
proti hnačke
• Monitorovať najmenej raz za týždeň až pokým neodznie
• Opätovne podať zníženú dávku (15 mg
dvakrát denne)
|
Abdominálna bolesť, stolica obsahujúca hlien alebo krv, zmeny
vo vyprázdňovaní, peritoneálne prejavy
ALEBO
Závažná hnačka (3. stupeň,
> 6 stolíc za deň oproti východiskovému stavu)
|
• Nepodať liek Copiktra až pokým neodznie
• Začať podpornú liečbu s enterickými steroidmi (napr. budezonid) alebo so systémovými steroidmi
• Monitorovať najmenej raz za týždeň až pokým neodznie
• Opätovne podať zníženú dávku (15 mg
dvakrát denne)
• V prípade opakovanej hnačky 3. stupňa alebo opakovanej kolitídy každého stupňa prerušiť podávanie lieku Copiktra
|
Život ohrozujúce
|
• Prerušiť podávanie lieku Copiktra
|
Kožné reakcie
|
1. – 2. stupeň
|
• Žiadna zmena dávky
• Začať podpornú starostlivosť s emolientami, antihistaminikami (pri svrbení) alebo lokálnymi steroidmi
• Dôkladne monitorovať
|
3. stupeň
|
• Nepodať liek Copiktra až pokým neodznie
• Preskúmať všetky súbežne podávané lieky
a prerušiť podávanie akéhokoľvek lieku, ktorý potenciálne prispieva k udalosti
• Začať podpornú starostlivosť pomocou steroidov (lokálnych alebo systémových) a antihistaminík pri svrbení
• Monitorovať najmenej raz za týždeň až pokým neodznie
• Opätovne podať zníženú dávku (15 mg
dvakrát denne)
• Ak sa závažné kožné reakcie nezlepšia, zhoršia sa alebo sa vrátia, prerušiť podávanie lieku Copiktra
|
Život ohrozujúce
|
• Prerušiť podávanie lieku Copiktra
|
SJS, TEN, DRESS (akéhokoľvek stupňa)
|
• Prerušiť podávanie lieku Copiktra pri každom stupni
|
Pneumonitída bez podozrenia na infekčný pôvod
|
Stredne závažná symptomatická pneumonitída (2. stupeň)
|
• Nepodať liek Copiktra
• Systémová liečba steroidmi
• Ak sa pneumonitída opäť zmierni na stupeň 0 alebo 1, môže sa opätovne podať znížená dávka lieku Copiktra (15 mg dvakrát denne)
• Ak sa neinfekčná pneumonitída vráti alebo pacient nereaguje na liečbu steroidmi, podávanie lieku Copiktra sa má prerušiť
|
Závažná alebo život ohrozujúca pneumonitída (3. stupeň)
|
• Prerušiť podávanie lieku Copiktra
• Liečiť systémovou liečbou steroidmi
|
Elevácia
ALT/AST
|
3 až 5 x horný limit normy
(ULN) (2. stupeň)
|
• Zachovať dávku lieku Copiktra
• Monitorovať najmenej raz za týždeň až do návratu k < 3 × ULN
|
T
oxicita
|
S
t
up
e
ň nežiaducej reakcie
|
O
dp
orúčané postupy
|
|
> 5 až 20 × ULN (3. stupeň)
|
• Nepodať liek Copiktra a monitorovať najmenej raz za týždeň až do návratu k < 3 × ULN
• Opätovne podať liek Copiktra v rovnakej dávke (25 mg dvakrát denne) pri prvom výskyte alebo v zníženej dávke (15 mg dvakrát denne) pri nasledujúcom výskyte
|
> 20 × ULN (4. stupeň)
|
• Prerušiť podávanie lieku Copiktra
|
H
e
m
atologické nežiaduce reakcie
|
Neutropénia
|
Absolútny počet
neutrofilov (ANC) 0,5 až
1,0 × 109/l
|
• Zachovať dávku lieku Copiktra
• Monitorovať ANC najmenej raz za týždeň
|
ANC menší než 0,5 × 109/l
|
• Nepodať liek Copiktra.
• Monitorovať ANC do > 0,5 × 109/l
• Opätovne podať liek Copiktra v rovnakej dávke (25 mg dvakrát denne) pri prvom výskyte alebo v zníženej dávke (15 mg dvakrát denne) pri nasledujúcom výskyte
|
Trombocytopénia
|
Počet krvných doštičiek 25
až < 50 × 109/l (3. stupeň)
s krvácaním 1. stupňa
|
• Žiadna zmena dávky
• Monitorovať počet krvných doštičiek najmenej raz za týždeň
|
Počet krvných doštičiek 25
až < 50 × 109/l (3. stupeň)
s krvácaním 2. stupňa alebo
Počet krvných doštičiek <
25 × 109/l (4. stupeň)
|
• Nepodať liek Copiktra
• Monitorovať počet krvných doštičiek pokým nebude ≥ 25 × 109/l a pokým neodznie krvácanie (ak sa uplatňuje)
• Opätovne podať liek Copiktra v rovnakej dávke (25 mg dvakrát denne) pri prvom výskyte alebo opätovne podať v zníženej dávke (15 mg dvakrát denne) pri nasledujúcom výskyte
|
Skratky: ALT = alanínaminotransferáza; ANC = absolútny počet neutrofilov; AST = aspartátaminotransferáza; CMV = cytomegalovírus; DRESS = lieková reakcia s eozinofíliou a systémovými symptómami; PCR = polymerázová reťazová reakcia; PJP = pneumónia vyvolaná
Pneumocystis jirovecii; SJS = Stevensov-Johnsonov syndróm; TEN = toxická epidermálna nekrolýza; ULN = horný limit normy
Poznámka: Vysadenie dávok na > 42 dní v dôsledku toxicity súvisiacej s liečbou bude viesť k trvalému ukončeniu liečby.
Osobitné populácieStarší pacientiU starších pacientov (vo veku ≥ 65 rokov) nie je potrebná žiadna osobitná úprava dávky (pozri časť 5.2).
Porucha funkcie obličiekV prípade pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek nie je potrebná úprava dávky. V prípade závažnej poruchy funkcie obličiek a terminálneho štádia poruchy funkcie obličiek s dialýzou alebo bez nej nie sú k dispozícii žiadne údaje (pozri časť 5.2).
Porucha funkcie pečeneU pacientov s poruchou funkcie pečene triedy A, B a C podľa Childovej-Pughovej klasifikácie nie je potrebná žiadna úprava úvodnej dávky (pozri časti 4.4 a 5.2).
Pediatrická populáciaBezpečnosť a účinnosť duvelizibu u detí vo veku 0 až 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
V pediatrickej populácii nie je relevantné použitie duvelizibu v indikácii CLL a FL.
Spôsob podávania
Copiktra je určená na perorálne použitie a môže sa užívať s jedlom alebo bez jedla. Kapsuly sa majú
prehltnúť celé. Pacienti majú byť poučení o tom, že kapsuly nemajú otvárať, lámať ani žuť.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Všeobecné
Bezpečnosť a účinnosť duvelizibu po predchádzajúcom užívaní idelalisibu neboli stanovené.
