COSENTYX 300 MG INJEKČNÝ ROZTOK V NAPLNENEJ INJEKČNEJ STRIEKAČKE sol inj 1x2 ml/300 mg (striek.inj.skl.)

(21,0 %)
|
(41,6 %)
|
(20,2 %)
|
(39,2 %)
|
Odpoveď IGA mod 2011
|
6
|
125
|
160
|
142
|
180
|
101
|
148
|
„bez prejavov“ alebo
|
(2,40 %)
|
(51,2 %)*
|
(65,3 %)*
|
(58,2 %)
|
(73,5 %)
|
(41,4 %)
|
(60,4 %)
|
„takmer bez prejavov“,
* *n (%)
Skúšanie 3Počet pacientov 59 59 58 - - - -
Odpoveď PASI 50, n (%) 3 (5,1 %) 51
(86,4 %)
Odpoveď PASI 75, n (%) 0 (0,0 %) 41
(69,5 %)*
*
Odpoveď PASI 90, n (%) 0 (0,0 %) 27
(45,8 %)
51
(87,9 %)
44
(75,9 %)*
*
35
(60,3 %)
- - - -
- - - -
- - - -
Odpoveď PASI 100,
n (%)
Odpoveď IGA mod 2011
„bez prejavov“ alebo
„takmer bez prejavov“,
n (%)
Skúšanie 4
0 (0,0 %) 5
(8,5 %)
0 (0,0 %) 31
(52,5 %)*
*
25
(43,1 %)
40
(69,0 %)*
*
- - - -
- - - -
Počet pacientov 61 60 60 - - - -
Odpoveď PASI 50, n (%) 5 (8,2 %) 48
(80,0 %)
Odpoveď PASI 75, n (%) 2 (3,3 %) 43
(71,7 %)*
*
Odpoveď PASI 90, n (%) 0 (0,0 %) 24
(40,0 %)
Odpoveď PASI 100, n (%) 0 (0,0 %) 10
(16,7 %)
58
(96,7 %)
52
(86,7 %)*
*
33
(55,0 %)
16
(26,7 %)
- - - -
- - - -
- - - -
- - - -
Odpoveď IGA mod 2011
„bez prejavov“ alebo
„takmer bez prejavov“,
n (%)
0 (0,0 %) 32
(53,3 %)*
*
44
(73,3 %)*
*
- - - -
* IGA mod 2011 je stupnica s 5 kategóriami, a to „0 = bez prejavov“, „1 = takmer bez prejavov“, „2 = mierna“, „3 = stredne závažná“ alebo „4 = závažná“, ktoré udávajú celkové hodnotenie závažnosti psoriázy lekárom, so zameraním na stvrdnutie, sčervenanie a šupinatosť. Úspešnosť liečby „bez prejavov“ alebo
„takmer bez prejavov“ znamenala žiadne prejavy psoriázy alebo normálne až ružovo sfarbené lézie, žiadne zhrubnutie ložísk a žiadna až minimálna ložisková šupinatosť.

** hodnoty p v porovnaní s placebom a upravené pre multiplicitu: p<0,0001.
 T
a
b
u
ľ
k
a 4 Súhrn klinickej odpovede v psoriatickom skúšaní 2 (FIXTURE)
12. týždeň 16. týždeň 52. týždeň
T
a
b
u
ľ
k
a 4 Súhrn klinickej odpovede v psoriatickom skúšaní 2 (FIXTURE)
12. týždeň 16. týždeň 52. týždeň
|
P
l
acebo
|
150 mg
|
300 mg
|
E
t
anercept
|
150 mg
|
300 mg
|
E
t
anercept
|
150 mg
|
300 mg
|
E
t
anercept
|
Počet
|
324
|
327
|
323
|
323
|
327
|
323
|
323
|
327
|
323
|
323
|
pacientov
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
49
|
266
|
296
|
226
|
290
|
302
|
257 (79,6 %)
|
249
|
274
|
234 (72,4 %)
|
PASI 50,
|
(15,1 %)
|
(81,3 %)
|
(91,6 %)
|
(70,0 %)
|
(88,7 %)
|
(93,5 %)
|
|
(76,1 %)
|
(84,8 %)
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
16
|
219
|
249
|
142
|
247
|
280
|
189 (58,5 %)
|
215
|
254
|
179 (55,4 %)
|
PASI 75,
|
(4,9 %)
|
(67,0 %)
|
(77,1 %)
|
(44,0 %)
|
(75,5 %)
|
(86,7 %)
|
|
(65,7 %)
|
(78,6 %)
|
|
n (%)
|
|
**
|
**
|
|
|
|
|
|
|
|
Odpoveď
|
5
|
137
|
175
|
67
|
176
|
234
|
101 (31,3 %)
|
147
|
210
|
108 (33,4 %)
|
PASI 90,
|
(1,5 %)
|
(41,9 %)
|
(54,2 %)
|
(20,7 %)
|
(53,8 %)
|
(72,4 %)
|
|
(45,0 %)
|
(65,0 %)
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
0 (0 %)
|
47
|
78
|
14 (4,3 %)
|
84
|
119
|
24 (7,4 %)
|
65
|
117
|
32 (9,9 %)
|
PASI 100,
|
|
(14,4 %)
|
(24,1 %)
|
|
(25,7 %)
|
(36,8 %)
|
|
(19,9 %)
|
(36,2 %)
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
9
|
167
|
202
|
88
|
200
|
244
|
127 (39,3 %)
|
168
|
219
|
120 (37,2 %)
|
IGA mod
|
(2,8 %)
|
(51,1 %)
|
(62,5 %)
|
(27,2 %)
|
(61,2 %)
|
(75,5 %)
|
|
(51,4 %)
|
(67,8 %)
|
|
2011 „bez
|
|
**
|
**
|
|
|
|
|
|
|
|
prejavov“
|
|
|
|
|
|
|
|
|
|
|
alebo
|
|
|
|
|
|
|
|
|
|
|
„takmer bez
|
|
|
|
|
|
|
|
|
|
|
prejavov“,
|
|
|
|
|
|
|
|
|
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
** hodnoty p v porovnaní s etanerceptom: p=0,0250
V ďalšom klinickom skúšaní pri psoriáze (CLEAR) sa vyhodnotilo 676 pacientov. Dávka sekukinumabu 300 mg splnila primárne a sekundárne koncové ukazovatele preukázaním superiority voči ustekinumabu na základe odpovede PASI 90 v 16. týždni (primárny koncový ukazovateľ), rýchlosti nástupu odpovede PASI 75 vo 4. týždni a dlhodobou odpoveďou PASI 90 v 52. týždni oproti ustekinumabu. Vyššia účinnosť sekukinumabu v porovnaní s ustekinumabom pre koncové ukazovatele PASI 75/90/100 a IGA mod 2011 s odpoveďou 0 alebo 1 („bez prejavov“ alebo „takmer
bez prejavov“) sa pozorovala od začiatku a pretrvávala do 52. týždňa.
 Tabuľka 5 Súhrn klinickej odpovede v skúšaní CLEAR
Tabuľka 5 Súhrn klinickej odpovede v skúšaní CLEAR 4. týždeň 16. týždeň 52. týždeň
4. týždeň 16. týždeň 52. týždeň
s
e
ku
k
inumab
u
s
t
e
k
inumab* sekukinumab
u
s
t
e
k
inumab* sekukinumab
u
s
t
e
k
inumab*
300 mg 300 mg 300 mg
Počet pacientov 334 335 334 335 334 335
Odpoveď PASI
75, n (%)
166 (49,7 %)** 69 (20,6 %) 311 (93,1 %) 276 (82,4 %) 306 (91,6 %) 262 (78,2 %)
Odpoveď PASI
90, n (%)
70 (21,0 %) 18 (5,4 %) 264 (79,0 %)** 192 (57,3 %) 250
(74,9 %)***
203 (60,6%)
Odpoveď PASI
100, n (%) Odpoveď IGA mod 2011 „bez prejavov“alebo
„takmer bez prejavov“, n (%)
14 (4,2 %) 3 (0,9 %) 148 (44,3 %) 95 (28,4 %) 150 (44,9 %) 123 (36,7 %)
128 (38,3 %) 41 (12,2 %) 278 (83,2 %) 226 (67,5 %) 261 (78,1 %) 213 (63,6 %)

* Pacienti liečení sekukinumabom dostávali dávku 300 mg v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala rovnaká dávka každé 4 týždne do 52. týždňa. Pacienti liečení ustekinumabom dostávali dávku 45 mg alebo 90 mg v 0. a 4. týždni, potom každých 12 týždňov do 52. týždňa (dávkované podľa telesnej hmotnosti na základe schváleného dávkovania)
** hodnoty p v porovnaní s ustekinumabom: p<0,0001 pre primárny koncový ukazovateľ PASI 90 v 16. týždni a sekundárny
koncový ukazovateľ PASI 75 vo 4. týždni
*** hodnoty p v porovnaní s ustekinumabom: p=0,0001 pre sekundárny koncový ukazovateľ PASI 90 v 52. týždni
Sekukinumab bol účinný u pacientov, ktorí predtým nedostali systémovú liečbu, u pacientov predtým neliečených biologickými liekmi, u pacientov s expozíciou biologickým liekom/anti-TNF a u pacientov po zlyhaní liečby biologickými liekmi/anti-TNF. Zlepšenie PASI 75 u pacientov, ktorí mali pri zaradení do skúšania aj psoriatickú artritídu, bolo podobné ako u celkovej populácie s ložiskovou psoriázou.
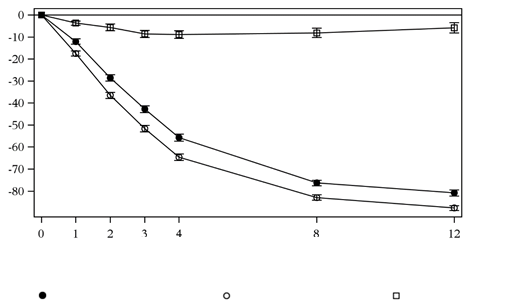
Sekukinumab mal rýchly nástup účinku so znížením strednej hodnoty PASI o 50 % do 3. týždňa pri
dávke 300 mg.
Obrázok 1 Časový priebeh percentuálnej zmeny stredného skóre PASI oproti východiskovej hodnote v skúšaní 1 (ERASURE)

Týždne liečby
N = počet vyhodnotiteľných pacientovsekukinumab 150 mg (n=243) sekukinumab 300 mg (n=245) Placebo (n=245)
Špecifické miesta/formy ložiskovej psoriázyV ďalších dvoch skúšaniach kontrolovaných placebom sa zaznamenalo zlepšenie pri psoriáze nechtov
(TRANSFIGURE, 198 pacientov), ako aj pri palmoplantárnej ložiskovej psoriáze (GESTURE,
205 pacientov). V skúšaní TRANSFIGURE bol v 16. týždni sekukinumab lepší ako placebo (46,1 %
pri 300 mg, 38,4 % pri 150 mg a 11,7 % pri placebe) pri hodnotení významného zlepšenia indexu závažnosti psoriázy nechtov (Nail Psoriasis Severity Index, NAPSI %) oproti východiskovým
hodnotám u pacientov so stredne ťažkou až ťažkou ložiskovou psoriázou s postihnutím nechtov. V
skúšaní GESTURE bol sekukinumab lepší ako placebo v 16. týždni (33,3 % pri 300 mg, 22,1 % pri
150 mg a 1,5 % pri placebe) pri hodnotení významného zlepšenia 0 alebo 1 odpovede pre ppIGA
(„bez prejavov“ alebo „takmer bez prejavov“) u pacientov so stredne ťažkou až ťažkou palmoplantárnou ložiskovou psoriázou.
V klinickom skúšaní kontrolovanom placebom bolo vyhodnotených 102 pacientov so stredne závažnou až závažnou psoriázou vlasatej časti hlavy, definovanou indexom závažnosti psoriázy vlasatej časti hlavy (Psoriasis Scalp Severity Index, PSSI) so skóre ≥12, odpoveďou IGA mod 2011 iba vlasatej časti hlavy so skóre 3 alebo vyšším a najmenej 30 % postihnutím povrchu vlasatej časti. Sekukinumab 300 mg bol v 12. týždni účinnejší ako placebo po 12. týždni, kedy boli vyhodnotené významné zlepšenia v oboch odpovediach PSSI 90 (52,9 % oproti 2,0 %) a IGA mod 2011 0 alebo 1 iba vo vlasatej časti hlavy (56,9 % oproti 5,9 %) oproti východiskovým hodnotám. Zlepšenie oboch koncových ukazovateľov pretrvávalo u pacientov, ktorí pokračovali v liečbe sekukinumabom, až do
24. týždňa.
Kvalita života/výsledky hlásené pacientmiŠtatisticky významné zlepšenie v 12. týždni (skúšania 1-4) oproti východiskovej hodnote v porovnaní s placebom sa prejavilo v DLQI (Dermatology Life Quality Index – Dermatologický index kvality života). Stredný pokles (zlepšenie) v DLQI oproti východiskovej hodnote v 12. týždni bol v rozmedzí
od -10,4 do -11,6 pri 300 mg sekukinumabu, od -7,7 do -10,1 pri 150 mg sekukinumabu, oproti -1,1
až -1,9 pri placebe. Toto zlepšenie pretrvalo 52 týždňov (skúšania 1 a 2).
V skúšaniach 1 a 2 vyplnilo 40 % účastníkov Denník psoriatických príznakov (Psoriasis Symptom Diary©). U účastníkov, ktorí v každom z týchto skúšaní vypĺňali denník, sa v 12. týždni prejavilo štatisticky významné zmiernenie svrbenia, bolesti a šupinatosti ako pacientmi hlásených prejavov
a príznakov oproti východiskovej hodnote v porovnaní s placebom.
Štatisticky významné zlepšenie po 4. týždni oproti východiskovej hodnote u pacientov liečených sekukinumabom v porovnaní s pacientmi liečenými ustekinumabom (CLEAR) sa prejavilo v DLQI a toto zlepšenie pretrvalo 52 týždňov.
Štatisticky významné zmiernenie svrbenia, bolesti a šupinatosti ako pacientmi hlásených prejavov
a príznakov sa preukázalo v Denníku psoriatických príznakov (Psoriasis Symptom Diary©)
po 16. týždni a 52. týždni (CLEAR) u pacientov liečených sekukinumabom v porovnaní s pacientmi
liečenými ustekinumabom.
Štatisticky významné zlepšenie (zníženie) v skúšaní pri psoriáze vlasatej časti hlavy sa preukázalo po 12. týždni pri pacientmi hlásených prejavoch a príznakoch svrbenia vlasatej časti hlavy, bolesti a šupinatosti oproti východiskovým hodnotám v porovnaní s placebom.
Pediatrická populácia
Ložiskovápsoriázau pediatrických pacientov
Preukázalo sa, že sekukinumab zlepšuje prejavy a príznaky, ako aj kvalitu života súvisiacu so zdravím
u pediatrických pacientov s ložiskovou psoriázou vo veku 6 rokov a starších (pozri Tabuľky 7 a 9).
Závažná ložisková psoriáza
Bezpečnosť a účinnosť sekukinumabu sa hodnotili v randomizovanom, dvojito zaslepenom klinickom skúšaní fázy III, kontrolovanom placebom a etanerceptom, u pediatrických pacientov so závažnou ložiskovou psoriázou vo veku 6 až <18 rokov, definovanou skóre PASI ≥20 a skóre IGA mod 2011 4
a rozsahom postihnutia kože s BSA ≥10%, ktorí boli kanditátmi na systémovú liečbu. Približne 43 % pacientov malo predchádzajúcu expozíciu fototerapii, 53 % konvenčnej systémovej terapii, 3 % biologickým látkam a 9 % malo sprievodnú psoriatickú artritídu.
V pediatrickom skúšaní 1 sa vyhodnotilo 162 pacientov, randomizovaných na podávanie nízkej dávky sekukinumabu (75 mg na telesnú hmotnosť <50 kg alebo 150 mg na telesnú hmotnosť ≥50 kg), vysokej dávky sekukinumabu (75 mg na telesnú hmotnosť <25 kg, 150 mg na telesnú hmotnosť medzi
≥25 kg a <50 kg, alebo 300 mg na telesnú hmotnosť ≥50 kg), alebo placeba v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala každé 4 týždne rovnaká dávka, alebo etanercept. Patienti radomizovaní na
etanercept dostávali 0,8 mg/kg týždenne (najviac však 50 mg). Rozdelenie pacientov podľa hmotnosti
a veku pri randomizácii je uvedené v Tabuľke 6.
T
a
b
u
ľ
k
a 6 Rozdelenie pacientov podľa hmotnosti a veku v pediatrickom skúšaní 1 pri psoriáze
R
a
n
d
o
m
i
z
a
č
n
á
v
r
stva
|
Popis
|
S
e
k
u
k
i
nu
m
a
b
n
í
z
k
a dávka n=40
|
S
e
k
u
k
i
nu
m
a
b
vy
soká dávka n=40
|
Placebo
n
=
4
1
|
E
t
a
n
e
r
ce
p
t
n
=
4
1
|
Sp
o
l
u
N
=
16
2
|
Vek
|
6-<12 rokov
|
8
|
9
|
10
|
10
|
37
|
≥12-
<18 rokov
|
32
|
31
|
31
|
31
|
125
|
Hmotnosť
|
<25 kg
|
2
|
3
|
3
|
4
|
12
|
≥25-<50 kg
|
17
|
15
|
17
|
16
|
65
|
≥50 kg
|
21
|
22
|
21
|
21
|
85
|
Pacienti randomizovaní na placebo, ktorí nedosiahli odpoveď do 12.týždňa prešli do skupiny s nízkou
alebo vysokou dávkou sekukinumabu (dávka na základe skupiny telesnej hmotnosti) a dostali skúšaný
liek v 12., 13., 14. a 15.týždni, po ktorom nasledovala každé 4 týždne rovnaká dávka, začínajúca
v 16.týždni. Spoločné primárne koncové ukazovatele boli podiely pacientov, ktorí dosiahli odpoveď
PASI 75 a IGA mod 2011 „bez prejavov“ alebo“takmer bez prejavov“ (0 alebo 1) v 12.týždni.
Účinnosť v oboch skupinách s nízkou a vysokou dávkou sekukinumabu počas placebom kontrolovaného obdobia do 12.týždňa bola porovnateľná pre spoločné primárne koncové ukazovatele. Odhadovaný pomer pravdepodobnosti v prospech oboch dávok sekukinumabu bol štatisticky významný pre odpovede PASI 75 a IGA mod 2011 0 alebo 1.
U všetkých pacientov sa sledovala účinnosť a bezpečnosť v priebehu 52 týždňov od prvého podania skúšaného lieku. Podiel pacientov, ktorí dosiahli PASI 75 a IGA mod 2011 „bez prejavov“ alebo
„takmer bez prejavov“ (0 alebo 1) preukázal rozdiel medzi skupinou liečenou so sekukinumabom
a skupinou s placebom pri prvej návšteve po východiskovej návšteve vo 4.týždni a výraznejší rozdiel nastal v 12.týždni. Odpoveď pretrvala počas 52-týždňového časového obdobia (pozri Tabuľku 7). Zlepšenie miery odpovede vyjadrené v hodnotách PASI 50, 90, 100 a Detského dermatologického
indexu kvality života (Children’s Dermatology Life Quality Index, (CDLQI)) zo skóre 0 alebo 1
taktiež pretrval počas 52-týždňového obdobia.
Naviac miera odpovede PASI 75, IGA 0 alebo 1, PASI 90 v 12. a 52. týždni pre obe skupiny s nízkou a vysokou dávkou sekukinumabu bola vyššia ako miera odpovede u pacientov liečených
s etanerceptom (pozri Tabuľku 7).
Po 12.týždni bola účinnosť oboch skupín s nízkou a vysokou dávkou sekukinumabu porovnateľná, hoci účinnosť s vyššou dávkou sekukinumabu bola vyššia u pacientov ≥50 kg. Profily bezpečnosti s nízkou dávkou a vysokou dávkou boli porovnateľné a v zhode s profilom bezpečnosti u dospelých pacientov.
T
a
b
u
ľ
k
a 7 Súhrn klinickej odpovede u pediatrických pacientov so závažnou psoriázou v 12. a 52.týždni (pediatrické skúšanie 1 pri psoriáze)*
K
r
i
t
ér
i
u
m odpovede
|
P
orovnanie liečby
|
„test“
|
„kontrola”
|
odhadovaný pomer pravdepodobnosti (95 % IS)
|
H
odnota p
|
„test“ vs. „kontrola“
|
n
**/m ( %)
|
n
**/m ( %)
|
v 12.týždni***
|
P
A
S
I 75
|
sekukinumab nízka dávka vs. placebo
sekukinumab vysoká dávka vs. placebo
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
|
32/40 (80,0)
31/40 (77,5)
32/40 (80,0)
31/40 (77,5)
|
6/41 (14,6)
6/41 (14,6)
26/41 (63,4)
26/41 (63,4)
|
25,78 (7,08; 114,66)
22,65 (6,31; 98,93)
2,25 (0,73; 7,38)
1,92 (0,64; 6,07)
|
<0,0001
<0,0001
|
I
G
A 0/1
|
sekukinumab nízka dávka vs. placebo
sekukinumab vysoká dávka vs. placebo
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
|
28/40 (70,0)
24/40 (60,0)
28/40 (70,0)
24/40 (60,0)
|
2/41 (4,9)
2/41 (4,9)
14/41 (34,1)
14/41 (34,1)
|
51,77 (10,02; 538,64)
32,52 (6,48; 329,52)
4,49 (1,60; 13,42)
2,86 (1,05; 8,13)
|
<0,0001
<0,0001
|
P
A
S
I 90
|
sekukinumab nízka dávka vs. placebo
sekukinumab vysoká dávka vs. placebo
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
|
29/40 (72,5)
27/40 (67,5)
29/40 (72,5)
27/40 (67,5)
|
1/41 (2,4)
1/41 (2,4)
12/41 (29,3)
12/41 (29,3)
|
133,67 (16,83; 6395,22)
102,86 (13,22; 4850,13)
7,03 (2,34; 23,19)
5,2 (1,82; 16,75)
|
<0,0001
<0,0001
|
v 52. týždni
|
P
A
S
I 75
|
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs.
etanercept
|
35/40 (87,5)
35/40 (87,5)
|
28/41 (68,3)
28/41 (68,3)
|
3,12 (0,91; 12,52)
3,09 (0,90; 12,39)
|
|
I
G
A 0/1
|
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs.
etanercept
|
29/40 (72,5)
30/40 (75,0)
|
23/41 (56,1)
23/41 (56,1)
|
2,02 (0,73; 5,77)
2,26 (0,81; 6,62)
|
|
P
A
S
I 90
|
sekukinumab nízka dávka vs.
etanercept
sekukinumab vysoká dávka vs. etanercept
|
30/40 (75,0)
32/40 (80,0)
|
21/41 (51,2)
21/41 (51,2)
|
2,85 (1,02; 8,38)
3,69 (1,27; 11,61)
|
|
* chýbajúce údaje boli vyhodnotené ako bez odpovede
** n je počet pacientov s odpoveďou, m = počet pacientov s hodnotiteľnou odpoveďou
*** rozšírené okno návštevy v 12.týždni
Pomer pravdepodobnosti, 95 % interval spoľahlivosti a hodnota p sú z presného modelu logistickej regresie s faktormi liečebná skupina, kategóriou východiskovej telesnej hmostnosti a vekovou kategóriou
|
Zlepšenie kvality života súvisiacej so zdravím meranej ako skóre CDLQI v hodnote 0 alebo 1
v porovnaní s placebom v 12.týždni hlásil vyšší podiel pediatrických pacientov liečených
sekukinumabom (nízka dávka 44,7 %, vysoká dávka 50 %, placebo 15 %). V čase až do 52.týždňa
v oboch skupinách s dávkou sekukinumabu bol počet pacientov s odpoveďou numericky vyšší ako
v skupine s etanerceptom (nízka dávka 60,6 %, vysoká dávka 66,7 %, etanercept 44,4 %).
Stredne závažná až závažná ložisková psoriázaNa základe preukázanej miery účinnosti a vzťahu odpovede na expozíciu u dospelých pacientov so stredne závažnou až závažnou ložiskovou psoriázou a podobnosti priebehu ochorenia, patofyziológie a účinku lieku u dospelých a pediatrických pacientov s rovnakými expozičnými hladinami sa
predpokladalo, že sekukinumab bude účinný aj pri liečbe pediatrických pacientov so stredne závažnou až závažnou ložiskovou psoriázou.
Okrem toho sa bezpečnosť a účinnosť sekukinumabu hodnotila v otvorenom multicentrickom skúšaní fázy III s dvomi ramenami a paralelnou skupinou u pediatrických pacientov so stredne závažnou až závažnou ložiskovou psoriázou vo veku 6 až <18 rokov, definovanej skóre PASI s ≥12, skóre IGA mod 2011 of ≥3 a postihnutím kože s BSA ≥10%, ktorí boli kandidátmi na systémovú liečbu.
V pediatrickom skúšaní 2 pri psoriáze sa vyhodnotilo 84 pacientov, randomizovaných na podávanie nízkej dávky sekukinumabu (75 mg na telesnú hmotnosť <50 kg alebo 150 mg na telesnú hmotnosť
≥50 kg) alebo vysokej dávky sekukinumabu (75 mg na telesnú hmotnosť <25 kg, 150 mg na telesnú
hmotnosť medzi ≥25 kg a <50 kg, alebo 300 mg na telesnú hmotnosť ≥50 kg) v 0., 1., 2., 3. a
4. týždni, po ktorých nasledovala každé 4 týždne rovnaká dávka. Rozdelenie pacientov podľa
hmotnosti a veku pri randomizácii je uvedené v Tabuľke 8.
Tabuľka 8 Rozdelenie pacientov podľa hmotnosti a veku v pediatrickom skúšaní 2 pri psoriázePodskupina
| Popis
| Sekukinumab nízka dávka n=42
| Sekukinumab vysoká dávka n=42
| Spolu
N=84
|
Vek
| 6-<12 rokov
| 17
| 16
| 33
|
≥12-<18 rokov
| 25
| 26
| 51
|
Hmotnosť
| <25 kg
| 4
| 4
| 8
|
≥25-<50 kg
| 13
| 12
| 25
|
≥50 kg
| 25
| 26
| 51
|
Spoločnými primárnymi koncovými ukazovateľmi boli podiely pacientov, ktorí dosiahli odpoveď
PASI 75 a IGA mod 2011 „bez prejavov“ alebo“takmer bez prejavov“ (0 alebo 1) v 12.týždni.
Účinnosť v oboch skupinách s nízkou a vysokou dávkou sekukinumabu bola porovnateľná
a preukázala štatistické zlepšenie v porovnaní s predchádzajúcim placebom v primárnych aj sekundárnych koncových ukazovateľoch. Odhadovaná posteriórna pravdepodobnosť pozitívneho liečebného účinku bola 100 %.
U všetkých pacientov sa sledovala účinnosť najmenej 24 týždňov po prvom podaní lieku (pozri
Tabuľku 9). Účinnosť (definovaná odpoveďou PASI 75 a IGA mod 2011 „bez prejavov“ alebo
„takmer bez prejavov“ [0 alebo 1]) sa pozorovala už pri prvej návšteve po východiskovej návšteve
v 2.týždni a podiel pacientov, ktorí dosiahli odpoveď PASI 75 a IGA mod 2011 „bez prejavov“ alebo
„takmer bez prejavov“ (0 alebo 1) sa zvýšil počas obdobia 24 týždňov.
Po 12 .týždni bola účinnosť oboch skupín s nízkou a vysokou dávkou sekukinumabu porovnateľná
. Profily bezpečnosti s nízkou dávkou a vysokou dávkou boli porovnateľné a v zhode s profilom bezpečnosti u dospelých pacientov.
Tabuľka 9 Súhrn klinickej odpovede v pediatrickom skúšaní so stredne závažnou až závažnoupsoriázou v 12.a 24.týždni (pediatrické skúšanie pri psoriáze 2)*
| 12 .týždeň
| 24 .týždeň
|
Sekukinumab nízka dávka
| Sekukinumab vysoká dávka
| Sekukinumab nízka dávka
| Sekukinumab vysoká dávka
|
Počet pacientov
| 42
| 42
| 42
| 42
|
Odpoveď PASI 75, n (%)
| 39 (92,9 %)
| 39 (92,9 %)
| 40 (95,2 %)
| 40 (95,2 %)
|
Odpoveď IGA mod 2011 „bez prejavov“ alebo “takmer bez prejavov“, n (%)
| 33 (78,6 %)
| 35 (83,3 %)
| 37 (88,1 %)
| 39 (92,9 %)
|
Odpoveď PASI 90, n (%)
| 29 (69 %)
| 32 (76,2 %)
| 37 (88,1 %)
| 37 (88,1 %)
|
Odpoveď PASI 100, n (%)
| 25 (59,5 %)
| 23 (54,8 %)
| 28 (66,7 %)
| 28 (66,7 %)
|
* chýbajúce údaje boli vyhodnotené ako bez odpovede
|
Tieto výsledky v pediatrickej populácii so stredne závažnou až závažnou ložiskovou psoriázou potvrdili vyššie uvedené prediktívne predpoklady, založené na základe účinnosti a odpovede na liečbu u dospelých pacientov, ktoré sú uvedené vyššie.
V skupine s nízkou dávkou dosiahlo 50 % a 70,7 % pacientov skóre CDLQI 0 alebo 1 v 12.a 24.týždni
v uvedenom poradí. V skupine s vysokou dávkou dosiahlo 61,9 % a 60,5 % pacientov skóre 0 alebo 1
v 12.a 24.týždni v uvedenom poradí.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Cosentyxom pre liečbu ložiskovej psoriázy u pediatrických pacientov od narodenia do menej ako 6 rokov a pre liečbu chronickej idiopatickej artritídy u pediatrických pacientov od narodenia do menej ako 2 rokov (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Cosentyxom pre
liečbu chronickej idiopatickej artritídy u pediatrických pacientov vo veku od 2 rokov do menej ako
18 rokov (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Väčšina farmakokinetických vlastností pozorovaných u pacientov s ložiskovou psoriázou, psoriatickou
artritídou a ankylozujúcou spondylitídou bola podobná.
Pediatrická populácia
V skupine dvoch pediatrických skúšaní pacienti so stredne závažnou až závažnou ložiskovou
psoriázou (vo veku 6 až menej ako 18 rokov) dostávali sekukinumab podľa odporúčaného pediatrického dávkovacieho režimu. Pacienti s hmotnosťou ≥25 a <50 kg mali v 24. týždni priemernú minimálnu koncentráciu v rovnovážnom stave (priemerné ± SD) v hodnote 19,8 ± 6,96 µg/ml (n=24)
po dávke 75 mg sekukinumabu a pacienti s hmotnosťou ≥50 kg mali priemernú minimálnu
koncentráciu v rovnovážnom stave 27,3 ± 10,1 µg/ml (n=36) po dávke 150 mg sekukinumabu. Priemerná minimálna koncentrácia v rovnovážnom stave (priemerné ± SD) u pacientov s hmotnosťou
<25 kg (n=8) v 24. týždni bola 32,6 ± 10,8 µg/ml po dávke 75 mg sekukinumabu.
Populácia dospelých
Absorpcia
Po jednorazovej subkutánnej dávke 300 mg v tekutej liekovej forme sa u zdravých dobrovoľníkov dosiahli maximálne koncentrácie sekukinumabu v sére 43,2±10,4 μg/ml medzi 2. a 14. dňom po podaní.
Podľa analýzy farmakokinetiky u populácie sa po jednorazovom subkutánnom podaní pacientom s ložiskovou psoriázou maximálne koncentrácie sekukinumabu v sére 13,7±4,8 µg/ml pri dávke
150 mg alebo 27,3±9,5 µg/ml pri dávke 300 mg dosiahli medzi 5. a 6. dňom po podaní.
Podľa analýzy farmakokinetiky u populácie bol po začiatočnom týždennom podávaní počas prvého mesiaca čas do dosiahnutia maximálnej koncentrácie medzi 31. a 34. dňom.
Podľa simulovaných údajov boli maximálne koncentrácie v rovnovážnom stave (Cmax,ss) po subkutánnom podaní 27,6 µg/ml pri 150 mg a 55,2 µg/ml pri 300 mg. Analýza farmakokinetiky u populácie naznačuje, že rovnovážny stav sa dosahuje pri režimoch mesačného podávania po
20 týždňoch.
V porovnaní s expozíciou po jednorazovej dávke ukázala analýza farmakokinetiky u populácie, že pacienti vykazovali po opakovanom mesačnom podávaní počas udržiavacej liečby zvýšenie maximálnych koncentrácií v sére a plochy pod krivkou (AUC) na dvojnásobok.
Analýza farmakokinetiky u populácie ukázala, že sekukinumab sa u pacientov s ložiskovou psoriázou absorboval s priemernou absolútnou biologickou dostupnosťou 73 %. Vo všetkých skúšaniach sa vypočítala absolútna biologická dostupnosť v rozmedzí od 60 do 77 %.
Biologická dostupnosť sekukinumabu u pacientov so PsA bola 85 % na základe farmakokinetického modelu populácie.


