
L
i
kv
i
dace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
BioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.de8. REGISTRAČNÍ ČÍSLO/REGISTRAČNÍ ČÍSLAEU/1/20/1528/001
9. DATUM REGISTRACE/PRODLOUŽENÍ REGISTRACEDatum první registrace: 21. prosince 2020
Datum posledního prodloužení registrace: 10. října 2022
10. DATUM REVIZE TEXTUPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky.
Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKUComirnaty 30 mikrogramů/dávku injekční disperze
mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍJedná se o jednodávkovou nebo vícedávkovou injekční lahvičku s šedým víčkem. Před použitím neřeďte.
Jedna jednodávková injekční lahvička obsahuje 1 dávku 0,3 ml, viz body 4.2 a 6.6.
Jedna vícedávková injekční lahvička (2,25 ml) obsahuje 6 dávek po 0,3 ml, viz body 4.2 a 6.6. Jedna dávka (0,3 ml) obsahuje 30 mikrogramů tozinameranu, mRNA vakcíny proti onemocnění
COVID-19 (zapouzdřené v lipidových nanočásticích).
Tozinameran je jednovláknová mediátorová (messenger) RNA (mRNA) s čepičkou na 5´ konci vyráběná
in vitro nebuněčnou transkripcí z příslušných matricí DNA a kódující spike (S) protein viru SARS-CoV-2.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMAInjekční disperze.
Vakcína je bílá až téměř bílá zmrazená disperze (pH: 6,9–7,9).
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikaceVakcína Comirnaty 30 mikrogramů/dávku injekční disperze je indikována pro aktivní imunizaci
k prevenci onemocnění COVID-19 způsobeného SARS-CoV-2 u osob ve věku 12 let a starších.
Tuto vakcínu je třeba používat v souladu s oficiálními doporučeními.
4.2 Dávkování a způsob podáníDávkováníZákladníočkováníOsoby ve věku 12 let a staršíVakcína Comirnaty se podává intramuskulárně jako základní očkování 2 dávkami (0,3 ml každá
dávka). Druhou dávku se doporučuje podat 3 týdny po první dávce (viz body 4.4 a 5.1).
Těžce imunokompromitované osoby ve věku 12 let a starší
Třetí dávka základního očkování může být podána intramuskulárně nejméně 28 dní po druhé dávce
jedincům, kteří jsou těžce imunokompromitovaní (viz bod 4.4).
Zaměnitelnost
Nebylo stanoveno, zda lze za účelem dokončení základního očkování zaměnit vakcínu Comirnaty za vakcíny proti onemocnění COVID-19 od jiných výrobců. Osoby, jimž byla podána dávka vakcíny Comirnaty, mají pokračovat v podávání vakcíny Comirnaty, aby bylo základní očkování dokončeno.
Dávky vakcíny Comirnaty 30 mikrogramů/dávku koncentrátu pro injekční disperzi po naředění (dodávané v injekční lahvičce s fialovým víčkem) a vakcíny Comirnaty 30 mikrogramů/dávku injekční disperze (dodávané v injekční lahvičce s šedým víčkem) jsou považovány za zaměnitelné.
Posilovací dávka
Posilovací dávka vakcíny Comirnaty 0,3 ml se podává intramuskulárně.
Posilovací dávka může být podána jedincům ve věku 12 let a starším. Mezi podáním vakcíny
Comirnaty a poslední předchozí dávkou vakcíny proti onemocnění COVID-19 má být interval alespoň
3 měsíce.
Pediatrická populace
K dispozici je pediatrická léková forma pro jedince ve věku od 6 měsíců do 4 let. Podrobné informace
naleznete v souhrnu údajů o přípravku pro vakcínu Comirnaty 3 mikrogramy/dávku koncentrát pro
injekční disperzi.
K dispozici je pediatrická léková forma pro jedince ve věku od 5 do 11 let (tj. od 5 do méně než
12 let). Podrobné informace naleznete v souhrnu údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi.
Bezpečnost a účinnost vakcíny Comirnaty u kojenců ve věku do 6 měsíců nebyly dosud stanoveny.
Starší populace
U osob ve věku ≥ 65 let není nutná žádná úprava dávkování.
Způsob podání
Vakcína Comirnaty 30 mikrogramů/dávku injekční disperze se podává intramuskulárně (viz bod 6.6).
Před použitím neřeďte.
Preferované místo je deltový sval horní části paže.
Vakcína se nesmí podávat intravaskulárně, subkutánně ani intradermálně.
Vakcína se nesmí mísit ve stejné injekční stříkačce s jinými vakcínami nebo léčivými přípravky. Pro opatření před podáním vakcíny viz bod 4.4.
Návod pro rozmrazení, zacházení s vakcínou a její likvidaci je uveden v bodě 6.6.
Jednodávkové injekčnílahvičky
Jednodávkové injekční lahvičky vakcíny Comirnaty obsahují 1 dávku vakcíny 0,3 ml.
• Natáhněte jednu 0,3ml dávku vakcíny Comirnaty.
• Injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Nekombinujte obsah z více injekčních lahviček vakcíny.
Vícedávkové injekčnílahvičky
Vícedávkové injekční lahvičky vakcíny Comirnaty obsahují 6 dávek po 0,3 ml vakcíny. K získání
6 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů. Pokud se používají standardní injekční stříkačky a jehly, nemusí být dostatečný objem k získání šesté dávky z jedné injekční lahvičky. Bez ohledu na typ injekční stříkačky a jehly:
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml,
injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Nekombinujte obsah z více injekčních lahviček vakcíny.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Sledovatelnost
Aby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Obecná doporučení
Hypersenzitivita a anafylaxe
Byly hlášeny případy anafylaxe. Pro případ, že by po podání vakcíny došlo k anafylaktické reakci, má
být zajištěna okamžitá lékařská péče a dohled.
Po vakcinaci se doporučuje pečlivé sledování po dobu minimálně 15 minut. Další dávka vakcíny nemá být podána osobám, které měly anafylaxi po předchozí dávce vakcíny Comirnaty.
Myokarditida a perikarditida
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy a perikarditidy. Tato
onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhé dávce vakcíny a častěji u mladších mužů a chlapců.
Dostupné údaje naznačují, že průběh myokarditidy a perikarditidy po vakcinaci se neliší od
myokarditidy nebo perikarditidy obecně (viz bod 4.8).
Zdravotničtí pracovníci mají pozorně sledovat známky a příznaky myokarditidy a perikarditidy. Očkovaní jedinci (včetně rodičů nebo pečovatelů) mají být poučeni, aby okamžitě vyhledali lékařskou pomoc, pokud se u nich po očkování objeví příznaky naznačující myokarditidu nebo perikarditidu, například bolest na hrudi (akutní a přetrvávající), dušnost nebo palpitace.
Zdravotničtí pracovníci mají k diagnostice a léčbě tohoto onemocnění používat návody a postupy
a/nebo se mají obrátit na specialisty.
Reakce spojené s úzkostí
V souvislosti se samotným procesem očkování se mohou objevit reakce spojené s úzkostí, včetně vazovagálních reakcí (synkopa), hyperventilace nebo reakcí spojených se stresem (např. závrať, palpitace, zvýšení srdeční frekvence, změny krevního tlaku, parestezie, hypestezie a pocení). Reakce spojené se stresem jsou dočasné a samy se upraví. Očkované osoby je třeba informovat o tom, aby na případné symptomy upozornily očkujícího zdravotníka, který je vyhodnotí. Je důležité, aby byla zavedena opatření, aby se zabránilo zranění v důsledku mdlob.
Současné onemocnění
U osob trpících závažným akutním horečnatým onemocněním nebo akutní infekcí se má podání
vakcíny Comirnaty odložit. Přítomnost mírné infekce a/nebo horečky nízkého stupně není důvod
k odložení vakcinace.
Trombocytopenie a poruchy koagulace
Stejně jako u jiných intramuskulárních injekcí je třeba vakcínu podávat opatrně osobám podstupujícím
léčbu antikoagulancii nebo osobám s trombocytopenií nebo poruchami koagulace (jako je hemofilie),
protože po intramuskulárním podání může u těchto osob dojít ke krvácení nebo tvorbě modřin.
Imunokompromitované osoby
Účinnost a bezpečnost vakcíny nebyly hodnoceny u imunokompromitovaných osob, včetně osob
podstupujících imunosupresivní léčbu. Účinnost vakcíny Comirnaty může být u
imunokompromitovaných osob nižší.
Doporučení zvážit podání třetí dávky u těžce imunokompromitovaných jedinců je založeno na omezených sérologických důkazech z případových studií z literatury z klinické léčby pacientů s iatrogenní imunosupresí po transplantaci solidních orgánů (viz bod 4.2).
Doba ochrany
Doba ochrany poskytovaná vakcínou není známa, protože je stále hodnocena v probíhajících
klinických studiích.
Omezení účinnosti vakcíny
Podobně jako u jiných vakcín je možné, že vakcinace vakcínou Comirnaty nebude chránit všechny její
příjemce. Osoby nemusí být plně chráněny po dobu 7 dnů po druhé dávce vakcíny.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Současné podání vakcíny Comirnaty s jinými vakcínami nebylo hodnoceno.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Velké množství observačních dat od těhotných žen očkovaných vakcínou Comirnaty během druhého a
třetího trimestru neprokázalo zvýšení nežádoucích výsledků těhotenství. Ačkoli údaje o výsledcích těhotenství po očkování během prvního trimestru jsou v současné době omezené, nebylo pozorováno zvýšené riziko potratu. Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na
těhotenství, vývoj embrya/plodu, porod nebo postnatální vývoj (viz bod 5.3). Vakcínu Comirnaty lze v těhotenství podávat.
Kojení
Systémová expozice vakcíně Comirnaty je u kojící matky zanedbatelná, a proto nejsou očekávány
žádné účinky na kojeného novorozence/kojence (skrze mateřské mléko). Observační údaje od žen, které po očkování kojily, neprokázaly riziko nežádoucích účinků u kojených novorozenců/kojenců.
Vakcínu Comirnaty lze během kojení podávat.
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky s ohledem na reprodukční toxicitu
(viz bod 5.3)
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vakcína Comirnaty nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Některé z účinků uvedených v bodě 4.8 však mohou schopnost řídit nebo obsluhovat stroje dočasně ovlivnit.
4.8 Nežádoucí účinky
Souhrn bezpečnostníhoprofilu
Účastnícivevěku16let a starší – po 2 dávkách
Ve studii 2 byla celkem 22 026 účastníkům ve věku 16 let nebo starším podána alespoň 1 dávka
vakcíny Comirnaty a celkem 22 021 účastníkům ve věku 16 let nebo starším bylo podáno placebo
(včetně 138 a 145 dospívajících ve věku 16 a 17 let ve skupinách vakcíny a placeba, v uvedeném
pořadí). Celkem 20 519 účastníků ve věku 16 let nebo starších dostalo 2 dávky vakcíny Comirnaty.
V době provedení analýzy studie 2 s datem ukončení sběru dat 13. března 2021 pro placebem kontrolované zaslepené období sledování až do dat odslepení účastníků bylo celkem 25 651 (58,2 %) účastníků (13 031 Comirnaty a 12 620 placebo) ve věku 16 let a starších sledováno po dobu ≥4 měsíce po podání druhé dávky. To zahrnovalo celkem 15 111 (7 704 Comirnaty a 7 407 placebo) účastníků ve věku 16 až 55 let a celkem 10 540 (5 327 Comirnaty a 5 213 placebo) účastníků ve věku 56 let a starších.
Nejčastějšími nežádoucími účinky byla u účastníků ve věku 16 let a starších, kteří dostali 2 dávky, bolest v místě injekce (> 80 %), únava (> 60 %), bolest hlavy (> 50 %), myalgie (> 40 %), zimnice
(> 30 %), artralgie (> 20 %), pyrexie a zduření v místě injekce (> 10 %). Tyto nežádoucí účinky byly
zpravidla mírné nebo střední intenzity a odezněly během několika dní po vakcinaci. Mírně nižší frekvence příhod reaktogenity souvisela s vyšším věkem.
Bezpečnostní profil u 545 účastníků ve věku 16 let a starších, kterým byla podána vakcína Comirnaty
a kteří byli séropozitivní na SARS-CoV-2 při výchozím stavu, byl podobný jako u obecné populace.
Dospívající ve věku 12až15let – po 2 dávkách
V analýze dlouhodobého následného sledování bezpečnosti ve studii 2 bylo 2 260 dospívajících (1 131
dostalo vakcínu Comirnaty a 1 129 dostalo placebo) ve věku 12 až 15 let. Z toho 1 559 dospívajících
(786 dostalo vakcínu Comirnaty a 773 dostalo placebo) bylo sledováno po dobu ≥ 4 měsíců po druhé dávce vakcíny Comirnaty. Hodnocení bezpečnosti ve studii 2 probíhá.
Celkový bezpečnostní profil vakcíny Comirnaty u dospívajících ve věku 12 až 15 let byl podobný jako
u účastníků ve věku 16 let a starších. Nejčastějšími nežádoucími účinky u dospívajících ve věku 12 až
15 let, kteří dostali 2 dávky, byly bolest v místě vpichu (> 90 %), únava a bolest hlavy (> 70 %), myalgie a zimnice (> 40 %), artralgie a pyrexie (> 20 %).
Účastníci ve věku 12 let a starší – po posilovací dávce
Bezpečnost posilovací dávky vakcíny Comirnaty u účastníků ve věku 12 let a starších je odvozena od
údajů o bezpečnosti ze studií posilovací dávky vakcíny Comirnaty u účastníků ve věku 16 let a starších.
Podskupina účastníků studie fáze 2/3, do které bylo zařazeno 306 dospělých ve věku 18 až 55 let, kteří dokončili původní cyklus 2 dávek vakcíny Comirnaty, dostala posilovací dávku vakcíny Comirnaty přibližně 6 měsíců (rozmezí 4,8 až 8,0 měsíců) po podání 2. dávky.
Celkový bezpečnostní profil posilovací dávky byl podobný jako po 2 dávkách. Nejčastějšími nežádoucími účinky u účastníků ve věku 18 až 55 let byly bolest v místě vpichu (> 80 %), únava (>
60 %), bolest hlavy (> 40 %), myalgie (> 30 %), zimnice a artralgie (> 20 %).
Ve studii 4, placebem kontrolované studii posilovací dávky, byla účastníkům ve věku 16 let a starším
zařazeným ze studie 2 podána posilovací dávka vakcíny Comirnaty (5 081 účastníků) nebo placebo (5 044 účastníků), a to nejméně 6 měsíců po druhé dávce vakcíny Comirnaty. Medián doby následného sledování účastníků, kterým byla posilovací dávka podána, činil celkem 2,5 měsíce po podání posilovací dávky do dne ukončení sběru dat (5. října 2021). Žádné nové nežádoucí účinky vakcíny Comirnaty nebyly zjištěny.
Účastníci ve věku 12 let a starší – po následných posilovacích dávkách
Bezpečnost posilovací dávky vakcíny Comirnaty u účastníků ve věku 12 let a starších je odvozena od
údajů o bezpečnosti ze studií posilovací dávky vakcíny Comirnaty u účastníků ve věku 18 let a starších.
Podmnožina 325 dospělých ve věku 18 až ≤ 55 let, kteří dokončili očkování třemi dávkami vakcíny
Comirnaty, obdržela posilovací dávku (čtvrtou dávku) vakcíny Comirnaty 90 až 180 dnů po podání
3. dávky. Účastníci, kteří obdrželi posilovací dávku (čtvrtou dávku) vakcíny Comirnaty, měli medián sledování do dne ukončení sběru dat 11. března 2022 nejméně 1,4 měsíce. Nejčastějšími nežádoucími účinky u těchto účastníků byly bolest v místě vpichu (> 70 %), únava (> 60 %), bolest hlavy (> 40 %), myalgie a zimnice (> 20 %) a artralgie (>10 %).
V podmnožině ze studie 4 (3. fáze) obdrželo 305 dospělých ve věku > 55 let, kteří dokončili očkování třemi dávkami vakcíny Comirnaty, posilovací dávku (čtvrtou dávku) vakcíny Comirnaty 5 až
12 měsíců po podání 3. dávky. Účastníci, kteří obdrželi posilovací dávku (čtvrtou dávku) vakcíny
Comirnaty, měli medián sledování do dne ukončení sběru dat 16. května 2022 nejméně 1,7 měsíce. Celkový bezpečnostní profil posilovací dávky (čtvrté dávky) vakcíny Comirnaty byl podobný profilu zjištěnému po posilovací dávce (třetí dávce) vakcíny Comirnaty. U účastníků ve věku vyšším než
55 let byly nejčastějšími nežádoucími účinky bolest v místě vpichu (> 50 %), únava (> 40 %), bolest hlavy (> 20 %), myalgie a zimnice (> 10 %).
Posilovací dávka poprimárnívakcinacijinouschválenouvakcínouprotionemocněníCOVID-19
V 5 nezávislých studiích o použití posilovací dávky vakcíny Comirnaty u osob, které absolvovaly
primární vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19 (heterologní posilovací
dávka), nebyly zjištěny žádné nové bezpečnostní problémy (viz bod 5.1).
T
abulkový
s
eznam
nežádoucích
účinků
z
klinických studií a po uvedení
na
trh
u
osob
ve
věku
12
let a
starších
Nežádoucí účinky pozorované z klinických studií jsou uvedeny níže podle následujících kategorií
frekvence:
Velmi časté (≥ 1/10)
Časté (≥ 1/100 až < 1/10)
Méně časté (≥ 1/1 000 až < 1/100) Vzácné (≥ 1/10 000 až < 1/1 000) Velmi vzácné (< 1/10 000)
Není známo (z dostupných údajů nelze určit)
T
abulka 1: Nežádoucí účinky vakcíny Comirnaty z klinických studií a po uvedení na trh u
osob ve věku 12 let a starších
T
řída
orgánového systému
|
V
elmi
časté
(
≥ 1/10)
|
Č
asté
(
≥ 1/100
až
< 1/10)
|
Méně časté
(
≥ 1/1 000 až
< 1/100)
|
V
z
ácné
(
≥ 1/10 000
až
< 1/1 000)
|
V
elmi vzácné
(
< 1/10 000
|
N
ení
z
námo (z dostupnýc h údajů
nelze určit)
|
Poruchy krve a
lymfatického systému
|
|
|
Lymfadenopatiea
|
|
|
|
Poruchy
imunitního systému
|
|
|
Hypersenzitivní
reakce (např.
vyrážka, pruritus, urtikarieb, angioedémb)
|
|
|
Anafylaxe
|
Poruchy
metabolismu a výživy
|
|
|
Snížená chuť k
jídlu
|
|
|
|
Psychiatrické
poruchy
|
|
|
Insomnie
|
|
|
|
Poruchy
nervového systému
|
Bolest
hlavy
|
|
Letargie
|
Akutní
periferní paralýza n. facialisc
|
|
Parestezied;
hypestezied
|
Srdeční poruchy
|
|
|
|
|
Myokarditidad;
Perikarditidad
|
|
Gastrointestinální
poruchy
|
Průjemd
|
Nauzea;
zvraceníd
|
|
|
|
|
Poruchy kůže a
podkožní tkáně
|
|
|
Hyperhidróza;
noční pocení
|
|
|
Erythema
multiformed
|
Poruchy svalové
a kosterní soustavy a
pojivové tkáně
|
Artralgie;
myalgie
|
|
Bolest
v končetiněe
|
|
|
|
Poruchy
reprodukčního
systému a prsu
|
|
|
|
|
|
Silné
menstruační
krváceníh
|
Celkové poruchy
a reakce v místě
aplikace
|
Bolest v
místě
injekce; únava; zimnice; pyrexief; zduření v místě injekce
|
Zarudnutí
v místě
injekce
|
Astenie;
malátnost;
svědění v místě
injekce
|
|
|
Rozsáhlý
otok
končetiny, do níž byla vakcína podánad; otok obličejeg
|
a. U účastníků, kteří dostali posilovací dávku ve studii 4, byla pozorována vyšší frekvence lymfadenopatie
(2,8 % oproti 0,4 %) ve srovnání s účastníky, kteří dostali dvě dávky.
b. Kategorie frekvence pro urtikarii a angioedém byla vzácná.
c. Během období sledování bezpečnosti v klinické studii do 14. listopadu 2020 byla hlášena akutní periferní paralýza
n. facialis u čtyř účastníků ve skupině mRNA vakcíny proti onemocnění COVID-19. Nástup obrny
obličeje byl 37. den po podání 1. dávky (účastník nedostal 2. dávku) a 3., 9. a 48. den po 2. dávce. Ve
skupině s placebem nebyly hlášeny žádné případy akutní periferní paralýzy
n. facialis. d. Nežádoucí účinek byl stanoven po registraci.
e. Týká se končetiny/paže, do které byla daná osoba očkována.
f. Vyšší frekvence pyrexie byla pozorována po druhé dávce v porovnání s první dávkou.
g. Po uvedení přípravku na trh byly hlášeny případy otoku obličeje u očkovaných osob, které v minulosti
podstoupily injekční aplikaci dermálních výplní do obličeje.
h. Většina případů se zdála být nezávažné a dočasné povahy.
Popis vybraných nežádoucíchúčinkůMyokarditida a perikarditidaZvýšené riziko myokarditidy po očkování vakcínou Comirnaty je nejvyšší u mladších mužů a chlapců
(viz bod 4.4).
Zvýšené riziko u mladších mužů a chlapců po podání druhé dávky vakcíny Comirnaty bylo blíže určeno ve dvou velkých evropských farmakoepidemiologických studiích. Z jedné studie vyplynulo, že v období 7 dnů po podání druhé dávky se u mužů a chlapců ve věku 12–29 let vyskytlo přibližně o
0,265 (95% CI 0,255–0,275) případů myokarditidy na 10 000 osob více než u neočkovaných osob. V
další studii se v období 28 dnů po podání druhé dávky u mužů a chlapců ve věku 16–24 let vyskytlo o
0,56 (95% CI 0,37–0,74) případů myokarditidy na 10 000 osob více než u neočkovaných osob.
Omezené údaje ukazují, že riziko myokarditidy a perikarditidy je po očkování vakcínou Comirnaty
u dětí ve věku od 5 do 11 let pravděpodobně nižší než u dospívajících ve věku od 12 do 17 let.
Hlášení podezření na nežádoucí účinkyHlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to

pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V a uvedli přitom číslo šarže, je-li k dispozici.
4.9 PředávkováníÚdaje o předávkování jsou k dispozici od 52 účastníků studie zařazených do klinického hodnocení, kterým bylo kvůli chybě v ředění podáno 58 mikrogramů vakcíny Comirnaty. Příjemci vakcíny nehlásili zvýšení reaktogenity ani nežádoucí účinky.
V případě předávkování se doporučuje sledovat základní životní funkce a případně zahájit symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: vakcíny, jiné virové vakcíny, ATC kód: J07BX03
Mechanismus účinkuMediátorová (messenger) RNA s modifikovanými nukleosidy je ve vakcíně Comirnaty zapouzdřená
v lipidových nanočásticích, což umožňuje transfer nereplikující RNA do hostitelských buněk pro přímou přechodnou expresi S antigenu viru SARS-CoV-2. mRNA kóduje v membráně ukotvený S v
plné délce se dvěma bodovými mutacemi v centrální šroubovici. Mutace těchto dvou aminokyselin na prolin uzamyká S v antigenně preferované prefuzní konformaci. Vakcína vyvolává jak odpověď
neutralizačních protilátek, tak i imunitní odpověď buněk na spike (S) antigen, což může přispívat
k ochraně před onemocněním COVID-19.
Ú
činnost
Studie 2 je multicentrická, mezinárodní, randomizovaná, placebem kontrolovaná studie fáze 1/2/3
zaslepená pro pozorovatele, která hodnotí dávku, výběr vakcíny a její účinnost u účastníků ve věku
12 let a starších. Randomizace byla stratifikována podle věku: věk 12 až 15 let, věk 16 až 55 let a věk
56 let a více; do věkové skupiny ≥ 56 let spadalo minimálně 40 % účastníků. Ze studie byli vyřazeni imunokompromitovaní účastníci a osoby, u nichž byla dříve stanovena klinická či mikrobiologická
diagnóza onemocnění COVID-19. Účastníci s preexistujícími stabilizovanými onemocněními, definovanými jako onemocnění, jež během 6 týdnů před zařazením nevyžadovala významnou změnu
léčby nebo hospitalizaci z důvodu zhoršení nemoci, byli do studie zařazeni stejně jako účastníci se známou stabilizovanou infekcí virem lidské imunodeficience (HIV), virem hepatitidy C (HCV) nebo virem hepatitidy B (HBV).
Účinnost u osobvevěku16let a starších – po 2 dávkách
Během fáze 2/3 Studie 2 bylo na základě dat získaných do 14. listopadu 2020 ve stejném poměru
randomizováno zhruba 44 000 účastníků a byly jim podány 2 dávky mRNA vakcíny proti onemocnění
COVID-19 nebo placeba. Analýzy účinnosti zahrnovaly účastníky, kteří dostali druhou dávku vakcíny v rozmezí 19 až 42 dnů po první dávce vakcíny. Většina (93,1 %) příjemců vakcíny dostala druhou dávku 19 dnů až 23 dnů po první dávce. Pro účely hodnocení bezpečnosti a účinnosti vakcíny proti onemocnění COVID-19 budou na základě připraveného plánu účastníci dále sledováni po dobu až
24 měsíců po 2. dávce. V klinické studii bylo od účastníků vyžadováno dodržení minimálního
intervalu 14 dní před a po podání vakcíny proti chřipce, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19. V klinické studii bylo od účastníků vyžadováno dodržení minimálního intervalu 60 dní před nebo po podání přípravků z krve/plazmy nebo imunoglobulinů v průběhu studie, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19.
Do populace pro analýzu primárního cílového parametru účinnosti bylo zařazeno 36 621 účastníků ve věku 12 let a starších (18 242 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 18 379 ve skupině s placebem), u nichž se během 7 dní po podání druhé dávky neprokázala předchozí infekce SARS-CoV-2. Kromě toho bylo 134 účastníků ve věku od 16 do 17 let (66 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 68 ve skupině s placebem) a 1 616 účastníků ve věku 75 let a starších (804 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 812 ve skupině s placebem).
V čase provedení primární analýzy účinnosti byli účastníci ve skupině mRNA vakcíny proti onemocnění COVID-19 sledováni z hlediska symptomatického onemocnění COVID-19 po celkovou dobu 2 214 osoboroků, účastníci ve skupině s placebem po dobu minimálně 2 222 osoboroků.
U účastníků s rizikem závažného onemocnění COVID-19, včetně osob s 1 nebo více komorbiditami,
které riziko závažného onemocnění COVID-19 zvyšují (např. astma, index tělesné hmotnosti (BMI)
≥ 30 kg/m2, chronické plicní onemocnění, diabetes mellitus, hypertenze), nebyly zjištěny žádné
významné klinické rozdíly v celkové účinnosti vakcíny.
Informace o účinnosti vakcíny jsou uvedeny v tabulce 2.
Tabulka 2: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů)První výskyt COVID-19 od 7 dnů po 2. dávce u účastníků bez důkazu předchozí infekce SARS- CoV-2 *
|
Podskupina
| Vakcína mRNA proti
onemocnění COVID-19
na=18 198
Případy
n1b
Doba sledováníc (n2d)
|
Placebo na=18 325
Případy
n1b
Doba sledováníc (n2d)
|
Účinnost vakcíny % (95% CI)e
|
Všichni účastníci
| 8
2,214 (17 411)
| 162
2,222 (17 511)
| 95,0
(90,0; 97,9)
|
16 až 64 let
| 7
1,706 (13 549)
| 143
1,710 (13 618)
| 95,1
(89,6; 98,1)
|
65 let a starší
| 1
0,508 (3 848)
| 19
0,511 (3 880)
| 94,7
(66,7; 99,9)
|
65 až 75 let
| 1
0,406 (3 074)
| 14
0,406 (3 095)
| 92,9
(53,1; 99,8)
|
75 let a starší
| 0
0,102 (774)
| 5
0,106 (785)
| 100,0
(-13,1; 100,0)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 [*Definice případu: (alespoň 1 z) horečka, nový nebo zhoršený kašel, nová nebo zhoršená dyspnoe, zimnice, nová nebo zhoršená bolest svalů, nová ztráta chuti nebo čichu, bolest v krku, průjem nebo zvracení.]
* Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (7 nebo více dnů před podáním poslední dávky) předcházející infekce SARS-CoV-2 (tj. N-vazebná protilátka [sérum] negativní při návštěvě 1 a SARS- CoV-2 nedetekován pomocí amplifikačních testů nukleových kyselin (NAAT) [výtěr nosu] při návštěvách 1 a 2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do analýzy.
a. n = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osobo-rocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy metody upravené podle doby sledování. CI není upraven na multiplicitu.
Účinnost vakcíny mRNA proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění COVID-19 po 7 dnech po 2. dávce ve srovnání s placebem 94,6 % (95% interval spolehlivosti 89,6 % až 97,6 %) u účastníků ve věku 16 let a starších s nebo bez důkazů o předchozí infekci virem SARS- CoV-2.
Analýzy podskupin primárního cílového parametru účinnosti navíc ukázaly podobné odhady bodů účinnosti u pohlaví, etnických skupin a účastníků s komorbiditami spojenými s vysokým rizikem závažného onemocnění COVID-19.
Byly provedeny aktualizované analýzy účinnosti s dalšími potvrzenými případy COVID-19, které se objevily během zaslepeného placebem kontrolovaného sledování, což v populaci s účinností představuje až 6 měsíců po podání 2. dávky.
Aktualizované informace o účinnosti vakcíny jsou uvedeny v tabulce 3.
Podskupina
|
V
akcína mRNA proti
onemocnění COVID-19
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Placebo
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Ú
č
i
n
nost vakcíny % (95% CI
e
)
|
Všichni účastnícif
|
77
6,247 (20 712)
|
850
6,003 (20 713)
|
91,3
(89,0; 93,2)
|
16 až 64 let
|
70
4,859 (15 519)
|
710
4,654 (15 515)
|
90,6
(87,9; 92,7)
|
65 let a starší
|
7
1,233 (4 192)
|
124
1,202 (4 226)
|
94,5
(88,3; 97,8)
|
65 až 74 let
|
6
0,994 (3 350)
|
98
0,966 (3 379)
|
94,1
(86,6; 97,9)
|
75 let a starší
|
1
0,239 (842)
|
26
0,237 (847)
|
96,2
(76,9; 99,9)
|
|
|
T
abulka 3: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce předchozí infekce virem SARS- CoV-2* před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů) během placebem kontrolovaného období sledování
N =20 998
N =21 096
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení).
* Účastníci, kteří neměli žádné důkazy předcházející infekce virem SARS-CoV-2 (tj. N-vazebná protilátka
[sérum] negativní při návštěvě 1 a virus SARS-CoV-2 nedetekován pomocí NAAT [výtěr nosu] při návštěvách 1 a
2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do
analýzy.
a. N = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný 95% interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a
Pearsonovy metody upravené podle doby sledování. CI není upraven na multiplicitu.
f. Zahrnuté potvrzené případy u účastníků ve věku 12 až 15 let: 0 ve skupině s mRNA vakcínou proti
onemocnění COVID-19; 16 ve skupině s placebem.
V aktualizované analýze účinnosti byla účinnost mRNA vakcíny proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění COVID-19 od 7 dnů po podání 2. dávky ve srovnání s placebem 91,1 % (95% CI 88,8 % až 93,0 %) u účastníků v hodnotitelné populaci účinnosti s průkazem nebo bez průkazu předchozí infekce SARS-CoV-2.
Aktualizované analýzy účinnosti podle podskupin navíc ukázaly podobné bodové odhady účinnosti u všech pohlaví, etnických skupin, zeměpisných oblastí a účastníků se zdravotními komorbiditami a obezitou spojenou s vysokým rizikem závažného onemocnění COVID-19.
Účinnost proti závažnému onemocnění COVID-19
Aktualizované analýzy účinnosti sekundárních cílových parametrů účinnosti podpořily přínos mRNA
vakcíny proti onemocnění COVID-19 v prevenci závažného onemocnění COVID-19.
Od 13. března 2021 je účinnost vakcíny proti závažnému onemocnění COVID-19 prezentována pouze pro účastníky s předchozí infekcí SARS-CoV-2 nebo bez ní (tabulka 4), protože počty případů onemocnění COVID-19 u účastníků bez předchozí infekce SARS-CoV-2 byly stejné jako u účastníků
s předchozí infekcí SARS-CoV-2 nebo bez ní jak ve skupině s mRNA vakcínou proti onemocnění
COVID-19, tak ve skupině s placebem.
| Vakcína mRNA proti
onemocnění COVID-19
Případy
n1a
Doba sledováníc (n2b)
|
Placebo Případy n1a
Doba sledováníc (n2b)
|
Účinnost vakcíny % (95% CIc)
| Po 1. dávced
| 1
8,439e (22 505)
| 30
8,288e (22 435)
| 96,7
(80,3; 99,9)
| 7 dnů po 2. dávcef
| 1
6,522g (21 649)
| 21
6,404g (21 730)
| 95,3
(70,9; 99,9)
|
|
|
Tabulka 4: Účinnost vakcíny – první závažný výskyt onemocnění COVID-19 u účastníků s nebo bez předchozí infekce virem SARS-CoV-2 na základě údajů Úřadu pro kontrolu potravin a léčiv (FDA)* po podání 1. dávky nebo od 7 dnů po podání 2. dávky v placebem kontrolovaném období sledováníPoznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení.)
* Závažné onemocnění COVID-19 podle definice FDA je potvrzené onemocnění COVID-19 a přítomnost alespoň 1 z následujících příznaků:
• Klinické příznaky v klidu svědčící pro závažné systémové onemocnění (dechová frekvence ≥ 30 dechů za minutu, srdeční frekvence ≥ 125 tepů za minutu, saturace kyslíkem ≤ 93 % na pokojovém vzduchu při hladině moře nebo poměr arteriálního parciálního tlaku kyslíku k frakčnímu inspirovanému kyslíku
< 300 mmHg).
• Respirační selhání [definované jako potřeba vysokého průtoku kyslíku, neinvazivní ventilace,
mechanické ventilace nebo extrakorporální membránové oxygenace (ECMO)].
• Průkaz šoku (systolický krevní tlak < 90 mmHg, diastolický krevní tlak < 60 mmHg nebo potřeba
podání vazopresorických látek).
• Významná akutní renální, jaterní nebo neurologická dysfunkce.
• Přijetí na jednotku intenzivní péče.
• Smrt.
a. n1 = Počet účastníků splňujících definici cílového parametru.
b. n2 = počet účastníků ohrožených daným cílovým parametrem.
c. Dvoustranný interval spolehlivosti (CI) pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování.
d. Účinnost hodnocená na základě veškeré populace pro hodnocení účinnosti s 1. dávkou (modifikovaná se záměrem léčit), která zahrnovala všechny randomizované účastníky, kteří dostali alespoň 1 dávku hodnocené léčby.
e. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od po 1. dávce do konce období sledování.
f. Účinnost hodnocená na základě populace hodnotitelné pro účinnost (7 dní), která zahrnovala všechny způsobilé randomizované účastníky, kteří dostali všechny dávky hodnocené léčby podle randomizace v rámci předem stanoveného časového období a neměli žádné další důležité odchylky od protokolu podle rozhodnutí lékaře.
g. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro daný cílový parametr. Časové období pro získání případu onemocnění COVID-19 je od
7 dnů po 2. dávce do konce období sledování.
Účinnost a imunogenicita udospívajícíchvevěku12až15let – po 2 dávkáchV úvodní analýze Studie 2 u dospívajících ve věku 12 až 15 let (jež reprezentovala medián doby
následného sledování v délce ˃ 2 měsíce po 2. dávce) bez průkazu prodělané infekce nebyly žádné případy u 1 005 účastníků, kteří dostali vakcínu, a 16 případů z 978 účastníků, kteří dostali placebo. Bodový odhad účinnosti je 100 % (95% interval spolehlivosti 75,3; 100,0). U účastníků s nebo bez průkazu prodělané infekce bylo 0 případů z 1 119 účastníků, kteří dostali vakcínu, a 18 případů
z 1 110 účastníků, kteří dostali placebo. To také naznačuje, že bodový odhad účinnosti je 100 % (95%
interval spolehlivosti 78,1; 100,0).
U dodatečných potvrzených případů onemocnění COVID-19, k nimž došlo během zaslepeného, placebem kontrolovaného následného sledování, byly provedeny aktualizované analýzy účinnosti, jež reprezentovaly dobu až 6 měsíců po 2. dávce v populaci pro hodnocení účinnosti.
V aktualizované analýze účinnosti ve studii 2, provedené u dospívajících ve věku 12 až 15 let bez průkazu prodělané infekce, se u 1 057 účastníků, kterým byla podána vakcína, nevyskytl žádný případ a u 1 030 účastníků, kterým bylo podáno placebo, došlo ke 28 případům. Odhad bodu účinnosti je
100 % (95% interval spolehlivosti 86,8; 100,0). U účastníků s průkazem prodělané infekce nebo bez tohoto průkazu nedošlo u 1 119 těch, kterým byla podána vakcína, k žádnému případu
a u 1 109 účastníků, jimž bylo podáno placebo, ke 30 případům. To rovněž naznačuje, že odhad bodu
účinnosti je 100 % (95% interval spolehlivosti 87,5; 100,0).
Ve Studii 2 byla provedena analýza neutralizačních titrů SARS-CoV-2 jeden měsíc po druhé dávce u náhodně vybrané podskupiny účastníků, kteří do 1 měsíce po dávce 2 neměli sérologické nebo virologické průkazy prodělané infekce SARS-CoV-2, která porovnávala odpověď u dospívajících ve věku 12 až 15 let (n = 190) s účastníky ve věku 16 až 25 let (n = 170).
Poměr geometrických průměrných titrů (GMT) ve skupině ve věku 12 až 15 let ke skupině ve věku 16 až 25 let byl 1,76; s oboustranným 95% CI 1,47 až 2,10. Proto bylo splněno 1,5násobné kritérium noninferiority, protože dolní mez 2stranného 95% CI pro poměr geometrického průměru [GMR] byla
> 0,67.
Imunogenicita u účastníkůvevěku18letastarších– po posilovací dávce
Účinnost posilovací dávky vakcíny Comirnaty byla ve studii 2 založena na hodnocení 50%
neutralizačních titrů protilátek (NT50) proti SARS-CoV-2 (USA_WA1/2020). V této studii byla posilovací dávka podána 5 až 8 měsíců (medián 7 měsíců) po druhé dávce. Ve studii 2 prokázaly
analýzy NT50 1 měsíc po posilovací dávce v porovnání s 1 měsícem po primární sérii u jedinců ve věku 18 až 55 let, kteří neměli žádné sérologické nebo virologické důkazy o prodělané infekci virem
SARS-CoV-2 do 1 měsíce po posilovací vakcinaci, noninferioritu jak pro geometrický průměrný poměr (GMR), tak pro rozdíl v četnostech sérologické odpovědi. Sérologická odpověď u účastníka byla definována jako dosažení ≥ 4násobného zvýšení NT50 oproti výchozímu stavu (před primární
sérií). Tyto analýzy jsou shrnuty v tabulce 5.
|
n
|
1 měsíc po posilovací dávce (95% CI)
|
1 měsíc po primární sérii (95% CI)
|
1 měsíc po
posilovací dávce / 1 měsíc po primární sérii
(
97,5% CI)
|
Splnila cíl noninferiority (A/N)
|
G
eometrický
průměr 50%
neutralizačního
ti
t
ru
|
212a
|
2466,0b
(2202,6; 2760,8)
|
750,6b
(656,2; 858,6)
|
3,29c
(2,77; 3,90)
|
Ad
|
Č
etnost sérologické odpovědi (%) pro
50% neutralizační
ti
t
r
†
|
200e
|
199f
99,5 %
(97,2 %; 100,0 %)
|
196f
98,0 %
(95,0 %; 99,5 %)
|
1,5 %g
(-0,7 %; 3,7 %h)
|
Ai
|
|
|
T
abulka 5: Neutralizační test SARS-CoV-2 – NT50 (titr)
†
(
SARS-CoV-2 USA_WA1/2020) – srovnání GMT a četnosti sérologické odpovědi 1 měsíc po posilovací dávce a 1 měsíc po primární sérii – účastníci ve věku 18 až 55 let bez známek infekce do 1 měsíce po posilovací dávce* – populace s hodnotitelnou imunogenicitou po posilovací dávce
±
Zkratky: CI = interval spolehlivosti; GMR = poměr geometrického průměru; GMT = titr geometrického
průměru; LLOQ = dolní limit kvantifikace; N-vazba = SARS-CoV-2 nukleoproteinová vazba; NAAT = test amplifikace nukleové kyseliny; NT50 = 50% neutralizační titr; SARS-CoV-2 = závažný akutní respirační syndrom vyvolaný koronavirem 2; A/N = ano/ne.
† NT50 SARS-CoV-2 byly stanoveny pomocí mikroneutralizačního testu viru SARS-CoV-2 mNeonGreen.
Test používá fluorescenční reportérový virus odvozený od kmene USA_WA1/2020 a neutralizace viru se odečítá na monovrstvách buněk Vero. Vzorek NT50 je definován jako vzájemné ředění séra, při kterém je neutralizováno 50 % viru.
* Do analýzy byli zahrnuti účastníci, kteří neměli žádný sérologický nebo virologický průkaz (do 1 měsíce po podání posilovací dávky vakcíny Comirnaty) o prodělané infekci virem SARS-CoV-2 (tj. N-vázané protilátky [sérum] negativní a virus SARS-CoV-2 nebyl detekován pomocí NAAT [nosní výtěr]) a měli negativní NAAT (nosní výtěr) při jakékoli neplánované návštěvě do 1 měsíce po posilovací dávce.
± Všichni způsobilí účastníci, kteří dostali 2 dávky vakcíny Comirnaty podle původní randomizace, přičemž
2. dávku dostali v rámci předem definovaného časového období (během 19 až 42 dnů po 1. dávce), dostali posilovací dávku vakcíny Comirnaty, měli alespoň 1 platný a určitelný výsledek imunogenicity po posilovací dávce z odběru krve v rámci příslušného časového období (během 28 až 42 dnů po posilovací dávce) a neměli žádné další důležité odchylky od protokolu podle rozhodnutí lékaře.
a. n = Počet účastníků s platnými a určitelnými výsledky testu v obou časových bodech odběru vzorků v rámci stanoveného časového období.
b. GMT a 2stranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a příslušných CI (na základě Studentova t rozdělení). Výsledky testů pod LLOQ byly stanoveny na
0,5násobku LLOQ.
c. GMR a 2stranné 97,5% CI byly vypočteny exponováním průměrných rozdílů logaritmů testu a odpovídajících CI (na základě Studentova t rozdělení).
d. Noninferiorita je deklarována, pokud je dolní hranice dvoustranného 97,5% CI pro GMR > 0,67 a bodový
odhad GMR činí ≥ 0,80.
e. n = počet účastníků s platnými a určitelnými výsledky testu pro daný test při výchozím stavu, 1 měsíc po
2. dávce a 1 měsíc po posilovací dávce v rámci stanoveného časového období. Tyto hodnoty jsou denominátory pro výpočty procent.
f. Počet účastníků se sérologickou odpovědí na daný test v daném časovém bodě odběru dávky / vzorku.
Přesný dvoustranný CI na základě Clopperovy a Pearsonovy metody.
g. Rozdíl v podílech vyjádřený v procentech (1 měsíc po posilovací dávce – 1 měsíc po 2. dávce).
h. Upravený dvoustranný Waldův CI pro rozdíl v podílech, vyjádřený v procentech.
i. Noninferiorita je deklarována, pokud je dolní hranice dvoustranného 97,5% CI pro procentuální rozdíl
> 10 %.
Relativníúčinnostvakcínyuúčastníkůvevěku16 let a starších – po posilovací dávcePrůběžná analýza účinnosti ve studii 4, placebem kontrolované studii posilovací dávky provedené
u přibližně 10 000 účastníků ve věku 16 let a starších, kteří byli zařazeni ze studie 2, hodnotila
potvrzené případy onemocnění COVID-19 zjištěné nejméně 7 dní po vakcinaci posilovací dávkou až
do dne ukončení sběru dat 5. října 2021, což představuje medián 2,5 měsíce následného sledování po
posilovací dávce. Posilovací dávka byla podána 5 až 13 měsíců (medián 11 měsíců) po druhé dávce. Hodnocena byla účinnost vakcinace posilovací dávkou vakcíny Comirnaty po základním očkování ve srovnání se skupinou, jíž bylo v rámci posilovací dávky podáno placebo a jež dostala pouze základní očkování.
Informace o relativní účinnosti vakcíny u účastníků ve věku 16 let a starších bez důkazu předchozí infekce SARS-CoV-2 uvádí tabulka 6. Relativní účinnost vakcíny u účastníků s důkazem předchozí infekce SARS-CoV-2 či bez takového důkazu byla 94,6 % (95% interval spolehlivosti 88,5 % až
97,9 %), podobně jako tomu bylo u účastníků bez důkazu předchozí infekce. Případy primárního onemocnění COVID-19, pozorované od 7 dní po podání posilovací dávky vakcíny, zahrnovaly
7 primárních případů ve skupině s vakcínou Comirnaty a 124 primárních případů ve skupině
s placebem.
První výskyt onemocnění COVID-19 od 7 dnů po posilovací dávce u účastníků bez důkazu
předchozí infekce SARS-CoV-2*
|
| Comirnaty na=4 695
Případy
n1b
Doba sledováníc (n2d)
| Placebo na=4 671
Případy
n1b
Doba sledováníc (n2d)
|
Relativní účinnost vakcínye % (95% CIf)
| První výskyt
onemocnění COVID-19 od 7 dnů po posilovací dávce vakcíny
|
6
0,823 (4 659)
|
123
0,792 (4 614)
|
95,3 (89,5; 98,3)
|
|
|
Tabulka 6: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po posilovací dávce vakcíny – účastníci ve věku 16 let a starší bez důkazu infekce – populace hodnotitelná pro účinnostPoznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení).
* Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (7 nebo více dnů před podáním posilovací
dávky vakcíny) předcházející infekce SARS-CoV-2 (tj. N-vazebná protilátka [sérum] negativní při návštěvě 1 a SARS-CoV-2 nedetekován pomocí testů NAAT [výtěr nosu] při návštěvě 1) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po posilovací dávce, byli zařazeni do analýzy.
a. n = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po posilovací dávce vakcíny do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Relativní účinnost vakcíny ve skupině s posilovací dávkou vakcíny Comirnaty ve srovnání se skupinou s placebem (bez posilovací dávky).
f. Dvoustranný interval spolehlivosti pro relativní účinnost vakcíny je odvozen na základě Clopperovy
a Pearsonovy metody upravené vzhledem k době sledování.
ImunogenicitaposilovacídávkypoprimárnívakcinacijinouschválenouvakcínouprotionemocněníCOVID-19Účinnost posilovací dávky vakcíny Comirnaty (30 mikrogramů) u osob, které absolvovaly primární
vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19 (heterologní posilovací dávka), je odvozena z údajů o imunogenicitě z nezávislé otevřené klinické studie fáze 1/2 National Institutes of Health (NIH) (NCT04889209) provedené ve Spojených státech. V této studii dostali dospělí (věkové rozmezí 19 až 80 let), kteří absolvovali primární vakcinaci vakcínou Moderna 100 mikrogramů v sérii
2 dávek (n=51, průměrný věk 54 ± 17 let), jednorázovou dávkou vakcíny Janssen (n=53, průměrný věk 48±14 let) nebo vakcínou Comirnaty 30 mikrogramů v sérii 2 dávek (n=50, průměrný věk
50±18 let) nejméně 12 týdnů před zařazením do studie a kteří neuvedli žádnou anamnézu infekce
virem SARS-CoV-2, posilovací dávku vakcíny Comirnaty (30 mikrogramů). Posilovací dávka vakcíny
Comirnaty vyvolala 36, 12 a 20násobné zvýšení GMR neutralizačních titrů po primárních dávkách
vakcín Janssen, Moderna a Comirnaty, v uvedeném pořadí.
Heterologní posilovací dávka vakcíny Comirnaty byla rovněž hodnocena ve studii CoV-BOOST (EudraCT 2021 002175-19), multicentrické, randomizované, kontrolované studii fáze 2 u třetí posilovací dávky vakcíny proti onemocnění COVID-19, ve které bylo 107 dospělých účastníků (medián věku 71 let, interkvartilové rozmezí 54 až 77 let) randomizováno nejméně 70 dní po podání
2 dávek vakcíny AstraZeneca proti onemocnění COVID-19. Po primární sérii vakcíny AstraZeneca
proti onemocnění COVID-19 se násobná změna GMR NT50 neutralizačních protilátek proti pseudoviru (divoký typ) zvýšila 21,6násobně při heterologní posilovací dávce vakcíny Comirnaty (n=95).
Imunogenicita u účastníkůstarších 55 let – po posilovací dávce (čtvrté dávce) vakcíny Comirnaty(30 mikrogramů)V dílčí analýze podmnožiny ze studie 4 (podstudie E) obdrželo 305 účastníků starších 55 let, kteří
dokončili řadu 3 dávek vakcíny Comirnaty, jako posilovací dávku (čtvrtou dávku) vakcínu Comirnaty
(30 mikrogramů) 5 až 12 měsíců po obdržení 3. dávky. Údaje o imunogenicitě pro podskupinu viz
tabulka 7.
Imunogenicita u účastníkůvevěku18 až ≤ 55 let – po posilovací dávce (čtvrtédávce) vakcínyComirnaty (30 mikrogramů)V podstudii D [podmnožina ze studie 2 (3. fáze) a studie 4 (3. fáze)] obdrželo 325 účastníků ve věku
18 až ≤ 55 let, kteří dokončili řadu 3 dávek vakcíny Comirnaty, jako posilovací dávku (čtvrtou dávku)
vakcínu Comirnaty (30 mikrogramů) 90 až 180 dnů po obdržení 3. dávky. Údaje o imunogenicitě pro podskupinu viz tabulka 7.
| Dávka /
čas odběru krevních vzorkůa
| Podstudie D
(věk 18 až < 55 let) Comirnaty
30 mikrogramů
| Podstudie E
(věk > 55 let) Comirnaty
30 mikrogramů
|
GMT
|
|
Nb
| GMT (95% CId)
|
Nb
| GMT (95% CId)
| Analýza neutralizace
SARS-CoV-2 – Omicron BA.1 –
NT50 (titr)
| 1/před
očkováním
|
226
| 315,0
(269,0; 368,9)
|
167
| 67,5
(52,9; 86,3)
|
1/1 měsíc
|
228
| 1 063,2
(935,8; 1 207,9)
|
163
| 455,8
(365,9; 567,6)
| Analýza neutralizace
SARS-CoV-2 –
referenční kmen – NT50 (titr)
| 1/před
očkováním
|
226
| 3 999,0
(3 529,5; 4 531,0)
|
179
| 1 389,1
(1 142,1; 1 689,5)
|
1/1 měsíc
|
227
| 12 009,9
(10 744,3; 13 424,6)
|
182
| 5 998,1
(5 223,6; 6 887,4)
| Četnost sérologické
odpovědi 1 měsíc po
4. dávce
|
|
Nc
|
ne (%) (95% CIf)
|
Nc
|
ne (%) (95% CIf)
| Analýza neutralizace
SARS-CoV-2 – Omicron BA.1 – NT50 (titr)
|
1/1 měsíc
|
226
|
91 (40,3 %) (33,8; 47,0)
|
149
|
85 (57,0 %) (48,7; 65,1)
|
|
|
Tabulka 7. Souhrn údajů o imunogenicitě od účastníků studie C4591031, podstudie D (množina úplné rozšířené kohorty 2) a podstudie E (rozšířená kohorta – podmnožina imunogenicity), kteří obdrželi jako posilovací dávku (čtvrtou dávku) vakcínu Comirnaty 30 mikrogramů – účastníci bez průkazu infekce až do 1 měsíce po posilovací dávce – populace s hodnotitelnou imunogenicitou
Analýza neutralizace
SARS-CoV-2 –
referenční kmen – NT50 (titr)
|
1/1 měsíc
|
225
|
76 (33,8 %) (27,6; 40,4)
|
179
|
88 (49,2 %) (41,6; 56,7)
|
Zkratky: CI = interval spolehlivosti; GMFR = geometrický průměr násobného nárůstu; GMT = titr geometrického
průměru; LLOQ = dolní limit kvantifikace; N-vazba = SARS-CoV-2 nukleoproteinová vazba; NAAT = test amplifikace nukleové kyseliny; NT50 = 50% neutralizační titr; SARS-CoV-2 = závažný akutní respirační syndrom vyvolaný koronavirem 2.
