
učení o tom, aby informovali svojho lekára, ak sa u nich vyskytne závažná alebo pretrvávajúca hnačka, alebo krvácanie v dolnej časti gastrointestinálneho traktu (pozri časť 4.8).
Ak sa vyskytne pretrvávajúca (napr. dlhšie než 1 týždeň) alebo závažná hnačka, má sa vyhľadať lekárska pomoc a zvážiť dočasné vysadenie linaklotidu, až kým epizóda hnačky neprejde. Osobitná opatrnosť je potrebná u pacientov, ktorí sú náchylní na poruchu rovnováhy vody alebo elektrolytov (napr. starší pacienti, pacienti s kardiovaskulárnymi (KV) ochoreniami, diabetom, hypertenziou) a má sa zvážiť kontrola elektrolytov.
U pacientov s chronickými zápalovými ochoreniami črevného traktu ako je Crohnova choroba
a ulcerózna kolitída sa linaklotid neskúmal; preto sa použitie Constelly u týchto pacientov neodporúča.
Starší pacienti
U starších pacientov sú k dispozícii len obmedzené údaje (pozri časť 5.1). U týchto pacientov je potrebná zvýšená pozornosť a starostlivé a pravidelné vyhodnotenie pomeru rizika voči prínosom liečby, pretože v klinických skúšaniach bolo pozorované vyššie riziko hnačky (pozri časť 4.8).
Pediatrická populácia
Constella sa nemá používať u detí a dospievajúcich, pretože sa v tejto skupine pacientov neskúmala. Keďže je známa nadmerná expresia GC-C receptora v ranom veku, deti mladšie ako 2 roky môžu byť obzvlášť citlivé na účinky linaklotidu.
4.5 Liekové a iné interakcie
Nevykonali sa žiadne interakčné štúdie. Linaklotid je po podaní odporučených klinických dávok zriedkavo v plazme detekovateľný a štúdie in vitro ukázali, že linaklotid nie je ani substrátom, ani inhibítorom/induktorom enzýmového systému cytochrómu P450 a nespôsobuje interakcie so sériou bežných efluxných transportérov a transportérov spätného vychytávania (pozri časť 5.2).
Klinická štúdia interakcií s jedlom so zdravými jedincami ukázala, že linaklotid nebol pri terapeutických dávkach v plazme detekovateľný ani v stave nasýtenia, ani v stave na lačno. Užívanie Constelly s jedlom spôsobilo častejšiu a redšiu stolicu a tiež viac gastrointestinálnych nežiaducich udalostí než užívanie na lačno (pozri časť 5.1). Kapsula by sa nemala užívať 30 minút pred jedlom (pozri časť 4.2).
Súbežná liečba inhibítormi protónovej pumpy, laxatívmi alebo nesteroidnými protizápalovými liekmi (NSAID) môže zvýšiť riziko hnačky. Pri súbežnom podávaní Constelly s takýmito liekmi je potrebná opatrnosť.
V prípade závažnej alebo pretrvávajúcej hnačky môže byť ovplyvnená absorpcia iných perorálnych liekov. Účinnosť perorálnej antikoncepcie môže byť znížená a preto ako prevencia možného zlyhania perorálnej antikoncepcie sa odporúča použitie dodatočnej metódy antikoncepcie (pozri informácie
na predpisovanie perorálnej antikoncepcie). Pri predpisovaní liekov, ktoré sa absorbujú v črevnom trakte s úzkym terapeutickým indexom, ako je levotyroxín, je potrebná zvýšená opatrnosť, keďže môže byť znížená účinnosť týchto liekov.
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Množstvo údajov ohľadne použitia linaklotidu u gravidných žien je obmedzené. Štúdie na zvieratách
nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3). Avšak ako preventívne opatrenie sa uprednostňuje nepoužívať Constellu počas gravidity.
