
ých ciest
menej časté akútne zlyhanie obličiek, zlyhanie obličiek, nefrolitiáza, zvýšená hladina kreatinínu v krvi, proteinúria, bilirubinúria, dyzúria, noktúria, polakizúria
zriedkavé znížený renálny klírens kreatinínu
Poruchy reprodukčného systému a prsníkov
menej časté erektilná dysfunkcia, gynekomastia
Celkové poruchy a reakcie v mieste podania
časté asténia, únava
menej časté pyrexia, bolesť na hrudi, periférne opuchy,
nevoľnosť, pocit horúčavy, podráždenie, bolesť
zriedkavé triaška, abnormálne pocity, xeróza
Opisvybranýchnežiaducichreakcií
Vyrážka
V klinických skúšaniach bola vyrážka najčastejšie miene až stredne závažná, často sa vyskytla v prvých štyroch týždňoch liečby a ustúpila pri ďalšom užívaní. Pre prípady závažných kožných reakcií
pozri upozornenie v časti 4.4.
Počas programu klinického vývoja raltegraviru sa u predtým liečených pacientov častejšie pozorovala vyrážka, bez ohľadu na kauzalitu, pri režimoch obsahujúcich darunavir + raltegravir v porovnaní
s režimami obsahujúcimi darunavir bez raltegraviru alebo raltegravir bez darunaviru. Vyrážka považovaná skúšajúcim za súvisiacu s liekom, sa vyskytla v podobnej miere. Expozícii prispôsobené miery vyrážky (všetky príčiny) boli 10,9;, 4,2 a 3,8 na 100 pacientorokov v danom poradí a u vyrážky súvisiacej s liekom boli 2,4,; 1,1 a 2,3 na 100 pacientorokov v danom poradí. Vyrážky pozorované
v klinických štúdiách boli mierne až stredne závažné a neviedli k prerušeniu liečby (pozri časť 4.4).
Metabolické parametre
Počas antiretrovírusovej liečby sa môže zvýšiť telesná hmotnosť a hladiny lipidov a glukózy v krvi
(pozri časť 4.4).
Poruchy kostrovej a svalovej sústavy
Počas liečby inhibítormi proteáz, najmä v kombinácii s NRTI, sa vyskytli prípady zvýšenia hladiny
kreatínfosfokinázy, myalgie, myozitídy a zriedkavo aj rabdomyolýzy.
Boli hlásené prípady osteonekrózy, najmä u pacientov so všeobecne potvrdenými rizikovými faktormi, pokročilým ochorením HIV alebo dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART). Frekvencia nie je známa (pozri časť 4.4).
Imunoreštitučný zápalový syndróm
U pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej
antiretrovírusovej liečby (CART) môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie. Boli tiež zaznamenané aj autoimunitné poruchy (ako je Gravesova choroba); avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.4).
Krvácanie u pacientov s hemofíliou
U pacientov s hemofíliou dostávajúcich antiretrovirálne inhibítory proteázy boli hlásené prípady
zvýšeného spontánneho krvácania (pozri časť 4.4).
Pediatrická populácia
Posúdenie bezpečnosti u pediatrických pacientov je založené na 48-týždňovej analýze údajov
o bezpečnosti z troch klinických skúšaní fázy II. Boli hodnotené nasledovné populácie pacientov
(pozri časť 5.1):
● 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17
rokov a s hmotnosťou najmenej 20 kg, ktorí užívali darunavir tablety s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku od 3 do < 6
rokov a s hmotnosťou 10 kg až < 20 kg (16 účastníkov od 15 kg do < 20 kg), ktorí užívali
darunavir perorálnu suspenziu s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 12 pediatrických pacientov infikovaných HIV-1 vo veku od 12 do 17 rokov a s hmotnosťou
najmenej 40 kg, ktorí neboli predtým liečení ART. Títo pacienti dostávali darunavir tablety
s nízkou dávkou ritonaviru jedenkrát denne spolu s inými antiretrovírusovými liekmi. (pozri
časť 5.1).
Celkovo bol bezpečnostný profil u týchto pediatrických pacientov podobný bezpečnostnému profilu
pozorovanému u dospelých.
Iné osobitné skupiny pacientov
Pacienti súčasne infikovaní vírusom hepatitídy B a/alebo hepatitídy C
Spomedzi 1968 predtým liečených pacientov, ktorí užívali darunavir spolu s ritonavirom 600/100 mg dvakrát denne, bolo 236 pacientov infikovaných zároveň hepatitídou B alebo C. U súčasne infikovaných pacientov bola vyššia pravdepodobnosť počiatočného a pri liečbe vznikajúceho zvýšenia hladiny pečeňových transamináz ako u pacientov bez chronickej vírusovej hepatitídy (pozri časť 4.4).
H
l
ásenie
podozrení
na
nežiaduce
r
eakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSkúsenosti s akútnym predávkovaním darunavirom užívaným s nízkou dávkou ritonaviru u ľudí sú obmedzené. Zdravým dobrovoľníkom sa darunavir podával vo forme perorálneho roztoku
v jednorazových dávkach do 3200 mg ako samotný, resp. vo forme tabliet v jednorazových dávkach
do 1600 mg v kombinácii s ritonavirom bez toho, že by sa u nich pozorovali nepriaznivé symptomatické účinky.
V prípade predávkovania darunavirom nie je k dispozícii žiadne špecifické antidotum. Liečba predávkovania darunavirom zahŕňa všeobecné podporné opatrenia, vrátane sledovania vitálnych prejavov a pozorovania klinického stavu pacienta. Ak je to indikované, eliminácia nevstrebanej aktívnej látky sa dosiahne pomocou vyvolania vracania.
Aj podanie aktívneho uhlia môže pomôcť pri eliminácii nevstrebanej aktívnej látky. Vzhľadom na vysokú väzbu darunaviru na plazmatické bielkoviny je nepravdepodobné, že by dialýza mala väčší význam pri eliminácii aktívnej látky z cirkulácie.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antivirotiká na systémové použitie, inhibítory proteáz, ATC kód: J05AE10
Mechanizmus účinkuDarunavir je inhibítor dimerizácie a katalytickej aktivity HIV-1 proteázy (KD 4,5 x 10-12M). Selektívne
inhibuje odštiepenie HIV kódovaných polyproteínov Gag-Pol v bunkách infikovaných vírusom, čím zabraňuje tvorbe zrelých infekčných vírusových častíc.
Antivírusová aktivita in vitroDarunavir vykazuje aktivitu proti laboratórnym kmeňom a klinickým izolátom HIV-1 a laboratórnym
kmeňom HIV-2 v akútne infikovaných líniách T-buniek, mononukleárnych bunkách z periférnej krvi
človeka a ľudských monocytov/makrofágov so strednými hodnotami EC50 v rozmedzí
1,2 až 8,5 nmol/l (0,7 až 5,0 ng/ml).
In vitro má darunavir antivírusovú aktivitu voči širokému spektru
primárnych izolátov zo skupiny M HIV-1 (A, B, C, D, E, F, G) a skupiny O s hodnotami EC50
v rozmedzí od < 0,1 do 4,3 nmol/l.
Tieto hodnoty EC50 sú výrazne nižšie ako je rozsah toxickej koncentrácie pre 50 % buniek od
87 µmol/l do > 100 µmol/l.
RezistenciaSelekcia vírusu rezistentného na darunavir z divokého typu HIV-1
in vitro trvala pomerne dlho
(> 3 roky). Vyselektované vírusy neboli schopné rásť v prítomnosti darunaviru v koncentráciách nad
400 nmol/l.
U vírusov vyselektovaných v týchto podmienkach a vykazujúcich zníženú citlivosť voči darunaviru
(rozmedzie: 23 až 50-násobne) sa v géne pre proteázu zistili substitúcie 2 až 4 aminokyselín. Zníženú citlivosť vznikajúcich vírusov na darunavir v selekcii nie je možné vysvetliť vznikom týchto mutácií proteázy.
Údaje z klinických štúdií s pacientmi liečenými ART (štúdia TITAN a zlúčená analýza štúdií POWER
1, 2 a 3 a štúdií DUET 1 a 2) preukázali, že virologická odpoveď na darunavir užívaný s nízkou
dávkou ritonaviru bola znížená, keď boli na začiatku prítomné 3 a viac mutácií spojených
s rezistenciou voči darunaviru (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V a
L89V) alebo keď sa tieto mutácie vyvinuli počas liečby.
Zvýšenie východiskovej násobnej zmeny darunaviru v EC50 (FC) súviselo s poklesom virologickej odpovede. Určila sa dolná a horná klinická hranica 10 a 40. Izoláty s východiskovou hodnotou FC
≤ 10 sú citlivé; izoláty s FC > 10 až 40 majú zníženú citlivosť; izoláty s FC > 40 sú rezistentné (pozri
Klinické výsledky).
Vírusy izolované u pacientov s dávkou darunavir/ritonavir 600/100 mg dvakrát denne, u ktorých sa vyskytlo spontánne virologické zlyhanie, ktoré boli citlivé na tipranavir na začiatku, zostali vo veľkej väčšine prípadov citlivé na tipranavir po liečbe.
Najnižšie miery rozvoja rezistencie vírusu HIV sú pozorované u pacientov doposiaľ neliečených ART, ktorí sú po prvýkrát liečení darunavirom v kombinácii s inou ART.
Tabuľka nižšie zobrazuje vývoj mutácií a stratu citlivosti na PI pri virologickom zlyhaní v závere
liečby v štúdiách ARTEMIS, ODIN a TITAN.
ARTEMIS ODIN TITAN
Darunavir /
ritonavir
800/100 mg jedenkrát denne N=343
Darunavir /
ritonavir
800/100 mg jedenkrát denne N=294
Darunavir /
ritonavir
600/100 mg dvakrát denne N=296
Darunavir /
ritonavir
600/100 mg dvakrát denne N=298
Celkový počet virologických zlyhanía, n (%)
55 (16,0 %) 65 (22,1 %) 54 (18,2 %) 31 (10,4 %)
Spontánne 39 (11,4 %) 11 (3,7 %) 11 (3,7 %) 16 (5,4 %)
Jedinci nereagujúci na
liečbu
16 (4,7 %) 54 (18,4 %) 43 (14,5 %) 15 (5,0 %)
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými genotypmi, u ktorých sa vyvinuli mutácieb v závere liečby, n/N
Primárne (významné)
mutácie PI
0/43 1/60 0/42 6/28

PI RAM 4/43 7/60 4/42 10/28
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými fenotypmi, u ktorých sa preukázala strata citlivosti na PI v závere liečby v porovnaní so začiatkom liečby, n/N
PI
darunavir 0/39 1/58 0/41 3/26 amprenavir 0/39 1/58 0/40 0/22 atazanavir 0/39 2/56 0/40 0/22 indinavir 0/39 2/57 0/40 1/24 lopinavir 0/39 1/58 0/40 0/23 sakvinavir 0/39 0/56 0/40 0/22 tipranavir 0/39 0/58 0/41 1/25
a TLOVR ne-VF cenzurovaný algoritmus založený na HIV-1 RNA < 50 kópií/ml, okrem
TITAN (HIV-1 RNA
< 400 kópií/ml)
b zoznamy IAS-USA
SkríženárezistenciaNásobná zmena (FC) darunaviru bola menej ako 10 pre 90 % z 3309 klinických izolátov rezistentných
voči amprenaviru, atazanaviru, indinaviru, lopinaviru, nelfinaviru, ritonaviru, sakvinaviru a/alebo
tipraniviru, čo naznačuje, že vírusy rezistentné voči väčšine PI zostávajú citlivé voči darunaviru.
V štúdii ARTEMIS sa v prípade virologického zlyhania nepozorovala žiadna skrížená rezistencia
s inými PI.
Klinické výsledky
Dospelí pacienti
Výsledky klinických štúdií u dospelých pacientov doposiaľ neliečených ART, pozri súhrn charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Úči nnosť darunaviru 600 mg dvakrát denne užívanej so 100 mg ritonaviru dvakrát denne u pacientov
pre dtým li ečený ch ART
Údaje o účinnosti kombinácie darunavir a ritonavir (600/100 mg dvakrát denne) u pacientov predtým
liečených ART sú založené na analýze 96 týždňovej štúdie fázy III TITAN u pacientov predtým
liečených ART, ktorí ešte neužívali lopinavir, na analýze 48 týždňovej štúdie fázy III ODIN
u pacientov predtým liečených ART bez DRV-RAM a na analýze údajov z 96 týždňov trvajúcich štúdií fázy IIb POWER 1 a 2 u pacientov predtým liečených ART s vysokou rezistenciou na PI.
TITAN je randomizovaná, kontrolovaná, otvorená štúdia fázy III porovnávajúca darunavir podávaný spolu s ritonavirom (600/100 mg dvakrát denne) verzus lopinavir/ritonavir (400/100 mg dvakrát denne) u dospelých pacientov predtým liečených ART infikovaných HIV-1, ktorí ešte neužívali
lopinavir. Obe ramená používali optimalizovaný základný režim (OBR, z angl. Optimised Background
Regimen), ktorý pozostával z minimálne 2 antiretrovirotík (NRTI s alebo bez NNRTI).
Nižšie uvedená tabuľka uvádza údaje 48-týždňovej analýzy účinnosti zo štúdie TITAN.

TITAN
Výsledky Darunavir /ritonavir
600/100 mg dvakrát denne + OBR N=298
lopinavir/ritonavir
400/100 mg dvakrát denne + OBR N=297
Rozdiel v liečbe
(95 % CI rozdielu)
HIV-1 RNA < 50 kópií/mla 70,8 % (211) 60,3 % (179) 10,5 % (2,9; 18,1)b
medián zmeny v počte buniek CD4+ oproti východiskovej hodnote (x 106/l)c
88 81
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede
c NC=F
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom,
definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA < 400 a < 50 kópií/ml, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat
(ITT) aj u pacientov sledovaných v protokole (On Protocol). Tieto výsledky sa potvrdili v analýze
údajov z 96 týždňov trvajúcej liečby v štúdii TITAN, pričom v 96. týždni malo 60,4 % pacientov v ramene s darunavirom/ritonavirom HIV-1 RNA < 50 kópií/ml v porovnaní s 55,2 % pacientov v ramene s lopinavirom/ritonavirom [rozdiel: 5,2 %, 95 % CI (-2,8; 13,1)].
ODIN je randomizovaná otvorená štúdia fázy III porovnávajúca darunavir/ritonavir 800/100 mg jedenkrát denne a darunavir/ritonavir 600/100 mg dvakrát denne u pacientov infikovaných HIV-1 predtým liečených ART, u ktorých skríningové vyšetrenie rezistencie genotypu nepreukázalo RAM voči darunaviru (t.j. V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V, L89V)
a skríningové vyšetrenie HIV-1 RNA bolo > 1 000 kópií/ml. Analýza účinnosti je založená na liečbe trvajúcej 48 týždňov (pozri tabuľku nižšie). Obe ramená použili optimalizovaný základný režim (OBR) ≥ 2 NRTI.
Výsledky Darunavir/ritonavir
800/100 mg jedenkrát denne + OBR
N=294
ODIN Darunavir/ritonavir
600/100 mg dvakrát
denne + OBR N=296
Rozdiel v liečbe
(95% CI rozdielu)
HIV-1 RNA
< 50 kópií/mla
S východiskovou HIV-1 RNA (kópie/ml)
72,1 % (212) 70,9 % (210) 1,2 % (-6,1; 8,5)b
< 100 000
≥ 100 000
S východiskovým počtom buniek CD4+ (x 106/l)
≥ 100
< 100
S kmeňom HIV-1
Typ B Typ AE Typ C Inéc
77,6 % (198/255)
35,9 % (14/39)
75,1 % (184/245)
57,1 % (28/49)
70,4 % (126/179)
90,5 % (38/42)
72,7 % (32/44)
55,2 % (16/29)
73,2 % (194/265)
51 6 % (16/31)
72,5 % (187/258)
60,5 % (23/38)
64,3 % (128/199)
91,2 % (31/34)
78,8 % (26/33)
83,3 % (25/30)
4,4 % (-3,0; 11,9)
-15,7 % (-39,2; 7,7)
2,6 % (-5,1; 10,3)
-3,4 % (-24,5; 17,8)
6,1 % (-3,4; 15,6)
-0,7 % (-14,0; 12,6)
-6,1 % (-2,6; 13,7)
-28,2 % (-51,0; -5,3)
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x 106/l)e
108 112 -5d (-25; 16)
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede c Kmene A1, D, F1, G, K, CRF02_AG, CRF12_BF, a CRF06_CPX d Rozdiel stredných hodnôt
e Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward)
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom
800/100 mg jedenkrát denne, definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA
< 50 kópií/ml v porovnaní s darunavirom/ritonavirom 600/100 mg dvakrát denne, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat (ITT) aj
u pacientov sledovaných v protokole (On Protocol).
Darunavir/ritonavir 800/100 mg jedenkrát denne sa nemá používať u tých pacientov liečených ART, ktorí majú jednu alebo viac mutácií spojených s rezistenciou voči darunaviru (DRV-RAM) alebo
HIV-1 RNA ≥ 100 000 kópií/ml alebo počet buniek CD4+ < 100 buniek x 106/l (pozri časti 4.2 a 4.4).
U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje.
POWER 1 a POWER 2 sú randomizované, kontrolované štúdie porovnávajúce darunavir užívaný spolu s ritonavirom (600/100 mg dvakrát denne) s kontrolnou skupinou užívajúcou inhibítor proteáz vybraný skúšajúcim u pacientov infikovaných HIV-1, ktorí predtým absolvovali viac ako jeden neúspešný liečebný režim zahŕňajúci inhibítor proteáz (PI). V oboch štúdiách bol použitý OBR zahŕňajúci aspoň 2 NRTI s enfuvirtidom (ENF) alebo bez neho.
V nasledujúcej tabuľke sú uvedené údaje 48-týždňovej a 96-týždňovej analýzy účinnosti zo zlúčených štúdií POWER 1 a POWER 2.
Spoločné údaje získané v štúdiách POWER 1 a POWER 2
48. týždeň 96. týždeň
Výsledky Darunavir/
ritonavir
600/100 mg
2x denne n=131
Kontrola n=124
Rozdiel medzi
liečbami
Darunavir/
ritonavir
600/100 mg 2x denne
n=131
Kontrol a
n=124
Rozdiel medzi liečbami
HIV RNA
< 50 kópií/ mla
45,0 % (59) 11,3 % (14)
33,7 % (23,4 %;

44,1 %)c
38,9 % (51) 8,9 % (11)
30,1 % (20,1; 40,0)c
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x
106/l)b
103 17 86
(57; 114)c
133 15 118 (83,9;
153,4)c
a Výpočet na základe algoritmu TLOVR.
b Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward).
c 95 % interval spoľahlivosti.
Analýza údajov z 96 týždňov trvajúcej liečby v štúdiách POWER preukázala zachovanú
antiretrovírusovú účinnosť a imunologický benefit.
Z 59 pacientov, ktorí reagovali na liečbu úplnou supresiou vírusu (< 50 kópií/ml) v 48. týždni,
47 pacientov (80 % respondérov na liečbu v 48. týždni) naďalej reagovalo na liečbu v 96. týždni.
Východiskový genotyp alebo fenotyp a virologický výsledok
Východiskový genotyp a FC darunaviru (zmena citlivosti vzhľadom k východiskovej hodnote) sa ukázal ako prediktívny faktor virologického výsledku.
Podiel (%) pacientov s odpoveďou (HIV-1 RNA < 50 kópii/ml v týždni 24) na darunavir užívaný spolu s ritonavirom (600/100 mg dvakrát denne) u východiskového genotypu*a a východiskovej FC darunaviru pri užívaní enfuvirtidu (ENF): podľa analýz liečby štúdií POWER a DUET.
Odpoveď (HIV-1
RNA < 50 kópií/ml
Počet východiskových mutáciía Východisková DRV FCb
Všetky
v 24. týždni)
%, n/N
Všetky
rozpätia
0-2 3 ³ 4
rozpätia £ 10 10-40 > 40
Všetci pacienti 45 %
455/1 014
54 %
359/660
39 %
67/172
12 %
20/171
45 %
455/1 014
55 %
364/659
29 %
59/203
8 %
9/118
Pacienti neužívajúci/opako- vane užívajúci ENFc Pacienti s de novo ENFd
39 %
290/741
60 %
165/273
50 %
238/477
66 %
121/183
29 %
35/120
62 %
32/52
7 %
10/135
28 %
10/36
39 %
290/741
60 %
165/273
51 %
244/477
66 %
120/182
17 %
25/147
61 %
34/56
5 %
5/94
17 %
4/24
a Počet mutácií zo zoznamu mutácií spojených so znížením odpovede na kombináciu darunavir/ritonavir (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V alebo L89V)
b násobná zmena v EC50
c “Pacienti neužívajúci/opakovane užívajúci ENF”, t.j. pacienti, ktorí neužívali ENF alebo tí, ktorí ENF užívali, ale nie
prvýkrát.
d “Pacienti s
de novo ENF”, t.j. pacienti, ktorí užívali ENF prvýkrát.
Pediatrickí pacientiVýsledky klinických štúdií u pediatrických pacientov vo veku 12 až 17 rokov, ktorí neboli predtým liečení ART, nájdete v Súhrne charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Pediatrickí pacienti od 6 do < 18 rokov a s hmotnosťou najmenej 20 kg, pre dt ým lieč ení ART
DE
LPHI
je otvorená štúdia fázy II hodnotiaca farmakokinetiku, bezpečnosť, toleranciu a účinnosť darunaviru s nízkou dávkou ritonaviru u 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg. Títo pacienti užívali darunavir/ritonavir dvakrát denne spolu s inými antiretrovírusovými látkami (pre odporúčané dávkovanie v závislosti od hmotnosti pacienta pozri časť 4.2). Virologická odpoveď bola definovaná ako pokles vírusového zaťaženia HIV-1 RNA v plazme o najmenej 1,0 log10 oproti východiskovej hodnote.
V tejto štúdii bolo pacientom so zvýšeným rizikom prerušenia liečby z dôvodu intolerancie perorálneho roztoku ritonaviru (napr. averzia voči chuti) umožnené prejsť na užívanie kapsúl. Zo 44 pacientov užívajúcich perorálny roztok ritonaviru, 27 prešlo na užívanie 100 mg kapsúl a prekročilo dávku ritonaviru odvodenú od hmotnosti bez zmien v sledovanej bezpečnosti.

DELPHI
Výsledky po 48. týždni Darunavir/ritonavir
N=80
HIV-1 RNA < 50 kópií/mla 47,5 % (38)
Priemerná zmena počtu buniek CD4+ oproti
východiskovej hodnoteb
a Výpočet na základe algoritmu TLOVR.
147
b Pacienti, ktorí neukončili liečbu, sú počítaní ako zlyhanie: pacientom, ktorí prerušili liečbu predčasne, sa priradí
zmena rovná nule.
Podľa cenzurovaného TLOVR algoritmu pre nevirologické zlyhanie u 24 (30,0 %) pacientov došlo
k virologickému zlyhaniu, z nich u 17 (21,3 %) pacientov sa objavil rebound fenomén a 7 (8,8 %)
pacientov neodpovedalo na liečbu.
Pediatrickí pacienti vo veku 3 až < 6 rokov predtým lieč ení ART
U 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART vo veku od 3 do < 6
rokov a s hmotnosťou 10 kg až < 20 kg bola farmakokinetika, bezpečnosť, tolerovateľnosť a účinnosť
darunaviru/ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami hodnotená v otvorenej štúdii fázy II, ARIEL. Pacienti dostávali dávku dvakrát denne v závislosti od ich hmotnosti, pacienti s hmotnosťou 10 kg až < 15 kg dostávali darunavir/ritonavir 25/3 mg/kg dvakrát denne a pacienti s hmotnosťou 15 kg až < 20 kg dostávali darunavir/ritonavir 375/50 mg dvakrát denne. Virologická odpoveď, definovaná ako percento pacientov s potvrdenou plazmatickou hladinou
< 50 HIV-1 RNA kópií/ml, bola v 48. týždni hodnotená u 16 pediatrických pacientov s hmotnosťou
15 kg až < 20 kg a u 5 pediatrických pacientov s hmotnosťou 10 kg až < 15 kg dostávajúcich darunavir/ritonavir v kombinácii s inými antiretrovírusovými látkami. (pozri časť 4.2 pre odporúčanie
dávkovania podľa telesnej hmotnosti).

ARIEL
Výsledky po 48. týždni Darunavir/ritonavir
10 kg až < 15 kg
N=5
15 kg až < 20 kg
N=16
HIV-1 RNA < 50 kópií/mla 80,0 % (4) 81,3 % (13)
CD4+ percentuálna zmena oproti východiskovej hodnoteb Priemerná zmena počtu buniek CD4+ oproti východiskovej hodnoteb
a Imputácie na základe algoritmu TLOVR.
b NC=F
4 4
16 241
Dostupné sú obmedzené údaje o účinnosti u pediatrických pacientov s hmotnosťou menej ako 15 kg,
a preto neumožňujú uviesť odporúčania pre dávkovanie.
G
ravidita a obdobie po pôrode
Darunavir/ritonavir (600/100 mg dvakrát denne alebo 800/100 mg jedenkrát denne) v kombinácii so základným režimom bol hodnotený v klinickej štúdii s 34 gravidnými ženami (17 v každej skupine) počas druhého a tretieho trimestra a po pôrode. Virologická odpoveď sa udržala počas trvania štúdie v oboch skupinách. U detí narodených 29 pacientkám, ktoré zotrvali na antivirotickej liečbe až do
pôrodu, sa nevyskytol prenos z matky na dieťa. Nevyskytli sa žiadne nové klinicky významné zistenia týkajúce sa bezpečnosti v porovnaní so známym profilom bezpečnosti darunaviru/ritonaviru
u dospelých infikovaných HIV-1 (pozri časti 4.2, 4.4 a 5.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti darunaviru podávaného v kombinácii s ritonavirom sa skúmali
u zdravých dospelých dobrovoľníkov a u pacientov infikovaných vírusom HIV-1. Expozícia voči
darunaviru u pacientov infikovaných vírusom HIV-1 bola vyššia ako u zdravých dobrovoľníkov. Vyššiu expozíciu voči darunaviru u pacientov infikovaných vírusom HIV-1 v porovnaní so zdravými dobrovoľníkmi je možné vysvetliť na základe vyšších koncentrácií α1-kyslého glykoproteínu (AAG) u pacientov infikovaných vírusom HIV-1, čo vedie k vyššej väzbe darunaviru na plazmatický AAG,
a tým aj k vyšším koncentráciám tejto látky v plazme.
Darunavir sa metabolizuje najmä prostredníctvom CYP3A. Ritonavir inhibuje CYP3A, v dôsledku
čoho sa výrazne zvyšujú plazmatické koncentrácie darunaviru.
Absorpcia
Darunavir sa rýchlo vstrebáva po perorálnom podaní. Pri podávaní v kombinácii s ritonavirom
v nízkej dávke sa maximálne plazmatické koncentrácie darunaviru obyčajne dosiahnu v priebehu 2,5 -
4 hodín.
Absolútna biologická dostupnosť po jednorazovom perorálnom podaní samotného darunaviru v dávke
600 mg bola približne 37 %, pričom pri podávaní ritonaviru v dávke 100 mg dvakrát denne sa zvýšila približne na 82 %. Po jednorazovom perorálnom podaní darunaviru v dávke 600 mg v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne sa systémová expozícia voči darunaviru
zvýšila 14-násobne (pozri časť 4.4).
Pri užívaní nalačno bola relatívna biologická dostupnosť darunaviru v prítomnosti nízko-dávkovaného ritonaviru o 30 % nižšia ako pri užívaní lieku s jedlom. Z toho dôvodu sa tablety darunaviru musia užívať spolu s ritonavirom a jedlom. Druh jedla nemá vplyv na expozíciu voči darunaviru.
Distribúcia
Približne 95 % darunaviru sa viaže na plazmatické bielkoviny, najmä na α1-kyslý glykoproteín.
Po intravenóznom podaní bol distribučný objem samotného darunaviru 88,1 ± 59,0 l (stredný ± SD)
a v prítomnosti 100 mg ritonaviru podávaného dvakrát denne bol zvýšený na 131 ± 49,9 l
(stredný ± SD).
Biotransformácia
In vitro štúdie využívajúce mikrozómy pečene človeka (HLM) naznačujú, že darunavir sa
metabolizuje najmä oxidatívnym metabolizmom. Darunavir sa výrazne metabolizuje prostredníctvom systému CYP v pečeni a takmer výlučne prostredníctvom izoenzýmu CYP3A4. V štúdii skúmajúcej aplikáciu darunaviru označeného rádioaktívnym 14C zdravým dobrovoľníkom sa zistilo, že väčšina rádioaktivity v plazme po jednorazovom podaní darunaviru s ritonavirom v dávke 400/100 mg pochádzala z pôvodného liečiva. U ľudí sa zistila prítomnosť najmenej 3 oxidatívnych metabolitov
darunaviru, pričom všetky z nich mali aktivitu proti divokému typu HIV najmenej 10x nižšiu ako
darunavir.
Eliminácia
Po podaní darunaviru s ritonavirom v dávke 400/100 mg označeného rádioaktívnym 14C sa 79,5 %
rádioaktivity zachytilo v stolici a 13,9 % rádioaktivity sa zachytilo v moči. Rádioaktivita nezmeneného darunaviru v stolici predstavovala 41,2 % a v moči predstavovala 7,7 % z podanej dávky. Terminálny polčas eliminácie darunaviru podávaného v kombinácii s ritonavirom bol približne
15 hodín.
Klírens darunaviru (150 mg) podaného intravenózne v monoterapii bol 32,8 l/h, kým pri kombinácii s nízko dávkovaným ritonavirom 5,9 l/h.
Osobitné skupiny pacientov
Pediatrická populácia
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 74 predtým liečených pediatrických pacientov vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg preukázala, že dávky darunaviru/ritonaviru podávané v závislosti od hmotnosti, mali za následok expozíciu darunaviru porovnateľnú s expozíciou u dospelých pacientov užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 14 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 15 kg až < 20 kg ukázala, že dávkovanie založené na hmotnosti viedlo k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 12 pediatrických pacientov vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení ART, ukázala, že dávka darunaviru/ritonaviru 800/100 mg jedenkrát denne viedla k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne. Z toho dôvodu sa rovnaké dávkovanie jedenkrát denne môže použiť u dospievajúcich vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg
s predchádzajúcou liečbou bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)*
a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+
≥ 100 buniek x 106/l (pozri časť 4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 10 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 14 kg až < 20 kg preukázala, že dávky odvodené od hmotnosti viedli k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne (pozri časť 4.2). Farmakokinetické modelovanie a simulácia expozície darunaviru u pediatrických pacientov vo veku 3 až < 18 rokov navyše potvrdili expozície darunaviru, ktoré boli pozorované v klinických štúdiách
a umožnili určenie dávkovania darunavir/ritonaviru jedenkrát denne v závislosti na hmotnosti
u pediatrických pacientov s hmotnosťou najmenej 15 kg, ktorí buď predtým nedostávali liečbu ART
alebo u predtým liečených pediatrických pacientov bez DRV-RAM* a s plazmatickou hladinou HIV-1
RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri časť 4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V.
Starší ľudia
Pri analýze farmakokinetiky v rôznych populáciách pacientov s infekciou HIV sa zistilo, že
farmakokinetika darunaviru sa výraznejšie nelíši u pacientov s infekciou HIV vo vekovom rozmedzí
od 18 do 75 rokov (n = 12, vek ≥ 65 rokov) (pozri časť 4.4). U pacientov starších ako 65 rokov boli
k dispozícii iba obmedzené údaje.
Pohlavie
Pri analýze farmakokinetiky v rôznych populáciách pacientov sa u žien s infekciou HIV zistila mierne vyššia expozícia voči darunaviru (o 16,8 %) ako u mužov infikovaných HIV. Tento rozdiel nemá žiadny klinický význam.
Porucha funkcie obličiek
Výsledky štúdie zachovania hmotnosti s 14C rádioaktívne označeným darunavirom s ritonavirom ukázali, že približne 7,7 % z podanej dávky darunaviru sa vylučuje do moču v nezmenenej forme.
Aj keď sa podávanie darunaviru nesledovalo u pacientov s poruchou funkcie obličiek, pri analýze farmakokinetiky v rôznych populáciách pacientov sa zistilo, že stredne závažná porucha funkcie obličiek (klírens kreatinínu: 30 - 60 ml/min, n=20) nemá väčší vplyv na farmakoniketiku darunaviru u pacientov s infekciou HIV (pozri časti 4.2 a 4.4).
Porucha funkcie pečene
Darunavir sa metabolizuje a eliminuje prevažne v pečeni. V štúdii s opakovaným podaním lieku darunavir užívaným s ritonavirom (600/100 mg dvakrát denne) sa preukázalo, že celkové koncentrácie
darunaviru v plazme u subjektov s miernou (Childova-Pughova trieda A, n=8) a stredne ťažkou
(Childova-Pughova trieda B, n=8) poruchou funkcie pečene boli porovnateľné s tými u zdravých
subjektov. Koncentrácie voľného darunaviru boli približne o 55 % (Childova-Pughova Trieda A)
a 100 % (Childova-Pughova trieda B) vyššie. Klinický význam tohto nárastu nie je známy, preto treba
darunavir užívať so zvýšenou opatrnosťou. Účinok ťažkej poruchy funkcie pečene na farmakokinetiku
darunaviru nebol dosiaľ študovaný (pozri časti 4.2, 4.3 a 4.4).
Gravidita a obdobie po pôrode
Expozícia celkovému darunaviru a ritonaviru po užití darunaviru/ritonaviru 600/100 mg dvakrát denne a darunaviru/ritonaviru 800/100 mg jedenkrát denne v rámci antivirotického režimu bola všeobecne
nižšia počas gravidity v porovnaní s obdobím po pôrode. V prípade neviazaného (t.j. aktívneho)
darunaviru však boli farmakokinetické parametre menej znížené počas gravidity v porovnaní
s obdobím po pôrode z dôvodu zvýšenia neviazanej frakcie darunaviru počas gravidity v porovnaní s obdobím po pôrode.
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru 600/100 mg dvakrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho trimestra gravidity a po pôrode
 F
armakokinetika celkového darunaviru
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=11)aTretí trimester gravidity (n=11)Obdobie po pôrode (6-12 týždňov) (n=11)
Cmax, ng/ml 4 601 ± 1 125 5 111 ± 1 517 6 499 ± 2 411
AUC12h, ng.h/ml 38 950 ± 10 010 43 700 ± 16 400 55 300 ± 27 020
Cmin, ng/mlb 1 980 ± 839,9 2 498 ± 1 193 2 711 ± 2 268
a n=10 pre AUC12h
b vylúčením hodnoty Cmin pod LLOQ, n=10 pre referenciu
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru 800/100 mgjedenkrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretiehotrimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=16)
Tretí trimester gravidity (n=14)
Obdobie po pôrode (6-12 týždňov) (n=15)
Cmax, ng/ml 4 988 ± 1 551 5 138 ± 1 243 7 445 ± 1 674
AUC12h, ng.h/ml 61 303 ± 16 232 60 439 ± 14 052 94 529 ± 28 572
Cmin, ng/mla 1 193 ± 509 1 098 ± 609 1 572 ± 1 108
a n=12 pre obdobie po pôrode, n=15 pre druhý trimester a n=14 pre tretí trimester
U žien užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 28 %, 24 % a 17 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 19 %, 17 % nižšie a o 2 % vyššie, v tomto poradí, v porovnaní s obdobím po pôrode.
U žien užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne počas druhého trimestra gravidity boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 34 %, 34 %
a 32 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity
boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 31 %, 35 % a 50 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode.
5.3 Predklinické údaje o bezpečnosti
Toxikologické štúdie so samotným darunavirom sa vykonali v dávkovaní až do klinických hladín
u myší, potkanov a psov, kým štúdie s jeho kombináciou s ritonavirom sa vykonali u potkanov a psov.
V štúdiách toxicity po opakovanej dávke u myší, potkanov a psov sa zistili len obmedzené účinky liečby darunavirom. Identifikovaným cieľovým orgánom u hlodavcov bol hemopoetický systém, koagulačný systém, pečeň a štítna žľaza. Pozoroval sa premenlivý, ale mierny pokles parametrov v súvislosti s erytrocytmi spolu s predĺžením aktivovaného parciálneho tromboplastínového času.
Zmeny boli pozorované v pečeni (hypertrofia hepatocytov, vakuolizácia, zvýšenie hladiny pečeňových enzýmov) a štítnej žľaze (folikulárna hypertrofia). U laboratórnych potkanov viedla kombinácia darunaviru s ritonavirom k miernemu zvýšeniu účinku na parametre červených krviniek, pečene
a štítnu žľazu a k zvýšeniu incidencie ostrovčekov fibrózy v pankrease (iba u samcov laboratórnych potkanov) v porovnaní so samotným darunavirom. U psov sa nepozorovali žiadne väčšie známky
toxicity ani cieľové orgány toxicity pri rovnakej expozícii ako u človeka, ktorý užíva odporučené
dávky.
V štúdii na potkanoch bol znížený počet corpora lutea a implantácií v prípade maternálnej toxicity. Na druhej strane sa nepozorovali žiadne účinky na párenie alebo plodnosť po podávaní darunaviru
v dávkach do 1000 mg/kg/deň a pri expozícii (AUC - 0,5-násobok) nižšej ako u človeka dostávajúceho odporučené dávky. Pri podávaní rovnako veľkých dávok samotného darunaviru sa u potkanov
a králikov nepozorovala teratogenita podobne ako u myší dostávajúcich darunavir v kombinácii
s ritonavirom. Stupeň expozície bol nižší ako u človeka dostávajúceho odporučené dávky. Pri
hodnotení prenatálneho a postnatálneho vývoja u potkanov, darunavir podávaný samostatne alebo
v kombinácii s ritonavirom viedol k prechodnému zníženiu prírastku telesnej hmotnosti u dojčených mláďat a došlo k miernemu oneskoreniu v otvorení očí a uší. Darunavir v kombinácii s ritonavirom
spôsobil zníženie prežívania mláďat, ktoré sa prejavilo nepriaznivou odpoveďou v 15. deň laktácie
a znížením počtu mláďat, ktoré prežili počas laktácie. Tieto účinky môžu byť prisúdené sekundárnej expozícii liečiva u mláďat prostredníctvom materského mlieka a/alebo maternálnej toxicity. Darunavir
podávaný ako samotný alebo v kombinácii s ritonavirom neovplyvnil žiadne funkcie po odstavení.
U juvenilných laboratórnych potkanov, ktorí dostávali darunavir 23. – 26. deň, sa pozorovala zvýšená
mortalita s kŕčmi u niektorých zvierat. Medzi 5. a 11. dňom života bola expozícia v plazme, pečeni
a mozgu výrazne vyššia ako u dospelých laboratórnych potkanov po porovnateľných dávkach
v mg/kg. Po 23. dni života bola expozícia porovnateľná s tou u dospelých potkanov. Zvýšená expozícia bola pravdepodobne aspoň čiastočne z dôvodu nezrelosti enzýmov metabolizujúcich liek
u juvenilných zvierat. U juvenilných potkanov neboli spozorované žiadne úmrtia v súvislosti s liečbou dávkami 1000 mg/kg darunaviru (jednorazová dávka) v 26. deň veku alebo 500 mg/kg (opakovaná dávka) od 23. do 50. dňa veku a expozícia a profil toxicity boli porovnateľné s tými, pozorovanými
u dospelých potkanov.
Darunavir s nízkou dávkou ritonaviru sa nemá používať u detí mladších ako 3 roky z dôvodu
nejasnosti ohľadom miery rozvoja hematoencefalickej bariéry a pečeňových enzýmov.
Karcinogénny potenciál darunaviru bol vyhodnotený pri podávaní lieku myšiam a potkanom sondou do žalúdka počas 104 týždňov. Myšiam boli podávané denné dávky 150, 450 a 1000 mg/kg a potkanom boli podávané dávky 50, 150 a 500 mg/kg. Výskyt hepatocelulárnych adenómov a karcinómov stúpal v závislosti od dávky a pozoroval sa u samcov a samíc oboch živočíšnych druhov. U samcov potkanov sa vyskytli adenómy folikulárnych buniek štítnej žľazy. Podávanie darunaviru nespôsobilo štatisticky významný nárast iných benígnych alebo malígnych novotvarov u myší alebo potkanov. Pozorované hepatocelulárné tumory a tumory štítnej žľazy u hlodavcov sa považujú za
obmedzené z hľadiska významnosti pre človeka. Opakované podávanie darunaviru potkanom spôsobilo indukciu mikrozomálnych enzýmov v pečeni a zvýšilo vylučovanie hormónov štítnej žľazy, ktoré predurčuje potkanov, ale nie ľudí, na výskyt neoplaziem štítnej žľazy. Pri najvyšších testovaných dávkach boli systémové expozície (založené na AUC) darunaviru 0,4- až 0,7-násobkom (myši) a 0,7- až 1-násobkom (potkany) v pomere k expozíciám pozorovaným u ľudí pri odporúčaných terapeutických dávkach.
Po dvoch rokoch podávania darunaviru pri expozíciách na úrovni alebo pod úrovňou expozícií u ľudí sa pozorovali zmeny na obličkách u myší (nefróza) a u potkanov (chronická progresívna nefropatia).
Darunavir nebol mutagénny ani genotoxický v súbore in vitro a in vivo testov vrátane bakteriálnej reverznej mutácie (Amesov test), chromozómovej aberácie v ľudských lymfocytoch
a mikronukleového testu u myší in vivo.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Oxid kremičitý, koloidný bezvodý
Mikrokryštalická celulóza
Krospovidón
Sodná soľ glykolátu škrobu
Hypromelóza
Magnéziumstearát
Obal tablety
Polyvinylalkohol, čiastočne hydrolyzovaný
Oxid titaničitý Makrogol Mastenec
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po prvom otvorení HDPE fľaše: 100 dní.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PVC/PE/PVDC-Al blistrové balenie s obsahom 480 tabliet a 480 x 1 tabliet.
Za studena spracované PVC/Al/OPAPE/PVDC-Al blisterové balenie s obsahom 480 tabliet a 480 x
1 tabliet.
Balenie s HDPE fľašou so PP skrutkovacím uzáverom s obsahom 480 tabliet. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky na likvidáciu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMylan S.A.S.
117 Allee des Parcs
69 800 Saint Priest
Francúzsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/16/1140/001
EU/1/16/1140/002
EU/1/16/1140/003
EU/1/16/1140/004
EU/1/16/1140/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: [DD. mesiac RRRR]
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
Darunavir Mylan 150 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Každá filmom obalená tableta obsahuje 150 mg darunaviru. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalené tablety.
Biele až sivobiele kapsulové obojstranne vypuklé filmom obalené tablety s rozmermi približne
12,75 mm x 6,3 mm, s označením „M“ na jednej strane a „DV2“ na druhej strane.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Darunavir podávaný súčasne s nízkou dávkou ritonaviru je indikovaná v kombinácii s inými antiretrovirálnymi liekmi na liečbu pacientov s infekciou vyvolanou vírusom ľudskej imunodeficiencie (HIV-1).
Darunavir Mylan 150 mg tablety sa môžu použit na zabezpečenie vhodného režimu dávkovania (pozri časť 4.2):
· Pri liečbe infekcie HIV- 1 u dospelých pacientov predtým liečených antiretrovírusovou liečbou
(ART, antiretroviral treatment), vrátane tých s intenzívnou predchádzajúcou liečbou.
· Pri liečbe infekcie HIV-1 u pediatrických pacientov od 3 rokov a s telesnou hmotnosťou
najmenej 15 kg.
Pri rozhodovaní o začatí liečby darunaviromužívaným spolu s nízkou dávkou ritonaviru je potrebné dôkladne zvážiť liekovú anamnézu jednotlivého pacienta a schémy mutácií súvisiace s rôznymi agensmi. Používanie darunaviru má byť odvodené od geno- a fenotypizácie (ak sú dostupné)
a liečebnej anamnézy.
4.2 Dávkovanie a spôsob podávania
Liečbu môže začať len lekár, ktorý má skúsenosti s liečbou infekcie HIV. Po začatí liečby darunavirom treba pacientov poučiť, aby nemenili dávku, liekovú formu ani neprerušovali liečbu bez toho, aby sa poradili so svojím lekárom.
Dávkovanie
Darunavir Mylan sa musí vždy podávať perorálne v kombinácii s nízkou dávkou ritonaviru, ktorý
zlepšuje jej farmakokinetiku, a s inými antiretrovírusovými liekmi. Z toho dôvodu je pred začatím kombinovanej liečby darunavirom a ritonavirom potrebné prečítať si súhrn charakteristických vlastností ritonaviru.
Dospelí pacienti liečení ART
Odporúčané dávkovanie je 600 mg dvakrát denne súčasne so 100 mg ritonaviru dvakrát denne a s jedlom. Darunavir 150 mg tablety sa môžu použiť na zostavenie režimu dávkovania 600 mg dvakrát
denne.
Užívanie 150 mg tabliet na dosiahnutie odporúčanej dávky je vhodné v prípade možnej precitlivenosti
na niektoré farbivá alebo v prípade ťažkostí s prehĺtaním 300 mg alebo 600 mg tabliet.
Dávkovací režim 800 mg jedenkrát denne s kobicistatom 150 mg jedenkrát denne alebo so 100 mg ritonaviru jedenkrát denne a s jedlom sa môže použiť u pacientov s predchádzajúcou liečbou antiretrovirotikami, ale bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM, z angl. darunavir-resistance associated mutations)* a s plazmatickou hladinou HIV-1 RNA
< 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri súhrn charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Dospelí pacienti predtým neliečení ARTOdporučené dávkovanie u pacientov predtým neliečených ART nájdete v súhrne charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Pediatrickí pacienti predtým neliečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 15 kg)Dávka darunaviru a ritonaviru u pediatrických pacientov odvodená od ich hmotnosti sa uvádza v tabuľke nižšie.
Odporúčaná dávka tabliet darunaviru
a ritonavirua na liečbu predtým neliečených pediatrickýchpacientov (3 až 17 rokov)Telesná hmotnosť (kg) Dávka (jedenkrát denne s jedlom)≥ 15 kg až < 30 kg 600 mg darunavir/100 mg ritonavir jedenkrát denne
≥ 30 kg až < 40 kg 675 mg darunavir/100 mg ritonavir jedenkrát denne
³ 40 kg 800 mg darunavir/100 mg ritonavir jedenkrát denne
a ritonavir perorálny roztok: 80 mg/ml
Pediatrickí pacienti liečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 15 kg)Zvyčajne sa odporúča darunavir dvakrát denne súčasne s ritonavirom a s jedlom.
Dávkovanie darunaviru užívaného s ritonaviromu jedenkrát denne a s jedlom sa môže použiť
u pacientov s predchádzajúcou liečbou antiretrovirotikami, ale bez mutácií spojených s rezistenciou
voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom
buniek CD4+ ³ 100 buniek x 106/l.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Dávka darunaviru a ritonaviru u pediatrických pacientov odvodená od ich hmotnosti sa uvádza
v tabuľke nižšie. Odporúčaná dávka darunaviru s nízkou dávkou ritonaviru nemá prekročiť dávku odporúčanú pre dospelých (600/100 mg dvakrát denne alebo 800/100 mg jedenkrát denne).
 Odporúčaná dávka tabliet
Odporúčaná dávka tabliet darunaviru
a ritonavirua u predtým liečenýchpediatrických pacientov (3 až 17 rokov)
Hm
o
t
nosť (kg) Dávka (jedenkrát denne s jedlom)
≥ 15 kg–< 30 kg 600 mg darunavir/100 mg ritonavir jedenkrát denne
≥ 30 kg–< 40 kg 675 mg darunavir/100 mg ritonavir jedenkrát denne
≥ 40 kg 800 mg darunavir/100 mg ritonavir jedenkrát denne
a s perorálnym roztokom ritonaviru: 80 mg/ml
Dávka (dvakrát denne s jedlom)
375 mg darunavir/50 mg ritonavir dvakrát denne
450 mg darunavir/60 mg ritonavir dvakrát denne
600 mg darunavir/100 mg ritonavir dvakrát denne

U pediatrických pacientov predtým liečených ART sa odporúča testovanie genotypu HIV. Ak
testovanie genotypu HIV nie je k dispozícii, dávkovanie darunaviru/ritonaviru jedenkrát denne sa odporúča u pediatrických pacientov, ktorí predtým neužívali HIV proteázové inhibítory, a dávkovanie dvakrát denne sa odporúča u pacientov, ktorí predtým užívali HIV proteázové inhibítory.
Použitie výlučne 75 mg a 150 mg tabliet na dosiahnutie odporúčanej dávky darunaviru môže byť vhodné, keď existuje riziko precitlivenosti na niektoré farbivo.
Odporúčanie pri vynechaní dávky
V prípade, že pacient zabudne užiť dávku darunaviru a/alebo ritonaviru do 6 hodín od doby, kedy liek zvyčajne užíva, treba ho poučiť, že má užiť predpísanú dávku s jedlom čo najskôr. Ak od zvyčajnej doby použitia uplynie viac ako 6 hodín, vynechaná dávka sa nemá užiť a pacient má pokračovať vo zvyčajnom dávkovacom režime.
Toto usmernenie vyplynulo z 15 hodinového polčasu darunaviru v prítomnosti ritonaviru a
odporúčaného dávkovacieho intervalu približne 12 hodín.
Osobitné skupiny pacientov
Starší ľudia
Informácie o tejto populácii sú obmedzené, a preto sa má darunavir používať v tejto vekovej skupine
so zvýšenou opatrnosťou (pozri časti 4.4 a 5.2).
Porucha funkcie pečene
Darunavir sa metabolizuje v pečeňovom systéme. U pacientov s miernou (Childova-Pughova Trieda A) alebo stredne ťažkou (Childova-Pughova Trieda B) poruchou funkcie pečene sa neodporúča žiadna úprava dávky, avšak týmto pacientom sa má darunavir podávať so zvýšenou opatrnosťou. U pacientov s ťažkou poruchou funkcie pečene nie sú k dispozícii žiadne farmakokinetické údaje. Ťažká porucha funkcie pečene môže viesť k zvýšeniu expozície darunaviru a k zhoršeniu jeho bezpečnostného
profilu. Z toho dôvodu sa darunavir nesmie podávať pacientom s ťažkou poruchou funkcie pečene
(Childova-Pughova Trieda C) (pozri časti 4.3, 4.4 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávkovania (pozri časti 4.4
a 5.2).
Pediatrickí pacienti
Darunavir/ritonavir sa nemá používať u detí s hmotnosťou menej ako 15 kg, pretože dávka pre túto populáciu nebola stanovená na dostatočnom počte pacientov (pozri časť 5.1). Darunavir/ritonavir sa
nemá používať u detí mladších ako 3 roky z dôvodu bezpečnosti (pozri časti 4.4 a 5.3).
Boli stanovené expozície darunaviru u dospievajúcich vo veku 12 až 17 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení a ktorí dostávali darunavir 800 mg jedenkrát denne a ukázalo sa, že sa nachádzali v rámci terapeutického rozsahu, ktorý bol stanovený u dospelých pacientov dostávajúcich darunavir 800 mg jedenkrát denne. Ako dôsledok vzhľadom na to, že
darunavir jedenkrát denne bol tiež zaregistrovaný na použitie u dospelých s predchádzajúcou liečbou
bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1
RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l, rovnaká indikácia darunaviru
jedenkrát denne sa vzťahuje na deti vo veku 3 až 17 rokov a s hmotnosťou najmenej 15 kg s predchádzajúcou liečbou.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Gravidita a obdobie po pôrode
Počas gravidity a po pôrode nie je potrebná úprava dávky darunaviru/ritonaviru. Darunavir sa má
užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko (pozri časti 4.4, 4.6 a 5.2).
Spôsob podávania
Pacientov treba poučiť, aby užívali Darunavir Mylan s nízkou dávkou ritonaviru do 30 minút po jedle.
Druh jedla neovplyvňuje expozíciu darunaviru (pozri časti 4.4, 4.5 a 5.2).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Pacienti s ťažkou (Childova-Pughova Trieda C) poruchou funkcie pečene.
Súčasné podávanie rifampicínu a darunaviru spolu s nízkou dávkou ritonaviru (pozri časť 4.5). Súčasné podávanie lieku s kombináciou lopinavir/ritonavir (pozri časť 4.5).
Súčasné podávanie rastlinných preparátov obsahujúcich výťažok z ľubovníka bodkovaného
(Hypericum perforatum) (pozri časť 4.5).
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru a liečiv, ktorých metabolizmus (klírens) je vysoko závislý na CYP3A, a ktorých zvýšené plazmatické koncentrácie sú spojené s vážnymi a/alebo život ohrozujúcimi udalosťami. Medzi tieto liečivá patria napr.:
- alfuzosín (antagonista alfa 1-adrenoreceptora)
- amiodarón, bepridil, dronedarón, chinidín, ranolazín, lidokaín (podávaný systémovo) (antiarytmiká/lieky proti angíne)
- astemizol, terfenadín (antihistaminiká)
- kolchicín, keď sa používa u pacientov s poruchou funkcie obličiek a/alebo pečene (liek proti dne) (pozri časť 4.5)
- námeľové alkaloidy (napr. dihydroergotamín, ergometrín, ergotamín, metylergonovín)
- cisaprid (prokinetiká)
- pimozid, kvetiapín, sertindol (antipsychotiká/neuroleptiká) (pozri časť 4.5)
- triazolam, midazolam podávaný perorálne (sedatíva/hypnotiká) (pre upozornenie na midazolam
podávaný parenterálne, pozri časť 4.5)
- sildenafil - keď sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, avanafil (inhibítory
PDE-5)
- simvastatín a lovastatín (inhibítory HMG-CoA reduktázy) (pozri časť 4.5)
- tikagrelor (inhibítor agregácie trombocytov) (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hoci sa preukázalo, že účinná vírusová supresia dosiahnutá pri antiretrovírusovej terapii značne znižuje riziko prenosu HIV pohlavným stykom, reziduálne riziko nie je možné vylúčiť. Je potrebné prijať opatrenia na zabránenie prenosu HIV v súlade s národnými odporúčaniami.
Odporúča sa pravidelné sledovanie virologickej odpovede. V prípade nedostatočnej alebo žiadnej virologickej odpovede sa má urobiť test na rezistenciu.
Darunavir sa má používať len v kombinácii s nízkou dávkou ritonaviru, ktorý zlepšuje
farmakokinetiku darunaviru (pozri časť 5.2).
Zvyšovanie dávkovania ritonaviru oproti odporúčaniam v časti 4.2 významnejšie neovplyvňuje
koncentrácie darunaviru a neodporúča sa.
Darunavir sa viaže prevažne na a1-kyslý glykoproteín. Táto proteínová väzba závisí od koncentrácie, ktorú určuje saturácia väzby. Preto lieky, ktoré sa vysoko viažu na a1-kyslý glykoproteín, môžu znemožniť túto väzbu (pozri časť 4.5).
Pacienti liečení ART – dávkovanie jedenkrát denne
Darunavir užívaný v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru jedenkrát denne
u pacientov liečených ART sa nemá používať u pacientov s jednou alebo viacerými mutáciami spojenými s rezistenciou voči darunaviru (DRV-RAM) alebo s hladinou HIV-1 RNA
≥ 100 000 kópií/ml alebo počtom buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2). V tejto populácii
sa neskúmali iné kombinácie s optimalizovaným základným režimom (OBR, optimised background regimen) ako ≥ 2 NRTI. U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje (pozri časť 5.1).
Pediatrická populácia
Darunavir sa neodporúča používať u pediatrických pacientov mladších ako 3 roky alebo s hmotnosťou
menej ako 15 kg (pozri časti 4.2 a 5.3).
Gravidita
Darunavir sa má užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko. Opatrnosť je potrebná u gravidných žien súbežne užívajúcich lieky, ktoré môžu ďalej znižovať expozíciu darunaviru (pozri časti 4.5 a 5.2).
Starší ľudia
Keďže k dispozícii sú len obmedzené údaje o používaní darunaviru u pacientov vo veku 65 rokov a
starších, pri podávaní darunaviru starším pacientom je potrebná opatrnosť vzhľadom k vyššiemu výskytu zhoršenej funkcie pečene a pridružených chorôb, alebo k inej liečbe (pozri časti 4.2 a 5.2).
Závažné kožnéreakcie
Počas programu klinického vývoja (N=3 063) boli u 0,4 % pacientov hlásené závažné kožné reakcie,
ktoré mohla sprevádzať horúčka a/alebo zvýšenie transamináz. Zriedkavo (< 0,1 %) bola hlásená lieková vyrážka s eozinofíliou a systémovými príznakmi (DRESS, drug rash with eosinophilia and systemic symptoms) a Stevensov-Johnsonov syndróm a počas postmarketingových skúseností bola hlásená toxická epidermálna nekrolýza a akútna generalizovaná exantematózna pustulóza. Ak sa objavia prejavy alebo príznaky závažnej kožnej reakcie, liečba darunavirom/ritonavirom sa má okamžite prerušiť. Príznaky môžu zahŕňať, ale nemusia byť obmedzené na, závažnú vyrážku alebo vyrážku sprevádzanú horúčkou, celkovú nevoľnosť, únavu, bolesť svalov alebo kĺbov, pľuzgiere, orálne lézie, konjunktivitídu, hepatitídu a/alebo eozinofíliu.
Vyrážka sa častejšie vyskytla u predtým liečených pacientov, ktorí boli liečení darunavirom spolu
s raltegravirom v porovnaní s pacientmi, ktorí užívali darunavir bez raltegraviru alebo raltegravir bez darunaviru (pozri časť 4.8).
Darunavir obsahuje sulfónamidovú funkčnú skupinu. Darunavir Mylan sa má podávať opatrne
pacientom so známou alergiou na sulfónamid.
Hepatotoxicita
Počas užívania darunaviru bola hlásená liekom indukovaná hepatitída (napr. akútna hepatitída,
cytolytická hepatitída). Počas programu klinického vývoja (N=3 063) bola hepatitída hlásená u 0,5 % pacientov, ktorí boli liečení kombinovanou antiretrovirálnou terapiou s darunavirom/ritonavirom. Pacienti s preexistujúcou dysfunkciou pečene, vrátane chronickej aktívnej hepatitídy B alebo C, majú zvýšené riziko porúch funkcie pečene, vrátane vážnych a potenciálne smrteľných hepatických nežiaducich reakcií. V prípade súčasnej antivírovej liečby hepatitídy B alebo C, obráťte sa na relevantnú informáciu o týchto liekoch.
Pred začatím liečby darunavirom/ritonavirom sa majú urobiť vhodné laboratórne vyšetrenia a pacienti majú byť sledovaní aj počas liečby. U pacientov s chronickou hepatitídou, cirhózou alebo u pacientov, ktorí mali pred liečbou zvýšenú hladinu transamináz, treba zvážiť zvýšené sledovanie AST/ALT, najmä počas prvých mesiacov po začatí liečby darunavirom/ritonavirom.
Ak sa vyskytne alebo sa zhorší dysfunkcia pečene (vrátane klinicky významného zvýšenia pečeňových enzýmov a/alebo príznakov ako únava, anorexia, nauzea, žltačka, tmavý moč, citlivosť pečene,
hepatomegália) u pacientov užívajúcich darunavir/ritonavir, treba okamžite zvážiť zastavenie alebo prerušenie liečby.
Pacienti s pridruženými ochoreniami
Porucha funkcie pečene
U pacientov so závažnými poruchami pečene sa účinnosť a bezpečnosť darunaviru nestanovovala, preto je darunavir u pacientov s ťažkou poruchou funkcie pečene kontraindikovaný. Vzhľadom na
nárast koncentrácie voľného darunaviru v plazme sa darunavir musí používať opatrne u pacientov
s miernou alebo stredne ťažkou poruchou funkcie pečene (pozri časti 4.2, 4.3 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa nevyžadujú žiadne osobitné opatrenia alebo úpravy dávkovania. Darunavir aj ritonavir sa výrazne viažu na plazmatické bielkoviny, a preto je
nepravdepodobné, že by sa dali z cirkulácie významnejšie odstrániť pomocou hemodialýzy alebo
peritoneálnej dialýzy. Preto sa u týchto pacientov nevyžadujú žiadne osobitné opatrenia alebo úpravy dávkovania (pozri časti 4.2 a 5.2).
Pacienti s hemofíliou
U pacientov s hemofíliou A a B liečených PI sa opísali prípady väčšieho krvácania, vrátane spontánnych kožných hematómov a krvácania do kĺbov. Niektorým pacientom sa podal dodatočný
faktor VIII. Vo viac ako polovici hlásených prípadov sa pokračovalo v liečbe PI, prípadne sa táto
liečba obnovila po jej dočasnom prerušení. Bola naznačená kauzálna súvislosť, hoci mechanizmus
účinku nebol zatiaľ objasnený. Z toho dôvodu je potrebné hemofilikov upozorniť na možnosť väčšieho krvácania.
Telesná hmotnosť a metabolické parametre
Počas antiretrovírusovej liečby môže dôjsť k zvýšeniu telesnej hmotnosti a hladín lipidov a glukózy
v krvi. Takéto zmeny môžu čiastočne súvisieť s kontrolou ochorenia a životným štýlom. Pokiaľ ide
o lipidy, v niektorých prípadoch sú dôkazy o vplyve liečby, kým pri prírastku telesnej hmotnosti nie sú
presvedčivé dôkazy o tom, že súvisí s niektorou konkrétnou liečbou. Pri monitorovaní hladín lipidov a glukózy v krvi sa treba riadiť zavedenými odporúčaniami na liečbu infekcie HIV. Poruchy metabolizmu lipidov majú byť klinicky vhodne liečené.
Osteonekróza
Napriek tomu, že etiológia ochorenia závisí od mnohých faktorov (vrátane užívania kortikosteroidov,
konzumácie alkoholu, vážnej imunosupresie, vyššieho BMI), prípady osteonekrózy boli hlásené najmä u pacientov s pokročilým ochorením HIV a/alebo s dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART, combination antiretroviral therapy). Pacientov treba poučiť, aby vyhľadali lekársku pomoc v prípade, že budú pociťovať bolesti kĺbov, stuhnutosť kĺbov alebo ťažkosti pri pohybe.
Imunoreštitučný zápalový syndróm
U pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej
antiretrovírusovej liečby môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie, čo môže byť spojené so závažným klinickým stavom resp. so
zhoršením symptómov. Tieto reakcie sa obyčajne pozorujú v priebehu prvých týždňov až mesiacov od
začatia podávania kombinovanej antiretrovírusovej liečby. Príkladom môže byť cytomegalovírusová
retinitída, generalizované alebo fokálne mykobaktériové infekcie a pneumónia vyvolaná Pneumocystis jirovecii (predtým známa ako Pneumocystis carinii). Je nutné zhodnotiť všetky príznaky zápalového ochorenia a v prípade potreby začať liečbu. Ďalej sa v klinických štúdiách s darunavirom užívaným s nízkou dávkou ritonaviru pozorovala reaktivácia herpesu simplex a herpesu zoster.
Boli tiež zaznamenané aj poruchy autoimunitného systému (ako je Gravesova choroba) objavujúce sa v dôsledku imunitnej reaktivácie; avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.8).
Liekové interakcie
Niektoré štúdie interakcií sa uskutočnili s darunavirom v nižších dávkach ako sú odporúčané. Účinky
súčasne podávaných liekov môžu byť preto podhodnotené a môže byť potrebné klinické sledovanie bezpečnosti. Pre úplnú informáciu o liekových interakciách pozri časť 4.5.
Efavirenz v kombinácii s darunavirom/ritonavirom 800/100 mg jedenkrát denne môže viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť v kombinácii s darunavirom/ritonavirom, treba použiť režim dávkovania darunavir/ritonavir 600/100 mg dvakrát denne. Pozri súhrn charakteristických vlastností lieku darunavir 75 mg, 300 mg alebo 600 mg tablety (pozri časť 4.5).
Život ohrozujúce a smrteľné liekové interakcie sa zaznamenali u pacientov liečených kolchicínom
a silnými inhibítormi CYP3A a P-glykoproteínu (P-gp; pozri časti 4.3 a 4.5).
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Darunavir a ritonavir sú inhibítormi CYP3A, CYP2D6 a P-gp. Súčasné podávanie darunaviru a ritonaviru s liekmi metabolizovanými najmä CYP3A a/alebo CYP2D6 alebo transportovanými P-gp môže viesť k zvýšeniu systémovej expozície týmto liekom, čo by mohlo zosilniť alebo predĺžiť ich terapeutický účinok a viesť k nežiaducim účinkom.
Darunavir užívaný s nízkou dávkou ritonaviru sa nesmie kombinovať s liekmi, ktorých klírens výrazne závisí od CYP3A, a pri ktorých zvýšená systémová expozícia je sprevádzaná závažnými a/alebo život ohrozujúcimi nežiaducimi udalosťami (t.j. ktoré majú úzky terapeutický index) (pozri časť 4.3).
Celkové zvýšenie farmakokinetického účinku ritonavirom viedlo približne k 14-násobnému zvýšeniu
systémovej expozície darunaviru, po jednorazovej 600 mg perorálnej dávke darunaviru v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne. Z toho dôvodu sa darunavir môže používať
len v kombinácii s nízkou dávkou ritonaviru, ktorý zlepšuje farmakokinetiku darunaviru (pozri časti
4.4 a 5.2).
Klinická štúdia s viacerými liekmi, ktoré sú metabolizované cytochrómami CYP2C9, CYP2C19 a CYP2D6 dokázala nárast aktivity CYP2C9 a CYP2C19 a inhibíciu aktivity CYP2D6 v prítomnosti darunaviru/ritonaviru, čo sa môže pripisovať prítomnosti nízkej dávky ritonaviru. Súčasné podávanie darunaviru a ritonaviru a liekov, ktoré sa predovšetkým metabolizujú prostredníctvom CYP2D6 (napríklad flekainid, propafenon, metoprolol), môže vyústiť do zvýšených plazmatických koncentrácií týchto liekov, čo môže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce reakcie. Súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C9 (napríklad warfarín) a CYP2C19 (napríklad metadon), môže vyústiť do zníženia systémových
expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Napriek tomu, že sa účinok na CYP2C8 sledoval iba in vitro, súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C8 (napríklad paklitaxel, rosiglitazón, repaglinid), môže vyústiť do zníženia systémových expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Lieky, ktoré ovplyvňujú expozíciu darunaviru/ritonaviru
Darunavir a ritonavir sa metabolizujú prostredníctvom CYP3A. Očakáva sa, že lieky indukujúce činnosť CYP3A zvýšia klírens darunaviru a ritonaviru, čo má za následok zníženie plazmatických koncentrácií darunaviru a ritonaviru (napr. rifampicín, ľubovník bodkovaný, lopinavir). Súčasné
podávanie darunaviru a ritonaviru v kombinácii s liekmi, ktoré inhibujú CYP3A môže znížiť klírens
darunaviru a ritonaviru a môže mať za následok zvýšenie plazmatických koncentrácií darunaviru
a ritonaviru (napr. indinavir, systémové azoly ako ketokonazol a klotrimazol). Tieto interakcie sú opísané v nižšie uvedenej tabuľke interakcií.
Tabuľkainterakcií
Interakcie medzi darunavirom/ritonavirom a antiretrovirotikami a ne-antiretrovírusovými liekmi sú uvedené v tabuľke nižšie (neurčené ako „ND“ (z angl. not determinated)). Smer šípky pri každom
farmakokinetickom parametri je založený na 90 % intervale spoľahlivosti stredného geometrického pomeru v rámci 80-125 % rozpätia (↔), pod ním (↓) alebo nad ním (↑).
Niekoľko štúdií interakcií (v tabuľke nižšie označené ako #) sa uskutočnili s nižšími ako odporúčanými dávkami darunaviru alebo s iným režimom dávkovania (pozri časť 4.2 Dávkovanie). Účinky súčasne podávaných liekov môžu byť preto podhodnotené a môže byť potrebné klinické sledovanie bezpečnosti.
INTERAKCIE A ODPORUČENÉ DÁVKOVANIE S INÝMI LIEKMI
L
ieky podľa
t
erapeutických oblastí
I
nterakcia
Z
m
e
na geometrického priemeru (%)
O
dporúčania pri súčasnom
podávaní
 ANTIRETROVIROTIKÁ NA HIV
I
nhibítory prenosu reťazcov integrázou
ANTIRETROVIROTIKÁ NA HIV
I
nhibítory prenosu reťazcov integrázou
Dolutegravir dolutegravir AUC ↓ 32 % dolutegravir C24h 38 % dolutegravir Cmax ↓ 11 % darunavir ↔*
* Použitím porovnania údajov zo skríženej štúdie
s historickými údajmi o farmakokinetike
Elvitegravir elvitegravir AUC ↔ elvitegravir Cmin ↔ elvitegravir Cmax ↔ darunavir AUC ↔ darunavir Cmin 17 % darunavir Cmax ↔
Raltegravir Niektoré klinické štúdie naznačujú, že raltegravir môže spôsobiť mierny pokles koncentrácie darunaviru v plazme.
Darunavir užívaný s nízkou dávkou ritonaviru a dolutegravir sa môžu užívať bez úpravy dávky.
Keď sa darunavir užívaný súčasne s nízkou dávkou ritonaviru (600/100 mg dvakrát denne) užíva spolu s elvitegravirom, dávka elvitegraviru má byť 150 mg jedenkrát denne.
Farmakokinetika a odporúčania na dávkovanie pre iné dávky darunaviru alebo
s elvitegravirom/kobicistatom neboli stanovené. Súčasné užívanie darunaviru s nízkou dávkou ritonaviru v iných dávkach ako
600/100 mg dvakrát denne
a elvitegraviru sa preto neodporúča. Súčasné užívanie darunaviru
s nízkou dávkou ritonaviru
a elvitegravirom za prítomnosti
kobicistatu sa neodporúča.
V súčasnosti sa účinok raltegraviru na plazmatické koncentrácie darunaviru nejaví ako klinicky významný. Darunavir súčasne podávaný s nízkou dávkou ritonaviru a raltegraviru sa môže užívať bez úpravy dávky.
Nukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, nucleo(s/t)ide reverse transcriptase inhibitors)
Didanozín
400 mg jedenkrát denne
Tenofovir disoproxil fumarát
300 mg jedenkrát denne
Abakavir Emtricitabín Lamivudín Stavudín Zidovudín
didanozín AUC ↓ 9 % didanozín Cmin ND didanozín Cmax ↓ 16 % darunavir AUC ↔ darunavir Cmin ↔ darunavir Cmax ↔
tenofovir AUC ↑ 22 % tenofovir Cmin ↑ 37 % tenofovir Cmax ↑ 24 %
#darunavir AUC ↑ 21 %
#darunavir Cmin ↑ 24 %
#darunavir Cmax ↑ 16 %
(↑ tenofoviru v dôsledku efektu na MDR-1
transport v renálnych tubuloch)
Neskúmalo sa. Vzhľadom na rôzne cesty eliminácie ostatných NRTI zidovudín, emtricitabín, stavudín, lamivudín, ktoré sa vylučujú prevažne renálnou cestou
a abakavir, ktorý nie je metabolizovaný prostredníctvom CYP450, sa pri ich kombinácii s darunavirom užívaným
s nízkou dávkou ritonaviru
nepredpokladajú žiadne liekové interakcie.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a didanozínu sa môže užívať bez úpravy dávok.
Didanozín sa má podávať nalačno, preto ho treba užiť 1 hodinu pred alebo 2 hodiny po užití darunaviru/ritonaviru s jedlom.
Pri súčasnom podávaní darunaviru a nízkej dávky ritonaviru
v kombinácii s tenofovirom môže byť indikované sledovanie renálnych funkcií, najmä
u pacientov so systémovým alebo renálnym ochorením, alebo
u pacientov užívajúcich
nefrotoxické lieky.
Darunavir užívaný s nízkou dávkou ritonaviru sa môže užívať s týmito NRTI bez úpravy dávky.
Nenukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, non-nucleo(s/t)ide reverse transcriptase inhibitors)

Efavirenz
600 mg jedenkrát denne
Etravirín
100 mg dvakrát denne
Nevirapín
200 mg dvakrát denne
efavirenz AUC ↑ 21 % efavirenz Cmin ↑ 17 % efavirenz Cmax ↑ 15 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↓ 31 %
#darunavir Cmax ↓ 15 %
(↑ efavirenzu v dôsledku inhibície
CYP3A)
(↓ darunaviru v dôsledku inhibície
CYP3A)
etravirín AUC ↓ 37 % etravirín Cmin ↓ 49 % etravirín Cmax ↓ 32 % darunavir AUC ↑ 15 % darunavir Cmin ↔ darunavir Cmax ↔ nevirapín AUC ↑ 27 % nevirapín Cmin ↑ 47 % nevirapín Cmax ↑ 18 %
#darunavir: koncentrácie
sa zhodovali s údajmi z minulosti. (↑ nevirapínu v dôsledku inhibície CYP3A)
Pri súčasnom podávaní darunaviru a nízkej dávky ritonaviru
v kombinácii s efavirenzom môže byť indikované klinické sledovanie toxicity centrálneho nervového systému spojené so zvýšenou expozíciou voči efavirenzu.
Efavirenz v kombinácii
s darunavirom/ritonavirom
800/100 mg jedenkrát denne môže viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť
v kombinácii
s darunavirom/ritonavirom, treba použiť režim dávkovania darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.4). Darunavir súčasne podávaný
s nízkou dávkou ritonaviru a
s etravirínom
200 mg dvakrátdennesa môže užívať bez úpravy dávky.
Darunavir súčasne podávaná
s nízkou dávkou ritonaviru a
s nevirapínom sa môže užívať bez
úpravy dávky.
Rilpivirín
150 mg jedenkrát denne
rilpivirín AUC ↑ 130 % rilpivirín Cmin ↑ 178 % rilpivirín Cmax ↑ 79 % darunavir AUC ↔ darunavir Cmin ↓ 11 % darunavir Cmax ↔
Darunavir súčasne podávaná
s nízkou dávkou ritonaviru
a s rilpivirínom sa môže užívať bez
úpravy dávky.
H
I
V Proteázové inhibítory (PI) - bez doplnkového súčasného podávania nízkej dávky ritonaviru
†
Atazanavir
300 mg jedenkrát denne
Indinavir
800 mg dvakrát denne
Sakvinavir
1 000 mg dvakrát denne
atazanavir AUC ↔ atazanavir Cmin ↑ 52 % atazanavir Cmax ↓ 11 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Atazanavir: porovnanie atazanaviru/ritonaviru 300/100 mg jedenkrát denne vs. atazanavir 300 mg jedenkrát denne v kombinácii
s darunavirom/ritonavirom 400/100 mg dvakrát denne.
Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg dvakrát denne v kombinácii s atazanavirom 300 mg jedenkrát denne.
indinavir AUC ↑ 23 % indinavir Cmin ↑ 125 % indinavir Cmax ↔
#darunavir AUC ↑ 24 %
#darunavir Cmin ↑ 44 %
#darunavir Cmax ↑ 11 %
Indinavir: porovnanie indinaviru/ritonaviru
800/100 mg dvakrát denne vs. indinavir/darunavir/ritonavir
800/400/100 mg dvakrát denne. Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii s indinavirom 800 mg dvakrát denne.
#darunavir AUC ↓ 26 %
#darunavir Cmin ↓ 42 %
#darunavir Cmax ↓ 17 %
#sakvinavir AUC ↓ 6 %
#sakvinavir Cmin ↓ 18 %
#sakvinavir Cmax ↓ 6 %
Sakvinavir: porovnanie sakvinaviru/ritonaviru 1000/100 mg dvakrát denne vs. sakvinavir/darunavir/ritonavir
1000/400/100 mg dvakrát denne Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii so sakvinavirom 1000 mg dvakrát denne.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s atazanavirom sa môže užívať bez
úpravy dávky.
Pri intolerancii súčasného podávania darunaviru užívaného s nízkou dávkou ritonaviru
s indinavirommôže byť indikované
zníženie dávkovania indinaviru
z 800 mg dvakrát denne na 600 mg dvakrát denne.
Neodporúča sa kombinovať darunavir užívaný s nízkou dávkou ritonaviru so sakvinavirom.
H
I
V Proteázové inhibítory (PI) - podávané s nízkou dávkou ritonaviru
†
Lopinavir/ritonavir
400/100 mg dvakrát denne
Lopinavir/ritonavir
533/133,3 mg dvakrát denne
CCR5-ANTAGONISTY
Maravirok
150 mg dvakrát denne
ANESTETIKÁ
lopinavir AUC ↑ 9 % lopinavir Cmin ↑ 23 % lopinavir Cmax ↓ 2 % darunavir AUC ↓ 38 %‡ darunavir Cmin ↓ 51 %‡ darunavir Cmax ↓ 21 %‡ lopinavir AUC ↔ lopinavir Cmin ↑ 13 % lopinavir Cmax ↑ 11 % darunavir AUC ↓ 41% darunavir Cmin ↓ 55 % darunavir Cmax ↓ 21 %
‡ na základe neštandardizovaných veľkostí dávky
maravirok AUC ↑ 305 % maravirok Cmin ND maravirok Cmax ↑ 129 %
koncentrácie darunaviru, ritonaviru sa
zhodovali s údajmi z minulosti
Vzhľadom na pokles AUC darunaviru o 40%, kombinácie vhodných dávok neboli stanovené. Preto je súčasné podávanie darunaviru užívaného s nízkou dávkou ritonaviru a lieku
s kombináciou lopinavir/ritonavir
kontraindikované (pozri časť 4.3).
Keď sa maravirok podáva
s darunavirom s nízkou dávkou
ritonaviru, jeho dávka má byť
150 mg dvakrát denne.
Alfentanil Neskúmalo sa. Metabolizmus alfentanilu je sprostredkovaný CYP3A a v podstate môže byť inhibovaný darunavirom užívaným s nízkou dávkou ritonaviru.
LIEKY PROTI ANGÍNE/ANTIARYTMIKÁ
Súčasné užívanie s darunavirom
a nízkou dávkou ritonaviru môže vyžadovať zníženie dávky alfentanilu a sledovanie rizík predĺženého alebo oneskoreného respiračného útlmu.

Disopyramid Flekainid Mexiletín Propafenón
Amiodarón Bepridil Dronedarón
Lidokaín (podávaný systémovo)
Chinidín Ranolazín Digoxín
0,4 mg jednorazová
dávka
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto antiarytmík.
(inhibícia CYP3A)
digoxín AUC ↑ 61 % digoxín Cmin ND digoxín Cmax ↑ 29 %
(↑ digoxínu v dôsledku možnej inhibície P-
gp)
Pri súčasnom užívaní týchto antiarytmík a darunaviru užívaného s nízkou dávkou ritonaviru sa vyžaduje opatrnosť a odporúča sa sledovanie terapeutických koncentrácií, ak je dostupné.
Súčasné užívanie darunaviru užívanej s nízkou dávkou ritonaviru a amiodarónu, bepridilu, dronedarónu, lidokaínu podávaného systémovo, chinidínu alebo ranolazínu je kontraindikované (pozri časť 4.3).
Vzhľadom na to, že digoxín má úzky terapeutický index, v prípade, že pacienti liečení darunavirom/ritonavirom užívajú digoxín, odporúča sa na začiatok predpísať čo najnižšiu možnú dávku digoxínu. Dávka digoxínu sa má opatrne titrovať, kým sa nedosiahne želaný klinický účinok pri sledovaní celkového klinického stavu
pacienta.
ANTIBIOTIKÁ
Klaritromycín
500 mg dvakrát denne
ANTIKOAGULANCIÁ
Apixaban Dabigatran etexilát Rivaroxaban
klaritromycín AUC ↑ 57 % klaritromycín Cmin ↑ 174 % klaritromycín Cmax ↑ 26 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↑ 1 %
#darunavir Cmax ↓ 17 %
Pri podávaní s darunavirom/ritonavirom neboli zistiteľné koncentrácie metabolitu
14-OH-klaritromycínu.
(↑ klaritromycínu v dôsledku inhibície
CYP3A a možnej inhibície P-gp)
Neskúmalo sa. Súčasné užívanie darunaviru a týchto antikoagulancií môže zvýšiť koncentrácie antikoagulantu. (inhibícia CYP3A a/alebo P-gp).
Vyžaduje sa opatrnosť, keď sa klaritromycín podáva v kombinácii s darunavirom užívaným spolu
s nízkou dávkou ritonaviru.
Užívanie darunaviru užívaného
s nízkou dávkou ritonaviru a týchto
antikoagulancií sa neodporúča.
Warfarín Neskúmalo sa. Pri súčasnom podávaní warfarínu a darunaviru s ritonavirom (nízkou dávkou) môže dôjsť k ovplyvneniu koncentrácií warfarínu.
ANTIKONVULZÍVA
Pri súčasnom podávaní warfarínu s darunavirom užívanou s nízkou dávkou ritonaviru sa odporúča monitorovať INR (International Normalized Ratio).