Infekcie
U pacientov užívajúcich duvelizib sa objavili závažné infekcie vrátane infekcií končiacich smrťou.
Najčastejšie závažné infekcie zahŕňali pneumóniu, sepsu a infekcie dolných dýchacích ciest. Medián času do nástupu infekcie akéhokoľvek stupňa bol 3 mesiace, pričom 75 % prípadov sa vyskytlo v priebehu 6 mesiacov (pozri časť 4.8).
Pred začatím podávania duvelizibu sa majú vyliečiť všetky infekcie. Pacienti majú byť počas liečby sledovaní z hľadiska možného výskytu infekcie vrátane respiračných prejavov a príznakov. Pacienti majú byť poučení o tom, aby bezodkladne hlásili nové alebo zhoršujúce sa prípady infekcie (odporúčané postupy pozri v tabuľke 1).
U pacientov užívajúcich duvelizib sa vyskytla závažná pneumónia PJP vrátane prípadov končiacich smrťou. Profylaxia pri PJP sa má preto poskytnúť všetkým pacientom (pozri tabuľku 1). U pacientov užívajúcich duvelizib sa vyskytla reaktivácia/infekcia CMV. Počas liečby sa má zvážiť podávanie profylaktických antivírusových liekov na prevenciu infekcie CMV vrátane reaktivácie CMV (pozri tabuľku 1).
Odporúčaná profylaxia
Pred začatím podávania duvelizibu sa majú vyliečiť všetky infekcie. Pacienti majú byť počas liečby sledovaní z hľadiska možného výskytu infekcie vrátane respiračných príznakov a symptómov. Pacienti majú byť poučení o tom, aby bezodkladne hlásili nové alebo zhoršujúce sa prípady infekcie (odporúčané postupy pozri v tabuľke 1).
Počas liečby duvelizibom sa má preto všetkým pacientom pri PJP poskytnúť profylaxia. Po ukončení liečby duvelizibom sa má v profylaxii pri PJP pokračovať, až kým absolútny počet CD4+ T buniek nie je vyšší než 200 buniek/µl.
Duvelizib sa má prestať podávať u pacientov s podozrením na PJP akéhokoľvek stupňa a pri potvrdení
PJP sa má vysadiť.
Počas liečby duvelizibom sa má zvážiť podávanie profylaktických antivírusových liekov na prevenciu infekcie CMV vrátane reaktivácie CMV.
Hnačka alebo kolitída
U pacientov užívajúcich duvelizib sa vyskytla závažná hnačka alebo kolitída vrátane vrátane prípadov
končiacich smrťou. Medián času do nástupu hnačky alebo kolitídy akéhokoľvek stupňa bol 4 mesiace, pričom 75 % prípadov sa vyskytlo v priebehu 8 mesiacov. Medián trvania udalosti bol 0,5 mesiaca.
Pacienti majú byť poučení o tom, aby bezodkladne hlásili nové alebo zhoršujúce sa prípady hnačky
(odporúčané postupy pozri v tabuľke 1) (pozri časť 4.8).
Kožné reakcie
U pacientov užívajúcich duvelizib sa vyskytli závažné kožné reakcie vrátane ich prípadov končiacich
smrťou. Prípady končiace smrťou zahŕňali liekovú reakciu s eozinofíliou a systémovými symptómami (DRESS) a toxickú epidermálnu nekrolýzu (TEN). Medián času do nástupu kožnej reakcie akéhokoľvek stupňa bol 3 mesiace a medián trvania udalosti bol 1 mesiac (pozri časť 4.8).
Príznaky závažných kožných udalostí boli opísané primárne ako pruritické, erytematózne alebo makulopapulózne. Menej časté príznaky zahŕňajú exantém, deskvamáciu, erytrodermiu, exfoliáciu kože, nekrózu keratínocytov a papulóznu vyrážku. Pacienti majú byť poučení o tom, aby hlásili akékoľvek nové alebo zhoršujúce sa prípady kožných reakcií (odporúčané postupy pozri v tabuľke 1). Majú sa preskúmať všetky súbežne podávané lieky a má sa ukončiť podávanie všetkých liekov, ktoré potenciálne prispievajú k danej udalosti.
Pneumonitída
U pacientov užívajúcich duvelizib sa vyskytla závažná pneumonitída bez zjavnej infekčnej príčiny
vrátane prípadov končiacich smrťou. Medián času do nástupu pneumonitídy akéhokoľvek stupňa bol 4 mesiace, pričom 75 % prípadov sa vyskytlo v priebehu 9 mesiacov (pozri časť 4.8). Medián trvania udalosti bol 1 mesiac, pričom 75 % prípadov odznelo do 2 mesiacov (odporúčané postupy pozri
v tabuľke 1).
Hepatotoxicita
U pacientov užívajúcich duvelizib sa objavila elevácia hladiny ALT a/alebo AST 3. a 4. stupňa. Dve
percentá pacientov mali hodnoty ALT aj AST vyššie než 3 x ULN a celková hladina bilirubínu bola vyššia než 2 x ULN. Medián času do nástupu zvýšenia hladiny transaminázy akéhokoľvek stupňa bol
2 mesiace s mediánom trvania udalosti 1 mesiac. Počas liečby duvelizibom sa má sledovať funkcia pečene, najmä počas prvých troch mesiacov liečby, a to raz za mesiac. Toto usmernenie sa týka pacientov, ktorí majú len eleváciu hladiny ALT a AST.
Neutropénia
U pacientov užívajúcich duvelizib sa vyskytla neutropénia 3. a 4. stupňa. Medián času do nástupu
neutropénie stupňa ≥ 3 bol 2 mesiace, pričom 75 % prípadov sa vyskytlo v priebehu 4 mesiacov. Počas prvých 2 mesiacov liečby duvelizibom sa najmenej každé 2 týždne má sledovať počet neutrofilov.
Induktory CYP3A4
Expozíciu duvelizibu je možné znížiť súbežným podávaním silných induktorov CYP3A. Keďže
zníženie plazmatických koncentrácií duvelizibu môže viesť k zníženej účinnosti, má sa predchádzať súbežnému podávaniu duvelizibu so silnými induktormi CYP3A (pozri časť 4.5).
Substráty CYP3A
Duvelizib a jeho hlavný metabolit IPI-656 sú silné inhibítory CYP3A4. Duvelizib má preto potenciál
vzájomne pôsobiť s liekmi, ktoré sú metabolizované prostredníctvom CYP3A, čo môže viesť k zvýšeným koncentráciám iného lieku v sére (pozri časť 4.5). Ak sa duvelizib podáva súbežne s inými liekmi, je nutné skontrolovať súhrn charakteristických vlastností lieku (SmPC) týchto liekov, pokiaľ ide o odporúčania týkajúce sa súbežného podávania s inhibítormi CYP3A4. Má sa predchádzať súbežnej liečbe duvelizibom a citlivými substrátmiCYP3A a podľa možnosti sa majú použiť alternatívne lieky, ktoré sú menej citlivé na inhibíciu CYP3A4.