Laktácia
Nie je známe, či sa linaklotid vylučuje do ľudského mlieka. Riziko u novorodencov/dojčiat nemôže
byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu Constellou sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Štúdie na zvieratách nepreukazujú žiadny vplyv na mužskú alebo ženskú fertilitu.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Constella nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
V kontrolovaných klinických štúdiách sa linaklotid perorálne podal 1 166 pacientom s IBS-C. Z týchto pacientov 892 pacientov dostalo linaklotid v odporúčanej dávke 290 mikrogramov denne. Celková expozícia v pláne klinického vývoja presiahla 1 500 paciento-rokov. Najčastejšie hlásená nežiaduca reakcia spojená s liečbou Constellou bola hnačka, najmä miernej až stredne závažnej intenzity, vyskytujúca sa u menej než 20 % pacientov. V zriedkavých a závažnejších prípadoch môže,
následkom tohto, dôjsť k vzniku dehydratácie, hypokaliémie, zníženiu hladiny bikarbonátu v krvi, závratom a ortostatickej hypotenzii.
Iné časté nežiaduce reakcie (> 1 %) boli bolesť brucha, abdominálna distenzia a plynatosť.
Zoznam nežiaducich reakcií v tabuľkovom formáte
V kontrolovaných klinických štúdiách sa pri odporúčanej dávke 290 mikrogramov denne hlásili nasledovné nežiaduce reakcie s frekvenciami zodpovedajúcimi: veľmi časté (≥ 1/10), časté (≥ 1/100 až
< 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000) a veľmi zriedkavé
(< 1/10 000) a neznáme (z dostupných údajov).
T
rieda orgánových systémov podľa databázy MedDRA
V
eľmi časté
Č
asté Menej časté Zriedkavé Neznáme
Infekcie a nákazy Vírusová gastroenteritída
Poruchy metabolizmu a výživy
Hypokaliémia Dehydratácia Znížená chuť do jedla
Poruchy nervového systému
Závrat
Poruchy ciev Ortostatická hypotenzia
Fekálna inkontinencia Naliehavé nutkanie na stolicu Krvácanie v dolnej
Poruchy gastrointestinálneho traktu
Poruchy kože
a podkožného tkaniva
Laboratórne
a funkčné vyšetrenia
Hnačka
Bolesť brucha
Plynatosť Abdominálna distenzia
časti
gastrointestinálneho traktu vrátane krvácania hemoroidov
a krvácania z konečníka
Nevoľnosť
Vracanie
Zníženiu hladiny bikarbonátu v krvi
Vyrážka
 Popis vybraných nežiaducich reakcií
Popis vybraných nežiaducich reakcií
Hnačka je najčastejšou nežiaducou reakciou a je v súlade s farmakologickou aktivitou liečiva. 2 %
liečených pacientov mali závažnú hnačku a 5 % pacientov prerušilo liečbu z dôvodu hnačky
v klinických štúdiách.
Väčšina hlásených prípadov hnačky bola mierna (43 %) až stredne závažná (47 %); 2 % liečených
pacientov mali závažnú hnačku. Približne polovica epizód hnačky sa začala v prvom týždni liečby.
Približne u jednej tretiny pacientov hnačka ustúpila do siedmich dní, avšak 80 pacientov (50 %) malo hnačku trvajúcu viac ako 28 dní (čo predstavuje 9,9 % všetkých pacientov liečených linaklotidom).
V klinických štúdiách došlo k prerušeniu liečby z dôvodu hnačky u piatich percent pacientov. U tých pacientov, u ktorých hnačka viedla k prerušeniu liečby, hnačka ustala po niekoľkých dňoch po
prerušení liečby.
U starších osôb (> 65 rokov), pacientov s hypertenziou a diabetom sa hnačka hlásila častejšie v
porovnaní s celkovou populáciou pacientov s IBS-C zaradenou v klinických skúšaniach.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávkovanie
Predávkovanie môže mať za následok vznik symptómov vyplývajúcich zo znásobenia známych farmakodynamických účinkov lieku, najmä hnačky. V štúdii so zdravými dobrovoľníkmi, ktorí užili jednorazovú dávku 2 897 mikrogramov (až 10-násobok odporúčanej terapeutickej dávky) bol bezpečnostný profil zhodný s profilom celkovej populácie, pričom najčastejšie hlásená nežiaduca udalosť bola hnačka.