Po jednorazovej subkutánnej injekcii 300 mg injekčného roztoku v naplnenej injekčnej striekačke u pacientov s ložiskovou psoriázou bola systémová expozícia sekukinumabu podobná tej, ktorá sa predtým pozorovala pri dvoch injekciách po 150 mg.
DistribúciaStredný objem distribúcie počas koncovej fázy (Vz) po jednorazovom intravenóznom podaní u pacientov s ložiskovou psoriázou bol v rozmedzí od 7,10 do 8,60 litrov, čo naznačuje, že
sekukinumab sa len obmedzene distribuuje do periférnych kompartmentov.
BiotransformáciaVäčšina eliminácie IgG sa uskutočňuje prostredníctvom intracelulárneho katabolizmu po endocytóze z
tekutej fázy alebo sprostredkovanej receptorom.
ElimináciaStredný systémový klírens (CL) po jednorazovom intravenóznom podaní pacientom s ložiskovou
psoriázou bol v rozmedzí od 0,13 do 0,36 l/deň. V analýze farmakokinetiky bola stredná hodnota
systémového klírensu (CL) u populácie pacientov s ložiskovou psoriázou 0,19 l/deň. CL nebol ovplyvnený pohlavím. Klírens nezávisel od dávky a času.
Stredný polčas eliminácie bol podľa odhadu analýzy farmakokinetiky u populácie pacientov
s ložiskovou psoriázou 27 dní, s rozmedzím od 18 do 46 dní, vo všetkých psoriatických skúšaniach
s intravenóznym podávaním.
Linearita/nelinearitaFarmakokinetika sekukinumabu po jednorazovom a opakovanom podávaní pacientom s ložiskovou
psoriázou sa stanovila v niekoľkých skúšaniach s intravenózne podanými dávkami v rozmedzí od
1x 0,3 mg/kg do 3x 10 mg/kg a so subkutánne podanými dávkami v rozmedzí od 1x 25 mg po opakované dávky 300 mg. Expozícia bola úmerná dávke pri všetkých dávkovacích režimoch.
Osobitné populáciePacienti s poruchoufunkcieobličiekalebopečeneNie sú dostupné farmakokinetické údaje o pacientoch s poruchou funkcie obličiek alebo pečene. Predpokladá sa, že pri intaktnom sekukinumabe, monoklonálnej protilátke IgG, je eliminácia obličkami nízka a málo významná. IgG sa eliminujú hlavne prostredníctvom katabolizmu
a nepredpokladá sa, že porucha funkcie pečene má vplyv na klírens sekukinumabu.
Vplyv telesnej hmotnosti na farmakokinetikuKlírens a distribučný objem sekukinumabu sa zvyšujú so zvyšujúcou sa telesnou hmotnosťou.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a reprodukčnej toxicity, alebo pri testovaní skríženej reaktivity tkanív, neodhalili žiadne osobitné riziko pre ľudí (dospelých a dospievajúcich).
Štúdie na zvieratách na vyhodnotenie karcinogénneho potenciálu sekukinumabu sa nevykonali.
6
. FARMACEUTICKÉ INFORMÁCIE
6
.
1 Zoznam pomocných látok
dihydrát trehalózy histidín
monohydrát histidínium-chloridu metionín
polysorbát 80
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
18 mesiacov
Ak je to nevyhnutné, Cosentyx sa môže jedenkrát uchovávať mimo chladničky pri izbovej teplote neprevyšujúcej 30°C, nie viac ako 4 dni.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Cosentyx 75 mg injekčný roztok v naplnenej injekčnej striekačke sa dodáva v 0,5 ml naplnenej sklenenej injekčnej striekačke s brómbutylovou gumenou piestovou zátkou potiahnutou silikónom, nasadenou ihlou 27G x ½″ a pevným krytom ihly zo styrén-butadiénového kaučuku, spojeným s automatickým chráničom ihly z polykarbonátu.
Cosentyx 75 mg injekčný roztok v naplnenej injekčnej striekačke je dostupný v jednotlivých baleniach obsahujúcich 1 naplnenú injekčnú striekačku a v multibaleniach obsahujúcich 3 (3 balenia po 1) naplnené injekčné striekačky.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Cosentyx 75 mg injekčný roztok sa dodáva v naplnenej injekčnej striekačke na jednorazové individuálne použitie. Injekčná striekačka sa má vybrať z chladničky 20 minút pred podaním, aby sa mohla ohriať na izbovú teplotu.
Pred použitím sa odporúča vizuálna kontrola naplnenej injekčnej striekačky. Tekutina má byť číra. Jej farba sa môže meniť od bezfarebnej po bledožltú. Môže sa v nej objaviť malá vzduchová bublina, čo je normálne. Nepoužite tekutinu, ak obsahuje ľahko viditeľné častice, ak je zakalená alebo výrazne hnedá.
Podrobný návod na použitie je uvedený v písomnej informácii pre používateľa.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7
. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLAEU/1/14/980/012-013
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 15.január 2015
Dátum posledného predĺženia registrácie: 03. september 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1
. NÁZOV LIEKU
Cosentyx 150 mg injekčný roztok v naplnenej injekčnej striekačke Cosentyx 300 mg injekčný roztok v naplnenej injekčnej striekačke Cosentyx 150 mg injekčný roztok v naplnenom pere
Cosentyx 300 mg injekčný roztok v naplnenom pere
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIECosentyx 150 mg injekčný roztok v naplnenej injekčnej striekačkeKaždá naplnená injekčná striekačka obsahuje 150 mg sekukinumabu v 1 ml.
Cosentyx 300 mg injekčný roztok v naplnenejinjekčnejstriekačkeKaždá naplnená injekčná striekačka obsahuje 300 mg sekukinumabu v 2 ml.
Cosentyx 150 mg injekčný roztok v naplnenompereKaždé naplnené pero obsahuje 150 mg sekukinumabu v 1 ml.
Cosentyx 300 mg injekčný roztok v naplnenompereKaždé naplnené pero obsahuje 300 mg sekukinumabu v 2 ml.
Sekukinumab je rekombinantná, plne ľudská monoklonálna protilátka, produkovaná v bunkách ovária
čínskeho škrečka (CHO).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInjekčný roztok (injekcia)
Roztok je číry a bezfarebný až bledožltý.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieLožisková psoriáza u dospelýchCosentyx je indikovaný na liečbu stredne závažnej až závažnej ložiskovej psoriázy u dospelých, ktorí
sú kandidátmi na systémovú liečbu.

Ložisková psoriáza u pediatrických pacientov

Cosentyx je indikovaný na liečbu stredne závažnej až závažnej ložiskovej psoriázy u detí a
dospievajúcich vo veku od 6 rokov, ktorí sú kandidátmi na systémovú liečbu.
P
s
o
r
ia
t
i
c
k
á artritída
Cosentyx, v monoterapii alebo v kombinácii s metotrexátom (MTX), je indikovaný na liečbu aktívnej
psoriatickej artritídy u dospelých pacientov, keď odpoveď na predchádzajúcu liečbu antireumatickým liekom modifikujúcim chorobu (
disease-modifying anti-rheumatic drug, DMARD) nebola dostatočná (pozri časť 5.1).
Axiálna spondyloartritída (axSpA)Ankylozujúca spondylitída (AS) / axiálna spondyloartritída s rádiografickým dôkazomCosentyx je indikovaný na liečbu aktívnej ankylozujúcej spondylitídy u dospelých, u ktorých odpoveď na konvenčnú liečbu nebola dostatočná.
Axiálna spondyloartritída bez rádiografického dôkazu (nr-axSpA)Cosentyx je indikovaný na liečbu aktívnej axiálnej spondyloartritídy bez rádiografického dôkazu s
objektívnymi prejavmi zápalu preukázanými zvýšenou hodnotou C-reaktívneho proteínu (CRP) a/alebo dôkazom podľa magnetickej rezonancie (MRI) u dospelých bez adekvátnej odpovede na nesteroidné protizápalové lieky (NSAID).
4.2 Dávkovanie a spôsob podávaniaCosentyx je určený na použitie pod vedením a dohľadom lekára so skúsenosťami v diagnostike a
liečbe chorôb, na ktoré je Cosentyx indikovaný.
DávkovanieLožiskovápsoriáza u dospelýchOdporúčaná dávka je 300 mg sekukinumabu podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4 týždni, po ktorých nasledujú mesačné udržiavacie dávky. Každá dávka 300 mg sa podáva ako jedna subkutánna injekcia po 300 mg alebo ako dve subkutánne injekcie po 150 mg.
Ložiskovápsoriázau pediatrických pacientov (dospievajúci a deti vo veku od 6 rokov)Odporúčaná dávka závisí od telesnej hmotnosti pacienta (Tabuľka 1) a podáva sa subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky. Každá dávka 75 mg sa podáva ako jedna subkutánna injekcia po 75 mg. Každá dávka 150 mg sa podáva ako jedna subkutánna injekcia po 150 mg. Každá dávka 300 mg sa podáva ako jedna subkutánna injekcia po 300 mg alebo ako dve subkutánne injekcie po 150 mg.
Tabuľka 1 Odporúčaná dávka pre pediatrických pacientov s ložiskovou psoriázouTelesná hmotnosť v čase dávkovania
| Odporúčaná dávka
|
<25 kg
| 75 mg
|
25 až <50 kg
| 75 mg
|
≥50 kg
| 150 mg (*môže sa zvýšiť na 300 mg)
|
*Niektorí pacienti môžu mať daľší prínos z vyššej dávky.
150 mg a 300 mg injekčný roztok v naplnenej injekčnej striekačke a v naplnenom pere nie sú indikované na použitie u pediatrických pacientov s hmotnosťou <50 kg. Cosentyx môže byť dostupný v ďalších silách a/alebo formách v závislosti od individuálnych liečebných potrieb.
Psoriatická artritídaU pacientov so sprievodnou stredne závažnou až závažnou ložiskovou psoriázou alebo u pacientov
bez adekvátnej odpovede na liečbu anti-TNFα (
inadequate responders, IR) je odporúčaná dávka
300 mg podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky. Každá dávka 300 mg sa podáva ako jedna subkutánna injekcia
po 300 mg alebo ako dve subkutánne injekcie po 150 mg.
U ostatných pacientov je odporúčaná dávka 150 mg podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky. Na základe klinickej odpovede sa dávka môže zvýšiť na 300 mg.
Axiálna spondyloartritída (axSpA)Ankylozujúca spondylitída (AS, axiálna spondyloartritída s rádiografickým dôkazom)Odporúčaná dávka je 150 mg podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3.
a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky. Na základe klinickej odpovede sa dávka

môže zvýšiť na 300 mg. Každá 300 mg dávka sa podáva ako jedna subkutánna injekcia po 300 mg alebo ako dve 150 mg subkutánne injekcie.
Axiálna spondyloartritída bez rádiografického dôkazu (nr-axSpA)Odporúčaná dávka je 150 mg podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky.
Údaje, ktoré sú k dispozícii, naznačujú, že pri všetkých horeuvedených indikáciách sa klinická odpoveď na liečbu dosiahne zvyčajne v priebehu 16 týždňov liečby. Ukončenie liečby sa má zvážiť u pacientov, u ktorých sa odpoveď neprejavila do 16 týždňov liečby. U niektorých pacientov, ktorí majú spočiatku čiastočnú odpoveď, sa môže stav následne zlepšiť pri pokračujúcej liečbe po
16. týždni.
Osobitné skupiny pacientovStaršípacienti(voveku65 rokov a viac)Nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Poruchafunkcieobličiek/poruchafunkciepečeneCosentyx sa neskúmal u týchto skupín pacientov. Odporúčania pre dávkovanie nemožno urobiť.
Pediatrická populáciaBezpečnosť a účinnosť Cosentyxu u detí s ložiskovou psoriázou vo veku menej ako 6 rokov neboli stanovené.
Bezpečnosť a účinnosť Cosentyxu u detí vo veku menej ako 18 rokov v iných indikáciách neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávaniaCosentyx sa má podávať subkutánnou injekciou. Ak je to možné, je potrebné vyhýbať sa podaniu
injekcií do oblastí kože s prejavmi psoriázy. Injekčná striekačka alebo pero sa nesmie pretrepávať.
Po primeranom zaškolení v technike podávania subkutánnej injekcie si sám pacient alebo opatrovateľ môže podávať Cosentyx, ak lekár rozhodne, že je to vhodné. Lekár však má zabezpečiť dostatočné sledovanie pacientov. Pacientov a opatrovateľov je potrebné poučiť, aby podávali celé množstvo Cosentyxu podľa pokynov uvedených v písomnej informácii pre používateľa. Podrobný návod na podanie obsahuje písomná informácia pre používateľa.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Klinicky významné aktívne infekcie, napr. aktívna tuberkulóza (pozri časť 4.4).
4
.
4 Osobitné upozornenia a opatrenia pri používaní
S
l
e
d
ov
a
t
e
ľ
no
s
ť
Aby sa zlepšila sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
InfekcieSekukinumab má potenciál zvyšovať riziko infekcií. Po uvedení lieku na trh sa zaznamenali
u pacientov používajúcich sekukinumab závažné infekcie. Opatrnosť je potrebná pri zvažovaní použitia sekukinumabu u pacientov s chronickou infekciou alebo s opakovanými infekciami v
anamnéze.
Pacientov treba poučiť, aby vyhľadali lekársku pomoc, ak sa u nich objavia prejavy alebo príznaky poukazujúce na infekciu. Ak u pacienta vznikne závažná infekcia, je potrebné pacienta dôsledne sledovať a sekukinumab sa mu až do vymiznutia infekcie nemá podať.
Infekcie sa pozorovali v klinických skúšaniach u pacientov, ktorí dostávali sekukinumab (pozri časť 4.8). Väčšinou to boli ľahké alebo stredne ťažké infekcie horných dýchacích ciest, napr. nazofaryngitída, a nevyžadovali prerušenie liečby.
V súvislosti s mechanizmom účinku sekukinumabu sa v klinických skúšaniach pri psoriáze nezávažné kandidové infekcie slizníc a kože zaznamenali častejšie pri sekukinumabe ako pri placebe (3,55 na
100 pacientorokov pri 300 mg sekukinumabu oproti 1,00 na 100 pacientorokov pri placebe) (pozri
časť 4.8).
V klinických skúšaniach nebola hlásená zvýšená náchylnosť na tuberkulózu. Sekukinumab sa však nemá podávať pacientom s aktívnou tuberkulózou. U pacientov s latentnou tuberkulózou sa má pred začiatkom liečby sekukinumabom zvážiť antituberkulózna liečba.
Zápalové ochorenie čriev (vrátane Crohnovej choroby a ulceróznej kolitídy)Boli hlásené prípady vzniku alebo exacerbácie zápalového ochorenia čriev pri používaní
sekukinumabu (pozri časť 4.8). Sekukinumab sa neodporúča u pacientov so zápalovým ochorením
čriev. Ak sa u pacienta objavia prejavy a príznaky zápalového ochorenia čriev alebo ak dôjde k exacerbácii už existujúceho zápalového ochorenia čriev, je potrebné ukončiť liečbu so sekukinumabom a začať s primeranou lekárskou starostlivosťou.
Reakcie z precitlivenostiV klinických skúšaniach sa u pacientov liečených sekukinumabom pozorovali zriedkavé prípady
anafylaktických reakcií. Ak sa vyskytnú anafylaktické alebo iné závažné alergické reakcie, podávanie
sekukinumabu sa má okamžite ukončiť a má sa začať primeraná liečba.
Osoby citlivé na latex –len Cosentyx 150 mg injekčnýroztokvnaplnenejinjekčnejstriekačkea Cosentyx 150 mg injekčný roztok v naplnenom pereSnímateľný kryt ihly Cosentyxu 150 mg injekčného roztoku v naplnenej injekčnej striekačke a


Cosentyxu 150 mg injekčného roztoku v naplnenom pere obsahuje derivát prírodného latexu. Doteraz sa v snímateľnom kryte ihly nenašiel žiadny prírodný latex. Používanie Cosentyxu 150 mg injekčného roztoku v naplnenej injekčnej striekačke a Cosentyxu 150 mg injekčného roztoku v naplnenom pere u
osôb citlivých na latex sa však neskúmalo, a preto existuje možné riziko reakcií z precitlivenosti, ktoré
sa nedajú úplne vylúčiť.
V
a
k
ci
n
á
c
i
e
Živé vakcíny sa nemajú podávať súbežne so sekukinumabom.
Pacienti, ktorí dostávajú sekukinumab, môžu súčasne dostať inaktivované alebo neživé vakcíny. V klinickom skúšaní po vakcinácii meningokokovou a inaktivovanou chrípkovou vakcínou podobné podiely zdravých dobrovoľníkov, ktorí dostávali 150 mg sekukinumabu a ktorí dostávali placebo, dokázali vyprodukovať dostatočnú imunitnú odpoveď, ktorá predstavovala najmenej 4 -násobné zvýšenie titrov protilátok proti meningokokovým a chrípkovým vakcínam. Údaje naznačujú, že sekukinumab nepotláča humorálnu imunitnú odpoveď na meningokokové alebo chrípkové vakcíny.
Pred začatím liečby Cosentyxom sa odporúča, aby pediatrickí pacienti absolvovali všetky potrebné imunizácie podľa veku v súlade s aktuálnymi usmerneniami pre imunizáciu.
Súbežná imunosupresívna liečba
Bezpečnosť a účinnosť sekukinumabu v kombinácii s imunosupresívami, vrátane biologických liekov,
alebo fototerapiou sa nehodnotila v skúšaniach pri psoriáze. Sekukinumab sa podával súbežne
s metotrexátom (MTX), sulfasalazínom a/alebo kortikosteroidmi v skúšaniach pri artritíde (vrátane pacientov so psoriatickou artritídou a ankylozujúcou spondylitídou). Opatrnosť je potrebná pri zvažovaní súbežného použitia iných imunosupresív a sekukinumabu (pozri tiež časť 4.5).
4.5 Liekové a iné interakcie
Živé vakcíny sa nemajú podať súbežne so sekukinumabom (pozri tiež časť 4.4).
V skúšaní u dospelých pacientov s ložiskovou psoriázou sa nepozorovala žiadna interakcia medzi
sekukinumabom a midazolamom (substrát CYP3A4).
Keď sa sekukinumab podával súbežne s metotrexátom (MTX) a/alebo kortikosteroidmi v skúšaniach pri artritíde (vrátane pacientov so psoriatickou artritídou a axiálnou spondyloartritídou), nepozorovali sa žiadne interakcie.
4.6 Fertilita, gravidita a laktácia
Ženy v plodnomveku
Ženy v plodnom veku majú počas liečby a najmenej 20 týždňov po skončení liečby používať účinnú
metódu antikoncepcie.
Gravidita
Nie sú k dispozícii dostatočné údaje o používaní sekukinumabu u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame účinky na reprodukčnú toxicitu (pozri
časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Cosentyxu počas gravidity.
D
o
jče
ni
e
Nie je známe, či sa sekukinumab vylučuje do ľudského mlieka. Imunoglobulíny sa vylučujú do
ľudského mlieka a nie je známe, či sa sekukinumab systémovo absorbuje po požití. Vzhľadom na možnosť nežiaducich reakcií na sekukinumab u dojčených detí sa rozhodnutie, či ukončiť dojčenie počas liečby a do 20 týždňov po skončení liečby, alebo ukončiť liečbu Cosentyxom, má urobiť po
zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Účinok sekukinumabu na fertilitu ľudí sa neskúmal. Štúdie na zvieratách nenaznačujú priame alebo
nepriame škodlivé účinky týkajúce sa fertility.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Cosentyx nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutie profilu bezpečnosti
Najčastejšie hlásenými nežiaducimi reakciami sú infekcie horných dýchacích ciest (17,7 %)
(najčastejšie nazofaryngitída a rinitída).
Tabuľkový zoznam nežiaducich reakcií
Nežiaduce reakcie v klinických skúšaniach a z hlásení po uvedení lieku na trh (Tabuľka 2) sú
zatriedené podľa orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, najčastejšie ako prvé. V rámci každej skupiny frekvencie sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti. Okrem toho zodpovedajúca
kategória frekvencie každej nežiaducej reakcie vychádza z nasledujúcej konvencie: veľmi časté
(≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až
<1/1 000); veľmi zriedkavé (<1/10 000); a neznáme (z dostupných údajov).
V zaslepených a otvorených klinických skúšaniach sa liečilo sekukinumabom viac ako
18 000 pacientov s rôznymi indikáciami (ložisková psoriáza, psoriatická artritída, axiálna spondyloartritída a iné autoimunitné choroby), čo predstavuje 30 565 pacientorokov expozície.
Najmenej jeden rok dostávalo sekukinumab viac ako 11 700 z týchto pacientov. Profil bezpečnosti sekukinumabu sa vo všetkých indikáciach zhoduje.
T
a
b
u
ľ
k
a 2 Zoznam nežiaducich reakcií v klinických skúšaniach
1
)
a pri používaní po uvedení lieku na trh
T
r
i
e
d
a orgánových
systémov
|
Frekvencia
|
N
e
ž
ia
d
u
c
a reakcia
|
Infekcie a nákazy
|
Veľmi časté
|
Infekcie horných dýchacích ciest
|
Časté
|
Orálny herpes
|
Tinea pedis
|
Menej časté
|
Orálna kandidóza
|
Otitis externa
|
Infekcie dolných dýchacích ciest
|
Neznáme
|
Kandidóza slizníc a kože (vrátane kandidózy ezofágu)
|
Poruchy krvi
a lymfatického systému
|
Menej časté
|
Neutropénia
|
Poruchy imunitného systému
|
Zriedkavé
|
Anafylaktické reakcie
|
Poruchy nervového systému
|
Časté
|
Bolesť hlavy
|
Poruchy oka
|
Menej časté
|
Konjunktivitída
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Časté
|
Rinorea
|
Poruchy
gastrointestinálneho traktu
|
Časté
|
Hnačka
|
Časté
|
Nauzea
|
Menej časté
|
Zápalové ochorenie čriev
|
Poruchy kože
a podkožného tkaniva
|
Menej časté
|
Urtikária
|
Zriedkavé
|
Exfoliatívna dermatitída2)
|
Hypersenzitívna vaskulitída
|
Celkové poruchy a
reakcie v mieste podania
|
Časté
|
Únava
|
1) Klinické skúšania kontrolované placebom (fáza III) u pacientov s ložiskovou psoriázou, PsA, AS
a nr-axSpA pri dávkach 300 mg, 150 mg, 75 mg alebo pri placebe s liečbou trvajúcou do
12 týždňov (pri psoriáze) alebo 16 týždňov (pri PsA, AS a nr-axSpA)
2) Prípady boli hlásené u pacientov s diagnózou psoriázy
|
P
op
i
s
vybraných
nežiaducich
reakcií
I
n
f
ek
c
i
e
Počas obdobia kontrolovaného placebom v klinických skúšaniach u pacientov s ložiskovou psoriázou
(spolu 1 382 pacientov liečených sekukinumabom a 694 pacientov liečených placebom do 12 týždňov) boli infekcie hlásené u 28,7 % pacientov liečených sekukinumabom v porovnaní s 18,9 % pacientov, ktorí dostávali placebo. Väčšina infekcií boli nie závažné a ľahké až stredne ťažké infekcie horných dýchacích ciest, napr. nazofaryngitída, ktoré nevyžadovali prerušenie liečby. V súlade s
mechanizmom účinku sa zvýšil výskyt kandidózy slizníc alebo kože, ale tieto prípady boli ľahké až
stredne ťažké, nie závažné, reagovali na štandardnú liečbu a nevyžadovali prerušenie liečby. Závažné
infekcie sa vyskytli u 0,14 % pacientov liečených sekukinumabom a u 0,3 % pacientov, ktorí dostávali
placebo (pozri časť 4.4).
Počas celého obdobia liečby (celkovo 3 430 pacientov liečených sekukinumabom, väčšina z nich až
do 52 týždňov) boli infekcie hlásené u 47,5 % pacientov liečených sekukinumabom (0,9 pripadajúcich na pacientorok sledovania). Závažné infekcie boli hlásené u 1,2 % pacientov liečených sekukinumabom (0,015 pripadajúcich na pacientorok sledovania).
Výskyt infekcií pozorovaný v klinických skúšaniach pri psoriatickej artritíde a axiálnej spondyloartritíde (ankylozujúcej spondylitíde a axiálnej spondyloartritíde bez rádiografického dôkazu) je podobný výskytu infekcií, ktorý sa pozoroval v psoriatických skúšaniach.
Neutropénia
V klinických skúšaniach fázy III pri psoriáze sa neutropénia pozorovala častejšie pri sekukinumabe ako pri placebe, ale vo väčšine prípadov bola mierna, prechodná a reverzibilná. Neutropénia
<1,0-0,5x109/l (CTCAE stupňa 3) bola hlásená u 18 z 3 430 (0,5 %) pacientov liečených
sekukinumabom, bez závislosti od dávky a bez časového vzťahu k infekciám v 15 z 18 prípadov. Prípady závažnejšej neutropénie sa nezaznamenali. Vo zvyšných 3 prípadoch boli hlásené nezávažné infekcie s obvyklou odpoveďou na štandardnú starostlivosť, ktoré si nevyžiadali vysadenie sekukinumabu.
Frekvencia výskytu neutropénie pri psoriatickej artritíde a axiálnej spondyloartritíde (ankylozujúcej spondylitíde a axiálnej spondyloartritíde bez rádiografického dôkazu) bola podobná frekvencii výskytu neutropénie pri psoriáze.
Boli hlásené zriedkavé prípady neutropénie <0,5x109/l (CTCAE stupňa 4).
Reakcie z precitlivenosti
V klinických skúšaniach sa pozorovala urtikária a zriedkavé prípady anafylaktickej reakcie na
sekukinumab (pozri aj časť 4.4).
Imunogenita
V klinických skúšaniach pri psoriáze, psoriatickej artritíde a axiálnej spondyloartritíde (ankylozujúcej spondylitíde a axiálnej spondyloartritíde bez rádiografického dôkazu) sa protilátky proti sekukinumabu vytvorili u menej ako 1 % pacientov liečených sekukinumabom počas liečby trvajúcej do 52 týždňov. Približne polovica protilátok proti liečivu, ktoré sa objavili počas liečby, bola neutralizujúca, ale nespájalo sa to so stratou účinnosti alebo farmakokinetickými abnormalitami.
Pediatrická populácia
Nežiaduceúčinkyupediatrickýchpacientovsložiskovoupsoriázouvovekuod6 rokov
Bezpečnosť sekukinumabu u pediatrických pacientov s ložiskovou psoriázou sa hodnotila v dvoch skúšaniach fázy III. Prvé skúšanie (pediatrické skúšanie 1) bolo dvojito zaslepené, placebom kontrolované skúšanie u 162 pacientov so závažnou ložiskovou psoriázou vo veku od 6 rokov do menej ako 18 rokov. Druhé skúšanie (pediatrické skúšanie 2) je otvorené klinické skúšanie u
84 pacientov so stredne závažnou až závažnou ložiskovou psoriázou vo veku od 6 rokov do menej ako
18 rokov. Profil bezpečnosti zaznamenaný v týchto dvoch skúšaniach bol zhodný s profilom bezpečnosti u dospelých pacientov s ložiskovou psoriázou.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 Predávkovanie
V klinických skúšaniach sa intravenózne podali dávky až do 30 mg/kg (približne 2 000 až 3 000 mg) bez toxicity obmedzujúcej dávku. V prípade predávkovania sa odporúča, aby sa u pacienta sledovali akékoľvek prejavy a príznaky nežiaducich reakcií a aby sa okamžite začala primeraná symptomatická liečba.
5
. FARMAKOLOGICKÉ VLASTNOSTI
5
.
1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunosupresíva, inhibítory interleukínu, ATC kód: L04AC10
Mechanizmus účinku
Sekukinumab je plne ľudská monoklonálna protilátka IgG1/κ, ktorá sa selektívne viaže na
prozápalový cytokín interleukín-17A (IL-17A) a neutralizuje ho. Sekukinumab sa zameriava na
IL-17A a inhibuje jeho interakciu s receptorom IL-17, ktorý je exprimovaný na rôznych typoch buniek
vrátane keratinocytov. Dôsledkom je, že sekukinumab inhibuje uvoľňovanie zápalových cytokínov, chemokínov a mediátorov poškodenia tkanív a znižuje vplyv sprostredkovaný IL-17A na autoimunitné a zápalové ochorenia. Klinicky významné koncentrácie sekukinumabu sa dostávajú do kože a znižujú lokálne zápalové markery. Priamym dôsledkom liečby sekukinumabom je zmiernenie erytému,
stvrdnutia a šupinatosti kože v léziách ložiskovej psoriázy.
IL-17A je prirodzene sa vyskytujúci cytokín, ktorý sa podieľa na normálnych zápalových a imunitných reakciách. IL-17A zohráva kľúčovú úlohu v patogenéze ložiskovej psoriázy, psoriatickej artritídy a axiálnej spondyloartritídy (ankylozujúcej spondylitídy a axiálnej spondyloartritídy bez rádiografického dôkazu) a jeho množstvo je zvýšené v kožných léziách na rozdiel od kože bez lézií u pacientov s ložiskovou psoriázou a v synoviálnom tkanive pacientov so psoriatickou artritídou. Výskyt buniek produkujúcich IL-17 bol tiež významne vyšší v subchondrálnej kostnej dreni facetových kĺbov u pacientov s ankylozujúcou spondylitídou. . U pacientov s axiálnou spondyloartritídou bez rádiografického dôkazu sa tiež zistil zvýšený počet lymfocytov produkujúcich IL-17A. Inhibícia
IL-17A sa preukázala ako účinná pri liečbe ankylozujúcej spondylitídy, čo stanovuje kľúčovú úlohu
tohto cytokínu pri axiálnej spondyloartritíde.
Farmakodynamické účinky
Sérové koncentrácie celkového IL-17A (voľného IL-17A a IL-17A viazaného na sekukinumab) sa
spočiatku zvýšia u pacientov, ktorí dostávajú sekukinumab. Potom nasleduje pomalý pokles v
dôsledku zníženia klírensu IL-17A viazaného na sekukinumab, čo ukazuje, že sekukinumab selektívne vychytáva voľný IL-17A, ktorý zohráva kľúčovú úlohu v patogenéze ložiskovej psoriázy.
V skúšaní so sekukinumabom sa po jednom až dvoch týždňoch liečby významne znížili hodnoty infiltrujúcich epidermálnych neutrofilov a rôznych markerov súvisiacich s neutrofilmi, ktoré sú zvýšené v kožných léziách pacientov s ložiskovou psoriázou.
Sekukinumab preukázal zníženie (v priebehu 1 až 2 týždňov liečby) koncentrácií C-reaktívneho proteínu, ktorý je zápalovým markerom.
K
li
ni
c
k
á účinnosť a bezpečnosť
Ložiskovápsoriáza u dospelých
Bezpečnosť a účinnosť sekukinumabu sa hodnotili v štyroch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických skúšaniach fázy III u pacientov so stredne ťažkou až ťažkou ložiskovou psoriázou, ktorí boli kandidátmi na fototerapiu alebo systémovú liečbu [ERASURE,
FIXTURE, FEATURE, JUNCTURE]. Účinnosť a bezpečnosť 150 mg a 300 mg sekukinumabu sa
hodnotili v porovnaní buď s placebom, alebo s etanerceptom. Okrem toho jedno skúšanie hodnotilo
chronický režim liečby v porovnaní s režimom „opakovania liečby podľa potreby“ [SCULPTURE].
Z 2 403 pacientov, ktorí sa zúčastnili klinických skúšaní kontrolovaných placebom, bolo 79 % pacientov v minulosti neliečených biologickými liekmi, 45 % pacientov po zlyhaní nebiologickej liečby a 8 % pacientov po zlyhaní biologickej liečby (6 % po zlyhaní liečby anti-TNF a 2 % po zlyhaní liečby anti-p40). Približne 15 až 25 % pacientov v klinických skúšaniach fázy III malo na začiatku psoriatickú artritídu (PsA).
Psoriatické skúšanie 1 (ERASURE) vyhodnotilo 738 pacientov. Pacienti randomizovaní na sekukinumab dostali v 0., 1., 2., 3. a 4. týždni dávky 150 mg alebo 300 mg, po ktorých nasledovala každý mesiac rovnaká dávka. Psoriatické skúšanie 2 (FIXTURE) vyhodnotilo 1 306 pacientov. Pacienti randomizovaní na sekukinumab dostali v 0., 1., 2., 3. a 4. týždni dávky 150 mg alebo 300 mg, po ktorých nasledovala každý mesiac rovnaká dávka. Pacienti randomizovaní na etanercept dostávali dvakrát týždenne dávky 50 mg počas 12 týždňov, po ktorých nasledovalo 50 mg každý týždeň. V skúšaní 1 aj v skúšaní 2 pacienti randomizovaní na placebo, ktorí nereagovali do 12. týždňa, potom
prešli na podávanie sekukinumabu (buď 150 mg, alebo 300 mg) v 12., 13., 14. a 15. týždni, po ktorých
nasledovala počnúc 16. týždňom každý mesiac rovnaká dávka. Všetci pacienti boli sledovaní do
52 týždňov od prvého podania skúšaného lieku.
Psoriatické skúšanie 3 (FEATURE) vyhodnotilo 177 pacientov používajúcich naplnenú injekčnú striekačku v porovnaní s placebom po 12 týždňoch liečby, aby sa stanovila bezpečnosť, znášanlivosť a použiteľnosť samopodávania sekukinumabu naplnenou injekčnou striekačkou. Psoriatické skúšanie 4 (JUNCTURE) vyhodnotilo 182 pacientov používajúcich naplnené pero v porovnaní s placebom po
12 týždňoch liečby, aby sa stanovila bezpečnosť, znášanlivosť a použiteľnosť samopodávania
sekukinumabu naplneným perom. V skúšaní 3 aj v skúšaní 4 pacienti randomizovaní na sekukinumab dostali v 0., 1., 2., 3. a 4. týždni dávky 150 mg alebo 300 mg, po ktorých nasledovala každý mesiac
rovnaká dávka. Pacienti boli tiež randomizovaní na placebo v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovalo každý mesiac rovnaké podanie.
Psoriatické skúšanie 5 (SCULPTURE) vyhodnotilo 966 pacientov. Všetci pacienti dostali v 0., 1., 2.,
3., 4., 8. a 12. týždni sekukinumab v dávke 150 mg alebo 300 mg a potom boli od 12. týždňa randomizovaní buď na udržiavací režim s rovnakou dávkou každý mesiac, alebo na režim
„opakovania liečby podľa potreby“ s rovnakou dávkou. Pacienti randomizovaní na „opakovanie liečby podľa potreby“ nedosahovali primeranú udržiavaciu odpoveď, preto sa odporúča pevný mesačný
režim udržiavacej liečby.
Spoločné primárne ukazovatele v klinických skúšaniach kontrolovaných placebom a účinným liekom boli podiely pacientov, ktorí dosiahli v 12. týždni odpoveď PASI 75 a odpoveď IGA mod 2011 „bez prejavov“ alebo „takmer bez prejavov“ v porovnaní s placebom (pozri Tabuľky 3 a 4). Dávkou
300 mg sa dosiahol lepší ústup prejavov na koži, najmä pre kožu „bez prejavov“ alebo „takmer bez prejavov“ celkovo pri ukazovateľoch účinnosti PASI 90, PASI 100 a IGA mod 2011, odpoveď 0 alebo
1 vo všetkých klinických skúšaniach s maximálnymi účinkami pozorovanými v 16. týždni, preto sa odporúča táto dávka.
T
a
b
u
ľ
k
a 3 Súhrn klinickej odpovede PASI 50/75/90/100 & IGA⃰ mod 2011 „bez prejavov“ alebo „takmer bez prejavov“ v psoriatických skúšaniach 1, 3 a 4 (ERASURE, FEATURE a JUNCTURE)
 12. týždeň 16. týždeň 52. týždeň
12. týždeň 16. týždeň 52. týždeň
|
P
l
acebo
|
150 mg
|
300 mg
|
150 mg
|
300 mg
|
150 mg
|
300 mg
|
S
k
ú
š
a
n
i
e 1
|
|
|
|
|
|
|
|
Počet pacientov
|
246
|
244
|
245
|
244
|
245
|
244
|
245
|
Odpoveď PASI 50, n (%)
|
22
|
203
|
222
|
212
|
224
|
187
|
207
|
|
(8,9 %)
|
(83,5 %)
|
(90,6 %)
|
(87,2 %)
|
(91,4 %)
|
(77 %)
|
(84,5 %)
|
Odpoveď PASI 75, n (%)
|
11
|
174
|
200
|
188
|
211
|
146
|
182
|
|
(4,5 %)
|
(71,6 %)*
|
(81,6 %)*
|
(77,4 %)
|
(86,1 %)
|
(60,1 %)
|
(74,3 %)
|
|
|
*
|
*
|
|
|
|
|
Odpoveď PASI 90, n (%)
|
3 (1,2 %)
|
95
|
145
|
130
|
171
|
88
|
147
|
|
|
(39,1 %)*
|
(59,2 %)*
|
(53,5 %)
|
(69,8 %)
|
(36,2 %)
|
(60,0 %)
|
|
|
*
|
*
|
|
|
|
|
Odpoveď PASI 100, n (%)
|
2 (0,8 %)
|
31
|
70
|
51
|
102
|
49
|
96
|
|
|
(12,8 %)
|
(28,6 %)
|
(21,0 %)
|
(41,6 %)
|
(20,2 %)
|
(39,2 %)
|
Odpoveď IGA mod 2011
|
6
|
125
|
160
|
142
|
180
|
101
|
148
|
„bez prejavov“ alebo
|
(2,40 %)
|
(51,2 %)*
|
(65,3 %)*
|
(58,2 %)
|
(73,5 %)
|
(41,4 %)
|
(60,4 %)
|
|
|
„takmer bez prejavov“,
* *n (%)
Skúšanie 3Počet pacientov 59 59 58 - - - -
Odpoveď PASI 50, n (%) 3 (5,1 %) 51
(86,4 %)
Odpoveď PASI 75, n (%) 0 (0,0 %) 41
(69,5 %)*
*
Odpoveď PASI 90, n (%) 0 (0,0 %) 27
(45,8 %)
51
(87,9 %)
44
(75,9 %)*
*
35
(60,3 %)
- - - -
- - - -
- - - -
Odpoveď PASI 100,
n (%)
Odpoveď IGA mod 2011
„bez prejavov“ alebo
„takmer bez prejavov“,
n (%)
Skúšanie 4
0 (0,0 %) 5
(8,5 %)
0 (0,0 %) 31
(52,5 %)*
*
25
(43,1 %)
40
(69,0 %)*
*
- - - -
- - - -
Počet pacientov 61 60 60 - - - -
Odpoveď PASI 50, n (%) 5 (8,2 %) 48
(80,0 %)
Odpoveď PASI 75, n (%) 2 (3,3 %) 43
(71,7 %)*
*
Odpoveď PASI 90, n (%) 0 (0,0 %) 24
(40,0 %)
Odpoveď PASI 100, n (%) 0 (0,0 %) 10
(16,7 %)
58
(96,7 %)
52
(86,7 %)*
*
33
(55,0 %)
16
(26,7 %)
- - - -
- - - -
- - - -
- - - -
Odpoveď IGA mod 2011
„bez prejavov“ alebo
„takmer bez prejavov“,
n (%)
0 (0,0 %) 32
(53,3 %)*
*
44
(73,3 %)*
*
- - - -
* IGA mod 2011 je stupnica s 5 kategóriami, a to „0 = bez prejavov“, „1 = takmer bez prejavov“, „2 = mierna“, „3 = stredne závažná“ alebo „4 = závažná“, ktoré udávajú celkové hodnotenie závažnosti psoriázy lekárom, so zameraním na stvrdnutie, sčervenanie a šupinatosť. Úspešnosť liečby „bez prejavov“ alebo
„takmer bez prejavov“ znamenala žiadne prejavy psoriázy alebo normálne až ružovo sfarbené lézie, žiadne zhrubnutie ložísk a žiadna až minimálna ložisková šupinatosť.
** hodnoty p v porovnaní s placebom a upravené pre multiplicitu: p<0,0001.
T
a
b
u
ľ
k
a 4 Súhrn klinickej odpovede v psoriatickom skúšaní 2 (FIXTURE)
12. týždeň 16. týždeň 52. týždeň
|
P
l
acebo
|
150 mg
|
300 mg
|
E
t
anercept
|
150 mg
|
300 mg
|
E
t
anercept
|
150 mg
|
300 mg
|
E
t
anercept
|
Počet
|
324
|
327
|
323
|
323
|
327
|
323
|
323
|
327
|
323
|
323
|
pacientov
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
49
|
266
|
296
|
226
|
290
|
302
|
257 (79,6 %)
|
249
|
274
|
234 (72,4 %)
|
PASI 50,
|
(15,1 %)
|
(81,3 %)
|
(91,6 %)
|
(70,0 %)
|
(88,7 %)
|
(93,5 %)
|
|
(76,1 %)
|
(84,8 %)
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
16
|
219
|
249
|
142
|
247
|
280
|
189 (58,5 %)
|
215
|
254
|
179 (55,4 %)
|
PASI 75,
|
(4,9 %)
|
(67,0 %)
|
(77,1 %)
|
(44,0 %)
|
(75,5 %)
|
(86,7 %)
|
|
(65,7 %)
|
(78,6 %)
|
|
n (%)
|
|
**
|
**
|
|
|
|
|
|
|
|
Odpoveď
|
5
|
137
|
175
|
67
|
176
|
234
|
101 (31,3 %)
|
147
|
210
|
108 (33,4 %)
|
PASI 90,
|
(1,5 %)
|
(41,9 %)
|
(54,2 %)
|
(20,7 %)
|
(53,8 %)
|
(72,4 %)
|
|
(45,0 %)
|
(65,0 %)
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
0 (0 %)
|
47
|
78
|
14 (4,3 %)
|
84
|
119
|
24 (7,4 %)
|
65
|
117
|
32 (9,9 %)
|
PASI 100,
|
|
(14,4 %)
|
(24,1 %)
|
|
(25,7 %)
|
(36,8 %)
|
|
(19,9 %)
|
(36,2 %)
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
9
|
167
|
202
|
88
|
200
|
244
|
127 (39,3 %)
|
168
|
219
|
120 (37,2 %)
|
IGA mod
|
(2,8 %)
|
(51,1 %)
|
(62,5 %)
|
(27,2 %)
|
(61,2 %)
|
(75,5 %)
|
|
(51,4 %)
|
(67,8 %)
|
|
2011 „bez
|
|
**
|
**
|
|
|
|
|
|
|
|
prejavov“
|
|
|
|
|
|
|
|
|
|
|
alebo
|
|
|
|
|
|
|
|
|
|
|
„takmer bez
|
|
|
|
|
|
|
|
|
|
|
prejavov“,
|
|
|
|
|
|
|
|
|
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
** hodnoty p v porovnaní s etanerceptom: p=0,0250
V ďalšom klinickom skúšaní pri psoriáze (CLEAR) sa vyhodnotilo 676 pacientov. Dávka sekukinumabu 300 mg splnila primárne a sekundárne koncové ukazovatele preukázaním superiority voči ustekinumabu na základe odpovede PASI 90 v 16. týždni (primárny koncový ukazovateľ), rýchlosti nástupu odpovede PASI 75 vo 4. týždni a dlhodobou odpoveďou PASI 90 v 52. týždni oproti ustekinumabu. Vyššia účinnosť sekukinumabu v porovnaní s ustekinumabom pre koncové ukazovatele PASI 75/90/100 a IGA mod 2011 s odpoveďou 0 alebo 1 („bez prejavov“ alebo „takmer
bez prejavov“) sa pozorovala od začiatku a pretrvávala do 52. týždňa.
Tabuľka 5 Súhrn klinickej odpovede v skúšaní CLEAR4. týždeň 16. týždeň 52. týždeň
s
e
ku
k
inumab
u
s
t
e
k
inumab* sekukinumab
u
s
t
e
k
inumab* sekukinumab
u
s
t
e
k
inumab*
300 mg 300 mg 300 mg
Počet pacientov 334 335 334 335 334 335
Odpoveď PASI
75, n (%)
166 (49,7 %)** 69 (20,6 %) 311 (93,1 %) 276 (82,4 %) 306 (91,6 %) 262 (78,2 %)
Odpoveď PASI
90, n (%)
70 (21,0 %) 18 (5,4 %) 264 (79,0 %)** 192 (57,3 %) 250
(74,9 %)***
203 (60,6%)
Odpoveď PASI
100, n (%) Odpoveď IGA mod 2011 „bez prejavov“alebo
„takmer bez prejavov“, n (%)
14 (4,2 %) 3 (0,9 %) 148 (44,3 %) 95 (28,4 %) 150 (44,9 %) 123 (36,7 %)
128 (38,3 %) 41 (12,2 %) 278 (83,2 %) 226 (67,5 %) 261 (78,1 %) 213 (63,6 %)