4.5 Liekové a iné interakcie
Ú
č
i
nky iných liekov na farmakokinetiku duvelizibu
Silné a stredne silné induktory CYP3A4
Súbežné podávanie silného induktora CYP3A, rifampicínu, v dávke 600 mg raz denne počas 7 dní
s jednorazovou perorálnou dávkou 25 mg duvelizibu u zdravých dospelých (n = 13) znížilo cmax duvelizibu o 66 % a AUC o 82 %. Súbežné podávanie so silným induktorom CYP3A znižuje plochu pod krivkou (AUC) duvelizibu (pozri časť 5.2), čo môže znížiť účinnosť duvelizibu. Má sa predchádzať súbežnému podávaniu duvelizibu so silnými induktormi CYP3A4 (napr. apalutamid, karbamezepín, enzalutamid, mitotán, fenytoín, rifampicín, ľubovník bodkovaný).
Súbežné podávanie etravirinu, stredne silného induktora CYP3A, v dávke 200 mg dvakrát denne počas 10 dní s jednorazovou perorálnou dávkou 25 mg duvelizibu u zdravých dospelých (n = 20) znížilo cmax duvelizibu o 16 % a AUC o 35 %. Súbežné podávanie duvelizibu so stredne silnými induktormi CYP3A znižuje AUC duvelizibu na menej než 1,5-násobok a zníženie dávky sa neodporúča. Príklady stredne silných induktorov CYP3A4 zahŕňajú bosentán, efavirenz, etravirin, fenobarbital, primidón. Ak je nevynutné použiť stredne silný induktor CYP3A4, pacient sa má dôkladne sledovať z hľadiska potenciálnej nedostatočnej účinnosti. Príklady: bosentán, efavirenz, etravirin, fenobarbital, primidón.
Silné a stredne silné inhibítory CYP3A
Súbežné podávanie ketokonazolu, silného inhibítora CYP3A, (v dávke 200 mg dvakrát denne (BID)
počas 5 dní) s jednorazovou perorálnou dávkou 10 mg duvelizibu u zdravých dospelých (n = 16) zvýšilo cmax duvelizibu 1,7-násobne a AUC 4-násobne. V dôsledku časovo závislej autoinhibície CYP3A4 je citlivosť duvelizibu na stredne silné a silné inhibítory CYP3A4 znížená v podmienkach rovnovážneho stavu. Na základe fyziologicky založeného farmakokinetického (physiologically-based pharmacokinetic, PBPK) modelovania a simulovania sa zvýšenie expozície duvelizibu odhaduje na
~1,6-násobok v rovnovážnom stave u pacientov s rakovinou, keď súbežne užívali silné inhibítory
CYP3A4, ako je ketokonazol a itrakonazol.
Dávka duvelizibu sa má znížiť na 15 mg dvakrát denne, keď sa podáva súbežne so silným inhibítorom CYP3A4 (pozri časť 4.2) (napr. ketokonazol, indinavir, nelfinavir, ritonavir, sachinavir, klaritromycín, telitromycín, itrakonazol, nefazodón, kobicistát, vorikonazol a posakonazol a grapefruitová šťava).
PBPK modelovanie a simulácia neposkytli odhad žiadneho klinicky významného účinku na expozície duvelizibu pri súbežnom podávaní stredne silných inhibítorov CYP3A4. Zníženie dávky duvelizibu nie je potrebné pri súbežnom podávaní so stredne silnými inhibítormi CYP3A4 (pozri časť 4.2) (napr. aprepitant, ciprofloxacín, konivaptán, krizotinib, cyklosporín, diltiazem, dronedarón, erytromycín, flukonazol, fluvoxamín, imatinib, tofizopam, verapamil).
Účinok duvelizibu na farmakokinetiku iných liekov
Substráty CYP3A4
Súbežné podávanie opakovaných dávok duvelizibu 25 mg dvakrát denne počas 5 dní s jednorazovou perorálnou dávkou 2 mg midazolamu, citlivého substrátu CYP3A4, u zdravých dospelých (n = 14) zvýšilo pri midalozame AUC 4,3-násobne a cmax 2,2-násobne. PBPK simulácie u pacientov
s rakovinou za podmienok rovnovážneho stavu preukázali, že cmax a AUC midazolamu by sa zvýšili približne 2,5-násobne a ≥ 5-násobne v uvedenom poradí. Má sa predchádzať súbežnému podávaniu midazolamu s duvelizibom.
Duvelizib a jeho hlavný metabolit IPI-656 sú silné inhibítory CYP3A4. Zníženie dávky substrátu CYP3A4 sa má zvážiť vtedy, keď sa podáva súbežne s duvelizibom, najmä v prípade liekov s úzkym terapeutickým indexom. Pacienti majú byť sledovaní z hľadiska prejavov toxicity súbežne podávaného citlivého substrátu CYP3A. Príklady citlivých substrátov zahŕňajú: alfentanil, avanafil, buspirón, konivaptán, darifenacín, darunavir, ebastín, everolimus, ibrutinib, lomitapid, lovastatín, midazolam, naloxegol, nisoldipín, sachinavir, simvastatín, sirolimus, takrolimus, tipranavir, triazolam, vardenafil,
budezonid, dasatinib, dronedarón, eletriptán, eplerenón, felodipín, indinavir, lurazidón, maravirok, kvetiapín, sildenafil, tikagrelor, tolvaptán. Príklady stredne citlivých substrátov zahŕňajú: alprazolam, aprepitant, atorvastatín, kolchicín, eliglustát, pimozid, rilpivirín, rivaroxabán, tadalafil. Tento zoznam nie je úplný a slúži len ako odporúčanie. Odporúčania týkajúce sa súbežného podávania s inhibítormi CYP3A4 (pozri časť 4.4) sa nachádzajú v SPC týchto liekov.
Hormonálna antikoncepcia
V súčasnosti nie je známe, či duvelizib znižuje účinnosť hormonálnej antikoncepcie. Ženy, ktoré užívajú hormonálnu antikoncepciu, majú preto byť poučené o tom , aby používali bariérovú metódu ako druhú formu antikoncepcie (pozri časť 4.6).
Inhibítory protónovej pumpy
Analýza populačnej farmakokinetiky (Population Pharmacokinetic, POPPK) ukázala, že inhibítory
protónovej pumpy (PPI) nemajú vplyv na expozíciu lieku Copiktra. PPI sa môžu podávať súbežne s duvelizibom
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii žiadne údaje o použití duvelizibu u gravidných žien. V štúdiách na zvieratách sa
v klinicky relevantných expozíciách nepreukázali priame alebo nepriame škodlivé účinky, pokiaľ ide o reprodukčnú toxicitu (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu lieku Copiktra počas tehotenstva.
Dojčenie
Nie je známe, či sa duvelizib alebo jeho metabolity vylučujú do ľudského mlieka. Riziko pre dojčené
deti nie je možné vylúčiť. Počas liečby liekom Copiktra a aspoň jeden mesiac po poslednej dávke sa má dojčenie prerušiť.