Poznámka: Medián doby od 3. dávky do 4. dávky vakcíny Comirnaty 30 mikrogramů je 4,0 měsíců pro podstudii D,
kohortu 2 a 6,3 měsíců pro podstudii E, rozšířenou kohortu.
Poznámka: Plná rozšířená množina podstudie D = kohorta 2 bez ověřovací skupiny; podmnožina imunogenicity podstudie E = náhodný vzorek 230 účastníků v každé skupině vakcíny vybraných z rozšířené kohorty.
Poznámka: Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (odběr vzorků krve dříve než 1 měsíc po
studijní vakcinaci) předcházející infekce SARS-CoV-2 (tj. negativní výsledek N-binding protilátky [sérum] na návštěvách při studijní vakcinaci a za 1 měsíc po vakcinaci, negativní výsledek NAAT [stěr z nosu] na návštěvě se studijní vakcinací
a jakékoliv neplánované návštěvě před odběrem vzorku krve 1 měsíc po studijní vakcinaci) a kteří neměli onemocnění
COVID-19 ve zdravotní anamnéze, byli zařazeni do analýzy.
Poznámka: Sérologická odpověď je definována jako dosažení ≥ 4násobného zvýšení NT50 oproti výchozímu stavu (před studijní vakcinací). Pokud je hodnota naměřená ve výchozím stavu pod hodnotou LLQQ, je považována za sérologickou odpověď naměřená hodnota po očkování ≥ 4 × LLOQ.
a. Protokolárně stanovené načasování odběru krevních vzorků.
b. N = počet účastníků s platnými a určitelnými výsledky testu pro daný test v daném časovém bodě odběru vzorků.
c. N = počet účastníků s platnými a určitelnými výsledky testu pro daný test jak v časovém bodě před očkováním, tak
i v daném časovém bodě odběru vzorků
d. GMT a 2stranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a příslušných CI (na základě Studentova t rozdělení). Výsledky testů pod LLOQ byly stanoveny na 0,5násobku LLOQ.
e. Počet účastníků se sérologickou odpovědí na daný test v daném časovém bodě odběru vzorku. f. Přesný dvoustranný CI na základě Clopperovy a Pearsonovy metody.
Pediatrická populaceEvropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s vakcínou
Comirnaty u pediatrické populace k prevenci onemocnění COVID-19 (informace o použití u pediatrické populace viz bod 4.2).
5.2 Farmakokinetické vlastnostiNeuplatňuje se.
5.3 Předklinické údaje vztahující se k bezpečnostiNeklinické údaje získané na základě konvenčních studií toxicity po opakovaném podávání a
reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
Obecná toxicitaU potkanů, kterým byla intramuskulárně podávána vakcína Comirnaty (dostali 3 plné dávky pro
člověka jednou týdně, které vedly k relativně vyšším hladinám u potkanů v důsledku rozdílů v tělesné
hmotnosti), se vyskytl edém a erytém v místě podání injekce a zvýšení počtu bílých krvinek (včetně bazofilů a eozinofilů) odpovídající zánětlivé odpovědi a rovněž vakuolizace portálních hepatocytů bez
známek poškození jater. Všechny účinky byly reverzibilní.
Genotoxicita / karcinogenitaStudie genotoxicity ani kancerogenity nebyly provedeny. U složek vakcíny (lipidy a mRNA) se
neočekává genotoxický potenciál.
Reprodukční toxicitaReprodukční a vývojová toxicita byla hodnocena na potkanech v kombinované studii fertility a
vývojové toxicity, ve které byla samicím potkanů podána intramuskulárně vakcína Comirnaty před
pářením a během březosti (dostaly 4 plné dávky pro člověka, které vedly k relativně vyšším hladinám u potkanů v důsledku rozdílů v tělesné hmotnosti mezi 21. dnem před pářením a 20. dnem březosti). Odpovědi na neutralizační protilátky SARS-CoV-2 byly přítomny u samic před pářením až do konce studie ve 21. postnatálním dni a rovněž u plodů a potomků. Nebyly zjištěny žádné účinky spojené s očkováním na plodnost samic, těhotenství nebo vývoj embrya/plodu nebo potomstva. Údaje o možném placentárním přenosu nebo vylučování do mateřského mléka pro vakcínu Comirnaty nejsou k dispozici.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159) Kolfosceryl-stearát
Cholesterol
Trometamol
Trometamol-hydrochlorid
Sacharosa
Voda pro injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Neotevřená injekční lahvička
Zmrazenáinjekčnílahvička
18 měsíců při teplotě -90 °C až -60 °C.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána buď při teplotě -90 °C až -60 °C nebo 2 °C až 8 °C.
Jednodávkové injekční lahvičky
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
10 jednodávkových injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 2 hodin nebo
mohou být jednotlivé injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Vícedávkové injekční lahvičky
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
10 vícedávkových injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 6 hodin nebo mohou
být jednotlivé injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Rozmrazená injekčnílahvička
10 týdnů uchovávání a transportu při teplotě 2 °C až 8 °C během 18měsíční doby použitelnosti.
• Po přenesení vakcíny pro uchovávání při teplotě 2 °C až 8 °C musí být na vnější obal zapsáno aktualizované datum použitelnosti a vakcína má být použita nebo zlikvidována do aktualizovaného data použitelnosti. Původní datum použitelnosti má být přeškrtnuto.
• Pokud je vakcína přijata při teplotě 2 °C až 8 °C, má být uchovávána při teplotě 2 °C až 8 °C.
Datum použitelnosti na vnějším obalu mělo být již dříve aktualizováno tak, aby odpovídalo datu použitelnosti v chladu, a původní datum použitelnosti mělo být již dříve přeškrtnuto.
Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Po rozmrazení vakcína nesmí být znovu zmrazena.
Manipulace při teplotních výkyvechpřiuchovávánívchladničce
• Údaje o stabilitě prokazují, že je neotevřená injekční lahvička stabilní až 10 týdnů, je-li
uchovávána při teplotě od -2 °C do 2 °C během 10týdenní doby uchovávání při teplotě 2 °C a
8 °C.
• Údaje o stabilitě prokazují, že injekční lahvička může být uchovávána až 24 hodin při teplotě
od 8 °C do 30 °C, včetně až 12 hodin po prvním vpichu.
Tyto informace slouží jako návod pro zdravotnické pracovníky pouze v případě dočasného výkyvu
teploty.
Otevřená injekční lahvička
Chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 12 hodin při teplotě
2 °C až 30 °C, která zahrnuje až 6 hodin transportu. Z mikrobiologického hlediska, pokud způsob otevření nevyloučí rizika mikrobiologické kontaminace, má být přípravek použit okamžitě. Pokud
není použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v
odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Během uchovávání je třeba minimalizovat vystavení přípravku světlu v místnosti a zabránit vystavení
přímému slunečnímu světlu a ultrafialovému světlu.
Podmínky uchovávání tohoto léčivého přípravku po prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Vakcína Comirnaty disperze se dodává v čiré injekční lahvičce (sklo třídy I) o objemu 2 ml se zátkou
(syntetická brombutylová pryž) a šedým odtrhovacím plastovým víčkem s hliníkovým krytem.
Jedna jednodávková injekční lahvička obsahuje 1 dávku 0,3 ml, viz body 4.2 a 6.6.
Jedna vícedávková injekční lahvička (2,25 ml) obsahuje 6 dávek po 0,3 ml, viz body 4.2 a 6.6.
Velikost balení jednodávkových injekčních lahviček: 10 injekčních lahviček.
Velikost balení vícedávkových injekčních lahviček: 10 nebo 195 injekčních lahviček. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Instrukce pro zacházení s vakcínou
Vakcína Comirnaty má být připravována zdravotnickým pracovníkem pomocí aseptické techniky, aby
byla zajištěna sterilita připravené disperze.
NÁV
O
D SE VZTAHUJE NA JEDNODÁVKOVÉ I VÍCEDÁVKOVÉ INJEKČNÍ LAHVIČKY
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY
30 MIKROGRAMŮ/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
• Zkontrolujte, zda má injekční lahvička
 Š
e
d
é víčko
Š
e
d
é víčko
 Neřeďte
Neřeďte
šedé plastové víčko a šedý okraj kolem štítku a název přípravku je Comirnaty
30 mikrogramů/dávku injekční
disperze.
• Zkontrolujte, zda se jedná
o jednodávkovou nebo vícedávkovou injekční lahvičku, a řiďte se příslušnými pokyny pro zacházení uvedenými níže.
• Pokud má injekční lahvička šedé plastové víčko a šedý okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.1
(15/15 mikrogramů)/dávku injekční
disperze nebo Comirnaty
Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční disperze, přečtěte si souhrn údajů o přípravku pro toto složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro
injekční disperzi nebo Comirnaty
Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte
si souhrn údajů o přípravku pro vakcínu Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi.


Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY 30 MIKROGRAMŮ/DÁVKU INJEKČNÍ
D
I
SPERZE (12 LET A STARŠÍ) PŘED POUŽITÍM
|
U
chovávejte po dobu až
10 týdnů při
t
eplotě 2 °C až
8 °C, aktualizujte datum použitelnosti na krabičce.
|
• Pokud se jednodávková nebo vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se
rozmrazily. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
o Jednodávkové injekční lahvičky:
Rozmrazení balení
10 jednodávkových injekčních lahviček může trvat 2 hodiny.
o Vícedávkové injekční lahvičky:
Rozmrazení balení
10 vícedávkových injekčních lahviček může trvat 6 hodin.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
Jemně
× 10
|
• Injekční lahvičky před použitím 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Před smícháním může rozmrazená disperze obsahovat bílé až téměř bílé, matné amorfní částice.
• Po smíchání se má vakcína jevit jako bílá až téměř bílá disperze bez viditelných částic. Jestliže jsou
v naředěné vakcíně patrné částice nebo vakcína změnila barvu, nepoužívejte ji.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,3 ml VAKCÍNY COMIRNATY
30 MIKROGRAMŮ/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
J
ednodávkové injekční
l
ahvičky
• Natáhněte jednu 0,3ml dávku vakcíny.
• Injekční lahvičku a veškerý přebytečný
objem zlikvidujte.
Vícedávkovéinjekčnílahvičky• Vícedávkové injekční lahvičky obsahují
6 dávek po 0,3 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím antiseptického tampónu.
• Natáhněte 0,3 ml vakcíny Comirnaty.
V
akcína o objemu 0,3 ml
K získání šesti dávek z jedné injekční
lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční
stříkačky a jehly s malým mrtvým
prostorem má mít mrtvý objem
maximálně 35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání šesté dávky z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Zaznamenejte příslušné datum/čas na injekční lahvičku. Veškerou vakcínu, jež nebyla použita do 12 hodin po prvním vpichu, zlikvidujte.
L
i
kv
i
dace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.de
8. REGISTRAČNÍ ČÍSLO/REGISTRAČNÍ ČÍSLA
J
ednodávkové injekční lahvičkyEU/1/20/1528/013
Vícedávkové injekční lahvičkyEU/1/20/1528/002
EU/1/20/1528/003
9. DATUM REGISTRACE/PRODLOUŽENÍ REGISTRACEDatum první registrace: 21. prosince 2020
Datum posledního prodloužení registrace: 10. října 2022
10. DATUM REVIZE TEXTUPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKUComirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍJedná se o vícedávkovou injekční lahvičku s oranžovým víčkem, jejíž obsah je nutno před použitím naředit.
Jedna injekční lahvička (1,3 ml) obsahuje 10 dávek po 0,2 ml po naředění, viz body 4.2 a 6.6.
Jedna dávka (0,2 ml) obsahuje 10 mikrogramů tozinameranu, mRNA vakcíny proti onemocnění
COVID-19 (zapouzdřené v lipidových nanočásticích).
Tozinameran je jednovláknová mediátorová (messenger) RNA (mRNA) s čepičkou na 5´ konci vyráběná
in vitro nebuněčnou transkripcí z příslušných matricí DNA a kódující spike (S) protein viru SARS-CoV-2.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMAKoncentrát pro injekční disperzi (sterilní koncentrát).
Vakcína je bílá až téměř bílá zmrazená disperze (pH: 6,9–7,9).
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikaceVakcína Comirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi je indikována pro aktivní
imunizaci k prevenci onemocnění COVID-19 způsobeného SARS-CoV-2 u dětí ve věku 5 až 11 let.
Tuto vakcínu je třeba používat v souladu s oficiálními doporučeními.
4.2 Dávkování a způsob podáníDávkováníZákladníočkováníDěti ve věku 5 až 11 let (tj. 5 až méně než 12 let)Vakcína Comirnaty 10 mikrogramů/dávku se podává intramuskulárně po naředění jako základní očkování 2 dávkami (0,2 ml každá dávka). Druhou dávku se doporučuje podat 3 týdny po první dávce (viz body 4.4 a 5.1).
Těžce imunokompromitované osoby ve věku 5 let a staršíTřetí dávka základního očkování může být podána intramuskulárně nejméně 28 dní po druhé dávce
jedincům, kteří jsou těžce imunokompromitovaní (viz bod 4.4).
Pokud dítě mezi jednotlivými dávkami základního očkování dosáhne věku 12 let, má sérii dokončit se
stejnou dávkou 10 mikrogramů.
Posilovací dávka
Posilovací dávka u dětí ve věku od 5 do 11 let
Posilovací dávka vakcíny Comirnaty 10 mikrogramů může být podána u dětí ve věku od 5 do 11 let
intramuskulárně za nejméně 6 měsíců po základním očkování.
Zaměnitelnost
Nebylo stanoveno, zda lze za účelem dokončení základního očkování zaměnit vakcínu Comirnaty za
vakcíny proti onemocnění COVID-19 od jiných výrobců. Osoby, jimž byla podána dávka vakcíny
Comirnaty, mají pokračovat v podávání vakcíny Comirnaty, aby bylo základní očkování dokončeno.
Pediatrická populace
K dispozici je pediatrická léková forma pro kojence a děti ve věku od 6 měsíců do 4 let. Podrobné
informace naleznete v souhrnu údajů o přípravku pro vakcínu Comirnaty 3 mikrogramy/dávku
koncentrát pro injekční disperzi.
Bezpečnost a účinnost vakcíny Comirnaty u kojenců ve věku do 6 měsíců nebyly dosud stanoveny.
Způsob podání
Vakcína Comirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi se podává intramuskulárně
po naředění (viz bod 6.6).
Po naředění obsahují injekční lahvičky vakcíny Comirnaty 10 dávek po 0,2 ml vakcíny. K získání
10 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů. Pokud se používají standardní injekční stříkačky a jehly, nemusí být dostatečný objem k získání 10 dávek z jedné injekční lahvičky. Bez ohledu na typ injekční stříkačky a jehly:
• Každá dávka musí obsahovat 0,2 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,2 ml,
injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Nekombinujte obsah z více injekčních lahviček vakcíny. Preferované místo je deltový sval horní části paže.
Vakcína se nesmí podávat intravaskulárně, subkutánně ani intradermálně.
Vakcína se nesmí mísit ve stejné injekční stříkačce s jinými vakcínami nebo léčivými přípravky. Pro opatření před podáním vakcíny viz bod 4.4.
Návod pro rozmrazení, zacházení s vakcínou a její likvidaci je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Sledovatelnost
Aby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Obecná doporučení
Hypersenzitivita a anafylaxe
Byly hlášeny případy anafylaxe. Pro případ, že by po podání vakcíny došlo k anafylaktické reakci, má
být zajištěna okamžitá lékařská péče a dohled.
Po vakcinaci se doporučuje pečlivé sledování po dobu minimálně 15 minut. Další dávka vakcíny nemá
být podána osobám, které měly anafylaxi po předchozí dávce vakcíny Comirnaty.
Myokarditida a perikarditida
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy a perikarditidy. Tato
onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhé dávce vakcíny a častěji u mladších mužů a chlapců. Dostupné údaje naznačují, že průběh myokarditidy a perikarditidy po vakcinaci se neliší od myokarditidy nebo perikarditidy obecně (viz bod 4.8).
Zdravotničtí pracovníci mají pozorně sledovat známky a příznaky myokarditidy a perikarditidy. Očkovaní jedinci (včetně rodičů a pečovatelů) mají být poučeni, aby okamžitě vyhledali lékařskou pomoc, pokud se u nich po očkování objeví příznaky naznačující myokarditidu nebo perikarditidu, například bolest na hrudi (akutní a přetrvávající), dušnost nebo palpitace.
Zdravotničtí pracovníci mají k diagnostice a léčbě tohoto onemocnění používat návody a postupy
a/nebo se mají obrátit na specialisty.
Reakce spojené s úzkostí
V souvislosti se samotným procesem očkování se mohou objevit reakce spojené s úzkostí, včetně
vazovagálních reakcí (synkopa), hyperventilace nebo reakcí spojených se stresem (např. závrať, palpitace, zvýšení srdeční frekvence, změny krevního tlaku, parestezie, hypestezie a pocení). Reakce
spojené se stresem jsou dočasné a samy se upraví. Očkované osoby je třeba informovat o tom, aby na
případné symptomy upozornily očkujícího zdravotníka, který je vyhodnotí. Je důležité, aby byla zavedena opatření, aby se zabránilo zranění v důsledku mdlob.
Současné onemocnění
U osob trpících závažným akutním horečnatým onemocněním nebo akutní infekcí se má podání
vakcíny Comirnaty odložit. Přítomnost mírné infekce a/nebo horečky nízkého stupně není důvod
k odložení vakcinace.
Trombocytopenie a poruchy koagulace
Stejně jako u jiných intramuskulárních injekcí je třeba vakcínu podávat opatrně osobám podstupujícím
léčbu antikoagulancii nebo osobám s trombocytopenií nebo poruchami koagulace (jako je hemofilie),
protože po intramuskulárním podání může u těchto osob dojít ke krvácení nebo tvorbě modřin.
I
m
unokompromitované osoby
Účinnost a bezpečnost vakcíny nebyly hodnoceny u imunokompromitovaných osob, včetně osob
podstupujících imunosupresivní léčbu. Účinnost vakcíny Comirnaty může být u
imunokompromitovaných osob nižší.
Doporučení zvážit podání třetí dávky u těžce imunokompromitovaných jedinců je založeno na omezených sérologických důkazech z případových studií z literatury z klinické léčby dospělých pacientů s iatrogenní imunosupresí po transplantaci solidních orgánů (viz bod 4.2).
Doba ochrany
Doba ochrany poskytovaná vakcínou není známa, protože je stále hodnocena v probíhajících
klinických studiích.
Omezení účinnosti vakcíny
Podobně jako u jiných vakcín je možné, že vakcinace vakcínou Comirnaty nebude chránit všechny její
příjemce. Osoby nemusí být plně chráněny po dobu 7 dnů po druhé dávce vakcíny.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Současné podání vakcíny Comirnaty s jinými vakcínami nebylo hodnoceno.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Velké množství observačních dat od těhotných žen očkovaných vakcínou Comirnaty během druhého a
třetího trimestru neprokázalo zvýšení nežádoucích výsledků těhotenství. Ačkoli údaje o výsledcích těhotenství po očkování během prvního trimestru jsou v současné době omezené, nebylo pozorováno
zvýšené riziko potratu. Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na
těhotenství, vývoj embrya/plodu, porod nebo postnatální vývoj (viz bod 5.3). Vakcínu Comirnaty lze v těhotenství podávat.
Kojení
Systémová expozice vakcíně Comirnaty je u kojící matky zanedbatelná, a proto nejsou očekávány
žádné účinky na kojeného novorozence/kojence (skrze mateřské mléko). Observační údaje od žen, které po očkování kojily, neprokázaly riziko nežádoucích účinků u kojených novorozenců/kojenců. Vakcínu Comirnaty lze během kojení podávat.
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky s ohledem na reprodukční toxicitu
(viz bod 5.3)
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vakcína Comirnaty nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Některé z účinků uvedených v bodě 4.8 však mohou schopnost řídit nebo obsluhovat stroje dočasně ovlivnit.
4.8 Nežádoucí účinky
Souhrn bezpečnostníhoprofilu
Dětivevěku5až11let(tj.vevěku5doméněnež12let)– po 2 dávkách
Ve studii 3 byla alespoň 1 dávka vakcíny Comirnaty 10 mikrogramů podána celkem 1 518 dětem ve
věku 5 až 11 let a celkem 750 dětem ve věku 5 až 11 let bylo podáno placebo. V době analýzy studie 3
fáze 2/3 s údaji do data ukončení sběru dat 6. září 2021 bylo 2 158 (95,1 %) (1 444 dětí, kterým byla podána vakcína Comirnaty 10 mikrogramů a 714 dětí, kterým bylo podáno placebo) sledováno po dobu nejméně 2 měsíců po druhé dávce vakcíny Comirnaty 10 mikrogramů. Analýza údajů o nežádoucích příhodách studie 3 fáze 2/3 zahrnovala také dalších 2 379 účastníků [1 591 účastníkům byla podána vakcína Comirnaty 10 mikrogramů a 788 účastníkům bylo podáno placebo], z nichž
71,2 % bylo sledováno po dobu nejméně 2 týdnů po 2. dávce až do data ukončení sběru dat
8. října 2021. Hodnocení bezpečnosti ve studii 3 probíhá.
Celkový bezpečnostní profil vakcíny Comirnaty u účastníků ve věku 5 až 15 let byl podobný jako u účastníků ve věku 16 let a starších. Nejčastějšími nežádoucími účinky u dětí ve věku 5 až 11 let, kterým byly podány 2 dávky, byly bolest v místě vpichu (> 80 %), únava (> 50 %), bolest hlavy (>
30 %), zarudnutí a otok v místě vpichu (> 20 %), myalgie a zimnice (> 10 %).
Děti ve věku 5až11let (tj. vevěku5doméněnež12 let) – po posilovací dávce
V podskupině účastníků studie 3 dostalo celkem 401 dětí ve věku 5 až 11 let posilovací dávku vakcíny
Comirnaty 10 mikrogramů za nejméně 5 měsíců (rozmezí 5 až 9 měsíců) po dokončení základního očkování. Analýza podskupiny ve studii 3 fáze 2/3 je založena na údajích s datem ukončení sběru dat
do 22. března 2022 (medián doby sledování 1,3 měsíce).
Celkový bezpečnostní profil posilovací dávky byl podobný jako po základním očkování. Nejčastějšími nežádoucími účinky u dětí ve věku 5 až 11 let byly bolest v místě vpichu (> 70 %), únava (> 40 %), bolest hlavy (> 30 %), myalgie, zimnice a zarudnutí a otok v místě vpichu (> 10 %).
Dospívající ve věku 12až15let – po 2 dávkách
V analýze dlouhodobého následného sledování bezpečnosti ve studii 2 bylo 2 260 dospívajících
(1 131 dostalo vakcínu Comirnaty a 1 129 dostalo placebo) ve věku 12 až 15 let. Z toho
1 559 dospívajících (786 dostalo vakcínu Comirnaty a 773 dostalo placebo) bylo sledováno po dobu
≥ 4 měsíců po druhé dávce vakcíny Comirnaty. Hodnocení bezpečnosti ve studii 2 probíhá.
Celkový bezpečnostní profil vakcíny Comirnaty u dospívajících ve věku 12 až 15 let byl podobný jako
u účastníků ve věku 16 let a starších. Nejčastějšími nežádoucími účinky u dospívajících ve věku 12 až
15 let, kteří dostali 2 dávky, byly bolest v místě injekce (> 90 %), únava a bolest hlavy (> 70 %), myalgie a zimnice (> 40 %), artralgie a pyrexie (> 20 %).
Účastníci ve věku 16let a starší – po 2 dávkách
Ve studii 2 byla celkem 22 026 účastníkům ve věku 16 let nebo starším podána alespoň 1 dávka
vakcíny Comirnaty 30 mikrogramů a celkem 22 021 účastníkům ve věku 16 let nebo starším bylo podáno placebo (včetně 138 a 145 dospívajících ve věku 16 a 17 let ve skupinách vakcíny a placeba, v uvedeném pořadí). Celkem 20 519 účastníků ve věku 16 let nebo starších dostalo 2 dávky vakcíny Comirnaty.
V době provedení analýzy studie 2 s datem ukončení sběru dat 13. března 2021 pro placebem kontrolované zaslepené období sledování až do dat odslepení účastníků bylo celkem 25 651 (58,2 %) účastníků (13 031 Comirnaty a 12 620 placebo) ve věku 16 let a starších sledováno po dobu ≥4 měsíce po podání druhé dávky. To zahrnovalo celkem 15 111 (7 704 Comirnaty a 7 407 placebo) účastníků ve
věku 16 až 55 let a celkem 10 540 (5 327 Comirnaty a 5 213 placebo) účastníků ve věku 56 let a starších.
Nejčastějšími nežádoucími účinky byla u účastníků ve věku 16 let a starších, kteří dostali 2 dávky, bolest v místě injekce (> 80 %), únava (> 60 %), bolest hlavy (> 50 %), myalgie (> 40 %), zimnice
(> 30 %), artralgie (> 20 %), pyrexie a zduření v místě injekce (> 10 %). Tyto nežádoucí účinky byly zpravidla mírné nebo střední intenzity a odezněly během několika dní po vakcinaci. Mírně nižší frekvence příhod reaktogenity souvisela s vyšším věkem.
Bezpečnostní profil u 545 účastníků ve věku 16 let a starších, kterým byla podána vakcína Comirnaty
a kteří byli séropozitivní na SARS-CoV-2 při výchozím stavu, byl podobný jako u obecné populace.
Účastníci ve věku 16let a starší – po posilovací dávce
Podskupina účastníků studie fáze 2/3, do které bylo zařazeno 306 dospělých ve věku 18 až 55 let, kteří
dokončili původní cyklus 2 dávek vakcíny Comirnaty, dostala posilovací dávku vakcíny Comirnaty
přibližně 6 měsíců (rozmezí 4,8 až 8,0 měsíců) po podání 2. dávky.
Celkový bezpečnostní profil posilovací dávky byl podobný jako po 2 dávkách. Nejčastějšími nežádoucími účinky u účastníků ve věku 18 až 55 let byly bolest v místě vpichu (> 80 %), únava (>
60 %), bolest hlavy (> 40 %), myalgie (> 30 %), zimnice a artralgie (> 20 %).
Ve studii 4, placebem kontrolované studii posilovací dávky, byla účastníkům ve věku 16 let a starším
zařazeným ze studie 2 podána posilovací dávka vakcíny Comirnaty (5 081 účastníků) nebo placebo (5 044 účastníků), a to nejméně 6 měsíců po druhé dávce vakcíny Comirnaty. Celkem činil medián doby následného sledování účastníků, kterým byla posilovací dávka podána, 2,5 měsíce po podání posilovací dávky do data ukončení sběru (5. října 2021). Žádné nové nežádoucí účinky vakcíny Comirnaty nebyly zjištěny.
Posilovací dávka poprimárnívakcinacijinouschválenouvakcínouprotionemocněníCOVID-19
V 5 nezávislých studiích o použití posilovací dávky vakcíny Comirnaty u osob, které absolvovaly
primární vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19 (heterologní posilovací
dávka), nebyly zjištěny žádné nové bezpečnostní problémy.
Tabulkovýseznamnežádoucíchúčinkůzklinickýchstudiíapouvedenínatrhuosobvevěku5let astarších
Nežádoucí účinky pozorované z klinických studií jsou uvedeny níže podle následujících kategorií
frekvence:
Velmi časté (≥ 1/10)
Časté (≥ 1/100 až < 1/10)
Méně časté (≥ 1/1 000 až < 1/100)
Vzácné (≥ 1/10 000 až < 1/1 000) Velmi vzácné (< 1/10 000)
Není známo (z dostupných údajů nelze určit)
T
abulka 1: Nežádoucí účinky vakcíny Comirnaty z klinických studií a po uvedení na trh u
osob ve věku 5 let a starších
T
řída
orgánového systému
|
V
elmi
časté
(
≥ 1/10)
|
Č
asté
(
≥ 1/100
až
< 1/10)
|
Méně časté
(
≥ 1/1 000 až
< 1/100)
|
V
z
ácné
(
≥ 1/10 000
až
< 1/1 000)
|
V
elmi vzácné
(
< 1/10 000
|
N
ení
z
námo (z dostupnýc h údajů
nelze určit)
|
Poruchy krve a
lymfatického systému
|
|
|
Lymfadenopatiea
|
|
|
|
Poruchy
imunitního systému
|
|
|
Hypersenzitivní
reakce (např.
vyrážka, pruritus, urtikarieb, angioedémb)
|
|
|
Anafylaxe
|
Poruchy
metabolismu a výživy
|
|
|
Snížená chuť k
jídlu
|
|
|
|
Psychiatrické
poruchy
|
|
|
Insomnie
|
|
|
|
Poruchy
nervového systému
|
Bolest
hlavy
|
|
Letargie
|
Akutní
periferní paralýza n. facialisc
|
|
Parestezied,
hypestezied
|
Srdeční poruchy
|
|
|
|
|
Myokarditidad;
Perikarditidad
|
|
Gastrointestinální
poruchy
|
Průjemd
|
Nauzea;
zvraceníd
|
|
|
|
|
Poruchy kůže a
podkožní tkáně
|
|
|
Hyperhidróza;
noční pocení
|
|
|
Erythema
multiformed
|
Poruchy svalové
a kosterní soustavy a
pojivové tkáně
|
Artralgie;
myalgie
|
|
Bolest
v končetiněe
|
|
|
|
Poruchy
reprodukčního
systému a prsu
|
|
|
|
|
|
Silné
menstruační
krváceníi
|
Celkové poruchy
a reakce v místě
aplikace
|
Bolest v
místě
injekce; únava; zimnice; pyrexief; zduření v místě injekce
|
Zarudnutí
v místě
injekceh
|
Astenie;
malátnost;
svědění v místě
injekce
|
|
|
Rozsáhlý
otok
končetiny, do níž byla vakcína podánad; otok obličejeg
|
a. U účastníků, kteří dostali posilovací dávku, byla pozorována vyšší frekvence lymfadenopatie ve srovnání
s účastníky ve věku 5 až 11 let ve studii 3 (2,5 % vs. 0,9 %) a s účastníky ve věku 16 let a starších ve studii 4 (2,8 % vs. 0,4 %), kteří dostali dvě dávky.
b. Kategorie frekvence pro urtikarii a angioedém byla vzácná.
c. Během období sledování bezpečnosti v klinické studii do 14. listopadu 2020 byla hlášena akutní periferní paralýza
n. facialis u čtyř účastníků ve skupině mRNA vakcíny proti onemocnění COVID-19. Nástup obrny
obličeje byl 37. den po podání 1. dávky (účastník nedostal 2. dávku) a 3., 9. a 48. den po 2. dávce. Ve
skupině s placebem nebyly hlášeny žádné případy akutní periferní paralýzy
n. facialis. d. Nežádoucí účinek byl stanoven po registraci.
e. Týká se končetiny/paže, do které byla daná osoba očkována.
f. Vyšší frekvence pyrexie byla pozorována po druhé dávce v porovnání s první dávkou.
g. Po uvedení přípravku na trh byly hlášeny případy otoku obličeje u očkovaných osob, které v minulosti
podstoupily injekční aplikaci dermálních výplní do obličeje.
h. Zarudnutí v místě injekce se vyskytovalo s vyšší frekvencí (velmi častou) u dětí ve věku 5 až 11 let. i. Většina případů se zdála být nezávažné a dočasné povahy.
Popis vybraných nežádoucíchúčinkůMyokarditida a perikarditidaZvýšené riziko myokarditidy po očkování vakcínou Comirnaty je nejvyšší u mladších mužů a chlapců
(viz bod 4.4).
Zvýšené riziko u mladších mužů a chlapců po podání druhé dávky vakcíny Comirnaty bylo blíže určeno ve dvou velkých evropských farmakoepidemiologických studiích. Z jedné studie vyplynulo, že v období 7 dnů po podání druhé dávky se u mužů a chlapců ve věku 12–29 let vyskytlo přibližně o
0,265 (95% CI 0,255–0,275) případů myokarditidy na 10 000 osob více než u neočkovaných osob.
V další studii se v období 28 dnů po podání druhé dávky u mužů a chlapců ve věku 16–24 let vyskytlo o 0,56 (95% CI 0,37–0,74) případů myokarditidy na 10 000 osob více než u neočkovaných osob.
Omezené údaje ukazují, že riziko myokarditidy a perikarditidy je po očkování vakcínou Comirnaty
u dětí ve věku od 5 do 11 let pravděpodobně nižší než u dospívajících ve věku od 12 do 17 let.
Hlášení podezření na nežádoucí účinkyHlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to

pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích
účinků uvedeného v Dodatku V a uvedli přitom číslo šarže, je-li k dispozici.
4.9 PředávkováníÚdaje o předávkování jsou k dispozici od 52 účastníků studie zařazených do klinického hodnocení, kterým bylo kvůli chybě v ředění podáno 58 mikrogramů vakcíny Comirnaty. Příjemci vakcíny nehlásili zvýšení reaktogenity ani nežádoucí účinky.
V případě předávkování se doporučuje sledovat základní životní funkce a případně zahájit symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: vakcíny, jiné virové vakcíny, ATC kód: J07BX03
Mechanismus účinkuMediátorová (messenger) RNA s modifikovanými nukleosidy je ve vakcíně Comirnaty zapouzdřená
v lipidových nanočásticích, což umožňuje transfer nereplikující RNA do hostitelských buněk pro přímou přechodnou expresi S antigenu viru SARS-CoV-2. mRNA kóduje v membráně ukotvený
S v plné délce se dvěma bodovými mutacemi v centrální šroubovici. Mutace těchto dvou aminokyselin
na prolin uzamyká S v antigenně preferované prefuzní konformaci. Vakcína vyvolává jak odpověď
neutralizačních protilátek, tak i imunitní odpověď buněk na spike (S) antigen, což může přispívat
k ochraně před onemocněním COVID-19.
Účinnost
Studie 2 je multicentrická, mezinárodní, randomizovaná, placebem kontrolovaná studie fáze 1/2/3
zaslepená pro pozorovatele, která hodnotí dávku, výběr vakcíny a její účinnost u účastníků ve věku
12 let a starších. Randomizace byla stratifikována podle věku: věk 12 až 15 let, věk 16 až 55 let a věk
56 let a více; do věkové skupiny ≥ 56 let spadalo minimálně 40 % účastníků. Ze studie byli vyřazeni imunokompromitovaní účastníci a osoby, u nichž byla dříve stanovena klinická či mikrobiologická diagnóza onemocnění COVID-19. Účastníci s preexistujícími stabilizovanými onemocněními, definovanými jako onemocnění, jež během 6 týdnů před zařazením nevyžadovala významnou změnu léčby nebo hospitalizaci z důvodu zhoršení nemoci, byli do studie zařazeni stejně jako účastníci se známou stabilizovanou infekcí virem lidské imunodeficience (HIV), virem hepatitidy C (HCV) nebo virem hepatitidy B (HBV).
Účinnost u osobvevěku16let a starších – po 2 dávkách
Během fáze 2/3 Studie 2 bylo na základě dat získaných do 14. listopadu 2020 ve stejném poměru
randomizováno zhruba 44 000 účastníků a byly jim podány 2 dávky mRNA vakcíny proti onemocnění
COVID-19 nebo placeba. Analýzy účinnosti zahrnovaly účastníky, kteří dostali druhou dávku vakcíny v rozmezí 19 až 42 dnů po první dávce vakcíny. Většina (93,1 %) příjemců vakcíny dostala druhou dávku 19 dnů až 23 dnů po první dávce. Pro účely hodnocení bezpečnosti a účinnosti vakcíny proti onemocnění COVID-19 budou na základě připraveného plánu účastníci dále sledováni po dobu až
24 měsíců po 2. dávce. V klinické studii bylo od účastníků vyžadováno dodržení minimálního
intervalu 14 dní před a po podání vakcíny proti chřipce, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19. V klinické studii bylo od účastníků vyžadováno dodržení minimálního intervalu 60 dní před nebo po podání přípravků z krve/plazmy nebo imunoglobulinů v průběhu studie, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19.
Do populace pro analýzu primárního cílového parametru účinnosti bylo zařazeno 36 621 účastníků ve věku 12 let a starších (18 242 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 18 379 ve skupině s placebem), u nichž se během 7 dní po podání druhé dávky neprokázala předchozí infekce SARS-CoV-2. Kromě toho bylo 134 účastníků ve věku od 16 do 17 let (66 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 68 ve skupině s placebem) a 1 616 účastníků ve věku 75 let a starších (804 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 812 ve skupině s placebem).
V čase provedení primární analýzy účinnosti byli účastníci ve skupině mRNA vakcíny proti onemocnění COVID-19 sledováni z hlediska symptomatického onemocnění COVID-19 po celkovou dobu 2 214 osoboroků, účastníci ve skupině s placebem po dobu minimálně 2 222 osoboroků.
U účastníků s rizikem závažného onemocnění COVID-19, včetně osob s 1 nebo více komorbiditami,
které riziko závažného onemocnění COVID-19 zvyšují (např. astma, index tělesné hmotnosti (BMI)
≥ 30 kg/m2, chronické plicní onemocnění, diabetes mellitus, hypertenze), nebyly zjištěny žádné
významné klinické rozdíly v celkové účinnosti vakcíny.
Informace o účinnosti vakcíny jsou uvedeny v tabulce 2.
Tabulka 2: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů)První výskyt COVID-19 od 7 dnů po 2. dávce u účastníků bez důkazu předchozí infekce SARS- CoV-2 *
|
Podskupina
| Vakcína mRNA proti
onemocnění COVID-19
na=18 198
Případy
n1b
Doba sledováníc (n2d)
|
Placebo na=18 325
Případy
n1b
Doba sledováníc (n2d)
|
Účinnost vakcíny % (95% CI)e
|
Všichni účastníci
| 8
2,214 (17 411)
| 162
2,222 (17 511)
| 95,0
(90,0; 97,9)
|
16 až 64 let
| 7
1,706 (13 549)
| 143
1,710 (13 618)
| 95,1
(89,6; 98,1)
|
65 let a starší
| 1
0,508 (3 848)
| 19
0,511 (3 880)
| 94,7
(66,7; 99,9)
|
65 až 75 let
| 1
0,406 (3 074)
| 14
0,406 (3 095)
| 92,9
(53,1; 99,8)
|
75 let a starší
| 0
0,102 (774)
| 5
0,106 (785)
| 100,0
(-13,1; 100,0)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 [*Definice případu: (alespoň 1 z) horečka, nový nebo zhoršený kašel, nová nebo zhoršená dyspnoe, zimnice, nová nebo zhoršená bolest svalů, nová ztráta chuti nebo čichu, bolest v krku, průjem nebo zvracení.]
* Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (7 nebo více dnů před podáním poslední dávky) předcházející infekce SARS-CoV-2 (tj. N-vazebná protilátka [sérum] negativní při návštěvě 1 a SARS- CoV-2 nedetekován pomocí amplifikačních testů nukleových kyselin (NAAT) [výtěr nosu] při návštěvách 1 a 2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do analýzy.
a. n = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování. CI není upraven na multiplicitu.
Účinnost vakcíny mRNA proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění COVID-19 po 7 dnech po 2. dávce ve srovnání s placebem 94,6 % (95% interval spolehlivosti 89,6 % až 97,6 %) u účastníků ve věku 16 let a starších s nebo bez důkazů o předchozí infekci virem SARS- CoV-2.
Analýzy podskupin primárního cílového parametru účinnosti navíc ukázaly podobné odhady bodů účinnosti u pohlaví, etnických skupin a účastníků s komorbiditami spojenými s vysokým rizikem závažného onemocnění COVID-19.
Byly provedeny aktualizované analýzy účinnosti s dalšími potvrzenými případy COVID-19, které se objevily během zaslepeného placebem kontrolovaného sledování, což v populaci s účinností představuje až 6 měsíců po podání 2. dávky.
Aktualizované informace o účinnosti vakcíny jsou uvedeny v tabulce 3.
Podskupina
|
V
akcína mRNA proti
onemocnění COVID-19
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Placebo
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Ú
č
i
n
nost vakcíny % (95% CI
e
)
|
Všichni účastnícif
|
77
6,247 (20 712)
|
850
6,003 (20 713)
|
91,3
(89,0; 93,2)
|
16 až 64 let
|
70
4,859 (15 519)
|
710
4,654 (15 515)
|
90,6
(87,9; 92,7)
|
65 let a starší
|
7
1,233 (4 192)
|
124
1,202 (4 226)
|
94,5
(88,3; 97,8)
|
65 až 74 let
|
6
0,994 (3 350)
|
98
0,966 (3 379)
|
94,1
(86,6; 97,9)
|
75 let a starší
|
1
0,239 (842)
|
26
0,237 (847)
|
96,2
(76,9; 99,9)
|
|
|
T
abulka 3: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce předchozí infekce virem SARS- CoV-2* před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů) během placebem kontrolovaného období sledování
N =20 998
N =21 096
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-
PCR) a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu
chuti nebo čichu, bolest v krku, průjem nebo zvracení).
* Účastníci, kteří neměli žádné důkazy předcházející infekce virem SARS-CoV-2 (tj. N-vazebná protilátka
[sérum] negativní při návštěvě 1 a virus SARS-CoV-2 nedetekován pomocí NAAT [výtěr nosu] při návštěvách 1 a
2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do
analýzy.
a. N = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný 95% interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a
Pearsonovy metody upravené podle doby sledování. CI není upraven na multiplicitu.
f. Zahrnuté potvrzené případy u účastníků ve věku 12 až 15 let: 0 ve skupině s mRNA vakcínou proti
onemocnění COVID-19; 16 ve skupině s placebem.
V aktualizované analýze účinnosti byla účinnost mRNA vakcíny proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění COVID-19 od 7 dnů po podání 2. dávky ve srovnání s placebem 91,1 % (95% CI 88,8 % až 93,0 %) u účastníků v hodnotitelné populaci účinnosti s průkazem nebo bez průkazu předchozí infekce SARS-CoV-2.
Aktualizované analýzy účinnosti podle podskupin navíc ukázaly podobné bodové odhady účinnosti u všech pohlaví, etnických skupin, zeměpisných oblastí a účastníků se zdravotními komorbiditami a obezitou spojenou s vysokým rizikem závažného onemocnění COVID-19.
Účinnost proti závažnému onemocnění COVID-19
Aktualizované analýzy účinnosti sekundárních cílových parametrů účinnosti podpořily přínos mRNA
vakcíny proti onemocnění COVID-19 v prevenci závažného onemocnění COVID-19.
Od 13. března 2021 je účinnost vakcíny proti závažnému onemocnění COVID-19 prezentována pouze pro účastníky s předchozí infekcí SARS-CoV-2 nebo bez ní (tabulka 4), protože počty případů onemocnění COVID-19 u účastníků bez předchozí infekce SARS-CoV-2 byly stejné jako u účastníků
s předchozí infekcí SARS-CoV-2 nebo bez ní jak ve skupině s mRNA vakcínou proti onemocnění
COVID-19, tak ve skupině s placebem.
| Vakcína mRNA proti
onemocnění COVID-19
Případy
n1a
Doba sledováníc (n2b)
|
Placebo Případy n1a
Doba sledováníc (n2b)
|
Účinnost vakcíny % (95% CIc)
| Po 1. dávced
| 1
8,439e (22 505)
| 30
8,288e (22 435)
| 96,7
(80,3; 99,9)
| 7 dnů po 2. dávcef
| 1
6,522g (21 649)
| 21
6,404g (21 730)
| 95,3
(70,9; 99,9)
|
|
|
Tabulka 4: Účinnost vakcíny – první závažný výskyt onemocnění COVID-19 u účastníků s nebo bez předchozí infekce virem SARS-CoV-2 na základě údajů Úřadu pro kontrolu potravin a léčiv (FDA)* po podání 1. dávky nebo od 7 dnů po podání 2. dávky v placebem kontrolovaném období sledováníPoznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení.)
* Závažné onemocnění COVID-19 podle definice FDA je potvrzené onemocnění COVID-19 a přítomnost alespoň 1 z následujících příznaků:
• Klinické příznaky v klidu svědčící pro závažné systémové onemocnění (dechová frekvence ≥ 30 dechů za minutu, srdeční frekvence ≥ 125 tepů za minutu, saturace kyslíkem ≤ 93 % na pokojovém vzduchu při hladině moře nebo poměr arteriálního parciálního tlaku kyslíku k frakčnímu inspirovanému kyslíku
< 300 mmHg).
• Respirační selhání [definované jako potřeba vysokého průtoku kyslíku, neinvazivní ventilace,
mechanické ventilace nebo extrakorporální membránové oxygenace (ECMO)].
• Průkaz šoku (systolický krevní tlak < 90 mmHg, diastolický krevní tlak < 60 mmHg nebo potřeba
podání vazopresorických látek).
• Významná akutní renální, jaterní nebo neurologická dysfunkce.
• Přijetí na jednotku intenzivní péče.
• Smrt.
a. n1 = Počet účastníků splňujících definici cílového parametru.
b. n2 = počet účastníků ohrožených daným cílovým parametrem.
c. Dvoustranný interval spolehlivosti (CI) pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování.
d. Účinnost hodnocená na základě veškeré populace pro hodnocení účinnosti s 1. dávkou (modifikovaná se záměrem léčit), která zahrnovala všechny randomizované účastníky, kteří dostali alespoň 1 dávku hodnocené léčby.
e. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od po 1. dávce do konce období sledování.
f. Účinnost hodnocená na základě populace hodnotitelné pro účinnost (7 dní), která zahrnovala všechny způsobilé randomizované účastníky, kteří dostali všechny dávky hodnocené léčby podle randomizace v rámci předem stanoveného časového období a neměli žádné další důležité odchylky od protokolu podle rozhodnutí
lékaře.
g. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro daný cílový parametr. Časové období pro získání případu onemocnění COVID-19 je od
7 dnů po 2. dávce do konce období sledování.
Účinnost a imunogenicita udospívajícíchvevěku12až15let – po 2 dávkáchV úvodní analýze Studie 2 u dospívajících ve věku 12 až 15 let (jež reprezentovala medián doby
následného sledování v délce ˃ 2 měsíce po 2. dávce) bez průkazu prodělané infekce nebyly žádné případy u 1 005 účastníků, kteří dostali vakcínu, a 16 případů z 978 účastníků, kteří dostali placebo.
Bodový odhad účinnosti je 100 % (95% interval spolehlivosti 75,3; 100,0). U účastníků s nebo bez
průkazu prodělané infekce bylo 0 případů z 1 119 účastníků, kteří dostali vakcínu, a 18 případů
z 1 110 účastníků, kteří dostali placebo. To také naznačuje, že bodový odhad účinnosti je 100 % (95%
interval spolehlivosti 78,1; 100,0).
U dodatečných potvrzených případů onemocnění COVID-19, k nimž došlo během zaslepeného, placebem kontrolovaného následného sledování, byly provedeny aktualizované analýzy účinnosti, jež reprezentovaly dobu až 6 měsíců po 2. dávce v populaci pro hodnocení účinnosti.
V aktualizované analýze účinnosti ve studii 2, provedené u dospívajících ve věku 12 až 15 let bez průkazu prodělané infekce, se u 1 057 účastníků, kterým byla podána vakcína, nevyskytl žádný případ a u 1 030 účastníků, kterým bylo podáno placebo, došlo ke 28 případům. Odhad bodu účinnosti je
100 % (95% interval spolehlivosti 86,8; 100,0). U účastníků s průkazem prodělané infekce nebo bez
tohoto průkazu nedošlo u 1 119 těch, kterým byla podána vakcína, k žádnému případu
a u 1 109 účastníků, jimž bylo podáno placebo, ke 30 případům. To rovněž naznačuje, že odhad bodu účinnosti je 100 % (95% interval spolehlivosti 87,5; 100,0).
Ve Studii 2 byla provedena analýza neutralizačních titrů SARS-CoV-2 jeden měsíc po druhé dávce u náhodně vybrané podskupiny účastníků, kteří do 1 měsíce po dávce 2 neměli sérologické nebo virologické průkazy prodělané infekce SARS-CoV-2, která porovnávala odpověď u dospívajících ve věku 12 až 15 let (n = 190) s účastníky ve věku 16 až 25 let (n = 170).
Poměr geometrických průměrných titrů (GMT) ve skupině ve věku 12 až 15 let ke skupině ve věku 16 až 25 let byl 1,76; s oboustranným 95% CI 1,47 až 2,10. Proto bylo splněno 1,5násobné kritérium noninferiority, protože dolní mez 2stranného 95% CI pro poměr geometrického průměru [GMR] byla
> 0,67.
Účinnost a imunogenicita udětívevěku5až11let(tj.vevěku5ažméněnež12let) – po 2 dávkáchStudie 3 je studie fáze 1/2/3, která se skládá z otevřené části zaměřené na zjištění dávky vakcíny
(fáze 1) a multicentrické, mezinárodní, randomizované, fyziologickým roztokem jako placebem
kontrolované části zaslepené pro pozorovatele hodnotící účinnost (fáze 2/3), do které byli zařazeni účastníci ve věku 5 až 11 let. Většina (94,4 %) randomizovaných příjemců vakcíny dostala druhou
dávku 19 dní až 23 dní po první dávce.
Deskriptivní výsledky účinnosti vakcíny u dětí ve věku 5 až 11 let bez známek předchozí infekce virem SARS-CoV-2 jsou uvedeny v tabulce 5. U účastníků s prokázanou předchozí infekcí virem SARS-CoV-2 nebyly pozorovány žádné případy onemocnění COVID-19 ani ve skupině s vakcínou, ani ve skupině s placebem.
Tabulka 5: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce: bez průkazu infekce před 7 dny po 2. dávce – fáze 2/3 – děti ve věku 5 až 11 let hodnotitelná populace pro účinnostPrvní výskyt onemocnění COVID-19 od 7 dnů po 2. dávce u dětí ve věku 5 až 11 let bez
průkazu předchozí infekce SARS-CoV-2*
|
| Vakcína mRNA proti
onemocnění
COVID-19
Dávka 10 mikrogramů/dávku Na=1 305
Případy
n1b
Doba sledováníc (n2d)
|
Placebo
Na=663
Případy
n1b
Doba sledováníc (n2d)
|
Účinnost vakcíny
% (95% CI)
|
Děti ve věku 5 až 11 let
| 3
0,322 (1273)
| 16
0,159 (637)
| 90,7
(67,7; 98,3)
|
Poznámka: Potvrzené případy byly určeny na základě reverzní transkripční polymerázové řetězové reakce (RT-
PCR) a alespoň 1 příznaku odpovídajícího onemocnění COVID-19 (příznaky zahrnovaly: horečku; nový nebo

zesílený kašel; novou nebo zesílenou dušnost; zimnici; novou nebo zesílenou bolest svalů; novou ztrátu chuti nebo čichu; bolest v krku; průjem; zvracení).
* Do analýzy byli zahrnuti účastníci, kteří neměli žádný průkaz o prodělané infekci virem SARS-CoV-2 (tj. negativní N-vázané protilátky [sérum] při návštěvě 1 a virus SARS-CoV-2 nezjištěný pomocí NAAT [nosní výtěr] při návštěvě 1 a 2) a měli negativní NAAT (nosní výtěr) při jakékoli neplánované návštěvě před 7 dny po 2. dávce
a. N = počet účastníků v dané skupině.
b. n1 = počet účastníků splňujících definici cílového ukazatele.
c. Celková doba sledování v 1 000 osoborocích pro daný cílový ukazatel u všech účastníků v každé skupině s rizikem pro cílový ukazatel. Časové období pro nárůst případů onemocnění COVID-19 je od 7 dnů po podání 2. dávky do konce období sledování.
d. n2 = počet účastníků s rizikem pro daný cílový ukazatel.
Ve studii 3 prokázala analýza 50% neutralizačních titrů viru SARS-CoV-2 (NT50) 1 měsíc po podání
2. dávky u náhodně vybrané podskupiny účastníků účinnost imunologickým přemostěním imunitních odpovědí při srovnání dětí ve věku 5 až 11 let (tj. ve věku 5 až méně než 12 let) v části studie 3 fáze
2/3 s účastníky ve věku 16 až 25 let v části studie 2 fáze 2/3, kteří neměli žádné sérologické nebo
virologické průkazy o prodělané infekci virem SARS-CoV-2 do 1 měsíce po podání 2. dávky, což splňuje předem stanovená kritéria imunologického přemostění jak pro poměr geometrického průměru (GMR), tak pro rozdíl sérologických odpovědí, přičemž sérologická odpověď byla definována jako dosažení alespoň čtyřnásobného zvýšení NT50 SARS-CoV-2 oproti výchozí hodnotě (před podáním 1. dávky).