Fenobarbital
Fenytoín
Karbamazepín
200 mg dvakrát denne
ANTIDEPRESÍVAParoxetín
20 mg jedenkrát denne
Sertralín
50 mg jedenkrát denne
Amitriptylín Desipramín Imipramín Nortriptylín Trazodón
Neskúmalo sa. Predpokladá sa, že fenobarbital a fenytoín znižujú plazmatické koncentrácie darunaviru. (indukcia enzýmov CYP450) karbamazepín AUC ↑ 45 % karbamazepín Cmin ↑ 54 % karbamazepín Cmax ↑ 43 %
darunavir AUC ↔
darunavir Cmin ↓ 15 %
darunavir Cmax ↔
paroxetín AUC ↓ 39 % paroxetín Cmin ↓ 37 % paroxetín Cmax ↓ 36 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
sertralín AUC ↓ 49 %
sertralín Cmin ↓ 49 %
sertralín Cmax ↓ 44 %
#darunavir AUC ↔
#darunavir Cmin ↓ 6 %
#darunavir Cmax ↔
Súčasné užívanie darunaviru užívaného s nízkou dávkou ritonaviru a týchto antidepresív môže zvýšiť koncentrácie antidepresíva.
(inhibícia CYP2D6 a/alebo CYP3A).
Darunavir užívaný s nízkou dávkou
ritonaviru sa nesmie používať
v kombinácii s týmito liečivami.
Neodporúča sa žiadna úprava dávky darunaviru/ritonaviru. V prípade potreby kombinovať darunavir/ritonavir a karbamazepín sa majú pacienti sledovať z dôvodu možných nežiaducich udalostí spojených s karbamazepínom. Je potrebné sledovať koncentrácie karbamazepínu a titrovať jeho
dávku až do dosiahnutia primeranej odpovede. Na základe zistení môže byť potrebné znížiť dávku karbamazepínu o 25% až 50%, ak
sa podáva s darunavirom
/ritonavirom.
Ak sa antidepresíva užívajú súčasne s darunavirom užívaným s nízkou dávkou ritonaviru, odporúčaný prístup je titrácia dávky antidepresíva na základe klinického posúdenia odpovede na antidepresívum. Navyše u pacientov na ustálenej dávke týchto antidepresív, ktorí začínajú liečbu darunavirom užívaným s nízkou dávkou ritonaviru, sa má sledovať odpoveď na antidepresívum.
Keď sa tieto antidepresíva užívajú s darunavirom s nízkou dávkou ritonaviru, odporúča sa klinické sledovanie a môže byť potrebná úprava dávky antidepresíva.
ANTIMYKOTIKÁ
Vorikonazol Neskúmalo sa. Ritonavir môže znížiť plazmatické koncentrácie vorikonazolu. (indukcia enzýmov CYP450 ritonavirom)
Vorikonazol sa nesmie používať
spolu s darunavirom užívaným
s nízkou dávkou ritonaviru, kým vyhodnotenie pomeru prínos/riziko neodôvodní použitie vorikonazolu.
Ketokonazol
200 mg dvakrát denne
ketokonazol AUC ↑ 212 % ketokonazol Cmin ↑ 868 % ketokonazol Cmax ↑ 111 %
#darunavir AUC ↑ 42 %
#darunavir Cmin ↑ 73 %
#darunavir Cmax ↑ 21 %
(inhibícia CYP3A)
Vyžaduje sa opatrnosť a odporúča sa klinické sledovanie. Ak je nutná súčasná liečba, denná dávka ketokonazolu nesmie presiahnuť
200 mg.
Posakonazol Neskúmalo sa. Darunavir môže zvýšiť plazmatické koncentrácie antimykotika (inhibícia P-gp) a posakonazol môže zvýšiť koncentrácie darunaviru. (inhibícia CYP3A)
Itrakonazol Neskúmalo sa. Súčasné systémové podanie
itrakonazolu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť plazmatické koncentrácie darunaviru. Zároveň súčasné podávanie darunaviru užívaného s nízkou dávkou ritonaviru
a itrakonazolu môže zvýšiť plazmatické koncentrácie itrakonazolu.
(inhibícia CYP3A)
Klotrimazol Neskúmalo sa. Súčasné systémové podanie
klotrimazolu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť plazmatické koncentrácie darunaviru. darunavir AUC24h ↑ 33 % (na základe farmakokinetického modelu populácie)
Vyžaduje sa opatrnosť a odporúča
sa klinické sledovanie.
Vyžaduje sa opatrnosť a odporúča sa klinické sledovanie. Ak je nutná súčasná liečba, denná dávka itrakonazolu nesmie presiahnuť
200 mg.
Ak je nutná súčasná liečba klotrimazolom, vyžaduje sa opatrnosť a odporúča sa klinické sledovanie.
ANTIURATIKÁ
Kolchicín Neskúmalo sa. Súčasné užívanie
kolchicínu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť
expozíciu kolchicínu.
ANTIMALARIKÁ
Ak je potrebná liečba darunavirom užívanou s nízkou dávkou ritonaviru, odporúča sa u pacientov s normálnou funkciou obličiek alebo pečene znížiť dávku
kolchicínu alebo liečbu kolchicínom prerušiť.
Pacientom s poruchou funkcie
obličiek alebo pečene sa nemá podávať kolchicín s darunavirom užívanou s nízkou dávkou ritonaviru (pozri časť 4.4).

Artemeter/
lumefantrín
80/480 mg, 6 dávok v 0., 8., 24., 36., 48. a 60. hodine
artemeter AUC ↓ 16 % artemeter Cmin ↔ artemeter Cmax ↓ 18 %
dihydroartemisinín AUC ↓ 18 %
dihydroartemisinín Cmin ↔ dihydroartemisinín Cmax ↓ 18 % lumefantrín AUC ↑ 175 % lumefantrín Cmin ↑ 126 % lumefantrín Cmax ↑ 65 % darunavir AUC ↔
darunavir Cmin ↓ 13 %
darunavir Cmax ↔
Kombinácia darunaviru a artemeteru/lumefantrínu sa môže podávať bez úpravy dávky;
z dôvodu zvýšenia expozície lumefantrínu však treba kombináciu používať s opatrnosťou.
 ANTITUBERKULOTIKÁ
ANTITUBERKULOTIKÁ
Rifampicín
Rifapentín
Rifabutin
150 mg jedenkrát každý druhý deň
ANTINEOPLASTIKÁDasatinib Nilotinib Vinblastín Vinkristín
Everolimus
Neskúmalo sa. Rifapentín a rifampicín sú silnými induktormi CYP3A a bolo dokázané, že spôsobujú signifikantné zníženie koncentrácií iných proteázových inhibítorov, čo môže vyústiť do zlyhania antivirologickej odpovede a rozvinutia rezistencie (indukcia enzýmu CYP450). Pri pokusoch prekonať zníženú expozíciu zvýšením dávky iných proteázových inhibítorov s nízkou dávkou ritonaviru, sa s rifampicínom zistila vysoká frekvencia pečeňových reakcií.
rifabutin AUC** ↑ 55 % rifabutin Cmin** ↑ ND rifabutin Cmax** ↔ darunavir AUC ↑ 53 % darunavir Cmin ↑ 68 % darunavir Cmax ↑ 39 %
** súčet účinných zložiek rifabutinu (materský liek
+ 25-
O-desacetyl metabolit)
Štúdia interakcií preukázala porovnateľnú
dennú systémovú expozíciu rifabutinu
v liečbe samotným rifabutinom 300 mg
jedenkrát denne a v liečbe rifabutinom
150 mg jedenkrát každý druhý deň spolu s darunavirom/ritonavirom (600/100 mg dvakrát denne) s približne 10-násobným zvýšením dennej expozície aktívnemu metabolitu 25-
O-desacetylrifabutinu. Okrem toho, AUC súčtu účinných zložiek rifabutinu (materský liek + 25-
O-desacetyl metabolit) sa zvýšila 1,6-násobne, pričom Cmax zostala porovnateľná.
Údaje o porovnaní s referenčnou dávkou
150 mg jedenkrát denne nie sú k dispozícii.
(Rifabutin je induktorom a substrátom enzýmov CYP3A.) Keď sa darunavir užívaný spolu s 100 mg ritonaviru podávala spolu s rifabutinom (150 mg jedenkrát každý druhý deň) pozorovalo sa
zvýšenie systémovej expozície darunaviru.
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto antineoplastík.
(inhibícia CYP3A)
Kombinácia rifapentínu
a darunaviru užívanej s nízkou
dávkou ritonaviru sa neodporúča.
Súčasné podávanie rifampicínu
a darunaviru spolu s nízkou dávkou ritonaviru je kontraindikované (pozri časť 4.3).
U pacientov užívajúcich tieto lieky súčasne je nevyhnutná redukcia zvyčajnej dávky rifabutinu
300 mg/deň o 75 % (t.j. rifabutin
150 mg jedenkrát každý druhý deň) a zvýšené sledovanie nežiaducich udalostí súvisiacich s rifabutinom. V prípade ohrozenia bezpečnosti sa má zvážiť ďalšie predĺženie dávkovacieho intervalu rifabutinu a/alebo sledovanie hladiny rifabutinu.
Treba brať do úvahy oficiálnu smernicu pre liečbu tuberkulózy u pacientov infikovaných HIV.
Na základe bezpečnostného profilu darunaviru/ritonaviru, toto zvýšenie expozície darunaviru v prítomnosti rifabutinu nevyžaduje úpravu dávky darunaviru/ritonaviru.
Na základe farmakokinetického modelu možno dávku zredukovať o 75 % aj u pacientov, ktorí
dostávajú inú dávku rifabutinu ako
300 mg/deň.
Pri súčasnom užívaní týchto liekov a darunavirom s nízkou dávkou ritonaviru môžu byť ich koncentrácie zvýšené, čo vedie k možnosti nárastu nežiaducich udalostí zvyčajne súvisiacich
s týmito liekmi.
Pri súčasnom užívani darunaviru s nízkou dávkou ritonaviru
a jedného z antineoplastík je
potrebná opatrnosť.
Súčasné užívanie everolimu a darunaviru užívaného s nízkou dávkou ritonaviru sa neodporúča.
I
NHIBÍTORY AGREGÁCIE TROMBOCYTOV
Tikagrelor Neskúmalo sa. Súčasné užívanie
s darunavirom posilneným nízkou dávkou ritonaviru môže viesť k značnému zvýšeniu expozície tikagreloru.
ANTIPSYCHOTIKÁ/NEUROLEPTIKÁ
Kvetiapín V dôsledku inhibície CYP3A darunavirom sa očakáva zvýšenie koncentrácií antipsychotík/neuroleptík.
Súčasné užívanie darunavirus nízkou dávkou ritonaviru a s tikagrelorom je kontraindikované.
Užívanie iných antitrombotík neovplyvnených inhibíciou alebo indukciou CYP (napr. prasugrel) sa odporúča.
Súčasné podávanie darunavirus nízkou dávkou ritonaviru a kvetiapínu je kontraindikované, pretože môže zvýšiť toxicitu súvisiacu
s kvetiapínom. Zvýšené koncentrácie kvetiapínu môžu viesť ku kóme.
Risperidón
Tioridazín
Pimozid
Sertindol
β-BLOKÁTORY Karvedilol Metoprolol Timolol
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto antipsychotík.
(inhibícia CYP2D6 a/alebo P-gp)
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto beta blokátorov.
(inhibícia CYP2D6)
Môže byť potrebné zníženie dávky týchto liekov, keď sa podávajú súčasne s darunavirom užívaným
s nízkou dávkou ritonaviru.
Súčasné podávanie darunaviru s nízkou dávkou ritonaviru a pimozidu alebo sertindolu je kontraindikované.
Pri súčasnom užívaní darunaviru s beta blokátormi sa odporúča klinické sledovanie. Má sa zvážiť zníženie dávky beta blokátora.
B
L
OK
ÁTORY KALCIOVÉHO KANÁLA
Amlodipín Diltiazem Felodipín Nikardipín Nifedipín Verapamil
Nesledovalo sa. Môže sa očakávať, že darunavir užívaný s nízkou dávkou ritonaviru bude zvyšovať plazmatické koncentrácie blokátorov kalciového kanála.
(inhibícia CYP3A a/alebo CYP2D6)
Ak sa tieto lieky podávajú súčasne s darunavirom a s nízkou dávkou ritonaviru, odporúča sa klinické sledovanie liečebných a nežiaducich udalostí.
KO
RTIKOSTEROIDY
Flutikazón
Budezonid
Dexametazón
(podávaný systémovo)
V klinickej štúdii, pri ktorej sa zdravým jedincom počas 7 dní podávali kapsuly ritonaviru 100 mg dvakrát denne spolu
s 50 µg flutikazónpropionátu intranazálne (štyrikrát denne), sa významne zvýšili sérové hladiny flutikazónpropionátu, zatiaľ čo prirodzená hladina kortizolu klesla približne o 86 % (pri 90 % intervale spoľahlivosti 82 % - 89 %). Vyššie účinky možno očakávať, keď sa flutikazón inhaluje. Systémové účinky kortikosteroidov, vrátane Cushingovho syndrómu a adrenálnej supresie, boli hlásené u pacientov, ktorí užívali ritonavir
a súčasne inhalovali alebo intranazálne užili flutikazón; podobná situácia môže vzniknúť aj pri súčasnom užívaní iných kortikosteroidov, ktoré sa metabolizujú prostredníctvom cytochrómu P4503A, napr. budezonidu. Účinky vysokej systémovej expozície flutikazónu na plazmatické hladiny ritonaviru nie sú dosiaľ známe.
Neskúmalo sa. Dexametazón môže znížiť plazmatickú koncentráciu darunaviru. (indukcia CYP3A)
Preto sa súčasná liečba darunavirom v kombinácii s nízkou dávkou ritonaviru a týmito
glukokortikoidmi neodporúča, pokiaľ možný prínos nepreváži riziko systémových kortikosteroidných účinkov. Má sa zvážiť zníženie dávky glukokortikoidov spolu
s dôkladným monitorovaním lokálnych a systémových účinkov, alebo prevod na iný glukokortikoid, ktorý nepatrí k substrátom pre CYP3A (napr. beklometazón).
V prípade vysadenia glukokortikoidov môže byť navyše potrebné, aby progresívne znižovanie dávky trvalo dlhší čas.
Dexametazón podávaný systémovo sa má podávať so zvýšenou opatrnosťou, ak sa užíva spolu
s darunavirom a nízkou dávkou ritonaviru.
Prednizón Neskúmalo sa. Darunavir môže zvýšiť plazmatické koncentrácie prednizónu. (inhibícia CYP3A)
ANTIENDOTELÍNY
Bosentan Neskúmalo sa. Súčasné užívanie
bosentanu a darunaviru užívaného s nízkou
dávkou ritonaviru môže zvýšiť
plazmatické koncentrácie bosentanu.
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru
a prednizónu môže zvýšiť riziko vzniku systémových kortikosteroidných účinkov, vrátane Cushingovho syndrómu
a adrenálnej supresie. Pri súčasnom užívaní darunaviru s nízkou dávkou ritonaviru a kortikosteroidov sa odporúča klinické sledovanie.
Ak sa podáva súčasne
s darunavirom a nízkou dávkou ritonaviru, má sa u pacienta
sledovať tolerovateľnosť bosentanu.
ANTIVIROTIKÁ PRIAMO ÚČINKUJÚCE PROTI VÍRUSU HEPATITÍDY C (HCV)
Proteázové inhibítory NS3-4A
Telaprevir
750 mg každých
8 hodín
Boceprevir
800 mg trikrát denne
telaprevir AUC ↓ 35 % telaprevir Cmin ↓ 32 % telaprevir Cmax ↓ 36 % darunavir AUC12 ↓ 40 % darunavir Cmin ↓ 42 % darunavir Cmax ↓ 40 % boceprevir AUC ↓ 32 % boceprevir Cmin ↓ 35 % boceprevir Cmax ↓ 25 % darunavir AUC ↓ 44 % darunavir Cmin ↓ 59 % darunavir Cmax ↓ 36 %
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru a
s telaprevirom sa neodporúča.
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru a
s boceprevirom sa neodporúča.
Simeprevir simeprevir AUC ↑ 159 % simeprevir Cmin ↑ 358 % simeprevir Cmax ↑ 79 % darunavir AUC ↑ 18 % darunavir Cmin ↑ 31 % darunavir Cmax «
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru
a simepreviru sa neodporúča.
RASTLINNÉ LIEČIVÁ Ľubovník bodkovaný (Hypericum perforatum)
Dávka simepreviru v tejto interakčnej
štúdii bola 50 mg, keď sa podával spolu
s darunavirom/ritonavirom, v porovnaní so
150 mg v skupine liečenej samotným
simeprevirom.
Neskúmalo sa. Predpokladá sa, že ľubovník bodkovaný spôsobuje pokles plazmatických koncentrácií darunaviru a ritonaviru.
(indukcia CYP450)
Darunavir užívaná s nízkou dávkou ritonaviru sa nesmie podávať
v kombinácii s liekmi obsahujúcimi výťažok z ľubovníka bodkovaného (Hypericum perforatum) (pozri časť
4.3). Ak už pacient užíva ľubovník bodkovaný, užívanie ukončite, a ak je to možné, skontrolujte vírusové hladiny. Expozícia darunaviru (a tiež expozícia ritonaviru) sa môže po ukončení užívania ľubovníka bodkovaného zvýšiť. Indukujúci účinok môže pretrvávať najmenej 2 týždne po ukončení liečby ľubovníkom bodkovaným.
I
NHIBÍTORY HMG CO-A REDUKTÁZY

Lovastatín
Simvastatín
Atorvastatín
10 mg jedenkrát denne
Pravastatín
40 mg jednorazová dávka
Rosuvastatín
10 mg jedenkrát denne
Neskúmalo sa. Predpokladá sa, že lovastatín a simvastatín budú mať zreteľne zvýšené plazmatické koncentrácie, ak sa budú podávať súčasne s darunavirom užívaným s nízkou dávkou ritonaviru. (inhibícia CYP3A)
atorvastatín AUC ↑ 3-4 krát atorvastatín Cmin ↑ ≈5,5-10 krát atorvastatín Cmax ↑ ≈2 krát
#darunavir
pravastatín AUC ↑ 81 %¶ pravastatín Cmin ND pravastatín Cmax ↑ 63 %
¶ u niektorých subjektov sa pozoroval až 5-násobný nárast
rosuvastatín AUC ↑ 48%║
rosuvastatín Cmax ↑ 144%║
║ na základe publikovaných údajov
Zvýšené plazmatické koncentrácie lovastatínu alebo simvastatínu môžu vyvolať myopatiu, vrátane rabdomyolýzy. Súčasné podávanie darunaviru užívanej s nízkou
dávkou ritonaviru s lovastatínom a simvastatínom je preto kontraindikované (pozri časť 4.3). Ak je potrebné súčasné podávanie atorvastatínu s darunavirom užívanou s nízkou dávkou ritonaviru, odporúča sa začať podávať atorvastatín v dávke 10 mg jedenkrát denne. Postupné zvyšovanie dávkovania
atorvastatínu je možné prispôsobiť
na základe klinickej odpovede.
Ak je potrebné súčasné podávanie pravastatínu s darunavirom užívanou s nízkou dávkou ritonaviru, odporúča sa začať
s najnižšou možnou dávkou pravastatínu a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
Ak je potrebné súčasné podávanie rosuvastatínu s darunavirom užívanou s nízkou dávkou ritonaviru, odporúča sa začať
s najnižšou možnou dávkou rosuvastatínu a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
I
NHIBÍTORY H
2
-
RECEPTORA
Ranitidín
150 mg dvakrát denne
IMUNOSUPRESÍVA Ciklosporín Sirolimus Takrolimus
Everolimus
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Neskúmalo sa. Pri súčasnom podávaní darunaviru užívaného s nízkou dávkou ritonaviru s týmito imunosupresívami sa zvýši expozícia voči týmto látkam. (inhibícia CYP3A)
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s inhibítormi H2-receptora sa môže užívať bez úpravy dávky.
Pri súčasnom podaní imunosupresív sa musia monitorovať terapeutické hladiny týchto liekov.
Súčasné užívanie everolimu a darunaviru užívanej s nízkou dávkou ritonaviru sa neodporúča.
I
NHALAČNÉ BETA-AGONISTY
Salmeterol Neskúmalo sa. Súčasné užívanie
salmeterolu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť
plazmatické koncentrácie salmeterolu.
Súčasné užívanie salmeterolu
a darunaviru užívanej s nízkou dávkou ritonaviru sa neodporúča. Táto kombinácia môže viesť
k zvýšenému riziku kardiovaskulárnych nežiaducich udalostí salmeterolu, vrátane predĺženia QT intervalu, palpitácií a sínusovej tachykardie.
NARKOTICKÉ ANALGETIKÁ / LIEČBA ZÁVISLOSTI NA OPIOIDOCH
Metadon individuálna dávka
v rozsahu od 55 mg do
150 mg jedenkrát denne
Buprenorfín/naloxón
8/2 mg–16/4 mg jedenkrát denne
R(-) metadon AUC ↓ 16 % R(-) metadon Cmin ↓ 15 % R(-) metadon Cmax ↓ 24 %
buprenorfín AUC ↓ 11 % buprenorfín Cmin ↔ buprenorfín Cmax ↓ 8 % norbuprenorfín AUC ↑ 46 % norbuprenorfín Cmin ↑ 71 % norbuprenorfín Cmax ↑ 36 % naloxón AUC ↔
naloxón Cmin ND
naloxón Cmax ↔
Na začiatku užívania spolu
s darunavirom/ritonavirom nie je potrebná úprava dávky metadonu. Avšak zvýšené dávky metadonu môžu byť potrebné kvôli indukcii
metabolizmu ritonavirom pri
súčasnom podávaní počas dlhšieho časového obdobia. Z toho dôvodu
sa odporúča klinické monitorovanie, pretože u niektorých pacientov
môže byť potrebná úprava udržiavacej liečby.
Klinický význam zvýšenia farmakokinetických parametrov norbuprenorfínu nebol stanovený. Úprava dávky buprenorfínu nemusí byť potrebná, ak sa podáva spolu
s darunavirom/ritonavirom, ale odporúča sa pozorné sledovanie prejavov toxicity opiátov.
ANTIKONCEPCIA NA BÁZE ESTROGÉNOV

Etinylestradiol
Noretisterón
35 mg/1 mg jedenkrát denne
etinylestradiol AUC ↓ 44 % etinylestradiol Cmin ↓ 62 % etinylestradiol Cmax ↓ 32 % noretisterón AUC ↓ 14 % noretisterón Cmin ↓ 30 % noretisterón Cmax ↔
Pacientkam užívajúcim antikoncepciu na báze estrogénov v kombinácii s darunavirom
a s nízkou dávkou ritonaviru sa odporúča zmeniť resp. rozšíriť formy antikoncepcie. U pacientok užívajúcich estrogén ako hormonálnu substitúciu majú byť klinicky sledované prejavy deficitu estrogénu.
FOSFODIESTERÁZA, INHIBÍTORY TYPU 5 (PDE-5)
Pri liečbe erektilnej
dysfunkcie Avanafil Sildenafil Tadalafil Vardenafil
Pri liečbe pulmonálnej
arteriálnej hypertenzie
Sildenafil
Tadalafil
V štúdii skúmajúcej interakcie# sa pozorovala porovnateľná systémová expozícia voči sildenafilu po jednorazovom podaní samotného sildenafilu v dávke 100 mg a po jednorazovom podaní sildenafilu v dávke
25 mg v kombinácii s darunavirom a nízkou dávkou ritonaviru.
Neskúmalo sa. Súčasné užívanie sildenafilu alebo tadalafilu pri liečbe pulmonálnej arteriálnej hypertenzie a darunaviru užívaného s nízkou dávkou ritonaviru môže zvýšiť plazmatické koncentrácie sildenafilu alebo tadalafilu.
Kombinácia avanafilu a darunaviru s nízkou dávkou ritonaviru je kontraindikovaná (pozri časť 4.3). Pri súčasnom podávaní iných inhibítorov PDE-5 na liečbu erektilnej dysfunkcie
s darunavirom, užívaným s nízkou dávkou ritonaviru, je potrebná opatrnosť. Ak je indikované súčasné podávanie darunaviru
a nízkej dávky ritonaviru so sildenafilom, vardenafilom alebo tadalafilom, jednorazová dávka sildenafilu nesmie prekročiť 25 mg za obdobie 48 hodín, resp. jednorazová dávka vardenafilu nesmie prekročiť 2,5 mg za obdobie
72 hodín a jednorazová dávka tadalafilu nesmie presiahnuť 10 mg za obdobie 72 hodín.
Bezpečná a účinná dávka sildenafilu pri liečbe pulmonálnej arteriálnej hypertenzie a pri súčasnom užívaní darunaviru a nízkej dávky ritonaviru nebola stanovená.
Existuje zvýšená možnosť vzniku nežiaducich udalostí súvisiacich so sildenafilom (vrátane porúch zraku,
hypotenzie, predĺženej erekcie a synkopy). Preto je súčasné užívanie darunaviru s nízkou
dávkou ritonaviru a sildenafilu, ak
sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, kontraindikované (pozri časť 4.3). Pri liečbe pulmonálnej arteriálnej hypertenzie sa súčasné užívanie tadalafilu s darunavirom a nízkou dávkou ritonaviru neodporúča.
I
NHIBÍTORY PROTÓNOVEJ PUMPY
Omeprazol
20 mg jedenkrát denne
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Darunavir užívaný s nízkou dávkou ritonaviru je možné súčasne podávať s inhibítormi protónovej pumpy bez nutnosti úpravy dávkovania.
SEDATÍVA/HYPNOTIKÁ
Buspirón Klorazepát Diazepam Estazolam Flurazepam Triazolam Zolpidem
Midazolam
Neskúmalo sa. Sedatíva/hypnotiká sú výrazne metabolizované prostredníctvom CYP3A. Súčasné užívanie
s darunavirom/ritonavirom môže spôsobiť významné zvýšenie koncentrácií týchto liekov.
Na základe údajov o iných inhibítoroch CYP3A sa očakáva, že plazmatické koncentrácie midazolamu budú výrazne vyššie, keď sa midazolam podáva perorálne s darunavirom užívaným
s nízkou dávkou ritonaviru.
Ak sa parenterálny midazolam podáva súčasne s darunavirom užívaným s nízkou dávkou ritonaviru, môže spôsobiť významné zvýšenie koncentrácie tohto benzodiazepínu. Údaje o súčasnom užívaní parenterálneho midazolamu s inými proteázovými inhibítormi naznačujú
možné 3-4-násobné zvýšenie
plazmatických hladín midazolamu.
Pri súčasnom užívaní darunaviru
s týmito sedatívami/hypnotikami sa odporúča klinické sledovanie a má sa zvážiť zníženie dávky sedatív/hypnotík. Súčasné užívanie darunaviru užívaného s nízkou dávkou ritonaviru a triazolamu je
kontraindikované.
Užívanie darunaviru s nízkou dávkou ritonaviru v kombinácii s perorálne podávaným
midazolamom je kontraindikované (pozri časť 4.3); pričom sa odporúča opatrnosť pri súčasnom užívaní darunaviru s nízkou dávkou ritonaviru a parenterálneho midazolamu.
Ak sa parenterálny midazolam podáva súčasne s darunavirom užívaným s nízkou dávkou ritonaviru, má sa tak uskutočniť na jednotke intenzívnej starostlivosti (JIS) alebo v podobnom zariadení, ktoré zabezpečuje dôkladné klinické sledovanie a vhodnú lekársku starostlivosť v prípade respiračného útlmu a/alebo predĺženého útlmu. Treba zvážiť úpravu dávkovania midazolamu, najmä ak sa podáva viac ako jednorazová dávka.
 †
† Účinnosť a bezpečnosť používania darunaviru so 100 mg ritonaviru a s niektorým iným HIV PI (napr. (fos)amprenavir, nelfinavir a tipranavir) neboli u pacientov s HIV stanové. Podľa súčasných liečebných postupov sa vo všeobecnosti duálna liečba proteázovými inhibítormi neodporúča.
4.6 Fertilita, gravidita a laktáciaGraviditaVo všeobecnosti, keď sa rozhoduje o použití antiretrovirotík na liečbu infekcie HIV u gravidných žien a následne na zníženie rizika vertikálneho prenosu HIV na novorodenca, treba vziať do úvahy údaje
o zvieratách ako aj klinické skúsenosti s gravidnými ženami.
S darunavirom sa nevykonali žiadne primerané ani dobre kontrolované štúdie o vplyve na graviditu u gravidných žien. Štúdie na zvieratách nepreukázali priame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3).
Darunavir užívaný s nízkou dávkou ritonaviru sa môže podávať gravidným ženám len vtedy, ak možný prínos liečby prevýši možné riziká.
DojčenieNie je známe, či sa darunavir vylučuje do ľudského mlieka. V štúdiách na potkanoch sa preukázalo vylučovanie darunaviru do mlieka a pri vysokých hladinách (1000 mg/kg/deň) spôsobilo toxicitu. Matky majú byť poučené, aby za žiadnych okolností nedojčili počas liečby darunavirom jednak kvôli možnosti prenosu HIV a aj možnosti výskytu nežiaducich reakcií u dojčených detí.
FertilitaNie sú k dispozícii žiadne údaje o účinku darunaviru na fertilitu u ľudí. Pri liečbe potkanov
darunavirom nemal darunavir vplyv na párenie a fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Darunavir spolu s ritonavirom nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U niektorých pacientov sa však počas liečby darunavirom užívaným s nízkou dávkou ritonaviru vyskytli závraty. Na túto možnosť je potrebné myslieť pri posudzovaní schopnosti pacienta viesť vozidlá a obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Počas programu klinického vývoja (N=2 613 predtým liečených jedincov, ktorí začali liečbu
s darunavirom/ritonavirom 600/100 mg dvakrát denne), 51,3 % jedincov malo aspoň jednu nežiaducu reakciu. Celkové priemerné trvanie liečby pacientov bolo 95,3 týždňov. Najčastejšími nežiaducimi
reakciami hlásenými v klinických skúšaniach a v spontánnych hláseniach sú hnačka,
nauzea, vyrážka, bolesť hlavy a vracanie. Najčastejšími závažnými reakciami sú akútne renálne zlyhanie, infarkt myokardu, imunoreštitučný zápalový syndróm, trombocytopénia, osteonekróza, hnačka, hepatitída a pyrexia.
V analýze po 96 týždňoch bol bezpečnostný profil darunaviru/ritonaviru 800/100 mg jedenkrát denne u jedincov, ktorí doteraz neboli liečení, podobný ako u darunaviru/ritonaviru 600/100 mg dvakrát denne u jedincov, ktorí boli predtým liečení, až na nauzeu, ktorá sa častejšie vyskytovala u doteraz neliečených jedincov. Nauzea bola miernej intenzity. V analýze po 192 týždňoch sa u doteraz neliečených jedincov, u ktorých liečba darunavirom/ritonavirom 800/100 mg jdenkrát denne trvala priemerne 162,5 týždňa, neidentifikovali žiadne nové nálezy týkajúce sa bezpečnosti.
Zoznam nežiaducichreakciívtabuľkách
Nežiaduce reakcie sa uvádzajú podľa postihnutia triedy orgánových systémov a podľa skupiny frekvencie. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti. Skupiny frekvencií sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000)
a neznáme (z dostupných údajov).
Nežiaduce reakcie v klinických skúšaniach a v postmarketingovom sledovaní
Trieda orgánových systémov podľa databázy
MedDRA
Kategória frekvencie
Infekcie a nákazy
Nežiaduca reakcia
menej časté herpes simplex
Poruchy krvi a lymfatického systémumenej časté trombocytopénia, neutropénia, anémia, leukopénia
zriedkavé zvýšenie počtu eozinofilov
Poruchy imunitného systémumenej časté imunoreštitučný zápalový syndróm, precitlivenosť
(na liek)
Poruchy endokrinného systémumenej časté hypotyreóza, zvýšenie hladiny hormónu stimulujúceho štítnu žľazu v krvi
Poruchy metabolizmu a výživyčasté diabetes mellitus, hypertriglyceridémia, hypercholesterolémia, hyperlipidémia
menej časté dna, anorexia, znížená chuť do jedla, zníženie hmotnosti, zvýšenie hmotnosti, hyperglykémia, rezistencia na inzulín, pokles lipoproteínu
s vysokou hustotou, zvýšená chuť do jedla, polydipsia, zvýšená hladina LDH v krvi
Psychické poruchyčasté insomnia
menej časté depresia, dezorientácia, úzkosť, poruchy spánku,
neprirodzené sny, nočné mory, pokles libida
zriedkavé stav zmätenosti, zmeny nálady, nepokoj
Poruchy nervového systémučasté bolesti hlavy, periférna neuropatia, závraty
menej časté letargia, parestézia, hypoestézia, dysgeúzia,
porucha pozornosti, poškodenie pamäti,
somnolencia
zriedkavé synkopa, konvulzia, ageuzia, porucha fázy spánkového rytmu
Poruchy okamenej časté konjunktiválna hyperémia, suché oči
zriedkavé porucha zraku
Poruchy ucha a labyrintumenej časté vertigo
Poruchy srdca a srdcovej činnostimenej časté infarkt myokardu, angína pektoris, predĺžený QT
elektrokardiogram, tachykardia
zriedkavé akútny infarkt myokardu, sínusová bradykardia,
palpitácie
Poruchy cievmenej časté hypertenzia, návaly horúčavy
Poruchy dýchacej sústavy, hrudníka a mediastínamenej časté dyspnoe, kašeľ, epistaxa, podráždenie hrdla
zriedkavé rinorea
Poruchy gastrointestinálneho traktuveľmi časté diarea
časté vracanie, nauzea, bolesti brucha, zvýšená hladina
amylázy v krvi, dyspepsia, abdominálna distenzia, flatulencia
menej časté pankreatitída, gastritída, gastroezofagálny reflux, aftózna stomatitída, napínanie na vracanie, sucho
v ústach, abdominálny diskomfort, zápcha, zvýšená
hladina lipázy, eruktácia, perorálna dyzestézia
zriedkavé stomatitída, hemateméza, cheilitída, suché pery, povlak na jazyku
Poruchy pečene a žlčových ciestčasté zvýšenie alanínaminotransferázy

menej časté hepatitída, cytolytická hepatitída, steatóza pečene, hepatomegália, zvýšenie transamináz, zvýšenie aspartátaminotransferázy, zvýšenie hladiny bilirubínu v krvi, zvýšenie alkalickej fosfatázy
v krvi, zvýšenie gamaglutamyltransferázy
Poruchy kože a podkožného tkanivačasté vyrážka (vrátane makulárnej, makulopapulárnej, papulárnej, erytematóznej a pruritickej vyrážky), pruritus
menej časté angioedém, generalizovaná vyrážka, alergická
dermatitída, urtikária, ekzém, erytém,
hyperhidróza, nočné potenie, alopécia, akné, suchá koža, sfarbenie nechtov
zriedkavé DRESS, Stevensov-Johnsonov syndróm, multiformný erytém, dermatitída, seboroická dermatitída, kožné lézie, xeroderma
neznáme toxická epidermálna nekrolýza, akútna generalizovaná exantematózna pustulóza
Poruchy kostrovej a svalovej sústavy a spojivového tkanivamenej časté myalgia, osteonekróza, svalové kŕče, svalová slabosť, artralgia, bolesti končatín, osteoporóza, zvýšená hladina kreatínfosfokinázy v krvi
zriedkavé muskuloskeletálna stuhnutosť, artritída, stuhnutie
kĺbov
Poruchy obličiek a močových ciestmenej časté akútne zlyhanie obličiek, zlyhanie obličiek, nefrolitiáza, zvýšená hladina kreatinínu v krvi, proteinúria, bilirubinúria, dyzúria, noktúria, polakizúria
zriedkavé znížený renálny klírens kreatinínu
Poruchy reprodukčného systému a prsníkovmenej časté erektilná dysfunkcia, gynekomastia
Celkové poruchy a reakcie v mieste podaniačasté asténia, únava
menej časté pyrexia, bolesť na hrudi, periférne opuchy,
nevoľnosť, pocit horúčavy, podráždenie, bolesť
zriedkavé triaška, abnormálne pocity, xeróza
OpisvybranýchnežiaducichreakciíVyrážkaV klinických skúšaniach bola vyrážka najčastejšie miene až stredne závažná, často sa vyskytla v prvých štyroch týždňoch liečby a ustúpila pri ďalšom užívaní. Pre prípady závažných kožných reakcií pozri upozornenie v časti 4.4.
Počas programu klinického vývoja raltegraviru sa u predtým liečených pacientov častejšie pozorovala vyrážka, bez ohľadu na kauzalitu, pri režimoch obsahujúcich darunavir + raltegravir v porovnaní
s režimami obsahujúcimi darunavir bez raltegraviru alebo raltegravir bez darunaviru. Vyrážka
považovaná skúšajúcim za súvisiacu s liekom, sa vyskytla v podobnej miere. Expozícii prispôsobené
miery vyrážky (všetky príčiny) boli 10,9; 4,2 a 3,8 na 100 pacientorokov v danom poradí a u vyrážky
súvisiacej s liekom boli 2,4; 1,1 a 2,3 na 100 pacientorokov v danom poradí. Vyrážky pozorované
v klinických štúdiách boli mierne až stredne závažné a neviedli k prerušeniu liečby (pozri časť 4.4).
Metabolické parametrePočas antiretrovírusovej liečby sa môže zvýšiť telesná hmotnosť a hladiny lipidov a glukózy v krvi
(pozri časť 4.4).
Poruchy kostrovej a svalovej sústavyPočas liečby inhibítormi proteáz, najmä v kombinácii s NRTI, sa vyskytli prípady zvýšenia hladiny
kreatínfosfokinázy, myalgie, myozitídy a zriedkavo aj rabdomyolýzy.
Boli hlásené prípady osteonekrózy, najmä u pacientov so všeobecne potvrdenými rizikovými faktormi, pokročilým ochorením HIV alebo dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART). Frekvencia nie je známa (pozri časť 4.4).
Imunoreštitučný zápalový syndrómU pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej
antiretrovírusovej liečby (CART) môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie. Boli tiež zaznamenané aj autoimunitné poruchy (ako je Gravesova
choroba); avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť
mnoho mesiacov po začatí liečby (pozri časť 4.4).
Krvácanie u pacientov s hemofíliouU pacientov s hemofíliou dostávajúcich antiretrovirálne inhibítory proteázy boli hlásené prípady
zvýšeného spontánneho krvácania (pozri časť 4.4).
Pediatrická populáciaPosúdenie bezpečnosti u pediatrických pacientov je založené na 48-týždňovej analýze údajov
o bezpečnosti z troch klinických skúšaní fázy II. Boli hodnotené nasledovné populácie pacientov
(pozri časť 5.1):
● 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg, ktorí užívali darunavir tablety s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku od 3 do < 6
rokov a s hmotnosťou 10 kg až < 20 kg (16 účastníkov od 15 kg do < 20 kg), ktorí užívali
darunavir perorálnu suspenziu s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 12 pediatrických pacientov infikovaných HIV-1 vo veku od 12 do 17 rokov a s hmotnosťou
najmenej 40 kg, ktorí neboli predtým liečení ART. Títo pacienti dostávali darunavir tablety
s nízkou dávkou ritonaviru jedenkrát denne spolu s inými antiretrovírusovými liekmi. (pozri
časť 5.1).
Celkovo bol bezpečnostný profil u týchto pediatrických pacientov podobný bezpečnostnému profilu
pozorovanému u dospelých.
Iné osobitné skupiny pacientovPacienti súčasne infikovaní vírusom hepatitídy B a/alebo hepatitídy CSpomedzi 1968 predtým liečených pacientov, ktorí užívali darunavir spolu s ritonavirom 600/100 mg dvakrát denne, bolo 236 pacientov infikovaných zároveň hepatitídou B alebo C. U súčasne infikovaných pacientov bola vyššia pravdepodobnosť počiatočného a pri liečbe vznikajúceho zvýšenia hladiny pečeňových transamináz ako u pacientov bez chronickej vírusovej hepatitídy (pozri časť 4.4).
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Skúsenosti s akútnym predávkovaním darunavirom užívaným s nízkou dávkou ritonaviru u ľudí sú obmedzené. Zdravým dobrovoľníkom sa darunavir podával vo forme perorálneho roztoku
v jednorazových dávkach do 3200 mg ako samotný, resp. vo forme tabliet v jednorazových dávkach
do 1600 mg v kombinácii s ritonavirom bez toho, že by sa u nich pozorovali nepriaznivé symptomatické účinky.
V prípade predávkovania darunavirom nie je k dispozícii žiadne špecifické antidotum. Liečba predávkovania darunavirom zahŕňa všeobecné podporné opatrenia, vrátane sledovania vitálnych prejavov a pozorovania klinického stavu pacienta. Ak je to indikované, eliminácia nevstrebanej aktívnej látky sa dosiahne pomocou vyvolania vracania.
Aj podanie aktívneho uhlia môže pomôcť pri eliminácii nevstrebanej aktívnej látky. Vzhľadom na vysokú väzbu darunaviru na plazmatické bielkoviny je nepravdepodobné, že by dialýza mala väčší význam pri eliminácii aktívnej látky z cirkulácie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antivirotiká na systémové použitie, inhibítory proteáz, ATC kód: J05AE10
Mechanizmus účinku
Darunavir je inhibítor dimerizácie a katalytickej aktivity HIV-1 proteázy (KD 4,5 x 10-12M). Selektívne
inhibuje odštiepenie HIV kódovaných polyproteínov Gag-Pol v bunkách infikovaných vírusom, čím zabraňuje tvorbe zrelých infekčných vírusových častíc.
Antivírusová aktivita in vitro
Darunavir vykazuje aktivitu proti laboratórnym kmeňom a klinickým izolátom HIV-1 a laboratórnym
kmeňom HIV-2 v akútne infikovaných líniách T-buniek, mononukleárnych bunkách z periférnej krvi
človeka a ľudských monocytov/makrofágov so strednými hodnotami EC50 v rozmedzí
1,2 až 8,5 nmol/l (0,7 až 5,0 ng/ml). In vitro má darunavir antivírusovú aktivitu voči širokému spektru
primárnych izolátov zo skupiny M HIV-1 (A, B, C, D, E, F, G) a skupiny O s hodnotami EC50
v rozmedzí od < 0,1 do 4,3 nmol/l.
Tieto hodnoty EC50 sú výrazne nižšie ako je rozsah toxickej koncentrácie pre 50 % buniek od
87 µmol/l do > 100 µmol/l.
Rezistencia
Selekcia vírusu rezistentného na darunavir z divokého typu HIV-1 in vitro trvala pomerne dlho
(> 3 roky). Vyselektované vírusy neboli schopné rásť v prítomnosti darunaviru v koncentráciách nad
400 nmol/l. U vírusov vyselektovaných v týchto podmienkach a vykazujúcich zníženú citlivosť voči
darunaviru (rozmedzie 23 až 50-násobne) sa v géne pre proteázu zistili substitúcie 2 až 4 aminokyselín. Zníženú citlivosť vznikajúcich vírusov na darunavir v selekcii nie je možné vysvetliť vznikom týchto mutácií proteázy.
Údaje z klinických štúdií s pacientmi liečenými ART (štúdia TITAN a zlúčená analýza štúdií POWER
1, 2 a 3 a štúdií DUET 1 a 2) preukázali, že virologická odpoveď na darunavir užívaný s nízkou dávkou ritonaviru bola znížená, keď boli na začiatku prítomné 3 a viac mutácií spojených s rezistenciou voči darunaviru (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V a L89V) alebo keď sa tieto mutácie vyvinuli počas liečby.
Zvýšenie východiskovej násobnej zmeny darunaviru v EC50 (FC) súviselo s poklesom virologickej odpovede. Určila sa dolná a horná klinická hranica 10 a 40. Izoláty s východiskovou hodnotou FC
≤ 10 sú citlivé; izoláty s FC > 10 až 40 majú zníženú citlivosť; izoláty s FC > 40 sú rezistentné (pozri
Klinické výsledky).
Vírusy izolované u pacientov s dávkou darunavir/ritonavir 600/100 mg dvakrát denne, u ktorých sa vyskytlo spontánne virologické zlyhanie, ktoré boli citlivé na tipranavir na začiatku, zostali vo veľkej väčšine prípadov citlivé na tipranavir po liečbe.
Najnižšie miery rozvoja rezistencie vírusu HIV sú pozorované u pacientov doposiaľ neliečených ART, ktorí sú po prvýkrát liečení darunavirom v kombinácii s inou ART.
Tabuľka nižšie zobrazuje vývoj mutácií a stratu citlivosti na PI pri virologickom zlyhaní v závere
liečby v štúdiách ARTEMIS, ODIN a TITAN.
ARTEMIS ODIN TITAN
Darunavir/
ritonavir
800/100 mg jedenkrát denne N=343
Darunavir/
ritonavir
800/100 mg jedenkrát denne N=294
Darunavir/
ritonavir
600/100 mg dvakrát denne N=296
Darunavir/
ritonavir
600/100 mg dvakrát denne N=298
Celkový počet virologických zlyhanía, n (%)
55 (16,0 %) 65 (22,1 %) 54 (18,2 %) 31 (10,4 %)
Spontánne 39 (11,4 %) 11 (3,7 %) 11 (3,7 %) 16 (5,4 %)
Jedinci nereagujúci na
liečbu
16 (4,7 %) 54 (18,4 %) 43 (14,5 %) 15 (5,0 %)
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými genotypmi, u ktorých sa vyvinuli mutácieb v závere liečby, n/N
Primárne (významné)
mutácie PI
0/43 1/60 0/42 6/28
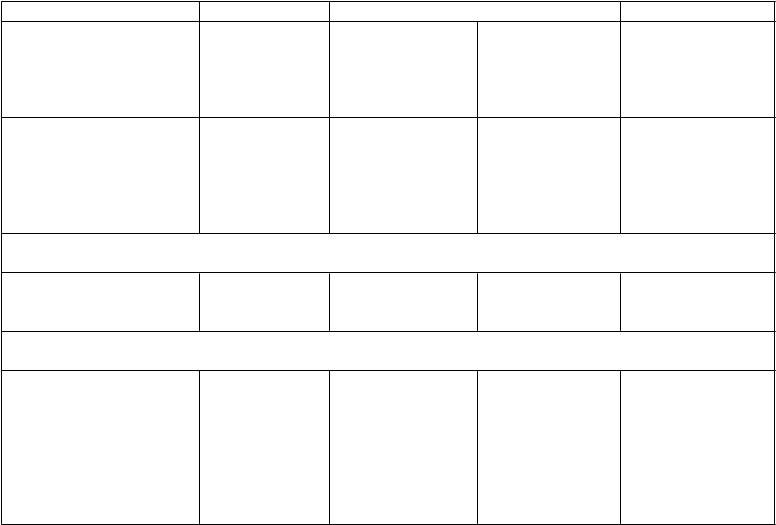
PI RAM 4/43 7/60 4/42 10/28
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými fenotypmi, u ktorých sa preukázala strata citlivosti na PI v závere liečby v porovnaní so začiatkom liečby, n/N
PI
darunavir 0/39 1/58 0/41 3/26 amprenavir 0/39 1/58 0/40 0/22 atazanavir 0/39 2/56 0/40 0/22 indinavir 0/39 2/57 0/40 1/24 lopinavir 0/39 1/58 0/40 0/23 sakvinavir 0/39 0/56 0/40 0/22 tipranavir 0/39 0/58 0/41 1/25
a TLOVR ne-VF cenzurovaný algoritmus založený na HIV-1 RNA < 50 kópií/ml, okrem
TITAN (HIV-1 RNA
< 400 kópií/ml)
b zoznamy IAS-USA
SkríženárezistenciaNásobná zmena (FC) darunaviru bola menej ako 10 pre 90 % z 3309 klinických izolátov rezistentných
voči amprenaviru, atazanaviru, indinaviru, lopinaviru, nelfinaviru, ritonaviru, sakvinaviru a/alebo
tipraniviru, čo naznačuje, že vírusy rezistentné voči väčšine PI zostávajú citlivé voči darunaviru.
V štúdii
ARTEMIS sa v prípade virologického zlyhania nepozorovala žiadna skrížená rezistencia
s inými PI.
Klinické výsledkyDospelí pacienti
Výsledky klinických štúdií u dospelých pacientov doposiaľ neliečených ART, pozri súhrn charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Úči nnosť darunaviru 600 mg dvakrát denne užívanej so 100 mg ritonaviru dvakrát denne u pacientov
pre dtým li ečený ch ART
Údaje o účinnosti kombinácie darunavir a ritonavir (600/100 mg dvakrát denne) u pacientov predtým
liečených ART sú založené na analýze 96 týždňovej štúdie fázy III TITAN u pacientov predtým
liečených ART, ktorí ešte neužívali lopinavir, na analýze 48 týždňovej štúdie fázy III ODIN
u pacientov predtým liečených ART bez DRV-RAM a na analýze údajov z 96 týždňov trvajúcich štúdií fázy IIb POWER 1 a 2 u pacientov predtým liečených ART s vysokou rezistenciou na PI.
TITAN je randomizovaná, kontrolovaná, otvorená štúdia fázy III porovnávajúca darunavir podávaný spolu s ritonavirom (600/100 mg dvakrát denne) verzus lopinavir/ritonavir (400/100 mg dvakrát denne) u dospelých pacientov predtým liečených ART infikovaných HIV-1, ktorí ešte neužívali
lopinavir. Obe ramená používali optimalizovaný základný režim (OBR, z angl. Optimised Background
Regimen), ktorý pozostával z minimálne 2 antiretrovirotík (NRTI s alebo bez NNRTI).
Nižšie uvedená tabuľka uvádza údaje 48-týždňovej analýzy účinnosti zo štúdie TITAN.

TITAN
Výsledky Darunavir/ritonavir
600/100 mg dvakrát denne + OBR N=298
lopinavir/ritonavir
400/100 mg dvakrát denne + OBR N=297
Rozdiel v liečbe
(95 % CI rozdielu)
HIV-1 RNA < 50 kópií/mla 70,8 % (211) 60,3 % (179) 10,5 % (2,9; 18,1)b
medián zmeny v počte buniek CD4+ oproti východiskovej hodnote (x 106/l)c
88 81
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede
c NC=F
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom,
definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA < 400 a < 50 kópií/ml, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat
(ITT) aj u pacientov sledovaných v protokole (On Protocol). Tieto výsledky sa potvrdili v analýze
údajov z 96 týždňov trvajúcej liečby v štúdii TITAN, pričom v 96. týždni malo 60,4 % pacientov v ramene s darunavirom/ritonavirom HIV-1 RNA < 50 kópií/ml v porovnaní s 55,2 % pacientov v ramene s lopinavirom/ritonavirom [rozdiel: 5,2 %, 95 % CI (-2,8; 13,1)].
ODIN je randomizovaná otvorená štúdia fázy III porovnávajúca darunavir/ritonavir 800/100 mg jedenkrát denne a darunavir/ritonavir 600/100 mg dvakrát denne u pacientov infikovaných HIV-1 predtým liečených ART, u ktorých skríningové vyšetrenie rezistencie genotypu nepreukázalo RAM voči darunaviru (t.j. V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V, L89V)
a skríningové vyšetrenie HIV-1 RNA bolo > 1 000 kópií/ml. Analýza účinnosti je založená na liečbe trvajúcej 48 týždňov (pozri tabuľku nižšie). Obe ramená použili optimalizovaný základný režim (OBR) ≥ 2 NRTI.
Výsledky Darunavir/ritonavir
800/100 mg jedenkrát denne + OBR
N=294
ODIN
Darunavir/ritonavir
600/100 mg dvakrát denne + OBR N=296
Rozdiel v liečbe
(95% CI rozdielu)
HIV-1 RNA
< 50 kópií/mla
S východiskovou HIV-1 RNA (kópie/ml)
72,1 % (212) 70,9 % (210) 1,2 % (-6,1; 8,5)b
< 100 000
≥ 100 000
77,6 % (198/255)
35,9 % (14/39)
73,2 % (194/265)

51 6 % (16/31)
4,4 % (-3,0; 11,9)
-15,7 % (-39,2; 7,7)
S východiskovým počtom buniek CD4+ (x 106/l)
≥ 100
< 100
S kmeňom HIV-1
Typ B Typ AE Typ C Inéc
75,1 % (184/245)
57,1 % (28/49)
70,4 % (126/179)
90,5 % (38/42)
72,7 % (32/44)
55,2 % (16/29)
72,5 % (187/258)
60,5 % (23/38)
64,3 % (128/199)

91,2 % (31/34)
78,8 % (26/33)
83,3 % (25/30)
2,6 % (-5,1; 10,3)
-3,4 % (-24,5; 17,8)
6,1 % (-3,4; 15,6)
-0,7 % (-14,0; 12,6)
-6,1 % (-2,6; 13,7)
-28,2 % (-51,0; -5,3)
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x 106/l)e
108 112 -5d (-25; 16)
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede
c Kmene A1, D, F1, G, K, CRF02_AG, CRF12_BF, a CRF06_CPX
d Rozdiel stredných hodnôt
e Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward)
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom
800/100 mg jedenkrát denne, definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA
< 50 kópií/ml v porovnaní s darunavirom/ritonavirom 600/100 mg dvakrát denne, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat (ITT) aj u pacientov sledovaných v protokole (On Protocol).
Darunavir/ritonavir 800/100 mg jedenkrát denne sa nemá používať u tých pacientov liečených ART,
ktorí majú jednu alebo viac mutácií spojených s rezistenciou voči darunaviru (DRV-RAM) alebo
HIV-1 RNA ≥ 100 000 kópií/ml alebo počet buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2 a 4.4).
U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje.
POWER 1 a POWER 2 sú randomizované, kontrolované štúdie porovnávajúce darunavir užívaný spolu s ritonavirom (600/100 mg dvakrát denne) s kontrolnou skupinou užívajúcou inhibítor proteáz vybraný skúšajúcim u pacientov infikovaných HIV-1, ktorí predtým absolvovali viac ako jeden neúspešný liečebný režim zahŕňajúci inhibítor proteáz (PI).
V oboch štúdiách bol použitý OBR zahŕňajúci aspoň 2 NRTI s enfuvirtidom (ENF) alebo bez neho.
V nasledujúcej tabuľke sú uvedené údaje 48-týždňovej a 96-týždňovej analýzy účinnosti zo zlúčených štúdií POWER 1 a POWER 2.
Spoločné údaje získané v štúdiách POWER 1 a POWER 2
48. týždeň 96. týždeň
Výsledky Darunavir/ritonavir
600/100 mg
2x denne n=131
Kontrola n=124
Rozdiel medzi liečbami
Darunavir/ritonavir
600/100 mg
2x denne n=131
Kontrola n=124
Rozdiel medzi liečbami
HIV RNA
< 50 kópií/
mla
45,0 % (59) 11,3 % (14)
33,7 % (23,4 %;

44,1 %)c
38,9 % (51) 8,9 % (11)
30,1 % (20,1;
40,0)c
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x
106/l)b
103 17 86
(57; 114)c
133 15 118 (83,9;
153,4)c
a Výpočet na základe algoritmu TLOVR.
b Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward).
c 95 % interval spoľahlivosti.
Analýza údajov z 96 týždňov trvajúcej liečby v štúdiách POWER preukázala zachovanú
antiretrovírusovú účinnosť a imunologický benefit.
Z 59 pacientov, ktorí reagovali na liečbu úplnou supresiou vírusu (< 50 kópií/ml) v 48. týždni,
47 pacientov (80 % respondérov na liečbu v 48. týždni) naďalej reagovalo na liečbu v 96. týždni.
Východiskový genotyp alebo fenotyp a virologický výsledok
Východiskový genotyp a FC darunaviru (zmena citlivosti vzhľadom k východiskovej hodnote) sa ukázal ako prediktívny faktor virologického výsledku.
Podiel (%) pacientov s odpoveďou (HIV-1 RNA < 50 kópii/ml v týždni 24) na darunavir užívaný spolu s ritonavirom (600/100 mg dvakrát denne) u východiskového genotypu*a a východiskovej FC darunaviru pri užívaní enfuvirtidu (ENF): podľa analýz liečby štúdií POWER a DUET.
Odpoveď (HIV-1
RNA < 50 kópií/ml
Počet východiskových mutáciía Východisková DRV FCb
Všetky
v 24. týždni)
%, n/N
Všetky
rozpätia
0-2 3 ³ 4
rozpätia £ 10 10-40 > 40
Všetci pacienti 45 %
455/1 014
54 %
359/660
39 %
67/172
12 %
20/171
45 %
455/1 014
55 %
364/659
29 %
59/203
8 %
9/118
Pacienti neužívajúci/opako- vane užívajúci ENFc Pacienti s de novo ENFd
39 %
290/741
60 %
165/273
50 %
238/477
66 %
121/183
29 %
35/120
62 %
32/52
7 %
10/135
28 %
10/36
39 %
290/741
60 %
165/273
51 %
244/477
66 %
120/182
17 %
25/147
61 %
34/56
5 %
5/94
17 %
4/24
a Počet mutácií zo zoznamu mutácií spojených so znížením odpovede na kombináciu darunavir/ritonavir (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V alebo L89V)
b násobná zmena v EC50
c “Pacienti neužívajúci/opakovane užívajúci ENF”, t.j. pacienti, ktorí neužívali ENF alebo tí, ktorí ENF užívali, ale nie
prvýkrát.
d “Pacienti s
de novo ENF”, t.j. pacienti, ktorí užívali ENF prvýkrát.
Pediatrickí pacientiVýsledky klinických štúdií u pediatrických pacientov vo veku 12 až 17 rokov, ktorí neboli predtým liečení ART, nájdete v Súhrne charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Pediatrickí pacienti od 6 do < 18 rokov a s hmotnosťou naj menej 20 kg pre dt ým li eče ní ART DELPHI je otvorená štúdia fázy II hodnotiaca farmakokinetiku, bezpečnosť, toleranciu a účinnosť darunaviru s nízkou dávkou ritonaviru u 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg. Títo pacienti užívali darunavir/ritonavir dvakrát denne spolu s inými antiretrovírusovými látkami (pre odporúčané dávkovanie v závislosti od hmotnosti pacienta pozri časť 4.2). Virologická odpoveď bola definovaná ako pokles vírusového zaťaženia HIV-1 RNA v plazme o najmenej 1,0 log10 oproti východiskovej hodnote.
V tejto štúdii bolo pacientom so zvýšeným rizikom prerušenia liečby z dôvodu intolerancie perorálneho roztoku ritonaviru (napr. averzia voči chuti) umožnené prejsť na užívanie kapsúl. Zo
44 pacientov užívajúcich perorálny roztok ritonaviru, 27 prešlo na užívanie 100 mg kapsúl
a prekročilo dávku ritonaviru odvodenú od hmotnosti bez zmien v sledovanej bezpečnosti.

DELPHI
Výsledky po 48. týždni Darunavir/ritonavir
N=80
HIV-1 RNA < 50 kópií/mla 47,5 % (38)
Priemerná zmena počtu buniek CD4+ oproti
východiskovej hodnoteb
147

a Výpočet na základe algoritmu TLOVR.
b Pacienti, ktorí neukončili liečbu, sú počítaní ako zlyhanie: pacientom, ktorí prerušili liečbu predčasne, sa priradí
zmena rovná nule.
Podľa cenzurovaného TLOVR algoritmu pre nevirologické zlyhanie u 24 (30,0 %) pacientov došlo
k virologickému zlyhaniu, z nich u 17 (21,3 %) pacientov sa objavil rebound fenomén a 7 (8,8 %)
pacientov neodpovedalo na liečbu.
Pedi at ri ckí paci ent i vo veku 3 až < 6 rokov predtým lieč ení ART U 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART vo veku od 3 do < 6
rokov a s hmotnosťou 10 kg až < 20 kg bola farmakokinetika, bezpečnosť, tolerovateľnosť a účinnosť
darunaviru/ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami hodnotená v otvorenej štúdii fázy II
ARIEL. Pacienti dostávali dávku dvakrát denne v závislosti od ich
hmotnosti, pacienti s hmotnosťou 10 kg až < 15 kg dostávali darunavir/ritonavir 25/3 mg/kg dvakrát denne a pacienti s hmotnosťou 15 kg až < 20 kg dostávali darunavir/ritonavir 375/50 mg dvakrát
denne. Virologická odpoveď, definovaná ako percento pacientov s potvrdenou plazmatickou hladinou
< 50 HIV-1 RNA kópií/ml, bola v 48. týždni hodnotená u 16 pediatrických pacientov s hmotnosťou
15 kg až < 20 kg a u 5 pediatrických pacientov s hmotnosťou 10 kg až < 15 kg dostávajúcich darunavir/ritonavir v kombinácii s inými antiretrovírusovými látkami. (pozri časť 4.2 pre odporúčanie
dávkovania podľa telesnej hmotnosti).

ARIEL
Výsledky po 48. týždni Darunavir/ritonavir
10 kg až < 15 kg
N=5
15 kg až < 20 kg
N=16
HIV-1 RNA < 50 kópií/mla 80,0 % (4) 81,3 % (13)
CD4+ percentuálna zmena oproti východiskovej hodnoteb Priemerná zmena počtu buniek CD4+ oproti východiskovej hodnoteb
a Imputácie na základe algoritmu TLOVR.
b NC=F
4 4
16 241
Dostupné sú obmedzené údaje o účinnosti u pediatrických pacientov s hmotnosťou menej ako 15 kg,
a preto neumožňujú uviesť odporúčania pre dávkovanie.
Gravidita a obdobie po pôrode
Darunavir/ritonavir (600/100 mg dvakrát denne alebo 800/100 mg jedenkrát denne) v kombinácii so
základným režimom bol hodnotený v klinickej štúdii s 34 gravidnými ženami (17 v každej skupine) počas druhého a tretieho trimestra a po pôrode. Virologická odpoveď sa udržala počas trvania štúdie v oboch skupinách. U detí narodených 29 pacientkám, ktoré zotrvali na antivirotickej liečbe až do
pôrodu, sa nevyskytol prenos z matky na dieťa. Nevyskytli sa žiadne nové klinicky významné zistenia týkajúce sa bezpečnosti v porovnaní so známym profilom bezpečnosti darunaviru/ritonaviru
u dospelých infikovaných HIV-1 (pozri časti 4.2, 4.4 a 5.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti darunaviru podávaného v kombinácii s ritonavirom sa skúmali
u zdravých dospelých dobrovoľníkov a u pacientov infikovaných vírusom HIV-1. Expozícia voči darunaviru u pacientov infikovaných vírusom HIV-1 bola vyššia ako u zdravých dobrovoľníkov. Vyššiu expozíciu voči darunaviru u pacientov infikovaných vírusom HIV-1 v porovnaní so zdravými dobrovoľníkmi je možné vysvetliť na základe vyšších koncentrácií α1-kyslého glykoproteínu (AAG) u pacientov infikovaných vírusom HIV-1, čo vedie k vyššej väzbe darunaviru na plazmatický AAG,
a tým aj k vyšším koncentráciám tejto látky v plazme.
Darunavir sa metabolizuje najmä prostredníctvom CYP3A. Ritonavir inhibuje CYP3A, v dôsledku
čoho sa výrazne zvyšujú plazmatické koncentrácie darunaviru.
Absorpcia
Darunavir sa rýchlo vstrebáva po perorálnom podaní. Pri podávaní v kombinácii s ritonavirom
v nízkej dávke sa maximálne plazmatické koncentrácie darunaviru obyčajne dosiahnu v priebehu 2,5 -
4 hodín.
Absolútna biologická dostupnosť po jednorazovom perorálnom podaní samotného darunaviru v dávke
600 mg bola približne 37 %, pričom pri podávaní ritonaviru v dávke 100 mg dvakrát denne sa zvýšila približne na 82 %. Po jednorazovom perorálnom podaní darunaviru v dávke 600 mg v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne sa systémová expozícia voči darunaviru
zvýšila 14-násobne (pozri časť 4.4).
Pri užívaní nalačno bola relatívna biologická dostupnosť darunaviru v prítomnosti nízko-dávkovaného ritonaviru o 30 % nižšia ako pri užívaní lieku s jedlom. Z toho dôvodu sa tablety darunaviru musia užívať spolu s ritonavirom a jedlom. Druh jedla nemá vplyv na expozíciu voči darunaviru.
Distribúcia
Približne 95 % darunaviru sa viaže na plazmatické bielkoviny, najmä na α1-kyslý glykoproteín.
Po intravenóznom podaní bol distribučný objem samotného darunaviru 88,1 ± 59,0 l (stredný ± SD)
a v prítomnosti 100 mg ritonaviru podávaného dvakrát denne bol zvýšený na 131 ± 49,9 l
(stredný ± SD).
Biotransformácia
In vitro štúdie využívajúce mikrozómy pečene človeka (HLM) naznačujú, že darunavir sa
metabolizuje najmä oxidatívnym metabolizmom. Darunavir sa výrazne metabolizuje prostredníctvom systému CYP v pečeni a takmer výlučne prostredníctvom izoenzýmu CYP3A4. V štúdii skúmajúcej aplikáciu darunaviru označeného rádioaktívnym 14C zdravým dobrovoľníkom sa zistilo, že väčšina rádioaktivity v plazme po jednorazovom podaní darunaviru s ritonavirom v dávke 400/100 mg pochádzala z pôvodného liečiva. U ľudí sa zistila prítomnosť najmenej 3 oxidatívnych metabolitov darunaviru, pričom všetky z nich mali aktivitu proti divokému typu HIV najmenej 10x nižšiu ako darunavir.
Eliminácia
Po podaní darunaviru s ritonavirom v dávke 400/100 mg označeného rádioaktívnym 14C sa 79,5 %
rádioaktivity zachytilo v stolici a 13,9 % rádioaktivity sa zachytilo v moči. Rádioaktivita
nezmeneného darunaviru v stolici predstavovala 41,2 % a v moči predstavovala 7,7 % z podanej
dávky. Terminálny polčas eliminácie darunaviru podávaného v kombinácii s ritonavirom bol približne
15 hodín.
Klírens darunaviru (150 mg) podaného intravenózne v monoterapii bol 32,8 l/h, kým pri kombinácii s nízko dávkovaným ritonavirom 5,9 l/h.
Osobitné skupiny pacientov
Pediatrická populácia
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 74 predtým liečených pediatrických pacientov vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg preukázala, že dávky
darunaviru/ritonaviru podávané v závislosti od hmotnosti, mali za následok expozíciu darunaviru
porovnateľnú s expozíciou u dospelých pacientov užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 14 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 15 kg až < 20 kg ukázala, že dávkovanie založené na hmotnosti viedlo k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 12 pediatrických pacientov vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení ART, ukázala, že dávka darunavir/ritonaviru 800/100 mg jedenkrát denne viedla k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne. Z toho dôvodu sa rovnaké dávkovanie jedenkrát denne môže použiť u dospievajúcich vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg
s predchádzajúcou liečbou bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)*
a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+
≥ 100 buniek x 106/l (pozri časť 4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 10 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 14 kg až < 20 kg preukázala, že dávky odvodené od hmotnosti viedli k expozícii darunaviru, ktorá bola porovnateľná
s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne (pozri
časť 4.2). Farmakokinetické modelovanie a simulácia expozície darunaviru u pediatrických pacientov vo veku 3 až < 18 rokov navyše potvrdili expozície darunaviru, ktoré boli pozorované v klinických štúdiách a umožnili určenie dávkovania darunavir/ritonaviru jedenkrát denne v závislosti na hmotnosti u pediatrických pacientov s hmotnosťou najmenej 15 kg, ktorí buď predtým nedostávali liečbu ART alebo u predtým liečených pediatrických pacientov bez DRV-RAM* a s plazmatickou hladinou
HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri časť 4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V.
Starší ľudia
Pri analýze farmakokinetiky v rôznych populáciách pacientov s infekciou HIV sa zistilo, že farmakokinetika darunaviru sa výraznejšie nelíši u pacientov s infekciou HIV vo vekovom rozmedzí
od 18 do 75 rokov (n = 12, vek ≥ 65 rokov) (pozri časť 4.4). U pacientov starších ako 65 rokov boli
k dispozícii iba obmedzené údaje.
Pohlavie
Pri analýze farmakokinetiky v rôznych populáciách pacientov sa u žien s infekciou HIV zistila mierne vyššia expozícia voči darunaviru (o 16,8 %) ako u mužov infikovaných HIV. Tento rozdiel nemá žiadny klinický význam.
Porucha funkcie obličiek
Výsledky štúdie zachovania hmotnosti s 14C rádioaktívne označeným darunavirom s ritonavirom ukázali, že približne 7,7 % z podanej dávky darunaviru sa vylučuje do moču v nezmenenej forme.
Aj keď sa podávanie darunaviru nesledovalo u pacientov s poruchou funkcie obličiek, pri analýze farmakokinetiky v rôznych populáciách pacientov sa zistilo, že stredne závažná porucha funkcie obličiek (klírens kreatinínu: 30 - 60 ml/min, n=20) nemá väčší vplyv na farmakoniketiku darunaviru u pacientov s infekciou HIV (pozri časti 4.2 a 4.4).
Porucha funkcie pečene
Darunavir sa metabolizuje a eliminuje prevažne v pečeni. V štúdii s opakovaným podaním lieku darunavir užívaným s ritonavirom (600/100 mg dvakrát denne) sa preukázalo, že celkové koncentrácie
darunaviru v plazme u subjektov s miernou (Childova-Pughova trieda A, n=8) a stredne ťažkou
(Childova-Pughova trieda B, n=8) poruchou funkcie pečene boli porovnateľné s tými u zdravých
subjektov. Koncentrácie voľného darunaviru boli približne o 55 % (Childova-Pughova Trieda A)
a 100 % (Childova-Pughova trieda B) vyššie. Klinický význam tohto nárastu nie je známy, preto treba darunavir užívať so zvýšenou opatrnosťou. Účinok ťažkej poruchy funkcie pečene na farmakokinetiku darunaviru nebol dosiaľ študovaný (pozri časti 4.2, 4.3 a 4.4).
G
ravidita a obdobie po pôrode
Expozícia celkovému darunaviru a ritonaviru po užití darunaviru/ritonaviru 600/100 mg dvakrát denne a darunaviru/ritonaviru 800/100 mg jedenkrát denne v rámci antivirotického režimu bola všeobecne
nižšia počas gravidity v porovnaní s obdobím po pôrode. V prípade neviazaného (t.j. aktívneho)
darunaviru však boli farmakokinetické parametre menej znížené počas gravidity v porovnaní
s obdobím po pôrode z dôvodu zvýšenia neviazanej frakcie darunaviru počas gravidity v porovnaní s obdobím po pôrode.
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru
600/100 mg dvakrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho trimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=11)aTretí trimester gravidity (n=11)Obdobie po pôrode (6-12 týždňov) (n=11)
Cmax, ng/ml 4 601 ± 1 125 5 111 ± 1 517 6 499 ± 2 411
AUC12h, ng.h/ml 38 950 ± 10 010 43 700 ± 16 400 55 300 ± 27 020
Cmin, ng/mlb 1 980 ± 839,9 2 498 ± 1 193 2 711 ± 2 268
a n=10 pre AUC12h
b vylúčením hodnoty Cmin pod LLOQ, n=10 pre referenciu
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru 800/100 mgjedenkrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho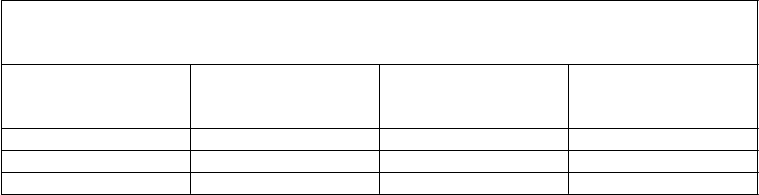 trimestra gravidity a po pôrode
trimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=16)
Tretí trimester gravidity (n=14)
Obdobie po pôrode (6-12 týždňov) (n=15)
Cmax, ng/ml 4 988 ± 1 551 5 138 ± 1 243 7 445 ± 1 674
AUC12h, ng.h/ml 61 303 ± 16 232 60 439 ± 14 052 94 529 ± 28 572
Cmin, ng/mla 1 193 ± 509 1 098 ± 609 1 572 ± 1 108
a n=12 pre obdobie po pôrode, n=15 pre druhý trimester a n=14 pre tretí trimester
U žien užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 28 %, 24 %
a 17 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity
boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 19 %, 17 % nižšie a o 2 % vyššie, v tomto poradí, v porovnaní s obdobím po pôrode.
U žien užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 34 %, 34 %
a 32 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 31 %, 35 % a 50 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode.
5.3 Predklinické údaje o bezpečnosti
Toxikologické štúdie so samotným darunavirom sa vykonali v dávkovaní až do klinických hladín
u myší, potkanov a psov, kým štúdie s jeho kombináciou s ritonavirom sa vykonali u potkanov a psov.
V štúdiách toxicity po opakovanej dávke u myší, potkanov a psov sa zistili len obmedzené účinky liečby darunavirom. Identifikovaným cieľovým orgánom u hlodavcov bol hemopoetický systém, koagulačný systém, pečeň a štítna žľaza. Pozoroval sa premenlivý, ale mierny pokles parametrov v súvislosti s erytrocytmi spolu s predĺžením aktivovaného parciálneho tromboplastínového času.
Zmeny boli pozorované v pečeni (hypertrofia hepatocytov, vakuolizácia, zvýšenie hladiny pečeňových enzýmov) a štítnej žľaze (folikulárna hypertrofia). U laboratórnych potkanov viedla kombinácia darunaviru s ritonavirom k miernemu zvýšeniu účinku na parametre červených krviniek, pečene
a štítnu žľazu a k zvýšeniu incidencie ostrovčekov fibrózy v pankrease (iba u samcov laboratórnych potkanov) v porovnaní so samotným darunavirom. U psov sa nepozorovali žiadne väčšie známky toxicity ani cieľové orgány toxicity pri rovnakej expozícii ako u človeka, ktorý užíva odporučené dávky.
V štúdii na potkanoch bol znížený počet corpora lutea a implantácií v prípade maternálnej toxicity. Na druhej strane sa nepozorovali žiadne účinky na párenie alebo plodnosť po podávaní darunaviru
v dávkach do 1000 mg/kg/deň a pri expozícii (AUC - 0,5-násobok) nižšej ako u človeka dostávajúceho
odporučené dávky. Pri podávaní rovnako veľkých dávok samotného darunaviru sa u potkanov
a králikov nepozorovala teratogenita podobne ako u myší dostávajúcich darunavir v kombinácii s ritonavirom. Stupeň expozície bol nižší ako u človeka dostávajúceho odporučené dávky. Pri
hodnotení prenatálneho a postnatálneho vývoja u potkanov, darunavir podávaný samostatne alebo
v kombinácii s ritonavirom viedol k prechodnému zníženiu prírastku telesnej hmotnosti u dojčených mláďat a došlo k miernemu oneskoreniu v otvorení očí a uší. Darunavir v kombinácii s ritonavirom
spôsobil zníženie prežívania mláďat, ktoré sa prejavilo nepriaznivou odpoveďou v 15. deň laktácie
a znížením počtu mláďat, ktoré prežili počas laktácie. Tieto účinky môžu byť prisúdené sekundárnej expozícii liečiva u mláďat prostredníctvom materského mlieka a/alebo maternálnej toxicity. Darunavir podávaný ako samotný alebo v kombinácii s ritonavirom neovplyvnil žiadne funkcie po odstavení.
U juvenilných laboratórnych potkanov, ktorí dostávali darunavir 23. – 26. deň, sa pozorovala zvýšená
mortalita s kŕčmi u niektorých zvierat. Medzi 5. a 11. dňom života bola expozícia v plazme, pečeni
a mozgu výrazne vyššia ako u dospelých laboratórnych potkanov po porovnateľných dávkach v mg/kg. Po 23. dni života bola expozícia porovnateľná s tou u dospelých potkanov. Zvýšená expozícia bola pravdepodobne aspoň čiastočne z dôvodu nezrelosti enzýmov metabolizujúcich liek
u juvenilných zvierat. U juvenilných potkanov neboli spozorované žiadne úmrtia v súvislosti s liečbou
dávkami 1000 mg/kg darunaviru (jednorazová dávka) v 26. deň veku alebo 500 mg/kg (opakovaná dávka) od 23. do 50. dňa veku a expozícia a profil toxicity boli porovnateľné s tými, pozorovanými
u dospelých potkanov.
Darunavir s nízkou dávkou ritonaviru sa nemá používať u detí mladších ako 3 roky z dôvodu
nejasnosti ohľadom miery rozvoja hematoencefalickej bariéry a pečeňových enzýmov.
Karcinogénny potenciál darunaviru bol vyhodnotený pri podávaní lieku myšiam a potkanom sondou do žalúdka počas 104 týždňov. Myšiam boli podávané denné dávky 150, 450 a 1000 mg/kg a potkanom boli podávané dávky 50, 150 a 500 mg/kg. Výskyt hepatocelulárnych adenómov a karcinómov stúpal v závislosti od dávky a pozoroval sa u samcov a samíc oboch živočíšnych druhov. U samcov potkanov sa vyskytli adenómy folikulárnych buniek štítnej žľazy. Podávanie darunaviru nespôsobilo štatisticky významný nárast iných benígnych alebo malígnych novotvarov u myší alebo potkanov. Pozorované hepatocelulárné tumory a tumory štítnej žľazy u hlodavcov sa považujú za obmedzené z hľadiska významnosti pre človeka. Opakované podávanie darunaviru potkanom spôsobilo indukciu mikrozomálnych enzýmov v pečeni a zvýšilo vylučovanie hormónov štítnej žľazy,
ktoré predurčuje potkanov, ale nie ľudí, na výskyt neoplaziem štítnej žľazy. Pri najvyšších testovaných dávkach boli systémové expozície (založené na AUC) darunaviru 0,4- až 0,7-násobkom (myši) a 0,7- až 1-násobkom (potkany) v pomere k expozíciám pozorovaným u ľudí pri odporúčaných terapeutických dávkach.
Po dvoch rokoch podávania darunaviru pri expozíciách na úrovni alebo pod úrovňou expozícií u ľudí sa pozorovali zmeny na obličkách u myší (nefróza) a u potkanov (chronická progresívna nefropatia).
Darunavir nebol mutagénny ani genotoxický v súbore in vitro a in vivo testov vrátane bakteriálnej reverznej mutácie (Amesov test), chromozómovej aberácie v ľudských lymfocytoch
a mikronukleového testu u myší in vivo.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Oxid kremičitý, koloidný bezvodý
Mikrokryštalická celulóza
Krospovidón
Sodná soľ glykolátu škrobu
Hypromelóza
Magnéziumstearát
Obal tablety
Polyvinylalkohol, čiastočne hydrolyzovaný
Oxid titaničitý Makrogol Mastenec
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po prvom otvorení HDPE fľaše: 100 dní.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
VC/PE/PVDC-Al blistrové balenie s obsahom 240 tabliet a 240 x 1 tabliet. Za studena spracované
PVC/Al/OPA-Al blistrové balenie s obsahom 240 tabliet a 240 x 1 tabliet.
Balenie s HDPE fľašou so skrutkovacím uzáverom s obsahom 60 a 240 tabliet.Balenie s HDPE fľašou
s PP skrutkovacím uzáverom s obsahom 60 a 240 tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Mylan S.A.S.
117 Allee des Parcs
69 800 Saint Priest
Francúzsko
8. REGISTRAČNÉ ČÍSLO
EU/1/16/1140/006
EU/1/16/1140/007
EU/1/16/1140/008
EU/1/16/1140/009
EU/1/16/1140/010
EU/1/16/1140/011
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: [DD. mesiac RRRR]
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
Darunavir Mylan 300 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Každá filmom obalená tableta obsahuje 300 mg darunaviru. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalené tablety.
Biele až sivobiele oválne obojstranne vypuklé filmom obalené tablety s rozmermi približne 16,5 mm x
8,2 mm, s označením „M“ na jednej strane tablety a „DV3“ na druhej strane.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Darunavir podávaný súčasne s nízkou dávkou ritonaviru je indikovaná v kombinácii s inými antiretrovirálnymi liekmi na liečbu pacientov s infekciou vyvolanou vírusom ľudskej imunodeficiencie (HIV-1).
Darunavir Mylan 300 mg tablety sa môžu použit na zabezpečenie vhodného režimu dávkovania (pozri časť 4.2):
· Pri liečbe infekcie HIV-1 u dospelých pacientov predtým liečených antiretrovírusovou liečbou
(ART, antiretroviral treatment), vrátane tých s intenzívnou predchádzajúcou liečbou.
· Pri liečbe infekcie HIV-1 u pediatrických pacientov od 3 rokov a s telesnou hmotnosťou
najmenej 15 kg.
Pri rozhodovaní o začatí liečby darunavirom užívaným spolu s nízkou dávkou ritonaviru je potrebné dôkladne zvážiť liekovú anamnézu jednotlivého pacienta a schémy mutácií súvisiace s rôznymi agensmi. Používanie darunaviru má byť odvodené od geno- alebo fenotypizácie (ak sú dostupné)
a liečebnej anamnézy.
4.2 Dávkovanie a spôsob podávania
Liečbu môže začať len lekár, ktorý má skúsenosti s liečbou infekcie HIV. Po začatí liečby darunavirom treba pacientov poučiť, aby nemenili dávku, liekovú formu ani neprerušovali liečbu bez toho, aby sa poradili so svojím lekárom.
Dávkovanie
Darunavir sa musí vždy podávať perorálne v kombinácii s nízkou dávkou ritonaviru, ktorý zlepšuje jej
farmakokinetiku, a s inými antiretrovírusovými liekmi. Z toho dôvodu je pred začatím kombinovanej
liečby darunavirom a ritonavirom potrebné prečítať si súhrn charakteristických vlastností ritonaviru.
Dospelí pacienti liečení ART
Odporúčané dávkovanie je 600 mg dvakrát denne súčasne so 100 mg ritonaviru dvakrát denne
a s jedlom. Darunavir Mylan 300 mg tablety sa môžu použiť na zostavenie režimu dávkovania 600 mg dvakrát denne.
Užívanie 75 a 150 mg tabliet na dosiahnutie odporúčanej dávky je vhodné v prípade možnej
precitlivenosti na niektoré farbivá alebo v prípade ťažkostí s prehĺtaním 300 mg tabliet.
Dávkovací režim 800 mg jedenkrát denne s kobicistatom 150 mg jedenkrát denne alebo so 100 mg ritonaviru jedenkrát denne a s jedlom sa môže použiť u pacientov s predchádzajúcou liečbou antiretrovirotikami, ale bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM, z angl. darunavir-resistance associated mutations)* a s plazmatickou hladinou HIV-1 RNA
< 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri súhrn charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Dospelí pacienti predtým neliečení ARTOdporučené dávkovanie u pacientov predtým neliečených ART nájdete v Súhrne charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Pediatrickí pacienti predtým neliečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 15 kg)Dávka darunaviru a ritonaviru u pediatrických pacientov odvodená od ich hmotnosti sa uvádza v tabuľke nižšie.
Odporúčaná dávka tabliet darunaviru
a ritonavirua na liečbu predtým neliečených pediatrickýchpacientov (3 až 17 rokov)Telesná hmotnosť (kg) Dávka (jedenkrát denne s jedlom)≥ 15 kg až < 30 kg 600 mg darunavir/100 mg ritonavir jedenkrát denne
≥ 30 kg až < 40 kg 675 mg darunavir/100 mg ritonavir jedenkrát denne
³ 40 kg 800 mg darunavir/100 mg ritonavir jedenkrát denne
a ritonavir perorálny roztok: 80 mg/ml
Pediatrickí pacienti liečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 15 kg)Zvyčajne sa odporúča darunavir dvakrát denne súčasne s ritonavirom a s jedlom.
Dávkovanie darunaviru užívaného s ritonavirom jedenkrát denne a s jedlom sa môže použiť
u pacientov s predchádzajúcou liečbou antiretrovirotikami, ale bez mutácií spojených s rezistenciou
voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom
buniek CD4+ ³ 100 buniek x 106/l.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
U pediatrických pacientov je odporúčaná dávka darunaviru s nízkou dávkou ritonaviru odvodená od hmotnosti. Dávka darunaviru/ritonaviru (600/100 mg dvakrát denne alebo 800/100 mg jedenkrát denne) vhodná pre dospelých sa môže použiť u pediatrických pacientov s hmotnosťou 40 kg alebo vyššou.
Odporúčaná dávka tabliet darunaviru a ritonavirua u predtým liečených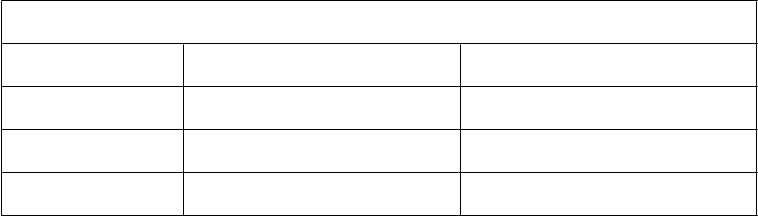 pediatrických pacientov (3 až 17 rokov)
pediatrických pacientov (3 až 17 rokov)
Hm
o
t
nosť (kg) Dávka (jedenkrát denne s jedlom)
≥ 15 kg–< 30 kg 600 mg darunavir/100 mg ritonavir jedenkrát denne
≥ 30 kg–< 40 kg 675 mg darunavir/100 mg ritonavir jedenkrát denne
≥ 40 kg 800 mg darunavir/100 mg ritonavir jedenkrát denne
a s perorálnym roztokom ritonaviru: 80 mg/ml
Dávka (dvakrát denne s jedlom)
375 mg darunavir/50 mg ritonavir dvakrát denne
450 mg darunavir/60 mg ritonavir dvakrát denne
600 mg darunavir/100 mg ritonavir dvakrát denne
U pediatrických pacientov predtým liečených ART sa odporúča testovanie genotypu HIV. Ak
testovanie genotypu HIV nie je k dispozícii, dávkovanie darunaviru/ritonaviru jedenkrát denne sa
odporúča u pediatrických pacientov, ktorí predtým neužívali HIV proteázové inhibítory, a dávkovanie
dvakrát denne sa odporúča u pacientov, ktorí predtým užívali HIV proteázové inhibítory.
Použitie výlučne 75 mg a 150 mg tabliet na dosiahnutie odporúčanej dávky darunaviru môže byť vhodné, keď existuje riziko precitlivenosti na niektoré farbivo.
Odporúčanie pri vynechaní dávky
V prípade, že pacient zabudne užiť dávku darunaviru a/alebo ritonaviru do 6 hodín od doby, kedy liek
zvyčajne užíva, treba ho poučiť, že má užiť predpísanú dávku s jedlom čo najskôr. Ak od zvyčajnej doby použitia uplynie viac ako 6 hodín, vynechaná dávka sa nemá užiť a pacient má pokračovať vo zvyčajnom dávkovacom režime.
Toto usmernenie vyplynulo z 15 hodinového polčasu darunaviru v prítomnosti ritonaviru a
odporúčaného dávkovacieho intervalu približne 12 hodín.
Osobitné skupiny pacientov
Starší ľudia
Informácie o tejto populácii sú obmedzené, a preto sa má darunavir používať v tejto vekovej skupine
so zvýšenou opatrnosťou (pozri časti 4.4 a 5.2).
Porucha funkcie pečene
Darunavir sa metabolizuje v pečeňovom systéme. U pacientov s miernou (Childova-Pughova Trieda
A) alebo stredne ťažkou (Childova-Pughova Trieda B) poruchou funkcie pečene sa neodporúča žiadna úprava dávky, avšak týmto pacientom sa má darunavir podávať so zvýšenou opatrnosťou. U pacientov s ťažkou poruchou funkcie pečene nie sú k dispozícii žiadne farmakokinetické údaje. Ťažká porucha funkcie pečene môže viesť k zvýšeniu expozície darunaviru a k zhoršeniu jeho
bezpečnostného profilu. Z toho dôvodu sa darunavir nesmie podávať pacientom s ťažkou poruchou
funkcie pečene (Childova-Pughova Trieda C) (pozri časti 4.3, 4.4 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávkovania (pozri časti 4.4
a 5.2).
Pediatrickí pacienti
Darunavir/ritonavir sa nemá používať u detí s hmotnosťou menej ako 15 kg, pretože dávka pre túto populáciu nebola stanovená na dostatočnom počte pacientov (pozri časť 5.1). Darunavir/ritonavir sa nemá používať u detí mladších ako 3 roky z dôvodu bezpečnosti (pozri časti 4.4 a 5.3).
Boli stanovené expozície darunaviru u dospievajúcich vo veku 12 až 17 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení a ktorí dostávali darunavir 800 mg jedenkrát denne a ukázalo sa, že sa nachádzali v rámci terapeutického rozsahu, ktorý bol stanovený u dospelých pacientov dostávajúcich darunavir 800 mg jedenkrát denne. Ako dôsledok vzhľadom na to, že
darunavir jedenkrát denne bol tiež zaregistrovaný na použitie u dospelých s predchádzajúcou liečbou
bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1
RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l, rovnaká indikácia darunaviru
jedenkrát denne sa vzťahuje na deti vo veku 3 až 17 rokov a s hmotnosťou najmenej 15 kg s predchádzajúcou liečbou.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Gravidita a obdobie po pôrode
Počas gravidity a po pôrode nie je potrebná úprava dávky darunaviru/ritonaviru. Darunavir sa má
užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko (pozri časti 4.4, 4.6 a 5.2).
Spôsob podávania
Pacientov treba poučiť, aby užívali Darunavir Mylan s nízkou dávkou ritonaviru do 30 minút po jedle.
Druh jedla neovplyvňuje expozíciu darunaviru (pozri časti 4.4, 4.5 a 5.2).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Pacienti s ťažkou (Childova-Pughova Trieda C) poruchou funkcie pečene.
Súčasné podávanie rifampicínu a darunavir spolu s nízkou dávkou ritonaviru (pozri časť 4.5). Súčasné podávanie lieku s kombináciou lopinavir/ritonavir (pozri časť 4.5).
Súčasné podávanie rastlinných preparátov obsahujúcich výťažok z ľubovníka bodkovaného
(Hypericum perforatum) (pozri časť 4.5).
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru a liečiv, ktorých metabolizmus (klírens) je vysoko závislý na CYP3A, a ktorých zvýšené plazmatické koncentrácie sú spojené s vážnymi a/alebo život ohrozujúcimi udalosťami. Medzi tieto liečivá patria napr.:
- alfuzosín (antagonista alfa 1-adrenoreceptora)
- amiodarón, bepridil, dronedarón, chinidín, ranolazín, lidokaín (podávaný systémovo) (antiarytmiká/lieky proti angíne)
- astemizol, terfenadín (antihistaminiká)
- kolchicín, keď sa používa u pacientov s poruchou funkcie obličiek a/alebo pečene (liek proti dne) (pozri časť 4.5)
- námeľové alkaloidy (napr. dihydroergotamín, ergometrín, ergotamín, metylergonovín)
- cisaprid (prokinetiká)
- pimozid, kvetiapín, sertindol (antipsychotiká/neuroleptiká) (pozri časť 4.5)
- triazolam, midazolam podávaný perorálne (sedatíva/hypnotiká) (pre upozornenie na midazolam
podávaný parenterálne, pozri časť 4.5)
- sildenafil - keď sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, avanafil (inhibítory
PDE-5)
- simvastatín a lovastatín (inhibítory HMG-CoA reduktázy) (pozri časť 4.5)
- tikagrelor (inhibítor agregácie trombocytov) (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hoci sa preukázalo, že účinná vírusová supresia dosiahnutá pri antiretrovírusovej terapii značne znižuje riziko prenosu HIV pohlavným stykom, reziduálne riziko nie je možné vylúčiť. Je potrebné prijať opatrenia na zabránenie prenosu HIV v súlade s národnými odporúčaniami.
Odporúča sa pravidelné sledovanie virologickej odpovede. V prípade nedostatočnej alebo žiadnej virologickej odpovede sa má urobiť test na rezistenciu.
Darunavir sa má používať len v kombinácii s nízkou dávkou ritonaviru, ktorý zlepšuje farmakokinetiku darunaviru (pozri časť 5.2).
Zvyšovanie dávkovania ritonaviru oproti odporúčaniam v časti 4.2 významnejšie neovplyvňuje
koncentrácie darunaviru a neodporúča sa.
Darunavir sa viaže prevažne na a1-kyslý glykoproteín. Táto proteínová väzba závisí od koncentrácie, ktorú určuje saturácia väzby. Preto lieky, ktoré sa vysoko viažu na a1-kyslý glykoproteín, môžu znemožniť túto väzbu (pozri časť 4.5).
PacientiliečeníART–dávkovanie jedenkrát denne
Darunavir užívaný v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru jedenkrát denne u pacientov liečených ART sa nemá používať u pacientov s jednou alebo viacerými
mutáciami spojenými s rezistenciou voči darunaviru (DRV-RAM) alebo s hladinou HIV-1 RNA
≥ 100 000 kópií/ml alebo počtom buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2). V tejto populácii sa neskúmali iné kombinácie s optimalizovaným základným režimom (OBR, optimised background
regimen) ako ≥ 2 NRTI. U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje
(pozri časť 5.1).
Pediatrická populácia
Darunavir sa neodporúča používať u pediatrických pacientov mladších ako 3 roky alebo s hmotnosťou
menej ako 15 kg (pozri časti 4.2 a 5.3).
Gravidita
Darunavir sa má užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko. Opatrnosť je potrebná u gravidných žien súbežne užívajúcich lieky, ktoré môžu ďalej znižovať expozíciu darunaviru (pozri časti 4.5 a 5.2).
Staršíľudia
Keďže k dispozícii sú len obmedzené údaje o používaní darunaviru u pacientov vo veku 65 rokov a starších, pri podávaní darunaviru starším pacientom je potrebná opatrnosť vzhľadom k vyššiemu výskytu zhoršenej funkcie pečene a pridružených chorôb, alebo k inej liečbe (pozri časti 4.2 a 5.2).
Závažnékožnéreakcie
Počas programu klinického vývoja (N=3 063) boli u 0,4 % pacientov hlásené závažné kožné reakcie, ktoré mohla sprevádzať horúčka a/alebo zvýšenie transamináz. Zriedkavo (< 0,1 %) bola hlásená lieková vyrážka s eozinofíliou a systémovými príznakmi (DRESS, drug rash with eosinophilia and systemic symptoms) a Stevensov-Johnsonov syndróm a počas postmarketingových skúseností bola hlásená toxická epidermálna nekrolýza a akútna generalizovaná exantematózna pustulóza. Ak sa objavia prejavy alebo príznaky závažnej kožnej reakcie liečba darunavirom/ritonavirom sa má okamžite prerušiť. Príznaky môžu zahŕňať, ale nemusia byť obmedzené na, závažnú vyrážku alebo vyrážku sprevádzanú horúčkou, celkovú nevoľnosť, únavu, bolesť svalov alebo kĺbov, pľuzgiere, orálne lézie, konjunktivitídu, hepatitídu a/alebo eozinofíliu.
Vyrážka sa častejšie vyskytla u predtým liečených pacientov, ktorí boli liečení darunavirom spolu
s raltegravirom v porovnaní s pacientmi, ktorí užívali darunavir bez raltegraviru alebo raltegravir bez darunaviru (pozri časť 4.8).
Darunavir obsahuje sulfónamidovú funkčnú skupinu. Darunavir sa má podávať opatrne pacientom so
známou alergiou na sulfónamid.
Hepatotoxicita
Počas užívania darunaviru bola hlásená liekom indukovaná hepatitída (napr. akútna hepatitída,
cytolytická hepatitída). Počas programu klinického vývoja (N=3 063) bola hepatitída hlásená u 0,5 % pacientov, ktorí boli liečení kombinovanou antiretrovirálnou terapiou s darunavirom/ritonavirom. Pacienti s preexistujúcou dysfunkciou pečene, vrátane chronickej aktívnej hepatitídy B alebo C, majú zvýšené riziko porúch funkcie pečene, vrátane vážnych a potenciálne smrteľných hepatických nežiaducich reakcií. V prípade súčasnej antivírovej liečby hepatitídy B alebo C, obráťte sa na relevantnú informáciu o týchto liekoch.
Pred začatím liečby darunavirom/ritonavirom sa majú urobiť vhodné laboratórne vyšetrenia a pacienti majú byť sledovaní aj počas liečby. U pacientov s chronickou hepatitídou, cirhózou alebo u pacientov, ktorí mali pred liečbou zvýšenú hladinu transamináz, treba zvážiť zvýšené sledovanie AST/ALT, najmä počas prvých mesiacov po začatí liečby darunavirom/ritonavirom.
Ak sa vyskytne alebo sa zhorší dysfunkcia pečene (vrátane klinicky významného zvýšenia pečeňových enzýmov a/alebo príznakov ako únava, anorexia, nauzea, žltačka, tmavý moč, citlivosť pečene, hepatomegália) u pacientov užívajúcich darunavir/ritonavir, treba okamžite zvážiť zastavenie alebo prerušenie liečby.
Pacienti s pridruženými ochoreniami
Porucha funkcie pečene
U pacientov so závažnými poruchami pečene sa účinnosť a bezpečnosť darunaviru nestanovovala, preto je darunavir u pacientov s ťažkou poruchou pečene kontraindikovaný. Vzhľadom na nárast koncentrácie voľného darunaviru v plazme sa darunavir musí používať opatrne u pacientov s miernou alebo stredne ťažkou poruchou funkcie pečene (pozri časti 4.2, 4.3 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa nevyžadujú žiadne osobitné opatrenia alebo úpravy dávkovania. Darunavir aj ritonavir sa výrazne viažu na plazmatické bielkoviny, a preto je
nepravdepodobné, že by sa dali z cirkulácie významnejšie odstrániť pomocou hemodialýzy alebo
peritoneálnej dialýzy. Preto sa u týchto pacientov nevyžadujú žiadne osobitné opatrenia alebo úpravy dávkovania (pozri časti 4.2 a 5.2).
Pacienti s hemofíliou
U pacientov s hemofíliou A a B liečených PI sa opísali prípady väčšieho krvácania, vrátane
spontánnych kožných hematómov a krvácania do kĺbov. Niektorým pacientom sa podal dodatočný
faktor VIII. Vo viac ako polovici hlásených prípadov sa pokračovalo v liečbe PI, prípadne sa táto liečba obnovila po jej dočasnom prerušení. Bola naznačená kauzálna súvislosť, hoci mechanizmus účinku nebol zatiaľ objasnený. Z toho dôvodu je potrebné hemofilikov upozorniť na možnosť väčšieho krvácania.
Telesná hmotnosť a metabolické parametre
Počas antiretrovírusovej liečby môže dôjsť k zvýšeniu telesnej hmotnosti a hladín lipidov a glukózy
v krvi. Takéto zmeny môžu čiastočne súvisieť s kontrolou ochorenia a životným štýlom. Pokiaľ ide
o lipidy, v niektorých prípadoch sú dôkazy o vplyve liečby, kým pri prírastku telesnej hmotnosti nie sú
presvedčivé dôkazy o tom, že súvisí s niektorou konkrétnou liečbou. Pri monitorovaní hladín lipidov a glukózy v krvi sa treba riadiť zavedenými odporúčaniami na liečbu infekcie HIV. Poruchy metabolizmu lipidov majú byť klinicky vhodne liečené.
Osteonekróza
Napriek tomu, že etiológia ochorenia závisí od mnohých faktorov (vrátane užívania kortikosteroidov, konzumácie alkoholu, vážnej imunosupresie, vyššieho BMI), prípady osteonekrózy boli hlásené najmä u pacientov s pokročilým ochorením HIV a/alebo s dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART, combination antiretroviral therapy). Pacientov treba poučiť, aby vyhľadali lekársku pomoc v prípade, že budú pociťovať bolesti kĺbov, stuhnutosť kĺbov alebo ťažkosti pri pohybe.
Imunoreštitučnýzápalovýsyndróm
U pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej antiretrovírusovej liečby môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie, čo môže byť spojené so závažným klinickým stavom resp. so
zhoršením symptómov. Tieto reakcie sa obyčajne pozorujú v priebehu prvých týždňov až mesiacov od začatia podávania kombinovanej antiretrovírusovej liečby. Príkladom môže byť cytomegalovírusová
retinitída, generalizované alebo fokálne mykobaktériové infekcie a pneumónia vyvolaná Pneumocystis jirovecii (predtým známa ako Pneumocystis carinii). Je nutné zhodnotiť všetky príznaky zápalového
ochorenia a v prípade potreby začať liečbu. Ďalej sa v klinických štúdiách s darunavirom užívaným s nízkou dávkou ritonaviru pozorovala reaktivácia herpesu simplex a herpesu zoster.
Boli tiež zaznamenané aj poruchy autoimunitného systému (ako je Gravesova choroba) objavujúce sa v dôsledku imunitnej reaktivácie; avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.8).
Liekové interakcie
Niektoré štúdie interakcií sa uskutočnili s darunavirom v nižších dávkach ako sú odporúčané. Účinky súčasne podávaných liekov môžu byť preto podhodnotené a môže byť potrebné klinické sledovanie bezpečnosti. Pre úplnú informáciu o liekových interakciách pozri časť 4.5.
Efavirenz v kombinácii s darunavirom/ritonavirom 800/100 mg jedenkrát denne môže viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť v kombinácii s darunavirom/ritonavirom, treba použiť režim dávkovania darunavir/ritonavir 600/100 mg dvakrát denne. Pozri Súhrn charakteristických vlastností lieku darunavir 75 mg, 150 mg, alebo 600 mg tablety (pozri časť 4.5).
Život ohrozujúce a smrteľné liekové interakcie sa zaznamenali u pacientov liečených kolchicínom
a silnými inhibítormi CYP3A a P-glykoproteínu (P-gp; pozri časti 4.3 a 4.5).
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Darunavir a ritonavir sú inhibítormi CYP3A, CYP2D6 a P-gp. Súčasné podávanie darunaviru a ritonaviru s liekmi metabolizovanými najmä CYP3A a/alebo CYP2D6 alebo transportovanými P-gp môže viesť k zvýšeniu systémovej expozície týmto liekom, čo by mohlo zosilniť alebo predĺžiť ich terapeutický účinok a viesť k nežiaducim účinkom.
Darunavir užívaný s nízkou dávkou ritonaviru sa nesmie kombinovať s liekmi, ktorých klírens výrazne závisí od CYP3A, a pri ktorých zvýšená systémová expozícia je sprevádzaná závažnými a/alebo život ohrozujúcimi nežiaducimi udalosťami (t.j. ktoré majú úzky terapeutický index) (pozri časť 4.3).
Celkové zvýšenie farmakokinetického účinku ritonavirom viedlo približne k 14-násobnému zvýšeniu
systémovej expozície darunaviru, po jednorazovej 600 mg perorálnej dávke darunaviru v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne. Z toho dôvodu sa darunavir môže používať
len v kombinácii s nízkou dávkou ritonaviru, ktorý zlepšuje farmakokinetiku darunaviru (pozri časti
4.4 a 5.2).
Klinická štúdia s viacerými liekmi, ktoré sú metabolizované cytochrómami CYP2C9, CYP2C19 a CYP2D6 dokázala nárast aktivity CYP2C9 a CYP2C19 a inhibíciu aktivity CYP2D6 v prítomnosti darunaviru/ritonaviru, čo sa môže pripisovať prítomnosti nízkej dávky ritonaviru. Súčasné podávanie darunaviru a ritonaviru a liekov, ktoré sa predovšetkým metabolizujú prostredníctvom CYP2D6 (napríklad flekainid, propafenón, metoprolol), môže vyústiť do zvýšených plazmatických koncentrácií týchto liekov, čo môže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce reakcie. Súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C9 (napríklad warfarín) a CYP2C19 (napríklad metadon), môže vyústiť do zníženia systémových
expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Napriek tomu, že sa účinok na CYP2C8 sledoval iba in vitro, súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C8 (napríklad paklitaxel, rosiglitazón, repaglinid), môže vyústiť do zníženia systémových expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Lieky, ktoré ovplyvňujú expozíciu darunaviru/ritonaviru
Darunavir a ritonavir sa metabolizujú prostredníctvom CYP3A. Očakáva sa, že lieky indukujúce činnosť CYP3A zvýšia klírens darunaviru a ritonaviru, čo má za následok zníženie plazmatických
koncentrácií darunaviru a ritonaviru (napr. rifampicín, ľubovník bodkovaný, lopinavir). Súčasné
podávanie darunaviru a ritonaviru v kombinácii s liekmi, ktoré inhibujú CYP3A môže znížiť klírens
darunaviru a ritonaviru a môže mať za následok zvýšenie plazmatických koncentrácií darunaviru
a ritonaviru (napr. indinavir, systémové azoly ako ketokonazol a klotrimazol). Tieto interakcie sú opísané v nižšie uvedenej tabuľke interakcií.
T
abuľka
interakcií
Interakcie medzi darunavirom/ritonavirom a antiretrovirotikami a ne-antiretrovírusovými liekmi sú uvedené v tabuľke nižšie (neurčené ako „ND“ (z angl. not determinated). Smer šípky pri každom farmakokinetickom parametri je založený na 90 % intervale spoľahlivosti stredného geometrického pomeru v rámci 80-125 % rozpätia (↔), pod ním (↓) alebo nad ním (↑).
Niekoľko štúdií interakcií (v tabuľke nižšie označené ako #) sa uskutočnili s nižšími ako odporúčanými dávkami darunaviru alebo s iným režimom dávkovania (pozri časť 4.2 Dávkovanie). Účinky súčasne podávaných liekov môžu byť preto podhodnotené a môže byť potrebné klinické sledovanie bezpečnosti.
INTERAKCIE A ODPORUČENÉ DÁVKOVANIE S INÝMI LIEKMI
L
ieky podľa
t
erapeutických oblastí
I
nterakcia
Z
m
e
na geometrického priemeru (%)
O
dporúčania pri súčasnom
podávaní
ANTIRETROVIROTIKÁ NA HIV
I
nhibítory prenosu reťazcov integrázou
Dolutegravir dolutegravir AUC ↓ 32 % dolutegravir C24h 38 % dolutegravir Cmax ↓ 11 % darunavir ↔*
* Použitím porovnania údajov zo skríženej štúdie
s historickými údajmi o farmakokinetike
Elvitegravir elvitegravir AUC ↔ elvitegravir Cmin ↔ elvitegravir Cmax ↔ darunavir AUC ↔ darunavir Cmin 17 % darunavir Cmax ↔
Raltegravir Niektoré klinické štúdie naznačujú, že raltegravir môže spôsobiť mierny pokles koncentrácie darunaviru v plazme.
Darunavir užívaný s nízkou dávkou ritonaviru a dolutegravir sa môžu užívať bez úpravy dávky.
Keď sa darunaviružívaný súčasne s nízkou dávkou ritonaviru (600/100 mg dvakrát denne) užíva spolu s elvitegravirom, dávka elvitegraviru má byť 150 mg jedenkrát denne.
Farmakokinetika a odporúčania na dávkovanie pre iné dávky darunaviru alebo
s elvitegravirom/kobicistatom neboli stanovené. Súčasné užívanie darunaviru s nízkou dávkou ritonaviru v iných dávkach ako
600/100 mg dvakrát denne
a elvitegraviru sa preto neodporúča. Súčasné užívanie darunaviru
s nízkou dávkou ritonaviru
a elvitegravirom za prítomnosti
kobicistatu sa neodporúča.
V súčasnosti sa účinok raltegraviru na plazmatické koncentrácie darunaviru nejaví ako klinicky významný. Darunavir súčasne podávaný s nízkou dávkou ritonaviru a raltegraviru sa môže užívať bez úpravy dávky.
Nukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, nucleo(s/t)ide reverse transcriptase inhibitors)