* Pacienti liečení sekukinumabom dostávali dávku 300 mg v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala rovnaká dávka každé 4 týždne do 52. týždňa. Pacienti liečení ustekinumabom dostávali dávku 45 mg alebo 90 mg v 0. a 4. týždni, potom každých 12 týždňov do 52. týždňa (dávkované podľa telesnej hmotnosti na základe schváleného dávkovania)
** hodnoty p v porovnaní s ustekinumabom: p<0,0001 pre primárny koncový ukazovateľ PASI 90 v 16. týždni a sekundárny
koncový ukazovateľ PASI 75 vo 4. týždni
*** hodnoty p v porovnaní s ustekinumabom: p=0,0001 pre sekundárny koncový ukazovateľ PASI 90 v 52. týždni
Sekukinumab bol účinný u pacientov, ktorí predtým nedostali systémovú liečbu, u pacientov predtým neliečených biologickými liekmi, u pacientov s expozíciou biologickým liekom/anti-TNF a u pacientov po zlyhaní liečby biologickými liekmi/anti-TNF. Zlepšenie PASI 75 u pacientov, ktorí mali pri zaradení do skúšania aj psoriatickú artritídu, bolo podobné ako u celkovej populácie s ložiskovou psoriázou.
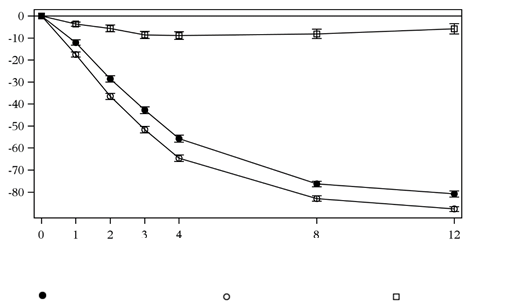
Sekukinumab mal rýchly nástup účinku so znížením strednej hodnoty PASI o 50 % do 3. týždňa pri
dávke 300 mg.
Obrázok 1 Časový priebeh percentuálnej zmeny stredného skóre PASI oproti východiskovej hodnote v skúšaní 1 (ERASURE)Týždne liečby
N = počet vyhodnotiteľných pacientovsekukinumab 150 mg (n=243) sekukinumab 300 mg (n=245) Placebo (n=245)
Špecifické miesta/formy ložiskovej psoriázyV ďalších dvoch skúšaniach kontrolovaných placebom sa zaznamenalo zlepšenie pri psoriáze nechtov
(TRANSFIGURE, 198 pacientov), ako aj pri palmoplantárnej ložiskovej psoriáze (GESTURE,
205 pacientov). V skúšaní TRANSFIGURE bol v 16. týždni sekukinumab lepší ako placebo (46,1 %
pri 300 mg, 38,4 % pri 150 mg a 11,7 % pri placebe) pri hodnotení významného zlepšenia indexu závažnosti psoriázy nechtov (Nail Psoriasis Severity Index, NAPSI %) oproti východiskovým
hodnotám u pacientov so stredne ťažkou až ťažkou ložiskovou psoriázou s postihnutím nechtov. V
skúšaní GESTURE bol sekukinumab lepší ako placebo v 16. týždni (33,3 % pri 300 mg, 22,1 % pri
150 mg a 1,5 % pri placebe) pri hodnotení významného zlepšenia 0 alebo 1 odpovede pre ppIGA
(„bez prejavov“ alebo „takmer bez prejavov“) u pacientov so stredne ťažkou až ťažkou palmoplantárnou ložiskovou psoriázou.
V klinickom skúšaní kontrolovanom placebom bolo vyhodnotených 102 pacientov so stredne závažnou až závažnou psoriázou vlasatej časti hlavy, definovanou indexom závažnosti psoriázy vlasatej časti hlavy (Psoriasis Scalp Severity Index, PSSI) so skóre ≥12, odpoveďou IGA mod 2011 iba vlasatej časti hlavy so skóre 3 alebo vyšším a najmenej 30 % postihnutím povrchu vlasatej časti. Sekukinumab 300 mg bol v 12. týždni účinnejší ako placebo po 12. týždni, kedy boli vyhodnotené významné zlepšenia v oboch odpovediach PSSI 90 (52,9 % oproti 2,0 %) a IGA mod 2011 0 alebo 1 iba vo vlasatej časti hlavy (56,9 % oproti 5,9 %) oproti východiskovým hodnotám. Zlepšenie oboch koncových ukazovateľov pretrvávalo u pacientov, ktorí pokračovali v liečbe sekukinumabom, až do
24. týždňa.
Kvalita života/výsledky hlásené pacientmiŠtatisticky významné zlepšenie v 12. týždni (skúšania 1-4) oproti východiskovej hodnote v porovnaní s placebom sa prejavilo v DLQI (Dermatology Life Quality Index – Dermatologický index kvality života). Stredný pokles (zlepšenie) v DLQI oproti východiskovej hodnote v 12. týždni bol v rozmedzí
od -10,4 do -11,6 pri 300 mg sekukinumabu, od -7,7 do -10,1 pri 150 mg sekukinumabu, oproti -1,1
až -1,9 pri placebe. Toto zlepšenie pretrvalo 52 týždňov (skúšania 1 a 2).
V skúšaniach 1 a 2 vyplnilo 40 % účastníkov Denník psoriatických príznakov (Psoriasis Symptom Diary©). U účastníkov, ktorí v každom z týchto skúšaní vypĺňali denník, sa v 12. týždni prejavilo štatisticky významné zmiernenie svrbenia, bolesti a šupinatosti ako pacientmi hlásených prejavov
a príznakov oproti východiskovej hodnote v porovnaní s placebom.
Štatisticky významné zlepšenie po 4. týždni oproti východiskovej hodnote u pacientov liečených sekukinumabom v porovnaní s pacientmi liečenými ustekinumabom (CLEAR) sa prejavilo v DLQI a toto zlepšenie pretrvalo 52 týždňov.
Štatisticky významné zmiernenie svrbenia, bolesti a šupinatosti ako pacientmi hlásených prejavov
a príznakov sa preukázalo v Denníku psoriatických príznakov (Psoriasis Symptom Diary©)
po 16. týždni a 52. týždni (CLEAR) u pacientov liečených sekukinumabom v porovnaní s pacientmi
liečenými ustekinumabom.
Štatisticky významné zlepšenie (zníženie) v skúšaní pri psoriáze vlasatej časti hlavy sa preukázalo po 12. týždni pri pacientmi hlásených prejavoch a príznakoch svrbenia vlasatej časti hlavy, bolesti a šupinatosti oproti východiskovým hodnotám v porovnaní s placebom.
Psoriatická artritída
Bezpečnosť a účinnosť sekukinumabu sa hodnotili u 1 999 pacientov v troch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických skúšaniach fázy III u pacientov s aktívnou psoriatickou artritídou (≥3 opuchnuté a ≥3 citlivé kĺby) aj napriek liečbe nesteroidným antiflogistikom
(NSAID), kortikosteroidom alebo antireumatickým liekom modifikujúcim chorobu (DMARD). Do
týchto klinických skúšaní boli zaradení pacienti s každým podtypom PsA, vrátane polyartikulárnej artritídy bez zjavných reumatoidných uzlíkov, spondylitídy s periférnou artritídou, asymetrickou periférnou artritídou, distálneho interfalangeálneho postihnutia a mutilujúcej artritídy. U pacientov v týchto klinických skúšaniach bola diagnostikovaná PsA trvajúca najmenej päť rokov. Väčšina pacientov mala aj aktívne psoriatické kožné lézie alebo zdokumentovanú psoriázu v anamnéze. Na začiatku liečby malo viac ako 61 % pacientov so PsA entezitídu a viac ako 42 % pacientov so PsA
daktylitídu. Primárnym koncovým ukazovateľom všetkých klinických skúšaní bola odpoveď na liečbu
ACR 20 podľa kritérií Americkej reumatologickej spoločnosti (American College of Rheumatology, ACR). V skúšaní 1 pri psoriatickej artritíde (skúšanie 1 pri PsA) a v skúšaní 2 pri psoriatickej artritíde (skúšanie 2 pri PsA) bol primárny koncový ukazovateľ ACR 20 vyhodnotený v 24. týždni.
V skúšaní 3 pri psoriatickej artritíde (skúšanie 3 pri PsA) bol primárny koncový ukazovateľ ACR 20 vyhodnotený v 16. týždni a kľúčový sekundárny koncový ukazovateľ, zmena modifikovaného celkového Sharpovho skóre (modified Total Sharp Score, mTSS) oproti východiskovému stavu, v
24. týždni.
V skúšaní 1 pri PsA, v skúšaní 2 pri PsA a v skúšaní 3 pri PsA bolo 29 %, 35 % a 30 % pacientov
v uvedenom poradí predtým liečených látkou anti-TNFα a ukončilo liečbu látkou anti-TNFα buď pre
nedostatočnú účinnosť, alebo pre intoleranciu (anti-TNFα-IR pacienti).
Skúšanie 1 pri PsA (FUTURE 1) vyhodnotilo 606 pacientov, z ktorých 60,7 % dostávalo súbežne
MTX. Pacienti randomizovaní na sekukinumab dostali intravenózne v 0., 2. a 4. týždni dávku
10 mg/kg, po ktorej nasledovali subkutánne dávky buď 75 mg, alebo 150 mg počnúc 8. týždňom
každý mesiac. Pacienti randomizovaní na placebo, ktorí nereagovali do 16. týždňa (včasná záchrana),
a ostatní pacienti, ktorí dostávali placebo, prešli na podávanie sekukinumabu (buď 75 mg, alebo
150 mg subkutánne) v 24. týždni, po ktorom nasledovala každý mesiac rovnaká dávka.
Skúšanie 2 pri PsA (FUTURE 2) vyhodnotilo 397 pacientov, z ktorých 46,6 % malo súbežne MTX.
Pacienti randomizovaní na sekukinumab dostali subkutánne v 0., 1., 2., 3. a 4. týždni dávku 75 mg,
150 mg alebo 300 mg, po ktorej nasledovala každý mesiac rovnaká dávka. Pacienti randomizovaní na
placebo, ktorí nereagovali do 16. týždňa (včasná záchrana), prešli na podávanie sekukinumabu (buď
150 mg, alebo 300 mg subkutánne) v 16. týždni, po ktorom nasledovala každý mesiac rovnaká dávka. Pacienti randomizovaní na placebo, ktorí nereagovali do 16. týždňa, prešli na podávanie sekukinumabu (buď 150 mg, alebo 300 mg subkutánne) v 24. týždni, po ktorom nasledovala každý
mesiac rovnaká dávka.
Skúšanie 3 pri PsA (FUTURE 5) vyhodnotilo 996 pacientov, z ktorých 50,1 % súbežne užívalo MTX.
Pacienti boli randomizovaní na subkutánne podávanie sekukinumabu 150 mg, 300 mg alebo placeba v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala každý mesiac rovnaká dávka, alebo injekcia sekukinumabu 150 mg raz za mesiac (bez nasycovania). Pacienti randomizovaní na placebo, ktorí nereagovali do 16. týždňa (včasná záchrana), prešli na podávanie sekukinumabu (buď 150 mg, alebo
300 mg subkutánne) v 16. týždni, po ktorom nasledovala každý mesiac rovnaká dávka. Pacienti
randomizovaní na placebo, ktorí reagovali do 16. týždňa, prešli na podávanie sekukinumabu (buď
150 mg, alebo 300 mg subkutánne) v 24. týždni, po ktorom nasledovala každý mesiac rovnaká dávka.
Prejavy a príznakyV 16. a 24. týždni mala liečba sekukinumabom za následok významné zlepšenie merané ako aktivita
ochorenia v porovnaní s placebom (pozri Tabuľku 6).
Tabuľka 6 Klinická odpoveď v skúšaní 2 pri PsA a skúšaní 3 pri PsA po 16. a 24. týždni
| Skúšanie 2 pri PsA Skúšanie 3 pri PsA
|
| Placebo 150 mg1 300 mg1
| Placebo 150 mg1 300 mg1
|
Počet
randomizovaných
pacientov
| 98 100 100
| 332 220 222
|
Odpoveď ACR20
n (% )
16. týždeň
24. týždeň
|
18 60 57
(18,4 %) (60,0 %***) (57,0 %***)
15◊ 51◊ 54◊
(15,3 %) (51,0 %***) (54,0 %***)
|
91◊ 122◊ 139◊
(27,4 %) (55,5 %***) (62,6 %***)
78 117 141
(23,5 %) (53,2 %***) (63,5 %***)
|
Odpoveď ACR50
n (% )
16. týždeň
24. týždeň
|
6 37 35
(6,1 %) (37,0 %***) (35,0 %***)
7 35 35
(7,1 %) (35,0 %) (35,0 %**)
|
27 79 88
(8,1 %) (35,9 %*) (39,6 %*)
29 86 97
(8,7 %) (39,1 %***) (43,7 %***)
|
Odpoveď ACR70
n (% )
16. týždeň
24. týždeň
|
2 17 15
(2,0 %) (17,0 %**) (15,0 %**)
1 21 20
(1,0 %) (21,0 %**) (20,0 %**)
|
14 40 45
(4,2 %) (18,2 %***) (20,3 %***)
13 53 57
(3,9 %) (24,1 %***) (25,7 %***)
|
DAS28-CRP
16. týždeň
24. týždeň
|
-0,50 -1,45*** -1,51***
-0,96 -1,58** -1,61**
|
-0,63 -1,29* -1,49*
-0,84 -1,57*** -1,68***
|
Počet pacientov
s kožou postihnutou psoriázou na ≥3 %
BSA na začiatku liečby
| 43 58 41
(43,9 %) (58,0 %) (41,0 %)
| 162 125 110
(48,8 %) (56,8 %) (49,5 %)
|
Odpoveď PASI 75
n (% )
16. týždeň
24. týždeň
|
3 33 27
(7,0 %) (56,9 %***) (65,9 %***)
7 28 26
(16,3 %) (48,3 %**) (63,4 %***)
|
20 75 77
(12,3 %) (60,0 %*) (70,0 %*)
29 80 78
(17,9 %) (64,0 %***) (70,9 %***)
|
O
dp
ov
e
ď PASI 90
n (% )
1
6
. týždeň
2
4
. týždeň
|
3 22 18
(7,0 %) (37,9 %***) (43,9 %***)
4 19 20
(9,3 %) (32,8 %**) (48,8 %***)
|
15 46 59
(9,3 %) (36,8 %*) (53,6 %*)
19 51 60
(11,7 %) (40,8 %***) (54,5 %***)
|
V
y
m
i
z
nut
i
e
d
a
k
t
y
l
i
t
í
d
y n (% ) †
1
6
. týždeň
2
4
. týždeň
|
10 21 26
(37 %) (65,6 %*) (56,5 %)
4 16 26
(14,8 %) (50,0 %**) (56,5 %**)
|
40 46 54
(32,3 %) (57,5 %*) (65,9 %*)
42 51 52
(33,9 %) (63,8 %***) (63,4 %***)
|
V
y
m
i
z
nut
i
e
e
n
t
e
z
i
t
í
d
y n (% ) ‡
1
6
. týždeň
2
4
. týždeň
|
17 32 32
(26,2 %) (50,0 %**) (57,1 %***)
14 27 27
(21,5 %) (42,2 %*) (48,2 %**)
|
68 77 78
(35,4 %) (54,6 %*) (55,7 %*)
66 77 86
(34,4 %) (54,6 %***) (61,4 %***)
|
* p<0,05; ** p<0,01; *** p<0,001; oproti placebu
Všetky hodnoty p sú upravené pre multiplicitu testovania na základe vopred definovanej hierarchie
v 24. týždni pre skúšanie 2 pri PsA, s výnimkou pre ACR70, daktylitídu a entezitídu, ktoré boli výskumné koncové ukazovatele, a všetky koncové ukazovatele v 16. týždni.
Všetky hodnoty p sú upravené pre multiplicitu testovania na základe vopred definovanej hierarchie
v 16. týždni pre skúšanie 3 pri PsA, s výnimkou pre ACR70, ktorá bola výskumný koncový ukazovateľ,
a všetky koncové ukazovatele v 24. týždni.
Chýbajúce údaje boli vyhodnotené ako bez odpovede pre binárny koncový ukazovateľ.
ACR: Americká reumatologická spoločnosť (American College of Rheumatology); PASI: index plochy a
závažnosti psoriázy (Psoriasis Area and Severity Index); DAS: skóre aktivity ochorenia (Disease Activity
Score); BSA: plocha povrchu tela (Body Surface Area)
◊Primárny koncový ukazovateľ
1Sekukinumab 150 mg alebo 300 mg s.c. v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala každý mesiac
rovnaká dávka
†U pacientov s daktylitídou na začiatku liečby (n=27, 32, 46, v uvedenom poradí, pre skúšanie 2 pri PsA, a n=124, 80, 82, v uvedenom poradí, pre skúšanie 3 pri PsA)
‡U pacientov s entezitídou na začiatku liečby (n=65, 64, 56, v uvedenom poradí, pre skúšanie 2 pri PsA, a
n=192, 141, 140, v uvedenom poradí, pre skúšanie 3 pri PsA)
|
K nástupu účinku sekukinumabu došlo už v 2. týždni. V 3. týždni sa dosiahol štatisticky významný
rozdiel v ACR 20 oproti placebu.
Percentuálny podiel pacientov, ktorí dosiahli odpoveď ACR 20 pri návšteve, je znázornený na
Obrázku 2.
O
b
r
á
z
o
k 2 Odpoveď ACR20 v skúšaní 2 pri PsA v čase až do 52. týždňa