GMR NT50 SARS-CoV-2 1 měsíc po 2. dávce u dětí ve věku 5 až 11 let (tj. ve věku 5 až méně než
12 let) proti mladým dospělým ve věku 16 až 25 let byl 1,04 (2stranný 95% CI: 0,93; 1,18). Mezi
účastníky bez předchozích známek infekce virem SARS-CoV-2 do 1 měsíce po podání 2. dávky mělo
99,2 % dětí ve věku 5 až 11 let a 99,2 % účastníků ve věku 16 až 25 let sérologickou odpověď 1 měsíc po podání 2. dávky V případě dětí ve věku 5 až 11 let a mladých lidí ve věku 16 až 25 let byla
sérologická odpověď prokázána až po 1 měsíci po podání 2. dávky Rozdíl v podílech účastníků, kteří
měli sérologickou odpověď mezi dvěma věkovými skupinami (děti – mladí dospělí), byl 0,0 % (2stranný 95% CI: -2,0 %; 2,2 %). Tyto informace jsou uvedeny v tabulce 6.
T
abulka 6: Souhrn geometrického průměrného poměru pro 50% neutralizační titr a rozdíl v procentech účastníků se sérologickou odpovědí – srovnání dětí ve věku 5 až 11 let (studie 3) s účastníky ve věku 16 až 25 let (studie 2) – účastníci bez známek infekce
do 1 měsíce po podání 2. dávky – podskupina imunologického přemostění – fáze 2/3 –
populace hodnotitelná na imunogenicitu
|
V
akcína mRNA proti onemocnění
COV
I
D
-19
|
5 až 11 let /
16 až 25 let
|
D
ávka
10 mikrogramů/dávku
5 až 11 let
N
a
=
264
|
D
ávka
30 mikrogramů/dáv
ku
16 až 25 let
N
a
=
253
|
|
Č
asový
bod
b
|
G
MT
c
(
95% CI
c
)
|
G
MT
c
(
95% CI
c
)
|
G
MR
d
(
95% CI
d
)
|
Splnila cíl
im
unologické ho přemostění
e
(
A
/
N)
|
G
eometrický
průměrný
50% neutralizační titr
f
(
G
MT
c
)
|
1 měsíc po 2. dávce
|
1 197,6
(1 106,1; 1 296,6)
|
1 146,5
(1 045,5; 1 257,2)
|
1,04 (0,93; 1,18)
|
A
|
|
Č
asový
bod
b
|
n
g
(
%)
(
95% CI
h
)
|
n
g
(
%)
(
95% CI
h
)
|
R
ozdíl %
i
(
95% CI
j
)
|
Splnila cíl imunologické
ho přemostění
k
(
A
/
N)
|
Č
etnost
sérologické
odpovědi (%) pro 50% neutralizační titr
f
|
1 měsíc po 2. dávce
|
262 (99,2) (97,3; 99,9)
|
251 (99,2) (97,2; 99,9)
|
0,0
(-2,0; 2,2)
|
A
|
Zkratky: CI = interval spolehlivosti; GMR = geometrický průměrný poměr; GMT = geometrický průměrný titr;
LLOQ = dolní mez kvantifikace; NAAT = test amplifikace nukleových kyselin; NT50 = 50% neutralizační titr;
virus SARS-CoV-2 = těžký akutní respirační syndrom vyvolaný koronavirem 2.
Poznámka: Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (do 1 měsíce po odběru vzorku krve po 2. dávce) o prodělané infekci virem SARS-CoV-2 (tj, N-vázané protilátky [sérum] negativní při návštěvě s podáním 1. dávky a 1 měsíc po 2. dávce, virus SARS-CoV-2 nezjištěn pomocí NAAT [nosní výtěr] při návštěvě s podáním 1. dávky a 2. dávky a negativní NAAT (nosní výtěr) při kterékoli neplánované návštěvě do 1 měsíce po odběru krve po 2. dávce) a neměli v anamnéze žádné onemocnění COVID-19, byli zahrnuti do analýzy.
Poznámka: Sérologická odpověď je definována jako dosažení ≥ 4násobného zvýšení oproti výchozí hodnotě (před 1. dávkou). Pokud je výchozí měření nižší než LLOQ, považuje se výsledek testu po očkování ≥4× LLOQ za sérologickou odpověď.
a. N = počet účastníků s platnými a určitelnými výsledky testu před očkováním a 1 měsíc po podání 2. dávky.
Tyto hodnoty jsou také jmenovateli použitými při výpočtech procentuální četnosti sérologické odpovědi.
b. Protokolárně stanovené načasování odběru krevních vzorků.
c. GMT a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a odpovídajících CI (na základě Studentova t rozložení). Výsledky testů pod LLOQ byly stanoveny na 0,5 × LLOQ.
d. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (věk 5 až 11 let minus věk 16 až 25 let) a odpovídajícího CI (na základě Studentova t rozložení).
e. Imunologické přemostění podle GMT je deklarováno, pokud je dolní hranice dvoustranného 95% CI pro
GMR větší než 0,67 a bodový odhad GMR je ≥ 0,8.
f. NT50 SARS-CoV-2 byly stanoveny pomocí mikroneutralizačního testu SARS-CoV-2 mNeonGreen Virus
Assay. Test používá fluorescenční reportérový virus odvozený od kmene USA_WA1/2020 a neutralizace
viru se odečítá na monovrstvách buněk Vero. Vzorek NT50 je definován jako vzájemné zředění séra, při
kterém je neutralizováno 50 % viru.
g. n = počet účastníků se sérologickou odpovědí na základě NT50 1 měsíc po podání 2. dávky.
h. Přesný dvoustranný CI na základě Clopperovy a Pearsonovy metody.
i. Rozdíl v podílech vyjádřený v procentech (věk 5 až 11 let minus věk 16 až 25 let).
j. Přesný 2stranný CI založený na metodě Miettinena a Nurminena pro rozdíl v podílech, vyjádřený v
procentech.
k. Imunologické přemostění založené na četnosti sérologické odpovědi je deklarováno, pokud je dolní hranice
2stranného 95% CI pro rozdíl sérologické odpovědi větší než 10,0 %.
Imunogenicita u dětívevěkuod5do11let(tj.vevěku5ažméněnež12let) – po posilovací dávcePosilovací dávka vakcíny Comirnaty byla ve studii 3 podána 401 náhodně vybraným účastníkům.
Účinnost posilovací dávky ve věku 5 až 11 let je odvozena z údajů o imunogenicitě. Imunogenicita byla hodnocena podle NT50 u referenčního kmene SARS-CoV-2 (USA_WA1/2020). Analýzy NT50 za 1 měsíc po posilovací dávce v porovnání se stavem před posilovací dávkou prokázaly podstatné zvýšení GMT u jedinců ve věku od 5 do 11 let včetně, kteří neměli sérologický ani virologický průkaz předchozí infekce virem SARS-CoV-2 do 1 měsíce po 2. dávce a po posilovací dávce. Tato analýza je shrnuta v tabulce 7.
| Časový bod odběrua
|
|
Test
|
1 měsíc po posilovací
dávce (nb=67)
GMTc
(95% CIc)
|
1 měsíc po 2. dávce
(nb=96)
GMTc
(95% CIc)
| 1 měsíc po posilovací
dávce /
1 měsíc po 2. dávce
GMRd
(95% CId)
| SARS-CoV-2
neutralizační test - NT50 (titr)
|
2 720,9
(2 280,1; 3 247,0)
|
1 253,9
(1 116,0; 1 408,9)
|
2,17 (1,76; 2,68)
| Zkratky: CI = interval spolehlivosti; GMR = geometrický průměr poměrů; GMT = geometrický průměr titrů;
LLOQ = dolní mez kvantifikace; NT50 = 50% neutralizační titr; virus SARS-CoV-2 = těžký akutní respirační
syndrom vyvolaný koronavirem 2.
a. Protokolárně stanovené načasování odběru krevních vzorků.
b. n = počet účastníků s platnými a konečnými výsledky testu pro specifický test v časovém bodě podání dávky/odběru.
c. GMT a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů
a odpovídajících CI (na základě Studentova t rozložení). Výsledky testů pod LLOQ byly stanoveny na
0,5 × LLOQ.
d. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (1 měsíc po posilovací dávce minus 1 měsíc po 2. dávce) a odpovídajícího CI (na základě Studentova t rozložení).
|
|
|
Tabulka 7: Souhrn geometrického průměru titrů – NT50 – účastníci bez průkazu infekce – fáze 2/3 – soubor imunogenicity – věk 5 až 11 let včetně – populace hodnotitelná na imunogenicituPediatrická populaceEvropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s vakcínou
Comirnaty u pediatrické populace k prevenci onemocnění COVID-19 (informace o použití u pediatrické populace viz bod 4.2).
5.2 Farmakokinetické vlastnostiNeuplatňuje se.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních studií toxicity po opakovaném podávání a reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
Obecná toxicita
U potkanů, kterým byla intramuskulárně podávána vakcína Comirnaty (dostali 3 plné dávky pro
člověka jednou týdně, které vedly k relativně vyšším hladinám u potkanů v důsledku rozdílů v tělesné hmotnosti), se vyskytl edém a erytém v místě podání injekce a zvýšení počtu bílých krvinek (včetně bazofilů a eozinofilů) odpovídající zánětlivé odpovědi a rovněž vakuolizace portálních hepatocytů bez známek poškození jater. Všechny účinky byly reverzibilní.
Genotoxicita / karcinogenita
Studie genotoxicity ani kancerogenity nebyly provedeny. U složek vakcíny (lipidy a mRNA) se
neočekává genotoxický potenciál.
Reprodukční toxicita
Reprodukční a vývojová toxicita byla hodnocena na potkanech v kombinované studii fertility a
vývojové toxicity, ve které byla samicím potkanů podána intramuskulárně vakcína Comirnaty před pářením a během březosti (dostaly 4 plné dávky pro člověka, které vedly k relativně vyšším hladinám
u potkanů v důsledku rozdílů v tělesné hmotnosti mezi 21. dnem před pářením a 20. dnem březosti). Odpovědi na neutralizační protilátky SARS-CoV-2 byly přítomny u samic před pářením až do konce
studie ve 21. postnatálním dni a rovněž u plodů a potomků. Nebyly zjištěny žádné účinky spojené s očkováním na plodnost samic, těhotenství nebo vývoj embrya/plodu nebo potomstva. Údaje o
možném placentárním přenosu nebo vylučování do mateřského mléka pro vakcínu Comirnaty nejsou k dispozici.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159) Kolfosceryl-stearát
Cholesterol
Trometamol
Trometamol-hydrochlorid
Sacharosa
Voda pro injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou
uvedeny v bodě 6.6.
6.3 Doba použitelnosti
N
eotevřená injekčnílahvička
Zmrazenáinjekčnílahvička
18 měsíců při teplotě -90 °C až -60 °C.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána při teplotě -90 °C až -60 °C nebo 2 °C až 8 °C.
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení
10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 4 hodin nebo mohou být jednotlivé
injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Rozmrazená injekčnílahvička
10 týdnů uchovávání a transportu při teplotě 2 °C až 8 °C během 18měsíční doby použitelnosti.
• Po přenesení vakcíny pro uchovávání při teplotě 2 °C až 8 °C musí být na vnější obal zapsáno aktualizované datum použitelnosti a vakcína má být použita nebo zlikvidována do aktualizovaného data použitelnosti. Původní datum použitelnosti má být přeškrtnuto.
• Pokud je vakcína přijata při teplotě 2 °C až 8 °C, má být uchovávána při teplotě 2 °C až 8 °C.
Datum použitelnosti na vnějším obalu má být aktualizováno tak, aby odráželo datum použitelnosti v chladu, a původní datum použitelnosti má být přeškrtnuto.
Neotevřené injekční lahvičky lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Po rozmrazení vakcína nesmí být znovu zmrazena.
Manipulace přiteplotníchvýkyvechpřiuchovávánívchladničce
• Údaje o stabilitě prokazují, že je neotevřená injekční lahvička stabilní až 10 týdnů, je-li
uchovávána při teplotě od -2 °C do 2 °C a během 10týdenního uchovávání při teplotě 2 °C až
8 °C.
• Údaje o stabilitě ukazují, že injekční lahvička může být uchovávána až 24 hodin při teplotě od
8 °C do 30 °C, včetně až 12 hodin po prvním vpichu.
Tyto informace slouží jako návod pro zdravotnické pracovníky pouze v případě dočasného výkyvu
teploty.
Zředěný léčivý přípravek
Chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 12 hodin při teplotě
2 °C až 30 °C po naředění injekčním roztokem chloridu sodného 9 mg/ml (0,9%), která zahrnuje až
6 hodin transportu. Z mikrobiologického hlediska, pokud způsob ředění nevyloučí riziko mikrobiologické kontaminace, má být přípravek použit okamžitě. Pokud není použit okamžitě, doba a
podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Během uchovávání je třeba minimalizovat vystavení přípravku světlu v místnosti a zabránit vystavení
přímému slunečnímu světlu a ultrafialovému světlu.
Podmínky uchovávání tohoto léčivého přípravku po jeho rozmrazení a naředění jsou uvedeny
v bodě 6.3.
6.5 Druh obalu a obsah balení
1,3 ml koncentrátu pro disperzi v čiré vícedávkové injekční lahvičce (sklo třídy I) o objemu 2 ml se zátkou (syntetická brombutylová pryž) a oranžovým odtrhovacím plastovým víčkem s hliníkovým krytem. Jedna injekční lahvička obsahuje 10 dávek, viz bod 6.6.
Velikosti balení: 10 injekčních lahviček nebo 195 injekčních lahviček
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Instrukce pro zacházení s vakcínou
Vakcína Comirnaty 10 mikrogramů/dávku má být připravována zdravotnickým pracovníkem pomocí
aseptické techniky, aby byla zajištěna sterilita připravené disperze.
KONTROLA INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY
10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
O
ranžové
víčko
 P
o naředění
10 mikrogramů
P
o naředění
10 mikrogramů
• Zkontrolujte, zda má injekční lahvička oranžové plastové víčko a oranžový okraj kolem štítku a název přípravku je Comirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko a oranžový okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro injekční disperzi, přečtěte si souhrn údajů
o přípravku pro toto složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička šedé
plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku injekční disperze, Comirnaty Original/Omicron BA.1
(15/15 mikrogramů)/dávku injekční
disperze nebo Comirnaty
Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi.
Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY 10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET) PŘED POUŽITÍM
|
U
chovávejte až
10 týdnů při teplotě 2 °C až
8 °C.
|
• Pokud se vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se rozmrazily; rozmrazení balení 10 injekčních lahviček může trvat 4 hodiny. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
MÍCHÁNÍ VAKCÍNY COMIRNATY 10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO
I
N
JEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET) PŘED ŘEDĚNÍM
|
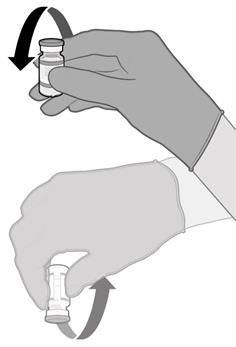
Jemně × 10
|
• Rozmrazenou injekční lahvičku nechte ohřát na pokojovou teplotu a před ředěním ji 10krát jemně převraťte. Netřepejte.
• Před ředěním může rozmrazená disperze obsahovat bílé až téměř bílé matné amorfní částice.
|
ŘEDĚN
Í VAKCÍNY COMIRNATY 10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
|
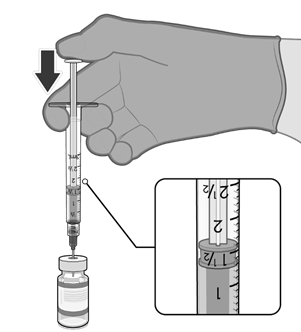
1,3 ml injekčního roztoku chloridu
sodného 9 mg/ml (0,9 %)
|
• Rozmrazená vakcína se musí naředit v původní injekční lahvičce 1,3 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%) za použití jehly
o velikosti 21 gauge (21G) nebo užší a uplatnění aseptických metod.
|

V
ytáhněte píst na značku 1,3 ml pro
odstranění vzduchu z injekční lahvičky.
|
• Nasátím 1,3 ml vzduchu do prázdné stříkačky na rozpouštědlo vyrovnejte před vyjmutím jehly z injekční lahvičky tlak v injekční lahvičce.
|

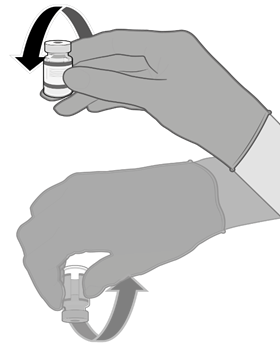
Jemně x 10
|
• Disperzi 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Naředěná vakcína se má jevit jako bílá až téměř bílá disperze bez viditelných
částic. Jestliže jsou v naředěné vakcíně
patrné částice nebo vakcína změnila
barvu, nepoužívejte ji.
|
Č
as likvidace
Z
a
z
namenejte příslušné datum a čas. Použijte během 12 hodin po naředění.
|
• Injekční lahvičky s naředěnou vakcínou je třeba označit příslušným datem
a časem.
• Po naředění uchovávejte vakcínu při teplotě 2 °C až 30 °C a použijte ji během
12 hodin.
• Naředěnou disperzi nezmrazujte ani s ní netřepejte. Pokud je v chladu, nechte naředěnou disperzi před použitím dosáhnout pokojové teploty.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,2 ml VAKCÍNY COMIRNATY
10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
 N
aředěná vakcína o objemu 0,2 ml
N
aředěná vakcína o objemu 0,2 ml
• Po naředění obsahuje injekční lahvička
2,6 ml, ze kterých lze získat 10 dávek po
0,2 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím
antiseptického tampónu.
• Natáhněte 0,2 ml vakcíny Comirnaty pro
děti ve věku 5 až 11 let.
K získání 10 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně
35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání deseti dávek z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,2 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,2 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Veškerou vakcínu, jež nebyla použita do
12 hodin po naředění, zlikvidujte.
L
i
kv
i
dace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.de8. REGISTRAČNÍ ČÍSLO/REGISTRAČNÍ ČÍSLAEU/1/20/1528/004
EU/1/20/1528/005
9. DATUM REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. prosince 2020
Datum posledního prodloužení registrace: 10. října 2022
10. DATUM REVIZE TEXTUPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky.
Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKUComirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi
mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍJedná se o vícedávkovou injekční lahvičku s hnědočerveným víčkem, jejíž obsah je nutno před použitím naředit.
Jedna injekční lahvička (0,4 ml) obsahuje 10 dávek po 0,2 ml po naředění, viz body 4.2 a 6.6.
Jedna dávka (0,2 ml) obsahuje 3 mikrogramy tozinameranu, mRNA vakcíny proti onemocnění
COVID-19 (zapouzdřené v lipidových nanočásticích).
Tozinameran je jednovláknová mediátorová (messenger) RNA (mRNA) s čepičkou na 5´ konci vyráběná
in vitro nebuněčnou transkripcí z příslušných matricí DNA a kódující spike (S) protein viru SARS-CoV-2.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMAKoncentrát pro injekční disperzi (sterilní koncentrát).
Vakcína je bílá až téměř bílá zmrazená disperze (pH: 6,9–7,9).
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikaceVakcína Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi je indikována pro aktivní imunizaci k prevenci onemocnění COVID-19 způsobeného SARS-CoV-2 u kojenců a dětí ve věku od
6 měsíců do 4 let.
Tuto vakcínu je třeba používat v souladu s oficiálními doporučeními.
4.2 Dávkování a způsob podáníDávkováníKojenci a dětivevěkuod6měsícůdo4letVakcína Comirnaty 3 mikrogramy/dávku se podává intramuskulárně po naředění jako základní
očkování 3 dávkami (0,2 ml každá dávka). Druhou dávku se doporučuje podat 3 týdny po první dávce a následně podat třetí dávku nejdříve 8 týdnů po druhé dávce (viz body 4.4 a 5.1).
Pokud dítě mezi jednotlivými dávkami základního očkování dosáhne věku 5 let, má sérii dokončit se
stejnou dávkou 3 mikrogramy.
Zaměnitelnost
Nebylo stanoveno, zda lze za účelem dokončení základního očkování zaměnit vakcínu Comirnaty za
vakcíny proti onemocnění COVID-19 od jiných výrobců. Osoby, jimž byla podána dávka vakcíny
Comirnaty, mají pokračovat v podávání vakcíny Comirnaty, aby bylo základní očkování dokončeno.
Pediatrická populace
K dispozici je pediatrická léková forma pro jedince ve věku od 5 do 11 let (tj. od 5 do méně než
12 let). Podrobné informace naleznete v souhrnu údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi.
Bezpečnost a účinnost vakcíny Comirnaty u kojenců ve věku do 6 měsíců nebyly dosud stanoveny.
Způsob podání
Vakcína Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi se podává intramuskulárně
po naředění (viz bod 6.6).
Po naředění obsahují injekční lahvičky vakcíny Comirnaty 10 dávek po 0,2 ml vakcíny. K získání
10 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů. Pokud se používají standardní injekční stříkačky a jehly, nemusí být dostatečný objem k získání 10 dávek z jedné injekční lahvičky. Bez ohledu na typ injekční stříkačky a jehly:
• Každá dávka musí obsahovat 0,2 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,2 ml,
injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Nekombinujte obsah z více injekčních lahviček vakcíny.
U kojenců ve věku od 6 do méně než 12 měsíců je doporučeným místem vpichu anterolaterální strana stehna. U jedinců od 1 roku věku je doporučeným místem vpichu anterolaterální strana stehna nebo deltový sval.
Vakcína se nesmí podávat intravaskulárně, subkutánně ani intradermálně.
Vakcína se nesmí mísit ve stejné injekční stříkačce s jinými vakcínami nebo léčivými přípravky. Pro opatření před podáním vakcíny viz bod 4.4.
Návod pro rozmrazení, zacházení s vakcínou a její likvidaci je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Sledovatelnost
Aby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
O
becná doporučení
Hypersenzitivita a anafylaxe
Byly hlášeny případy anafylaxe. Pro případ, že by po podání vakcíny došlo k anafylaktické reakci, má
být zajištěna okamžitá lékařská péče a dohled.
Po vakcinaci se doporučuje pečlivé sledování po dobu minimálně 15 minut. Další dávka vakcíny nemá
být podána osobám, které měly anafylaxi po předchozí dávce vakcíny Comirnaty.
Myokarditida a perikarditida
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy a perikarditidy. Tato
onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhé dávce vakcíny a častěji u mladších mužů a chlapců. Dostupné údaje naznačují, že průběh myokarditidy a perikarditidy po vakcinaci se neliší od myokarditidy nebo perikarditidy obecně (viz bod 4.8).
Zdravotničtí pracovníci mají pozorně sledovat známky a příznaky myokarditidy a perikarditidy. Očkovaní jedinci (včetně rodičů a pečovatelů) mají být poučeni, aby okamžitě vyhledali lékařskou pomoc, pokud se u nich po očkování objeví příznaky naznačující myokarditidu nebo perikarditidu, například bolest na hrudi (akutní a přetrvávající), dušnost nebo palpitace.
Zdravotničtí pracovníci mají k diagnostice a léčbě tohoto onemocnění používat návody a postupy
a/nebo se mají obrátit na specialisty.
Reakce spojené s úzkostí
V souvislosti se samotným procesem očkování se mohou objevit reakce spojené s úzkostí, včetně
vazovagálních reakcí (synkopa), hyperventilace nebo reakcí spojených se stresem (např. závrať, palpitace, zvýšení srdeční frekvence, změny krevního tlaku, parestezie, hypestezie a pocení). Reakce
spojené se stresem jsou dočasné a samy se upraví. Očkované osoby je třeba informovat o tom, aby na
případné symptomy upozornily očkujícího zdravotníka, který je vyhodnotí. Je důležité, aby byla zavedena opatření, aby se zabránilo zranění v důsledku mdlob.
Současné onemocnění
U osob trpících závažným akutním horečnatým onemocněním nebo akutní infekcí se má podání
vakcíny Comirnaty odložit. Přítomnost mírné infekce a/nebo horečky nízkého stupně není důvod
k odložení vakcinace.
Trombocytopenie a poruchy koagulace
Stejně jako u jiných intramuskulárních injekcí je třeba vakcínu podávat opatrně osobám podstupujícím
léčbu antikoagulancii nebo osobám s trombocytopenií nebo poruchami koagulace (jako je hemofilie),
protože po intramuskulárním podání může u těchto osob dojít ke krvácení nebo tvorbě modřin.
Imunokompromitované osoby
Účinnost a bezpečnost vakcíny nebyly hodnoceny u imunokompromitovaných osob, včetně osob
podstupujících imunosupresivní léčbu. Účinnost vakcíny Comirnaty může být u
imunokompromitovaných osob nižší.
Doba ochrany
Doba ochrany poskytovaná vakcínou není známa, protože je stále hodnocena v probíhajících
klinických studiích.
Om
ezení
ú
činnosti
vakcíny
Podobně jako u jiných vakcín je možné, že vakcinace vakcínou Comirnaty nebude chránit všechny její
příjemce. Osoby nemusí být plně chráněny po dobu 7 dnů po základním očkování 3 dávkami vakcíny.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Současné podání vakcíny Comirnaty s jinými vakcínami nebylo hodnoceno.
4.6 Fertilita, těhotenství a kojení
Vakcína Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi není určena pro jedince
starší 5 let věku.
Podrobnosti o použití u jedinců starších 5 let věku jsou uvedeny v Souhrnu údajů o přípravku pro Comirnaty 30 mikrogramů/dávku koncentrát pro injekční disperzi, Comirnaty 30 mikrogramů/dávku injekční disperze nebo Comirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vakcína Comirnaty nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Některé z účinků uvedených v bodě 4.8 však mohou schopnost řídit nebo obsluhovat stroje dočasně ovlivnit.
4.8 Nežádoucí účinky
Souhrn bezpečnostníhoprofilu
Dětivevěku6až 23 měsíců– po 3 dávkách
V analýze studie 3 (fáze 2/3) bylo 1 776 dětí (1 178 dětí, kterým byla podána vakcína Comirnaty
3 mikrogramy a 598 dětí, kterým bylo podáno placebo) ve věku od 6 do 23 měsíců. Na základě údajů ze zaslepeného placebem kontrolovaného období sledování až do data ukončení sběru dat 29. dubna
2022 bylo 570 dětí ve věku 6 až 23 měsíců, které obdržely 3 dávky základního očkování (386, kterým
byla podána vakcína Comirnaty 3 mikrogramy a 184, kterým bylo podáno placebo), sledováno po dobu s mediánem 1,3 měsíce po třetí dávce.
Nejčastějšími nežádoucími účinky u dětí ve věku od 6 do 23 měsíců, kterým byla podána jakákoli dávka základního očkování, byly podrážděnost (> 60 %), ospalost (> 40 %), snížená chuť k jídlu (> 30 %), citlivost v místě injekce (> 20 %), zarudnutí a otok v místě vpichu (> 10 %).
Děti ve věku od2do 4 let – po 3 dávkách
V analýze studie 3 (fáze 2/3) bylo 2 750 dětí (1 835 dětí, kterým byla podána vakcína Comirnaty
3 mikrogramy a 915 dětí, kterým bylo podáno placebo) ve věku od 2 do 4 let. Na základě údajů ze zaslepeného placebem kontrolovaného období sledování až do data ukončení sběru dat 29. dubna 2022 bylo 886 dětí ve věku 2 až 4 roky, které obdržely 3 dávky základního očkování (606, kterým byla
podána vakcína Comirnaty 3 mikrogramy a 280, kterým bylo podáno placebo) sledováno po dobu s mediánem 1,4 měsíce po třetí dávce.
Nejčastějšími nežádoucími účinky u dětí ve věku od 2 do 4 let, kterým byla podána jakákoli dávka základního očkování, byly bolest v místě injekce a únava (> 40 %), zarudnutí v místě vpichu a horečka (> 10 %).
Děti ve věku 5až11let(tj.vevěku5doméněnež12let)– po 2 dávkách
Ve studii 3 byla alespoň 1 dávka vakcíny Comirnaty 10 mikrogramů podána celkem 1 518 dětem ve
věku 5 až 11 let a celkem 750 dětem ve věku 5 až 11 let bylo podáno placebo. V době analýzy studie 3 fáze 2/3 s údaji do data ukončení sběru dat 6. září 2021 bylo 2 158 (95,1 %) (1 444 dětí, kterým byla podána vakcína Comirnaty 10 mikrogramů a 714 dětí, kterým bylo podáno placebo) sledováno po
dobu nejméně 2 měsíců po druhé dávce vakcíny Comirnaty 10 mikrogramů. Analýza údajů o nežádoucích příhodách studie 3 fáze 2/3 zahrnovala také dalších 2 379 účastníků [1 591 účastníkům
byla podána vakcína Comirnaty 10 mikrogramů a 788 účastníkům bylo podáno placebo], z nichž
71,2 % bylo sledováno po dobu nejméně 2 týdnů po 2. dávce až do data ukončení sběru dat
8. října 2021. Hodnocení bezpečnosti ve studii 3 probíhá.
Celkový bezpečnostní profil vakcíny Comirnaty u účastníků ve věku 5 až 15 let byl podobný jako u účastníků ve věku 16 let a starších. Nejčastějšími nežádoucími účinky u dětí ve věku 5 až 11 let, kterým byly podány 2 dávky, byly bolest v místě vpichu (> 80 %), únava (> 50 %), bolest hlavy (>
30 %), zarudnutí a otok v místě vpichu (> 20 %), myalgie a zimnice (> 10 %).
Děti ve věku 5až11let (tj. vevěku5doméněnež12let) – po posilovací dávce
V podskupině účastníků studie 3 dostalo celkem 401 dětí ve věku 5 až 11 let posilovací dávku vakcíny
Comirnaty 10 mikrogramů za nejméně 5 měsíců (rozmezí 5 až 9 měsíců) po dokončení základního očkování. Analýza podskupiny ve studii 3 fáze 2/3 je založena na údajích s datem ukončení sběru dat do 22. března 2022 (medián doby sledování 1,3 měsíce).
Celkový bezpečnostní profil posilovací dávky byl podobný jako po základním očkování. Nejčastějšími nežádoucími účinky u dětí ve věku 5 až 11 let byly bolest v místě vpichu (> 70 %), únava (> 40 %), bolest hlavy (> 30 %), myalgie, zimnice a zarudnutí a otok v místě vpichu (> 10 %).
Dospívající ve věku 12až15let – po 2 dávkách
V analýze dlouhodobého následného sledování bezpečnosti ve studii 2 bylo 2 260 dospívajících
(1 131 dostalo vakcínu Comirnaty a 1 129 dostalo placebo) ve věku 12 až 15 let. Z toho
1 559 dospívajících (786 dostalo vakcínu Comirnaty a 773 dostalo placebo) bylo sledováno po dobu
≥ 4 měsíců po druhé dávce vakcíny Comirnaty. Hodnocení bezpečnosti ve studii 2 probíhá.
Celkový bezpečnostní profil vakcíny Comirnaty u dospívajících ve věku 12 až 15 let byl podobný jako
u účastníků ve věku 16 let a starších. Nejčastějšími nežádoucími účinky u dospívajících ve věku 12 až
15 let, kteří dostali 2 dávky, byly bolest v místě injekce (> 90 %), únava a bolest hlavy (> 70 %), myalgie a zimnice (> 40 %), artralgie a pyrexie (> 20 %).
Účastníci ve věku 16let a starší – po 2 dávkách
Ve studii 2 byla celkem 22 026 účastníkům ve věku 16 let nebo starším podána alespoň 1 dávka
vakcíny Comirnaty 30 mikrogramů a celkem 22 021 účastníkům ve věku 16 let nebo starším bylo podáno placebo (včetně 138 a 145 dospívajících ve věku 16 a 17 let ve skupinách vakcíny a placeba, v uvedeném pořadí). Celkem 20 519 účastníků ve věku 16 let nebo starších dostalo 2 dávky vakcíny Comirnaty.
V době provedení analýzy studie 2 s datem ukončení sběru dat 13. března 2021 pro placebem
kontrolované zaslepené období sledování až do dat odslepení účastníků bylo celkem 25 651 (58,2 %)
účastníků (13 031 Comirnaty a 12 620 placebo) ve věku 16 let a starších sledováno po dobu ≥4 měsíce po podání druhé dávky. To zahrnovalo celkem 15 111 (7 704 Comirnaty a 7 407 placebo) účastníků ve věku 16 až 55 let a celkem 10 540 (5 327 Comirnaty a 5 213 placebo) účastníků ve věku 56 let a starších.
Nejčastějšími nežádoucími účinky byla u účastníků ve věku 16 let a starších, kteří dostali 2 dávky, bolest v místě injekce (> 80 %), únava (> 60 %), bolest hlavy (> 50 %), myalgie (> 40 %), zimnice
(> 30 %), artralgie (> 20 %), pyrexie a zduření v místě injekce (> 10 %). Tyto nežádoucí účinky byly
zpravidla mírné nebo střední intenzity a odezněly během několika dní po vakcinaci. Mírně nižší frekvence příhod reaktogenity souvisela s vyšším věkem.
Bezpečnostní profil u 545 účastníků ve věku 16 let a starších, kterým byla podána vakcína Comirnaty
a kteří byli séropozitivní na SARS-CoV-2 při výchozím stavu, byl podobný jako u obecné populace.
Účastníci ve věku 16let a starší – po posilovací dávce
Podskupina účastníků studie fáze 2/3, do které bylo zařazeno 306 dospělých ve věku 18 až 55 let, kteří
dokončili původní cyklus 2 dávek vakcíny Comirnaty, dostala posilovací dávku vakcíny Comirnaty
přibližně 6 měsíců (rozmezí 4,8 až 8,0 měsíců) po podání 2. dávky.
Celkový bezpečnostní profil posilovací dávky byl podobný jako po 2 dávkách. Nejčastějšími nežádoucími účinky u účastníků ve věku 18 až 55 let byly bolest v místě vpichu (> 80 %), únava (>
60 %), bolest hlavy (> 40 %), myalgie (> 30 %), zimnice a artralgie (> 20 %).
Ve studii 4, placebem kontrolované studii posilovací dávky, byla účastníkům ve věku 16 let a starším
zařazeným ze studie 2 podána posilovací dávka vakcíny Comirnaty (5 081 účastníků) nebo placebo (5 044 účastníků), a to nejméně 6 měsíců po druhé dávce vakcíny Comirnaty. Celkem činil medián doby následného sledování účastníků, kterým byla posilovací dávka podána, 2,5 měsíce po podání posilovací dávky do data ukončení sběru (5. října 2021). Žádné nové nežádoucí účinky vakcíny Comirnaty nebyly zjištěny.
Posilovací dávka poprimárnívakcinacijinouschválenouvakcínouprotionemocněníCOVID-19
V 5 nezávislých studiích o použití posilovací dávky vakcíny Comirnaty u osob, které absolvovaly
primární vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19 (heterologní posilovací
dávka), nebyly zjištěny žádné nové bezpečnostní problémy.
Tabulkovýseznamnežádoucíchúčinkůzklinickýchstudiíapouvedenínatrhuosobvevěku
6 měsíců astarších
Nežádoucí účinky pozorované z klinických studií jsou uvedeny níže podle následujících kategorií
frekvence:
Velmi časté (≥ 1/10)
Časté (≥ 1/100 až < 1/10)
Méně časté (≥ 1/1 000 až < 1/100) Vzácné (≥ 1/10 000 až < 1/1 000) Velmi vzácné (< 1/10 000)
Není známo (z dostupných údajů nelze určit)
T
abulka 1: Nežádoucí účinky vakcíny Comirnaty z klinických studií a po uvedení na trh u
osob ve věku 6 měsíců a starších
T
řída
orgánového systému
|
V
elmi
časté
(
≥ 1/10)
|
Č
asté
(
≥ 1/100
až
< 1/10)
|
Méně časté
(
≥ 1/1 000 až
< 1/100)
|
V
z
ácné
(
≥ 1/10 000
až
< 1/1 000)
|
V
elmi vzácné
(
< 1/10 000
|
N
ení
z
námo (z dostupnýc h údajů
nelze určit)
|
Poruchy krve a
lymfatického systému
|
|
|
Lymfadenopatiea
|
|
|
|
Poruchy
imunitního systému
|
|
|
Hypersenzitivní
reakce (např. vyrážkai, pruritus, urtikarie, angioedémb)
|
|
|
Anafylaxe
|
Poruchy
metabolismu a výživy
|
|
|
Snížená chuť k
jídluj
|
|
|
|
Psychiatrické
poruchy
|
Podráždě
nostk
|
|
Insomnie
|
|
|
|
Poruchy
nervového systému
|
Bolest
hlavy;
ospalostk
|
|
Letargie
|
Akutní
periferní paralýza n. facialisc
|
|
Parestezied,
hypestezied
|
Srdeční poruchy
|
|
|
|
|
Myokarditidad;
Perikarditidad
|
|
Gastrointestinální
poruchy
|
Průjemd
|
Nauzea;
zvraceníd
|
|
|
|
|
Poruchy kůže a
podkožní tkáně
|
|
|
Hyperhidróza;
noční pocení
|
|
|
Erythema
multiformed
|
Poruchy svalové
a kosterní soustavy a
pojivové tkáně
|
Artralgie;
myalgie
|
|
Bolest
v končetiněe
|
|
|
|
Poruchy
reprodukčního
systému a prsu
|
|
|
|
|
|
Silné
menstruační
krváceníl
|
Celkové poruchy
a reakce v místě
aplikace
|
Bolest v
místě
injekce; citlivost v místě
injekcek;ú nava;
zimnice; pyrexief; zduření v místě injekce
|
Zarudnutí
v místě
injekceh
|
Astenie;
malátnost;
svědění v místě
injekce
|
|
|
Rozsáhlý
otok
končetiny, do níž byla vakcína podánad; otok obličejeg
|
a. U účastníků, kteří dostali posilovací dávku, byla pozorována vyšší frekvence lymfadenopatie ve srovnání
s účastníky ve věku 5 až 11 let ve studii 3 (2,5 % vs. 0,9 %) a s účastníky ve věku 16 let a starších ve studii 4 (2,8 % vs. 0,4 %), kteří dostali dvě dávky.
b. Kategorie frekvence pro angioedém byla vzácná.
c. Během období sledování bezpečnosti v klinické studii do 14. listopadu 2020 byla hlášena akutní periferní
paralýza n. facialis u čtyř účastníků ve skupině mRNA vakcíny proti onemocnění COVID-19. Nástup obrny
obličeje byl 37. den po podání 1. dávky (účastník nedostal 2. dávku) a 3., 9. a 48. den po 2. dávce. Ve
skupině s placebem nebyly hlášeny žádné případy akutní periferní paralýzy n. facialis.
d. Nežádoucí účinek byl stanoven po registraci.
e. Týká se končetiny/paže, do které byla daná osoba očkována.
f. Vyšší frekvence pyrexie byla pozorována po druhé dávce v porovnání s první dávkou.
g. Po uvedení přípravku na trh byly hlášeny případy otoku obličeje u očkovaných osob, které v minulosti
podstoupily injekční aplikaci dermálních výplní do obličeje.
h. Zarudnutí v místě injekce se vyskytovalo s vyšší frekvencí (velmi častou) u 5 účastníků ve věku 6 měsíců až
11 let.
i. Kategorie frekvence pro vyrážku byla častá u účastníků ve věku 6 až 23 měsíců.
j. Kategorie frekvence pro sníženou chuť k jídlu byla velmi častá u účastníků ve věku 6 až 23 měsíců. k. Podrážděnost, citlivost v místě injekce a ospalost se vztahují k účastníkům ve věku 6 až 23 měsíců. l. Většina případů se zdála být nezávažné a dočasné povahy.
Popis vybraných nežádoucíchúčinkůMyokarditida a perikarditidaZvýšené riziko myokarditidy po očkování vakcínou Comirnaty je nejvyšší u mladších mužů a chlapců
(viz bod 4.4).
Zvýšené riziko u mladších mužů a chlapců po podání druhé dávky vakcíny Comirnaty bylo blíže určeno ve dvou velkých evropských farmakoepidemiologických studiích. Z jedné studie vyplynulo, že v období 7 dnů po podání druhé dávky se u mužů a chlapců ve věku 12–29 let vyskytlo přibližně o
0,265 (95% CI 0,255–0,275) případů myokarditidy na 10 000 osob více než u neočkovaných osob.
V další studii se v období 28 dnů po podání druhé dávky u mužů a chlapců ve věku 16–24 let vyskytlo o 0,56 (95% CI 0,37–0,74) případů myokarditidy na 10 000 osob více než u neočkovaných osob.
Omezené údaje ukazují, že riziko myokarditidy a perikarditidy je po očkování vakcínou Comirnaty
u dětí ve věku od 5 do 11 let pravděpodobně nižší než u dospívajících ve věku od 12 do 17 let.
Hlášení podezření na nežádoucí účinkyHlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to
pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V a uvedli přitom číslo šarže, je-li k dispozici.
4.9 PředávkováníÚdaje o předávkování jsou k dispozici od 52 účastníků studie zařazených do klinického hodnocení, kterým bylo kvůli chybě v ředění podáno 58 mikrogramů vakcíny Comirnaty. Příjemci vakcíny nehlásili zvýšení reaktogenity ani nežádoucí účinky.
V případě předávkování se doporučuje sledovat základní životní funkce a případně zahájit symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: vakcíny, jiné virové vakcíny, ATC kód: J07BX03
Mechanismus účinkuMediátorová (messenger) RNA s modifikovanými nukleosidy je ve vakcíně Comirnaty zapouzdřená
v lipidových nanočásticích, což umožňuje transfer nereplikující RNA do hostitelských buněk pro
přímou přechodnou expresi S antigenu viru SARS-CoV-2. mRNA kóduje v membráně ukotvený
S v plné délce se dvěma bodovými mutacemi v centrální šroubovici. Mutace těchto dvou aminokyselin na prolin uzamyká S v antigenně preferované prefuzní konformaci. Vakcína vyvolává jak odpověď neutralizačních protilátek, tak i imunitní odpověď buněk na spike (S) antigen, což může přispívat
k ochraně před onemocněním COVID-19.
Účinnost
Studie 2 je multicentrická, mezinárodní, randomizovaná, placebem kontrolovaná studie fáze ½/3
zaslepená pro pozorovatele, která hodnotí dávku, výběr vakcíny a její účinnost u účastníků ve věku
12 let a starších. Randomizace byla stratifikována podle věku: věk 12 až 15 let, věk 16 až 55 let a věk
56 let a více; do věkové skupiny ≥ 56 let spadalo minimálně 40 % účastníků. Ze studie byli vyřazeni
imunokompromitovaní účastníci a osoby, u nichž byla dříve stanovena klinická či mikrobiologická diagnóza onemocnění COVID-19. Účastníci s preexistujícími stabilizovanými onemocněními,
definovanými jako onemocnění, jež během 6 týdnů před zařazením nevyžadovala významnou změnu
léčby nebo hospitalizaci z důvodu zhoršení nemoci, byli do studie zařazeni stejně jako účastníci se známou stabilizovanou infekcí virem lidské imunodeficience (HIV), virem hepatitidy C (HCV) nebo virem hepatitidy B (HBV).
Účinnost u osobvevěku16 let a starších – po 2 dávkách
Během fáze 2/3 Studie 2 bylo na základě dat získaných do 14. listopadu 2020 ve stejném poměru
randomizováno zhruba 44 000 účastníků a byly jim podány 2 dávky mRNA vakcíny proti onemocnění
COVID-19 nebo placeba. Analýzy účinnosti zahrnovaly účastníky, kteří dostali druhou dávku vakcíny v rozmezí 19 až 42 dnů po první dávce vakcíny. Většina (93,1 %) příjemců vakcíny dostala druhou dávku 19 dnů až 23 dnů po první dávce. Pro účely hodnocení bezpečnosti a účinnosti vakcíny proti onemocnění COVID-19 budou na základě připraveného plánu účastníci dále sledováni po dobu až
24 měsíců po 2. dávce. V klinické studii bylo od účastníků vyžadováno dodržení minimálního intervalu 14 dní před a po podání vakcíny proti chřipce, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19. V klinické studii bylo od účastníků vyžadováno dodržení minimálního intervalu 60 dní před nebo po podání přípravků z krve/plazmy nebo imunoglobulinů v průběhu studie, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19.
Do populace pro analýzu primárního cílového parametru účinnosti bylo zařazeno 36 621 účastníků ve věku 12 let a starších (18 242 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 18 379 ve skupině s placebem), u nichž se během 7 dní po podání druhé dávky neprokázala předchozí infekce SARS-CoV-2. Kromě toho bylo 134 účastníků ve věku od 16 do 17 let (66 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 68 ve skupině s placebem) a 1 616 účastníků ve věku 75 let a starších (804 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 812 ve skupině s placebem).
V čase provedení primární analýzy účinnosti byli účastníci ve skupině mRNA vakcíny proti onemocnění COVID-19 sledováni z hlediska symptomatického onemocnění COVID-19 po celkovou dobu 2 214 osoboroků, účastníci ve skupině s placebem po dobu minimálně 2 222 osoboroků.
U účastníků s rizikem závažného onemocnění COVID-19, včetně osob s 1 nebo více komorbiditami,
které riziko závažného onemocnění COVID-19 zvyšují (např. astma, index tělesné hmotnosti (BMI)
≥ 30 kg/m2, chronické plicní onemocnění, diabetes mellitus, hypertenze), nebyly zjištěny žádné
významné klinické rozdíly v celkové účinnosti vakcíny.
Informace o účinnosti vakcíny jsou uvedeny v tabulce 2.
Tabulka 2: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů)První výskyt COVID-19 od 7 dnů po 2. dávce u účastníků bez důkazu předchozí infekce SARS- CoV-2 *
|
Podskupina
| Vakcína mRNA proti
onemocnění COVID-19
na=18 198
Případy
n1b
Doba sledováníc (n2d)
|
Placebo na=18 325
Případy
n1b
Doba sledováníc (n2d)
|
Účinnost vakcíny % (95% CI)e
|
Všichni účastníci
| 8
2,214 (17 411)
| 162
2,222 (17 511)
| 95,0
(90,0; 97,9)
|
16 až 64 let
| 7
1,706 (13 549)
| 143
1,710 (13 618)
| 95,1
(89,6; 98,1)
|
65 let a starší
| 1
0,508 (3 848)
| 19
0,511 (3 880)
| 94,7
(66,7; 99,9)
|
65 až 75 let
| 1
0,406 (3 074)
| 14
0,406 (3 095)
| 92,9
(53,1; 99,8)
|
75 let a starší
| 0
0,102 (774)
| 5
0,106 (785)
| 100,0
(-13,1; 100,0)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 [*Definice případu: (alespoň 1 z) horečka, nový nebo zhoršený kašel, nová nebo zhoršená dyspnoe, zimnice, nová nebo zhoršená bolest svalů, nová ztráta chuti nebo čichu, bolest v krku, průjem nebo zvracení.]
* Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (7 nebo více dnů před podáním poslední dávky) předcházející infekce SARS-CoV-2 (tj. N-vazebná protilátka [sérum] negativní při návštěvě 1 a SARS- CoV-2 nedetekován pomocí amplifikačních testů nukleových kyselin (NAAT) [výtěr nosu] při návštěvách 1 a 2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do analýzy.
a. n = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování. CI není upraven na multiplicitu.
Účinnost vakcíny mRNA proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění COVID-19 po 7 dnech po 2. dávce ve srovnání s placebem 94,6 % (95% interval spolehlivosti 89,6 % až 97,6 %) u účastníků ve věku 16 let a starších s nebo bez důkazů o předchozí infekci virem SARS- CoV-2.
Analýzy podskupin primárního cílového parametru účinnosti navíc ukázaly podobné odhady bodů účinnosti u pohlaví, etnických skupin a účastníků s komorbiditami spojenými s vysokým rizikem závažného onemocnění COVID-19.
Byly provedeny aktualizované analýzy účinnosti s dalšími potvrzenými případy COVID-19, které se objevily během zaslepeného placebem kontrolovaného sledování, což v populaci s účinností představuje až 6 měsíců po podání 2. dávky.
Aktualizované informace o účinnosti vakcíny jsou uvedeny v tabulce 3.
Tabulka 3: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podlevěkové podskupiny – účastníci bez důkazu infekce předchozí infekce virem SARS-
C
oV-2* před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů) během
placebem kontrolovaného období sledování
Podskupina
|
V
akcína mRNA proti
onemocnění COVID-19
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Placebo
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Ú
č
i
n
nost vakcíny % (95% CI
e
)
|
Všichni účastnícif
|
77
6,247 (20 712)
|
850
6,003 (20 713)
|
91,3
(89,0; 93,2)
|
16 až 64 let
|
70
4,859 (15 519)
|
710
4,654 (15 515)
|
90,6
(87,9; 92,7)
|
65 let a starší
|
7
1,233 (4 192)
|
124
1,202 (4 226)
|
94,5
(88,3; 97,8)
|
65 až 74 let
|
6
0,994 (3 350)
|
98
0,966 (3 379)
|
94,1
(86,6; 97,9)
|
75 let a starší
|
1
0,239 (842)
|
26
0,237 (847)
|
96,2
(76,9; 99,9)
|
|
|
N =20 998
N =21 096
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-
PCR) a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo
zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení).
* Účastníci, kteří neměli žádné důkazy předcházející infekce virem SARS-CoV-2 (tj. N-vazebná protilátka
[sérum] negativní při návštěvě 1 a virus SARS-CoV-2 nedetekován pomocí NAAT [výtěr nosu] při návštěvách 1 a
2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do
analýzy.
a. N = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný 95% interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a
Pearsonovy metody upravené podle doby sledování. CI není upraven na multiplicitu.
f. Zahrnuté potvrzené případy u účastníků ve věku 12 až 15 let: 0 ve skupině s mRNA vakcínou proti
onemocnění COVID-19; 16 ve skupině s placebem.
V aktualizované analýze účinnosti byla účinnost mRNA vakcíny proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění COVID-19 od 7 dnů po podání 2. dávky ve srovnání s placebem 91,1 % (95% CI 88,8 % až 93,0 %) u účastníků v hodnotitelné populaci účinnosti s průkazem nebo bez průkazu předchozí infekce SARS-CoV-2.
Aktualizované analýzy účinnosti podle podskupin navíc ukázaly podobné bodové odhady účinnosti u všech pohlaví, etnických skupin, zeměpisných oblastí a účastníků se zdravotními komorbiditami a obezitou spojenou s vysokým rizikem závažného onemocnění COVID-19.
Účinnost proti závažnému onemocnění COVID-19
Aktualizované analýzy účinnosti sekundárních cílových parametrů účinnosti podpořily přínos mRNA
vakcíny proti onemocnění COVID-19 v prevenci závažného onemocnění COVID-19.
Od 13. března 2021 je účinnost vakcíny proti závažnému onemocnění COVID-19 prezentována pouze pro účastníky s předchozí infekcí SARS-CoV-2 nebo bez ní (tabulka 4), protože počty případů onemocnění COVID-19 u účastníků bez předchozí infekce SARS-CoV-2 byly stejné jako u účastníků
s předchozí infekcí SARS-CoV-2 nebo bez ní jak ve skupině s mRNA vakcínou proti onemocnění
COVID-19, tak ve skupině s placebem.
| Vakcína mRNA proti
onemocnění COVID-19
Případy
n1a
Doba sledováníc (n2b)
|
Placebo Případy n1a
Doba sledováníc (n2b)
|
Účinnost vakcíny % (95% CIc)
| Po 1. dávced
| 1
8,439e (22 505)
| 30
8,288e (22 435)
| 96,7
(80,3; 99,9)
| 7 dnů po 2. dávcef
| 1
6,522g (21 649)
| 21
6,404g (21 730)
| 95,3
(70,9; 99,9)
|
|
|
Tabulka 4: Účinnost vakcíny – první závažný výskyt onemocnění COVID-19 u účastníků s nebo bez předchozí infekce virem SARS-CoV-2 na základě údajů Úřadu pro kontrolu potravin a léčiv (FDA)* po podání 1. dávky nebo od 7 dnů po podání 2. dávky v placebem kontrolovaném období sledováníPoznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení.)
* Závažné onemocnění COVID-19 podle definice FDA je potvrzené onemocnění COVID-19 a přítomnost alespoň 1 z následujících příznaků:
• Klinické příznaky v klidu svědčící pro závažné systémové onemocnění (dechová frekvence ≥ 30 dechů za minutu, srdeční frekvence ≥ 125 tepů za minutu, saturace kyslíkem ≤ 93 % na pokojovém vzduchu při hladině moře nebo poměr arteriálního parciálního tlaku kyslíku k frakčnímu inspirovanému kyslíku
< 300 mmHg).