Didanozín
400 mg jedenkrát denne
didanozín AUC ↓ 9 % didanozín Cmin ND didanozín Cmax ↓ 16 % darunavir AUC ↔ darunavir Cmin ↔ darunavir Cmax ↔
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a didanozínu sa môže užívať bez úpravy dávok.
Didanozín sa má podávať nalačno, preto ho treba užiť 1 hodinu pred alebo 2 hodiny po užití darunaviru/ritonaviru s jedlom.
Tenofovir disoproxil fumarát
300 mg jedenkrát denne
Abakavir Emtricitabín Lamivudín Stavudín Zidovudín
tenofovir AUC ↑ 22 % tenofovir Cmin ↑ 37 % tenofovir Cmax ↑ 24 %
#darunavir AUC ↑ 21 %
#darunavir Cmin ↑ 24 %
#darunavir Cmax ↑ 16 %
(↑ tenofoviru v dôsledku efektu na MDR-1
transport v renálnych tubuloch)
Neskúmalo sa. Vzhľadom na rôzne cesty eliminácie ostatných NRTI zidovudín, emtricitabín, stavudín, lamivudín, ktoré sa vylučujú prevažne renálnou cestou
a abakavir, ktorý nie je metabolizovaný prostredníctvom CYP450, sa pri ich kombinácii s darunaviromužívaným
s nízkou dávkou ritonaviru
nepredpokladajú žiadne liekové interakcie.
Pri súčasnom podávaní darunaviru a nízkej dávky ritonaviru
v kombinácii s tenofovirom môže byť indikované sledovanie renálnych funkcií, najmä
u pacientov so systémovým alebo renálnym ochorením, alebo
u pacientov užívajúcich
nefrotoxické lieky.
Darunavir užívaný s nízkou dávkou ritonaviru sa môže užívať s týmito NRTI bez úpravy dávky.
Nenukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, non-nucleo(s/t)ide reverse transcriptase inhibitors)
Efavirenz
600 mg jedenkrát denne
Etravirín
100 mg dvakrát denne
Nevirapín
200 mg dvakrát denne
Rilpivirín
150 mg jedenkrát denne
efavirenz AUC ↑ 21 % efavirenz Cmin ↑ 17 % efavirenz Cmax ↑ 15 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↓ 31 %
#darunavir Cmax ↓ 15 %
(↑ efavirenzu v dôsledku inhibície
CYP3A)
(↓ darunaviru v dôsledku inhibície
CYP3A)
etravirín AUC ↓ 37 % etravirín Cmin ↓ 49 % etravirín Cmax ↓ 32 % darunavir AUC ↑ 15 % darunavir Cmin ↔ darunavir Cmax ↔ nevirapín AUC ↑ 27 % nevirapín Cmin ↑ 47 % nevirapín Cmax ↑ 18 %
#darunavir: koncentrácie
sa zhodovali s údajmi z minulosti. (↑ nevirapínu v dôsledku inhibície CYP3A)
rilpivirín AUC ↑ 130 % rilpivirín Cmin ↑ 178 % rilpivirín Cmax ↑ 79 % darunavir AUC ↔ darunavir Cmin ↓ 11 % darunavir Cmax ↔
Pri súčasnom podávaní darunaviru a nízkej dávky ritonaviru
v kombinácii s efavirenzom môže byť indikované klinické sledovanie toxicity centrálneho nervového systému spojené so zvýšenou expozíciou voči efavirenzu.
Efavirenz v kombinácii
s darunavirom/ritonavirom
800/100 mg jedenkrát denne môže viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť
v kombinácii
s darunavirom/ritonavirom, treba použiť režim dávkovania darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.4). Darunavir súčasne podávaný
s nízkou dávkou ritonaviru a
s etravirínom
200 mg dvakrátdennesa môže užívať bez úpravy dávky.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s nevirapínom sa môže užívať bez
úpravy dávky.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru
a s rilpivirínom sa môže užívať bez
úpravy dávky.
H
I
V Proteázové inhibítory (PI) - bez doplnkového súčasného podávania nízkej dávky ritonaviru
†
Atazanavir
300 mg jedenkrát denne
Indinavir
800 mg dvakrát denne
Sakvinavir
1 000 mg dvakrát denne
atazanavir AUC ↔ atazanavir Cmin ↑ 52 % atazanavir Cmax ↓ 11 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Atazanavir: porovnanie atazanaviru/ritonaviru 300/100 mg jedenkrát denne vs. atazanavir 300 mg jedenkrát denne v kombinácii
s darunavirom/ritonavirom 400/100 mg dvakrát denne.
Darunavir: porovnanie
darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg dvakrát denne v kombinácii s atazanavirom 300 mg jedenkrát denne. indinavir AUC ↑ 23 %
indinavir Cmin ↑ 125 %
indinavir Cmax ↔
#darunavir AUC ↑ 24 %
#darunavir Cmin ↑ 44 %
#darunavir Cmax ↑ 11 %
Indinavir: porovnanie indinaviru/ritonaviru
800/100 mg dvakrát denne vs. indinavir/darunavir/ritonavir
800/400/100 mg dvakrát denne.
Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii s indinavirom 800 mg dvakrát denne.
#darunavir AUC ↓ 26 %
#darunavir Cmin ↓ 42 %
#darunavir Cmax ↓ 17 %
#sakvinavir AUC ↓ 6 %
#sakvinavir Cmin ↓ 18 %
#sakvinavir Cmax ↓ 6 %
Sakvinavir: porovnanie sakvinaviru/ritonaviru 1000/100 mg dvakrát denne vs. sakvinavir/darunavir/ritonavir
1000/400/100 mg dvakrát denne Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii so sakvinavirom 1000 mg dvakrát denne.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s atazanavirom sa môže užívať bez úpravy dávky.
Pri intolerancii súčasného podávania darunaviru užívaného s nízkou dávkou ritonaviru
s indinavirom môže byť indikované
zníženie dávkovania indinaviru
z 800 mg dvakrát denne na 600 mg dvakrát denne.
Neodporúča sa kombinovať darunavir užívaný s nízkou dávkou ritonaviru so sakvinavirom.
H
I
V Proteázové inhibítory (PI) - podávané s nízkou dávkou ritonaviru
†
Lopinavir/ritonavir
400/100 mg dvakrát denne
Lopinavir/ritonavir
533/133,3 mg dvakrát denne
CCR5-ANTAGONISTY
Maravirok
150 mg dvakrát denne
ANESTETIKÁ
lopinavir AUC ↑ 9 % lopinavir Cmin ↑ 23 % lopinavir Cmax ↓ 2 % darunavir AUC ↓ 38 %‡ darunavir Cmin ↓ 51 %‡ darunavir Cmax ↓ 21 %‡ lopinavir AUC ↔ lopinavir Cmin ↑ 13 % lopinavir Cmax ↑ 11 % darunavir AUC ↓ 41 % darunavir Cmin ↓ 55 % darunavir Cmax ↓ 21 %
‡ na základe neštandardizovaných veľkostí dávky
maravirok AUC ↑ 305 % maravirok Cmin ND maravirok Cmax ↑ 129 %
koncentrácie darunaviru, ritonaviru sa
zhodovali s údajmi z minulosti
Vzhľadom na pokles AUC darunaviru o 40 %, kombinácie vhodných dávok neboli stanovené. Preto je súčasné podávanie darunaviru užívaného s nízkou dávkou ritonaviru a lieku
s kombináciou lopinavir/ritonavir
kontraindikované (pozri časť 4.3).
Keď sa maravirok podáva
s darunavirom s nízkou dávkou
ritonaviru, jeho dávka má byť
150 mg dvakrát denne.
Alfentanil Neskúmalo sa. Metabolizmus alfentanilu je sprostredkovaný CYP3A a v podstate môže byť inhibovaný darunavirom užívaným s nízkou dávkou ritonaviru.
LIEKY PROTI ANGÍNE/ANTIARYTMIKÁ
Súčasné užívanie s darunavirom
a nízkou dávkou ritonaviru môže vyžadovať zníženie dávky alfentanilu a sledovanie rizík predĺženého alebo oneskoreného respiračného útlmu.
Disopyramid Flekainid Mexiletín Propafenón
Amiodarón Bepridil Dronedarón
Lidokaín (podávaný systémovo)
Chinidín Ranolazín Digoxín
0,4 mg jednorazová
dávka
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto antiarytmík.
(inhibícia CYP3A)
digoxín AUC ↑ 61 % digoxín Cmin ND digoxín Cmax ↑ 29 %
(↑ digoxínu v dôsledku možnej inhibície P-
gp)
Pri súčasnom užívaní týchto antiarytmík a darunaviru užívanej s nízkou dávkou ritonaviru sa vyžaduje opatrnosť a odporúča sa sledovanie terapeutických koncentrácií, ak je dostupné.
Súčasné užívanie darunaviru užívanej s nízkou dávkou ritonaviru a amiodarónu, bepridilu, dronedarónu, lidokaínu podávaného systémovo, chinidínu alebo ranolazínu je kontraindikované (pozri časť 4.3).
Vzhľadom na to, že digoxín má úzky terapeutický index, v prípade, že pacienti liečení darunavirom/ritonavirom užívajú digoxín, odporúča sa na začiatok predpísať čo najnižšiu možnú dávku digoxínu. Dávka digoxínu sa má opatrne titrovať, kým sa nedosiahne želaný klinický účinok pri sledovaní celkového klinického stavu
pacienta.
ANTIBIOTIKÁ
Klaritromycín
500 mg dvakrát denne
ANTIKOAGULANCIÁ
Apixaban Dabigatran etexilát Rivaroxaban
klaritromycín AUC ↑ 57 % klaritromycín Cmin ↑ 174 % klaritromycín Cmax ↑ 26 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↑ 1 %
#darunavir Cmax ↓ 17 %
Pri podávaní s PREZISTOU/ritonavirom neboli zistiteľné koncentrácie metabolitu
14-OH-klaritromycínu.
(↑ klaritromycínu v dôsledku inhibície
CYP3A a možnej inhibície P-gp)
Neskúmalo sa. Súčasné užívanie darunaviru a týchto antikoagulancií môže zvýšiť koncentrácie antikoagulantu. (inhibícia CYP3A a/alebo P-gp).
Vyžaduje sa opatrnosť, keď sa klaritromycín podáva v kombinácii s PREZISTOU užívanou spolu
s nízkou dávkou ritonaviru.
Užívanie darunaviru užívaného
s nízkou dávkou ritonaviru a týchto
antikoagulancií sa neodporúča.
Warfarín Neskúmalo sa. Pri súčasnom podávaní
warfarínu a darunaviru
s ritonavirom(nízkou dávkou) môže dôjsť
k ovplyvneniu koncentrácií warfarínu.
ANTIKONVULZÍVA
Pri súčasnom podávaní warfarínu
s darunavirom užívanýmu s nízkou dávkou ritonaviru sa odporúča monitorovať INR (International Normalized Ratio).
Fenobarbital
Fenytoín
Karbamazepín
200 mg dvakrát denne
ANTIDEPRESÍVAParoxetín
20 mg jedenkrát denne
Sertralín
50 mg jedenkrát denne
Amitriptylín Desipramín Imipramín Nortriptylín Trazodón
Neskúmalo sa. Predpokladá sa, že fenobarbital a fenytoín znižujú plazmatické koncentrácie darunaviru. (indukcia enzýmov CYP450) karbamazepín AUC ↑ 45 % karbamazepín Cmin ↑ 54 % karbamazepín Cmax ↑ 43 %
darunavir AUC ↔
darunavir Cmin ↓ 15 %
darunavir Cmax ↔
paroxetín AUC ↓ 39 % paroxetín Cmin ↓ 37 % paroxetín Cmax ↓ 36 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
sertralín AUC ↓ 49 %
sertralín Cmin ↓ 49 %
sertralín Cmax ↓ 44 %
#darunavir AUC ↔
#darunavir Cmin ↓ 6 %
#darunavir Cmax ↔
Súčasné užívanie darunaviru užívanéhoj s nízkou dávkou ritonaviru a týchto antidepresív môže zvýšiť koncentrácie antidepresíva.
(inhibícia CYP2D6 a/alebo CYP3A).
Darunavir užívaný s nízkou dávkou
ritonaviru sa nesmie používať
v kombinácii s týmito liečivami.
Neodporúča sa žiadna úprava dávky darunaviru/ritonaviru. V prípade potreby kombinovať darunavir/ritonavir a karbamazepín sa majú pacienti sledovať z dôvodu možných nežiaducich udalostí spojených s karbamazepínom. Je potrebné sledovať koncentrácie karbamazepínu a titrovať jeho
dávku až do dosiahnutia primeranej odpovede. Na základe zistení môže byť potrebné znížiť dávku karbamazepínu o 25% až 50%, ak
sa podáva s darunavirom
/ritonavirom.
Ak sa antidepresíva užívajú súčasne s darunavirom užívaným s nízkou dávkou ritonaviru, odporúčaný prístup je titrácia dávky antidepresíva na základe klinického posúdenia odpovede na antidepresívum. Navyše u pacientov na ustálenej dávke týchto antidepresív, ktorí začínajú liečbu darunavirom užívaným s nízkou dávkou ritonaviru, sa má sledovať odpoveď na antidepresívum.
Keď sa tieto antidepresíva užívajú s darunavirom s nízkou dávkou ritonaviru, odporúča sa klinické sledovanie a môže byť potrebná úprava dávky antidepresíva.
ANTIMYKOTIKÁ
Vorikonazol Neskúmalo sa. Ritonavir môže znížiť plazmatické koncentrácie vorikonazolu. (indukcia enzýmov CYP450 ritonavirom)
Vorikonazol sa nesmie používať
spolu s darunavirom užívanou
s nízkou dávkou ritonaviru, kým vyhodnotenie pomeru prínos/riziko neodôvodní použitie vorikonazolu.
Ketokonazol
200 mg dvakrát denne
ketokonazol AUC ↑ 212 % ketokonazol Cmin ↑ 868 % ketokonazol Cmax ↑ 111 %
#darunavir AUC ↑ 42 %
#darunavir Cmin ↑ 73 %
#darunavir Cmax ↑ 21 % (inhibícia CYP3A)
Vyžaduje sa opatrnosť a odporúča sa klinické sledovanie. Ak je nutná súčasná liečba, denná dávka ketokonazolu nesmie presiahnuť
200 mg.
Posakonazol Neskúmalo sa. Darunavir môže zvýšiť plazmatické koncentrácie antimykotika (inhibícia P-gp) a posakonazol môže zvýšiť koncentrácie darunaviru. (inhibícia CYP3A)
Itrakonazol Neskúmalo sa. Súčasné systémové podanie
itrakonazolu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť plazmatické koncentrácie darunaviru. Zároveň súčasné podávanie darunaviru užívaného s nízkou dávkou ritonaviru
a itrakonazolu môže zvýšiť plazmatické koncentrácie itrakonazolu.
(inhibícia CYP3A)
Klotrimazol Neskúmalo sa. Súčasné systémové podanie
klotrimazolu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť plazmatické koncentrácie darunaviru. darunavir AUC24h ↑ 33 % (na základe farmakokinetického modelu populácie)
Vyžaduje sa opatrnosť a odporúča
sa klinické sledovanie.
Vyžaduje sa opatrnosť a odporúča sa klinické sledovanie. Ak je nutná súčasná liečba, denná dávka itrakonazolu nesmie presiahnuť
200 mg.
Ak je nutná súčasná liečba klotrimazolom, vyžaduje sa opatrnosť a odporúča sa klinické sledovanie.
ANTIURATIKÁ
Kolchicín Neskúmalo sa. Súčasné užívanie
kolchicínu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť
expozíciu kolchicínu.
ANTIMALARIKÁ
Ak je potrebná liečba darunavirom užívaným s nízkou dávkou ritonaviru, odporúča sa u pacientov s normálnou funkciou obličiek alebo pečene znížiť dávku
kolchicínu alebo liečbu kolchicínom prerušiť.
Pacientom s poruchou funkcie
obličiek alebo pečene sa nemá podávať kolchicín s darunavirom užívaným s nízkou dávkou ritonaviru (pozri časť 4.4).
Artemeter/
lumefantrín
80/480 mg, 6 dávok v 0., 8., 24., 36., 48. a 60. hodine
artemeter AUC ↓ 16 % artemeter Cmin ↔ artemeter Cmax ↓ 18 %
dihydroartemisinín AUC ↓ 18 %
dihydroartemisinín Cmin ↔ dihydroartemisinín Cmax ↓ 18 % lumefantrín AUC ↑ 175 % lumefantrín Cmin ↑ 126 % lumefantrín Cmax ↑ 65 % darunavir AUC ↔
darunavir Cmin ↓ 13 %
darunavir Cmax ↔
Kombinácia darunaviru a artemeteru/lumefantrínu sa môže podávať bez úpravy dávky;
z dôvodu zvýšenia expozície lumefantrínu však treba kombináciu používať s opatrnosťou.
ANTITUBERKULOTIKÁ
Rifampicín
Rifapentín
Rifabutin
150 mg jedenkrát každý druhý deň
ANTINEOPLASTIKÁDasatinib Nilotinib Vinblastín Vinkristín
Everolimus
Neskúmalo sa. Rifapentín a rifampicín sú silnými induktormi CYP3A a bolo dokázané, že spôsobujú signifikantné zníženie koncentrácií iných proteázových inhibítorov, čo môže vyústiť do zlyhania antivirologickej odpovede a rozvinutia rezistencie (indukcia enzýmu CYP450). Pri pokusoch prekonať zníženú expozíciu zvýšením dávky iných proteázových inhibítorov s nízkou dávkou ritonaviru, sa s rifampicínom zistila vysoká frekvencia pečeňových reakcií.
rifabutin AUC** ↑ 55 % rifabutin Cmin** ↑ ND rifabutin Cmax** ↔ darunavir AUC ↑ 53 % darunavir Cmin ↑ 68 % darunavir Cmax ↑ 39 %
** súčet účinných zložiek rifabutinu (materský liek
+ 25-
O-desacetyl metabolit)
Štúdia interakcií preukázala porovnateľnú
dennú systémovú expozíciu rifabutinu
v liečbe samotným rifabutinom 300 mg
jedenkrát denne a v liečbe rifabutinom
150 mg jedenkrát každý druhý deň spolu s darunavirom/ritonavirom (600/100 mg dvakrát denne) s približne 10-násobným zvýšením dennej expozície aktívnemu metabolitu 25-
O-desacetylrifabutinu. Okrem toho, AUC súčtu účinných zložiek rifabutinu (materský liek + 25-
O-desacetyl metabolit) sa zvýšila 1,6-násobne, pričom Cmax zostala porovnateľná.
Údaje o porovnaní s referenčnou dávkou
150 mg jedenkrát denne nie sú k dispozícii.
(Rifabutin je induktorom a substrátom enzýmov CYP3A.) Keď sa darunavir užívaný spolu s 100 mg ritonaviru podávala spolu s rifabutinom (150 mg jedenkrát každý druhý deň) pozorovalo sa
zvýšenie systémovej expozície darunaviru.
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto antineoplastík.
(inhibícia CYP3A)
Kombinácia rifapentínu
a darunaviru užívaného s nízkou
dávkou ritonaviru sa neodporúča.
Súčasné podávanie rifampicínu
a darunaviru spolu s nízkou dávkou ritonaviru je kontraindikované (pozri časť 4.3).
U pacientov užívajúcich tieto lieky súčasne je nevyhnutná redukcia zvyčajnej dávky rifabutinu
300 mg/deň o 75 % (t.j. rifabutin
150 mg jedenkrát každý druhý deň) a zvýšené sledovanie nežiaducich udalostí súvisiacich s rifabutinom. V prípade ohrozenia bezpečnosti sa má zvážiť ďalšie predĺženie dávkovacieho intervalu rifabutinu a/alebo sledovanie hladiny rifabutinu.
Treba brať do úvahy oficiálnu smernicu pre liečbu tuberkulózy u pacientov infikovaných HIV.
Na základe bezpečnostného profilu darunaviru/ritonaviru, toto zvýšenie expozície darunaviru v prítomnosti rifabutinu nevyžaduje úpravu dávky darunaviru /ritonaviru.
Na základe farmakokinetického modelu možno dávku zredukovať o 75 % aj u pacientov, ktorí
dostávajú inú dávku rifabutinu ako
300 mg/deň.
Pri súčasnom užívaní týchto liekov a darunaviru s nízkou dávkou ritonaviru môžu byť ich koncentrácie zvýšené, čo vedie k možnosti nárastu nežiaducich udalostí zvyčajne súvisiacich
s týmito liekmi.
Pri súčasnom užívani darunaviru s nízkou dávkou ritonaviru
a jedného z antineoplastík je
potrebná opatrnosť.
Súčasné užívanie everolimu a darunaviru užívaného s nízkou dávkou ritonaviru sa neodporúča.
I
NHIBÍTORY AGREGÁCIE TROMBOCYTOV
Tikagrelor Neskúmalo sa. Súčasné užívanie
s darunavirom posilneným nízkou dávkou ritonaviru môže viesť k značnému zvýšeniu expozície tikagreloru.
ANTIPSYCHOTIKÁ/NEUROLEPTIKÁ
Kvetiapín V dôsledku inhibície CYP3A darunavirom sa očakáva zvýšenie koncentrácií antipsychotík/neuroleptík.
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru
a s tikagrelorom je kontraindikované.
Užívanie iných antitrombotík neovplyvnených inhibíciou alebo indukciou CYP (napr. prasugrel) sa odporúča.
Súčasné podávanie darunaviru s nízkou dávkou ritonaviru
a kvetiapínu je kontraindikované, pretože môže zvýšiť toxicitu súvisiacu s kvetiapínom. Zvýšené koncentrácie kvetiapínu môžu viesť ku kóme.
Risperidón
Tioridazín
Pimozid
Sertindol
β-BLOKÁTORY Karvedilol Metoprolol Timolol
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto antipsychotík.
(inhibícia CYP2D6 a/alebo P-gp)
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto beta blokátorov.
(inhibícia CYP2D6)
Môže byť potrebné zníženie dávky týchto liekov, keď sa podávajú súčasne s darunavirom užívaným
s nízkou dávkou ritonaviru.
Súčasné podávanie darunaviru s nízkou dávkou ritonaviru a pimozidu alebo sertindolu je kontraindikované.
Pri súčasnom užívaní darunaviru s beta blokátormi sa odporúča klinické sledovanie. Má sa zvážiť zníženie dávky beta blokátora.
B
L
OK
ÁTORY KALCIOVÉHO KANÁLA
Amlodipín Diltiazem Felodipín Nikardipín Nifedipín Verapamil
Nesledovalo sa. Môže sa očakávať, že darunavir užívaný s nízkou dávkou ritonaviru bude zvyšovať plazmatické koncentrácie blokátorov kalciového kanála.
(inhibícia CYP3A a/alebo CYP2D6)
Ak sa tieto lieky podávajú súčasne s darunavirom a s nízkou dávkou ritonaviru, odporúča sa klinické sledovanie liečebných a nežiaducich udalostí.
KO
RTIKOSTEROIDY
Flutikazón
Budezonid
Dexametazón
(podávaný systémovo)
V klinickej štúdii, pri ktorej sa zdravým jedincom počas 7 dní podávali kapsuly ritonaviru 100 mg dvakrát denne spolu
s 50 µg flutikazónpropionátu intranazálne (štyrikrát denne), sa významne zvýšili sérové hladiny flutikazónpropionátu, zatiaľ čo prirodzená hladina kortizolu klesla približne o 86 % (pri 90 % intervale spoľahlivosti 82 % - 89 %). Vyššie účinky možno očakávať, keď sa flutikazón inhaluje. Systémové účinky kortikosteroidov, vrátane Cushingovho syndrómu a adrenálnej supresie, boli hlásené u pacientov, ktorí užívali ritonavir
a súčasne inhalovali alebo intranazálne užili flutikazón; podobná situácia môže vzniknúť aj pri súčasnom užívaní iných kortikosteroidov, ktoré sa metabolizujú prostredníctvom cytochrómu P4503A, napr. budezonidu. Účinky vysokej systémovej expozície flutikazónu na plazmatické hladiny ritonaviru nie sú dosiaľ známe.
Neskúmalo sa. Dexametazón môže znížiť plazmatickú koncentráciu darunaviru. (indukcia CYP3A)
Preto sa súčasná liečba darunavirom v kombinácii s nízkou dávkou ritonaviru a týmito
glukokortikoidmi neodporúča, pokiaľ možný prínos nepreváži riziko systémových kortikosteroidných účinkov. Má sa zvážiť zníženie dávky glukokortikoidov spolu
s dôkladným monitorovaním lokálnych a systémových účinkov, alebo prevod na iný glukokortikoid, ktorý nepatrí k substrátom pre CYP3A (napr. beklometazón).
V prípade vysadenia glukokortikoidov môže byť navyše potrebné, aby progresívne znižovanie dávky trvalo dlhší čas.
Dexametazón podávaný systémovo sa má podávať so zvýšenou opatrnosťou, ak sa užíva spolu
s darunavirom a nízkou dávkou ritonaviru.
Prednizón Neskúmalo sa. Darunavir môže zvýšiť plazmatické koncentrácie prednizónu. (inhibícia CYP3A)
ANTIENDOTELÍNY
Bosentan Neskúmalo sa. Súčasné užívanie
bosentanu a darunaviru užívaného s nízkou
dávkou ritonaviru môže zvýšiť
plazmatické koncentrácie bosentanu.
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru
a prednizónu môže zvýšiť riziko vzniku systémových kortikosteroidných účinkov, vrátane Cushingovho syndrómu
a adrenálnej supresie. Pri súčasnom užívaní darunaviru s nízkou dávkou ritonaviru a kortikosteroidov sa odporúča klinické sledovanie.
Ak sa podáva súčasne
s darunavirom a nízkou dávkou ritonaviru, má sa u pacienta
sledovať tolerovateľnosť bosentanu.
ANTIVIROTIKÁ PRIAMO ÚČINKUJÚCE PROTI VÍRUSU HEPATITÍDY C (HCV)
Proteázové inhibítory NS3-4A
Telaprevir
750 mg každých
8 hodín
Boceprevir
800 mg trikrát denne
telaprevir AUC ↓ 35 % telaprevir Cmin ↓ 32 % telaprevir Cmax ↓ 36 % darunavir AUC12 ↓ 40 % darunavir Cmin ↓ 42 % darunavir Cmax ↓ 40 % boceprevir AUC ↓ 32 % boceprevir Cmin ↓ 35 % boceprevir Cmax ↓ 25 % darunavir AUC ↓ 44 % darunavir Cmin ↓ 59 % darunavir Cmax ↓ 36 %
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru a
s telaprevirom sa neodporúča.
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru a
s boceprevirom sa neodporúča.
Simeprevir simeprevir AUC ↑ 159 % simeprevir Cmin ↑ 358 % simeprevir Cmax ↑ 79 % darunavir AUC ↑ 18 % darunavir Cmin ↑ 31 % darunavir Cmax «
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru
a simepreviru sa neodporúča.
RASTLINNÉ LIEČIVÁ Ľubovník bodkovaný (Hypericum perforatum)
Dávka simepreviru v tejto interakčnej
štúdii bola 50 mg, keď sa podával spolu
s darunavirom/ritonavirom, v porovnaní so
150 mg v skupine liečenej samotným
simeprevirom.
Neskúmalo sa. Predpokladá sa, že ľubovník bodkovaný spôsobuje pokles plazmatických koncentrácií darunaviru a ritonaviru.
(indukcia CYP450)
Darunavir užívaný s nízkou dávkou
ritonaviru sa nesmie podávať
v kombinácii s liekmi obsahujúcimi výťažok z ľubovníka bodkovaného (Hypericum perforatum) (pozri časť
4.3). Ak už pacient užíva ľubovník bodkovaný, užívanie ukončite, a ak je to možné, skontrolujte vírusové hladiny. Expozícia darunaviru (a tiež expozícia ritonaviru) sa môže po ukončení užívania ľubovníka bodkovaného zvýšiť. Indukujúci účinok môže pretrvávať najmenej 2 týždne po ukončení liečby ľubovníkom bodkovaným.
I
NHIBÍTORY HMG CO-A REDUKTÁZY
Lovastatín
Simvastatín
Atorvastatín
10 mg jedenkrát denne
Pravastatín
40 mg jednorazová dávka
Rosuvastatín
10 mg jedenkrát denne
Neskúmalo sa. Predpokladá sa, že lovastatín a simvastatín budú mať zreteľne zvýšené plazmatické koncentrácie, ak sa budú podávať súčasne s darunavirom užívaným s nízkou dávkou ritonaviru. (inhibícia CYP3A)
atorvastatín AUC ↑ 3-4 krát atorvastatín Cmin ↑ ≈5,5-10 krát atorvastatín Cmax ↑ ≈2 krát
#darunavir
pravastatín AUC ↑ 81 %¶ pravastatín Cmin ND pravastatín Cmax ↑ 63 %
¶ u niektorých subjektov sa pozoroval až 5-násobný nárast
rosuvastatín AUC ↑ 48%║
rosuvastatín Cmax ↑ 144%║
║ na základe publikovaných údajov
Zvýšené plazmatické koncentrácie lovastatínu alebo simvastatínu môžu vyvolať myopatiu, vrátane rabdomyolýzy. Súčasné podávanie darunaviru užívaného s nízkou dávkou ritonaviru s lovastatínom
a simvastatínom je preto
kontraindikované (pozri časť 4.3). Ak je potrebné súčasné podávanie atorvastatínu s darunavirom užívanou s nízkou dávkou ritonaviru, odporúča sa začať podávať atorvastatín v dávke 10 mg jedenkrát denne. Postupné zvyšovanie dávkovania
atorvastatínu je možné prispôsobiť
na základe klinickej odpovede.
Ak je potrebné súčasné podávanie pravastatínu s darunavirom užívaným s nízkou dávkou ritonaviru, odporúča sa začať
s najnižšou možnou dávkou pravastatínu a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
Ak je potrebné súčasné podávanie rosuvastatínu s darunavirom užívaným s nízkou dávkou ritonaviru, odporúča sa začať
s najnižšou možnou dávkou rosuvastatín a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
I
NHIBÍTORY H
2
-
RECEPTORA
Ranitidín
150 mg dvakrát denne
IMUNOSUPRESÍVA Ciklosporín Sirolimus Takrolimus
Everolimus
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Neskúmalo sa. Pri súčasnom podávaní darunaviru užívaného s nízkou dávkou ritonaviru s týmito imunosupresívami sa zvýši expozícia voči týmto látkam. (inhibícia CYP3A)
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s inhibítormi H2-receptora sa môže užívať bez úpravy dávky.
Pri súčasnom podaní imunosupresív sa musia monitorovať terapeutické hladiny týchto liekov.
Súčasné užívanie everolimu a darunaviru užívaného s nízkou dávkou ritonaviru sa neodporúča.
I
NHALAČNÉ BETA-AGONISTY
Salmeterol Neskúmalo sa. Súčasné užívanie
salmeterolu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť
plazmatické koncentrácie salmeterolu.
Súčasné užívanie salmeterolu
a darunaviru užívanej s nízkou dávkou ritonaviru sa neodporúča. Táto kombinácia môže viesť
k zvýšenému riziku kardiovaskulárnych nežiaducich udalostí salmeterolu, vrátane predĺženia QT intervalu, palpitácií a sínusovej tachykardie.
NARKOTICKÉ ANALGETIKÁ / LIEČBA ZÁVISLOSTI NA OPIOIDOCH
Metadon individuálna dávka
v rozsahu od 55 mg do
150 mg jedenkrát denne
Buprenorfín/naloxón
8/2 mg–16/4 mg jedenkrát denne
R(-) metadon AUC ↓ 16 % R(-) metadon Cmin ↓ 15 % R(-) metadon Cmax ↓ 24 %
buprenorfín AUC ↓ 11 % buprenorfín Cmin ↔ buprenorfín Cmax ↓ 8 % norbuprenorfín AUC ↑ 46 % norbuprenorfín Cmin ↑ 71 % norbuprenorfín Cmax ↑ 36 % naloxón AUC ↔
naloxón Cmin ND
naloxón Cmax ↔
Na začiatku užívania spolu
s darunavirom/ritonavirom nie je potrebná úprava dávky metadonu. Avšak zvýšené dávky metadonu môžu byť potrebné kvôli indukcii
metabolizmu ritonavirom pri
súčasnom podávaní počas dlhšieho časového obdobia. Z toho dôvodu
sa odporúča klinické monitorovanie, pretože u niektorých pacientov
môže byť potrebná úprava udržiavacej liečby.
Klinický význam zvýšenia farmakokinetických parametrov norbuprenorfínu nebol stanovený. Úprava dávky buprenorfínu nemusí byť potrebná, ak sa podáva spolu
s darunavirom/ritonavirom, ale odporúča sa pozorné sledovanie prejavov toxicity opiátov.
ANTIKONCEPCIA NA BÁZE ESTROGÉNOV
Etinylestradiol
Noretisterón
35 mg/1 mg jedenkrát denne
etinylestradiol AUC ↓ 44 % etinylestradiol Cmin ↓ 62 % etinylestradiol Cmax ↓ 32 % noretisterón AUC ↓ 14 % noretisterón Cmin ↓ 30 % noretisterón Cmax ↔
Pacientkam užívajúcim antikoncepciu na báze estrogénov v kombinácii
s darunaviroma s nízkou dávkou ritonaviru sa odporúča zmeniť resp. rozšíriť formy antikoncepcie. U pacientok užívajúcich estrogén ako hormonálnu substitúciu majú byť klinicky sledované prejavy deficitu estrogénu.
FOSFODIESTERÁZA, INHIBÍTORY TYPU 5 (PDE-5)
Pri liečbe erektilnej
dysfunkcie Avanafil Sildenafil Tadalafil Vardenafil
Pri liečbe pulmonálnej
arteriálnej hypertenzie
Sildenafil
Tadalafil
V štúdii skúmajúcej interakcie# sa pozorovala porovnateľná systémová expozícia voči sildenafilu po jednorazovom podaní samotného sildenafilu v dávke 100 mg a po jednorazovom podaní sildenafilu v dávke
25 mg v kombinácii s darunavirom a nízkou dávkou ritonaviru.
Neskúmalo sa. Súčasné užívanie sildenafilu alebo tadalafilu pri liečbe pulmonálnej arteriálnej hypertenzie a darunaviru užívaného s nízkou dávkou ritonaviru môže zvýšiť plazmatické koncentrácie sildenafilu alebo tadalafilu.
Kombinácia avanafilu a darunaviru s nízkou dávkou ritonaviru je kontraindikovaná (pozri časť 4.3). Pri súčasnom podávaní iných inhibítorov PDE-5 na liečbu erektilnej dysfunkcie
s darunavirom, užívaným s nízkou dávkou ritonaviru, je potrebná opatrnosť. Ak je indikované súčasné podávanie darunaviru
a nízkej dávky ritonaviru so sildenafilom, vardenafilom alebo tadalafilom, jednorazová dávka sildenafilu nesmie prekročiť 25 mg za obdobie 48 hodín, resp. jednorazová dávka vardenafilu nesmie prekročiť 2,5 mg za obdobie
72 hodín a jednorazová dávka tadalafilu nesmie presiahnuť 10 mg za obdobie 72 hodín.
Bezpečná a účinná dávka sildenafilu pri liečbe pulmonálnej arteriálnej hypertenzie a pri súčasnom užívaní darunaviru a nízkej dávky ritonaviru nebola stanovená.
Existuje zvýšená možnosť vzniku nežiaducich udalostí súvisiacich so sildenafilom (vrátane porúch zraku,
hypotenzie, predĺženej erekcie a synkopy). Preto je súčasné užívanie darunaviru s nízkou
dávkou ritonaviru a sildenafilu, ak
sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, kontraindikované (pozri časť 4.3). Pri liečbe pulmonálnej arteriálnej hypertenzie sa súčasné užívanie tadalafilu s darunavirom a nízkou dávkou ritonaviru neodporúča.
I
NHIBÍTORY PROTÓNOVEJ PUMPY
Omeprazol
20 mg jedenkrát denne
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Darunavir užívaný s nízkou dávkou ritonaviru je možné súčasne podávať s inhibítormi protónovej pumpy bez nutnosti úpravy dávkovania.
SEDATÍVA/HYPNOTIKÁ
Buspirón Klorazepát Diazepam Estazolam Flurazepam Triazolam Zolpidem
Midazolam
Neskúmalo sa. Sedatíva/hypnotiká sú výrazne metabolizované prostredníctvom CYP3A. Súčasné užívanie s darunavirom
/ritonavirom môže spôsobiť významné zvýšenie koncentrácií týchto liekov.
Na základe údajov o iných inhibítoroch CYP3A sa očakáva, že plazmatické koncentrácie midazolamu budú výrazne vyššie, keď sa midazolam podáva perorálne s darunavirom užívaným
s nízkou dávkou ritonaviru.
Ak sa parenterálny midazolam podáva súčasne s darunavirom užívaným s nízkou dávkou ritonaviru, môže spôsobiť významné zvýšenie koncentrácie tohto benzodiazepínu. Údaje o súčasnom užívaní parenterálneho midazolamu s inými proteázovými inhibítormi naznačujú
možné 3-4-násobné zvýšenie
plazmatických hladín midazolamu.
Pri súčasnom užívaní darunaviru
s týmito sedatívami/hypnotikami sa odporúča klinické sledovanie a má sa zvážiť zníženie dávky sedatív/hypnotík. Súčasné užívanie darunaviru užívaného s nízkou dávkou ritonaviru a triazolamu je
kontraindikované.
Užívanie darunaviru s nízkou dávkou ritonaviru v kombinácii s perorálne podávaným
midazolamom je kontraindikované (pozri časť 4.3); pričom sa odporúča opatrnosť pri súčasnom užívaní darunaviru s nízkou dávkou ritonaviru a parenterálneho midazolamu.
Ak sa parenterálny midazolam podáva súčasne s darunavirom užívaným s nízkou dávkou ritonaviru, má sa tak uskutočniť na jednotke intenzívnej starostlivosti (JIS) alebo v podobnom zariadení, ktoré zabezpečuje dôkladné klinické sledovanie a vhodnú lekársku starostlivosť v prípade respiračného útlmu a/alebo predĺženého útlmu. Treba zvážiť úpravu dávkovania midazolamu, najmä ak sa podáva viac ako jednorazová dávka.
† Účinnosť a bezpečnosť používania darunaviru so 100 mg ritonaviru a s niektorým iným HIV PI (napr. (fos)amprenavir, nelfinavir a tipranavir) neboli u pacientov s HIV stanovené. Podľa súčasných liečebných postupov sa vo všeobecnosti duálna liečba proteázovými inhibítormi neodporúča.
4.6 Fertilita, gravidita a laktáciaGraviditaVo všeobecnosti, keď sa rozhoduje o použití antiretrovirotík na liečbu infekcie HIV u gravidných žien a následne na zníženie rizika vertikálneho prenosu HIV na novorodenca, treba vziať do úvahy údaje
o zvieratách ako aj klinické skúsenosti s gravidnými ženami.
S darunavirom sa nevykonali žiadne primerané ani dobre kontrolované štúdie o vplyve na graviditu u gravidných žien. Štúdie na zvieratách nepreukázali priame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3).
Darunavir užívaný s nízkou dávkou ritonaviru sa môže podávať gravidným ženám len vtedy, ak možný prínos liečby prevýši možné riziká.
DojčenieNie je známe, či sa darunavir vylučuje do ľudského mlieka. V štúdiách na potkanoch sa preukázalo vylučovanie darunaviru do mlieka a pri vysokých hladinách (1000 mg/kg/deň) spôsobilo toxicitu.
Matky majú byť poučené, aby za žiadnych okolností nedojčili počas liečby darunavirom jednak kvôli
možnosti prenosu HIV a aj možnosti výskytu nežiaducich reakcií u dojčených detí.
FertilitaNie sú k dispozícii žiadne údaje o účinku darunaviru na fertilitu u ľudí. Pri liečbe potkanov
darunavirom nemal darunavir vplyv na párenie a fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Darunavir spolu s ritonavirom nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U niektorých pacientov sa však počas liečby darunavirom užívaným s nízkou dávkou ritonaviru vyskytli závraty. Na túto možnosť je potrebné myslieť pri posudzovaní schopnosti pacienta viesť vozidlá a obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Počas programu klinického vývoja (N=2 613 predtým liečených jedincov, ktorí začali liečbu
s darunavirom/ritonavirom 600/100 mg dvakrát denne), 51,3 % jedincovmalo aspoň jednu nežiaducu reakciu. Celkové priemerné trvanie liečby pacientov bolo 95,3 týždňov. Najčastejšími nežiaducimi
reakciami hlásenými v klinických skúšaniach a v spontánnych hláseniach sú hnačka,
nauzea, vyrážka, bolesť hlavy a vracanie. Najčastejšími závažnými reakciami sú akútne renálne
zlyhanie, infarkt myokardu, imunoreštitučný zápalový syndróm, trombocytopénia, osteonekróza,
hnačka, hepatitída a pyrexia.
V analýze po 96 týždňoch bol bezpečnostný profil darunaviru/ritonaviru 800/100 mg jedenkrát denne u jedincov, ktorí doteraz neboli liečení, podobný ako u darunaviru /ritonaviru 600/100 mg dvakrát denne u jedincov, ktorí boli predtým liečení, až na nauzeu, ktorá sa častejšie vyskytovala u doteraz neliečených jedincov. Nauzea bola miernej intenzity. V analýze po 192 týždňoch sa u doteraz neliečených pacientov, u ktorých liečba darunavirom/ritonavirom 800/100 mg jedenkrát denne trvala priemerne 162,5 týždňa, neidentifikovali žiadne nové nálezy týkajúce sa bezpečnosti.
Zoznam nežiaducichreakciívtabuľkách
Nežiaduce reakcie sa uvádzajú podľa postihnutia triedy orgánových systémov a podľa skupiny frekvencie. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí
klesajúcej závažnosti. Skupiny frekvencií sú definované nasledovne: veľmi časté (≥ 1/10), časté
(≥ 1/100 až < 1/10), menej časté (≥ 1/1000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000)
a neznáme (z dostupných údajov).
Nežiaduce reakcie v klinických skúšaniach a v postmarketingovom sledovaní
Trieda orgánových systémov podľa databázy
MedDRA
Kategória frekvencie
Infekcie a nákazy
Nežiaduca reakcia
menej časté herpes simplex
Poruchy krvi a lymfatického systémumenej časté trombocytopénia, neutropénia, anémia, leukopénia
zriedkavé zvýšenie počtu eozinofilov
Poruchy imunitného systémumenej časté imunoreštitučný zápalový syndróm, precitlivenosť
(na liek)
Poruchy endokrinného systémumenej časté hypotyreóza, zvýšenie hladiny hormónu stimulujúceho štítnu žľazu v krvi
Poruchy metabolizmu a výživyčasté diabetes mellitus, hypertriglyceridémia, hypercholesterolémia, hyperlipidémia
menej časté dna, anorexia, znížená chuť do jedla, zníženie hmotnosti, zvýšenie hmotnosti, hyperglykémia, rezistencia na inzulín, pokles lipoproteínu
s vysokou hustotou, zvýšená chuť do jedla, polydipsia, zvýšená hladina LDH v krvi
Psychické poruchyčasté insomnia
menej časté depresia, dezorientácia, úzkosť, poruchy spánku,
neprirodzené sny, nočné mory, pokles libida
zriedkavé stav zmätenosti, zmeny nálady, nepokoj
Poruchy nervového systémučasté bolesti hlavy, periférna neuropatia, závraty
menej časté letargia, parestézia, hypoestézia, dysgeúzia,
porucha pozornosti, poškodenie pamäti,
somnolencia
zriedkavé synkopa, konvulzia, ageuzia, porucha fázy spánkového rytmu
Poruchy okamenej časté konjunktiválna hyperémia, suché oči
zriedkavé porucha zraku
Poruchy ucha a labyrintumenej časté vertigo
Poruchy srdca a srdcovej činnostimenej časté infarkt myokardu, angína pektoris, predĺžený QT
elektrokardiogram, tachykardia
zriedkavé akútny infarkt myokardu, sínusová bradykardia,
palpitácie
Poruchy cievmenej časté hypertenzia, návaly horúčavy
Poruchy dýchacej sústavy, hrudníka a mediastínamenej časté dyspnoe, kašeľ, epistaxa, podráždenie hrdla
zriedkavé rinorea
Poruchy gastrointestinálneho traktuveľmi časté diarea
časté vracanie, nauzea, bolesti brucha, zvýšená hladina
amylázy v krvi, dyspepsia, abdominálna distenzia, flatulencia
menej časté pankreatitída, gastritída, gastroezofagálny reflux, aftózna stomatitída, napínanie na vracanie, sucho
v ústach, abdominálny diskomfort, zápcha, zvýšená
hladina lipázy, eruktácia, perorálna dyzestézia
zriedkavé stomatitída, hemateméza, cheilitída, suché pery, povlak na jazyku
Poruchy pečene a žlčových ciestčasté zvýšenie alanínaminotransferázy
menej časté hepatitída, cytolytická hepatitída, steatóza pečene, hepatomegália, zvýšenie transamináz, zvýšenie aspartátaminotransferázy, zvýšenie hladiny bilirubínu v krvi, zvýšenie alkalickej fosfatázy
v krvi, zvýšenie gamaglutamyltransferázy
Poruchy kože a podkožného tkanivačasté vyrážka (vrátane makulárnej, makulopapulárnej, papulárnej, erytematóznej a pruritickej vyrážky), pruritus
menej časté angioedém, generalizovaná vyrážka, alergická
dermatitída, urtikária, ekzém, erytém,
hyperhidróza, nočné potenie, alopécia, akné, suchá
koža, sfarbenie nechtov
zriedkavé DRESS, Stevensov-Johnsonov syndróm, multiformný erytém, dermatitída, seboroická dermatitída, kožné lézie, xeroderma
neznáme toxická epidermálna nekrolýza, akútna generalizovaná exantematózna pustulóza
Poruchy kostrovej a svalovej sústavy a spojivového tkanivamenej časté myalgia, osteonekróza, svalové kŕče, svalová slabosť, artralgia, bolesti končatín, osteoporóza, zvýšená hladina kreatínfosfokinázy v krvi
zriedkavé muskuloskeletálna stuhnutosť, artritída, stuhnutie
kĺbov
Poruchy obličiek a močových ciestmenej časté akútne zlyhanie obličiek, zlyhanie obličiek, nefrolitiáza, zvýšená hladina kreatinínu v krvi, proteinúria, bilirubinúria, dyzúria, noktúria, polakizúria
zriedkavé znížený renálny klírens kreatinínu
Poruchy reprodukčného systému a prsníkovmenej časté erektilná dysfunkcia, gynekomastia
Celkové poruchy a reakcie v mieste podaniačasté asténia, únava
menej časté pyrexia, bolesť na hrudi, periférne opuchy,
nevoľnosť, pocit horúčavy, podráždenie, bolesť
zriedkavé triaška, abnormálne pocity, xeróza
OpisvybranýchnežiaducichreakciíVyrážkaV klinických skúšaniach bola vyrážka najčastejšie miene až stredne závažná, často sa vyskytla v prvých štyroch týždňoch liečby a ustúpila pri ďalšom užívaní. Pre prípady závažných kožných reakcií pozri upozornenie v časti 4.4.
Počas programu klinického vývoja raltegraviru sa u predtým liečených pacientov častejšie pozorovala vyrážka, bez ohľadu na kauzalitu, pri režimoch obsahujúcich darunavir + raltegravir v porovnaní
s režimami obsahujúcimi darunavir bez raltegraviru alebo raltegravir bez darunaviru. Vyrážka
považovaná skúšajúcim za súvisiacu s liekom, sa vyskytla v podobnej miere. Expozícii prispôsobené
miery vyrážky (všetky príčiny) boli 10,9; 4,2 a 3,8 na 100 pacientorokov v danom poradí a u vyrážky
súvisiacej s liekom boli 2,4; 1,1 a 2,3 na 100 pacientorokov v danom poradí. Vyrážky pozorované
v klinických štúdiách boli mierne až stredne závažné a neviedli k prerušeniu liečby (pozri časť 4.4).
Metabolické parametrePočas antiretrovírusovej liečby sa môže zvýšiť telesná hmotnosť a hladiny lipidov a glukózy v krvi
(pozri časť 4.4).
Poruchy kostrovej a svalovej sústavyPočas liečby inhibítormi proteáz, najmä v kombinácii s NRTI, sa vyskytli prípady zvýšenia hladiny
kreatínfosfokinázy, myalgie, myozitídy a zriedkavo aj rabdomyolýzy.
Boli hlásené prípady osteonekrózy, najmä u pacientov so všeobecne potvrdenými rizikovými faktormi, pokročilým ochorením HIV alebo dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART). Frekvencia nie je známa (pozri časť 4.4).
Imunoreštitučný zápalový syndrómU pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej
antiretrovírusovej liečby (CART) môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie. Boli tiež zaznamenané aj autoimunitné poruchy (ako je Gravesova
choroba); avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť
mnoho mesiacov po začatí liečby (pozri časť 4.4).
Krvácanie u pacientov s hemofíliouU pacientov s hemofíliou dostávajúcich antiretrovirálne inhibítory proteázy boli hlásené prípady
zvýšeného spontánneho krvácania (pozri časť 4.4).
Pediatrická populáciaPosúdenie bezpečnosti u pediatrických pacientov je založené na 48-týždňovej analýze údajov
o bezpečnosti z troch klinických skúšaní fázy II. Boli hodnotené nasledovné populácie pacientov
(pozri časť 5.1):
● 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg, ktorí užívali darunavir tablety s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku od 3 do < 6
rokov a s hmotnosťou 10 kg až < 20 kg (16 účastníkov od 15 kg do < 20 kg), ktorí užívali
darunavir perorálnu suspenziu s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 12 pediatrických pacientov infikovaných HIV-1 vo veku od 12 do 17 rokov a s hmotnosťou
najmenej 40 kg, ktorí neboli predtým liečení ART. Títo pacienti dostávali darunavir tablety
s nízkou dávkou ritonaviru jedenkrát denne spolu s inými antiretrovírusovými liekmi. (pozri
časť 5.1).
Celkovo bol bezpečnostný profil u týchto pediatrických pacientov podobný bezpečnostnému profilu
pozorovanému u dospelých.
Iné osobitné skupiny pacientovPacienti súčasne infikovaní vírusom hepatitídy B a/alebo hepatitídy CSpomedzi 1968 predtým liečených pacientov, ktorí užívali darunavir spolu s ritonavirom 600/100 mg dvakrát denne, bolo 236 pacientov infikovaných zároveň hepatitídou B alebo C. U súčasne infikovaných pacientov bola vyššia pravdepodobnosť počiatočného a pri liečbe vznikajúceho zvýšenia hladiny pečeňových transamináz ako u pacientov bez chronickej vírusovej hepatitídy (pozri časť 4.4).
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Skúsenosti s akútnym predávkovaním darunavirom užívaným s nízkou dávkou ritonaviru u ľudí sú obmedzené. Zdravým dobrovoľníkom sa darunavir podával vo forme perorálneho roztoku
v jednorazových dávkach do 3200 mg ako samotný, resp. vo forme tabliet v jednorazových dávkach
do 1600 mg v kombinácii s ritonavirom bez toho, že by sa u nich pozorovali nepriaznivé symptomatické účinky.
V prípade predávkovania darunavirom nie je k dispozícii žiadne špecifické antidotum. Liečba predávkovania darunavirom zahŕňa všeobecné podporné opatrenia, vrátane sledovania vitálnych znakov a pozorovania klinického stavu pacienta. Ak je to indikované, eliminácia nevstrebanej aktívnej látky sa dosiahne pomocou vyvolania vracania.
Aj podanie aktívneho uhlia môže pomôcť pri eliminácii nevstrebanej aktívnej látky. Vzhľadom na vysokú väzbu darunaviru na plazmatické bielkoviny je nepravdepodobné, že by dialýza mala väčší význam pri eliminácii aktívnej látky z cirkulácie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antivirotiká na systémové použitie, inhibítory proteázy, ATC kód: J05AE10
Mechanizmus účinku
Darunavir je inhibítor dimerizácie a katalytickej aktivity HIV-1 proteázy (KD 4,5 x 10-12M). Selektívne
inhibuje odštiepenie HIV kódovaných polyproteínov Gag-Pol v bunkách infikovaných vírusom, čím zabraňuje tvorbe zrelých infekčných vírusových častíc.
Antivírusová aktivita in vitro
Darunavir vykazuje aktivitu proti laboratórnym kmeňom a klinickým izolátom HIV-1 a laboratórnym
kmeňom HIV-2 v akútne infikovaných líniách T-buniek, mononukleárnych bunkách z periférnej krvi
človeka a ľudských monocytov/makrofágov so strednými hodnotami EC50 v rozmedzí
1,2 až 8,5 nmol/l (0,7 až 5,0 ng/ml). In vitro má darunavir antivírusovú aktivitu voči širokému spektru
primárnych izolátov zo skupiny M HIV-1 (A, B, C, D, E, F, G) a skupiny O s hodnotami EC50
v rozmedzí od < 0,1 do 4,3 nmol/l.
Tieto hodnoty EC50 sú výrazne nižšie ako je rozsah toxickej koncentrácie pre 50 % buniek od
87 µmol/l do > 100 µmol/l.
Rezistencia
Selekcia vírusu rezistentného na darunavir z divokého typu HIV-1 in vitro trvala pomerne dlho
(> 3 roky). Vyselektované vírusy neboli schopné rásť v prítomnosti darunaviru v koncentráciách nad
400 nmol/l. U vírusov vyselektovaných v týchto podmienkach a vykazujúcich zníženú citlivosť voči
darunaviru (rozmedzie 23 až 50-násobne) sa v géne pre proteázu zistili substitúcie 2 až 4 aminokyselín. Zníženú citlivosť vznikajúcich vírusov na darunavir v selekcii nie je možné vysvetliť vznikom týchto mutácií proteázy.
Údaje z klinických štúdií s pacientmi liečenými ART (štúdia TITAN a zlúčená analýza štúdií POWER
1, 2 a 3 a štúdií DUET 1 a 2) preukázali, že virologická odpoveď na darunavir užívaný s nízkou dávkou ritonaviru bola znížená, keď boli na začiatku prítomné 3 a viac mutácií spojených s rezistenciou voči darunaviru (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V a L89V) alebo keď sa tieto mutácie vyvinuli počas liečby.
Zvýšenie východiskovej násobnej zmeny darunaviru v EC50 (FC) súviselo s poklesom virologickej odpovede. Určila sa dolná a horná klinická hranica 10 a 40. Izoláty s východiskovou hodnotou FC
≤ 10 sú citlivé; izoláty s FC > 10 až 40 majú zníženú citlivosť; izoláty s FC > 40 sú rezistentné (pozri
Klinické výsledky).
Vírusy izolované u pacientov s dávkou darunavir/ritonavir 600/100 mg dvakrát denne, u ktorých sa vyskytlo spontánne virologické zlyhanie, ktoré boli citlivé na tipranavir na začiatku, zostali vo veľkej väčšine prípadov citlivé na tipranavir po liečbe.
Najnižšie miery rozvoja rezistencie vírusu HIV sú pozorované u pacientov doposiaľ neliečených ART, ktorí sú po prvýkrát liečení darunavirom v kombinácii s inou ART.
Tabuľka nižšie zobrazuje vývoj mutácií a stratu citlivosti na PI pri virologickom zlyhaní v závere
liečby v štúdiách ARTEMIS, ODIN a TITAN.
ARTEMIS ODIN TITAN
Darunavir/
ritonavir
800/100 mg jedenkrát denne N=343
Darunavir/
ritonavir
800/100 mg jedenkrát denne N=294
Darunavir/
ritonavir
600/100 mg dvakrát denne N=296
Darunavir/
ritonavir
600/100 mg dvakrát denne N=298
Celkový počet virologických zlyhanía, n (%)
55 (16,0 %) 65 (22,1 %) 54 (18,2 %) 31 (10,4 %)
Spontánne 39 (11,4 %) 11 (3,7 %) 11 (3,7 %) 16 (5,4 %)
Jedinci nereagujúci na
liečbu
16 (4,7 %) 54 (18,4 %) 43 (14,5 %) 15 (5,0 %)
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými genotypmi, u ktorých sa vyvinuli mutácieb v závere liečby, n/N
Primárne (významné)
mutácie PI
0/43 1/60 0/42 6/28
PI RAM 4/43 7/60 4/42 10/28
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými genotypmi, u ktorých sa preukázala strata citlivosti na PI v závere liečby v porovnaní so začiatkom liečby, n/N
PI
darunavir 0/39 1/58 0/41 3/26 amprenavir 0/39 1/58 0/40 0/22 atazanavir 0/39 2/56 0/40 0/22 indinavir 0/39 2/57 0/40 1/24 lopinavir 0/39 1/58 0/40 0/23 sakvinavir 0/39 0/56 0/40 0/22 tipranavir 0/39 0/58 0/41 1/25
a TLOVR ne-VF cenzurovaný algoritmus založený na HIV-1 RNA < 50 kópií/ml, okrem
TITAN (HIV-1 RNA
< 400 kópií/ml)
b zoznamy IAS-USA
SkríženárezistenciaNásobná zmena (FC) darunaviru bola menej ako 10 pre 90 % z 3309 klinických izolátov rezistentných
voči amprenaviru, atazanaviru, indinaviru, lopinaviru, nelfinaviru, ritonaviru, sakvinaviru a/alebo tipraniviru, čo naznačuje, že vírusy rezistentné voči väčšine PI zostávajú citlivé voči darunaviru.
V štúdii
ARTEMIS sa pri virologických zlyhaniach nepozorovala žiadna skrížená rezistencia s inými
PI.
Klinické výsledkyDospelí pacienti
Výsledky klinických štúdií u dospelých pacientov doposiaľ neliečených ART, pozri Súhrn
charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Úči nnosť darunaviru 600 mg dvakrát denne užívanej so 100 mg ritonaviru dvakrát denne u pacientov
pre dtým li ečený ch ART
Údaje o účinnosti kombinácie darunavir a ritonavir (600/100 mg dvakrát denne) u pacientov predtým
liečených ART sú založené na analýze 96 týždňovej štúdie fázy III TITAN u pacientov predtým
liečených ART, ktorí ešte neužívali lopinavir, na analýze 48 týždňovej štúdie fázy III ODIN
u pacientov predtým liečených ART bez DRV-RAM a na analýze údajov z 96 týždňov trvajúcich štúdií fázy IIb POWER 1 a 2 u pacientov predtým liečených ART s vysokou rezistenciou na PI.
TITAN je randomizovaná, kontrolovaná, otvorená štúdia fázy III porovnávajúca darunavir podávaný spolu s ritonavirom (600/100 mg dvakrát denne) verzus lopinavir/ritonavir (400/100 mg dvakrát denne) u dospelých pacientov predtým liečených ART infikovaných HIV-1, ktorí ešte neužívali
lopinavir. Obe ramená používali optimalizovaný základný režim (OBR, z angl. Optimised Background
Regimen), ktorý pozostával z minimálne 2 antiretrovirotík (NRTI s alebo bez NNRTI).
Nižšie uvedená tabuľka uvádza údaje 48-týždňovej analýzy účinnosti zo štúdie TITAN.

