
minút. Nemá byť
podávaná ako intravenózny bolus alebo push. Na dosiahnutie potrebného trvania infúzie v dĺžke
približne 60 minút sa nemá prekročiť najvyššia rýchlosť infúzie 25 mg/min, naopak, trvanie infúzie sa
má predĺžiť. Počas podávania infúzie je potrebné pacienta monitorovať na prejavy reakcií súvisiacich s infúziou (pozri časť 4.4) a treba zabezpečiť dostupnosť vhodného resuscitačného vybavenia.
Pokyny na riedenie lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Cievne tromboembolické príhody
V klinických štúdiách boli hlásené závažné, niekedy smrteľné cievne tromboembolické príhody (ATE
- arterial thromboembolic events), vrátane infarktu myokardu, zástavy srdca, mozgovej mŕtvice a
mozgovej ischémie. Podávanie ramucirumabu sa má natrvalo prerušiť u pacientov, u ktorých sa vyskytli závažné ATE (pozri časť 4.2).
Gastrointestinálne perforácie
Ramucirumab je antiangiogénny liek a môže zvyšovať riziko gastrointestinálnych perforácií. Podávanie ramucirumabu sa má natrvalo prerušiť u pacientov, u ktorých sa objavila gastrointestinálna
perforácia (pozri časť 4.2).
Závažné krvácanie
Ramucirumab je antiangiogénny liek a môže zvyšovať riziko závažného krvácania. Podávanie
ramucirumabu sa má natrvalo prerušiť u pacientov, u ktorých sa vyskytlo krvácanie 3. alebo 4. stupňa (pozri časť 4.2). U pacientov s náchylnosťou na krvácanie a u pacientov na liečbe antikoagulantami alebo inými súčasne podávanými liekmi, ktoré zvyšujú riziko krvácania, sa má monitorovať krvný obraz a koagulačné parametre .
U pacientov s rakovinou žalúdka užívajúcich ramucirumab v kombinácii s paklitaxelom bolo hlásené závažné gastrointestinálne krvácanie vrátane smrteľných prípadov .
Reakcie súvisiace s infúziou
V klinických skúšaniach s ramucirumabom boli hlásené reakcie súvisiace s podaním infúzie . Väčšina prípadov sa vyskytla počas alebo po prvom alebo druhom podaní infúzie. V priebehu podávania
infúzie majú byť pacienti monitorovaní na prejavy precitlivenosti. K príznakom patrí
stuhnutosť/triaška, bolesť chrbta/kŕče, bolesť a/alebo stuhnutosť na hrudi, zimnica, návaly tepla, dýchavičnosť, chripot, hypoxia a parestézia. V závažných prípadoch príznaky zahŕňajú
bronchospazmus, supraventrikulárnu tachykardiu a hypotenziu. Podávanie ramucirumabu sa má
okamžite a natrvalo prerušiť u pacientov, u ktorých sa vyskytuje 3. alebo 4. stupeň IRR (pozri časť
4.2).
Hypertenzia
U pacientov užívajúcich ramucirumab bol v porovnaní s pacientmi užívajúcimi placebo hlásený zvýšený výskyt závažnej hypertenzie. Vo väčšine prípadov sa hypertenzia liečila pomocou štandardnej
antihypertenzívnej liečby. Pacienti s nekontrolovanou hypertenziou boli zo štúdií vylúčení: liečba
ramucirumabom sa u takýchto pacientov nemá začať, kým a pokiaľ ich preexistujúca hypertenzia nie je pod kontrolou. U pacientov, ktorí sa liečia ramucirumabom, sa má monitorovať krvný tlak. Podávanie ramucirumabu sa má v prípade závažnej hypertenzie dočasne prerušiť dovtedy, kým bude
kontrolovaná liečbou. V prípade, ak medicínsky závažná hypertenzia nemôže byť kontrolovaná
antihypertenzívnou liečbou, podávanie ramucirumabu sa má natrvalo prerušiť (pozri časť 4.2).
Poruchy hojenia rán
U pacientov so závažnými alebo zle sa hojacimi ranami nebol účinok ramucirumabu vyhodnocovaný. V štúdii so zvieratami ramucirumab nenarúšal hojenie rán. Keďže je však ramucirumab
antiangiogénna liečba a môže mať nežiaduce účinky pri hojení rán, najmenej 4 týždne pred
plánovanou operáciou sa liečba ramucirumabom nemá podávať. Rozhodnutie znovu začať s
podávaním ramucirumabu po chirurgickom zákroku sa má zakladať na klinickom posúdení vhodného hojenia rán.
Ak sa u pacienta počas liečby vyskytnú komplikácie s hojením rán, podávanie ramucirumabu sa má prerušiť dovtedy, kým sa rana úplne nezahojí (pozri časť 4.2).
Poruchyfunkciepečene
Pacientom so závažnou cirhózou pečene (Childovo-Pughovo skóre B alebo C), cirhózou s pečeňovou
encefalopatiou, klinicky závažným ascitom spôsobeným cirhózou alebo hepatorenálnym syndrómom sa má ramucirumab podávať opatrne. U týchto pacientov sa má ramucirumab používať iba v prípade, ak sa usúdi, že možný prínos liečby prevýši možné riziko progresívneho zlyhania pečene.
Fistula
Ak sa pacientom podáva Cyramza, môže sa u ich vyskytnúť zvýšené riziko vzniku fistuly. Podávanie
ramucirumabu sa má prerušiť u pacientov, u ktorých sa objavila fistula (pozri časť 4.2).