• Respirační selhání [definované jako potřeba vysokého průtoku kyslíku, neinvazivní ventilace,
mechanické ventilace nebo extrakorporální membránové oxygenace (ECMO)].
• Průkaz šoku (systolický krevní tlak < 90 mmHg, diastolický krevní tlak < 60 mmHg nebo potřeba
podání vazopresorických látek).
• Významná akutní renální, jaterní nebo neurologická dysfunkce.
• Přijetí na jednotku intenzivní péče.
• Smrt.
a. n1 = Počet účastníků splňujících definici cílového parametru.
b. n2 = počet účastníků ohrožených daným cílovým parametrem.
c. Dvoustranný interval spolehlivosti (CI) pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování.
d. Účinnost hodnocená na základě veškeré populace pro hodnocení účinnosti s 1. dávkou (modifikovaná se záměrem léčit), která zahrnovala všechny randomizované účastníky, kteří dostali alespoň 1 dávku hodnocené léčby.
e. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od po 1. dávce do konce období sledování.
f. Účinnost hodnocená na základě populace hodnotitelné pro účinnost (7 dní), která zahrnovala všechny způsobilé randomizované účastníky, kteří dostali všechny dávky hodnocené léčby podle randomizace v rámci předem stanoveného časového období a neměli žádné další důležité odchylky od protokolu podle rozhodnutí
lékaře.
g. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro daný cílový parametr. Časové období pro získání případu onemocnění COVID-19 je od
7 dnů po 2. dávce do konce období sledování.
Účinnost a imunogenicita udospívajícíchvevěku12až15let – po 2 dávkáchV úvodní analýze Studie 2 u dospívajících ve věku 12 až 15 let (jež reprezentovala medián doby
následného sledování v délce ˃ 2 měsíce po 2. dávce) bez průkazu prodělané infekce nebyly žádné případy u 1 005 účastníků, kteří dostali vakcínu, a 16 případů z 978 účastníků, kteří dostali placebo.
Bodový odhad účinnosti je 100 % (95% interval spolehlivosti 75,3; 100,0). U účastníků s nebo bez
průkazu prodělané infekce bylo 0 případů z 1 119 účastníků, kteří dostali vakcínu, a 18 případů
z 1 110 účastníků, kteří dostali placebo. To také naznačuje, že bodový odhad účinnosti je 100 % (95%
interval spolehlivosti 78,1; 100,0).
U dodatečných potvrzených případů onemocnění COVID-19, k nimž došlo během zaslepeného, placebem kontrolovaného následného sledování, byly provedeny aktualizované analýzy účinnosti, jež reprezentovaly dobu až 6 měsíců po 2. dávce v populaci pro hodnocení účinnosti.
V aktualizované analýze účinnosti ve studii 2, provedené u dospívajících ve věku 12 až 15 let bez průkazu prodělané infekce, se u 1 057 účastníků, kterým byla podána vakcína, nevyskytl žádný případ a u 1 030 účastníků, kterým bylo podáno placebo, došlo ke 28 případům. Odhad bodu účinnosti je
100 % (95% interval spolehlivosti 86,8; 100,0). U účastníků s průkazem prodělané infekce nebo bez
tohoto průkazu nedošlo u 1 119 těch, kterým byla podána vakcína, k žádnému případu
a u 1 109 účastníků, jimž bylo podáno placebo, ke 30 případům. To rovněž naznačuje, že odhad bodu účinnosti je 100 % (95% interval spolehlivosti 87,5; 100,0).
Ve Studii 2 byla provedena analýza neutralizačních titrů SARS-CoV-2 jeden měsíc po druhé dávce u náhodně vybrané podskupiny účastníků, kteří do 1 měsíce po dávce 2 neměli sérologické nebo virologické průkazy prodělané infekce SARS-CoV-2, která porovnávala odpověď u dospívajících ve věku 12 až 15 let (n = 190) s účastníky ve věku 16 až 25 let (n = 170).
Poměr geometrických průměrných titrů (GMT) ve skupině ve věku 12 až 15 let ke skupině ve věku 16 až 25 let byl 1,76; s oboustranným 95% CI 1,47 až 2,10. Proto bylo splněno 1,5násobné kritérium noninferiority, protože dolní mez 2stranného 95% CI pro poměr geometrického průměru [GMR] byla
> 0,67.
Účinnost a imunogenicita udětívevěku5až11let(tj.vevěku5ažméněnež12let) – po 2 dávkáchStudie 3 je studie fáze 1/2/3, která se skládá z otevřené části zaměřené na zjištění dávky vakcíny
(fáze 1) a multicentrické, mezinárodní, randomizované, fyziologickým roztokem jako placebem
kontrolované části zaslepené pro pozorovatele hodnotící účinnost (fáze 2/3), do které byli zařazeni účastníci ve věku 5 až 11 let. Většina (94,4 %) randomizovaných příjemců vakcíny dostala druhou
dávku 19 dní až 23 dní po první dávce.
Deskriptivní výsledky účinnosti vakcíny u dětí ve věku 5 až 11 let bez známek předchozí infekce virem SARS-CoV-2 jsou uvedeny v tabulce 5. U účastníků s prokázanou předchozí infekcí virem SARS-CoV-2 nebyly pozorovány žádné případy onemocnění COVID-19 ani ve skupině s vakcínou, ani ve skupině s placebem.
Tabulka 5: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce: bez průkazu infekce před 7 dny po 2. dávce – fáze 2/3 – děti ve věku 5 až 11 let hodnotitelná populace pro účinnostPrvní výskyt onemocnění COVID-19 od 7 dnů po 2. dávce u dětí ve věku 5 až 11 let bez
průkazu předchozí infekce SARS-CoV-2*
|
| Vakcína mRNA proti
onemocnění
COVID-19
Dávka 10 mikrogramů/dávku Na=1 305
Případy
n1b
Doba sledováníc (n2d)
|
Placebo
Na=663
Případy
n1b
Doba sledováníc (n2d)
|
Účinnost vakcíny
% (95% CI)
|
Děti ve věku 5 až 11 let
| 3
0,322 (1273)
| 16
0,159 (637)
| 90,7
(67,7; 98,3)
|
Poznámka: Potvrzené případy byly určeny na základě reverzní transkripční polymerázové řetězové reakce (RT-
PCR) a alespoň 1 příznaku odpovídajícího onemocnění COVID-19 (příznaky zahrnovaly: horečku; nový nebo
zesílený kašel; novou nebo zesílenou dušnost; zimnici; novou nebo zesílenou bolest svalů; novou ztrátu chuti nebo čichu; bolest v krku; průjem; zvracení).
* Do analýzy byli zahrnuti účastníci, kteří neměli žádný průkaz o prodělané infekci virem SARS-CoV-2 (tj. negativní N-vázané protilátky [sérum] při návštěvě 1 a virus SARS-CoV-2 nezjištěný pomocí NAAT [nosní výtěr] při návštěvě 1 a 2) a měli negativní NAAT (nosní výtěr) při jakékoli neplánované návštěvě před 7 dny po 2. dávce
a. N = počet účastníků v dané skupině.
b. n1 = počet účastníků splňujících definici cílového ukazatele.
c. Celková doba sledování v 1 000 osoborocích pro daný cílový ukazatel u všech účastníků v každé skupině s rizikem pro cílový ukazatel. Časové období pro nárůst případů onemocnění COVID-19 je od 7 dnů po podání 2. dávky do konce období sledování.
d. n2 = počet účastníků s rizikem pro daný cílový ukazatel.
Ve studii 3 prokázala analýza 50% neutralizačních titrů viru SARS-CoV-2 (NT50) 1 měsíc po podání
2. dávky u náhodně vybrané podskupiny účastníků účinnost imunologickým přemostěním imunitních odpovědí při srovnání dětí ve věku 5 až 11 let (tj. ve věku 5 až méně než 12 let) v části studie 3 fáze
2/3 s účastníky ve věku 16 až 25 let v části studie 2 fáze 2/3, kteří neměli žádné sérologické nebo
virologické průkazy o prodělané infekci virem SARS-CoV-2 do 1 měsíce po podání 2. dávky, což splňuje předem stanovená kritéria imunologického přemostění jak pro poměr geometrického průměru (GMR), tak pro rozdíl sérologických odpovědí, přičemž sérologická odpověď byla definována jako dosažení alespoň čtyřnásobného zvýšení NT50 SARS-CoV-2 oproti výchozí hodnotě (před podáním 1. dávky).
GMR NT50 SARS-CoV-2 1 měsíc po 2. dávce u dětí ve věku 5 až 11 let (tj. ve věku 5 až méně než
12 let) proti mladým dospělým ve věku 16 až 25 let byl 1,04 (2stranný 95% CI: 0,93; 1,18). Mezi
účastníky bez předchozích známek infekce virem SARS-CoV-2 do 1 měsíce po podání 2. dávky mělo
99,2 % dětí ve věku 5 až 11 let a 99,2 % účastníků ve věku 16 až 25 let sérologickou odpověď 1 měsíc po podání 2. dávky V případě dětí ve věku 5 až 11 let a mladých lidí ve věku 16 až 25 let byla
sérologická odpověď prokázána až po 1 měsíci po podání 2. dávky Rozdíl v podílech účastníků, kteří
měli sérologickou odpověď mezi dvěma věkovými skupinami (děti – mladí dospělí), byl 0,0 % (2stranný 95% CI: -2,0 %; 2,2 %). Tyto informace jsou uvedeny v tabulce 6.
T
abulka 6: Souhrn geometrického průměrného poměru pro 50% neutralizační titr a rozdíl v procentech účastníků se sérologickou odpovědí – srovnání dětí ve věku 5 až 11 let (studie 3) s účastníky ve věku 16 až 25 let (studie 2) – účastníci bez známek infekce
do 1 měsíce po podání 2. dávky – podskupina imunologického přemostění – fáze 2/3 –
populace hodnotitelná na imunogenicitu
|
V
akcína mRNA proti onemocnění
COV
I
D
-19
|
5 až 11 let /
16 až 25 let
|
D
ávka
10 mikrogramů/dávku
5 až 11 let
N
a
=
264
|
D
ávka
30 mikrogramů/dáv
ku
16 až 25 let
N
a
=
253
|
|
Č
asový
bod
b
|
G
MT
c
(
95% CI
c
)
|
G
MT
c
(
95% CI
c
)
|
G
MR
d
(
95% CI
d
)
|
Splnila cíl
im
unologické ho přemostění
e
(
A
/
N)
|
G
eometrický
průměrný
50% neutralizační titr
f
(
G
MT
c
)
|
1 měsíc po 2. dávce
|
1 197,6
(1 106,1; 1 296,6)
|
1 146,5
(1 045,5; 1 257,2)
|
1,04 (0,93; 1,18)
|
A
|
|
Č
asový
bod
b
|
n
g
(
%)
(
95% CI
h
)
|
n
g
(
%)
(
95% CI
h
)
|
R
ozdíl %
i
(
95% CI
j
)
|
Splnila cíl imunologické
ho přemostění
k
(
A
/
N)
|
Č
etnost
sérologické
odpovědi (%) pro 50% neutralizační titr
f
|
1 měsíc po 2. dávce
|
262 (99,2) (97,3; 99,9)
|
251 (99,2) (97,2; 99,9)
|
0,0
(-2,0; 2,2)
|
A
|
Zkratky: CI = interval spolehlivosti; GMR = geometrický průměrný poměr; GMT = geometrický průměrný titr;
LLOQ = dolní mez kvantifikace; NAAT = test amplifikace nukleových kyselin; NT50 = 50% neutralizační titr;
virus SARS-CoV-2 = těžký akutní respirační syndrom vyvolaný koronavirem 2.
Poznámka: Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (do 1 měsíce po odběru vzorku krve po 2. dávce) o prodělané infekci virem SARS-CoV-2 (tj, N-vázané protilátky [sérum] negativní při návštěvě s podáním 1. dávky a 1 měsíc po 2. dávce, virus SARS-CoV-2 nezjištěn pomocí NAAT [nosní výtěr] při návštěvě s podáním 1. dávky a 2. dávky a negativní NAAT (nosní výtěr) při kterékoli neplánované návštěvě do 1 měsíce po odběru krve po 2. dávce) a neměli v anamnéze žádné onemocnění COVID-19, byli zahrnuti do analýzy.
Poznámka: Sérologická odpověď je definována jako dosažení ≥ 4násobného zvýšení oproti výchozí hodnotě (před 1. dávkou). Pokud je výchozí měření nižší než LLOQ, považuje se výsledek testu po očkování ≥4× LLOQ za sérologickou odpověď.
a. N = počet účastníků s platnými a určitelnými výsledky testu před očkováním a 1 měsíc po podání 2. dávky.
Tyto hodnoty jsou také jmenovateli použitými při výpočtech procentuální četnosti sérologické odpovědi.
b. Protokolárně stanovené načasování odběru krevních vzorků.
c. GMT a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a odpovídajících CI (na základě Studentova t rozložení). Výsledky testů pod LLOQ byly stanoveny na 0,5 × LLOQ.
d. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (věk 5 až 11 let minus věk 16 až 25 let) a odpovídajícího CI (na základě Studentova t rozložení).
e. Imunologické přemostění podle GMT je deklarováno, pokud je dolní hranice dvoustranného 95% CI pro
GMR větší než 0,67 a bodový odhad GMR je ≥ 0,8.
f. NT50 SARS-CoV-2 byly stanoveny pomocí mikroneutralizačního testu SARS-CoV-2 mNeonGreen Virus
Assay. Test používá fluorescenční reportérový virus odvozený od kmene USA_WA1/2020 a neutralizace
viru se odečítá na monovrstvách buněk Vero. Vzorek NT50 je definován jako vzájemné zředění séra, při
kterém je neutralizováno 50 % viru.
g. n = počet účastníků se sérologickou odpovědí na základě NT50 1 měsíc po podání 2. dávky.
h. Přesný dvoustranný CI na základě Clopperovy a Pearsonovy metody.
i. Rozdíl v podílech vyjádřený v procentech (věk 5 až 11 let minus věk 16 až 25 let).
j. Přesný 2stranný CI založený na metodě Miettinena a Nurminena pro rozdíl v podílech, vyjádřený v
procentech.
k. Imunologické přemostění založené na četnosti sérologické odpovědi je deklarováno, pokud je dolní hranice
2stranného 95% CI pro rozdíl sérologické odpovědi větší než 10,0 %.
Imunogenicita u dětívevěkuod5do11let(tj.vevěku5ažméněnež12let) – po posilovací dávcePosilovací dávka vakcíny Comirnaty byla ve studii 3 podána 401 náhodně vybraným účastníkům.
Účinnost posilovací dávky ve věku 5 až 11 let je odvozena z údajů o imunogenicitě. Imunogenicita byla hodnocena podle NT50 u referenčního kmene SARS-CoV-2 (USA_WA1/2020). Analýzy NT50 za 1 měsíc po posilovací dávce v porovnání se stavem před posilovací dávkou prokázaly podstatné zvýšení GMT u jedinců ve věku od 5 do 11 let včetně, kteří neměli sérologický ani virologický průkaz předchozí infekce virem SARS-CoV-2 do 1 měsíce po 2. dávce a po posilovací dávce. Tato analýza je shrnuta v tabulce 7.
| Časový bod odběrua
|
|
Test
|
1 měsíc po posilovací
dávce (nb=67)
GMTc
(95% CIc)
|
1 měsíc po 2. dávce
(nb=96)
GMTc
(95% CIc)
| 1 měsíc po posilovací
dávce /
1 měsíc po 2. dávce
GMRd
(95% CId)
| SARS-CoV-2
neutralizační test - NT50 (titr)
|
2 720,9
(2 280,1; 3 247,0)
|
1 253,9
(1 116,0; 1 408,9)
|
2,17 (1,76; 2,68)
| Zkratky: CI = interval spolehlivosti; GMR = geometrický průměr poměrů; GMT = geometrický průměr titrů;
LLOQ = dolní mez kvantifikace; NT50 = 50% neutralizační titr; virus SARS-CoV-2 = těžký akutní respirační
syndrom vyvolaný koronavirem 2.
a. Protokolárně stanovené načasování odběru krevních vzorků.
b. n = počet účastníků s platnými a konečnými výsledky testu pro specifický test v časovém bodě podání dávky/odběru.
c. GMT a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů
a odpovídajících CI (na základě Studentova t rozložení). Výsledky testů pod LLOQ byly stanoveny na
0,5 × LLOQ.
d. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (1 měsíc po posilovací dávce minus 1 měsíc po 2. dávce) a odpovídajícího CI (na základě Studentova t rozložení).
|
|
|
Tabulka 7: Souhrn geometrického průměru titrů – NT50 – účastníci bez průkazu infekce – fáze 2/3 – soubor imunogenicity – věk 5 až 11 let včetně – populace hodnotitelná na imunogenicituÚčinnostaimunogenicitazákladníhoočkování3dávkami u dětívevěkuod6měsícůdo4letAnalýza účinnosti ve studii 3 byla provedena v rámci kombinované populace účastníků ve věku od
6 měsíců do 4 let na základě potvrzených případů mezi 873 účastníky ve skupině očkované mRNA
vakcínou proti onemocnění COVID-19 a 381 účastníky ve skupině, která obdržela placebo (poměr
randomizace 2 : 1), kteří během zaslepeného období sledování, když byla převládající variantou SARS-CoV-2 v populaci varianta Omikron (BA.1), obdrželi všechny 3 dávky hodnocené intervence (datum konce sběru dat 17. června 2022).
Výsledky účinnosti vakcíny po 3. dávce u účastníků ve věku od 6 měsíců do 4 let jsou uvedeny v tabulce .
P
r
vní výskyt onemocnění COVID-19 od 7 dnů po 3. dávce u účastníků bez průkazu prodělané
i
n
f
e
k
c
e SARS-CoV-2*
|
Podskupina
|
V
akcína mRNA proti
onemocnění COVID-19 3 μg/dávku N
a
=
8
73
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Placebo
N
a
=
3
81
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Ú
č
i
n
nost vakcíny % (95% CI
e
)
|
6 měsíců až 4 rokye
|
13
0,124 (794)
|
21
0,054 (351)
|
73,2
(43,8; 87,6)
|
2 až 4 roky
|
9
0,081 (498)
|
13
0,033 (204)
|
71,8
(28,6; 89,4)
|
6 měsíců až
23 měsíců
|
4
0,042 (296)
|
8
0,020 (147)
|
75,8
(9,7; 94,7)
|
|
|
T
abulka 8: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 3. dávce – zaslepené období sledování – účastníci bez průkazu infekce před 7 dny po 3. dávce – fáze 2/3 – účastníci ve věku od 6 měsíců do 4 let – populace hodnotitelná pro účinnost (3 dávky)
Zkratky: NAAT = test amplifikace nukleové kyseliny; N-vazba = SARS-CoV-2 nukleoproteinová vazba;
SARS-CoV-2 = závažný akutní respirační syndrom vyvolaný koronavirem 2; VE = účinnost vakcíny.
* Do analýzy byli zahrnuti účastníci, kteří neměli žádný sérologický nebo virologický průkaz (dříve než
7 dnů po obdržení 3. dávky) prodělané infekce SARS-CoV-2 (tj. N-vázané protilátky [sérum] negativní při návštěvách v rámci studie při 1. dávce, 1 měsíc po 2. dávce (je-li to relevantní) a při 3. dávce (je-li to relevantní), virus SARS-CoV-2 nebyl detekován pomocí NAAT [nosní výtěr] při návštěvách v rámci
studie při 1. dávce, 2. dávce a 3. dávce a měli negativní výsledek NAAT [nosní výtěr] při jakékoli neplánované návštěvě do 7 dnů po obdržení 3. dávky) a neměli v anamnéze žádné onemocnění COVID-19.
a. N = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v
každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po
3. dávce do konce období sledování.
d. Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný 95% interval spolehlivosti (CI) pro účinnost vakcíny je odvozen na základě Clopperovy
a Pearsonovy metody upravené podle doby sledování.
Účinnost vakcíny u účastníků s prodělanou infekcí SARS-CoV-2 nebo bez ní byla podobná jako
u účastníků bez prodělané infekce SARS-CoV-2.
Kritéria závažného onemocnění COVID-19 (jak je popsáno v protokolu podle definice FDA
a upraveno pro děti) byla splněna u 12 případů (8 z nich dostalo vakcínu mRNA proti onemocnění
COVID-19 a 4 placebo) mezi účastníky ve věku od 6 měsíců do 4 let. Mezi účastníky ve věku
6 měsíců až 23 měsíců věku byla splněna kritéria závažného onemocnění COVID-19 u 3 případů
(2 z nich dostali vakcínu mRNA proti onemocnění COVID-19 a 1 placebo).
Byly provedeny analýzy imunogenicity u 82 účastníků studie 3 v podskupině s imunologickým přemostěním ve věku od 6 do 23 měsíců a u 143 účastníků studie 3 ve věku od 2 do 4 let bez průkazu infekce až do 1 měsíce po podání 3. dávky (datum konce sběru dat 29 dubna 2022).
Byly porovnány 50% neutralizační titry protilátek (NT50) proti SARS-CoV-2 mezi účastníky
v podskupině s imunogenicitou fáze 2/3 ve věku od 6 do 23 měsíců a ve věku od 2 do 4 let ze studie 3
1 měsíc po absolvování 3dávkového základního očkovacího schématu a účastníky v náhodně vybrané podskupině ze studie 2 fáze 2/3 ve věku 16 až 25 let 1 měsíc po absolvování 2dávkového základního očkovacího schématu. Tyto neutralizační titry byly stanoveny pomocí mikroneutralizačního testu
u referenčního kmene (USA_WA1/2020).
Primární analýzy imunologického přemostění porovnávaly geometrické průměry titrů (s využitím
geometrického průměru poměrů [GMR]) a četnost sérologické odpovědi (definované jako dosažení
≥ 4 násobného nárůstu SARS-CoV-2 NT50 oproti výchozí hodnotě před podáním 1. dávky)
u populace hodnotitelné na imunogenicitu bez průkazu prodělané infekce SARS-CoV-2 až do
1 měsíce po podání 3. dávky v případě účastníků ve věku od 6 do 23 měsíců a ve věku od 2 do 4 let a až do 1 měsíce po podání 2. dávky v případě účastníků ve věku 16 až 25 let. U obou věkových skupin
byla splněna předem stanovená kritéria imunologického přemostění jak pro poměr geometrického
průměru (GMR), tak pro rozdíl sérologických odpovědí (tabulka 9).
Tabulka 9: GMT SARS-CoV-2 (NT50) a rozdíly v procentuálních počtech účastníků se sérologickou odpovědí 1 měsíc po absolvování očkovacího schématu – podskupina s imunologickým přemostěním – účastníci ve věku od 6 měsíců do 4 let věku (studie 3) 1 měsíc po podání 3. dávky a účastníci ve věku od 16 do 25 let (studie 2)Neutralizační test SARS-CoV-2 (NT50) – GMT 1 měsíc po očkovacím schématu
| Neutralizační test SARS-CoV-2 – NT50 (titr)e
|
Věk
|
Na
| GMTb
(95% CIb) (1 měsíc po
3. dávce)
|
Věk
|
Na
| GMTb
(95% CIb) (1 měsíc po
2. dávce)
|
Věk
|
GMRc,d
(95% CI)
|
2 až
4 roky
|
143
|
1535,2 (1388,2; 1697,8)
|
16 až
25 let
|
170
| 1180,0
(1066,6;
1305,4)
| 2 až
4 roky /
16 až 25 let
|
1,30 (1,13,;1,50)
|
6 měsíců
až
23 měsíců
|
82
|
1406,5 (1211,3; 1633,1)
|
16 až
25 let
|
170
|
1180,0 (1066,6;
1305,4)
| 6 měsíců
až
23 měsíců /
16 až 25 let
|
1,19 (1,00; 1,42)
| Rozdíl v procentuálních počtech účastníků se sérologickou odpovědí 1 měsíc po očkovacím
schématu
| Neutralizační test SARS-CoV-2 – NT50 (titr)e
|
Věk
|
Na
|
nf (%) (95% CIg) (1 měsíc po
3. dávce)
|
Věk
|
Na
|
nf (%) (95% CIg) (1 měsíc po
2. dávce)
|
Věk
| Rozdíl
v četnostech sérologické odpovědi v %h (95% CIi)j
|
2 až
4 roky
|
141
|
141 (100,0) (97,4; 100,0)
|
16 až
25 let
|
170
|
168 (98,8) (95,8; 99,9)
| 2 až
4 roky /
16 až 25 let
|
1,2 (1,5; 4,2)
|
6 měsíců
až
23 měsíců
|
80
|
80 (100,0) (95,5; 100,0)
|
16 až
25 let
|
170
|
168 (98,8) (95,8; 99,9)
| 6 měsíců
až
23 měsíců /
16 až 25 let
|
1,2 (3,4; 4,2)
|
|
|
1 měsíc po podání 2. dávky – bez průkazu infekce SARS-CoV-2 – populace s hodnotitelnou imunogenicitouZkratky: CI = interval spolehlivosti; GMR = poměr geometrického průměru; GMT = titr geometrického
průměru; LLOQ = dolní limit kvantifikace; NAAT = test amplifikace nukleové kyseliny; N-vazba = SARS- CoV-2 nukleoproteinová vazba; NT50 = 50% neutralizační titr; SARS CoV-2 = závažný akutní respirační syndrom vyvolaný koronavirem 2.
Poznámka: Do analýzy byli zahrnuti účastníci, kteří neměli žádný sérologický nebo virologický průkaz [(odběr krve do 1 měsíce po podání 2. dávky (studie 2) nebo 1 měsíc po podání 3. dávky (studie 3)] prodělané infekce virem SARS-CoV-2 (tj. N-vázané protilátky [sérum] negativní při podání 1. dávky, 3. dávky (studie 3)
a 1 měsíc po podání 2. dávky (studie 2) nebo 1 měsíc po podání 3. dávky (studie 3) a virus SARS-CoV-2 nebyl detekován pomocí NAAT [nosní výtěr] při návštěvách s podáním 1. dávky, 2. dávky a 3. dávky (studie 3) a měli negativní NAAT (nosní výtěr) při kterékoli neplánované návštěvě do 1 měsíce po odběru krve po 2. dávce
(studie 2) nebo 1 měsíc po odběru krve po 3. dávce (studie 3)] a neměli v anamnéze žádné onemocnění COVID-
19.
Poznámka: Sérologická odpověď je definována jako dosažení ≥ 4násobného zvýšení oproti výchozí hodnotě (před 1. dávkou). Pokud je výchozí měření nižší než LLOQ, považuje se výsledek testu po očkování ≥4× LLOQ za sérologickou odpověď.
a. N = Počet účastníků s platnými a určitelnými výsledky testů pro daný test v daném časovém bodě dávky / odběru vzorků pro GMT a počet účastníků s platnými a určitelnými výsledky testů pro daný test jak ve výchozím stavu, tak v daném časovém bodě dávky / odběru vzorků pro četnost sérologické odpovědi.
b. GMT a 2stranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů
a příslušných CI (na základě Studentova t rozdělení). Výsledky testů pod LLOQ byly stanoveny na
0,5násobku LLOQ.
c. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (mladší věková skupina minus věk 16–25 let) a odpovídajícího CI (na základě Studentova t rozložení).
d. Pro každou mladší věkovou skupinu (2–4 roky, 6–23 měsíců) je imunologické přemostění podle GMT
deklarováno, pokud je dolní hranice 2stranného 95% CI pro GMR větší než 0,67 a bodový odhad GMR je
≥0,8.
e. NT50 SARS-CoV-2 byly stanoveny pomocí mikroneutralizačního testu SARS-CoV-2 mNeonGreen Virus Assay. Test používá fluorescenční reportérový virus odvozený od kmene USA_WA1/2020 a neutralizace viru se odečítá na monovrstvách buněk Vero. Vzorek NT50 je definován jako vzájemné zředění séra, při kterém je neutralizováno 50 % viru.
f. n = počet účastníků se sérologickou odpovědí na základě NT50 1 měsíc po podání 2. dávky. g. Přesný dvoustranný CI na základě Clopperovy a Pearsonovy metody.
h. Rozdíl v podílech vyjádřený v procentech (věk 5 až 11 let minus věk 16 až 25 let).
i. Přesný 2stranný CI založený na metodě Miettinena a Nurminena pro rozdíl v podílech, vyjádřený
v procentech.
j. Pro každou mladší věkovou skupinu (2 až 4 roky, 6 až 23 měsíců) je imunologické přemostění založené na četnosti sérologické odpovědi deklarováno, pokud je dolní hranice dvoustranného 95% CI pro rozdíl
v podílech větší než -10,0 % za předpokladu, že byla splněna kritéria imunologického přemostění na základě GMR.
Pediatrická populaceEvropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s vakcínou
Comirnaty u pediatrické populace k prevenci onemocnění COVID-19 (informace o použití u pediatrické populace viz bod 4.2).
5.2 Farmakokinetické vlastnostiNeuplatňuje se.
5.3 Předklinické údaje vztahující se k bezpečnostiNeklinické údaje získané na základě konvenčních studií toxicity po opakovaném podávání a
reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
Obecná toxicitaU potkanů, kterým byla intramuskulárně podávána vakcína Comirnaty (dostali 3 plné dávky pro
člověka jednou týdně, které vedly k relativně vyšším hladinám u potkanů v důsledku rozdílů v tělesné
hmotnosti), se vyskytl edém a erytém v místě podání injekce a zvýšení počtu bílých krvinek (včetně bazofilů a eozinofilů) odpovídající zánětlivé odpovědi a rovněž vakuolizace portálních hepatocytů bez známek poškození jater. Všechny účinky byly reverzibilní.
Genotoxicita / karcinogenitaStudie genotoxicity ani kancerogenity nebyly provedeny. U složek vakcíny (lipidy a mRNA) se
neočekává genotoxický potenciál.
Reprodukční toxicitaReprodukční a vývojová toxicita byla hodnocena na potkanech v kombinované studii fertility a
vývojové toxicity, ve které byla samicím potkanů podána intramuskulárně vakcína Comirnaty před pářením a během březosti (dostaly 4 plné dávky pro člověka, které vedly k relativně vyšším hladinám u potkanů v důsledku rozdílů v tělesné hmotnosti mezi 21. dnem před pářením a 20. dnem březosti).
Odpovědi na neutralizační protilátky SARS-CoV-2 byly přítomny u samic před pářením až do konce studie ve 21. postnatálním dni a rovněž u plodů a potomků. Nebyly zjištěny žádné účinky spojené s očkováním na plodnost samic, těhotenství nebo vývoj embrya/plodu nebo potomstva. Údaje o možném placentárním přenosu nebo vylučování do mateřského mléka pro vakcínu Comirnaty nejsou k dispozici.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159) Kolfosceryl-stearát
Cholesterol
Trometamol
Trometamol-hydrochlorid
Sacharosa
Voda pro injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou
uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřená injekční lahvička
Zmrazená injekčnílahvička
18 měsíců při teplotě -90 °C až -60 °C.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána při teplotě -90 °C až -60 °C nebo 2 °C až 8 °C.
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení
10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 2 hodin nebo mohou být jednotlivé
injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Rozmrazená injekční lahvička
10 týdnů uchovávání a transportu při teplotě 2 °C až 8 °C během 18měsíční doby použitelnosti.
• Po přenesení vakcíny pro uchovávání při teplotě 2 °C až 8 °C musí být na vnější obal zapsáno aktualizované datum použitelnosti a vakcína má být použita nebo zlikvidována do aktualizovaného data použitelnosti. Původní datum použitelnosti má být přeškrtnuto.
• Pokud je vakcína přijata při teplotě 2 °C až 8 °C, má být uchovávána při teplotě 2 °C až 8 °C.
Datum použitelnosti na vnějším obalu má být aktualizováno tak, aby odráželo datum
použitelnosti v chladu, a původní datum použitelnosti má být přeškrtnuto.
Neotevřené injekční lahvičky lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
P
o rozmrazení vakcína nesmí být znovu zmrazena.
M
anipulace
přiteplotníchvýkyvechpřiuchovávánívchladničce
• Údaje o stabilitě prokazují, že je neotevřená injekční lahvička stabilní až 10 týdnů, je-li
uchovávána při teplotě od -2 °C do 2 °C a během 10týdenního uchovávání při teplotě 2 °C až
8 °C.
• Údaje o stabilitě ukazují, že injekční lahvička může být uchovávána až 24 hodin při teplotě od
8 °C do 30 °C, včetně až 12 hodin po prvním vpichu.
Tyto informace slouží jako návod pro zdravotnické pracovníky pouze v případě dočasného výkyvu
teploty.
Zředěný léčivý přípravek
Chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 12 hodin při teplotě
2 °C až 30 °C po naředění injekčním roztokem chloridu sodného 9 mg/ml (0,9%), která zahrnuje až
6 hodin transportu. Z mikrobiologického hlediska, pokud způsob ředění nevyloučí riziko mikrobiologické kontaminace, má být přípravek použit okamžitě. Pokud není použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Během uchovávání je třeba minimalizovat vystavení přípravku světlu v místnosti a zabránit vystavení
přímému slunečnímu světlu a ultrafialovému světlu.
Podmínky uchovávání tohoto léčivého přípravku po jeho rozmrazení a naředění jsou uvedeny
v bodě 6.3.
6.5 Druh obalu a obsah balení
0,4 ml koncentrátu pro disperzi v čiré vícedávkové injekční lahvičce (sklo třídy I) o objemu 2 ml se zátkou (syntetická brombutylová pryž) a hnědočerveným odtrhovacím plastovým víčkem
s hliníkovým krytem. Jedna injekční lahvička obsahuje 10 dávek, viz bod 6.6.
Velikosti balení: 10 injekčních lahviček
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Instrukce pro zacházení s vakcínou
Vakcína Comirnaty 3 mikrogramy/dávku má být připravována zdravotnickým pracovníkem pomocí
aseptické techniky, aby byla zajištěna sterilita připravené disperze.
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY
3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (KOJENCI A
DĚT
I VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY)
H
nědočervené víčko
 P
o naředění
3 mikrogramy
P
o naředění
3 mikrogramy
• Zkontrolujte, zda má injekční lahvička hnědočervené plastové víčko.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička šedé
plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku injekční disperze, Comirnaty Original/Omicron BA.1
(15/15 mikrogramů)/dávku injekční
disperze nebo Comirnaty
Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze.
• Pokud má injekční lahvička oranžové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi nebo Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro injekční disperzi.
Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY 3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (KOJENCI A DĚTI VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY) PŘED POUŽITÍM
|
U
chovávejte až
10 týdnů při teplotě 2 °C až
8 °C.
|
• Pokud se vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se rozmrazily;
rozmrazení balení 10 injekčních lahviček může trvat 2 hodiny. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
MÍCHÁNÍ VAKCÍNY COMIRNATY 3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO
I
N
JEKČNÍ DISPERZI (KOJENCI A DĚTI VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY) PŘED ŘEDĚNÍM
|
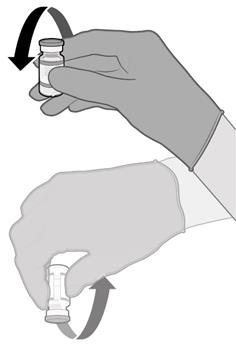
Jemně × 10
|
• Rozmrazenou injekční lahvičku nechte ohřát na pokojovou teplotu a před ředěním ji 10krát jemně převraťte. Netřepejte.
• Před ředěním může rozmrazená disperze obsahovat bílé až téměř bílé matné amorfní částice.
|
ŘEDĚN
Í VAKCÍNY COMIRNATY 3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO
I
N
JEKČNÍ DISPERZI (KOJENCI A DĚTI VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY)
|
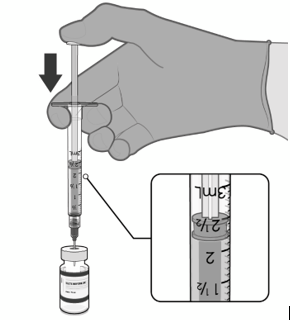
2,2 ml injekčního roztoku chloridu
sodného 9 mg/ml (0,9 %)
|
• Rozmrazená vakcína se musí naředit v původní injekční lahvičce 2,2 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%) za použití jehly
o velikosti 21 gauge (21G) nebo užší a uplatnění aseptických metod.
|

V
ytáhněte píst na značku 2,2 ml pro
odstranění vzduchu z injekční lahvičky.
|
• Nasátím 2,2 ml vzduchu do prázdné stříkačky na rozpouštědlo vyrovnejte před vyjmutím jehly z injekční lahvičky tlak v injekční lahvičce.
|
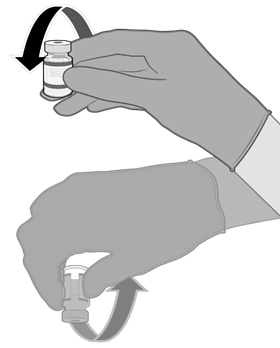
Jemně x 10
|
• Disperzi 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Naředěná vakcína se má jevit jako bílá až téměř bílá disperze bez viditelných
částic. Jestliže jsou v naředěné vakcíně
patrné částice nebo vakcína změnila
barvu, nepoužívejte ji.
|
Č
as likvidace
Z
a
z
namenejte příslušné datum a čas. Použijte během 12 hodin po naředění.
|
• Injekční lahvičky s naředěnou vakcínou je třeba označit příslušným datem
a časem.
• Po naředění uchovávejte vakcínu při teplotě 2 °C až 30 °C a použijte ji během
12 hodin.
• Naředěnou disperzi nezmrazujte ani s ní netřepejte. Pokud je v chladu, nechte naředěnou disperzi před použitím dosáhnout pokojové teploty.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,2 ml VAKCÍNY COMIRNATY
3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (KOJENCI A
DĚT
I VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY)
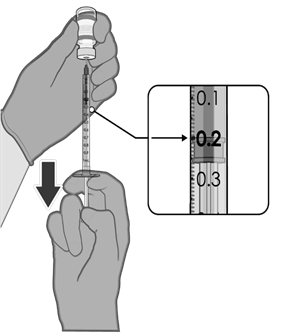 N
aředěná vakcína o objemu 0,2 ml
N
aředěná vakcína o objemu 0,2 ml
• Po naředění obsahuje injekční lahvička
2,6 ml, ze kterých lze získat 10 dávek po
0,2 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím
antiseptického tampónu.
• Natáhněte 0,2 ml vakcíny Comirnaty pro kojence a děti ve věku 6 měsíců až
4 roky.
K získání 10 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční stříkačky
a jehly s malým mrtvým prostorem má
mít mrtvý objem maximálně
35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání deseti dávek z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,2 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,2 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Veškerou vakcínu, jež nebyla použita do
12 hodin po naředění, zlikvidujte.
L
i
kv
i
dace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.de8. REGISTRAČNÍ ČÍSLO/REGISTRAČNÍ ČÍSLAEU/1/20/1528/010
9. DATUM REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. prosince 2020
Datum posledního prodloužení registrace: 10. října 2022
10. DATUM REVIZE TEXTUPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKUComirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční disperze mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍJedná se o vícedávkovou injekční lahvičku s šedým víčkem. Před použitím neřeďte. Jedna injekční lahvička (2,25 ml) obsahuje 6 dávek po 0,3 ml, viz body 4.2 a 6.6.
Jedna dávka (0,3 ml) obsahuje 15 mikrogramů tozinameranu a 15 mikrogramů riltozinameranu,
mRNA vakcíny proti onemocnění COVID-19 (zapouzdřené v lipidových nanočásticích).
Tozinameran je jednovláknová mediátorová (messenger) RNA (mRNA) s čepičkou na 5´ konci vyráběná
in vitro nebuněčnou transkripcí z příslušných matricí DNA a kódující spike (S) protein viru SARS-CoV-2 (Original). Riltozinameran je jednovláknová mediátorová (messenger) RNA (mRNA)
s čepičkou na 5´ konci vyráběná
in vitro nebuněčnou transkripcí z příslušných matricí DNA a kódující spike (S) protein viru SARS-CoV-2 (Omikron BA.1).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMAInjekční disperze.
Vakcína je bílá až téměř bílá zmrazená disperze (pH: 6,9–7,9).
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikaceVakcína Comirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční disperze je indikována pro aktivní imunizaci k prevenci onemocnění COVID-19 způsobeného SARS-CoV-2 u osob ve věku 12 let a starších, kteří již absolvovali alespoň základní očkování proti onemocnění COVID-19 (viz body 4.2 a 5.1).
Tuto vakcínu je třeba používat v souladu s oficiálními doporučeními.
4.2 Dávkování a způsob podáníDávkováníDávka vakcíny Comirnaty Original/Omicron BA.1 je 0,3 ml podávaná intramuskulárně.
Mezi podáním vakcíny Comirnaty Original/Omicron BA.1 a poslední předchozí dávkou vakcíny proti onemocnění COVID-19 má uplynout interval nejméně 3 měsíce.
Vakcína Comirnaty Original/Omicron BA.1 je indikována pouze u jedinců, kteří již dříve podstoupili alespoň základní očkování proti onemocnění COVID-19.
Podrobnosti ohledně základního očkování u osob ve věku 12 let a starších naleznete v souhrnu údajů o přípravku pro vakcínu Comirnaty 30 mikrogramů/dávku koncentrátu pro injekční disperzi a vakcínu Comirnaty 30 mikrogramů/dávku injekční disperze.
Pediatrická populace
Bezpečnost a účinnost vakcíny Comirnaty Original/Omicron BA.1 u dětí ve věku do 12 let nebyly
dosud stanoveny. Nejsou dostupné žádné údaje.
Starší populace
U osob ve věku ≥ 65 let není nutná žádná úprava dávkování.
Způsob podání
Vakcína Comirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční disperze se podává
intramuskulárně (viz bod 6.6). Před použitím neřeďte.
Injekční lahvičky vakcíny Comirnaty Original/Omicron BA.1 obsahují 6 dávek po 0,3 ml vakcíny. K získání 6 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů. Pokud se používají standardní injekční stříkačky a jehly, nemusí být dostatečný objem k získání šesté dávky z jedné injekční lahvičky. Bez ohledu na typ injekční stříkačky a jehly:
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml,
injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Nekombinujte obsah z více injekčních lahviček vakcíny. Preferované místo je deltový sval horní části paže.
Vakcína se nesmí podávat intravaskulárně, subkutánně ani intradermálně.
Vakcína se nesmí mísit ve stejné injekční stříkačce s jinými vakcínami nebo léčivými přípravky. Pro opatření před podáním vakcíny viz bod 4.4.
Návod pro rozmrazení, zacházení s vakcínou a její likvidaci je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Sledovatelnost
Aby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
O
becná doporučení
Hypersenzitivita a anafylaxe
Byly hlášeny případy anafylaxe. Pro případ, že by po podání vakcíny došlo k anafylaktické reakci, má
být zajištěna okamžitá lékařská péče a dohled.
Po vakcinaci se doporučuje pečlivé sledování po dobu minimálně 15 minut. Další dávka vakcíny nemá být podána osobám, které měly anafylaxi po předchozí dávce vakcíny Comirnaty.
Myokarditida a perikarditida
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy a perikarditidy. Tato
onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhé dávce vakcíny a častěji u mladších mužů a chlapců. Dostupné údaje naznačují, že průběh myokarditidy a perikarditidy po vakcinaci se neliší od myokarditidy nebo perikarditidy obecně (viz bod 4.8).
Zdravotničtí pracovníci mají pozorně sledovat známky a příznaky myokarditidy a perikarditidy. Očkovaní jedinci (včetně rodičů nebo pečovatelů) mají být poučeni, aby okamžitě vyhledali lékařskou pomoc, pokud se u nich po očkování objeví příznaky naznačující myokarditidu nebo perikarditidu, například bolest na hrudi (akutní a přetrvávající), dušnost nebo palpitace.
Zdravotničtí pracovníci mají k diagnostice a léčbě tohoto onemocnění používat návody a postupy
a/nebo se mají obrátit na specialisty.
Reakce spojené s úzkostí
V souvislosti se samotným procesem očkování se mohou objevit reakce spojené s úzkostí, včetně
vazovagálních reakcí (synkopa), hyperventilace nebo reakcí spojených se stresem (např. závrať, palpitace, zvýšení srdeční frekvence, změny krevního tlaku, parestezie, hypestezie a pocení). Reakce
spojené se stresem jsou dočasné a samy se upraví. Očkované osoby je třeba informovat o tom, aby na
případné symptomy upozornily očkujícího zdravotníka, který je vyhodnotí. Je důležité, aby byla
zavedena opatření, aby se zabránilo zranění v důsledku mdlob.
Současné onemocnění
U osob trpících závažným akutním horečnatým onemocněním nebo akutní infekcí se má podání
vakcíny Comirnaty odložit. Přítomnost mírné infekce a/nebo horečky nízkého stupně není důvod
k odložení vakcinace.
Trombocytopenie a poruchy koagulace
Stejně jako u jiných intramuskulárních injekcí je třeba vakcínu podávat opatrně osobám podstupujícím
léčbu antikoagulancii nebo osobám s trombocytopenií nebo poruchami koagulace (jako je hemofilie),
protože po intramuskulárním podání může u těchto osob dojít ke krvácení nebo tvorbě modřin.
Imunokompromitované osoby
Účinnost a bezpečnost vakcíny nebyly hodnoceny u imunokompromitovaných osob, včetně osob
podstupujících imunosupresivní léčbu. Účinnost vakcíny Comirnaty Original/Omicron BA.1 může být
u imunokompromitovaných osob nižší.
Doba ochrany
Doba ochrany poskytovaná vakcínou není známa, protože je stále hodnocena v probíhajících
klinických studiích.
Om
ezení
ú
činnosti vakcíny
Podobně jako u jiných vakcín je možné, že vakcinace vakcínou Comirnaty Original/Omicron BA.1
nebude chránit všechny její příjemce.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Současné podání vakcíny Comirnaty Original/Omicron BA.1 s jinými vakcínami nebylo hodnoceno.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Údaje o použití vakcíny Comirnaty Original/Omicron BA.1 během těhotenství nejsou k dispozici.
Nicméně velké množství observačních dat od těhotných žen očkovaných původně schválenou vakcínou Comirnaty během druhého a třetího trimestru neprokázalo zvýšení nežádoucích výsledků těhotenství. Ačkoli údaje o výsledcích těhotenství po očkování během prvního trimestru jsou v současné době omezené, nebylo pozorováno zvýšené riziko potratu. Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na těhotenství, vývoj embrya/plodu, porod nebo postnatální vývoj (viz bod 5.3). Protože rozdíly mezi přípravky jsou omezeny na sekvenci spike proteinu a neexistují klinicky významné rozdíly v reaktogenicitě, vakcínu Comirnaty Original/Omicron BA.1 lze
v těhotenství podávat.
Kojení
Údaje o použití vakcíny Comirnaty Original/Omicron BA.1 během kojení nejsou k dispozici.
Systémová expozice vakcíně je u kojící matky nicméně zanedbatelná, a proto nejsou očekávány žádné účinky na kojeného novorozence/kojence (skrze mateřské mléko). Observační údaje od žen, které po očkování původně schválenou vakcínou Comirnaty kojily, neprokázaly riziko nežádoucích účinků u kojených novorozenců/kojenců. Vakcínu Comirnaty Original/Omicron BA.1 lze během kojení podávat.
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky s ohledem na reprodukční toxicitu
(viz bod 5.3)
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vakcína Comirnaty Original/Omicron BA.1 nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Některé z účinků uvedených v bodě 4.8 však mohou schopnost řídit nebo obsluhovat stroje dočasně ovlivnit.
4.8 Nežádoucí účinky
Souhrn bezpečnostníhoprofilu
Comirnaty Original/Omicron BA.1
Účastníci ve věku > 55 let – po posilovací dávce vakcíny Comirnaty Original/Omicron BA.1 (čtvrtá
dávka)
V podmnožině ze studie 4 (3. fáze) obdrželo 305 dospělých ve věku > 55 let, kteří dokončili očkování třemi dávkami vakcíny Comirnaty, posilovací dávku (čtvrtou dávku) vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) 4,7 až 11,5 měsíců po podání 3. dávky. Účastníci, kteří obdrželi posilovací dávku (čtvrtou dávku) vakcíny Comirnaty Original/Omicron BA.1 měli medián sledování nejméně 1,7 měsíce.
Celkový bezpečnostní profil posilovací dávky (čtvrté dávky) vakcíny Comirnaty Original/Omicron BA.1 byl podobný profilu zjištěnému po posilovací dávce (třetí dávce) vakcíny Comirnaty. U účastníků ve věku vyšším než 55 let byly nejčastějšími nežádoucími účinky bolest v místě vpichu (> 50 %), únava (> 40 %), bolest hlavy (> 30 %), myalgie (> 20 %), zimnice a artralgie (>10 %). U vakcíny Comirnaty Original/Omicron BA.1 nebyly zjištěny žádné nové nežádoucí účinky.
Účastníci ve věku 18 až ≤ 55 let – po posilovací dávce monovalentní vakcíny Omicron BA.1 (čtvrtá
dávka)
Bezpečnost posilovací dávky vakcíny Comirnaty Original/Omicron BA.1 u jednotlivců ve věku
18 až ≤ 55 let je extrapolována z bezpečnostních údajů z podmnožiny 315 dospělých ve věku
18 až ≤ 55 let, kteří obdrželi 30 mikrogramů posilovací dávky (čtvrté dávky) vakcíny Omicron BA.1 (monovalentní) po absolvování 3 dávek vakcíny Comirnaty. Nejčastějšími nežádoucími účinky u těchto účastníků ve věku 18 až ≤ 55 let byla bolest v místě vpichu (> 70 %), únava (> 60 %), bolest hlavy (> 40 %), myalgie (> 30 %), zimnice (> 30 %) a artralgie (>20 %).
Vakcína Comirnaty 30 mikrogramů
Účastníci ve věku 16 let a starší – po 2 dávkách
Ve studii 2 byla celkem 22 026 účastníkům ve věku 16 let nebo starším podána alespoň 1 dávka vakcíny Comirnaty a celkem 22 021 účastníkům ve věku 16 let nebo starším bylo podáno placebo
(včetně 138 a 145 dospívajících ve věku 16 a 17 let ve skupinách vakcíny a placeba, v uvedeném
pořadí). Celkem 20 519 účastníků ve věku 16 let nebo starších dostalo 2 dávky vakcíny Comirnaty.
V době provedení analýzy studie 2 s datem ukončení sběru dat 13. března 2021 pro placebem kontrolované zaslepené období sledování až do dat odslepení účastníků bylo celkem 25 651 (58,2 %) účastníků (13 031 Comirnaty a 12 620 placebo) ve věku 16 let a starších sledováno po dobu ≥4 měsíce po podání druhé dávky. To zahrnovalo celkem 15 111 (7 704 Comirnaty a 7 407 placebo) účastníků ve věku 16 až 55 let a celkem 10 540 (5 327 Comirnaty a 5 213 placebo) účastníků ve věku 56 let a starších.
Nejčastějšími nežádoucími účinky byla u účastníků ve věku 16 let a starších, kteří dostali 2 dávky, bolest v místě injekce (> 80 %), únava (> 60 %), bolest hlavy (> 50 %), myalgie (> 40 %), zimnice
(> 30 %), artralgie (> 20 %), pyrexie a zduření v místě injekce (> 10 %). Tyto nežádoucí účinky byly zpravidla mírné nebo střední intenzity a odezněly během několika dní po vakcinaci. Mírně nižší frekvence příhod reaktogenity souvisela s vyšším věkem.
Bezpečnostní profil u 545 účastníků ve věku 16 let a starších, kterým byla podána vakcína Comirnaty
a kteří byli séropozitivní na SARS-CoV-2 při výchozím stavu, byl podobný jako u obecné populace.
Dospívající ve věku 12 až 15 let – po 2 dávkách
V analýze dlouhodobého následného sledování bezpečnosti ve studii 2 bylo 2 260 dospívajících (1 131
dostalo vakcínu Comirnaty a 1 129 dostalo placebo) ve věku 12 až 15 let. Z toho 1 559 dospívajících
(786 dostalo vakcínu Comirnaty a 773 dostalo placebo) bylo sledováno po dobu ≥ 4 měsíců po druhé dávce vakcíny Comirnaty. Hodnocení bezpečnosti ve studii 2 probíhá.
Celkový bezpečnostní profil vakcíny Comirnaty u dospívajících ve věku 12 až 15 let byl podobný jako
u účastníků ve věku 16 let a starších. Nejčastějšími nežádoucími účinky u dospívajících ve věku 12 až
15 let, kteří dostali 2 dávky, byly bolest v místě vpichu (> 90 %), únava a bolest hlavy (> 70 %), myalgie a zimnice (> 40 %), artralgie a pyrexie (> 20 %).