TITAN
Výsledky darunavir/ritonavir
600/100 mg dvakrát denne + OBR N=298
lopinavir/ritonavir
400/100 mg dvakrát denne + OBR N=297
Rozdiel v liečbe
(95 % CI rozdielu)
HIV-1 RNA < 50 kópií/mla 70,8 % (211) 60,3 % (179) 10,5 % (2,9; 18,1)b
medián zmeny v počte buniek CD4+ oproti východiskovej hodnote (x 106/l)c
88 81
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede
c NC=F
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom,
definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA < 400 a < 50 kópií/ml, sa dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat
(ITT) aj u pacientov sledovaných v protokole (On Protocol). Tieto výsledky sa potvrdili v analýze
údajov z 96 týždňov trvajúcej liečby v štúdii TITAN, pričom v 96. týždni malo 60,4 % pacientov v ramene s darunavirom/ritonavirom HIV-1 RNA < 50 kópií/ml v porovnaní s 55,2 % pacientov v ramene s lopinavirom/ritonavirom [rozdiel: 5,2 %, 95 % CI (-2,8;13,1)].
ODIN je randomizovaná otvorená štúdia fázy III porovnávajúca darunavir/ritonavir 800/100 mg jedenkrát denne a darunavir/ritonavir 600/100 mg dvakrát denne u pacientov infikovaných HIV-1 predtým liečených ART, u ktorých skríningové vyšetrenie rezistencie genotypu nepreukázalo RAM voči darunaviru (t.j. V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V, L89V)
a skríningové vyšetrenie HIV-1 RNA bolo > 1 000 kópií/ml. Analýza účinnosti je založená na liečbe trvajúcej 48 týždňov (pozri tabuľku nižšie). Obe ramená použili optimalizovaný základný režim (OBR) ≥ 2 NRTI.
Výsledky Darunavir/ritonavir
800/100 mg jedenkrát denne + OBR
N=294
ODIN
Darunavir/ritonavir
600/100 mg dvakrát denne + OBR N=296
Rozdiel v liečbe
(95% CI rozdielu)
HIV-1 RNA
< 50 kópií/mla
S východiskovou HIV-1 RNA (kópie/ml)
72,1 % (212) 70,9 % (210) 1,2 % (-6,1; 8,5)b
< 100 000
≥ 100 000
77,6 % (198/255)
35,9 % (14/39)
73,2 % (194/265)
51 6 % (16/31)
4,4 % (-3,0; 11,9)
-15,7 % (-39,2; 7,7)
S východiskovým počtom buniek CD4+ (x 106/l)
≥ 100
< 100
S kmeňom HIV-1
Typ B Typ AE Typ C Inéc
75,1 % (184/245)
57,1 % (28/49)
70,4 % (126/179)
90,5 % (38/42)
72,7 % (32/44)
55,2 % (16/29)
72,5 % (187/258)
60,5 % (23/38)
64,3 % (128/199)
91,2 % (31/34)
78,8 % (26/33)
83,3 % (25/30)
2,6 % (-5,1; 10,3)
-3,4 % (-24,5; 17,8)
6,1 % (-3,4; 15,6)
-0,7 % (-14,0; 12,6)
-6,1 % (-2,6; 13,7)
-28,2 % (-51,0; -5,3)
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x 106/l)e
108 112 -5d (-25; 16)
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede c Kmene A1, D, F1, G, K, CRF02_AG, CRF12_BF, a CRF06_CPX d Rozdiel stredných hodnôt
e Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward)
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom
800/100 mg jedenkrát denne, definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA
< 50 kópií/ml v porovnaní s darunavirom/ritonavirom 600/100 mg dvakrát denne, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat (ITT) aj u pacientov sledovaných v protokole (On Protocol).
Darunavir/ritonavir 800/100 mg jedenkrát denne sa nemá používať u tých pacientov liečených ART,
ktorí majú jednu alebo viac mutácií spojených s rezistenciou voči darunaviru (DRV-RAM) alebo
HIV-1 RNA ≥ 100 000 kópií/ml alebo počet buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2 a 4.4).
U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje.
POWER 1 a POWER 2 sú randomizované, kontrolované štúdie porovnávajúce darunavir užívaný spolu s ritonavirom (600/100 mg dvakrát denne) s kontrolnou skupinou užívajúcou inhibítor proteáz vybraný skúšajúcim u acientov infikovaných HIV-1, ktorí predtým absolvovali viac ako jeden neúspešný liečebný režim zahŕňajúci inhibítor proteáz (PI). V oboch štúdiách bol použitý OBR zahŕňajúci aspoň 2 NRTI s enfuvirtidom (ENF) alebo bez neho.
V nasledujúcej tabuľke sú uvedené údaje 48-týždňovej a 96-týždňovej analýzy účinnosti zo zlúčených štúdií POWER 1 a POWER 2.
Spoločné údaje získané v štúdiách POWER 1 a POWER 2
48. týždeň 96. týždeň
Výsledky Darunavir/ritonavir
600/100 mg 2x denne
n=131
Kontrola n=124
Rozdiel medzi liečbami
Darunavir/ritonavir
600/100 mg 2x denne
n=131
Kontrola n=124
Rozdiel medzi liečbami
HIV RNA
< 50 kópií/
mla
45,0 % (59) 11,3 % (14)
33,7 % (23,4 %;
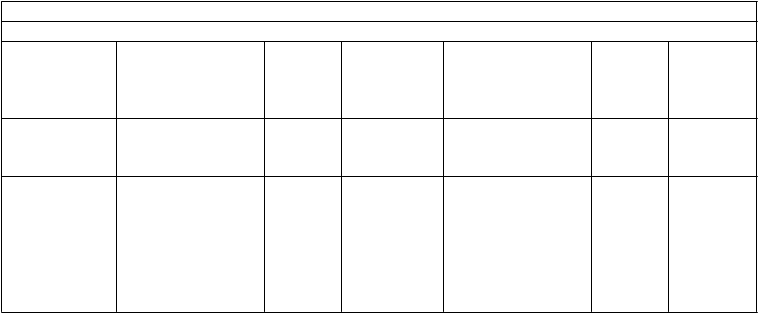
44,1 %)c
38,9 % (51) 8,9 % (11)
30,1 % (20,1;
40,0)c
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x
106/l)b
103 17 86
(57; 114)c
133 15 118 (83,9;
153,4)c
a Výpočet na základe algoritmu TLOVR.
b Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward).
c 95 % interval spoľahlivosti.
Analýza údajov z 96 týždňov trvajúcej liečby v štúdiách POWER preukázala zachovanú
antiretrovírusovú účinnosť a imunologický benefit.
Z 59 pacientov, ktorí reagovali na liečbu úplnou supresiou vírusu (< 50 kópií/ml) v 48. týždni,
47 pacientov (80 % respondérov na liečbu v 48. týždni) naďalej reagovalo na liečbu v 96. týždni.
Východiskový genotyp alebo fenotyp a virologický výsledok
Východiskový genotyp a FC darunaviru (zmena citlivosti vzhľadom k východiskovej hodnote) sa ukázal ako prediktívny faktor virologického výsledku.
Podiel (%) pacientov s odpoveďou (HIV-1 RNA < 50 kópii/ml v týždni 24) na darunaviružívaný spolu s ritonavirom (600/100 mg dvakrát denne) u východiskového genotypu*a a východiskovej FC darunaviru pri užívaní enfuvirtidu (ENF): podľa analýz liečby štúdií POWER a DUET.
Odpoveď (HIV-1
Počet východiskových mutáciía Východisková DRV FCb
RNA < 50 kópií/ml v 24. týždni)
%, n/N
Všetky
rozpätia 0-2 3 ³ 4
Všetky
rozpätia £ 10 10-40 > 40
Všetci pacienti 45 %
455/1 014
54 %
359/660
39 %
67/172
12 %
20/171
45 %
455/1 014
55 %
364/659
29 %
59/203
8 %
9/118
Pacienti neužívajúci/opako- vane užívajúci ENFc Pacienti s de novo ENFd
39 %
290/741
60 %
165/273
50 %
238/477
66 %
121/183
29 %
35/120
62 %
32/52
7 %
10/135
28 %
10/36
39 %
290/741
60 %
165/273
51 %
244/477
66 %
120/182
17 %
25/147
61 %
34/56
5 %
5/94
17 %
4/24
a Počet mutácií zo zoznamu mutácií spojených so znížením odpovede na kombináciu darunavir/ritonavir (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V alebo L89V)
b násobná zmena v EC50
c “Pacienti neužívajúci/opakovane užívajúci ENF”, t.j. pacienti, ktorí neužívali ENF alebo tí, ktorí ENF užívali, ale nie
prvýkrát.
d “Pacienti s de novo ENF”, t.j. pacienti, ktorí užívali ENF prvýkrát.
Pediatrickí pacienti
Výsledky klinických štúdií u pediatrických pacientov vo veku 12 až 17 rokov, ktorí neboli predtým liečení ART, nájdete v Súhrne charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Pediatrickí pacienti od 6 do < 18 rokov a s hmotnosťou naj menej 20 kg dot eraz neli eč ení ART DELPHI je otvorená štúdia fázy II hodnotiaca farmakokinetiku, bezpečnosť, toleranciu a účinnosť darunaviru s nízkou dávkou ritonaviru u 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg. Títo pacienti užívali darunavir/ritonavir dvakrát denne spolu s inými antiretrovírusovými látkami (pre odporúčané dávkovanie v závislosti od hmotnosti pacienta pozri časť 4.2). Virologická odpoveď bola definovaná ako pokles vírusového zaťaženia HIV-1 RNA v plazme o najmenej 1,0 log10 oproti východiskovej hodnote.
V tejto štúdii bolo pacientom so zvýšeným rizikom prerušenia liečby z dôvodu intolerancie perorálneho roztoku ritonaviru (napr. averzia voči chuti) umožnené prejsť na užívanie kapsúl. Zo
44 pacientov užívajúcich perorálny roztok ritonaviru, 27 prešlo na užívanie 100 mg kapsúl
a prekročilo dávku ritonaviru odvodenú od hmotnosti bez zmien v sledovanej bezpečnosti.
DELPHI
Výsledky po 48. týždni Darunavir/ritonavir
N=80
HIV-1 RNA < 50 kópií/mla 47,5 % (38)
Priemerná zmena počtu buniek CD4+ oproti
východiskovej hodnoteb
147
a Výpočet na základe algoritmu TLOVR.
b Pacienti, ktorí neukončili liečbu, sú počítaní ako zlyhanie: pacientom, ktorí prerušili liečbu predčasne, sa priradí
zmena rovná nule.
Podľa cenzurovaného TLOVR algoritmu pre nevirologické zlyhanie u 24 (30,0 %) pacientov došlo
k virologickému zlyhaniu, z nich u 17 (21,3 %) pacientov sa objavil rebound fenomén a 7 (8,8 %)
pacientov neodpovedalo na liečbu.
Pedi at ri ckí paci ent i vo veku 3 až < 6 rokov predtým lieč ení ARTU 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART vo veku od 3 do < 6
rokov a s hmotnosťou 10 kg až < 20 kg bola farmakokinetika, bezpečnosť, tolerovateľnosť a účinnosť
darunaviru/ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami hodnotená v otvorenej štúdii fázy II
ARIEL. Pacienti dostávali dávku dvakrát denne v závislosti od ich
hmotnosti, pacienti s hmotnosťou 10 kg až < 15 kg dostávali darunavir/ritonavir 25/3 mg/kg dvakrát denne a pacienti s hmotnosťou 15 kg až < 20 kg dostávali darunavir/ritonavir 375/50 mg dvakrát
denne. Virologická odpoveď, definovaná ako percento pacientov s potvrdenou plazmatickou hladinou
< 50 HIV-1 RNA kópií/ml, bola v 48. týždni hodnotená u 16 pediatrických pacientov s hmotnosťou
15 kg až < 20 kg a u 5 pediatrických pacientov s hmotnosťou 10 kg až < 15 kg dostávajúcich darunavir/ritonavir v kombinácii s inými antiretrovírusovými látkami. (pozri časť 4.2 pre odporúčanie
dávkovania podľa telesnej hmotnosti).
ARIEL
Výsledky po 48. týždni Darunavir/ritonavir
10 kg až < 15 kg
N=5
15 kg až < 20 kg
N=16
HIV-1 RNA < 50 kópií/mla 80,0 % (4) 81,3 % (13)
CD4+ percentuálna zmena oproti východiskovej hodnoteb Priemerná zmena počtu buniek CD4+ oproti východiskovej hodnoteb
a Imputácie na základe algoritmu TLOVR.
b NC=F
4 4
16 241
Dostupné sú obmedzené údaje o účinnosti u pediatrických pacientov s hmotnosťou menej ako 15 kg,
a preto neumožňujú uviesť odporúčania pre dávkovanie.
Gravidita a obdobie po pôrode
Darunavir/ritonavir (600/100 mg dvakrát denne alebo 800/100 mg jedenkrát denne) v kombinácii so
základným režimom bol hodnotený v klinickej štúdii s 34 gravidnými ženami (17 v každej skupine) počas druhého a tretieho trimestra a po pôrode. Virologická odpoveď sa udržala počas trvania štúdie v oboch skupinách. U detí narodených 29 pacientkám, ktoré zotrvali na antivirotickej liečbe až do
pôrodu, sa nevyskytol prenos z matky na dieťa. Nevyskytli sa žiadne nové klinicky významné zistenia týkajúce sa bezpečnosti v porovnaní so známym profilom bezpečnosti darunaviru/ritonaviru
u dospelých infikovaných HIV-1 (pozri časti 4.2, 4.4 a 5.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti darunaviru podávaného v kombinácii s ritonavirom sa skúmali
u zdravých dospelých dobrovoľníkov a u pacientov infikovaných vírusom HIV-1. Expozícia voči darunaviru u pacientov infikovaných vírusom HIV-1 bola vyššia ako u zdravých dobrovoľníkov. Vyššiu expozíciu voči darunaviru u pacientov infikovaných vírusom HIV-1 v porovnaní so zdravými dobrovoľníkmi je možné vysvetliť na základe vyšších koncentrácií α1-kyslého glykoproteínu (AAG) u pacientov infikovaných vírusom HIV-1, čo vedie k vyššej väzbe darunaviru na plazmatický AAG,
a tým aj k vyšším koncentráciám tejto látky v plazme.
Darunavir sa metabolizuje najmä prostredníctvom CYP3A. Ritonavir inhibuje CYP3A, v dôsledku
čoho sa výrazne zvyšujú plazmatické koncentrácie darunaviru.
Absorpcia
Darunavir sa rýchlo vstrebáva po perorálnom podaní. Pri podávaní v kombinácii s ritonavirom
v nízkej dávke sa maximálne plazmatické koncentrácie darunaviru obyčajne dosiahnu v priebehu 2,5 -
4 hodín.
Absolútna biologická dostupnosť po jednorazovom perorálnom podaní samotného darunaviru v dávke
600 mg bola približne 37 %, pričom pri podávaní ritonaviru v dávke 100 mg dvakrát denne sa zvýšila približne na 82 %. Po jednorazovom perorálnom podaní darunaviru v dávke 600 mg v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne sa systémová expozícia voči darunaviru
zvýšila 14-násobne (pozri časť 4.4).
Pri užívaní nalačno bola relatívna biologická dostupnosť darunaviru v prítomnosti nízko-dávkovaného ritonaviru o 30 % nižšia ako pri užívaní lieku s jedlom. Z toho dôvodu sa tablety darunavir musia užívať spolu s ritonavirom a jedlom. Druh jedla nemá vplyv na expozíciu voči darunaviru.
Distribúcia
Približne 95 % darunaviru sa viaže na plazmatické bielkoviny, najmä na α1-kyslý glykoproteín.
Po intravenóznom podaní bol distribučný objem samotného darunaviru 88,1 ± 59,0 l (stredný ± SD)
a v prítomnosti 100 mg ritonaviru podávaného dvakrát denne bol zvýšený na 131 ± 49,9 l
(stredný ± SD).
Biotransformácia
In vitro štúdie využívajúce mikrozómy pečene človeka (HLM) naznačujú, že darunavir sa
metabolizuje najmä oxidatívnym metabolizmom. Darunavir sa výrazne metabolizuje prostredníctvom systému CYP v pečeni a takmer výlučne prostredníctvom izoenzýmu CYP3A4. V štúdii skúmajúcej aplikáciu darunaviru označeného rádioaktívnym 14C zdravým dobrovoľníkom sa zistilo, že väčšina rádioaktivity v plazme po jednorazovom podaní darunaviru s ritonavirom v dávke 400/100 mg pochádzala z pôvodného liečiva. U ľudí sa zistila prítomnosť najmenej 3 oxidatívnych metabolitov darunaviru, pričom všetky z nich mali aktivitu proti divokému typu HIV najmenej 10x nižšiu ako darunavir.
Eliminácia
Po podaní darunaviru s ritonavirom v dávke 400/100 mg označeného rádioaktívnym 14C sa 79,5 %
rádioaktivity zachytilo v stolici a 13,9 % rádioaktivity sa zachytilo v moči. Rádioaktivita
nezmeneného darunaviru v stolici predstavovala 41,2 % a v moči predstavovala 7,7 % z podanej
dávky. Terminálny polčas eliminácie darunaviru podávaného v kombinácii s ritonavirom bol približne
15 hodín.
Klírens darunaviru (150 mg) podaného intravenózne v monoterapii bol 32,8 l/h, kým pri kombinácii s nízko dávkovaným ritonavirom 5,9 l/h.
Osobitné skupiny pacientov
Pediatrická populácia
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 74 predtým liečených pediatrických pacientov vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg preukázala, že dávky darunaviru/ritonaviru podávané v závislosti od hmotnosti, mali za následok expozíciu darunaviru porovnateľnú s expozíciou u dospelých pacientov užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 14 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 15 kg až < 20 kg ukázala, že dávkovanie založené na hmotnosti viedlo k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 12 pediatrických pacientov vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení ART, ukázala, že dávka darunaviru/ritonaviru 800/100 mg jedenkrát denne viedla k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne. Z toho dôvodu sa rovnaké dávkovanie jedenkrát denne môže použiť u dospievajúcich vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg
s predchádzajúcou liečbou bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)*
a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+
≥ 100 buniek x 106/l (pozri časť 4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 10 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 14 kg až < 20 kg preukázala, že dávky odvodené od hmotnosti viedli k expozícii darunaviru, ktorá bola porovnateľná
s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne (pozri časť 4.2). Farmakokinetické modelovanie a simulácia expozície darunaviru u pediatrických pacientov vo veku 3 až < 18 rokov navyše potvrdili expozície darunaviru, ktoré boli pozorované v klinických štúdiách a umožnili určenie dávkovania darunaviru/ritonaviru jedenkrát denne v závislosti na hmotnosti u pediatrických pacientov s hmotnosťou najmenej 15 kg, ktorí buď predtým nedostávali liečbu ART alebo u predtým liečených pediatrických pacientov bez DRV-RAM* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri časť
4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V.
Starší ľudia
Pri analýze farmakokinetiky v rôznych populáciách pacientov s infekciou HIV sa zistilo, že farmakokinetika darunaviru sa výraznejšie nelíši u pacientov s infekciou HIV vo vekovom rozmedzí
od 18 do 75 rokov (n = 12, vek ≥ 65 rokov) (pozri časť 4.4). U pacientov starších ako 65 rokov boli
k dispozícii iba obmedzené údaje.
Pohlavie
Pri analýze farmakokinetiky v rôznych populáciách pacientov sa u žien s infekciou HIV zistila mierne vyššia expozícia voči darunaviru (o 16,8 %) ako u mužov infikovaných HIV. Tento rozdiel nemá žiadny klinický význam.
Porucha funkcie obličiek
Výsledky štúdie zachovania hmotnosti s 14C rádioaktívne označeným darunavirom s ritonavirom ukázali, že približne 7,7 % z podanej dávky darunaviru sa vylučuje do moču v nezmenenej forme.
Aj keď sa podávanie darunaviru nesledovalo u pacientov s poruchou funkcie obličiek, pri analýze farmakokinetiky v rôznych populáciách pacientov sa zistilo, že stredne závažná porucha funkcie obličiek (klírens kreatinínu: 30 - 60 ml/min, n=20) nemá väčší vplyv na farmakoniketiku darunaviru u pacientov s infekciou HIV (pozri časti 4.2 a 4.4).
Porucha funkcie pečene
Darunavir sa metabolizuje a eliminuje prevažne v pečeni. V štúdii s opakovaným podaním lieku darunavir užívaného s ritonavirom (600/100 mg dvakrát denne) sa preukázalo, že celkové
koncentrácie darunaviru v plazme u subjektov s miernou (Childova-Pughova trieda A, n=8) a stredne
ťažkou (Childova-Pughova trieda B, n=8) poruchou funkcie pečene boli porovnateľné s tými u zdravých subjektov. Koncentrácie voľného darunaviru boli približne o 55 % (Childova-Pughova Trieda A) a 100 % (Childova-Pughova trieda B) vyššie. Klinický význam tohto nárastu nie je známy,
preto treba darunavir užívať so zvýšenou opatrnosťou. Účinok ťažkej poruchy funkcie pečene na
farmakokinetiku darunaviru nebol dosiaľ študovaný (pozri časti 4.2, 4.3 a 4.4).
Gravidita a obdobie po pôrode
Expozícia celkovému darunaviru a ritonaviru po užití darunaviru/ritonaviru 600/100 mg dvakrát denne a darunaviru/ritonaviru 800/100 mg jedenkrát denne v rámci antivirotického režimu bola všeobecne
nižšia počas gravidity v porovnaní s obdobím po pôrode. V prípade neviazaného (t.j. aktívneho)
darunaviru však boli farmakokinetické parametre menej znížené počas gravidity v porovnaní
s obdobím po pôrode z dôvodu zvýšenia neviazanej frakcie darunaviru počas gravidity v porovnaní s obdobím po pôrode.
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru
600/100 mg dvakrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho trimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=11)aTretí trimester gravidity (n=11)Obdobie po pôrode (6-12 týždňov) (n=11)
Cmax, ng/ml 4 601 ± 1 125 5 111 ± 1 517 6 499 ± 2 411
AUC12h, ng.h/ml 38 950 ± 10 010 43 700 ± 16 400 55 300 ± 27 020
Cmin, ng/mlb 1 980 ± 839,9 2 498 ± 1 193 2 711 ± 2 268
a n=10 pre AUC12h
b vylúčením hodnoty Cmin pod LLOQ, n=10 pre referenciu
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru 800/100 mgjedenkrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho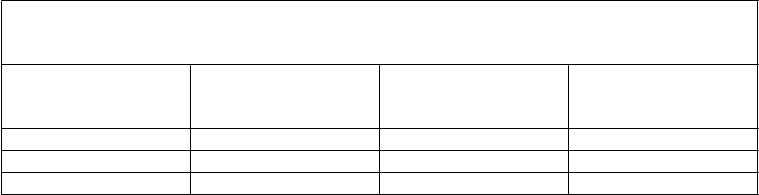 trimestra gravidity a po pôrode
trimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=16)
Tretí trimester gravidity (n=14)
Obdobie po pôrode (6-12 týždňov) (n=15)
Cmax, ng/ml 4 988 ± 1 551 5 138 ± 1 243 7 445 ± 1 674
AUC12h, ng.h/ml 61 303 ± 16 232 60 439 ± 14 052 94 529 ± 28 572
Cmin, ng/mla 1 193 ± 509 1 098 ± 609 1 572 ± 1 108
a n=12 pre obdobie po pôrode, n=15 pre druhý trimester a n=14 pre tretí trimester
U žien užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 28 %, 24 % a 17 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 19 %, 17 % nižšie a o 2 % vyššie, v tomto poradí, v porovnaní s obdobím po pôrode.
U žien užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 34 %, 34 %
a 32 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 31 %, 35 % a 50 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode.
5.3 Predklinické údaje o bezpečnosti
Toxikologické štúdie so samotným darunavirom sa vykonali v dávkovaní až do klinických hladín
u myší, potkanov a psov, kým štúdie s jeho kombináciou s ritonavirom sa vykonali u potkanov a psov.
V štúdiách toxicity po opakovanej dávke u myší, potkanov a psov sa zistili len obmedzené účinky liečby darunavirom. Identifikovaným cieľovým orgánom u hlodavcov bol hemopoetický systém, koagulačný systém, pečeň a štítna žľaza. Pozoroval sa premenlivý, ale mierny pokles parametrov v súvislosti s erytrocytmi spolu s predĺžením aktivovaného parciálneho tromboplastínového času.
Zmeny boli pozorované v pečeni (hypertrofia hepatocytov, vakuolizácia, zvýšenie hladiny pečeňových enzýmov) a štítnej žľaze (folikulárna hypertrofia). U laboratórnych potkanov viedla kombinácia darunaviru s ritonavirom k miernemu zvýšeniu účinku na parametre červených krviniek, pečene
a štítnu žľazu a k zvýšeniu incidencie ostrovčekov fibrózy v pankrease (iba u samcov laboratórnych potkanov) v porovnaní so samotným darunavirom. U psov sa nepozorovali žiadne väčšie známky toxicity ani cieľové orgány toxicity pri rovnakej expozícii ako u človeka, ktorý užíva odporučené dávky.
V štúdii na potkanoch bol znížený počet corpora lutea a implantácií v prípade maternálnej toxicity. Na druhej strane sa nepozorovali žiadne účinky na párenie alebo plodnosť po podávaní darunaviru
v dávkach do 1000 mg/kg/deň a pri expozícii (AUC - 0,5-násobok) nižšej ako u človeka dostávajúceho odporučené dávky. Pri podávaní rovnako veľkých dávok samotného darunaviru sa u potkanov
a králikov nepozorovala teratogenita podobne ako u myší dostávajúcich darunavir v kombinácii
s ritonavirom. Stupeň expozície bol nižší ako u človeka dostávajúceho odporučené dávky. Pri
hodnotení prenatálneho a postnatálneho vývoja u potkanov, darunavir podávaný samostatne alebo
v kombinácii s ritonavirom viedol k prechodnému zníženiu prírastku telesnej hmotnosti u dojčených mláďat a došlo k miernemu oneskoreniu v otvorení očí a uší. Darunavir v kombinácii s ritonavirom spôsobil zníženie prežívania mláďat, ktoré sa prejavilo nepriaznivou odpoveďou v 15. deň laktácie
a znížením počtu mláďat, ktoré prežili počas laktácie. Tieto účinky môžu byť prisúdené sekundárnej expozícii liečiva u mláďat prostredníctvom materského mlieka a/alebo maternálnej toxicity. Darunavir
podávaný ako samotný alebo v kombinácii s ritonavirom neovplyvnil žiadne funkcie po odstavení.
U juvenilných laboratórnych potkanov, ktorí dostávali darunavir 23. – 26. deň, sa pozorovala zvýšená
mortalita s kŕčmi u niektorých zvierat. Medzi 5. a 11. dňom života bola expozícia v plazme, pečeni
a mozgu výrazne vyššia ako u dospelých laboratórnych potkanov po porovnateľných dávkach v mg/kg. Po 23. dni života bola expozícia porovnateľná s tou u dospelých potkanov. Zvýšená expozícia bola pravdepodobne aspoň čiastočne z dôvodu nezrelosti enzýmov metabolizujúcich liek
u juvenilných zvierat. U juvenilných potkanov neboli spozorované žiadne úmrtia v súvislosti s liečbou
dávkami 1000 mg/kg darunaviru (jednorazová dávka) v 26. deň veku alebo 500 mg/kg (opakovaná dávka) od 23. do 50. dňa veku a expozícia a profil toxicity boli porovnateľné s tými, pozorovanými u dospelých potkanov.
Darunavir s nízkou dávkou ritonaviru sa nemá používať u detí mladších ako 3 roky z dôvodu
nejasnosti ohľadom miery rozvoja hematoencefalickej bariéry a pečeňových enzýmov.
Karcinogénny potenciál darunaviru bol vyhodnotený pri podávaní lieku myšiam a potkanom sondou do žalúdka počas 104 týždňov. Myšiam boli podávané denné dávky 150, 450 a 1000 mg/kg a potkanom boli podávané dávky 50, 150 a 500 mg/kg. Výskyt hepatocelulárnych adenómov a karcinómov stúpal v závislosti od dávky a pozoroval sa u samcov a samíc oboch živočíšnych druhov. U samcov potkanov sa vyskytli adenómy folikulárnych buniek štítnej žľazy. Podávanie darunaviru nespôsobilo štatisticky významný nárast iných benígnych alebo malígnych novotvarov u myší alebo potkanov. Pozorované hepatocelulárné tumory a tumory štítnej žľazy u hlodavcov sa považujú za obmedzené z hľadiska významnosti pre človeka. Opakované podávanie darunaviru potkanom spôsobilo indukciu mikrozomálnych enzýmov v pečeni a zvýšilo vylučovanie hormónov štítnej žľazy,
ktoré predurčuje potkanov, ale nie ľudí, na výskyt neoplaziem štítnej žľazy. Pri najvyšších testovaných
dávkach boli systémové expozície (založené na AUC) darunaviru 0,4- až 0,7-násobkom (myši) a 0,7-
až 1-násobkom (potkany) v pomere k expozíciám pozorovaným u ľudí pri odporúčaných
terapeutických dávkach.
Po dvoch rokoch podávania darunaviru pri expozíciách na úrovni alebo pod úrovňou expozícií u ľudí sa pozorovali zmeny na obličkách u myší (nefróza) a u potkanov (chronická progresívna nefropatia).
Darunavir nebol mutagénny ani genotoxický v súbore in vitro a in vivo testov vrátane bakteriálnej reverznej mutácie (Amesov test), chromozómovej aberácie v ľudských lymfocytoch
a mikronukleového testu u myší in vivo.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Oxid kremičitý, koloidný bezvodý
Mikrokryštalická celulóza
Krospovidón
Sodná soľ glykolátu škrobu
Hypromelóza
Magnéziumstearát
Obal tablety
Polyvinylalkohol, čiastočne hydrolyzovaný
Oxid titaničitý Makrogol Mastenec
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po prvom otvorení HDPE fľaše: 100 dní.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PVC/PE/PVDC-Al blistrové balenie s obsahom 30, 60 a 120 tabliet a 120 x 1 tabliet.
Za studena spracované PVC/Al/OPA-Al blistrové balenie s obsahom 30, 60 a 120 tabliet a 120 x
1 tabliet.
Balenie s HDPE fľašou s PP skrutkovacím uzáverom s obsahom 30 a 120 tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky na likvidáciu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Mylan S.A.S.
117 Allee des Parcs
69 800 Saint Priest
Francúzsko
8. REGISTRAČNÉ ČÍSLO
EU/1/16/1140/012
EU/1/16/1140/013
EU/1/16/1140/014
EU/1/16/1140/015
EU/1/16/1140/016
EU/1/16/1140/017
EU/1/16/1140/018
EU/1/16/1140/019
EU/1/16/1140/020
EU/1/16/1140/021
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: [DD. mesiac RRRR]
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/ema/.
1. NÁZOV LIEKU
Darunavir Mylan 400 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Každá filmom obalená tableta obsahuje 400 mg darunaviru. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalené tablety.
Biele až sivobiele oválne obojstranne vypuklé filmom obalené tablety s rozmermi približne 19,2 mm x
9,6 mm, s označením „M“ na jednej strane a „DV4“ na druhej strane.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Darunavir podávaný súčasne s nízkou dávkou ritonaviru je indikovaná v kombinácii s inými antiretrovirálnymi liekmi na liečbu pacientov s infekciou vyvolanou vírusom ľudskej imunodeficiencie (HIV-1).
Darunavir podávaný súčasne s kobicistatom je indikovaná v kombinácii s inými antiretrovirálnymi liekmi na liečbu infekcie vyvolanej vírusom ľudskej imunodeficiencie (HIV-1) u dospelých pacientov (pozri časť 4.2).
Darunavir Mylan 400 mg tablety sa môžu použiť na zabezpečenie vhodného režimu dávkovania pri
liečbe infekcie HIV-1 u dospelých a pediatrických pacientov od 3 rokov a s hmotnosťou najmenej
40 kg, ktorí:
· neboli predtým liečení antiretrovírusovou liečbou (ART) (pozri časť 4.2).
· boli predtým liečení ART bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM, z angl. darunavir-resistance associated mutations) a s plazmatickou hladinou HIV-1 RNA
< 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l. Pri rozhodovaní o začiatku liečby liekom darunavir u pacientov, ktorí uz boli predtým liečení ART, by malo vyšetrenie
genotypu usmerniť použitie lieku darunavir (pozri časti 4.2, 4.3, 4.4 a 5.1).
4.2 Dávkovanie a spôsob podávania
Liečbu môže začať len lekár, ktorý má skúsenosti s liečbou infekcie HIV. Po začatí liečby darunavirom treba pacientov poučiť, aby nemenili dávku, liekovú formu ani neprerušovali liečbu bez toho, aby sa poradili so svojím lekárom.
Interakčný profil darunaviru závisí od toho, či sa na zlepšenie farmakokinetiky použije ritonavir alebo kobicistat. Darunavir môže mať preto rôzne kontraindikácie a odporúčania pre súčasne podávané lieky v závislosti od toho, či je zlúčenina posilnená ritonavirom alebo kobicistatom (pozri časti 4.3, 4.4 a
4.5).
Dávkovanie
Darunavir sa musí vždy podávať perorálne v kombinácii s kobicistatom alebo s nízkou dávkou ritonaviru, ktorý zlepšuje jej farmakokinetiku, a s inými antiretrovírusovými liekmi. Z toho dôvodu je pred začatím kombinovanej liečby darunavirom a ritonavirom potrebné prečítať si Súhrn
charakteristických vlastností kobicistatu alebo ritonaviru, podľa toho, čo je náležité. Kobicistat nie je
indikovaný na užívanie dvakrát denne ani na použitie v pediatrickej populácii.
Dospelí pacienti predtým neliečení ART
Odporúčaný režim dávkovania je 800 mg jedenkrát denne súčasne so 150 mg kobicistatu jedenkrát denne alebo so 100 mg ritonaviru jedenkrát denne a s jedlom. Darunavir 400 mg tablety sa môžu
použiť na zostavenie režimu dávkovania 800 mg jedenkrát denne.
Dospelí pacienti predtým liečení ART
Odporúčané sú nasledujúce režimy dávkovania:
· U pacientov liečených ART bez mutácií spojených s rezistenciou voči darunaviru (DRV- RAM)* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+
≥ 100 buniek x 106/l (pozri časť 4.1) sa môže použiť dávkovanie 800 mg jedenkrát denne
súčasne so 150 mg kobicistatu jedenkrát denne alebo so 100 mg ritovariru jedenkrát denne
a s jedlom. Darunavir 400 mg tablety sa môžu použiť na zostavenie režimu dávkovania 800 mg jedenkrát denne.
· V prípade ostatných pacientov liečených ART alebo v prípade, že nie je k dispozícii test genotypu HIV-1, je odporúčané dávkovanie darunaviru 600 mg dvakrát denne súčasne so
100 mg ritonaviru dvakrát denne a s jedlom. Pozri Súhrn charakteristických vlastností lieku
darunavir 75 mg, 150 mg, 300 mg alebo 600 mg tablety.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Pediatrickí pacienti (vo veku 3 až 17 rokov a s hmotnosťou najmenej 40 kg) predtým neliečení ART Odporúčaný režim dávkovania je 800 mg jedenkrát denne súčasne so 100 mg ritonaviru jedenkrát denne a s jedlom. U detí mladších ako 18 rokov nebola stanovená dávka kobicistatu, ktorá má byť užitá s darunavirom.
Pediatrickí pacienti predtým liečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 40 kg)
U detí mladších ako 18 rokov nebola stanovená dávka kobicistatu, ktorá má byť užitá s darunavirom.
Odporúčané sú nasledujúce režimy dávkovania:
● U pacientov predtým liečených ART bez DRV-RAM* a s plazmatickou hladinou HIV-1 RNA
< 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri časť 4.1) sa môže použiť dávkovací režim 800 mg jedenkrát denne so 100 mg ritonaviru jedenkrát denne s jedlom.
Darunavir 400 mg tablety sa môžu použiť na zostavenie režimu dávkovania 800 mg jedenkrát denne.
● V prípade ostatných pacientov liečených ART alebo v prípade, že nie je k dispozícii test
genotypu HIV-1, je odporúčaný režim dávkovania opísaný v Súhrne charakteristických vlastností lieku darunavir 75 mg, 150 mg, 300 mg alebo 600 mg tablety.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Odporúčanie pri vynechaní dávky
V prípade, že pacient zabudne užiť jedenkrát denne dávku darunaviru a/alebo kobicistatu alebo ritonaviru do 12 hodín od doby, kedy liek zvyčajne užíva, treba ho poučiť, že má užiť predpísanú dávku darunaviru a kobicistatu alebo ritonaviru s jedlom čo najskôr. Ak od zvyčajnej doby použitia uplynie viac ako 12 hodín, vynechaná dávka sa nemá užiť a pacient má pokračovať vo zvyčajnom dávkovacom režime.
Toto usmernenie vyplynulo z polčasu darunaviru v prítomnosti kobicistatu alebo ritonaviru a
odporúčaného dávkovacieho intervalu približne 24 hodín.
Osobitné skupiny pacientov
Starší ľudia
Informácie o tejto populácii sú obmedzené, a preto sa má darunavir používať v tejto vekovej skupine
so zvýšenou opatrnosťou (pozri časti 4.4 a 5.2).
Porucha funkcie pečene
Darunavir sa metabolizuje v pečeňovom systéme. U pacientov s miernou (Childova-Pughova Trieda A) alebo stredne ťažkou (Childova-Pughova Trieda B) poruchou funkcie pečene sa neodporúča žiadna úprava dávky, avšak týmto pacientom sa má darunavir podávať so zvýšenou opatrnosťou. U pacientov s ťažkou poruchou funkcie pečene nie sú k dispozícii žiadne farmakokinetické údaje. Ťažká porucha funkcie pečene môže viesť k zvýšeniu expozície darunaviru a k zhoršeniu jeho bezpečnostného
profilu. Z toho dôvodu sa darunavir nesmie podávať pacientom s ťažkou poruchou funkcie pečene
(Childova-Pughova Trieda C) (pozri časti 4.3, 4.4 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávkovania darunaviru/ritonaviru (pozri časti 4.4 a 5.2). Kobicistat sa neskúmal u pacientov na dialýze, a preto nie je možné stanoviť žiadne odporúčanie ohľadom užívania darunaviru/kobicistatu u týchto pacientov.
Kobicistat inhibuje tubulárnu sekréciu kreatinínu a môže spôsobiť mierne zvýšenie sérového
kreatinínu a mierny pokles klírensu kreatinínu. Použitie klírensu kreatinínu za účelom odhadu kapacity renálneho vylučovania môže byť preto zavádzajúce. Z toho dôvodu sa kobicistat na zlepšenie farmakokinetiky darunaviru nemá podávať pacientom s klírensom kreatinínu nižším ako 70 ml/min, ak si niektorá súčasne užívaná látka vyžaduje úpravu dávkovania v závislosti od klírensu kreatinínu: napr. emtricitabín, lamivudín, tenofovir disoproxil fumarát alebo adefovir dipovoxil.
Informácie o kobicistate si prečítajte v Súhrne charakteristických vlastností kobicistatu.
Pediatrická populácia
Darunavir sa nemá používať u pediatrických pacientov mladších ako 3 roky alebo s telesnou
hmotnosťou menej ako 15 kg (pozri časti 4.4 a 5.3).
Pediatrickí pacienti doteraz neliečení ART (mladší ako 3 roky alebo s hmotnosťou menej ako 15 kg)
V tejto populácii nemožno stanoviť odporúčania pre dávkovanie.
Pediatrickí pacienti predtým liečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 40 kg) Boli stanovené expozície darunaviru u dospievajúcich vo veku 12 až 17 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení a ktorí dostávali darunavir/ritoanvir 800 mg/100 mg jedenkrát denne a ukázalo sa, že sa nachádzali v rámci terapeutického rozsahu, ktorý bol stanovený u dospelých pacientov dostávajúcich darunavir/ritonavir 800 mg/100 mg jedenkrát denne. Ako dôsledok vzhľadom na to, že darunavir/ritonavir 800 mg/100 mg jedenkrát denne bola tiež zaregistrovaná na použitie u dospelých s predchádzajúcou liečbou bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1 RNA
< 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l, rovnaká indikácia darunaviru 800 mg
jedenkrát denne sa vzťahuje na deti vo veku 3 až 17 rokov a s hmotnosťou najmenej 40 kg
s predchádzajúcou liečbou. Dávka darunaviru s kobicistatom nebola v tejto skupine pacientov stanovená.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Pre odporučené dávkovanie u detí, pozrite Súhrn charakteristických vlastností lieku darunavir 75 mg,
150 mg, 300 mg, 600 mg tablety.
Darunavir sa nemá používať u detí s hmotnosťou menej ako 15 kg, pretože dávka pre túto populáciu nebola stanovená na dostatočnom počte pacientov. Darunavir sa nemá používať u detí mladších ako 3 roky z dôvodu bezpečnosti.
Gravidita a obdobie po pôrode
Počas gravidity a po pôrode nie je potrebná úprava dávky darunaviru/ritonaviru. Darunavir sa má
užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko (pozri časti 4.4, 4.6 a 5.2).
Spôsob podávania
Pacientov treba poučiť, aby užívali Darunavir Mylans kobicistatom alebo nízkou dávkou ritonaviru do
30 minút po jedle. Druh jedla neovplyvňuje expozíciu darunaviru (pozri časti 4.4, 4.5 a 5.2).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Použitie u pacientov s ťažkou (Childova-Pughova Trieda C) poruchou funkcie pečene.
Súčasná liečba niektorým z nasledujúcich liekov je kontraindikovaná z dôvodu očakávaného poklesu plazmatických koncentrácií darunaviru, ritonaviru a kobicistatu a potenciálnej straty terapeutického účinku (pozri časti 4.4 a 4.5).
Relevantné pre darunavir posilnený ritonavirom alebo kobicistatom:
- Liek s kombináciou lopinavir/ritonavir (pozri časť 4.5).
- Silné induktory CYP3A, rifampicín a rastlinné preparáty obsahujúce výťažok z ľubovníka bodkovaného (Hypericum perforatum). Očakáva sa, že súčasné podávanie zníži plazmatické koncentrácie darunaviru, ritonaviru a kobicistatu, čo môže viesť k strate terapeutického účinku a možnému rozvoju rezistencie (pozri časti 4.4 a 4.5).
Relevantné pre darunavir posilnený kobicistatom, nie prípady, kedy je posilnený ritonavirom:
- Darunavir posilnený kobicistatom je citlivejší na indukciu CYP3A ako darunavir posilnený ritonavirom. Súčasné užívanie so silnými induktormi CYP3A je kontraindikované, pretože tieto
môžu znížiť expozíciu kobicistatu a darunaviru a spôsobiť stratu terapeutického účinku. Silné
induktory CYP3A zahŕňajú napr. karbamazepín, fenobarbital a fenytoín (pozri časti 4.4 a 4.5).
Darunavir posilnený ritonavirom alebo kobicistatom inhibuje elimináciu liečiv, ktorých metabolizmus (klírens) je vysoko závislý na CYP3A, čo vedie k zvýšenej expozícii súčasne užívanému lieku. Z toho dôvodu je súčasná liečba takými liekmi, ktorých zvýšené plazmatické koncentrácie sú spojené
s vážnymi a/alebo život ohrozujúcimi udalosťami, kontraindikovaná (relevantné pre darunavir posilnený ritonavirom alebo kobicistatom). Medzi tieto liečivá patria napr.:
- alfuzosín (antagonista alfa 1-adrenoreceptora)
- amiodarón, bepridil, dronedarón, chinidín, ranolazín, lidokaín (podávaný systémovo) (antiarytmiká/lieky proti angíne)
- astemizol, terfenadín (antihistaminiká)
- kolchicín, keď sa používa u pacientov s poruchou funkcie obličiek a/alebo pečene (liek proti dne) (pozri časť 4.5)
- námeľové alkaloidy (napr. dihydroergotamín, ergometrín, ergotamín, metylergonovín)
- cisaprid (prokinetiká)
- pimozid, kvetiapín, sertindol (antipsychotiká/neuroleptiká) (pozri časť 4.5)
- triazolam, midazolam podávaný perorálne (sedatíva/hypnotiká) (pre upozornenie na midazolam
podávaný parenterálne, pozri časť 4.5)
- sildenafil - keď sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, avanafil (inhibítory
PDE-5)
- simvastatín a lovastatín (inhibítory HMG-CoA reduktázy) (pozri časť 4.5)
- tikagrelor (inhibítor agregácie trombocytov) (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hoci sa preukázalo, že účinná vírusová supresia dosiahnutá pri antiretrovírusovej terapii značne znižuje riziko prenosu HIV pohlavným stykom, reziduálne riziko nie je možné vylúčiť. Je potrebné prijať opatrenia na zabránenie prenosu HIV v súlade s národnými odporúčaniami.
Odporúča sa pravidelné sledovanie virologickej odpovede. V prípade nedostatočnej alebo žiadnej virologickej odpovede sa má urobiť test na rezistenciu.
Darunavir sa musí vždy podávať perorálne s kobicistatom alebo s nízkou dávkou ritonaviru na zlepšenie farmakokinetiky a v kombinácii s inými antiretrovirotikami (pozri časť 5.2). Pred začatím liečby darunavirom si preto pozrite Súhrn charakteristických vlastností kobicistatu alebo ritonaviru.
Zvyšovanie dávkovania ritonaviru oproti odporúčaniam v časti 4.2 významnejšie neovplyvňuje
koncentrácie darunaviru. Neodporúča sa meniť dávku kobicistatu alebo ritonaviru.
Darunavir sa viaže prevažne na a1-kyslý glykoproteín. Táto proteínová väzba závisí od koncentrácie, ktorú určuje saturácia väzby. Preto lieky, ktoré sa vysoko viažu na a1-kyslý glykoproteín, môžu znemožniť túto väzbu (pozri časť 4.5).
Pacienti liečení ART –dávkovanie jedenkrát denne
Darunavir užívaný v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru jedenkrát denne
u pacientov liečených ART sa nemá používať u pacientov s jednou alebo viacerými mutáciami spojenými s rezistenciou voči darunaviru (DRV-RAM) alebo s hladinou HIV-1 RNA
≥ 100 000 kópií/ml alebo počtom buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2). V tejto populácii sa neskúmali iné kombinácie s optimalizovaným základným režimom (OBR, optimised background
regimen) ako ≥ 2 NRTI. U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje
(pozri časť 5.1).
Pediatrická populácia
Darunavir sa neodporúča používať u pediatrických pacientov mladších ako 3 roky alebo s hmotnosťou
menej ako 15 kg (pozri časti 4.2 a 5.3).
Gravidita
Darunavir sa má užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko.
Opatrnosť je potrebná u gravidných žien súbežne užívajúcich lieky, ktoré môžu ďalej znižovať
expozíciu darunaviru (pozri časti 4.5 a 5.2).
Starší ľudia
Keďže k dispozícii sú len obmedzené údaje o používaní darunaviru u pacientov starších ako 65 rokov,
pri podávaní darunaviru starším pacientom je potrebná opatrnosť vzhľadom k vyššiemu výskytu zhoršenej funkcie pečene a pridružených chorôb, alebo k inej liečbe (pozri časti 4.2 a 5.2).
Závažné kožnéreakcie
Počas programu klinického vývoja darunaviru/ritonaviru (N=3 063) boli u 0,4 % pacientov hlásené
závažné kožné reakcie, ktoré mohla sprevádzať horúčka a/alebo zvýšenie transamináz. Zriedkavo
(<0,1%) bola hlásená lieková vyrážka s eozinofíliou a systémovými príznakmi (DRESS, drug rash with eosinophilia and systemic symptoms) a Stevensov-Johnsonov syndróm a počas postmarketingových skúseností bola hlásená toxická epidermálna nekrolýza a akútna generalizovaná exantematózna pustulóza. Ak sa objavia prejavy alebo príznaky závažnej kožnej reakcie, liečba darunaviru sa má okamžite prerušiť. Príznaky môžu zahŕňať, ale nemusia byť obmedzené na, závažnú vyrážku alebo vyrážku sprevádzanú horúčkou, celkovú nevoľnosť, únavu, bolesť svalov alebo kĺbov, pľuzgiere, orálne lézie, konjunktivitídu, hepatitídu a/alebo eozinofíliu.
Vyrážka sa častejšie vyskytla u predtým liečených pacientov, ktorí boli liečení darunavirom/ritonavirom spolu s raltegravirom v porovnaní s pacientmi, ktorí užívali darunavir/ritonavir bez raltegraviru alebo raltegravir bez darunaviru (pozri časť 4.8).
Darunavir obsahuje sulfónamidovú funkčnú skupinu. Darunavir sa má podávať opatrne pacientom so
známou alergiou na sulfónamid.
Hepatotoxicita
Počas užívania darunaviru bola hlásená liekom indukovaná hepatitída (napr. akútna hepatitída,
cytolytická hepatitída). Počas programu klinického vývoja darunaviru/ritonaviru (N=3 063) bola
hepatitída hlásená u 0,5 % pacientov, ktorí boli liečení kombinovanou antiretrovirálnou terapiou s darunavirom/ritonavirom. Pacienti s preexistujúcou dysfunkciou pečene, vrátane chronickej aktívnej hepatitídy B alebo C, majú zvýšené riziko porúch funkcie pečene, vrátane vážnych a potenciálne smrteľných hepatických nežiaducich reakcií. V prípade súčasnej antivírovej liečby hepatitídy B alebo C, obráťte sa na relevantnú informáciu o týchto liekoch.
Pred začatím liečby darunavirom užívaným v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru sa majú urobiť vhodné laboratórne vyšetrenia a pacienti majú byť sledovaní aj počas liečby. U pacientov s chronickou hepatitídou, cirhózou alebo u pacientov, ktorí mali pred liečbou zvýšenú hladinu transamináz, treba zvážiť zvýšené sledovanie AST/ALT, najmä počas prvých mesiacov po začatí liečby darunavirom užívaným v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru.
Ak sa vyskytne alebo sa zhorší dysfunkcia pečene (vrátane klinicky významného zvýšenia pečeňových enzýmov a/alebo príznakov ako únava, anorexia, nauzea, žltačka, tmavý moč, citlivosť pečene, hepatomegália) u pacientov užívajúcich darunavir užívaný v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru, treba okamžite zvážiť zastavenie alebo prerušenie liečby.
Pacienti s pridruženými ochoreniami
Porucha funkcie pečene
U pacientov so závažnými poruchami pečene sa účinnosť a bezpečnosť darunaviru nestanovovala, preto je darunavir u pacientov s ťažkou poruchou funkcie pečene kontraindikovaný. Vzhľadom na nárast koncentrácie voľného darunaviru v plazme sa darunavir musí používať opatrne u pacientov s miernou alebo stredne ťažkou poruchou funkcie pečene (pozri časti 4.2, 4.3 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa nevyžadujú žiadne osobitné opatrenia alebo úpravy
dávkovania darunaviru/ritonaviru. Darunavir aj ritonavir sa výrazne viažu na plazmatické bielkoviny, a preto je nepravdepodobné, že by sa dali z cirkulácie významnejšie odstrániť pomocou hemodialýzy alebo peritoneálnej dialýzy. Preto sa u týchto pacientov nevyžadujú žiadne osobitné opatrenia alebo úpravy dávkovania (pozri časti 4.2 a 5.2). Kobicistat sa neskúmal u pacientov na dialýze, a preto nie je možné stanoviť žiadne odporúčanie ohľadom užívania darunaviru/kobicistatu u týchto pacientov
(pozri časť 4.2).
Kobicistat znižuje odhadovaný klírens kreatinínu z dôvodu inhibície tubulárnej sekrécie kreatinínu. Treba to vziať do úvahy, ak sa darunavir s kobicistatom podáva pacientom, u ktorých sa odhadovaný klírens kreatinínu používa na úpravu dávok súčasne užívaných liekov (pozri časť 4.2 a SPC kobicistatu).
V súčasnosti nie sú k dispozícii údaje postačujúce na určenie, či súčasné užívanie tenofovir disoproxil fumarátu a kobicistatu súvisí s vyšším rizikom renálnych nežiaducich reakcií v porovnaní s režimami, ktoré obsahujú tenofovir disoproxil fumarát bez kobicistatu.
Pacienti s hemofíliou
U pacientov s hemofíliou A a B liečených PI sa opísali prípady väčšieho krvácania, vrátane spontánnych kožných hematómov a krvácania do kĺbov. Niektorým pacientom sa podal faktor VIII. Vo viac ako polovici hlásených prípadov sa pokračovalo v liečbe PI, prípadne sa táto liečba obnovila
po jej dočasnom prerušení. Bola naznačená kauzálna súvislosť, hoci mechanizmus účinku nebol zatiaľ
objasnený. Z toho dôvodu je potrebné hemofilikov upozorniť na možnosť väčšieho krvácania.
Telesná hmotnosť a metabolické parametre
Počas antiretrovírusovej liečby môže dôjsť k zvýšeniu telesnej hmotnosti a hladín lipidov a glukózy
v krvi. Takéto zmeny môžu čiastočne súvisieť s kontrolou ochorenia a životným štýlom. Pokiaľ ide
o lipidy, v niektorých prípadoch sú dôkazy o vplyve liečby, kým pri prírastku telesnej hmotnosti nie sú
presvedčivé dôkazy o tom, že súvisí s niektorou konkrétnou liečbou. Pri monitorovaní hladín lipidov a glukózy v krvi sa treba riadiť zavedenými odporúčaniami na liečbu infekcie HIV. Poruchy metabolizmu lipidov majú byť klinicky vhodne liečené.
O
steonekróza
Napriek tomu, že etiológia ochorenia závisí od mnohých faktorov (vrátane užívania kortikosteroidov,
konzumácie alkoholu, vážnej imunosupresie, vyššieho BMI), prípady osteonekrózy boli hlásené najmä
u pacientov s pokročilým ochorením HIV a/alebo s dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART, combination antiretroviral therapy). Pacientov treba poučiť, aby vyhľadali lekársku pomoc v prípade, že budú pociťovať bolesti kĺbov, stuhnutosť kĺbov alebo ťažkosti pri pohybe.
Imunoreštitučný zápalový syndróm
U pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej
antiretrovírusovej liečby môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie, čo môže byť spojené so závažným klinickým stavom resp. so
zhoršením symptómov. Tieto reakcie sa obyčajne pozorujú v priebehu prvých týždňov až mesiacov od začatia podávania kombinovanej antiretrovírusovej liečby. Príkladom môže byť cytomegalovírusová retinitída, generalizované alebo fokálne mykobaktériové infekcie a pneumónia vyvolaná Pneumocystis
jirovecii (predtým známa ako Pneumocystis carinii). Je nutné zhodnotiť všetky prejavy zápalového
ochorenia a v prípade potreby začať liečbu. Ďalej sa v klinických štúdiách s darunavirom užívaným s nízkou dávkou ritonaviru pozorovala reaktivácia herpesu simplex a herpesu zoster.
Boli tiež zaznamenané aj poruchy autoimunitného systému (ako je Gravesova choroba) objavujúce sa v dôsledku imunitnej reaktivácie; avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.8).
Liekové interakcie
Liek na zlepšenie farmakokinetiky a súčasne užívané lieky
Darunavir má rôzne interakčné profily v závislosti od toho, či je zlúčenina posilnená ritonavirom alebo
kobicistatom:
- Darunavir posilnený kobicistatom je citlivejší na indukciu CYP3A: súčasné užívanie
darunaviru/kobicistatu a silných induktorov CYP3A je preto kontraindikované (pozri časť 4.3)
a súčasné užívanie so slabými a stredne silnými induktormi CYP3A sa neodporúča (pozri časť
4.5). Súčasné užívanie darunaviru/ritonaviru a darunaviru/kobicistatu
s lopinavirom/ritonavirom, rifampicínom a rastlinnými prípravkami obsahujúcimi ľubovník
bodkovaný, Hypericum perforatum, je kontraindikované (pozri časť 4.5).
- Na rozdiel od ritonaviru, kobicistat nemá žiadny indukujúci vplyv na enzýmy alebo transportné proteíny (pozri časť 4.5). Pri zmene lieku zlepšujúceho farmakokinetiku z ritonaviru na kobicistat sa počas prvých dvoch týždňov liečby darunavirom/kobicistatom vyžaduje opatrnosť, obzvlášť ak boli počas užívania ritonaviru za účelom zlepšenia farmakokinetiky titrované alebo upravené dávky akýchkoľvek súčasne podávaných liekov. V týchto prípadoch môže byť potrebné zníženie dávky súčasne podávaného lieku.
Efavirenz v kombinácii s darunavirom/ritonavirom 800/100 mg jedenkrát denne môže viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť v kombinácii s darunavirom/ritonavirom, treba použiť dávkovací režim darunavir/ritonavir 600/100 mg dvakrát
denne. Pozri Súhrn charakteristických vlastností lieku darunavir 75 mg, 150 mg, 300 mg alebo 600 mg
tablety (pozri časť 4.5).
Život ohrozujúce a smrteľné liekové interakcie sa zaznamenali u pacientov liečených kolchicínom
a silnými inhibítormi CYP3A a P-glykoproteínu (P-gp; pozri časti 4.3 a 4.5).
4.5 Liekové a iné interakcie
Interakčný profil darunaviru sa môže líšiť v závislosti od toho, či sa na zlepšenie farmakokinetiky použije ritonavir alebo kobicistat. Odporúčania dané pre súčasné užívanie darunaviru a iných liekov sa
môžu preto líšiť v závislosti od toho, či je darunavir posilnený ritonavirom alebo kobicistatom (pozri časti 4.3 a 4.4). Vyžaduje sa tiež opatrnosť na začiatku liečby, ak došlo ku zmene lieku na zlepšenie farmakokinetiky z ritonaviru na kobicistat (pozri časť 4.4).
Lieky,ktoréovplyvňujúexpozíciudarunaviru(ritonavirakolieknazlepšeniefarmakokinetiky)Darunavir a ritonavir sa metabolizujú prostredníctvom CYP3A. Očakáva sa, že lieky indukujúce činnosť CYP3A zvýšia klírens darunaviru a ritonaviru, čo má za následok zníženie plazmatických koncentrácií týchto zlúčenín a následne darunaviru a vedie k strate terapeutického účinku a možnému rozvoju rezistencie (pozri časti 4.3 a 4.4). Induktory CYP3A, ktoré sú kontraindikované, zahŕňajú napr. rifampicín, ľubovník bodkovaný a lopinavir.
Súčasné užívanie darunaviru a ritonaviru s inými liekmi, ktoré inhibujú CYP3A, môže znížiť klírens
darunaviru a ritonaviru a môže mať za následok zvýšenie plazmatických koncentrácií darunaviru
a ritonaviru. Súčasné užívanie so silnými inhibítormi CYP3A4 sa neodporúča a vyžaduje opatrnosť. Tieto interakcie sú opísané v nižšie uvedenej tabuľke interakcií (napr. indinavir, systémové azoly ako ketokonazol a klotrimazol).
Lieky,ktoréovplyvňujúexpozíciudarunaviru(kobicistatakolieknazlepšeniefarmakokinetiky)
Darunavir a kobicistat sa metabolizujú prostredníctvom CYP3A, a preto môže súčasné užívanie
s induktormi CYP3A viesť k subterapeutickým plazmatickým expozíciám darunaviru. Darunavir
posilnený kobicistatom je citlivejší na indukciu CYP3A ako darunavir posilnený ritonavirom: súčasné užívanie darunaviru/kobicistatu s liekmi, ktoré sú silnými induktormi CYP3A (napr. ľubovník bodkovaný, rifampicín, karbamazepín, fenobarbital a fenytoín) je kontraindikované (pozri časť 4.3). Súčasné užívanie darunaviru/kobicistatu so slabými až stredne silnými induktormi CYP3A (napr. efavirenz, etravirín, nevirapín, boceprevir, telaprevir, flutikazón a bosentan) sa neodporúča (pozri nižšie uvedenú tabuľku interakcií).
Na súčasné užívanie so silnými inhibítormi CYP3A4 sa vzťahujú rovnaké odporúčania, nezávisle od toho, či je darunavir posilnený ritonavirom alebo kobicistatom (pozri časť vyššie).
Lieky,ktorémôžubyťovplyvnenédarunaviromposilnenýmritonaviromDarunavir a ritonavir sú inhibítormi CYP3A, CYP2D6 a P-gp. Súčasné podávanie darunaviru/ritonaviru s liekmi metabolizovanými najmä CYP3A a/alebo CYP2D6 alebo transportovanými P-gp môže viesť k zvýšeniu systémovej expozície týmto liekom, čo by mohlo zosilniť alebo predĺžiť ich terapeutický účinok a viesť k nežiaducim účinkom.
Darunavir užívaný s nízkou dávkou ritonaviru sa nesmie kombinovať s liekmi, ktorých metabolizmus výrazne závisí od CYP3A, a ktorých zvýšená systémová expozícia je sprevádzaná závažnými a/alebo život ohrozujúcimi nežiaducimi účinkami (t.j. ktoré majú úzky terapeutický index) (pozri časť 4.3).
Celkové zvýšenie farmakokinetického účinku ritonavirom viedlo približne k 14-násobnému zvýšeniu systémovej expozície darunaviru, po jednorazovej 600 mg perorálnej dávke darunaviru v kombinácii s ritonavirom podávaným v dávke 100 mg dvakrát denne. Kobicistat 150 mg podávaný s 800 mg darunaviru jedenkrát denne zlepšuje farmakokinetické parametre darunaviru porovnateľným
spôsobom ako ritonavir (pozri časť 5.2). Z toho dôvodu sa darunavir môže používať len v kombinácii s liekom, ktorý zlepšuje farmakokinetiku darunaviru (pozri časť 5.2).
Klinická štúdia s viacerými liekmi, ktoré sú metabolizované cytochrómami CYP2C9, CYP2C19 a CYP2D6 dokázala nárast aktivity CYP2C9 a CYP2C19 a inhibíciu aktivity CYP2D6 v prítomnosti darunaviru/ritonaviru, čo sa môže pripisovať prítomnosti nízkej dávky ritonaviru. Súčasné podávanie darunaviru a ritonaviru a liekov, ktoré sa predovšetkým metabolizujú prostredníctvom CYP2D6 (napríklad flekainid, propafenon, metoprolol), môže vyústiť do zvýšených plazmatických koncentrácií týchto liekov, čo môže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce reakcie. Súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C9 (napríklad warfarín) a CYP2C19 (napríklad metadon), môže vyústiť do zníženia systémových
expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Napriek tomu, že sa účinok na CYP2C8 sledoval iba in vitro, súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C8 (napríklad paklitaxel, rosiglitazón, repaglinid), môže vyústiť do zníženia systémových expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Ritonavir inhibuje trasportérov P-glykoproteín, OATP1B1 a OATP1B3 a súčasné užívanie so substrátmi týchto transportérov môže viesť k zvýšeniu plazmatických koncentrácií týchto zlúčenín (napr. dabigatran etexilát, digoxín, statíny a bosentan; pozri nižšie uvedenú tabuľku interakcií).
Lieky,ktorémôžubyťovplyvnené darunavirom posilneným kobicistatom
Odporúčania pre darunavir posilnený ritonavirom s ohľadom na substráty CYP3A4, CYP2D6,
P-glykoproteín, OATP1B1 a OATP1B3 sú adekvátne aj pre darunavir posilnený kobicistatom (pozri kontraindikácie a odporúčania uvedené v časti vyššie). Kobicistat 150 mg podávaný s 800 mg
darunaviru jedenkrát denne zlepšuje farmakokinetické parametre darunaviru porovnateľným spôsobom ako ritonavir (pozri časť 5.2).
Na rozdiel od ritonaviru, kobicistat neindukuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ani
UGT1A1. Ďalšie informácie o kobicistate si pozrite v Súhrne charakteristických vlastností kobicistatu.
Tabuľkainterakcií
Interakčné štúdie sa uskutočnili len u dospelých.
Niekoľko interakčných štúdií (v tabuľke nižšie sú označené #) sa uskutočnilo s nižšími ako sú odporúčané dávky darunaviru alebo s inými režimami dávkovania (pozri časť 4.2 Dávkovanie). Účinky na súčasne užívané lieky môžu byť preto podhodnotené a môže byť indikované klinické sledovanie bezpečnosti.
Interakčný profil darunaviru závisí od toho, či sa na zlepšenie farmakokinetiky použije ritonavir alebo kobicistat. Darunavir môže mať preto rôzne odporúčania pre súčasne podávané lieky v závislosti od toho, či je zlúčenina posilnená ritonavirom alebo kobicistatom. Interakčné štúdie uvedené v tabuľke sa neuskutočnili s darunavirom posilneným kobicistatom. Odporúčania sú rovnaké, ak sa neuvádza inak. Ďalšie informácie o kobicistate si pozrite v Súhrne charakteristických vlastností kobicistatu.
Interakcie medzi darunavirom/ritonavirom a antiretrovirotikami a ne-antiretrovírusovými liekmi sú uvedené v tabuľkách nižšie (neurčené ako „ND“ (z angl. not determinated)). Smer šípky pri každom farmakokinetickom parametri je založený na 90 % intervale spoľahlivosti stredného geometrického pomeru v rámci 80-125 % rozpätia (↔), pod ním (↓) alebo nad ním (↑).
V nižšie uvedenej tabuľke sa uvádza konkrétny liek na zlepšenie farmakokinetiky, keď sa odporúčania líšia. V prípade, že sú odporúčania rovnaké pre darunavir súčasne užívaný s nízkou dávkou ritonaviru alebo s kobicistatom, používa sa označenie „posilnený darunavir“.
INTERAKCIE A ODPORUČENÉ DÁVKOVANIE S INÝMI LIEKMI
L
ieky podľa
t
erapeutických oblastí
I
nterakcia
Z
m
e
na geometrického priemeru (%)
O
dporúčania pri súčasnom
podávaní
 ANTIRETROVIROTIKÁ NA HIV
I
nhibítory prenosu reťazcov integrázou
ANTIRETROVIROTIKÁ NA HIV
I
nhibítory prenosu reťazcov integrázou
Dolutegravir dolutegravir AUC ↓ 32 % dolutegravir C24h 38 % dolutegravir Cmax ↓ 11 % darunavir ↔*
* Použitím porovnania údajov zo skríženej štúdie
s historickými údajmi o farmakokinetike
Posilnený darunavir a dolutegravir
sa môžu užívať bez úpravy dávky.
Elvitegravir elvitegravir AUC ↔ elvitegravir Cmin ↔ elvitegravir Cmax ↔ darunavir AUC ↔ darunavir Cmin 17 % darunavir Cmax ↔
Raltegravir Niektoré klinické štúdie naznačujú, že raltegravir môže spôsobiť mierny pokles koncentrácie darunaviru v plazme.
Keď sa darunavir užívaný súčasne s nízkou dávkou ritonaviru (600/100 mg dvakrát denne) užíva spolu s elvitegravirom, dávka elvitegraviru má byť 150 mg jedenkrát denne.
Darunavir súčasne užívaný
s kobicistatom sa nemá užívať spolu s inými antiretrovirotikami, ktoré si vyžadujú farmakokinetickú
podporu, pretože neboli stanovené odporúčania na dávkovanie takej kombinácie.
Farmakokinetika a odporúčania na dávkovanie pre iné dávky darunaviru alebo
s elvitegravirom/kobicistatom neboli stanovené. Súčasné užívanie darunaviru s nízkou dávkou ritonaviru v iných dávkach ako
600/100 mg dvakrát denne
a elvitegraviru sa preto neodporúča. Súčasné užívanie darunaviru
s nízkou dávkou ritonaviru
a elvitegravirom za prítomnosti
kobicistatu sa neodporúča.
V súčasnosti sa účinok raltegraviru na plazmatické koncentrácie darunaviru nejaví ako klinicky významný. Posilnený darunavir
a raltegravir sa môže užívať bez
úpravy dávky.
Nukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, nucleo(s/t)ide reverse transcriptase inhibitors)
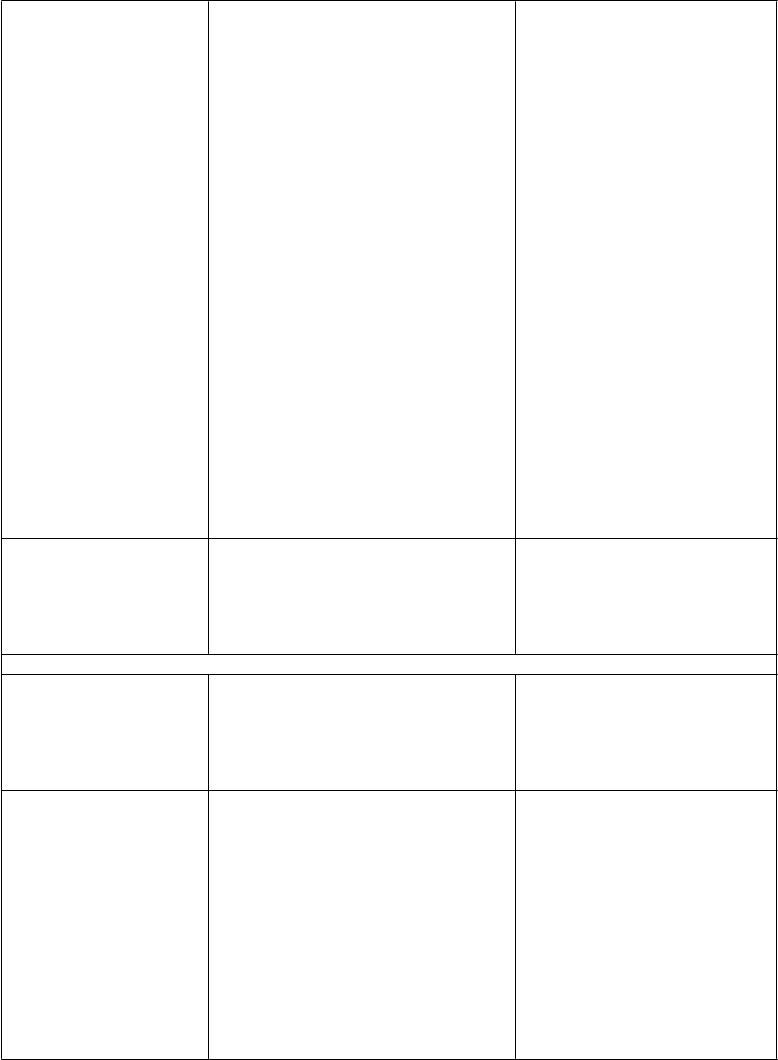
Didanozín
400 mg jedenkrát denne
Tenofovir disoproxil fumarát
300 mg jedenkrát denne
didanozín AUC ↓ 9 % didanozín Cmin ND didanozín Cmax ↓ 16 % darunavir AUC ↔ darunavir Cmin ↔ darunavir Cmax ↔ tenofovir AUC ↑ 22 % tenofovir Cmin ↑ 37 % tenofovir Cmax ↑ 24 %
#darunavir AUC ↑ 21 %
#darunavir Cmin ↑ 24 %
#darunavir Cmax ↑ 16 %
(↑ tenofoviru v dôsledku efektu na MDR-1
transport v renálnych tubuloch)
Posilnený darunavir a didanozín sa môže užívať bez úpravy dávok. Didanozín sa má podávať nalačno, preto ho treba užiť 1 hodinu pred alebo 2 hodiny po užití posilneného darunaviru s jedlom.
Pri súčasnom podávaní posilneného darunavir v kombinácii
s tenofovirom môže byť indikované sledovanie renálnych funkcií, najmä u pacientov so systémovým alebo renálnym ochorením, alebo
u pacientov užívajúcich
nefrotoxické lieky.
Darunavir súčasne užívaný
s kobicistatom znižuje klírens kreatinínu. Ak sa klírens kreatinínu používa na úpravu dávky tenofoviru, pozri časť 4.4.
Abakavir Emtricitabín Lamivudín Stavudín Zidovudín
Neskúmalo sa. Vzhľadom na rôzne cesty eliminácie ostatných NRTI zidovudín, emtricitabín, stavudín, lamivudín, ktoré sa vylučujú prevažne renálnou cestou
a abakavir, ktorý nie je metabolizovaný prostredníctvom CYP450, sa pri ich kombinácii s posilneným darunavirom nepredpokladajú žiadne liekové interakcie.
Posilnený darunavir sa môže užívať
s týmito NRTI bez úpravy dávky.
Darunavir súčasne užívaný
s kobicistatom znižuje klírens kreatinínu. Ak sa klírens kreatinínu používa na úpravu dávky emtricitabínu alebo lamivudínu, pozri časť 4.4.
Nenukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, non-nucleo(s/t)ide reverse transcriptase inhibitors)

Efavirenz
600 mg jedenkrát denne
Etravirín
100 mg dvakrát denne
Nevirapín
200 mg dvakrát denne
Rilpivirín
150 mg jedenkrát denne
efavirenz AUC ↑ 21 % efavirenz Cmin ↑ 17 % efavirenz Cmax ↑ 15 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↓ 31 %
#darunavir Cmax ↓ 15 %
(↑ efavirenzu v dôsledku inhibície
CYP3A)
(↓ darunaviru v dôsledku inhibície
CYP3A)
etravirín AUC ↓ 37 % etravirín Cmin ↓ 49 % etravirín Cmax ↓ 32 % darunavir AUC ↑ 15 % darunavir Cmin ↔ darunavir Cmax ↔
nevirapín AUC ↑ 27 % nevirapín Cmin ↑ 47 % nevirapín Cmax ↑ 18 %
#darunavir: koncentrácie
sa zhodovali s údajmi z minulosti. (↑ nevirapínu v dôsledku inhibície CYP3A)
rilpivirín AUC ↑ 130 % rilpivirín Cmin ↑ 178 % rilpivirín Cmax ↑ 79 % darunavir AUC ↔ darunavir Cmin ↓ 11 % darunavir Cmax ↔
Pri súčasnom podávaní darunaviru a nízkej dávky ritonaviru
v kombinácii s efavirenzom môže byť indikované klinické sledovanie toxicity centrálneho nervového systému spojené so zvýšenou expozíciou voči efavirenzu.
Efavirenz v kombinácii
s darunavirom/ritonavirom
800/100 mg jedenkrát denne môže
viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť
v kombinácii
s darunavirom/ritonavirom, treba použiť režim dávkovania darunavir/ritonavir 600/100 mg
dvakrát denne (pozri časť 4.4).
Súčasné úžívanie s darunavirom užívaným súčasne s kobicistatom sa neodporúča (pozri časť 4.4). Darunavir súčasne podávaný
s nízkou dávkou ritonaviru a
s etravirínom
200 mg dvakrátdennesa môže užívať bez úpravy dávky.
Súčasné úžívanie s darunavirom užívaným súčasne s kobicistatom sa neodporúča (pozri časť 4.4). Darunavir súčasne podávaný
s nízkou dávkou ritonaviru a
s nevirapínom sa môže užívať bez
úpravy dávky.
Súčasné úžívanie s darunavirom užívaným súčasne s kobicistatom sa neodporúča (pozri časť 4.4). Posilnený darunavir a rilpivirínsa môže užívať bez úpravy dávky.
H
I
V Proteázové inhibítory (PI) - bez doplnkového súčasného podávania nízkej dávky ritonaviru
†
Atazanavir
300 mg jedenkrát denne
Indinavir
800 mg dvakrát denne
Sakvinavir
1 000 mg dvakrát denne
atazanavir AUC ↔ atazanavir Cmin ↑ 52 % atazanavir Cmax ↓ 11 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Atazanavir: porovnanie atazanaviru/ritonaviru 300/100 mg jedenkrát denne vs. atazanavir 300 mg jedenkrát denne v kombinácii
s darunavirom/ritonavirom 400/100 mg dvakrát denne.
Darunavir: porovnanie
darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg dvakrát denne v kombinácii s atazanavirom 300 mg jedenkrát denne. indinavir AUC ↑ 23 %
indinavir Cmin ↑ 125 %
indinavir Cmax ↔
#darunavir AUC ↑ 24 %
#darunavir Cmin ↑ 44 %
#darunavir Cmax ↑ 11 %
Indinavir: porovnanie indinaviru/ritonaviru
800/100 mg dvakrát denne vs. indinavir/darunavir/ritonavir
800/400/100 mg dvakrát denne.
Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii s indinavirom 800 mg dvakrát denne.
#darunavir AUC ↓ 26 %
#darunavir Cmin ↓ 42 %
#darunavir Cmax ↓ 17 %
#sakvinavir AUC ↓ 6 %
#sakvinavir Cmin ↓ 18 %
#sakvinavir Cmax ↓ 6 %
Sakvinavir: porovnanie sakvinaviru/ritonaviru 1000/100 mg dvakrát denne vs. sakvinavir/darunavir/ritonavir
1000/400/100 mg dvakrát denne Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii so sakvinavirom 1000 mg dvakrát denne.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s atazanavirom sa môže užívať bez
úpravy dávky.
Darunavir užívaný súčasne
s kobicistatom sa nemá používať v kombinácii s inou antiretrovírusovou látkou, ktorá si vyžaduje zlepšenie farmakokinetiky pomocou súčasného užívania inhibítora CYP3A4 (pozri časť 4.5).
Pri intolerancii súčasného podávania darunaviru užívaného s nízkou dávkou ritonaviru
s indinavirom môže byť indikované
zníženie dávkovania indinaviru
z 800 mg dvakrát denne na 600 mg dvakrát denne.
Darunavir užívaný súčasne
s kobicistatom sa nemá používať v kombinácii s inou antiretrovírusovou látkou, ktorá si vyžaduje zlepšenie farmakokinetiky pomocou súčasného užívania inhibítora CYP3A4 (pozri časť 4.5).
Neodporúča sa kombinovať darunavir užívaný s nízkou dávkou ritonaviru so sakvinavirom.
Darunavir užívaný súčasne
s kobicistatom sa nemá používať v kombinácii s inou antiretrovírusovou látkou, ktorá si vyžaduje zlepšenie farmakokinetiky pomocou súčasného užívania inhibítora CYP3A4 (pozri časť 4.5).
H
I
V Proteázové inhibítory (PI) - podávané s nízkou dávkou ritonaviru
†
Lopinavir/ritonavir
400/100 mg dvakrát denne
Lopinavir/ritonavir
533/133,3 mg dvakrát denne
CCR5-ANTAGONISTY
Maravirok
150 mg dvakrát denne
ANESTETIKÁ
lopinavir AUC ↑ 9 % lopinavir Cmin ↑ 23 % lopinavir Cmax ↓ 2 % darunavir AUC ↓ 38 %‡ darunavir Cmin ↓ 51 %‡ darunavir Cmax ↓ 21 %‡ lopinavir AUC ↔ lopinavir Cmin ↑ 13 % lopinavir Cmax ↑ 11 % darunavir AUC ↓ 41 % darunavir Cmin ↓ 55 % darunavir Cmax ↓ 21 %
‡ na základe neštandardizovaných veľkostí dávky
maravirok AUC ↑ 305 % maravirok Cmin ND maravirok Cmax ↑ 129 %
koncentrácie darunaviru, ritonaviru sa
zhodovali s údajmi z minulosti
Vzhľadom na pokles AUC darunaviru o 40 %, kombinácie vhodných dávok neboli stanovené. Preto je súčasné podávanie posilneného darunaviru a lieku
s kombináciou lopinavir/ritonavir
kontraindikované (pozri časť 4.3).
Keď sa maravirok podáva
s posilneným darunavirom, jeho dávka má byť 150 mg dvakrát denne.
Alfentanil Neskúmalo sa. Metabolizmus alfentanilu je sprostredkovaný CYP3A a v podstate môže byť inhibovaný posilneným darunavirom.
LIEKY PROTI ANGÍNE/ANTIARYTMIKÁ
Súčasné užívanie s posilneným darunavirom môže vyžadovať zníženie dávky alfentanilu
a sledovanie rizík predĺženého alebo oneskoreného respiračného útlmu.
Disopyramid Flekainid Mexiletín Propafenón
Amiodarón Bepridil Dronedarón
Lidokaín (podávaný systémovo)
Chinidín
Ranolazín
Digoxín
0,4 mg jednorazová dávka
Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto antiarytmík.
(inhibícia CYP3A)
digoxín AUC ↑ 61 % digoxín Cmin ND digoxín Cmax ↑ 29 %
(↑ digoxínu v dôsledku možnej inhibície P-
gp)
Pri súčasnom užívaní týchto antiarytmík a posilneného darunaviru sa vyžaduje opatrnosť a odporúča sa sledovanie terapeutických koncentrácií, ak je dostupné.
Súčasné užívanie posilneného darunaviru a amiodarónu, bepridilu, dronedarónu, lidokaínu podávaného systémovo, chinidínu alebo ranolazínu je kontraindikované (pozri časť 4.3).
Vzhľadom na to, že digoxín má úzky terapeutický index, v prípade, že pacienti liečení posilneným darunavirom užívajú digoxín, odporúča sa na začiatok predpísať
čo najnižšiu možnú dávku digoxínu. Dávka digoxínu sa má opatrne titrovať, kým sa nedosiahne želaný klinický účinok pri sledovaní celkového klinického stavu
pacienta.
ANTIBIOTIKÁ
Klaritromycín
500 mg dvakrát denne
ANTIKOAGULANCIÁ
Apixaban Dabigatran etexilát Rivaroxaban
klaritromycín AUC ↑ 57 % klaritromycín Cmin ↑ 174 % klaritromycín Cmax ↑ 26 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↑ 1 %
#darunavir Cmax ↓ 17 %
Pri podávaní s darunavirom/ritonavirom neboli zistiteľné koncentrácie metabolitu
14-OH-klaritromycínu.
(↑ klaritromycínu v dôsledku inhibície
CYP3A a možnej inhibície P-gp)
Neskúmalo sa. Súčasné užívanie posilneného darunaviru a týchto antikoagulancií môže zvýšiť koncentrácie antikoagulantu.
(inhibícia CYP3A a/alebo P-gp)
Vyžaduje sa opatrnosť, keď sa klaritromycín podáva v kombinácii s posilneným darunavirom.
Odporúčanú dávku u pacientov s poruchou funkcie obličiek si pozrite v Súhrne charakteristických vlastností klaritromycínu.
Užívanie posilneného darunaviru a týchto antikoagulancií sa neodporúča.
Warfarín Neskúmalo sa. Pri súčasnom podávaní warfarínu a posilneného darunaviru môže dôjsť k ovplyvneniu koncentrácií warfarínu.
Pri súčasnom podávaní warfarínu s posilneným darunavirom sa odporúča monitorovať INR (International Normalized Ratio).
 ANTIKONVULZÍVA
ANTIKONVULZÍVA Fenobarbital Fenytoín
Karbamazepín
200 mg dvakrát denne
Neskúmalo sa. Predpokladá sa, že fenobarbital a fenytoín znižujú plazmatické koncentrácie darunaviru
a lieku na zlepšenie jeho farmakokinetiky. (indukcia enzýmov CYP450)
karbamazepín AUC ↑ 45 % karbamazepín Cmin ↑ 54 % karbamazepín Cmax ↑ 43 % darunavir AUC ↔ darunavir Cmin ↓ 15 % darunavir Cmax ↔
Darunavir užívaný s nízkou dávkou
ritonaviru sa nesmie používať
v kombinácii s týmito liečivami.
Užívanie týchto liekov
s darunavirom/kobicistatom je kontraindikované (pozri časť 4.3). Neodporúča sa žiadna úprava dávky darunaviru/ritonaviru. V prípade potreby kombinovať darunavir/ritonavir a karbamazepín sa majú pacienti sledovať z dôvodu možných nežiaducich udalostí spojených s karbamazepínom. Je potrebné sledovať koncentrácie karbamazepínu a titrovať jeho
dávku až do dosiahnutia primeranej odpovede. Na základe zistení môže byť potrebné znížiť dávku
karbamazepínu o 25% až 50%, ak
sa podáva
s darunavirom/ritonavirom.
Užívanie karbamazepínu
s darunavirom súčasne užívanou
s kobicistatom je kontraindikované
(pozri časť 4.3).
ANTIDEPRESÍVA
Paroxetín
20 mg jedenkrát denne
Sertralín
50 mg jedenkrát denne
Amitriptylín Desipramín Imipramín Nortriptylín Trazodón
ANTIDIABETIKÁ
paroxetín AUC ↓ 39 % paroxetín Cmin ↓ 37 % paroxetín Cmax ↓ 36 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
sertralín AUC ↓ 49 %
sertralín Cmin ↓ 49 %
sertralín Cmax ↓ 44 %
#darunavir AUC ↔
#darunavir Cmin ↓ 6 %
#darunavir Cmax ↔
Na rozdiel od týchto údajov s darunaviru/ritonavirom, darunavir/kobicistat môže zvýšiť plazmatické koncentrácie týchto antidepresív (inhibícia CYP2D6 a/alebo CYP3A).
Súčasné užívanie posilneného darunaviru a týchto antidepresív môže zvýšiť koncentrácie antidepresíva.
(inhibícia CYP2D6 a/alebo CYP3A)
Ak sa antidepresíva užívajú súčasne s posilneným darunaviru, odporúčaný prístup je titrácia dávky antidepresíva na základe klinického posúdenia odpovede na antidepresívum. Navyše u pacientov na ustálenej dávke týchto antidepresív, ktorí začínajú liečbu posilneným darunavirom, sa má sledovať odpoveď na antidepresívum.
Keď sa tieto antidepresíva užívajú s posilneným darunavirom, odporúča sa klinické sledovanie
a môže byť potrebná úprava dávky
antidepresíva.
Metformín Neskúmalo sa. Na základe teoretických
úvah sa očakáva, že darunavir užívaný s kobicistatom zvýši plazmatické koncentrácie metformínu.
(inhibícia MATE1)
ANTIMYKOTIKÁ
Vorikonazol Neskúmalo sa. Ritonavir môže znížiť plazmatické koncentrácie vorikonazolu. (indukcia enzýmov CYP450)
Koncentrácia vorikonazolu sa môže zvýšiť alebo znížiť, keď sa užíva súčasne
s darunavirom užívanou spolu
s kobicistatom.
(inhibícia enzýmov CYP450)
Odporúča sa starostlivé sledovanie
pacienta a úprava dávky
metformínu u pacientov užívajúcich darunavir spolu s kobicistatom. (netýka sa darunaviru užívaného spolu s ritonavirom)
Vorikonazol sa nesmie používať spolu s posilneným darunavirom, kým vyhodnotenie pomeru prínos/riziko neodôvodní použitie vorikonazolu.
Ketokonazol
200 mg dvakrát denne
Flukonazol
Posakonazol
ketokonazol AUC ↑ 212 % ketokonazol Cmin ↑ 868 % ketokonazol Cmax ↑ 111 %
#darunavir AUC ↑ 42 %
#darunavir Cmin ↑ 73 %
#darunavir Cmax ↑ 21 % (inhibícia CYP3A)
Neskúmalo sa. Posilnený darunavir môže zvýšiť plazmatické koncentrácie antimykotika (inhibícia P-gp) a posakonazol alebo flukonazol môže zvýšiť koncentrácie darunaviru.
(inhibícia CYP3A)
Vyžaduje sa opatrnosť a odporúča
sa klinické sledovanie, keď sa užíva v kombinácii s posilneným darunavirom. Ak je nutná súčasná liečba, denná dávka ketokonazolu nesmie presiahnuť 200 mg.
Vyžaduje sa opatrnosť a odporúča
sa klinické sledovanie.
Itrakonazol Neskúmalo sa. Súčasné systémové podanie itrakonazolu a posilneného darunaviru môže zvýšiť plazmatické koncentrácie darunaviru a itrakonazolu.
(inhibícia CYP3A)
Klotrimazol Neskúmalo sa. Súčasné systémové podanie klotrimazolu a posilneného darunaviru môže zvýšiť plazmatické koncentrácie darunaviru a/alebo klotrimazolu.
darunavir AUC24h ↑ 33 % (na základe farmakokinetického modelu populácie)
Vyžaduje sa opatrnosť a odporúča
sa klinické sledovanie, keď sa užíva v kombinácii s posilnenými darunavirom. Ak je nutná súčasná
liečba, denná dávka itrakonazolu nesmie presiahnuť 200 mg.
Ak je nutná súčasná liečba klotrimazolom, vyžaduje sa opatrnosť a odporúča sa klinické sledovanie.
ANTIURATIKÁ
Kolchicín Neskúmalo sa. Súčasné užívanie kolchicínu a posilneného darunaviru môže zvýšiť expozíciu kolchicínu.
(inhibícia CYP3A a/alebo
P-glykoproteínu)
ANTIMALARIKÁ
Ak je potrebná liečba posilneným darunavirom, odporúča sa
u pacientov s normálnou funkciou obličiek alebo pečene znížiť dávku kolchicínu alebo liečbu kolchicínom prerušiť.
U pacientov s poruchou funkcie obličiek alebo pečene je kolchicín s posilneným darunavirom kontraindikovaný (pozri časť 4.3).
Artemeter/
lumefantrín
80/480 mg, 6 dávok v 0., 8., 24., 36., 48. a 60. hodine
ANTITUBERKULOTIKÁRifampicín
Rifapentín
artemeter AUC ↓ 16 % artemeter Cmin ↔ artemeter Cmax ↓ 18 %
dihydroartemisinín AUC ↓ 18 %
dihydroartemisinín Cmin ↔ dihydroartemisinín Cmax ↓ 18 % lumefantrín AUC ↑ 175 % lumefantrín Cmin ↑ 126 % lumefantrín Cmax ↑ 65 % darunavir AUC ↔
darunavir Cmin ↓ 13 %
darunavir Cmax ↔
Neskúmalo sa. Rifapentín a rifampicín sú silnými induktormi CYP3A a bolo dokázané, že spôsobujú signifikantné zníženie koncentrácií iných proteázových inhibítorov, čo môže vyústiť do zlyhania antivirologickej odpovede a rozvinutia rezistencie (indukcia enzýmu CYP450). Pri pokusoch prekonať zníženú expozíciu zvýšením dávky iných proteázových inhibítorov s nízkou dávkou ritonaviru, sa s rifampicínom zistila vysoká frekvencia pečeňových reakcií.
Kombinácia posilneného darunaviru a artemeteru/lumefantrínu sa môže podávať bez úpravy dávky;
z dôvodu zvýšenia expozície lumefantrínu však treba kombináciu používať s opatrnosťou.
Kombinácia rifapentínu
a posilneného darunaviru sa
neodporúča.
Súčasné podávanie rifampicínu a posilneného darunaviru je kontraindikované (pozri časť 4.3).
Rifabutin
150 mg jedenkrát každý druhý deň
ANTINEOPLASTIKÁ
Dasatinib Nilotinib Vinblastín Vinkristín
rifabutin AUC** ↑ 55 % rifabutin Cmin** ↑ ND rifabutin Cmax** ↔ darunavir AUC ↑ 53 % darunavir Cmin ↑ 68 % darunavir Cmax ↑ 39 %
** súčet účinných zložiek rifabutinu (materský liek
+ 25-O-desacetyl metabolit)
Štúdia interakcií preukázala porovnateľnú
dennú systémovú expozíciu rifabutinu
v liečbe samotným rifabutinom 300 mg
jedenkrát denne a v liečbe rifabutinom
150 mg jedenkrát každý druhý deň spolu s darunavirom/ritonavirom (600/100 mg dvakrát denne) s približne 10-násobným zvýšením dennej expozície aktívnemu metabolitu 25-O-desacetylrifabutinu. Okrem toho, AUC súčtu účinných zložiek rifabutinu (materský liek + 25-O-desacetyl metabolit) sa zvýšila 1,6-násobne, pričom Cmax zostala porovnateľná.
Údaje o porovnaní s referenčnou dávkou
150 mg jedenkrát denne nie sú k dispozícii.
(Rifabutin je induktorom a substrátom enzýmov CYP3A.) Keď sa darunavir užívaný spolu s 100 mg ritonaviru podávala spolu s rifabutinom (150 mg jedenkrát každý druhý deň) pozorovalo sa
zvýšenie systémovej expozície darunaviru.
Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto antineoplastík.
(inhibícia CYP3A)
U pacientov užívajúcich kombináciu s darunavirom súčasne s ritonavirom je nevyhnutná
redukcia zvyčajnej dávky rifabutinu
300 mg/deň o 75 % (t.j. rifabutin
150 mg jedenkrát každý druhý deň) a zvýšené sledovanie nežiaducich udalostí súvisiacich s rifabutinom. V prípade ohrozenia bezpečnosti sa má zvážiť ďalšie predĺženie dávkovacieho intervalu rifabutinu a/alebo sledovanie hladiny rifabutinu.
Treba brať do úvahy oficiálnu smernicu pre liečbu tuberkulózy
u pacientov infikovaných HIV.
Na základe bezpečnostného profilu darunaviru/ritonaviru, toto zvýšenie expozície darunaviru v prítomnosti rifabutinu nevyžaduje úpravu dávky darunaviru/ritonaviru.
Na základe farmakokinetického modelu možno dávku zredukovať o 75 % aj u pacientov, ktorí
dostávajú inú dávku rifabutinu ako
300 mg/deň.
Súčasné užívanie darunaviru
užívaného s kobicistatom
a rifabutínu sa neodporúča.
Pri súčasnom užívaní týchto liekov a posilneného darunaviru môžu byť ich koncentrácie zvýšené, čo vedie
k možnosti nárastu nežiaducich
udalostí zvyčajne súvisiacich
s týmito liekmi.
Pri súčasnom užívaní posilneného darunaviru a jedného
z antineoplastík je potrebná
opatrnosť.