Proteinúria
Bola hlásená zvýšená incidencia proteinúrie u pacientov užívajúcich ramucirumab v porovnaní
s placebom. Pacienti majú byť počas liečby ramucirumabom monitorovaní na vznik alebo zhoršenie
proteinúrie. Ak bude bielkovina v moči na diagnostickom prúžku ≥2+, má sa vykonať 24-
hodinový zber moču. Ak bude hladina bielkovín v moči ≥2 g/24 hod., liečba ramucirumabom sa má dočasne prerušiť. Ak sa hladina bielkovín v moči vráti na <2 g/24 hod., má sa opäť začať s liečbou so zníženou úrovňou dávky (6 mg/kg každé 2 týždne). Druhé zníženie dávky (na 5 mg/kg každé 2 týždne) sa odporúča v prípade, ak sa znovu objaví hladina bielkovín v moči ≥2 g/24 hod. Ak bude hladina bielkovín v moči >3 g/24 hod. alebo v prípade výskytu nefrotického syndrómu , sa má liečba ramucirumabom natrvalo prerušiť.
Poruchyfunkcieobličiek
Nemáme k dispozícii žiadne údaje o bezpečnosti pre pacientov so závažnou poruchou funkcie obličiek
(odhadovaný klírens kreatinínu < 30 ml/min), ktorí boli liečení ramucirumabom (pozri časti 4.2 a 5.2).
Diéta so zníženým obsahom sodíka
Každá 10 ml injekčná liekovka obsahuje približne 17 mg sodíka a každá 50 ml injekčná liekovka
obsahuje približne 85 mg sodíka. Treba to vziať do úvahy u pacientov, ktorí majú diétu s obmedzeným obsahom sodíka.
4.5 Liekové a iné interakcie
Medzi ramucirumabom a paklitaxelom neboli pozorované žiadne liekové interakcie. Farmakokinetické vlastnosti paklitaxelu neboli ovplyvnené pri súbežnom podávaní s ramucirumabom, ani farmakokinetické vlastnosti ramucirumabu neboli ovplyvnené pri súbežnom podávaní s paklitaxelom.
4.6 Fertilita, gravidita a laktácia
Ženy v plodnom veku/ženská antikoncepcia
Ženám v plodnom veku je potrebné odporučiť, aby sa počas užívania Cyramzy vyhli otehotneniu
a informovať ich o možných rizikách pre graviditu a plod. Ženy v plodnom veku majú počas užívania
ramucirumabu a tri mesiace po užití poslednej dávky ramucirumabu používať účinnú antikoncepciu.
G
r
avidita
Nie sú známe žiadne údaje o podávaní ramucirumabu gravidným ženám. Pokiaľ ide o reprodukčnú toxicitu, sú štúdie na zvieratách nedostatočné (pozri časť 5.3). Pretože je angiogenéza pre udržanie gravidity a vývoj plodu dôležitá, inhibícia angiogenézy po podaní ramucirumabu môže mať za následok nežiaduce účinky na graviditu aj na plod. Cyramza sa má podávať iba vtedy, ak sa možný prínos liečby pre matku odôvodní oproti možnému riziku počas gravidity. Ak pacientka otehotnie počas liečby ramucirumabom, musí byť informovaná o možných rizikách pri udržaní gravidity a o riziku pre plod. Cyramza sa neodporúča počas gravidity ani na podávanie ženám v plodnom veku, ktoré nepoužívajú antikoncepciu.
Laktácia
Nie je známe, či sa ramucirumab vylučuje do ľudského mlieka. Očakáva sa, že vylučovanie do mlieka a perorálna absorpcia budú nízke. Keďže riziká pre novorodencov/dojčatá sa nedajú vylúčiť, počas
užívania Cyramzy a najmenej 3 mesiace po podaní poslednej dávky sa má laktácia prerušiť.
Fertilita
Nie sú dostupné žiadne informácie o účinku ramucirumabu na ľudskú fertilitu. Podľa štúdií na zvieratách môže byť ženská fertilita počas liečby ramucirumabom ohrozená (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Cyramza nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Ak sa u pacientov vyskytnú príznaky ovplyvňujúce ich schopnosť sústrediť sa a reagovať, odporúča sa, aby neviedli vozidlá a neobsluhovali stroje až do ústupu týchto účinkov.
4.8 Nežiaduce účinky
Zhrnutiebezpečnostnéhoprofilu
Najzávažnejšie nežiaduce reakcie súvisiace s liečbou ramucirumabom (v monoterapii alebo v kombinácii s cytotoxickou chemoterapiou) boli:
gastrointestinálna perforácia (pozri časť 4.4)
závažné gastrointestinálne krvácanie (pozri časť 4.4)
cievne tromboembolické príhody (pozri časť 4.4)
Najčastejšími nežiaducimi reakciami pozorovanými u pacientov liečených ramucirumabom sú:
únava/asténia, neutropénia, leukopénia, hnačka, krvácanie z nosa a hypertenzia.
Tabuľkový súhrn nežiaducich reakcií
Nežiaduce liekové reakcie (ADR), ktoré boli hlásené u pacientov s pokročilou rakovinou žalúdka, sú
uvedené nižšie podľa triedy orgánových systémov MedDRA, frekvencie výskytu a stupňa závažnosti. Na klasifikáciu frekvencie boli použité nasledujúc dohodnuté termíny:
veľmi časté (≥1/10)
časté (≥1/100 to <1/10)
menej časté (≥1/1000 - <1/100) zriedkavé (≥1/10 000 - <1/1000) veľmi zriedkavé (<1/10 000)
V rámci každej skupiny miery výskytu sú ADR uvedené v poradí podľa klesajúcej závažnosti.
Ramucirumab v kombinácii s paklitaxelom
Nasledujúca tabuľka uvádza mieru výskytu a závažnosti ADR (ADR - Adverse reactions = nežiaduce reakcie) na základe výsledkov štúdie RAINBOW, štúdie fázy 3, zameranej na dospelých pacientov
s pokročilou rakovinou žalúdka, randomizovaných na liečbu ramucirumabom v kombinácii s
paklitaxelom alebo na liečbu placebom a paklitaxelom.