Účastníci ve věku 16 let a starší – po posilovací dávce
Podskupina účastníků studie fáze 2/3, do které bylo zařazeno 306 dospělých ve věku 18 až 55 let, kteří dokončili původní cyklus 2 dávek vakcíny Comirnaty, dostala posilovací dávku vakcíny Comirnaty
přibližně 6 měsíců (rozmezí 4,8 až 8,0 měsíců) po podání 2. dávky.
Celkový bezpečnostní profil posilovací dávky byl podobný jako po 2 dávkách. Nejčastějšími nežádoucími účinky u účastníků ve věku 18 až 55 let byly bolest v místě vpichu (> 80 %), únava (>
60 %), bolest hlavy (> 40 %), myalgie (> 30 %), zimnice a artralgie (> 20 %).
Ve studii 4, placebem kontrolované studii posilovací dávky, byla účastníkům ve věku 16 let a starším
zařazeným ze studie 2 podána posilovací dávka vakcíny Comirnaty (5 081 účastníků) nebo placebo (5 044 účastníků), a to nejméně 6 měsíců po druhé dávce vakcíny Comirnaty. Medián doby následného sledování účastníků, kterým byla posilovací dávka podána, činil celkem 2,5 měsíce po podání posilovací dávky do dne ukončení sběru dat (5. října 2021). Žádné nové nežádoucí účinky vakcíny Comirnaty nebyly zjištěny.
Posilovací dávka po primární vakcinaci jinou schválenou vakcínou protionemocněníCOVID-19
V 5 nezávislých studiích o použití posilovací dávky vakcíny Comirnaty u osob, které absolvovaly
primární vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19 (heterologní posilovací
dávka), nebyly zjištěny žádné nové bezpečnostní problémy (viz bod 5.1).
Tabulkovýseznamnežádoucíchúčinkůzklinických studií vakcíny Comirnaty a Comirnaty
Original/Omicron BA.1apouvedenívakcínyComirnatynatrhuosobvevěku12let a starších
Nežádoucí účinky pozorované z klinických studií jsou uvedeny níže podle následujících kategorií
frekvence:
Velmi časté (≥ 1/10)
Časté (≥ 1/100 až < 1/10)
Méně časté (≥ 1/1 000 až < 1/100)
Vzácné (≥ 1/10 000 až < 1/1 000) Velmi vzácné (< 1/10 000)
Není známo (z dostupných údajů nelze určit)
T
abulka 1: Nežádoucí účinky vakcíny Comirnaty a Comirnaty Original/Omicron BA.1
T
řída
orgánového systému
|
V
elmi
časté
(
≥ 1/10)
|
Č
asté
(
≥ 1/100
až
< 1/10)
|
Méně časté
(
≥ 1/1 000 až
< 1/100)
|
V
z
ácné
(
≥ 1/10 000
až
< 1/1 000)
|
V
elmi vzácné
(
< 1/10 000
|
N
ení známo (z
dostupnýc
h údajů nelze určit)
|
Poruchy krve a
lymfatického systému
|
|
|
Lymfadenopatiea
|
|
|
|
Poruchy
imunitního systému
|
|
|
Hypersenzitivní
reakce (např. vyrážka, pruritus, urtikarieb, angioedémb)
|
|
|
Anafylaxe
|
Poruchy
metabolismu a výživy
|
|
|
Snížená chuť k
jídlu
|
|
|
|
Psychiatrické
poruchy
|
|
|
Insomnie
|
|
|
|
Poruchy
nervového systému
|
Bolest
hlavy
|
|
Letargie
|
Akutní
periferní paralýza n. facialisc
|
|
Parestezied;
hypestezied
|
Srdeční poruchy
|
|
|
|
|
Myokarditidad;
Perikarditidad
|
|
Gastrointestinální
poruchy
|
Průjemd
|
Nauzea;
zvraceníd
|
|
|
|
|
Poruchy kůže a
podkožní tkáně
|
|
|
Hyperhidróza;
noční pocení
|
|
|
Erythema
multiformed
|
Poruchy svalové
a kosterní soustavy a pojivové tkáně
|
Artralgie;
myalgie
|
|
Bolest
v končetiněe
|
|
|
|
Poruchy
reprodukčního
systému a prsu
|
|
|
|
|
|
Silné
menstruační
krváceníh
|
Celkové poruchy
a reakce v místě
aplikace
|
Bolest v
místě injekce; únava; zimnice; pyrexief; zduření v místě injekce
|
Zarudnutí
v místě
injekce
|
Astenie;
malátnost; svědění v místě injekce
|
|
|
Rozsáhlý
otok končetiny, do níž byla vakcína podánad; otok obličejeg
|
|
|
z klinických studií a po uvedení vakcíny Comirnaty na trh u osob ve věku 12 let a starších
a. U účastníků, kteří dostali posilovací dávku ve studii 4, byla pozorována vyšší frekvence lymfadenopatie
(2,8 % oproti 0,4 %) ve srovnání s účastníky, kteří dostali dvě dávky.
b. Kategorie frekvence pro urtikarii a angioedém byla vzácná.
c. Během období sledování bezpečnosti v klinické studii do 14. listopadu 2020 byla hlášena akutní periferní paralýza
n. facialis u čtyř účastníků ve skupině mRNA vakcíny proti onemocnění COVID-19. Nástup obrny
obličeje byl 37. den po podání 1. dávky (účastník nedostal 2. dávku) a 3., 9. a 48. den po 2. dávce. Ve
skupině s placebem nebyly hlášeny žádné případy akutní periferní paralýzy
n. facialis. d. Nežádoucí účinek byl stanoven po registraci.
e. Týká se končetiny/paže, do které byla daná osoba očkována.
f. Vyšší frekvence pyrexie byla pozorována po druhé dávce v porovnání s první dávkou.
g. Po uvedení přípravku na trh byly hlášeny případy otoku obličeje u očkovaných osob, které v minulosti
podstoupily injekční aplikaci dermálních výplní do obličeje.
h. Většina případů se zdála být nezávažné a dočasné povahy.
Popis vybraných nežádoucíchúčinkůMyokarditida a perikarditidaZvýšené riziko myokarditidy po očkování vakcínou Comirnaty je nejvyšší u mladších mužů a chlapců
(viz bod 4.4).
Zvýšené riziko u mladších mužů a chlapců po podání druhé dávky vakcíny Comirnaty bylo blíže určeno ve dvou velkých evropských farmakoepidemiologických studiích. Z jedné studie vyplynulo, že v období 7 dnů po podání druhé dávky se u mužů a chlapců ve věku 12–29 let vyskytlo přibližně o
0,265 (95% CI 0,255–0,275) případů myokarditidy na 10 000 osob více než u neočkovaných osob. V
další studii se v období 28 dnů po podání druhé dávky u mužů a chlapců ve věku 16–24 let vyskytlo o
0,56 (95% CI 0,37–0,74) případů myokarditidy na 10 000 osob více než u neočkovaných osob.
Omezené údaje ukazují, že riziko myokarditidy a perikarditidy je po očkování vakcínou Comirnaty
u dětí ve věku od 5 do 11 let pravděpodobně nižší než u dospívajících ve věku od 12 do 17 let.
Hlášení podezření na nežádoucí účinkyHlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to
pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V a uvedli přitom číslo šarže, je-li k dispozici.
4.9 PředávkováníÚdaje o předávkování jsou k dispozici od 52 účastníků studie zařazených do klinického hodnocení, kterým bylo kvůli chybě v ředění podáno 58 mikrogramů vakcíny Comirnaty. Příjemci vakcíny nehlásili zvýšení reaktogenity ani nežádoucí účinky.
V případě předávkování se doporučuje sledovat základní životní funkce a případně zahájit symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: vakcíny, jiné virové vakcíny, ATC kód: J07BX03
Mechanismus účinkuMediátorová (messenger) RNA s modifikovanými nukleosidy je ve vakcíně Comirnaty zapouzdřená
v lipidových nanočásticích, což umožňuje transfer nereplikující RNA do hostitelských buněk pro přímou přechodnou expresi S antigenu viru SARS-CoV-2. mRNA kóduje v membráně ukotvený S v
plné délce se dvěma bodovými mutacemi v centrální šroubovici. Mutace těchto dvou aminokyselin na
prolin uzamyká S v antigenně preferované prefuzní konformaci. Vakcína vyvolává jak odpověď neutralizačních protilátek, tak i imunitní odpověď buněk na spike (S) antigen, což může přispívat
k ochraně před onemocněním COVID-19.
Ú
činnost
V
akcína Comirnaty Original/Omicron BA.1
R
elativní imunogenicita vakcíny u účastníků ve věku > 55 let – po posilovací dávce vakcíny Comirnaty
O
ri
ginal/Omicron BA.1 (čtvrtá dávka)
V dílčí analýze množiny ze studie 4 (podstudie E) obdrželo 610 dospělých starších 55 let, kteří dokončili řadu 3 dávek vakcíny Comirnaty, následující dávku jako posilovací dávku (čtvrtou dávku):
vakcína Comirnaty (30 mikrogramů) nebo vakcína Comirnaty Original/Omicron BA.1
(15/15 mikrogramů). GMR a míry odpovědi v séru byly hodnoceny 1 měsíc po posilovací vakcinaci vakcínou Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) až do data ukončení
16. května 2022, což představuje medián nejméně 1,7 měsíce sledování po posilovací dávce.
Posilovací dávka vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) byla podána v intervalu 4,7 až 11,5 měsíce (medián 6,3 měsíce) po třetí dávce.
Primárním cílem analýzy bylo vyhodnotit superioritu vzhledem k úrovni neutralizačního titru a non- inferioritu ohledně míry odpovědi v séru imunitní odpovědi proti viru Omikron indukované dávkou vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) v porovnání s odpovědí vyvolanou dávkou vakcíny Comirnaty (30 mikrogramů) podanou jako čtvrtá dávka u účastníků starších 55 let, kteří již byli vakcínou Comirnaty očkováni.
Superiorita vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) vůči vakcíně Comirnaty
(30 mikrogramů) byla splněna, protože dolní hranice 2stranného 95% CI pro GMR byla > 1 (tabulka 2).
Sérová odpověď je definována jako dosažení ≥ 4násobný nárůst vůči výchozímu stavu (před studijním očkování). Pokud je výchozí měření nižší než LLOQ, měření po vakcinaci ≥ 4 × LLOQ se považuje za sérovou odpověď.
Rozdíl v procentech účastníků, kteří dosáhli sérové odpovědi na variantu Omikron, mezi skupinou Comirnaty Original/Omicron BA.1 (71,6 %) a skupinou Comirnaty (57 %) byl 14,6 % (2stranný 95% CI: 4,0 %, 24,9 %). Tak byla splněna podmínka non-inferiority.
Tabulka 2: Podstudie E – poměry geometrických průměrů porovnání mezi skupinami vakcín – účastníci bez prokázané infekce až do 1 měsíce po 4. dávce – rozšířená kohorta – podmnožina imunogenicity – účastníci starší 55 let – populace s vyhodnotitelnou imunogenicitou
Analýza
|
Skupina s vakcínou
(podle randomizace)
| Časový
bod odběru vzorkůa
|
nb
|
GMT (95% CIc)
|
GMR (95% CId)
|
Analýza neutralizace SARS-CoV-2 – Omikron BA.1 - NT50 (titr)
| Comirnaty
(30 mikrogramů)
|
1 měsíc
|
163
| 455,8
(365,9; 567,6)
|
|
Comirnaty
Original/Omicron BA.1 (15/15 mikrogramů)
|
1 měsíc
|
178
|
711,0 (588,3; 859,2)
|
1,56 (1,17; 2,08)
|
Analýza neutralizace SARS-CoV-2 – referenční kmen – NT50 (titr)
| Comirnaty
(30 mikrogramů)
|
1 měsíc
|
182
| 5 998,1
(5 223,6;
6 887,4)
|
|
Comirnaty
Original/Omicron BA.1 (15/15 mikrogramů)
|
1 měsíc
|
186
| 5 933,2
(5 188,2;
6 785,2)
|
0,99 (0,82; 1,20)
|
Zkratky: CI = interval spolehlivosti; GMR = poměr geometrického průměru; GMT = titr geometrického průměru; LLOQ = dolní limit kvantifikace; N-vazba = SARS-CoV-2 nukleoproteinová vazba; NAAT = test amplifikace nukleové kyseliny; NT50 = 50% neutralizační titr; SARS-CoV-2 = závažný akutní respirační syndrom vyvolaný koronavirem 2.
Poznámka: Podmnožina imunogenicity = náhodný vzorek 230 účastníků v každé skupině vakcíny vybraných
z rozšířené kohorty.
Poznámka: Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (odběr vzorků krve dříve než
1 měsíc po studijní vakcinaci) předcházející infekce SARS-CoV-2 (tj. negativní výsledek N-binding protilátky
[sérum] na návštěvách při studijní vakcinaci a za 1 měsíc po vakcinaci, negativní výsledek NAAT [stěr z nosu] na návštěvě se studijní vakcinací a jakékoliv neplánované návštěvě před odběrem vzorku krve 1 měsíc po studijní vakcinaci) a kteří neměli onemocnění COVID-19 ve zdravotní anamnéze, byli zařazeni do analýzy.
a. Protokolárně stanovené načasování odběru krevních vzorků.
b. n = počet účastníků s platnými a určitelnými výsledky testu pro daný test při výchozím stavu, v daném
časovém bodě odběru vzorků.
c. GMT a 2stranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a příslušných CI (na základě Studentova t rozdělení). Výsledky testů pod LLOQ byly stanoveny na
0,5násobku LLOQ.
d. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (skupina vakcíny v příslušném řádku Comirnaty [30 mikrogramů]) a odpovídajícího CI (na základě Studentova t rozložení).
Vakcína Comirnaty 30 mikrogramůStudie 2 je multicentrická, mezinárodní, randomizovaná, placebem kontrolovaná studie fáze 1/2/3
zaslepená pro pozorovatele, která hodnotí dávku, výběr vakcíny a její účinnost u účastníků ve věku
12 let a starších. Randomizace byla stratifikována podle věku: věk 12 až 15 let, věk 16 až 55 let a věk
56 let a více; do věkové skupiny ≥ 56 let spadalo minimálně 40 % účastníků. Ze studie byli vyřazeni imunokompromitovaní účastníci a osoby, u nichž byla dříve stanovena klinická či mikrobiologická diagnóza onemocnění COVID-19. Účastníci s preexistujícími stabilizovanými onemocněními, definovanými jako onemocnění, jež během 6 týdnů před zařazením nevyžadovala významnou změnu léčby nebo hospitalizaci z důvodu zhoršení nemoci, byli do studie zařazeni stejně jako účastníci se známou stabilizovanou infekcí virem lidské imunodeficience (HIV), virem hepatitidy C (HCV) nebo virem hepatitidy B (HBV).
Účinnost u osob ve věku 16 let a starších – po 2 dávkáchBěhem fáze 2/3 Studie 2 bylo na základě dat získaných do 14. listopadu 2020 ve stejném poměru
randomizováno zhruba 44 000 účastníků a byly jim podány 2 dávky mRNA vakcíny proti onemocnění COVID-19 nebo placeba. Analýzy účinnosti zahrnovaly účastníky, kteří dostali druhou dávku vakcíny v rozmezí 19 až 42 dnů po první dávce vakcíny. Většina (93,1 %) příjemců vakcíny dostala druhou dávku 19 dnů až 23 dnů po první dávce. Pro účely hodnocení bezpečnosti a účinnosti vakcíny proti onemocnění COVID-19 budou na základě připraveného plánu účastníci dále sledováni po dobu až
24 měsíců po 2. dávce. V klinické studii bylo od účastníků vyžadováno dodržení minimálního intervalu 14 dní před a po podání vakcíny proti chřipce, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19. V klinické studii bylo od účastníků vyžadováno dodržení minimálního intervalu 60 dní před nebo po podání přípravků z krve/plazmy nebo imunoglobulinů v průběhu studie, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19.
Do populace pro analýzu primárního cílového parametru účinnosti bylo zařazeno 36 621 účastníků ve věku 12 let a starších (18 242 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 18 379 ve skupině s placebem), u nichž se během 7 dní po podání druhé dávky neprokázala předchozí infekce SARS-CoV-2. Kromě toho bylo 134 účastníků ve věku od 16 do 17 let (66 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 68 ve skupině s placebem) a 1 616 účastníků ve věku 75 let a starších (804 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 812 ve skupině s placebem).
V čase provedení primární analýzy účinnosti byli účastníci ve skupině mRNA vakcíny proti onemocnění COVID-19 sledováni z hlediska symptomatického onemocnění COVID-19 po celkovou dobu 2 214 osoboroků, účastníci ve skupině s placebem po dobu minimálně 2 222 osoboroků.
U účastníků s rizikem závažného onemocnění COVID-19, včetně osob s 1 nebo více komorbiditami,
které riziko závažného onemocnění COVID-19 zvyšují (např. astma, index tělesné hmotnosti (BMI)
≥ 30 kg/m2, chronické plicní onemocnění, diabetes mellitus, hypertenze), nebyly zjištěny žádné
významné klinické rozdíly v celkové účinnosti vakcíny. Informace o účinnosti vakcíny jsou uvedeny v tabulce 3.
Tabulka 3: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů)První výskyt COVID-19 od 7 dnů po 2. dávce u účastníků bez důkazu předchozí infekce SARS- CoV-2 *
|
Podskupina
| Vakcína mRNA proti
onemocnění COVID-19
na=18 198
Případy
n1b
Doba sledováníc (n2d)
|
Placebo na=18 325
Případy
n1b
Doba sledováníc (n2d)
|
Účinnost vakcíny % (95% CI)e
|
Všichni účastníci
| 8
2,214 (17 411)
| 162
2,222 (17 511)
| 95,0
(90,0; 97,9)
|
16 až 64 let
| 7
1,706 (13 549)
| 143
1,710 (13 618)
| 95,1
(89,6; 98,1)
|
65 let a starší
| 1
0,508 (3 848)
| 19
0,511 (3 880)
| 94,7
(66,7; 99,9)
|
65 až 75 let
| 1
0,406 (3 074)
| 14
0,406 (3 095)
| 92,9
(53,1; 99,8)
|
75 let a starší
| 0
0,102 (774)
| 5
0,106 (785)
| 100,0
(-13,1; 100,0)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR) a
alespoň 1 příznakem odpovídajícím onemocnění COVID-19 [*Definice případu: (alespoň 1 z) horečka, nový nebo zhoršený kašel, nová nebo zhoršená dyspnoe, zimnice, nová nebo zhoršená bolest svalů, nová ztráta chuti nebo čichu, bolest v krku, průjem nebo zvracení.]
* Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (7 nebo více dnů před podáním poslední dávky) předcházející infekce SARS-CoV-2 (tj. N-vazebná protilátka [sérum] negativní při návštěvě 1 a SARS-CoV-2 nedetekován pomocí amplifikačních testů nukleových kyselin (NAAT) [výtěr nosu] při návštěvách 1 a 2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do analýzy.
a. n = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy metody upravené podle doby sledování. CI není upraven na multiplicitu.
Účinnost vakcíny mRNA proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění
COVID-19 po 7 dnech po 2. dávce ve srovnání s placebem 94,6 % (95% interval spolehlivosti 89,6 %
až 97,6 %) u účastníků ve věku 16 let a starších s nebo bez důkazů o předchozí infekci virem SARS- CoV-2.
Analýzy podskupin primárního cílového parametru účinnosti navíc ukázaly podobné odhady bodů účinnosti u pohlaví, etnických skupin a účastníků s komorbiditami spojenými s vysokým rizikem závažného onemocnění COVID-19.
Byly provedeny aktualizované analýzy účinnosti s dalšími potvrzenými případy COVID-19, které se objevily během zaslepeného placebem kontrolovaného sledování, což v populaci s účinností představuje až 6 měsíců po podání 2. dávky.
Aktualizované informace o účinnosti vakcíny jsou uvedeny v tabulce 4.
Tabulka 4: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce předchozí infekce virem SARS- CoV-2* před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů) během placebem kontrolovaného období sledování
Podskupina
|
V
akcína mRNA proti
onemocnění COVID-19
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Placebo
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Ú
č
i
n
nost vakcíny % (95% CI
e
)
|
Všichni účastnícif
|
77
6,247 (20 712)
|
850
6,003 (20 713)
|
91,3
(89,0; 93,2)
|
16 až 64 let
|
70
4,859 (15 519)
|
710
4,654 (15 515)
|
90,6
(87,9; 92,7)
|
65 let a starší
|
7
1,233 (4 192)
|
124
1,202 (4 226)
|
94,5
(88,3; 97,8)
|
65 až 74 let
|
6
0,994 (3 350)
|
98
0,966 (3 379)
|
94,1
(86,6; 97,9)
|
75 let a starší
|
1
0,239 (842)
|
26
0,237 (847)
|
96,2
(76,9; 99,9)
|
|
|
N =20 998
N =21 096
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR) a
alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení).
* Účastníci, kteří neměli žádné důkazy předcházející infekce virem SARS-CoV-2 (tj. N-vazebná protilátka [sérum]
negativní při návštěvě 1 a virus SARS-CoV-2 nedetekován pomocí NAAT [výtěr nosu] při návštěvách 1 a 2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do analýzy.
a. N = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný 95% interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování. CI není upraven na multiplicitu.
f. Zahrnuté potvrzené případy u účastníků ve věku 12 až 15 let: 0 ve skupině s mRNA vakcínou proti onemocnění
COVID-19; 16 ve skupině s placebem.
V aktualizované analýze účinnosti byla účinnost mRNA vakcíny proti onemocnění COVID-19 v
prevenci prvního výskytu onemocnění COVID-19 od 7 dnů po podání 2. dávky ve srovnání s
placebem 91,1 % (95% CI 88,8 % až 93,0 %) u účastníků v hodnotitelné populaci účinnosti s průkazem nebo bez průkazu předchozí infekce SARS-CoV-2.
Aktualizované analýzy účinnosti podle podskupin navíc ukázaly podobné bodové odhady účinnosti u všech pohlaví, etnických skupin, zeměpisných oblastí a účastníků se zdravotními komorbiditami a obezitou spojenou s vysokým rizikem závažného onemocnění COVID-19.
Účinnost proti závažnému onemocnění COVID-19Aktualizované analýzy účinnosti sekundárních cílových parametrů účinnosti podpořily přínos mRNA
vakcíny proti onemocnění COVID-19 v prevenci závažného onemocnění COVID-19.
Od 13. března 2021 je účinnost vakcíny proti závažnému onemocnění COVID-19 prezentována pouze pro účastníky s předchozí infekcí SARS-CoV-2 nebo bez ní (tabulka 5), protože počty případů onemocnění COVID-19 u účastníků bez předchozí infekce SARS-CoV-2 byly stejné jako u účastníků s předchozí infekcí SARS-CoV-2 nebo bez ní jak ve skupině s mRNA vakcínou proti onemocnění COVID-19, tak ve skupině s placebem.
Tabulka 5: Účinnost vakcíny – první závažný výskyt onemocnění COVID-19 u účastníků s nebo bez předchozí infekce virem SARS-CoV-2 na základě údajů Úřadu pro kontrolu potravin a léčiv (FDA)* po podání 1. dávky nebo od 7 dnů po podání 2. dávky v placebem kontrolovaném období sledování
| Vakcína mRNA proti
onemocnění COVID-19
Případy
n1a
Doba sledováníc (n2b)
|
Placebo Případy n1a
Doba sledováníc (n2b)
|
Účinnost vakcíny % (95% CIc)
|
Po 1. dávced
| 1
8,439e (22 505)
| 30
8,288e (22 435)
| 96,7
(80,3; 99,9)
|
7 dnů po 2. dávcef
| 1
6,522g (21 649)
| 21
6,404g (21 730)
| 95,3
(70,9; 99,9)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR) a
alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení.)
* Závažné onemocnění COVID-19 podle definice FDA je potvrzené onemocnění COVID-19 a přítomnost alespoň 1
z následujících příznaků:
• Klinické příznaky v klidu svědčící pro závažné systémové onemocnění (dechová frekvence ≥ 30 dechů za minutu, srdeční frekvence ≥ 125 tepů za minutu, saturace kyslíkem ≤ 93 % na pokojovém vzduchu při hladině moře nebo poměr arteriálního parciálního tlaku kyslíku k frakčnímu inspirovanému kyslíku
< 300 mmHg).
• Respirační selhání [definované jako potřeba vysokého průtoku kyslíku, neinvazivní ventilace, mechanické
ventilace nebo extrakorporální membránové oxygenace (ECMO)].
• Průkaz šoku (systolický krevní tlak < 90 mmHg, diastolický krevní tlak < 60 mmHg nebo potřeba podání
vazopresorických látek).
• Významná akutní renální, jaterní nebo neurologická dysfunkce.
• Přijetí na jednotku intenzivní péče.
• Smrt.
a. n1 = Počet účastníků splňujících definici cílového parametru.
b. n2 = počet účastníků ohrožených daným cílovým parametrem.
c. Dvoustranný interval spolehlivosti (CI) pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování.
d. Účinnost hodnocená na základě veškeré populace pro hodnocení účinnosti s 1. dávkou (modifikovaná se záměrem léčit), která zahrnovala všechny randomizované účastníky, kteří dostali alespoň 1 dávku hodnocené léčby.
e. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od po 1. dávce do konce období sledování.
f. Účinnost hodnocená na základě populace hodnotitelné pro účinnost (7 dní), která zahrnovala všechny způsobilé
randomizované účastníky, kteří dostali všechny dávky hodnocené léčby podle randomizace v rámci předem stanoveného časového období a neměli žádné další důležité odchylky od protokolu podle rozhodnutí lékaře.
g. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro daný cílový parametr. Časové období pro získání případu onemocnění COVID-19 je od
7 dnů po 2. dávce do konce období sledování.
Ú
činnost a imunogenicita u dospívajících ve věku 12 až 15 let – po 2 dávkách
V úvodní analýze Studie 2 u dospívajících ve věku 12 až 15 let (jež reprezentovala medián doby následného sledování v délce ˃ 2 měsíce po 2. dávce) bez průkazu prodělané infekce nebyly žádné
případy u 1 005 účastníků, kteří dostali vakcínu, a 16 případů z 978 účastníků, kteří dostali placebo.
Bodový odhad účinnosti je 100 % (95% interval spolehlivosti 75,3; 100,0). U účastníků s nebo bez průkazu prodělané infekce bylo 0 případů z 1 119 účastníků, kteří dostali vakcínu, a 18 případů
z 1 110 účastníků, kteří dostali placebo. To také naznačuje, že bodový odhad účinnosti je 100 % (95%
interval spolehlivosti 78,1; 100,0).
U dodatečných potvrzených případů onemocnění COVID-19, k nimž došlo během zaslepeného, placebem kontrolovaného následného sledování, byly provedeny aktualizované analýzy účinnosti, jež reprezentovaly dobu až 6 měsíců po 2. dávce v populaci pro hodnocení účinnosti.
V aktualizované analýze účinnosti ve studii 2, provedené u dospívajících ve věku 12 až 15 let bez průkazu prodělané infekce, se u 1 057 účastníků, kterým byla podána vakcína, nevyskytl žádný případ a u 1 030 účastníků, kterým bylo podáno placebo, došlo ke 28 případům. Odhad bodu účinnosti je
100 % (95% interval spolehlivosti 86,8; 100,0). U účastníků s průkazem prodělané infekce nebo bez tohoto průkazu nedošlo u 1 119 těch, kterým byla podána vakcína, k žádnému případu
a u 1 109 účastníků, jimž bylo podáno placebo, ke 30 případům. To rovněž naznačuje, že odhad bodu
účinnosti je 100 % (95% interval spolehlivosti 87,5; 100,0).
Ve Studii 2 byla provedena analýza neutralizačních titrů SARS-CoV-2 jeden měsíc po druhé dávce u náhodně vybrané podskupiny účastníků, kteří do 1 měsíce po dávce 2 neměli sérologické nebo virologické průkazy prodělané infekce SARS-CoV-2, která porovnávala odpověď u dospívajících ve věku 12 až 15 let (n = 190) s účastníky ve věku 16 až 25 let (n = 170).
Poměr geometrických průměrných titrů (GMT) ve skupině ve věku 12 až 15 let ke skupině ve věku 16 až 25 let byl 1,76; s oboustranným 95% CI 1,47 až 2,10. Proto bylo splněno 1,5násobné kritérium noninferiority, protože dolní mez 2stranného 95% CI pro poměr geometrického průměru [GMR] byla
> 0,67.
Imunogenicita u účastníků ve věku 18 let a starších – po posilovací dávce
Účinnost posilovací dávky vakcíny Comirnaty byla ve studii 2 založena na hodnocení 50%
neutralizačních titrů protilátek (NT50) proti SARS-CoV-2 (USA_WA1/2020). V této studii byla posilovací dávka podána 5 až 8 měsíců (medián 7 měsíců) po druhé dávce. Ve studii 2 prokázaly
analýzy NT50 1 měsíc po posilovací dávce v porovnání s 1 měsícem po primární sérii u jedinců ve
věku 18 až 55 let, kteří neměli žádné sérologické nebo virologické důkazy o prodělané infekci virem SARS-CoV-2 do 1 měsíce po posilovací vakcinaci, noninferioritu jak pro geometrický průměrný poměr (GMR), tak pro rozdíl v četnostech sérologické odpovědi. Sérologická odpověď u účastníka byla definována jako dosažení ≥ 4násobného zvýšení NT50 oproti výchozímu stavu (před primární sérií). Tyto analýzy jsou shrnuty v tabulce 6.
|
n
|
1 měsíc po posilovací dávce (95% CI)
|
1 měsíc po primární sérii (95% CI)
|
1 měsíc po
posilovací dávce / 1 měsíc po primární sérii
(
97,5% CI)
|
Splnila cíl noninferiority (A/N)
|
G
eometrický
průměr 50%
neutralizačního
ti
t
ru
|
212a
|
2466,0b
(2202,6; 2760,8)
|
750,6b
(656,2; 858,6)
|
3,29c
(2,77; 3,90)
|
Ad
|
Č
etnost sérologické odpovědi (%) pro
50% neutralizační
ti
t
r
†
|
200e
|
199f
99,5 %
(97,2 %; 100,0 %)
|
196f
98,0 %
(95,0 %; 99,5 %)
|
1,5 %g
(-0,7 %; 3,7 %h)
|
Ai
|
|
|
T
abulka 6: Neutralizační test SARS-CoV-2 – NT50 (titr)
†
(
SARS-CoV-2 USA_WA1/2020) – srovnání GMT a četnosti sérologické odpovědi 1 měsíc po posilovací dávce a 1 měsíc po primární sérii – účastníci ve věku 18 až 55 let bez známek infekce do 1 měsíce po posilovací dávce* – populace s hodnotitelnou imunogenicitou po posilovací dávce
±
Zkratky: CI = interval spolehlivosti; GMR = poměr geometrického průměru; GMT = titr geometrického
průměru; LLOQ = dolní limit kvantifikace; N-vazba = SARS-CoV-2 nukleoproteinová vazba; NAAT = test amplifikace nukleové kyseliny; NT50 = 50% neutralizační titr; SARS-CoV-2 = závažný akutní respirační syndrom vyvolaný koronavirem 2; A/N = ano/ne.
† NT50 SARS-CoV-2 byly stanoveny pomocí mikroneutralizačního testu viru SARS-CoV-2 mNeonGreen.
Test používá fluorescenční reportérový virus odvozený od kmene USA_WA1/2020 a neutralizace viru se odečítá na monovrstvách buněk Vero. Vzorek NT50 je definován jako vzájemné ředění séra, při kterém je neutralizováno 50 % viru.
* Do analýzy byli zahrnuti účastníci, kteří neměli žádný sérologický nebo virologický průkaz (do 1 měsíce po podání posilovací dávky vakcíny Comirnaty) o prodělané infekci virem SARS-CoV-2 (tj. N-vázané protilátky [sérum] negativní a virus SARS-CoV-2 nebyl detekován pomocí NAAT [nosní výtěr]) a měli negativní NAAT (nosní výtěr) při jakékoli neplánované návštěvě do 1 měsíce po posilovací dávce.
± Všichni způsobilí účastníci, kteří dostali 2 dávky vakcíny Comirnaty podle původní randomizace, přičemž
2. dávku dostali v rámci předem definovaného časového období (během 19 až 42 dnů po 1. dávce), dostali posilovací dávku vakcíny Comirnaty, měli alespoň 1 platný a určitelný výsledek imunogenicity po posilovací dávce z odběru krve v rámci příslušného časového období (během 28 až 42 dnů po posilovací dávce) a neměli žádné další důležité odchylky od protokolu podle rozhodnutí lékaře.
a. n = Počet účastníků s platnými a určitelnými výsledky testu v obou časových bodech odběru vzorků v
rámci stanoveného časového období.
b. GMT a 2stranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a příslušných CI (na základě Studentova t rozdělení). Výsledky testů pod LLOQ byly stanoveny na
0,5násobku LLOQ.
c. GMR a 2stranné 97,5% CI byly vypočteny exponováním průměrných rozdílů logaritmů testu a odpovídajících CI (na základě Studentova t rozdělení).
d. Noninferiorita je deklarována, pokud je dolní hranice dvoustranného 97,5% CI pro GMR > 0,67 a bodový
odhad GMR činí ≥ 0,80.
e. n = počet účastníků s platnými a určitelnými výsledky testu pro daný test při výchozím stavu, 1 měsíc po
2. dávce a 1 měsíc po posilovací dávce v rámci stanoveného časového období. Tyto hodnoty jsou denominátory pro výpočty procent.
f. Počet účastníků se sérologickou odpovědí na daný test v daném časovém bodě odběru dávky / vzorku.
Přesný dvoustranný CI na základě Clopperovy a Pearsonovy metody.
g. Rozdíl v podílech vyjádřený v procentech (1 měsíc po posilovací dávce – 1 měsíc po 2. dávce).
h. Upravený dvoustranný Waldův CI pro rozdíl v podílech, vyjádřený v procentech.
i. Noninferiorita je deklarována, pokud je dolní hranice dvoustranného 97,5% CI pro procentuální rozdíl
> 10 %.
Relativní účinnost vakcíny u účastníků ve věku 16 let a starších – po posilovací dávcePrůběžná analýza účinnosti ve studii 4, placebem kontrolované studii posilovací dávky provedené u přibližně 10 000 účastníků ve věku 16 let a starších, kteří byli zařazeni ze studie 2, hodnotila
potvrzené případy onemocnění COVID-19 zjištěné nejméně 7 dní po vakcinaci posilovací dávkou až
do dne ukončení sběru dat 5. října 2021, což představuje medián 2,5 měsíce následného sledování po
posilovací dávce. Posilovací dávka byla podána 5 až 13 měsíců (medián 11 měsíců) po druhé dávce.
Hodnocena byla účinnost vakcinace posilovací dávkou vakcíny Comirnaty po základním očkování ve srovnání se skupinou, jíž bylo v rámci posilovací dávky podáno placebo a jež dostala pouze základní očkování.
Informace o relativní účinnosti vakcíny u účastníků ve věku 16 let a starších bez důkazu předchozí infekce SARS-CoV-2 uvádí tabulka 7. Relativní účinnost vakcíny u účastníků s důkazem předchozí infekce SARS-CoV-2 či bez takového důkazu byla 94,6 % (95% interval spolehlivosti 88,5 % až
97,9 %), podobně jako tomu bylo u účastníků bez důkazu předchozí infekce. Případy primárního onemocnění COVID-19, pozorované od 7 dní po podání posilovací dávky vakcíny, zahrnovaly
7 primárních případů ve skupině s vakcínou Comirnaty a 124 primárních případů ve skupině
s placebem.
Tabulka 7: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po posilovací dávce vakcíny – účastníci ve věku 16 let a starší bez důkazu infekce – populace hodnotitelná pro účinnostPrvní výskyt onemocnění COVID-19 od 7 dnů po posilovací dávce u účastníků bez důkazu
předchozí infekce SARS-CoV-2*
|
| Comirnaty na=4 695
Případy
n1b
Doba sledováníc (n2d)
| Placebo na=4 671
Případy
n1b
Doba sledováníc (n2d)
|
Relativní účinnost vakcínye % (95% CIf)
|
První výskyt
onemocnění COVID-19
od 7 dnů po posilovací
dávce vakcíny
|
6
0,823 (4 659)
|
123
0,792 (4 614)
|
95,3 (89,5; 98,3)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení).
* Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (7 nebo více dnů před podáním posilovací
dávky vakcíny) předcházející infekce SARS-CoV-2 (tj. N-vazebná protilátka [sérum] negativní při návštěvě 1 a SARS-CoV-2 nedetekován pomocí testů NAAT [výtěr nosu] při návštěvě 1) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po posilovací dávce, byli zařazeni do analýzy.
a. n = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po posilovací dávce vakcíny do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Relativní účinnost vakcíny ve skupině s posilovací dávkou vakcíny Comirnaty ve srovnání se skupinou s placebem (bez posilovací dávky).
f. Dvoustranný interval spolehlivosti pro relativní účinnost vakcíny je odvozen na základě Clopperovy
a Pearsonovy metody upravené vzhledem k době sledování.
Imunogenicita posilovací dávky po primární vakcinaci jinou schválenou vakcínou proti onemocněníCOVID-19Účinnost posilovací dávky vakcíny Comirnaty (30 mikrogramů) u osob, které absolvovaly primární vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19 (heterologní posilovací dávka), je odvozena z údajů o imunogenicitě z nezávislé otevřené klinické studie fáze 1/2 National Institutes of Health (NIH) (NCT04889209) provedené ve Spojených státech. V této studii dostali dospělí (věkové rozmezí 19 až 80 let), kteří absolvovali primární vakcinaci vakcínou Moderna 100 mikrogramů v sérii
2 dávek (n=51, průměrný věk 54 ± 17 let), jednorázovou dávkou vakcíny Janssen (n=53, průměrný věk 48±14 let) nebo vakcínou Comirnaty 30 mikrogramů v sérii 2 dávek (n=50, průměrný věk
50±18 let) nejméně 12 týdnů před zařazením do studie a kteří neuvedli žádnou anamnézu infekce
virem SARS-CoV-2, posilovací dávku vakcíny Comirnaty (30 mikrogramů). Posilovací dávka vakcíny
Comirnaty vyvolala 36, 12 a 20násobné zvýšení GMR neutralizačních titrů po primárních dávkách
vakcín Janssen, Moderna a Comirnaty, v uvedeném pořadí.
Heterologní posilovací dávka vakcíny Comirnaty byla rovněž hodnocena ve studii CoV-BOOST (EudraCT 2021 002175-19), multicentrické, randomizované, kontrolované studii fáze 2 u třetí posilovací dávky vakcíny proti onemocnění COVID-19, ve které bylo 107 dospělých účastníků (medián věku 71 let, interkvartilové rozmezí 54 až 77 let) randomizováno nejméně 70 dní po podání
2 dávek vakcíny AstraZeneca proti onemocnění COVID-19. Po primární sérii vakcíny AstraZeneca
proti onemocnění COVID-19 se násobná změna GMR NT50 neutralizačních protilátek proti pseudoviru (divoký typ) zvýšila 21,6násobně při heterologní posilovací dávce vakcíny Comirnaty (n=95).
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s vakcínou
Comirnaty u pediatrické populace k prevenci onemocnění COVID-19 (informace o použití u pediatrické populace viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Neuplatňuje se.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních studií toxicity po opakovaném podávání a reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
Obecná toxicita
U potkanů, kterým byla intramuskulárně podávána vakcína Comirnaty (dostali 3 plné dávky pro
člověka jednou týdně, které vedly k relativně vyšším hladinám u potkanů v důsledku rozdílů v tělesné hmotnosti), se vyskytl edém a erytém v místě podání injekce a zvýšení počtu bílých krvinek (včetně bazofilů a eozinofilů) odpovídající zánětlivé odpovědi a rovněž vakuolizace portálních hepatocytů bez známek poškození jater. Všechny účinky byly reverzibilní.
Genotoxicita / karcinogenita
Studie genotoxicity ani kancerogenity nebyly provedeny. U složek vakcíny (lipidy a mRNA) se
neočekává genotoxický potenciál.
Reprodukční toxicita
Reprodukční a vývojová toxicita byla hodnocena na potkanech v kombinované studii fertility a
vývojové toxicity, ve které byla samicím potkanů podána intramuskulárně vakcína Comirnaty před pářením a během březosti (dostaly 4 plné dávky pro člověka, které vedly k relativně vyšším hladinám
u potkanů v důsledku rozdílů v tělesné hmotnosti mezi 21. dnem před pářením a 20. dnem březosti). Odpovědi na neutralizační protilátky SARS-CoV-2 byly přítomny u samic před pářením až do konce
studie ve 21. postnatálním dni a rovněž u plodů a potomků. Nebyly zjištěny žádné účinky spojené s očkováním na plodnost samic, těhotenství nebo vývoj embrya/plodu nebo potomstva. Údaje o možném placentárním přenosu nebo vylučování do mateřského mléka pro vakcínu Comirnaty nejsou
k dispozici.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159) Kolfosceryl-stearát
Cholesterol
Trometamol
Trometamol-hydrochlorid
Sacharosa
Voda pro injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Neotevřená injekční lahvička
Zmrazenáinjekčnílahvička
18 měsíců při teplotě -90 °C až -60 °C.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána buď při teplotě -90 °C až -60 °C nebo 2 °C až 8 °C.
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 6 hodin nebo mohou být jednotlivé
injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Rozmrazená injekčnílahvička
10 týdnů uchovávání a transportu při teplotě 2 °C až 8 °C během 18měsíční doby použitelnosti.
• Po přenesení vakcíny pro uchovávání při teplotě 2 °C až 8 °C musí být na vnější obal zapsáno aktualizované datum použitelnosti a vakcína má být použita nebo zlikvidována do aktualizovaného data použitelnosti. Původní datum použitelnosti má být přeškrtnuto.
• Pokud je vakcína přijata při teplotě 2 °C až 8 °C, má být uchovávána při teplotě 2 °C až 8 °C.
Datum použitelnosti na vnějším obalu mělo být již dříve aktualizováno tak, aby odpovídalo datu
použitelnosti v chladu, a původní datum použitelnosti mělo být již dříve přeškrtnuto.
Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Po rozmrazení vakcína nesmí být znovu zmrazena.
Manipulace při teplotních výkyvechpřiuchovávánívchladničce
• Údaje o stabilitě prokazují, že je neotevřená injekční lahvička stabilní až 10 týdnů, je-li
uchovávána při teplotě od -2 °C do 2 °C během 10týdenní doby uchovávání při teplotě 2 °C a
8 °C.
• Údaje o stabilitě prokazují, že injekční lahvička může být uchovávána až 24 hodin při teplotě
od 8 °C do 30 °C, včetně až 12 hodin po prvním vpichu.
Tyto informace slouží jako návod pro zdravotnické pracovníky pouze v případě dočasného výkyvu
teploty.
Otevřená injekční lahvička
Chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 12 hodin při teplotě
2 °C až 30 °C, která zahrnuje až 6 hodin transportu. Z mikrobiologického hlediska, pokud způsob otevření nevyloučí rizika mikrobiologické kontaminace, má být přípravek použit okamžitě. Pokud
není použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v
odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Během uchovávání je třeba minimalizovat vystavení přípravku světlu v místnosti a zabránit vystavení
přímému slunečnímu světlu a ultrafialovému světlu.
Podmínky uchovávání tohoto léčivého přípravku po prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
2,25 ml disperze v čiré vícedávkové injekční lahvičce (sklo třídy I) o objemu 2 ml se zátkou (syntetická brombutylová pryž) a šedým odtrhovacím plastovým víčkem s hliníkovým krytem. Jedna injekční lahvička obsahuje 6 dávek, viz bod 6.6.
Velikost balení: 10 nebo 195 injekčních lahviček
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Instrukce pro zacházení s vakcínou
Vakcína Comirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku má být připravována
zdravotnickým pracovníkem pomocí aseptické techniky, aby byla zajištěna sterilita připravené
disperze.
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.1 (15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
Š
e
d
é víčko
 C
omirnaty Original/ Omicron BA.1
N
eřeďte
C
omirnaty Original/ Omicron BA.1
N
eřeďte
• Zkontrolujte, zda má injekční lahvička šedé plastové víčko a šedý okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.1
(15/15 mikrogramů)/dávku injekční
disperze.
• Pokud má injekční lahvička šedé plastové víčko a šedý okraj a název přípravku je Comirnaty
30 mikrogramů/dávku injekční disperze
nebo Comirnaty Original/Omicron
BA.4-5 (15/15 mikrogramů)/dávku injekční disperze, přečtěte si, prosím, souhrn údajů o přípravku pro příslušné složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi nebo Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte
si souhrn údajů o přípravku pro vakcínu
Comirnaty 3 mikrogramy/dávku
koncentrát pro injekční disperzi.

Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.1
(
15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ) PŘED
P
O
U
Ž
ITÍM
|
U
chovávejte po dobu až
10 týdnů při teplotě 2 °C až
8 °C,
aktualizujte datum použitelnosti na krabičce.
|
• Pokud se vícedávková injekční lahvička uchovává zmrazená, musí
se před použitím rozmrazit. Zmrazené
injekční lahvičky je třeba přenést do prostředí o teplotě 2 °C až 8 °C, aby se rozmrazily; rozmrazení balení
10 injekčních lahviček může trvat
6 hodin. Ujistěte se, že jsou injekční lahvičky před použitím úplně
rozmrazené.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené
injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
Jemně
× 10
|
• Injekční lahvičky před použitím 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Před smícháním může rozmrazená disperze obsahovat bílé až téměř bílé, matné amorfní částice.
• Po smíchání se má vakcína jevit jako bílá až téměř bílá disperze bez viditelných částic. Jestliže jsou
v naředěné vakcíně patrné částice nebo vakcína změnila barvu, nepoužívejte ji.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,3 ml VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.1 (15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
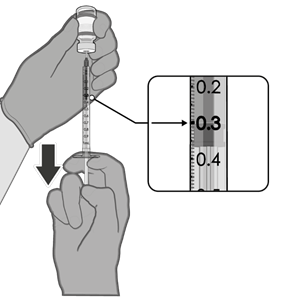 V
akcína o objemu 0,3 ml
V
akcína o objemu 0,3 ml
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím antiseptického tampónu.
• Natáhněte 0,3 ml vakcíny Comirnaty
Original/Omicron BA.1.
K získání šesti dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání šesté dávky z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Zaznamenejte příslušné datum/čas na injekční lahvičku. Veškerou vakcínu, jež nebyla použita do 12 hodin po prvním vpichu, zlikvidujte.
L
i
kv
i
dace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.de8. REGISTRAČNÍ ČÍSLO/REGISTRAČNÍ ČÍSLAEU/1/20/1528/006
EU/1/20/1528/007
9. DATUM REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. prosince 2020
Datum posledního prodloužení registrace: 10. října 2022
10. DATUM REVIZE TEXTUPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky
http://www.ema.europa.eu/
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky.
Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKUComirnaty Original/Omicron BA.4-5 (15/15 mikrogramů)/dávku injekční disperze mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍJedná se o jednodávkovou nebo vícedávkovou injekční lahvičku s šedým víčkem. Před použitím neřeďte.
Jedna jednodávková injekční lahvička obsahuje 1 dávku 0,3 ml, viz body 4.2 a 6.6.
Jedna vícedávková injekční lahvička (2,25 ml) obsahuje 6 dávek po 0,3 ml, viz body 4.2 a 6.6. Jedna dávka (0,3 ml) obsahuje 15 mikrogramů tozinameranu a 15 mikrogramů famtozinameranu,
mRNA vakcíny proti onemocnění COVID-19 (zapouzdřené v lipidových nanočásticích).
Tozinameran je jednovláknová mediátorová (messenger) RNA (mRNA) s čepičkou na 5´ konci vyráběná
in vitro nebuněčnou transkripcí z příslušných matricí DNA a kódující spike (S) protein viru SARS-CoV-2 (Original). Famtozinameran je jednovláknová mediátorová (messenger) RNA (mRNA)
s čepičkou na 5´ konci vyráběná
in vitro nebuněčnou transkripcí z příslušných matricí DNA a kódující spike (S) protein viru SARS-CoV-2 (Omikron BA.4-5).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMAInjekční disperze.
Vakcína je bílá až téměř bílá zmrazená disperze (pH: 6,9–7,9).
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikaceVakcína Comirnaty Original/Omicron BA.4-5 (15/15 mikrogramů)/dávku injekční disperze je indikována pro aktivní imunizaci k prevenci onemocnění COVID-19 způsobeného SARS-CoV-2 u osob ve věku 12 let a starších, kteří již absolvovali alespoň základní očkování proti onemocnění COVID-19 (viz body 4.2 a 5.1).
Tuto vakcínu je třeba používat v souladu s oficiálními doporučeními.
4.2 Dávkování a způsob podáníDávkováníDávka vakcíny Comirnaty Original/Omicron BA.4-5 je 0,3 ml podávaná intramuskulárně.
Mezi podáním vakcíny Comirnaty Original/Omicron BA.4-5 a poslední předchozí dávkou vakcíny proti onemocnění COVID-19 má uplynout interval nejméně 3 měsíce.
Vakcína Comirnaty Original/Omicron BA.4-5 je indikována pouze u jedinců, kteří již dříve
podstoupili alespoň základní očkování proti onemocnění COVID-19.
Podrobnosti ohledně základního očkování u osob ve věku 12 let a starších naleznete v souhrnu údajů o přípravku pro vakcínu Comirnaty 30 mikrogramů/dávku koncentrátu pro injekční disperzi a vakcínu Comirnaty 30 mikrogramů/dávku injekční disperze.
Pediatrická populace
K dispozici je pediatrická léková forma pro děti ve věku od 5 do 11 let (tj. od 5 do méně než 12 let).
Podrobné informace naleznete v souhrnu údajů o přípravku pro vakcínu Comirnaty Original/Omicron
BA.4-5 (5/5 mikrogramů)/dávku koncentrát pro injekční disperzi.
Bezpečnost a účinnost vakcíny Comirnaty Original/Omicron BA.4-5 u dětí ve věku do 12 let nebyly dosud stanoveny. Nejsou dostupné žádné údaje.
Starší populace
U osob ve věku ≥ 65 let není nutná žádná úprava dávkování.
Způsob podání
Vakcína Comirnaty Original/Omicron BA.4-5 (15/15 mikrogramů)/dávku injekční disperze se podává
intramuskulárně (viz bod 6.6). Před použitím neřeďte.
Preferované místo je deltový sval horní části paže.
Vakcína se nesmí podávat intravaskulárně, subkutánně ani intradermálně.
Vakcína se nesmí mísit ve stejné injekční stříkačce s jinými vakcínami nebo léčivými přípravky. Pro opatření před podáním vakcíny viz bod 4.4.
Návod pro rozmrazení, zacházení s vakcínou a její likvidaci je uveden v bodě 6.6.
Jednodávkové injekční lahvičky
Jednodávkové injekční lahvičky vakcíny Comirnaty obsahují 1 dávku vakcíny 0,3 ml.
• Natáhněte jednu 0,3ml dávku vakcíny Comirnaty.
• Injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Nekombinujte obsah z více injekčních lahviček vakcíny.
Vícedávkové injekčnílahvičky
Vícedávkové injekční lahvičky vakcíny Comirnaty Original/Omicron BA.4-5 obsahují 6 dávek po
0,3 ml vakcíny. K získání 6 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo
jehly s malým mrtvým prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů. Pokud se používají standardní injekční stříkačky a jehly, nemusí být dostatečný objem k získání šesté dávky z jedné injekční lahvičky. Bez ohledu na typ injekční stříkačky a jehly:
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml,
injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Nekombinujte obsah z více injekčních lahviček vakcíny.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Sledovatelnost
Aby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Obecná doporučení
Hypersenzitivita a anafylaxe
Byly hlášeny případy anafylaxe. Pro případ, že by po podání vakcíny došlo k anafylaktické reakci, má
být zajištěna okamžitá lékařská péče a dohled.
Po vakcinaci se doporučuje pečlivé sledování po dobu minimálně 15 minut. Další dávka vakcíny nemá být podána osobám, které měly anafylaxi po předchozí dávce vakcíny Comirnaty.
Myokarditida a perikarditida
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy a perikarditidy. Tato
onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhé dávce vakcíny a častěji u mladších mužů a chlapců. Dostupné údaje naznačují, že průběh myokarditidy a perikarditidy po vakcinaci se neliší od myokarditidy nebo perikarditidy obecně (viz bod 4.8).
