP s počiatočnou hodnotou (pred podaním bronchodilatátora) FEV1 < 50% predpokladaných normálnych a minulých častých exacerbácií. Štúdia vyhodnocovala účinok roflumilastu na mieru exacerbácií CHOCHP u pacientov liečených pevnou kombináciou LABA a inhalovaných kortikosteroidov, v porovnaní s placebom. Celkom 1 935 pacientov bolo randomizovaných do dvojito zaslepenej liečby a približne 70% tiež používalo v priebehu štúdie dlhodobo pôsobiace antagonisty muskarínových receptorov (LAMA). Primárnym cieľom bolo zníženie miery stredných alebo závažných exacerbácií CHOCHP na pacienta na rok. Miera závažných exacerbácií CHOCHP a zmeny hodnôt FEV1 sa vyhodnocovali ako kľúčové sekundárne ciele.
Tabuľka 2. Súhrn cieľov týkajúcich sa exacerbácií CHOCHP v štúdii RO-2455-404-RD
Roflumilast
(N=969)
Placebo
(N=966)
P
o
me
r Roflumilast/Placebo
2-
stranná
Kategória
exacerbácie
Stredná alebo
ModelanalýzyPoissono
Miera(n)0,805
Miera (n)
(n)0,927
PomermierZmena(%) 95% IShodnot a p
závažná
vá regresia
(380)
(432) 0,868 -13,2 0,753;
1,002
0,0529
Stredná Poissono vá
regresia
Závažná negatívna binominá
0,574 (287)
0,239 (151)
0,627
(333) 0,914 -8,6 0,775;
1,078
0,315
(192) 0,757 -24,3 0,601;
0,2875
lna regresia
0,952 0,0175
Vyskytol sa trend smerom k zníženiu stredných a závažných exacerbácií u pacientov liečených
roflumilastom v porovnaní s placebom po dobu 52 týždňov, ktorý však nedosiahol štatistický význam (tabuľka 2). Predurčená analýza citlivosti s použitím modelu binominálnej regresie preukázala štatistický významný rozdiel -14,2% (pomer mier: 0,86; 95% IS: 0,74 až 0,99).
Pomery mier analýzy s Poissonovou regresiou podľa protokolu a analýzy pôvodného zámeru liečby vylučujúcej nevýznamnú citlivosť s Poissonovou regresiou boli 0,81 (95% IS: 0,69 až 0,94) a 0,89 (95% IS: 0,77 až 1,02), v uvedenom poradí.
Zníženia sa dosiahli v podskupine pacientov súbežne liečených LAMA (pomer mier: 0,88; 95% IS:
0,75 až 1,04) a v podskupine súbežne neliečenej LAMA (pomer mier: 0,83; 95% IS: 0,62 až 1,12).
Miera závažných exacerbácií sa znížila v celkovej skupine pacientov (pomer mier: 0,76; 95% IS: 0,60 až 0,95) s mierou 0,24 na pacienta/rok v porovnaní s mierou 0,32 na pacienta/rok u pacientov užívajúcich placebo. Podobné zníženie sa dosiahlo v podskupine pacientov súbežne liečenej LAMA (pomer mier: 0,77; 95% IS: 0,60 až 0,99) a v podskupine súbežne neliečenej LAMA (pomer mier:
0,71; 95% IS: 0,42 až 1,20).
Roflumilast zlepšil funkciu pľúc po 4 týždňoch (ktorá sa zachovala po dobu 52 týždňov). Hodnoty
FEV1 po podaní bronchodilatátora sa zvýšili v skupine s roflumilastom o 52 ml (95% IS: 40, 65 ml)
a znížili v skupine s placebom o 4 ml (95% IS: -16, 9 ml). Hodnoty FEV1 po podaní bronchodilatátora
ukázali klinicky významné zlepšenie v prospech roflumilastu o 56 ml nad placebom (95% IS: 38,
73 ml).
Sedemnásť (1,8%) pacientov v skupine s roflumilastom a 18 (1,9%) pacientov v skupine s placebom zomrelo počas dvojito zaslepenej fázy z akýchkoľvek dôvodov a 7 (0,7%) pacientov v každej skupine zomrelo z dôvodu exacerbácie CHOCHP. Pomer pacientov, u ktorých sa vyskytol aspoň 1 nežiaduci účinok počas dvojito zaslepenej fázy bol 648 (66,9%) pacientov a 572 (59,2%) pacientov v skupinách
s roflumilastom a placebom, v uvedenom poradí. Pozorované nežiaduce účinky pre roflumilast v štúdii
RO-2455-404-RD boli v súlade s nežiaducimi účinkami už zahrnutými v časti 4.8.