T
abuľka č. 2: ADR hlásené u ≥ 5 % pacientov liečených ramucirumabom v štúdii RAINBOW
T
rieda orgánových systémov
|
Miera výskytu
|
ADR
|
C
yramza
a paklitaxel (N=327)
|
|
Placebo
a paklitaxel (N=329)
|
|
|
|
|
V
šetky
stupne toxicity (%)
|
T
oxicita
≥ 3. stupňa (%)
|
V
šetky
stupne toxicity (%)
|
T
oxicita
≥ 3. stupňa (%)
|
Poruchy krvi
a lymfatického systému
|
veľmi
časté
|
neutropénia
|
54,4
|
40,7
|
31,0
|
18,8
|
veľmi
časté
|
leukopénia
|
33,9
|
17,4
|
21,0
|
6,7
|
veľmi
časté
|
trombocytopénia
|
13,1
|
1,5
|
6,1
|
1,8
|
Poruchy
metabolizmu a výživy
|
veľmi
časté
|
hypoalbuminémia
|
11,0
|
1,2
|
4,9
|
0,9
|
Poruchy ciev
|
veľmi
časté
|
hypertenziaa
|
25,1
|
14,7
|
5,8
|
2,7
|
Poruchy dýchacej
sústavy, hrudníka a mediastína
|
veľmi
časté
|
epistaxa
|
30,6
|
0,0
|
7,0
|
0,0
|
Poruchy gastrointestinálneho traktu
|
veľmi
časté
|
prípady
gastrointestinálneho krvácaniab
|
10,1
|
3,7
|
6,1
|
1,5
|
veľmi
časté
|
stomatitída
|
19,6
|
0,6
|
7,3
|
0,6
|
veľmi
časté
|
hnačka
|
32,4
|
3,7
|
23,1
|
1,5
|
Poruchy obličiek a
močových ciest
|
veľmi
časté
|
proteinúria
|
16,8
|
1,2
|
6,1
|
0,0
|
Celkové poruchy a reakcie v mieste podania
|
veľmi
časté
|
únava/asténia
|
56,9
|
11,9
|
43,8
|
5,5
|
veľmi
časté
|
periférny edém
|
25,1
|
1,5
|
13,7
|
0,6
|
|
|
a Zahŕňa hypertenzívnu kardiomyopatiu.
b Zahŕňa tieto najvhodnejšie termíny MedDRA: análne krvácanie, krvácanie pri hnačke,
žalúdočné krvácanie, gastrointestinálne krvácanie, hematoméza, hematochézia, hemoroidálne krvácanie, Mallory-Weissov syndróm, meléna, ezofágové krvácanie, rektálne krvácanie a horné gastrointestinálne krvácanie.
Klinicky relevantné ADR hlásené u ≥ 1% a < 5 % pacientov liečených ramucirumabom a paklitaxelom v štúdii RAINBOW boli gastrointestinálna perforácia (1,2 % pri ramucirumabe a paklitaxeli v porovnaní s 0,3 % pri placebe a paklitaxeli) a sepsa (3,1 % pri ramucirumabe
a paklitaxeli v porovnaní s 1,8 % pri placebe a paklitaxeli).
Ramucirumab v monoterapiiNasledujúca tabuľka uvádza mieru výskytu a závažnosti ADR na základe štúdie REGARD, štúdie
fázy 3, zameranej na dospelých pacientov s pokročilou rakovinou žalúdka, randomizovaných na liečbu
ramucirumabom v monoterapii spolu s najlepšou podpornou liečbou (BSC) alebo na liečbu placebom
a BSC.
T
abuľka č. 3: ADR hlásené u ≥ 5 % pacientov liečených ramucirumabom v štúdii REGARD
T
rieda orgánových
systémov
|
Miera
výskytu
|
ADR
a,b
|
C
yramza
(
N=
236)
|
Placebo
(
N=
115)
|
V
šetky stupne
c
t
oxicity (%)
|
T
oxicita
3.-4. stupňa (%)
|
V
šetky
stupne toxicity (%)
|
T
oxicita
3.-4. stupňa (%)
|
Poruchy metabolizmu a výživy
|
časté
|
hypokalémiad
|
5,9
|
2,1
|
5,2
|
0,9
|
časté
|
hyponatrémia
|
5,5
|
3,4
|
1,7
|
0,9
|
Poruchy nervového
systému
|
časté
|
bolesť hlavy
|
9,3
|
0
|
3,5
|
0
|
Poruchy ciev
|
veľmi
časté
|
hypertenziae
|
16,1
|
7,6
|
7,8
|
2,6
|
Poruchy gastrointestinálneho traktu
|
veľmi
časté
|
bolesť
brucha
|
28,8
|
5,9
|
27,8
|
2,6
|
veľmi
časté
|
hnačka
|
14,4
|
0,8
|
8,7
|
1,7
|
|
|
f
a najvhodnejší termín MedDRA (verzia 15.0)
b U Cyramzy sa nevyskytli žiadne ADR 5. stupňa. Vyskytla sa iba jedna ADR hypokalémie 4. stupňa
a jedna ADR hyponatrémie 4. stupňa.
c Každý stupeň toxicity pozri v kritériách NCI CTCAE (verzia 4.0).
d Zahŕňa tieto najvhodnejšie termíny MedDRA: znížený obsah draslíka v krvi a hypokalémia.
e Zahŕňa tieto najvhodnejšie termíny MedDRA: zvýšený krvný tlak a hypertenzia.
f Zahŕňa tieto najvhodnejšie termíny MedDRA: bolesť brucha, bolesť v dolnej časti brucha, bolesť
v hornej časti brucha a bolesť v oblasti pečene.