Zdravotničtí pracovníci mají pozorně sledovat známky a příznaky myokarditidy a perikarditidy. Očkovaní jedinci (včetně rodičů nebo pečovatelů) mají být poučeni, aby okamžitě vyhledali lékařskou pomoc, pokud se u nich po očkování objeví příznaky naznačující myokarditidu nebo perikarditidu, například bolest na hrudi (akutní a přetrvávající), dušnost nebo palpitace.
Zdravotničtí pracovníci mají k diagnostice a léčbě tohoto onemocnění používat návody a postupy
a/nebo se mají obrátit na specialisty.
Reakce spojené s úzkostí
V souvislosti se samotným procesem očkování se mohou objevit reakce spojené s úzkostí, včetně
vazovagálních reakcí (synkopa), hyperventilace nebo reakcí spojených se stresem (např. závrať, palpitace, zvýšení srdeční frekvence, změny krevního tlaku, parestezie, hypestezie a pocení). Reakce
spojené se stresem jsou dočasné a samy se upraví. Očkované osoby je třeba informovat o tom, aby na
případné symptomy upozornily očkujícího zdravotníka, který je vyhodnotí. Je důležité, aby byla
zavedena opatření, aby se zabránilo zranění v důsledku mdlob.
Současné onemocnění
U osob trpících závažným akutním horečnatým onemocněním nebo akutní infekcí se má podání
vakcíny Comirnaty odložit. Přítomnost mírné infekce a/nebo horečky nízkého stupně není důvod
k odložení vakcinace.
Trombocytopenie a poruchy koagulace
Stejně jako u jiných intramuskulárních injekcí je třeba vakcínu podávat opatrně osobám podstupujícím léčbu antikoagulancii nebo osobám s trombocytopenií nebo poruchami koagulace (jako je hemofilie), protože po intramuskulárním podání může u těchto osob dojít ke krvácení nebo tvorbě modřin.
Imunokompromitované osoby
Účinnost a bezpečnost vakcíny nebyly hodnoceny u imunokompromitovaných osob, včetně osob
podstupujících imunosupresivní léčbu. Účinnost vakcíny Comirnaty Original/Omicron BA.4-5 může
být u imunokompromitovaných osob nižší.
Doba ochrany
Doba ochrany poskytovaná vakcínou není známa, protože je stále hodnocena v probíhajících
klinických studiích.
Omezení účinnosti vakcíny
Podobně jako u jiných vakcín je možné, že vakcinace vakcínou Comirnaty Original/Omicron BA.4-5
nebude chránit všechny její příjemce.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Současné podání vakcíny Comirnaty Original/Omicron BA.4-5 s jinými vakcínami nebylo hodnoceno.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Údaje o použití vakcíny Comirnaty Original/Omicron BA.4-5 během těhotenství nejsou k dispozici.
Nicméně velké množství observačních dat od těhotných žen očkovaných původně schválenou vakcínou Comirnaty během druhého a třetího trimestru neprokázalo zvýšení nežádoucích výsledků těhotenství. Ačkoli údaje o výsledcích těhotenství po očkování během prvního trimestru jsou v současné době omezené, nebylo pozorováno zvýšené riziko potratu. Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na těhotenství, vývoj embrya/plodu, porod nebo postnatální vývoj (viz bod 5.3). Na základě dostupných údajů o jiných variantách vakcíny lze vakcínu Comirnaty Original/Omicron BA.4-5 v těhotenství podávat.
Kojení
Údaje o použití vakcíny Comirnaty Original/Omicron BA.4-5 během kojení nejsou k dispozici.
Systémová expozice vakcíně je u kojící matky nicméně zanedbatelná, a proto nejsou očekávány žádné účinky na kojeného novorozence/kojence (skrze mateřské mléko). Observační údaje od žen, které po očkování původně schválenou vakcínou Comirnaty kojily, neprokázaly riziko nežádoucích účinků u kojených novorozenců/kojenců. Vakcínu Comirnaty Original/Omicron BA.4-5 lze během kojení podávat.
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky s ohledem na reprodukční toxicitu
(viz bod 5.3)
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vakcína Comirnaty Original/Omicron BA.4-5 nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Některé z účinků uvedených v bodě 4.8 však mohou schopnost řídit nebo obsluhovat stroje dočasně ovlivnit.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnost posilovací dávky vakcíny Comirnaty Original/Omicron BA.4-5 se odvozuje od
bezpečnostních údajů posilovací dávky vakcíny adaptované na variantu Omikron BA.1 u jedinců ve věku 18 let a starších a také od posilovací dávky původně schválené vakcíny Comirnaty u jedinců ve
věku 5 let a starších.
Vakcína Comirnaty 30 mikrogramů
Účastníci ve věku 16 let a starší – po 2 dávkách
Ve studii 2 byla celkem 22 026 účastníkům ve věku 16 let nebo starším podána alespoň 1 dávka vakcíny Comirnaty a celkem 22 021 účastníkům ve věku 16 let nebo starším bylo podáno placebo (včetně 138 a 145 dospívajících ve věku 16 a 17 let ve skupinách vakcíny a placeba, v uvedeném pořadí). Celkem 20 519 účastníků ve věku 16 let nebo starších dostalo 2 dávky vakcíny Comirnaty.
V době provedení analýzy studie 2 s datem ukončení sběru dat 13. března 2021 pro placebem kontrolované zaslepené období sledování až do dat odslepení účastníků bylo celkem 25 651 (58,2 %) účastníků (13 031 Comirnaty a 12 620 placebo) ve věku 16 let a starších sledováno po dobu ≥4 měsíce po podání druhé dávky. To zahrnovalo celkem 15 111 (7 704 Comirnaty a 7 407 placebo) účastníků ve věku 16 až 55 let a celkem 10 540 (5 327 Comirnaty a 5 213 placebo) účastníků ve věku 56 let a starších.
Nejčastějšími nežádoucími účinky byla u účastníků ve věku 16 let a starších, kteří dostali 2 dávky, bolest v místě injekce (> 80 %), únava (> 60 %), bolest hlavy (> 50 %), myalgie (> 40 %), zimnice
(> 30 %), artralgie (> 20 %), pyrexie a zduření v místě injekce (> 10 %). Tyto nežádoucí účinky byly zpravidla mírné nebo střední intenzity a odezněly během několika dní po vakcinaci. Mírně nižší frekvence příhod reaktogenity souvisela s vyšším věkem.
Bezpečnostní profil u 545 účastníků ve věku 16 let a starších, kterým byla podána vakcína Comirnaty
a kteří byli séropozitivní na SARS-CoV-2 při výchozím stavu, byl podobný jako u obecné populace.
Dospívající ve věku 12 až 15 let – po 2 dávkách
V analýze dlouhodobého následného sledování bezpečnosti ve studii 2 bylo 2 260 dospívajících (1 131 dostalo vakcínu Comirnaty a 1 129 dostalo placebo) ve věku 12 až 15 let. Z toho 1 559 dospívajících (786 dostalo vakcínu Comirnaty a 773 dostalo placebo) bylo sledováno po dobu ≥ 4 měsíců po druhé dávce vakcíny Comirnaty. Hodnocení bezpečnosti ve studii 2 probíhá.
Celkový bezpečnostní profil vakcíny Comirnaty u dospívajících ve věku 12 až 15 let byl podobný jako
u účastníků ve věku 16 let a starších. Nejčastějšími nežádoucími účinky u dospívajících ve věku 12 až
15 let, kteří dostali 2 dávky, byly bolest v místě vpichu (> 90 %), únava a bolest hlavy (> 70 %), myalgie a zimnice (> 40 %), artralgie a pyrexie (> 20 %).
Účastníci ve věku 16 let a starší – po posilovací dávce
Podskupina účastníků studie fáze 2/3, do které bylo zařazeno 306 dospělých ve věku 18 až 55 let, kteří dokončili původní cyklus 2 dávek vakcíny Comirnaty, dostala posilovací dávku vakcíny Comirnaty
přibližně 6 měsíců (rozmezí 4,8 až 8,0 měsíců) po podání 2. dávky.
Celkový bezpečnostní profil posilovací dávky byl podobný jako po 2 dávkách. Nejčastějšími nežádoucími účinky u účastníků ve věku 18 až 55 let byly bolest v místě vpichu (> 80 %), únava (>
60 %), bolest hlavy (> 40 %), myalgie (> 30 %), zimnice a artralgie (> 20 %).
Ve studii 4, placebem kontrolované studii posilovací dávky, byla účastníkům ve věku 16 let a starším
zařazeným ze studie 2 podána posilovací dávka vakcíny Comirnaty (5 081 účastníků) nebo placebo (5 044 účastníků), a to nejméně 6 měsíců po druhé dávce vakcíny Comirnaty. Medián doby následného sledování účastníků, kterým byla posilovací dávka podána, činil celkem 2,5 měsíce po podání posilovací dávky do dne ukončení sběru dat (5. října 2021). Žádné nové nežádoucí účinky vakcíny Comirnaty nebyly zjištěny.
Posilovací dávka poprimárnívakcinacijinouschválenouvakcínouprotionemocněníCOVID-19
V 5 nezávislých studiích o použití posilovací dávky vakcíny Comirnaty u osob, které absolvovaly
primární vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19 (heterologní posilovací
dávka), nebyly zjištěny žádné nové bezpečnostní problémy (viz bod 5.1).
Vakcína Comirnaty adaptovaná na variantu Omikron
Účastníci ve věku > 55 let – po posilovací dávce vakcíny Comirnaty Original/Omicron BA.1 (čtvrtá
dávka)
V podmnožině ze studie 4 (3. fáze) obdrželo 305 dospělých ve věku > 55 let, kteří dokončili očkování třemi dávkami vakcíny Comirnaty, posilovací dávku (čtvrtou dávku) vakcíny Comirnaty
Original/Omicron BA.1 (15/15 mikrogramů) 4,7 až 11,5 měsíců po podání 3. dávky. Účastníci, kteří
obdrželi posilovací dávku (čtvrtou dávku) vakcíny Comirnaty Original/Omicron BA.1 měli medián sledování nejméně 1,7 měsíce.
Celkový bezpečnostní profil posilovací dávky (čtvrté dávky) vakcíny Comirnaty Original/Omicron BA.1 byl podobný profilu zjištěnému po posilovací dávce (třetí dávce) vakcíny Comirnaty. U účastníků ve věku vyšším než 55 let byly nejčastějšími nežádoucími účinky bolest v místě vpichu (> 50 %), únava (> 40 %), bolest hlavy (> 30 %), myalgie (> 20 %), zimnice a artralgie (>10 %). U vakcíny Comirnaty Original/Omicron BA.1 nebyly zjištěny žádné nové nežádoucí účinky.
Účastníci ve věku 18 až ≤ 55 let – po posilovací dávce monovalentní vakcíny Omicron BA.1 (čtvrtá
dávka)
Bezpečnost posilovací dávky vakcíny Comirnaty Original/Omicron BA.1 u jednotlivců ve věku
18 až ≤ 55 let je extrapolována z bezpečnostních údajů z podmnožiny 315 dospělých ve věku
18 až ≤ 55 let, kteří obdrželi 30 mikrogramů posilovací dávky (čtvrté dávky) vakcíny Omicron BA.1 (monovalentní) po absolvování 3 dávek vakcíny Comirnaty. Nejčastějšími nežádoucími účinky u těchto účastníků ve věku 18 až ≤ 55 let byla bolest v místě vpichu (> 70 %), únava (> 60 %), bolest hlavy (> 40 %), myalgie (> 30 %), zimnice (> 30 %) a artralgie (>20 %).
Tabulkovýseznamnežádoucíchúčinkůzklinických studií vakcíny Comirnaty a Comirnaty
Original/Omicron BA.1apouvedenívakcínyComirnatynatrhuosobvevěku12let a starších
Nežádoucí účinky pozorované z klinických studií jsou uvedeny níže podle následujících kategorií
frekvence:
Velmi časté (≥ 1/10)
Časté (≥ 1/100 až < 1/10)
Méně časté (≥ 1/1 000 až < 1/100) Vzácné (≥ 1/10 000 až < 1/1 000) Velmi vzácné (< 1/10 000)
Není známo (z dostupných údajů nelze určit)
T
abulka 1: Nežádoucí účinky vakcíny Comirnaty a Comirnaty Original/Omicron BA.1
T
řída
orgánového systému
|
V
elmi
časté
(
≥ 1/10)
|
Č
asté
(
≥ 1/100
až
< 1/10)
|
Méně časté
(
≥ 1/1 000 až
< 1/100)
|
V
z
ácné
(
≥ 1/10 000
až
< 1/1 000)
|
V
elmi vzácné
(
< 1/10 000
|
N
ení známo (z
dostupnýc
h údajů nelze určit)
|
Poruchy krve a
lymfatického systému
|
|
|
Lymfadenopatiea
|
|
|
|
Poruchy
imunitního systému
|
|
|
Hypersenzitivní
reakce (např. vyrážka, pruritus, urtikarieb, angioedémb)
|
|
|
Anafylaxe
|
Poruchy
metabolismu a výživy
|
|
|
Snížená chuť k
jídlu
|
|
|
|
Psychiatrické
poruchy
|
|
|
Insomnie
|
|
|
|
Poruchy
nervového systému
|
Bolest
hlavy
|
|
Letargie
|
Akutní
periferní paralýza n. facialisc
|
|
Parestezied;
hypestezied
|
Srdeční poruchy
|
|
|
|
|
Myokarditidad;
Perikarditidad
|
|
Gastrointestinální
poruchy
|
Průjemd
|
Nauzea;
zvraceníd
|
|
|
|
|
Poruchy kůže a
podkožní tkáně
|
|
|
Hyperhidróza;
noční pocení
|
|
|
Erythema
multiformed
|
Poruchy svalové
a kosterní soustavy a pojivové tkáně
|
Artralgie;
myalgie
|
|
Bolest
v končetiněe
|
|
|
|
Poruchy
reprodukčního
systému a prsu
|
|
|
|
|
|
Silné
menstruační
krváceníh
|
Celkové poruchy
a reakce v místě
aplikace
|
Bolest v
místě injekce; únava; zimnice; pyrexief; zduření v místě injekce
|
Zarudnutí
v místě
injekce
|
Astenie;
malátnost; svědění v místě injekce
|
|
|
Rozsáhlý
otok končetiny, do níž byla vakcína podánad; otok obličejeg
|
|
|
z klinických studií a po uvedení vakcíny Comirnaty na trh u osob ve věku 12 let a starších
a. U účastníků, kteří dostali posilovací dávku ve studii 4, byla pozorována vyšší frekvence lymfadenopatie
(2,8 % oproti 0,4 %) ve srovnání s účastníky, kteří dostali dvě dávky.
b. Kategorie frekvence pro urtikarii a angioedém byla vzácná.
c. Během období sledování bezpečnosti v klinické studii do 14. listopadu 2020 byla hlášena akutní periferní paralýza
n. facialis u čtyř účastníků ve skupině mRNA vakcíny proti onemocnění COVID-19. Nástup obrny
obličeje byl 37. den po podání 1. dávky (účastník nedostal 2. dávku) a 3., 9. a 48. den po 2. dávce. Ve
skupině s placebem nebyly hlášeny žádné případy akutní periferní paralýzy
n. facialis. d. Nežádoucí účinek byl stanoven po registraci.
e. Týká se končetiny/paže, do které byla daná osoba očkována.
f. Vyšší frekvence pyrexie byla pozorována po druhé dávce v porovnání s první dávkou.
g. Po uvedení přípravku na trh byly hlášeny případy otoku obličeje u očkovaných osob, které v minulosti
podstoupily injekční aplikaci dermálních výplní do obličeje.
h. Většina případů se zdála být nezávažné a dočasné povahy.
Popis vybraných nežádoucíchúčinkůMyokarditida a perikarditidaZvýšené riziko myokarditidy po očkování vakcínou Comirnaty je nejvyšší u mladších mužů a chlapců
(viz bod 4.4).
Zvýšené riziko u mladších mužů a chlapců po podání druhé dávky vakcíny Comirnaty bylo blíže určeno ve dvou velkých evropských farmakoepidemiologických studiích. Z jedné studie vyplynulo, že v období 7 dnů po podání druhé dávky se u mužů a chlapců ve věku 12–29 let vyskytlo přibližně o
0,265 (95% CI 0,255–0,275) případů myokarditidy na 10 000 osob více než u neočkovaných osob. V
další studii se v období 28 dnů po podání druhé dávky u mužů a chlapců ve věku 16–24 let vyskytlo o
0,56 (95% CI 0,37–0,74) případů myokarditidy na 10 000 osob více než u neočkovaných osob.
Omezené údaje ukazují, že riziko myokarditidy a perikarditidy je po očkování vakcínou Comirnaty
u dětí ve věku od 5 do 11 let pravděpodobně nižší než u dospívajících ve věku od 12 do 17 let.
Hlášení podezření na nežádoucí účinkyHlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to
pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V a uvedli přitom číslo šarže, je-li k dispozici.
4.9 PředávkováníÚdaje o předávkování jsou k dispozici od 52 účastníků studie zařazených do klinického hodnocení, kterým bylo kvůli chybě v ředění podáno 58 mikrogramů vakcíny Comirnaty. Příjemci vakcíny nehlásili zvýšení reaktogenity ani nežádoucí účinky.
V případě předávkování se doporučuje sledovat základní životní funkce a případně zahájit symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: vakcíny, jiné virové vakcíny, ATC kód: J07BX03
Mechanismus účinkuMediátorová (messenger) RNA s modifikovanými nukleosidy je ve vakcíně Comirnaty zapouzdřená
v lipidových nanočásticích, což umožňuje transfer nereplikující RNA do hostitelských buněk pro přímou přechodnou expresi S antigenu viru SARS-CoV-2. mRNA kóduje v membráně ukotvený S v
plné délce se dvěma bodovými mutacemi v centrální šroubovici. Mutace těchto dvou aminokyselin na prolin uzamyká S v antigenně preferované prefuzní konformaci. Vakcína vyvolává jak odpověď
neutralizačních protilátek, tak i imunitní odpověď buněk na spike (S) antigen, což může přispívat
k ochraně před onemocněním COVID-19.
Ú
činnost
V
akcína Comirnaty adaptovaná na variantu Omikron
Účinnost posilovací dávky vakcíny Comirnaty Original/Omicron BA.4-5 se odvozuje od
imunogenicity vakcíny adaptované na variantu Omikron BA.1 u jedinců ve věku 55 let a starších.
Vakcína Comirnaty Original/Omicron BA.1Relativní imunogenicita vakcíny u účastníků ve věku > 55 let – po posilovací dávce vakcíny ComirnatyOriginal/Omicron BA.1 (čtvrtá dávka)V dílčí analýze množiny ze studie 4 (podstudie E) obdrželo 610 dospělých starších 55 let, kteří dokončili řadu 3 dávek vakcíny Comirnaty, následující dávku jako posilovací dávku (čtvrtou dávku):
vakcína Comirnaty (30 mikrogramů) nebo vakcína Comirnaty Original/Omicron BA.1
(15/15 mikrogramů). GMR a míry odpovědi v séru byly hodnoceny 1 měsíc po posilovací vakcinaci
vakcínou Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) až do data ukončení
16. května 2022, což představuje medián nejméně 1,7 měsíce sledování po posilovací dávce. Posilovací dávka vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) byla podána
v intervalu 4,7 až 11,5 měsíce (medián 6,3 měsíce) po třetí dávce.
Primárním cílem analýzy bylo vyhodnotit superioritu vzhledem k úrovni neutralizačního titru a non- inferioritu ohledně míry odpovědi v séru imunitní odpovědi proti viru Omikron indukované dávkou vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) v porovnání s odpovědí vyvolanou dávkou vakcíny Comirnaty (30 mikrogramů) podanou jako čtvrtá dávka u účastníků starších 55 let, kteří již byli vakcínou Comirnaty očkováni.
Superiorita vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) vůči vakcíně Comirnaty
(30 mikrogramů) byla splněna, protože dolní hranice 2stranného 95% CI pro GMR byla > 1 (tabulka 2).
Sérová odpověď je definována jako dosažení ≥ 4násobný nárůst vůči výchozímu stavu (před studijním očkování). Pokud je výchozí měření nižší než LLOQ, měření po vakcinaci ≥ 4 × LLOQ se považuje za sérovou odpověď.
Rozdíl v procentech účastníků, kteří dosáhli sérové odpovědi na variantu Omikron, mezi skupinou Comirnaty Original/Omicron BA.1 (71,6 %) a skupinou Comirnaty (57 %) byl 14,6 % (2stranný 95% CI: 4,0 %, 24,9 %). Tak byla splněna podmínka non-inferiority.
Tabulka 2: Podstudie E – poměry geometrických průměrů porovnání mezi skupinami vakcín – účastníci bez prokázané infekce až do 1 měsíce po 4. dávce – rozšířená kohorta – podmnožina imunogenicity – účastníci starší 55 let – populace s vyhodnotitelnou imunogenicitou
Analýza
| Skupina s vakcínou (podle
randomizace)
| Časový bod
odběru vzorkůa
|
nb
|
GMT (95% CIc)
|
GMR (95% CId)
|
Analýza neutralizace SARS-CoV-2 – Omikron BA.1 - NT50 (titr)
| Comirnaty
(30 mikrogramů)
|
1 měsíc
|
163
| 455,8
(365,9; 567,6)
|
|
Comirnaty Original/Omicron
BA.1 (15/15 mikrogramů)
|
1 měsíc
|
178
|
711,0 (588,3; 859,2)
| 1,56
(1,17;
2,08)
|
Analýza neutralizace SARS-CoV-2 – referenční kmen
– NT50 (titr)
| Comirnaty (30 mikrogramů)
|
1 měsíc
|
182
| 5 998,1
(5 223,6; 6 887,4)
|
|
Comirnaty Original/Omicron
BA.1 (15/15 mikrogramů)
|
1 měsíc
|
186
|
5 933,2
(5 188,2; 6 785,2)
| 0,99
(0,82;
1,20)
|
Zkratky: CI = interval spolehlivosti; GMR = poměr geometrického průměru; GMT = titr geometrického průměru; LLOQ = dolní limit kvantifikace; N-vazba = SARS-CoV-2 nukleoproteinová vazba; NAAT = test amplifikace nukleové kyseliny; NT50 = 50% neutralizační titr; SARS-CoV-2 = závažný akutní respirační syndrom vyvolaný koronavirem 2.
Poznámka: Podmnožina imunogenicity = náhodný vzorek 230 účastníků v každé skupině vakcíny vybraných
z rozšířené kohorty.
Poznámka: Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (odběr vzorků krve dříve než
1 měsíc po studijní vakcinaci) předcházející infekce SARS-CoV-2 (tj. negativní výsledek N-binding protilátky
[sérum] na návštěvách při studijní vakcinaci a za 1 měsíc po vakcinaci, negativní výsledek NAAT [stěr z nosu] na návštěvě se studijní vakcinací a jakékoliv neplánované návštěvě před odběrem vzorku krve 1 měsíc po studijní vakcinaci) a kteří neměli onemocnění COVID-19 ve zdravotní anamnéze, byli zařazeni do analýzy.
a. Protokolárně stanovené načasování odběru krevních vzorků.
b. n = počet účastníků s platnými a určitelnými výsledky testu pro daný test v daném časovém bodě odběru vzorků.
c. GMT a 2stranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a příslušných CI (na základě Studentova t rozdělení). Výsledky testů pod LLOQ byly stanoveny na
0,5násobku LLOQ.
d. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (skupina vakcíny v příslušném řádku Comirnaty [30 mikrogramů]) a odpovídajícího CI (na základě Studentova t rozložení).
Vakcína Comirnaty 30 mikrogramůStudie 2 je multicentrická, mezinárodní, randomizovaná, placebem kontrolovaná studie fáze 1/2/3
zaslepená pro pozorovatele, která hodnotí dávku, výběr vakcíny a její účinnost u účastníků ve věku
12 let a starších. Randomizace byla stratifikována podle věku: věk 12 až 15 let, věk 16 až 55 let a věk
56 let a více; do věkové skupiny ≥ 56 let spadalo minimálně 40 % účastníků. Ze studie byli vyřazeni imunokompromitovaní účastníci a osoby, u nichž byla dříve stanovena klinická či mikrobiologická diagnóza onemocnění COVID-19. Účastníci s preexistujícími stabilizovanými onemocněními, definovanými jako onemocnění, jež během 6 týdnů před zařazením nevyžadovala významnou změnu léčby nebo hospitalizaci z důvodu zhoršení nemoci, byli do studie zařazeni stejně jako účastníci se známou stabilizovanou infekcí virem lidské imunodeficience (HIV), virem hepatitidy C (HCV) nebo virem hepatitidy B (HBV).
Účinnost u osob ve věku 16 let a starších – po 2 dávkáchBěhem fáze 2/3 Studie 2 bylo na základě dat získaných do 14. listopadu 2020 ve stejném poměru
randomizováno zhruba 44 000 účastníků a byly jim podány 2 dávky mRNA vakcíny proti onemocnění COVID-19 nebo placeba. Analýzy účinnosti zahrnovaly účastníky, kteří dostali druhou dávku vakcíny v rozmezí 19 až 42 dnů po první dávce vakcíny. Většina (93,1 %) příjemců vakcíny dostala druhou dávku 19 dnů až 23 dnů po první dávce. Pro účely hodnocení bezpečnosti a účinnosti vakcíny proti onemocnění COVID-19 budou na základě připraveného plánu účastníci dále sledováni po dobu až
24 měsíců po 2. dávce. V klinické studii bylo od účastníků vyžadováno dodržení minimálního intervalu 14 dní před a po podání vakcíny proti chřipce, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19. V klinické studii bylo od účastníků vyžadováno dodržení minimálního intervalu 60 dní před nebo po podání přípravků z krve/plazmy nebo imunoglobulinů v průběhu studie, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19.
Do populace pro analýzu primárního cílového parametru účinnosti bylo zařazeno 36 621 účastníků ve věku 12 let a starších (18 242 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 18 379 ve skupině s placebem), u nichž se během 7 dní po podání druhé dávky neprokázala předchozí infekce SARS-CoV-2. Kromě toho bylo 134 účastníků ve věku od 16 do 17 let (66 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 68 ve skupině s placebem) a 1 616 účastníků ve věku 75 let a starších (804 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 812 ve skupině s placebem).
V čase provedení primární analýzy účinnosti byli účastníci ve skupině mRNA vakcíny proti onemocnění COVID-19 sledováni z hlediska symptomatického onemocnění COVID-19 po celkovou dobu 2 214 osoboroků, účastníci ve skupině s placebem po dobu minimálně 2 222 osoboroků.
U účastníků s rizikem závažného onemocnění COVID-19, včetně osob s 1 nebo více komorbiditami,
které riziko závažného onemocnění COVID-19 zvyšují (např. astma, index tělesné hmotnosti (BMI)
≥ 30 kg/m2, chronické plicní onemocnění, diabetes mellitus, hypertenze), nebyly zjištěny žádné
významné klinické rozdíly v celkové účinnosti vakcíny. Informace o účinnosti vakcíny jsou uvedeny v tabulce 3.
Tabulka 3: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů)První výskyt COVID-19 od 7 dnů po 2. dávce u účastníků bez důkazu předchozí infekce SARS- CoV-2 *
|
Podskupina
| Vakcína mRNA proti
onemocnění COVID-19
na=18 198
Případy
n1b
Doba sledováníc (n2d)
|
Placebo na=18 325
Případy
n1b
Doba sledováníc (n2d)
|
Účinnost vakcíny % (95% CI)e
|
Všichni účastníci
| 8
2,214 (17 411)
| 162
2,222 (17 511)
| 95,0
(90,0; 97,9)
|
16 až 64 let
| 7
1,706 (13 549)
| 143
1,710 (13 618)
| 95,1
(89,6; 98,1)
|
65 let a starší
| 1
0,508 (3 848)
| 19
0,511 (3 880)
| 94,7
(66,7; 99,9)
|
65 až 75 let
| 1
0,406 (3 074)
| 14
0,406 (3 095)
| 92,9
(53,1; 99,8)
|
75 let a starší
| 0
0,102 (774)
| 5
0,106 (785)
| 100,0
(-13,1; 100,0)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR) a
alespoň 1 příznakem odpovídajícím onemocnění COVID-19 [*Definice případu: (alespoň 1 z) horečka, nový nebo zhoršený kašel, nová nebo zhoršená dyspnoe, zimnice, nová nebo zhoršená bolest svalů, nová ztráta chuti nebo čichu, bolest v krku, průjem nebo zvracení.]
* Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (7 nebo více dnů před podáním poslední dávky) předcházející infekce SARS-CoV-2 (tj. N-vazebná protilátka [sérum] negativní při návštěvě 1 a SARS-CoV-2 nedetekován pomocí amplifikačních testů nukleových kyselin (NAAT) [výtěr nosu] při návštěvách 1 a 2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do analýzy.
a. n = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování. CI není upraven na multiplicitu.
Účinnost vakcíny mRNA proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění
COVID-19 po 7 dnech po 2. dávce ve srovnání s placebem 94,6 % (95% interval spolehlivosti 89,6 %
až 97,6 %) u účastníků ve věku 16 let a starších s nebo bez důkazů o předchozí infekci virem SARS- CoV-2.
Analýzy podskupin primárního cílového parametru účinnosti navíc ukázaly podobné odhady bodů účinnosti u pohlaví, etnických skupin a účastníků s komorbiditami spojenými s vysokým rizikem závažného onemocnění COVID-19.
Byly provedeny aktualizované analýzy účinnosti s dalšími potvrzenými případy COVID-19, které se objevily během zaslepeného placebem kontrolovaného sledování, což v populaci s účinností představuje až 6 měsíců po podání 2. dávky.
Aktualizované informace o účinnosti vakcíny jsou uvedeny v tabulce 4.
Tabulka 4: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce předchozí infekce virem SARS- CoV-2* před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů) během placebem kontrolovaného období sledování
Podskupina
|
V
akcína mRNA proti
onemocnění COVID-19
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Placebo
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Ú
č
i
n
nost vakcíny % (95% CI
e
)
|
Všichni účastnícif
|
77
6,247 (20 712)
|
850
6,003 (20 713)
|
91,3
(89,0; 93,2)
|
16 až 64 let
|
70
4,859 (15 519)
|
710
4,654 (15 515)
|
90,6
(87,9; 92,7)
|
65 let a starší
|
7
1,233 (4 192)
|
124
1,202 (4 226)
|
94,5
(88,3; 97,8)
|
65 až 74 let
|
6
0,994 (3 350)
|
98
0,966 (3 379)
|
94,1
(86,6; 97,9)
|
75 let a starší
|
1
0,239 (842)
|
26
0,237 (847)
|
96,2
(76,9; 99,9)
|
|
|
N =20 998
N =21 096
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR) a
alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení).
* Účastníci, kteří neměli žádné důkazy předcházející infekce virem SARS-CoV-2 (tj. N-vazebná protilátka [sérum]
negativní při návštěvě 1 a virus SARS-CoV-2 nedetekován pomocí NAAT [výtěr nosu] při návštěvách 1 a 2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do analýzy.
a. N = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný 95% interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování. CI není upraven na multiplicitu.
f. Zahrnuté potvrzené případy u účastníků ve věku 12 až 15 let: 0 ve skupině s mRNA vakcínou proti onemocnění
COVID-19; 16 ve skupině s placebem.
V aktualizované analýze účinnosti byla účinnost mRNA vakcíny proti onemocnění COVID-19 v
prevenci prvního výskytu onemocnění COVID-19 od 7 dnů po podání 2. dávky ve srovnání s
placebem 91,1 % (95% CI 88,8 % až 93,0 %) u účastníků v hodnotitelné populaci účinnosti s průkazem nebo bez průkazu předchozí infekce SARS-CoV-2.
Aktualizované analýzy účinnosti podle podskupin navíc ukázaly podobné bodové odhady účinnosti u všech pohlaví, etnických skupin, zeměpisných oblastí a účastníků se zdravotními komorbiditami a obezitou spojenou s vysokým rizikem závažného onemocnění COVID-19.
Účinnost proti závažnému onemocnění COVID-19Aktualizované analýzy účinnosti sekundárních cílových parametrů účinnosti podpořily přínos mRNA
vakcíny proti onemocnění COVID-19 v prevenci závažného onemocnění COVID-19.
Od 13. března 2021 je účinnost vakcíny proti závažnému onemocnění COVID-19 prezentována pouze pro účastníky s předchozí infekcí SARS-CoV-2 nebo bez ní (tabulka 5), protože počty případů onemocnění COVID-19 u účastníků bez předchozí infekce SARS-CoV-2 byly stejné jako u účastníků s předchozí infekcí SARS-CoV-2 nebo bez ní jak ve skupině s mRNA vakcínou proti onemocnění COVID-19, tak ve skupině s placebem.
Tabulka 5: Účinnost vakcíny – první závažný výskyt onemocnění COVID-19 u účastníků s nebo bez předchozí infekce virem SARS-CoV-2 na základě údajů Úřadu pro kontrolu potravin a léčiv (FDA)* po podání 1. dávky nebo od 7 dnů po podání 2. dávky v placebem kontrolovaném období sledování
| Vakcína mRNA proti
onemocnění COVID-19
Případy
n1a
Doba sledováníc (n2b)
|
Placebo Případy n1a
Doba sledováníc (n2b)
|
Účinnost vakcíny % (95% CIc)
|
Po 1. dávced
| 1
8,439e (22 505)
| 30
8,288e (22 435)
| 96,7
(80,3; 99,9)
|
7 dnů po 2. dávcef
| 1
6,522g (21 649)
| 21
6,404g (21 730)
| 95,3
(70,9; 99,9)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR) a
alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení.)
* Závažné onemocnění COVID-19 podle definice FDA je potvrzené onemocnění COVID-19 a přítomnost alespoň 1
z následujících příznaků:
• Klinické příznaky v klidu svědčící pro závažné systémové onemocnění (dechová frekvence ≥ 30 dechů za minutu, srdeční frekvence ≥ 125 tepů za minutu, saturace kyslíkem ≤ 93 % na pokojovém vzduchu při hladině moře nebo poměr arteriálního parciálního tlaku kyslíku k frakčnímu inspirovanému kyslíku
< 300 mmHg).
• Respirační selhání [definované jako potřeba vysokého průtoku kyslíku, neinvazivní ventilace, mechanické ventilace nebo extrakorporální membránové oxygenace (ECMO)].
• Průkaz šoku (systolický krevní tlak < 90 mmHg, diastolický krevní tlak < 60 mmHg nebo potřeba podání
vazopresorických látek).
• Významná akutní renální, jaterní nebo neurologická dysfunkce.
• Přijetí na jednotku intenzivní péče.
• Smrt.
a. n1 = Počet účastníků splňujících definici cílového parametru.
b. n2 = počet účastníků ohrožených daným cílovým parametrem.
c. Dvoustranný interval spolehlivosti (CI) pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování.
d. Účinnost hodnocená na základě veškeré populace pro hodnocení účinnosti s 1. dávkou (modifikovaná se záměrem léčit), která zahrnovala všechny randomizované účastníky, kteří dostali alespoň 1 dávku hodnocené léčby.
e. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od po 1. dávce do konce období sledování.
f. Účinnost hodnocená na základě populace hodnotitelné pro účinnost (7 dní), která zahrnovala všechny způsobilé
randomizované účastníky, kteří dostali všechny dávky hodnocené léčby podle randomizace v rámci předem stanoveného časového období a neměli žádné další důležité odchylky od protokolu podle rozhodnutí lékaře.
g. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro daný cílový parametr. Časové období pro získání případu onemocnění COVID-19 je od
7 dnů po 2. dávce do konce období sledování.
Ú
činnost a imunogenicita u dospívajících ve věku 12 až 15 let – po 2 dávkách
V úvodní analýze Studie 2 u dospívajících ve věku 12 až 15 let (jež reprezentovala medián doby následného sledování v délce ˃ 2 měsíce po 2. dávce) bez průkazu prodělané infekce nebyly žádné
případy u 1 005 účastníků, kteří dostali vakcínu, a 16 případů z 978 účastníků, kteří dostali placebo.
Bodový odhad účinnosti je 100 % (95% interval spolehlivosti 75,3; 100,0). U účastníků s nebo bez průkazu prodělané infekce bylo 0 případů z 1 119 účastníků, kteří dostali vakcínu, a 18 případů
z 1 110 účastníků, kteří dostali placebo. To také naznačuje, že bodový odhad účinnosti je 100 % (95%
interval spolehlivosti 78,1; 100,0).
U dodatečných potvrzených případů onemocnění COVID-19, k nimž došlo během zaslepeného, placebem kontrolovaného následného sledování, byly provedeny aktualizované analýzy účinnosti, jež reprezentovaly dobu až 6 měsíců po 2. dávce v populaci pro hodnocení účinnosti.
V aktualizované analýze účinnosti ve studii 2, provedené u dospívajících ve věku 12 až 15 let bez průkazu prodělané infekce, se u 1 057 účastníků, kterým byla podána vakcína, nevyskytl žádný případ a u 1 030 účastníků, kterým bylo podáno placebo, došlo ke 28 případům. Odhad bodu účinnosti je
100 % (95% interval spolehlivosti 86,8; 100,0). U účastníků s průkazem prodělané infekce nebo bez tohoto průkazu nedošlo u 1 119 těch, kterým byla podána vakcína, k žádnému případu
a u 1 109 účastníků, jimž bylo podáno placebo, ke 30 případům. To rovněž naznačuje, že odhad bodu
účinnosti je 100 % (95% interval spolehlivosti 87,5; 100,0).
Ve Studii 2 byla provedena analýza neutralizačních titrů SARS-CoV-2 jeden měsíc po druhé dávce u náhodně vybrané podskupiny účastníků, kteří do 1 měsíce po dávce 2 neměli sérologické nebo virologické průkazy prodělané infekce SARS-CoV-2, která porovnávala odpověď u dospívajících ve věku 12 až 15 let (n = 190) s účastníky ve věku 16 až 25 let (n = 170).
Poměr geometrických průměrných titrů (GMT) ve skupině ve věku 12 až 15 let ke skupině ve věku 16 až 25 let byl 1,76; s oboustranným 95% CI 1,47 až 2,10. Proto bylo splněno 1,5násobné kritérium noninferiority, protože dolní mez 2stranného 95% CI pro poměr geometrického průměru [GMR] byla
> 0,67.
Imunogenicita u účastníků ve věku 18 let a starších – po posilovací dávce
Účinnost posilovací dávky vakcíny Comirnaty byla ve studii 2 založena na hodnocení 50%
neutralizačních titrů protilátek (NT50) proti SARS-CoV-2 (USA_WA1/2020). V této studii byla posilovací dávka podána 5 až 8 měsíců (medián 7 měsíců) po druhé dávce. Ve studii 2 prokázaly
analýzy NT50 1 měsíc po posilovací dávce v porovnání s 1 měsícem po primární sérii u jedinců ve
věku 18 až 55 let, kteří neměli žádné sérologické nebo virologické důkazy o prodělané infekci virem SARS-CoV-2 do 1 měsíce po posilovací vakcinaci, noninferioritu jak pro geometrický průměrný poměr (GMR), tak pro rozdíl v četnostech sérologické odpovědi. Sérologická odpověď u účastníka byla definována jako dosažení ≥ 4násobného zvýšení NT50 oproti výchozímu stavu (před primární sérií). Tyto analýzy jsou shrnuty v tabulce 6.
|
n
|
1 měsíc po posilovací dávce (95% CI)
|
1 měsíc po primární sérii (95% CI)
|
1 měsíc po
posilovací dávce / 1 měsíc po primární sérii
(
97,5% CI)
|
Splnila cíl noninferiority (A/N)
|
G
eometrický
průměr 50%
neutralizačního
ti
t
ru
|
212a
|
2466,0b
(2202,6; 2760,8)
|
750,6b
(656,2; 858,6)
|
3,29c
(2,77; 3,90)
|
Ad
|
Č
etnost sérologické odpovědi (%) pro
50% neutralizační
ti
t
r
†
|
200e
|
199f
99,5 %
(97,2 %; 100,0 %)
|
196f
98,0 %
(95,0 %; 99,5 %)
|
1,5 %g
(-0,7 %; 3,7 %h)
|
Ai
|
|
|
T
abulka 6: Neutralizační test SARS-CoV-2 – NT50 (titr)
†
(
SARS-CoV-2 USA_WA1/2020) – srovnání GMT a četnosti sérologické odpovědi 1 měsíc po posilovací dávce a 1 měsíc po primární sérii – účastníci ve věku 18 až 55 let bez známek infekce do 1 měsíce po posilovací dávce* – populace s hodnotitelnou imunogenicitou po posilovací dávce
±
Zkratky: CI = interval spolehlivosti; GMR = poměr geometrického průměru; GMT = titr geometrického
průměru; LLOQ = dolní limit kvantifikace; N-vazba = SARS-CoV-2 nukleoproteinová vazba; NAAT = test amplifikace nukleové kyseliny; NT50 = 50% neutralizační titr; SARS-CoV-2 = závažný akutní respirační syndrom vyvolaný koronavirem 2; A/N = ano/ne.
† NT50 SARS-CoV-2 byly stanoveny pomocí mikroneutralizačního testu viru SARS-CoV-2 mNeonGreen.
Test používá fluorescenční reportérový virus odvozený od kmene USA_WA1/2020 a neutralizace viru se odečítá na monovrstvách buněk Vero. Vzorek NT50 je definován jako vzájemné ředění séra, při kterém je neutralizováno 50 % viru.
* Do analýzy byli zahrnuti účastníci, kteří neměli žádný sérologický nebo virologický průkaz (do 1 měsíce po podání posilovací dávky vakcíny Comirnaty) o prodělané infekci virem SARS-CoV-2 (tj. N-vázané protilátky [sérum] negativní a virus SARS-CoV-2 nebyl detekován pomocí NAAT [nosní výtěr]) a měli negativní NAAT (nosní výtěr) při jakékoli neplánované návštěvě do 1 měsíce po posilovací dávce.
± Všichni způsobilí účastníci, kteří dostali 2 dávky vakcíny Comirnaty podle původní randomizace, přičemž
2. dávku dostali v rámci předem definovaného časového období (během 19 až 42 dnů po 1. dávce), dostali posilovací dávku vakcíny Comirnaty, měli alespoň 1 platný a určitelný výsledek imunogenicity po posilovací dávce z odběru krve v rámci příslušného časového období (během 28 až 42 dnů po posilovací dávce) a neměli žádné další důležité odchylky od protokolu podle rozhodnutí lékaře.
a. n = Počet účastníků s platnými a určitelnými výsledky testu v obou časových bodech odběru vzorků v rámci stanoveného časového období.
b. GMT a 2stranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a příslušných CI (na základě Studentova t rozdělení). Výsledky testů pod LLOQ byly stanoveny na
0,5násobku LLOQ.
c. GMR a 2stranné 97,5% CI byly vypočteny exponováním průměrných rozdílů logaritmů testu a odpovídajících CI (na základě Studentova t rozdělení).
d. Noninferiorita je deklarována, pokud je dolní hranice dvoustranného 97,5% CI pro GMR > 0,67 a bodový
odhad GMR činí ≥ 0,80.
e. n = počet účastníků s platnými a určitelnými výsledky testu pro daný test při výchozím stavu, 1 měsíc po
2. dávce a 1 měsíc po posilovací dávce v rámci stanoveného časového období. Tyto hodnoty jsou denominátory pro výpočty procent.
f. Počet účastníků se sérologickou odpovědí na daný test v daném časovém bodě odběru dávky / vzorku.
Přesný dvoustranný CI na základě Clopperovy a Pearsonovy metody.
g. Rozdíl v podílech vyjádřený v procentech (1 měsíc po posilovací dávce – 1 měsíc po 2. dávce). h. Upravený dvoustranný Waldův CI pro rozdíl v podílech, vyjádřený v procentech.
i. Noninferiorita je deklarována, pokud je dolní hranice dvoustranného 97,5% CI pro procentuální rozdíl
> 10 %.
Relativní účinnost vakcíny u účastníků ve věku 16 let a starších – po posilovací dávcePrůběžná analýza účinnosti ve studii 4, placebem kontrolované studii posilovací dávky provedené u přibližně 10 000 účastníků ve věku 16 let a starších, kteří byli zařazeni ze studie 2, hodnotila
potvrzené případy onemocnění COVID-19 zjištěné nejméně 7 dní po vakcinaci posilovací dávkou až
do dne ukončení sběru dat 5. října 2021, což představuje medián 2,5 měsíce následného sledování po
posilovací dávce. Posilovací dávka byla podána 5 až 13 měsíců (medián 11 měsíců) po druhé dávce.
Hodnocena byla účinnost vakcinace posilovací dávkou vakcíny Comirnaty po základním očkování ve srovnání se skupinou, jíž bylo v rámci posilovací dávky podáno placebo a jež dostala pouze základní očkování.
Informace o relativní účinnosti vakcíny u účastníků ve věku 16 let a starších bez důkazu předchozí infekce SARS-CoV-2 uvádí tabulka 7. Relativní účinnost vakcíny u účastníků s důkazem předchozí infekce SARS-CoV-2 či bez takového důkazu byla 94,6 % (95% interval spolehlivosti 88,5 % až
97,9 %), podobně jako tomu bylo u účastníků bez důkazu předchozí infekce. Případy primárního onemocnění COVID-19, pozorované od 7 dní po podání posilovací dávky vakcíny, zahrnovaly
7 primárních případů ve skupině s vakcínou Comirnaty a 124 primárních případů ve skupině
s placebem.
Tabulka 7: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po posilovací dávce vakcíny – účastníci ve věku 16 let a starší bez důkazu infekce – populace hodnotitelná pro účinnostPrvní výskyt onemocnění COVID-19 od 7 dnů po posilovací dávce u účastníků bez důkazu předchozí infekce SARS-CoV-2*
|
| Comirnaty na=4 695
Případy
n1b
Doba sledováníc (n2d)
| Placebo na=4 671
Případy
n1b
Doba sledováníc (n2d)
|
Relativní účinnost vakcínye % (95% CIf)
|
První výskyt
onemocnění COVID-19
od 7 dnů po posilovací
dávce vakcíny
|
6
0,823 (4 659)
|
123
0,792 (4 614)
|
95,3 (89,5; 98,3)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení).
* Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (7 nebo více dnů před podáním posilovací dávky vakcíny) předcházející infekce SARS-CoV-2 (tj. N-vazebná protilátka [sérum] negativní při
návštěvě 1 a SARS-CoV-2 nedetekován pomocí testů NAAT [výtěr nosu] při návštěvě 1) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po posilovací dávce, byli zařazeni do analýzy.
a. n = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osoborocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po posilovací dávce vakcíny do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Relativní účinnost vakcíny ve skupině s posilovací dávkou vakcíny Comirnaty ve srovnání se skupinou s placebem (bez posilovací dávky).
f. Dvoustranný interval spolehlivosti pro relativní účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy metody upravené vzhledem k době sledování.
Imunogenicita posilovací dávky po primární vakcinaci jinou schválenou vakcínou proti onemocněníCOVID-19Účinnost posilovací dávky vakcíny Comirnaty (30 mikrogramů) u osob, které absolvovaly primární vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19 (heterologní posilovací dávka), je odvozena z údajů o imunogenicitě z nezávislé otevřené klinické studie fáze 1/2 National Institutes of Health (NIH) (NCT04889209) provedené ve Spojených státech. V této studii dostali dospělí (věkové rozmezí 19 až 80 let), kteří absolvovali primární vakcinaci vakcínou Moderna 100 mikrogramů v sérii
2 dávek (n=51, průměrný věk 54 ± 17 let), jednorázovou dávkou vakcíny Janssen (n=53, průměrný věk 48±14 let) nebo vakcínou Comirnaty 30 mikrogramů v sérii 2 dávek (n=50, průměrný věk
50±18 let) nejméně 12 týdnů před zařazením do studie a kteří neuvedli žádnou anamnézu infekce
virem SARS-CoV-2, posilovací dávku vakcíny Comirnaty (30 mikrogramů). Posilovací dávka vakcíny
Comirnaty vyvolala 36, 12 a 20násobné zvýšení GMR neutralizačních titrů po primárních dávkách
vakcín Janssen, Moderna a Comirnaty, v uvedeném pořadí.
Heterologní posilovací dávka vakcíny Comirnaty byla rovněž hodnocena ve studii CoV-BOOST (EudraCT 2021 002175-19), multicentrické, randomizované, kontrolované studii fáze 2 u třetí posilovací dávky vakcíny proti onemocnění COVID-19, ve které bylo 107 dospělých účastníků (medián věku 71 let, interkvartilové rozmezí 54 až 77 let) randomizováno nejméně 70 dní po podání
2 dávek vakcíny AstraZeneca proti onemocnění COVID-19. Po primární sérii vakcíny AstraZeneca
proti onemocnění COVID-19 se násobná změna GMR NT50 neutralizačních protilátek proti pseudoviru (divoký typ) zvýšila 21,6násobně při heterologní posilovací dávce vakcíny Comirnaty (n=95).
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s vakcínou
Comirnaty u pediatrické populace k prevenci onemocnění COVID-19 (informace o použití u pediatrické populace viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Neuplatňuje se.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě konvenčních studií toxicity po opakovaném podávání a reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
Obecná toxicita
U potkanů, kterým byla intramuskulárně podávána vakcína Comirnaty (dostali 3 plné dávky pro
člověka jednou týdně, které vedly k relativně vyšším hladinám u potkanů v důsledku rozdílů v tělesné hmotnosti), se vyskytl edém a erytém v místě podání injekce a zvýšení počtu bílých krvinek (včetně bazofilů a eozinofilů) odpovídající zánětlivé odpovědi a rovněž vakuolizace portálních hepatocytů bez známek poškození jater. Všechny účinky byly reverzibilní.
Genotoxicita / karcinogenita
Studie genotoxicity ani kancerogenity nebyly provedeny. U složek vakcíny (lipidy a mRNA) se
neočekává genotoxický potenciál.
Reprodukční toxicita
Reprodukční a vývojová toxicita byla hodnocena na potkanech v kombinované studii fertility a
vývojové toxicity, ve které byla samicím potkanů podána intramuskulárně vakcína Comirnaty před pářením a během březosti (dostaly 4 plné dávky pro člověka, které vedly k relativně vyšším hladinám
u potkanů v důsledku rozdílů v tělesné hmotnosti mezi 21. dnem před pářením a 20. dnem březosti). Odpovědi na neutralizační protilátky SARS-CoV-2 byly přítomny u samic před pářením až do konce
studie ve 21. postnatálním dni a rovněž u plodů a potomků. Nebyly zjištěny žádné účinky spojené s očkováním na plodnost samic, těhotenství nebo vývoj embrya/plodu nebo potomstva. Údaje o možném placentárním přenosu nebo vylučování do mateřského mléka pro vakcínu Comirnaty nejsou
k dispozici.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159) Kolfosceryl-stearát
Cholesterol
Trometamol
Trometamol-hydrochlorid
Sacharosa
Voda pro injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky.
6.3 Doba použitelnosti
Neotevřená injekční lahvička
Zmrazenáinjekčnílahvička
18 měsíců při teplotě -90 °C až -60 °C.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána buď při teplotě -90 °C až -60 °C nebo 2 °C až 8 °C.
Jednodávkové injekční lahvičky
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
10 jednodávkových injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 2 hodin nebo
mohou být jednotlivé injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Vícedávkové injekční lahvičky
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
10 vícedávkových injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 6 hodin nebo mohou být jednotlivé injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Rozmrazená injekčnílahvička
10 týdnů uchovávání a transportu při teplotě 2 °C až 8 °C během 18měsíční doby použitelnosti.
• Po přenesení vakcíny pro uchovávání při teplotě 2 °C až 8 °C musí být na vnější obal zapsáno aktualizované datum použitelnosti a vakcína má být použita nebo zlikvidována do aktualizovaného data použitelnosti. Původní datum použitelnosti má být přeškrtnuto.
• Pokud je vakcína přijata při teplotě 2 °C až 8 °C, má být uchovávána při teplotě 2 °C až 8 °C.
Datum použitelnosti na vnějším obalu mělo být již dříve aktualizováno tak, aby odpovídalo datu použitelnosti v chladu, a původní datum použitelnosti mělo být již dříve přeškrtnuto.
Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
P
o rozmrazení vakcína nesmí být znovu zmrazena.
M
anipulace
p
ř
i
t
eplotních
výkyvechpřiuchovávání v chladničce
• Údaje o stabilitě prokazují, že je neotevřená injekční lahvička stabilní až 10 týdnů, je-li
uchovávána při teplotě od -2 °C do 2 °C během 10týdenní doby uchovávání při teplotě 2 °C a
8 °C.
• Údaje o stabilitě prokazují, že injekční lahvička může být uchovávána až 24 hodin při teplotě
od 8 °C do 30 °C, včetně až 12 hodin po prvním vpichu.