Viac pacientov v skupine s roflumilastom (27,6%) v porovnaní s placebom (19,8%) ukončilo užívanie skúmaného lieku z akéhokoľvek dôvodu (pomer rizika: 1,40; 95% IS: 1,19 až 1,65). Najhlavnejšími dôvodmi ukončenia účasti na štúdii boli stiahnutie súhlasu a hlásenie nežiaducich účinkov.
Štúdiatitrácieúvodnejdávky
Znášanlivosť roflumilastu bola hodnotená v 12-týždňovej randomizovanej, dvojito zaslepenej štúdii s paralelnými skupinami (RO-2455-302-RD) u pacientov so závažnou CHOCHP súvisiacou
s chronickou bronchitídou. Počas skríningu museli mať pacienti aspoň jednu exacerbáciu
v predchádzajúcom roku a museli dostávať štandardnú udržiavaciu liečbu CHOCHP počas minimálne
12 týždňov. Celkovo 1 323 pacientov bolo randomizovaných, aby dostávali 500 mikrogramov roflumilastu raz denne počas 12 týždňov (n=443), 500 mikrogramov roflumilastu každý druhý deň počas 4 týždňov a potom 500 mikrogramov roflumilastu raz denne počas 8 týždňov (n=439) alebo
250 mikrogramov roflumilastu raz denne počas 4 týždňov a potom 500 mikrogramov roflumilastu raz denne počas 8 týždňov (n=441).
Počas celého obdobia štúdie 12 týždňov bolo percento pacientov, ktorí vysadili liečbu z akéhokoľvek dôvodu, štatisticky významne nižšie u pacientov, ktorí na začiatku užívali 250 mikrogramov roflumilastu raz denne počas 4 týždňov a potom 500 mikrogramov roflumilastu raz denne počas 8 týždňov (18,4%) v porovnaní s pacientmi, ktorí dostávali 500 mikrogramov roflumilastu raz denne počas 12 týždňov (24,6%; pomer pravdepodobnosti 0,66, 95% IS [0,47; 0,93], p=0,017). Miera vysadenia u pacientov, ktorí užívali 500 mikrogramov každý druhý deň počas 4 týždňov a potom
500 mikrogramov raz denne počas 8 týždňov, nebola štatisticky významne odlišná ako u pacientov, ktorí užívali 500 mikrogramov raz denne počas 12 týždňov. Percento pacientov, u ktorých sa vyskytovali sledované nežiaduce udalosti súvisiace s liečbou (Treatment Emergent Adverse Event, TEAE), definované ako hnačka, nauzea, bolesť hlavy, znížená chuť do jedla, nespavosť a bolesť brucha (sekundárny cieľ), bolo štatisticky významne nižšie u pacientov, ktorí na začiatku užívali
250 mikrogramov roflumilastu raz denne počas 4 týždňov a potom 500 mikrogramov roflumilastu raz denne počas 8 týždňov (45,4%) v porovnaní s pacientmi, ktorí užívali 500 mikrogramov roflumilastu raz denne počas 12 týždňov (54,2%, pomer pravdepodobnosti 0,63, 95% IS [0,47; 0,83], p=0,001). Miera výskytu sledovaných TEAE u pacientov užívajúcich 500 mikrogramov každý druhý deň počas
4 týždňov a potom 500 mikrogramov raz denne počas 8 týždňov nebola štatisticky významne odlišná
ako u pacientov užívajúcich 500 mikrogramov raz denne počas 12 týždňov.
Pacienti užívajúci 500 mikrogramov jedenkrát denne mali medián inhibičnej aktivity PDE4 1,2 (0,35;
2,03) a pacienti užívajúci 250 mikrogramov jedenkrát denne mali medián inhibičnej aktivity PDE4 0,6
(0,20; 1,24). Dlhodobé podávanie v dávke 250 mikrogramov nemusí indukovať dostatočnú inhibíciu PDE4 potrebnú na vyvolanie klinickej účinnosti. 250 mikrogramov raz denne je subterapeutická dávka a má sa používať len ako úvodná dávka počas prvých 28 dní (pozri časti 4.2 a 5.2).