Čas (týždne)
Podobné odpovede pre primárne a kľúčové sekundárne koncové ukazovatele sa zaznamenali
u pacientov so PsA bez ohľadu na to, či boli súbežne liečení MTX, alebo nie. V skúšaní 2 pri PsA v 24. týždni mali pacienti liečení sekukinumabom so súbežne použitým MTX vyššiu odpoveď ACR 20 (47,7 % pri 150 mg a 54,4 % pri 300 mg v porovnaní s 20,0 % pri placebe) a odpoveď
ACR 50 (31,8 % pri 150 mg a 38,6 % pri 300 mg v porovnaní s 8,0 % pri placebe). Pacienti liečení
sekukinumabom bez súbežne použitého MTX mali vyššiu odpoveď ACR 20 (53,6 % pri 150 mg a 53,6 % pri 300 mg v porovnaní s 10,4 % pri placebe) a odpoveď ACR 50 (37,5 % pri 150 mg
a 32,1 % pri 300 mg v porovnaní s 6,3 % pri placebe).
V skúšaní 2 pri PsA anti-TNFα-naivní pacienti liečení sekukinumabom, ako aj anti-TNFα-IR pacienti liečení sekukinumabom mali v 24. týždni významne vyššiu odpoveď ACR 20 v porovnaní s placebom, s mierne vyššou odpoveďou v skupine anti-TNFα-naivných pacientov (anti-TNFα-naivní pacienti:
64 % pri 150 mg a 58 % pri 300 mg v porovnaní s 15,9 % pri placebe; anti-TNFα-IR: 30 % pri 150 mg a 46 % pri 300 mg v porovnaní so 14,3 % pri placebe). V podskupine anti-TNFα-IR pacientov
vykázala len dávka 300 mg významne vyššiu mieru odpovede pre ACR 20 v porovnaní s placebom (p<0,05) a preukázala klinicky významný prínos oproti dávke 150 mg pre viaceré sekundárne koncové ukazovatele. Zlepšenia v odpovedi PASI 75 sa pozorovali v oboch podskupinách a pri dávke 300 mg
sa preukázal štatisticky významný prínos u anti-TNFα-IR pacientov.
Zlepšenie sa preukázalo vo všetkých zložkách skóre ACR, vrátane hodnotenia bolesti pacientom. V skúšaní 2 pri PsA bol podiel pacientov, ktorí dosiahli odpoveď podľa modifikovaného kritéria odpovede na liečbu PsA (PsA Response Criteria, PsARC), v 24. týždni väčší u pacientov liečených sekukinumabom (59,0 % pri 150 mg a 61,0 % a pri 300 mg) v porovnaní s placebom (26,5 %).
V skúšaní 1 pri PsA a v skúšaní 2 pri PsA sa účinnosť zachovala do 104. týždňa. V skúšaní 2 pri PsA
bolo spomedzi 200 pacientov, ktorí boli pôvodne randomizovaní na sekukinumab 150 mg a 300 mg, v 52. týždni 178 (89 %) pacientov stále liečených. Zo 100 pacientov randomizovaných na sekukinumab 150 mg malo 64 pacientov odpoveď ACR 20, 39 pacientov malo odpoveď ACR 50
a 20 pacientov malo odpoveď ACR 70. Zo 100 pacientov randomizovaných na sekukinumab 300 mg malo 64 pacientov odpoveď ACR 20, 44 pacientov malo odpoveď ACR 50 a 24 pacientov malo odpoveď ACR 70.
Rádiografická odpoveďV skúšaní 3 pri PsA sa inhibícia progresie štrukturálneho poškodenia hodnotila rádiograficky
a vyjadrila sa ako zmena modifikovaného celkového Sharpovho skóre (mTSS) a jeho zložiek, skóre
erózie (Erosion Score, ES) a skóre zúženia kĺbovej štrbiny (Joint Space Narrowing, JSN). Röntgenové snímky rúk, zápästí a nôh sa urobili na začiatku liečby, v 16. a/alebo 24. týždni a vyhodnotili sa nezávisle najmenej dvomi hodnotiteľmi, pre ktorých boli zaslepené skupina liečby a číslo návštevy. Liečba sekukinumabom 150 mg a 300 mg významne znížila mieru progresie poškodenia periférnych
kĺbov v porovnaní s liečbou placebom, stanovenú ako zmena mTSS v 24. týždni oproti východiskovému stavu (Tabuľka 7).
Inhibícia progresie štrukturálneho poškodenia sa hodnotila aj v skúšaní 1 pri PsA v 24. a 52. týždni
oproti východiskovému stavu. Údaje v 24. týždni sú uvedené v Tabuľke 7.
Tabuľka 7 Zmena modifikovaného celkového Sharpovho skóre pri psoriatickej artritíde
| Skúšanie 3 pri PsA
Placebo sekukinumab sekukinumab n=296 150 mg1 300 mg1
n=213 n=217
| Skúšanie 1 pri PsA
Placebo sekukinumab n=179 150 mg2
n=185
|
Celkové skóre
|
Východiskový stav
(SD)
| 15,0 13,5 12,9
(38,2) (25,6) (23,8)
| 28,4 22,3
(63,5) (48,0)
|
Priemerná
zmena
v 24. týždni
| 0,50 0,13* 0,02*
| 0,57 0,13*
|
*p<0,05 na základe nominálnej, ale nie upravenej hodnoty p
1sekukinumab 150 mg alebo 300 mg s.c. v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala rovnaká dávka každý mesiac
210 mg/kg v 0., 2. a 4. týždni, po ktorých nasledovali subkutánne dávky 75 mg alebo 150 mg
|
V skúšaní 1 pri PsA sa inhibícia štrukturálneho poškodenia zachovala pri liečbe sekukinumabom až do
52. týždňa.
V skúšaní 3 pri PsA bol percentuálny podiel pacientov bez progresie ochorenia (definované ako zmena mTSS oproti východiskovému stavu ≤0,5) od randomizácie do 24. týždňa 80,3 % pri 150 mg a 88,5 % pri 300 mg sekukinumabu a 73,6 % pri placebe. Inhibičný účinok na štrukturálne poškodenie sa pozoroval u pacientov bez predchádzajúcej liečby anti-TNFα a u pacientov anti-TNFα-IR a
u pacientov so súbežnou liečbou MTX, ako aj bez nej.
V skúšaní 1 pri PsA percentuálny podiel pacientov bez progresie ochorenia (definované ako zmena mTSS oproti východiskovému stavu ≤0,5) od randomizácie do 24. týždňa bol 82,3 % pri intravenóznej bolusovej dávke sekukinumabu 10 mg/kg – udržiavacia subkutánna dávka 150 mg, a 75,7 % pri placebe. Percentuálny podiel pacientov bez progresie ochorenia od 24. týždňa do 52. týždňa pri intravenóznej nasycovacej dávke sekukinumabu 10 mg/kg – po ktorej nasledovala subkutánna udržiavacia liečba 150 mg, a u pacientov, ktorí prešli z placeba na 75 mg alebo 150 mg subkutánne každé 4 týždne po 16. týždni alebo 24. týždni, bol 85,7 % a 86,8 % v uvedenom poradí.
Axiálne prejavy pri PsA
Randomizované, dvojito zaslepené, placebom kontrolované klinické skúšanie (MAXIMISE) hodnotilo účinnosť sekukinumabu u 485 pacientov s PsA s axiálnymi prejavmi, ktorí v minulosti neboli liečení biologickými liekmi a nemali dostatočnú odpoveď na NSAID. Primárny cieľ skúšania, ktorým bolo minimálne 20 % zlepšenie stavu pacientov hodnotené podľa kritérií Medzinárodnej spoločnosti pre spondyloartritídu (ASAS 20) po 12 týždňoch liečby, bol splnený. V porovnaní s placebom viedla
liečba sekukinumabom v dávkach 300 mg a 150 mg k výraznejšiemu zlepšeniu prejavov a príznakov
(vrátane zníženia bolesti chrbta oproti východiskovému stavu) a zlepšeniu fyzickej kondície (pozri
Tabuľku 8).
Tabuľka 8 Klinická odpoveď v skúšaní MAXIMISE v 12. týždni
| Placebo
(n=164)
| 150 mg
(n=157)
| 300 mg
(n=164)
|
Odpoveď ASAS 20, % (95% IS)
| 31,2 (24,6; 38,7)
| 66,3 (58,4; 73,3)*
| 62,9 (55,2; 70,0)*
|
Odpoveď ASAS 40, %
(95% IS)
| 12,2 (7,8; 18,4)
| 39,5 (32,1; 47,4)**
| 43,6 (36,2; 51,3)**
|
BASDAI 50, %
(95% IS)
| 9,8 (5,9; 15,6)
| 32,7 (25,8; 40,5)**
| 37,4 (30,1; 45,4)**
|
Bolesť chrbta, VAS
(95% IS)
| -13,6 (-17,2; -10,0)
| -28,5 (-32,2; -24,8)**
| -26,5 (-30,1; -22,9)**
|
Fyzická kondícia,
HAQ-DI (95% IS)
| -0,155 (-0,224; -0,086)
| -0,330 (-0,401;
-0,259)**
| -0,389 (-0,458;
-0,320)**
|
* p<0,0001; oproti placebu s využitím viacnásobnej imputácie.
** Porovnanie oproti placebu nebolo upravené z dôvodu multiplicity.
ASAS: kritériá pre hodnotenie podľa Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society Criteria); BASDAI: index aktivity ankylozujúcej
spondylitídy z Bathu (Bath Ankylosing Spondylitis Disease Activity Index); VAS: vizuálna analógová stupnica (Visual Analog Scale); HAQ-DI: dotazník na hodnotenie zdravia podľa indexu invalidity (Health Assessment Questionnaire – Disability Index).
|
Pre obe dávky sekukinumabu sa pozorovalo zlepšenie odpovedí ASAS 20 a ASAS 40 v 4. týždni a
zachovalo sa až do 52. týždňa.
Fyzická kondícia a kvalita života súvisiaca so zdravímV skúšaní 2 pri PsA a v skúšaní 3 pri PsA sa v 24. týždni a 16. týždni, v uvedenom poradí, pomocou
dotazníka na hodnotenie zdravia podľa indexu invalidity (Health Assessment Questionnaire -Disability
Index, HAQ-DI) preukázalo u pacientov liečených sekukinumabom 150 mg (p=0,0555 a p<0,0001)
a 300 mg (p=0,0040 a p<0,0001) zlepšenie fyzickej kondície v porovnaní s pacientmi, ktorí dostávali
placebo. Zlepšenia skóre HAQ-DI sa pozorovali bez ohľadu na predchádzajúcu expozíciu anti-TNFα.
Podobné odpovede sa pozorovali v skúšaní 1 pri PsA.
Pacienti liečení sekukinumabom hlásili významné zlepšenia kvality života súvisiacej so zdravím podľa sumárneho skóre stručného formulára prieskumu fyzického zdravia (Short Form-36 Health Survey Physical Component Summary, SF-36 PCS) (p<0,001). Došlo tiež k štatisticky významnému
zlepšeniu preukázanému výskumnými ukazovateľmi, stanovenému prostredníctvom skóre funkčného posúdenia liečby chronického ochorenia – únava (Functional Assessment of Chronic Illness Therapy – Fatigue, FACIT-F) pri 150 mg a 300 mg v porovnaní s placebom (7,97 a 5,97 oproti 1,63) a toto zlepšenie sa zachovalo až do 104. týždňa v skúšaní 2 pri PsA.
Podobné odpovede sa pozorovali v skúšaní 1 pri PsA a účinnosť sa zachovala až do 52. týždňa.
Axiálna spondyloartritída (axSpA)
Ankylozujúca spondylitída (AS) / Axiálna spondyloartritída s rádiografickým dôkazom
Bezpečnosť a účinnosť sekukinumabu sa hodnotili u 816 pacientov v troch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických skúšaniach fázy III u pacientov s aktívnou ankylozujúcou spondylitídou (AS) s indexom aktivity ankylozujúcej spondylitídy z Bathu (Bath Ankylosing Spondylitis Disease Activity Index, BASDAI) ≥4 aj napriek liečbe nesteroidným antiflogistikom (NSAID), kortikosteroidom alebo antireumatickým liekom modifikujúcim chorobu (DMARD). U pacientov v skúšaní 1 pri ankylozujúcej spondylitíde (skúšanie 1 pri AS)
a v skúšaní 2 pri ankylozujúcej spondylitíde (skúšanie 2 pri AS) bola diagnostikovaná AS so strednou hodnotou trvania 2,7 až 5,8 rokov. Primárnym koncovým ukazovateľom oboch klinických skúšaní bolo zlepšenie hodnotené podľa kritérií Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society, ASAS 20) o najmenej 20% v 16. týždni.
V skúšaní 1 pri ankylozujúcej spondylitíde (skúšanie 1 pri AS), v skúšaní 2 pri ankylozujúcej
spondylitíde (skúšanie 2 pri AS) a v skúšaní 3 pri ankylozujúcej spondylitíde (skúšanie 3 pri AS) bolo
27,0 %, 38,8 % a 23,5 % pacientov v uvedenom poradí predtým liečených anti-TNFα látkou
a ukončilo liečbu látkou anti-TNFα buď pre nedostatočnú účinnosť, alebo intoleranciu (anti-TNFα-IR
pacienti).
Skúšanie 1 pri AS (MEASURE 1) vyhodnotilo 371 pacientov, z ktorých 14,8 % dostávalo súbežne MTX a 33,4 % dostávalo súbežne sulfasalazín. Pacienti randomizovaní na sekukinumab dostali intravenózne v 0., 2. a 4. týždni dávku 10 mg/kg, po ktorej nasledovali každý mesiac subkutánne dávky buď 75 mg, alebo 150 mg počnúc 8. týždňom. Pacienti randomizovaní na placebo, ktorí nereagovali do 16. týždňa (včasná záchrana), a všetci ostatní pacienti, ktorí dostávali placebo, prešli v 24. týždni na podávanie sekukinumabu (buď 75 mg, alebo 150 mg subkutánne), po ktorom nasledovala každý mesiac rovnaká dávka.
Skúšanie 2 pri AS (MEASURE 2) vyhodnotilo 219 pacientov, z ktorých 11,9 % dostávalo súbežne MTX a 14,2 % dostávalo súbežne sulfasalazín. Pacienti randomizovaní na sekukinumab dostali subkutánne v 0., 1., 2., 3. a 4. týždni dávku buď 75 mg, alebo 150 mg, po ktorej nasledovala každý mesiac rovnaká dávka. Pacienti randomizovaní na placebo pri zaradení do skúšania boli opäť randomizovaní v 16. týždni na podávanie sekukinumabu (buď 75 mg, alebo 150 mg subkutánne) každý mesiac.
Skúšanie 3 pri AS (MEASURE 3) vyhodnotilo 226 pacientov, z ktorých 13,3 % a 23,5 % dostávalo súbežne MTX alebo sulfasalazín, v uvedenom poradí. Pacienti randomizovaní na sekukinumab dostali intravenózne dávku 10 mg/kg v 0., 2. a 4. týždni, po ktorej nasledovala buď 150 mg alebo 300 mg dávka subkutánne každý mesiac. Pacienti randomizovaní na placebo pri zaradení do skúšania boli
opäť randomizovaní v 16. týždni na podávanie sekukinumabu (buď 150 mg, alebo 300 mg subkutánne) každý mesiac. Primárnym koncovým ukazovateľom bolo ASAS 20 v 16. týždni. Pacienti boli zaslepení na liečebný režim až do 52. týždňa a štúdia pokračovala do 156 týždňa.
Prejavy a príznaky:
V skúšaní 2 pri AS mala v 16. týždni liečba sekukinumabom 150 mg za následok väčšie zlepšenie
merané ako aktivita ochorenia v porovnaní s placebom (pozri Tabuľku 9).
 T
a
b
u
ľ
k
a 9 Klinická odpoveď v skúšaní 2 pri AS v 16. týždni
T
a
b
u
ľ
k
a 9 Klinická odpoveď v skúšaní 2 pri AS v 16. týždni
V
ý
sledok (hodnota p oproti placebu)
|
Placebo
(
n = 74)
|
7
5 mg
(
n = 73)
|
1
5
0 mg
(
n = 72)
|
Odpoveď ASAS 20, %
|
28,4
|
41,1
|
61,1***
|
Odpoveď ASAS 40, %
|
10,8
|
26,0
|
36,1***
|
hsCRP, (pomer post-BSL/BSL)
|
1,13
|
0,61
|
0,55***
|
ASAS 5/6, %
|
8,1
|
34,2
|
43,1***
|
Parciálna remisia ASAS, %
|
4,1
|
15,1
|
13,9
|
BASDAI 50, %
|
10,8
|
24,7*
|
30,6**
|
Výrazné zlepšenie ASDAS-CRP
|
4,1
|
15,1*
|
25,0***
|
* p<0,05; ** p<0,01; *** p<0,001; oproti placebu
Všetky hodnoty p sú upravené pre multiplicitu testovania na základe vopred definovanej hierarchie,
s výnimkou BASDAI 50 a ASDAS-CRP
Chýbajúce údaje boli vyhodnotené ako bez odpovede pre binárny koncový ukazovateľ
ASAS: kritériá pre hodnotenie podľa Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society Criteria); BASDAI: index aktivity ankylozujúcej spondylitídy z Bathu (Bath Ankylosing Spondylitis Disease Activity Index); hsCRP: vysoko citlivý C-reaktívny proteín (high-sensitivity C-reactive protein); ASDAS: skóre aktivity ankylozujúcej spondylitídy (Ankylosing Spondylitis Disease Activity Score); BSL: východiskový stav (baseli ne)
|
K nástupu účinku sekukinumabu 150 mg došlo už v 1. týždni pre ASAS 20 a v 2. týždni pre ASAS 40
(účinnejší ako placebo) v skúšaní 2 pri AS.
V 16. týždni sa u anti-TNFα-naivných pacientov (68,2 % oproti 31,1 %; p<0,05) a u anti-TNFα-IR
pacientov (50,0 % oproti 24,1 %; p<0,05) zlepšili odpovede ASAS 20 pre sekukinumab 150 mg v porovnaní s placebom, v uvedenom poradí.
V skúšaniach 1 pri AS a 2 pri AS sa u pacientov liečených sekukinumabom (150 mg v skúšaní 2 pri
AS a oba režimy v skúšaní 1 pri AS) preukázalo v 16. týždni významné zlepšenie prejavov
a príznakov, s porovnateľnou veľkosťou odpovede a účinnosťou zachovanou až do 52. týždňa
u anti-TNFα-naivných pacientov, ako aj u anti-TNFα-IR pacientov. V skúšaní 2 pri AS bolo spomedzi
72 pacientov, ktorí boli na začiatku randomizovaní na sekukinumab 150 mg, v 52. týždni 61 (84,7 %)
pacientov stále liečených. Zo 72 pacientov randomizovaných na sekukinumab 150 mg malo
45 pacientov odpoveď ASAS 20 a 35 pacientov malo odpoveď ASAS 40.
V skúšaní 3 pri AS preukázali pacienti liečení sekukinumabom (150 mg a 300 mg) zlepšenie prejavov a symptómov a mali porovnateľné výsledky účinnosti bez ohľadu na dávku, ktorá bola v primárnom cieľovom ukazovateli (ASAS 20) v 16. týždni lepšia ako placebo. Celkovo bola miera účinnosti odpovede pre sekundárne cieľové ukazovatele konzistentne vyššia v skupine s 300 mg v porovnaní so skupinou so 150 mg. Počas zaslepého obdobia boli v 52. týždni odpovede ASAS 20 a ASAS 40
69,7 % a 47,6 % pre 150 mg a 74,3 % a 57,4 % pre 300 mg. Odpovede ASAS 20 a ASAS 40 sa
udržiavali až do 156.týždňa (69,5 % a 47,6 % pre 150 mg oproti 74,8 % a 55,6 % pre 300 mg). Vyššia
miera odpovede v prospech 300 mg sa pozorovala aj pri odpovedi na čiastočnú remisiu ASAS (ASAS PR) v 16. týždni a udržala sa až do týždňa 156. U pacientov s anti-TNFα-IR (n=36) sa v porovnaní s pacientmi bez predchádzajúcej liečby TNFα (n=114) pozorovali väčšie rozdiely v miere odpovede
v prospech 300 mg oproti 150 mg.
Mobilita chrbtice:
U pacientov liečených sekukinumabom 150 mg sa preukázalo zlepšenie mobility chrbtice stanovené ako zmena oproti východiskovému stavu pomocou BASMI po 16. týždni v skúšaní 1 pri AS (-0,40 oproti -0,12 pri placebe; p=0,0114) a v skúšaní 2 pri AS (-0,51 oproti -0,22 pri placebe; p=0,0533). Tieto zlepšenia sa zachovali až do 52. týždňa.
Fyzická kondícia a kvalita života súvisiaca so zdravím:
V skúšaní 1 a skúšaní 2 pri AS sa u pacientov liečených sekukinumabom 150 mg preukázalo zlepšenie
kvality života súvisiacej so zdravím hodnotenej podľa dotazníka o kvalite života pri AS (AS Quality of Life Questionnaire, AS QoL) (p=0,001) a podľa sumárneho skóre fyzickej zložky SF-36 (SF-36
Physical Component Summary, SF-36 PCS) (p<0,001). U pacientov liečených sekukinumabom
150 mg sa tiež preukázalo štatisticky významné zlepšenie výskumných ukazovateľov týkajúcich sa fyzickej kondície, hodnotené bathskym funkčným indexom ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Functional Index, BASFI) v porovnaní s placebom (-2,15 oproti -0,68) a škály únavy pri liečbe chronického ochorenia (Functional Assessment of Chronic Illness Therapy-Fatigue,
FACIT-Fatigue) v porovnaní s placebom (8,10 oproti 3,30). Tieto zlepšenia sa zachovali až do
52. týždňa.
Axiálna spondyloartritída bez rádiografického dôkazu (nr-axSpA)
Bezpečnosť a účinnosť sekukinamubu sa hodnotili u 555 pacientov v jednom randomizovanom, dvojito zaslepenom, placebom kontrolovanom klinickom skúšaní fázy III (PREVENT), pozostávajúcom z 2-ročnej základnej fázy a 2-ročnej fázy predĺženia, u pacientov s aktívnou axiálnou
spondyloartritídou bez rádiografického dôkazu (nr-axSpA), ktorí spĺňali kritériá klasifikácie
Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society, ASAS) pre axiálnu spondyloartritídu bez rádiografického dôkazu zmien sakroiliakálnych kĺbov, ktoré by spĺňali modifikované newyorské kritériá pre ankylozujúcu spondylitídu (AS). Zaradení pacienti mali aktívne ochorenie, definované indexom aktivity ankylozujúcej spondylitídy z Bathu (Bath Ankylosing Spondylitis Disease Activity Index, BASDAI) ≥4 a celkovou bolesťou chrbta ≥40 (na škále 0-100 mm) vizuálnej analógovej stupnice (Visual Analogue Scale,VAS) napriek prebiehajúcej alebo predchádzajúcej liečbe nesteroidnými protizápalovými liekmi (NSAID) a zvýšený C-reaktívny proteín (CRP) a/alebo dôkaz sakroiliitídy pri zobrazení magnetickou rezonanciou (MRI). Pacienti v tomto klinickom skúšaní mali diagnózu axSpA priemerne 2,1 až 3,0 roka a 54 % účastníkov klinického skúšania boli ženy.
V klinickom skúšaní PREVENT bolo 9,7 % pacientov predtým liečených anti-TNFα látkou a ukončilo liečbu anti-TNFα látkou buď pre nedostatočnú účinnosť, alebo intoleranciu (anti-TNFα-IR pacienti).
V klinickom skúšaní PREVENT dostávalo 9,9 % pacientov súbežne MTX a 14,8 % pacientov
sulfasalazín. V období dvojitého zaslepenia dostávali pacienti buď placebo alebo sekukinumab počas
52 týždňov. Pacienti randomizovaní na sekukinumab dostali subkutánne v 0., 1., 2., 3. a 4. týždni
dávku 150 mg, po ktorej nasledovala, počnúc 4. týždňom, každý mesiac rovnaká dávka, alebo raz
mesačne 150 mg injekcia sekukinumabu. Primárnym koncovým ukazovateľom bolo najmenej 40 % zlepšenie stavu hodnotené podľa kritérií Medzinárodnej spoločnosti pre spondyloartritíd u (ASAS 40) v 16. týždni u anti-TNFα-naivných pacientov.
Prejavy a príznaky:
V klinickom skúšaní PREVENT viedla liečba sekukinumabom 150 mg v 16. týždni k významnému zlepšeniu miery aktivity ochorenia v porovnaní s placebom. Tieto merania zahŕňajú ASAS 40,
ASAS 5/6, skóre BASDAI, BASDAI 50, vysoko citlivý CRP (hsCRP), ASAS 20 a dosiahnutie
čiastočnej remisie ASAS v porovnaní s placebom (Tabuľka 10). Odpovede pretrvali až do 52. týždňa.
 T
a
b
u
ľ
k
a 10 Klinická odpoveď v skúšaní PREVENT v 16. týždni
T
a
b
u
ľ
k
a 10 Klinická odpoveď v skúšaní PREVENT v 16. týždni
V
ý
sledok (hodnota oproti placebu)
|
Placebo
|
1
5
0 mg
1
|
Počet zaradených anti-TNFα-naivných pacientov
|
1
7
1
|
1
6
4
|
Odpoveď ASAS 40, %
|
29,2
|
41,5*
|
C
el
k
o
v
ý počet zaradených pacientov
|
1
8
6
|
1
8
5
|
Odpoveď ASAS 40, %
|
28,0
|
40,0*
|
ASAS 5/6, %
|
23,7
|
40,0*
|
BASDAI, LS priemerná zmena oproti východiskovej hodnote
|
-1,46
|
-2,35*
|
BASDAI 50, %
|
21,0
|
37,3*
|
hsCRP, (pomer post-BSL/BSL)
|
0,91
|
0,64*
|
Odpoveď ASAS 20, %
|
45,7
|
56,8*
|
Čiastočná remisia ASAS, %
|
7,0
|
21,6*
|
*p<0,05 oproti placebu
Všetky hodnoty p sú upravené pre multiplicitu testovania na základe vopred definovanej hierarchie
Chýbajúce údaje boli vyhodnotené ako bez odpovede pre binárny koncový ukazovateľ
1sekukinumab 150 mg s.c. v 0., 1., 2., 3., a 4. týždni po ktorých nasledovala každý mesiac rovnaká
dávka.
ASAS: kritériá pre hodnotenie podľa Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society Criteria); BASDAI: index aktivity ankylozujúcej spondylitídy z Bathu (Bath Ankylosing Spondylitis Disease Activity Index); hsCRP: vysoko citlivý C-reaktívny proteín (high-sensitivity C-reactive protein); BSL: východiskový stav (baseline); LS: metóda najmenších štvorcov (Least square)
|
V klinickom skúšaní PREVENT sa u anti-TNFα-naivných pacientov prejavil nástup účinku
sekukinumabu v dávke150 mg v hodnotách ASAS 40 už v 3. týždni (lepší účinok ako placebo). Percentuálny podiel anti-TNFα-naivných pacientov, ktorí dosiahli odpoveď ASAS 40 je znázornený na obrázku 3.
O
b
r
á
z
o
k 3 OdpoveďASAS 40 v skúšaní PREVENT u anti-TNFα-naivných pacientov čase až
d
o 16.týždňa
Percentuálny podiel pacientov s odpoveďou na liečbu
Čas (týždne)