Tyto informace slouží jako návod pro zdravotnické pracovníky pouze v případě dočasného výkyvu
teploty.
Otevřená injekční lahvička
Chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 12 hodin při teplotě
2 °C až 30 °C, která zahrnuje až 6 hodin transportu. Z mikrobiologického hlediska, pokud způsob otevření nevyloučí rizika mikrobiologické kontaminace, má být přípravek použit okamžitě. Pokud
není použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v
odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Během uchovávání je třeba minimalizovat vystavení přípravku světlu v místnosti a zabránit vystavení
přímému slunečnímu světlu a ultrafialovému světlu.
Podmínky uchovávání tohoto léčivého přípravku po prvním otevření jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Vakcína Comirnaty disperze se dodává v čiré injekční lahvičce (sklo třídy I) o objemu 2 ml se zátkou
(syntetická brombutylová pryž) a šedým odtrhovacím plastovým víčkem s hliníkovým krytem.
Jedna jednodávková injekční lahvička obsahuje 1 dávku 0,3 ml, viz body 4.2 a 6.6.
Jedna vícedávková injekční lahvička (2,25 ml) obsahuje 6 dávek po 0,3 ml, viz body 4.2 a 6.6.
Velikost balení jednodávkových injekčních lahviček: 10 injekčních lahviček.
Velikost balení vícedávkových injekčních lahviček: 10 nebo 195 injekčních lahviček. Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Instrukce pro zacházení s vakcínou
Vakcína Comirnaty Original/Omicron BA.4-5 (15/15 mikrogramů)/dávku má být připravována
zdravotnickým pracovníkem pomocí aseptické techniky, aby byla zajištěna sterilita připravené
disperze.
NÁV
O
D SE VZTAHUJE NA JEDNODÁVKOVÉ I VÍCEDÁVKOVÉ INJEKČNÍ LAHVIČKY
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.4-5 (15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
Š
e
d
é víčko
 C
omirnaty Original/ Omicron BA.4-5
N
eřeďte
C
omirnaty Original/ Omicron BA.4-5
N
eřeďte
• Zkontrolujte, zda má injekční lahvička šedé plastové víčko a šedý okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze.
• Zkontrolujte, zda se jedná
o jednodávkovou nebo vícedávkovou injekční lahvičku, a řiďte se příslušnými pokyny pro zacházení uvedenými níže.
• Pokud má injekční lahvička šedé plastové víčko a šedý okraj a název přípravku je Comirnaty
30 mikrogramů/dávku injekční disperze
nebo Comirnaty Original/Omicron
BA.1 (15/15 mikrogramů)/dávku injekční disperze, přečtěte si, prosím, souhrn údajů o přípravku pro příslušné složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi nebo Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte
si souhrn údajů o přípravku pro vakcínu
Comirnaty 3 mikrogramy/dávku
koncentrát pro injekční disperzi.
Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.4-5
(
15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ) PŘED
P
O
U
Ž
ITÍM
|
U
chovávejte po dobu až
10 týdnů při teplotě 2 °C až
8 °C,
aktualizujte datum použitelnosti na krabičce.
|
• Pokud se jednodávková nebo vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se rozmrazily. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
o Jednodávkové injekční lahvičky:
Rozmrazení balení
10 jednodávkových injekčních lahviček může trvat 2 hodiny.
o Vícedávkové injekční lahvičky:
Rozmrazení balení
10 vícedávkových injekčních lahviček může trvat 6 hodin.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
Jemně
× 10
|
• Injekční lahvičky před použitím 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Před smícháním může rozmrazená disperze obsahovat bílé až téměř bílé, matné amorfní částice.
• Po smíchání se má vakcína jevit jako bílá až téměř bílá disperze bez viditelných částic. Jestliže jsou
v naředěné vakcíně patrné částice nebo vakcína změnila barvu, nepoužívejte ji.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,3 ml VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.4-5 (15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
 V
akcína o objemu 0,3 ml
J
ednodávkové
i
n
j
ekční
l
ahvičky
V
akcína o objemu 0,3 ml
J
ednodávkové
i
n
j
ekční
l
ahvičky
• Natáhněte jednu 0,3ml dávku vakcíny.
• Injekční lahvičku a veškerý přebytečný
objem zlikvidujte.
Vícedávkovéinjekčnílahvičky• Vícedávkové injekční lahvičky obsahují
6 dávek po 0,3 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím antiseptického tampónu.
• Natáhněte 0,3 ml vakcíny Comirnaty
Original/Omicron BA.4-5.
K získání šesti dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání šesté dávky z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Zaznamenejte příslušné datum/čas na injekční lahvičku. Veškerou vakcínu, jež nebyla použita do 12 hodin po prvním vpichu, zlikvidujte.
L
i
kv
i
dace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
BioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.de8. REGISTRAČNÍ ČÍSLO/REGISTRAČNÍ ČÍSLAJednodávkové injekční lahvičkyEU/1/20/1528/014
Vícedávkové injekční lahvičkyEU/1/20/1528/008
EU/1/20/1528/009
9. DATUM REGISTRACE/PRODLOUŽENÍ REGISTRACEDatum první registrace: 21. prosince 2020
Datum posledního prodloužení registrace: 10. října 2022
10. DATUM REVIZE TEXTUPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky.
Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKUComirnaty Original/Omicron BA.4-5 (5/5 mikrogramů/dávku koncentrát pro injekční disperzi mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍJedná se o vícedávkovou injekční lahvičku s oranžovým víčkem, jejíž obsah je nutno před použitím naředit.
Jedna injekční lahvička (1,3 ml) obsahuje 10 dávek po 0,2 ml po naředění, viz body 4.2 a 6.6.
Jedna dávka (0,2 ml) obsahuje 5 mikrogramů tozinameranu a 5 mikrogramů famtozinameranu, mRNA
vakcíny proti onemocnění COVID-19 (zapouzdřené v lipidových nanočásticích).
Tozinameran je jednovláknová mediátorová (messenger) RNA (mRNA) s čepičkou na 5´ konci vyráběná
in vitro nebuněčnou transkripcí z příslušných matricí DNA a kódující spike (S) protein viru SARS-CoV-2 (Original). Famtozinameran je jednovláknová mediátorová (messenger) RNA (mRNA)
s čepičkou na 5´ konci vyráběná
in vitro nebuněčnou transkripcí z příslušných matricí DNA a kódující spike (S) protein viru SARS-CoV-2 (Omikron BA.4-5).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMAKoncentrát pro injekční disperzi (sterilní koncentrát).
Vakcína je bílá až téměř bílá zmrazená disperze (pH: 6,9–7,9).
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikaceVakcína Comirnaty Original/Omicron BA.4-5 (5/5 mikrogramů)/dávku koncentrát pro injekční disperzi je indikována pro aktivní imunizaci k prevenci onemocnění COVID-19 způsobeného SARS- CoV-2 u dětí ve věku 5 až 11 let, které podstoupily alespoň základní očkování proti onemocnění COVID-19 (viz body 4.2 a 5.1).
Tuto vakcínu je třeba používat v souladu s oficiálními doporučeními.
4.2 Dávkování a způsob podáníDávkováníDětivevěku5až11let(tj.5ažméněnež12let)Dávka vakcíny Comirnaty Original/Omicron BA.4-5 je 0,2 ml podávaná intramuskulárně.
Mezi podáním vakcíny Comirnaty Original/Omicron BA.4-5 a poslední předchozí dávkou vakcíny proti onemocnění COVID-19 má uplynout interval nejméně 3 měsíce.
Vakcína Comirnaty Original/Omicron BA.4-5 je indikována pouze u jedinců, kteří již dříve
podstoupili alespoň základní očkování proti onemocnění COVID-19.
Podrobnosti ohledně základního očkování u dětí ve věku od 5 do 11 let naleznete v souhrnu údajů o přípravku pro vakcínu Comirnaty 10 mikrogramů/dávku koncentrátu pro injekční disperzi.
Vakcína Comirnaty Original/Omicron BA.4-5 (5/5 mikrogramů)/dávku se má používat pouze u dětí ve věku od 5 do 11 let.
Pediatrická populace
Bezpečnost a účinnost vakcíny Comirnaty Original/Omicron BA.4-5 u dětí mladších než 5 let nebyly
dosud stanoveny. Nejsou dostupné žádné údaje.
Způsob podání
Vakcína Comirnaty Original/Omicron BA.4-5 (5/5 mikrogramů)/dávku koncentrát pro injekční
disperzi se podává intramuskulárně po naředění (viz bod 6.6).
Po naředění obsahují injekční lahvičky vakcíny Comirnaty Original/Omicron BA.4-5 10 dávek po
0,2 ml vakcíny. K získání 10 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo
jehly s malým mrtvým prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů. Pokud se používají standardní injekční stříkačky a
jehly, nemusí být dostatečný objem k získání 10 dávek z jedné injekční lahvičky. Bez ohledu na typ
injekční stříkačky a jehly:
• Každá dávka musí obsahovat 0,2 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,2 ml,
injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Nekombinujte obsah z více injekčních lahviček vakcíny. Preferované místo je deltový sval horní části paže.
Vakcína se nesmí podávat intravaskulárně, subkutánně ani intradermálně.
Vakcína se nesmí mísit ve stejné injekční stříkačce s jinými vakcínami nebo léčivými přípravky. Pro opatření před podáním vakcíny viz bod 4.4.
Návod pro rozmrazení, zacházení s vakcínou a její likvidaci je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Sledovatelnost
Aby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
O
becná doporučení
Hypersenzitivita a anafylaxe
Byly hlášeny případy anafylaxe. Pro případ, že by po podání vakcíny došlo k anafylaktické reakci, má
být zajištěna okamžitá lékařská péče a dohled.
Po vakcinaci se doporučuje pečlivé sledování po dobu minimálně 15 minut. Další dávka vakcíny nemá být podána osobám, které měly anafylaxi po předchozí dávce vakcíny Comirnaty.
Myokarditida a perikarditida
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy a perikarditidy. Tato
onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhé dávce vakcíny a častěji u mladších mužů a chlapců. Dostupné údaje naznačují, že průběh myokarditidy a perikarditidy po vakcinaci se neliší od myokarditidy nebo perikarditidy obecně (viz bod 4.8).
Zdravotničtí pracovníci mají pozorně sledovat známky a příznaky myokarditidy a perikarditidy. Očkovaní jedinci (včetně rodičů a pečovatelů) mají být poučeni, aby okamžitě vyhledali lékařskou pomoc, pokud se u nich po očkování objeví příznaky naznačující myokarditidu nebo perikarditidu, například bolest na hrudi (akutní a přetrvávající), dušnost nebo palpitace.
Zdravotničtí pracovníci mají k diagnostice a léčbě tohoto onemocnění používat návody a postupy
a/nebo se mají obrátit na specialisty.
Reakce spojené s úzkostí
V souvislosti se samotným procesem očkování se mohou objevit reakce spojené s úzkostí, včetně
vazovagálních reakcí (synkopa), hyperventilace nebo reakcí spojených se stresem (např. závrať, palpitace, zvýšení srdeční frekvence, změny krevního tlaku, parestezie, hypestezie a pocení). Reakce spojené se stresem jsou dočasné a samy se upraví. Očkované osoby je třeba informovat o tom, aby na případné symptomy upozornily očkujícího zdravotníka, který je vyhodnotí. Je důležité, aby byla zavedena opatření, aby se zabránilo zranění v důsledku mdlob.
Současné onemocnění
U osob trpících závažným akutním horečnatým onemocněním nebo akutní infekcí se má podání
vakcíny Comirnaty odložit. Přítomnost mírné infekce a/nebo horečky nízkého stupně není důvod
k odložení vakcinace.
Trombocytopenie a poruchy koagulace
Stejně jako u jiných intramuskulárních injekcí je třeba vakcínu podávat opatrně osobám podstupujícím
léčbu antikoagulancii nebo osobám s trombocytopenií nebo poruchami koagulace (jako je hemofilie),
protože po intramuskulárním podání může u těchto osob dojít ke krvácení nebo tvorbě modřin.
Imunokompromitované osoby
Účinnost a bezpečnost vakcíny nebyly hodnoceny u imunokompromitovaných osob, včetně osob
podstupujících imunosupresivní léčbu. Účinnost vakcíny Comirnaty Original/Omicron BA.4-5 může
být u imunokompromitovaných osob nižší.
D
oba ochrany
Doba ochrany poskytovaná vakcínou není známa, protože je stále hodnocena v probíhajících
klinických studiích.
Omezení účinnosti vakcíny
Podobně jako u jiných vakcín je možné, že vakcinace vakcínou Comirnaty Original/Omicron BA.4-5
nebude chránit všechny její příjemce.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí.
Současné podání vakcíny Comirnaty Original/Omicron BA.4-5 s jinými vakcínami nebylo hodnoceno.
4.6 Fertilita, těhotenství a kojení
Těhotenství
Údaje o použití vakcíny Comirnaty Original/Omicron BA.4-5 během těhotenství nejsou k dispozici.
Nicméně velké množství observačních dat od těhotných žen očkovaných původně schválenou vakcínou Comirnaty během druhého a třetího trimestru neprokázalo zvýšení nežádoucích výsledků těhotenství. Ačkoli údaje o výsledcích těhotenství po očkování během prvního trimestru jsou v současné době omezené, nebylo pozorováno zvýšené riziko potratu. Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky na těhotenství, vývoj embrya/plodu, porod nebo postnatální vývoj (viz bod 5.3). Na základě dostupných údajů o jiných variantách vakcíny lze vakcínu Comirnaty Original/Omicron BA.4-5 v těhotenství podávat.
Kojení
Údaje o použití vakcíny Comirnaty Original/Omicron BA.4-5 během kojení nejsou k dispozici.
Systémová expozice vakcíně je u kojící matky nicméně zanedbatelná, a proto nejsou očekávány žádné účinky na kojeného novorozence/kojence (skrze mateřské mléko). Observační údaje od žen, které po očkování původně schválenou vakcínou Comirnaty kojily, neprokázaly riziko nežádoucích účinků u kojených novorozenců/kojenců. Vakcínu Comirnaty Original/Omicron BA.4-5 lze během kojení podávat.
Fertilita
Studie na zvířatech nenaznačují přímé ani nepřímé škodlivé účinky s ohledem na reprodukční toxicitu
(viz bod 5.3)
4.7 Účinky na schopnost řídit a obsluhovat stroje
Vakcína Comirnaty Original/Omicron BA.4-5 nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje. Některé z účinků uvedených v bodě 4.8 však mohou schopnost řídit nebo obsluhovat stroje dočasně ovlivnit.
4.8 Nežádoucí účinky
Souhrn bezpečnostníhoprofilu
Bezpečnost posilovací dávky vakcíny Comirnaty Original/Omicron BA.4-5 se odvozuje od
bezpečnostních údajů posilovací dávky vakcíny adaptované na variantu Omikron BA.1 u jedinců ve
věku 18 let a starších a také od posilovací dávky původně schválené vakcíny Comirnaty u jedinců ve věku 5 let a starších.
Vakcína Comirnaty
Děti ve věku 5 až 11 let (tj. ve věku 5 do méně než 12 let) – po 2 dávkách
Ve studii 3 byla alespoň 1 dávka vakcíny Comirnaty 10 mikrogramů podána celkem 1 518 dětem ve věku 5 až 11 let a celkem 750 dětem ve věku 5 až 11 let bylo podáno placebo. V době analýzy studie 3
fáze 2/3 s údaji do data ukončení sběru dat 6. září 2021 bylo 2 158 (95,1 %) (1 444 dětí, kterým byla podána vakcína Comirnaty 10 mikrogramů a 714 dětí, kterým bylo podáno placebo) sledováno po
dobu nejméně 2 měsíců po druhé dávce vakcíny Comirnaty 10 mikrogramů. Analýza údajů o nežádoucích příhodách studie 3 fáze 2/3 zahrnovala také dalších 2 379 účastníků [1 591 účastníkům byla podána vakcína Comirnaty 10 mikrogramů a 788 účastníkům bylo podáno placebo], z nichž
71,2 % bylo sledováno po dobu nejméně 2 týdnů po 2. dávce až do data ukončení sběru dat
8. října 2021. Hodnocení bezpečnosti ve studii 3 probíhá.
Celkový bezpečnostní profil vakcíny Comirnaty u účastníků ve věku 5 až 15 let byl podobný jako u účastníků ve věku 16 let a starších. Nejčastějšími nežádoucími účinky u dětí ve věku 5 až 11 let, kterým byly podány 2 dávky, byly bolest v místě vpichu (> 80 %), únava (> 50 %), bolest hlavy (>
30 %), zarudnutí a otok v místě vpichu (> 20 %), myalgie a zimnice (> 10 %).
Děti ve věku 5 až 11 let (tj. ve věku 5 do méně než 12 let) – po posilovací dávce
V podskupině účastníků studie 3 dostalo celkem 401 dětí ve věku 5 až 11 let posilovací dávku vakcíny
Comirnaty 10 mikrogramů za nejméně 5 měsíců (rozmezí 5 až 9 měsíců) po dokončení základního očkování. Analýza podskupiny ve studii 3 fáze 2/3 je založena na údajích s datem ukončení sběru dat do 22. března 2022 (medián doby sledování 1,3 měsíce).
Celkový bezpečnostní profil posilovací dávky byl podobný jako po základním očkování. Nejčastějšími nežádoucími účinky u dětí ve věku 5 až 11 let byly bolest v místě vpichu (> 70 %), únava (> 40 %), bolest hlavy (> 30 %), myalgie, zimnice a zarudnutí a otok v místě vpichu (> 10 %).
Dospívající ve věku 12 až 15 let – po 2 dávkách
V analýze dlouhodobého následného sledování bezpečnosti ve studii 2 bylo 2 260 dospívajících
(1 131 dostalo vakcínu Comirnaty a 1 129 dostalo placebo) ve věku 12 až 15 let. Z toho
1 559 dospívajících (786 dostalo vakcínu Comirnaty a 773 dostalo placebo) bylo sledováno po dobu
≥ 4 měsíců po druhé dávce vakcíny Comirnaty. Hodnocení bezpečnosti ve studii 2 probíhá.
Celkový bezpečnostní profil vakcíny Comirnaty u dospívajících ve věku 12 až 15 let byl podobný jako u účastníků ve věku 16 let a starších. Nejčastějšími nežádoucími účinky u dospívajících ve věku 12 až
15 let, kteří dostali 2 dávky, byly bolest v místě injekce (> 90 %), únava a bolest hlavy (> 70 %),
myalgie a zimnice (> 40 %), artralgie a pyrexie (> 20 %).
Účastníci ve věku 16 let a starší – po 2 dávkách
Ve studii 2 byla celkem 22 026 účastníkům ve věku 16 let nebo starším podána alespoň 1 dávka vakcíny Comirnaty 30 mikrogramů a celkem 22 021 účastníkům ve věku 16 let nebo starším bylo
podáno placebo (včetně 138 a 145 dospívajících ve věku 16 a 17 let ve skupinách vakcíny a placeba, v uvedeném pořadí). Celkem 20 519 účastníků ve věku 16 let nebo starších dostalo 2 dávky vakcíny
Comirnaty.
V době provedení analýzy studie 2 s datem ukončení sběru dat 13. března 2021 pro placebem kontrolované zaslepené období sledování až do dat odslepení účastníků bylo celkem 25 651 (58,2 %) účastníků (13 031 Comirnaty a 12 620 placebo) ve věku 16 let a starších sledováno po dobu ≥4 měsíce po podání druhé dávky. To zahrnovalo celkem 15 111 (7 704 Comirnaty a 7 407 placebo) účastníků ve věku 16 až 55 let a celkem 10 540 (5 327 Comirnaty a 5 213 placebo) účastníků ve věku 56 let a starších.
Nejčastějšími nežádoucími účinky byla u účastníků ve věku 16 let a starších, kteří dostali 2 dávky, bolest v místě injekce (> 80 %), únava (> 60 %), bolest hlavy (> 50 %), myalgie (> 40 %), zimnice
(> 30 %), artralgie (> 20 %), pyrexie a zduření v místě injekce (> 10 %). Tyto nežádoucí účinky byly
zpravidla mírné nebo střední intenzity a odezněly během několika dní po vakcinaci. Mírně nižší frekvence příhod reaktogenity souvisela s vyšším věkem.
Bezpečnostní profil u 545 účastníků ve věku 16 let a starších, kterým byla podána vakcína Comirnaty
a kteří byli séropozitivní na SARS-CoV-2 při výchozím stavu, byl podobný jako u obecné populace.
Účastníci ve věku 16 let a starší – po posilovací dávce
Podskupina účastníků studie fáze 2/3, do které bylo zařazeno 306 dospělých ve věku 18 až 55 let, kteří dokončili původní cyklus 2 dávek vakcíny Comirnaty, dostala posilovací dávku vakcíny Comirnaty přibližně 6 měsíců (rozmezí 4,8 až 8,0 měsíců) po podání 2. dávky.
Celkový bezpečnostní profil posilovací dávky byl podobný jako po 2 dávkách. Nejčastějšími nežádoucími účinky u účastníků ve věku 18 až 55 let byly bolest v místě vpichu (> 80 %), únava (>
60 %), bolest hlavy (> 40 %), myalgie (> 30 %), zimnice a artralgie (> 20 %).
Ve studii 4, placebem kontrolované studii posilovací dávky, byla účastníkům ve věku 16 let a starším
zařazeným ze studie 2 podána posilovací dávka vakcíny Comirnaty (5 081 účastníků) nebo placebo
(5 044 účastníků), a to nejméně 6 měsíců po druhé dávce vakcíny Comirnaty. Celkem činil medián doby následného sledování účastníků, kterým byla posilovací dávka podána, 2,5 měsíce po podání posilovací dávky do data ukončení sběru (5. října 2021). Žádné nové nežádoucí účinky vakcíny Comirnaty nebyly zjištěny.
Posilovací dávka po primární vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19
V 5 nezávislých studiích o použití posilovací dávky vakcíny Comirnaty u osob, které absolvovaly
primární vakcinaci jinou schválenou vakcínou proti onemocnění COVID-19 (heterologní posilovací
dávka), nebyly zjištěny žádné nové bezpečnostní problémy.
Vakcína Comirnaty adaptovaná na variantu Omikron
Účastníci ve věku > 55 let – po posilovací dávce vakcíny Comirnaty Original/Omicron BA.1 (čtvrtá
dávka)
V podmnožině ze studie 4 (3. fáze) obdrželo 305 dospělých ve věku > 55 let, kteří dokončili očkování třemi dávkami vakcíny Comirnaty, posilovací dávku (čtvrtou dávku) vakcíny Comirnaty
Original/Omicron BA.1 (15/15 mikrogramů) 4,7 až 11,5 měsíců po podání 3. dávky. Účastníci, kteří
obdrželi posilovací dávku (čtvrtou dávku) vakcíny Comirnaty Original/Omicron BA.1 měli medián sledování nejméně 1,7 měsíce.
Celkový bezpečnostní profil posilovací dávky (čtvrté dávky) vakcíny Comirnaty Original/Omicron
BA.1 byl podobný profilu zjištěnému po posilovací dávce (třetí dávce) vakcíny Comirnaty.
U účastníků ve věku vyšším než 55 let byly nejčastějšími nežádoucími účinky bolest v místě vpichu
(> 50 %), únava (> 40 %), bolest hlavy (> 30 %), myalgie (> 20 %), zimnice a artralgie (>10 %). U vakcíny Comirnaty Original/Omicron BA.1 nebyly zjištěny žádné nové nežádoucí účinky.
Účastníci ve věku 18 až ≤ 55 let – po posilovací dávce monovalentní vakcíny Omicron BA.1 (čtvrtá
dávka)
Bezpečnost posilovací dávky vakcíny Comirnaty Original/Omicron BA.1 u jednotlivců ve věku
18 až ≤ 55 let je extrapolována z bezpečnostních údajů z podmnožiny 315 dospělých ve věku
18 až ≤ 55 let, kteří obdrželi 30 mikrogramů posilovací dávky (čtvrté dávky) vakcíny Omicron BA.1
(monovalentní) po absolvování 3 dávek vakcíny Comirnaty. Nejčastějšími nežádoucími účinky u
těchto účastníků ve věku 18 až ≤ 55 let byla bolest v místě vpichu (> 70 %), únava (> 60 %), bolest hlavy (> 40 %), myalgie (> 30 %), zimnice (> 30 %) a artralgie (>20 %).
Tabulkovýseznamnežádoucíchúčinkůzklinických studií vakcíny Comirnaty a Comirnaty
Original/Omicron BA.1 a po uvedení vakcíny Comirnaty na trh u osob ve věku 5 let a starších
Nežádoucí účinky pozorované z klinických studií jsou uvedeny níže podle následujících kategorií
frekvence:
Velmi časté (≥ 1/10)
Časté (≥ 1/100 až < 1/10)
Méně časté (≥ 1/1 000 až < 1/100)
Vzácné (≥ 1/10 000 až < 1/1 000) Velmi vzácné (< 1/10 000)
Není známo (z dostupných údajů nelze určit)
T
abulka 1: Nežádoucí účinky vakcíny Comirnaty a Comirnaty Original/Omicron BA.1
T
řída
orgánového systému
|
V
elmi
časté
(
≥ 1/10)
|
Č
asté
(
≥ 1/100
až
< 1/10)
|
Méně časté
(
≥ 1/1 000 až
< 1/100)
|
V
z
ácné
(
≥ 1/10 000
až
< 1/1 000)
|
V
elmi vzácné
(
< 1/10 000
|
N
ení známo (z
dostupnýc
h údajů nelze určit)
|
Poruchy krve a
lymfatického systému
|
|
|
Lymfadenopatiea
|
|
|
|
Poruchy
imunitního systému
|
|
|
Hypersenzitivní
reakce (např. vyrážka, pruritus, urtikarieb, angioedémb)
|
|
|
Anafylaxe
|
Poruchy
metabolismu a výživy
|
|
|
Snížená chuť k
jídlu
|
|
|
|
Psychiatrické
poruchy
|
|
|
Insomnie
|
|
|
|
Poruchy
nervového systému
|
Bolest
hlavy
|
|
Letargie
|
Akutní
periferní paralýza n. facialisc
|
|
Parestezied,
hypestezied
|
Srdeční poruchy
|
|
|
|
|
Myokarditidad;
Perikarditidad
|
|
Gastrointestinální
poruchy
|
Průjemd
|
Nauzea;
zvraceníd
|
|
|
|
|
Poruchy kůže a
podkožní tkáně
|
|
|
Hyperhidróza;
noční pocení
|
|
|
Erythema
multiformed
|
Poruchy svalové
a kosterní soustavy a pojivové tkáně
|
Artralgie;
myalgie
|
|
Bolest
v končetiněe
|
|
|
|
Poruchy
reprodukčního
systému a prsu
|
|
|
|
|
|
Silné
menstruační
krváceníi
|
Celkové poruchy
a reakce v místě
aplikace
|
Bolest v
místě injekce; únava; zimnice; pyrexief; zduření v místě injekce
|
Zarudnutí
v místě
injekceh
|
Astenie;
malátnost; svědění v místě injekce
|
|
|
Rozsáhlý
otok končetiny, do níž byla vakcína podánad; otok obličejeg
|
|
|
z klinických studií a po uvedení vakcíny Comirnaty na trh u osob ve věku 5 let a starších
a. U účastníků, kteří dostali posilovací dávku, byla pozorována vyšší frekvence lymfadenopatie ve srovnání
s účastníky ve věku 5 až 11 let ve studii 3 (2,5 % vs. 0,9 %) a s účastníky ve věku 16 let a starších ve studii 4 (2,8 % vs. 0,4 %), kteří dostali dvě dávky.
b. Kategorie frekvence pro urtikarii a angioedém byla vzácná.
c. Během období sledování bezpečnosti v klinické studii do 14. listopadu 2020 byla hlášena akutní periferní paralýza
n. facialis u čtyř účastníků ve skupině mRNA vakcíny proti onemocnění COVID-19. Nástup obrny
obličeje byl 37. den po podání 1. dávky (účastník nedostal 2. dávku) a 3., 9. a 48. den po 2. dávce. Ve
skupině s placebem nebyly hlášeny žádné případy akutní periferní paralýzy
n. facialis. d. Nežádoucí účinek byl stanoven po registraci.
e. Týká se končetiny/paže, do které byla daná osoba očkována.
f. Vyšší frekvence pyrexie byla pozorována po druhé dávce v porovnání s první dávkou.
g. Po uvedení přípravku na trh byly hlášeny případy otoku obličeje u očkovaných osob, které v minulosti
podstoupily injekční aplikaci dermálních výplní do obličeje.
h. Zarudnutí v místě injekce se vyskytovalo s vyšší frekvencí (velmi častou) u dětí ve věku 5 až 11 let. i. Většina případů se zdála být nezávažné a dočasné povahy.
Popis vybraných nežádoucíchúčinkůMyokarditida a perikarditidaZvýšené riziko myokarditidy po očkování vakcínou Comirnaty je nejvyšší u mladších mužů a chlapců
(viz bod 4.4).
Zvýšené riziko u mladších mužů a chlapců po podání druhé dávky vakcíny Comirnaty bylo blíže určeno ve dvou velkých evropských farmakoepidemiologických studiích. Z jedné studie vyplynulo, že v období 7 dnů po podání druhé dávky se u mužů a chlapců ve věku 12–29 let vyskytlo přibližně o
0,265 (95% CI 0,255–0,275) případů myokarditidy na 10 000 osob více než u neočkovaných osob.
V další studii se v období 28 dnů po podání druhé dávky u mužů a chlapců ve věku 16–24 let vyskytlo o 0,56 (95% CI 0,37–0,74) případů myokarditidy na 10 000 osob více než u neočkovaných osob.
Omezené údaje ukazují, že riziko myokarditidy a perikarditidy je po očkování vakcínou Comirnaty
u dětí ve věku od 5 do 11 let pravděpodobně nižší než u dětí ve věku od 12 do 17 let.
Hlášení podezření na nežádoucí účinkyHlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to
pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích
účinků uvedeného v Dodatku V a uvedli přitom číslo šarže, je-li k dispozici.
4.9 PředávkováníÚdaje o předávkování jsou k dispozici od 52 účastníků studie zařazených do klinického hodnocení, kterým bylo kvůli chybě v ředění podáno 58 mikrogramů vakcíny Comirnaty. Příjemci vakcíny nehlásili zvýšení reaktogenity ani nežádoucí účinky.
V případě předávkování se doporučuje sledovat základní životní funkce a případně zahájit symptomatickou léčbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: vakcíny, jiné virové vakcíny, ATC kód: J07BX03
Mechanismus účinkuMediátorová (messenger) RNA s modifikovanými nukleosidy je ve vakcíně Comirnaty zapouzdřená
v lipidových nanočásticích, což umožňuje transfer nereplikující RNA do hostitelských buněk pro přímou přechodnou expresi S antigenu viru SARS-CoV-2. mRNA kóduje v membráně ukotvený
S v plné délce se dvěma bodovými mutacemi v centrální šroubovici. Mutace těchto dvou aminokyselin
na prolin uzamyká S v antigenně preferované prefuzní konformaci. Vakcína vyvolává jak odpověď
neutralizačních protilátek, tak i imunitní odpověď buněk na spike (S) antigen, což může přispívat
k ochraně před onemocněním COVID-19.
ÚčinnostVakcína Comirnaty adaptovaná na variantu OmikronÚčinnost posilovací dávky vakcíny Comirnaty Original/Omicron BA.4-5 se odvozuje od
imunogenicity vakcíny adaptované na variantu Omikron BA.1 u jedinců ve věku 55 let a starších.
Vakcína Comirnaty Original/Omicron BA.1Relativní imunogenicita vakcíny u účastníků ve věku > 55 let – po posilovací dávce vakcíny ComirnatyOriginal/Omicron BA.1 (čtvrtá dávka)V dílčí analýze množiny ze studie 4 (podstudie E) obdrželo 610 dospělých starších 55 let, kteří dokončili řadu 3 dávek vakcíny Comirnaty, následující dávku jako posilovací dávku (čtvrtou dávku):
vakcína Comirnaty (30 mikrogramů) nebo vakcína Comirnaty Original/Omicron BA.1
(15/15 mikrogramů). GMR a míry odpovědi v séru byly hodnoceny 1 měsíc po posilovací vakcinaci
vakcínou Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) až do data ukončení
16. května 2022, což představuje medián nejméně 1,7 měsíce sledování po posilovací dávce.
Posilovací dávka vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) byla podána
v intervalu 4,7 až 11,5 měsíce (medián 6,3 měsíce) po třetí dávce.
Primárním cílem analýzy bylo vyhodnotit superioritu vzhledem k úrovni neutralizačního titru a non- inferioritu ohledně míry odpovědi v séru imunitní odpovědi proti viru Omikron indukované dávkou vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) v porovnání s odpovědí vyvolanou dávkou vakcíny Comirnaty (30 mikrogramů) podanou jako čtvrtá dávka u účastníků starších 55 let, kteří již byli vakcínou Comirnaty očkováni.
Superiorita vakcíny Comirnaty Original/Omicron BA.1 (15/15 mikrogramů) vůči vakcíně Comirnaty
(30 mikrogramů) byla splněna, protože dolní hranice 2stranného 95% CI pro GMR byla > 1 (tabulka 2).
Sérová odpověď je definována jako dosažení ≥ 4násobný nárůst vůči výchozímu stavu (před studijním očkování). Pokud je výchozí měření nižší než LLOQ, měření po vakcinaci ≥ 4 × LLOQ se považuje za sérovou odpověď.
Rozdíl v procentech účastníků, kteří dosáhli sérové odpovědi na variantu Omikron, mezi skupinou Comirnaty Original/Omicron BA.1 (71,6 %) a skupinou Comirnaty (57 %) byl 14,6 % (2stranný 95% CI: 4,0 %, 24,9 %). Tak byla splněna podmínka non-inferiority.
Tabulka 2: Podstudie E – poměry geometrických průměrů porovnání mezi skupinami vakcín – účastníci bez prokázané infekce až do 1 měsíce po 4. dávce – rozšířená kohorta – podmnožina imunogenicity – účastníci starší 55 let – populace s vyhodnotitelnou imunogenicitou
Analýza
| Skupina s vakcínou (podle randomizace)
| Časový bod odběru
vzorkůa
|
nb
|
GMT (95% CIc)
|
GMR (95% CId)
|
Analýza
neutralizace SARS-CoV-2 – Omikron BA.1 - NT50 (titr)
| Comirnaty
(30 mikrogramů)
|
1 měsíc
|
163
| 455,8
(365,9; 567,6)
|
|
Comirnaty Original/Omicron
BA.1 (15/15 mikrogramů)
|
1 měsíc
|
178
|
711,0 (588,3; 859,2)
| 1,56
(1,17;
2,08)
|
Analýza
neutralizace
| Comirnaty (30 mikrogramů)
|
1 měsíc
|
182
| 5 998,1
(5 223,6; 6 887,4)
|
|
SARS-CoV-2 –
referenční kmen
– NT50 (titr)
|
Comirnaty Original/Omicron
BA.1 (15/15 mikrogramů)
|
1 měsíc
|
186
|
5 933,2
(5 188,2; 6 785,2)
|
0,99
(0,82;
1,20)
|
Zkratky: CI = interval spolehlivosti; GMR = poměr geometrického průměru; GMT = titr geometrického
průměru; LLOQ = dolní limit kvantifikace; N-vazba = SARS-CoV-2 nukleoproteinová vazba; NAAT = test amplifikace nukleové kyseliny; NT50 = 50% neutralizační titr; SARS-CoV-2 = závažný akutní respirační syndrom vyvolaný koronavirem 2.
Poznámka: Podmnožina imunogenicity = náhodný vzorek 230 účastníků v každé skupině vakcíny vybraných
z rozšířené kohorty.
Poznámka: Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (odběr vzorků krve dříve než
1 měsíc po studijní vakcinaci) předcházející infekce SARS-CoV-2 (tj. negativní výsledek N-binding protilátky
[sérum] na návštěvách při studijní vakcinaci a za 1 měsíc po vakcinaci, negativní výsledek NAAT [stěr z nosu] na návštěvě se studijní vakcinací a jakékoliv neplánované návštěvě před odběrem vzorku krve 1 měsíc po studijní vakcinaci) a kteří neměli onemocnění COVID-19 ve zdravotní anamnéze, byli zařazeni do analýzy.
a. Protokolárně stanovené načasování odběru krevních vzorků.
b. n = počet účastníků s platnými a určitelnými výsledky testu pro daný test v daném časovém bodě odběru vzorků.
c. GMT a 2stranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a příslušných CI (na základě Studentova t rozdělení). Výsledky testů pod LLOQ byly stanoveny na
0,5násobku LLOQ.
d. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (skupina vakcíny v příslušném řádku Comirnaty [30 mikrogramů]) a odpovídajícího CI (na základě Studentova t rozložení).
Vakcína ComirnatyStudie 2 je multicentrická, mezinárodní, randomizovaná, placebem kontrolovaná studie fáze 1/2/3
zaslepená pro pozorovatele, která hodnotí dávku, výběr vakcíny a její účinnost u účastníků ve věku
12 let a starších. Randomizace byla stratifikována podle věku: věk 12 až 15 let, věk 16 až 55 let a věk
56 let a více; do věkové skupiny ≥ 56 let spadalo minimálně 40 % účastníků. Ze studie byli vyřazeni imunokompromitovaní účastníci a osoby, u nichž byla dříve stanovena klinická či mikrobiologická
diagnóza onemocnění COVID-19. Účastníci s preexistujícími stabilizovanými onemocněními,
definovanými jako onemocnění, jež během 6 týdnů před zařazením nevyžadovala významnou změnu léčby nebo hospitalizaci z důvodu zhoršení nemoci, byli do studie zařazeni stejně jako účastníci se
známou stabilizovanou infekcí virem lidské imunodeficience (HIV), virem hepatitidy C (HCV) nebo
virem hepatitidy B (HBV).
Účinnost u osob ve věku 16 let a starších – po 2 dávkáchBěhem fáze 2/3 Studie 2 bylo na základě dat získaných do 14. listopadu 2020 ve stejném poměru randomizováno zhruba 44 000 účastníků a byly jim podány 2 dávky mRNA vakcíny proti onemocnění COVID-19 nebo placeba. Analýzy účinnosti zahrnovaly účastníky, kteří dostali druhou dávku vakcíny v rozmezí 19 až 42 dnů po první dávce vakcíny. Většina (93,1 %) příjemců vakcíny dostala druhou dávku 19 dnů až 23 dnů po první dávce. Pro účely hodnocení bezpečnosti a účinnosti vakcíny proti onemocnění COVID-19 budou na základě připraveného plánu účastníci dále sledováni po dobu až
24 měsíců po 2. dávce. V klinické studii bylo od účastníků vyžadováno dodržení minimálního intervalu 14 dní před a po podání vakcíny proti chřipce, aby dostali buď placebo nebo mRNA vakcínu
proti onemocnění COVID-19. V klinické studii bylo od účastníků vyžadováno dodržení minimálního
intervalu 60 dní před nebo po podání přípravků z krve/plazmy nebo imunoglobulinů v průběhu studie, aby dostali buď placebo nebo mRNA vakcínu proti onemocnění COVID-19.
Do populace pro analýzu primárního cílového parametru účinnosti bylo zařazeno 36 621 účastníků ve věku 12 let a starších (18 242 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 18 379 ve skupině s placebem), u nichž se během 7 dní po podání druhé dávky neprokázala předchozí infekce SARS-CoV-2. Kromě toho bylo 134 účastníků ve věku od 16 do 17 let (66 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 68 ve skupině s placebem) a 1 616 účastníků ve věku 75 let a starších (804 ve skupině s mRNA vakcínou proti onemocnění COVID-19 a 812 ve skupině s placebem).
V čase provedení primární analýzy účinnosti byli účastníci ve skupině mRNA vakcíny proti onemocnění COVID-19 sledováni z hlediska symptomatického onemocnění COVID-19 po celkovou dobu 2 214 osoboroků, účastníci ve skupině s placebem po dobu minimálně 2 222 osoboroků.
U účastníků s rizikem závažného onemocnění COVID-19, včetně osob s 1 nebo více komorbiditami,
které riziko závažného onemocnění COVID-19 zvyšují (např. astma, index tělesné hmotnosti (BMI)
≥ 30 kg/m2, chronické plicní onemocnění, diabetes mellitus, hypertenze), nebyly zjištěny žádné
významné klinické rozdíly v celkové účinnosti vakcíny.
Informace o účinnosti vakcíny jsou uvedeny v tabulce 3.
První výskyt COVID-19 od 7 dnů po 2. dávce u účastníků bez důkazu předchozí infekce SARS-
CoV-2 *
|
Podskupina
| Vakcína mRNA proti
onemocnění COVID-19
a
Případy
n1b
Doba sledováníc (n2d)
|
Placebo
a
Případy
n1b
Doba sledováníc (n2d)
|
Účinnost vakcíny % (95% CI)e
| Všichni účastníci
| 8
2,214 (17 411)
| 162
2,222 (17 511)
| 95,0
(90,0; 97,9)
| 16 až 64 let
| 7
1,706 (13 549)
| 143
1,710 (13 618)
| 95,1
(89,6; 98,1)
| 65 let a starší
| 1
0,508 (3 848)
| 19
0,511 (3 880)
| 94,7
(66,7; 99,9)
| 65 až 75 let
| 1
0,406 (3 074)
| 14
0,406 (3 095)
| 92,9
(53,1; 99,8)
| 75 let a starší
| 0
0,102 (774)
| 5
0,106 (785)
| 100,0
(-13,1; 100,0)
|
|
|
Tabulka 3: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů)
n =18 198
n =18 325
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 [*Definice případu: (alespoň 1 z) horečka, nový nebo zhoršený kašel, nová nebo zhoršená dyspnoe, zimnice, nová nebo zhoršená bolest svalů, nová ztráta chuti nebo čichu, bolest v krku, průjem nebo zvracení.]
* Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (7 nebo více dnů před podáním poslední dávky) předcházející infekce SARS-CoV-2 (tj. N-vazebná protilátka [sérum] negativní při návštěvě 1 a SARS- CoV-2 nedetekován pomocí amplifikačních testů nukleových kyselin (NAAT) [výtěr nosu] při návštěvách 1 a 2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do analýzy.
a. n = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osobo-rocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování. CI není upraven na multiplicitu.
Účinnost vakcíny mRNA proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění
COVID-19 po 7 dnech po 2. dávce ve srovnání s placebem 94,6 % (95% interval spolehlivosti 89,6 %
až 97,6 %) u účastníků ve věku 16 let a starších s nebo bez důkazů o předchozí infekci virem SARS- CoV-2.
Analýzy podskupin primárního cílového parametru účinnosti navíc ukázaly podobné odhady bodů účinnosti u pohlaví, etnických skupin a účastníků s komorbiditami spojenými s vysokým rizikem závažného onemocnění COVID-19.
Byly provedeny aktualizované analýzy účinnosti s dalšími potvrzenými případy COVID-19, které se objevily během zaslepeného placebem kontrolovaného sledování, což v populaci s účinností představuje až 6 měsíců po podání 2. dávky.
Aktualizované informace o účinnosti vakcíny jsou uvedeny v tabulce 4.
Tabulka 4: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce, podle věkové podskupiny – účastníci bez důkazu infekce předchozí infekce virem SARS- CoV-2* před 7 dny po 2. dávce – populace hodnotitelná pro účinnost (7 dnů) během placebem kontrolovaného období sledování
Podskupina
|
V
akcína mRNA proti
onemocnění COVID-19
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Placebo
a
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Ú
č
i
n
nost vakcíny % (95% CI
e
)
|
Všichni účastnícif
|
77
6,247 (20 712)
|
850
6,003 (20 713)
|
91,3
(89,0; 93,2)
|
16 až 64 let
|
70
4,859 (15 519)
|
710
4,654 (15 515)
|
90,6
(87,9; 92,7)
|
65 let a starší
|
7
1,233 (4 192)
|
124
1,202 (4 226)
|
94,5
(88,3; 97,8)
|
65 až 74 let
|
6
0,994 (3 350)
|
98
0,966 (3 379)
|
94,1
(86,6; 97,9)
|
75 let a starší
|
1
0,239 (842)
|
26
0,237 (847)
|
96,2
(76,9; 99,9)
|
|
|
N =20 998
N =21 096
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-
PCR) a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo
zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení).
* Účastníci, kteří neměli žádné důkazy předcházející infekce virem SARS-CoV-2 (tj. N-vazebná protilátka
[sérum] negativní při návštěvě 1 a virus SARS-CoV-2 nedetekován pomocí NAAT [výtěr nosu] při návštěvách 1 a
2) a měli negativní NAAT (výtěr nosu) při jakékoli neplánované návštěvě před 7 dny po 2. dávce, byli zařazeni do
analýzy.
a. N = Počet účastníků ve specifikované skupině.
b. n1 = Počet účastníků splňujících definici cílového parametru.
c. Celková doba sledování vyjádřená v 1 000 osobo-rocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od 7 dnů po 2. dávce do konce období sledování.
d. n2 = Počet účastníků s rizikem pro cílový parametr.
e. Dvoustranný 95% interval spolehlivosti pro účinnost vakcíny je odvozen na základě Clopperovy a
Pearsonovy metody upravené podle doby sledování. CI není upraven na multiplicitu.
f. Zahrnuté potvrzené případy u účastníků ve věku 12 až 15 let: 0 ve skupině s mRNA vakcínou proti
onemocnění COVID-19; 16 ve skupině s placebem.
V aktualizované analýze účinnosti byla účinnost mRNA vakcíny proti onemocnění COVID-19 v prevenci prvního výskytu onemocnění COVID-19 od 7 dnů po podání 2. dávky ve srovnání s placebem 91,1 % (95% CI 88,8 % až 93,0 %) u účastníků v hodnotitelné populaci účinnosti s průkazem nebo bez průkazu předchozí infekce SARS-CoV-2.
Aktualizované analýzy účinnosti podle podskupin navíc ukázaly podobné bodové odhady účinnosti u všech pohlaví, etnických skupin, zeměpisných oblastí a účastníků se zdravotními komorbiditami a obezitou spojenou s vysokým rizikem závažného onemocnění COVID-19.
Účinnost proti závažnému onemocnění COVID-19Aktualizované analýzy účinnosti sekundárních cílových parametrů účinnosti podpořily přínos mRNA
vakcíny proti onemocnění COVID-19 v prevenci závažného onemocnění COVID-19.
Od 13. března 2021 je účinnost vakcíny proti závažnému onemocnění COVID-19 prezentována pouze pro účastníky s předchozí infekcí SARS-CoV-2 nebo bez ní (tabulka 5), protože počty případů onemocnění COVID-19 u účastníků bez předchozí infekce SARS-CoV-2 byly stejné jako u účastníků s předchozí infekcí SARS-CoV-2 nebo bez ní jak ve skupině s mRNA vakcínou proti onemocnění COVID-19, tak ve skupině s placebem.
Tabulka 5: Účinnost vakcíny – první závažný výskyt onemocnění COVID-19 u účastníků s nebo bez předchozí infekce virem SARS-CoV-2 na základě údajů Úřadu pro kontrolu potravin a léčiv (FDA)* po podání 1. dávky nebo od 7 dnů po podání 2. dávky v placebem kontrolovaném období sledování
| Vakcína mRNA proti
onemocnění COVID-19
Případy
n1a
Doba sledováníc (n2b)
|
Placebo Případy n1a
Doba sledováníc (n2b)
|
Účinnost vakcíny % (95% CIc)
|
Po 1. dávced
| 1
8,439e (22 505)
| 30
8,288e (22 435)
| 96,7
(80,3; 99,9)
|
7 dnů po 2. dávcef
| 1
6,522g (21 649)
| 21
6,404g (21 730)
| 95,3
(70,9; 99,9)
|
Poznámka: Potvrzené případy byly stanoveny reverzní transkripčně-polymerázovou řetězovou reakcí (RT-PCR)
a alespoň 1 příznakem odpovídajícím onemocnění COVID-19 (příznaky zahrnovaly: horečku, nový nebo zhoršený kašel, novou nebo zhoršenou dyspnoe, zimnici, novou nebo zhoršenou bolest svalů, novou ztrátu chuti nebo čichu, bolest v krku, průjem nebo zvracení.)
* Závažné onemocnění COVID-19 podle definice FDA je potvrzené onemocnění COVID-19 a přítomnost alespoň 1 z následujících příznaků:
• Klinické příznaky v klidu svědčící pro závažné systémové onemocnění (dechová frekvence ≥ 30 dechů za minutu, srdeční frekvence ≥ 125 tepů za minutu, saturace kyslíkem ≤ 93 % na pokojovém vzduchu při hladině moře nebo poměr arteriálního parciálního tlaku kyslíku k frakčnímu inspirovanému kyslíku
< 300 mmHg).
• Respirační selhání [definované jako potřeba vysokého průtoku kyslíku, neinvazivní ventilace, mechanické ventilace nebo extrakorporální membránové oxygenace (ECMO)].
• Průkaz šoku (systolický krevní tlak < 90 mmHg, diastolický krevní tlak < 60 mmHg nebo potřeba
podání vazopresorických látek).
• Významná akutní renální, jaterní nebo neurologická dysfunkce.
• Přijetí na jednotku intenzivní péče.
• Smrt.
a. n1 = Počet účastníků splňujících definici cílového parametru.
b. n2 = počet účastníků ohrožených daným cílovým parametrem.
c. Dvoustranný interval spolehlivosti (CI) pro účinnost vakcíny je odvozen na základě Clopperovy a Pearsonovy
metody upravené podle doby sledování.
d. Účinnost hodnocená na základě veškeré populace pro hodnocení účinnosti s 1. dávkou (modifikovaná se záměrem léčit), která zahrnovala všechny randomizované účastníky, kteří dostali alespoň 1 dávku hodnocené léčby.
e. Celková doba sledování vyjádřená v 1 000 osobo-rocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro cílový parametr. Časové období pro získání případu COVID-19 je od po 1. dávce do konce období sledování.
f. Účinnost hodnocená na základě populace hodnotitelné pro účinnost (7 dní), která zahrnovala všechny způsobilé randomizované účastníky, kteří dostali všechny dávky hodnocené léčby podle randomizace v rámci předem stanoveného časového období a neměli žádné další důležité odchylky od protokolu podle rozhodnutí lékaře.
g. Celková doba sledování vyjádřená v 1 000 osobo-rocích pro daný cílový parametr u všech účastníků v každé skupině s rizikem pro daný cílový parametr. Časové období pro získání případu onemocnění COVID-19 je od
7 dnů po 2. dávce do konce období sledování.
Účinnost a imunogenicita u dospívajících ve věku 12 až 15 let – po 2 dávkách
V úvodní analýze Studie 2 u dospívajících ve věku 12 až 15 let (jež reprezentovala medián doby následného sledování v délce ˃ 2 měsíce po 2. dávce) bez průkazu prodělané infekce nebyly žádné
případy u 1 005 účastníků, kteří dostali vakcínu, a 16 případů z 978 účastníků, kteří dostali placebo. Bodový odhad účinnosti je 100 % (95% interval spolehlivosti 75,3; 100,0). U účastníků s nebo bez
průkazu prodělané infekce bylo 0 případů z 1 119 účastníků, kteří dostali vakcínu, a 18 případů
z 1 110 účastníků, kteří dostali placebo. To také naznačuje, že bodový odhad účinnosti je 100 % (95%
interval spolehlivosti 78,1; 100,0).
U dodatečných potvrzených případů onemocnění COVID-19, k nimž došlo během zaslepeného, placebem kontrolovaného následného sledování, byly provedeny aktualizované analýzy účinnosti, jež reprezentovaly dobu až 6 měsíců po 2. dávce v populaci pro hodnocení účinnosti.
V aktualizované analýze účinnosti ve studii 2, provedené u dospívajících ve věku 12 až 15 let bez průkazu prodělané infekce, se u 1 057 účastníků, kterým byla podána vakcína, nevyskytl žádný případ a u 1 030 účastníků, kterým bylo podáno placebo, došlo ke 28 případům. Odhad bodu účinnosti je
100 % (95% interval spolehlivosti 86,8; 100,0). U účastníků s průkazem prodělané infekce nebo bez tohoto průkazu nedošlo u 1 119 těch, kterým byla podána vakcína, k žádnému případu
a u 1 109 účastníků, jimž bylo podáno placebo, ke 30 případům. To rovněž naznačuje, že odhad bodu
účinnosti je 100 % (95% interval spolehlivosti 87,5; 100,0).
Ve Studii 2 byla provedena analýza neutralizačních titrů SARS-CoV-2 jeden měsíc po druhé dávce u náhodně vybrané podskupiny účastníků, kteří do 1 měsíce po dávce 2 neměli sérologické nebo virologické průkazy prodělané infekce SARS-CoV-2, která porovnávala odpověď u dospívajících ve věku 12 až 15 let (n = 190) s účastníky ve věku 16 až 25 let (n = 170).