Klinicky relevantné ADR hlásené u ≥ 1% a < 5% pacientov liečených ramucirumabom v štúdii REGARD boli: neutropénia, cievne tromboembolické príhody (pozri časti 4.2 a 4.4), intestinálna obštrukcia, krvácanie z nosa a vyrážka.
Klinicky relevantné reakcie (vrátane ≥ 3. stupňa) súvisiace s antiangiogenickou liečbou pozorované u pacientov liečených ramucirumabom naprieč klinickými skúšaniami boli: gastrointestinálne perforácie, reakcie spojené s podávaním infúzie a proteinúria (pozri časti 4.2 a 4.4).
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieK dispozícii nie sú žiadne informácie o predávkovaní u ľudí. Cyramza bola podávaná vo fáze 1 štúdie v dávke najviac 10 mg/kg každé dva týždne, bez dosiahnutia najvyššej tolerovanej dávky. V prípade predávkovania sa má použiť podporná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antineoplastické látky , monoklonálne protilátky, ATC kód: ešte
nestanovený
Mechanizmus účinku
Receptor VEGF-2 (VEGF = Vascular Endothelial Growth Factor) je kľúčovým sprostredkovateľom VEGF-indukovanej angiogenézy. Ramucirumab je ľudská protilátka zacielená na receptor, ktorá sa špecificky viaže na receptor VEGF-2 a blokuje väzby VEGF-A, VEGF-C a VEGF-D. Následkom toho ramucirumab inhibuje ligandom stimulovanú aktiváciu receptora VEGF-2 a jeho nadväzné signálne komponenty, vrátane p44/p42 mitogénom aktivovaných proteínkináz, neutralizuje ligandom indukovanú proliferáciu a migráciu ľudských endotelových buniek.
Farmakodynamické účinky
RAINBOW
Štúdie RAINBOW, celosvetového randomizovaného dvojito zaslepeného klinického skúšania
s Cyramzou a paklitaxelom v porovnaní s placebom a paklitaxelom, sa zúčastnilo 665 pacientov s lokálne relapsujúcou a neresekovateľnou alebo metastázujúcou rakovinou žalúdka (vrátane
adenokarcinómu GEJ ) po chemoterapii na báze platiny a fluoropyrimidínu, s antracyklínom alebo bez
neho. Primárnym koncovým ukazovateľom bolo celkové prežitie (OS) a sekundárne koncové ukazovatele zahŕňali prežitie bez progresie (PFS) a celkovú mieru odpovede (ORR). Pacienti museli mať progresiu ochorenia v priebehu prvej línie liečby alebo do 4 mesiacov po podaní poslednej dávky prvej línie liečby a spĺňať kritériá pre ECOG PS 0-1. Pacienti boli randomizovaní v pomere 1:1 a bola im podávaná Cyramza s paklitaxelom (n = 330) alebo placebo s paklitaxelom (n = 335). Randomizácia bola stratifikovaná podľa geografickej oblasti, času do progresie od začiatku prvej línie liečby (< 6 mesiacov oproti ≥ 6 mesiacom) a merateľnosti ochorenia. Cyramza v dávke 8 mg/kg alebo placebo
boli podávané vo forme intravenóznej infúzie každé 2 týždne (v 1. a 15. deň) 28-denného cyklu. Paklitaxel v dávke 80 mg/m2 bol podávaný vo forme intravenóznej infúzie 1., 8., a 15. deň každého
28-denného cyklu.
Väčšine (75 %) pacientov randomizovaných v tomto klinickom skúšaní bola predtým podávaná kombinovaná liečba s platinou a fluoropyrimidínom bez antracyklínu. Zvyšok pacientov (25 %) predtým užíval kombinovanú liečbu na báze platiny a fluoropyrimidínu s antracyklínom. Dve tretiny pacientov mali progresiu ochorenia ešte v priebehu liečby prvej línie (66,8 %). Vstupné demografické údaje pacientov a charakteristika ich ochorenia boli obvykle vyrovnané v oboch liečebných skupinách: stredný vek bol 61 rokov; 71 % pacientov boli muži; 61 % boli belosi, 35 % Ázijci; ECOG PS bolo
u 39 % pacientov 0, u 61 % pacientov bolo ECOG PS 1; 81 % pacientov malo merateľné ochorenie a 79 % malo rakovinu žalúdka; 21 % malo adenokarcinóm GEJ. U väčšiny pacientov (76 %) sa do 6 mesiacov od začiatku liečby prvej línie vyskytla progresia ochorenia. U pacientov užívajúcich Cyramzu s paklitaxelom bola stredná dĺžka liečby 19 týždňov a u pacientov užívajúcich placebo
s paklitaxelom bola stredná dĺžka liečby 12 týždňov. Stredná relatívna intenzita dávky Cyramzy bola
98,6 % a placeba 99,6 %. Stredná relatívna intenzita dávky paklitaxelu bola 87,7 % v liečebnej skupine s Cyramzou a paklitaxelom a 93,2 % v skupine s placebom a paklitaxelom. Liečbu kvôli nežiaducim udalostiam prerušilo podobné percento pacientov: 12 % pacientov užívajúcich Cyramzu s paklitaxelom, v porovnaní s 11 % pacientov užívajúcich placebo s paklitaxelom. Následná systémová protirakovinová liečba po prerušení liečby bola podaná 47,9 % pacientov, ktorí užívali Cyramzu s paklitaxelom a 46,0 % pacientov užívajúcich placebo s paklitaxelom.