Ak sa vyskytne predávkovanie, pacient má byť liečený symptomaticky a v prípade potreby sa majú
zaviesť podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: liečivá na zápchu, iné liečivá na zápchu, ATC kód: A06AX04
Mechanizmus účinku
Linaklotid je agonistom receptora guanylát cyklázy C (GC-C) s viscerálnymi analgetickými a
sekrečnými činnosťami.
Linaklotid je syntetický peptid pozostávajúci zo 14-aminokyselín štrukturálne príbuzný rodine endogénnych peptidov guanylínu. Linaklotid a aj jeho aktívny metabolit sa viažu na receptor GC-C na luminálnom povrchu črevného epitelu. Preukázalo sa, že linaklotid prostredníctvom jeho pôsobenia na GC-C znižuje viscerálnu bolesť a urýchľuje pasáž potravy cez gastrointestinálny trakt na modeloch zvierat a urýchľuje pasáž potravy cez hrubé črevo u ľudí. Aktivácia GC-C vedie k zvýšeniu koncentrácií cyklického guanozínmonofosfátu (cGMP), a to extracelulárne aj intracelulárne. Extracelulárny cGMP znižuje aktivitu vlákien vedúcich bolesť, v dôsledku čoho sa na modeloch zvierat znižuje viscerálna bolesť. Intracelulárny cGMP spôsobuje vylučovanie chloridu a bikarbonátu do lumenu čreva prostredníctvom aktivácie transmembránového regulátora vodivosti cystickej fibrózy (Cystic fibrosis transmembrane conductance regulator, CFTR), čo má za následok zvýšenie črevnej tekutiny a urýchlenie pasáže potravy.
Farmakodynamickéúčinky
V krížovej štúdii interakcií s jedlom sa Constella 290 mikrogramov podávala 18 zdravým jedincom počas 7 dní na lačno aj v stave nasýtenia. Užívanie Constelly ihneď po raňajkách s vysokým obsahom
tukov viedlo k častejšej a redšej stolici a tiež k vzniku viac gastrointestinálnych nežiaducich udalostí
v porovnaní s jej užívaním v stave na lačno.
Klinickáúčinnosťabezpečnosť
Účinnosť linaklotidu sa stanovila v dvoch randomizovaných, dvojito zaslepených, placebom kontrolovaných klinických štúdiách fázy 3 s pacientmi s IBS-C. V jednej klinickej štúdii (štúdia 1) sa Constellou 290 mikrogramov alebo placebom liečilo jedenkrát denne počas 26 týždňov 804 pacientov. V druhej klinickej štúdii (štúdia 2) sa liečilo počas 12 týždňov 800 pacientov a potom boli opätovne randomizovaní na obdobie ďalších 4 týždňov liečby. Počas dvojtýždňového východiskového obdobia pred liečbou mali pacienti priemerné skóre bolesti brucha 5,6 (na stupnici 0 – 10) s 2,2 % dní bez bolestí brucha, priemerné skóre plynatosti 6,6 (na stupnici 0 – 10) a priemerne 1,8 spontánnych defekácií (spontaneous bowel movements, SBM) za týždeň.
Charakteristiky populácie pacientov zaradenej do klinických skúšaní fázy 3 boli nasledovné: priemerný vek 43,9 roka [rozsah 18 – 87 rokov s 5,3 % ≥ 65 rokov veku], 90,1 % žien. Všetci pacienti spĺňali Rímske kritériá II pre IBS-C a boli požiadaní o hlásenie priemerného skóre bolesti brucha ≥ 3 na bodovej numerickej hodnotiacej stupnici 0 – 10 (kritériá zodpovedajúce populácii s miernym až závažným IBS), < 3 úplných spontánnych defekácií a ≤ 5 spontánnych defekácií za týždeň počas dvojtýždňového východiskového obdobia.