Sekukinumab 150 mg dávka

Placebo
V 16. týždni sa zlepšila odpoveď ASAS 40 aj u anti-TNFα-IR pacientov liečených sekukinumabom
150 mg v porovnaní s placebom.
Fyzická kondícia a kvalita života súvisiaca so zdravím:
Pacienti liečení sekukinumabom 150 mg preukázali v 16. týždni štatisticky významné zlepšenia telesnej funkcie, na základe zmeny hodnoty indexu BASFI, v porovnaní s pacientami užívajúcimi placebo (16. týždeň: -1,75 oproti -1,01, p<0,05). Pacienti liečení sekukinumabom preukázali v
porovnaní s pacientami užívajúcimi placebo v 16. týždni štatisticky významné zlepšenia kvality života
súvisiacej so zdravím, meranej pomocou ASQoL (priemerná zmena LS v 16. týždni: -3,45
oproti -1,84, p<0,05) a podľa sumárneho skóre fyzickej zložky SF-36 (Physical Component Summary,
SF-36 PCS) (priemerná zmena LS v 16. týždni: 5,71 oproti 2,93, p<0,05). Tieto zlepšenia sa zachovali
až do 52.týždňa.
Mobilita chrbtice:
Mobilita chrbtice sa hodnotila pomocou indexu BASMI až do 16. týždňa. Vo 4., 8., 12. a 16. týždni sa preukázali numericky vyššie hodnoty zlepšenia u pacientov užívajúcich sekukinumab v porovnaní s pacientami užívajúcimi placebo.
Inhibícia zápalu pri zobrazení magnetickou rezonanciou (MRI):
Prejavy zápalu sa hodnotili MRI pri zaradení do skúšania a v 16. týždni a vyjadrili sa zmenou skóre Berlin SI-joint oedema pre sakroiliakálne kĺby, skóre ASspiMRI-a a Berlin spine skóre pre chrbticu oproti východiskovému stavu. U pacientov liečených sekukinumabom sa pozorovala inhibícia
zápalových prejavov pri sakroiliakálnych kĺboch aj pri chrbtici. Priemerná zmena voči východiskovej hodnote Berlin SI-joint oedema skóre bola -1,68 u pacientov liečených sekukinumabom 150 mg (n=180) oproti -0,39 u pacientov (n=174) (p<0,05).
Pediatrická populáciaLožiskovápsoriázau pediatrických pacientovPreukázalo sa, že sekukinumab zlepšuje prejavy a príznaky, ako aj kvalitu života súvisiacu so zdravím
u pediatrických pacientov s ložiskovou psoriázou vo veku 6 rokov a starších (pozri Tabuľky 12 a 14).
Z
á
v
a
ž
n
á ložisková psoriáza
Bezpečnosť a účinnosť sekukinumabu sa hodnotili v randomizovanom, dvojito zaslepenom klinickom skúšaní fázy III, kontrolovanom placebom a etanerceptom, u pediatrických pacientov so závažnou ložiskovou psoriázou vo veku 6 až <18 rokov, definovanou skóre PASI ≥20 a skóre IGA mod 2011 4
a rozsahom postihnutia kože s BSA ≥10%, ktorí boli kanditátmi na systémovú liečbu. Približne 43 % pacientov malo predchádzajúcu expozíciu fototerapii, 53 % konvenčnej systémovej terapii, 3 % biologickým látkam a 9 % malo sprievodnú psoriatickú artritídu.
V pediatrickom skúšaní 1 sa vyhodnotilo 162 pacientov, randomizovaných na podávanie nízkej dávky sekukinumabu (75 mg na telesnú hmotnosť <50 kg alebo 150 mg na telesnú hmotnosť ≥50 kg), vysokej dávky sekukinumabu (75 mg na telesnú hmotnosť <25 kg, 150 mg na telesnú hmotnosť medzi
≥25 kg a <50 kg, alebo 300 mg na telesnú hmotnosť ≥50 kg), alebo placeba v 0., 1., 2., 3. a 4. týždni,
po ktorých nasledovala každé 4 týždne rovnaká dávka, alebo etanercept. Patienti radomizovaní na etanercept dostávali 0,8 mg/kg týždenne (najviac však 50 mg). Rozdelenie pacientov podľa hmotnosti a veku pri randomizácii je uvedené v Tabuľke 11.
Tabuľka 11 Rozdelenie pacientov podľa hmotnosti a veku v pediatrickom skúšaní 1 pri psoriázeRandomizačná
vrstva
| Popis
| Sekukinumab nízka dávka n=40
| Sekukinumab vysoká dávka n=40
| Placebo
n=41
| Etanercept
n=41
| Spolu
N=162
|
Vek
| 6-<12 rokov
| 8
| 9
| 10
| 10
| 37
|
≥12-
<18 rokov
| 32
| 31
| 31
| 31
| 125
|
Hmotnosť
| <25 kg
| 2
| 3
| 3
| 4
| 12
|
≥25-<50 kg
| 17
| 15
| 17
| 16
| 65
|
≥50 kg
| 21
| 22
| 21
| 21
| 85
|
Pacienti randomizovaní na placebo, ktorí nedosiahli odpoveď do 12.týždňa prešli do skupiny s nízkou
alebo vysokou dávkou sekukinumabu (dávka na základe skupiny telesnej hmotnosti) a dostali skúšaný
liek v 12., 13., 14. a 15.týždni, po ktorom nasledovala každé 4 týždne rovnaká dávka, začínajúca
v 16.týždni. Spoločné primárne koncové ukazovatele boli podiely pacientov, ktorí dosiahli odpoveď
PASI 75 a IGA mod 2011 „bez prejavov“ alebo“takmer bez prejavov“ (0 alebo 1) v 12.týždni.
Účinnosť v oboch skupinách s nízkou a vysokou dávkou sekukinumabu počas placebom kontrolovaného obdobia do 12.týždňa bola porovnateľná pre spoločné primárne koncové ukazovatele. Odhadovaný pomer pravdepodobnosti v prospech oboch dávok sekukinumabu bol štatisticky významný pre odpovede PASI 75 a IGA mod 2011 0 alebo 1.
U všetkých pacientov sa sledovala účinnosť a bezpečnosť v priebehu 52 týždňov od prvého podania skúšaného lieku. Podiel pacientov, ktorí dosiahli PASI 75 a IGA mod 2011 „bez prejavov“ alebo
„takmer bez prejavov“ (0 alebo 1) preukázal rozdiel medzi skupinou liečenou so sekukinumabom
a skupinou s placebom pri prvej návšteve po východiskovej návšteve vo 4.týždni a výraznejší rozdiel nastal v 12.týždni. Odpoveď pretrvala počas 52-týždňového časového obdobia (pozri Tabuľku 12). Zlepšenie miery odpovede vyjadrené v hodnotách PASI 50, 90, 100 a Detského dermatologického
indexu kvality života (Children’s Dermatology Life Quality Index, (CDLQI)) zo skóre 0 alebo 1
taktiež pretrval počas 52-týždňového obdobia.
Naviac miera odpovede PASI 75, IGA 0 alebo 1, PASI 90 v 12. a 52. týždni pre obe skupiny s nízkou a vysokou dávkou sekukinumabu bola vyššia ako miera odpovede u pacientov liečených
s etanerceptom (pozri Tabuľku 12).
Po 12.týždni bola účinnosť oboch skupín s nízkou a vysokou dávkou sekukinumabu porovnateľná, hoci účinnosť s vyššou dávkou sekukinumabu bola vyššia u pacientov ≥50 kg. Profily bezpečnosti s nízkou dávkou a vysokou dávkou boli porovnateľné a v zhode s profilom bezpečnosti u dospelých pacientov.
Tabuľka 12 Súhrn klinickej odpovede u pediatrických pacientov so závažnou psoriázou v 12. a 52.týždni (pediatrické skúšanie 1 pri psoriáze)*
Kritérium odpovede
| Porovnanie liečby
| „test“
| „kontrola”
| odhadovaný pomer pravdepodobnosti (95 % IS)
|
Hodnota p
|
„test“ vs. „kontrola“
|
n**/m ( %)
|
n**/m ( %)
|
v 12.týždni***
|
PASI 75
| sekukinumab nízka dávka vs. placebo
sekukinumab vysoká dávka vs. placebo
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
| 32/40 (80,0)
31/40 (77,5)
32/40 (80,0)
31/40 (77,5)
| 6/41 (14,6)
6/41 (14,6)
26/41 (63,4)
26/41 (63,4)
| 25,78 (7,08; 114,66)
22,65 (6,31; 98,93)
2,25 (0,73; 7,38)
1,92 (0,64; 6,07)
| <0,0001
<0,0001
|
IGA 0/1
| sekukinumab nízka dávka vs. placebo
sekukinumab vysoká dávka vs. placebo
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
| 28/40 (70,0)
24/40 (60,0)
28/40 (70,0)
24/40 (60,0)
| 2/41 (4,9)
2/41 (4,9)
14/41 (34,1)
14/41 (34,1)
| 51,77 (10,02; 538,64)
32,52 (6,48; 329,52)
4,49 (1,60; 13,42)
2,86 (1,05; 8,13)
| <0,0001
<0,0001
|
PASI 90
| sekukinumab nízka dávka vs. placebo
sekukinumab vysoká dávka vs. placebo
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
| 29/40 (72,5)
27/40 (67,5)
29/40 (72,5)
27/40 (67,5)
| 1/41 (2,4)
1/41 (2,4)
12/41 (29,3)
12/41 (29,3)
| 133,67 (16,83; 6395,22)
102,86 (13,22; 4850,13)
7,03 (2,34; 23,19)
5,2 (1,82; 16,75)
| <0,0001
<0,0001
|
v 52. týždni
|
PASI 75
| sekukinumab nízka dávka vs.
etanercept
sekukinumab vysoká dávka vs. etanercept
| 35/40 (87,5)
35/40 (87,5)
| 28/41 (68,3)
28/41 (68,3)
| 3,12 (0,91; 12,52)
3,09 (0,90; 12,39)
|
|
IGA 0/1
| sekukinumab nízka dávka vs.
etanercept
sekukinumab vysoká dávka vs. etanercept
| 29/40 (72,5)
30/40 (75,0)
| 23/41 (56,1)
23/41 (56,1)
| 2,02 (0,73; 5,77)
2,26 (0,81; 6,62)
|
|
PASI 90
| sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
| 30/40 (75,0)
32/40 (80,0)
| 21/41 (51,2)
21/41 (51,2)
| 2,85 (1,02; 8,38)
3,69 (1,27; 11,61)
|
|
* chýbajúce údaje boli vyhodnotené ako bez odpovede
** n je počet pacientov s odpoveďou, m = počet pacientov s hodnotiteľnou odpoveďou
*** rozšírené okno návštevy v 12.týždni
Pomer pravdepodobnosti, 95 % interval spoľahlivosti a hodnota p sú z presného modelu logistickej regresie s faktormi liečebná skupina, kategóriou východiskovej telesnej hmostnosti a vekovou kategóriou
|
Zlepšenie kvality života súvisiacej so zdravím meranej ako skóre CDLQI v hodnote 0 alebo 1
v porovnaní s placebom v 12.týždni hlásil vyšší podiel pediatrických pacientov liečených
sekukinumabom (nízka dávka 44,7 %, vysoká dávka 50 %, placebo 15 %). V čase až do 52.týždňa v oboch skupinách s dávkou sekukinumabu bol počet pacientov s odpoveďou numericky vyšší ako v skupine s etanerceptom (nízka dávka 60,6 %, vysoká dávka 66,7 %, etanercept 44,4 %).
S
t
r
e
d
n
e závažná až závažná ložisková psoriáza
Na základe preukázanej miery účinnosti a vzťahu odpovede na expozíciu u dospelých pacientov so stredne závažnou až závažnou ložiskovou psoriázou a podobnosti priebehu ochorenia, patofyziológie a účinku lieku u dospelých a pediatrických pacientov s rovnakými expozičnými hladinami sa
predpokladalo, že sekukinumab bude účinný aj pri liečbe pediatrických pacientov so stredne závažnou až závažnou ložiskovou psoriázou.
Okrem toho sa bezpečnosť a účinnosť sekukinumabu hodnotila v otvorenom multicentrickom skúšaní fázy III s dvomi ramenami a paralelnou skupinou u pediatrických pacientov so stredne závažnou až závažnou ložiskovou psoriázou vo veku 6 až <18 rokov, definovanej skóre PASI s ≥12, skóre IGA mod 2011 of ≥3 a postihnutím kože s BSA ≥10%, ktorí boli kandidátmi na systémovú liečbu.
V pediatrickom skúšaní 2 pri psoriáze sa vyhodnotilo 84 pacientov, randomizovaných na podávanie nízkej dávky sekukinumabu (75 mg na telesnú hmotnosť <50 kg alebo 150 mg na telesnú hmotnosť
≥50 kg) alebo vysokej dávky sekukinumabu (75 mg na telesnú hmotnosť <25 kg, 150 mg na telesnú
hmotnosť medzi ≥25 kg a <50 kg, alebo 300 mg na telesnú hmotnosť ≥50 kg) v 0., 1., 2., 3. a
4. týždni, po ktorých nasledovala každé 4 týždne rovnaká dávka. Rozdelenie pacientov podľa
hmotnosti a veku pri randomizácii je uvedené v Tabuľke 13.
Tabuľka 13 Rozdelenie pacientov podľa hmotnosti a veku v pediatrickom skúšaní 2 pri psoriázePodskupina
| Popis
| Sekukinumab nízka dávka n=42
| Sekukinumab vysoká dávka n=42
| Spolu
N=84
|
Vek
| 6-<12 rokov
| 17
| 16
| 33
|
≥12-<18 rokov
| 25
| 26
| 51
|
Hmotnosť
| <25 kg
| 4
| 4
| 8
|
≥25-<50 kg
| 13
| 12
| 25
|
≥50 kg
| 25
| 26
| 51
|
Spoločnými primárnymi koncovými ukazovateľmi boli podiely pacientov, ktorí dosiahli odpoveď
PASI 75 a IGA mod 2011 „bez prejavov“ alebo“takmer bez prejavov“ (0 alebo 1) v 12.týždni.
Účinnosť v oboch skupinách s nízkou a vysokou dávkou sekukinumabu bola porovnateľná
a preukázala štatistické zlepšenie v porovnaní s predchádzajúcim placebom v primárnych aj sekundárnych koncových ukazovateľoch. Odhadovaná posteriórna pravdepodobnosť pozitívneho liečebného účinku bola 100 %.
U všetkých pacientov sa sledovala účinnosť najmenej 24 týždňov po prvom podaní lieku (pozri
Tabuľku 14). Účinnosť (definovaná odpoveďou PASI 75 a IGA mod 2011 „bez prejavov“ alebo
„takmer bez prejavov“ [0 alebo 1]) sa pozorovala už pri prvej návšteve po východiskovej návšteve
v 2.týždni a podiel pacientov, ktorí dosiahli odpoveď PASI 75 a IGA mod 2011 „bez prejavov“ alebo
„takmer bez prejavov“ (0 alebo 1) sa zvýšil počas obdobia 24 týždňov.
Po 12 .týždni bola účinnosť oboch skupín s nízkou a vysokou dávkou sekukinumabu porovnateľná
. Profily bezpečnosti s nízkou dávkou a vysokou dávkou boli porovnateľné a v zhode s profilom bezpečnosti u dospelých pacientov.
T
a
b
u
ľ
k
a 14 Súhrn klinickej odpovede v pediatrickom skúšaní so stredne závažnou až závažnou
p
soriázou v 12.a 24.týždni (pediatrické skúšanie pri psoriáze 2)*
|
1
2 .týždeň
|
2
4 .týždeň
|
S
e
k
u
k
i
nu
m
a
b nízka dávka
|
S
e
k
u
k
i
nu
m
a
b vysoká dávka
|
S
e
k
u
k
i
nu
m
a
b nízka dávka
|
S
e
k
u
k
i
nu
m
a
b vysoká dávka
|
Počet pacientov
|
42
|
42
|
42
|
42
|
Odpoveď PASI 75, n (%)
|
39 (92,9 %)
|
39 (92,9 %)
|
40 (95,2 %)
|
40 (95,2 %)
|
Odpoveď IGA mod 2011 „bez prejavov“ alebo “takmer bez prejavov“, n (%)
|
33 (78,6 %)
|
35 (83,3 %)
|
37 (88,1 %)
|
39 (92,9 %)
|
Odpoveď PASI 90, n (%)
|
29 (69 %)
|
32 (76,2 %)
|
37 (88,1 %)
|
37 (88,1 %)
|
Odpoveď PASI 100, n (%)
|
25 (59,5 %)
|
23 (54,8 %)
|
28 (66,7 %)
|
28 (66,7 %)
|
* chýbajúce údaje boli vyhodnotené ako bez odpovede
|
Tieto výsledky v pediatrickej populácii so stredne závažnou až závažnou ložiskovou psoriázou
potvrdili vyššie uvedené prediktívne predpoklady, založené na základe účinnosti a odpovede na liečbu
u dospelých pacientov, ktoré sú uvedené vyššie.
V skupine s nízkou dávkou dosiahlo 50 % a 70,7 % pacientov skóre CDLQI 0 alebo 1 v 12.a 24.týždni
v uvedenom poradí. V skupine s vysokou dávkou dosiahlo 61,9 % a 60,5 % pacientov skóre 0 alebo 1
v 12.a 24.týždni v uvedenom poradí.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Cosentyxom pre liečbu ložiskovej psoriázy u pediatrických pacientov od narodenia do menej ako 6 rokov a pre liečbu chronickej idiopatickej artritídy u pediatrických pacientov od narodenia do menej ako 2 rokov (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Cosentyxom pre
liečbu chronickej idiopatickej artritídy u pediatrických pacientov vo veku od 2 rokov do menej ako
18 rokov (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiVäčšina farmakokinetických vlastností pozorovaných u pacientov s ložiskovou psoriázou, psoriatickou
artritídou a ankylozujúcou spondylitídou bola podobná.
AbsorpciaPo jednorazovej subkutánnej dávke 300 mg v tekutej liekovej forme sa u zdravých dobrovoľníkov
dosiahli maximálne koncentrácie sekukinumabu v sére 43,2±10,4 μg/ml medzi 2. a 14. dňom po
podaní.
Podľa analýzy farmakokinetiky u populácie sa po jednorazovom subkutánnom podaní pacientom s ložiskovou psoriázou maximálne koncentrácie sekukinumabu v sére 13,7±4,8 µg/ml pri dávke
150 mg alebo 27,3±9,5 µg/ml pri dávke 300 mg dosiahli medzi 5. a 6. dňom po podaní.
Podľa analýzy farmakokinetiky u populácie bol po začiatočnom týždennom podávaní počas prvého mesiaca čas do dosiahnutia maximálnej koncentrácie medzi 31. a 34. dňom.
Podľa simulovaných údajov boli maximálne koncentrácie v rovnovážnom stave (Cmax,ss) po subkutánnom podaní 27,6 µg/ml pri 150 mg a 55,2 µg/ml pri 300 mg. Analýza farmakokinetiky u populácie naznačuje, že rovnovážny stav sa dosahuje pri režimoch mesačného podávania po
20 týždňoch.
V porovnaní s expozíciou po jednorazovej dávke ukázala analýza farmakokinetiky u populácie, že pacienti vykazovali po opakovanom mesačnom podávaní počas udržiavacej liečby zvýšenie maximálnych koncentrácií v sére a plochy pod krivkou (AUC) na dvojnásobok.
Analýza farmakokinetiky u populácie ukázala, že sekukinumab sa u pacientov s ložiskovou psoriázou absorboval s priemernou absolútnou biologickou dostupnosťou 73 %. Vo všetkých skúšaniach sa vypočítala absolútna biologická dostupnosť v rozmedzí od 60 do 77 %.
Biologická dostupnosť sekukinumabu u pacientov so PsA bola 85 % na základe farmakokinetického modelu populácie.

Po jednorazovej subkutánnej injekcii 300 mg injekčného roztoku v naplnenej injekčnej striekačke u pacientov s ložiskovou psoriázou bola systémová expozícia sekukinumabu podobná tej, ktorá sa predtým pozorovala pri dvoch injekciách po 150 mg.
DistribúciaStredný objem distribúcie počas koncovej fázy (Vz) po jednorazovom intravenóznom podaní
u pacientov s ložiskovou psoriázou bol v rozmedzí od 7,10 do 8,60 litrov, čo naznačuje, že
sekukinumab sa len obmedzene distribuuje do periférnych kompartmentov.
BiotransformáciaVäčšina eliminácie IgG sa uskutočňuje prostredníctvom intracelulárneho katabolizmu po endocytóze z
tekutej fázy alebo sprostredkovanej receptorom.
ElimináciaStredný systémový klírens (CL) po jednorazovom intravenóznom podaní pacientom s ložiskovou
psoriázou bol v rozmedzí od 0,13 do 0,36 l/deň. V analýze farmakokinetiky bola stredná hodnota
systémového klírensu (CL) u populácie pacientov s ložiskovou psoriázou 0,19 l/deň. CL nebol ovplyvnený pohlavím. Klírens nezávisel od dávky a času.
Stredný polčas eliminácie bol podľa odhadu analýzy farmakokinetiky u populácie pacientov
s ložiskovou psoriázou 27 dní, s rozmedzím od 18 do 46 dní, vo všetkých psoriatických skúšaniach
s intravenóznym podávaním.
Linearita/nelinearitaFarmakokinetika sekukinumabu po jednorazovom a opakovanom podávaní pacientom s ložiskovou
psoriázou sa stanovila v niekoľkých skúšaniach s intravenózne podanými dávkami v rozmedzí od
1x 0,3 mg/kg do 3x 10 mg/kg a so subkutánne podanými dávkami v rozmedzí od 1x 25 mg po opakované dávky 300 mg. Expozícia bola úmerná dávke pri všetkých dávkovacích režimoch.
Osobitné populácieStaršípacientiPodľa analýzy farmakokinetiky u populácie obmedzeného počtu starších pacientov (n=71 vo veku
≥65 rokov a n=7 vo veku ≥75 rokov) bol klírens podobný u starších pacientov a u pacientov mladších
ako 65 rokov.
Pacienti s poruchoufunkcieobličiekalebopečeneNie sú dostupné farmakokinetické údaje o pacientoch s poruchou funkcie obličiek alebo pečene. Predpokladá sa, že pri intaktnom sekukinumabe, monoklonálnej protilátke IgG, je eliminácia obličkami nízka a málo významná. IgG sa eliminujú hlavne prostredníctvom katabolizmu
a nepredpokladá sa, že porucha funkcie pečene má vplyv na klírens sekukinumabu.
Vplyv telesnej hmotnosti na farmakokinetiku
Klírens a distribučný objem sekukinumabu sa zvyšujú so zvyšujúcou sa telesnou hmotnosťou.
Pediatrická populácia
V skupine dvoch pediatrických skúšaní pacienti so stredne závažnou až závažnou ložiskovou psoriázou (vo veku 6 až menej ako 18 rokov) dostávali sekukinumab podľa odporúčaného pediatrického dávkovacieho režimu. Pacienti s hmotnosťou ≥25 a <50 kg mali v 24. týždni priemernú
minimálnu koncentráciu v rovnovážnom stave (priemerné ± SD) v hodnote 19,8 ± 6,96 µg/ml (n=24)
po dávke 75 mg sekukinumabu a pacienti s hmotnosťou ≥50 kg mali priemernú minimálnu koncentráciu v rovnovážnom stave 27,3 ± 10,1 µg/ml (n=36) po dávke 150 mg sekukinumabu. Priemerná minimálna koncentrácia v rovnovážnom stave (priemerné ± SD) u pacientov s hmotnosťou
<25 kg (n=8) v 24. týždni bola 32,6 ± 10,8 µg/ml po dávke 75 mg sekukinumabu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a reprodukčnej toxicity, alebo pri testovaní skríženej reaktivity tkanív, neodhalili žiadne osobitné riziko pre ľudí (dospelých a dospievajúcich).
Štúdie na zvieratách na vyhodnotenie karcinogénneho potenciálu sekukinumabu sa nevykonali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
dihydrát trehalózy histidín
monohydrát histidínium-chloridu
metionín
polysorbát 80
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
18 mesiacov
Ak je to nevyhnutné, Cosentyx sa môže jedenkrát uchovávať mimo chladničky pri izbovej teplote neprevyšujúcej 30°C, nie viac ako 4 dni.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Cosentyx 150 mg injekčný roztok v naplnenej injekčnejstriekačke
Cosentyx 150 mg injekčný roztok v naplnenej injekčnej striekačke sa dodáva v 1 ml naplnenej
sklenenej injekčnej striekačke s brómbutylovou gumenou piestovou zátkou potiahnutou silikónom, nasadenou ihlou 27G x ½″ a pevným krytom ihly zo styrén-butadiénového kaučuku, spojeným s automatickým chráničom ihly z polykarbonátu.
Cosentyx 150 mg injekčný roztok v naplnenej injekčnej striekačke je dostupný v jednotlivých baleniach obsahujúcich 1 alebo 2 naplnené injekčné striekačky a v multibaleniach obsahujúcich
6 (3 balenia po 2) naplnených injekčných striekačiek.
Cosentyx 300 mg injekčný roztok v naplnenejinjekčnejstriekačke
Cosentyx 300 mg injekčný roztok v naplnenej injekčnej striekačke sa dodáva v 2,25 ml naplnenej
sklenenej injekčnej striekačke s brómbutylovou gumenou piestovou zátkou potiahnutou silikónom,
nasadenou ihlou 27G x ½″ a pevným krytom ihly zo syntetického polyizoprénového kaučuku, spojeným s automatickým chráničom ihly z polykarbonátu.
Cosentyx 300 mg injekčný roztok v naplnenej injekčnej striekačke je dostupný v jednotlivých baleniach obsahujúcich 1 naplnenú injekčnú striekačku a v multibaleniach obsahujúcich 3 (3 balenia po 1) naplnené injekčné striekačky.
Cosentyx 150 mg injekčný roztok v naplnenom pere
Cosentyx 150 mg injekčný roztok v naplnenom pere sa dodáva v naplnenej injekčnej striekačke na
jednorazové použitie, ktorá je súčasťou trojuholníkového pera s priehľadným okienkom a štítkom. Naplnená injekčná striekačka vo vnútri pera je 1 ml sklenená injekčná striekačka s brómbutylovou gumenou piestovou zátkou potiahnutou silikónom, nasadenou ihlou 27G x ½″ a pevným krytom ihly
zo styrén butadiénového kaučuku.
Cosentyx 150 mg injekčný roztok v naplnenom pere je dostupný v jednotlivých baleniach obsahujúcich 1 alebo 2 naplnené perá a v multibaleniach obsahujúcich 6 (3 balenia po 2) naplnených pier.
Cosentyx 300 mg injekčný roztok v naplnenompere
Cosentyx 300 mg injekčný roztok v naplnenom pere sa dodáva v naplnenej injekčnej striekačke na
jednorazové použitie, ktorá je súčasťou štvorhranného pera s priehľadným okienkom a štítkom. Naplnená injekčná striekačka vo vnútri pera je 2,25 ml sklenená, injekčná striekačka s brómbutylovou gumenou piestovou zátkou potiahnutou silikónom, nasadenou ihlou 27G x ½″ a pevným krytom ihly zo syntetického polyizoprénového kaučuku.
Cosentyx 300 mg injekčný roztok v naplnenom pere je dostupný v jednotlivých baleniach obsahujúcich 1 naplnené pero a v multibaleniach obsahujúcich 3 (3 balenia po 1) naplnené perá.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Cosentyx 150 mg injekčný roztok v naplnenejinjekčnejstriekačke
Cosentyx 150 mg injekčný roztok sa dodáva v naplnenej injekčnej striekačke na jednorazové
individuálne použitie. Injekčná striekačka sa má vybrať z chladničky 20 minút pred podaním, aby sa
mohla ohriať na izbovú teplotu.
Cosentyx 300 mg injekčný roztok v naplnenejinjekčnejstriekačke
Cosentyx 300 mg injekčný roztok sa dodáva v naplnenej injekčnej striekačke na jednorazové
individuálne použitie. Injekčná striekačka sa má vybrať z chladničky 30-45 minút pred podaním, aby
sa mohla ohriať na izbovú teplotu.
C
o
s
e
n
t
y
x 150 mg injekčný roztok v naplnenom pere
Cosentyx 150 mg injekčný roztok sa dodáva v naplnenom pere na jednorazové individuálne použitie.
Pero sa má vybrať z chladničky 20 minút pred podaním, aby sa mohlo ohriať na izbovú teplotu.
Cosentyx 300 mg injekčný roztok v naplnenompere
Cosentyx 300 mg injekčný roztok sa dodáva v naplnenom pere na jednorazové individuálne použitie.
Pero sa má vybrať z chladničky 30-45 minút pred podaním, aby sa mohlo ohriať na izbovú teplotu.
Pred použitím sa odporúča vizuálna kontrola naplnenej injekčnej striekačky alebo naplneného pera. Tekutina má byť číra. Jej farba sa môže meniť od bezfarebnej po bledožltú. Môže sa v nej objaviť malá vzduchová bublina, čo je normálne. Nepoužite tekutinu, ak obsahuje ľahko viditeľné častice, ak je zakalená alebo výrazne hnedá.
Podrobný návod na použitie je uvedený v písomnej informácii pre používateľa.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLA
Cosentyx 150 mg injekčný roztok v naplnenejinjekčnejstriekačke
EU/1/14/980/002
EU/1/14/980/003
EU/1/14/980/006
Cosentyx 300 mg injekčný roztok v naplnenejinjekčnej striekačke
EU/1/14/980/008-009
Cosentyx 150 mg injekčný roztok v naplnenompere
EU/1/14/980/004
EU/1/14/980/005
EU/1/14/980/007
Cosentyx 300 mg injekčný roztok v naplnenompere
EU/1/14/980/010-011
9
. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 15.január 2015
Dátum posledného predĺženia registrácie: 03. september 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1
. NÁZOV LIEKU
Cosentyx 150 mg prášok na injekčný roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Každá injekčná liekovka s práškom obsahuje 150 mg sekukinumabu. Po rekonštitúcii 1 ml roztoku obsahuje 150 mg sekukinumabu.
Sekukinumab je rekombinantná, plne ľudská monoklonálna protilátka, produkovaná v bunkách ovária čínskeho škrečka (CHO).
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA Prášok na injekčný roztok Prášok je biely pevný lyofilizát.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Ložisková psoriáza u dospelých
Cosentyx je indikovaný na liečbu stredne závažnej až závažnej ložiskovej psoriázy u dospelých, ktorí
sú kandidátmi na systémovú liečbu.
Ložisková psoriáza u pediatrických pacientov
Cosentyx je indikovaný na liečbu stredne závažnej až závažnej ložiskovej psoriázy u detí
a dospievajúcich vo veku od 6 rokov, ktorí sú kandidátmi na systémovú liečbu.
Psoriatická artritída
Cosentyx, v monoterapii alebo v kombinácii s metotrexátom (MTX), je indikovaný na liečbu aktívnej
psoriatickej artritídy u dospelých pacientov, keď odpoveď na predchádzajúcu liečbu antireumatickým liekom modifikujúcim chorobu (disease-modifying anti-rheumatic drug, DMARD) nebola dostatočná (pozri časť 5.1).
Axiálna spondyloartritída (axSpA)
Ankylozujúca spondylitída (AS) / axiálna spondyloartritída s rádiografickým dôkazom
Cosentyx je indikovaný na liečbu aktívnej ankylozujúcej spondylitídy u dospelých, u ktorých odpoveď na konvenčnú liečbu nebola dostatočná.
Axiálna spondyloartritída bez rádiografického dôkazu (nr-axSpA)
Cosentyx je indikovaný na liečbu aktívnej axiálnej spondyloartritídy bez rádiografického dôkazu s objektívnymi prejavmi zápalu preukázanými zvýšenou hodnotou C-reaktívneho proteínu (CRP) a/alebo dôkazom podľa magnetickej rezonancie (MRI) u dospelých bez adekvátnej odpovede na nesteroidné protizápalové lieky (NSAID).
4
.
2 Dávkovanie a spôsob podávania
Cosentyx je určený na použitie pod vedením a dohľadom lekára so skúsenosťami v diagnostike a liečbe chorôb, na ktoré je Cosentyx indikovaný.
DávkovanieLožiskovápsoriázaudospelýchOdporúčaná dávka je 300 mg sekukinumabu podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky. Každá dávka 300 mg sa podáva ako dve subkutánne injekcie po 150 mg.
Ložiskovápsoriázau pediatrických pacientov (dospievajúci a deti vo veku od 6 rokov)Odporúčaná dávka závisí od telesnej hmotnosti pacienta (Tabuľka 1) a podáva sa subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky. Každá dávka 75 mg sa podáva ako jedna subkutánna injekcia po 75 mg. Každá dávka 150 mg sa podáva ako jedna subkutánna injekcia po 150 mg. Každá dávka 300 mg sa podáva ako dve subkutánne injekcie po 150 mg.
Tabuľka 1 Odporúčaná dávka pre pediatrických pacientov s ložiskovou psoriázouTelesná hmotnosť v čase dávkovania
| Odporúčaná dávka
|
<25 kg
| 75 mg
|
25 až <50 kg
| 75 mg
|
≥50 kg
| 150 mg (*môže sa zvýšiť na 300 mg)
|
*Niektorí pacienti môžu mať daľší prínos z vyššej dávky.
Cosentyx môže byť dostupný v ďalších silách a/alebo formách v závislosti od individuálnych liečebných potrieb.
Psoriatická artritídaU pacientov so sprievodnou stredne závažnou až závažnou ložiskovou psoriázou alebo u pacientov
bez adekvátnej odpovede na liečbu anti-TNFα (
inadequate responders, IR) je odporúčaná dávka
300 mg podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky. Každá dávka 300 mg sa podáva ako dve subkutánne injekcie po
150 mg.
U ostatných pacientov je odporúčaná dávka 150 mg podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky. Na základe klinickej odpovede sa dávka môže zvýšiť na 300 mg.
Axiálna spondyloartritída (axSpA)Ankylozujúca spondylitída (AS, axiálna spondyloartritída s rádiografickým dôkazom)Odporúčaná dávka je 150 mg podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3.
a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky. Na základe klinickej odpovede sa dávka
môže zvýšiť na 300 mg. Každá 300 mg dávka sa podáva ako dve 150 mg subkutánne injekcie.
Axiálna spondyloartritída bez rádiografického dôkazu (nr-axSpA)Odporúčaná dávka je 150 mg podaná subkutánnou injekciou so začiatočnými dávkami v 0., 1., 2., 3. a 4. týždni, po ktorých nasledujú mesačné udržiavacie dávky.
Údaje, ktoré sú k dispozícii, naznačujú, že pri všetkých horeuvedených indikáciách sa klinická odpoveď na liečbu dosiahne zvyčajne v priebehu 16 týždňov liečby. Ukončenie liečby sa má zvážiť u pacientov, u ktorých sa odpoveď neprejavila do 16 týždňov liečby. U niektorých pacientov, ktorí majú spočiatku čiastočnú odpoveď, sa môže stav následne zlepšiť pri pokračujúcej liečbe po
16. týždni.
O
s
o
b
it
n
é skupiny pacientov
S
t
a
rší
p
a
c
i
e
n
t
i
(
v
o
v
e
k
u
6
5
rokov a viac)
Nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Poruchafunkcieobličiek/poruchafunkciepečene
Cosentyx sa neskúmal u týchto skupín pacientov. Odporúčania pre dávkovanie nemožno urobiť.
Pediatrická populácia
Bezpečnosť a účinnosť Cosentyxu u detí s ložiskovou psoriázou vo veku menej ako 6 rokov neboli stanovené.
Bezpečnosť a účinnosť Cosentyxu u detí vo veku menej ako 18 rokov v iných indikáciách neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Cosentyx sa má podávať subkutánnou injekciou. Ak je to možné, je potrebné vyhýbať sa podaniu
injekcií do oblastí kože s prejavmi psoriázy. Prášok na injekčný roztok sa musí pred použitím rekonštituovať.
Rekonštitúciu, prípravu dávky a podanie prášku na injekčný roztok má vykonať zdravotnícky pracovník. Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6 a Pokyny na použitie v informácii pre používateľa.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Klinicky významné aktívne infekcie, napr. aktívna tuberkulóza (pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Infekcie
Sekukinumab má potenciál zvyšovať riziko infekcií. Po uvedení lieku na trh sa zaznamenali
u pacientov používajúcich sekukinumab závažné infekcie. Opatrnosť je potrebná pri zvažovaní použitia sekukinumabu u pacientov s chronickou infekciou alebo s opakovanými infekciami v
anamnéze.
Pacientov treba poučiť, aby vyhľadali lekársku pomoc, ak sa u nich objavia prejavy alebo príznaky poukazujúce na infekciu. Ak u pacienta vznikne závažná infekcia, je potrebné pacienta dôsledne sledovať a sekukinumab sa mu až do vymiznutia infekcie nemá podať.
Infekcie sa pozorovali v klinických skúšaniach u pacientov, ktorí dostávali sekukinumab (pozri časť 4.8). Väčšinou to boli ľahké alebo stredne ťažké infekcie horných dýchacích ciest, napr. nazofaryngitída, a nevyžadovali prerušenie liečby.
V súvislosti s mechanizmom účinku sekukinumabu sa v klinických skúšaniach pri psoriáze nezávažné kandidové infekcie slizníc a kože zaznamenali častejšie pri sekukinumabe ako pri placebe (3,55 na
100 pacientorokov pri 300 mg sekukinumabu oproti 1,00 na 100 pacientorokov pri placebe) (pozri
časť 4.8).
V klinických skúšaniach nebola hlásená zvýšená náchylnosť na tuberkulózu. Sekukinumab sa však nemá podávať pacientom s aktívnou tuberkulózou. U pacientov s latentnou tuberkulózou sa má pred začiatkom liečby sekukinumabom zvážiť antituberkulózna liečba.
Zápalové ochorenie čriev (vrátane Crohnovej choroby a ulceróznej kolitídy)
Boli hlásené prípady vzniku alebo exacerbácie zápalového ochorenia čriev pri používaní
sekukinumabu (pozri časť 4.8). Sekukinumab sa neodporúča u pacientov so zápalovým ochorením
čriev. Ak sa u pacienta objavia prejavy a príznaky zápalového ochorenia čriev alebo ak dôjde
k exacerbácii už existujúceho zápalového ochorenia čriev, je potrebné ukončiť liečbu so
sekukinumabom a začať s primeranou lekárskou starostlivosťou.
Reakcie z precitlivenosti
V klinických skúšaniach sa u pacientov liečených sekukinumabom pozorovali zriedkavé prípady
anafylaktických reakcií. Ak sa vyskytnú anafylaktické alebo iné závažné alergické reakcie, podávanie sekukinumabu sa má okamžite ukončiť a má sa začať primeraná liečba.
Vakcinácie
Živé vakcíny sa nemajú podávať súbežne so sekukinumabom.
Pacienti, ktorí dostávajú sekukinumab, môžu súčasne dostať inaktivované alebo neživé vakcíny. V klinickom skúšaní po vakcinácii meningokokovou a inaktivovanou chrípkovou vakcínou podobné podiely zdravých dobrovoľníkov, ktorí dostávali 150 mg sekukinumabu a ktorí dostávali placebo, dokázali vyprodukovať dostatočnú imunitnú odpoveď, ktorá predstavovala najmenej 4 -násobné zvýšenie titrov protilátok proti meningokokovým a chrípkovým vakcínam. Údaje naznačujú, že sekukinumab nepotláča humorálnu imunitnú odpoveď na meningokokové alebo chrípkové vakcíny.
Pred začatím liečby Cosentyxom sa odporúča, aby pediatrickí pacienti absolvovali všetky potrebné imunizácie podľa veku v súlade s aktuálnymi usmerneniami pre imunizáciu.
Súbežná imunosupresívna liečba
Bezpečnosť a účinnosť sekukinumabu v kombinácii s imunosupresívami, vrátane biologických liekov,
alebo fototerapiou sa nehodnotila v skúšaniach pri psoriáze. Sekukinumab sa podával súbežne
s metotrexátom (MTX), sulfasalazínom a/alebo kortikosteroidmi v skúšaniach pri artritíde (vrátane pacientov so psoriatickou artritídou a ankylozujúcou spondylitídou). Opatrnosť je potrebná pri zvažovaní súbežného použitia iných imunosupresív a sekukinumabu (pozri tiež časť 4.5).
4.5 Liekové a iné interakcie
Živé vakcíny sa nemajú podať súbežne so sekukinumabom (pozri tiež časť 4.4).
V skúšaní u dospelých pacientov s ložiskovou psoriázou sa nepozorovala žiadna interakcia medzi
sekukinumabom a midazolamom (substrát CYP3A4).
Keď sa sekukinumab podával súbežne s metotrexátom (MTX) a/alebo kortikosteroidmi v skúšaniach pri artritíde (vrátane pacientov so psoriatickou artritídou a axiálnou spondyloartritídou), nepozorovali sa žiadne interakcie.
4
.
6 Fertilita, gravidita a laktácia
Ženy v plodnomveku
Ženy v plodnom veku majú počas liečby a najmenej 20 týždňov po skončení liečby používať účinnú
metódu antikoncepcie.
Gravidita
Nie sú k dispozícii dostatočné údaje o používaní sekukinumabu u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame účinky na reprodukčnú toxicitu (pozri
časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Cosentyxu počas gravidity.
Dojčenie
Nie je známe, či sa sekukinumab vylučuje do ľudského mlieka. Imunoglobulíny sa vylučujú do
ľudského mlieka a nie je známe, či sa sekukinumab systémovo absorbuje po požití. Vzhľadom na možnosť nežiaducich reakcií na sekukinumab u dojčených detí sa rozhodnutie, či ukončiť dojčenie počas liečby a do 20 týždňov po skončení liečby, alebo ukončiť liečbu Cosentyxom, má urobiť po
zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Účinok sekukinumabu na fertilitu ľudí sa neskúmal. Štúdie na zvieratách nenaznačujú priame alebo
nepriame škodlivé účinky týkajúce sa fertility.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Cosentyx nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutie profilu bezpečnosti
Najčastejšie hlásenými nežiaducimi reakciami sú infekcie horných dýchacích ciest (17,7 %)
(najčastejšie nazofaryngitída a rinitída).
Tabuľkový zoznam nežiaducich reakcií
Nežiaduce reakcie v klinických skúšaniach a z hlásení po uvedení lieku na trh (Tabuľka 2) sú
zatriedené podľa orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, najčastejšie ako prvé. V rámci každej skupiny
frekvencie sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti. Okrem toho zodpovedajúca
kategória frekvencie každej nežiaducej reakcie vychádza z nasledujúcej konvencie: veľmi časté
(≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až
<1/1 000); veľmi zriedkavé (<1/10 000); a neznáme (z dostupných údajov).
V zaslepených a otvorených klinických skúšaniach sa liečilo sekukinumabom viac ako
18 000 pacientov s rôznymi indikáciami (ložisková psoriáza, psoriatická artritída, axiálna spondyloartritída a iné autoimunitné choroby), čo predstavuje 30 565 pacientorokov expozície.
Najmenej jeden rok dostávalo sekukinumab viac ako 11 700 z týchto pacientov. Profil bezpečnosti
sekukinumabu sa vo všetkých indikáciach zhoduje.
T
a
b
u
ľ
k
a 2 Zoznam nežiaducich reakcií v klinických skúšaniach
1
)
a pri používaní po uvedení lieku na trh
T
r
i
e
d
a orgánových
systémov
|
Frekvencia
|
N
e
ž
ia
d
u
c
a reakcia
|
Infekcie a nákazy
|
Veľmi časté
|
Infekcie horných dýchacích ciest
|
Časté
|
Orálny herpes
|
Tinea pedis
|
Menej časté
|
Orálna kandidóza
|
Otitis externa
|
Infekcie dolných dýchacích ciest
|
Neznáme
|
Kandidóza slizníc a kože (vrátane kandidózy ezofágu)
|
Poruchy krvi
a lymfatického systému
|
Menej časté
|
Neutropénia
|
Poruchy imunitného systému
|
Zriedkavé
|
Anafylaktické reakcie
|
Poruchy nervového
systému
|
Časté
|
Bolesť hlavy
|
Poruchy oka
|
Menej časté
|
Konjunktivitída
|
Poruchy dýchacej
sústavy, hrudníka a mediastína
|
Časté
|
Rinorea
|
Poruchy
gastrointestinálneho traktu
|
Časté
|
Hnačka
|
Časté
|
Nauzea
|
Menej časté
|
Zápalové ochorenie čriev
|
Poruchy kože
a podkožného tkaniva
|
Menej časté
|
Urtikária
|
Zriedkavé
|
Exfoliatívna dermatitída2)
|
Hypersenzitívna vaskulitída
|
Celkové poruchy a
reakcie v mieste podania
|
Časté
|
Únava
|
1) Klinické skúšania kontrolované placebom (fáza III) u pacientov s ložiskovou psoriázou, PsA, AS
a nr-axSpA pri dávkach 300 mg, 150 mg, 75 mg alebo pri placebe s liečbou trvajúcou do
12 týždňov (pri psoriáze) alebo 16 týždňov (pri PsA, AS a nr-axSpA)
2) Prípady boli hlásené u pacientov s diagnózou psoriázy
|
P
op
i
s
vybraných
nežiaducich
reakcií
I
n
f
ek
c
i
e
Počas obdobia kontrolovaného placebom v klinických skúšaniach u pacientov s ložiskovou psoriázou
(spolu 1 382 pacientov liečených sekukinumabom a 694 pacientov liečených placebom do 12 týždňov) boli infekcie hlásené u 28,7 % pacientov liečených sekukinumabom v porovnaní s 18,9 % pacientov, ktorí dostávali placebo. Väčšina infekcií boli nie závažné a ľahké až stredne ťažké infekcie horných dýchacích ciest, napr. nazofaryngitída, ktoré nevyžadovali prerušenie liečby. V súlade s
mechanizmom účinku sa zvýšil výskyt kandidózy slizníc alebo kože, ale tieto prípady boli ľahké až
stredne ťažké, nie závažné, reagovali na štandardnú liečbu a nevyžadovali prerušenie liečby. Závažné infekcie sa vyskytli u 0,14 % pacientov liečených sekukinumabom a u 0,3 % pacientov, ktorí dostávali placebo (pozri časť 4.4).
Počas celého obdobia liečby (celkovo 3 430 pacientov liečených sekukinumabom, väčšina z nich až
do 52 týždňov) boli infekcie hlásené u 47,5 % pacientov liečených sekukinumabom (0,9 pripadajúcich na pacientorok sledovania). Závažné infekcie boli hlásené u 1,2 % pacientov liečených sekukinumabom (0,015 pripadajúcich na pacientorok sledovania).
Výskyt infekcií pozorovaný v klinických skúšaniach pri psoriatickej artritíde a axiálnej spondyloartritíde (ankylozujúcej spondylitíde a axiálnej spondyloartritíde bez rádiografického dôkazu) je podobný výskytu infekcií, ktorý sa pozoroval v psoriatických skúšaniach.
Neutropénia
V klinických skúšaniach fázy III pri psoriáze sa neutropénia pozorovala častejšie pri sekukinumabe ako pri placebe, ale vo väčšine prípadov bola mierna, prechodná a reverzibilná. Neutropénia
<1,0-0,5x109/l (CTCAE stupňa 3) bola hlásená u 18 z 3 430 (0,5 %) pacientov liečených
sekukinumabom, bez závislosti od dávky a bez časového vzťahu k infekciám v 15 z 18 prípadov. Prípady závažnejšej neutropénie sa nezaznamenali. Vo zvyšných 3 prípadoch boli hlásené nezávažné infekcie s obvyklou odpoveďou na štandardnú starostlivosť, ktoré si nevyžiadali vysadenie sekukinumabu.
Frekvencia výskytu neutropénie pri psoriatickej artritíde a axiálnej spondyloartritíde (ankylozujúcej spondylitíde a axiálnej spondyloartritíde bez rádiografického dôkazu) bola podobná frekvencii výskytu neutropénie pri psoriáze.
Boli hlásené zriedkavé prípady neutropénie <0,5x109/l (CTCAE stupňa 4).
Reakcie z precitlivenosti
V klinických skúšaniach sa pozorovala urtikária a zriedkavé prípady anafylaktickej reakcie na sekukinumab (pozri aj časť 4.4).
Imunogenita
V klinických skúšaniach pri psoriáze, psoriatickej artritíde a axiálnej spondyloartritíde (ankylozujúcej spondylitíde a axiálnej spondyloartritíde bez rádiografického dôkazu) sa protilátky proti sekukinumabu vytvorili u menej ako 1 % pacientov liečených sekukinumabom počas liečby trvajúcej do 52 týždňov. Približne polovica protilátok proti liečivu, ktoré sa objavili počas liečby, bola neutralizujúca, ale nespájalo sa to so stratou účinnosti alebo farmakokinetickými abnormalitami.
Pediatrická populácia
Nežiaduceúčinkyupediatrickýchpacientovsložiskovoupsoriázouvovekuod6 rokov
Bezpečnosť sekukinumabu u pediatrických pacientov s ložiskovou psoriázou sa hodnotila v dvoch skúšaniach fázy III. Prvé skúšanie (pediatrické skúšanie 1) bolo dvojito zaslepené, placebom kontrolované skúšanie u 162 pacientov so závažnou ložiskovou psoriázou vo veku od 6 rokov do menej ako 18 rokov. Druhé skúšanie (pediatrické skúšanie 2) je otvorené klinické skúšanie u'
84 pacientov so stredne závažnou až závažnou ložiskovou psoriázou vo veku od 6 rokov do menej ako
18 rokov. Profil bezpečnosti zaznamenaný v týchto dvoch skúšaniach bol zhodný s profilom bezpečnosti u dospelých pacientov s ložiskovou psoriázou.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 Predávkovanie
V klinických skúšaniach sa intravenózne podali dávky až do 30 mg/kg (približne 2 000 až 3 000 mg) bez toxicity obmedzujúcej dávku. V prípade predávkovania sa odporúča, aby sa u pacienta sledovali akékoľvek prejavy a príznaky nežiaducich reakcií a aby sa okamžite začala primeraná symptomatická liečba.
5
. FARMAKOLOGICKÉ VLASTNOSTI
5
.
1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: imunosupresíva, inhibítory interleukínu, ATC kód: L04AC10
Mechanizmus účinku
Sekukinumab je plne ľudská monoklonálna protilátka IgG1/κ, ktorá sa selektívne viaže na
prozápalový cytokín interleukín-17A (IL-17A) a neutralizuje ho. Sekukinumab sa zameriava na
IL-17A a inhibuje jeho interakciu s receptorom IL-17, ktorý je exprimovaný na rôznych typoch buniek
vrátane keratinocytov. Dôsledkom je, že sekukinumab inhibuje uvoľňovanie zápalových cytokínov, chemokínov a mediátorov poškodenia tkanív a znižuje vplyv sprostredkovaný IL-17A na autoimunitné a zápalové ochorenia. Klinicky významné koncentrácie sekukinumabu sa dostávajú do kože a znižujú lokálne zápalové markery. Priamym dôsledkom liečby sekukinumabom je zmiernenie erytému,
stvrdnutia a šupinatosti kože v léziách ložiskovej psoriázy.
IL-17A je prirodzene sa vyskytujúci cytokín, ktorý sa podieľa na normálnych zápalových a imunitných reakciách. IL-17A zohráva kľúčovú úlohu v patogenéze ložiskovej psoriázy, psoriatickej artritídy a axiálnej spondyloartritídy (ankylozujúcej spondylitídy a axiálnej spondyloartritídy bez rádiografického dôkazu) a jeho množstvo je zvýšené v kožných léziách na rozdiel od kože bez lézií u pacientov s ložiskovou psoriázou a v synoviálnom tkanive pacientov so psoriatickou artritídou. Výskyt buniek produkujúcich IL-17 bol tiež významne vyšší v subchondrálnej kostnej dreni facetových kĺbov u pacientov s ankylozujúcou spondylitídou. U pacientov s axiálnou spondyloartritídou bez rádiografického dôkazu sa tiež zistil zvýšený počet lymfocytov produkujúcich IL-17A. Inhibícia
IL-17A sa preukázala ako účinná pri liečbe ankylozujúcej spondylitídy, čo stanovuje kľúčovú úlohu
tohto cytokínu pri axiálnej spondyloartritíde.
Farmakodynamické účinky
Sérové koncentrácie celkového IL-17A (voľného IL-17A a IL-17A viazaného na sekukinumab) sa
spočiatku zvýšia u pacientov, ktorí dostávajú sekukinumab. Potom nasleduje pomalý pokles v
dôsledku zníženia klírensu IL-17A viazaného na sekukinumab, čo ukazuje, že sekukinumab selektívne
vychytáva voľný IL-17A, ktorý zohráva kľúčovú úlohu v patogenéze ložiskovej psoriázy.
V skúšaní so sekukinumabom sa po jednom až dvoch týždňoch liečby významne znížili hodnoty infiltrujúcich epidermálnych neutrofilov a rôznych markerov súvisiacich s neutrofilmi, ktoré sú zvýšené v kožných léziách pacientov s ložiskovou psoriázou.
Sekukinumab preukázal zníženie (v priebehu 1 až 2 týždňov liečby) koncentrácií C-reaktívneho proteínu, ktorý je zápalovým markerom.
K
li
ni
c
k
á účinnosť a bezpečnosť
Ložiskovápsoriázaudospelých
Bezpečnosť a účinnosť sekukinumabu sa hodnotili v štyroch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických skúšaniach fázy III u pacientov so stredne ťažkou až ťažkou ložiskovou psoriázou, ktorí boli kandidátmi na fototerapiu alebo systémovú liečbu [ERASURE,
FIXTURE, FEATURE, JUNCTURE]. Účinnosť a bezpečnosť 150 mg a 300 mg sekukinumabu sa
hodnotili v porovnaní buď s placebom, alebo s etanerceptom. Okrem toho jedno skúšanie hodnotilo
chronický režim liečby v porovnaní s režimom „opakovania liečby podľa potreby“ [SCULPTURE].
Z 2 403 pacientov, ktorí sa zúčastnili klinických skúšaní kontrolovaných placebom, bolo 79 % pacientov v minulosti neliečených biologickými liekmi, 45 % pacientov po zlyhaní nebiologickej liečby a 8 % pacientov po zlyhaní biologickej liečby (6 % po zlyhaní liečby anti-TNF a 2 % po zlyhaní liečby anti-p40). Približne 15 až 25 % pacientov v klinických skúšaniach fázy III malo na začiatku psoriatickú artritídu (PsA).
Psoriatické skúšanie 1 (ERASURE) vyhodnotilo 738 pacientov. Pacienti randomizovaní na sekukinumab dostali v 0., 1., 2., 3. a 4. týždni dávky 150 mg alebo 300 mg, po ktorých nasledovala každý mesiac rovnaká dávka. Psoriatické skúšanie 2 (FIXTURE) vyhodnotilo 1 306 pacientov. Pacienti randomizovaní na sekukinumab dostali v 0., 1., 2., 3. a 4. týždni dávky 150 mg alebo 300 mg, po ktorých nasledovala každý mesiac rovnaká dávka. Pacienti randomizovaní na etanercept dostávali dvakrát týždenne dávky 50 mg počas 12 týždňov, po ktorých nasledovalo 50 mg každý týždeň. V skúšaní 1 aj v skúšaní 2 pacienti randomizovaní na placebo, ktorí nereagovali do 12. týždňa, potom
prešli na podávanie sekukinumabu (buď 150 mg, alebo 300 mg) v 12., 13., 14. a 15. týždni, po ktorých nasledovala počnúc 16. týždňom každý mesiac rovnaká dávka. Všetci pacienti boli sledovaní do
52 týždňov od prvého podania skúšaného lieku.
Psoriatické skúšanie 3 (FEATURE) vyhodnotilo 177 pacientov používajúcich naplnenú injekčnú striekačku v porovnaní s placebom po 12 týždňoch liečby, aby sa stanovila bezpečnosť, znášanlivosť a použiteľnosť samopodávania sekukinumabu naplnenou injekčnou striekačkou. Psoriatické skúšanie 4 (JUNCTURE) vyhodnotilo 182 pacientov používajúcich naplnené pero v porovnaní s placebom po
12 týždňoch liečby, aby sa stanovila bezpečnosť, znášanlivosť a použiteľnosť samopodávania
sekukinumabu naplneným perom. V skúšaní 3 aj v skúšaní 4 pacienti randomizovaní na sekukinumab dostali v 0., 1., 2., 3. a 4. týždni dávky 150 mg alebo 300 mg, po ktorých nasledovala každý mesiac
rovnaká dávka. Pacienti boli tiež randomizovaní na placebo v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovalo každý mesiac rovnaké podanie.
Psoriatické skúšanie 5 (SCULPTURE) vyhodnotilo 966 pacientov. Všetci pacienti dostali v 0., 1., 2.,
3., 4., 8. a 12. týždni sekukinumab v dávke 150 mg alebo 300 mg a potom boli od 12. týždňa randomizovaní buď na udržiavací režim s rovnakou dávkou každý mesiac, alebo na režim
„opakovania liečby podľa potreby“ s rovnakou dávkou. Pacienti randomizovaní na „opakovanie liečby podľa potreby“ nedosahovali primeranú udržiavaciu odpoveď, preto sa odporúča pevný mesačný
režim udržiavacej liečby.
Spoločné primárne ukazovatele v klinických skúšaniach kontrolovaných placebom a účinným liekom boli podiely pacientov, ktorí dosiahli v 12. týždni odpoveď PASI 75 a odpoveď IGA mod 2011 „bez prejavov“ alebo „takmer bez prejavov“ v porovnaní s placebom (pozri Tabuľky 3 a 4). Dávkou
300 mg sa dosiahol lepší ústup prejavov na koži, najmä pre kožu „bez prejavov“ alebo „takmer bez prejavov“ celkovo pri ukazovateľoch účinnosti PASI 90, PASI 100 a IGA mod 2011, odpoveď 0 alebo
1 vo všetkých klinických skúšaniach s maximálnymi účinkami pozorovanými v 16. týždni, preto sa odporúča táto dávka.
T
a
b
u
ľ
k
a 3 Súhrn klinickej odpovede PASI 50/75/90/100 & IGA⃰ mod 2011 „bez prejavov“ alebo „takmer bez prejavov“ v psoriatických skúšaniach 1, 3 a 4 (ERASURE, FEATURE a JUNCTURE)
12. týždeň 16. týždeň 52. týždeň
|
P
l
acebo
|
150 mg
|
300 mg
|
150 mg
|
300 mg
|
150 mg
|
300 mg
|
S
k
ú
š
a
n
i
e 1
|
|
|
|
|
|
|
|
Počet pacientov
|
246
|
244
|
245
|
244
|
245
|
244
|
245
|
Odpoveď PASI 50, n (%)
|
22
|
203
|
222
|
212
|
224
|
187
|
207
|
|
(8,9 %)
|
(83,5 %)
|
(90,6 %)
|
(87,2 %)
|
(91,4 %)
|
(77 %)
|
(84,5 %)
|
Odpoveď PASI 75, n (%)
|
11
|
174
|
200
|
188
|
211
|
146
|
182
|
|
(4,5 %)
|
(71,6 %)*
|
(81,6 %)*
|
(77,4 %)
|
(86,1 %)
|
(60,1 %)
|
(74,3 %)
|
|
|
*
|
*
|
|
|
|
|
Odpoveď PASI 90, n (%)
|
3 (1,2 %)
|
95
|
145
|
130
|
171
|
88
|
147
|
|
|
(39,1 %)*
|
(59,2 %)*
|
(53,5 %)
|
(69,8 %)
|
(36,2 %)
|
(60,0 %)
|
|
|
*
|
*
|
|
|
|
|
Odpoveď PASI 100, n (%)
|
2 (0,8 %)
|
31
|
70
|
51
|
102
|
49
|
96
|
|
|
(12,8 %)
|
(28,6 %)
|
(21,0 %)
|
(41,6 %)
|
(20,2 %)
|
(39,2 %)
|
Odpoveď IGA mod 2011
|
6
|
125
|
160
|
142
|
180
|
101
|
148
|
„bez prejavov“ alebo
|
(2,40 %)
|
(51,2 %)*
|
(65,3 %)*
|
(58,2 %)
|
(73,5 %)
|
(41,4 %)
|
(60,4 %)
|
|
|
„takmer bez prejavov“,
* *n (%)
Skúšanie 3Počet pacientov 59 59 58 - - - -
Odpoveď PASI 50, n (%) 3 (5,1 %) 51
(86,4 %)
Odpoveď PASI 75, n (%) 0 (0,0 %) 41
(69,5 %)*
*
Odpoveď PASI 90, n (%) 0 (0,0 %) 27
(45,8 %)
51
(87,9 %)
44
(75,9 %)*
*
35
(60,3 %)
- - - -
- - - -
- - - -
Odpoveď PASI 100,
n (%)
Odpoveď IGA mod 2011
„bez prejavov“ alebo
„takmer bez prejavov“,
n (%)
Skúšanie 4
0 (0,0 %) 5
(8,5 %)
0 (0,0 %) 31
(52,5 %)*
*
25
(43,1 %)
40
(69,0 %)*
*
- - - -
- - - -
Počet pacientov 61 60 60 - - - -
Odpoveď PASI 50, n (%) 5 (8,2 %) 48
(80,0 %)
Odpoveď PASI 75, n (%) 2 (3,3 %) 43
(71,7 %)*
*
Odpoveď PASI 90, n (%) 0 (0,0 %) 24
(40,0 %)
Odpoveď PASI 100, n (%) 0 (0,0 %) 10
(16,7 %)
58
(96,7 %)
52
(86,7 %)*
*
33
(55,0 %)
16
(26,7 %)
- - - -
- - - -
- - - -
- - - -
Odpoveď IGA mod 2011
„bez prejavov“ alebo
„takmer bez prejavov“,
n (%)
0 (0,0 %) 32
(53,3 %)*
*
44
(73,3 %)*
*
- - - -
* IGA mod 2011 je stupnica s 5 kategóriami, a to „0 = bez prejavov“, „1 = takmer bez prejavov“, „2 = mierna“, „3 = stredne závažná“ alebo „4 = závažná“, ktoré udávajú celkové hodnotenie závažnosti psoriázy lekárom, so zameraním na stvrdnutie, sčervenanie a šupinatosť. Úspešnosť liečby „bez prejavov“ alebo
„takmer bez prejavov“ znamenala žiadne prejavy psoriázy alebo normálne až ružovo sfarbené lézie, žiadne zhrubnutie ložísk a žiadna až minimálna ložisková šupinatosť.