Everolimus
INHIBÍTORY AGREGÁCIE TROMBOCYTOVTikagrelor Neskúmalo sa. Súčasné užívanie
s posilneným darunavirom môže viesť k značnému zvýšeniu expozície tikagreloru.
Súčasné užívanie everolimu
a posilneného darunaviru sa
neodporúča.
Súčasné užívanie posilneného darunaviru a tikagreloru je kontraindikované.
Užívanie iných antitrombotík neovplyvnených inhibíciou alebo indukciou CYP (napr. prasugrel) sa odporúča.
ANTIPSYCHOTIKÁ/NEUROLEPTIKÁ
Kvetiapín Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto antipsychotík.
(inhibícia CYP3A)
Súčasné podávanie posilneného darunaviru a kvetiapínu je kontraindikované, pretože môže zvýšiť toxicitu súvisiacu
s kvetiapínom. Zvýšené koncentrácie kvetiapínu môžu viesť ku kóme.
Perfenazín Risperidón Tioridazín
Pimozid
Sertindol
β-BLOKÁTORY Karvedilol Metoprolol Timolol
Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto antipsychotík.
(inhibícia CYP2D6 a/alebo P-gp)
Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto beta blokátorov.
(inhibícia CYP2D6)
Môže byť potrebné zníženie dávky týchto liekov, keď sa podávajú súčasne s posilneným darunavirom.
Súčasné podávanie posilneného darunaviru a pimozidu alebo sertindolu je kontraindikované.
Pri súčasnom užívaní posilneného darunaviru s beta blokátormi sa odporúča klinické sledovanie. Má sa zvážiť zníženie dávky beta blokátora.
B
L
OK
ÁTORY KALCIOVÉHO KANÁLA
Amlodipín Diltiazem Felodipín Nikardipín Nifedipín Verapamil
Neskúmalo sa. Môže sa očakávať, že posilnený darunavir bude zvyšovať plazmatické koncentrácie blokátorov kalciového kanála.
(inhibícia CYP3A a/alebo CYP2D6)
Ak sa tieto lieky podávajú súčasne s posilneným darunavirom, odporúča sa klinické sledovanie liečebných a nežiaducich udalostí.
 KO
RTIKOSTEROIDY
KO
RTIKOSTEROIDY
Flutikazón
Budezonid
Dexametazón
(podávaný systémovo)
V klinickej štúdii, pri ktorej sa zdravým jedincom počas 7 dní podávali kapsuly ritonaviru 100 mg dvakrát denne spolu
s 50 µg flutikazónpropionátu intranazálne (štyrikrát denne), sa významne zvýšili sérové hladiny flutikazónpropionátu, zatiaľ čo prirodzená hladina kortizolu klesla približne o 86 % (pri 90 % intervale spoľahlivosti 82 % - 89 %). Vyššie účinky možno očakávať, keď sa flutikazón inhaluje. Systémové účinky kortikosteroidov, vrátane Cushingovho syndrómu a adrenálnej supresie, boli hlásené u pacientov, ktorí užívali ritonavir
a súčasne inhalovali alebo intranazálne užili flutikazón; podobná situácia môže vzniknúť aj pri súčasnom užívaní iných kortikosteroidov, ktoré sa metabolizujú prostredníctvom cytochrómu P4503A, napr. budezonidu. Účinky vysokej systémovej expozície flutikazónu na plazmatické hladiny ritonaviru nie sú dosiaľ známe.
Neskúmalo sa. Dexametazón môže znížiť plazmatickú koncentráciu darunaviru. (indukcia CYP3A)
Preto sa súčasná liečba posilneným darunavirom a týmito glukokortikoidmi neodporúča, pokiaľ možný prínos nepreváži riziko systémových kortikosteroidných účinkov. Má sa zvážiť zníženie dávky glukokortikoidov spolu
s dôkladným monitorovaním lokálnych a systémových účinkov, alebo prevod na iný glukokortikoid, ktorý nepatrí k substrátom pre
CYP3A (napr. beklometazón). V prípade vysadenia
glukokortikoidov môže byť navyše potrebné, aby progresívne znižovanie dávky trvalo dlhší čas.
Dexametazón podávaný systémovo sa má podávať so zvýšenou opatrnosťou, ak sa užíva spolu
s posilneným darunavirom.
Prednizón Neskúmalo sa. Posilnený darunavir môže zvýšiť plazmatické koncentrácie prednizónu.
(inhibícia CYP3A)
ANTIENDOTELÍNY
Bosentan Neskúmalo sa. Súčasné užívanie bosentanu a posilneného darunaviru môže zvýšiť plazmatické koncentrácie bosentanu.
Očakáva sa, že bosentan zníži plazmatické koncentrácie darunaviru a/alebo lieku na zlepšenie jeho farmakokinetiky.
(indukcia CYP3A)
Súčasné užívanie posilneného darunaviru s nízkou dávkou ritonaviru a prednizónom môže zvýšiť riziko vzniku systémových kortikosteroidných účinkov, vrátane Cushingovho syndrómu
a adrenálnej supresie. Pri súčasnom užívaní posilneného darunaviru
a kortikosteroidov sa odporúča
klinické sledovanie.
Ak sa podáva súčasne
s darunavirom a nízkou dávkou ritonaviru, má sa u pacienta
sledovať tolerovateľnosť bosentanu.
Súčasné užívanie darunaviru
užívaného s kobicistatom
a bosentanu sa neodporúča.
ANTIVIROTIKÁ PRIAMO ÚČINKUJÚCE PROTI VÍRUSU HEPATITÍDY C (HCV)
Proteázové inhibítory NS3-4A
Telaprevir
750 mg každých
8 hodín
Boceprevir
800 mg trikrát denne
telaprevir AUC ↓ 35 % telaprevir Cmin ↓ 32 % telaprevir Cmax ↓ 36 % darunavir AUC12 ↓ 40 % darunavir Cmin ↓ 42 % darunavir Cmax ↓ 40 % boceprevir AUC ↓ 32 % boceprevir Cmin ↓ 35 % boceprevir Cmax ↓ 25 % darunavir AUC ↓ 44 % darunavir Cmin ↓ 59 % darunavir Cmax ↓ 36 %
Súčasné užívanie posilneného darunaviru a telapreviru sa neodporúča.
Súčasné užívanie posilneného darunaviru a bocepreviru sa neodporúča.
Simeprevir simeprevir AUC ↑ 159 %
simeprevir Cmin ↑ 358 % simeprevir Cmax ↑ 79 % darunavir AUC ↑ 18 % darunavir Cmin ↑ 31 % darunavir Cmax «
Súčasné užívanie posilneného
darunaviru a simepreviru sa
neodporúča.
 RASTLINNÉ LIEČIVÁ
RASTLINNÉ LIEČIVÁ Ľubovník bodkovaný
(Hypericum perforatum)Dávka simepreviru v tejto interakčnej
štúdii bola 50 mg, keď sa podával spolu
s darunavirom/ritonavirom, v porovnaní so
150 mg v skupine liečenej samotným
simeprevirom.
Neskúmalo sa. Predpokladá sa, že ľubovník bodkovaný spôsobuje pokles plazmatických koncentrácií darunaviru alebo lieku na zlepšenie jeho farmakokinetiky.
(indukcia CYP450)
Posilnený darunavir sa nesmie podávať v kombinácii s liekmi obsahujúcimi výťažok z ľubovníka bodkovaného (
Hypericum perforatum) (pozri časť 4.3). Ak už pacient užíva ľubovník bodkovaný, užívanie ukončite, a ak je to možné, skontrolujte vírusové hladiny. Expozícia darunaviru (a tiež expozícia ritonaviru) sa môže po ukončení užívania ľubovníka bodkovaného zvýšiť. Indukujúci účinok môže pretrvávať najmenej 2 týždne po ukončení liečby ľubovníkom bodkovaným.
I
NHIBÍTORY HMG CO-A REDUKTÁZY
Lovastatín
Simvastatín
Atorvastatín
10 mg jedenkrát denne
Pravastatín
40 mg jednorazová dávka
Rosuvastatín
10 mg jedenkrát denne
Neskúmalo sa. Predpokladá sa, že lovastatín a simvastatín budú mať zreteľne zvýšené plazmatické koncentrácie, ak sa budú podávať súčasne s posilneným darunavirom.
(inhibícia CYP3A)
atorvastatín AUC ↑ 3-4 krát atorvastatín Cmin ↑ ≈5,5-10 krát atorvastatín Cmax ↑ ≈2 krát
#darunavir
pravastatín AUC ↑ 81 %¶ pravastatín Cmin ND pravastatín Cmax ↑ 63 %
¶ u niektorých subjektov sa pozoroval až 5-násobný nárast
rosuvastatín AUC ↑ 48 %║
rosuvastatín Cmax ↑ 144 %║
║ na základe publikovaných údajov
Zvýšené plazmatické koncentrácie lovastatínu alebo simvastatínu môžu vyvolať myopatiu, vrátane rabdomyolýzy. Súčasné podávanie posilneného darunaviru
s lovastatínom a simvastatínom je
preto kontraindikované (pozri časť
4.3).
Ak je potrebné súčasné podávanie atorvastatínu s posilneným darunavirom, odporúča sa začať podávať atorvastatín v dávke 10 mg jedenkrát denne. Postupné zvyšovanie dávkovania
atorvastatínu je možné prispôsobiť
na základe klinickej odpovede.
Ak je potrebné súčasné podávanie pravastatínu s posilneným darunavirom, odporúča sa začať
s najnižšou možnou dávkou pravastatínu a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
Ak je potrebné súčasné podávanie rosuvastatínu s posilneným darunavirom, odporúča sa začať
s najnižšou možnou dávkou rosuvastatín a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
I
NHIBÍTORY H
2
-
RECEPTORA
Ranitidín
150 mg dvakrát denne
IMUNOSUPRESÍVA Ciklosporín Sirolimus Takrolimus
Everolimus
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Neskúmalo sa. Pri súčasnom podávaní posilneného darunaviru s týmito imunosupresívami sa zvýši expozícia voči týmto látkam.
(inhibícia CYP3A)
Posilnený darunavir a inhibítory H2-receptora sa môžu užívať bez úpravy dávky.
Pri súčasnom podaní imunosupresív sa musia monitorovať terapeutické hladiny týchto liekov.
Súčasné užívanie everolimu a posilneného darunaviru sa neodporúča.
 I
NHALAČNÉ BETA-AGONISTY
I
NHALAČNÉ BETA-AGONISTY
Salmeterol Neskúmalo sa. Súčasné užívanie salmeterolu a posilneného darunaviru môže zvýšiť plazmatické koncentrácie salmeterolu.
Súčasné užívanie salmeterolu a posilneného darunaviru sa neodporúča. Táto kombinácia môže viesť k zvýšenému riziku kardiovaskulárnych nežiaducich udalostí salmeterolu, vrátane predĺženia QT intervalu, palpitácií a sínusovej tachykardie.
NARKOTICKÉ ANALGETIKÁ / LIEČBA ZÁVISLOSTI NA OPIOIDOCH
Metadon individuálna dávka
v rozsahu od 55 mg do
150 mg jedenkrát denne
Buprenorfín/naloxón
8/2 mg–16/4 mg jedenkrát denne
R(-) metadon AUC ↓ 16 % R(-) metadon Cmin ↓ 15 % R(-) metadon Cmax ↓ 24 %
Darunavir /kobicistat môže, naopak, zvýšiť plazmatické koncentrácie metadonu (pozri SPC kobicistatu).
buprenorfín AUC ↓ 11 % buprenorfín Cmin ↔ buprenorfín Cmax ↓ 8 % norbuprenorfín AUC ↑ 46 % norbuprenorfín Cmin ↑ 71 % norbuprenorfín Cmax ↑ 36 % naloxón AUC ↔
naloxón Cmin ND
naloxón Cmax ↔
Na začiatku užívania spolu
s posilneným darunavirom nie je potrebná úprava dávky metadonu. Avšak úprava dávky metadonu môže byť potrebná pri súčasnom podávaní počas dlhšieho časového obdobia. Z toho dôvodu sa
odporúča klinické monitorovanie,
pretože u niektorých pacientov môže byť potrebná úprava udržiavacej liečby.
Klinický význam zvýšenia farmakokinetických parametrov norbuprenorfínu nebol stanovený. Úprava dávky buprenorfínu nemusí byť potrebná, ak sa podáva spolu
s posilneným darunavirom, ale odporúča sa pozorné sledovanie prejavov toxicity opiátov.
ANTIKONCEPCIA NA BÁZE ESTROGÉNOV
Etinylestradiol
Noretisterón
35 mg/1 mg jedenkrát denne
etinylestradiol AUC ↓ 44 % etinylestradiol Cmin ↓ 62 % etinylestradiol Cmax ↓ 32 % noretisterón AUC ↓ 14 % noretisterón Cmin ↓ 30 % noretisterón Cmax ↔
Pacientkam užívajúcim antikoncepciu na báze estrogénov v kombinácii s posilneným darunavirom sa odporúča zmeniť
resp. rozšíriť formy antikoncepcie. U pacientok užívajúcich estrogén ako hormonálnu substitúciu majú byť klinicky sledované prejavy deficitu estrogénu.
FOSFODIESTERÁZA, INHIBÍTORY TYPU 5 (PDE-5)

Pri liečbe erektilnej
dysfunkcie Avanafil Sildenafil Tadalafil Vardenafil
V štúdii skúmajúcej interakcie# sa pozorovala porovnateľná systémová expozícia voči sildenafilu po jednorazovom podaní samotného sildenafilu v dávke 100 mg a po jednorazovom podaní sildenafilu v dávke
25 mg v kombinácii s darunavirom a nízkou dávkou ritonaviru.
Kombinácia avanafilu
a posilneného darunaviru je kontraindikovaná (pozri časť 4.3). Pri súčasnom podávaní iných inhibítorov PDE-5 na liečbu erektilnej dysfunkcie s posilneným darunavirom je potrebná opatrnosť. Ak je indikované súčasné podávanie posilneného darunaviru a
sildenafilu, vardenafilu alebo tadalafilu, jednorazová dávka
sildenafilu nesmie prekročiť 25 mg
za obdobie 48 hodín, resp. jednorazová dávka vardenafilu nesmie prekročiť 2,5 mg za obdobie
72 hodín a jednorazová dávka tadalafilu nesmie presiahnuť 10 mg za obdobie 72 hodín.
Pri liečbe pulmonálnej
arteriálnej hypertenzie
Sildenafil
Tadalafil
Neskúmalo sa. Súčasné užívanie sildenafilu alebo tadalafilu pri liečbe pulmonálnej arteriálnej hypertenzie a posilneného darunaviru môže zvýšiť plazmatické koncentrácie sildenafilu alebo tadalafilu.
(inhibícia CYP3A)
Bezpečná a účinná dávka sildenafilu pri liečbe pulmonálnej arteriálnej hypertenzie a pri súčasnom užívaní posilneného darunaviru nebola stanovená.
Existuje zvýšená možnosť vzniku nežiaducich udalostí súvisiacich so sildenafilom (vrátane porúch zraku, hypotenzie, predĺženej erekcie
a synkopy). Preto je súčasné užívanie posilneného darunaviru a sildenafilu, ak sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, kontraindikované (pozri časť 4.3).
Pri liečbe pulmonálnej arteriálnej hypertenzie sa súčasné užívanie tadalafilu s posilneným darunavirom neodporúča.
I
NHIBÍTORY PROTÓNOVEJ PUMPY
Omeprazol
20 mg jedenkrát denne
SEDATÍVA/HYPNOTIKÁ
Buspirón Klorazepát Diazepam Estazolam Flurazepam Midazolam (parenterálny) Zolpidem
Midazolam (perorálny) Triazolam
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Neskúmalo sa. Sedatíva/hypnotiká sú výrazne metabolizované prostredníctvom CYP3A. Súčasné užívanie s posilneným darunavirom môže spôsobiť významné zvýšenie koncentrácií týchto liekov.
Ak sa parenterálny midazolam podáva súčasne s posilneným darunavirom, môže spôsobiť významné zvýšenie koncentrácie tohto benzodiazepínu. Údaje o súčasnom užívaní parenterálneho midazolamu
s inými proteázovými inhibítormi naznačujú možné 3-4-násobné zvýšenie plazmatických hladín midazolamu.
Posilnený darunavir je možné súčasne podávať s inhibítormi protónovej pumpy bez nutnosti úpravy dávkovania.
Pri súčasnom užívaní posilneného darunaviru s týmito sedatívami/hypnotikami sa odporúča klinické sledovanie a má sa zvážiť zníženie dávky sedatív/hypnotík.
Ak sa parenterálny midazolam podáva súčasne s posilneným darunavirom, má sa tak uskutočniť na jednotke intenzívnej starostlivosti (JIS) alebo
v podobnom zariadení, ktoré zabezpečuje dôkladné klinické sledovanie a vhodnú lekársku starostlivosť v prípade respiračného útlmu a/alebo predĺženého útlmu. Treba zvážiť úpravu dávkovania midazolamu, najmä ak sa podáva viac ako jedna dávka.
Súčasné užívanie posilneného darunaviru a triazolamu alebo perorálneho midazolamu je kontraindikované (pozri časť 4.3)
 †
† Účinnosť a bezpečnosť používania darunaviru so 100 mg ritonaviru a s niektorým iným HIV PI (napr. (fos)amprenavir, nelfinavir a tipranavir) neboli u pacientov s HIV stanovené. Podľa súčasných liečebných postupov sa vo všeobecnosti duálna liečba proteázovými inhibítormi neodporúča.
4.6 Fertilita, gravidita a laktáciaGraviditaVo všeobecnosti, keď sa rozhoduje o použití antiretrovirotík na liečbu infekcie HIV u gravidných žien a následne na zníženie rizika vertikálneho prenosu HIV na novorodenca, treba vziať do úvahy údaje
o zvieratách ako aj klinické skúsenosti s gravidnými ženami.
S darunavirom sa nevykonali žiadne primerané ani dobre kontrolované štúdie o vplyve na graviditu u gravidných žien. Štúdie na zvieratách nepreukázali priame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3).
Darunavir užívaný s kobicistatom alebo s nízkou dávkou ritonaviru sa môže podávať gravidným ženám len vtedy, ak možný prínos liečby prevýši možné riziká.
DojčenieNie je známe, či sa darunavir vylučuje do ľudského mlieka. V štúdiách na potkanoch sa preukázalo vylučovanie darunaviru do mlieka a pri vysokých hladinách (1000 mg/kg/deň) spôsobilo toxicitu. Matky majú byť poučené, aby za žiadnych okolností nedojčili počas liečby darunavirom kvôli jednak možnosti prenosu HIV a aj možnosti výskytu nežiaducich reakcií u dojčených detí.
FertilitaNie sú k dispozícii žiadne údaje o účinku darunaviru na fertilitu u ľudí. Pri liečbe potkanov
darunavirom nemal darunavir vplyv na párenie a fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeDarunavir spolu s kobicistatom alebo s ritonavirom nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U niektorých pacientov sa však počas liečby darunavirom užívaným s kobicistatom alebo s nízkou dávkou ritonaviru vyskytli závraty. Na túto možnosť je potrebné myslieť pri posudzovaní schopnosti pacienta viesť vozidlá a obsluhovať stroje (pozri časť
4.8).
4.8 Nežiaduce účinkySúhrnbezpečnostnéhoprofiluPočas programu klinického vývoja (N=2 613 predtým liečených jedincov, ktorí začali liečbu
s darunavirom/ritonavirom 600/100 mg dvakrát denne), 51,3 % jedincov malo aspoň jednu nežiaducu reakciu. Celkové priemerné trvanie liečby pacientov bolo 95,3 týždňov. Najčastejšími nežiaducimi
reakciami hlásenými v klinických skúšaniach a v spontánnych hláseniach sú hnačka,
nauzea, vyrážka, bolesť hlavy a vracanie. Najčastejšími závažnými reakciami sú akútne renálne zlyhanie, infarkt myokardu, imunoreštitučný zápalový syndróm, trombocytopénia, osteonekróza, hnačka, hepatitída a pyrexia.
V analýze po 96 týždňoch bol bezpečnostný profil darunaviru/ritonaviru 800/100 mg jedenkrát denne u jedincov, ktorí doteraz neboli liečení, podobný ako u darunaviru/ritonaviru 600/100 mg dvakrát denne u jedincov, ktorí boli predtým liečení, až na nauzeu, ktorá sa častejšie vyskytovala u doteraz neliečených jedincov. Nauzea bola miernej intenzity. V analýze po 192 týždňoch sa u doteraz neliečených jedincov, u ktorých liečba darunavirom/ritonavirom 800/100 mg jedenkrát denne trvala priemerne 162,5 týždňa, neidentifikovali žiadne nové nálezy týkajúce sa bezpečnosti.

Počas klinickej štúdie fázy III GS-US-216-130 s darunavirom/kobicistatom (N = 313 doposiaľ neliečených a predtým liečených pacientov), 66,5 % osôb zaznamenalo aspoň jednu nežiaducu reakciu. Liečba trvala priemerne 58,4 týždňa. Najčastejšie hlásené nežiaduce reakcie boli diarea (28 %), nauzea (23 %) a vyrážka (16 %). Závažné nežiaduce reakcie sú diabetes melitus, precitlivenosť (na liek), imunoreštitučný zápalový syndróm, vyrážka a vracanie.
Informácie o kobicistate si pozrite v Súhrne charakteristických vlastností kobicistatu.
Zoznam nežiaducichreakciívtabuľkáchNežiaduce reakcie sa uvádzajú podľa postihnutia triedy orgánových systémov a podľa skupiny frekvencie. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti. Skupiny frekvencií sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000)
a neznáme (z dostupných údajov).
N
ežiaduce reakcie pozorované s darunavirom/ritonavirom v klinických skúšaniach
a v postmarketingovom sledovaní
Trieda orgánových systémov podľa databázy
MedDRA
Kategória frekvencie
Infekcie a nákazy
Nežiaduca reakcia
menej časté herpes simplex
Poruchy krvi a lymfatického systémumenej časté trombocytopénia, neutropénia, anémia, leukopénia
zriedkavé zvýšenie počtu eozinofilov
Poruchy imunitného systémumenej časté imunoreštitučný zápalový syndróm, precitlivenosť
(na liek)
Poruchy endokrinného systémumenej časté hypotyreóza, zvýšenie hladiny hormónu stimulujúceho štítnu žľazu v krvi
Poruchy metabolizmu a výživyčasté diabetes mellitus, hypertriglyceridémia, hypercholesterolémia, hyperlipidémia
menej časté dna, anorexia, znížená chuť do jedla, zníženie
hmotnosti, zvýšenie hmotnosti, hyperglykémia,
rezistencia na inzulín, pokles lipoproteínu
s vysokou hustotou, zvýšená chuť do jedla, polydipsia, zvýšená hladina LDH v krvi
Psychické poruchyčasté insomnia
menej časté depresia, dezorientácia, úzkosť, poruchy spánku,
neprirodzené sny, nočné mory, pokles libida
zriedkavé stav zmätenosti, zmeny nálady, nepokoj
Poruchy nervového systémučasté bolesti hlavy, periférna neuropatia, závraty
menej časté letargia, parestézia, hypestézia, dysgeuzia, porucha
pozornosti, poškodenie pamäti, somnolencia
zriedkavé synkopa, konvulzia, ageuzia, porucha fázy spánkového rytmu
Poruchy okamenej časté konjunktiválna hyperémia, suché oči
zriedkavé porucha zraku
Poruchy ucha a labyrintumenej časté vertigo
Poruchy srdca a srdcovej činnostimenej časté infarkt myokardu, angína pektoris, predĺžený QT
elektrokardiogram, tachykardia
zriedkavé akútny infarkt myokardu, sínusová bradykardia,
palpitácie
 Poruchy ciev
Poruchy ciev
menej časté hypertenzia, návaly horúčavy
Poruchy dýchacej sústavy, hrudníka a mediastínamenej časté dyspnoe, kašeľ, epistaxa, podráždenie hrdla
zriedkavé rinorea
Poruchy gastrointestinálneho traktuveľmi časté diarea
časté vracanie, nauzea, bolesti brucha, zvýšená hladina
amylázy v krvi, dyspepsia, abdominálna distenzia, flatulencia
menej časté pankreatitída, gastritída, gastroezofagálny reflux, aftózna stomatitída, napínanie na vracanie, sucho
v ústach, abdominálny diskomfort, zápcha, zvýšená
hladina lipázy, eruktácia, perorálna dyzestézia
zriedkavé stomatitída, hemateméza, cheilitída, suché pery, povlak na jazyku
Poruchy pečene a žlčových ciestčasté zvýšenie alanínaminotransferázy
menej časté hepatitída, cytolytická hepatitída, steatóza pečene,
hepatomegália, zvýšenie transamináz, zvýšenie aspartátaminotransferázy, zvýšenie hladiny bilirubínu v krvi, zvýšenie alkalickej fosfatázy v krvi, zvýšenie gamaglutamyltransferázy
Poruchy kože a podkožného tkanivačasté vyrážka (vrátane makulárnej, makulopapulárnej, papulárnej, erytematóznej a pruritickej vyrážky), pruritus
menej časté angioedém, generalizovaná vyrážka, alergická
dermatitída, urtikária, ekzém, erytém,
hyperhidróza, nočné potenie, alopécia, akné, suchá koža, sfarbenie nechtov
zriedkavé DRESS, Stevensov-Johnsonov syndróm, multiformný erytém, dermatitída, seboroická dermatitída, kožné lézie, xeroderma
neznáme toxická epidermálna nekrolýza, akútna generalizovaná exantematózna pustulóza
Poruchy kostrovej a svalovej sústavy a spojivového tkanivamenej časté myalgia, osteonekróza, svalové kŕče, svalová slabosť, artralgia, bolesti končatín, osteoporóza, zvýšená hladina kreatínfosfokinázy v krvi
zriedkavé muskuloskeletálna stuhnutosť, artritída, stuhnutie
kĺbov
Poruchy obličiek a močových ciestmenej časté akútne zlyhanie obličiek, zlyhanie obličiek, nefrolitiáza, zvýšená hladina kreatinínu v krvi, proteinúria, bilirubinúria, dyzúria, noktúria, polakizúria
zriedkavé znížený renálny klírens kreatinínu
Poruchy reprodukčného systému a prsníkov
menej časté erektilná dysfunkcia, gynekomastia
Celkové poruchy a reakcie v mieste podania
časté asténia, únava
menej časté pyrexia, bolesť na hrudi, periférne opuchy,
nevoľnosť, pocit horúčavy, podráždenie, bolesť
zriedkavé triaška, abnormálne pocity, xeróza
Nežiaduce reakcie s darunavirom/kobicistatom u dospelých pacientov
Trieda orgánových systémov podľa databázy
MedDRA
Kategória frekvencie
Poruchy imunitného systému
Nežiaduca reakcia


časté precitlivenosť (na liek)
menej časté imunoreštitučný zápalový syndróm
Poruchy metabolizmu a výživyčasté anorexia, diabetes mellitus, hypercholesterolémia, hypertriglyceridémia, hyperlipidémia
Psychické poruchyčasté neprirodzené sny
Poruchy nervového systémuveľmi časté bolesť hlavy
Poruchy gastrointestinálneho traktuveľmi časté diarea, nauzea
časté vracanie, bolesti brucha, abdominálna distenzia,
dyspepsia, flatulencia, zvýšená hladina
pankreatických enzýmov
menej časté akútna pankreatitída
Poruchy pečene a žlčových ciestčasté zvýšená hladina hepatálnych enzýmov
menej časté hepatitída*, cytolytická hepatitída*
Poruchy kože a podkožného tkanivaveľmi časté vyrážka (vrátane makulárnej, makulopapulárnej, papulárnej, erytematóznej, pruritickej vyrážky, generalizovanej vyrážky a alergickej dermatitídy)
časté angioedém, pruritus, urtikária
zriedkavé lieková reakcia s eozinofíliou a systémovými symptómami*, Stevensov-Johnsonov syndróm*
neznáme toxická epidermálna nekrolýza*, akútna generalizovaná exantematózna pustulóza*
Poruchy kostrovej a svalovej sústavy a spojivového tkanivačasté myalgia, osteonekróza*
Poruchy reprodukčného systému a prsníkovmenej časté gynekomastia*
 C
elkové poruchy a reakcie v mieste podania
C
elkové poruchy a reakcie v mieste podania
časté únava
menej časté asténia
Laboratórne a funkčné vyšetreniačasté zvýšená hladina kreatinínu v krvi
* tieto nežiaduce reakcie na liek neboli hlásené v klinických štúdiách s darunavirom/kobicistatom, ale zaznamenali sa pri liečbe darunavirom/ritonavirom a možno ich očakávať aj s darunavirom/kobicistatom.
Opisvybranýchnežiaducich reakciíVyrážkaV klinických skúšaniach bola vyrážka najčastejšie miene až stredne závažná, často sa vyskytla v prvých štyroch týždňoch liečby a ustúpila pri ďalšom užívaní. Pre prípady závažných kožných reakcií
pozri upozornenie v časti 4.4. V štúdii s jednou skupinou, ktorá skúmala darunavir 800 mg jedenkrát denne v kombinácii s kobicistatom 150 mg jedenkrát denne a s iným antiretrovirotikami, 2,2 %
pacientov ukončilo liečbu kvôli vyrážke.
Počas programu klinického vývoja raltegraviru sa u predtým liečených pacientov častejšie pozorovala vyrážka, bez ohľadu na kauzalitu, pri režimoch obsahujúcich darunavir/ritonavir + raltegravir
v porovnaní s režimami obsahujúcimi darunavir/ritonavir bez raltegraviru alebo raltegravir bez
darunaviru/ritonaviru. Vyrážka považovaná skúšajúcim za súvisiacu s liekom, sa vyskytla v podobnej
miere. Expozícii prispôsobené miery vyrážky (všetky príčiny) boli 10,9, 4,2 a 3,8 na 100
pacientorokov v danom poradí a u vyrážky súvisiacej s liekom boli 2,4, 1,1 a 2,3 na 100 pacientorokov v danom poradí. Vyrážky pozorované v klinických štúdiách boli mierne až stredne závažné a neviedli
k prerušeniu liečby (pozri časť 4.4).
Metabolické parametrePočas antiretrovírusovej liečby sa môže zvýšiť telesná hmotnosť a hladiny lipidov a glukózy v krvi
(pozri časť 4.4).
Poruchy kostrovej a svalovej sústavyPočas liečby inhibítormi proteáz, najmä v kombinácii s NRTI, sa vyskytli prípady zvýšenia hladiny
kreatínfosfokinázy, myalgie, myozitídy a zriedkavo aj rabdomyolýzy.
Boli hlásené prípady osteonekrózy, najmä u pacientov so všeobecne potvrdenými rizikovými faktormi, pokročilým ochorením HIV alebo dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART). Frekvencia nie je známa (pozri časť 4.4).
Imunoreštitučný zápalový syndrómU pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej
antiretrovírusovej liečby (CART) môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie. Boli tiež zaznamenané aj poruchy imunitného systému (ako je Gravesova choroba); avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.4).
Krvácanie u pacientov s hemofíliouU pacientov s hemofíliou dostávajúcich antiretrovirálne inhibítory proteázy boli hlásené prípady
zvýšeného spontánneho krvácania (pozri časť 4.4).
Pediatrická populáciaPosúdenie bezpečnosti u pediatrických pacientov je založené na 48-týždňovej analýze údajov
o bezpečnosti z troch klinických štúdií fázy II. Boli hodnotené nasledovné populácie pacientov (pozri časť 5.1):
● 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17
rokov a s hmotnosťou najmenej 20 kg, ktorí užívali darunavir tablety s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku od 3 do < 6 rokov a s hmotnosťou 10 kg až < 20 kg (16 účastníkov od 15 kg do < 20 kg), ktorí užívali darunavir perorálnu suspenziu s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 12 pediatrických pacientov infikovaných HIV-1 vo veku od 12 do 17 rokov a s hmotnosťou
najmenej 40 kg, ktorí neboli predtým liečení ART. Títo pacienti dostávali darunavir tablety
s nízkou dávkou ritonaviru jedenkrát denne spolu s inými antiretrovírusovými liekmi. (pozri
časť 5.1).
Celkovo bol bezpečnostný profil u týchto pediatrických pacientov podobný bezpečnostnému profilu
pozorovanému u dospelých.
Iné osobitné skupiny pacientovPacienti súčasne infikovaní vírusom hepatitídy B a/alebo hepatitídy CSpomedzi 1968 predtým liečených pacientov, ktorí užívali darunavir spolu s ritonavirom 600/100 mg dvakrát denne, bolo 236 pacientov infikovaných zároveň hepatitídou B alebo C. U súčasne
infikovaných pacientov bola vyššia pravdepodobnosť počiatočného a pri liečbe vznikajúceho zvýšenia
hladiny pečeňových transamináz ako u pacientov bez chronickej vírusovej hepatitídy (pozri časť 4.4).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v
Prílohe V.4.9 PredávkovanieSkúsenosti s akútnym predávkovaním darunavirom užívaným s kobicistatom alebo s nízkou dávkou ritonaviru u ľudí sú obmedzené. Zdravým dobrovoľníkom sa darunavir podával vo forme perorálneho roztoku v jednorazových dávkach do 3200 mg ako samotný, resp. vo forme tabliet
v jednorazových dávkach do 1600 mg v kombinácii s ritonavirom bez toho, že by sa u nich pozorovali nežiaduce účinky.
V prípade predávkovania darunavirom nie je k dispozícii žiadne špecifické antidotum. Liečba predávkovania darunavirom zahŕňa všeobecné podporné opatrenia, vrátane sledovania vitálnych znakov a pozorovania klinického stavu pacienta. Ak je to indikované, eliminácia nevstrebanej aktívnej látky sa dosiahne pomocou vyvolania vracania.
Aj podanie aktívneho uhlia môže pomôcť pri eliminácii nevstrebanej aktívnej látky. Vzhľadom na vysokú väzbu darunaviru na plazmatické bielkoviny je nepravdepodobné, že by dialýza mala väčší význam pri eliminácii aktívnej látky z cirkulácie.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antivirotiká na systémové použitie, inhibítory proteáz, ATC kód: J05AE10
Mechanizmus účinkuDarunavir je inhibítor dimerizácie a katalytickej aktivity HIV-1 proteázy (KD 4,5 x 10-12M). Selektívne
inhibuje odštiepenie HIV kódovaných polyproteínov Gag-Pol v bunkách infikovaných vírusom, čím zabraňuje tvorbe zrelých infekčných vírusových častíc.
A
ntivírusová aktivita in vitro
Darunavir vykazuje aktivitu proti laboratórnym kmeňom a klinickým izolátom HIV-1 a laboratórnym kmeňom HIV-2 v akútne infikovaných líniách T-buniek, mononukleárnych bunkách z periférnej krvi človeka a ľudských monocytov/makrofágov so strednými hodnotami EC50 v rozmedzí
1,2 až 8,5 nmol/l (0,7 až 5,0 ng/ml). In vitro má darunavir antivírusovú aktivitu voči širokému spektru
primárnych izolátov zo skupiny M HIV-1 (A, B, C, D, E, F, G) a skupiny O s hodnotami EC50
v rozmedzí od < 0,1 do 4,3 nmol/l.
Tieto hodnoty EC50 sú výrazne nižšie ako je rozsah toxickej koncentrácie pre 50 % buniek od
87 µmol/l do > 100 µmol/l.
Rezistencia
Selekcia vírusu rezistentného na darunavir z divokého typu HIV-1 in vitro trvala pomerne dlho
(> 3 roky). Vyselektované vírusy neboli schopné rásť v prítomnosti darunaviru v koncentráciách nad
400 nmol/l. U vírusov vyselektovaných v týchto podmienkach a vykazujúcich zníženú citlivosť voči
darunaviru (rozmedzie: 23 až 50-násobne) sa v géne pre proteázu zistili substitúcie 2 až 4 aminokyselín. Zníženú citlivosť vznikajúcich vírusov na darunavir v selekcii nie je možné vysvetliť vznikom týchto mutácií proteázy.
Údaje z klinických štúdií s pacientmi liečenými ART (štúdia TITAN a zlúčená analýza štúdií POWER
1, 2 a 3 a štúdií DUET 1 a 2) preukázali, že virologická odpoveď na darunavir užívaný s nízkou dávkou ritonaviru bola znížená, keď boli na začiatku prítomné 3 a viac mutácií spojených s rezistenciou voči darunaviru (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V a L89V) alebo keď sa tieto mutácie vyvinuli počas liečby.
Zvýšenie východiskovej násobnej zmeny darunaviru v EC50 (FC) súviselo s poklesom virologickej odpovede. Určila sa dolná a horná klinická hranica 10 a 40. Izoláty s východiskovou hodnotou FC
≤ 10 sú citlivé; izoláty s FC > 10 až 40 majú zníženú citlivosť; izoláty s FC > 40 sú rezistentné (pozri
Klinické výsledky).
Vírusy izolované u pacientov s dávkou darunavir/ritonavir 600/100 mg dvakrát denne, u ktorých sa vyskytlo spontánne virologické zlyhanie, ktoré boli citlivé na tipranavir na začiatku, zostali vo veľkej väčšine prípadov citlivé na tipranavir po liečbe.
Najnižšie miery rozvoja rezistencie vírusu HIV sú pozorované u pacientov predtým neliečených ART, ktorí sú po prvýkrát liečení darunavirom v kombinácii s inou ART.
Tabuľka nižšie zobrazuje vývoj HIV-1 proteázových mutácií a stratu citlivosti na PI pri virologickom zlyhaní v závere liečby v štúdiách ARTEMIS, ODIN a TITAN.
ARTEMIS
192. týždeň darunavir / ritonavir
ODIN
48. týždeň
darunavir /
ritonavir
darunavir /
ritonavir
TITAN
48. týždeň darunavir / ritonavir
800/100 mg
jedenkrát denne
N=343
800/100 mg
jedenkrát denne
N=294
600/100 mg
dvakrát denne
N=296
600/100 mg
dvakrát denne
N=298
Celkový počet virologických zlyhanía, n (%)
55 (16,0 %) 65 (22,1 %) 54 (18,2 %) 31 (10,4 %)
Spontánne 39 (11,4 %) 11 (3,7 %) 11 (3,7 %) 16 (5,4 %)
Jedinci nereagujúci na
liečbu
16 (4,7 %) 54 (18,4 %) 43 (14,5 %) 15 (5,0 %)
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými genotypmi, u ktorých sa vyvinuli mutácieb v závere liečby, n/N
Primárne (významné)
mutácie PI
0/43 1/60 0/42 6/28
PI RAM 4/43 7/60 4/42 10/28
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými fenotypmi, u ktorých sa preukázala strata citlivosti na PI v závere v porovnaní so začiatkom liečby, n/N
PI
darunavir 0/39 1/58 0/41 3/26 amprenavir 0/39 1/58 0/40 0/22 atazanavir 0/39 2/56 0/40 0/22 indinavir 0/39 2/57 0/40 1/24 lopinavir 0/39 1/58 0/40 0/23 sakvinavir 0/39 0/56 0/40 0/22 tipranavir 0/39 0/58 0/41 1/25
a TLOVR ne-VF cenzurovaný algoritmus založený na HIV-1 RNA < 50 kópií/ml, okrem TITAN (HIV-1 RNA
< 400 kópií/ml)
b zoznamy IAS-USA
Nízke miery rozvoja rezistencie vírusu HIV-1 boli pozorované u pacientov doposiaľ neliečených
ART, ktorí sú po prvýkrát liečení darunavirom/kobicistatom jedenkrát denne v kombinácii s inou ART
a u pacientov predtým liečených ART bez RAM voči darunaviru, ktorí dostávali darunavir/kobicistat v kombinácii s inou ART. Tabuľka nižšie zobrazuje vývoj HIV-1 proteázových mutácií a rezistenciu voči PI pri virologickom zlyhaní v závere liečby v štúdii GS-US-216-130.
Doposiaľ neliečení pacienti
GS-US-216-130
48. týždeň
Predtým liečení pacienti
darunavir/kobicistat 800/150 mg jedenkrát denne
N=295
darunavir/kobicistat 800/150 mg jedenkrát denne
N=18
Počet pacientov s virologickým zlyhaníma a údajmi o genotype, u ktorých sa vyvinuli mutácie b v závere
liečby, n/N
Primárne (významné)
mutácie PI
0/8 1/7

PI RAM 2/8 1/7
Počet pacientov s virologickým zlyhaníma a údajmi o fenotype, ktoré poukazujú na rezistenciu na PI v závere
liečbyc, n/N
HIV PI
darunavir 0/8 0/7 amprenavir 0/8 0/7 atazanavir 0/8 0/7 indinavir 0/8 0/7 lopinavir 0/8 0/7 sakvinavir 0/8 0/7 tipranavir 0/8 0/7
a Virologické zlyhanie bolo definované ako: nikdy nepotlačené: potvrdené zníženie HIV-1 RNA < 1 log10 oproti východiskovej hodnote a ≥ 50 kópií/ml v 8. týždni; rebound: HIV-1 RNA < 50 kópií/ml s následným potvrdením HIV-1 RNA na ≥ 400 kópií/ml alebo potvrdený nárast > 1 log10 HIV-1 RNA z najnižšieho bodu; ukončenie s HIV-1
RNA ≥ 400 kópií/ml pri poslednej návšteve
b zoznamy IAS-USA
c V GS-US-216-130 nebol úvodný fenotyp dostupný.
SkríženárezistenciaNásobná zmena (FC) darunaviru bola menej ako 10 pre 90 % z 3309 klinických izolátov rezistentných
voči amprenaviru, atazanaviru, indinaviru, lopinaviru, nelfinaviru, ritonaviru, sakvinaviru a/alebo tipraniviru, čo naznačuje, že vírusy rezistentné voči väčšine PI zostávajú citlivé voči darunaviru.
V štúdii
ARTEMIS sa v prípade virologického zlyhania nepozorovala žiadna skrížená rezistencia
s inými PI. V štúdii GS-US-216-130 sa v prípade virologického zlyhania nepozorovala žiadna skrížená
rezistencia s inými HIV PI.
Klinické výsledky
Vplyv kobicistatu na zlepšenie farmakokinetiky darunaviru sa hodnotil v štúdii fázy I u zdravých osôb, ktorým sa podával darunavir 800 mg buď so 150 mg kobicistatu alebo so 100 mg ritonaviru jedenkrát denne. Farmakokinetické parametre darunaviru boli v ustálenom stave porovnateľné, keď bol posilnený kobicistatom alebo ritonavirom. Informácie o kobicistate si pozrite v Súhrne charakteristických vlastností kobicistatu.
Dospelí pacienti
Úči nnosť darunavi ru 800 mg j edenkr át denne uží vaného s o 150 mg kobicistatu jedenkrát denne
u doposi aľ nel i eče nýc h paci ent ov a u paci ent ov pre dt ým l i eče ných ART
GS-US-216-130 je otvorená štúdia fázy III s jednou skupinou, ktorá hodnotila farmakokinetiku, bezpečnosť, tolerovateľnosť a účinnosť darunaviru s kobicistatom u 313 dospelých pacientov infikovaných HIV-1 (295 doposiaľ neliečených pacientov a 18 predtým liečených pacientov). Títo pacienti dostávali darunavir 800 mg jedenkrát denne v kombinácii s kobicistatom 150 mg jedenkrát denne so základným režimom zvoleným skúšajúcim, ktorý obsahoval 2 aktívne NRTI.
U pacientov infikovaných HIV-1, ktorí boli vhodní pre túto štúdiu, skríningové vyšetrenie rezistencie genotypu nepreukázalo RAM voči darunaviru a skríningové vyšetrenie plazmatickej HIV-1 RNA bolo
≥ 1000 kópií/ml. Tabuľka nižšie uvádza údaje o účinnosti z analýzy v 48. týždni štúdie
GS-US-216-130:
Výsledky v 48. týždni
Doposiaľ neliečení pacienti darunavir/kobicistat
800/150 mg jedenkrát denne + OBR N=295
GS-US-216-130
Predtým liečení pacienti darunavir/kobicistat
800/150 mg jedenkrát denne + OBR N=18
Všetci pacienti
darunavir/kobicistat
800/150 mg jedenkrát denne + OBR N=313
HIV-1 RNA < 50 kópií/mla 245 (83,1 %) 8 (44,4 %) 253 (80,8 %)
priemerná zmena
v HIV-1 RNA log oproti východiskovej hodnote (log10 kópií/ml) Priemerná zmena v počte buniek CD4+ oproti východiskovej hodnoteb
a Imputácie na základe algoritmu TLOVR
b Posledné pozorovanie sa dopočítalo LOCF
-3,01 -2,39 -2,97
+174 +102 +170
Úči nnosť darunaviru 800 mg jedenkrát denne uží vanej so 100 mg ritonaviru jedenkrát denne
u pacientov doposiaľ neli ečený ch ART
Dôkaz účinnosti darunaviru/ritonaviru 800/100 mg jedenkrát denne je založený na analýze 192-
týždňových údajov z randomizovanej, kontrolovanej, otvorenej štúdie fázy III ARTEMIS u pacientov infikovaných HIV-1, ktorí doposiaľ neboli liečení antiretrovírusovou liečbou v porovnaní
s darunavirom/ritonavirom 800/100 mg jedenkrát denne s lopinavirom/ritonavirom 800/200 mg denne
(užívané dvakrát denne, alebo jedenkrát denne). Obe ramená používali fixný základný režim, ktorý
pozostával z tenofovir disoproxil fumarátu 300 mg jedenkrát denne a emtricitabinu 200 mg jedenkrát denne.
Nižšie uvedená tabuľka uvádza údaje 48-týždňovej a 96-týždňovej analýzy účinnosti zo štúdie
ARTEMIS:
ARTEMIS
Týždeň 48a Týždeň 96b
Výsledky Darunavir/
ritonavir
800/100 mg jedenkrát denne N=343
Lopinavir/
ritonavir
800/200 mg denne N=346
Rozdiel v liečbe (95 % CI rozdielu)
Darunavir/
ritonavir
800/100 mg jedenkrát denne N=343
Lopinavir/
ritonavir
800/200 mg denne N=346
Rozdiel v liečbe (95 % CI rozdielu)
HIV-1 RNA
< 50 kópií/mlc
Všetci pacienti 83,7 % (287)
78,3 % (271)
5,3 %
(-0,5; 11,2)d
79,0 % (271)
70,8 % (245)
8,2 %
(1,7; 14,7)d
S východiskovou
HIV-RNA
< 100 000
S východiskovou
HIV-RNA
≥ 100 000
S východiskovým počtom buniek CD4+ < 200
S východiskovým počtom buniek CD4+ ≥ 200
85,8 %
(194/226)
79,5 % (93/117)
79,4 % (112/141)
86,6 % (175/202)
84,5 %
(191/226)
66,7 % (80/120)
70,3 % (104/148)
84,3 % (167/198)
1,3 %
(-5,2; 7,9)d
12,8 % (1,6; 24,1)d
9,2 %
(-0,8; 19,2)d
2,3 %
(-4,6; 9,2)d
80,5 %
(182/226)
76,1 % (89/117)
78,7 % (111/141)
79,2 % (160/202)
75,2 %
(170/226)
62,5 % (75/120)
64,9 % (96/148)
75,3 % (149/198)
5,3 %
(-2,3; 13,0)d
13,6 % (1,9; 25,3)d
13,9 % (3,5; 24,2)d
4,0 %
(-4,3; 12,2)d
medián zmeny v počte buniek CD4+ oproti východiskovej hodnote (x 106/l)e
137 141 171 188

a Údaje založené na analýzach v 48. týždni b Údaje založené na analýzach v 96. týždni c Imputácie na základe algoritmu TLOVR
d Na základe obvyklého približného odhadu rozdielu v % odpovede
e Pacienti, ktorí neukončili liečbu, sú počítaní ako zlyhanie: pacientom, ktorí prerušili liečbu predčasne, sa priradí zmena
rovná nule.
V 48-týždňovej analýze sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom
/ritonavirom, definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA < 50 kópií/ml, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat
(ITT) aj u pacientov sledovaných v protokole (OP, z angl. On Protocol). Tieto výsledky sa potvrdili v
analýze údajov 96-týždňovej liečby v štúdii
ARTEMIS. Tieto výsledky boli zachované do 192. týždňa liečby v štúdii
ARTEMIS.
Úči nnosť darunaviru 800 mg j edenkr át denne uží vanej so 100 mg ritonaviru jedenkrát denneu pacientov predtým li ečenýc h A RTODIN je randomizovaná otvorená štúdia fázy III porovnávajúca darunavir/ritonavir 800/100 mg
jedenkrát denne a darunavir/ritonavir 600/100 mg dvakrát denne u pacientov infikovaných HIV-1 predtým liečených ART, u ktorých skríningové vyšetrenie rezistencie genotypu nepreukázalo RAM voči darunaviru (t.j. V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V, L89V)
a skríningové vyšetrenie HIV-1 RNA bolo > 1 000 kópií/ml. Analýza účinnosti je založená na liečbe trvajúcej 48 týždňov (pozri tabuľku nižšie). Obe ramená použili optimalizovaný základný režim
(OBR) ≥ 2 NRTI.

ODIN
Výsledky Darunavir/ritonavir
800/100 mg jedenkrát denne + OBR
N=294
Darunavir/ritonavir
600/100 mg dvakrát denne + OBR N=296
Rozdiel v liečbe
(95% CI rozdielu)
HIV-1 RNA
< 50 kópií/mla
S východiskovou HIV-1 RNA (kópie/ml)
72,1 % (212) 70,9 % (210) 1,2 % (-6,1; 8,5)b
< 100,000
≥ 100,000
S východiskovým počtom buniek CD4+ (x 106/l)
≥ 100
< 100
S kmeňom HIV-1
Typ B Typ AE Typ C Inéc
77,6 % (198/255)
35,9 % (14/39)
75,1 % (184/245)
57,1 % (28/49)
70,4 % (126/179)
90,5 % (38/42)
72,7 % (32/44)
55,2 % (16/29)
73,2 % (194/265)
51 6 % (16/31)
72,5 % (187/258)
60,5 % (23/38)
64,3 % (128/199)
91,2 % (31/34)
78,8 % (26/33)
83,3 % (25/30)
4,4 % (-3,0; 11,9)
-15,7 % (-39,2; 7,7)
2,6 % (-5,1; 10,3)
-3,4 % (-24,5; 17,8)
6,1 % (-3,4; 15,6)
-0,7 % (-14,0; 12,6)
-6,1 % (-2,6; 13,7)
-28,2 % (-51,0; -5,3)
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x 106/l)e
108 112 -5d (-25; 16)
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede
c Kmene A1, D, F1, G, K, CRF02_AG, CRF12_BF, a CRF06_CPX
d Rozdiel stredných hodnôt
e Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward)
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom
800/100 mg jedenkrát denne, definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA
< 50 kópií/ml v porovnaní s darunavirom/ritonavirom 600/100 mg dvakrát denne, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat (ITT) aj
u pacientov sledovaných v protokole (On Protocol).
Darunavir/ritonavir 800/100 mg jedenkrát denne sa nemá používať u tých pacientov liečených ART,
ktorí majú jednu alebo viac mutácií spojených s rezistenciou voči darunaviru (DRV-RAM) alebo
HIV-1 RNA ≥ 100 000 kópií/ml alebo počet buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2 a 4.4).
U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje.
Pediatrickí pacientiPediatrickí pacienti vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtýmliečení ARTDIONE je otvorená štúdia fázy II hodnotiaca farmakokinetiku, bezpečnosť, toleranciu a účinnosť
darunaviru s nízkou dávkou ritonaviru u 12 pediatrických pacientov infikovaných HIV-1 vo veku od
12 do menej ako 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení ART. Títo
pacienti dostávali darunavir/ritonavir 800/100 mg jedenkrát denne spolu s inými antiretrovírusovými
látkami. Virologická odpoveď bola definovaná ako pokles vírusového zaťaženia HIV-1 RNA
v plazme o najmenej 1,0 log10 oproti východiskovej hodnote.
DIONE
Výsledky v 48. týždni Darunavir/ritonavir
N = 12
HIV-1 RNA < 50 kópií/mla 83,3 % (10)
CD4+ percentuálna zmena oproti východiskovej 14
hodnoteb
Priemerná zmena počtu buniek CD4+ oproti
východiskovej hodnoteb
≥ 1,0 log10 pokles vírusového zaťaženia v plazme
oproti východiskovej hodnote
a Imputácie na základe algoritmu TLOVR.
221