Celkové prežitie sa štatisticky významne zlepšilo u pacientov užívajúcich Cyramzu s paklitaxelom v porovnaní s pacientmi užívajúcimi placebo s paklitaxelom (HR 0,807; 95 % CI: 0,678 - 0,962;
p = 0,0169). Stredné prežitie sa zvýšilo o 2,3 mesiacov v prospech skupiny s Cyramzou a
paklitaxelom: 9,63 mesiacov v skupine s Cyramzou a paklitaxelom a 7,36 mesiacov v skupine s placebom a paklitaxelom. Prežitie bez progresie sa štatisticky významne zlepšilo u pacientov užívajúcich Cyramzu s paklitaxelom v porovnaní s pacientmi užívajúcimi placebo s paklitaxelom (HR 0,635; 95 % CI: 0,536 - 0,752; p < 0,0001). Stredné PFS sa zvýšilo o 1,5 mesiaca v prospech
skupiny s Cyramzou a paklitaxelom: 4,4 mesiaca v skupine s Cyramzou a paklitaxelom a 2,9 mesiaca v skupine s placebom a paklitaxelom. Miera objektívnej odpovede (úplná odpoveď [CR] + čiastočná
odpoveď [PR]) sa významne zlepšili u pacientov užívajúcich Cyramzu s paklitaxelom v porovnaní
s pacientmi užívajúcimi placebo s paklitaxelom (pomer šancí [Odds ratio] 2,140; 95 % CI: 1,499 -
3,160; p = 0,0001). ORR v skupine s Cyramzou a paklitaxelom bola 27,9 % a v skupine s placebom a paklitaxelom bola 16,1 %. Zlepšenie OS a PFS boli konzistentne pozorované u predšpecifikovaných
podskupín rozdelených podľa veku, pohlavia, rasy a vo väčšine ďalších predšpecifikovaných podskupín. Výsledky účinnosti sú uvedené v tabuľke č. 4.
Tabuľka č. 4: Zhrnutie údajov o účinnosti – populácia ITT (Intent to Treat)
| Cyramza a paklitaxel
N=330
| Placebo a paklitaxel
N=335
|
Celkové prežitie, v mesiacoch
|
|
|
Stredná hodnota (95% CI)
| 9,6 (8,5; 10,8)
| 7,4 (6,3; 8,4)
|
Pomer rizík (95% CI)
| 0,807 (0,678; 0,962)
|
Stratifikovaná log-rank p-hodnota
| 0,0169
|
Prežitie bez progresie, v mesiacoch
|
|
|
Stredná hodnota (95% CI)
| 4,4 (4,2; 5,3)
| 2,9 (2,8; 3,0)
|
Pomer rizík (95% CI)
| 0,635 (0,536; 0,752)
|
Stratifikovaná log-rank p-hodnota
| <0,0001
|
Miera objektívnej odpovede (CR +PR)
|
|
|
Miera- v percentách (95% CI)
|
27,9 (23,3; 33,0)
| 16,1 (12,6; 20,4)
|
Pomer šancí
| 2,140 (1,449; 3,160)
|
Stratifikovaná CMH p-hodnota
| 0,0001
|
Skratky: CI = interval spoľahlivosti, CR= úplná odpoveď, PR= čiastočná odpoveď, CMH= Cochran-
Mantel-Haenszelov test
 Obrázok č. 1: Kaplan-Meierove krivky celkového prežitia pre Cyramzu s paklitaxelom v porovnaní s placebom a paklitaxelom v štúdii RAINBOW
Obrázok č. 1: Kaplan-Meierove krivky celkového prežitia pre Cyramzu s paklitaxelom v porovnaní s placebom a paklitaxelom v štúdii RAINBOW
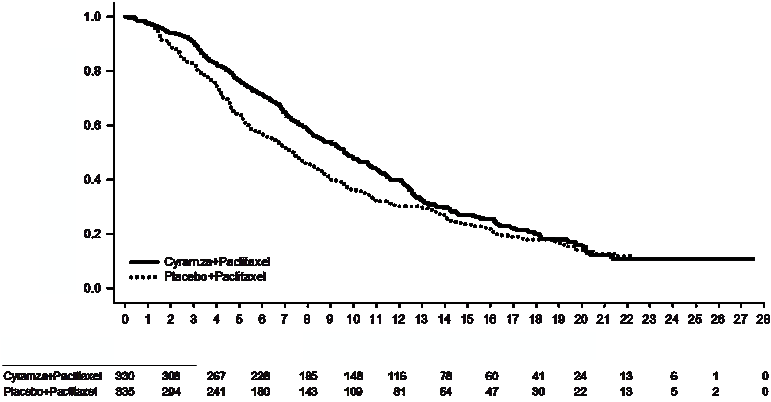
Počet pacientov na úrovni rizika
Čas od randomizácie (mesiace)
Cyramza+Paclitaxel 330
|
259
|
188
|
104
|
|
70
|
43
|
28
|
15
|
11
|
7
|
3
|
1
|
Placebo+Paclitaxel 335
|
214
|
124
|
50
|
|
34
|
21
|
12
|
8
|
5
|
3
|
3
|
3
|
REGARD

Štúdie REGARD, multinárodného randomizovaného dvojito zaslepeného klinického skúšania Cyramzy s najlepšou podpornou liečbou (BSC = best supportive care) v porovnaní s placebom a BSC, sa zúčastnilo 355 pacientov s lokálne relapsujúcou a neresekovateľnou alebo metastázujúcou rakovinou žalúdka (vrátane adenokarcinómu gastroezofageálneho spojenia [GEJ]) po chemoterapii na báze platiny alebo fluoropyrimidínu. Primárnym koncovým ukazovateľom bolo celkové prežitie (OS) a k sekundárnym koncovým ukazovateľom patrilo prežitie bez progresie (PFS) a 12-týždňová miera PFS. Zaradení pacienti museli mať progresiu ochorenia v priebehu prvej línie liečby alebo do
4 mesiacov od podania poslednej dávky prvej línie liečby metastázujúceho ochorenia, alebo počas adjuvantnej liečby alebo do 6 mesiacov od podania poslednej dávky adjuvantnej liečby a museli mať PS ECOG 0-1. Do klinického skúšania mohli byť zaradení iba tí pacienti, ktorí mali hodnoty celkového bilirubínu ≤ 1,5 mg/dl, AST a ALT ≤ 3-krát ULN alebo ≤ 5-krát ULN v prípade, ak boli prítomné aj metastázy v pečeni.