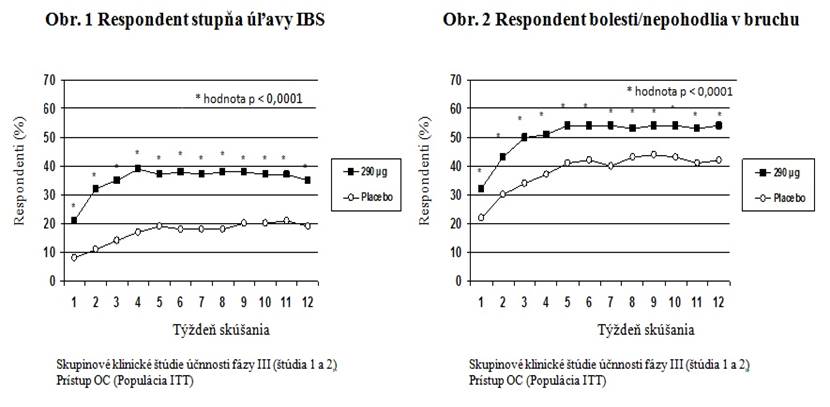
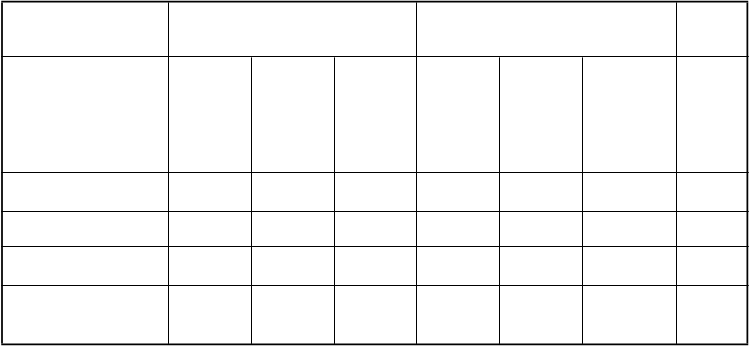
Spoločné primárne koncové ukazovatele v oboch klinických štúdiách boli 12-týždňové hodnotenie stupňa úľavy IBS u respondentov a 12-týždňové hodnotenie bolesti/nepohodlia v bruchu u respondentov. Respondent stupňa úľavy IBS bol pacient, ktorému sa značne alebo úplne uľavilo na obdobie aspoň 50 % obdobia liečby; respondent bolesti/nepohodlia v bruchu bol pacient, ktorý mal zlepšenie o 30 % alebo viac na obdobie aspoň 50 % obdobia liečby.
Pre 12-týždňové údaje štúdia 1 ukazuje, že sa u 39 % pacientov liečených linaklotidom v porovnaní so
17 % pacientov liečenými placebom potvrdila reakcia stupňa úľavy IBS (p < 0,0001) a u 54 % pacientov liečených linaklotidom v porovnaní s 39 % pacientov liečenými placebom sa potvrdila reakcia na bolesť/nepohodlie v bruchu (p < 0,0001). Štúdia 2 ukazuje, že 37 % pacientov liečených linaklotidom v porovnaní s 19 % pacientov liečenými placebom zaznamenalo reakciu stupňa úľavy IBS (p < 0,0001) a 55 % pacientov liečených linaklotidom v porovnaní so 42 % pacientov liečenými placebom reakciu na bolesť/nepohodlie v bruchu (p = 0,0002).
Pre 26-týždňové údaje štúdia 1 ukazuje, že 37 % pacientov liečených linaklotidom v porovnaní so
17 % pacientov liečenými placebom zaznamenalo reakciu stupňa úľavy IBS (p < 0,0001) a 54 %
pacientov liečených linaklotidom v porovnaní s 36 % pacientov liečenými placebom reakciu na bolesť/nepohodlie v bruchu (p < 0,0001).
V oboch štúdiách boli tieto zlepšenia viditeľné do 1. týždňa a zachovali sa počas celého obdobia liečby (Obrázky 1 a 2). Pri linaklotide sa nepreukázalo, že spôsobuje rebound fenomén, keď
k prerušeniu došlo po 3 mesiacoch nepretržitej liečby.
Ostatné prejavy a symptómy IBS-C vrátane plynatosti, frekvencie úplnej spontánnej defekácie (complete spontaneous bowel movements, CSBM), námahy pri defekácii, konzistencie stolice sa zlepšili u pacientov liečených linaklotidom voči placebu (p < 0,0001), ako ukazuje nasledovná tabuľka. Tieto účinky sa dosiahli v 1. týždni a zachovali sa počas celého obdobia liečby.