100 %
b Pacienti, ktorí neukončili liečbu, sú počítaní ako zlyhanie: pacientom, ktorí prerušili liečbu predčasne, sa priradí
zmena rovná nule.
Pre ďalšie výsledky klinických štúdií u dospelých pacientov liečených ART a pediatrických pacientov,
pozri Súhrn charakteristických vlastností liekov darunavir 75 mg, 150 mg, 300 mg.
Gravidita a obdobie po pôrode
Darunavir/ritonavir (600/100 mg dvakrát denne alebo 800/100 mg jedenkrát denne) v kombinácii so základným režimom bol hodnotený v klinickej štúdii s 34 gravidnými ženami (17 v každej skupine) počas druhého a tretieho trimestra a po pôrode. Virologická odpoveď sa udržala počas trvania štúdie v oboch skupinách. U detí narodených 29 pacientkám, ktoré zotrvali na antivirotickej liečbe až do
pôrodu, sa nevyskytol prenos z matky na dieťa. Nevyskytli sa žiadne nové klinicky významné zistenia
týkajúce sa bezpečnosti v porovnaní so známym profilom bezpečnosti darunaviru/ritonaviru u dospelých infikovaných HIV-1 (pozri časti 4.2, 4.4 a 5.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti darunaviru podávaného v kombinácii s kobicistatom alebo s ritonavirom sa skúmali u zdravých dospelých dobrovoľníkov a u pacientov infikovaných vírusom HIV-1. Expozícia voči darunaviru u pacientov infikovaných vírusom HIV-1 bola vyššia ako u zdravých dobrovoľníkov. Vyššiu expozíciu voči darunaviru u pacientov infikovaných vírusom HIV-1
v porovnaní so zdravými dobrovoľníkmi je možné vysvetliť na základe vyšších koncentrácií α1- kyslého glykoproteínu (AAG) u pacientov infikovaných vírusom HIV-1, čo vedie k vyššej väzbe
darunaviru na plazmatický AAG, a tým aj k vyšším koncentráciám tejto látky v plazme.
Darunavir sa metabolizuje najmä prostredníctvom CYP3A. Kobicistat a ritonavir inhibujú CYP3A, v dôsledku čoho sa výrazne zvyšujú plazmatické koncentrácie darunaviru.
Informácie o farmakokinetických vlastnostiach kobicistatu si pozrite v Súhrne charakteristických vlastností kobicistatu.
Absorpcia
Darunavir sa rýchlo vstrebáva po perorálnom podaní. Pri podávaní v kombinácii s ritonavirom
v nízkej dávke sa maximálne plazmatické koncentrácie darunaviru obyčajne dosiahnu v priebehu 2,5 -
4 hodín.
Absolútna biologická dostupnosť po jednorazovom perorálnom podaní samotného darunaviru v dávke
600 mg bola približne 37 %, pričom pri podávaní ritonaviru v dávke 100 mg dvakrát denne sa zvýšila približne na 82 %. Po jednorazovom perorálnom podaní darunaviru v dávke 600 mg v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne sa systémová expozícia voči darunaviru
zvýšila 14-násobne (pozri časť 4.4).
Pri užívaní nalačno bola relatívna biologická dostupnosť darunaviru v prítomnosti kobicistatu alebo nízkej dávky ritonaviru nižšia ako pri užívaní lieku s jedlom. Z toho dôvodu sa tablety darunaviru musia užívať spolu s kobicistatom alebo s ritonavirom a jedlom. Druh jedla nemá vplyv na expozíciu voči darunaviru.
Distribúcia
Približne 95 % darunaviru sa viaže na plazmatické bielkoviny, najmä na α1-kyslý glykoproteín.
Po intravenóznom podaní bol distribučný objem samotného darunaviru 88,1 ± 59,0 l (stredný ± SD)
a v prítomnosti 100 mg ritonaviru podávaného dvakrát denne bol zvýšený na 131 ± 49,9 l
(stredný ± SD).
Biotransformácia
In vitro štúdie využívajúce mikrozómy pečene človeka (HLM) naznačujú, že darunavir sa
metabolizuje najmä oxidatívnym metabolizmom. Darunavir sa výrazne metabolizuje prostredníctvom systému CYP v pečeni a takmer výlučne prostredníctvom izoenzýmu CYP3A4. V štúdii skúmajúcej aplikáciu darunaviru označeného rádioaktívnym 14C zdravým dobrovoľníkom sa zistilo, že väčšina rádioaktivity v plazme po jednorazovom podaní darunaviru s ritonavirom v dávke 400/100 mg pochádzala z pôvodného liečiva. U ľudí sa zistila prítomnosť najmenej 3 oxidatívnych metabolitov darunaviru, pričom všetky z nich mali aktivitu proti divokému typu HIV najmenej 10x nižšiu ako
darunavir.
Eliminácia
Po podaní darunaviru s ritonavirom v dávke 400/100 mg označeného rádioaktívnym 14C sa 79,5 %
rádioaktivity zachytilo v stolici a 13,9 % rádioaktivity sa zachytilo v moči. Rádioaktivita nezmeneného darunaviru v stolici predstavovala 41,2 % a v moči predstavovala 7,7 % z podanej dávky. Terminálny polčas eliminácie darunaviru podávaného v kombinácii s ritonavirom bol približne
15 hodín.
Klírens darunaviru (150 mg) podaného intravenózne v monoterapii bol 32,8 l/h, kým pri kombinácii s nízko dávkovaným ritonavirom 5,9 l/h.
Osobitné skupiny pacientov
Pediatrická populácia
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 74 predtým liečených pediatrických pacientov vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg preukázala, že dávky
darunaviru/ritonaviru podávané v závislosti od hmotnosti, mali za následok expozíciu darunaviru
porovnateľnú s expozíciou u dospelých pacientov užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 14 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 15 kg až < 20 kg ukázala, že dávkovanie založené na hmotnosti viedlo k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 12 pediatrických pacientov vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení ART, ukázala, že dávka darunaviru/ritonaviru 800/100 mg jedenkrát denne viedla k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne. Z toho dôvodu sa rovnaké dávkovanie jedenkrát denne môže použiť u dospievajúcich vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg
s predchádzajúcou liečbou bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)*
a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+
≥ 100 buniek x 106/l (pozri časť 4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 10 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 14 kg až < 20 kg preukázala, že dávky odvodené od hmotnosti viedli k expozícii darunaviru, ktorá bola porovnateľná
s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne (pozri časť 4.2). Farmakokinetické modelovanie a simulácia expozície darunaviru u pediatrických pacientov vo veku 3 až < 18 rokov navyše potvrdili expozície darunaviru, ktoré boli pozorované v klinických
štúdiách a umožnili určenie dávkovania darunaviru/ritonaviru jedenkrát denne v závislosti na hmotnosti u pediatrických pacientov s hmotnosťou najmenej 15 kg, ktorí buď predtým nedostávali liečbu ART alebo u predtým liečených pediatrických pacientov bez DRV-RAM* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri časť
4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V.
Starší ľudia
Pri analýze farmakokinetiky v rôznych populáciách pacientov s infekciou HIV sa zistilo, že farmakokinetika darunaviru sa výraznejšie nelíši u pacientov s infekciou HIV vo vekovom rozmedzí od 18 do 75 rokov (n = 12, vek ³ 65 rokov) (pozri časť 4.4). U pacientov starších ako 65 rokov boli
k dispozícii iba obmedzené údaje.
Pohlavie
Pri analýze farmakokinetiky v rôznych populáciách pacientov sa u žien s infekciou HIV zistila mierne
vyššia expozícia voči darunaviru (o 16,8 %) ako u mužov infikovaných HIV. Tento rozdiel nemá žiadny klinický význam.
Porucha funkcie obličiek
Výsledky štúdie zachovania hmotnosti s 14C rádioaktívne označeným darunavirom s ritonavirom ukázali, že približne 7,7 % z podanej dávky darunaviru sa vylučuje do moču v nezmenenej forme.
Aj keď sa podávanie darunaviru nesledovalo u pacientov s poruchou funkcie obličiek, pri analýze farmakokinetiky v rôznych populáciách pacientov sa zistilo, že stredne závažná porucha funkcie obličiek (klírens kreatinínu: 30 - 60 ml/min, n=20) nemá väčší vplyv na farmakoniketiku darunaviru u pacientov s infekciou HIV (pozri časti 4.2 a 4.4).
Porucha funkcie pečene
Darunavir sa metabolizuje a eliminuje prevažne v pečeni. V štúdii s opakovaným podaním lieku darunavir užívaného s ritonavirom (600/100 mg dvakrát denne) sa preukázalo, že celkové koncentrácie darunaviru v plazme u subjektov s miernou (Childova-Pughova trieda A, n=8) a stredne ťažkou (Childova-Pughova trieda B, n=8) poruchou funkcie pečene boli porovnateľné s tými u zdravých subjektov. Koncentrácie voľného darunaviru boli približne o 55 % (Childova-Pughova Trieda A) a 100 % (Childova-Pughova trieda B) vyššie. Klinický význam tohto nárastu nie je známy, preto treba darunavir užívať so zvýšenou opatrnosťou. Účinok ťažkej poruchy funkcie pečene na farmakokinetiku darunaviru nebol dosiaľ študovaný (pozri časti 4.2, 4.3 a 4.4).
Gravidita a obdobie po pôrode
Expozícia celkovému darunaviru a ritonaviru po užití darunaviru/ritonaviru 600/100 mg dvakrát denne a darunaviru/ritonaviru 800/100 mg jedenkrát denne v rámci antivirotického režimu bola všeobecne nižšia počas gravidity v porovnaní s obdobím po pôrode. V prípade neviazaného (t.j. aktívneho) darunaviru však boli farmakokinetické parametre menej znížené počas gravidity v porovnaní
s obdobím po pôrode z dôvodu zvýšenia neviazanej frakcie darunaviru počas gravidity v porovnaní s obdobím po pôrode.
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru
600/100 mg dvakrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho trimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=11)aTretí trimester gravidity (n=11)Obdobie po pôrode (6-12 týždňov) (n=11)
Cmax, ng/ml 4 601 ± 1 125 5 111 ± 1 517 6 499 ± 2 411
AUC12h, ng.h/ml 38 950 ± 10 010 43 700 ± 16 400 55 300 ± 27 020
Cmin, ng/mlb 1 980 ± 839,9 2 498 ± 1 193 2 711 ± 2 268
a n=10 pre AUC12h
b vylúčením hodnoty Cmin pod LLOQ, n=10 pre referenciu
F
armakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru 800/100 mg jedenkrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho trimestra gravidity a po pôrode
 F
armakokinetika celkového darunaviru
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=16)Tretí trimester gravidity (n=14)Obdobie po pôrode (6-12 týždňov) (n=15)
Cmax, ng/ml 4 988 ± 1 551 5 138 ± 1 243 7 445 ± 1 674
AUC12h, ng.h/ml 61 303 ± 16 232 60 439 ± 14 052 94 529 ± 28 572
Cmin, ng/mla 1 193 ± 509 1 098 ± 609 1 572 ± 1 108
a n=12 pre obdobie po pôrode, n=15 pre druhý trimester a n=14 pre tretí trimester
U žien užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 28 %, 24 %
a 17 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 19 %, 17 % nižšie a o 2 % vyššie, v tomto poradí, v porovnaní s obdobím po pôrode.
U žien užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 34 %, 34 % a 32 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 31 %, 35 % a 50 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode.
5.3 Predklinické údaje o bezpečnosti
Toxikologické štúdie so samotným darunavirom sa vykonali v dávkovaní až do klinických hladín
u myší, potkanov a psov, kým štúdie s jeho kombináciou s ritonavirom sa vykonali u potkanov a psov.
V štúdiách toxicity po opakovanej dávke u myší, potkanov a psov sa zistili len obmedzené účinky liečby darunavirom. Identifikovaným cieľovým orgánom u hlodavcov bol hemopoetický systém, koagulačný systém, pečeň a štítna žľaza. Pozoroval sa premenlivý, ale mierny pokles parametrov v súvislosti s erytrocytmi spolu s predĺžením aktivovaného parciálneho tromboplastínového času.
Zmeny boli pozorované v pečeni (hypertrofia hepatocytov, vakuolizácia, zvýšenie hladiny pečeňových enzýmov) a štítnej žľaze (folikulárna hypertrofia). U laboratórnych potkanov viedla kombinácia darunaviru s ritonavirom k miernemu zvýšeniu účinku na parametre červených krviniek, pečene
a štítnu žľazu a k zvýšeniu incidencie ostrovčekov fibrózy v pankrease (iba u samcov laboratórnych potkanov) v porovnaní so samotným darunavirom. U psov sa nepozorovali žiadne väčšie známky
toxicity ani cieľové orgány toxicity pri rovnakej expozícii ako u človeka, ktorý užíva odporučené
dávky.
V štúdii na potkanoch bol znížený počet corpora lutea a implantácií v prípade maternálnej toxicity. Na druhej strane sa nepozorovali žiadne účinky na párenie alebo plodnosť po podávaní darunaviru
v dávkach do 1000 mg/kg/deň a pri expozícii (AUC - 0,5-násobok) nižšej ako u človeka dostávajúceho odporučené dávky. Pri podávaní rovnako veľkých dávok samotného darunaviru sa u potkanov
a králikov nepozorovala teratogenita podobne ako u myší dostávajúcich darunavir v kombinácii s ritonavirom. Stupeň expozície bol nižší ako u človeka dostávajúceho odporučené dávky. Pri
hodnotení prenatálneho a postnatálneho vývoja u potkanov, darunavir podávaný samostatne alebo
v kombinácii s ritonavirom viedol k prechodnému zníženiu prírastku telesnej hmotnosti u dojčených mláďat a došlo k miernemu oneskoreniu v otvorení očí a uší. Darunavir v kombinácii s ritonavirom
spôsobil zníženie prežívania mláďat, ktoré sa prejavilo nepriaznivou odpoveďou v 15. deň laktácie
a znížením počtu mláďat, ktoré prežili počas laktácie. Tieto účinky môžu byť prisúdené sekundárnej expozícii liečiva u mláďat prostredníctvom materského mlieka a/alebo maternálnej toxicity. Darunavir
podávaný ako samotný alebo v kombinácii s ritonavirom neovplyvnil žiadne funkcie po odstavení.
U juvenilných laboratórnych potkanov, ktorí dostávali darunavir 23. – 26. deň, sa pozorovala zvýšená
mortalita s kŕčmi u niektorých zvierat. Medzi 5. a 11. dňom života bola expozícia v plazme, pečeni
a mozgu výrazne vyššia ako u dospelých laboratórnych potkanov po porovnateľných dávkach
v mg/kg. Po 23. dni života bola expozícia porovnateľná s tou u dospelých potkanov. Zvýšená
expozícia bola pravdepodobne aspoň čiastočne z dôvodu nezrelosti enzýmov metabolizujúcich liek
u juvenilných zvierat. U juvenilných potkanov neboli spozorované žiadne úmrtia v súvislosti s liečbou
dávkami 1000 mg/kg darunaviru (jednorazová dávka) v 26. deň veku alebo 500 mg/kg (opakovaná dávka) od 23. do 50. dňa veku a expozícia a profil toxicity boli porovnateľné s tými, pozorovanými u dospelých potkanov.
Karcinogénny potenciál darunaviru bol vyhodnotený pri podávaní lieku myšiam a potkanom sondou do žalúdka počas 104 týždňov. Myšiam boli podávané denné dávky 150, 450 a 1000 mg/kg a potkanom boli podávané dávky 50, 150 a 500 mg/kg. Výskyt hepatocelulárnych adenómov a karcinómov stúpal v závislosti od dávky a pozoroval sa u samcov a samíc oboch živočíšnych druhov. U samcov potkanov sa vyskytli adenómy folikulárnych buniek štítnej žľazy. Podávanie darunaviru nespôsobilo štatisticky významný nárast iných benígnych alebo malígnych novotvarov u myší alebo potkanov. Pozorované hepatocelulárné tumory a tumory štítnej žľazy u hlodavcov sa považujú za obmedzené z hľadiska významnosti pre človeka. Opakované podávanie darunaviru potkanom spôsobilo indukciu mikrozomálnych enzýmov v pečeni a zvýšilo vylučovanie hormónov štítnej žľazy,
ktoré predurčuje potkanov, ale nie ľudí, na výskyt neoplaziem štítnej žľazy. Pri najvyšších testovaných dávkach boli systémové expozície (založené na AUC) darunaviru 0,4- až 0,7-násobkom (myši) a 0,7-
až 1-násobkom (potkany) v pomere k expozíciám pozorovaným u ľudí pri odporúčaných
terapeutických dávkach.
Po dvoch rokoch podávania darunaviru pri expozíciách na úrovni alebo pod úrovňou expozícií u ľudí sa pozorovali zmeny na obličkách u myší (nefróza) a u potkanov (chronická progresívna nefropatia).
Darunavir nebol mutagénny ani genotoxický v súbore in vitro a in vivo testov vrátane bakteriálnej reverznej mutácie (Amesov test), chromozómovej aberácie v ľudských lymfocytoch
a mikronukleového testu u myší in vivo.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Oxid kremičitý, koloidný bezvodý
Mikrokryštalická celulóza
Krospovidón
Sodná soľ glykolátu škrobu Hypromelóza Magnéziumstearát
Obal tablety
Polyvinylalkohol, čiastočne hydrolyzovaný
Oxid titaničitý
Makrogol
Mastenec
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po prvom otvorení HDPE fľaše: 100 dní.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaPVC/PE/PVDC-Al blistrové balenie s obsahom 30 a 60 tabliet a 60 x 1 tabliet.
Za studena spracované PVC/Al/OPA-Al blistrové balenie s obsahom 30 a 60 tabliet a 60 x 1 tabliet.
Balenie s HDPE fľašou s PP skrutkovacím uzáverom s obsahom 60 a 100 tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomŽiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMylan S.A.S.
117 Allee des Parcs
69 800 Saint Priest
Francúzsko
8. REGISTRAČNÉ ČÍSLOEU/1/16/1140/022
EU/1/16/1140/023
EU/1/16/1140/024
EU/1/16/1140/025
EU/1/16/1140/026
EU/1/16/1140/027
EU/1/16/1140/028
EU/1/16/1140/029
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: [DD. mesiac RRRR]
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/ema/.
1. NÁZOV LIEKU
Darunavir Mylan 600 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Každá filmom obalená tableta obsahuje 600 mg darunaviru. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalené tablety.
Biele až sivobiele oválne obojstranne vypuklé filmom obalené tablety s rozmermi približne 21,2 mm x
10,6 mm, s označením „M“ na jednej strane a „DV5“ na druhej strane.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Darunavir podávaný súčasne s nízkou dávkou ritonaviru je indikovaná v kombinácii s inými antiretrovirálnymi liekmi na liečbu pacientov s infekciou vyvolanou vírusom ľudskej imunodeficiencie (HIV-1).
Darunavir 600 mg tablety sa môžu použiť na zabezpečenie vhodného režimu dávkovania (pozri časť
4.2):
· Pri liečbe infekcie HIV-1 u dospelých pacientov predtým liečených antiretrovírusovou liečbou
(ART, antiretroviral treatment), vrátane tých s intenzívnou predchádzajúcou liečbou.
· Pri liečbe infekcie HIV-1 u detí a dospievajúcich od 3 rokov a s telesnou hmotnosťou najmenej
15 kg.
Pri rozhodovaní o začatí liečby darunavirom užívaným spolu s nízkou dávkou ritonaviru je potrebné dôkladne zvážiť liekovú anamnézu jednotlivého pacienta a schémy mutácií súvisiace s rôznymi agensmi. Používanie darunaviru má byť odvodené od geno- alebo fenotypizácie (ak sú dostupné)
a liečebnej anamnézy.
4.2 Dávkovanie a spôsob podávania
Liečbu môže začať len lekár, ktorý má skúsenosti s liečbou infekcie HIV. Po začatí liečby darunavirom treba pacientov poučiť, aby nemenili dávku, liekovú formu ani neprerušovali liečbu bez toho, aby sa poradili so svojím lekárom.
Dávkovanie
Darunavir Mylan sa musí vždy podávať perorálne v kombinácii s nízkou dávkou ritonaviru, ktorý
zlepšuje jej farmakokinetiku, a s inými antiretrovírusovými liekmi. Z toho dôvodu je pred začatím kombinovanej liečby darunavirom a ritonavirom potrebné prečítať si súhrn charakteristických vlastností ritonaviru.
Dospelí pacienti liečení ART
Odporúčané dávkovanie je 600 mg dvakrát denne súčasne so 100 mg ritonaviru dvakrát denne
a s jedlom. darunavir Mylan 75 mg, 150 mg a 600 mg tablety sa môžu použiť na zostavenie režimu
dávkovania 600 mg dvakrát denne.
Užívanie 75 a 150 mg tabliet na dosiahnutie odporúčanej dávky je vhodné v prípade možnej
precitlivenosti na niektoré farbivá alebo v prípade ťažkostí s prehĺtaním 600 mg tabliet.
Dávkovací režim 800 mg jedenkrát denne so 150 mg kobicistatu jedenkrát denne alebo 100 mg ritonaviru jedenkrát denne a s jedlom sa môže použiť u pacientov s predchádzajúcou liečbou antiretrovirotikami, ale bez mutácií spojených s rezistenciouspojených s rezistenciou voči darunaviru (DRV-RAM, z angl. darunavir-resistance associated mutations)* a s plazmatickou hladinou HIV-1
RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri Súhrn charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Dospelí pacienti predtým neliečení ARTOdporučené dávkovanie u pacientov predtým neliečených ART nájdete v Súhrne charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Pediatrickí pacienti predtým neliečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 15 kg)Dávka darunaviru a ritonaviru u pediatrických pacientov odvodená od ich hmotnosti sa uvádza v tabuľke nižšie.
Odporúčaná dávka tabliet darunaviru a ritonavirua na liečbu predtým neliečenýchpediatrických pacientov (3 až 17 rokov)Telesná hmotnosť (kg) Dávka (jedenkrát denne s jedlom)≥ 15 kg až < 30 kg 600 mg darunavir/100 mg ritonavir jedenkrát denne
≥ 30 kg až < 40 kg 675 mg darunavir/100 mg ritonavir jedenkrát denne
³ 40 kg 800 mg darunavir/100 mg ritonavir jedenkrát denne
a ritonavir perorálny roztok: 80 mg/ml
Pediatrickí pacienti liečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 15 kg)Zvyčajne sa odporúča darunavir dvakrát denne súčasne s ritonavirom a s jedlom.
Dávkovanie darunaviru užívaného s ritonavirom jedenkrát denne a s jedlom sa môže použiť
u pacientov s predchádzajúcou liečbou antiretrovirotikami, ale bez mutácií spojených s rezistenciou
voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom
buniek CD4+ ³ 100 buniek x 106/l.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
U pediatrických pacientov je odporúčaná dávka darunaviru s nízkou dávkou ritonaviru odvodená od hmotnosti a nemá presiahnuť odporúčanú dávku pre dospelých (600/100 mg dvakrát denne alebo
800/100 mg jedenkrát denne).
Odporúčaná dávka tabliet darunaviru a ritonavirua u predtým liečenýchpediatrických pacientov (3 až 17 rokov)
Hm
o
t
nosť (kg) Dávka (jedenkrát denne s jedlom)
≥ 15 kg–< 30 kg 600 mg darunavir/100 mg ritonavir jedenkrát denne
≥ 30 kg–< 40 kg 675 mg darunavir/100 mg ritonavir jedenkrát denne
≥ 40 kg 800 mg darunavir/100 mg ritonavir jedenkrát denne
a s perorálnym roztokom ritonaviru: 80 mg/ml
Dávka (dvakrát denne s jedlom)
375 mg darunavir/50 mg ritonavir dvakrát denne
450 mg darunavir/60 mg ritonavir dvakrát denne
600 mg darunavir/100 mg ritonavir dvakrát denne
U pediatrických pacientov predtým liečených ART sa odporúča testovanie genotypu HIV. Ak
testovanie genotypu HIV nie je k dispozícii, dávkovanie darunaviru/ritonaviru jedenkrát denne sa
odporúča u pediatrických pacientov, ktorí predtým neužívali HIV proteázové inhibítory, a dávkovanie
dvakrát denne sa odporúča u pacientov, ktorí predtým užívali HIV proteázové inhibítory.
O
dporúčanie pri vynechaní dávky
V prípade, že pacient zabudne užiť dávku darunaviru a/alebo ritonaviru do 6 hodín od doby, kedy liek zvyčajne užíva, treba ho poučiť, že má užiť predpísanú dávku s jedlom čo najskôr. Ak od zvyčajnej doby použitia uplynie viac ako 6 hodín, vynechaná dávka sa nemá užiť a pacient má pokračovať vo zvyčajnom dávkovacom režime.
Toto usmernenie vyplynulo z 15 hodinového polčasu darunaviru v prítomnosti ritonaviru a
odporúčaného dávkovacieho intervalu približne 12 hodín.
Osobitné skupiny pacientov
Starší ľudia
Informácie o tejto populácii sú obmedzené, a preto sa má darunavir používať v tejto vekovej skupine
so zvýšenou opatrnosťou (pozri časti 4.4 a 5.2).
Porucha funkcie pečene
Darunavir sa metabolizuje v pečeňovom systéme. U pacientov s miernou (Childova-Pughova Trieda
A) alebo stredne ťažkou (Childova-Pughova Trieda B) poruchou funkcie pečene sa neodporúča žiadna úprava dávky, avšak týmto pacientom sa má darunavir podávať so zvýšenou opatrnosťou. U pacientov
s ťažkou poruchou funkcie pečene nie sú k dispozícii žiadne farmakokinetické údaje. Ťažká porucha
funkcie pečene môže viesť k zvýšeniu expozície darunaviru a k zhoršeniu jeho bezpečnostného profilu. Z toho dôvodu sa darunavir nesmie podávať pacientom s ťažkou poruchou funkcie pečene (Childova-Pughova Trieda C) (pozri časti 4.3, 4.4 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávkovania (pozri časti 4.4
a 5.2).
Pediatrickí pacienti
Darunavir/ritonavir sa nemá používať u detí s hmotnosťou menej ako 15 kg, pretože dávka pre túto populáciu nebola stanovená na dostatočnom počte pacientov (pozri časť 5.1). Darunavir/ritonavir sa
nemá používať u detí mladších ako 3 roky z dôvodu bezpečnosti (pozri časti 4.4 a 5.3).
Boli stanovené expozície darunaviru u dospievajúcich vo veku 12 až 17 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení a ktorí dostávali darunavir 800 mg jedenkrát denne a ukázalo sa, že sa nachádzali v rámci terapeutického rozsahu, ktorý bol stanovený u dospelých pacientov dostávajúcich darunavir 800 mg jedenkrát denne. Ako dôsledok vzhľadom na to, že
darunavir jedenkrát denne bol tiež zaregistrovaný na použitie u dospelých s predchádzajúcou liečbou
bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1
RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l, rovnaká indikácia darunaviru
jedenkrát denne sa vzťahuje na deti vo veku 3 až 17 rokov a s hmotnosťou najmenej 15 kg s predchádzajúcou liečbou.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Gravidita a obdobie po pôrode
Počas gravidity a po pôrode nie je potrebná úprava dávky darunaviru/ritonaviru. Darunavir sa má
užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko (pozri časti 4.4, 4.6 a 5.2).
Spôsob podávania
Pacientov treba poučiť, aby užívali Darunavir Mylan s nízkou dávkou ritonaviru do 30 minút po jedle.
Druh jedla neovplyvňuje expozíciu darunaviru (pozri časti 4.4, 4.5 a 5.2).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Pacienti s ťažkou (Childova-Pughova Trieda C) poruchou funkcie pečene.
Súčasné podávanie rifampicínu a darunaviru spolu s nízkou dávkou ritonaviru (pozri časť 4.5). Súčasné podávanie lieku s kombináciou lopinavir/ritonavir (pozri časť 4.5).
Súčasné podávanie rastlinných preparátov obsahujúcich výťažok z ľubovníka bodkovaného
(Hypericum perforatum) (pozri časť 4.5).
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru a liečiv, ktorých metabolizmus (klírens) je vysoko závislý na CYP3A, a ktorých zvýšené plazmatické koncentrácie sú spojené s vážnymi a/alebo život ohrozujúcimi udalosťami. Medzi tieto liečivá patria napr.:
- alfuzosín (antagonista alfa 1-adrenoreceptora)
- amiodarón, bepridil, dronedarón, chinidín, ranolazín, lidokaín (podávaný systémovo) (antiarytmiká/lieky proti angíne)
- astemizol, terfenadín (antihistaminiká)
- kolchicín, keď sa používa u pacientov s poruchou funkcie obličiek a/alebo pečene (liek proti dne) (pozri časť 4.5)
- námeľové alkaloidy (napr. dihydroergotamín, ergometrín, ergotamín, metylergonovín)
- cisaprid (prokinetiká)
- pimozid, kvetiapín, sertindol (antipsychotiká/neuroleptiká) (pozri časť 4.5)
- triazolam, midazolam podávaný perorálne (sedatíva/hypnotiká) (pre upozornenie na midazolam
podávaný parenterálne, pozri časť 4.5)
- sildenafil - keď sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, avanafil (inhibítory
PDE-5)
- simvastatín a lovastatín (inhibítory HMG-CoA reduktázy) (pozri časť 4.5)
- tikagrelor (inhibítor agregácie trombocytov) (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hoci sa preukázalo, že účinná vírusová supresia dosiahnutá pri antiretrovírusovej terapii značne znižuje riziko prenosu HIV pohlavným stykom, reziduálne riziko nie je možné vylúčiť. Je potrebné prijať opatrenia na zabránenie prenosu HIV v súlade s národnými odporúčaniami.
Odporúča sa pravidelné sledovanie virologickej odpovede. V prípade nedostatočnej alebo žiadnej virologickej odpovede sa má urobiť test na rezistenciu.
Darunavir sa má používať len v kombinácii s nízkou dávkou ritonaviru, ktorý zlepšuje farmakokinetiku darunaviru (pozri časť 5.2).
Zvyšovanie dávkovania ritonaviru oproti odporúčaniam v časti 4.2 významnejšie neovplyvňuje
koncentrácie darunaviru a neodporúča sa.
Darunavir sa viaže prevažne na a1-kyslý glykoproteín. Táto proteínová väzba závisí od koncentrácie, ktorú určuje saturácia väzby. Preto lieky, ktoré sa vysoko viažu na a1-kyslý glykoproteín, môžu znemožniť túto väzbu (pozri časť 4.5).
Pacienti liečení ART – dávkovanie jedenkrát denne
Darunavir užívaný v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru jedenkrát denne
u pacientov liečených ART sa nemá používať u pacientov s jednou alebo viacerými
mutáciami spojenými s rezistenciou voči darunaviru (DRV-RAM) alebo s hladinou HIV-1 RNA
≥ 100 000 kópií/ml alebo počtom buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2). V tejto populácii sa neskúmali iné kombinácie s optimalizovaným základným režimom (OBR, optimised background
regimen) ako ≥ 2 NRTI. U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje
(pozri časť 5.1).
Pediatrická populácia
Darunavir sa neodporúča používať u pediatrických pacientov mladších ako 3 roky alebo s hmotnosťou
menej ako 15 kg (pozri časti 4.2 a 5.3).
Gravidita
Darunavir sa má užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko.
Opatrnosť je potrebná u gravidných žien súbežne užívajúcich lieky, ktoré môžu ďalej znižovať
expozíciu darunaviru (pozri časti 4.5 a 5.2).
Starší ľudia
Keďže k dispozícii sú len obmedzené údaje o používaní darunaviru u pacientov vo veku 65 rokov a
starších, pri podávaní darunaviru starším pacientom je potrebná opatrnosť vzhľadom k vyššiemu výskytu zhoršenej funkcie pečene a pridružených chorôb, alebo k inej liečbe (pozri časti 4.2 a 5.2).
Závažné kožnéreakcie
Počas programu klinického vývoja (N=3 063) boli u 0,4 % pacientov hlásené závažné kožné reakcie,
ktoré mohla sprevádzať horúčka a/alebo zvýšenie transamináz. Zriedkavo (< 0,1 %) bola hlásená
lieková vyrážka s eozinofíliou a systémovými príznakmi (DRESS, drug rash with eosinophilia and systemic symptoms) a Stevensov-Johnsonov syndróm a počas postmarketingových skúseností bola hlásená toxická epidermálna nekrolýza a akútna generalizovaná exantematózna pustulóza. Ak sa objavia prejavy alebo príznaky závažnej kožnej reakcie, liečba darunavirom/ritonavirom sa má okamžite prerušiť. Príznaky môžu zahŕňať, ale nemusia byť obmedzené na, závažnú vyrážku alebo vyrážku sprevádzanú horúčkou, celkovú nevoľnosť, únavu, bolesť svalov alebo kĺbov, pľuzgiere, orálne lézie, konjunktivitídu, hepatitídu a/alebo eozinofíliu
Vyrážka sa častejšie vyskytla u predtým liečených pacientov, ktorí boli liečení darunavirom spolu
s raltegravirom v porovnaní s pacientmi, ktorí užívali darunavir bez raltegraviru alebo raltegravir bez darunaviru (pozri časť 4.8).
Darunavir obsahuje sulfónamidovú funkčnú skupinu. Darunavir sa má podávať opatrne pacientom so
známou alergiou na sulfónamid.
Hepatotoxicita
Počas užívania darunaviru bola hlásená liekom indukovaná hepatitída (napr. akútna hepatitída,
cytolytická hepatitída). Počas programu klinického vývoja (N=3 063) bola hepatitída hlásená u 0,5 %
pacientov, ktorí boli liečení kombinovanou antiretrovirálnou terapiou s darunavirom/ritonavirom. Pacienti s preexistujúcou dysfunkciou pečene, vrátane chronickej aktívnej hepatitídy B alebo C, majú
zvýšené riziko porúch funkcie pečene, vrátane vážnych a potenciálne smrteľných hepatických
nežiaducich reakcií. V prípade súčasnej antivírovej liečby hepatitídy B alebo C, obráťte sa na
relevantnú informáciu o týchto liekoch.
Pred začatím liečby darunavirom/ritonavirom sa majú urobiť vhodné laboratórne vyšetrenia a pacienti majú byť sledovaní aj počas liečby. U pacientov s chronickou hepatitídou, cirhózou alebo u pacientov, ktorí mali pred liečbou zvýšenú hladinu transamináz, treba zvážiť zvýšené sledovanie AST/ALT, najmä počas prvých mesiacov po začatí liečby darunavirom/ritonavirom.
Ak sa vyskytne alebo sa zhorší dysfunkcia pečene (vrátane klinicky významného zvýšenia pečeňových enzýmov a/alebo príznakov ako únava, anorexia, nauzea, žltačka, tmavý moč, citlivosť pečene, hepatomegália) u pacientov užívajúcich darunavir/ritonavir, treba okamžite zvážiť zastavenie alebo prerušenie liečby.
Pacienti s pridruženými ochoreniami
Porucha funkcie pečene
U pacientov so závažnými poruchami pečene sa účinnosť a bezpečnosť darunaviru nestanovovala, preto je darunavir u pacientov s ťažkou poruchou funkcie pečene kontraindikovaný. Vzhľadom na nárast koncentrácie voľného darunaviru v plazme sa darunavir musí používať opatrne u pacientov s miernou alebo stredne ťažkou poruchou funkcie pečene (pozri časti 4.2, 4.3 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa nevyžadujú žiadne osobitné opatrenia alebo úpravy dávkovania. Darunavir aj ritonavir sa výrazne viažu na plazmatické bielkoviny, a preto je
nepravdepodobné, že by sa dali z cirkulácie významnejšie odstrániť pomocou hemodialýzy alebo
peritoneálnej dialýzy. Preto sa u týchto pacientov nevyžadujú žiadne osobitné opatrenia alebo úpravy dávkovania (pozri časti 4.2 a 5.2).
Pacienti s hemofíliou
U pacientov s hemofíliou A a B liečených PI sa opísali prípady väčšieho krvácania, vrátane spontánnych kožných hematómov a krvácania do kĺbov. Niektorým pacientom sa podal dodatočný
faktor VIII. Vo viac ako polovici prípadov sa pokračovalo v liečbe PI, prípadne sa táto liečba obnovila po jej dočasnom prerušení. Bola naznačená kauzálna súvislosť, hoci mechanizmus účinku nebol zatiaľ
objasnený. Z toho dôvodu je potrebné hemofilikov upozorniť na možnosť väčšieho krvácania.
Telesná hmotnosť a metabolické parametre
Počas antiretrovírusovej liečby môže dôjsť k zvýšeniu telesnej hmotnosti a hladín lipidov a glukózy
v krvi. Takéto zmeny môžu čiastočne súvisieť s kontrolou ochorenia a životným štýlom. Pokiaľ ide
o lipidy, v niektorých prípadoch sú dôkazy o vplyve liečby, kým pri prírastku telesnej hmotnosti nie sú
presvedčivé dôkazy o tom, že súvisí s niektorou konkrétnou liečbou. Pri monitorovaní hladín lipidov
a glukózy v krvi sa treba riadiť zavedenými odporúčaniami na liečbu infekcie HIV. Poruchy
metabolizmu lipidov majú byť klinicky vhodne liečené.
Osteonekróza
Napriek tomu, že etiológia ochorenia závisí od mnohých faktorov (vrátane užívania kortikosteroidov,
konzumácie alkoholu, vážnej imunosupresie, vyššieho BMI), prípady osteonekrózy boli hlásené najmä
u pacientov s pokročilým ochorením HIV a/alebo s dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART, combination antiretroviral therapy). Pacientov treba poučiť, aby
vyhľadali lekársku pomoc v prípade, že budú pociťovať bolesti kĺbov, stuhnutosť kĺbov alebo ťažkosti
pri pohybe.
Imunoreštitučný zápalový syndróm
U pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej
antiretrovírusovej liečby môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie, čo môže byť spojené so závažným klinickým stavom resp. so
zhoršením symptómov. Tieto reakcie sa obyčajne pozorujú v priebehu prvých týždňov až mesiacov od
začatia podávania kombinovanej antiretrovírusovej liečby. Príkladom môže byť cytomegalovírusová retinitída, generalizované alebo fokálne mykobaktériové infekcie a pneumónia vyvolaná Pneumocystis jirovecii (predtým známa ako Pneumocystis carinii). Je nutné zhodnotiť všetky príznaky zápalového ochorenia a v prípade potreby začať liečbu. Ďalej sa v klinických štúdiách s darunavirom užívaným s nízkou dávkou ritonaviru pozorovala reaktivácia herpesu simplex a herpesu zoster.
Boli tiež zaznamenané aj poruchy autoimunitného systému (ako je Gravesova choroba) objavujúce sa v dôsledku imunitnej reaktivácie; avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.8).
Liekové interakcie
Niektoré štúdie interakcií sa uskutočnili s darunavirom v nižších dávkach ako sú odporúčané. Účinky
súčasne podávaných liekov môžu byť preto podhodnotené a môže byť potrebné klinické sledovanie bezpečnosti. Pre úplnú informáciu o liekových interakciách pozri časť 4.5.
Efavirenz v kombinácii s darunavirom/ritonavirom 800/100 mg jedenkrát denne môže viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť v kombinácii s darunavirom/ritonavirom, treba použiť režim dávkovania darunavir/ritonavir 600/100 mg dvakrát denne. Pozri súhrn charakteristických vlastností lieku darunavir 75 mg, 150 mg alebo 300 mg tablety (pozri časť 4.5).
Život ohrozujúce a smrteľné liekové interakcie sa zaznamenali u pacientov liečených kolchicínom
a silnými inhibítormi CYP3A a P-glykoproteínu (P-gp; pozri časti 4.3 a 4.5).
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Darunavir a ritonavir sú inhibítormi CYP3A, CYP2D6 a P-gp. Súčasné podávanie darunaviru a ritonaviru s liekmi metabolizovanými najmä CYP3A a/alebo CYP2D6 alebo transportovanými P-gp môže viesť k zvýšeniu systémovej expozície týmto liekom, čo by mohlo zosilniť alebo predĺžiť ich terapeutický účinok a viesť k nežiaducim účinkom.
Darunavir užívaný s nízkou dávkou ritonaviru sa nesmie kombinovať s liekmi, ktorých klírens výrazne závisí od CYP3A, a pri ktorých zvýšená systémová expozícia je sprevádzaná závažnými a/alebo život ohrozujúcimi nežiaducimi udalosťami (t.j. ktoré majú úzky terapeutický index) (pozri časť 4.3).
Celkové zvýšenie farmakokinetického účinku ritonavirom viedlo približne k 14-násobnému zvýšeniu
systémovej expozície darunaviru, po jednorazovej 600 mg perorálnej dávke darunaviru v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne. Z toho dôvodu sa darunavir môže používať
len v kombinácii s nízkou dávkou ritonaviru, ktorý zlepšuje farmakokinetiku darunaviru (pozri časti
4.4 a 5.2).
Klinická štúdia s viacerými liekmi, ktoré sú metabolizované cytochrómami CYP2C9, CYP2C19 a CYP2D6 dokázala nárast aktivity CYP2C9 a CYP2C19 a inhibíciu aktivity CYP2D6 v prítomnosti darunaviru/ritonaviru, čo sa môže pripisovať prítomnosti nízkej dávky ritonaviru. Súčasné podávanie darunaviru a ritonaviru a liekov, ktoré sa predovšetkým metabolizujú prostredníctvom CYP2D6 (napríklad flekainid, propafenon, metoprolol), môže vyústiť do zvýšených plazmatických koncentrácií týchto liekov, čo môže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce reakcie. Súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C9 (napríklad warfarín) a CYP2C19 (napríklad metadon), môže vyústiť do zníženia systémových
expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Napriek tomu, že sa účinok na CYP2C8 sledoval iba in vitro, súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C8 (napríklad paklitaxel, rosiglitazón, repaglinid), môže vyústiť do zníženia systémových expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Lieky, ktoré ovplyvňujú expozíciu darunaviru/ritonaviru
Darunavir a ritonavir sa metabolizujú prostredníctvom CYP3A. Očakáva sa, že lieky indukujúce činnosť CYP3A zvýšia klírens darunaviru a ritonaviru, čo má za následok zníženie plazmatických koncentrácií darunaviru a ritonaviru (napr. rifampicín, ľubovník bodkovaný, lopinavir). Súčasné podávanie darunaviru a ritonaviru v kombinácii s liekmi, ktoré inhibujú CYP3A môže znížiť klírens darunaviru a ritonaviru a môže mať za následok zvýšenie plazmatických koncentrácií darunaviru
a ritonaviru (napr. indinavir, systémové azoly ako ketokonazol a klotrimazol). Tieto interakcie sú opísané v nižšie uvedenej tabuľke interakcií.
Tabuľkainterakcií
Interakcie medzi darunavirom/ritonavirom a antiretrovirotikami a ne-antiretrovírusovými liekmi sú uvedené v tabuľkách nižšie (neurčené ako „ND“ (z angl. not determinated)). Smer šípky pri každom
farmakokinetickom parametri je založený na 90 % intervale spoľahlivosti stredného geometrického
pomeru v rámci 80-125 % rozpätia (↔), pod ním (↓) alebo nad ním (↑).
Niekoľko štúdií interakcií (v tabuľke nižšie označené ako #) sa uskutočnili s nižšími ako odporúčanými dávkami darunaviru alebo s iným režimom dávkovania (pozri časť 4.2 Dávkovanie). Účinky súčasne podávaných liekov môžu byť preto podhodnotené a môže byť potrebné klinické sledovanie bezpečnosti.
INTERAKCIE A ODPORUČENÉ DÁVKOVANIE S INÝMI LIEKMI
L
ieky podľa
t
erapeutických oblastí
I
nterakcia
Z
m
e
na geometrického priemeru (%)
O
dporúčania pri súčasnom
podávaní
ANTIRETROVIROTIKÁ NA HIV
I
nhibítory prenosu reťazcov integrázou
Dolutegravir dolutegravir AUC ↓ 32 % dolutegravir C24h 38 % dolutegravir Cmax ↓ 11 % darunavir ↔*
* Použitím porovnania údajov zo skríženej štúdie
s historickými údajmi o farmakokinetike
Elvitegravir elvitegravir AUC ↔ elvitegravir Cmin ↔ elvitegravir Cmax ↔ darunavir AUC ↔ darunavir Cmin 17 % darunavir Cmax ↔
Raltegravir Niektoré klinické štúdie naznačujú, že raltegravir môže spôsobiť mierny pokles koncentrácie darunaviru v plazme.
Darunavir užívaná s nízkou dávkou ritonaviru a dolutegravir sa môžu užívať bez úpravy dávky.
Keď sa darunavir užívaná súčasne s nízkou dávkou ritonaviru (600/100 mg dvakrát denne) užíva spolu s elvitegravirom, dávka elvitegraviru má byť 150 mg jedenkrát denne.
Farmakokinetika a odporúčania na dávkovanie pre iné dávky darunaviru alebo
s elvitegravirom/kobicistatom neboli stanovené. Súčasné užívanie darunaviru s nízkou dávkou ritonaviru v iných dávkach ako
600/100 mg dvakrát denne
a elvitegraviru sa preto neodporúča. Súčasné užívanie darunaviru
s nízkou dávkou ritonaviru
a elvitegravirom za prítomnosti
kobicistatu sa neodporúča.
V súčasnosti sa účinok raltegraviru na plazmatické koncentrácie darunaviru nejaví ako klinicky významný. darunavir súčasne podávaný s nízkou dávkou ritonaviru a raltegraviru sa môže užívať bez úpravy dávky.
Nukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, nucleo(s/t)ide reverse transcriptase inhibitors)
Didanozín
400 mg jedenkrát denne
didanozín AUC ↓ 9 % didanozín Cmin ND didanozín Cmax ↓ 16 % darunavir AUC ↔ darunavir Cmin ↔ darunavir Cmax ↔
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a didanozínu sa môže užívať bez úpravy dávok.
Didanozín sa má podávať nalačno, preto ho treba užiť 1 hodinu pred alebo 2 hodiny po užití darunaviru/ritonaviru s jedlom.
Tenofovir disoproxil fumarát
300 mg jedenkrát denne
Abakavir Emtricitabín Lamivudín Stavudín Zidovudín
tenofovir AUC ↑ 22 % tenofovir Cmin ↑ 37 % tenofovir Cmax ↑ 24 %
#darunavir AUC ↑ 21 %
#darunavir Cmin ↑ 24 %
#darunavir Cmax ↑ 16 %
(↑ tenofoviru v dôsledku efektu na MDR-1
transport v renálnych tubuloch)
Neskúmalo sa. Vzhľadom na rôzne cesty eliminácie ostatných NRTI zidovudín, emtricitabín, stavudín, lamivudín, ktoré sa vylučujú prevažne renálnou cestou
a abakavir, ktorý nie je metabolizovaný prostredníctvom CYP450, sa pri ich kombinácii s darunavirom užívaným'
s nízkou dávkou ritonaviru
nepredpokladajú žiadne liekové interakcie.
Pri súčasnom podávaní darunaviru a nízkej dávky ritonaviru
v kombinácii s tenofovirom môže byť indikované sledovanie renálnych funkcií, najmä
u pacientov so systémovým alebo renálnym ochorením, alebo
u pacientov užívajúcich
nefrotoxické lieky.
Darunavir užívaný s nízkou dávkou ritonaviru sa môže užívať s týmito NRTI bez úpravy dávky.
Nenukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, non-nucleo(s/t)ide reverse transcriptase inhibitors)
Efavirenz
600 mg jedenkrát denne
Etravirín
100 mg dvakrát denne
Nevirapín
200 mg dvakrát denne
Rilpivirín
150 mg jedenkrát denne
efavirenz AUC ↑ 21 % efavirenz Cmin ↑ 17 % efavirenz Cmax ↑ 15 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↓ 31 %
#darunavir Cmax ↓ 15 %
(↑ efavirenzu v dôsledku inhibície
CYP3A)
(↓ darunaviru v dôsledku inhibície
CYP3A)
etravirín AUC ↓ 37 % etravirín Cmin ↓ 49 % etravirín Cmax ↓ 32 % darunavir AUC ↑ 15 % darunavir Cmin ↔ darunavir Cmax ↔ nevirapín AUC ↑ 27 % nevirapín Cmin ↑ 47 % nevirapín Cmax ↑ 18 %
#darunavir: koncentrácie
sa zhodovali s údajmi z minulosti. (↑ nevirapínu v dôsledku inhibície CYP3A)
rilpivirín AUC ↑ 130 % rilpivirín Cmin ↑ 178 % rilpivirín Cmax ↑ 79 % darunavir AUC ↔ darunavir Cmin ↓ 11 % darunavir Cmax ↔
Pri súčasnom podávaní darunaviru a nízkej dávky ritonaviru
v kombinácii s efavirenzom môže byť indikované klinické sledovanie toxicity centrálneho nervového systému spojené so zvýšenou expozíciou voči efavirenzu.
Efavirenz v kombinácii
s darunavirom/ritonavirom
800/100 mg jedenkrát denne môže viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť
v kombinácii
s darunavirom/ritonavirom, treba použiť režim dávkovania darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.4). Darunavir súčasne podávaný
s nízkou dávkou ritonaviru a
s etravirínom
200 mg dvakrátdennesa môže užívať bez úpravy dávky.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s nevirapínom sa môže užívať bez
úpravy dávky.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru
a s rilpivirínom sa môže užívať bez
úpravy dávky.
H
I
V Proteázové inhibítory (PI) - bez doplnkového súčasného podávania nízkej dávky ritonaviru
†
Atazanavir
300 mg jedenkrát denne
Indinavir
800 mg dvakrát denne
Sakvinavir
1 000 mg dvakrát denne
atazanavir AUC ↔ atazanavir Cmin ↑ 52 % atazanavir Cmax ↓ 11 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Atazanavir: porovnanie atazanaviru/ritonaviru 300/100 mg jedenkrát denne vs. atazanavir 300 mg jedenkrát denne v kombinácii
s darunavirom/ritonavirom 400/100 mg dvakrát denne.
Darunavir: porovnanie
darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg dvakrát denne v kombinácii s atazanavirom 300 mg jedenkrát denne. indinavir AUC ↑ 23 %
indinavir Cmin ↑ 125 %
indinavir Cmax ↔
#darunavir AUC ↑ 24 %
#darunavir Cmin ↑ 44 %
#darunavir Cmax ↑ 11 %
Indinavir: porovnanie indinaviru/ritonaviru
800/100 mg dvakrát denne vs. indinavir/darunavir/ritonavir
800/400/100 mg dvakrát denne.
Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii s indinavirom 800 mg dvakrát denne.
#darunavir AUC ↓ 26 %
#darunavir Cmin ↓ 42 %
#darunavir Cmax ↓ 17 %
#sakvinavir AUC ↓ 6 %
#sakvinavir Cmin ↓ 18 %
#sakvinavir Cmax ↓ 6 %
Sakvinavir: porovnanie sakvinaviru/ritonaviru 1000/100 mg dvakrát denne vs. sakvinavir/darunavir/ritonavir
1000/400/100 mg dvakrát denne Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii so sakvinavirom 1000 mg dvakrát denne.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s atazanavirom sa môže užívať bez úpravy dávky.
Pri intolerancii súčasného podávania darunaviru užívaného s nízkou dávkou ritonaviru
s indinavirom môže byť indikované
zníženie dávkovania indinaviru
z 800 mg dvakrát denne na 600 mg dvakrát denne.
Neodporúča sa kombinovať darunavir užívaný s nízkou dávkou ritonaviru so sakvinavirom.
H
I
V Proteázové inhibítory (PI) - podávané s nízkou dávkou ritonaviru
†
Lopinavir/ritonavir
400/100 mg dvakrát denne
Lopinavir/ritonavir
533/133,3 mg dvakrát denne
CCR5-ANTAGONISTY
Maravirok
150 mg dvakrát denne
ANESTETIKÁ
lopinavir AUC ↑ 9 % lopinavir Cmin ↑ 23 % lopinavir Cmax ↓ 2 % darunavir AUC ↓ 38 %‡ darunavir Cmin ↓ 51 %‡ darunavir Cmax ↓ 21 %‡ lopinavir AUC ↔ lopinavir Cmin ↑ 13 % lopinavir Cmax ↑ 11 % darunavir AUC ↓ 41 % darunavir Cmin ↓ 55 % darunavir Cmax ↓ 21 %
‡ na základe neštandardizovaných veľkostí dávky
maravirok AUC ↑ 305 % maravirok Cmin ND maravirok Cmax ↑ 129 %
koncentrácie darunaviru, ritonaviru sa
zhodovali s údajmi z minulosti
Vzhľadom na pokles AUC darunaviru o 40 %, kombinácie vhodných dávok neboli stanovené. Preto je súčasné podávanie darunaviru užívaného s nízkou dávkou ritonaviru a lieku
s kombináciou lopinavir/ritonavir
kontraindikované (pozri časť 4.3).
Keď sa maravirok podáva
s darunavirom s nízkou dávkou
ritonaviru, jeho dávka má byť
150 mg dvakrát denne.
Alfentanil Neskúmalo sa. Metabolizmus alfentanilu je sprostredkovaný CYP3A a v podstate môže byť inhibovaný darunavirom užívaným s nízkou dávkou ritonaviru.
LIEKY PROTI ANGÍNE/ANTIARYTMIKÁ
Súčasné užívanie s darunavirom
a nízkou dávkou ritonaviru môže vyžadovať zníženie dávky alfentanilu a sledovanie rizík predĺženého alebo oneskoreného respiračného útlmu.
Disopyramid Flekainid Mexiletín Propafenón
Amiodarón Bepridil Dronedarón
Lidokaín (podávaný systémovo)
Chinidín Ranolazín Digoxín
0,4 mg jednorazová
dávka
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto antiarytmík.
(inhibícia CYP3A)
digoxín AUC ↑ 61 % digoxín Cmin ND digoxín Cmax ↑ 29 %
(↑ digoxínu v dôsledku možnej inhibície P-
gp)
Pri súčasnom užívaní týchto antiarytmík a darunaviru užívaného s nízkou dávkou ritonaviru sa vyžaduje opatrnosť a odporúča sa sledovanie terapeutických koncentrácií, ak je dostupné.
Súčasné užívanie darunaviru užívaného s nízkou dávkou ritonaviru a amiodarónu, bepridilu, dronedarónu, lidokaínu podávaného systémovo, chinidínu alebo ranolazínu je kontraindikované (pozri časť 4.3).
Vzhľadom na to, že digoxín má úzky terapeutický index, v prípade, že pacienti liečení darunavirom/ritonavirom užívajú digoxín, odporúča sa na začiatok predpísať čo najnižšiu možnú dávku digoxínu. Dávka digoxínu sa má opatrne titrovať, kým sa nedosiahne želaný klinický účinok pri sledovaní celkového klinického stavu
pacienta.
ANTIBIOTIKÁ
Klaritromycín
500 mg dvakrát denne
ANTIKOAGULANCIÁ
Apixaban Dabigatran etexilát Rivaroxaban
klaritromycín AUC ↑ 57 % klaritromycín Cmin ↑ 174 % klaritromycín Cmax ↑ 26 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↑ 1 %
#darunavir Cmax ↓ 17 %
Pri podávaní s darunavirom/ritonavirom neboli zistiteľné koncentrácie metabolitu
14-OH-klaritromycínu.
(↑ klaritromycínu v dôsledku inhibície
CYP3A a možnej inhibície P-gp)
Neskúmalo sa. Súčasné užívanie darunaviru a týchto antikoagulancií môže zvýšiť koncentrácie antikoagulantu. (inhibícia CYP3A a/alebo P-gp).
Vyžaduje sa opatrnosť, keď sa klaritromycín podáva v kombinácii s darunavirom užívaným spolu
s nízkou dávkou ritonaviru.
Užívanie darunaviru užívaného
s nízkou dávkou ritonaviru a týchto
antikoagulancií sa neodporúča.
Warfarín Neskúmalo sa. Pri súčasnom podávaní
warfarínu a darunaviru
s ritonavirom(nízkou dávkou) môže dôjsť
k ovplyvneniu koncentrácií warfarínu.
ANTIKONVULZÍVA
Pri súčasnom podávaní warfarínu s darunavirom užívaným s nízkou dávkou ritonaviru sa odporúča monitorovať INR (International Normalized Ratio).
Fenobarbital
Fenytoín
Karbamazepín
200 mg dvakrát denne
ANTIDEPRESÍVAParoxetín
20 mg jedenkrát denne
Sertralín
50 mg jedenkrát denne
Amitriptylín Desipramín Imipramín Nortriptylín Trazodón
Neskúmalo sa. Predpokladá sa, že fenobarbital a fenytoín znižujú plazmatické koncentrácie darunaviru. (indukcia enzýmov CYP450) karbamazepín AUC ↑ 45 % karbamazepín Cmin ↑ 54 % karbamazepín Cmax ↑ 43 %
darunavir AUC ↔
darunavir Cmin ↓ 15 %
darunavir Cmax ↔
paroxetín AUC ↓ 39 % paroxetín Cmin ↓ 37 % paroxetín Cmax ↓ 36 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
sertralín AUC ↓ 49 %
sertralín Cmin ↓ 49 %
sertralín Cmax ↓ 44 %
#darunavir AUC ↔
#darunavir Cmin ↓ 6 %
#darunavir Cmax ↔
Súčasné užívanie darunaviru užívaného s nízkou dávkou ritonaviru a týchto antidepresív môže zvýšiť koncentrácie antidepresíva.
(inhibícia CYP2D6 a/alebo CYP3A).
Darunavir užívaný s nízkou dávkou
ritonaviru sa nesmie používať
v kombinácii s týmito liečivami.
Neodporúča sa žiadna úprava dávky darunaviru/ritonaviru. V prípade potreby kombinovať darunavir/ritonavir a karbamazepín sa majú pacienti sledovať z dôvodu možných nežiaducich udalostí spojených s karbamazepínom. Je potrebné sledovať koncentrácie karbamazepínu a titrovať jeho
dávku až do dosiahnutia primeranej odpovede. Na základe zistení môže byť potrebné znížiť dávku karbamazepínu o 25% až 50%, ak
sa podáva
s darunavirom/ritonavirom.
Ak sa antidepresíva užívajú súčasne s darunavirom užívaným s nízkou dávkou ritonaviru, odporúčaný prístup je titrácia dávky antidepresíva na základe klinického posúdenia odpovede na antidepresívum. Navyše u pacientov na ustálenej dávke týchto antidepresív, ktorí začínajú liečbu darunavirom užívanou s nízkou dávkou ritonaviru, sa má sledovať odpoveď na antidepresívum.
Keď sa tieto antidepresíva užívajú s darunavirom s nízkou dávkou ritonaviru, odporúča sa klinické sledovanie a môže byť potrebná úprava dávky antidepresíva.
ANTIMYKOTIKÁ
Vorikonazol Neskúmalo sa. Ritonavir môže znížiť plazmatické koncentrácie vorikonazolu. (indukcia enzýmov CYP450 ritonavirom)
Vorikonazol sa nesmie používať
spolu s darunavirom užívanou
s nízkou dávkou ritonaviru, kým vyhodnotenie pomeru prínos/riziko neodôvodní použitie vorikonazolu.
Ketokonazol
200 mg dvakrát denne
ketokonazol AUC ↑ 212 % ketokonazol Cmin ↑ 868 % ketokonazol Cmax ↑ 111 %
#darunavir AUC ↑ 42 %
#darunavir Cmin ↑ 73 %
#darunavir Cmax ↑ 21 % (inhibícia CYP3A)
Vyžaduje sa opatrnosť a odporúča sa klinické sledovanie. Ak je nutná súčasná liečba, denná dávka ketokonazolu nesmie presiahnuť
200 mg.
Posakonazol Neskúmalo sa. darunavir môže zvýšiť plazmatické koncentrácie antimykotika (inhibícia P-gp) a posakonazol môže zvýšiť koncentrácie darunaviru. (inhibícia CYP3A)
Itrakonazol Neskúmalo sa. Súčasné systémové podanie
itrakonazolu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť plazmatické koncentrácie darunaviru. Zároveň súčasné podávanie darunaviru užívaného s nízkou dávkou ritonaviru
a itrakonazolu môže zvýšiť plazmatické koncentrácie itrakonazolu.
(inhibícia CYP3A)
Klotrimazol Neskúmalo sa. Súčasné systémové podanie
klotrimazolu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť plazmatické koncentrácie darunaviru. darunavir AUC24h ↑ 33 % (na základe farmakokinetického modelu populácie)
Vyžaduje sa opatrnosť a odporúča
sa klinické sledovanie.
Vyžaduje sa opatrnosť a odporúča sa klinické sledovanie. Ak je nutná súčasná liečba, denná dávka itrakonazolu nesmie presiahnuť
200 mg.
Ak je nutná súčasná liečba klotrimazolom, vyžaduje sa opatrnosť a odporúča sa klinické sledovanie.
ANTIURATIKÁ
Kolchicín Neskúmalo sa. Súčasné užívanie
kolchicínu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť
expozíciu kolchicínu.
ANTIMALARIKÁ
Ak je potrebná liečba darunavirom užívaným s nízkou dávkou ritonaviru, odporúča sa u pacientov s normálnou funkciou obličiek alebo pečene znížiť dávku
kolchicínu alebo liečbu kolchicínom prerušiť.
Pacientom s poruchou funkcie
obličiek alebo pečene sa nemá podávať kolchicín s darunavirom užívaným s nízkou dávkou ritonaviru (pozri časť 4.4).
Artemeter/
lumefantrín
80/480 mg, 6 dávok v 0., 8., 24., 36., 48. a 60. hodine
artemeter AUC ↓ 16 % artemeter Cmin ↔ artemeter Cmax ↓ 18 %
dihydroartemisinín AUC ↓ 18 %
dihydroartemisinín Cmin ↔ dihydroartemisinín Cmax ↓ 18 % lumefantrín AUC ↑ 175 % lumefantrín Cmin ↑ 126 % lumefantrín Cmax ↑ 65 % darunavir AUC ↔
darunavir Cmin ↓ 13 %
darunavir Cmax ↔
Kombinácia darunaviru a artemeteru/lumefantrínu sa môže podávať bez úpravy dávky;
z dôvodu zvýšenia expozície lumefantrínu však treba kombináciu používať s opatrnosťou.
ANTITUBERKULOTIKÁ
Rifampicín
Rifapentín
Rifabutin
150 mg jedenkrát každý druhý deň
ANTINEOPLASTIKÁDasatinib Nilotinib Vinblastín Vinkristín
Everolimus
Neskúmalo sa. Rifapentín a rifampicín sú silnými induktormi CYP3A a bolo dokázané, že spôsobujú signifikantné zníženie koncentrácií iných proteázových inhibítorov, čo môže vyústiť do zlyhania antivirologickej odpovede a rozvinutia rezistencie (indukcia enzýmu CYP450). Pri pokusoch prekonať zníženú expozíciu zvýšením dávky iných proteázových inhibítorov s nízkou dávkou ritonaviru, sa s rifampicínom zistila vysoká frekvencia pečeňových reakcií.
rifabutin AUC** ↑ 55 % rifabutin Cmin** ↑ ND rifabutin Cmax** ↔ darunavir AUC ↑ 53 % darunavir Cmin ↑ 68 % darunavir Cmax ↑ 39 %
** súčet účinných zložiek rifabutinu (materský liek
+ 25-
O-desacetyl metabolit)
Štúdia interakcií preukázala porovnateľnú
dennú systémovú expozíciu rifabutinu
v liečbe samotným rifabutinom 300 mg
jedenkrát denne a v liečbe rifabutinom
150 mg jedenkrát každý druhý deň spolu s darunavirom/ritonavirom (600/100 mg dvakrát denne) s približne 10-násobným zvýšením dennej expozície aktívnemu metabolitu 25-
O-desacetylrifabutinu. Okrem toho, AUC súčtu účinných zložiek rifabutinu (materský liek + 25-
O-desacetyl metabolit) sa zvýšila 1,6-násobne, pričom Cmax zostala porovnateľná.
Údaje o porovnaní s referenčnou dávkou
150 mg jedenkrát denne nie sú k dispozícii.
(Rifabutin je induktorom a substrátom enzýmov CYP3A.) Keď sa darunavir užívaný spolu s 100 mg ritonaviru podávala spolu s rifabutinom (150 mg jedenkrát každý druhý deň) pozorovalo sa
zvýšenie systémovej expozície darunaviru.
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto antineoplastík.
(inhibícia CYP3A)
Kombinácia rifapentínu
a darunaviru užívaného s nízkou
dávkou ritonaviru sa neodporúča.
Súčasné podávanie rifampicínu
a darunaviru spolu s nízkou dávkou ritonaviru je kontraindikované (pozri časť 4.3).
U pacientov užívajúcich tieto lieky súčasne je nevyhnutná redukcia zvyčajnej dávky rifabutinu
300 mg/deň o 75 % (t.j. rifabutin
150 mg jedenkrát každý druhý deň) a zvýšené sledovanie nežiaducich udalostí súvisiacich s rifabutinom. V prípade ohrozenia bezpečnosti sa má zvážiť ďalšie predĺženie dávkovacieho intervalu rifabutinu a/alebo sledovanie hladiny rifabutinu.
Treba brať do úvahy oficiálnu smernicu pre liečbu tuberkulózy u pacientov infikovaných HIV.
Na základe bezpečnostného profilu darunaviru/ritonaviru, toto zvýšenie expozície darunaviru v prítomnosti rifabutinu nevyžaduje úpravu dávky darunaviru/ritonaviru.
Na základe farmakokinetického modelu možno dávku zredukovať o 75 % aj u pacientov, ktorí
dostávajú inú dávku rifabutinu ako
300 mg/deň.
Pri súčasnom užívaní týchto liekov a darunaviru s nízkou dávkou ritonaviru môžu byť ich koncentrácie zvýšené, čo vedie k možnosti nárastu nežiaducich udalostí zvyčajne súvisiacich
s týmito liekmi.
Pri súčasnom užívani darunaviru s nízkou dávkou ritonaviru
a jedného z antineoplastík je
potrebná opatrnosť.
Súčasné užívanie everolimu a darunaviru užívaného s nízkou dávkou ritonaviru sa neodporúča.
I
NHIBÍTORY AGREGÁCIE TROMBOCYTOV
Tikagrelor Neskúmalo sa. Súčasné užívanie
s darunavirom posilneným nízkou dávkou ritonaviru môže viesť k značnému zvýšeniu expozície tikagreloru.
ANTIPSYCHOTIKÁ/NEUROLEPTIKÁ
Kvetiapín V dôsledku inhibície CYP3A darunavirom sa očakáva zvýšenie koncentrácií antipsychotík/neuroleptík.
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru
a s tikagrelorom je kontraindikované.
Užívanie iných antitrombotík neovplyvnených inhibíciou alebo indukciou CYP (napr. prasugrel) sa odporúča.
Súčasné podávanie darunaviru s nízkou dávkou ritonaviru
a kvetiapínu je kontraindikované, pretože môže zvýšiť toxicitu súvisiacu s kvetiapínom. Zvýšené koncentrácie kvetiapínu môžu viesť ku kóme.
Risperidón
Tioridazín
Pimozid
Sertindol
β-BLOKÁTORY Karvedilol Metoprolol Timolol
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto antipsychotík.
(inhibícia CYP2D6 a/alebo P-gp)
Neskúmalo sa. Očakáva sa, že darunavir zvýši plazmatické koncentrácie týchto beta blokátorov.
(inhibícia CYP2D6)
Môže byť potrebné zníženie dávky týchto liekov, keď sa podávajú súčasne s darunavirom užívaným
s nízkou dávkou ritonaviru.
Súčasné podávanie darunaviru s nízkou dávkou ritonaviru a pimozidu alebo sertindolu je kontraindikované.
Pri súčasnom užívaní darunaviru s beta blokátormi sa odporúča klinické sledovanie. Má sa zvážiť zníženie dávky beta blokátora.
B
L
OK
ÁTORY KALCIOVÉHO KANÁLA
Amlodipín Diltiazem Felodipín Nikardipín Nifedipín Verapamil
Nesledovalo sa. Môže sa očakávať, že darunavir užívaný s nízkou dávkou ritonaviru bude zvyšovať plazmatické koncentrácie blokátorov kalciového kanála.
(inhibícia CYP3A a/alebo CYP2D6)
Ak sa tieto lieky podávajú súčasne s darunavirom a s nízkou dávkou ritonaviru, odporúča sa klinické sledovanie liečebných a nežiaducich udalostí.
KO
RTIKOSTEROIDY
Flutikazón
Budezonid
Dexametazón
(podávaný systémovo)
V klinickej štúdii, pri ktorej sa zdravým jedincom počas 7 dní podávali kapsuly ritonaviru 100 mg dvakrát denne spolu
s 50 µg flutikazónpropionátu intranazálne (štyrikrát denne), sa významne zvýšili sérové hladiny flutikazónpropionátu, zatiaľ čo prirodzená hladina kortizolu klesla približne o 86 % (pri 90 % intervale spoľahlivosti 82 % - 89 %). Vyššie účinky možno očakávať, keď sa flutikazón inhaluje. Systémové účinky kortikosteroidov, vrátane Cushingovho syndrómu a adrenálnej supresie, boli hlásené u pacientov, ktorí užívali ritonavir
a súčasne inhalovali alebo intranazálne užili flutikazón; podobná situácia môže vzniknúť aj pri súčasnom užívaní iných kortikosteroidov, ktoré sa metabolizujú prostredníctvom cytochrómu P4503A, napr. budezonidu. Účinky vysokej systémovej expozície flutikazónu na plazmatické hladiny ritonaviru nie sú dosiaľ známe.
Neskúmalo sa. Dexametazón môže znížiť plazmatickú koncentráciu darunaviru. (indukcia CYP3A)
Preto sa súčasná liečba darunavirom v kombinácii s nízkou dávkou ritonaviru a týmito
glukokortikoidmi neodporúča, pokiaľ možný prínos nepreváži riziko systémových kortikosteroidných účinkov. Má sa zvážiť zníženie dávky glukokortikoidov spolu
s dôkladným monitorovaním lokálnych a systémových účinkov, alebo prevod na iný glukokortikoid, ktorý nepatrí k substrátom pre CYP3A (napr. beklometazón).
V prípade vysadenia glukokortikoidov môže byť navyše potrebné, aby progresívne znižovanie dávky trvalo dlhší čas.
Dexametazón podávaný systémovo sa má podávať so zvýšenou opatrnosťou, ak sa užíva spolu
s darunavirom a nízkou dávkou ritonaviru.
Prednizón Neskúmalo sa. Darunavir môže zvýšiť plazmatické koncentrácie prednizónu. (inhibícia CYP3A)
ANTIENDOTELÍNY
Bosentan Neskúmalo sa. Súčasné užívanie
bosentanu a darunaviru užívaného s nízkou
dávkou ritonaviru môže zvýšiť
plazmatické koncentrácie bosentanu.
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru
a prednizónu môže zvýšiť riziko vzniku systémových kortikosteroidných účinkov, vrátane Cushingovho syndrómu
a adrenálnej supresie. Pri súčasnom užívaní darunaviru s nízkou dávkou ritonaviru a kortikosteroidov sa odporúča klinické sledovanie.
Ak sa podáva súčasne
s darunavirom a nízkou dávkou ritonaviru, má sa u pacienta
sledovať tolerovateľnosť bosentanu.
ANTIVIROTIKÁ PRIAMO ÚČINKUJÚCE PROTI VÍRUSU HEPATITÍDY C (HCV)
Proteázové inhibítory NS3-4A
Telaprevir
750 mg každých
8 hodín
Boceprevir
800 mg trikrát denne
telaprevir AUC ↓ 35 % telaprevir Cmin ↓ 32 % telaprevir Cmax ↓ 36 % darunavir AUC12 ↓ 40 % darunavir Cmin ↓ 42 % darunavir Cmax ↓ 40 % boceprevir AUC ↓ 32 % boceprevir Cmin ↓ 35 % boceprevir Cmax ↓ 25 % darunavir AUC ↓ 44 % darunavir Cmin ↓ 59 % darunavir Cmax ↓ 36 %
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru a
s telaprevirom sa neodporúča.
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru a
s boceprevirom sa neodporúča.
Simeprevir simeprevir AUC ↑ 159 % simeprevir Cmin ↑ 358 % simeprevir Cmax ↑ 79 % darunavir AUC ↑ 18 % darunavir Cmin ↑ 31 % darunavir Cmax «
Súčasné užívanie darunaviru s nízkou dávkou ritonaviru
a simepreviru sa neodporúča.
RASTLINNÉ LIEČIVÁ Ľubovník bodkovaný (Hypericum perforatum)
Dávka simepreviru v tejto interakčnej
štúdii bola 50 mg, keď sa podával spolu
s darunavirom/ritonavirom, v porovnaní so
150 mg v skupine liečenej samotným
simeprevirom.
Neskúmalo sa. Predpokladá sa, že ľubovník bodkovaný spôsobuje pokles plazmatických koncentrácií darunaviru a ritonaviru.
(indukcia CYP450)
Darunavir užívaný s nízkou dávkou
ritonaviru sa nesmie podávať
v kombinácii s liekmi obsahujúcimi výťažok z ľubovníka bodkovaného (Hypericum perforatum) (pozri časť
4.3). Ak už pacient užíva ľubovník bodkovaný, užívanie ukončite, a ak je to možné, skontrolujte vírusové hladiny. Expozícia darunaviru (a tiež expozícia ritonaviru) sa môže po ukončení užívania ľubovníka bodkovaného zvýšiť. Indukujúci účinok môže pretrvávať najmenej 2 týždne po ukončení liečby ľubovníkom bodkovaným.
I
NHIBÍTORY HMG CO-A REDUKTÁZY
Lovastatín
Simvastatín
Atorvastatín
10 mg jedenkrát denne
Pravastatín
40 mg jednorazová dávka
Rosuvastatín
10 mg jedenkrát denne
Neskúmalo sa. Predpokladá sa, že lovastatín a simvastatín budú mať zreteľne zvýšené plazmatické koncentrácie, ak sa budú podávať súčasne s darunavirom užívaným s nízkou dávkou ritonaviru. (inhibícia CYP3A)
atorvastatín AUC ↑ 3-4 krát atorvastatín Cmin ↑ ≈5,5-10 krát atorvastatín Cmax ↑ ≈2 krát
#darunavir
pravastatín AUC ↑ 81 %¶ pravastatín Cmin ND pravastatín Cmax ↑ 63 %
¶ u niektorých subjektov sa pozoroval až 5-násobný nárast
rosuvastatín AUC ↑ 48%║
rosuvastatín Cmax ↑ 144%║
║ na základe publikovaných údajov
Zvýšené plazmatické koncentrácie lovastatínu alebo simvastatínu môžu vyvolať myopatiu, vrátane rabdomyolýzy. Súčasné podávanie darunaviru užívaného s nízkou dávkou ritonaviru s lovastatínom
a simvastatínom je preto
kontraindikované (pozri časť 4.3). Ak je potrebné súčasné podávanie atorvastatínu s darunavirom užívaným s nízkou dávkou ritonaviru, odporúča sa začať podávať atorvastatín v dávke 10 mg jedenkrát denne. Postupné zvyšovanie dávkovania
atorvastatínu je možné prispôsobiť
na základe klinickej odpovede.
Ak je potrebné súčasné podávanie pravastatínu s darunavirom užívaným s nízkou dávkou ritonaviru, odporúča sa začať
s najnižšou možnou dávkou pravastatínu a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
Ak je potrebné súčasné podávanie rosuvastatínu s darunavirom užívaným s nízkou dávkou ritonaviru, odporúča sa začať
s najnižšou možnou dávkou rosuvastatín a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
I
NHIBÍTORY H
2
-
RECEPTORA
Ranitidín
150 mg dvakrát denne
IMUNOSUPRESÍVA Ciklosporín Sirolimus Takrolimus
Everolimus
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Neskúmalo sa. Pri súčasnom podávaní darunaviru užívaného s nízkou dávkou ritonaviru s týmito imunosupresívami sa zvýši expozícia voči týmto látkam. (inhibícia CYP3A)
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s inhibítormi H2-receptora sa môže užívať bez úpravy dávky.
Pri súčasnom podaní imunosupresív sa musia monitorovať terapeutické hladiny týchto liekov.
Súčasné užívanie everolimu a darunaviru užívaného s nízkou dávkou ritonaviru sa neodporúča.
I
NHALAČNÉ BETA-AGONISTY
Salmeterol Neskúmalo sa. Súčasné užívanie
salmeterolu a darunaviru užívaného
s nízkou dávkou ritonaviru môže zvýšiť
plazmatické koncentrácie salmeterolu.
Súčasné užívanie salmeterolu
a darunaviru užívaného s nízkou dávkou ritonaviru sa neodporúča. Táto kombinácia môže viesť
k zvýšenému riziku kardiovaskulárnych nežiaducich udalostí salmeterolu, vrátane predĺženia QT intervalu, palpitácií a sínusovej tachykardie.
NARKOTICKÉ ANALGETIKÁ / LIEČBA ZÁVISLOSTI NA OPIOIDOCH
Metadon individuálna dávka
v rozsahu od 55 mg do
150 mg jedenkrát denne
Buprenorfín/naloxón
8/2 mg–16/4 mg jedenkrát denne
R(-) metadon AUC ↓ 16 % R(-) metadon Cmin ↓ 15 % R(-) metadon Cmax ↓ 24 %
buprenorfín AUC ↓ 11 % buprenorfín Cmin ↔ buprenorfín Cmax ↓ 8 % norbuprenorfín AUC ↑ 46 % norbuprenorfín Cmin ↑ 71 % norbuprenorfín Cmax ↑ 36 % naloxón AUC ↔
naloxón Cmin ND
naloxón Cmax ↔
Na začiatku užívania spolu
s darunavirom/ritonavirom nie je potrebná úprava dávky metadonu. Avšak zvýšené dávky metadonu môžu byť potrebné kvôli indukcii
metabolizmu ritonavirom pri
súčasnom podávaní počas dlhšieho časového obdobia. Z toho dôvodu
sa odporúča klinické monitorovanie,
pretože u niektorých pacientov môže byť potrebná úprava udržiavacej liečby.
Klinický význam zvýšenia farmakokinetických parametrov norbuprenorfínu nebol stanovený. Úprava dávky buprenorfínu nemusí byť potrebná, ak sa podáva spolu
s darunavirom/ritonavirom, ale odporúča sa pozorné sledovanie prejavov toxicity opiátov.
ANTIKONCEPCIA NA BÁZE ESTROGÉNOV
Etinylestradiol
Noretisterón
35 mg/1 mg jedenkrát denne
etinylestradiol AUC ↓ 44 % etinylestradiol Cmin ↓ 62 % etinylestradiol Cmax ↓ 32 % noretisterón AUC ↓ 14 % noretisterón Cmin ↓ 30 % noretisterón Cmax ↔
Pacientkam užívajúcim antikoncepciu na báze estrogénov v kombinácii s darunavirom
a s nízkou dávkou ritonaviru sa odporúča zmeniť resp. rozšíriť formy antikoncepcie. U pacientok užívajúcich estrogén ako hormonálnu substitúciu majú byť klinicky sledované prejavy deficitu estrogénu.
FOSFODIESTERÁZA, INHIBÍTORY TYPU 5 (PDE-5)
Pri liečbe erektilnej
dysfunkcie Avanafil Sildenafil Tadalafil Vardenafil
Pri liečbe pulmonálnej
arteriálnej hypertenzie
Sildenafil
Tadalafil
V štúdii skúmajúcej interakcie# sa pozorovala porovnateľná systémová expozícia voči sildenafilu po jednorazovom podaní samotného sildenafilu v dávke 100 mg a po jednorazovom podaní sildenafilu v dávke
25 mg v kombinácii s darunavirom a nízkou dávkou ritonaviru.
Neskúmalo sa. Súčasné užívanie sildenafilu alebo tadalafilu pri liečbe pulmonálnej arteriálnej hypertenzie a darunaviru užívaného s nízkou dávkou ritonaviru môže zvýšiť plazmatické koncentrácie sildenafilu alebo tadalafilu.
Kombinácia avanafilu a darunaviru s nízkou dávkou ritonaviru je kontraindikovaná (pozri časť 4.3). Pri súčasnom podávaní iných inhibítorov PDE-5 na liečbu erektilnej dysfunkcie
s darunavirom, užívaným s nízkou dávkou ritonaviru, je potrebná opatrnosť. Ak je indikované súčasné podávanie darunaviru
a nízkej dávky ritonaviru so sildenafilom, vardenafilom alebo tadalafilom, jednorazová dávka sildenafilu nesmie prekročiť 25 mg za obdobie 48 hodín, resp. jednorazová dávka vardenafilu nesmie prekročiť 2,5 mg za obdobie
72 hodín a jednorazová dávka tadalafilu nesmie presiahnuť 10 mg za obdobie 72 hodín.
Bezpečná a účinná dávka sildenafilu pri liečbe pulmonálnej arteriálnej hypertenzie a pri súčasnom užívaní darunaviru a nízkej dávky ritonaviru nebola stanovená.
Existuje zvýšená možnosť vzniku nežiaducich udalostí súvisiacich so sildenafilom (vrátane porúch zraku,
hypotenzie, predĺženej erekcie a synkopy). Preto je súčasné užívanie darunaviru s nízkou
dávkou ritonaviru a sildenafilu, ak
sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, kontraindikované (pozri časť 4.3). Pri liečbe pulmonálnej arteriálnej hypertenzie sa súčasné užívanie tadalafilu s darunavirom a nízkou dávkou ritonaviru neodporúča.
I
NHIBÍTORY PROTÓNOVEJ PUMPY
Omeprazol
20 mg jedenkrát denne
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Darunavir užívaný s nízkou dávkou ritonaviru je možné súčasne podávať s inhibítormi protónovej pumpy bez nutnosti úpravy dávkovania.
SEDATÍVA/HYPNOTIKÁ
Buspirón Klorazepát Diazepam Estazolam Flurazepam Triazolam Zolpidem
Midazolam
Neskúmalo sa. Sedatíva/hypnotiká sú výrazne metabolizované prostredníctvom CYP3A. Súčasné užívanie
s darunavirom/ritonavirom môže spôsobiť významné zvýšenie koncentrácií týchto liekov.
Na základe údajov o iných inhibítoroch CYP3A sa očakáva, že plazmatické koncentrácie midazolamu budú výrazne vyššie, keď sa midazolam podáva perorálne s darunavirom užívaným
s nízkou dávkou ritonaviru.
Ak sa parenterálny midazolam podáva súčasne s darunavirom užívaným s nízkou dávkou ritonaviru, môže spôsobiť významné zvýšenie koncentrácie tohto benzodiazepínu. Údaje o súčasnom užívaní parenterálneho midazolamu s inými proteázovými inhibítormi naznačujú
možné 3-4-násobné zvýšenie
plazmatických hladín midazolamu.
Pri súčasnom užívaní darunaviru
s týmito sedatívami/hypnotikami sa odporúča klinické sledovanie a má sa zvážiť zníženie dávky sedatív/hypnotík. Súčasné užívanie darunaviru užívaného s nízkou dávkou ritonaviru a triazolamu je
kontraindikované.
Užívanie darunaviru s nízkou dávkou ritonaviru v kombinácii s perorálne podávaným
midazolamom je kontraindikované (pozri časť 4.3); pričom sa odporúča opatrnosť pri súčasnom užívaní darunaviru s nízkou dávkou ritonaviru a parenterálneho midazolamu.
Ak sa parenterálny midazolam podáva súčasne s darunavirom užívaným s nízkou dávkou ritonaviru, má sa tak uskutočniť na jednotke intenzívnej starostlivosti (JIS) alebo v podobnom zariadení, ktoré zabezpečuje dôkladné klinické sledovanie a vhodnú lekársku starostlivosť v prípade respiračného útlmu a/alebo predĺženého útlmu. Treba zvážiť úpravu dávkovania midazolamu, najmä ak sa podáva viac ako jednorazová dávka.
† Účinnosť a bezpečnosť používania darunaviru so 100 mg ritonaviru a s niektorým iným HIV PI (napr. (fos)amprenavir, nelfinavir a tipranavir) neboli u pacientov s HIV stanovené. Podľa súčasných liečebných postupov sa vo všeobecnosti duálna liečba proteázovými inhibítormi neodporúča.
4.6 Fertilita, gravidita a laktáciaGraviditaVo všeobecnosti, keď sa rozhoduje o použití antiretrovirotík na liečbu infekcie HIV u gravidných žien
a následne na zníženie rizika vertikálneho prenosu HIV na novorodenca, treba vziať do úvahy údaje
o zvieratách ako aj klinické skúsenosti s gravidnými ženami.
S darunavirom sa nevykonali žiadne primerané ani dobre kontrolované štúdie o vplyve na graviditu u gravidných žien. Štúdie na zvieratách nepreukázali priame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3).
Darunavir užívaný s nízkou dávkou ritonaviru sa môže podávať gravidným ženám len vtedy, ak
možný prínos liečby prevýši možné riziká.
DojčenieNie je známe, či sa darunavir vylučuje do ľudského mlieka. V štúdiách na potkanoch sa preukázalo
vylučovanie darunaviru do mlieka a pri vysokých hladinách (1000 mg/kg/deň) spôsobilo toxicitu. Matky majú byť poučené, aby za žiadnych okolností nedojčili počas liečby darunavirom jednak kvôli možnosti prenosu HIV a aj možnosti výskytu nežiaducich reakcií u dojčených detí.
Fertilita
Nie sú k dispozícii žiadne údaje o účinku darunaviru na fertilitu u ľudí. Pri liečbe potkanov
darunavirom nemal darunavir vplyv na párenie a fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Darunavir spolu s ritonavirom nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U niektorých pacientov sa však počas liečby darunavirom užívaným s nízkou dávkou ritonaviru vyskytli závraty. Na túto možnosť je potrebné myslieť pri posudzovaní schopnosti pacienta viesť vozidlá a obsluhovať stroje (pozri časť 4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Počas programu klinického vývoja (N=2 613 predtým liečených jedincov, ktorí začali liečbu
s darunavirom/ritonavirom 600/100 mg dvakrát denne), 51,3 % jedincovmalo aspoň jednu nežiaducu reakciu. Celkové priemerné trvanie liečby jedincovbolo 95,3 týždňov. Najčastejšími nežiaducimi
reakciami hlásenými v klinických štúdiách a v spontánnych hláseniach sú hnačka, nauzea, vyrážka,
bolesť hlavy a vracanie. Najčastejšími závažnými reakciami sú akútne renálne zlyhanie, infarkt myokardu, imunoreštitučný zápalový syndróm, trombocytopénia, osteonekróza, hnačka, hepatitída a pyrexia.
V analýze po 96 týždňoch bol bezpečnostný profil darunaviru/ritonaviru 800/100 mg jedenkrát denne u jedincov, ktorí doteraz neboli liečení, podobný ako u darunaviru/ritonaviru 600/100 mg dvakrát denne u jedincov, ktorí boli predtým liečení, až na nauzeu, ktorá sa častejšie vyskytovala u doteraz neliečených jedincov. Nauzea bola miernej intenzity. V analýze po 192 týždňoch sa u doteraz neliečených pacientov, u ktorých liečba darunavirom/ritonavirom 800/100 mg jedenkrát denne trvala priemerne 162,5 týždňa, neidentifikovali žiadne nové nálezy týkajúce sa bezpečnosti.
Zoznam nežiaducich reakcií v tabuľkách
Nežiaduce reakcie sa uvádzajú podľa postihnutia triedy orgánových systémov a podľa skupiny
frekvencie. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí
klesajúcej závažnosti. Skupiny frekvencií sú definované nasledovne: veľmi časté (≥ 1/10), časté
(≥ 1/100 až < 1/10), menej časté (≥ 1/1000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000)
a neznáme (z dostupných údajov).
Nežiaduce reakcie v klinických skúšaniach a v postmarketingovom sledovaní
Trieda orgánových systémov podľa databázy
MedDRA
Kategória frekvencie
Infekcie a nákazy
Nežiaduca reakcia
menej časté herpes simplex
Poruchy krvi a lymfatického systémumenej časté trombocytopénia, neutropénia, anémia, leukopénia
zriedkavé zvýšenie počtu eozinofilov
Poruchy imunitného systémumenej časté imunoreštitučný zápalový syndróm, precitlivenosť
(na liek)
Poruchy endokrinného systémumenej časté hypotyreóza, zvýšenie hladiny hormónu stimulujúceho štítnu žľazu v krvi
 Poruchy metabolizmu a výživy
Poruchy metabolizmu a výživy
časté diabetes mellitus, hypertriglyceridémia, hypercholesterolémia, hyperlipidémia
menej časté dna, anorexia, znížená chuť do jedla, zníženie
hmotnosti, zvýšenie hmotnosti, hyperglykémia,
rezistencia na inzulín, pokles lipoproteínu
s vysokou hustotou, zvýšená chuť do jedla, polydipsia, zvýšená hladina LDH v krvi
Psychické poruchyčasté insomnia
menej časté depresia, dezorientácia, úzkosť, poruchy spánku,
neprirodzené sny, nočné mory, pokles libida
zriedkavé stav zmätenosti, zmeny nálady, nepokoj
Poruchy nervového systémučasté bolesti hlavy, periférna neuropatia, závraty
menej časté letargia, parestézia, hypestézia, dysgeuzia, porucha
pozornosti, poškodenie pamäti, somnolencia
zriedkavé synkopa, konvulzia, ageuzia, porucha fázy spánkového rytmu
Poruchy okamenej časté konjunktiválna hyperémia, suché oči
zriedkavé porucha zraku
Poruchy ucha a labyrintumenej časté vertigo
Poruchy srdca a srdcovej činnostimenej časté infarkt myokardu, angína pektoris, predĺžený QT
elektrokardiogram, tachykardia
zriedkavé akútny infarkt myokardu, sínusová bradykardia,
palpitácie
Poruchy cievmenej časté hypertenzia, návaly horúčavy
Poruchy dýchacej sústavy, hrudníka a mediastínamenej časté dyspnoe, kašeľ, epistaxa, podráždenie hrdla
zriedkavé rinorea
Poruchy gastrointestinálneho traktuveľmi časté diarea
časté vracanie, nauzea, bolesti brucha, zvýšená hladina
amylázy v krvi, dyspepsia, abdominálna distenzia, flatulencia
menej časté pankreatitída, gastritída, gastroezofagálny reflux, aftózna stomatitída, napínanie na vracanie, sucho
v ústach, abdominálny diskomfort, zápcha, zvýšená
hladina lipázy, eruktácia, perorálna dyzestézia
zriedkavé stomatitída, hemateméza, cheilitída, suché pery, povlak na jazyku
 Poruchy pečene a žlčových ciest
Poruchy pečene a žlčových ciest
časté zvýšenie alanínaminotransferázy
menej časté hepatitída, cytolytická hepatitída, steatóza pečene,
hepatomegália, zvýšenie transamináz, zvýšenie aspartátaminotransferázy, zvýšenie hladiny
bilirubínu v krvi, zvýšenie alkalickej fosfatázy
v krvi, zvýšenie gamaglutamyltransferázy
Poruchy kože a podkožného tkanivačasté vyrážka (vrátane makulárnej, makulopapulárnej, papulárnej, erytematóznej a pruritickej vyrážky), pruritus
menej časté angioedém, generalizovaná vyrážka, alergická
dermatitída, urtikária, ekzém, erytém, hyperhidróza, nočné potenie, alopécia, akné, suchá koža, sfarbenie nechtov
zriedkavé DRESS, Stevensov-Johnsonov syndróm, multiformný erytém, dermatitída, seboroická dermatitída, kožné lézie, xeroderma
neznáme toxická epidermálna nekrolýza, akútna generalizovaná exantematózna pustulóza
Poruchy kostrovej a svalovej sústavy a spojivového tkanivamenej časté myalgia, osteonekróza, svalové kŕče, svalová slabosť, artralgia, bolesti končatín, osteoporóza, zvýšená hladina kreatínfosfokinázy v krvi
zriedkavé muskuloskeletálna stuhnutosť, artritída, stuhnutie
kĺbov
Poruchy obličiek a močových ciestmenej časté akútne zlyhanie obličiek, zlyhanie obličiek, nefrolitiáza, zvýšená hladina kreatinínu v krvi, proteinúria, bilirubinúria, dyzúria, noktúria, polakizúria
zriedkavé znížený renálny klírens kreatinínu
Poruchy reprodukčného systému a prsníkovmenej časté erektilná dysfunkcia, gynekomastia
Celkové poruchy a reakcie v mieste podaniačasté asténia, únava
menej časté pyrexia, bolesť na hrudi, periférne opuchy,
nevoľnosť, pocit horúčavy, podráždenie, bolesť
zriedkavé triaška, abnormálne pocity, xeróza
OpisvybranýchnežiaducichreakciíVyrážkaV klinických skúšaniach bola vyrážka najčastejšie miene až stredne závažná, často sa vyskytla v prvých štyroch týždňoch liečby a ustúpila pri ďalšom užívaní. Pre prípady závažných kožných reakcií
pozri varovanie v časti 4.4.
Počas programu klinického vývoja raltegraviru sa u predtým liečených pacientov častejšie pozorovala vyrážka, bez ohľadu na kauzalitu, pri režimoch obsahujúcich darunavir + raltegravir v porovnaní
s režimami obsahujúcimi darunavir bez raltegraviru alebo raltegravir bez darunaviru. Vyrážka považovaná skúšajúcim za súvisiacu s liekom, sa vyskytla v podobnej miere. Expozícii prispôsobené miery vyrážky (všetky príčiny) boli 10,9; 4,2 a 3,8 na 100 pacientorokov v danom poradí a u vyrážky súvisiacej s liekom boli 2,4; 1,1 a 2,3 na 100 pacientorokov v danom poradí. Vyrážky pozorované
v klinických štúdiách boli mierne až stredne závažné a neviedli k prerušeniu liečby (pozri časť 4.4).
Metabolické parametre
Počas antiretrovírusovej liečby sa môže zvýšiť telesná hmotnosť a hladiny lipidov a glukózy v krvi
(pozri časť 4.4).
Poruchy kostrovej a svalovej sústavy
Počas liečby inhibítormi proteáz, najmä v kombinácii s NRTI, sa vyskytli prípady zvýšenia hladiny
kreatínfosfokinázy, myalgie, myozitídy a zriedkavo aj rabdomyolýzy.
Boli hlásené prípady osteonekrózy, najmä u pacientov so všeobecne potvrdenými rizikovými faktormi, pokročilým ochorením HIV alebo dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART). Frekvencia nie je známa (pozri časť 4.4).
Imunoreštitučný zápalový syndróm
U pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej
antiretrovírusovej liečby (CART) môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie. Boli tiež zaznamenané aj autoimunitné poruchy (ako je Gravesova choroba); avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.4).
Krvácanie u pacientov s hemofíliou
U pacientov s hemofíliou dostávajúcich antiretrovirálne inhibítory proteázy boli hlásené prípady
zvýšeného spontánneho krvácania (pozri časť 4.4).
Pediatrická populácia
Posúdenie bezpečnosti u pediatrických pacientov je založené na 48-týždňovej analýze údajov
o bezpečnosti z troch klinických skúšaní fázy II. Boli hodnotené nasledovné populácie pacientov
(pozri časť 5.1):
● 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17
rokov a s hmotnosťou najmenej 20 kg, ktorí užívali darunavir tablety s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku od 3 do < 6
rokov a s hmotnosťou 10 kg až < 20 kg (16 účastníkov od 15 kg do < 20 kg), ktorí užívali darunavir perorálnu suspenziu s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 12 pediatrických pacientov infikovaných HIV-1 vo veku od 12 do 17 rokov a s hmotnosťou
najmenej 40 kg, ktorí neboli predtým liečení ART. Títo pacienti dostávali darunavir tablety
s nízkou dávkou ritonaviru jedenkrát denne spolu s inými antiretrovírusovými liekmi. (pozri
časť 5.1).
Celkovo bol bezpečnostný profil u týchto pediatrických pacientov podobný bezpečnostnému profilu
pozorovanému u dospelých.
Iné osobitné skupiny pacientov
Pacienti súčasne infikovaní vírusom hepatitídy B a/alebo hepatitídy C
Spomedzi 1968 predtým liečených pacientov, ktorí užívali darunavir spolu s ritonavirom 600/100 mg dvakrát denne, bolo 236 pacientov infikovaných zároveň hepatitídou B alebo C. U súčasne infikovaných pacientov bola vyššia pravdepodobnosť počiatočného a pri liečbe vznikajúceho zvýšenia hladiny pečeňových transamináz ako u pacientov bez chronickej vírusovej hepatitídy (pozri časť 4.4).
H
l
ásenie
podozrení
na
nežiaduce
r
eakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSkúsenosti s akútnym predávkovaním darunavirom užívaným s nízkou dávkou ritonaviru u ľudí sú obmedzené. Zdravým dobrovoľníkom sa darunavir podával vo forme perorálneho roztoku
v jednorazových dávkach do 3200 mg ako samotný, resp. vo forme tabliet v jednorazových dávkach
do 1600 mg v kombinácii s ritonavirom bez toho, že by sa u nich pozorovali nepriaznivé symptomatické účinky.
V prípade predávkovania darunavirom nie je k dispozícii žiadne špecifické antidotum. Liečba predávkovania darunavirom zahŕňa všeobecné podporné opatrenia, vrátane sledovania vitálnych prejavov a pozorovania klinického stavu pacienta. Ak je to indikované, eliminácia nevstrebanej aktívnej látky sa dosiahne pomocou vyvolania vracania.
Aj podanie aktívneho uhlia môže pomôcť pri eliminácii nevstrebanej aktívnej látky. Vzhľadom na vysokú väzbu darunaviru na plazmatické bielkoviny je nepravdepodobné, že by dialýza mala väčší význam pri eliminácii aktívnej látky z cirkulácie.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antivirotiká na systémové použitie, inhibítory proteázy, ATC kód: J05AE10
Mechanizmus účinkuDarunavir je inhibítor dimerizácie a katalytickej aktivity HIV-1 proteázy (KD 4,5 x 10-12M). Selektívne
inhibuje odštiepenie HIV kódovaných polyproteínov Gag-Pol v bunkách infikovaných vírusom, čím zabraňuje tvorbe zrelých infekčných vírusových častíc.
Antivírusová aktivita in vitroDarunavir vykazuje aktivitu proti laboratórnym kmeňom a klinickým izolátom HIV-1 a laboratórnym
kmeňom HIV-2 v akútne infikovaných líniách T-buniek, mononukleárnych bunkách z periférnej krvi
človeka a ľudských monocytov/makrofágov so strednými hodnotami EC50 v rozmedzí
1,2 až 8,5 nmol/l (0,7 až 5,0 ng/ml).
In vitro má darunavir antivírusovú aktivitu voči širokému spektru
primárnych izolátov zo skupiny M HIV-1 (A, B, C, D, E, F, G) a skupiny O s hodnotami EC50
v rozmedzí od < 0,1 do 4,3 nmol/l.
Tieto hodnoty EC50 sú výrazne nižšie ako je rozsah toxickej koncentrácie pre 50 % buniek od
87 µmol/l do > 100 µmol/l.
RezistenciaSelekcia vírusu rezistentného na darunavir z divokého typu HIV-1
in vitro trvala pomerne dlho
(> 3 roky). Vyselektované vírusy neboli schopné rásť v prítomnosti darunaviru v koncentráciách nad
400 nmol/l. U vírusov vyselektovaných v týchto podmienkach a vykazujúcich zníženú citlivosť voči darunaviru (rozmedzie 23 až 50-násobne) sa v géne pre proteázu zistili substitúcie 2 až 4 aminokyselín. Zníženú citlivosť vznikajúcich vírusov na darunavir v selekcii nie je možné vysvetliť vznikom týchto mutácií proteázy.
Údaje z klinických štúdií s pacientmi liečenými ART (štúdia TITAN a zlúčená analýza štúdií POWER
1, 2 a 3 a štúdií DUET 1 a 2) preukázali, že virologická odpoveď na darunavir užívaný s nízkou dávkou ritonaviru bola znížená, keď boli na začiatku prítomné 3 a viac mutácií spojených s rezistenciou voči darunaviru (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V a L89V) alebo keď sa tieto mutácie vyvinuli počas liečby.
Zvýšenie východiskovej násobnej zmeny darunaviru v EC50 (FC) súviselo s poklesom virologickej odpovede. Určila sa dolná a horná klinická hranica 10 a 40. Izoláty s východiskovou hodnotou FC
≤ 10 sú citlivé; izoláty s FC > 10 až 40 majú zníženú citlivosť; izoláty s FC > 40 sú rezistentné (pozri
Klinické výsledky).
Vírusy izolované u pacientov s dávkou darunavir/ritonavir 600/100 mg dvakrát denne, u ktorých sa vyskytlo spontánne virologické zlyhanie, ktoré boli citlivé na tipranavir na začiatku, zostali vo veľkej väčšine prípadov citlivé na tipranavir po liečbe.
Najnižšie miery rozvoja rezistencie vírusu HIV sú pozorované u pacientov doposiaľ neliečených ART, ktorí sú po prvýkrát liečení darunavirom v kombinácii s inou ART.
Tabuľka nižšie zobrazuje vývoj mutácií a stratu citlivosti na PI pri virologickom zlyhaní v závere
liečby v štúdiách ARTEMIS, ODIN a TITAN.
ARTEMIS ODIN TITAN
Darunavir/
ritonavir
800/100 mg jedenkrát denne N=343
Darunavir/
ritonavir
800/100 mg jedenkrát denne N=294
Darunavir/
ritonavir
600/100 mg dvakrát denne N=296
Darunavir/
ritonavir
600/100 mg dvakrát denne N=298
Celkový počet virologických zlyhanía, n (%)
55 (16,0 %) 65 (22,1 %) 54 (18,2 %) 31 (10,4 %)
Spontánne 39 (11,4 %) 11 (3,7 %) 11 (3,7 %) 16 (5,4 %)
Jedinci nereagujúci na
liečbu
16 (4,7 %) 54 (18,4 %) 43 (14,5 %) 15 (5,0 %)
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými genotypmi, u ktorých sa vyvinuli mutácieb v závere liečby, n/N
Primárne (významné)
mutácie PI
0/43 1/60 0/42 6/28
PI RAM 4/43 7/60 4/42 10/28
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými fenotypmi, u ktorých sa preukázala strata citlivosti na PI v závere liečby v porovnaní so začiatkom liečby, n/N
PI
darunavir 0/39 1/58 0/41 3/26 amprenavir 0/39 1/58 0/40 0/22 atazanavir 0/39 2/56 0/40 0/22 indinavir 0/39 2/57 0/40 1/24 lopinavir 0/39 1/58 0/40 0/23 sakvinavir 0/39 0/56 0/40 0/22 tipranavir 0/39 0/58 0/41 1/25
a TLOVR ne-VF cenzurovaný algoritmus založený na HIV-1 RNA < 50 kópií/ml, okrem
TITAN (HIV-1 RNA
< 400 kópií/ml)
b zoznamy IAS-USA
SkríženárezistenciaNásobná zmena (FC) darunaviru bola menej ako 10 pre 90 % z 3309 klinických izolátov rezistentných
voči amprenaviru, atazanaviru, indinaviru, lopinaviru, nelfinaviru, ritonaviru, sakvinaviru a/alebo
tipraniviru, čo naznačuje, že vírusy rezistentné voči väčšine PI zostávajú citlivé voči darunaviru.
V štúdii
ARTEMIS sa v prípade virologického zlyhania nepozorovala žiadna skrížená rezistencia
s inými PI.
K
l
i
n
i
cké výsledky
Dospelí pacienti
Výsledky klinických štúdií u dospelých pacientov doposiaľ neliečených ART, pozri súhrn charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Úči nnosť darunaviru 600 mg dvakrát denne užívanej so 100 mg ritonaviru dvakrát denne u pacientov
pre dtým li ečený ch ART
Údaje o účinnosti kombinácie darunavir a ritonavir (600/100 mg dvakrát denne) u pacientov predtým
liečených ART sú založené na analýze 96 týždňovej štúdie fázy III TITAN u pacientov predtým
liečených ART, ktorí ešte neužívali lopinavir, na analýze 48 týždňovej štúdie fázy III ODIN
u pacientov predtým liečených ART bez DRV-RAM a na analýze údajov z 96 týždňov trvajúcich štúdií fázy IIb POWER 1 a 2 u pacientov predtým liečených ART s vysokou rezistenciou na PI.
TITAN je randomizovaná, kontrolovaná, otvorená štúdia fázy III porovnávajúca darunavir podávaný spolu s ritonavirom (600/100 mg dvakrát denne) verzus lopinavir/ritonavir (400/100 mg dvakrát denne) u dospelých pacientov predtým liečených ART infikovaných HIV-1, ktorí ešte neužívali
lopinavir. Obe ramená používali optimalizovaný základný režim (OBR, z angl. Optimised Background
Regimen), ktorý pozostával z minimálne 2 antiretrovirotík (NRTI s alebo bez NNRTI).
Nižšie uvedená tabuľka uvádza údaje 48-týždňovej analýzy účinnosti zo štúdie TITAN.
TITAN
Výsledky Darunavir/ritonavir
600/100 mg dvakrát denne + OBR N=298
lopinavir/ritonavir
400/100 mg dvakrát denne + OBR N=297
Rozdiel v liečbe
(95 % CI rozdielu)
HIV-1 RNA < 50 kópií/mla 70,8 % (211) 60,3 % (179) 10,5 % (2,9; 18,1)b
medián zmeny v počte buniek CD4+ oproti východiskovej hodnote (x 106/l)c
88 81
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede
c NC=F
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom,
definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA < 400 a < 50 kópií/ml, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat
(ITT) aj u pacientov sledovaných v protokole (On Protocol). Tieto výsledky sa potvrdili v analýze údajov z 96 týždňov trvajúcej liečby v štúdii TITAN, pričom v 96. týždni malo 60,4 % pacientov
v ramene s darunavirom/ritonavirom HIV-1 RNA < 50 kópií/ml v porovnaní s 55,2 % pacientov v ramene s lopinavirom/ritonavirom [rozdiel: 5,2 %, 95 % CI (-2,8; 13,1)].
ODIN je randomizovaná otvorená štúdia fázy III porovnávajúca darunavir/ritonavir 800/100 mg jedenkrát denne a darunavir/ritonavir 600/100 mg dvakrát denne u pacientov infikovaných HIV-1 predtým liečených ART, u ktorých skríningové vyšetrenie rezistencie genotypu nepreukázalo RAM voči darunaviru (t.j. V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V, L89V)
a skríningové vyšetrenie HIV-1 RNA bolo > 1 000 kópií/ml. Analýza účinnosti je založená na liečbe trvajúcej 48 týždňov (pozri tabuľku nižšie). Obe ramená použili optimalizovaný základný režim (OBR) ≥ 2 NRTI.
Výsledky Darunavir/ritonavir
800/100 mg jedenkrát denne + OBR
N=294
ODIN Darunavir/ritonavir
600/100 mg dvakrát
denne + OBR N=296
Rozdiel v liečbe
(95% CI rozdielu)
HIV-1 RNA
< 50 kópií/mla
S východiskovou HIV-1 RNA (kópie/ml)
72,1 % (212) 70,9 % (210) 1,2 % (-6,1; 8,5)b
< 100 000
≥ 100 000
S východiskovým počtom buniek CD4+ (x 106/l)
≥ 100
< 100
S kmeňom HIV-1
Typ B Typ AE Typ C Inéc
77,6 % (198/255)
35,9 % (14/39)
75,1 % (184/245)
57,1 % (28/49)
70,4 % (126/179)
90,5 % (38/42)
72,7 % (32/44)
55,2 % (16/29)
73,2 % (194/265)
51 6 % (16/31)
72,5 % (187/258)
60,5 % (23/38)
64,3 % (128/199)
91,2 % (31/34)
78,8 % (26/33)
83,3 % (25/30)
4,4 % (-3,0; 11,9)
-15,7 % (-39,2; 7,7)
2,6 % (-5,1; 10,3)
-3,4 % (-24,5; 17,8)
6,1 % (-3,4; 15,6)
-0,7 % (-14,0; 12,6)
-6,1 % (-2,6; 13,7)
-28,2 % (-51,0; -5,3)
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x 106/l)e
108 112 -5d (-25; 16)
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede c Kmene A1, D, F1, G, K, CRF02_AG, CRF12_BF, a CRF06_CPX d Rozdiel stredných hodnôt
e Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward)
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom
800/100 mg jedenkrát denne, definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA
< 50 kópií/ml v porovnaní s darunavirom/ritonavirom 600/100 mg dvakrát denne, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat (ITT) aj
u pacientov sledovaných v protokole (On Protocol).
Darunavir/ritonavir 800/100 mg jedenkrát denne sa nemá používať u tých pacientov liečených ART,
ktorí majú jednu alebo viac mutácií spojených s rezistenciou voči darunaviru (DRV-RAM) alebo
HIV-1 RNA ≥ 100 000 kópií/ml alebo počet buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2 a 4.4).
U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje.
POWER 1 a POWER 2 sú randomizované, kontrolované porovnávajúce darunavir užívaný spolu
s ritonavirom (600/100 mg dvakrát denne) s kontrolnou skupinou užívajúcou inhibítor proteáz vybraný skúšajúcim u pacientov infikovaných HIV-1, ktorí predtým absolvovali viac ako jeden neúspešný
liečebný režim zahŕňajúci inhibítor proteáz (PI). V oboch štúdiách bol použitý OBR zahŕňajúci aspoň
2 NRTI s enfuvirtidom (ENF) alebo bez neho.
V nasledujúcej tabuľke sú uvedené údaje 48-týždňovej a 96-týždňovej analýzy účinnosti zo zlúčených štúdií POWER 1 a POWER 2.
Spoločné údaje získané v štúdiách POWER 1 a POWER 2
48. týždeň 96. týždeň
Výsledky Darunavir/ritonavir
600/100 mg 2x denne
n=131
Kontrola n=124
Rozdiel medzi liečbami
Darunavir/ritonavir;
600/100 mg 2x denne
n=131
Kontrola n=124
Rozdiel medzi liečbami
HIV RNA
< 50 kópií/
mla
45,0 % (59) 11,3 % (14)
33,7 % (23,4 %;
44,1 %)c
38,9 % (51) 8,9 % (11)
30,1 % (20,1;
40,0)c
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x
106/l)b
103 17 86
(57; 114)c
133 15 118 (83,9;
153,4)c
a Výpočet na základe algoritmu TLOVR.
b Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward).
c 95 % interval spoľahlivosti.
Analýza údajov z 96 týždňov trvajúcej liečby v štúdiách POWER preukázala zachovanú
antiretrovírusovú účinnosť a imunologický benefit.
Z 59 pacientov, ktorí reagovali na liečbu úplnou supresiou vírusu (< 50 kópií/ml) v 48. týždni,
47 pacientov (80 % respondérov na liečbu v 48. týždni) naďalej reagovalo na liečbu v 96. týždni.
Východiskový genotyp alebo fenotyp a virologický výsledok
Východiskový genotyp a FC darunaviru (zmena citlivosti vzhľadom k východiskovej hodnote) sa ukázal ako prediktívny faktor virologického výsledku.
Podiel (%) pacientov s odpoveďou (HIV-1 RNA < 50 kópii/ml v týždni 24) na darunavir užívaný spolu s ritonavirom (600/100 mg dvakrát denne) u východiskového genotypu*a a východiskovej FC darunaviru pri užívaní enfuvirtidu (ENF): podľa analýz liečby štúdií POWER a DUET.
Odpoveď (HIV-1
Počet východiskových mutáciía Východisková DRV FCb
RNA < 50 kópií/ml v 24. týždni)
%, n/N
Všetky
rozpätia 0-2 3 ³ 4
Všetky
rozpätia £ 10 10-40 > 40
Všetci pacienti 45 %
455/1 014
54 %
359/660
39 %
67/172
12 %
20/171
45 %
455/1 014
55 %
364/659
29 %
59/203
8 %
9/118
Pacienti neužívajúci/opako- vane užívajúci ENFc Pacienti s de novo ENFd
39 %
290/741
60 %
165/273
50 %
238/477
66 %
121/183
29 %
35/120
62 %
32/52
7 %
10/135
28 %
10/36
39 %
290/741
60 %
165/273
51 %
244/477
66 %
120/182
17 %
25/147
61 %
34/56
5 %
5/94
17 %
4/24
a Počet mutácií zo zoznamu mutácií spojených so znížením odpovede na kombináciu darunavir/ritonavir (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V alebo L89V)
b násobná zmena v EC50
c “Pacienti neužívajúci/opakovane užívajúci ENF”, t.j. pacienti, ktorí neužívali ENF alebo tí, ktorí ENF užívali, ale nie
prvýkrát.
d “Pacienti s
de novo ENF”, t.j. pacienti, ktorí užívali ENF prvýkrát.
Pediatrickí pacientiVýsledky klinických štúdií u pediatrických pacientov vo veku 12 až 17 rokov, ktorí neboli predtým
liečení ART, nájdete v Súhrne charakteristických vlastností lieku darunavir 400 mg a 800 mg tablety.
Pediatrickí pacienti od 6 do < 18 rokov a s hmotnosťou naj menej 20 kg pre dt ým li eče ní ART
DE
LPHI
je otvorená štúdia fázy II hodnotiaca farmakokinetiku, bezpečnosť, toleranciu a účinnosť darunaviru s nízkou dávkou ritonaviru u 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg. Títo pacienti užívali darunavir/ritonavir dvakrát denne spolu s inými antiretrovírusovými látkami (pre odporúčané dávkovanie v závislosti od hmotnosti pacienta pozri časť 4.2). Virologická odpoveď bola definovaná ako pokles vírusového zaťaženia HIV-1 RNA v plazme o najmenej 1,0 log10 oproti východiskovej hodnote.
V tejto štúdii bolo pacientom so zvýšeným rizikom prerušenia liečby z dôvodu intolerancie perorálneho roztoku ritonaviru (napr. averzia voči chuti) umožnené prejsť na užívanie kapsúl. Zo
44 pacientov užívajúcich perorálny roztok ritonaviru, 27 prešlo na užívanie 100 mg kapsúl a prekročilo dávku ritonaviru odvodenú od hmotnosti bez zmien v sledovanej bezpečnosti.
DELPHI
Výsledky po 48. týždni Darunavir/ritonavir
N=80
HIV-1 RNA < 50 kópií/mla 47,5 % (38)
Priemerná zmena počtu buniek CD4+ oproti
východiskovej hodnoteb
a Výpočet na základe algoritmu TLOVR.
147
b Pacienti, ktorí neukončili liečbu, sú počítaní ako zlyhanie: pacientom, ktorí prerušili liečbu predčasne, sa priradí
zmena rovná nule.
Podľa cenzurovaného TLOVR algoritmu pre nevirologické zlyhanie u 24 (30,0 %) pacientov došlo
k virologickému zlyhaniu, z nich u 17 (21,3 %) pacientov sa objavil rebound fenomén a 7 (8,8 %)
pacientov neodpovedalo na liečbu.
Pediatrickí pacienti vo veku 3 až < 6 rokov predtým lieč ení ART
U 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART vo veku od 3 do < 6
rokov a s hmotnosťou 10 kg až < 20 kg bola farmakokinetika, bezpečnosť, tolerovateľnosť a účinnosť
darunaviru/ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami hodnotená v otvorenej štúdii fázy II ARIEL. Pacienti dostávali dávku dvakrát denne v závislosti od ich hmotnosti, pacienti s hmotnosťou 10 kg až < 15 kg dostávali darunavir/ritonavir 25/3 mg/kg dvakrát denne a pacienti s hmotnosťou 15 kg až < 20 kg dostávali darunavir/ritonavir 375/50 mg dvakrát denne Virologická odpoveď, definovaná ako percento pacientov s potvrdenou plazmatickou hladinou
< 50 HIV-1 RNA kópií/ml, bola v 48. týždni hodnotená u 16 pediatrických pacientov s hmotnosťou
15 kg až < 20 kg a u 5 pediatrických pacientov s hmotnosťou 10 kg až < 15 kg dostávajúcich darunavir/ritonavir v kombinácii s inými antiretrovírusovými látkami. (pozri časť 4.2 pre odporúčanie
dávkovania podľa telesnej hmotnosti).
ARIEL
Výsledky po 48. týždni Darunavir/ritonavir
10 kg až < 15 kg
N=5
15 kg až < 20 kg
N=16
HIV-1 RNA < 50 kópií/mla 80,0 % (4) 81,3 % (13)
CD4+ percentuálna zmena oproti východiskovej hodnoteb Priemerná zmena počtu buniek CD4+ oproti východiskovej hodnoteb
a Imputácie na základe algoritmu TLOVR.
b NC=F
4 4
16 241
Dostupné sú obmedzené údaje o účinnosti u pediatrických pacientov s hmotnosťou menej ako 15 kg,
a preto neumožňujú uviesť odporúčania pre dávkovanie.
G
ravidita a obdobie po pôrode
Darunavir/ritonavir (600/100 mg dvakrát denne alebo 800/100 mg jedenkrát denne) v kombinácii so základným režimom bol hodnotený v klinickej štúdii s 34 gravidnými ženami (17 v každej skupine) počas druhého a tretieho trimestra a po pôrode. Virologická odpoveď sa udržala počas trvania štúdie v oboch skupinách. U detí narodených 29 pacientkám, ktoré zotrvali na antivirotickej liečbe až do
pôrodu, sa nevyskytol prenos z matky na dieťa. Nevyskytli sa žiadne nové klinicky významné zistenia týkajúce sa bezpečnosti v porovnaní so známym profilom bezpečnosti darunaviru/ritonaviru
u dospelých infikovaných HIV-1 (pozri časti 4.2, 4.4 a 5.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti darunaviru podávaného v kombinácii s ritonavirom sa skúmali
u zdravých dospelých dobrovoľníkov a u pacientov infikovaných vírusom HIV-1. Expozícia voči
darunaviru u pacientov infikovaných vírusom HIV-1 bola vyššia ako u zdravých dobrovoľníkov. Vyššiu expozíciu voči darunaviru u pacientov infikovaných vírusom HIV-1 v porovnaní so zdravými dobrovoľníkmi je možné vysvetliť na základe vyšších koncentrácií α1-kyslého glykoproteínu (AAG) u pacientov infikovaných vírusom HIV-1, čo vedie k vyššej väzbe darunaviru na plazmatický AAG,
a tým aj k vyšším koncentráciám tejto látky v plazme.
Darunavir sa metabolizuje najmä prostredníctvom CYP3A. Ritonavir inhibuje CYP3A, v dôsledku
čoho sa výrazne zvyšujú plazmatické koncentrácie darunaviru.
Absorpcia
Darunavir sa rýchlo vstrebáva po perorálnom podaní. Pri podávaní v kombinácii s ritonavirom
v nízkej dávke sa maximálne plazmatické koncentrácie darunaviru obyčajne dosiahnu v priebehu 2,5 -
4 hodín.
Absolútna biologická dostupnosť po jednorazovom perorálnom podaní samotného darunaviru v dávke
600 mg bola približne 37 %, pričom pri podávaní ritonaviru v dávke 100 mg dvakrát denne sa zvýšila približne na 82 %. Po jednorazovom perorálnom podaní darunaviru v dávke 600 mg v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne sa systémová expozícia voči darunaviru
zvýšila 14-násobne (pozri časť 4.4).
Pri užívaní nalačno bola relatívna biologická dostupnosť darunaviru v prítomnosti nízko-dávkovaného ritonaviru o 30 % nižšia ako pri užívaní lieku s jedlom. Z toho dôvodu sa tablety darunaviru musia užívať spolu s ritonavirom a jedlom. Druh jedla nemá vplyv na expozíciu voči darunaviru.
Distribúcia
Približne 95 % darunaviru sa viaže na plazmatické bielkoviny, najmä na α1-kyslý glykoproteín.
Po intravenóznom podaní bol distribučný objem samotného darunaviru 88,1 ± 59,0 l (stredný ± SD)
a v prítomnosti 100 mg ritonaviru podávaného dvakrát denne bol zvýšený na 131 ± 49,9 l
(stredný ± SD).
Biotransformácia
In vitro štúdie využívajúce mikrozómy pečene človeka (HLM) naznačujú, že darunavir sa
metabolizuje najmä oxidatívnym metabolizmom. Darunavir sa výrazne metabolizuje prostredníctvom systému CYP v pečeni a takmer výlučne prostredníctvom izoenzýmu CYP3A4. V štúdii skúmajúcej aplikáciu darunaviru označeného rádioaktívnym 14C zdravým dobrovoľníkom sa zistilo, že väčšina rádioaktivity v plazme po jednorazovom podaní darunaviru s ritonavirom v dávke 400/100 mg pochádzala z pôvodného liečiva. U ľudí sa zistila prítomnosť najmenej 3 oxidatívnych metabolitov
darunaviru, pričom všetky z nich mali aktivitu proti divokému typu HIV najmenej 10x nižšiu ako
darunavir.
Eliminácia
Po podaní darunaviru s ritonavirom v dávke 400/100 mg označeného rádioaktívnym 14C sa 79,5 %
rádioaktivity zachytilo v stolici a 13,9 % rádioaktivity sa zachytilo v moči. Rádioaktivita nezmeneného darunaviru v stolici predstavovala 41,2 % a v moči predstavovala 7,7 % z podanej dávky. Terminálny polčas eliminácie darunaviru podávaného v kombinácii s ritonavirom bol približne
15 hodín.
Klírens darunaviru (150 mg) podaného intravenózne v monoterapii bol 32,8 l/h, kým pri kombinácii s nízko dávkovaným ritonavirom 5,9 l/h.
Osobitné skupiny pacientov
Pediatrická populácia
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 74 predtým liečených pediatrických pacientov vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg preukázala, že dávky darunaviru/ritonaviru podávané v závislosti od hmotnosti, mali za následok expozíciu darunaviru porovnateľnú s expozíciou u dospelých pacientov užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 14 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 15 kg až < 20 kg ukázala, že dávkovanie založené na hmotnosti viedlo k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 12 pediatrických pacientov vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení ART, ukázala, že dávka darunaviru/ritonaviru 800/100 mg jedenkrát denne viedla k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne. Z toho dôvodu sa rovnaké dávkovanie jedenkrát denne môže použiť u dospievajúcich vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg
s predchádzajúcou liečbou bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)*
a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+
≥ 100 buniek x 106/l (pozri časť 4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 10 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 14 kg až < 20 kg preukázala, že dávky odvodené od hmotnosti viedli k expozícii darunaviru, ktorá bola porovnateľná
s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne (pozri
časť 4.2). Farmakokinetické modelovanie a simulácia expozície darunaviru u pediatrických pacientov
vo veku 3 až < 18 rokov navyše potvrdili expozície darunaviru, ktoré boli pozorované v klinických štúdiách a umožnili určenie dávkovania darunaviru/ritonaviru jedenkrát denne v závislosti na hmotnosti u pediatrických pacientov s hmotnosťou najmenej 15 kg, ktorí buď predtým nedostávali liečbu ART alebo u predtým liečených pediatrických pacientov bez DRV-RAM* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri časť
4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V.
Starší ľudia
Pri analýze farmakokinetiky v rôznych populáciách pacientov s infekciou HIV sa zistilo, že
farmakokinetika darunaviru sa výraznejšie nelíši u pacientov s infekciou HIV vo vekovom rozmedzí
od 18 do 75 rokov (n = 12, vek ≥ 65 rokov) (pozri časť 4.4). U pacientov starších ako 65 rokov boli
k dispozícii iba obmedzené údaje.
Pohlavie
Pri analýze farmakokinetiky v rôznych populáciách pacientov sa u žien s infekciou HIV zistila mierne vyššia expozícia voči darunaviru (o 16,8 %) ako u mužov infikovaných HIV. Tento rozdiel nemá žiadny klinický význam.
Porucha funkcie obličiek
Výsledky štúdie zachovania hmotnosti s 14C rádioaktívne označeným darunavirom s ritonavirom ukázali, že približne 7,7 % z podanej dávky darunaviru sa vylučuje do moču v nezmenenej forme.
Aj keď sa podávanie darunaviru nesledovalo u pacientov s poruchou funkcie obličiek, pri analýze farmakokinetiky v rôznych populáciách pacientov sa zistilo, že stredne závažná porucha funkcie obličiek (klírens kreatinínu: 30 - 60 ml/min, n=20) nemá väčší vplyv na farmakoniketiku darunaviru u pacientov s infekciou HIV (pozri časti 4.2 a 4.4).
Porucha funkcie pečene
Darunavir sa metabolizuje a eliminuje prevažne v pečeni. V štúdii s opakovaným podaním lieku darunavir užívaná s ritonavirom (600/100 mg dvakrát denne) sa preukázalo, že celkové koncentrácie darunaviru v plazme u subjektov s miernou (Childova-Pughova trieda A, n=8) a stredne ťažkou (Childova-Pughova trieda B, n=8) poruchou funkcie pečene boli porovnateľné s tými u zdravých subjektov. Koncentrácie voľného darunaviru boli približne o 55 % (Childova-Pughova Trieda A)
a 100 % (Childova-Pughova trieda B) vyššie. Klinický význam tohto nárastu nie je známy, preto treba darunavir užívať so zvýšenou opatrnosťou. Účinok ťažkej poruchy funkcie pečene na farmakokinetiku darunaviru nebol dosiaľ študovaný (pozri časti 4.2, 4.3 a 4.4).
Gravidita a obdobie po pôrode
Expozícia celkovému darunaviru a ritonaviru po užití darunaviru/ritonaviru 600/100 mg dvakrát denne a darunaviru/ritonaviru 800/100 mg jedenkrát denne v rámci antivirotického režimu bola všeobecne nižšia počas gravidity v porovnaní s obdobím po pôrode. V prípade neviazaného (t.j. aktívneho) darunaviru však boli farmakokinetické parametre menej znížené počas gravidity v porovnaní
s obdobím po pôrode z dôvodu zvýšenia neviazanej frakcie darunaviru počas gravidity v porovnaní s obdobím po pôrode.
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru
600/100 mg dvakrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho trimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=11)aTretí trimester gravidity (n=11)Obdobie po pôrode (6-12 týždňov) (n=11)
Cmax, ng/ml 4 601 ± 1 125 5 111 ± 1 517 6 499 ± 2 411
AUC12h, ng.h/ml 38 950 ± 10 010 43 700 ± 16 400 55 300 ± 27 020
Cmin, ng/mlb 1 980 ± 839,9 2 498 ± 1 193 2 711 ± 2 268
a n=10 pre AUC12h
b vylúčením hodnoty Cmin pod LLOQ, n=10 pre referenciu
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru 800/100 mgjedenkrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho trimestra gravidity a po pôrode
trimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=16)
Tretí trimester gravidity (n=14)
Obdobie po pôrode (6-12 týždňov) (n=15)
Cmax, ng/ml 4 988 ± 1 551 5 138 ± 1 243 7 445 ± 1 674
AUC12h, ng.h/ml 61 303 ± 16 232 60 439 ± 14 052 94 529 ± 28 572
Cmin, ng/mla 1 193 ± 509 1 098 ± 609 1 572 ± 1 108
a n=12 pre obdobie po pôrode, n=15 pre druhý trimester a n=14 pre tretí trimester
U žien užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 28 %, 24 %
a 17 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 19 %, 17 % nižšie a o 2 % vyššie, v tomto poradí, v porovnaní s obdobím po pôrode.
U žien užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne počas druhého trimestra gravidity boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 34 %, 34 %
a 32 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity
boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 31 %, 35 % a 50 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode.
5.3 Predklinické údaje o bezpečnosti
Toxikologické štúdie so samotným darunavirom sa vykonali v dávkovaní až do klinických hladín
u myší, potkanov a psov, kým štúdie s jeho kombináciou s ritonavirom sa vykonali u potkanov a psov.
V štúdiách toxicity po opakovanej dávke u myší, potkanov a psov sa zistili len obmedzené účinky liečby darunavirom. Identifikovaným cieľovým orgánom u hlodavcov bol hemopoetický systém, koagulačný systém, pečeň a štítna žľaza. Pozoroval sa premenlivý, ale mierny pokles parametrov v súvislosti s erytrocytmi spolu s predĺžením aktivovaného parciálneho tromboplastínového času.
Zmeny boli pozorované v pečeni (hypertrofia hepatocytov, vakuolizácia, zvýšenie hladiny pečeňových enzýmov) a štítnej žľaze (folikulárna hypertrofia). U laboratórnych potkanov viedla kombinácia darunaviru s ritonavirom k miernemu zvýšeniu účinku na parametre červených krviniek, pečene
a štítnu žľazu a k zvýšeniu incidencie ostrovčekov fibrózy v pankrease (iba u samcov laboratórnych potkanov) v porovnaní so samotným darunavirom. U psov sa nepozorovali žiadne väčšie známky
toxicity ani cieľové orgány toxicity pri rovnakej expozícii ako u človeka, ktorý užíva odporučené
dávky.
V štúdii na potkanoch bol znížený počet corpora lutea a implantácií v prípade maternálnej toxicity. Na druhej strane sa nepozorovali žiadne účinky na párenie alebo plodnosť po podávaní darunaviru
v dávkach do 1000 mg/kg/deň a pri expozícii (AUC - 0,5-násobok) nižšej ako u človeka dostávajúceho odporučené dávky. Pri podávaní rovnako veľkých dávok samotného darunaviru sa u potkanov
a králikov nepozorovala teratogenita podobne ako u myší dostávajúcich darunavir v kombinácii s ritonavirom. Stupeň expozície bol nižší ako u človeka dostávajúceho odporučené dávky. Pri hodnotení prenatálneho a postnatálneho vývoja u potkanov, darunavir podávaný samostatne alebo
v kombinácii s ritonavirom viedol k prechodnému zníženiu prírastku telesnej hmotnosti u dojčených mláďat a došlo k miernemu oneskoreniu v otvorení očí a uší. Darunavir v kombinácii s ritonavirom
spôsobil zníženie prežívania mláďat, ktoré sa prejavilo nepriaznivou odpoveďou v 15. deň laktácie
a znížením počtu mláďat, ktoré prežili počas laktácie. Tieto účinky môžu byť prisúdené sekundárnej expozícii liečiva u mláďat prostredníctvom materského mlieka a/alebo maternálnej toxicity. Darunavir
podávaný ako samotný alebo v kombinácii s ritonavirom neovplyvnil žiadne funkcie po odstavení.
U juvenilných laboratórnych potkanov, ktorí dostávali darunavir 23. – 26. deň, sa pozorovala zvýšená
mortalita s kŕčmi u niektorých zvierat. Medzi 5. a 11. dňom života bola expozícia v plazme, pečeni
a mozgu výrazne vyššia ako u dospelých laboratórnych potkanov po porovnateľných dávkach
v mg/kg. Po 23. dni života bola expozícia porovnateľná s tou u dospelých potkanov. Zvýšená expozícia bola pravdepodobne aspoň čiastočne z dôvodu nezrelosti enzýmov metabolizujúcich liek
u juvenilných zvierat. U juvenilných potkanov neboli spozorované žiadne úmrtia v súvislosti s liečbou
dávkami 1000 mg/kg darunaviru (jednorazová dávka) v 26. deň veku alebo 500 mg/kg (opakovaná dávka) od 23. do 50. dňa veku a expozícia a profil toxicity boli porovnateľné s tými, pozorovanými u dospelých potkanov.
Darunavir s nízkou dávkou ritonaviru sa nemá používať u detí mladších ako 3 roky z dôvodu
nejasnosti ohľadom miery rozvoja hematoencefalickej bariéry a pečeňových enzýmov.
Karcinogénny potenciál darunaviru bol vyhodnotený pri podávaní lieku myšiam a potkanom sondou do žalúdka počas 104 týždňov. Myšiam boli podávané denné dávky 150, 450 a 1000 mg/kg a potkanom boli podávané dávky 50, 150 a 500 mg/kg. Výskyt hepatocelulárnych adenómov a
karcinómov stúpal v závislosti od dávky a pozoroval sa u samcov a samíc oboch živočíšnych druhov. U samcov potkanov sa vyskytli adenómy folikulárnych buniek štítnej žľazy. Podávanie darunaviru nespôsobilo štatisticky významný nárast iných benígnych alebo malígnych novotvarov u myší alebo potkanov. Pozorované hepatocelulárné tumory a tumory štítnej žľazy u hlodavcov sa považujú za obmedzené z hľadiska významnosti pre človeka. Opakované podávanie darunaviru potkanom spôsobilo indukciu mikrozomálnych enzýmov v pečeni a zvýšilo vylučovanie hormónov štítnej žľazy,
ktoré predurčuje potkanov, ale nie ľudí, na výskyt neoplaziem štítnej žľazy. Pri najvyšších testovaných dávkach boli systémové expozície (založené na AUC) darunaviru 0,4- až 0,7-násobkom (myši) a 0,7-
až 1-násobkom (potkany) v pomere k expozíciám pozorovaným u ľudí pri odporúčaných
terapeutických dávkach.
Po dvoch rokoch podávania darunaviru pri expozíciách na úrovni alebo pod úrovňou expozícií u ľudí sa pozorovali zmeny na obličkách u myší (nefróza) a u potkanov (chronická progresívna nefropatia).
Darunavir nebol mutagénny ani genotoxický v súbore in vitro a in vivo testov vrátane bakteriálnej reverznej mutácie (Amesov test), chromozómovej aberácie v ľudských lymfocytoch
a mikronukleového testu u myší in vivo.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Oxid kremičitý, koloidný bezvodý
Mikrokryštalická celulóza
Krospovidón
Sodná soľ glykolátu škrobu
Hypromelóza
Magnéziumstearát
Obal tablety
Polyvinylalkohol, čiastočne hydrolyzovaný
Oxid titaničitý
Makrogol
Mastenec
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po prvom otvorení HDPE fľaše: 100 dní.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PVC/PE/PVDC-Al blistrové balenie s obsahom 30 a 60 tabliet a 60 x 1 tabliet.
Za studena spracované PVC/Al/OPA-Al blistrové balenie s obsahom 30 a 60 tabliet a 60 x 1 tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMylan S.A.S.
117 Allee des Parcs
69 800 Saint Priest
Francúzsko
8. REGISTRAČNÉ ČÍSLOEU/1/16/1140/030
EU/1/16/1140/031
EU/1/16/1140/032
EU/1/16/1140/033
EU/1/16/1140/034
EU/1/16/1140/035
EU/1/16/1140/036
EU/1/16/1140/037
EU/1/16/1140/038
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: [DD. mesiac RRRR]
10. DÁTUM REVÍZIE TEXTU{MM/RRRR}
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.
1. NÁZOV LIEKU
Darunavir Mylan 800 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE Každá filmom obalená tableta obsahuje 800 mg darunaviru. Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalené tablety.
Biele až sivobiele oválne obojstranne vypuklé filmom obalené tablety s rozmermi približne 21,2 mm x
10,6 mm, s označením „M“ na jednej strane a „DV8“ na druhej strane..
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Darunavir podávaný súčasne s nízkou dávkou ritonaviru je indikovaná v kombinácii s inými antiretrovirálnymi liekmi na liečbu pacientov s infekciou vyvolanou vírusom ľudskej imunodeficiencie (HIV-1).
Darunavir podávaný súčasne s kobicistatom je indikovaná v kombinácii s inými antiretrovirálnymi liekmi na liečbu infekcie vyvolanej vírusom ľudskej imunodeficiencie (HIV-1) u dospelých pacientov (pozri časť 4.2).
Darunavir Mylan 800 mg tablety sa môžu použiť na zabezpečenie vhodného režimu dávkovania pri
liečbe infekcie HIV-1 u dospelých a pediatrických pacientov od 3 rokov a s hmotnosťou najmenej
40 kg, ktorí:
· neboli predtým liečení antiretrovírusovou liečbou (ART) (pozri časť 4.2).
· boli predtým liečení ART bez mutácií vedúcich k rezistencii voči darunaviru (DRV-RAM, z angl. darunavir-resistance associated mutations) a s plazmatickou hladinou HIV-1 RNA
< 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l. Pri rozhodovaní o začiatku liečby liekom darunavir u pacientov, ktorí uz boli predtým liečení ART, by malo vyšetrenie
genotypu usmerniť použitie lieku darunavir (pozri časti 4.2, 4.3, 4.4 a 5.1).
4.2 Dávkovanie a spôsob podávania
Liečbu môže začať len lekár, ktorý má skúsenosti s liečbou infekcie HIV. Po začatí liečby darunavirom treba pacientov poučiť, aby nemenili dávku, liekovú formu ani neprerušovali liečbu bez toho, aby sa poradili so svojím lekárom.
Interakčný profil darunaviru závisí od toho, či sa na zlepšenie farmakokinetiky použije ritonavir alebo kobicistat. Darunavir môže mať preto rôzne kontraindikácie a odporúčania pre súčasne podávané lieky v závislosti od toho, či je zlúčenina posilnená ritonavirom alebo kobicistatom (pozri časti 4.3, 4.4 a
4.5).
Dávkovanie
Darunavir sa musí vždy podávať perorálne v kombinácii s kobicistatom alebo s nízkou dávkou
ritonaviru, ktorý zlepšuje jej farmakokinetiku, a s inými antiretrovírusovými liekmi. Z toho dôvodu je
pred začatím kombinovanej liečby darunavirom a ritonavirom potrebné prečítať si súhrn
charakteristických vlastností kobicistatu alebo ritonaviru, podľa toho, čo je náležité. Kobicistat nie je
indikovaný na užívanie dvakrát denne ani na použitie v pediatrickej populácii.
Dospelí pacienti predtým neliečení ART
Odporúčaný režim dávkovania je 800 mg jedenkrát denne súčasne so 150 mg kobicistatu jedenkrát denne alebo 100 mg ritonaviru jedenkrát denne a s jedlom. Darunavir 800 mg tablety sa môžu použiť
na zostavenie režimu dávkovania 800 mg jedenkrát denne.
Dospelí pacienti predtým liečení ART
Odporúčané sú nasledujúce režimy dávkovania:
· U pacientov liečených ART bez mutácií vedúcich k rezistencii voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri časť 4.1) sa môže použiť dávkovanie 800 mg jedenkrát denne súčasne so
150 mg kobicistatu jedenkrát denne alebo 100 mg ritovariru jedenkrát denne a s jedlom. Darunavir 800 mg tablety sa môžu použiť na zostavenie režimu dávkovania 800 mg jedenkrát
denne.
· V prípade ostatných pacientov liečených ART alebo v prípade, že nie je k dispozícii test genotypu HIV-1, je odporúčané dávkovanie darunaviru 600 mg dvakrát denne súčasne so
100 mg ritonaviru dvakrát denne a s jedlom. Pozri Súhrn charakteristických vlastností lieku darunavir 100 mg/ml perorálna suspenzia, 75 mg, 150 mg, 300 mg alebo 600 mg tablety.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Pediatrickí pacienti (vo veku 3 až 17 rokov a s hmotnosťou najmenej 40 kg) predtým neliečení ART Odporúčaný režim dávkovania je 800 mg jedenkrát denne súčasne so 100 mg ritonaviru jedenkrát denne a s jedlom. U detí mladších ako 18 rokov nebola stanovená dávka kobicistatu, ktorá má byť užitá s darunavirom.
Pediatrickí pacienti predtým liečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 40 kg)
U detí mladších ako 18 rokov nebola stanovená dávka kobicistatu, ktorá má byť užitá s darunavirom.
Odporúčané sú nasledujúce režimy dávkovania:
● U pacientov predtým liečených ART bez DRV-RAM* a s plazmatickou hladinou HIV-1 RNA
< 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri časť 4.1) sa môže použiť dávkovací režim 800 mg jedenkrát denne so 100 mg ritonaviru jedenkrát denne s jedlom.
Darunavir 800 mg tablety sa môžu použiť na zostavenie režimu dávkovania 800 mg jedenkrát denne.
● V prípade ostatných pacientov liečených ART alebo v prípade, že nie je k dispozícii test
genotypu HIV-1, je odporúčaný režim dávkovania opísaný v Súhrne charakteristických vlastností lieku darunavir 75 mg, 150 mg, 300 mg alebo 600 mg tablety.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Odporúčanie pri vynechaní dávky
V prípade, že pacient zabudne užiť jedenkrát denne dávku darunaviru a/alebo kobicistatu alebo ritonaviru do 12 hodín od doby, kedy liek zvyčajne užíva, treba ho poučiť, že má užiť predpísanú dávku darunaviru a kobicistatu alebo ritonaviru s jedlom čo najskôr. Ak od zvyčajnej doby použitia uplynie viac ako 12 hodín, vynechaná dávka sa nemá užiť a pacient má pokračovať vo zvyčajnom dávkovacom režime.
Toto usmernenie vyplynulo z polčasu darunaviru v prítomnosti kobicistatu alebo ritonaviru a
odporúčaného dávkovacieho intervalu približne 24 hodín.
Osobitné skupiny pacientov
Starší ľudia
Informácie o tejto populácii sú obmedzené, a preto sa má darunavir používať v tejto vekovej skupine
so zvýšenou opatrnosťou (pozri časti 4.4 a 5.2).
Porucha funkcie pečene
Darunavir sa metabolizuje v pečeňovom systéme. U pacientov s miernou (Childova-Pughova Trieda A) alebo stredne ťažkou (Childova-Pughova Trieda B) poruchou funkcie pečene sa neodporúča žiadna úprava dávky, avšak týmto pacientom sa má darunavir podávať so zvýšenou opatrnosťou. U pacientov s ťažkou poruchou funkcie pečene nie sú k dispozícii žiadne farmakokinetické údaje. Ťažká porucha funkcie pečene môže viesť k zvýšeniu expozície darunaviru a k zhoršeniu jeho bezpečnostného
profilu. Z toho dôvodu sa darunavir nesmie podávať pacientom s ťažkou poruchou funkcie pečene
(Childova-Pughova Trieda C) (pozri časti 4.3, 4.4 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávkovania darunaviru/ritonaviru (pozri časti 4.4 a 5.2). Kobicistat sa neskúmal u pacientov na dialýze, a preto nie je možné stanoviť žiadne odporúčanie ohľadom užívania darunaviru/kobicistatu u týchto pacientov.
Kobicistat inhibuje tubulárnu sekréciu kreatinínu a môže spôsobiť mierne zvýšenie sérového
kreatinínu a mierny pokles klírensu kreatinínu. Použitie klírensu kreatinínu za účelom odhadu kapacity renálneho vylučovania môže byť preto zavádzajúce. Z toho dôvodu sa kobicistat na zlepšenie farmakokinetiky darunaviru nemá podávať pacientom s klírensom kreatinínu nižším ako 70 ml/min, ak si niektorá súčasne užívaná látka vyžaduje úpravu dávkovania v závislosti od klírensu kreatinínu: napr. emtricitabín, lamivudín, tenofovir disoproxil fumarát alebo adefovir dipovoxil.
Informácie o kobicistate si prečítajte v Súhrne charakteristických vlastností kobicistatu.
Pediatrická populácia
Darunavir sa nemá používať u pediatrických pacientov mladších ako 3 roky alebo s hmotnosťou menej
ako 15 kg (pozri časti 4.4 a 5.3).
Pediatrickí pacienti doteraz neliečení ART (mladší ako 3 roky alebo s hmotnosťou menej ako 15 kg)
V tejto populácii nemožno stanoviť odporúčania pre dávkovanie.
Pediatrickí pacienti predtým liečení ART (vo veku 3 až 17 rokov a s hmotnosťou najmenej 40 kg) Boli stanovené expozície darunaviru u dospievajúcich vo veku 12 až 17 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení a ktorí dostávali darunavir/ritonavir 800 mg/100 mg jedenkrát denne a ukázalo sa, že sa nachádzali v rámci terapeutického rozsahu, ktorý bol stanovený u dospelých pacientov dostávajúcich darunavir/ritonavir 800 mg/100 mg jedenkrát denne. Ako dôsledok vzhľadom na to, že darunavir/ritonavir 800 mg/100 mg jedenkrát denne bola tiež zaregistrovaná na použitie u dospelých s predchádzajúcou liečbou bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1 RNA
< 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l, rovnaká indikácia darunaviru 800 mg
jedenkrát denne sa vzťahuje na deti vo veku 3 až 17 rokov a s hmotnosťou najmenej 40 kg
s predchádzajúcou liečbou. Dávka darunaviru s kobicistatom nebola v tejto skupine pacientov stanovená.
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Pre odporučené dávkovanie u detí, pozrite Súhrn charakteristických vlastností lieku darunavir 75 mg,
150 mg, 300 mg, 600 mg tablety.
Darunavir sa nemá používať u detí s hmotnosťou menej ako 15 kg, pretože dávka pre túto populáciu nebola stanovená na dostatočnom počte pacientov. Darunavir sa nemá používať u detí mladších ako 3 roky z dôvodu bezpečnosti.
Gravidita a obdobie po pôrode
Počas gravidity a po pôrode nie je potrebná úprava dávky darunaviru/ritonaviru. Darunavir sa má
užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko (pozri časti 4.4, 4.6 a 5.2).
Spôsob podávania
Pacientov treba poučiť, aby užívali Darunavir Mylan s kobicistatom alebo s nízkou dávkou ritonaviru
do 30 minút po jedle. Druh jedla neovplyvňuje expozíciu darunaviru (pozri časti 4.4, 4.5 a 5.2).
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Použitie u pacientov s ťažkou (Childova-Pughova Trieda C) poruchou funkcie pečene.
Súčasná liečba niektorým z nasledujúcich liekov je kontraindikovaná z dôvodu očakávaného poklesu plazmatických koncentrácií darunaviru, ritonaviru a kobicistatu a potenciálnej straty terapeutického účinku (pozri časti 4.4 a 4.5).
Relevantné pre darunavir posilnený ritonavirom alebo kobicistatom:
- Liek s kombináciou lopinavir/ritonavir (pozri časť 4.5).
- Silné induktory CYP3A, rifampicín a rastlinné preparáty obsahujúce výťažok z ľubovníka
bodkovaného (Hypericum perforatum). Očakáva sa, že súčasné podávanie zníži plazmatické
koncentrácie darunaviru, ritonaviru a kobicistatu, čo môže viesť k strate terapeutického účinku
a možnému rozvoju rezistencie (pozri časti 4.4 a 4.5).
Relevantné pre darunavir posilnený kobicistatom, nie prípady, kedy je posilnený ritonavirom:
- Darunavir posilnený kobicistatom je citlivejší na indukciu CYP3A ako darunavir posilnený ritonavirom. Súčasné užívanie so silnými induktormi CYP3A je kontraindikované, pretože tieto
môžu znížiť expozíciu kobicistatu a darunaviru a spôsobiť stratu terapeutického účinku. Silné
induktory CYP3A zahŕňajú napr. karbamazepín, fenobarbital a fenytoín (pozri časti 4.4 a 4.5).
Darunavir posilnený ritonavirom alebo kobicistatom inhibuje elimináciu liečiv, ktorých metabolizmus (klírens) je vysoko závislý na CYP3A, čo vedie k zvýšenej expozícii súčasne užívanému lieku. Z toho dôvodu je súčasná liečba takými liekmi, ktorých zvýšené plazmatické koncentrácie sú spojené
s vážnymi a/alebo život ohrozujúcimi udalosťami, kontraindikovaná (relevantné pre darunavir posilnený ritonavirom alebo kobicistatom). Medzi tieto liečivá patria napr.:
- alfuzosín (antagonista alfa1-adrenoreceptora)
- amiodarón, bepridil, dronedarón, chinidín, ranolazín, lidokaín (podávaný systémovo) (antiarytmiká/lieky proti angíne)
- astemizol, terfenadín (antihistaminiká)
- kolchicín, keď sa používa u pacientov s poruchou funkcie obličiek a/alebo pečene (liek proti dne) (pozri časť 4.5)
- námeľové alkaloidy (napr. dihydroergotamín, ergometrín, ergotamín, metylergonovín)
- cisaprid (prokinetiká)
- pimozid, kvetiapín, sertindol (antipsychotiká/neuroleptiká)(pozri časť 4.5)
- triazolam, midazolam podávaný perorálne (sedatíva/hypnotiká) (pre upozornenie na midazolam
podávaný parenterálne, pozri časť 4.5)
- sildenafil - keď sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, avanafil (inhibítory
PDE-5)
- simvastatín a lovastatín (inhibítory HMG-CoA reduktázy) (pozri časť 4.5)
- tikagrelor (inhibítor agregácie trombocytov) (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaní
Hoci sa preukázalo, že účinná vírusová supresia dosiahnutá pri antiretrovírusovej terapii značne znižuje riziko prenosu HIV pohlavným stykom, reziduálne riziko nie je možné vylúčiť. Je potrebné prijať opatrenia na zabránenie prenosu HIV v súlade s národnými odporúčaniami.
Odporúča sa pravidelné sledovanie virologickej odpovede. V prípade nedostatočnej alebo žiadnej virologickej odpovede sa má urobiť test na rezistenciu.
Darunavir sa musí vždy podávať perorálne s kobicistatom alebo s nízkou dávkou ritonaviru na zlepšenie farmakokinetiky a v kombinácii s inými antiretrovirotikami (pozri časť 5.2). Pred začatím liečby darunavirom je preto nevyhnutné prečítať si Súhrn charakteristických vlastností kobicistatu alebo ritonaviru.
Zvyšovanie dávkovania ritonaviru oproti odporúčaniam v časti 4.2 významnejšie neovplyvňuje
koncentrácie darunaviru. Neodporúča sa meniť dávku kobicistatu alebo ritonaviru.
Darunavir sa viaže prevažne na a1-kyslý glykoproteín. Táto proteínová väzba závisí od koncentrácie, ktorú určuje saturácia väzby. Preto lieky, ktoré sa vysoko viažu na a1-kyslý glykoproteín, môžu znemožniť túto väzbu (pozri časť 4.5).
Pacienti liečení ART – dávkovanie jedenkrát denne
Darunavir užívaný v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru jedenkrát denne
u pacientov liečených ART sa nemá používať u pacientov s jednou alebo viacerými
mutáciami spojenými s rezistenciou voči darunaviru (DRV-RAM) alebo s hladinou HIV-1 RNA
≥ 100 000 kópií/ml alebo počtom buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2). V tejto populácii sa neskúmali iné kombinácie s optimalizovaným základným režimom (OBR, optimised background
regimen) ako ≥ 2 NRTI. U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje
(pozri časť 5.1).
Pediatrická populácia
Darunavir sa neodporúča používať u pediatrických pacientov mladších ako 3 roky alebo s hmotnosťou
menej ako 15 kg (pozri časti 4.2 a 5.3).
Gravidita
Darunavir sa má užívať počas gravidity, len ak potenciálny prínos prevýši potenciálne riziko.
Opatrnosť je potrebná u gravidných žien súbežne užívajúcich lieky, ktoré môžu ďalej znižovať
expozíciu darunaviru (pozri časti 4.5 a 5.2).
Starší ľudia
Keďže k dispozícii sú len obmedzené údaje o používaní darunaviru u pacientov vo veku 65 rokov a
starších, pri podávaní darunaviru starším pacientom je potrebná opatrnosť vzhľadom k vyššiemu výskytu zhoršenej funkcie pečene a pridružených chorôb, alebo k inej liečbe (pozri časti 4.2 a 5.2).
Závažnékožnéreakcie
Počas programu klinického vývoja darunaviru/ritonaviru (N=3 063) boli u 0,4 % pacientov hlásené
závažné kožné reakcie, ktoré mohla sprevádzať horúčka a/alebo zvýšenie transamináz.
Zriedkavo(< 0,1 %) bola hlásená lieková vyrážka s eozinofíliou a systémovými príznakmi (DRESS, drug rash with eosinophilia and systemic symptoms) a Stevensov-Johnsonov syndróm a počas
postmarketingových skúseností bola hlásená toxická epidermálna nekrolýza a akútna generalizovaná
exantematózna pustulóza. Ak sa objavia prejavy alebo príznaky závažnej kožnej reakcie, liečba darunavirom sa má okamžite prerušiť. Príznaky môžu zahŕňať, ale nemusia byť obmedzené na, závažnú vyrážku alebo vyrážku sprevádzanú horúčkou, celkovú nevoľnosť, únavu, bolesť svalov alebo kĺbov, pľuzgiere, orálne lézie, konjunktivitídu, hepatitídu a/alebo eozinofíliu.
Vyrážka sa častejšie vyskytla u predtým liečených pacientov, ktorí boli liečení darunavirom/ritonavirom spolu s raltegravirom v porovnaní s pacientmi, ktorí užívali darunavir/ritonavir bez raltegraviru alebo raltegravir bez darunaviru (pozri časť 4.8).
Darunavir obsahuje sulfónamidovú funkčnú skupinu. Darunavir sa má podávať opatrne pacientom so
známou alergiou na sulfónamid.
H
epatotoxicita
Počas užívania darunaviru bola hlásená liekom indukovaná hepatitída (napr. akútna hepatitída, cytolytická hepatitída). Počas programu klinického vývoja darunaviru/ritonaviru (N=3 063) bola hepatitída hlásená u 0,5 % pacientov, ktorí boli liečení kombinovanou antiretrovirálnou terapiou s darunavirom/ritonavirom. Pacienti s preexistujúcou dysfunkciou pečene, vrátane chronickej aktívnej hepatitídy B alebo C, majú zvýšené riziko porúch funkcie pečene, vrátane vážnych a potenciálne smrteľných hepatických nežiaducich reakcií. V prípade súčasnej antivírovej liečby hepatitídy B alebo C, obráťte sa na relevantnú informáciu o týchto liekoch.
Pred začatím liečby darunavirom užívaným v kombinácii s kobicistatom alebo s nízkou dávkou ritonaviru sa majú urobiť vhodné laboratórne vyšetrenia a pacienti majú byť sledovaní aj počas liečby. U pacientov s chronickou hepatitídou, cirhózou alebo u pacientov, ktorí mali pred liečbou zvýšenú hladinu transamináz, treba zvážiť zvýšené sledovanie AST/ALT, najmä počas prvých mesiacov po začatí liečby darunavirom užívaným v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru.
Ak sa vyskytne alebo sa zhorší dysfunkcia pečene (vrátane klinicky významného zvýšenia pečeňových enzýmov a/alebo príznakov ako únava, anorexia, nauzea, žltačka, tmavý moč, citlivosť pečene, hepatomegália) u pacientov užívajúcich darunavir v kombinácii s kobicistatom alebo nízkou dávkou ritonaviru, treba okamžite zvážiť zastavenie alebo prerušenie liečby.
Pacienti s pridruženými ochoreniami
Porucha funkcie pečene
U pacientov so závažnými poruchami pečene sa účinnosť a bezpečnosť darunaviru nestanovovala, preto je darunavir u pacientov s ťažkou poruchou funkcie pečene kontraindikovaný. Vzhľadom na nárast koncentrácie voľného darunaviru v plazme sa darunavir musí používať opatrne u pacientov s miernou alebo stredne ťažkou poruchou funkcie pečene (pozri časti 4.2, 4.3 a 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa nevyžadujú žiadne osobitné opatrenia alebo úpravy dávkovania darunaviru/ritonaviru. Darunavir aj ritonavir sa výrazne viažu na plazmatické bielkoviny,
a preto je nepravdepodobné, že by sa dali z cirkulácie významnejšie odstrániť pomocou hemodialýzy
alebo peritoneálnej dialýzy. Preto sa u týchto pacientov nevyžadujú žiadne osobitné opatrenia alebo úpravy dávkovania (pozri časti 4.2 a 5.2). Kobicistat sa neskúmal u pacientov na dialýze, a preto nie je možné stanoviť žiadne odporúčanie ohľadom užívania darunaviru/kobicistatu u týchto pacientov
(pozri časť 4.2).
Kobicistat znižuje odhadovaný klírens kreatinínu z dôvodu inhibície tubulárnej sekrécie kreatinínu. Treba to vziať do úvahy, ak sa darunavir s kobicistatom podáva pacientom, u ktorých sa odhadovaný klírens kreatinínu používa na úpravu dávok súčasne užívaných liekov (pozri časť 4.2 a SPC kobicistatu).
V súčasnosti nie sú k dispozícii údaje postačujúce na určenie, či súčasné užívanie tenofovir disoproxil fumarátu a kobicistatu súvisí s vyšším rizikom renálnych nežiaducich reakcií v porovnaní s režimami, ktoré obsahujú tenofovir disoproxil fumarát bez kobicistatu.
Pacienti s hemofíliou
U pacientov s hemofíliou A a B liečených PI sa opísali prípady väčšieho krvácania, vrátane spontánnych kožných hematómov a krvácania do kĺbov. Niektorým pacientom sa podal dodatočný faktor VIII. Vo viac ako polovici hlásených prípadov sa pokračovalo v liečbe PI, prípadne sa táto liečba obnovila po jej dočasnom prerušení. Bola naznačená kauzálna súvislosť, hoci mechanizmus účinku nebol zatiaľ objasnený. Z toho dôvodu je potrebné hemofilikov upozorniť na možnosť väčšieho krvácania.
Telesná hmotnosť a metabolické parametre
Počas antiretrovírusovej liečby môže dôjsť k zvýšeniu telesnej hmotnosti a hladín lipidov a glukózy
v krvi. Takéto zmeny môžu čiastočne súvisieť s kontrolou ochorenia a životným štýlom. Pokiaľ ide
o lipidy, v niektorých prípadoch sú dôkazy o vplyve liečby, kým pri prírastku telesnej hmotnosti nie sú
presvedčivé dôkazy o tom, že súvisí s niektorou konkrétnou liečbou. Pri monitorovaní hladín lipidov a glukózy v krvi sa treba riadiť zavedenými odporúčaniami na liečbu infekcie HIV. Poruchy metabolizmu lipidov majú byť klinicky vhodne liečené.
Osteonekróza
Napriek tomu, že etiológia ochorenia závisí od mnohých faktorov (vrátane užívania kortikosteroidov, konzumácie alkoholu, vážnej imunosupresie, vyššieho BMI), prípady osteonekrózy boli hlásené najmä
u pacientov s pokročilým ochorením HIV a/alebo s dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART, combination antiretroviral therapy). Pacientov treba poučiť, aby
vyhľadali lekársku pomoc v prípade, že budú pociťovať bolesti kĺbov, stuhnutosť kĺbov alebo ťažkosti
pri pohybe.
Imunoreštitučnýzápalovýsyndróm
U pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej
antiretrovírusovej liečby môže vzniknúť zápalová reakcia na asymptomatické patogény alebo na reziduálne oportúnne patogény, čo môže byť spojené so závažným klinickým stavom resp. so zhoršením symptómov. Tieto reakcie sa obyčajne pozorujú v priebehu prvých týždňov až mesiacov od začatia podávania kombinovanej antiretrovírusovej liečby. Príkladom môže byť cytomegalovírusová retinitída, generalizované alebo fokálne mykobaktériové infekcie a pneumónia vyvolaná Pneumocystis jirovecii (predtým známa ako Pneumocystis carinii). Je nutné zhodnotiť všetky príznaky zápalového ochorenia a v prípade potreby začať liečbu. Ďalej sa v klinických štúdiách s darunavirom užívaným s nízkou dávkou ritonaviru pozorovala reaktivácia herpesu simplex a herpesu zoster.
Boli tiež zaznamenané aj poruchy autoimunitného systému (ako je Gravesova choroba) objavujúce sa v dôsledku imunitnej reaktivácie; avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.8).
Liekové interakcie
Liek na zlepšenie farmakokinetiky a súčasne užívané lieky
Darunavir má rôzne interakčné profily v závislosti od toho, či je zlúčenina posilnená ritonavirom alebo kobicistatom:
- Darunavir posilnený kobicistatom je citlivejší na indukciu CYP3A: súčasné užívanie darunaviru/kobicistatu a silných induktorov CYP3A je preto kontraindikované (pozri časť 4.3) a súčasné užívanie so slabými a stredne silnými induktormi CYP3A sa neodporúča (pozri časť
4.5). Súčasné užívanie darunaviru/ritonaviru a darunaviru/kobicistatu
s lopinavirom/ritonavirom, rifampicínom a rastlinnými prípravkami obsahujúcimi ľubovník
bodkovaný, Hypericum perforatum, je kontraindikované (pozri časť 4.5).
- Na rozdiel od ritonaviru, kobicistat nemá indukujúci vplyv na enzýmy alebo transportné
proteíny (pozri časť 4.5). Pri zmene lieku zlepšujúceho farmakokinetiku z ritonaviru na kobicistat sa počas prvých dvoch týždňov liečby darunavirom/kobicistatom vyžaduje opatrnosť,
obzvlášť ak boli počas užívania ritonaviru za účelom zlepšenia farmakokinetiky titrované alebo
upravené dávky akýchkoľvek súčasne podávaných liekov. V týchto prípadoch môže byť potrebné zníženie dávky súčasne podávaného lieku.
Efavirenz v kombinácii s darunavirom/ritonavirom 800/100 mg jedenkrát denne môže viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť v kombinácii s darunavirom/ritonavirom, treba použiť dávkovací režim darunavir/ritonavir 600/100 mg dvakrát
denne. Pozri Súhrn charakteristických vlastností lieku darunavir 75 mg, 150 mg, 300 mg alebo 600 mg
tablety (pozri časť 4.5).
Život ohrozujúce a smrteľné liekové interakcie sa zaznamenali u pacientov liečených kolchicínom
a silnými inhibítormi CYP3A a P-glykoproteínu (P-gp; pozri časti 4.3 a 4.5).
4.5 Liekové a iné interakcie
Interakčný profil darunaviru sa môže líšiť v závislosti od toho, či sa na zlepšenie farmakokinetiky použije ritonavir alebo kobicistat. Odporúčania dané pre súčasné užívanie darunaviru a iných liekov sa môžu preto líšiť v závislosti od toho, či je darunavir posilnený ritonavirom alebo kobicistatom (pozri časti 4.3 a 4.4). Vyžaduje sa tiež opatrnosť na začiatku liečby, ak došlo ku zmene lieku na zlepšenie farmakokinetiky z ritonaviru na kobicistat (pozri časť 4.4).
Lieky,ktoréovplyvňujúexpozíciudarunaviru(ritonavirakolieknazlepšeniefarmakokinetiky)Darunavir a ritonavir sa metabolizujú prostredníctvom CYP3A. Očakáva sa, že lieky indukujúce činnosť CYP3A zvýšia klírens darunaviru a ritonaviru, čo má za následok zníženie plazmatických koncentrácií týchto zlúčenín a následne darunaviru a vedie k strate terapeutického účinku a možnému rozvoju rezistencie (pozri časti 4.3 a 4.4). Induktory CYP3A, ktoré sú kontraindikované, zahŕňajú napr. rifampicín, ľubovník bodkovaný a lopinavir.
Súčasné užívanie darunaviru a ritonaviru s inými liekmi, ktoré inhibujú CYP3A, môže znížiť klírens
darunaviru a ritonaviru a môže mať za následok zvýšenie plazmatických koncentrácií darunaviru
a ritonaviru. Súčasné užívanie so silnými inhibítormi CYP3A4 sa neodporúča a vyžaduje opatrnosť. Tieto interakcie sú opísané v nižšie uvedenej tabuľke interakcií (napr. indinavir, systémové azoly ako
ketokonazol a klotrimazol).
Lieky,ktoréovplyvňujúexpozíciudarunaviru(kobicistatakolieknazlepšeniefarmakokinetiky)
Darunavir a kobicistat sa metabolizujú prostredníctvom CYP3A, a preto môže súčasné užívanie
s induktormi CYP3A viesť k subterapeutickým plazmatickým expozíciám darunaviru. Darunavir posilnený kobicistatom je citlivejší na indukciu CYP3A ako darunavir posilnený ritonavirom: súčasné užívanie darunaviru/kobicistatu s liekmi, ktoré sú silnými induktormi CYP3A (napr. ľubovník bodkovaný, rifampicín, karbamazepín, fenobarbital a fenytoín) je kontraindikované (pozri časť 4.3). Súčasné užívanie darunaviru/kobicistatu so slabými až stredne silnými induktormi CYP3A (napr. efavirenz, etravirín, nevirapín, boceprevir, telaprevir, flutikazón a bosentan) sa neodporúča (pozri nižšie uvedenú tabuľku interakcií).
Na súčasné užívanie so silnými inhibítormi CYP3A4 sa vzťahujú rovnaké odporúčania, nezávisle od toho, či je darunavir posilnený ritonavirom alebo kobicistatom (pozri časť vyššie).
Lieky,ktorémôžubyťovplyvnenédarunaviromposilnenýmritonaviromDarunavir a ritonavir sú inhibítormi CYP3A, CYP2D6 a P-gp. Súčasné podávanie darunaviru/ritonaviru s liekmi metabolizovanými najmä CYP3A a/alebo CYP2D6 alebo transportovanými P-gp môže viesť k zvýšeniu systémovej expozície týmto liekom, čo by mohlo zosilniť alebo predĺžiť ich terapeutický účinok a viesť k nežiaducim účinkom.
Darunavir užívaný s nízkou dávkou ritonaviru sa nesmie kombinovať s liekmi, ktorých metabolizmus výrazne závisí od CYP3A, a ktorých zvýšená systémová expozícia je sprevádzaná závažnými a/alebo život ohrozujúcimi nežiaducimi účinkami (t.j. ktoré majú úzky terapeutický index) (pozri časť 4.3). Celkové zvýšenie farmakokinetického účinku ritonavirom viedlo približne k 14-násobnému zvýšeniu systémovej expozície darunaviru, po jednorazovej 600 mg perorálnej dávke darunaviru v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne. Kobicistat 150 mg podávaný s 800 mg
darunaviru jedenkrát denne zlepšuje farmakokinetické parametre darunaviru porovnateľným
spôsobom ako ritonavir (pozri časť 5.2). Z toho dôvodu sa darunavir môže používať len v kombinácii s liekom, ktorý zlepšuje farmakokinetiku darunaviru (pozri časť 5.2).
Klinická štúdia s viacerými liekmi, ktoré sú metabolizované cytochrómami CYP2C9, CYP2C19 a CYP2D6 dokázala nárast aktivity CYP2C9 a CYP2C19 a inhibíciu aktivity CYP2D6 v prítomnosti darunaviru/ritonaviru, čo sa môže pripisovať prítomnosti nízkej dávky ritonaviru. Súčasné podávanie darunaviru a ritonaviru s liekmi, ktoré sa predovšetkým metabolizujú prostredníctvom CYP2D6 (napríklad flekainid, propafenon, metoprolol), môže vyústiť do zvýšených plazmatických koncentrácií týchto liekov, čo môže zvýšiť alebo predĺžiť ich terapeutický účinok a nežiaduce reakcie. Súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C9
(napríklad warfarín) a CYP2C19 (napríklad metadon), môže vyústiť do zníženia systémových
expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Napriek tomu, že sa účinok na CYP2C8 sledoval iba in vitro, súčasné podávanie darunaviru a ritonaviru a liekov predovšetkým metabolizovaných prostredníctvom CYP2C8 (napríklad paklitaxel, rosiglitazón, repaglinid), môže vyústiť do zníženia systémových expozícií týmto liekom, čo môže znížiť alebo skrátiť ich terapeutický účinok.
Ritonavir inhibuje trasportérov P-glykoproteín, OATP1B1 a OATP1B3 a súčasné užívanie so substrátmi týchto transportérov môže viesť k zvýšeniu plazmatických koncentrácií týchto zlúčenín (napr. dabigatran etexilát, digoxín, statíny a bosentan; pozri nižšie uvedenú tabuľku interakcií).
Lieky,ktorémôžubyťovplyvnenédarunaviromposilnenýmkobicistatom
Odporúčania pre darunavir posilnený ritonavirom s ohľadom na substráty CYP3A4, CYP2D6,
P-glykoproteín, OATP1B1 a OATP1B3 sú adekvátne aj pre darunavir posilnený kobicistatom (pozri kontraindikácie a odporúčania uvedené v časti vyššie). Kobicistat 150 mg podávaný s 800 mg
darunaviru jedenkrát denne zlepšuje farmakokinetické parametre darunaviru porovnateľným
spôsobom ako ritonavir (pozri časť 5.2).
Na rozdiel od ritonaviru, kobicistat neindukuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19 ani
UGT1A1. Ďalšie informácie o kobicistate si pozrite v Súhrne charakteristických vlastností kobicistatu.
Tabuľkainterakcií
Interakčné štúdie sa uskutočnili len u dospelých.
Niekoľko interakčných štúdií (v tabuľke nižšie sú označené #) sa uskutočnilo s nižšími ako sú odporúčané dávky darunaviru alebo s inými režimami dávkovania (pozri časť 4.2 Dávkovanie). Účinky na súčasne užívané lieky môžu byť preto podhodnotené a môže byť indikované klinické sledovanie bezpečnosti.
Interakčný profil darunaviru závisí od toho, či sa na zlepšenie farmakokinetiky použije ritonavir alebo kobicistat. Darunavir môže mať preto rôzne odporúčania pre súčasne podávané lieky v závislosti od toho, či je zlúčenina posilnená ritonavirom alebo kobicistatom. Interakčné štúdie uvedené nižšie sa neuskutočnili s darunavirom posilneným kobicistatom. Odporúčania sú rovnaké, ak sa neuvádza inak. Ďalšie informácie o kobicistate si pozrite v Súhrne charakteristických vlastností kobicistatu.
Interakcie medzi darunavirom/ritonavirom a antiretrovirotikami a ne-antiretrovírusovými liekmi sú uvedené v tabuľkách nižšie (neurčené ako „ND“ (z angl. not determinated)). Smer šípky pri každom farmakokinetickom parametri je založený na 90 % intervale spoľahlivosti stredného geometrického pomeru v rámci 80-125 % rozpätia (↔), pod ním (↓) alebo nad ním (↑).
V nižšie uvedenej tabuľke sa uvádza konkrétny liek na zlepšenie farmakokinetiky, keď sa odporúčania líšia. V prípade, že sú odporúčania rovnaké pre darunavir súčasne užívaný s nízkou dávkou ritonaviru alebo s kobicistatom, používa sa označenie „posilnený darunavir“.
INTERAKCIE A ODPORUČENÉ DÁVKOVANIE S INÝMI LIEKMI
L
ieky podľa
t
erapeutických oblastí
I
nterakcia
Z
m
e
na geometrického priemeru (%)
O
dporúčania pri súčasnom
podávaní
 ANTIRETROVIROTIKÁ NA HIV
I
nhibítory prenosu reťazcov integrázou
ANTIRETROVIROTIKÁ NA HIV
I
nhibítory prenosu reťazcov integrázou
Dolutegravir dolutegravir AUC ↓ 32 % dolutegravir C24h 38 % dolutegravir Cmax ↓ 11 % darunavir ↔*
* Použitím porovnania údajov zo skríženej štúdie
s historickými údajmi o farmakokinetike
Posilnený darunavir a dolutegravir
sa môžu užívať bez úpravy dávky.
Elvitegravir elvitegravir AUC ↔ elvitegravir Cmin ↔ elvitegravir Cmax ↔ darunavir AUC ↔ darunavir Cmin 17% darunavir Cmax ↔
Raltegravir Niektoré klinické štúdie naznačujú, že raltegravir môže spôsobiť mierny pokles koncentrácie darunaviru v plazme.
Keď sa darunavir užívaný súčasne s nízkou dávkou ritonaviru (600/100 mg dvakrát denne) užíva spolu s elvitegravirom, dávka elvitegraviru má byť 150 mg jedenkrát denne.
Darunavir súčasne užívaný
s kobicistatom sa nemá užívať spolu s inými antiretrovirotikami, ktoré si vyžadujú farmakokinetickú
podporu, pretože neboli stanovené odporúčania na dávkovanie takej kombinácie.
Farmakokinetika a odporúčania na dávkovanie pre iné dávky darunaviru alebo
s elvitegravirom/kobicistatom neboli stanovené. Súčasné užívanie darunaviru s nízkou dávkou ritonaviru v iných dávkach ako
600/100 mg dvakrát denne
a elvitegraviru sa preto neodporúča. Súčasné užívanie darunaviru
s nízkou dávkou ritonaviru
a elvitegravirom za prítomnosti
kobicistatu sa neodporúča.
V súčasnosti sa účinok raltegraviru na plazmatické koncentrácie darunaviru nejaví ako klinicky významný. Posilnený darunavir
a raltegravir sa môže užívať bez
úpravy dávky.
Nukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, nucleo(s/t)ide reverse transcriptase inhibitors)
Didanozín
400 mg jedenkrát denne
Tenofovir disoproxil fumarát
300 mg jedenkrát denne
didanozín AUC ↓ 9 % didanozín Cmin ND didanozín Cmax ↓ 16 % darunavir AUC ↔ darunavir Cmin ↔ darunavir Cmax ↔ tenofovir AUC ↑ 22 % tenofovir Cmin ↑ 37 % tenofovir Cmax ↑ 24 %
#darunavir AUC ↑ 21 %
#darunavir Cmin ↑ 24 %
#darunavir Cmax ↑ 16 %
(↑ tenofoviru v dôsledku efektu na MDR-1
transport v renálnych tubuloch)
Posilnený darunavir a didanozín sa môže užívať bez úpravy dávok. Didanozín sa má podávať nalačno, preto ho treba užiť 1 hodinu pred alebo 2 hodiny po užití posilneného darunaviru s jedlom.
Pri súčasnom podávaní posilneného darunaviru v kombinácii
s tenofovirom môže byť indikované sledovanie renálnych funkcií, najmä u pacientov so systémovým alebo renálnym ochorením, alebo
u pacientov užívajúcich
nefrotoxické lieky.
Darunavir súčasne užívaný
s kobicistatom znižuje klírens kreatinínu. Ak sa klírens kreatinínu používa na úpravu dávky tenofoviru, pozri časť 4.4.
Abakavir Emtricitabín Lamivudín Stavudín Zidovudín
Neskúmalo sa. Vzhľadom na rôzne cesty eliminácie ostatných NRTI zidovudín, emtricitabín, stavudín, lamivudín, ktoré sa vylučujú prevažne renálnou cestou
a abakavir, ktorý nie je metabolizovaný prostredníctvom CYP450, sa pri ich kombinácii s posilneným darunavirom nepredpokladajú žiadne liekové interakcie.
Posilnený darunavir sa môže užívať s týmito NRTI bez úpravy dávky.
Darunavir súčasne užívaná
s kobicistatom znižuje klírens kreatinínu. Ak sa klírens kreatinínu používa na úpravu dávky emtricitabínu alebo lamivudínu, pozri časť 4.4.
Nenukleo(z/t)idové inhibítory reverznej transkriptázy (NRTI, non-nucleo(s/t)ide reverse transcriptase inhibitors)