Fertilita
K dispozícii nie sú žiadne údaje o účinku duvelizibu na fertilitu u ľudí. U potkanov, nie však u opíc, sa
pozorovali účinky na semenníky.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Copiktra nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najčastejšie hlásené nežiaduce reakcie (incidencia ≥ 20 %) sú hnačka alebo kolitída, neutropénia,
vyrážka, únava, pyrexia, kašeľ, nauzea, infekcia horných dýchacích ciest, pneumónia, muskuloskeletálna bolesť a anémia.
Najčastejšie hlásené závažné nežiaduce reakcie boli pneumónia, kolitída a hnačka. Tabuľkovýzoznamnežiaducichreakcií
Nežiaduce reakcie hlásené preduvelizib sú uvedené podľa triedy orgánových systémov a frekvencie
v tabuľke 2. Frekvencie sú definované ako veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté
(≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000) a veľmi zriedkavé (< 1/10 000) a neznáme (nedá sa odhadnúť z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie uvedené v poradí podľa klesajúcej závažnosti.
T
abuľka 2: Nežiaduce reakcie na liek hlásené u pacientov s hematologickými malignitami
u
ž
í
vajúcich duvelizib (n = 442)
T
r
i
e
d
a orgánových systémov/preferovaný
výraz alebo nežiaduca reakcia
|
V
š
et
k
y stupne
|
3. stupeň alebo vyšší
|
In
fekcie a nákazy
|
|
|
Infekcia dolných dýchacích ciest1
|
Veľmi časté
|
Časté
|
Sepsa
|
Časté
|
Časté
|
Infekcia horných dýchacích ciest1
|
Veľmi časté
|
Menej časté
|
P
oruchy krvi a lymfatického systému
|
|
|
Neutropénia1
|
Veľmi časté
|
Veľmi časté
|
Anémia1
|
Veľmi časté
|
Veľmi časté
|
Trombocytopénia1
|
Veľmi časté
|
Veľmi časté
|
P
oruchy metabolizmu a výživy
|
|
|
Znížená chuť do jedla
|
Veľmi časté
|
Menej časté
|
P
oruchy nervového systému
|
|
|
Bolesť hlavy1
|
Veľmi časté
|
Menej časté
|
P
oruchy dýchacej sústavy, hrudníka a mediastína
|
|
|
Dyspnoe1
|
Veľmi časté
|
Časté
|
Pneumonitída2
|
Časté
|
Časté
|
Kašeľ1
|
Veľmi časté
|
Menej časté
|
P
oruchy gastrointestinálneho traktu
|
|
|
Hnačka/kolitída3
|
Veľmi časté
|
Veľmi časté
|
Nauzea1
|
Veľmi časté
|
Menej časté
|
Vracanie
|
Veľmi časté
|
Časté
|
Abdominálna bolesť1
|
Veľmi časté
|
Časté
|
Zápcha
|
Veľmi časté
|
Menej časté
|
P
oruchy kože a podkožného tkaniva
|
|
|
Vyrážka4
|
Veľmi časté
|
Časté
|
Svrbenie1
|
Časté
|
Menej časté
|
P
oruchy kostrovej a svalovej sústavy a spojivového
tkaniva
|
|
|
Muskuloskeletálna bolesť1
|
Veľmi časté
|
Časté
|
Bolesť kĺbov
|
Veľmi časté
|
Menej časté
|
C
el
k
ové poruchy a reakcie v mieste podania
|
|
|
Pyrexia
|
Veľmi časté
|
Časté
|
Únava1
|
Veľmi časté
|
Časté
|
Laboratórne a funkčné vyšetrenia
|
|
|
Zvýšená hladina lipázy
|
Časté
|
Časté
|
Zvýšené hladiny transamináz5
|
Veľmi časté
|
Časté
|
1 Skupinový výraz pre reakcie s viacerými preferovanými výrazmi
2 Pneumonitída zahŕňa preferované výrazy: pneumonitída, intersticiálna choroba pľúc, infiltrácia pľúc
3 Hnačka alebo kolitída zahŕňa preferované výrazy: kolitída, enterokolitída, mikroskopická kolitída, ulcerózna kolitída,
hnačka, hemoragická hnačka
4 Vyrážka zahŕňa preferované výrazy: dermatitída (vrátane alergickej, exfoliatívnej, perivaskulárnej), erytém (vrátane multiformného), vyrážka (vrátane exfoliatívnej, erytematóznej, folikulárnej, generalizovanej, makulopapulóznej, pruritickej, pustulárnej), toxická epidermálna nekrolýza a toxická kožná erupcia, lieková reakcia s eozinofíliou a systémovými symptómami, lieková erupcia, Stevensov-Johnsonov syndróm.
5 Elevácia transamináz zahŕňa preferované výrazy: zvýšená hladina alanínaminotransferázy, zvýšená hladina
aspartátaminotransferázy, zvýšené hladiny transaminázy, hypertransaminazémia, hepatocelulárne poškodenie, hepatotoxicita
Poznámka: Vysadenie dávok na > 42 dní v dôsledku toxicity súvisiacej s liečbou bude viesť k trvalému ukončeniu liečby.
Opis vybraných nežiaducich reakciíInfekcie
Najčastejšie závažné infekcie zahŕňali pneumóniu, sepsu a infekcie dolných dýchacích ciest. Medián času do nástupu infekcie akéhokoľvek stupňa bol 3 mesiace (rozsah: 1 deň až 32 mesiacov),
pričom 75 % prípadov sa vyskytlo v priebehu 6 mesiacov. Pred začatím podávania duvelizibu sa majú infekcie vyliečiť. Pacienti majú byť poučení o tom, aby hlásili nové alebo zhoršujúce sa prejavy
a príznaky infekcie.
Odporúčané postupy na liečbu infekcií pozri v časti 4.2 (tabuľka 1) a 4.4.
Hnačka a kolitídaMedián času do nástupu hnačky alebo kolitídy akéhokoľvek stupňa bol 4 mesiace (rozsah: 1 deň až 33 mesiacov), pričom 75 % prípadov sa vyskytlo v priebehu 8 mesiacov. Medián trvania udalosti bol 0,5 mesiaca (rozsah: 1 deň až 29 mesiacov; 75. percentil: 1 mesiac). Pacienti majú byť poučení o tom, aby hlásili nové alebo zhoršujúce sa prípady hnačky.
Neinfekčná pneumonitídaMedián času do nástupu pneumonitídy akéhokoľvek stupňa bol 4 mesiace (rozsah: 9 dní až 27 mesiacov), pričom 75 % prípadov sa vyskytlo do 9 mesiacov. Medián trvania udalosti bol 1 mesiac, pričom 75 % prípadov odznelo do 2 mesiacov.
Duvelizib sa má vysadiť u pacientov, u ktorých sa pri rádiologickom vyšetrení objavia nové alebo progresívne pulmonárne prejavy a príznaky, ako sú kašeľ, dyspnoe, hypoxia, intersticiálne infiltráty alebo znížená saturácia kyslíkom o viac než 5 %, a ktorí sú hodnotení na potvrdenie etiológie. Ak je pneumonitída infekčná, pacientom sa smie duvelizib opätovne podať v predchádzajúcej dávke, len čo infekcia, pulmonárne prejavy a príznaky odznejú.