** hodnoty p v porovnaní s placebom a upravené pre multiplicitu: p<0,0001.
 T
a
b
u
ľ
k
a 4 Súhrn klinickej odpovede v psoriatickom skúšaní 2 (FIXTURE)
12. týždeň 16. týždeň 52. týždeň
T
a
b
u
ľ
k
a 4 Súhrn klinickej odpovede v psoriatickom skúšaní 2 (FIXTURE)
12. týždeň 16. týždeň 52. týždeň
|
P
l
acebo
|
150 mg
|
300 mg
|
E
t
anercept
|
150 mg
|
300 mg
|
E
t
anercept
|
150 mg
|
300 mg
|
E
t
anercept
|
Počet
|
324
|
327
|
323
|
323
|
327
|
323
|
323
|
327
|
323
|
323
|
pacientov
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
49
|
266
|
296
|
226
|
290
|
302
|
257 (79,6 %)
|
249
|
274
|
234 (72,4 %)
|
PASI 50,
|
(15,1 %)
|
(81,3 %)
|
(91,6 %)
|
(70,0 %)
|
(88,7 %)
|
(93,5 %)
|
|
(76,1 %)
|
(84,8 %)
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
16
|
219
|
249
|
142
|
247
|
280
|
189 (58,5 %)
|
215
|
254
|
179 (55,4 %)
|
PASI 75,
|
(4,9 %)
|
(67,0 %)
|
(77,1 %)
|
(44,0 %)
|
(75,5 %)
|
(86,7 %)
|
|
(65,7 %)
|
(78,6 %)
|
|
n (%)
|
|
**
|
**
|
|
|
|
|
|
|
|
Odpoveď
|
5
|
137
|
175
|
67
|
176
|
234
|
101 (31,3 %)
|
147
|
210
|
108 (33,4 %)
|
PASI 90,
|
(1,5 %)
|
(41,9 %)
|
(54,2 %)
|
(20,7 %)
|
(53,8 %)
|
(72,4 %)
|
|
(45,0 %)
|
(65,0 %)
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
0 (0 %)
|
47
|
78
|
14 (4,3 %)
|
84
|
119
|
24 (7,4 %)
|
65
|
117
|
32 (9,9 %)
|
PASI 100,
|
|
(14,4 %)
|
(24,1 %)
|
|
(25,7 %)
|
(36,8 %)
|
|
(19,9 %)
|
(36,2 %)
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
Odpoveď
|
9
|
167
|
202
|
88
|
200
|
244
|
127 (39,3 %)
|
168
|
219
|
120 (37,2 %)
|
IGA mod
|
(2,8 %)
|
(51,1 %)
|
(62,5 %)
|
(27,2 %)
|
(61,2 %)
|
(75,5 %)
|
|
(51,4 %)
|
(67,8 %)
|
|
2011 „bez
|
|
**
|
**
|
|
|
|
|
|
|
|
prejavov“
|
|
|
|
|
|
|
|
|
|
|
alebo
|
|
|
|
|
|
|
|
|
|
|
„takmer bez
|
|
|
|
|
|
|
|
|
|
|
prejavov“,
|
|
|
|
|
|
|
|
|
|
|
n (%)
|
|
|
|
|
|
|
|
|
|
|
** hodnoty p v porovnaní s etanerceptom: p=0,0250
V ďalšom klinickom skúšaní pri psoriáze (CLEAR) sa vyhodnotilo 676 pacientov. Dávka sekukinumabu 300 mg splnila primárne a sekundárne koncové ukazovatele preukázaním superiority voči ustekinumabu na základe odpovede PASI 90 v 16. týždni (primárny koncový ukazovateľ), rýchlosti nástupu odpovede PASI 75 vo 4. týždni a dlhodobou odpoveďou PASI 90 v 52. týždni oproti ustekinumabu. Vyššia účinnosť sekukinumabu v porovnaní s ustekinumabom pre koncové ukazovatele PASI 75/90/100 a IGA mod 2011 s odpoveďou 0 alebo 1 („bez prejavov“ alebo „takmer
bez prejavov“) sa pozorovala od začiatku a pretrvávala do 52. týždňa.
Tabuľka 5 Súhrn klinickej odpovede v skúšaní CLEAR4. týždeň 16. týždeň 52. týždeň
s
e
ku
k
inumab
u
s
t
e
k
inumab* sekukinumab
u
s
t
e
k
inumab* sekukinumab
u
s
t
e
k
inumab*
300 mg 300 mg 300 mg
Počet pacientov 334 335 334 335 334 335
Odpoveď PASI
75, n (%)
166 (49,7 %)** 69 (20,6 %) 311 (93,1 %) 276 (82,4 %) 306 (91,6 %) 262 (78,2 %)
Odpoveď PASI
90, n (%)
70 (21,0 %) 18 (5,4 %) 264 (79,0 %)** 192 (57,3 %) 250
(74,9 %)***
203 (60,6%)
Odpoveď PASI
100, n (%) Odpoveď IGA mod 2011 „bez prejavov“alebo
„takmer bez prejavov“, n (%)
14 (4,2 %) 3 (0,9 %) 148 (44,3 %) 95 (28,4 %) 150 (44,9 %) 123 (36,7 %)
128 (38,3 %) 41 (12,2 %) 278 (83,2 %) 226 (67,5 %) 261 (78,1 %) 213 (63,6 %)
* Pacienti liečení sekukinumabom dostávali dávku 300 mg v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala rovnaká dávka každé 4 týždne do 52. týždňa. Pacienti liečení ustekinumabom dostávali dávku 45 mg alebo 90 mg v 0. a 4. týždni, potom každých 12 týždňov do 52. týždňa (dávkované podľa telesnej hmotnosti na základe schváleného dávkovania)
** hodnoty p v porovnaní s ustekinumabom: p<0,0001 pre primárny koncový ukazovateľ PASI 90 v 16. týždni a sekundárny
koncový ukazovateľ PASI 75 vo 4. týždni
*** hodnoty p v porovnaní s ustekinumabom: p=0,0001 pre sekundárny koncový ukazovateľ PASI 90 v 52. týždni
Sekukinumab bol účinný u pacientov, ktorí predtým nedostali systémovú liečbu, u pacientov predtým neliečených biologickými liekmi, u pacientov s expozíciou biologickým liekom/anti-TNF a u pacientov po zlyhaní liečby biologickými liekmi/anti-TNF. Zlepšenie PASI 75 u pacientov, ktorí mali pri zaradení do skúšania aj psoriatickú artritídu, bolo podobné ako u celkovej populácie s ložiskovou psoriázou.