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s roflumilastom
vo všetkých podskupinách pediatrickej populácie s chronickým obštrukčným ochorením pľúc
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Roflumilast sa u ľudí extenzívne metabolizuje a tvorí sa hlavný farmakodynamicky aktívny metabolit roflumilast N-oxid. Vzhľadom na to, že roflumilast aj roflumilast N-oxid sa podieľajú na PDE4 inhibičnej aktivite in vivo, farmakokinetické pozorovania sú založené na celkovej PDE4 inhibičnej aktivite (teda celkovej expozícii roflumilastu aj roflumilast N-oxidu).
Absorpcia
Absolútna biologická dostupnosť roflumilastu po perorálnej dávke 500 mikrogramov je približne 80%.
Maximálne plazmatické koncentrácie roflumilastu sa dosahujú približne 1 hodinu po užití dávky (od
0,5 h po 2 h) nalačno. Maximálne koncentrácie metabolitu N-oxidu sa dosiahnu približne po 8 hodinách (od 4 h po 13 h). Príjem potravy neovplyvňuje PDE4 inhibičnú aktivitu, ale spomaľuje čas na dosiahnutie maximálnej koncentrácie (tmax) roflumilastu o 1 hodinu a znižuje Cmax približne o 40%. Cmax a tmax roflumilast N-oxidu však nie sú ovplyvnené.
Distribúcia
Na bielkoviny sa viaže približne 99% roflumilastu a 97% roflumilast N-oxidu. Distribučný objem pre jednorazovú dávku 500 mikrogramov roflumilastu je okolo 2,9 l/kg. Vzhľadom na fyzikálno-chemické vlastnosti sa roflumilast rýchlo distribuuje do orgánov a tkanív včítane tukových tkanív myší,
škrečkov a potkanov. Po skorej distribučnej fáze so zjavnou penetráciou do tkanív dochádza k zjavnej
eliminačnej fáze z tukových tkanív, najpravdepodobnejšie ako dôsledok rozkladu pôvodnej zlúčeniny na roflumilast N-oxid. Tieto štúdie na potkanoch s rádioaktívne značeným roflumilastom indikujú aj nízku penetráciu cez krvno-mozgovú bariéru. Nie je žiaden dôkaz špecifickej akumulácie alebo zadržiavania roflumilastu alebo jeho metabolitov v orgánoch a tukových tkanivách.
Biotransformácia
Roflumilast sa extenzívne metabolizuje reakciami fázy I (cytochróm P450) a fázy II (konjugácia).
N-oxid je hlavným metabolitom zisteným v ľudskej plazme. Plazmatické AUC metabolitu N-oxidu je priemerne 10-násobne vyššie ako plazmatické AUC roflumilastu. Preto sa predpokladá, že N-oxid sa významne podieľa na celkovej PDE4 inhibičnej aktivite in vivo.
Štúdie in vitro a štúdie klinických interakcií naznačujú, že k metabolizovaniu roflumilastu na jeho N-oxid dochádza prostredníctvom CYP1A2 a 3A4. Z ďalších výsledkov in vitro v mikrozómoch ľudskej pečene vyplýva, že terapeutické plazmatické koncentrácie roflumilastu a roflumilast N-oxidu neinhibujú CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, 3A4/5 alebo 4A9/11. Z toho vyplýva, že je malá pravdepodobnosť relevantných interakcií so zlúčeninami metabolizovanými týmito P450 enzýmami. Okrem toho sa v in vitro štúdiách nepreukázala žiadna indukcia CYP1A2, 2A6, 2a9, 2C19 alebo 3A4/5 a len slabá indukcia CYP2B6 roflumilastom.
Eliminácia
Plazmatický klírens po krátkodobej intravenóznej infúzii roflumilastu je približne 9,6 l/h. Po perorálnej dávke je stredný účinný plazmatický polčas roflumilastu a jeho metabolitu N-oxidu
približne 17 a 30 hodín. Rovnovážne plazmatické koncentrácie roflumilastu a jeho metabolitu N-oxidu
sa dosiahnu približne po 4 dňoch u roflumilastu a po 6 dňoch u jeho metabolitu N-oxidu po podaní jednej dávky denne. Po intravenóznom alebo perorálnom podaní rádioaktívne značeného roflumilastu sa vylúči stolicou okolo 20% rádioaktivity a 70% močom ako neaktívne metabolity.
Linearita/nelinearita
Farmakokinetika roflumilastu a jeho metabolitu N-oxidu sú úmerné dávke v rozsahu dávok od
250 mikrogramov po 1 000 mikrogramov.
Osobitnépopulácie
U starších ľudí, žien a osôb inej ako bielej pleti bola celková inhibičná aktivita PDE4 zvýšená.