Závažné kožné reakcieMedián času do nástupu kožnej reakcie akéhokoľvek stupňa bol 3 mesiace (rozsah: 1 deň až 29 mesiacov, 75. percentil: 6 mesiacov) s mediánom trvania udalosti 1 mesiac (rozsah: 1 deň až 37 mesiacov, 75. percentil: 2 mesiace). Závažné kožné reakcie zahŕňajú vyrážku, Stevensov-Johnsonov syndróm (SJS), toxickú epidermálnu nekrolýzu (TEN) a liekovú reakciu s eozinofíliou a systémovými symptómami (DRESS).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieAk dôjde k predávkovaniu, u pacienta sa musia sledovať príznaky toxicity (pozri časť 4.8). V prípade predávkovania sa majú zabezpečiť všeobecné podporné opatrenia a liečba. U pacienta sa majú sledovať prejavy a príznaky, laboratórne parametre a vitálne funkcie.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostis
Farmakoterapeutická skupina: cytostatiká, inhibítory fosfatidylinozitol-3-kinázy (Pi3K), ATC kód: L01EM04
Mechanizmus účinkuDuvelizib je duálny inhibítor fosfatidylinozitol-3-kinázy p110δ (PI3K-δ) a PI3K-γ. Inhibícia PI3K-δ
priamo znižuje proliferáciu a prežitie malígnych B-bunkových línií a primárnych CLL nádorových
buniek, zatiaľ čo inhibícia PI3K-γ znižuje aktivitu CD4+ T buniek a makrofágov v mikroprostredí
nádoru na podporu malígnych B-buniek. Pri dávke 25 mg dvakrát denne nemusia byť plazmatické koncentrácie duvelizibu dostatočne vysoké, aby spôsobili nepretržitú inhibíciu PI3K-γ, a prínos inhibície PI3K-γ k účinnosti môže byť limitovaný.
Elektrofyziológia srdca
Účinnosť opakovaných dávok duvelizibu 25 a 75 mg dvakrát denne na korigovaný interval QT (QTc)
bola hodnotená u pacientov s predtým liečenými hematologickými malignitami. Nepozorovali sa zvýšenia intervalu QTc o > 20 ms.
Klinická účinnosť pri relapsujúcej alebo refraktérnej CLL/SLL
IPI-145-07
V randomizovanom, multicentrovom, otvorenom skúšaní (skúšanie IPI-145-07) sa duvelizib porovnával s ofatumumabom u 319 dospelých pacientov s CLL (n = 312) alebo SLL (n = 7)
po najmenej jednej predchádzajúcej liečbe. Pacienti neboli vhodní na liečbu režimom analógami na báze purínov [podľa usmernení National Comprehensive Cancer Network (Národnej komplexnej siete pre rakovinu) alebo (Európskej spoločnosti pre lekársku onkológiu)] vrátane relapsu ≤ 36 mesiacov od režimu chemoimunoterapie na báze purínov alebo relapsu ≤ 24 mesiacov od režimu monoterapie na báze purínov. Pacienti, ktorí predtým dostávali inhibítory BTK alebo PI3K boli zo skúšania vylúčení. ŽiadnyzozúčastnenýchpacientovnedostávalpredtýmliečbuinhibítoromBCL-2.
Skúšanie randomizovalo pacientov v pomere 1:1. V skupine dostávali buď duvelizib v dávke 25 mg dvakrát denne až do progresie ochorenia alebo neprijateľnej toxicity, alebo ofatumumab počas 7 cyklov. Ofatumumab bol podávaný intravenózne v úvodnej dávke 300 mg, po ktorej bola o týždeň neskôr podaná dávka 2 000 mg raz za týždeň spolu v 7 dávkach, a potom 2 000 mg raz za 4 týždne v 4 ďalších dávkach. Liečba ofatumumabom nebola nad rámec 7 cyklov povolená a žiadny z pacientov nedostal viac než 7 cyklov ofatumumabu.
V celkovej populácii skúšania (160 randomizovaných do skupiny s duvelizibom, 159 do skupiny s ofatumumabom) bol medián veku pacienta 69 rokov (rozsah: 39 až 90 rokov), pričom 68 % pacientov bolo starších ako 65 rokov, 60 % bolo mužov a 92 % malo podľa Eastern Cooperative Oncology Group (Východnej spolupracujúcej onkologickej skupiny, ECOG) výkonnostný stav 0 alebo 1. 61 %
pacientov malo Rai štádium ≥ I a 39 % malo štádium podľa Bineta ≥ B. Percentuálny podiel pacientov
s nemutovanou IGHV (Ig ťažký reťazec V-111) bol 71 %. Tridsaťosem percent (38 %) dostalo 1 predchádzajúcu líniu liečby a 62 % dostalo 2 alebo viac predchádzajúcich línií liečby. Deväťdesiatštyri percent (94 %) pacientov dostalo predchádzajúcu alkylačnú liečbu, pričom 38 % pacientov dostalo predchádzajúcu liečbu bendamustínom; 80 % pacientov dostalo predchádzajúcu liečbu rituximabom. 60 % v skupine s duvelizibom a 71 % v skupine s ofatumumabom dostalo
predchádzajúcu liečbu purínovým analógom (ale neboli refraktérni, ako definuje IwCLL). Na začiatku skúšania malo 46 % pacientov minimálne jeden nádor ≥ 5 cm, 24 % pacientov malo zdokumentovanú deléciu 17p, 32 % pacientov malo zdokumentovanú deléciu 17p a/alebo mutáciu TP53 a 23 % malo zdokumentovanú deléciu 11q. Medián času od počiatočnej diagnózy bol 7 rokov (rozsah: 0,3 až 34,7 roka). Medián času od posledného relapsu/refraktérnej diagnózy bol 2,4 mesiaca (rozsah: 0,2 až 80,2 mesiaca). Medián času od poslednej systémovej liečby bol 19,5 mesiaca (rozsah: 0,5 až 148,8 mesiaca).
Počas randomizovanej liečby bol medián trvania expozície duvelizibu 12 mesiacov (rozsah: 0,2 až
37), pričom 72 % pacientov dostávalo duvelizib aspoň 6 mesiacov a 49 % pacientov dostávalo duvelizib aspoň 12 mesiacov. Medián trvania expozície ofatumumabu bol 5 mesiacov (rozsah: < 0,1 až 6).
Schválenie lieku Copiktra je založené na analýze účinnosti a bezpečnosti u pacientov s aspoň 2 predchádzajúcimi líniami liečby, keď sa pomer prínosu a rizika v tejto populácii s intenzívnou predchádzajúcou liečbou javil väčší v porovnaní s celkovou populáciou v skúšaní.