Ú
činok linaklotidu na symptómy IBS-C počas prvých 12 týždňov liečby v súhrnných klinických
št
údiách účinnosti fázy 3 (Štúdie 1 a 2).
H
lavné sekundárne
parametre účinnosti
Plynatosť (11
-bodová
Placebo(N = 797)Východis ková hodnota Priemer
12 týždňov Priemer
 Zmena voči východis kovej hodnote
Zmena voči východis kovej hodnote Priemer
Linaklotid(N = 805)Východis ková hodnota Priemer
12 týždňov Priemer
Zmena voči východisko vejhodnotePriemer
Priemer ný rozdiel LS
stupnica hodnotenia) 6,5 5,4 -1,0 6,7 4,6 -1,9 -0,9*
CSBM/týždeň 0,2 1,0 0,7 0,2 2,5 2,2 1,6* Konzistencia stolice'
(skóre BSFS) 2,3 3,0 0,6 2,3 4,4 2,0 1,4*
Námaha pri defekácii
(5-bodová poradová stupnica)
3,5 2,8 -0,6 3,6 2,2 -1,3 -0,6*
*p < 0,0001, linaklotid verzus placebo. LS (Least Square): Najmenšie štvorce
CSBM (complete spontaneous bowel movements): Úplná spontánna defekácia
Liečba linaklotidom viedla aj k značným zlepšeniam vo validácii a v miere kvality života špecifickej
pre ochorenie (IBS-QoL; p < 0,0001) a EuroQoL (p = 0,001). Klinicky významná reakcia v celkovej miere IBS-QoL (rozdiel > 14 bodov) sa dosiahla u 54 % pacientov liečených linaklotidom voči 39 %
pacientov liečených placebom.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Constellou
v jednej alebo vo viacerých podskupinách pediatrickej populácie s funkčnou zápchou (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Vo všeobecnosti je linaklotid po terapeutických perorálnych dávkach minimálne detekovateľný
v plazme, a preto sa štandardné farmakokinetické parametre nedajú vypočítať.
Po jednorazových dávkach do 966 mikrogramov a po opakovaných dávkach do 290 mikrogramov linaklotidu sa v plazme nezistili žiadne hladiny základnej zlúčeniny ani aktívneho metabolitu (dez- tyrozínu). Pri podaní 2 897 mikrogramov na 8. deň po 7-dňovom období podávania 290 mikrogramov denne sa linaklotid detekoval iba u 2 z 18 jedincov v koncentráciách tesne nad dolnou hranicou kvantifikácie 0,2 ng/ml (koncentrácie boli v rozsahu od 0,212 do 0,735 ng/ml). V dvoch pivotných štúdiách fázy 3, v ktorých pacienti užívali 290 mikrogramov linaklotidu raz denne, sa linaklotid detekoval iba u 2 zo 162 pacientov približne 2 hodiny po úvodnej dávke linaklotidu (koncentrácie boli od 0,241 ng/ml do 0,239 ng/ml) a u žiadneho zo 162 pacientov po 4 týždňoch liečby. Aktívny metabolit nebol detekovaný u žiadneho zo 162 pacientov v žiadnom časovom období.
Distribúcia
Keďže je linaklotid po terapeutických dávkach v plazme zriedkavo detekovateľný, štandardné štúdie distribúcie sa nevykonali. Predpokladá sa, že distribúcia linaklotidu je zanedbateľná alebo nesystémová.
Biotransformácia
Linaklotid sa metabolizuje lokálne v gastrointestinálnom trakte na svoj aktívny primárny metabolit dez-tyrozín. Linaklotid aj aktívny metabolit dez-tyrozín sa redukujú a enzymaticky štiepia
v gastrointestinálnom trakte na menšie peptidy a prirodzene sa vyskytujúce aminokyseliny.
Potenciálna inhibičná aktivita linaklotidu a jeho aktívneho primárneho metabolitu MM-419447 na ľudských efluxných transportéroch BCRP, MRP2, MRP3 a MRP4 a ľudských transportéroch vychytávania OATP1B1, OATP1B3, OATP2B1, PEPT1 a OCTN1 sa zisťovala in vitro. Výsledky tejto štúdie ukázali, že žiaden peptid nie je inhibítorom bežných efluxných transportérov
a transportérov vychytávania skúmaných pri klinicky relevantných koncentráciách.