Pacienti boli randomizovaní v pomere 2:1 a každé dva týždne im bola podávaná intravenózna infúzia Cyramzy 8 mg/kg (n = 238) alebo placeba (n = 117). Randomizácia bola stratifikovaná podľa úbytku hmotnosti v priebehu prvých 3 mesiacov (≥ 10 % oproti < 10 %), geografickej oblasti a polohy primárneho nádoru (žalúdka oproti GEJ).
Vstupné demografické hodnoty a charakteristiky ochorenia boli vyvážené. ECOG PS bolo u 72 %
pacientov. Do štúdie REGARD neboli zaradení pacienti s Childovou-Pughovou cirhózou pečene stupňa B alebo C. 11 % pacientov užívajúcich Cyramzu a 6 % pacientov užívajúcich placebo ukončilo liečbu kvôli nežiaducim udalostiam. Celkové prežitie sa v porovnaní s pacientmi užívajúcimi placebo (pomer rizík [HR] 0,776; 95 % CI: 0,603 - 0,998; p = 0,0473) významne štatisticky zlepšilo
u pacientov užívajúcich Cyramzu , čo zodpovedá 22 % zníženiu rizika úmrtia a zvýšeniu stredného prežitia na 5,2 mesiaca pri Cyramze oproti 3,8 mesiaca pri placebe. Prežitie bez progresie sa štatisticky
významne zvýšilo u pacientov užívajúcich Cyramzu v porovnaní s pacientmi užívajúcim placebo
(HR 0,483; 95 % CI: 0,376 – 0,620; p <0,0001), čo zodpovedá 52 % zníženiu rizika progresie alebo úmrtia a zvýšeniu stredného PFS na 2,1 mesiaca pri Cyramze z 1,3 mesiaca pri placebe. Výsledky účinnosti sú uvedené v tabuľke č. 5.
 T
abuľka č. 5: Zhrnutie údajov o účinnosti – populácia Intent to Treat (ITT)
T
abuľka č. 5: Zhrnutie údajov o účinnosti – populácia Intent to Treat (ITT)
|
Cyramza
N=238
|
Placebo
N=117
|
Celkové prežitie, v mesiacoch
|
|
|
Stredná hodnota (95 % CI)
|
5,2 (4,4; 5,7)
|
3,8 (2,8; 4,7)
|
Pomer rizík (95 % CI)
|
0,776 (0,603; 0,998)
|
Stratifikovaná log-rank p-hodnota
|
0,0473
|
Prežitie bez progresie, v mesiacoch
|
|
|
Stredná hodnota (95 % CI)
|
2,1 (1,5; 2,7)
|
1,3 (1,3; 1,4)
|
Pomer rizík (95 % CI)
|
0,483 (0,376; 0,620)
|
Stratifikovaná log-rank p-hodnota
|
<0,0001
|
12-týždňová miera PFS v % (95 % CI)
|
40,1 (33,6; 46,4)
|
15,8 (9,7; 23,3)
|



Skratky: CI = interval spoľahlivosti

 Obrázok č. 3: Kaplan-Meierove krivky celkového prežitia s Cyramzou v porovnaní s placebom v štúdii REGARD
Obrázok č. 3: Kaplan-Meierove krivky celkového prežitia s Cyramzou v porovnaní s placebom v štúdii REGARD
1.0'
l a v
i
v r
Su ll a
r
e v
O f o
y t i
l
i
ab b o
r
P
|
|

0.8
0.6
0.4
Pacienti s výkonnostným stavom ECOG ≥2 (Eastern Cooperative Oncology Group)
Pacienti s ECOG skóre ≥2 boli z pivotných štúdií vyradení, a preto bezpečnosť a účinnosť Cyramzy
u tejto skupiny pacientov nie je známa.
Na základe obmedzených údajov pacientov s pozitívnym HER2 adenokarcinómom žalúdka alebo GEJ zo štúdie REGARD, ktorí boli pôvodne liečení trastuzumabom (RAINBOW) je nepravdepodobné, že Cyramza má nepriaznivý alebo žiadny účinok u pacientov s HER2-pozitívnou rakovinou žalúdka. Nerozvrstvená
post hoc analýza podskupiny pacientov zo štúdie RAINBOW, pôvodne liečených trastuzumabom (n = 39), naznačila prospech prežitia u týchto pacientov (HR 0,679, 95 % CI 0,327,
1,419) a vykázala prospech v prežití bez progresie (PFS) (HR 0,399, 95 % CI 0,194, 0,822).