V tejto podskupine pacientov s aspoň 2 predchádzajúcimi líniami liečby (95 randomizovaných do skupiny s duvelizibom, 101 do skupiny s ofatumumabom) bol medián veku pacientov 69 rokov
(rozsah: 40 až 90 rokov), pričom 70 % pacientov bolo starších ako 65 rokov, 59 % bolo mužov a 88 %
malo stav výkonnosti podľa ECOG rovný 0 alebo 1. 62 % pacientov malo štádium podľa Raia ≥ I a 38 % malo štádium podľa Bineta ≥ B. Percentuálny podiel pacientov s nemutovanou IGHV (Ig ťažký reťazec V-111) bol 69 %. Štyridsaťšesť percent (46 %) dostávalo 2 predchádzajúce línie liečby
a 54 % dostávalo 3 alebo viac predchádzajúcich línií liečby. Deväťdesiatšesť percent (96 %) pacientov dostávalo predchádzajúcu alkylačnú liečbu, pričom 51 % pacientov dostávalo predchádzajúcu liečbu bendamustínom; 86 % pacientov dostávalo predchádzajúcu liečbu rituximabom. 70 % v skupine
s duvelizibom a 77 % v skupine s ofatumumabom dostávalo predchádzajúcu liečbu purínovým analógom (ale neboli refraktérni, ako definuje IwCLL). Na začiatku skúšania malo 52 % pacientov minimálne jeden nádor ≥ 5 cm, 22 % pacientov malo zdokumentovanú deléciu 17p, 31 % pacientov malo zdokumentovanú deléciu 17p a/alebo mutáciu
TP53 a 27 % malo zdokumentovanú deléciu 11q. Medián času od počiatočnej diagnózy bol 8 rokov (rozsah: 0,9 až 34,7 roka). Medián času od poslednéhorelapsu/refraktérnej diagnózy bol 2,6 mesiaca (rozsah: 0,2 až 69 mesiaca). Medián času od poslednej systémovej liečby bol 15,5 mesiaca (rozsah: 0,5 až 107,2 mesiaca).
Počas randomizovanej liečby bol medián trvania expozície duvelizibu 13 mesiacov (rozsah: 0,2 až
37), pričom 80 % pacientov dostávalo duvelizib aspoň 6 mesiacov a 52 % pacientov dostávalo
duvelizib aspoň 12 mesiacov. Medián trvania expozície ofatumumabu bol 5 mesiacov (rozsah: < 0,1
až 6).
Účinnosť bola založená na primárnom cieľovomukazovateli prežitia bez progresie (progression-free survival, PFS) podľa hodnotenia nezávislej hodnotiacej komisie (Independent Review Committee, IRC). Pacienti v oboch skupinách boli po prerušení randomizovanej liečby naďalej sledovaní z hľadiska progresie ochorenia, až kým sa nezačala následná protirakovinová liečba. Ďalšie opatrenia v oblasti účinnosti zahŕňali celkovú mieru odpovede. Cieľové ukazovatele účinnosti týkajúce sa celkovej miery odpovede a celkového prežitia boli navrhnuté ako kľúčové sekundárne cieľové ukazovatele účinnosti a mali sa overiť postupne, len ak bol primárny cieľový ukazovateľ PFS významný.'
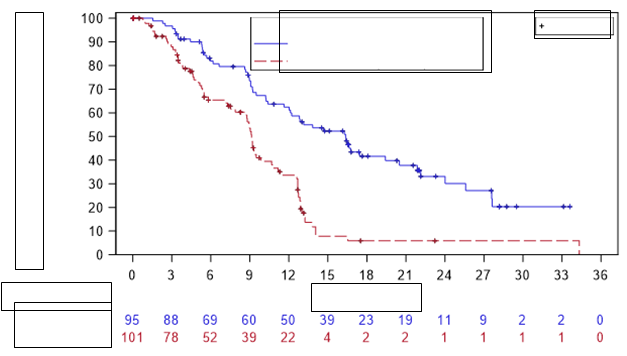
Výsledky sú uvedené v tabuľke 3 a na obrázku 1 pre podskupinu pacientov aspoň s dvoma predchádzajúcimi liečbami.
Tabuľka 3: Účinnosť pri CLL po aspoň dvoch predchádzajúcich liečbach (IPI-145-07)
Výsledok podľa IRC
|
Duvelizib n = 95
| Ofatumumab n = 101
|
PFS
|
Medián PFS (95 % IS)
v mesiacocha
| 16,4 (12,0; 20,5)
|
9,1 (7,9; 10,7)
|
Pomer rizika (95 % IS),b
Duvelizib/ofatumumab
| 0,4 (0,27; 0,59)
|
Hodnota p
| < 0,0001
|
Miera odpovede
|
ORR, n (%)c (95 % IS)
| 75 (78,9) (70,7; 87,1)
| 39 (38,6) (29,1; 48,1)
|
CR, n (%)
| 0
| 0
|
PR, n (%)
| 75 (78,9)
| 39 (38,6)
|
Hodnota p
| < 0,0001
|
Celkové prežitie (OSd)
|
Medián OS (95 % IS)
v mesiacocha
| NE
| NE
|
Pomer rizika (95 % IS),b
Duvelizib/ofatumumab hodnota
p
|
0,82 (0,49; 1,37)
0,4397
|
Skratky: IS = interval spoľahlivosti; CR = úplná odpoveď; IRC = nezávislá hodnotiaca komisia; PFS = prežitie bez progresie; PR = čiastočná odpoveď
a Odhad podľa Kaplanovej-Meierovej metódy
b Stratifikovaný Coxov model proporcionálneho rizika s použitím randomizačnej vrstvy, ako sa používa na randomizáciu
c Kritériá odpovede podľa IWCLL alebo revidované kritériá odpovede podľa IWG, s úpravou pre lymfocytózu súvisiacu
s liečbou
d Analýza OS zahŕňa údaje od osôb, ktoré v štúdii dostávali ofatumumab a v rozšírenej štúdii následne dostávali duvelizib na základe analýzy populácie so zámerom liečiť (intent-to-treat). Osoby v oboch skupinách boli po prerušení randomizovanej liečby naďalej sledovaní z hľadiska OS, a to bez ohľadu na nasledujúcu liečbu, ktorú dostávali.