Účinok linaklotidu a jeho metabolitov inhibovať bežné črevné enzýmy (CYP2C9 a CYP3A4)
a enzýmy pečene (CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 a 3A4) alebo indukovať enzýmy pečene
(CYP1A2, 2B6 a 3A4/5) sa zisťoval in vitro. Výsledky týchto štúdií ukázali, že linaklotid ani metabolit dez-tyrozín nie sú inhibítory ani induktory enzýmového systému cytochrómu P450.
Eliminácia
Po perorálnom podaní jednorazovej dávky 2 897 mikrogramov linaklotidu na 8. deň po 7-dňovom
období podávania 290 mikrogramov denne 18 zdravým dobrovoľníkom sa približne 3 až 5 % dávky
vylúčilo v stolici, prakticky celé množstvo vo forme aktívneho metabolitu dez-tyrozínu.
Vek a pohlavie
Klinické štúdie na stanovenie vplyvu veku a pohlavia na klinickú farmakokinetiku linaklotidu sa nevykonali, pretože linaklotid je zriedkavo detekovateľný v plazme. Nepredpokladá sa, že pohlavie
má nejaký vplyv na dávkovanie. Informácie vzťahujúce sa na vek pozrite v častiach 4.2, 4.4 a 4.8.
Porucha funkcieobličiek
Constella sa neskúmala u pacientov, ktorí majú poruchu funkcie obličiek. Linaklotid je zriedkavo detekovateľný v plazme, preto sa nepredpokladá, že porucha funkcie obličiek môže ovplyvniť klírens
základnej zlúčeniny alebo jej metabolitu.
Porucha funkciepečene
Constella sa neskúmala u pacientov, ktorí majú poruchu funkcie pečene. Linaklotid je zriedkavo detekovateľný v plazme a nie je metabolizovaný enzýmami cytochrómu P450 pečene, a preto sa nepredpokladá, že porucha funkcie pečene môže ovplyvniť metabolizmus alebo klírens základného liečiva alebo jeho metabolitu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, karcinogénneho potenciálu, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsuly
Mikrokryštalická celulóza
Hypromelóza 4-6 mPa´s – substitučný typ 2910
Dihydrát chloridu vápenatého
Leucín
Obal kapsuly
Oxid titaničitý (E171)
Želatína
Červený oxid železitý (E172) Žltý oxid železitý (E172)
A
tr
ament kapsuly Šelak Propylénglykol
Koncentrovaný roztok amoniaku
Hydroxid draselný
Oxid titaničitý (E171) Čierny oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
Neotvorená fľaška fľaška pre 28, 90 kapsúl a multibalenie s viacerými samostatnými baleniami, ktoré
obsahuje 112 kapsúl (4 balenia po 28 kapsúl): 3 roky.
Neotvorená fľaška pre 10 kapsúl: 24 mesiacov. Po prvom otvorení: 18 týždňov.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 ºC. Fľašku uchovávajte dôkladne uzatvorenú na ochranu
pred vlhkosťou.
Fľaška obsahuje jednu alebo viaceré utesnené nádobky obsahujúce silikagél na uchovanie kapsúl v suchu. Nádobky nechajte vo fľaške.
6.5 Druh obalu a obsah balenia
Biela fľaška z polyetylénu s vysokou hustotou (HDPE) s poistným tesnením a s uzáverom bezpečným
pred deťmi, spolu s jednou alebo viacerými nádobkami s desikantom obsahujúcimi silikagél.
Veľkosti balenia: 10, 28 alebo 90 kapsúl a multibalenia obsahujúce 112 (4 balenia po 28) kapsúl. Na
trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Allergan Pharmaceuticals International Limited
Clonshaugh Industrial Estate
Coolock
Dublin 17
Írsko
8. REGISTRAČNÉ ČÍSLA
EU/1/12/801/001
EU/1/12/801/002
EU/1/12/801/004
EU/1/12/801/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 26. novembra 2012
Dátum posledného predĺženia registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.