ImunogenitaPacientom vo dvoch štúdiách fázy 3, RAINBOW a REGARD, bola v rôznom čase vyšetrená
prítomnosť protiliekových protilátok (ADA, anti-drug antibodies). Vyšetrovali sa vzorky 956 pacientov: 527 pacientov liečených ramucirumabom a 429 pacientov liečených kontrolným liekom. U jedenástich (2,2 %) pacientov liečených ramucirumabom a dvoch (0,5 %) pacientov liečených
kontrolným liekom sa vyskytli ADA. U žiadneho z pacientov s ADA sa neobjavila IRR. U žiadneho pacienta sa nevyskytli neutralizujúce protilátky proti ramucirumabu. Nemáme dostatok informácií na vyhodnotenie vplyvu ADA na účinnosť a bezpečnosť ramucirumabu.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Cyramzou vo všetkých podskupinách pediatrickej populácie pre schválenú indikáciu adenokarcinómu žalúdka (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Po dávkovacom režime 8 mg/kg každé 2 týždne, bol geometrický priemer ramucirumabu Cmin v sére pacientov s pokročilou rakovinou žalúdka 49,5 μg/ml (v rozsahu 6,3 - 228 μg/ml) a 74,4 μg/ml
(v rozsahu 13,8 - 234 μg/ml) pred podaním štvrtej a siedmej dávky ramucirumabu (v uvedenom poradí) podávaného ako jediná liečba.
Absorpcia
Cyramza sa podáva vo forme intravenóznej infúzie. Neuskutočnili sa žiadne štúdie s inými spôsobmi podania.
Distribúcia
Podľa populačného farmakokinetického prístupu PopPK (PopPK = Population pharmacokinetic approach) bol stredný objem distribúcie ramucirumabu v stabilnom stave 5,5 l.
Biotransformácia
Metabolizmus ramucirumabu nebol skúmaný. Protilátky sa vylučujú hlavne prostredníctvom katabolizmu.
Eliminácia
Podľa PopPK bol stredný klírens ramucirumabu 0,014l/hod a stredný polčas rozpadu bol 15 dní.
Závislosťodčasuadávkovania
Vo farmakokinetike ramucirumabu nebola nájdená jasná odchýlka od proporcionality dávky od
6 mg/kg do 20 mg/kg. Pri dávkovaní každé 2 týždne bol pozorovaný akumulačný pomer
ramucirumabu 1,5. Na základe simulácií použitím PopPK modelu, ustálený stav by sa dosiahol šiestou dávkou.
Starší pacienti
Podľa PopPK nebol medzi pacientmi ≥ 65 rokov a pacientmi < 65 rokov rozdiel v expozícii ramucirumabu.
Porucha funkcieobličiek
Na vyhodnotenie vplyvu poruchy obličiek na farmakokinetiku ramucirumabu sa neuskutočnili žiadne formálne štúdie. Podľa PopPK bola expozícia ramucirumabu podobná u pacientov so slabou poruchou funkcie obličiek (odhadovaný klírens kreatinínu [CrCl] ≥ 60 až < 90 ml/min) aj u pacientov s miernou poruchou funkcie obličiek (CrCl ≥ 30 až <60 ml/min) na rozdiel od pacientov s normálnou funkciou obličiek (CrCl ≥ 90 ml/min). O pacientoch so závažnou poruchou funkcie obličiek nemáme žiadne informácie (CrCl < 30 ml/min).
Porucha funkciepečene
Na vyhodnotenie vplyvu poruchy pečene na farmakokinetiku ramucirumabu sa neuskutočnili žiadne formálne štúdie. Podľa PopPK bola expozícia ramucirumabu u pacientov s miernou poruchou funkcie pečene (celkový bilirubín 1,0-1,5 horná hranica normálu (ULN) alebo AST > ULN, ako je to definované kritériami NCI) podobná expozícii u pacientov s normálnou funkciou pečene (celkový bilirubín a AST ≤ ULN). U pacientov so strednou až závažnou poruchou funkcie pečene (celkový bilirubín > 1,5 až ≤ 3,0 ULN a akákoľvek hodnota AST; a celkový bilirubín > 3,0 ULN a akákoľvek hodnota AST, v uvedenom poradí) nebol ramucirumab skúmaný .
O
statné zvláštne populácie
Podľa PopPK za zistilo, že nasledujúce spolučinitele nemajú vplyv na dostupnosť ramucirumabu: vek
(rozpätie 19 - 86 rokov), pohlavie (316 mužov, 181 žien), rasa (337 belochov a 139 aziatov), telesná
hmotnosť (rozpätie 31,9 - 133 kg), hladina albumínu (rozpätie 15,5 - 64,8 g/l).
Vzťahmedzi expozíciou a odpoveďou:RAINBOW
Analýza vzťahu expozície a odpovede ukázala, že účinnosť a osobitné bezpečnostné parametre korelovali s expozíciou ramucirumabu. Účinnosť vyjadrená pomocou vyššieho OS a PFS bola spojená
s narastajúcou expozíciou ramucirumabu podávaného v dávke 8 mg/kg v 1., 15. a 28. deň cyklu.
Výskyt hypertenzie 3. stupňa, neutropénie a leukopénie sa tiež zvyšoval s vyššou expozíciou ramucirumabu.
Vzťahmedzi expozíciou a odpoveďou:REGARD
Obmedzené PK údaje a analýza vzťahu expozície a odpovede naznačujú, že účinnosť ramucirumabu
súvisí s expozíciou ramucirumabu.
5.3 Predklinické údaje o bezpečnosti
Neuskutočnili sa žiadne štúdie na zvieratách zamerané na testovanie ramucirumabu na možnú karcinogenicitu alebo genotoxicitu.
Cieľovými orgánmi identifikovanými v štúdiách s opakovanými dávkami u makakov jávskych zameraných na toxicitu boli obličky (glomerulonefritída), kosti (zhrubnutie a abnormálna endochondrálna osifikácia epifyzeálnej rastovej platničky) a ženské reprodukčné orgány (zníženie hmotnosti vaječníkov a maternice). U niektorých orgánov bol pozorovaný minimálny stupeň zápalu a/alebo infiltrácia mononukleárnych buniek.