Výsledok podľa IRC
| Duvelizib
| Ofatumumab
| Delécia 17p/mutácia TP53
| n = 29
| n = 30
| Medián PFS (95 % IS)
v mesiacocha
|
12,8 (8,9; 22,1)
|
8,7 (5,3; 12,6)
| Pomer rizika (95 % IS),b
Duvelizib/ofatumumab
| 0,36 (0,18; 0,72)
| ORR, (95 % IS)c
| 72,4 (56,1; 88,7)
| 36,7 (19,4; 53,9)
| Vek ≥ 65
| n = 68
| n = 69
| Medián PFS (95 % IS)
v mesiacocha
| 16,4 (10,4; 24,0)
| 9,2 (8,7; 10,8)
| Pomer rizika (95 % IS),b
Duvelizib/ofatumumab
| 0,38 (0,24; 0,61)
| ORR, (95 % IS)c
| 77,9 (68,1; 87,8)
| 39,1 (27,6; 50,6)
| Nemutovaná IGHV
| n = 65
| n = 70
| Medián PFS (95 % IS)
v mesiacocha
| 17,4 (12,0; 24,0)
| 9,0 (7,3; 10,7)
| Pomer rizika (95 % IS),b
Duvelizib/ofatumumab
| 0,27 (0,17; 0,45)
| ORR, (95 % IS)c
| 86,2 (77,8; 94,6)
| 40 (28,5; 51,5)
|
|
|
Tabuľka 4: Súhrn PFS a miery odpovede pri liečbe podskupín u pacientov s aspoň dvomi predchádzajúcimi liečbami – (IPI-145-07)Skratky: IS = interval spoľahlivosti; IRC = nezávislá hodnotiaca komisia; PFS = prežitie bez progresie
a Odhad podľa Kaplanovej-Meierovej metódy
b Coxov model proporcionálneho rizika
c Kritériá odpovede podľa IWCLL alebo revidované kritériá odpovede podľa IWG, s úpravou pre lymfocytózu súvisiacu s liečbou
O
b
r
ázok 1: Kaplanova-Meierova krivka PFS podľa IRC u pacientov s aspoň dvoma predchádzajúcimi liečbami (IPI-145-07)
Liečba
Copiktra 25 mg dvakrát denne (n= 95)
Ofatumumab (n= 101)
Cenzorované

Počet v ohrození
Copiktra
Ofatumumab
Čas (mesiace)
 K
li
nická
účinnosť
pri
re
l
a
psujúcom
a
l
e
bo
refra
ktérnom
f
olikulovom
l
ymfóme
(
F
L
)
I
P
I
-
145-06
K
li
nická
účinnosť
pri
re
l
a
psujúcom
a
l
e
bo
refra
ktérnom
f
olikulovom
l
ymfóme
(
F
L
)
I
P
I
-
145-06
Účinnosť duvelizibu u pacientov s predtým liečeným FL je založená na jednoskupinovom, multicentrovom skúšaní (skúšanie IPI-145-06). V tomto skúšaní bol duvelizib v dávke 25 mg dvakrát denne podávaný 129 pacientom s indolentným B-bunkovým non-Hodgkinovým lymfómom (iNHL vrátane: FL, n = 83; SLL, n = 28; a lymfómom marginálnej zóny [MZL], n = 18), ktorí boli refraktérni na rituximab a buď na chemoterapiu alebo rádioimunoterapiu. Refraktérne ochorenie bolo definované ako menšia než čiastočná remisia alebo relaps do 6 mesiacov po podaní poslednej dávky. Zo skúšania boli vylúčení pacienti s FL stupňa 3b, s veľkobunkovou transformáciou, predchádzajúcim
alogenickým transplantátom a predchádzajúcou expozíciou inhibítoru P13K alebo inhibítoru
Brutonovej tyrozinkinázy.
Medián veku bol 65 rokov (rozsah: 30 až 90 rokov), pričom 50 % osôb malo viac ako 65 rokov a 14 % osôb bolo vo veku 75 rokov alebo starších, 68 % boli muži a 40 % malo na začiatku skúšania veľkú nádorovú masu (cieľová lézia ≥ 5 cm). Pacienti mali medián 3 predchádzajúcich línií liečby (rozsah: 1 až 18), pričom 96 % pacientov bolo refraktérnych na poslednú liečbu a 77 % pacientov bolo refraktérnych na 2 alebo viac predchádzajúcich línií liečby. Deväťdesiatosem percent (98 %)
pacientov bolo refraktérnych na rituximab a 91 % bolo refraktérnych alkylačné liečivá. Väčšina pacientov (približne 75 %) mala skúsenosti s včasným relapsom [žiadna odpoveď na liečbu alebo progresívne ochorenie (progressive disease, PD), alebo čas do ďalšej liečby kratší než 2 roky] po prvom režime liečby. Medián času od počiatočnej diagnózy bol 4,5 roka (rozsah 4 mesiace až 27 rokov). Väčšina pacientov (95 %) mala stav výkonnosti podľa ECOG rovný 0 alebo 1.
Medián trvania expozície duvelizibu bol 7 mesiacov (rozsah: 0,4 až 45,5), pričom 53 % pacientov dostávalo duvelizib aspoň 6 mesiacov a 26 % pacientov dostávalo duvelizib aspoň 12 mesiacov.
Účinnosť bola založená na primárnom cieľovom ukazovateli celkovej miery odpovede. Sekundárne cieľové ukazovatele zahŕňali prežitie bez progresie, trvanie odpovede podľa hodnotenia IRC a celkové prežitie (Tabuľka 5).
T
abuľka 5: Účinnosť u pacientov s aspoň dvoma predchádzajúcimi liečbami, relapsujúcim
alebo refraktérnym FL (IPI-145-06)
C
i
e
ľ
ový ukazovateľ
|
|
F
olikulový lymfóm
|
n = 73
|
ORR, n (%)a
|
29 (40)
|
95 % CI
|
(31, 54)
|
CR, n (%)
|
0
|
PR, n (%)
|
29 (40)
|
Trvanie odpovede
|
|
Rozsah, mesiace
|
0,0+ až 41,9
|
Medián DOR (95 % IS) v mesiacochb
|
10,01 (6,3, NE)
|
Skratky: IS = interval spoľahlivosti; CR = úplná odpoveď; IRC = nezávislá hodnotiaca komisia; ORR = celková miera odpovede; PR = čiastočná odpoveď
a Podľa IRC v súlade s revidovanými kritériami medzinárodnej pracovnej skupiny
a Odhad podľa Kaplanovej-Meierovej metódy
+ Označuje cenzorovanú hodnotu
Starší pacientiKlinické skúšania duvelizibu zahŕňali 270 pacientov (61 %), ktorí boli vo veku 65 rokov a starší, a 104
pacientov (24 %), ktorí boli vo veku 75 rokov a starší. Medzi pacientmi mladšími ako 65 rokov
a pacientmi vo veku 65 rokov a staršími neboli pozorované žiadne veľké rozdiely v účinnosti alebo bezpečnosti. U starších pacientov (vo veku ≥ 65 rokov) sa nevyžaduje žiadna osobitná úprava dávky (pozri časť 5.2).
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s duvelizibom v
liečbe malignít zrelých B-buniekvo všetkých podskupinách pediatrickej populácie od narodenia do menej ako 18 rokov (informácie o použití v pediatrickej populácii pozri časť 4.2).
5.2 Farmakokinetické vlastnostiExpozícia duvelizibu sa zvýšila úmerne s dávkou v rámci rozsahu dávky od 8 mg do 75 mg (0,3- násobok až 3-násobok odporúčanej dávky) po jednorazovej dávke. Proporcionalita dávky nebola po opakovaných dávkach stanovená.
V rovnovážnom stave po podaní 25 mg duvelizibu pacientom dvakrát denne bol geometrický priemer
(CV %) maximálnej koncentrácie (cmax) 1,5 (64 %) µg/ml a hodnota AUC bola 7,9 (77 %) µg.h/ml.
AbsorpciaAbsolútna biologická dostupnosť 25 mg duvelizibu po jednorazovej perorálnej dávke u zdravých
dobrovoľníkov bola 42 %. Medián času do dosiahnutia maximálnej koncentrácie (tmax) bol u pacientov pozorovaný v rozmedzí 1 až 2 hodín.