Efavirenz
600 mg jedenkrát denne
Etravirín
100 mg dvakrát denne
Nevirapín
200 mg dvakrát denne
Rilpivirín
150 mg jedenkrát denne
efavirenz AUC ↑ 21 % efavirenz Cmin ↑ 17 % efavirenz Cmax ↑ 15 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↓ 31 %
#darunavir Cmax ↓ 15 %
(↑ efavirenzu v dôsledku inhibície
CYP3A)
(↓ darunaviru v dôsledku inhibície
CYP3A)
etravirín AUC ↓ 37 % etravirín Cmin ↓ 49 % etravirín Cmax ↓ 32 % darunavir AUC ↑ 15 % darunavir Cmin ↔ darunavir Cmax ↔
nevirapín AUC ↑ 27 % nevirapín Cmin ↑ 47 % nevirapín Cmax ↑ 18 %
#darunavir: koncentrácie
sa zhodovali s údajmi z minulosti. (↑ nevirapínu v dôsledku inhibície CYP3A)
rilpivirín AUC ↑ 130 % rilpivirín Cmin ↑ 178 % rilpivirín Cmax ↑ 79 % darunavir AUC ↔ darunavir Cmin ↓ 11 % darunavir Cmax ↔
Pri súčasnom podávaní darunaviru a nízkej dávky ritonaviru
v kombinácii s efavirenzom môže byť indikované klinické sledovanie toxicity centrálneho nervového systému spojené so zvýšenou expozíciou voči efavirenzu.
Efavirenz v kombinácii
s darunavirom/ritonavirom
800/100 mg jedenkrát denne môže viesť k zníženiu Cmin darunaviru pod optimálnu hodnotu. Ak sa efavirenz plánuje použiť
v kombinácii
s darunavirom/ritonavirom, treba použiť režim dávkovania darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.4).
Súčasné úžívanie s darunavirom užívanou súčasne s kobicistatom sa neodporúča (pozri časť 4.4). Darunavir súčasne podávaný
s nízkou dávkou ritonaviru a
s etravirínom
200 mg dvakrátdennesa môže užívať bez úpravy dávky.
Súčasné úžívanie s darunavirom užívaným súčasne s kobicistatom sa neodporúča (pozri časť 4.4). Darunavir súčasne podávaná
s nízkou dávkou ritonaviru a
s nevirapínom sa môže užívať bez
úpravy dávky.
Súčasné úžívanie s darunavirom užívanou súčasne s kobicistatom sa neodporúča (pozri časť 4.4). Posilnená darunavir a rilpivirín sa môže užívať bez úpravy dávky.
H
I
V Proteázové inhibítory (PI) - bez doplnkového súčasného podávania nízkej dávky ritonaviru
†
Atazanavir
300 mg jedenkrát denne
Indinavir
800 mg dvakrát denne
Sakvinavir
1 000 mg dvakrát denne
atazanavir AUC ↔ atazanavir Cmin ↑ 52 % atazanavir Cmax ↓ 11 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Atazanavir: porovnanie atazanaviru/ritonaviru 300/100 mg jedenkrát denne vs. atazanavir 300 mg jedenkrát denne v kombinácii
s darunavirom/ritonavirom 400/100 mg dvakrát denne.
Darunavir: porovnanie
darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg dvakrát denne v kombinácii s atazanavirom 300 mg jedenkrát denne. indinavir AUC ↑ 23 %
indinavir Cmin ↑ 125 %
indinavir Cmax ↔
#darunavir AUC ↑ 24 %
#darunavir Cmin ↑ 44 %
#darunavir Cmax ↑ 11 %
Indinavir: porovnanie indinaviru/ritonaviru
800/100 mg dvakrát denne vs. indinavir/darunavir/ritonavir
800/400/100 mg dvakrát denne.
Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii s indinavirom 800 mg dvakrát denne.
#darunavir AUC ↓ 26 %
#darunavir Cmin ↓ 42 %
#darunavir Cmax ↓ 17 %
#sakvinavir AUC ↓ 6 %
#sakvinavir Cmin ↓ 18 %
#sakvinavir Cmax ↓ 6 %
Sakvinavir: porovnanie sakvinaviru/ritonaviru 1000/100 mg dvakrát denne vs. sakvinavir/darunavir/ritonavir
1000/400/100 mg dvakrát denne Darunavir: porovnanie darunaviru/ritonaviru 400/100 mg dvakrát denne vs. darunavir/ritonavir 400/100 mg v kombinácii so sakvinavirom 1000 mg dvakrát denne.
Darunavir súčasne podávaný s nízkou dávkou ritonaviru a
s atazanavirom sa môže užívať bez
úpravy dávky.
Darunavir užívaný súčasne
s kobicistatom sa nemá používať v kombinácii s inou antiretrovírusovou látkou, ktorá si vyžaduje zlepšenie farmakokinetiky pomocou súčasného užívania inhibítora CYP3A4 (pozri časť 4.5).
Pri intolerancii súčasného podávania darunaviru užívaného s nízkou dávkou ritonaviru
s indinavirom môže byť indikované
zníženie dávkovania indinaviru
z 800 mg dvakrát denne na 600 mg dvakrát denne.
Darunavir užívaný súčasne
s kobicistatom sa nemá používať v kombinácii s inou antiretrovírusovou látkou, ktorá si vyžaduje zlepšenie farmakokinetiky pomocou súčasného užívania inhibítora CYP3A4 (pozri časť 4.5).
Neodporúča sa kombinovať darunavir užívaný s nízkou dávkou ritonaviru so sakvinavirom.
Darunavir užívaný súčasne
s kobicistatom sa nemá používať v kombinácii s inou antiretrovírusovou látkou, ktorá si vyžaduje zlepšenie farmakokinetiky pomocou súčasného užívania inhibítora CYP3A4 (pozri časť 4.5).
H
I
V Proteázové inhibítory (PI) - podávané s nízkou dávkou ritonaviru
†
Lopinavir/ritonavir
400/100 mg dvakrát denne
Lopinavir/ritonavir
533/133,3 mg dvakrát denne
CCR5-ANTAGONISTY
Maravirok
150 mg dvakrát denne
ANESTETIKÁ
lopinavir AUC ↑ 9 % lopinavir Cmin ↑ 23 % lopinavir Cmax ↓ 2 % darunavir AUC ↓ 38 %‡ darunavir Cmin ↓ 51 %‡ darunavir Cmax ↓ 21 %‡ lopinavir AUC ↔ lopinavir Cmin ↑ 13 % lopinavir Cmax ↑ 11 % darunavir AUC ↓ 41 % darunavir Cmin ↓ 55 % darunavir Cmax ↓ 21 %
‡ na základe neštandardizovaných veľkostí dávky
maravirok AUC ↑ 305 % maravirok Cmin ND maravirok Cmax ↑ 129 %
koncentrácie darunaviru, ritonaviru sa
zhodovali s údajmi z minulosti
Vzhľadom na pokles AUC darunaviru o 40 %, kombinácie vhodných dávok neboli stanovené. Preto je súčasné podávanie posilneného darunaviru a lieku
s kombináciou lopinavir/ritonavir
kontraindikované (pozri časť 4.3).
Keď sa maravirok podáva
s posilneným darunavirom, jeho dávka má byť 150 mg dvakrát denne.
Alfentanil Neskúmalo sa. Metabolizmus alfentanilu je sprostredkovaný CYP3A a v podstate môže byť inhibovaný posilneným darunavirom.
LIEKY PROTI ANGÍNE/ANTIARYTMIKÁ
Súčasné užívanie s posilneným darunavirom môže vyžadovať zníženie dávky alfentanilu
a sledovanie rizík predĺženého alebo
oneskoreného respiračného útlmu.
Disopyramid Flekainid Mexiletín Propafenón
Amiodarón Bepridil Dronedarón
Lidokaín (podávaný systémovo)
Chinidín
Ranolazín
Digoxín
0,4 mg jednorazová dávka
Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto antiarytmík.
(inhibícia CYP3A)
digoxín AUC ↑ 61 % digoxín Cmin ND digoxín Cmax ↑ 29 %
(↑ digoxínu v dôsledku možnej inhibície P-
gp)
Pri súčasnom užívaní týchto antiarytmík a posilneného darunaviru sa vyžaduje opatrnosť a odporúča sa sledovanie terapeutických koncentrácií, ak je dostupné.
Súčasné užívanie posilneného darunaviru a amiodarónu, bepridilu, dronedarónu, lidokaínu podávaného systémovo, chinidínu alebo ranolazínu je kontraindikované (pozri časť 4.3).
Vzhľadom na to, že digoxín má úzky terapeutický index, v prípade, že pacienti liečení posilneným darunavirom užívajú digoxín, odporúča sa na začiatok predpísať
čo najnižšiu možnú dávku digoxínu. Dávka digoxínu sa má opatrne titrovať, kým sa nedosiahne želaný klinický účinok pri sledovaní celkového klinického stavu
pacienta.
ANTIBIOTIKÁ
Klaritromycín
500 mg dvakrát denne
ANTIKOAGULANCIÁ
Apixaban Dabigatran etexilát Rivaroxaban
klaritromycín AUC ↑ 57 % klaritromycín Cmin ↑ 174 % klaritromycín Cmax ↑ 26 %
#darunavir AUC ↓ 13 %
#darunavir Cmin ↑ 1 %
#darunavir Cmax ↓ 17 %
Pri podávaní s darunavirom/ritonavirom neboli zistiteľné koncentrácie metabolitu
14-OH-klaritromycínu.
(↑ klaritromycínu v dôsledku inhibície
CYP3A a možnej inhibície P-gp)
Neskúmalo sa. Súčasné užívanie posilneného darunaviru a týchto antikoagulancií môže zvýšiť koncentrácie antikoagulantu.
(inhibícia CYP3A a/alebo P-gp)
Vyžaduje sa opatrnosť, keď sa klaritromycín podáva v kombinácii s posilneným darunavirom.
Odporúčanú dávku u pacientov s poruchou funkcie obličiek si pozrite v Súhrne charakteristických vlastností klaritromycínu.
Užívanie posilneného darunaviru a týchto antikoagulancií sa neodporúča.
Warfarín Neskúmalo sa. Pri súčasnom podávaní
warfarínu a posilneného darunaviru
(nízkou dávkou) môže dôjsť k ovplyvneniu koncentrácií warfarínu.
Pri súčasnom podávaní warfarínu s posilneným darunavirom sa odporúča monitorovať INR (International Normalized Ratio).
ANTIKONVULZÍVA Fenobarbital Fenytoín
Karbamazepín
200 mg dvakrát denne
Neskúmalo sa. Predpokladá sa, že fenobarbital a fenytoín znižujú plazmatické koncentrácie darunaviru
a lieku na zlepšenie jeho farmakokinetiky. (indukcia enzýmov CYP450)
karbamazepín AUC ↑ 45 % karbamazepín Cmin ↑ 54 % karbamazepín Cmax ↑ 43 % darunavir AUC ↔ darunavir Cmin ↓ 15 % darunavir Cmax ↔
Darunavir užívaný s nízkou dávkou
ritonaviru sa nesmie používať
v kombinácii s týmito liečivami.
Užívanie týchto liekov
s darunavirom/kobicistatom je kontraindikované (pozri časť 4.3). Neodporúča sa žiadna úprava dávky darunaviru/ritonaviru. V prípade potreby kombinovať darunavir/ritonavir a karbamazepín sa majú pacienti sledovať z dôvodu možných nežiaducich udalostí spojených s karbamazepínom. Je potrebné sledovať koncentrácie karbamazepínu a titrovať jeho
dávku až do dosiahnutia primeranej odpovede. Na základe zistení môže byť potrebné znížiť dávku
karbamazepínu o 25% až 50%, ak
sa podáva
s darunavirom/ritonavirom.
Užívanie karbamazepínu
s darunavirom súčasne užívaným
s kobicistatom je kontraindikované
(pozri časť 4.3).
ANTIDEPRESÍVA
Paroxetín
20 mg jedenkrát denne
Sertralín
50 mg jedenkrát denne
Amitriptylín Desipramín Imipramín Nortriptylín Trazodón
ANTIDIABETIKÁ
paroxetín AUC ↓ 39 % paroxetín Cmin ↓ 37 % paroxetín Cmax ↓ 36 %
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
sertralín AUC ↓ 49 %
sertralín Cmin ↓ 49 %
sertralín Cmax ↓ 44 %
#darunavir AUC ↔
#darunavir Cmin ↓ 6 %
#darunavir Cmax ↔
Na rozdiel od týchto údajov s darunavirom/ritonavirom, darunavir/kobicistat môže zvýšiť plazmatické koncentrácie týchto antidepresív (inhibícia CYP2D6 a/alebo CYP3A).
Súčasné užívanie posilneného darunaviru a týchto antidepresív môže zvýšiť koncentrácie antidepresíva.
(inhibícia CYP2D6 a/alebo CYP3A)
Ak sa antidepresíva užívajú súčasne s posilneným darunavirom, odporúčaný prístup je titrácia dávky antidepresíva na základe klinického posúdenia odpovede na antidepresívum. Navyše u pacientov na ustálenej dávke týchto antidepresív, ktorí začínajú liečbu posilneným darunavirom, sa má sledovať odpoveď na antidepresívum.
Keď sa tieto antidepresíva užívajú s posilneným darunavirom, odporúča sa klinické sledovanie
a môže byť potrebná úprava dávky
antidepresíva.
Metformín Neskúmalo sa. Na základe teoretických
úvah sa očakáva, že darunavir užívaná s kobicistatom zvýši plazmatické koncentrácie metformínu.
(inhibícia MATE1)
ANTIMYKOTIKÁ
Vorikonazol Neskúmalo sa. Ritonavir môže znížiť plazmatické koncentrácie vorikonazolu. (indukcia enzýmov CYP450)
Koncentrácia vorikonazolu sa môže zvýšiť alebo znížiť, keď sa užíva súčasne
s darunavirom užívaným spolu s kobicistatom.
(inhibícia enzýmov CYP450)
Odporúča sa starostlivé sledovanie
pacienta a úprava dávky
metformínu u pacientov užívajúcich darunavir spolu s kobicistatom. (netýka sa darunaviru užívaného spolu s ritonavirom)
Vorikonazol sa nesmie používať spolu s posilneným darunavirom, kým vyhodnotenie pomeru prínos/riziko neodôvodní použitie vorikonazolu.
Ketokonazol
200 mg dvakrát denne
Flukonazol
Posakonazol
ketokonazol AUC ↑ 212 % ketokonazol Cmin ↑ 868 % ketokonazol Cmax ↑ 111 %
#darunavir AUC ↑ 42 %
#darunavir Cmin ↑ 73 %
#darunavir Cmax ↑ 21 % (inhibícia CYP3A)
Neskúmalo sa. Posilnený darunavir môže zvýšiť plazmatické koncentrácie antimykotika (inhibícia P-gp) a posakonazol alebo flukonazol môže zvýšiť koncentrácie darunaviru.
(inhibícia CYP3A)
Vyžaduje sa opatrnosť a odporúča
sa klinické sledovanie, keď sa užíva v kombinácii s posilneným darunavirom. Ak je nutná súčasná liečba, denná dávka ketokonazolu nesmie presiahnuť 200 mg.
Vyžaduje sa opatrnosť a odporúča
sa klinické sledovanie.
Itrakonazol Neskúmalo sa. Súčasné systémové podanie itrakonazolu a posilneného darunaviru môže zvýšiť plazmatické koncentrácie darunaviru a itrakonazolu.
(inhibícia CYP3A)
Klotrimazol Neskúmalo sa. Súčasné systémové podanie klotrimazolu a posilneného darunaviru môže zvýšiť plazmatické koncentrácie darunaviru a/alebo klotrimazolu.
darunavir AUC24h ↑ 33 % (na základe farmakokinetického modelu populácie)
Vyžaduje sa opatrnosť a odporúča
sa klinické sledovanie, keď sa užíva v kombinácii s posilneným darunavirom. Ak je nutná súčasná
liečba, denná dávka itrakonazolu nesmie presiahnuť 200 mg.
Ak je nutná súčasná liečba klotrimazolom, vyžaduje sa opatrnosť a odporúča sa klinické sledovanie.
ANTIURATIKÁ
Kolchicín Neskúmalo sa. Súčasné užívanie kolchicínu a posilneného darunaviru môže zvýšiť expozíciu kolchicínu.
(inhibícia CYP3A a/alebo
P-glykoproteínu)
ANTIMALARIKÁ
Ak je potrebná liečba posilneným darunavirom, odporúča sa
u pacientov s normálnou funkciou obličiek alebo pečene znížiť dávku kolchicínu alebo liečbu kolchicínom prerušiť.
U pacientov s poruchou funkcie obličiek alebo pečene je kolchicín s posilneným darunavirom kontraindikovaný (pozri časť 4.3).