Neuskutočnili sa žiadne štúdie s ramucirumabom zamerané na reprodukčnú toxicitu, ale zvieracie modely spájajú angiogenézu, VEGF a VEGF receptor 2 s kritickými aspektmi ženskej reprodukcie, embryofetálnym a postnatálnym vývojom. Na základe mechanizmu účinku ramucirumabu je pravdepodobné, že u zvierat bude ramucirumab blokovať angiogenézu a mať za následok nežiaduce účinky na fertilitu (ovuláciu), vývoj placenty, fetálny a postnatálny vývoj.
Keď sa u opíc použil model incízie v rozsahu celej hrúbky kože, jednorazová dávka ramucirumabu nespôsobovala poruchu hojenia rán.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
histidín
monohydrát histidíniumchloridu chlorid sodný
glycín (E640)
polysorbát 80 (E433)
voda na injekciu
6.2 Inkompatibility
Cyramza sa nesmie podávať ani miešať s roztokmi dextrózy.
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
N
epoužitá injekčná
l
i
ekovka
3 roky
Po zriedení
Ak sa infúzny roztok Cyramzy pripraví podľa pokynov, neobsahuje žiadne antimikrobiálne
konzervačné látky.
Chemická a fyzikálna stabilita Cyramzy po otvorení v 9 mg/ml (0,9 %) injekčného roztoku chloridu sodného bola preukázaná počas: 24 hodín pri 2 ºC - 8 ºC alebo počas 4 hodín pri 25 ºC.
Z mikrobiologického hľadiska sa má liek podať okamžite. Ak sa nepodá okamžite, za čas skladovania
po jeho otvorení a podmienky pred použitím zodpovedá používateľ a tento čas by nemal byť dlhší ako
24 hodín pri teplote 2 ºC - 8 ºC pokiaľ sa riedenie neuskutočnilo v kontrolovaných a validovaných aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Skladujte v chladničke (2 ºC - 8 ºC ).
Neskladujte v mrazničke.
Na ochranu proti svetlu uchovávajte injekčnú liekovku vo vonkajšom obale . Podmienky na uchovávanie po zriedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
10 ml roztok v injekčnej liekovke (sklo typu I) s chlórbutylovým gumeným uzáverom, hliníkovým tesnením a polypropylénovým viečkom.
50 ml roztok v injekčnej liekovke (sklo typu I) s chlórbutylovým gumeným uzáverom, hliníkovým tesnením a polypropylénovým viečkom.
Balenie s 1 injekčnou liekovkou s obsahom 10 ml. Balenie s 2 injekčnými liekovkami s obsahom 10 ml. Balenie s 1 liekovkou s obsahom 50 ml.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Netraste injekčnou liekovkou.
Infúzny roztok pripravujte sterilným spôsobom, aby ste zaistili sterilnosť pripravovaného roztoku. Každá injekčná liekovka je určená iba na jednorazové použitie. Skontrolujte obsah injekčnej liekovky,
či v ňom nie sú častice alebo či pred riedením nezmenil farbu (infúzny koncentrát má byť číry až
mierne opalizujúci a bezfarebný až žltkastý bez viditeľných častíc). Ak sa objavia nejaké častice alebo
zafarbenie, injekčnú liekovku zlikvidujte.
Pred prípravou infúzneho roztoku vypočítajte potrebnú dávku a množstvo ramucirumabu. Injekčné liekovky obsahujú 100 mg alebo 500 mg roztoku ramucirumabu s koncentráciou 10 mg/ml. Ako riedidlo používajte iba 9 mg/ml (0,9%) injekčný roztok chloridu sodného.
V prípade, ak použijete vopred naplnenú nádobku s intravenóznou infúziou:
Na základe vypočítaného objemu ramucirumabu odoberte z 250 ml naplnenej nádobky s intravenóznou infúziou patričné množstvo 9 mg/ml (0,9%) injekčného roztoku chloridu sodného.
Sterilným spôsobom preneste vypočítaný objem ramucirumabu do nádobky na intravenóznu infúziu.
Celkové množstvo v nádobke má byť nakoniec 250 ml. Na zabezpečenie primeraného zmiešania sa
má nádobka jemne obracať. Infúzny roztok NEMRAZTE A NETRASTE ním. NERIEĎTE inými
roztokmi a nemiešajte s inými elektrolytmi ani liekmi.
V prípade, ak použijete nenaplnenú nádobku na infúzny roztokAsepticky preneste vypočítaný objem ramucirumabu do prázdnej nádob
ky na infúzny roztok. Do nádob
ky pridajte dostatočné množstvo 0,9 % (9 mg/ml) roztoku chloridu sodného na injekciu, tak aby
celkový objem infúzie bol 250 ml.. Na zabezpečenie primeraného zmiešania sa nádobka má jemne
obracať. Infúzny roztok NEMRAZTE A NETRASTE ním. NERIEĎTE inými roztokmi a nemiešajte s inými elektrolytmi ani liekmi.
Parenterálne lieky sa majú pred podaním vizuálne skontrolovať, či neobsahujú častice. Ak objavíte
nejaké častice, infúzny roztok zlikvidujte.
Zlikvidujte všetok nepoužitý ramucirumab, ktorý ostal v injekčnej liekovke, pretože liek neobsahuje žiadne antimikrobiálne konzervačné látky.
Podávajte pomocou infúznej pumpy. Na podanie infúzie sa má použiť osobitná infúzna hadička s
0,22-mikrónovým bielkoviny šetriacim filtrom a hadička sa má na konci infúzie prepláchnuť 9 mg/ml
(0,9 %) injekčným roztokom chloridu sodného.
Nepoužitý liek alebo odpad vzniknutý z lieku zlikvidujte podľa národných požiadaviek.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIEli Lilly Nederland B.V. Grootslag 1-5
NL-3991 RA, Houten
Holandsko
8. REGISTRAČNÉ ČÍSLOEU/1/14/957/001
EU/1/14/957/002
EU/1/14/957/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. decembra 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.