Sekukinumab mal rýchly nástup účinku so znížením strednej hodnoty PASI o 50 % do 3. týždňa pri
dávke 300 mg.
Obrázok 1 Časový priebeh percentuálnej zmeny stredného skóre PASI oproti východiskovej hodnote v skúšaní 1 (ERASURE)
Týždne liečby
N = počet vyhodnotiteľných pacientovsekukinimab 150 mg (n=243) sekukinumab 300 mg (n=245) Placebo (n=245)
Špecifické miesta/formy ložiskovej psoriázyV ďalších dvoch skúšaniach kontrolovaných placebom sa zaznamenalo zlepšenie pri psoriáze nechtov
(TRANSFIGURE, 198 pacientov), ako aj pri palmoplantárnej ložiskovej psoriáze (GESTURE,
205 pacientov). V skúšaní TRANSFIGURE bol v 16. týždni sekukinumab lepší ako placebo (46,1 %
pri 300 mg, 38,4 % pri 150 mg a 11,7 % pri placebe) pri hodnotení významného zlepšenia indexu závažnosti psoriázy nechtov (Nail Psoriasis Severity Index, NAPSI %) oproti východiskovým
hodnotám u pacientov so stredne ťažkou až ťažkou ložiskovou psoriázou s postihnutím nechtov. V
skúšaní GESTURE bol sekukinumab lepší ako placebo v 16. týždni (33,3 % pri 300 mg, 22,1 % pri
150 mg a 1,5 % pri placebe) pri hodnotení významného zlepšenia 0 alebo 1 odpovede pre ppIGA
(„bez prejavov“ alebo „takmer bez prejavov“) u pacientov so stredne ťažkou až ťažkou palmoplantárnou ložiskovou psoriázou.
V klinickom skúšaní kontrolovanom placebom bolo vyhodnotených 102 pacientov so stredne závažnou až závažnou psoriázou vlasatej časti hlavy, definovanou indexom závažnosti psoriázy vlasatej časti hlavy (Psoriasis Scalp Severity Index, PSSI) so skóre ≥12, odpoveďou IGA mod 2011 iba vlasatej časti hlavy so skóre 3 alebo vyšším a najmenej 30 % postihnutím povrchu vlasatej časti. Sekukinumab 300 mg bol v 12. týždni účinnejší ako placebo po 12. týždni, kedy boli vyhodnotené významné zlepšenia v oboch odpovediach PSSI 90 (52,9 % oproti 2,0 %) a IGA mod 2011 0 alebo 1 iba vo vlasatej časti hlavy (56,9 % oproti 5,9 %) oproti východiskovým hodnotám. Zlepšenie oboch koncových ukazovateľov pretrvávalo u pacientov, ktorí pokračovali v liečbe sekukinumabom, až do
24. týždňa.
Kvalita života/výsledky hlásené pacientmiŠtatisticky významné zlepšenie v 12. týždni (skúšania 1-4) oproti východiskovej hodnote v porovnaní s placebom sa prejavilo v DLQI (Dermatology Life Quality Index – Dermatologický index kvality života). Stredný pokles (zlepšenie) v DLQI oproti východiskovej hodnote v 12. týždni bol v rozmedzí
od -10,4 do -11,6 pri 300 mg sekukinumabu, od -7,7 do -10,1 pri 150 mg sekukinumabu, oproti -1,1
až -1,9 pri placebe. Toto zlepšenie pretrvalo 52 týždňov (skúšania 1 a 2).
V skúšaniach 1 a 2 vyplnilo 40 % účastníkov Denník psoriatických príznakov (Psoriasis Symptom Diary©). U účastníkov, ktorí v každom z týchto skúšaní vypĺňali denník, sa v 12. týždni prejavilo štatisticky významné zmiernenie svrbenia, bolesti a šupinatosti ako pacientmi hlásených prejavov
a príznakov oproti východiskovej hodnote v porovnaní s placebom.
Štatisticky významné zlepšenie po 4. týždni oproti východiskovej hodnote u pacientov liečených sekukinumabom v porovnaní s pacientmi liečenými ustekinumabom (CLEAR) sa prejavilo v DLQI a toto zlepšenie pretrvalo 52 týždňov.
Štatisticky významné zmiernenie svrbenia, bolesti a šupinatosti ako pacientmi hlásených prejavov
a príznakov sa preukázalo v Denníku psoriatických príznakov (Psoriasis Symptom Diary©)
po 16. týždni a 52. týždni (CLEAR) u pacientov liečených sekukinumabom v porovnaní s pacientmi
liečenými ustekinumabom.
Štatisticky významné zlepšenie (zníženie) v skúšaní pri psoriáze vlasatej časti hlavy sa preukázalo po 12. týždni pri pacientmi hlásených prejavoch a príznakoch svrbenia vlasatej časti hlavy, bolesti a šupinatosti oproti východiskovým hodnotám v porovnaní s placebom.
Psoriatická artritída
Bezpečnosť a účinnosť sekukinumabu sa hodnotili u 1 999 pacientov v troch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických skúšaniach fázy III u pacientov s aktívnou psoriatickou artritídou (≥3 opuchnuté a ≥3 citlivé kĺby) aj napriek liečbe nesteroidným antiflogistikom
(NSAID), kortikosteroidom alebo antireumatickým liekom modifikujúcim chorobu (DMARD). Do
týchto klinických skúšaní boli zaradení pacienti s každým podtypom PsA, vrátane polyartikulárnej artritídy bez zjavných reumatoidných uzlíkov, spondylitídy s periférnou artritídou, asymetrickou periférnou artritídou, distálneho interfalangeálneho postihnutia a mutilujúcej artritídy. U pacientov v týchto klinických skúšaniach bola diagnostikovaná PsA trvajúca najmenej päť rokov. Väčšina pacientov mala aj aktívne psoriatické kožné lézie alebo zdokumentovanú psoriázu v anamnéze. Na začiatku liečby malo viac ako 61 % pacientov so PsA entezitídu a viac ako 42 % pacientov so PsA
daktylitídu. Primárnym koncovým ukazovateľom všetkých klinických skúšaní bola odpoveď na liečbu
ACR 20 podľa kritérií Americkej reumatologickej spoločnosti (American College of Rheumatology, ACR). V skúšaní 1 pri psoriatickej artritíde (skúšanie 1 pri PsA) a v skúšaní 2 pri psoriatickej artritíde (skúšanie 2 pri PsA) bol primárny koncový ukazovateľ ACR 20 vyhodnotený v 24. týždni.
V skúšaní 3 pri psoriatickej artritíde (skúšanie 3 pri PsA) bol primárny koncový ukazovateľ ACR 20 vyhodnotený v 16. týždni a kľúčový sekundárny koncový ukazovateľ, zmena modifikovaného celkového Sharpovho skóre (modified Total Sharp Score, mTSS) oproti východiskovému stavu, v
24. týždni.
V skúšaní 1 pri PsA, v skúšaní 2 pri PsA a v skúšaní 3 pri PsA bolo 29 %, 35 % a 30 % pacientov
v uvedenom poradí predtým liečených látkou anti-TNFα a ukončilo liečbu látkou anti-TNFα buď pre nedostatočnú účinnosť, alebo pre intoleranciu (anti-TNFα-IR pacienti).
Skúšanie 1 pri PsA (FUTURE 1) vyhodnotilo 606 pacientov, z ktorých 60,7 % dostávalo súbežne
MTX. Pacienti randomizovaní na sekukinumab dostali intravenózne v 0., 2. a 4. týždni dávku
10 mg/kg, po ktorej nasledovali subkutánne dávky buď 75 mg, alebo 150 mg počnúc 8. týždňom
každý mesiac. Pacienti randomizovaní na placebo, ktorí nereagovali do 16. týždňa (včasná záchrana),
a ostatní pacienti, ktorí dostávali placebo, prešli na podávanie sekukinumabu (buď 75 mg, alebo
150 mg subkutánne) v 24. týždni, po ktorom nasledovala každý mesiac rovnaká dávka.
Skúšanie 2 pri PsA (FUTURE 2) vyhodnotilo 397 pacientov, z ktorých 46,6 % malo súbežne MTX.
Pacienti randomizovaní na sekukinumab dostali subkutánne v 0., 1., 2., 3. a 4. týždni dávku 75 mg,
150 mg alebo 300 mg, po ktorej nasledovala každý mesiac rovnaká dávka. Pacienti randomizovaní na
placebo, ktorí nereagovali do 16. týždňa (včasná záchrana), prešli na podávanie sekukinumabu (buď
150 mg, alebo 300 mg subkutánne) v 16. týždni, po ktorom nasledovala každý mesiac rovnaká dávka. Pacienti randomizovaní na placebo, ktorí nereagovali do 16. týždňa, prešli na podávanie sekukinumabu (buď 150 mg, alebo 300 mg subkutánne) v 24. týždni, po ktorom nasledovala každý
mesiac rovnaká dávka.
Skúšanie 3 pri PsA (FUTURE 5) vyhodnotilo 996 pacientov, z ktorých 50,1 % súbežne užívalo MTX.
Pacienti boli randomizovaní na subkutánne podávanie sekukinumabu 150 mg, 300 mg alebo placeba v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala každý mesiac rovnaká dávka, alebo injekcia sekukinumabu 150 mg raz za mesiac (bez nasycovania). Pacienti randomizovaní na placebo, ktorí nereagovali do 16. týždňa (včasná záchrana), prešli na podávanie sekukinumabu (buď 150 mg, alebo
300 mg subkutánne) v 16. týždni, po ktorom nasledovala každý mesiac rovnaká dávka. Pacienti
randomizovaní na placebo, ktorí reagovali do 16. týždňa, prešli na podávanie sekukinumabu (buď
150 mg, alebo 300 mg subkutánne) v 24. týždni, po ktorom nasledovala každý mesiac rovnaká dávka.
Prejavy a príznakyV 16. a 24. týždni mala liečba sekukinumabom za následok významné zlepšenie merané ako aktivita
ochorenia v porovnaní s placebom (pozri Tabuľku 6).
Tabuľka 6 Klinická odpoveď v skúšaní 2 pri PsA a skúšaní 3 pri PsA po 16. a 24. týždni
| Skúšanie 2 pri PsA Skúšanie 3 pri PsA
|
| Placebo 150 mg1 300 mg1
| Placebo 150 mg1 300 mg1
|
Počet
randomizovaných
pacientov
| 98 100 100
| 332 220 222
|
Odpoveď ACR20
n (% )
16. týždeň
24. týždeň
|
18 60 57
(18,4 %) (60,0 %***) (57,0 %***)
15◊ 51◊ 54◊
(15,3 %) (51,0 %***) (54,0 %***)
|
91◊ 122◊ 139◊
(27,4 %) (55,5 %***) (62,6 %***)
78 117 141
(23,5 %) (53,2 %***) (63,5 %***)
|
Odpoveď ACR50
n (% )
16. týždeň
24. týždeň
|
6 37 35
(6,1 %) (37,0 %***) (35,0 %***)
7 35 35
(7,1 %) (35,0 %) (35,0 %**)
|
27 79 88
(8,1 %) (35,9 %*) (39,6 %*)
29 86 97
(8,7 %) (39,1 %***) (43,7 %***)
|
Odpoveď ACR70
n (% )
16. týždeň
24. týždeň
|
2 17 15
(2,0 %) (17,0 %**) (15,0 %**)
1 21 20
(1,0 %) (21,0 %**) (20,0 %**)
|
14 40 45
(4,2 %) (18,2 %***) (20,3 %***)
13 53 57
(3,9 %) (24,1 %***) (25,7 %***)
|
DAS28-CRP
16. týždeň
24. týždeň
|
-0,50 -1,45*** -1,51***
-0,96 -1,58** -1,61**
|
-0,63 -1,29* -1,49*
-0,84 -1,57*** -1,68***
|
Počet pacientov
s kožou postihnutou psoriázou na ≥3 %
BSA na začiatku liečby
| 43 58 41
(43,9 %) (58,0 %) (41,0 %)
| 162 125 110
(48,8 %) (56,8 %) (49,5 %)
|
Odpoveď PASI 75
n (% )
16. týždeň
24. týždeň
|
3 33 27
(7,0 %) (56,9 %***) (65,9 %***)
7 28 26
(16,3 %) (48,3 %**) (63,4 %***)
|
20 75 77
(12,3 %) (60,0 %*) (70,0 %*)
29 80 78
(17,9 %) (64,0 %***) (70,9 %***)
|
O
dp
ov
e
ď PASI 90
n (% )
1
6
. týždeň
2
4
. týždeň
|
3 22 18
(7,0 %) (37,9 %***) (43,9 %***)
4 19 20
(9,3 %) (32,8 %**) (48,8 %***)
|
15 46 59
(9,3 %) (36,8 %*) (53,6 %*)
19 51 60
(11,7 %) (40,8 %***) (54,5 %***)
|
V
y
m
i
z
nut
i
e
d
a
k
t
y
l
i
t
í
d
y n (% ) †
1
6
. týždeň
2
4
. týždeň
|
10 21 26
(37 %) (65,6 %*) (56,5 %)
4 16 26
(14,8 %) (50,0 %**) (56,5 %**)
|
40 46 54
(32,3 %) (57,5 %*) (65,9 %*)
42 51 52
(33,9 %) (63,8 %***) (63,4 %***)
|
V
y
m
i
z
nut
i
e
e
n
t
e
z
i
t
í
d
y n (% ) ‡
1
6
. týždeň
2
4
. týždeň
|
17 32 32
(26,2 %) (50,0 %**) (57,1 %***)
14 27 27
(21,5 %) (42,2 %*) (48,2 %**)
|
68 77 78
(35,4 %) (54,6 %*) (55,7 %*)
66 77 86
(34,4 %) (54,6 %***) (61,4 %***)
|
* p<0,05; ** p<0,01; *** p<0,001; oproti placebu
Všetky hodnoty p sú upravené pre multiplicitu testovania na základe vopred definovanej hierarchie
v 24. týždni pre skúšanie 2 pri PsA, s výnimkou pre ACR70, daktylitídu a entezitídu, ktoré boli výskumné koncové ukazovatele, a všetky koncové ukazovatele v 16. týždni.
Všetky hodnoty p sú upravené pre multiplicitu testovania na základe vopred definovanej hierarchie
v 16. týždni pre skúšanie 3 pri PsA, s výnimkou pre ACR70, ktorá bola výskumný koncový ukazovateľ,
a všetky koncové ukazovatele v 24. týždni.
Chýbajúce údaje boli vyhodnotené ako bez odpovede pre binárny koncový ukazovateľ.
ACR: Americká reumatologická spoločnosť (American College of Rheumatology); PASI: index plochy a
závažnosti psoriázy (Psoriasis Area and Severity Index); DAS: skóre aktivity ochorenia (Disease Activity
Score); BSA: plocha povrchu tela (Body Surface Area)
◊Primárny koncový ukazovateľ
1 Sekukinumab 150 mg alebo 300 mg s.c. v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala každý mesiac
rovnaká dávka
†U pacientov s daktylitídou na začiatku liečby (n=27, 32, 46, v uvedenom poradí, pre skúšanie 2 pri PsA, a n=124, 80, 82, v uvedenom poradí, pre skúšanie 3 pri PsA)
‡U pacientov s entezitídou na začiatku liečby (n=65, 64, 56, v uvedenom poradí, pre skúšanie 2 pri PsA, a
n=192, 141, 140, v uvedenom poradí, pre skúšanie 3 pri PsA)
|
K nástupu účinku sekukinumabu došlo už v 2. týždni. V 3. týždni sa dosiahol štatisticky významný
rozdiel v ACR 20 oproti placebu.
Percentuálny podiel pacientov, ktorí dosiahli odpoveď ACR 20 pri návšteve, je znázornený na
Obrázku 2.
O
b
r
á
z
o
k 2 Odpoveď ACR20 v skúšaní 2 pri PsA v čase až do 52. týždňa

Čas (týždne)
Podobné odpovede pre primárne a kľúčové sekundárne koncové ukazovatele sa zaznamenali
u pacientov so PsA bez ohľadu na to, či boli súbežne liečení MTX, alebo nie. V skúšaní 2 pri PsA v 24. týždni mali pacienti liečení sekukinumabom so súbežne použitým MTX vyššiu odpoveď ACR 20 (47,7 % pri 150 mg a 54,4 % pri 300 mg v porovnaní s 20,0 % pri placebe) a odpoveď
ACR 50 (31,8 % pri 150 mg a 38,6 % pri 300 mg v porovnaní s 8,0 % pri placebe). Pacienti liečení
sekukinumabom bez súbežne použitého MTX mali vyššiu odpoveď ACR 20 (53,6 % pri 150 mg a 53,6 % pri 300 mg v porovnaní s 10,4 % pri placebe) a odpoveď ACR 50 (37,5 % pri 150 mg
a 32,1 % pri 300 mg v porovnaní s 6,3 % pri placebe).
V skúšaní 2 pri PsA anti-TNFα-naivní pacienti liečení sekukinumabom, ako aj anti-TNFα-IR pacienti liečení sekukinumabom mali v 24. týždni významne vyššiu odpoveď ACR 20 v porovnaní s placebom, s mierne vyššou odpoveďou v skupine anti-TNFα-naivných pacientov (anti-TNFα-naivní pacienti:
64 % pri 150 mg a 58 % pri 300 mg v porovnaní s 15,9 % pri placebe; anti-TNFα-IR: 30 % pri 150 mg a 46 % pri 300 mg v porovnaní so 14,3 % pri placebe). V podskupine anti-TNFα-IR pacientov
vykázala len dávka 300 mg významne vyššiu mieru odpovede pre ACR 20 v porovnaní s placebom (p<0,05) a preukázala klinicky významný prínos oproti dávke 150 mg pre viaceré sekundárne koncové ukazovatele. Zlepšenia v odpovedi PASI 75 sa pozorovali v oboch podskupinách a pri dávke 300 mg
sa preukázal štatisticky významný prínos u anti-TNFα-IR pacientov.
Zlepšenie sa preukázalo vo všetkých zložkách skóre ACR, vrátane hodnotenia bolesti pacientom. V skúšaní 2 pri PsA bol podiel pacientov, ktorí dosiahli odpoveď podľa modifikovaného kritéria odpovede na liečbu PsA (PsA Response Criteria, PsARC), v 24. týždni väčší u pacientov liečených sekukinumabom (59,0 % pri 150 mg a 61,0 % a pri 300 mg) v porovnaní s placebom (26,5 %).
V skúšaní 1 pri PsA a v skúšaní 2 pri PsA sa účinnosť zachovala do 104. týždňa. V skúšaní 2 pri PsA
bolo spomedzi 200 pacientov, ktorí boli pôvodne randomizovaní na sekukinumab 150 mg a 300 mg, v 52. týždni 178 (89 %) pacientov stále liečených. Zo 100 pacientov randomizovaných na sekukinumab 150 mg malo 64 pacientov odpoveď ACR 20, 39 pacientov malo odpoveď ACR 50
a 20 pacientov malo odpoveď ACR 70. Zo 100 pacientov randomizovaných na sekukinumab 300 mg malo 64 pacientov odpoveď ACR 20, 44 pacientov malo odpoveď ACR 50 a 24 pacientov malo odpoveď ACR 70.
Rádiografická odpoveďV skúšaní 3 pri PsA sa inhibícia progresie štrukturálneho poškodenia hodnotila rádiograficky
a vyjadrila sa ako zmena modifikovaného celkového Sharpovho skóre (mTSS) a jeho zložiek, skóre
erózie (Erosion Score, ES) a skóre zúženia kĺbovej štrbiny (Joint Space Narrowing, JSN). Röntgenové snímky rúk, zápästí a nôh sa urobili na začiatku liečby, v 16. a/alebo 24. týždni a vyhodnotili sa nezávisle najmenej dvomi hodnotiteľmi, pre ktorých boli zaslepené skupina liečby a číslo návštevy. Liečba sekukinumabom 150 mg a 300 mg významne znížila mieru progresie poškodenia periférnych
kĺbov v porovnaní s liečbou placebom, stanovenú ako zmena mTSS v 24. týždni oproti východiskovému stavu (Tabuľka 7).
Inhibícia progresie štrukturálneho poškodenia sa hodnotila aj v skúšaní 1 pri PsA v 24. a 52. týždni
oproti východiskovému stavu. Údaje v 24. týždni sú uvedené v Tabuľke 7.
Tabuľka 7 Zmena modifikovaného celkového Sharpovho skóre pri psoriatickej artritíde
| Skúšanie 3 pri PsA
Placebo sekukinumab sekukinumab n=296 150 mg1 300 mg1
n=213 n=217
| Skúšanie 1 pri PsA
Placebo sekukinumab n=179 150 mg2
n=185
|
Celkové skóre
|
Východiskový stav
(SD)
| 15,0 13,5 12,9
(38,2) (25,6) (23,8)
| 28,4 22,3
(63,5) (48,0)
|
Priemerná
zmena
v 24. týždni
| 0,50 0,13* 0,02*
| 0,57 0,13*
|
*p<0,05 na základe nominálnej, ale nie upravenej hodnoty p
1sekukinumab 150 mg alebo 300 mg s.c. v 0., 1., 2., 3. a 4. týždni, po ktorých nasledovala rovnaká
dávka každý mesiac
210 mg/kg v 0., 2. a 4. týždni, po ktorých nasledovali subkutánne dávky 75 mg alebo 150 mg
|
V skúšaní 1 pri PsA sa inhibícia štrukturálneho poškodenia zachovala pri liečbe sekukinumabom až do
52. týždňa.
V skúšaní 3 pri PsA bol percentuálny podiel pacientov bez progresie ochorenia (definované ako zmena mTSS oproti východiskovému stavu ≤0,5) od randomizácie do 24. týždňa 80,3 % pri 150 mg a 88,5 % pri 300 mg sekukinumabu a 73,6 % pri placebe. Inhibičný účinok na štrukturálne poškodenie sa pozoroval u pacientov bez predchádzajúcej liečby anti-TNFα a u pacientov anti-TNFα-IR a
u pacientov so súbežnou liečbou MTX, ako aj bez nej.
V skúšaní 1 pri PsA percentuálny podiel pacientov bez progresie ochorenia (definované ako zmena mTSS oproti východiskovému stavu ≤0,5) od randomizácie do 24. týždňa bol 82,3 % pri intravenóznej bolusovej dávke sekukinumabu 10 mg/kg – udržiavacia subkutánna dávka 150 mg, a 75,7 % pri placebe. Percentuálny podiel pacientov bez progresie ochorenia od 24. týždňa do 52. týždňa pri intravenóznej nasycovacej dávke sekukinumabu 10 mg/kg – po ktorej nasledovala subkutánna udržiavacia liečba 150 mg, a u pacientov, ktorí prešli z placeba na 75 mg alebo 150 mg subkutánne každé 4 týždne po 16. týždni alebo 24. týždni, bol 85,7 % a 86,8 % v uvedenom poradí.
Axiálne prejavy pri PsA
Randomizované, dvojito zaslepené, placebom kontrolované klinické skúšanie (MAXIMISE) hodnotilo účinnosť sekukinumabu u 485 pacientov s PsA s axiálnymi prejavmi, ktorí v minulosti neboli liečení biologickými liekmi a nemali dostatočnú odpoveď na NSAID. Primárny cieľ skúšania, ktorým bolo minimálne 20 % zlepšenie stavu pacientov hodnotené podľa kritérií Medzinárodnej spoločnosti pre spondyloartritídu (ASAS 20) po 12 týždňoch liečby, bol splnený. V porovnaní s placebom viedla liečba sekukinumabom v dávkach 300 mg a 150 mg k výraznejšiemu zlepšeniu prejavov a príznakov (vrátane zníženia bolesti chrbta oproti východiskovému stavu) a zlepšeniu fyzickej kondície (pozri Tabuľku 8).
Tabuľka 8 Klinická odpoveď v skúšaní MAXIMISE v 12. týždni
| Placebo
(n=164)
| 150 mg
(n=157)
| 300 mg
(n=164)
|
Odpoveď ASAS 20, % (95% IS)
| 31,2 (24,6; 38,7)
| 66,3 (58,4; 73,3)*
| 62,9 (55,2; 70,0)*
|
Odpoveď ASAS 40, %
(95% IS)
| 12,2 (7,8; 18,4)
| 39,5 (32,1; 47,4)**
| 43,6 (36,2; 51,3)**
|
BASDAI 50, %
(95% IS)
| 9,8 (5,9; 15,6)
| 32,7 (25,8; 40,5)**
| 37,4 (30,1; 45,4)**
|
Bolesť chrbta, VAS
(95% IS)
| -13,6 (-17,2; -10,0)
| -28,5 (-32,2; -24,8)**
| -26,5 (-30,1; -22,9)**
|
Fyzická kondícia,
HAQ-DI (95% IS)
| -0,155 (-0,224; -0,086)
| -0,330 (-0,401;
-0,259)**
| -0,389 (-0,458;
-0,320)**
|
* p<0,0001; oproti placebu s využitím viacnásobnej imputácie.
** Porovnanie oproti placebu nebolo upravené z dôvodu multiplicity.
ASAS: kritériá pre hodnotenie podľa Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society Criteria); BASDAI: index aktivity ankylozujúcej
spondylitídy z Bathu (Bath Ankylosing Spondylitis Disease Activity Index); VAS: vizuálna analógová stupnica (Visual Analog Scale); HAQ-DI: dotazník na hodnotenie zdravia podľa indexu invalidity (Health Assessment Questionnaire – Disability Index).
|
Pre obe dávky sekukinumabu sa pozorovalo zlepšenie odpovedí ASAS 20 a ASAS 40 v 4. týždni a
zachovalo sa až do 52. týždňa.
Fyzická kondícia a kvalita života súvisiaca so zdravímV skúšaní 2 pri PsA a v skúšaní 3 pri PsA sa v 24. týždni a 16. týždni, v uvedenom poradí, pomocou
dotazníka na hodnotenie zdravia podľa indexu invalidity (Health Assessment Questionnaire -Disability
Index, HAQ-DI) preukázalo u pacientov liečených sekukinumabom 150 mg (p=0,0555 a p<0,0001)
a 300 mg (p=0,0040 a p<0,0001) zlepšenie fyzickej kondície v porovnaní s pacientmi, ktorí dostávali
placebo. Zlepšenia skóre HAQ-DI sa pozorovali bez ohľadu na predchádzajúcu expozíciu anti-TNFα.
Podobné odpovede sa pozorovali v skúšaní 1 pri PsA.
Pacienti liečení sekukinumabom hlásili významné zlepšenia kvality života súvisiacej so zdravím podľa sumárneho skóre stručného formulára prieskumu fyzického zdravia (Short Form-36 Health Survey Physical Component Summary, SF-36 PCS) (p<0,001). Došlo tiež k štatisticky významnému
zlepšeniu preukázanému výskumnými ukazovateľmi, stanovenému prostredníctvom skóre funkčného posúdenia liečby chronického ochorenia – únava (Functional Assessment of Chronic Illness Therapy – Fatigue, FACIT-F) pri 150 mg a 300 mg v porovnaní s placebom (7,97 a 5,97 oproti 1,63) a toto zlepšenie sa zachovalo až do 104. týždňa v skúšaní 2 pri PsA.
Podobné odpovede sa pozorovali v skúšaní 1 pri PsA a účinnosť sa zachovala až do 52. týždňa.
Axiálna spondyloartritída (axSpA)
Ankylozujúca spondylitída (AS) / Axiálna spondyloartritída s rádiografickým dôkazom
Bezpečnosť a účinnosť sekukinumabu sa hodnotili u 816 pacientov v troch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických skúšaniach fázy III u pacientov s aktívnou ankylozujúcou spondylitídou (AS) s indexom aktivity ankylozujúcej spondylitídy z Bathu (Bath Ankylosing Spondylitis Disease Activity Index, BASDAI) ≥4 aj napriek liečbe nesteroidným antiflogistikom (NSAID), kortikosteroidom alebo antireumatickým liekom modifikujúcim chorobu (DMARD). U pacientov v skúšaní 1 pri ankylozujúcej spondylitíde (skúšanie 1 pri AS)
a v skúšaní 2 pri ankylozujúcej spondylitíde (skúšanie 2 pri AS) bola diagnostikovaná AS so strednou hodnotou trvania 2,7 až 5,8 rokov. Primárnym koncovým ukazovateľom oboch klinických skúšaní bolo zlepšenie hodnotené podľa kritérií Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society, ASAS 20) o najmenej 20% v 16. týždni.
V skúšaní 1 pri ankylozujúcej spondylitíde (skúšanie 1 pri AS), v skúšaní 2 pri ankylozujúcej spondylitíde (skúšanie 2 pri AS) a v skúšaní 3 pri ankylozujúcej spondylitíde (skúšanie 3 pri AS) bolo
27,0 % , 38,8 % a 23,5 % pacientov v uvedenom poradí predtým liečených anti-TNFα látkou
a ukončilo liečbu látkou anti-TNFα buď pre nedostatočnú účinnosť, alebo intoleranciu (anti-TNFα-IR
pacienti).
Skúšanie 1 pri AS (MEASURE 1) vyhodnotilo 371 pacientov, z ktorých 14,8 % dostávalo súbežne MTX a 33,4 % dostávalo súbežne sulfasalazín. Pacienti randomizovaní na sekukinumab dostali intravenózne v 0., 2. a 4. týždni dávku 10 mg/kg, po ktorej nasledovali každý mesiac subkutánne dávky buď 75 mg, alebo 150 mg počnúc 8. týždňom. Pacienti randomizovaní na placebo, ktorí nereagovali do 16. týždňa (včasná záchrana), a všetci ostatní pacienti, ktorí dostávali placebo, prešli v 24. týždni na podávanie sekukinumabu (buď 75 mg, alebo 150 mg subkutánne), po ktorom nasledovala každý mesiac rovnaká dávka.
Skúšanie 2 pri AS (MEASURE 2) vyhodnotilo 219 pacientov, z ktorých 11,9 % dostávalo súbežne MTX a 14,2 % dostávalo súbežne sulfasalazín. Pacienti randomizovaní na sekukinumab dostali subkutánne v 0., 1., 2., 3. a 4. týždni dávku buď 75 mg, alebo 150 mg, po ktorej nasledovala každý mesiac rovnaká dávka. Pacienti randomizovaní na placebo pri zaradení do skúšania boli opäť randomizovaní v 16. týždni na podávanie sekukinumabu (buď 75 mg, alebo 150 mg subkutánne) každý mesiac.
Skúšanie 3 pri AS (MEASURE 3) vyhodnotilo 226 pacientov, z ktorých 13,3 % a 23,5 % dostávalo súbežne MTX alebo sulfasalazín, v uvedenom poradí. Pacienti randomizovaní na sekukinumab dostali intravenózne dávku 10 mg/kg v 0., 2. a 4. týždni, po ktorej nasledovala buď 150 mg alebo 300 mg dávka subkutánne každý mesiac. Pacienti randomizovaní na placebo pri zaradení do skúšania boli
opäť randomizovaní v 16. týždni na podávanie sekukinumabu (buď 150 mg, alebo 300 mg subkutánne) každý mesiac. Primárnym koncovým ukazovateľom bolo ASAS 20 v 16. týždni. Pacienti boli zaslepení na liečebný režim až do 52. týždňa a štúdia pokračovala do 156 týždňa.
Prejavy a príznaky:
V skúšaní 2 pri AS mala v 16. týždni liečba sekukinumabom 150 mg za následok väčšie zlepšenie
merané ako aktivita ochorenia v porovnaní s placebom (pozri Tabuľku 9).
T
a
b
u
ľ
k
a 9 Klinická odpoveď v skúšaní 2 pri AS v 16. týždni
V
ý
sledok (hodnota p oproti placebu)
|
Placebo
(
n = 74)
|
7
5 mg
(
n = 73)
|
1
5
0 mg
(
n = 72)
|
Odpoveď ASAS 20, %
|
28,4
|
41,1
|
61,1***
|
Odpoveď ASAS 40, %
|
10,8
|
26,0
|
36,1***
|
hsCRP, (pomer post-BSL/BSL)
|
1,13
|
0,61
|
0,55***
|
ASAS 5/6, %
|
8,1
|
34,2
|
43,1***
|
Parciálna remisia ASAS, %
|
4,1
|
15,1
|
13,9
|
BASDAI 50, %
|
10,8
|
24,7*
|
30,6**
|
Výrazné zlepšenie ASDAS-CRP
|
4,1
|
15,1*
|
25,0***
|
* p<0,05; ** p<0,01; *** p<0,001; oproti placebu
Všetky hodnoty p sú upravené pre multiplicitu testovania na základe vopred definovanej hierarchie,
s výnimkou BASDAI 50 a ASDAS-CRP
Chýbajúce údaje boli vyhodnotené ako bez odpovede pre binárny koncový ukazovateľ
ASAS: kritériá pre hodnotenie podľa Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society Criteria); BASDAI: index aktivity ankylozujúcej spondylitídy z Bathu (Bath Ankylosing Spondylitis Disease Activity Index); hsCRP: vysoko citlivý C-reaktívny proteín (high-sensitivity C-reactive protein); ASDAS: skóre aktivity ankylozujúcej spondylitídy (Ankylosing Spondylitis Disease Activity Score); BSL: východiskový stav (baseline)
|
K nástupu účinku sekukinumabu 150 mg došlo už v 1. týždni pre ASAS 20 a v 2. týždni pre ASAS 40
(účinnejší ako placebo) v skúšaní 2 pri AS.
V 16. týždni sa u anti-TNFα-naivných pacientov (68,2 % oproti 31,1 %; p<0,05) a u anti-TNFα-IR
pacientov (50,0 % oproti 24,1 %; p<0,05) zlepšili odpovede ASAS 20 pre sekukinumab 150 mg v porovnaní s placebom, v uvedenom poradí.
V skúšaniach 1 pri AS a 2 pri AS sa u pacientov liečených sekukinumabom (150 mg v skúšaní 2 pri
AS a oba režimy v skúšaní 1 pri AS) preukázalo v 16. týždni významné zlepšenie prejavov
a príznakov, s porovnateľnou veľkosťou odpovede a účinnosťou zachovanou až do 52. týždňa
u anti-TNFα-naivných pacientov, ako aj u anti-TNFα-IR pacientov. V skúšaní 2 pri AS bolo spomedzi
72 pacientov, ktorí boli na začiatku randomizovaní na sekukinumab 150 mg, v 52. týždni 61 (84,7 %)
pacientov stále liečených. Zo 72 pacientov randomizovaných na sekukinumab 150 mg malo
45 pacientov odpoveď ASAS 20 a 35 pacientov malo odpoveď ASAS 40.
V skúšaní 3 pri AS preukázali pacienti liečení sekukinumabom (150 mg a 300 mg) zlepšenie prejavov a symptómov a mali porovnateľné výsledky účinnosti bez ohľadu na dávku, ktorá bola v primárnom cieľovom ukazovateli (ASAS 20) v 16. týždni lepšia ako placebo. Celkovo bola miera účinnosti odpovede pre sekundárne cieľové ukazovatele konzistentne vyššia v skupine s 300 mg v porovnaní so skupinou so 150 mg. Počas zaslepého obdobia boli v 52. týždni odpovede ASAS 20 a ASAS 40
69,7 % a 47,6 % pre 150 mg a 74,3 % a 57,4 % pre 300 mg. Odpovede ASAS 20 a ASAS 40 sa
udržiavali až do 156.týždňa (69,5 % a 47,6 % pre 150 mg oproti 74,8 % a 55,6 % pre 300 mg). Vyššia
miera odpovede v prospech 300 mg sa pozorovala aj pri odpovedi na čiastočnú remisiu ASAS (ASAS PR) v 16. týždni a udržala sa až do týždňa 156. U pacientov s anti-TNFα-IR (n=36) sa v porovnaní s pacientmi bez predchádzajúcej liečby TNFα (n=114) pozorovali väčšie rozdiely v miere odpovede
v prospech 300 mg oproti 150 mg.
Mobilita chrbtice:
U pacientov liečených sekukinumabom 150 mg sa preukázalo zlepšenie mobility chrbtice stanovené ako zmena oproti východiskovému stavu pomocou BASMI po 16. týždni v skúšaní 1 pri AS (-0,40 oproti -0,12 pri placebe; p=0,0114) a v skúšaní 2 pri AS (-0,51 oproti -0,22 pri placebe; p=0,0533). Tieto zlepšenia sa zachovali až do 52. týždňa.
Fyzická kondícia a kvalita života súvisiaca so zdravím:
V skúšaní 1 a skúšaní 2 pri AS sa u pacientov liečených sekukinumabom 150 mg preukázalo zlepšenie
kvality života súvisiacej so zdravím hodnotenej podľa dotazníka o kvalite života pri AS (AS Quality of Life Questionnaire, AS QoL) (p=0,001) a podľa sumárneho skóre fyzickej zložky SF-36 (SF-36
Physical Component Summary, SF-36 PCS) (p<0,001). U pacientov liečených sekukinumabom
150 mg sa tiež preukázalo štatisticky významné zlepšenie výskumných ukazovateľov týkajúcich sa fyzickej kondície, hodnotené bathskym funkčným indexom ankylozujúcej spondylitídy (Bath Ankylosing Spondylitis Functional Index, BASFI) v porovnaní s placebom (-2,15 oproti -0,68) a škály únavy pri liečbe chronického ochorenia (Functional Assessment of Chronic Illness Therapy-Fatigue,
FACIT-Fatigue) v porovnaní s placebom (8,10 oproti 3,30). Tieto zlepšenia sa zachovali až do
52. týždňa.
Axiálna spondyloartritída bez rádiografického dôkazu (nr-axSpA)
Bezpečnosť a účinnosť sekukinamubu sa hodnotili u 555 pacientov v jednom randomizovanom, dvojito zaslepenom, placebom kontrolovanom klinickom skúšaní fázy III (PREVENT), pozostávajúcom z 2-ročnej základnej fázy a 2-ročnej fázy predĺženia, u pacientov s aktívnou axiálnou
spondyloartritídou bez rádiografického dôkazu (nr-axSpA), ktorí spĺňali kritériá klasifikácie
Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society, ASAS) pre axiálnu spondyloartritídu bez rádiografického dôkazu zmien sakroiliakálnych kĺbov, ktoré by spĺňali modifikované newyorské kritériá pre ankylozujúcu spondylitídu (AS). Zaradení pacienti mali aktívne ochorenie, definované indexom aktivity ankylozujúcej spondylitídy z Bathu
(Bath Ankylosing Spondylitis Disease Activity Index, BASDAI) ≥4 a celkovou bolesťou chrbta ≥40 (na škále 0-100 mm) vizuálnej analógovej stupnice (Visual Analogue Scale,VAS) napriek prebiehajúcej alebo predchádzajúcej liečbe nesteroidnými protizápalovými liekmi (NSAID) a zvýšený C-reaktívny proteín (CRP) a/alebo dôkaz sakroiliitídy pri zobrazení magnetickou rezonanciou (MRI). Pacienti v tomto klinickom skúšaní mali diagnózu axSpA priemerne 2,1 až 3,0 roka a 54 % účastníkov klinického skúšania boli ženy.
V klinickom skúšaní PREVENT bolo 9,7 % pacientov predtým liečených anti-TNFα látkou a ukončilo liečbu anti-TNFα látkou buď pre nedostatočnú účinnosť, alebo intoleranciu (anti-TNFα-IR pacienti).
V klinickom skúšaní PREVENT dostávalo 9,9 % pacientov súbežne MTX a 14,8 % pacientov
sulfasalazín. V období dvojitého zaslepenia dostávali pacienti buď placebo alebo sekukinumab počas
52 týždňov. Pacienti randomizovaní na sekukinumab dostali subkutánne v 0., 1., 2., 3. a 4. týždni dávku 150 mg, po ktorej nasledovala, počnúc 4. týždňom, každý mesiac rovnaká dávka, alebo raz mesačne 150 mg injekcia sekukinumabu. Primárnym koncovým ukazovateľom bolo najmenej 40 %
zlepšenie stavu hodnotené podľa kritérií Medzinárodnej spoločnosti pre spondyloartritídu (ASAS 40)
v 16. týždni u anti-TNFα-naivných pacientov.
Prejavy a príznaky:
V klinickom skúšaní PREVENT viedla liečba sekukinumabom 150 mg v 16. týždni k významnému zlepšeniu miery aktivity ochorenia v porovnaní s placebom. Tieto merania zahŕňajú ASAS 40,
ASAS 5/6, skóre BASDAI, BASDAI 50, vysoko citlivý CRP (hsCRP), ASAS 20 a dosiahnutie
čiastočnej remisie ASAS v porovnaní s placebom (Tabuľka 10). Odpovede pretrvali až do 52. týždňa.
T
a
b
u
ľ
k
a 10 Klinická odpoveď v skúšaní PREVENT v 16. týždni
V
ý
sledok (hodnota oproti placebu)
|
Placebo
|
1
5
0 mg
1
|
Počet zaradených anti-TNFα-naivných pacientov
|
1
7
1
|
1
6
4
|
Odpoveď ASAS 40, %
|
29,2
|
41,5*
|
C
el
k
o
v
ý počet zaradených pacientov
|
1
8
6
|
1
8
5
|
Odpoveď ASAS 40, %
|
28,0
|
40,0*
|
ASAS 5/6, %
|
23,7
|
40,0*
|
BASDAI, LS priemerná zmena oproti východiskovej hodnote
|
-1,46
|
-2,35*
|
BASDAI 50, %
|
21,0
|
37,3*
|
hsCRP, (pomer post-BSL/BSL)
|
0,91
|
0,64*
|
Odpoveď ASAS 20, %
|
45,7
|
56,8*
|
Čiastočná remisia ASAS, %
|
7,0
|
21,6*
|
*p<0,05 oproti placebu
Všetky hodnoty p sú upravené pre multiplicitu testovania na základe vopred definovanej hierarchie
Chýbajúce údaje boli vyhodnotené ako bez odpovede pre binárny koncový ukazovateľ
1sekukinumab 150 mg s.c. v 0., 1., 2., 3., a 4. týždni po ktorých nasledovala každý mesiac rovnaká
dávka.
ASAS: kritériá pre hodnotenie podľa Medzinárodnej spoločnosti pre spondyloartritídu (Assessment of SpondyloArthritis International Society Criteria); BASDAI: index aktivity ankylozujúcej spondylitídy z Bathu (Bath Ankylosing Spondylitis Disease Activity Index); hsCRP: vysoko citlivý C-reaktívny proteín (high-sensitivity C-reactive protein); BSL: východiskový stav (baseline); LS: metóda najmenších štvorcov (Least square)
|
V klinickom skúšaní PREVENT sa u anti-TNFα-naivných pacientov prejavil nástup účinku
sekukinumabu v dávke150 mg v hodnotách ASAS 40 už v 3. týždni (lepší účinok ako placebo). Percentuálny podiel anti-TNFα-naivných pacientov, ktorí dosiahli odpoveď ASAS 40 je znázornený na obrázku 3.
O
b
r
á
z
o
k 3 OdpoveďASAS 40 v skúšaní PREVENT u anti-TNFα-naivných pacientov čase až
d
o 16.týždňa
Percentuálny podiel pacientov s odpoveďou na liečbu
Čas (týždne)