Artemeter/
lumefantrín
80/480 mg, 6 dávok v 0., 8., 24., 36., 48. a 60. hodine
ANTITUBERKULOTIKÁRifampicín
Rifapentín
artemeter AUC ↓ 16 % artemeter Cmin ↔ artemeter Cmax ↓ 18 %
dihydroartemisinín AUC ↓ 18 %
dihydroartemisinín Cmin ↔ dihydroartemisinín Cmax ↓ 18 % lumefantrín AUC ↑ 175 % lumefantrín Cmin ↑ 126 % lumefantrín Cmax ↑ 65 % darunavir AUC ↔
darunavir Cmin ↓ 13 %
darunavir Cmax ↔
Neskúmalo sa. Rifapentín a rifampicín sú silnými induktormi CYP3A a bolo dokázané, že spôsobujú signifikantné zníženie koncentrácií iných proteázových inhibítorov, čo môže vyústiť do zlyhania antivirologickej odpovede a rozvinutia rezistencie (indukcia enzýmu CYP450). Pri pokusoch prekonať zníženú expozíciu zvýšením dávky iných proteázových inhibítorov s nízkou dávkou ritonaviru, sa s rifampicínom zistila vysoká frekvencia pečeňových reakcií.
Kombinácia posilneného darunaviru a artemeteru/lumefantrínu sa môže podávať bez úpravy dávky;
z dôvodu zvýšenia expozície lumefantrínu však treba kombináciu používať s opatrnosťou.
Kombinácia rifapentínu
a posilneného darunaviru sa
neodporúča.
Súčasné podávanie rifampicínu a posilneného darunaviru je kontraindikované (pozri časť 4.3).
Rifabutin
150 mg jedenkrát každý druhý deň
ANTINEOPLASTIKÁ
Dasatinib Nilotinib Vinblastín Vinkristín
rifabutin AUC** ↑ 55 % rifabutin Cmin** ↑ ND rifabutin Cmax** ↔ darunavir AUC ↑ 53 % darunavir Cmin ↑ 68 % darunavir Cmax ↑ 39 %
** súčet účinných zložiek rifabutinu (materský liek
+ 25-O-desacetyl metabolit)
Štúdia interakcií preukázala porovnateľnú
dennú systémovú expozíciu rifabutinu
v liečbe samotným rifabutinom 300 mg
jedenkrát denne a v liečbe rifabutinom
150 mg jedenkrát každý druhý deň spolu s darunavirom/ritonavirom (600/100 mg dvakrát denne) s približne 10-násobným zvýšením dennej expozície aktívnemu metabolitu 25-O-desacetylrifabutinu. Okrem toho, AUC súčtu účinných zložiek rifabutinu (materský liek + 25-O-desacetyl metabolit) sa zvýšila 1,6-násobne, pričom Cmax zostala porovnateľná.
Údaje o porovnaní s referenčnou dávkou
150 mg jedenkrát denne nie sú k dispozícii.
(Rifabutin je induktorom a substrátom enzýmov CYP3A.) Keď sa darunavir užívaný spolu s 100 mg ritonaviru podával spolu s rifabutinom (150 mg jedenkrát každý druhý deň) pozorovalo sa zvýšenie systémovej expozície darunaviru.
Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto antineoplastík.
(inhibícia CYP3A)
U pacientov užívajúcich kombináciu s darunavirom súčasne s ritonavirom je nevyhnutná
redukcia zvyčajnej dávky rifabutinu
300 mg/deň o 75 % (t.j. rifabutin
150 mg jedenkrát každý druhý deň) a zvýšené sledovanie nežiaducich udalostí súvisiacich s rifabutinom. V prípade ohrozenia bezpečnosti sa má zvážiť ďalšie predĺženie dávkovacieho intervalu rifabutinu a/alebo sledovanie hladiny rifabutinu.
Treba brať do úvahy oficiálnu smernicu pre liečbu tuberkulózy
u pacientov infikovaných HIV.
Na základe bezpečnostného profilu darunaviru/ritonaviru, toto zvýšenie expozície darunaviru v prítomnosti rifabutinu nevyžaduje úpravu dávky darunaviru/ritonaviru.
Na základe farmakokinetického modelu možno dávku zredukovať o 75 % aj u pacientov, ktorí
dostávajú inú dávku rifabutinu ako
300 mg/deň.
Súčasné užívanie darunaviru
užívaného s kobicistatom
a rifabutínu sa neodporúča.
Pri súčasnom užívaní týchto liekov a posilneného darunaviru môžu byť ich koncentrácie zvýšené, čo vedie
k možnosti nárastu nežiaducich
udalostí zvyčajne súvisiacich
s týmito liekmi.
Pri súčasnom užívaní posilneného darunaviru a jedného
z antineoplastík je potrebná
opatrnosť.
Everolimus
INHIBÍTORY AGREGÁCIE TROMBOCYTOVTikagrelor Neskúmalo sa. Súčasné užívanie
s posilneným darunavirom môže viesť k značnému zvýšeniu expozície tikagreloru.
Súčasné užívanie everolimu
a posilneného darunaviru sa
neodporúča.
Súčasné užívanie posilneného darunaviru a tikagreloru je kontraindikované.
Užívanie iných antitrombotík neovplyvnených inhibíciou alebo indukciou CYP (napr. prasugrel) sa odporúča.
ANTIPSYCHOTIKÁ/NEUROLEPTIKÁ
Kvetiapín Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto antipsychotík.
(inhibícia CYP3A)
Súčasné podávanie posilneného darunaviru a kvetiapínu je kontraindikované, pretože môže zvýšiť toxicitu súvisiacu
s kvetiapínom. Zvýšené koncentrácie kvetiapínu môžu viesť ku kóme.
Perfenazín Risperidón Tioridazín
Pimozid
Sertindol
β-BLOKÁTORY Karvedilol Metoprolol Timolol
Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto antipsychotík.
(inhibícia CYP2D6 a/alebo P-gp)
Neskúmalo sa. Očakáva sa, že posilnený darunavir zvýši plazmatické koncentrácie týchto beta blokátorov.
(inhibícia CYP2D6)
Môže byť potrebné zníženie dávky týchto liekov, keď sa podávajú súčasne s posilneným darunavirom.
Súčasné podávanie posilneného darunaviru a pimozidu alebo sertindolu je kontraindikované.
Pri súčasnom užívaní posilneného darunaviru s beta blokátormi sa odporúča klinické sledovanie. Má sa zvážiť zníženie dávky beta blokátora.
B
L
OK
ÁTORY KALCIOVÉHO KANÁLA
Amlodipín Diltiazem Felodipín Nikardipín Nifedipín Verapamil
Neskúmalo sa. Môže sa očakávať, že posilnený darunavir bude zvyšovať plazmatické koncentrácie blokátorov kalciového kanála.
(inhibícia CYP3A a/alebo CYP2D6)
Ak sa tieto lieky podávajú súčasne s posilneným darunavirom, odporúča sa klinické sledovanie liečebných a nežiaducich udalostí.
KO
RTIKOSTEROIDY
Flutikazón
Budezonid
Dexametazón
(podávaný systémovo)
V klinickej štúdii, pri ktorej sa zdravým jedincom počas 7 dní podávali kapsuly ritonaviru 100 mg dvakrát denne spolu
s 50 µg flutikazónpropionátu intranazálne (štyrikrát denne), sa významne zvýšili sérové hladiny flutikazónpropionátu, zatiaľ čo prirodzená hladina kortizolu klesla približne o 86 % (pri 90 % intervale spoľahlivosti 82 % - 89 %). Vyššie účinky možno očakávať, keď sa flutikazón inhaluje. Systémové účinky kortikosteroidov, vrátane Cushingovho syndrómu a adrenálnej supresie, boli hlásené u pacientov, ktorí užívali ritonavir
a súčasne inhalovali alebo intranazálne užili flutikazón; podobná situácia môže vzniknúť aj pri súčasnom užívaní iných kortikosteroidov, ktoré sa metabolizujú prostredníctvom cytochrómu P4503A, napr. budezonidu. Účinky vysokej systémovej expozície flutikazónu na plazmatické hladiny ritonaviru nie sú dosiaľ známe.
Neskúmalo sa. Dexametazón môže znížiť plazmatickú koncentráciu darunaviru. (indukcia CYP3A)
Preto sa súčasná liečba posilneným darunavirom a týmito glukokortikoidmi neodporúča, pokiaľ možný prínos nepreváži riziko systémových kortikosteroidných účinkov. Má sa zvážiť zníženie dávky glukokortikoidov spolu
s dôkladným monitorovaním lokálnych a systémových účinkov, alebo prevod na iný glukokortikoid, ktorý nepatrí k substrátom pre
CYP3A (napr. beklometazón). V prípade vysadenia
glukokortikoidov môže byť navyše potrebné, aby progresívne znižovanie dávky trvalo dlhší čas.
Dexametazón podávaný systémovo sa má podávať so zvýšenou opatrnosťou, ak sa užíva spolu
s posilneným darunavirom.
Prednizón Neskúmalo sa. Posilnený darunavir môže zvýšiť plazmatické koncentrácie prednizónu.
(inhibícia CYP3A)
ANTIENDOTELÍNY
Bosentan Neskúmalo sa. Súčasné užívanie bosentanu a posilneného darunaviru môže zvýšiť plazmatické koncentrácie bosentanu.
Očakáva sa, že bosentan zníži plazmatické koncentrácie darunaviru a/alebo lieku na zlepšenie jeho farmakokinetiky.
(indukcia CYP3A)
Súčasné užívanie posilneného darunaviru s nízkou dávkou ritonaviru a prednizónom môže zvýšiť riziko vzniku systémových kortikosteroidných účinkov, vrátane Cushingovho syndrómu
a adrenálnej supresie. Pri súčasnom užívaní posilneného darunaviru
a kortikosteroidov sa odporúča
klinické sledovanie.
Ak sa podáva súčasne
s darunavirom a nízkou dávkou ritonaviru, má sa u pacienta
sledovať tolerovateľnosť bosentanu.
Súčasné užívanie darunaviru
užívaného s kobicistatom
a bosentanu sa neodporúča.
ANTIVIROTIKÁ PRIAMO ÚČINKUJÚCE PROTI VÍRUSU HEPATITÍDY C (HCV)
Proteázové inhibítory NS3-4A
Telaprevir
750 mg každých
8 hodín
Boceprevir
800 mg trikrát denne
telaprevir AUC ↓ 35 % telaprevir Cmin ↓ 32 % telaprevir Cmax ↓ 36 % darunavir AUC12 ↓ 40 % darunavir Cmin ↓ 42 % darunavir Cmax ↓ 40 % boceprevir AUC ↓ 32 % boceprevir Cmin ↓ 35 % boceprevir Cmax ↓ 25 % darunavir AUC ↓ 44 % darunavir Cmin ↓ 59 % darunavir Cmax ↓ 36 %
Súčasné užívanie posilneného darunaviru a telapreviru sa neodporúča.
Súčasné užívanie posilneného darunaviru a bocepreviru sa neodporúča.
Simeprevir simeprevir AUC ↑ 159 %
simeprevir Cmin ↑ 358 % simeprevir Cmax ↑ 79 % darunavir AUC ↑ 18 % darunavir Cmin ↑ 31 % darunavir Cmax «
Súčasné užívanie posilneného
darunaviru a simepreviru sa
neodporúča.
RASTLINNÉ LIEČIVÁ Ľubovník bodkovaný
(Hypericum perforatum)Dávka simepreviru v tejto interakčnej
štúdii bola 50 mg, keď sa podával spolu
s darunavirom/ritonavirom, v porovnaní so
150 mg v skupine liečenej samotným
simeprevirom.
Neskúmalo sa. Predpokladá sa, že ľubovník bodkovaný spôsobuje pokles plazmatických koncentrácií darunaviru alebo lieku na zlepšenie jeho farmakokinetiky.
(indukcia CYP450)
Posilnený darunavir sa nesmie podávať v kombinácii s liekmi obsahujúcimi výťažok z ľubovníka bodkovaného (
Hypericum perforatum) (pozri časť 4.3). Ak už pacient užíva ľubovník bodkovaný, užívanie ukončite, a ak je to možné, skontrolujte vírusové hladiny. Expozícia darunaviru (a tiež expozícia ritonaviru) sa môže po ukončení užívania ľubovníka bodkovaného zvýšiť. Indukujúci účinok môže pretrvávať najmenej 2 týždne po ukončení liečby ľubovníkom bodkovaným.
I
NHIBÍTORY HMG CO-A REDUKTÁZY
Lovastatín
Simvastatín
Atorvastatín
10 mg jedenkrát denne
Pravastatín
40 mg jednorazová dávka
Rosuvastatín
10 mg jedenkrát denne
Neskúmalo sa. Predpokladá sa, že lovastatín a simvastatín budú mať zreteľne zvýšené plazmatické koncentrácie, ak sa budú podávať súčasne s posilneným darunavirom.
(inhibícia CYP3A)
atorvastatín AUC ↑ 3-4 krát atorvastatín Cmin ↑ ≈5,5-10 krát atorvastatín Cmax ↑ ≈2 krát
#darunavir
pravastatín AUC ↑ 81 %¶ pravastatín Cmin ND pravastatín Cmax ↑ 63 %
¶ u niektorých subjektov sa pozoroval až 5-násobný nárast
rosuvastatín AUC ↑ 48 %║
rosuvastatín Cmax ↑ 144 %║
║ na základe publikovaných údajov
Zvýšené plazmatické koncentrácie lovastatínu alebo simvastatínu môžu vyvolať myopatiu, vrátane rabdomyolýzy. Súčasné podávanie posilneného darunaviru
s lovastatínom a simvastatínom je
preto kontraindikované (pozri časť
4.3).
Ak je potrebné súčasné podávanie atorvastatínu s posilneným darunavirom, odporúča sa začať podávať atorvastatín v dávke 10 mg jedenkrát denne. Postupné zvyšovanie dávkovania
atorvastatínu je možné prispôsobiť
na základe klinickej odpovede.
Ak je potrebné súčasné podávanie pravastatínu s posilneným darunavirom, odporúča sa začať
s najnižšou možnou dávkou pravastatínu a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
Ak je potrebné súčasné podávanie rosuvastatínu s posilneným darunavirom, odporúča sa začať
s najnižšou možnou dávkou rosuvastatínu a titrovať ju, kým sa nedosiahne želaný klinický účinok pri sledovaní bezpečnosti.
I
NHIBÍTORY H
2
-
RECEPTORA
Ranitidín
150 mg dvakrát denne
IMUNOSUPRESÍVA Ciklosporín Sirolimus Takrolimus
Everolimus
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Neskúmalo sa. Pri súčasnom podávaní posilneného darunaviru s týmito imunosupresívami sa zvýši expozícia voči týmto látkam.
(inhibícia CYP3A)
Posilnená darunavir a inhibítory H2-receptora sa môžu užívať bez úpravy dávky.
Pri súčasnom podaní imunosupresív sa musia monitorovať terapeutické hladiny týchto liekov.
Súčasné užívanie everolimu a posilneného darunaviru sa neodporúča.
I
NHALAČNÉ BETA-AGONISTY
Salmeterol Neskúmalo sa. Súčasné užívanie salmeterolu a posilneného darunaviru môže zvýšiť plazmatické koncentrácie salmeterolu.
Súčasné užívanie salmeterolu a posilneného darunaviru sa neodporúča. Táto kombinácia môže viesť k zvýšenému riziku kardiovaskulárnych nežiaducich udalostí salmeterolu, vrátane predĺženia QT intervalu, palpitácií a sínusovej tachykardie.
NARKOTICKÉ ANALGETIKÁ / LIEČBA ZÁVISLOSTI NA OPIOIDOCH
Metadon individuálna dávka
v rozsahu od 55 mg do
150 mg jedenkrát denne
Buprenorfín/naloxón
8/2 mg–16/4 mg jedenkrát denne
R(-) metadon AUC ↓ 16 % R(-) metadon Cmin ↓ 15 % R(-) metadon Cmax ↓ 24 %
Darunavir/kobicistat môže, naopak, zvýšiť plazmatické koncentrácie metadonu (pozri SPC kobicistatu).
buprenorfín AUC ↓ 11 % buprenorfín Cmin ↔ buprenorfín Cmax ↓ 8 % norbuprenorfín AUC ↑ 46 % norbuprenorfín Cmin ↑ 71 % norbuprenorfín Cmax ↑ 36 % naloxón AUC ↔
naloxón Cmin ND
naloxón Cmax ↔
Na začiatku užívania spolu
s posilneným darunavirom nie je potrebná úprava dávky metadonu. Avšak úprava dávky metadonu môže byť potrebná pri súčasnom podávaní počas dlhšieho časového obdobia. Z toho dôvodu sa
odporúča klinické monitorovanie,
pretože u niektorých pacientov môže byť potrebná úprava udržiavacej liečby.
Klinický význam zvýšenia farmakokinetických parametrov norbuprenorfínu nebol stanovený. Úprava dávky buprenorfínu nemusí byť potrebná, ak sa podáva spolu
s posilneným darunavirom, ale odporúča sa pozorné sledovanie prejavov toxicity opiátov.
ANTIKONCEPCIA NA BÁZE ESTROGÉNOV
Etinylestradiol
Noretisterón
35 mg/1 mg jedenkrát denne
etinylestradiol AUC ↓ 44 % etinylestradiol Cmin ↓ 62 % etinylestradiol Cmax ↓ 32 % noretisterón AUC ↓ 14 % noretisterón Cmin ↓ 30 % noretisterón Cmax ↔
Pacientkam užívajúcim antikoncepciu na báze estrogénov v kombinácii s posilneným darunavirom sa odporúča zmeniť
resp. rozšíriť formy antikoncepcie. U pacientok užívajúcich estrogén ako hormonálnu substitúciu majú byť klinicky sledované prejavy deficitu estrogénu.
FOSFODIESTERÁZA, INHIBÍTORY TYPU 5 (PDE-5)
Pri liečbe erektilnej
dysfunkcie Avanafil Sildenafil Tadalafil Vardenafil
V štúdii skúmajúcej interakcie# sa pozorovala porovnateľná systémová expozícia voči sildenafilu po jednorazovom podaní samotného sildenafilu v dávke 100 mg a po jednorazovom podaní sildenafilu v dávke
25 mg v kombinácii s darunavirom a nízkou dávkou ritonaviru.
Kombinácia avanafilu
a posilneného darunaviru je kontraindikovaná (pozri časť 4.3). Pri súčasnom podávaní iných inhibítorov PDE-5 na liečbu erektilnej dysfunkcie s posilneným darunavirom je potrebná opatrnosť. Ak je indikované súčasné podávanie posilneného darunaviru a
sildenafilu, vardenafilu alebo tadalafilu, jednorazová dávka
sildenafilu nesmie prekročiť 25 mg
za obdobie 48 hodín, resp. jednorazová dávka vardenafilu nesmie prekročiť 2,5 mg za obdobie
72 hodín a jednorazová dávka tadalafilu nesmie presiahnuť 10 mg za obdobie 72 hodín.
Pri liečbe pulmonálnej
arteriálnej hypertenzie
Sildenafil
Tadalafil
Neskúmalo sa. Súčasné užívanie sildenafilu alebo tadalafilu pri liečbe pulmonálnej arteriálnej hypertenzie a posilneného darunaviru môže zvýšiť plazmatické koncentrácie sildenafilu alebo tadalafilu.
(inhibícia CYP3A)
Bezpečná a účinná dávka sildenafilu pri liečbe pulmonálnej arteriálnej hypertenzie a pri súčasnom užívaní posilneného darunaviru nebola stanovená.
Existuje zvýšená možnosť vzniku nežiaducich udalostí súvisiacich so sildenafilom (vrátane porúch zraku, hypotenzie, predĺženej erekcie
a synkopy). Preto je súčasné užívanie posilneného darunaviru a sildenafilu, ak sa používa pri liečbe pulmonálnej arteriálnej hypertenzie, kontraindikované (pozri časť 4.3).
Pri liečbe pulmonálnej arteriálnej hypertenzie sa súčasné užívanie tadalafilu s posilneným darunavirom neodporúča.
I
NHIBÍTORY PROTÓNOVEJ PUMPY
Omeprazol
20 mg jedenkrát denne
SEDATÍVA/HYPNOTIKÁ
Buspirón Klorazepát Diazepam Estazolam Flurazepam Midazolam (parenterálny) Zolpidem
Midazolam (perorálny) Triazolam
#darunavir AUC ↔
#darunavir Cmin ↔
#darunavir Cmax ↔
Neskúmalo sa. Sedatíva/hypnotiká sú výrazne metabolizované prostredníctvom CYP3A. Súčasné užívanie s posilneným darunavirom môže spôsobiť významné zvýšenie koncentrácií týchto liekov.
Ak sa parenterálny midazolam podáva súčasne s posilneným darunavirom, môže spôsobiť významné zvýšenie koncentrácie tohto benzodiazepínu. Údaje o súčasnom užívaní parenterálneho midazolamu
s inými proteázovými inhibítormi naznačujú možné 3-4-násobné zvýšenie plazmatických hladín midazolamu.
Posilnenú darunavir je možné súčasne podávať s inhibítormi protónovej pumpy bez nutnosti úpravy dávkovania.
Pri súčasnom užívaní posilneného darunaviru s týmito sedatívami/hypnotikami sa odporúča klinické sledovanie a má sa zvážiť zníženie dávky sedatív/hypnotík.
Ak sa parenterálny midazolam podáva súčasne s posilneným darunavirom, má sa tak uskutočniť na jednotke intenzívnej starostlivosti (JIS) alebo
v podobnom zariadení, ktoré zabezpečuje dôkladné klinické sledovanie a vhodnú lekársku starostlivosť v prípade respiračného útlmu a/alebo predĺženého útlmu. Treba zvážiť úpravu dávkovania midazolamu, najmä ak sa podáva viac ako jednorazová dávka.
Súčasné užívanie posilneného darunaviru a triazolamu alebo perorálneho midazolamu je kontraindikované (pozri časť 4.3)
† Účinnosť a bezpečnosť používania darunaviru so 100 mg ritonaviru a s niektorým iným HIV PI (napr. (fos)amprenavir, nelfinavir a tipranavir) neboli u pacientov s HIV stanovené. Podľa súčasných liečebných postupov sa vo všeobecnosti duálna liečba proteázovými inhibítormi neodporúča.
4.6 Fertilita, gravidita a laktáciaGraviditaVo všeobecnosti, keď sa rozhoduje o použití antiretrovirotík na liečbu infekcie HIV u gravidných žien
a následne na zníženie rizika vertikálneho prenosu HIV na novorodenca, treba vziať do úvahy údaje
o zvieratách ako aj klinické skúsenosti s gravidnými ženami.
S darunavirom sa nevykonali žiadne primerané ani dobre kontrolované štúdie o vplyve na graviditu u gravidných žien. Štúdie na zvieratách nepreukázali priame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3).
Darunavir užívaný s kobicistatom a s nízkou dávkou ritonaviru sa môže podávať gravidným ženám len
vtedy, ak možný prínos liečby prevýši možné riziká.
Dojčenie
Nie je známe, či sa darunavir vylučuje do ľudského mlieka. V štúdiách na potkanoch sa preukázalo
vylučovanie darunaviru do mlieka a pri vysokých hladinách (1000 mg/kg/deň) spôsobilo toxicitu. Matky majú byť poučené, aby za žiadnych okolností nedojčili počas liečby darunavirom jednak kvôli možnosti prenosu HIV a aj možnosti výskytu nežiaducich reakcií u dojčených detí.
Fertilita
Nie sú k dispozícii žiadne údaje o účinku darunaviru na fertilitu u ľudí. Pri liečbe potkanov
darunavirom nemal darunavir vplyv na párenie a fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Darunavir spolu s kobicistatom alebo s ritonavirom nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U niektorých pacientov sa však počas liečby darunavirom užívanou s kobicistatom alebo s nízkou dávkou ritonaviru vyskytli závraty. Na túto možnosť je potrebné myslieť pri posudzovaní schopnosti pacienta viesť vozidlá a obsluhovať stroje (pozri časť
4.8).
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Počas programu klinického vývoja (N=2 613 predtým liečených jedincov, ktorí začali liečbu
s darunavirom/ritonavirom 600/100 mg dvakrát denne), 51,3 % jedincovmalo aspoň jednu nežiaducu reakciu. Celkové priemerné trvanie liečby pacientov bolo 95,3 týždňov. Najčastejšími nežiaducimi reakciami hlásenými v klinických skúšaniach a v spontánnych hláseniach sú hnačka,
nauzea, vyrážka, bolesť hlavy a vracanie. Najčastejšími závažnými reakciami sú akútne renálne
zlyhanie, infarkt myokardu, imunoreštitučný zápalový syndróm, trombocytopénia, osteonekróza,
hnačka, hepatitída a pyrexia.
V analýze po 96 týždňoch bol bezpečnostný profil darunaviru/ritonaviru 800/100 mg jedenkrát denne u jedincov, ktorí doteraz neboli liečení, podobný ako u darunaviru/ritonaviru 600/100 mg dvakrát denne u jedincov, ktorí boli predtým liečení, až na nauzeu, ktorá sa častejšie vyskytovala u doteraz neliečených jedincov. Nauzea bola miernej intenzity. V analýze po 192 týždňoch sa u doteraz neliečených pacientov, u ktorých liečba darunavirom/ritonavirom 800/100 mg jedenkrát denne trvala priemerne 162,5 týždňa, neidentifikovali žiadne nové nálezy týkajúce sa bezpečnosti.
Počas klinickej štúdie fázy III GS-US-216-130 s darunavirom/kobicistatom (N = 313 doposiaľ neliečených a predtým liečených pacientov), 66,5 % osôb zaznamenalo aspoň jednu nežiaducu reakciu. Liečba trvala priemerne 58,4 týždňa. Najčastejšie hlásené nežiaduce reakcie boli diarea (28 %), nauzea (23 %) a vyrážka (16 %). Závažné nežiaduce reakcie sú diabetes melitus, precitlivenosť (na liek), imunoreštitučnýzápalovýsyndróm, vyrážka a vracanie.
Informácie o kobicistate si pozrite v Súhrne charakteristických vlastností kobicistatu.
Z
o
z
nam nežiaducich reakcií v tabuľkách
Nežiaduce reakcie sa uvádzajú podľa postihnutia triedy orgánových systémov a podľa skupiny
frekvencie. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti. Skupiny frekvencií sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000)
a neznáme (z dostupných údajov).
Nežiaduce reakcie pozorované s darunavirom/ritonavirom v klinických skúšaniach
a v postmarketingovom sledovaní
Trieda orgánových systémov podľa databázy
MedDRA
Kategória frekvencie
Infekcie a nákazy
Nežiaduca reakcia

menej časté herpes simplex
Poruchy krvi a lymfatického systémumenej časté trombocytopénia, neutropénia, anémia, leukopénia
zriedkavé zvýšenie počtu eozinofilov
Poruchy imunitného systémumenej časté imunoreštitučný zápalový syndróm, precitlivenosť
(na liek)
Poruchy endokrinného systémumenej časté hypotyreóza, zvýšenie hladiny hormónu stimulujúceho štítnu žľazu v krvi
Poruchy metabolizmu a výživyčasté diabetes mellitus, hypertriglyceridémia, hypercholesterolémia, hyperlipidémia
menej časté dna, anorexia, znížená chuť do jedla, zníženie
hmotnosti, zvýšenie hmotnosti, hyperglykémia,
rezistencia na inzulín, pokles lipoproteínu
s vysokou hustotou, zvýšená chuť do jedla, polydipsia, zvýšená hladina LDH v krvi
Psychické poruchyčasté insomnia
menej časté depresia, dezorientácia, úzkosť, poruchy spánku,
neprirodzené sny, nočné mory, pokles libida
zriedkavé stav zmätenosti, zmeny nálady, nepokoj
Poruchy nervového systémučasté bolesti hlavy, periférna neuropatia, závraty
menej časté letargia, parestézia, hypoestézia, dysgeúzia,
porucha pozornosti, poškodenie pamäti,
somnolencia
zriedkavé synkopa, konvulzia, ageuzia, porucha fázy spánkového rytmu
Poruchy okamenej časté konjunktiválna hyperémia, suché oči
zriedkavé porucha zraku
 Poruchy ucha a labyrintu
Poruchy ucha a labyrintu
menej časté vertigo
Poruchy srdca a srdcovej činnostimenej časté infarkt myokardu, angína pektoris, predĺžený QT
elektrokardiogram, tachykardia
zriedkavé akútny infarkt myokardu, sínusová bradykardia,
palpitácie
Poruchy cievmenej časté hypertenzia, návaly horúčavy
Poruchy dýchacej sústavy, hrudníka a mediastínamenej časté dyspnoe, kašeľ, epistaxa, podráždenie hrdla
zriedkavé rinorea
Poruchy gastrointestinálneho traktuveľmi časté diarea
časté vracanie, nauzea, bolesti brucha, zvýšená hladina
amylázy v krvi, dyspepsia, abdominálna distenzia, flatulencia
menej časté pankreatitída, gastritída, gastroezofagálny reflux, aftózna stomatitída, napínanie na vracanie, sucho
v ústach, abdominálny diskomfort, zápcha, zvýšená
hladina lipázy, eruktácia, perorálna dyzestézia
zriedkavé stomatitída, hemateméza, cheilitída, suché pery, povlak na jazyku
Poruchy pečene a žlčových ciestčasté zvýšenie alanínaminotransferázy
menej časté hepatitída, cytolytická hepatitída, steatóza pečene,
hepatomegália, zvýšenie transamináz, zvýšenie aspartátaminotransferázy, zvýšenie hladiny bilirubínu v krvi, zvýšenie alkalickej fosfatázy v krvi, zvýšenie gamaglutamyltransferázy
Poruchy kože a podkožného tkanivačasté vyrážka (vrátane makulárnej, makulopapulárnej, papulárnej, erytematóznej a pruritickej vyrážky), pruritus
menej časté angioedém, generalizovaná vyrážka, alergická
dermatitída, urtikária, ekzém, erytém,
hyperhidróza, nočné potenie, alopécia, akné, suchá
koža, sfarbenie nechtov
zriedkavé DRESS, Stevensov-Johnsonov syndróm, multiformný erytém, dermatitída, seboroická dermatitída, kožné lézie, xeroderma
neznáme toxická epidermálna nekrolýza, akútna generalizovaná exantematózna pustulóza
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
menej časté myalgia, osteonekróza, svalové kŕče, svalová slabosť, artralgia, bolesti končatín, osteoporóza, zvýšená hladina kreatínfosfokinázy v krvi
zriedkavé muskuloskeletálna stuhnutosť, artritída, stuhnutie
kĺbov
Poruchy obličiek a močových ciest
menej časté akútne zlyhanie obličiek, zlyhanie obličiek, nefrolitiáza, zvýšená hladina kreatinínu v krvi, proteinúria, bilirubinúria, dyzúria, noktúria, polakizúria
zriedkavé znížený renálny klírens kreatinínu
Poruchy reprodukčného systému a prsníkov
menej časté erektilná dysfunkcia, gynekomastia
Celkové poruchy a reakcie v mieste podania
časté asténia, únava
menej časté pyrexia, bolesť na hrudi, periférne opuchy,
nevoľnosť, pocit horúčavy, podráždenie, bolesť
zriedkavé triaška, abnormálne pocity, xeróza
Nežiaduce reakcie s darunavirom/kobicistatom u dospelých pacientov
Trieda orgánových systémov podľa databázy
MedDRA
Kategória frekvencie
Poruchy imunitného systému
Nežiaduca reakcia


časté precitlivenosť (na liek)
menej časté imunoreštitučný zápalový syndróm
Poruchy metabolizmu a výživyčasté anorexia, diabetes mellitus, hypercholesterolémia, hypertriglyceridémia, hyperlipidémia
Psychické poruchyčasté neprirodzené sny
Poruchy nervového systémuveľmi časté bolesť hlavy
Poruchy gastrointestinálneho traktuveľmi časté diarea, nauzea
časté vracanie, bolesti brucha, abdominálna distenzia,
dyspepsia, flatulencia, zvýšená hladina
pankreatických enzýmov
menej časté akútna pankreatitída
Poruchy pečene a žlčových ciestčasté zvýšená hladina hepatálnych enzýmov
menej časté hepatitída*, cytolytická hepatitída*
 Poruchy kože a podkožného tkaniva
Poruchy kože a podkožného tkaniva
veľmi časté vyrážka (vrátane makulárnej, makulopapulárnej, papulárnej, erytematóznej, pruritickej vyrážky, generalizovanej vyrážky a alergickej dermatitídy)
časté angioedém, pruritus, urtikária
zriedkavé lieková reakcia s eozinofíliou a systémovými symptómami*, Stevensov-Johnsonov syndróm*
neznáme toxická epidermálna nekrolýza*, akútna generalizovaná exantematózna pustulóza*
Poruchy kostrovej a svalovej sústavy a spojivového tkanivačasté myalgia, osteonekróza*
Poruchy reprodukčného systému a prsníkovmenej časté gynekomastia*
Celkové poruchy a reakcie v mieste podaniačasté únava
menej časté asténia
Laboratórne a funkčné vyšetreniačasté zvýšená hladina kreatinínu v krvi
* tieto nežiaduce reakcie na liek neboli hlásené v klinických štúdiách s darunavirom/kobicistatom, ale zaznamenali sa pri
liečbe darunavirom/ritonavirom a možno ich očakávať aj s darunavirom/kobicistatom.
OpisvybranýchnežiaducichreakciíVyrážkaV klinických skúšaniach bola najčastejšie vyrážka miene až stredne závažná, často sa vyskytla v prvých štyroch týždňoch liečby a ustúpila pri ďalšom užívaní. Pre prípady závažných kožných reakcií
pozri upozornenie v časti 4.4. V štúdii s jednou skupinou, ktorá skúmala darunavir 800 mg jedenkrát
denne v kombinácii s kobicistatom 150 mg jedenkrát denne a s iným antiretrovirotikami, 2,2 %
pacientov ukončilo liečbu kvôli vyrážke.
Počas programu klinického vývoja raltegraviru sa u predtým liečených pacientov častejšie pozorovala vyrážka, bez ohľadu na kauzalitu, pri režimoch obsahujúcich darunavir/ritonavir + raltegravir
v porovnaní s režimami obsahujúcimi darunavir/ritonavir bez raltegraviru alebo raltegravir bez darunaviru/ritonaviru. Vyrážka považovaná skúšajúcim za súvisiacu s liekom, sa vyskytla v podobnej miere. Expozícii prispôsobené miery vyrážky (všetky príčiny) boli 10,9; 4,2 a 3,8 na 100
pacientorokov v danom poradí a u vyrážky súvisiacej s liekom boli 2,4; 1,1 a 2,3 na 100 pacientorokov v danom poradí. Vyrážky pozorované v klinických štúdiách boli mierne až stredne závažné a neviedli
k prerušeniu liečby (pozri časť 4.4).
Metabolické parametrePočas antiretrovírusovej liečby sa môže zvýšiť telesná hmotnosť a hladiny lipidov a glukózy v krvi
(pozri časť 4.4).
Poruchy kostrovej a svalovej sústavyPočas liečby inhibítormi proteáz, najmä v kombinácii s NRTI, sa vyskytli prípady zvýšenia hladiny
kreatínfosfokinázy, myalgie, myozitídy a zriedkavo aj rabdomyolýzy.
Boli hlásené prípady osteonekrózy, najmä u pacientov so všeobecne potvrdenými rizikovými faktormi, pokročilým ochorením HIV alebo dlhodobou expozíciou voči kombinovanej antiretrovírusovej liečbe (CART). Frekvencia nie je známa (pozri časť 4.4).
I
m
unoreštitučný zápalový syndróm
U pacientov s infekciou HIV a ťažkým deficitom imunity v čase začatia podávania kombinovanej antiretrovírusovej liečby (CART) môže vzniknúť zápalová reakcia na asymptomatické infekcie alebo na reziduálne oportúnne infekcie. Boli tiež zaznamenané aj autoimunitné poruchy (ako je Gravesova choroba); avšak zaznamenaný čas do ich nástupu je rôznorodejší a tieto udalosti sa môžu vyskytnúť mnoho mesiacov po začatí liečby (pozri časť 4.4).
Krvácanie u pacientov s hemofíliouU pacientov s hemofíliou dostávajúcich antiretrovirálne inhibítory proteázy boli hlásené prípady
zvýšeného spontánneho krvácania (pozri časť 4.4).
Pediatrická populáciaPosúdenie bezpečnosti u pediatrických pacientov je založené na 48-týždňovej analýze údajov
o bezpečnosti z troch klinických skúšaní fázy II. Boli hodnotené nasledovné populácie pacientov
(pozri časť 5.1):
● 80 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg, ktorí užívali darunavir tablety s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 21 pediatrických pacientov infikovaných HIV-1 a predtým liečených ART, vo veku od 3 do < 6
rokov a s hmotnosťou 10 kg až < 20 kg (16 účastníkov od 15 kg do < 20 kg), ktorí užívali
darunavir perorálnu suspenziu s nízkou dávkou ritonaviru dvakrát denne v kombinácii s inými antiretrovírusovými látkami.
● 12 pediatrických pacientov infikovaných HIV-1 vo veku od 12 do 17 rokov a s hmotnosťou
najmenej 40 kg, ktorí neboli predtým liečení ART. Títo pacienti dostávali darunavir tablety
s nízkou dávkou ritonaviru jedenkrát denne spolu s inými antiretrovírusovými liekmi. (pozri
časť 5.1).
Celkovo bol bezpečnostný profil u týchto pediatrických pacientov podobný bezpečnostnému profilu
pozorovanému u dospelých.
Iné osobitné skupiny pacientovPacienti súčasne infikovaní vírusom hepatitídy B a/alebo hepatitídy CSpomedzi 1968 predtým liečených pacientov, ktorí užívali darunavir spolu s ritonavirom 600/100 mg dvakrát denne, bolo 236 pacientov infikovaných zároveň hepatitídou B alebo C. U súčasne infikovaných pacientov bola vyššia pravdepodobnosť počiatočného a pri liečbe vznikajúceho zvýšenia hladiny pečeňových transamináz ako u pacientov bez chronickej vírusovej hepatitídy (pozri časť 4.4).
HláseniepodozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSkúsenosti s akútnym predávkovaním darunavirom užívaným s kobicistatom alebo s nízkou dávkou ritonaviru u ľudí sú obmedzené. Zdravým dobrovoľníkom sa darunavir podával vo forme perorálneho roztoku v jednorazových dávkach do 3200 mg ako samotný, resp. vo forme tabliet
v jednorazových dávkach do 1600 mg v kombinácii s ritonavirom bez toho, že by sa u nich pozorovali nepriaznivé symptomatické účinky.
V prípade predávkovania darunavirom nie je k dispozícii žiadne špecifické antidotum. Liečba predávkovania darunavirom zahŕňa všeobecné podporné opatrenia, vrátane sledovania vitálnych znakov a pozorovania klinického stavu pacienta. Ak je to indikované, eliminácia nevstrebanej aktívnej látky sa dosiahne pomocou vyvolania vracania.
Aj podanie aktívneho uhlia môže pomôcť pri eliminácii nevstrebanej aktívnej látky. Vzhľadom na vysokú väzbu darunaviru na plazmatické bielkoviny je nepravdepodobné, že by dialýza mala väčší význam pri eliminácii aktívnej látky z cirkulácie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antivirotiká na systémové použitie, inhibítory proteázy, ATC kód: J05AE10
Mechanizmus účinku
Darunavir je inhibítor dimerizácie a katalytickej aktivity HIV-1 proteázy (KD 4,5 x 10-12M). Selektívne
inhibuje odštiepenie HIV kódovaných polyproteínov Gag-Pol v bunkách infikovaných vírusom, čím
zabraňuje tvorbe zrelých infekčných vírusových častíc.
Antivírusová aktivita in vitro
Darunavir vykazuje aktivitu proti laboratórnym kmeňom a klinickým izolátom HIV-1 a laboratórnym kmeňom HIV-2 v akútne infikovaných líniách T-buniek, mononukleárnych bunkách z periférnej krvi človeka a ľudských monocytov/makrofágov so strednými hodnotami EC50 v rozmedzí
1,2 až 8,5 nmol/l (0,7 až 5,0 ng/ml). In vitro má darunavir antivírusovú aktivitu voči širokému spektru
primárnych izolátov zo skupiny M HIV-1 (A, B, C, D, E, F, G) a skupiny O s hodnotami EC50
v rozmedzí od < 0,1 do 4,3 nmol/l.
Tieto hodnoty EC50 sú výrazne nižšie ako je rozsah toxickej koncentrácie pre 50 % buniek od
87 µmol/l do > 100 µmol/l.
Rezistencia
Selekcia vírusu rezistentného na darunavir z divokého typu HIV-1 in vitro trvala pomerne dlho
(> 3 roky). Vyselektované vírusy neboli schopné rásť v prítomnosti darunaviru v koncentráciách nad
400 nmol/l. U vírusov vyselektovaných v týchto podmienkach a vykazujúcich zníženú citlivosť voči
darunaviru (rozmedzie 23 až 50-násobne) sa v géne pre proteázu zistili substitúcie 2 až 4 aminokyselín. Zníženú citlivosť vznikajúcich vírusov na darunavir v selekcii nie je možné vysvetliť vznikom týchto mutácií proteázy.
Údaje z klinických štúdií s pacientmi liečenými ART (štúdia TITAN a zlúčená analýza štúdií POWER
1, 2 a 3 a štúdií DUET 1 a 2) preukázali, že virologická odpoveď na darunavir užívaný s nízkou dávkou ritonaviru bola znížená, keď boli na začiatku prítomné 3 a viac mutácií spojených s rezistenciou voči darunaviru (V11I, V32I, L33F, I47V, I50V, I54L alebo M, T74P, L76V, I84V a L89V) alebo keď sa tieto mutácie vyvinuli počas liečby.
Zvýšenie východiskovej násobnej zmeny darunaviru v EC50 (FC) súviselo s poklesom virologickej
odpovede. Určila sa dolná a horná klinická hranica 10 a 40. Izoláty s východiskovou hodnotou FC
≤ 10 sú citlivé; izoláty s FC > 10 až 40 majú zníženú citlivosť; izoláty s FC > 40 sú rezistentné (pozri
Klinické výsledky).
Vírusy izolované u pacientov s dávkou darunavir/ritonavir 600/100 mg dvakrát denne, u ktorých sa vyskytlo spontánne virologické zlyhanie, ktoré boli citlivé na tipranavir na začiatku, zostali vo veľkej väčšine prípadov citlivé na tipranavir po liečbe.
Najnižšie miery rozvoja rezistencie vírusu HIV sú pozorované u pacientov predtým neliečených ART, ktorí sú po prvýkrát liečení darunavirom v kombinácii s inou ART.
Tabuľka nižšie zobrazuje vývoj HIV-1 proteázových mutácií a stratu citlivosti na PI pri virologickom zlyhaní v závere liečby v štúdiách ARTEMIS, ODIN a TITAN.
ARTEMIS
192. týždeň
Darunavir/
ritonavir
Darunavir/
ritonavir
ODIN
48. týždeň
Darunavir/
ritonavir
TITAN
48. týždeň
Darunavir/
ritonavir
800/100 mg jedenkrát denne N=343
800/100 mg jedenkrát denne N=294
600/100 mg dvakrát denne N=296
600/100 mg dvakrát denne N=298
Celkový počet virologických zlyhanía, n (%)
55 (16,0 %) 65 (22,1 %) 54 (18,2 %) 31 (10,4 %)
Spontánne 39 (11,4 %) 11 (3,7 %) 11 (3,7 %) 16 (5,4 %)
Jedinci nereagujúci na
liečbu
16 (4,7 %) 54 (18,4 %) 43 (14,5 %) 15 (5,0 %)
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými genotypmi, u ktorých sa vyvinuli mutácieb v závere liečby, n/N
Primárne (významné)
mutácie PI
0/43 1/60 0/42 6/28
PI RAM 4/43 7/60 4/42 10/28
Počet jedincov s virologickým zlyhaním a párovými východiskovými/záverečnými fenotypmi, u ktorých sa preukázala strata citlivosti na PI v závere v porovnaní so začiatkom liečby, n/N
PI
darunavir 0/39 1/58 0/41 3/26 amprenavir 0/39 1/58 0/40 0/22 atazanavir 0/39 2/56 0/40 0/22 indinavir 0/39 2/57 0/40 1/24 lopinavir 0/39 1/58 0/40 0/23 sakvinavir 0/39 0/56 0/40 0/22 tipranavir 0/39 0/58 0/41 1/25
a TLOVR ne-VF cenzurovaný algoritmus založený na HIV-1 RNA < 50 kópií/ml, okrem TITAN (HIV-1 RNA
< 400 kópií/ml)
b zoznamy IAS-USA
Nízke miery rozvoja rezistencie vírusu HIV-1 boli pozorované u pacientov doposiaľ neliečených
ART, ktorí sú po prvýkrát liečení darunavirom/kobicistatom jedenkrát denne v kombinácii s inou ART
a u pacientov predtým liečených ART bez RAM voči darunaviru, ktorí dostávali darunavir/kobicistat
v kombinácii s inou ART. Tabuľka nižšie zobrazuje vývoj HIV-1 proteázových mutácií a rezistenciu
voči PI pri virologickom zlyhaní v závere liečby v štúdii GS-US-216-130.
Doposiaľ neliečení pacienti
GS-US-216-130
48. týždeň
Predtým liečení pacienti
darunavir/kobicistat 800/150 mg jedenkrát denne
N=295
darunavir/kobicistat 800/150 mg jedenkrát denne
N=18
Počet pacientov s virologickým zlyhaníma a údajmi o genotype, u ktorých sa vyvinuli mutácie b v závere
liečby, n/N
Primárne (významné)
mutácie PI
0/8 1/7

PI RAM 2/8 1/7
Počet pacientov s virologickým zlyhaníma a údajmi o fenotype, ktoré poukazujú na rezistenciu na PI v závere
liečbyc, n/N
HIV PI
darunavir 0/8 0/7 amprenavir 0/8 0/7 atazanavir 0/8 0/7
indinavir 0/8 0/7 lopinavir 0/8 0/7 sakvinavir 0/8 0/7
tipranavir 0/8 0/7

a Virologické zlyhanie bolo definované ako: nikdy nepotlačené: potvrdené zníženie HIV-1 RNA < 1 log10 oproti východiskovej hodnote a ≥ 50 kópií/ml v 8. týždni; rebound: HIV-1 RNA < 50 kópií/ml s následným potvrdením HIV-1 RNA na ≥ 400 kópií/ml alebo potvrdený nárast > 1 log10 HIV-1 RNA z najnižšieho bodu; ukončenie s HIV-1
RNA ≥ 400 kópií/ml pri poslednej návšteve
b zoznamy IAS-USA
c V GS-US-216-130 nebol úvodný fenotyp dostupný.
SkríženárezistenciaNásobná zmena (FC) darunaviru bola menej ako 10 pre 90 % z 3309 klinických izolátov rezistentných
voči amprenaviru, atazanaviru, indinaviru, lopinaviru, nelfinaviru, ritonaviru, sakvinaviru a/alebo tipraniviru, čo naznačuje, že vírusy rezistentné voči väčšine PI zostávajú citlivé voči darunaviru.
V štúdii
ARTEMIS sa v prípade virologického zlyhania nepozorovala žiadna skrížená rezistencia
s inými PI.
V štúdii GS-US-216-130 sa v prípade virologického zlyhania nepozorovala žiadna skrížená rezistencia
s inými HIV PI.
Klinické výsledkyVplyv kobicistatu na zlepšenie farmakokinetiky darunaviru sa hodnotil v štúdii fázy I u zdravých
osôb, ktorým sa podával darunavir 800 mg buď so 150 mg kobicistatu alebo so 100 mg ritonaviru jedenkrát denne. Farmakokinetické parametre darunaviru boli v ustálenom stave porovnateľné, keď
bol posilnený kobicistatom alebo ritonavirom. Informácie o kobicistate si pozrite v súhrne
charakteristických vlastností kobicistatu.
Dospelí pacienti Úči nnosť darunavi ru 800 mg j edenkr át denne uží vaného s o 150 mg kobicistatu jedenkrát denneu doposi aľ nel i eče nýc h paci ent ov a u paci ent ov pre dt ým l i eče ných ART GS-US-216-130 je otvorená štúdia fázy III s jednou skupinou, ktorá hodnotila farmakokinetiku,
bezpečnosť, tolerovateľnosť a účinnosť darunaviru s kobicistatom u 313 dospelých pacientov infikovaných HIV-1 (295 doposiaľ neliečených pacientov a 18 predtým liečených pacientov). Títo
pacienti dostávali darunavir 800 mg jedenkrát denne v kombinácii s kobicistatom 150 mg jedenkrát
denne so základným režimom zvoleným skúšajúcim, ktorý obsahoval 2 aktívne NRTI.
U pacientov infikovaných HIV-1, ktorí boli vhodní pre túto štúdiu, skríningové vyšetrenie rezistencie genotypu nepreukázalo RAM voči darunaviru a skríningové vyšetrenie plazmatickej HIV-1 RNA bolo
≥ 1000 kópií/ml. Tabuľka nižšie uvádza údaje o účinnosti z analýzy v 48. týždni štúdie
GS-US-216-130:
Výsledky v 48. týždni
Doposiaľ neliečení pacienti darunavir/kobicistat
800/150 mg jedenkrát denne + OBR N=295
GS-US-216-130
Predtým liečení pacienti darunavir/kobicistat
800/150 mg jedenkrát denne + OBR N=18
Všetci pacienti
darunavir/kobicistat
800/150 mg jedenkrát denne + OBR N=313
HIV-1 RNA < 50 kópií/mla 245 (83,1 %) 8 (44,4 %) 253 (80,8 %)
priemerná zmena
v HIV-1 RNA log oproti východiskovej hodnote (log10 kópií/ml) Priemerná zmena v počte buniek CD4+ oproti východiskovej hodnoteb
-3,01 -2,39 -2,97
+174 +102 +170
a Imputácie na základe algoritmu TLOVR
b Posledné pozorovanie sa dopočítalo LOCF
Úči nnosť darunaviru 800 mg j edenkr át denne uží vanej so 100 mg ritonaviru jedenkrát denne
u pacientov doposiaľ neli ečený ch ART
Dôkaz účinnosti darunaviru/ritonaviru 800/100 mg jedenkrát denne je založený na analýze 192-
týždňových údajov z randomizovanej, kontrolovanej, otvorenej štúdie fázy III ARTEMIS u pacientov infikovaných HIV-1, ktorí doposiaľ neboli liečení antiretrovírusovou liečbou v porovnaní
s darunavirom/ritonavirom 800/100 mg jedenkrát denne s lopinavirom/ritonavirom 800/200 mg denne (užívané dvakrát denne, alebo jedenkrát denne). Obe ramená používali fixný základný režim, ktorý pozostával z tenofovir disoproxil fumarátu 300 mg jedenkrát denne a emtricitabinu 200 mg jedenkrát
denne.
Nižšie uvedená tabuľka uvádza údaje 48-týždňovej a 96-týždňovej analýzy účinnosti zo štúdie
ARTEMIS:
ARTEMIS
Týždeň 48a Týždeň 96b
Výsledky Darunavir/
ritonavir
800/100 mg jedenkrát denne N=343
Lopinavir/
ritonavir
800/200 mg denne N=346
Rozdiel v liečbe (95 % CI rozdielu)
Darunavir/
ritonavir
800/100 mg jedenkrát denne N=343
Lopinavir/
ritonavir
800/200 mg denne N=346
Rozdiel v liečbe (95 % CI rozdielu)
HIV-1 RNA
< 50 kópií/mlc
Všetci pacienti 83,7 % (287)
78,3 % (271)
5,3 %
(-0,5; 11,2)d
79,0 % (271)
70,8 % (245)
8,2 %
(1,7; 14,7)d
S východiskovou
HIV-RNA
< 100 000
S východiskovou
HIV-RNA
≥ 100 000
S východiskovým počtom buniek CD4+ < 200
S východiskovým počtom buniek CD4+ ≥ 200
85,8 %
(194/226)
79,5 % (93/117)
79,4 % (112/141)
86,6 % (175/202)
84,5 %
(191/226)
66,7 % (80/120)
70,3 % (104/148)
84,3 % (167/198)
1,3 %
(-5,2; 7,9)d
12,8 % (1,6; 24,1)d
9,2 %
(-0,8; 19,2)d
2,3 %
(-4,6; 9,2)d
80,5 %
(182/226)
76,1 % (89/117)
78,7 % (111/141)
79,2 % (160/202)
75,2 %
(170/226)
62,5 % (75/120)
64,9 % (96/148)
75,3 % (149/198)
5,3 %
(-2,3; 13,0)d
13,6 % (1,9; 25,3)d
13,9 % (3,5; 24,2)d
4,0 %
(-4,3; 12,2)d
medián zmeny v počte buniek CD4+ oproti východiskovej hodnote (x 106/l)e
137 141 171 188

a Údaje založené na analýzach v 48. týždni b Údaje založené na analýzach v 96. týždni c Imputácie na základe algoritmu TLOVR
d Na základe obvyklého približného odhadu rozdielu v % odpovede
e Pacienti, ktorí neukončili liečbu, sú počítaní ako zlyhanie: pacientom, ktorí prerušili liečbu predčasne, sa priradí zmena
rovná nule.
V 48-týždňovej analýze sa non-inferiorita vo virologickej odpovedi na liečbu
darunavirom/ritonavirom, definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA
< 50 kópií/ml, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat (ITT) aj u pacientov sledovaných v protokole (OP, z angl. On Protocol). Tieto výsledky sa potvrdili v analýze údajov 96-týždňovej liečby v štúdii
ARTEMIS. Tieto výsledky boli zachované do 192. týždňa liečby v štúdii
ARTEMIS.
Úči nnosť darunaviru 800 mg j edenkr át denne uží vanej so 100 mg ritonaviru jedenkrát denne
u pacientov predtým li ečenýc h ART
OD
IN
je randomizovaná otvorená štúdia fázy III porovnávajúca darunavir/ritonavir 800/100 mg
jedenkrát denne a darunavir/ritonavir 600/100 mg dvakrát denne u pacientov infikovaných HIV-1 predtým liečených ART, u ktorých skríningové vyšetrenie rezistencie genotypu nepreukázalo RAM voči darunaviru (t.j. V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V, L89V)
a skríningové vyšetrenie HIV-1 RNA bolo > 1 000 kópií/ml. Analýza účinnosti je založená na liečbe trvajúcej 48 týždňov (pozri tabuľku nižšie). Obe ramená použili optimalizovaný základný režim
(OBR) ≥ 2 NRTI.
Výsledky Darunavir/ritonavir
800/100 mg jedenkrát denne + OBR
N=294
ODIN
Darunavir/ritonavir
600/100 mg dvakrát denne + OBR N=296
Rozdiel v liečbe
(95% CI rozdielu)
HIV-1 RNA
< 50 kópií/mla
S východiskovou HIV-1 RNA (kópie/ml)
72,1 % (212) 70,9 % (210) 1,2 % (-6,1; 8,5)b
< 100,000
≥ 100,000
S východiskovým počtom buniek CD4+ (x 106/l)
≥ 100
< 100
S kmeňom HIV-1
Typ B Typ AE Typ C Inéc
77,6 % (198/255)
35,9 % (14/39)
75,1 % (184/245)
57,1 % (28/49)
70,4 % (126/179)
90,5 % (38/42)
72,7 % (32/44)
55,2 % (16/29)
73,2 % (194/265)
51 6 % (16/31)
72,5 % (187/258)
60,5 % (23/38)
64,3 % (128/199)
91,2 % (31/34)
78,8 % (26/33)
83,3 % (25/30)
4,4 % (-3,0; 11,9)
-15,7 % (-39,2; 7,7)
2,6 % (-5,1; 10,3)
-3,4 % (-24,5; 17,8)
6,1 % (-3,4; 15,6)
-0,7 % (-14,0; 12,6)
-6,1 % (-2,6; 13,7)
-28,2 % (-51,0; -5,3)
priemerná zmena počtu buniek CD4+ oproti východiskovej hodnote (x 106/l)e
108 112 -5d (-25; 16)
a Imputácie na základe algoritmu TLOVR
b Na základe obvyklého približného odhadu rozdielu v % odpovede
c Kmene A1, D, F1, G, K, CRF02_AG, CRF12_BF, a CRF06_CPX
d Rozdiel stredných hodnôt
e Posledné pozorovanie sa dopočítalo LOCF (Last Observation Carried Forward)
V 48. týždni sa non-inferiorita vo virologickej odpovedi na liečbu darunavirom/ritonavirom
800/100 mg jedenkrát denne, definovaná ako percento pacientov s plazmatickou hladinou HIV-1 RNA
< 50 kópií/ml v porovnaní s darunavirom/ritonavirom 600/100 mg dvakrát denne, dokázala (pri dopredu definovanej hranici non-inferiority 12 %) v oboch populáciách, Intent-To-Treat (ITT) aj
u pacientov sledovaných v protokole (On Protocol).
Darunavir/ritonavir 800/100 mg jedenkrát denne sa nemá používať u tých pacientov liečených ART,
ktorí majú jednu alebo viac mutácií spojených s rezistenciou voči darunaviru (DRV-RAM) alebo
HIV-1 RNA ≥ 100 000 kópií/ml alebo počet buniek CD4+ < 100 buniek x 106/l (pozri časť 4.2 a 4.4).
U pacientov s kmeňmi HIV-1 inými ako B sú dostupné obmedzené údaje.
Pediatrickí pacienti
Pediatrickí pacienti vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým
liečení ART
DIONE je otvorená štúdia fázy II hodnotiaca farmakokinetiku, bezpečnosť, toleranciu a účinnosť
darunaviru s nízkou dávkou ritonaviru u 12 pediatrických pacientov infikovaných HIV-1 vo veku od
12 do menej ako 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení ART. Títo pacienti dostávali darunavir/ritonavir 800/100 mg jedenkrát denne spolu s inými antiretrovírusovými látkami. Virologická odpoveď bola definovaná ako pokles vírusového zaťaženia HIV-1 RNA
v plazme o najmenej 1,0 log10 oproti východiskovej hodnote.
DIONE
Výsledky v 48. týždni Darunavir/ritonavir
N = 12
HIV-1 RNA < 50 kópií/mla 83,3 % (10)
CD4+ percentuálna zmena oproti východiskovej hodnoteb
Priemerná zmena počtu buniek CD4+ oproti východiskovej hodnoteb
≥ 1,0 log10 pokles vírusového zaťaženia v plazme
oproti východiskovej hodnote
a Imputácie na základe algoritmu TLOVR.
14
221
100 %
b Pacienti, ktorí neukončili liečbu, sú počítaní ako zlyhanie: pacientom, ktorí prerušili liečbu predčasne, sa priradí
zmena rovná nule.
Pre ďalšie výsledky klinických štúdií u dospelých pacientov liečených ART a pediatrických pacientov,
pozri Súhrn charakteristických vlastností liekov darunavir 75 mg, 150 mg, 300 mg alebo 600 mg tablety.
Gravidita a obdobie po pôrode
Darunavir/ritonavir (600/100 mg dvakrát denne alebo 800/100 mg jedenkrát denne) v kombinácii so
základným režimom bol hodnotený v klinickej štúdii s 34 gravidnými ženami (17 v každej skupine) počas druhého a tretieho trimestra a po pôrode. Virologická odpoveď sa udržala počas trvania štúdie v oboch skupinách. U detí narodených 29 pacientkám, ktoré zotrvali na antivirotickej liečbe až do
pôrodu, sa nevyskytol prenos z matky na dieťa. Nevyskytli sa žiadne nové klinicky významné zistenia týkajúce sa bezpečnosti v porovnaní so známym profilom bezpečnosti darunaviru/ritonaviru
u dospelých infikovaných HIV-1 (pozri časti 4.2, 4.4 a 5.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti darunaviru podávaného v kombinácii s kobicistatom alebo s ritonavirom sa skúmali u zdravých dospelých dobrovoľníkov a u pacientov infikovaných vírusom HIV-1. Expozícia voči darunaviru u pacientov infikovaných vírusom HIV-1 bola vyššia ako u zdravých dobrovoľníkov. Vyššiu expozíciu voči darunaviru u pacientov infikovaných vírusom HIV-1
v porovnaní so zdravými dobrovoľníkmi je možné vysvetliť na základe vyšších koncentrácií α1- kyslého glykoproteínu (AAG) u pacientov infikovaných vírusom HIV-1, čo vedie k vyššej väzbe
darunaviru na plazmatický AAG, a tým aj k vyšším koncentráciám tejto látky v plazme.
Darunavir sa metabolizuje najmä prostredníctvom CYP3A. Kobicistat a ritonavir inhibujú CYP3A, v dôsledku čoho sa výrazne zvyšujú plazmatické koncentrácie darunaviru.
Informácie o farmakokinetických vlastnostiach kobicistatu si pozrite v Súhrne charakteristických vlastností kobicistatu.
Absorpcia
Darunavir sa rýchlo vstrebáva po perorálnom podaní. Pri podávaní v kombinácii s ritonavirom
v nízkej dávke sa maximálne plazmatické koncentrácie darunaviru obyčajne dosiahnu v priebehu 2,5 -
4 hodín.
Absolútna biologická dostupnosť po jednorazovom perorálnom podaní samotného darunaviru v dávke
600 mg bola približne 37 %, pričom pri podávaní ritonaviru v dávke 100 mg dvakrát denne sa zvýšila približne na 82 %. Po jednorazovom perorálnom podaní darunaviru v dávke 600 mg v kombinácii
s ritonavirom podávaným v dávke 100 mg dvakrát denne sa systémová expozícia voči darunaviru zvýšila 14-násobne (pozri časť 4.4).
Pri užívaní nalačno bola relatívna biologická dostupnosť darunaviru v prítomnosti kobicistatu alebo nízkej dávky ritonaviru nižšia ako pri užívaní lieku s jedlom. Z toho dôvodu sa tablety darunaviru musia užívať spolu s kobicistatom alebo ritonavirom a jedlom. Druh jedla nemá vplyv na expozíciu voči darunaviru.
Distribúcia
Približne 95 % darunaviru sa viaže na plazmatické bielkoviny, najmä na α1-kyslý glykoproteín.
Po intravenóznom podaní bol distribučný objem samotného darunaviru 88,1 ± 59,0 l (stredný ± SD)
a v prítomnosti 100 mg ritonaviru podávaného dvakrát denne bol zvýšený na 131 ± 49,9 l
(stredný ± SD).
Biotransformácia
In vitro štúdie využívajúce mikrozómy pečene človeka (HLM) naznačujú, že darunavir sa
metabolizuje najmä oxidatívnym metabolizmom. Darunavir sa výrazne metabolizuje prostredníctvom systému CYP v pečeni a takmer výlučne prostredníctvom izoenzýmu CYP3A4. V štúdii skúmajúcej aplikáciu darunaviru označeného rádioaktívnym 14C zdravým dobrovoľníkom sa zistilo, že väčšina rádioaktivity v plazme po jednorazovom podaní darunaviru s ritonavirom v dávke 400/100 mg pochádzala z pôvodného liečiva. U ľudí sa zistila prítomnosť najmenej 3 oxidatívnych metabolitov darunaviru, pričom všetky z nich mali aktivitu proti divokému typu HIV najmenej 10x nižšiu ako
darunavir.
Eliminácia
Po podaní darunaviru s ritonavirom v dávke 400/100 mg označeného rádioaktívnym 14C sa 79,5 %
rádioaktivity zachytilo v stolici a 13,9 % rádioaktivity sa zachytilo v moči. Rádioaktivita nezmeneného darunaviru v stolici predstavovala 41,2 % a v moči predstavovala 7,7 % z podanej dávky. Terminálny polčas eliminácie darunaviru podávaného v kombinácii s ritonavirom bol približne
15 hodín.
Klírens darunaviru (150 mg) podaného intravenózne v monoterapii bol 32,8 l/h, kým pri kombinácii s nízko dávkovaným ritonavirom 5,9 l/h.
Osobitné skupiny pacientov
Pediatrická populácia
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 74 predtým liečených pediatrických pacientov vo veku 6 až 17 rokov a s hmotnosťou najmenej 20 kg preukázala, že dávky
darunaviru/ritonaviru podávané v závislosti od hmotnosti, mali za následok expozíciu darunaviru
porovnateľnú s expozíciou u dospelých pacientov užívajúcich darunavir/ritonavir 600/100 mg dvakrát
denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom dvakrát denne u 14 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 15 kg až < 20 kg ukázala, že dávkovanie založené na hmotnosti viedlo k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne (pozri časť 4.2).
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 12 pediatrických pacientov vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg, ktorí neboli predtým liečení ART, ukázala, že dávka darunaviru/ritonaviru 800/100 mg jedenkrát denne viedla k expozícii darunaviru, ktorá bola porovnateľná s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne. Z toho dôvodu sa rovnaké dávkovanie jedenkrát
denne môže použiť u dospievajúcich vo veku 12 až < 18 rokov a s hmotnosťou najmenej 40 kg s predchádzajúcou liečbou bez mutácií spojených s rezistenciou voči darunaviru (DRV-RAM)* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+
≥ 100 buniek x 106/l (pozri časť 4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V
Farmakokinetika darunaviru užívaného spolu s ritonavirom jedenkrát denne u 10 predtým liečených pediatrických pacientov vo veku 3 až < 6 rokov a s hmotnosťou najmenej 14 kg až < 20 kg preukázala, že dávky odvodené od hmotnosti viedli k expozícii darunaviru, ktorá bola porovnateľná
s expozíciou získanou u dospelých užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne (pozri
časť 4.2). Farmakokinetické modelovanie a simulácia expozície darunaviru u pediatrických pacientov vo veku 3 až < 18 rokov navyše potvrdili expozície darunaviru, ktoré boli pozorované v klinických štúdiách a umožnili určenie dávkovania darunaviru/ritonaviru jedenkrát denne v závislosti na hmotnosti u pediatrických pacientov s hmotnosťou najmenej 15 kg, ktorí buď predtým nedostávali liečbu ART alebo u predtým liečených pediatrických pacientov bez DRV-RAM* a s plazmatickou hladinou HIV-1 RNA < 100 000 kópií/ml a počtom buniek CD4+ ≥ 100 buniek x 106/l (pozri časť
4.2).
* DRV-RAM: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V a L89V.
Starší ľudia
Pri analýze farmakokinetiky v rôznych populáciách pacientov s infekciou HIV sa zistilo, že farmakokinetika darunaviru sa výraznejšie nelíši u pacientov s infekciou HIV vo vekovom rozmedzí od 18 do 75 rokov (n = 12, vek ³ 65 rokov) (pozri časť 4.4). U pacientov starších ako 65 rokov boli
k dispozícii iba obmedzené údaje.
Pohlavie
Pri analýze farmakokinetiky v rôznych populáciách pacientov sa u žien s infekciou HIV zistila mierne
vyššia expozícia voči darunaviru (o 16,8 %) ako u mužov infikovaných HIV. Tento rozdiel nemá žiadny klinický význam.
Porucha funkcie obličiek
Výsledky štúdie zachovania hmotnosti s 14C rádioaktívne označeným darunavirom s ritonavirom ukázali, že približne 7,7 % z podanej dávky darunaviru sa vylučuje do moču v nezmenenej forme.
Aj keď sa podávanie darunaviru nesledovalo u pacientov s poruchou funkcie obličiek, pri analýze farmakokinetiky v rôznych populáciách pacientov sa zistilo, že stredne závažná porucha funkcie obličiek (klírens kreatinínu: 30 - 60 ml/min, n=20) nemá väčší vplyv na farmakoniketiku darunaviru u pacientov s infekciou HIV (pozri časti 4.2 a 4.4).
Porucha funkcie pečene
Darunavir sa metabolizuje a eliminuje prevažne v pečeni. V štúdii s opakovaným podaním lieku darunavir užívaný s ritonavirom (600/100 mg dvakrát denne) sa preukázalo, že celkové koncentrácie darunaviru v plazme u subjektov s miernou (Childova-Pughova trieda A, n=8) a stredne ťažkou (Childova-Pughova trieda B, n=8) poruchou funkcie pečene boli porovnateľné s tými u zdravých subjektov. Koncentrácie voľného darunaviru boli približne o 55 % (Childova-Pughova Trieda A)
a 100 % (Childova-Pughova trieda B) vyššie. Klinický význam tohto nárastu nie je známy, preto treba darunavir užívať so zvýšenou opatrnosťou. Účinok ťažkej poruchy funkcie pečene na farmakokinetiku darunaviru nebol dosiaľ študovaný (pozri časti 4.2, 4.3 a 4.4).
Gravidita a obdobie po pôrode
Expozícia celkovému darunaviru a ritonaviru po užití darunaviru/ritonaviru 600/100 mg dvakrát denne a darunaviru/ritonaviru 800/100 mg jedenkrát denne v rámci antivirotického režimu bola všeobecne nižšia počas gravidity v porovnaní s obdobím po pôrode. V prípade neviazaného (t.j. aktívneho) darunaviru však boli farmakokinetické parametre menej znížené počas gravidity v porovnaní
s obdobím po pôrode z dôvodu zvýšenia neviazanej frakcie darunaviru počas gravidity v porovnaní s obdobím po pôrode.
F
armakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru
600/100 mg dvakrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho trimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=11)aTretí trimester gravidity (n=11)Obdobie po pôrode (6-12 týždňov) (n=11)
Cmax, ng/ml 4 601 ± 1 125 5 111 ± 1 517 6 499 ± 2 411
AUC12h, ng.h/ml 38 950 ± 10 010 43 700 ± 16 400 55 300 ± 27 020
Cmin, ng/mlb 1 980 ± 839,9 2 498 ± 1 193 2 711 ± 2 268
a n=10 pre AUC12h
b vylúčením hodnoty Cmin pod LLOQ, n=10 pre referenciu
Farmakokinetické výsledky celkového darunaviru po podaní darunaviru/ritonaviru 800/100 mgjedenkrát denne v rámci antivirotického režimu, počas druhého trimestra gravidity, tretieho trimestra gravidity a po pôrode
trimestra gravidity a po pôrode
F
armakokinetika celkového darunaviru (priemer ± SD)
Druhý trimester gravidity (n=16)
Tretí trimester gravidity (n=14)
Obdobie po pôrode (6-12 týždňov) (n=15)
Cmax, ng/ml 4 988 ± 1 551 5 138 ± 1 243 7 445 ± 1 674
AUC12h, ng.h/ml 61 303 ± 16 232 60 439 ± 14 052 94 529 ± 28 572
Cmin, ng/mla 1 193 ± 509 1 098 ± 609 1 572 ± 1 108
a n=12 pre obdobie po pôrode, n=15 pre druhý trimester a n=14 pre tretí trimester
U žien užívajúcich darunavir/ritonavir 600/100 mg dvakrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 28 %, 24 %
a 17 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 19 %, 17 % nižšie a o 2 % vyššie, v tomto poradí, v porovnaní s obdobím po pôrode.
U žien užívajúcich darunavir/ritonavir 800/100 mg jedenkrát denne počas druhého trimestra gravidity
boli priemerné intra-individuálne hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 34 %, 34 % a 32 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode; počas tretieho trimestra gravidity boli hodnoty Cmax, AUC12h a Cmin celkového darunaviru o 31 %, 35 % a 50 % nižšie, v tomto poradí, v porovnaní s obdobím po pôrode.
5.3 Predklinické údaje o bezpečnosti
Toxikologické štúdie so samotným darunavirom sa vykonali v dávkovaní až do klinických hladín
u myší, potkanov a psov, kým štúdie s jeho kombináciou s ritonavirom sa vykonali u potkanov a psov.
V štúdiách toxicity po opakovanej dávke u myší, potkanov a psov sa zistili len obmedzené účinky liečby darunavirom. Identifikovaným cieľovým orgánom u hlodavcov bol hemopoetický systém, koagulačný systém, pečeň a štítna žľaza. Pozoroval sa premenlivý, ale mierny pokles parametrov v súvislosti s erytrocytmi spolu s predĺžením aktivovaného parciálneho tromboplastínového času.
Zmeny boli pozorované v pečeni (hypertrofia hepatocytov, vakuolizácia, zvýšenie hladiny pečeňových enzýmov) a štítnej žľaze (folikulárna hypertrofia). U laboratórnych potkanov viedla kombinácia darunaviru s ritonavirom k miernemu zvýšeniu účinku na parametre červených krviniek, pečene
a štítnu žľazu a k zvýšeniu incidencie ostrovčekov fibrózy v pankrease (iba u samcov laboratórnych potkanov) v porovnaní so samotným darunavirom. U psov sa nepozorovali žiadne väčšie známky
toxicity ani cieľové orgány toxicity pri rovnakej expozícii ako u človeka, ktorý užíva odporučené
dávky.
V štúdii na potkanoch bol znížený počet corpora lutea a implantácií v prípade maternálnej toxicity. Na druhej strane sa nepozorovali žiadne účinky na párenie alebo plodnosť po podávaní darunaviru
v dávkach do 1000 mg/kg/deň a pri expozícii (AUC - 0,5-násobok) nižšej ako u človeka dostávajúceho odporučené dávky. Pri podávaní rovnako veľkých dávok samotného darunaviru sa u potkanov
a králikov nepozorovala teratogenita podobne ako u myší dostávajúcich darunavir v kombinácii s ritonavirom. Stupeň expozície bol nižší ako u človeka dostávajúceho odporučené dávky. Pri hodnotení prenatálneho a postnatálneho vývoja u potkanov, darunavir podávaný samostatne alebo
v kombinácii s ritonavirom viedol k prechodnému zníženiu prírastku telesnej hmotnosti u dojčených mláďat a došlo k miernemu oneskoreniu v otvorení očí a uší. Darunavir v kombinácii s ritonavirom spôsobil zníženie prežívania mláďat, ktoré sa prejavilo nepriaznivou odpoveďou v 15. deň laktácie
a znížením počtu mláďat, ktoré prežili počas laktácie. Tieto účinky môžu byť prisúdené sekundárnej expozícii liečiva u mláďat prostredníctvom materského mlieka a/alebo maternálnej toxicity. Darunavir
podávaný ako samotný alebo v kombinácii s ritonavirom neovplyvnil žiadne funkcie po odstavení.
U juvenilných laboratórnych potkanov, ktorí dostávali darunavir 23. – 26. deň, sa pozorovala zvýšená
mortalita s kŕčmi u niektorých zvierat. Medzi 5. a 11. dňom života bola expozícia v plazme, pečeni
a mozgu výrazne vyššia ako u dospelých laboratórnych potkanov po porovnateľných dávkach
v mg/kg. Po 23. dni života bola expozícia porovnateľná s tou u dospelých potkanov. Zvýšená
expozícia bola pravdepodobne aspoň čiastočne z dôvodu nezrelosti enzýmov metabolizujúcich liek
u juvenilných zvierat. U juvenilných potkanov neboli spozorované žiadne úmrtia v súvislosti s liečbou
dávkami 1000 mg/kg darunaviru (jednorazová dávka) v 26. deň veku alebo 500 mg/kg (opakovaná dávka) od 23. do 50. dňa veku a expozícia a profil toxicity boli porovnateľné s tými, pozorovanými u dospelých potkanov.
Karcinogénny potenciál darunaviru bol vyhodnotený pri podávaní lieku myšiam a potkanom sondou do žalúdka počas 104 týždňov. Myšiam boli podávané denné dávky 150, 450 a 1000 mg/kg a potkanom boli podávané dávky 50, 150 a 500 mg/kg. Výskyt hepatocelulárnych adenómov a karcinómov stúpal v závislosti od dávky a pozoroval sa u samcov a samíc oboch živočíšnych druhov. U samcov potkanov sa vyskytli adenómy folikulárnych buniek štítnej žľazy. Podávanie darunaviru nespôsobilo štatisticky významný nárast iných benígnych alebo malígnych novotvarov u myší alebo potkanov. Pozorované hepatocelulárné tumory a tumory štítnej žľazy u hlodavcov sa považujú za obmedzené z hľadiska významnosti pre človeka. Opakované podávanie darunaviru potkanom spôsobilo indukciu mikrozomálnych enzýmov v pečeni a zvýšilo vylučovanie hormónov štítnej žľazy,
ktoré predurčuje potkanov, ale nie ľudí, na výskyt neoplaziem štítnej žľazy. Pri najvyšších testovaných dávkach boli systémové expozície (založené na AUC) darunaviru 0,4- až 0,7-násobkom (myši) a 0,7-
až 1-násobkom (potkany) v pomere k expozíciám pozorovaným u ľudí pri odporúčaných
terapeutických dávkach.
Po dvoch rokoch podávania darunaviru pri expozíciách na úrovni alebo pod úrovňou expozícií u ľudí sa pozorovali zmeny na obličkách u myší (nefróza) a u potkanov (chronická progresívna nefropatia).
Darunavir nebol mutagénny ani genotoxický v súbore in vitro a in vivo testov vrátane bakteriálnej reverznej mutácie (Amesov test), chromozómovej aberácie v ľudských lymfocytoch
a mikronukleového testu u myší in vivo.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Oxid kremičitý, koloidný bezvodý
Mikrokryštalická celulóza
Krospovidón
Sodná soľ glykolátu škrobu
Hypromelóza
Magnéziumstearát
Obal tablety
Polyvinylalkohol, čiastočne hydrolyzovaný
Oxid titaničitý
Makrogol
Mastenec
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky
Čas použiteľnosti po prvom otvorení HDPE fľaše: 90 dní.
6.4 Špeciálne upozornenia na uchovávanie
PVC/PE/PVDC-hliníkové blistrové balenie
Uchovávajte pri teplote neprevyšujúcej 25 °C.
Za studena spracované PVC/hliník/OPA-hliníkové blistrové balenie
Tento liek nevyžaduje žiadne osobitné podmienky na skladovanie.
HDPEfľaša
Tento liek nevyžaduje žiadne osobitné podmienky na skladovanie.
6.5 Druh obalu a obsah balenia
PVC/PE/PVDC-Al blistrové balenie s obsahom 30 tabliet a 30 x 1 tabliet.
Za studena spracované PVC/Al/OPA-Al blistrové balenie s obsahom 30 tabliet a 30 x 1 tabliet.
Balenie s HDPE fľašou s PP skrutkovacím uzáverom s obsahom 30, 60 a 90 tabliet.
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Mylan S.A.S.
117 Allee des Parcs
69 800 Saint Priest
Francúzsko
8. REGISTRAČNÉ ČÍSLO
EU/1/16/1140/039
EU/1/16/1140/040
EU/1/16/1140/041
EU/1/16/1140/042
EU/1/16/1140/043
EU/1/16/1140/044
EU/1/16/1140/045
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: [DD. mesiac RRRR]
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.