Celková inhibičná aktivita PDE4 bola mierne znížená u fajčiarov. Žiadna z týchto zmien sa nepovažovala za klinicky významnú. U týchto pacientov nie je potrebné upravovať dávku. Kombinácia faktorov ako nefajčiarka čiernej pleti môže viesť k zvýšenej expozícii a pretrvávajúcej netolerovateľnosti. V takomto prípade je potrebné liečbu roflumilastom prehodnotiť (pozri časť 4.4).
V štúdii RO-2455-404-RD, pri porovnaní s celkovou populáciou bola zistená celková inhibičná aktivita PDE4 určená z ex vivo neviazaných frakcií, o 15% vyššia u pacientov vo veku ≥ 75 rokov a o 11% vyššia u pacientov so základnou telesnou hmotnosťou < 60 kg (pozri časť 4.4).
Porucha funkcie obličiek
U pacientov s ťažkou poruchou funkcie obličiek (klírens kreatinínu 10-30 ml/min) bola celková inhibičná aktivita PDE4 znížená o 9%. Nie je potrebná úprava dávky.
Porucha funkcie pečene
Farmakokinetika roflumilastu v dávke 250 mikrogramov jedenkrát denne sa hodnotila u 16 pacientov
s miernou až stredne ťažkou poruchou funkcie pečene klasifikovanou ako Child-Pugh A a B. U týchto
pacientov sa celková inhibičná aktivita PDE4 zvýšila približne o 20% u pacientov s Child-Pugh
A a približne o 90% u pacientov s Child-Pugh B. Simulácie podporujú proporcionalitu dávok medzi roflumilastom 250 mikrogramov a 500 mikrogramov u pacientov s miernou až stredne ťažkou poruchou funkcie pečene. U pacientov s Child-Pugh A je potrebná opatrnosť (pozri časť 4.2). Pacienti so stredne ťažkou alebo ťažkou poruchou funkcie pečene klasifikovanou ako Child-Pugh B alebo C nemajú užívať roflumilast (pozri časť 4.3).
5.3 Predklinické údaje o bezpečnosti
Nie je žiaden dôkaz pre imunotoxický, kožu senzibilizujúci alebo fototoxický potenciál.
V spojení epididymálnou toxicitou u potkanov bolo popísané mierne zníženie fertility samcov. Epididymálna toxicita alebo zmeny parametrov semena sa nezistili u hlodavcov alebo iných druhov včítane opíc ani pri vysokej expozícii.
V jednej z dvoch štúdií embryofetálneho vývoja u potkanov je popísaná vyššia incidencia nekompletnej osifikácie kostí lebky pri dávkach, ktoré spôsobili toxicitu u matky. V jednej z troch štúdií fertility a embryofetálneho vývoja u potkanov sa pozorovali poimplantačné straty. Poimplantačné straty sa nepozorovali u králikov. Predĺžená gestácia sa pozorovala u myší.
Závažnosť týchto pozorovaní pre človeka nie je známa.
Najzávažnejšie zistenia v štúdiách bezpečnej farmakológie a toxikológie sa pozorovali pri vysokých dávkach a expozícii dlhších ako bola predpokladaná dávka pre klinické použitie. Tieto zistenia sa týkali gastrointestinálnych nálezov (napr. vracanie, zvýšená žalúdočná sekrécia, erózie žalúdka, zápal čriev) a kardiálnych nálezov (napr. fokálnych krvácaní, hemosiderického depozitu a infiltrácie
lymfo-histiocytárnych buniek do pravého atria psov, zníženie krvného tlaku a zvýšenie frekvencie
srdca u potkanov, morčiat a psov).
Toxicita špecifická pre hlodavce sa pozorovala na sliznici nosa v štúdiách toxicity po opakovaných dávkach a v štúdiách karcinogenity. Predpokladá sa, že tento efekt je dôsledkom ADCP
(4-amino-3,5-dichloropyridín) N-oxidu, medziproduktu, ktorý sa tvorí na očnej sliznici hlodavcov a má špecifickú väzbovú afinitu u týchto druhov (napr. myš, potkan a škrečok).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro
Monohydrát laktózy Kukuričný škrob Povidón Magnéziumstearát
Filmovávrstva
Hypromelóza
Makrogol 4000
Oxid titaničitý (E171) Žltý oxid železitý (172)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnosti3 roky
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaPVC/PVDC hliníkové blistre v balení po 10, 14, 28, 30, 84, 90 alebo 98 filmom obalených tabliet. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLOEU/1/10/636/001-007
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 5. júl 2010
Dátum posledného predĺženia registrácie: 24. apríl 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.