Sekukinumab 150 mg dávka

Placebo
V 16. týždni sa zlepšila odpoveď ASAS 40 aj u anti-TNFα-IR pacientov liečených sekukinumabom
150 mg v porovnaní s placebom.
Fyzická kondícia a kvalita života súvisiaca so zdravím:
Pacienti liečení sekukinumabom 150 mg preukázali v 16. týždni štatisticky významné zlepšenia telesnej funkcie, na základe zmeny hodnoty indexu BASFI, v porovnaní s pacientami užívajúcimi placebo (16. týždeň: -1,75 oproti -1,01, p<0,05). Pacienti liečení sekukinumabom preukázali v
porovnaní s pacientami užívajúcimi placebo v 16. týždni štatisticky významné zlepšenia kvality života
súvisiacej so zdravím, meranej pomocou ASQoL (priemerná zmena LS v 16. týždni: -3,45
oproti -1,84, p<0,05) a podľa sumárneho skóre fyzickej zložky SF-36 (Physical Component Summary,
SF-36 PCS) (priemerná zmena LS v 16. týždni: 5,71 oproti 2,93, p<0,05). Tieto zlepšenia sa zachovali
až do 52.týždňa.
Mobilita chrbtice:
Mobilita chrbtice sa hodnotila pomocou indexu BASMI až do 16. týždňa. Vo 4., 8., 12. a 16. týždni sa preukázali numericky vyššie hodnoty zlepšenia u pacientov užívajúcich sekukinumab v porovnaní s pacientami užívajúcimi placebo.
Inhibícia zápalu pri zobrazení magnetickou rezonanciou (MRI):
Prejavy zápalu sa hodnotili MRI pri zaradení do skúšania a v 16. týždni a vyjadrili sa zmenou skóre Berlin SI-joint oedema pre sakroiliakálne kĺby, skóre ASspiMRI-a a Berlin spine skóre pre chrbticu oproti východiskovému stavu. U pacientov liečených sekukinumabom sa pozorovala inhibícia
zápalových prejavov pri sakroiliakálnych kĺboch aj pri chrbtici. Priemerná zmena voči východiskovej hodnote Berlin SI-joint oedema skóre bola -1,68 u pacientov liečených sekukinumabom 150 mg (n=180) oproti -0,39 u pacientov (n=174) (p<0,05).
Pediatrická populáciaLožiskovápsoriázau pediatrických pacientovPreukázalo sa, že sekukinumab zlepšuje prejavy a príznaky, ako aj kvalitu života súvisiacu so zdravím
u pediatrických pacientov s ložiskovou psoriázou vo veku 6 rokov a starších (pozri Tabuľky 12 a 14).
Z
á
v
a
ž
n
á ložisková psoriáza
Bezpečnosť a účinnosť sekukinumabu sa hodnotili v randomizovanom, dvojito zaslepenom klinickom skúšaní fázy III, kontrolovanom placebom a etanerceptom, u pediatrických pacientov so závažnou ložiskovou psoriázou vo veku 6 až <18 rokov, definovanou skóre PASI ≥20 a skóre IGA mod 2011 4
a rozsahom postihnutia kože s BSA ≥10%, ktorí boli kanditátmi na systémovú liečbu. Približne 43 % pacientov malo predchádzajúcu expozíciu fototerapii, 53 % konvenčnej systémovej terapii, 3 % biologickým látkam a 9 % malo sprievodnú psoriatickú artritídu.
V pediatrickom skúšaní 1 sa vyhodnotilo 162 pacientov, randomizovaných na podávanie nízkej dávky sekukinumabu (75 mg na telesnú hmotnosť <50 kg alebo 150 mg na telesnú hmotnosť ≥50 kg), vysokej dávky sekukinumabu (75 mg na telesnú hmotnosť <25 kg, 150 mg na telesnú hmotnosť medzi
≥25 kg a <50 kg, alebo 300 mg na telesnú hmotnosť ≥50 kg), alebo placeba v 0., 1., 2., 3. a 4. týždni,
po ktorých nasledovala každé 4 týždne rovnaká dávka, alebo etanercept. Patienti radomizovaní na etanercept dostávali 0,8 mg/kg týždenne (najviac však 50 mg). Rozdelenie pacientov podľa hmotnosti a veku pri randomizácii je uvedené v Tabuľke 11.
Tabuľka 11 Rozdelenie pacientov podľa hmotnosti a veku v pediatrickom skúšaní 1 pri psoriázeRandomizačná
vrstva
| Popis
| Sekukinumab nízka dávka n=40
| Sekukinumab vysoká dávka n=40
| Placebo
n=41
| Etanercept
n=41
| Spolu
N=162
|
Vek
| 6-<12 rokov
| 8
| 9
| 10
| 10
| 37
|
≥12-
<18 rokov
| 32
| 31
| 31
| 31
| 125
|
Hmotnosť
| <25 kg
| 2
| 3
| 3
| 4
| 12
|
≥25-<50 kg
| 17
| 15
| 17
| 16
| 65
|
≥50 kg
| 21
| 22
| 21
| 21
| 85
|
Pacienti randomizovaní na placebo, ktorí nedosiahli odpoveď do 12.týždňa prešli do skupiny s nízkou
alebo vysokou dávkou sekukinumabu (dávka na základe skupiny telesnej hmotnosti) a dostali skúšaný
liek v 12., 13., 14. a 15.týždni, po ktorom nasledovala každé 4 týždne rovnaká dávka, začínajúca
v 16.týždni. Spoločné primárne koncové ukazovatele boli podiely pacientov, ktorí dosiahli odpoveď
PASI 75 a IGA mod 2011 „bez prejavov“ alebo“takmer bez prejavov“ (0 alebo 1) v 12.týždni.
Účinnosť v oboch skupinách s nízkou a vysokou dávkou sekukinumabu počas placebom kontrolovaného obdobia do 12.týždňa bola porovnateľná pre spoločné primárne koncové ukazovatele. Odhadovaný pomer pravdepodobnosti v prospech oboch dávok sekukinumabu bol štatisticky významný pre odpovede PASI 75 a IGA mod 2011 0 alebo 1.
U všetkých pacientov sa sledovala účinnosť a bezpečnosť v priebehu 52 týždňov od prvého podania skúšaného lieku. Podiel pacientov, ktorí dosiahli PASI 75 a IGA mod 2011 „bez prejavov“ alebo
„takmer bez prejavov“ (0 alebo 1) preukázal rozdiel medzi skupinou liečenou so sekukinumabom
a skupinou s placebom pri prvej návšteve po východiskovej návšteve vo 4.týždni a výraznejší rozdiel nastal v 12.týždni. Odpoveď pretrvala počas 52-týždňového časového obdobia (pozri Tabuľku 12). Zlepšenie miery odpovede vyjadrené v hodnotách PASI 50, 90, 100 a Detského dermatologického
indexu kvality života (Children’s Dermatology Life Quality Index, (CDLQI)) zo skóre 0 alebo 1
taktiež pretrval počas 52-týždňového obdobia.
Naviac miera odpovede PASI 75, IGA 0 alebo 1, PASI 90 v 12. a 52. týždni pre obe skupiny s nízkou a vysokou dávkou sekukinumabu bola vyššia ako miera odpovede u pacientov liečených
s etanerceptom (pozri Tabuľku 12).
Po 12.týždni bola účinnosť oboch skupín s nízkou a vysokou dávkou sekukinumabu porovnateľná, hoci účinnosť s vyššou dávkou sekukinumabu bola vyššia u pacientov ≥50 kg. Profily bezpečnosti s nízkou dávkou a vysokou dávkou boli porovnateľné a v zhode s profilom bezpečnosti u dospelých pacientov.
Tabuľka 12 Súhrn klinickej odpovede u pediatrických pacientov so závažnou psoriázou v 12. a 52.týždni (pediatrické skúšanie 1 pri psoriáze)*
Kritérium odpovede
| Porovnanie liečby
| „test“
| „kontrola”
| odhadovaný pomer pravdepodobnosti (95 % IS)
|
Hodnota p
|
„test“ vs. „kontrola“
|
n**/m ( %)
|
n**/m ( %)
|
v 12.týždni***
|
PASI 75
| sekukinumab nízka dávka vs. placebo
sekukinumab vysoká dávka vs. placebo
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
| 32/40 (80,0)
31/40 (77,5)
32/40 (80,0)
31/40 (77,5)
| 6/41 (14,6)
6/41 (14,6)
26/41 (63,4)
26/41 (63,4)
| 25,78 (7,08; 114,66)
22,65 (6,31; 98,93)
2,25 (0,73; 7,38)
1,92 (0,64; 6,07)
| <0,0001
<0,0001
|
IGA 0/1
| sekukinumab nízka dávka vs. placebo
sekukinumab vysoká dávka vs. placebo
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
| 28/40 (70,0)
24/40 (60,0)
28/40 (70,0)
24/40 (60,0)
| 2/41 (4,9)
2/41 (4,9)
14/41 (34,1)
14/41 (34,1)
| 51,77 (10,02; 538,64)
32,52 (6,48; 329,52)
4,49 (1,60; 13,42)
2,86 (1,05; 8,13)
| <0,0001
<0,0001
|
PASI 90
| sekukinumab nízka dávka vs. placebo
sekukinumab vysoká dávka vs. placebo
sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
| 29/40 (72,5)
27/40 (67,5)
29/40 (72,5)
27/40 (67,5)
| 1/41 (2,4)
1/41 (2,4)
12/41 (29,3)
12/41 (29,3)
| 133,67 (16,83; 6395,22)
102,86 (13,22; 4850,13)
7,03 (2,34; 23,19)
5,2 (1,82; 16,75)
| <0,0001
<0,0001
|
v 52. týždni
|
PASI 75
| sekukinumab nízka dávka vs.
etanercept
sekukinumab vysoká dávka vs. etanercept
| 35/40 (87,5)
35/40 (87,5)
| 28/41 (68,3)
28/41 (68,3)
| 3,12 (0,91; 12,52)
3,09 (0,90; 12,39)
|
|
IGA 0/1
| sekukinumab nízka dávka vs.
etanercept
sekukinumab vysoká dávka vs. etanercept
| 29/40 (72,5)
30/40 (75,0)
| 23/41 (56,1)
23/41 (56,1)
| 2,02 (0,73; 5,77)
2,26 (0,81; 6,62)
|
|
PASI 90
| sekukinumab nízka dávka vs. etanercept
sekukinumab vysoká dávka vs. etanercept
| 30/40 (75,0)
32/40 (80,0)
| 21/41 (51,2)
21/41 (51,2)
| 2,85 (1,02; 8,38)
3,69 (1,27; 11,61)
|
|
* chýbajúce údaje boli vyhodnotené ako bez odpovede
** n je počet pacientov s odpoveďou, m = počet pacientov s hodnotiteľnou odpoveďou
*** rozšírené okno návštevy v 12.týždni
Pomer pravdepodobnosti, 95 % interval spoľahlivosti a hodnota p sú z presného modelu logistickej regresie s faktormi liečebná skupina, kategóriou východiskovej telesnej hmostnosti a vekovou kategóriou
|
Zlepšenie kvality života súvisiacej so zdravím meranej ako skóre CDLQI v hodnote 0 alebo 1
v porovnaní s placebom v 12.týždni hlásil vyšší podiel pediatrických pacientov liečených
sekukinumabom (nízka dávka 44,7 %, vysoká dávka 50 %, placebo 15 %). V čase až do 52.týždňa v oboch skupinách s dávkou sekukinumabu bol počet pacientov s odpoveďou numericky vyšší ako v skupine s etanerceptom (nízka dávka 60,6 %, vysoká dávka 66,7 %, etanercept 44,4 %).
S
t
r
e
d
n
e závažná až závažná ložisková psoriáza
Na základe preukázanej miery účinnosti a vzťahu odpovede na expozíciu u dospelých pacientov so stredne závažnou až závažnou ložiskovou psoriázou a podobnosti priebehu ochorenia, patofyziológie a účinku lieku u dospelých a pediatrických pacientov s rovnakými expozičnými hladinami sa
predpokladalo, že sekukinumab bude účinný aj pri liečbe pediatrických pacientov so stredne závažnou až závažnou ložiskovou psoriázou.
Okrem toho sa bezpečnosť a účinnosť sekukinumabu hodnotila v otvorenom multicentrickom skúšaní fázy III s dvomi ramenami a paralelnou skupinou u pediatrických pacientov so stredne závažnou až závažnou ložiskovou psoriázou vo veku 6 až <18 rokov, definovanej skóre PASI s ≥12, skóre IGA mod 2011 of ≥3 a postihnutím kože s BSA ≥10%, ktorí boli kandidátmi na systémovú liečbu.
V pediatrickom skúšaní 2 pri psoriáze sa vyhodnotilo 84 pacientov, randomizovaných na podávanie nízkej dávky sekukinumabu (75 mg na telesnú hmotnosť <50 kg alebo 150 mg na telesnú hmotnosť
≥50 kg) alebo vysokej dávky sekukinumabu (75 mg na telesnú hmotnosť <25 kg, 150 mg na telesnú
hmotnosť medzi ≥25 kg a <50 kg, alebo 300 mg na telesnú hmotnosť ≥50 kg) v 0., 1., 2., 3. a
4. týždni, po ktorých nasledovala každé 4 týždne rovnaká dávka. Rozdelenie pacientov podľa
hmotnosti a veku pri randomizácii je uvedené v Tabuľke 13.
Tabuľka 13 Rozdelenie pacientov podľa hmotnosti a veku v pediatrickom skúšaní 2 pri psoriázePodskupina
| Popis
| Sekukinumab nízka dávka n=42
| Sekukinumab vysoká dávka n=42
| Spolu
N=84
|
Vek
| 6-<12 rokov
| 17
| 16
| 33
|
≥12-<18 rokov
| 25
| 26
| 51
|
Hmotnosť
| <25 kg
| 4
| 4
| 8
|
≥25-<50 kg
| 13
| 12
| 25
|
≥50 kg
| 25
| 26
| 51
|
Spoločnými primárnymi koncovými ukazovateľmi boli podiely pacientov, ktorí dosiahli odpoveď
PASI 75 a IGA mod 2011 „bez prejavov“ alebo“takmer bez prejavov“ (0 alebo 1) v 12.týždni.
Účinnosť v oboch skupinách s nízkou a vysokou dávkou sekukinumabu bola porovnateľná
a preukázala štatistické zlepšenie v porovnaní s predchádzajúcim placebom v primárnych aj sekundárnych koncových ukazovateľoch. Odhadovaná posteriórna pravdepodobnosť pozitívneho liečebného účinku bola 100 %.
U všetkých pacientov sa sledovala účinnosť najmenej 24 týždňov po prvom podaní lieku (pozri
Tabuľku 14). Účinnosť (definovaná odpoveďou PASI 75 a IGA mod 2011 „bez prejavov“ alebo
„takmer bez prejavov“ [0 alebo 1]) sa pozorovala už pri prvej návšteve po východiskovej návšteve
v 2.týždni a podiel pacientov, ktorí dosiahli odpoveď PASI 75 a IGA mod 2011 „bez prejavov“ alebo
„takmer bez prejavov“ (0 alebo 1) sa zvýšil počas obdobia 24 týždňov.
Po 12 .týždni bola účinnosť oboch skupín s nízkou a vysokou dávkou sekukinumabu porovnateľná
. Profily bezpečnosti s nízkou dávkou a vysokou dávkou boli porovnateľné a v zhode s profilom bezpečnosti u dospelých pacientov.
T
a
b
u
ľ
k
a 14 Súhrn klinickej odpovede v pediatrickom skúšaní so stredne závažnou až závažnou
p
soriázou v 12.a 24.týždni (pediatrické skúšanie pri psoriáze 2)*
|
1
2 .týždeň
|
2
4 .týždeň
|
S
e
k
u
k
i
nu
m
a
b nízka dávka
|
S
e
k
u
k
i
nu
m
a
b vysoká dávka
|
S
e
k
u
k
i
nu
m
a
b nízka dávka
|
S
e
k
u
k
i
nu
m
a
b vysoká dávka
|
Počet pacientov
|
42
|
42
|
42
|
42
|
Odpoveď PASI 75, n (%)
|
39 (92,9 %)
|
39 (92,9 %)
|
40 (95,2 %)
|
40 (95,2 %)
|
Odpoveď IGA mod 2011 „bez prejavov“ alebo “takmer bez prejavov“, n (%)
|
33 (78,6 %)
|
35 (83,3 %)
|
37 (88,1 %)
|
39 (92,9 %)
|
Odpoveď PASI 90, n (%)
|
29 (69 %)
|
32 (76,2 %)
|
37 (88,1 %)
|
37 (88,1 %)
|
Odpoveď PASI 100, n (%)
|
25 (59,5 %)
|
23 (54,8 %)
|
28 (66,7 %)
|
28 (66,7 %)
|
* chýbajúce údaje boli vyhodnotené ako bez odpovede
|
Tieto výsledky v pediatrickej populácii so stredne závažnou až závažnou ložiskovou psoriázou
potvrdili vyššie uvedené prediktívne predpoklady, založené na základe účinnosti a odpovede na liečbu
u dospelých pacientov, ktoré sú uvedené vyššie.
V skupine s nízkou dávkou dosiahlo 50 % a 70,7 % pacientov skóre CDLQI 0 alebo 1 v 12.a 24.týždni
v uvedenom poradí. V skupine s vysokou dávkou dosiahlo 61,9 % a 60,5 % pacientov skóre 0 alebo 1
v 12.a 24.týždni v uvedenom poradí.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Cosentyxom pre liečbu ložiskovej psoriázy u pediatrických pacientov od narodenia do menej ako 6 rokov a pre liečbu chronickej idiopatickej artritídy u pediatrických pacientov od narodenia do menej ako 2 rokov (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Cosentyxom pre
liečbu chronickej idiopatickej artritídy u pediatrických pacientov vo veku od 2 rokov do menej ako
18 rokov (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiVäčšina farmakokinetických vlastností pozorovaných u pacientov s ložiskovou psoriázou, psoriatickou
artritídou a ankylozujúcou spondylitídou bola podobná.
AbsorpciaPo jednorazovej subkutánnej dávke 300 mg v tekutej liekovej forme sa u zdravých dobrovoľníkov
dosiahli maximálne koncentrácie sekukinumabu v sére 43,2±10,4 μg/ml medzi 2. a 14. dňom po
podaní.
Podľa analýzy farmakokinetiky u populácie sa po jednorazovom subkutánnom podaní pacientom s ložiskovou psoriázou maximálne koncentrácie sekukinumabu v sére 13,7±4,8 µg/ml pri dávke
150 mg alebo 27,3±9,5 µg/ml pri dávke 300 mg dosiahli medzi 5. a 6. dňom po podaní.
Podľa analýzy farmakokinetiky u populácie bol po začiatočnom týždennom podávaní počas prvého mesiaca čas do dosiahnutia maximálnej koncentrácie medzi 31. a 34. dňom.
Podľa simulovaných údajov boli maximálne koncentrácie v rovnovážnom stave (Cmax,ss) po subkutánnom podaní 27,6 µg/ml pri 150 mg a 55,2 µg/ml pri 300 mg. Analýza farmakokinetiky u populácie naznačuje, že rovnovážny stav sa dosahuje pri režimoch mesačného podávania po
20 týždňoch.
V porovnaní s expozíciou po jednorazovej dávke ukázala analýza farmakokinetiky u populácie, že pacienti vykazovali po opakovanom mesačnom podávaní počas udržiavacej liečby zvýšenie maximálnych koncentrácií v sére a plochy pod krivkou (AUC) na dvojnásobok.
Analýza farmakokinetiky u populácie ukázala, že sekukinumab sa u pacientov s ložiskovou psoriázou absorboval s priemernou absolútnou biologickou dostupnosťou 73 %. Vo všetkých skúšaniach sa vypočítala absolútna biologická dostupnosť v rozmedzí od 60 do 77 %.
Biologická dostupnosť sekukinumabu u pacientov so PsA bola 85 % na základe farmakokinetického modelu populácie.
Distribúcia
Stredný objem distribúcie počas koncovej fázy (Vz) po jednorazovom intravenóznom podaní
u pacientov s ložiskovou psoriázou bol v rozmedzí od 7,10 do 8,60 litrov, čo naznačuje, že
sekukinumab sa len obmedzene distribuuje do periférnych kompartmentov.
Biotransformácia
Väčšina eliminácie IgG sa uskutočňuje prostredníctvom intracelulárneho katabolizmu po endocytóze z
tekutej fázy alebo sprostredkovanej receptorom.
Eliminácia
Stredný systémový klírens (CL) po jednorazovom intravenóznom podaní pacientom s ložiskovou
psoriázou bol v rozmedzí od 0,13 do 0,36 l/deň. V analýze farmakokinetiky bola stredná hodnota systémového klírensu (CL) u populácie pacientov s ložiskovou psoriázou 0,19 l/deň. CL nebol ovplyvnený pohlavím. Klírens nezávisel od dávky a času.
Stredný polčas eliminácie bol podľa odhadu analýzy farmakokinetiky u populácie pacientov
s ložiskovou psoriázou 27 dní, s rozmedzím od 18 do 46 dní, vo všetkých psoriatických skúšaniach
s intravenóznym podávaním.
Linearita/nelinearita
Farmakokinetika sekukinumabu po jednorazovom a opakovanom podávaní pacientom s ložiskovou
psoriázou sa stanovila v niekoľkých skúšaniach s intravenózne podanými dávkami v rozmedzí od
1x 0,3 mg/kg do 3x 10 mg/kg a so subkutánne podanými dávkami v rozmedzí od 1x 25 mg po opakované dávky 300 mg. Expozícia bola úmerná dávke pri všetkých dávkovacích režimoch.
Osobitné populácie
Staršípacienti
Podľa analýzy farmakokinetiky u populácie obmedzeného počtu starších pacientov (n=71 vo veku
≥65 rokov a n=7 vo veku ≥75 rokov) bol klírens podobný u starších pacientov a u pacientov mladších
ako 65 rokov.
Pacienti s poruchoufunkcieobličiekalebopečene
Nie sú dostupné farmakokinetické údaje o pacientoch s poruchou funkcie obličiek alebo pečene. Predpokladá sa, že pri intaktnom sekukinumabe, monoklonálnej protilátke IgG, je eliminácia obličkami nízka a málo významná. IgG sa eliminujú hlavne prostredníctvom katabolizmu
a nepredpokladá sa, že porucha funkcie pečene má vplyv na klírens sekukinumabu.
Vplyv telesnej hmotnosti na farmakokinetiku
Klírens a distribučný objem sekukinumabu sa zvyšujú so zvyšujúcou sa telesnou hmotnosťou.
P
e
d
i
a
t
r
i
c
k
á populácia
V skupine dvoch pediatrických skúšaní pacienti so stredne závažnou až závažnou ložiskovou psoriázou (vo veku 6 až menej ako 18 rokov) dostávali sekukinumab podľa odporúčaného pediatrického dávkovacieho režimu. Pacienti s hmotnosťou ≥25 a <50 kg mali v 24. týždni priemernú minimálnu koncentráciu v rovnovážnom stave (priemerné ± SD) v hodnote 19,8 ± 6,96 µg/ml (n=24) po dávke 75 mg sekukinumabu a pacienti s hmotnosťou ≥50 kg mali priemernú minimálnu koncentráciu v rovnovážnom stave 27,3 ± 10,1 µg/ml (n=36) po dávke 150 mg sekukinumabu. Priemerná minimálna koncentrácia v rovnovážnom stave (priemerné ± SD) u pacientov s hmotnosťou
<25 kg (n=8) v 24. týždni bola 32,6 ± 10,8 µg/ml po dávke 75 mg sekukinumabu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní a reprodukčnej toxicity, alebo pri testovaní skríženej reaktivity tkanív, neodhalili žiadne osobitné riziko pre ľudí (dospelých a dospievajúcich).
Štúdie na zvieratách na vyhodnotenie karcinogénneho potenciálu sekukinumabu sa nevykonali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
sacharóza histidín
monohydrát histidínium-chloridu polysorbát 80
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
3 roky
Po rekonštitúcii
Chemická a fyzikálna stabilita počas použitia sa preukázala počas 24 hodín pri teplote 2ºC až 8ºC.
Z mikrobiologického hľadiska, pokiaľ spôsob rekonštitúcie nevylučuje riziko mikrobiálnej kontaminácie, liek sa má okamžite použiť.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C).
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Cosentyx sa dodáva v injekčnej liekovke z bezfarebného skla so sivou potiahnutou gumenou zátkou a hliníkovým uzáverom s bielym odlamovacím krytom, ktorá obsahuje 150 mg sekukinumabu.
Cosentyx je dostupný v baleniach obsahujúcich jednu injekčnú liekovku.
6
.
6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Injekčná liekovka na jednorazové použitie obsahuje 150 mg sekukinumabu na rekonštitúciu sterilnou vodou na injekcie. Výsledný roztok má byť číry a bezfarebný až bledožltý. Nepoužite ho, ak sa lyofilizovaný prášok úplne nerozpustil alebo ak tekutina obsahuje ľahko viditeľné častice, ak je zakalená alebo ak je výrazne hnedá.
Rekonštitúcia
Cosentyx 150 mg prášok na injekčný roztok musí byť pripravený zdravotníckym pracovníkom. Príprava roztoku na subkutánnu injekciu musí prebiehať bez prerušenia a musí sa zabezpečiť aseptický spôsob jeho prípravy. Čas prípravy od prepichnutia zátky až do ukončenia rekonštitúcie je v priemere
20 minút a nemá byť dlhší ako 90 minút.
1. Umiestnite injekčnú liekovku s práškom a sterilnú vodu na injekcie do prostredia s izbovou teplotou, aby sa ohriali na izbovú teplotu.
2. Odoberte do 1 ml injekčnej striekačky na jednorazové použitie s vyznačenou stupnicou o niečo
viac ako 1,0 ml sterilnej vody na injekcie a nastavte objem na 1,0 ml.
3. Odstráňte z injekčnej liekovky plastový uzáver.
4. Zaveďte ihlu injekčnej striekačky do injekčnej liekovky obsahujúcej prášok cez stred gumenej zátky a rekonštituujte prášok pomalým vstreknutím 1,0 ml sterilnej vody na injekcie do injekčnej liekovky. Prúd sterilnej vody na injekcie má smerovať na prášok.
5. Nakloňte injekčnú liekovku pod uhlom približne 45° a opatrne ju otáčajte prstami približne
1°minútu. Liekovku nepretrepávajte, ani ju neprevracajte.
6. Nechajte injekčnú liekovku stáť pri izbovej teplote najmenej 10 minút, aby sa jej obsah mohol
rozpustiť. Môže dôjsť k speneniu roztoku.
7. Nakloňte injekčnú liekovku pod uhlom približne 45° a opatrne ju otáčajte prstami približne
1 minútu. Liekovku nepretrepávajte, ani ju neprevracajte.
8. Nechajte injekčnú liekovku stáť pri izbovej teplote približne 5 minút. Výsledný roztok má byť číry. Jeho farba sa môže meniť od bezfarebnej po bledožltú. Nepoužite ho, ak sa lyofilizovaný prášok úplne nerozpustil alebo ak tekutina obsahuje ľahko viditeľné častice, ak je zakalená
alebo ak je výrazne hnedá.
9. Pripravte si potrebný počet injekčných liekoviek (2 injekčné liekovky na dávku 300 mg).
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
Použitie v pediatrickej populácii
Pre pediatrických pacientov, ktorí dostávajú dávku 75 mg z injekčnej liekovky na jednorazové
použitie, obsahujúcej 150 mg sekukinumabu na rekonštitúciu sterilnou vodou na injekcie, sa musí odobrať o niečo viac ako 0,5 ml rekonštituovaného roztoku na subkutánnu injekciu a zvyšok roztoku sa musí ihneď zlikvidovať. Podrobné pokyny na použitie sú uvedené v písomnej informácii pre používateľa.
7
. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLOEU/1/14/980/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 15.január 2015
Dátum posledného predĺženia registrácie: 03. september 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.