Vplyv jedlaDuvelizib sa môže podávať bez ohľadu na jedlo. Po podaní jednorazovej dávky duvelizibu s jedlom
s vysokým obsahom tukov (tuk predstavoval približne 50 % celkového kalorického obsahu jedla) sa cmax znížila o približne 37 % a AUC sa znížila približne o 6 % v porovnaní s užívaním nalačno.
DistribúciaVäzba duvelizibu na bielkoviny je väčšia než 95 %. Priemerný pomer koncentrácie v krvi/plazme bol
0,5. Geometrický priemer (CV %) zdanlivého distribučného objemu v rovnovážnom stave (Vss/F) je
28,5 l (62 %).
B
i
otransformácia
Duvelizib sa primárne metabolizuje cytochrómom P450 CYP3A4. Hlavným metabolitom je IPI-656,
ktorý je farmakologicky neaktívny pri klinicky pozorovaných hladinách expozície.
Eliminácia
Geometrický priemer (CV %) zdanlivého systémového klírensu v rovnovážnom stave je 4,2 l/h.
(56 %) u pacientov s lymfómom alebo leukémiou. Geometrický priemer (CV %) eliminačného polčasu duvelizibu je 4,7 hodiny (57 %) počas 0 až 8 hodín po dávke.
Vylučovanie
Po podaní jednorazovej 25 mg perorálnej dávky rádioaktívne označeného duvelizibu sa 79 %
rádioaktivity vylúčilo v stolici (11 % v nezmenenej podobe) a 14 % sa vylúčilo v moči (1 %
v nezmenenej podobe). Tieto údaje boli zistené u zdravých osôb.
Štúdie liekových interakcií invitro
Duvelizib je substrát P-glykoproteínu (P-gp) a proteínu rezistencie rakoviny prsníka (breast cancer-
resistant protein, BCRP). Duvelizib sa po perorálnej dávke vo vysokej miere absorbuje, a preto sa
žiadny klinicky relevantný účinok inhibítorov P-gp a BCRP neočakáva.
Štúdie in vitro kombinované s farmakokinetickými (PK) údajmi in vivo u ľudí naznačovali, že klinicky relevantné liekové interakcie duvelizibu a jeho hlavného metabolitu IPI-656 so substrátmi OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, BCRP alebo P-gp nie sú pravdepodobné. Z tohto
dôvodu sa štúdie interakcie s Pgp, BCRP a CYP2C8 nepovažujú za potrebné.
Duvelizib aj IPI-656 boli identifikované ako priame inhibítory CYP2C8 a CYP3A4, ako aj inhibítory CYP3A4 závislé od metabolizmu (pozri časť 4.5). Simulácie ukázali, že pri supraterapeutických dávkach môže byť duvalisib mierny inhibítor CYP2C8, čo pravdepodobne nepovedie ku klinicky relevantným interakciám.
Osobitné populácie
Vek (18 až 90 rokov), pohlavie, rasa, porucha funkcie obličiek (klírens kreatinínu 23 až 80 ml/min),
porucha funkcie pečene (trieda A, B alebo C podľa Childa-Pugha) a telesná hmotnosť (40 až 154 kg)
nemali žiadny klinicky významný účinok na expozíciu duvelizibu.
Farmakokinetika duvelizibu bola veľmi variabilná u osôb so stredne závažnou a závažnou poruchou funkcie pečene. Geometrický priemer duvelizibu AUC0-∞ u osôb s miernou, stredne závažnou
a závažnou poruchou funkcie pečene bol nižší (do 20 %) v porovnaní s expozíciou pozorovanou
u zdravých osôb a predstavoval 89 %, 94 % a 81 % expozície pozorovanej u zdravých
osôba nepovažuje sa za klinicky významný. Expozície u osôb so stredne závažnou a závažnou poruchou funkcie boli veľmi variabilné (CV % 46 – 67 %) a u týchto pacientov sa majú dôkladne sledovať nežiaduce udalosti (pozri časť 4.4).
Expozície zistené u pacientov s rakovinou boli približne 2-násobne vyššie ako expozície pozorované u zdravých osôb.
5.3 Predklinické údaje o bezpečnosti
V štúdiách toxicity po opakovaných dávkach u potkanov a makakoch dlhochvostých súviseli nežiaduce účinky najmä s očakávaným vystupňovaným farmakologickým účinkom vrátane nežiaducich účinkov na lymfatické tkanivá, kostnú dreň a hematologické parametre pri expozíciách voľnému duvelizibu pri 8- až 16-násobku, čo zodpovedá celkovému duvelizibu pri 2- až 11-násobku maximálnej odporúčanej dávky pre ľudí (MRHD) 25 mg dvakrát denne.
Duvelizib nespôsobil genetické poškodenie v testoch in vitro ani in vivo.
V zisteniach o rozsahu dávky a pivotných štúdiách embryo-fetálnej vývinovej toxicity u potkanov
a králikov duvelizib (voľná frakcia) indukoval embryo-fetálnu vývinovú toxicitu len pri expozíciách
voľnej frakcie v plazme v rozsahu > 25-násobku dávky 25 mg dvakrát denne u človeka (MRHD), čo zodpovedá 4- až 5-násobku celkových plazmatických koncentrácií.
S duvelizibom sa neuskutočnili žiadne štúdie fertility. Histologické nálezy u samcov a samíc potkanov sa pozorovali v štúdiách toxicity po opakovanom podaní a zahŕňali semenníky (atrofia epitelu semenovodov, znížená hmotnosť, mäkké semenníky) a nadsemenníky (malá veľkosť, oligo/aspermia) u samcov a vaječníky (znížená hmotnosť) a maternicu (atrofia) u samíc.
S duvelizibom sa neuskutočnili štúdie karcinogenity.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsahkapsuly
koloidný oxid kremičitý
krospovidón stearát horečnatý mikrokryštalická celulóza
Obalkapsuly želatína
oxid titaničitý (E 171)
červený oxid železitý (E 172)
Čiernafarbapotlače šelaková glazúra
čierny oxid železitý (E 172)
propylénglykol hydroxid amónny
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
Copiktra 15 mg tvrdé kapsuly
4 roky
Copiktra 25 mg tvrdé kapsuly
5 rokov
6.4 Špeciálne upozornenia na uchovávanie
Uschovávajte pri teplote do 30 °C.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
C
opiktra
15
m
g
t
vrdé
kapsuly
Detské bezpečnostné PVC/PE/PCTFE/hliníkové blistre.
Veľkosť balenia: Škatuľa na 28 dní obsahujúca 56 kapsúl (2 blistre, každý s 28 kapsulami).
Copiktra 25 mg tvrdé kapsulyDetské bezpečnostné PVC/PE/PCTFE/hliníkové blistre.
Veľkosť balenia: Škatuľa na 28 dní obsahujúca 56 kapsúl (2 blistre, každý s 28 kapsulami).
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIVerastem Europe GmbH Lange Strasse 70
29664 Walsrode
Nemecko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/21/1542/001
EU/1/21/1542/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.