Poměr geometrických průměrných titrů (GMT) ve skupině ve věku 12 až 15 let ke skupině ve věku 16 až 25 let byl 1,76; s oboustranným 95% CI 1,47 až 2,10. Proto bylo splněno 1,5násobné kritérium noninferiority, protože dolní mez 2stranného 95% CI pro poměr geometrického průměru [GMR] byla
> 0,67.
Účinnost a imunogenicita u dětí ve věku 5 až 11 let (tj. ve věku 5 až méně než 12 let) – po 2 dávkách Studie 3 je studie fáze 1/2/3, která se skládá z otevřené části zaměřené na zjištění dávky vakcíny (fáze 1) a multicentrické, mezinárodní, randomizované, fyziologickým roztokem jako placebem kontrolované části zaslepené pro pozorovatele hodnotící účinnost (fáze 2/3), do které byli zařazeni účastníci ve věku 5 až 11 let. Většina (94,4 %) randomizovaných příjemců vakcíny dostala druhou dávku 19 dní až 23 dní po první dávce.
Deskriptivní výsledky účinnosti vakcíny u dětí ve věku 5 až 11 let bez známek předchozí infekce virem SARS-CoV-2 jsou uvedeny v tabulce 6. U účastníků s prokázanou předchozí infekcí virem SARS-CoV-2 nebyly pozorovány žádné případy onemocnění COVID-19 ani ve skupině s vakcínou, ani ve skupině s placebem.
T
abulka 6: Účinnost vakcíny – první výskyt onemocnění COVID-19 od 7 dnů po 2. dávce: bez průkazu infekce před 7 dny po 2. dávce – fáze 2/3 – děti ve věku 5 až 11 let hodnotitelná populace pro účinnost
P
r
vní výskyt onemocnění COVID-19 od 7 dnů po 2. dávce u dětí ve věku 5 až 11 let bez
průkazu předchozí infekce SARS-CoV-2*
|
|
V
akcína mRNA proti
onemocnění
COV
I
D
-19
D
ávka 10 mikrogramů/dávku N
a
=
1 305
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Placebo
N
a
=
6
63
Případy
n1
b
D
oba sledování
c
(
n2
d
)
|
Ú
č
i
n
nost vakcíny
% (95% CI)
|
Děti ve věku 5 až 11 let
|
3
0,322 (1273)
|
16
0,159 (637)
|
90,7
(67,7; 98,3)
|
Poznámka: Potvrzené případy byly určeny na základě reverzní transkripční polymerázové řetězové reakce (RT-
PCR) a alespoň 1 příznaku odpovídajícího onemocnění COVID-19 (příznaky zahrnovaly: horečku; nový nebo zesílený kašel; novou nebo zesílenou dušnost; zimnici; novou nebo zesílenou bolest svalů; novou ztrátu chuti nebo čichu; bolest v krku; průjem; zvracení).
* Do analýzy byli zahrnuti účastníci, kteří neměli žádný průkaz o prodělané infekci virem SARS-CoV-2 (tj.
negativní N-vázané protilátky [sérum] při návštěvě 1 a virus SARS-CoV-2 nezjištěný pomocí NAAT [nosní výtěr] při návštěvě 1 a 2) a měli negativní NAAT (nosní výtěr) při jakékoli neplánované návštěvě před 7 dny po 2. dávce
a. N = počet účastníků v dané skupině.
b. n1 = počet účastníků splňujících definici cílového ukazatele.
c. Celková doba sledování v 1 000 osobo-rocích pro daný cílový ukazatel u všech účastníků v každé skupině s rizikem pro cílový ukazatel. Časové období pro nárůst případů onemocnění COVID-19 je od 7 dnů po podání 2. dávky do konce období sledování.
d. n2 = počet účastníků s rizikem pro daný cílový ukazatel.
Ve studii 3 prokázala analýza 50% neutralizačních titrů viru SARS-CoV-2 (NT50) 1 měsíc po podání
2. dávky u náhodně vybrané podskupiny účastníků účinnost imunologickým přemostěním imunitních odpovědí při srovnání dětí ve věku 5 až 11 let (tj. ve věku 5 až méně než 12 let) v části studie 3 fáze
2/3 s účastníky ve věku 16 až 25 let v části studie 2 fáze 2/3, kteří neměli žádné sérologické nebo
virologické průkazy o prodělané infekci virem SARS-CoV-2 do 1 měsíce po podání 2. dávky, což splňuje předem stanovená kritéria imunologického přemostění jak pro poměr geometrického průměru (GMR), tak pro rozdíl sérologických odpovědí, přičemž sérologická odpověď byla definována jako dosažení alespoň čtyřnásobného zvýšení NT50 SARS-CoV-2 oproti výchozí hodnotě (před podáním 1. dávky).
GMR NT50 SARS-CoV-2 1 měsíc po 2. dávce u dětí ve věku 5 až 11 let (tj. ve věku 5 až méně než
12 let) proti mladým dospělým ve věku 16 až 25 let byl 1,04 (2stranný 95% CI: 0,93; 1,18). Mezi
účastníky bez předchozích známek infekce virem SARS-CoV-2 do 1 měsíce po podání 2. dávky mělo
99,2 % dětí ve věku 5 až 11 let a 99,2 % účastníků ve věku 16 až 25 let sérologickou odpověď 1 měsíc po podání 2. dávky V případě dětí ve věku 5 až 11 let a mladých lidí ve věku 16 až 25 let byla
sérologická odpověď prokázána až po 1 měsíci po podání 2. dávky Rozdíl v podílech účastníků, kteří
měli sérologickou odpověď mezi dvěma věkovými skupinami (děti – mladí dospělí), byl 0,0 % (2stranný 95% CI: -2,0 %; 2,2 %). Tyto informace jsou uvedeny v tabulce 7.
T
abulka 7: Souhrn geometrického průměrného poměru pro 50% neutralizační titr a rozdíl v procentech účastníků se sérologickou odpovědí – srovnání dětí ve věku 5 až 11 let (studie 3) s účastníky ve věku 16 až 25 let (studie 2) – účastníci bez známek infekce
do 1 měsíce po podání 2. dávky – podskupina imunologického přemostění – fáze 2/3 –
populace hodnotitelná na imunogenicitu
|
V
akcína mRNA proti onemocnění
COV
I
D
-19
|
5 až 11 let /
16 až 25 let
|
D
ávka
10 mikrogramů/dávku
5 až 11 let
N
a
=
264
|
D
ávka
30 mikrogramů/dáv
ku
16 až 25 let
N
a
=
253
|
|
Č
asový
bod
b
|
G
MT
c
(
95% CI
c
)
|
G
MT
c
(
95% CI
c
)
|
G
MR
d
(
95% CI
d
)
|
Splnila cíl
im
unologické ho přemostění
e
(
A
/
N)
|
G
eometrický
průměrný
50% neutralizační titr
f
(
G
MT
c
)
|
1 měsíc po 2. dávce
|
1 197,6
(1 106,1; 1 296,6)
|
1 146,5
(1 045,5; 1 257,2)
|
1,04 (0,93; 1,18)
|
A
|
|
Č
asový
bod
b
|
n
g
(
%)
(
95% CI
h
)
|
n
g
(
%)
(
95% CI
h
)
|
R
ozdíl %
i
(
95% CI
j
)
|
Splnila cíl imunologické
ho přemostění
k
(
A
/
N)
|
Č
etnost
sérologické
odpovědi (%) pro 50% neutralizační titr
f
|
1 měsíc po 2. dávce
|
262 (99,2) (97,3; 99,9)
|
251 (99,2) (97,2; 99,9)
|
0,0
(-2,0; 2,2)
|
A
|
Zkratky: CI = interval spolehlivosti; GMR = geometrický průměrný poměr; GMT = geometrický průměrný titr;
LLOQ = dolní mez kvantifikace; NAAT = test amplifikace nukleových kyselin; NT50 = 50% neutralizační titr;
virus SARS-CoV-2 = těžký akutní respirační syndrom vyvolaný koronavirem 2.
Poznámka: Účastníci, kteří neměli žádné sérologické nebo virologické důkazy (do 1 měsíce po odběru vzorku krve po 2. dávce) o prodělané infekci virem SARS-CoV-2 (tj, N-vázané protilátky [sérum] negativní při návštěvě s podáním 1. dávky a 1 měsíc po 2. dávce, virus SARS-CoV-2 nezjištěn pomocí NAAT [nosní výtěr] při návštěvě s podáním 1. dávky a 2. dávky a negativní NAAT (nosní výtěr) při kterékoli neplánované návštěvě do 1 měsíce po odběru krve po 2. dávce) a neměli v anamnéze žádné onemocnění COVID-19, byli zahrnuti do analýzy.
Poznámka: Sérologická odpověď je definována jako dosažení ≥ 4násobného zvýšení oproti výchozí hodnotě (před 1. dávkou). Pokud je výchozí měření nižší než LLOQ, považuje se výsledek testu po očkování ≥4× LLOQ za sérologickou odpověď.
a. N = počet účastníků s platnými a určitelnými výsledky testu před očkováním a 1 měsíc po podání 2. dávky.
Tyto hodnoty jsou také jmenovateli použitými při výpočtech procentuální četnosti sérologické odpovědi.
b. Protokolárně stanovené načasování odběru krevních vzorků.
c. GMT a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů a odpovídajících CI (na základě Studentova t rozložení). Výsledky testů pod LLOQ byly stanoveny na 0,5 × LLOQ.
d. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (věk 5 až 11 let minus věk 16 až 25 let) a odpovídajícího CI (na základě Studentova t rozložení).
e. Imunologické přemostění podle GMT je deklarováno, pokud je dolní hranice dvoustranného 95% CI pro
GMR větší než 0,67 a bodový odhad GMR je ≥ 0,8.
f. NT50 SARS-CoV-2 byly stanoveny pomocí mikroneutralizačního testu SARS-CoV-2 mNeonGreen Virus
Assay. Test používá fluorescenční reportérový virus odvozený od kmene USA_WA1/2020 a neutralizace
viru se odečítá na monovrstvách buněk Vero. Vzorek NT50 je definován jako vzájemné zředění séra, při
kterém je neutralizováno 50 % viru.
g. n = počet účastníků se sérologickou odpovědí na základě NT50 1 měsíc po podání 2. dávky.
h. Přesný dvoustranný CI na základě Clopperovy a Pearsonovy metody.
i. Rozdíl v podílech vyjádřený v procentech (věk 5 až 11 let minus věk 16 až 25 let).
j. Přesný 2stranný CI založený na metodě Miettinena a Nurminena pro rozdíl v podílech, vyjádřený v
procentech.
k. Imunologické přemostění založené na četnosti sérologické odpovědi je deklarováno, pokud je dolní hranice
2stranného 95% CI pro rozdíl sérologické odpovědi větší než 10,0 %.
Imunogenicita u dětí ve věku od 5 do 11 let (tj. ve věku 5 až méně než 12 let) – po posilovací dávcePosilovací dávka vakcíny Comirnaty byla ve studii 3 podána 401 náhodně vybraným účastníkům. Účinnost posilovací dávky ve věku 5 až 11 let je odvozena z údajů o imunogenicitě. Imunogenicita
byla hodnocena podle NT50 u referenčního kmene SARS-CoV-2 (USA_WA1/2020). Analýzy NT50
za 1 měsíc po posilovací dávce v porovnání se stavem před posilovací dávkou prokázaly podstatné
zvýšení GMT u jedinců ve věku od 5 do 11 let včetně, kteří neměli sérologický ani virologický průkaz předchozí infekce virem SARS-CoV-2 do 1 měsíce po 2. dávce a po posilovací dávce. Tato analýza je shrnuta v tabulce 8.
| Časový bod odběrua
|
|
Test
|
1 měsíc po posilovací
dávce (nb=67)
GMTc
(95% CIc)
|
1 měsíc po 2. dávce
(nb=96)
GMTc
(95% CIc)
| 1 měsíc po posilovací
dávce /
1 měsíc po 2. dávce
GMRd
(95% CId)
| SARS-CoV-2
neutralizační test - NT50 (titr)
|
2 720,9
(2 280,1; 3 247,0)
|
1 253,9
(1 116,0; 1 408,9)
|
2,17 (1,76; 2,68)
| Zkratky: CI = interval spolehlivosti; GMR = geometrický průměr poměrů; GMT = geometrický průměr titrů;
LLOQ = dolní mez kvantifikace; NT50 = 50% neutralizační titr; virus SARS-CoV-2 = těžký akutní respirační
syndrom vyvolaný koronavirem 2.
a. Protokolárně stanovené načasování odběru krevních vzorků.
b. n = počet účastníků s platnými a konečnými výsledky testu pro specifický test v časovém bodě podání dávky/odběru.
c. GMT a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného logaritmu titrů
a odpovídajících CI (na základě Studentova t rozložení). Výsledky testů pod LLOQ byly stanoveny na
0,5 × LLOQ.
d. GMR a dvoustranné 95% CI byly vypočteny exponenciálním vyjádřením průměrného rozdílu logaritmů titrů (1 měsíc po posilovací dávce minus 1 měsíc po 2. dávce) a odpovídajícího CI (na základě Studentova t rozložení).
|
|
|
Tabulka 8: Souhrn geometrického průměru titrů – NT50 – účastníci bez průkazu infekce – fáze 2/3 – soubor imunogenicity – věk 5 až 11 let včetně – populace hodnotitelná na imunogenicituPediatrická populaceEvropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s vakcínou
Comirnaty u pediatrické populace k prevenci onemocnění COVID-19 (informace o použití u pediatrické populace viz bod 4.2).
5.2 Farmakokinetické vlastnostiNeuplatňuje se.
5.3 Předklinické údaje vztahující se k bezpečnostiNeklinické údaje získané na základě konvenčních studií toxicity po opakovaném podávání a reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka.
O
becná toxicita
U potkanů, kterým byla intramuskulárně podávána vakcína Comirnaty (dostali 3 plné dávky pro
člověka jednou týdně, které vedly k relativně vyšším hladinám u potkanů v důsledku rozdílů v tělesné
hmotnosti), se vyskytl edém a erytém v místě podání injekce a zvýšení počtu bílých krvinek (včetně bazofilů a eozinofilů) odpovídající zánětlivé odpovědi a rovněž vakuolizace portálních hepatocytů bez známek poškození jater. Všechny účinky byly reverzibilní.
Genotoxicita / karcinogenita
Studie genotoxicity ani kancerogenity nebyly provedeny. U složek vakcíny (lipidy a mRNA) se
neočekává genotoxický potenciál.
Reprodukční toxicita
Reprodukční a vývojová toxicita byla hodnocena na potkanech v kombinované studii fertility a
vývojové toxicity, ve které byla samicím potkanů podána intramuskulárně vakcína Comirnaty před pářením a během březosti (dostaly 4 plné dávky pro člověka, které vedly k relativně vyšším hladinám
u potkanů v důsledku rozdílů v tělesné hmotnosti mezi 21. dnem před pářením a 20. dnem březosti).
Odpovědi na neutralizační protilátky SARS-CoV-2 byly přítomny u samic před pářením až do konce studie ve 21. postnatálním dni a rovněž u plodů a potomků. Nebyly zjištěny žádné účinky spojené s očkováním na plodnost samic, těhotenství nebo vývoj embrya/plodu nebo potomstva. Údaje o možném placentárním přenosu nebo vylučování do mateřského mléka pro vakcínu Comirnaty nejsou k dispozici.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159) Kolfosceryl-stearát
Cholesterol
Trometamol
Trometamol-hydrochlorid
Sacharosa
Voda pro injekci
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou
uvedeny v bodě 6.6.
6.3 Doba použitelnosti
N
eotevřená injekčnílahvička
Zmrazenáinjekčnílahvička
18 měsíců při teplotě -90 °C až -60 °C.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána při teplotě -90 °C až -60 °C nebo 2 °C až 8 °C.
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení
10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 4 hodin nebo mohou být jednotlivé
injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Rozmrazená injekčnílahvička
10 týdnů uchovávání a transportu při teplotě 2 °C až 8 °C během 18měsíční doby použitelnosti.
• Po přenesení vakcíny pro uchovávání při teplotě 2 °C až 8 °C musí být na vnější obal zapsáno aktualizované datum použitelnosti a vakcína má být použita nebo zlikvidována do aktualizovaného data použitelnosti. Původní datum použitelnosti má být přeškrtnuto.
• Pokud je vakcína přijata při teplotě 2 °C až 8 °C, má být uchovávána při teplotě 2 °C až 8 °C.
Datum použitelnosti na vnějším obalu má být aktualizováno tak, aby odráželo datum použitelnosti v chladu, a původní datum použitelnosti má být přeškrtnuto.
Neotevřené injekční lahvičky lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Po rozmrazení vakcína nesmí být znovu zmrazena.
Manipulace přiteplotníchvýkyvechpřiuchovávánívchladničce
• Údaje o stabilitě prokazují, že je neotevřená injekční lahvička stabilní až 10 týdnů, je-li
uchovávána při teplotě od -2 °C do 2 °C a během 10týdenního uchovávání při teplotě 2 °C až
8 °C.
• Údaje o stabilitě ukazují, že injekční lahvička může být uchovávána až 24 hodin při teplotě od
8 °C do 30 °C, včetně až 12 hodin po prvním vpichu.
Tyto informace slouží jako návod pro zdravotnické pracovníky pouze v případě dočasného výkyvu
teploty.
Zředěný léčivý přípravek
Chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 12 hodin při teplotě
2 °C až 30 °C po naředění injekčním roztokem chloridu sodného 9 mg/ml (0,9%), která zahrnuje až
6 hodin transportu. Z mikrobiologického hlediska, pokud způsob ředění nevyloučí riziko mikrobiologické kontaminace, má být přípravek použit okamžitě. Pokud není použit okamžitě, doba a
podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Během uchovávání je třeba minimalizovat vystavení přípravku světlu v místnosti a zabránit vystavení
přímému slunečnímu světlu a ultrafialovému světlu.
Podmínky uchovávání tohoto léčivého přípravku po jeho rozmrazení a naředění jsou uvedeny
v bodě 6.3.
6.5 Druh obalu a obsah balení
1,3 ml koncentrátu pro disperzi v čiré vícedávkové injekční lahvičce (sklo třídy I) o objemu 2 ml se zátkou (syntetická brombutylová pryž) a oranžovým odtrhovacím plastovým víčkem s hliníkovým krytem. Jedna injekční lahvička obsahuje 10 dávek, viz bod 6.6.'
Velikosti balení: 10 injekčních lahviček nebo 195 injekčních lahviček
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Instrukce pro zacházení s vakcínou
Vakcína Comirnaty Original/Omicron BA.4-5 (5/5 mikrogramů)/dávku má být připravována
zdravotnickým pracovníkem pomocí aseptické techniky, aby byla zajištěna sterilita připravené
disperze.
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.4-5 (5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
• Zkontrolujte, zda má injekční lahvička
O
ranžové
víčko
C
omirnaty Original/ Omicron BA.4-5
P
o naředění
5/5 mikrogramů
oranžové plastové víčko a oranžový okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko a oranžový okraj kolem štítku a název přípravku je Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi, přečtěte si souhrn údajů o přípravku pro toto složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička šedé
plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku injekční disperze, Comirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční
disperze nebo Comirnaty
Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi.
Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.4-5
(
5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET) PŘED POUŽITÍM
|
U
chovávejte až
10 týdnů při teplotě 2 °C až
8 °C.
|
• Pokud se vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se rozmrazily;
rozmrazení balení 10 injekčních lahviček může trvat 4 hodiny. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
MÍCHÁNÍ VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.4-5
(
5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET) PŘED ŘEDĚNÍM
|

Jemně × 10
|
• Rozmrazenou injekční lahvičku nechte ohřát na pokojovou teplotu a před ředěním ji 10krát jemně převraťte. Netřepejte.
• Před ředěním může rozmrazená disperze obsahovat bílé až téměř bílé matné amorfní částice.
|
ŘEDĚN
Í VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.4-5
(
5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
|
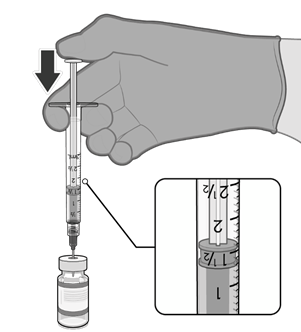
1,3 ml injekčního roztoku chloridu
sodného 9 mg/ml (0,9 %)
|
• Rozmrazená vakcína se musí naředit v původní injekční lahvičce 1,3 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%) za použití jehly
o velikosti 21 gauge (21G) nebo užší a uplatnění aseptických metod.
|
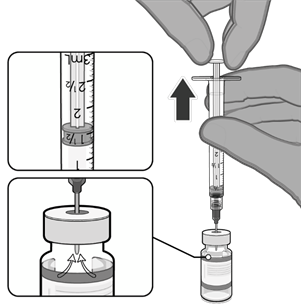
V
ytáhněte píst na značku 1,3 ml pro
odstranění vzduchu z injekční lahvičky.
|
• Nasátím 1,3 ml vzduchu do prázdné stříkačky na rozpouštědlo vyrovnejte před vyjmutím jehly z injekční lahvičky tlak v injekční lahvičce.
|
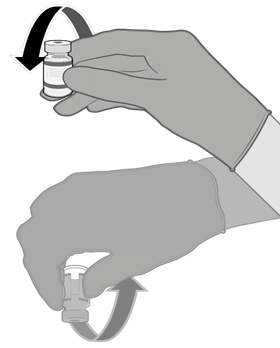
Jemně x 10
|
• Disperzi 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Naředěná vakcína se má jevit jako bílá až téměř bílá disperze bez viditelných
částic. Jestliže jsou v naředěné vakcíně
patrné částice nebo vakcína změnila
barvu, nepoužívejte ji.
|
Č
as likvidace
Z
a
z
namenejte příslušné datum a čas. Použijte během 12 hodin po naředění.
|
• Injekční lahvičky s naředěnou vakcínou je třeba označit příslušným datem
a časem.
• Po naředění uchovávejte vakcínu při teplotě 2 °C až 30 °C a použijte ji během
12 hodin.
• Naředěnou disperzi nezmrazujte ani s ní netřepejte. Pokud je v chladu, nechte naředěnou disperzi před použitím dosáhnout pokojové teploty.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,2 ml VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.4-5 (5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
• Po naředění obsahuje injekční lahvička
2,6 ml, ze kterých lze získat 10 dávek po
0,2 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím
antiseptického tampónu.
• Natáhněte 0,2 ml vakcíny Comirnaty Original/Omicron BA.4-5 pro děti ve věku 5 až 11 let.
K získání 10 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční stříkačky
a jehly s malým mrtvým prostorem má
mít mrtvý objem maximálně
35 mikrolitrů.
N
aředěná vakcína o objemu 0,2 ml
Pokud se používají standardní injekční
stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání deseti dávek z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,2 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,2 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Veškerou vakcínu, jež nebyla použita do
12 hodin po naředění, zlikvidujte.
L
i
kv
i
dace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.de8. REGISTRAČNÍ ČÍSLO/REGISTRAČNÍ ČÍSLAEU/1/20/1528/011
EU/1/20/1528/012
9. DATUM REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. prosince 2020
Datum posledního prodloužení registrace: 10. října 2022
10. DATUM REVIZE TEXTUPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/
P
Ř
Í
L
OH
A II
A
. VÝROBCI BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B
. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA
B
E
Z
P
EČN
É A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO
P
Ř
Í
P
RAV
K
U
A
. VÝROBCI BIOLOGICKÝCH LÉČIVÝCH LÁTEK A VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
N
ázev a adresa výrobců biologickéléčivélátky/biologickýchléčivýchlátek
BioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz
Německo
BioNTech Manufacturing Marburg GmbH Emil-von-Behring-Strasse 76
35041 Marburg
Německo
Pfizer Ireland Pharmaceuticals Grange Castle Business Park Clondalkin
Dublin 22
Irsko
Rentschler Biopharma SE Erwin-Rentschler-Strasse 21
88471 Laupheim
Německo
Wyeth BioPharma Division of Wyeth Pharmaceuticals LLC
1 Burtt Road
Andover, MA 01810
USA
Název a adresa výrobců odpovědnýchzapropouštěníšarží
BioNTech Manufacturing GmbH
Kupferbergterrasse 17-19
55116 Mainz
Německo
Pfizer Manufacturing Belgium NV Rijksweg 12
2870 Puurs
Belgie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
• Úřední propouštění šarží
Podle článku 114 směrnice 2001/83/ES bude úřední propouštění šarží provádět některá státní laboratoř nebo laboratoř k tomuto účelu určená.
C
. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti (PSUR)
Požadavky pro předkládání PSUR pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci (MAH) předloží první PSUR pro tento léčivý přípravek do 6 měsíců od
jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci (MAH) uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
P
Ř
Í
L
OH
A III
O
Z
NAČEN
Í NA OBALU A PŘÍBALOVÁ INFORMACE
A
. OZNAČENÍ NA OBALU
 ÚDA
JE UVÁDĚNÉ NA VNĚJŠÍM OBALU
ŠTÍTEK NA KRABIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ÚDA
JE UVÁDĚNÉ NA VNĚJŠÍM OBALU
ŠTÍTEK NA KRABIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
COMIRNATY 30 mikrogramů/dávku koncentrát pro injekční disperzi
Dospělí a dospívající od 12 let
mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEKJedna injekční lahvička obsahuje po naředění 6 dávek po 0,3 ml.
3. SEZNAM POMOCNÝCH LÁTEKPomocné látky: ALC-0315, ALC-0159, kolfosceryl-stearát, cholesterol, chlorid draselný,
dihydrogenfosforečnan draselný, chlorid sodný, dihydrát hydrogenfosforečnanu sodného, sacharosa,
voda pro injekci, hydroxid sodný, kyselina chlorovodíková
4. LÉKOVÁ FORMA A OBSAH BALENÍKoncentrát pro injekční disperzi
195 vícedávkových injekčních lahviček
5. ZPŮSOB A CESTA/CESTY PODÁNÍIntramuskulární podání po naředění.
Před použitím si přečtěte příbalovou informaci.
Více informací je k dispozici po sejmutí QR kódu.
Před použitím nařeďte: Jednu injekční lahvičku nařeďte 1,8 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%).
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁNMIMO DOHLED A DOSAH DĚTÍUchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ8. POUŽITELNOSTEXP (při -90 °C až -60 °C)
Použitelnost při teplotě 2 °C až 8 °C: …………………..
(Maximálně 1 měsíc. Přeškrtněte dřívější datum použitelnosti.)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávání: Před naředěním uchovávejte při teplotě -90 °C až -60 °C v původním obalu, aby byl
přípravek chráněn před světlem.
Po naředění vakcínu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 6 hodin.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮNEBO ODPADU Z NICH, POKUD JE TO VHODNÉ11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLAEU/1/20/1528/001
13. ČÍSLO ŠARŽELot
14. KLASIFIKACE PRO VÝDEJ15. NÁVOD K POUŽITÍ16. INFORMACE V BRAILLOVĚ PÍSMUNevyžaduje se – odůvodnění přijato.
17. JEDINEČNÝ IDENTIFIKÁTOR – 2D ČÁROVÝ KÓD2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR – DATA ČITELNÁ OKEMPC
SN NN
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU
ŠTÍTEK NA INJEKČNÍ LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
COMIRNATY 30 μg sterilní koncentrát
mRNA vakcína proti onemocnění COVID-19
tozinameran i.m.
2. ZPŮSOB PODÁNÍ3. POUŽITELNOSTEXP
4. ČÍSLO ŠARŽELot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET6 dávek po 30 mikrogramech po naředění
6. JINÉČas likvidace:
ÚDA
JE UVÁDĚNÉ NA VNĚJŠÍM OBALU
K
RA
B
I
Č
K
A
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
COMIRNATY 30 mikrogramů/dávku injekční disperze
dospělí a dospívající od 12 let
mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jednodávkové injekční lahvičky
Jedna injekční lahvička obsahuje 1 dávku 0,3 ml.

Vícedávkové injekční lahvičky
Jedna injekční lahvička obsahuje 6 dávek po 0,3 ml.
3. SEZNAM POMOCNÝCH LÁTEKPomocné látky: ALC-0315, ALC-0159, kolfosceryl-stearát, cholesterol, trometamol, trometamol-
hydrochlorid, sacharosa, voda pro injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍInjekční disperze
Jednodávkové injekční lahvičky
10 jednodávkových injekčních lahviček
Vícedávkové injekční lahvičky
10 vícedávkových injekčních lahviček
195 vícedávkových injekčních lahviček
5. ZPŮSOB A CESTA/CESTY PODÁNÍIntramuskulární podání.
Před použitím neřeďte.

Více informací je k dispozici po sejmutí QR kódu.
URL:
www.comirnatyglobal.com6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁNMIMO DOHLED A DOSAH DĚTÍUchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ8. POUŽITELNOSTEXP (při -90 °C až -60 °C)
Použitelnost při teplotě 2 °C až 8 °C: …………………..
(Maximálně 10 týdnů. Přeškrtněte dřívější datum použitelnosti.)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍUchovávání:
Po přijetí uchovávejte při teplotě 2 °C až 8 °C. Po rozmrazení znovu nezmrazujte. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Před použitím a pro další informace o uchovávání si přečtěte příbalovou informaci.
Vícedávkové injekční lahvičky
Po prvním vpichu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮNEBO ODPADU Z NICH, POKUD JE TO VHODNÉ11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLAJednodávkové injekční lahvičky
EU/1/20/1528/013
Vícedávkové injekční lahvičky
|
|
EU/1/20/1528/002
| 10 vícedávkových injekčních lahviček
|
|
EU/1/20/1528/003 195 vícedávkových injekčních lahviček
|
| | | |
13. ČÍSLO ŠARŽELot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se – odůvodnění přijato.
17. JEDINEČNÝ IDENTIFIKÁTOR – 2D ČÁROVÝ KÓD2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR – DATA ČITELNÁ OKEMPC
SN NN
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU
ŠTÍTEK NA INJEKČNÍ LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
COMIRNATY 30 μg injekce
mRNA vakcína proti onemocnění COVID-19
tozinameran i.m.
2. ZPŮSOB PODÁNÍNeřeďte
3. POUŽITELNOSTEXP
4. ČÍSLO ŠARŽELot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČETJednodávkové injekční lahvičky
1 dávka
Vícedávkové injekční lahvičky
6 dávek po 30 mikrogramech
6. JINÉVícedávkové injekční lahvičky
Čas likvidace:
ÚDA
JE UVÁDĚNÉ NA VNĚJŠÍM OBALU
K
RA
B
I
Č
K
A
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
COMIRNATY 10 mikrogramů/dávku koncentrát pro injekční disperzi
Děti ve věku 5 až 11 let
mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEKJedna injekční lahvička obsahuje po naředění 10 dávek po 0,2 ml.
3. SEZNAM POMOCNÝCH LÁTEKPomocné látky: ALC-0315, ALC-0159, kolfosceryl-stearát, cholesterol, trometamol, trometamol-
hydrochlorid, sacharosa, voda pro injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍKoncentrát pro injekční disperzi
10 vícedávkových injekčních stříkaček
195 vícedávkových injekčních lahviček
5. ZPŮSOB A CESTA/CESTY PODÁNÍIntramuskulární podání po naředění.
Před použitím si přečtěte příbalovou informaci a další informace o uchovávání.
Více informací je k dispozici po sejmutí QR kódu.
Před použitím nařeďte: Jednu injekční lahvičku nařeďte 1,3 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%).
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁNMIMO DOHLED A DOSAH DĚTÍUchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ8. POUŽITELNOSTEXP (při -90 °C až -60 °C)
Použitelnost při teplotě 2 °C až 8 °C: …………………..
(Maximálně 10 týdnů. Přeškrtněte dřívější datum použitelnosti.)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍUchovávání:
Po přijetí uchovávejte při teplotě 2 °C až 8 °C. Po rozmrazení znovu nezmrazujte. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Po naředění vakcínu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮNEBO ODPADU Z NICH, POKUD JE TO VHODNÉ11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLAEU/1/20/1528/004 10 vícedávkových injekčních lahviček
EU/1/20/1528/005 195 vícedávkových injekčních lahviček
13. ČÍSLO ŠARŽELot
14. KLASIFIKACE PRO VÝDEJ15. NÁVOD K POUŽITÍ16. INFORMACE V BRAILLOVĚ PÍSMUNevyžaduje se – odůvodnění přijato.
17. JEDINEČNÝ IDENTIFIKÁTOR – 2D ČÁROVÝ KÓD2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR – DATA ČITELNÁ OKEMPC
SN NN
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU
ŠTÍTEK NA INJEKČNÍ LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
COMIRNATY 10 μg sterilní koncentrát
mRNA vakcína proti onemocnění COVID-19
tozinameran i.m.
2. ZPŮSOB PODÁNÍ3. POUŽITELNOSTEXP
4. ČÍSLO ŠARŽELot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET10 dávek po 10 mikrogramech po naředění
6. JINÉČas likvidace:
ÚDA
JE UVÁDĚNÉ NA VNĚJŠÍM OBALU
K
RA
B
I
Č
K
A
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
COMIRNATY 3 mikrogramy/dávku koncentrát pro injekční disperzi
Děti ve věku 6 měsíců až 4 roky
mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEKJedna injekční lahvička obsahuje po naředění 10 dávek po 0,2 ml.
3. SEZNAM POMOCNÝCH LÁTEKPomocné látky: ALC-0315, ALC-0159, kolfosceryl-stearát, cholesterol, trometamol, trometamol-
hydrochlorid, sacharosa, voda pro injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍKoncentrát pro injekční disperzi
10 vícedávkových injekčních stříkaček
5. ZPŮSOB A CESTA/CESTY PODÁNÍIntramuskulární podání po naředění.
Před použitím si přečtěte příbalovou informaci a další informace o uchovávání.
Více informací je k dispozici po sejmutí QR kódu.
Před použitím nařeďte: Jednu injekční lahvičku nařeďte 2,2 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%).
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁNMIMO DOHLED A DOSAH DĚTÍUchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ8. POUŽITELNOSTEXP (při -90 °C až -60 °C)
Použitelnost při teplotě 2 °C až 8 °C: …………………..
(Maximálně 10 týdnů. Přeškrtněte dřívější datum použitelnosti.)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávání:
Po přijetí uchovávejte při teplotě 2 °C až 8 °C. Po rozmrazení znovu nezmrazujte. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Po naředění vakcínu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮNEBO ODPADU Z NICH, POKUD JE TO VHODNÉ11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLAEU/1/20/1528/010
13. ČÍSLO ŠARŽELot
14. KLASIFIKACE PRO VÝDEJ15. NÁVOD K POUŽITÍ16. INFORMACE V BRAILLOVĚ PÍSMUNevyžaduje se – odůvodnění přijato.
17. JEDINEČNÝ IDENTIFIKÁTOR – 2D ČÁROVÝ KÓD2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR – DATA ČITELNÁ OKEMPC
SN NN
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU
ŠTÍTEK NA INJEKČNÍ LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
COMIRNATY 3 μg sterilní koncentrát
mRNA vakcína proti onemocnění COVID-19
tozinameran i.m.
2. ZPŮSOB PODÁNÍ3. POUŽITELNOSTEXP
4. ČÍSLO ŠARŽELot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET10 dávek po 3 mikrogramech po naředění
6. JINÉČas likvidace:
ÚDA
JE UVÁDĚNÉ NA VNĚJŠÍM OBALU
K
RA
B
I
Č
K
A
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
COMIRNATY Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční disperze
Dospělí a dospívající od 12 let
mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran/riltozinameran
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEKJedna injekční lahvička obsahuje 6 dávek po 0,3 ml.
Jedna dávka obsahuje 15 mikrogramů tozinameranu a 15 mikrogramů riltozinameranu.
3. SEZNAM POMOCNÝCH LÁTEKPomocné látky: ALC-0315, ALC-0159, kolfosceryl-stearát, cholesterol, trometamol, trometamol-
hydrochlorid, sacharosa, voda pro injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍInjekční disperze
10 vícedávkových injekčních lahviček
195 vícedávkových injekčních lahviček
5. ZPŮSOB A CESTA/CESTY PODÁNÍIntramuskulární podání.
Před použitím neřeďte.
Více informací je k dispozici po sejmutí QR kódu.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁNMIMO DOHLED A DOSAH DĚTÍUchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ8. POUŽITELNOSTEXP (při -90 °C až -60 °C)
Použitelnost při teplotě 2 °C až 8 °C: …………………..
(Maximálně 10 týdnů. Přeškrtněte dřívější datum použitelnosti.)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávání:
Po přijetí uchovávejte při teplotě 2 °C až 8 °C. Po rozmrazení znovu nezmrazujte. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Před použitím a pro další informace o uchovávání si přečtěte příbalovou informaci.
Po prvním vpichu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮNEBO ODPADU Z NICH, POKUD JE TO VHODNÉ11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLAEU/1/20/1528/006 10 vícedávkových injekčních lahviček
EU/1/20/1528/007 195 vícedávkových injekčních lahviček
13. ČÍSLO ŠARŽELot
14. KLASIFIKACE PRO VÝDEJ15. NÁVOD K POUŽITÍ16. INFORMACE V BRAILLOVĚ PÍSMUNevyžaduje se – odůvodnění přijato.
17. JEDINEČNÝ IDENTIFIKÁTOR – 2D ČÁROVÝ KÓD2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR – DATA ČITELNÁ OKEMPC
SN NN
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU
ŠTÍTEK NA INJEKČNÍ LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
COMIRNATY Original/Omicron BA.1 15/15 μg injekce
mRNA vakcína proti onemocnění COVID-19
tozinameran/riltozinameran i.m.
2. ZPŮSOB PODÁNÍNeřeďte
3. POUŽITELNOSTEXP
4. ČÍSLO ŠARŽELot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET6 dávek po 15/15 mikrogramech
6. JINÉČas likvidace:
ÚDA
JE UVÁDĚNÉ NA VNĚJŠÍM OBALU
K
RA
B
I
Č
K
A
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
COMIRNATY Original/Omicron BA.4-5 (15/15 mikrogramů)/dávku injekční disperze
Dospělí a dospívající od 12 let
mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran/famtozinameran
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEKJedna dávka obsahuje 15 mikrogramů tozinameranu a 15 mikrogramů famtozinameranu.
Jednodávkové injekční lahvičky
Jedna injekční lahvička obsahuje 1 dávku 0,3 ml.
Vícedávkové injekční lahvičky
Jedna injekční lahvička obsahuje 6 dávek po 0,3 ml.
3. SEZNAM POMOCNÝCH LÁTEKPomocné látky: ALC-0315, ALC-0159, kolfosceryl-stearát, cholesterol, trometamol, trometamol-
hydrochlorid, sacharosa, voda pro injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍInjekční disperze
Jednodávkové injekční lahvičky
10 jednodávkových injekčních lahviček
Vícedávkové injekční lahvičky
10 vícedávkových injekčních lahviček
195 vícedávkových injekčních lahviček
5. ZPŮSOB A CESTA/CESTY PODÁNÍIntramuskulární podání.
Před použitím neřeďte.

Více informací je k dispozici po sejmutí QR kódu. URL:
www.comirnatyglobal.com6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁNMIMO DOHLED A DOSAH DĚTÍUchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ8. POUŽITELNOSTEXP (při -90 °C až -60 °C)
Použitelnost při teplotě 2 °C až 8 °C: …………………..
(Maximálně 10 týdnů. Přeškrtněte dřívější datum použitelnosti.)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍUchovávání:
Po přijetí uchovávejte při teplotě 2 °C až 8 °C. Po rozmrazení znovu nezmrazujte. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Před použitím a pro další informace o uchovávání si přečtěte příbalovou informaci.
Vícedávkové injekční lahvičky
Po prvním vpichu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮNEBO ODPADU Z NICH, POKUD JE TO VHODNÉ11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLAJednodávkové injekční lahvičky
EU/1/20/1528/014
Vícedávkové injekční lahvičky
|
|
EU/1/20/1528/008
| 10 vícedávkových injekčních lahviček
|
|
EU/1/20/1528/009 195 vícedávkových injekčních lahviček
|
| | | |
13. ČÍSLO ŠARŽELot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se – odůvodnění přijato.
17. JEDINEČNÝ IDENTIFIKÁTOR – 2D ČÁROVÝ KÓD2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR – DATA ČITELNÁ OKEMPC
SN NN
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU
ŠTÍTEK NA INJEKČNÍ LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
COMIRNATY Original/Omicron BA.4-5 15/15 μg injekce
mRNA vakcína proti onemocnění COVID-19
tozinameran/famtozinameran i.m.
2. ZPŮSOB PODÁNÍNeřeďte
3. POUŽITELNOSTEXP
4. ČÍSLO ŠARŽELot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČETJednodávkové injekční lahvičky
1 dávka
Vícedávkové injekční lahvičky
6 dávek po 15/15 mikrogramech
6. JINÉVícedávkové injekční lahvičky
Čas likvidace:
ÚDA
JE UVÁDĚNÉ NA VNĚJŠÍM OBALU
K
RA
B
I
Č
K
A
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
COMIRNATY Original/Omicron BA.4-5 (5/5 mikrogramů)/dávku koncentrát pro injekční disperzi
Děti ve věku 5 až 11 let
mRNA vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran/famtozinameran
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEKJedna injekční lahvička obsahuje po naředění 10 dávek po 0,2 ml.
Jedna dávka obsahuje 5 mikrogramů tozinameranu a 5 mikrogramů famtozinameranu.
3. SEZNAM POMOCNÝCH LÁTEKPomocné látky: ALC-0315, ALC-0159, kolfosceryl-stearát, cholesterol, trometamol, trometamol-
hydrochlorid, sacharosa, voda pro injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍKoncentrát pro injekční disperzi
10 vícedávkových injekčních stříkaček
195 vícedávkových injekčních lahviček
5. ZPŮSOB A CESTA/CESTY PODÁNÍIntramuskulární podání po naředění.
Před použitím si přečtěte příbalovou informaci a další informace o uchovávání.
Více informací je k dispozici po sejmutí QR kódu.
Před použitím nařeďte: Jednu injekční lahvičku nařeďte 1,3 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%).
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁNMIMO DOHLED A DOSAH DĚTÍUchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ8. POUŽITELNOSTEXP (při -90 °C až -60 °C)
Použitelnost při teplotě 2 °C až 8 °C: …………………..
(Maximálně 10 týdnů. Přeškrtněte dřívější datum použitelnosti.)
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍUchovávání:
Po přijetí uchovávejte při teplotě 2 °C až 8 °C. Po rozmrazení znovu nezmrazujte. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Po naředění vakcínu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮNEBO ODPADU Z NICH, POKUD JE TO VHODNÉ11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACIBioNTech Manufacturing GmbH
An der Goldgrube 12
55131 Mainz, Německo
12. REGISTRAČNÍ ČÍSLO/ČÍSLAEU/1/20/1528/011 10 vícedávkových injekčních lahviček
EU/1/20/1528/012 195 vícedávkových injekčních lahviček
13. ČÍSLO ŠARŽELot
14. KLASIFIKACE PRO VÝDEJ15. NÁVOD K POUŽITÍ16. INFORMACE V BRAILLOVĚ PÍSMUNevyžaduje se – odůvodnění přijato.
17. JEDINEČNÝ IDENTIFIKÁTOR – 2D ČÁROVÝ KÓD2D čárový kód s jedinečným identifikátorem.
18. JEDINEČNÝ IDENTIFIKÁTOR – DATA ČITELNÁ OKEMPC
SN NN
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU
ŠTÍTEK NA INJEKČNÍ LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
COMIRNATY Original/Omicron BA.4-5 5/5 μg sterilní koncentrát
mRNA vakcína proti onemocnění COVID-19
tozinameran/famtozinameran i.m.
2. ZPŮSOB PODÁNÍ3. POUŽITELNOSTEXP
4. ČÍSLO ŠARŽELot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET10 dávek po 5/5 mikrogramech po naředění
6. JINÉČas likvidace:
B
. PŘÍBALOVÁ INFORMACE
P
ř
í
balová informace: informace pro uživatele
C
omirnaty 30 mikrogramů/dávku koncentrát pro injekční disperzi
D
ospělí a dospívající od 12 let
m
RN
A vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou.
Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tato vakcína podána, protože obsahuje pro Vás důležité údaje.• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci1. Co je vakcína Comirnaty a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude vakcína Comirnaty podána
3. Jak se vakcína Comirnaty podává
4. Možné nežádoucí účinky
5. Jak vakcínu Comirnaty uchovávat
6. Obsah balení a další informace
1. Co je vakcína Comirnaty a k čemu se používáComirnaty je vakcína používaná pro prevenci onemocnění COVID-19, které způsobuje SARS-CoV-2. Vakcína Comirnaty 30 mikrogramů/dávku koncentrát pro injekční disperzi se podává dospělým
a dospívajícím ve věku 12 let a starším.
Vakcína způsobuje, že imunitní systém (přirozená obrana těla) vytváří protilátky a krevní buňky, které působí proti viru, a tím chrání před onemocněním COVID-19.
Jelikož vakcína Comirnaty neobsahuje virus k vyvolání imunity, nemůže u Vás vyvolat onemocnění
COVID-19.
2. Čemu musíte věnovat pozornost, než Vám bude vakcína Comirnaty podánaVakcína Comirnaty Vám nesmí být podána• jestliže jste alergický(á) na léčivou látku nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Upozornění a opatřeníPřed podáním vakcíny se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou, jestliže:
• jste někdy měl(a) závažnou alergickou reakci nebo potíže s dýcháním po jakékoli jiné vakcíně
podané v injekci nebo poté, co Vám byla v minulosti podána vakcína Comirnaty,
• jste v souvislosti s očkováním nervózní nebo jste někdy po jakékoli injekci omdlel(a),
• máte závažné onemocnění nebo infekci s vysokou horečkou. Avšak očkování můžete podstoupit, pokud máte mírně zvýšenou teplotu nebo lehkou infekci horních cest dýchacích, jako je nachlazení,
• máte krvácivé potíže, lehce se Vám tvoří modřiny nebo užíváte lék, který zabraňuje srážení
krve,
• máte oslabený imunitní systém, kvůli onemocnění, jako je infekce virem HIV, nebo užíváte lék,
jako je kortikosteroid, který imunitní systém ovlivňuje.
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy (zánětu srdečního svalu) a perikarditidy (zánětu osrdečníku (perikardu)) (viz bod 4). Tato onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhém očkování a častěji u mladších mužů a chlapců. Riziko myokarditidy a perikarditidy je pravděpodobně nižší u dětí ve věku od 5 do 11 let v porovnání s dospívajícími ve věku od 12 do
17 let. Po očkování u Vašeho dítěte pozorně sledujte příznaky myokarditidy a perikarditidy, například dušnost, silný tlukot srdce a bolest na hrudi, a pokud se u Vás vyskytnou, ihned vyhledejte lékařskou pomoc.
Podobně jako u jiných vakcín je možné, že vakcína Comirnaty nebude plně chránit všechny osoby,
kterým bude vakcína podána a není známo, jak dlouho budou chráněny.
Můžete dostat posilovací dávku vakcíny Comirnaty. I po posilovací dávce může být účinnost vakcíny Comirnaty nižší u osob s oslabenou imunitou. V těchto případech byste měl(a) i nadále dodržovat preventivní opatření, která pomáhají předcházet onemocnění COVID-19. Kromě toho by měly být podle potřeby očkovány i Vaše blízké kontakty. Proberte příslušná individuální doporučení se svým lékařem.
Děti
Vakcína Comirnaty 30 mikrogramů/dávku koncentrát pro injekční disperzi není určena pro děti ve
věku do 12 let.
Pro kojence a děti ve věku 6 měsíců až 4 roky je k dispozici léková forma pro děti. Podrobnosti naleznete v příbalové informaci vakcíny Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi.
Pro děti ve věku 5 až 11 let (tj. 5 až méně než 12 let) je k dispozici léková forma pro děti. Podrobnosti naleznete v příbalové informaci vakcíny Comirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi.
Vakcína Comirnaty není určena pro kojence mladší 6 měsíců.
Další léčivé přípravky a vakcína Comirnaty
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době
užíval(a) nebo které možná budete užívat, a o tom, zda Vám byla v nedávné době podána jiná vakcína.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem, zdravotní sestrou nebo lékárníkem dříve, než Vám bude tato vakcína podána.
Vakcínu Comirnaty lze podávat během těhotenství. Velké množství informací o těhotných ženách očkovaných vakcínou Comirnaty během druhého a třetího trimestru neprokázalo negativní účinky na těhotenství nebo novorozence. Informace o účincích na těhotenství nebo novorozence po očkování během prvního trimestru jsou sice omezené, ale nebylo pozorováno zvýšení rizika potratu.
Vakcínu Comirnaty lze podávat během kojení.
Řízení dopravních prostředků a obsluha strojů
Některé účinky očkování uvedené v bodě 4 (Možné nežádoucí účinky) mohou dočasně ovlivnit Vaši
schopnost řídit nebo obsluhovat stroje. Před řízením nebo obsluhou strojů počkejte, dokud tyto účinky
neodezní.
V
akcína Comirnaty obsahuje draslík a sodík
Tato vakcína obsahuje méně než 1 mmol (39 mg) draslíku v jedné dávce, to znamená, že je v podstatě
„bez draslíku“.
Tato vakcína obsahuje méně než 1 mmol (23 mg) sodíku v jedné dávce, to znamená, že je v podstatě
„bez sodíku“.
3. Jak se vakcína Comirnaty podává
Vakcína Comirnaty se podává po naředění jako injekce o objemu 0,3 ml do svalu v horní části paže.
Budou Vám podány 2 injekce.
Doporučuje se, abyste druhou dávku téže vakcíny dostal(a) 3 týdny po první dávce pro dokončení očkovacího cyklu.
Pokud máte oslabenou imunitu, můžete dostat třetí dávku vakcíny Comirnaty nejméně 28 dní po druhé dávce.
Posilovací dávka vakcíny Comirnaty smí být podána nejdříve 3 měsíce po poslední podané dávce
vakcíny proti onemocnění COVID-19 u osob ve věku 12 let a starších.
O způsobilosti a načasování posilovací dávky se prosím poraďte se svým zdravotnickým pracovníkem.
Máte-li jakékoli další otázky týkající se používání vakcíny Comirnaty, zeptejte se svého lékaře,
lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny vakcíny může mít i vakcína Comirnaty nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Nežádoucí účinky se mohou vyskytnout s následující četností:
Velmi časté nežádoucí účinky: mohou postihnout více než 1 z 10 lidí
• v místě injekce: bolest, zduření
• únava
• bolest hlavy
• bolest svalů
• zimnice
• bolest kloubů
• průjem
• horečka
Některé z těchto nežádoucích účinků se o něco častěji vyskytovaly u dospívajících ve věku 12 až
15 let než u dospělých.
Časté nežádoucí účinky: mohou postihnout až 1 z 10 lidí
• zarudnutí v místě injekce
• pocit na zvracení
• zvracení
Méně časté nežádoucí účinky: mohou postihnout až 1 ze 100 lidí
• zvětšené mízní uzliny (častěji pozorované po posilovací dávce)
• malátnost
• bolest paže
• nespavost
• svědění v místě injekce
• alergické reakce, jako je vyrážka nebo svědění
• pocit slabosti nebo nedostatku energie/ospalost
• snížená chuť k jídlu
• nadměrné pocení
• noční pocení
Vzácné nežádoucí účinky: mohou postihnout až 1 z 1 000 lidí
• dočasná jednostranná obrna lícního nervu
• alergické reakce, jako je kopřivka nebo otok obličeje
Velmi vzácné nežádoucí účinky: mohou postihnout až 1 z 10 000 lidí
• zánět srdečního svalu (myokarditida) nebo zánět osrdečníku (perikarditida), který může vést
k dušnosti, silnému tlukotu srdce nebo bolesti na hrudi
Není známo (z dostupných údajů nelze určit)
• závažná alergická reakce
• rozsáhlý otok končetiny, do které byla vakcína podána
• otok obličeje (otok obličeje se může objevit u pacientů, kterým byly do obličeje aplikovány
injekce kožních výplní)
• kožní reakce způsobující červené skvrny nebo fleky na kůži, které mohou vypadat jako terč (nebo střed terče) s tmavě červeným středem, který je obklopen kruhy světlejší červené barvy (erythema multiforme)
• neobvyklý pocit na kůži, jako je brnění nebo pocit šimrání (parestezie)
• snížený pocit nebo citlivost, zejména v kůži (hypestezie)
• silné menstruační krvácení (většina případů se zdála být nezávažné a dočasné povahy)
Hlášení nežádoucích účinkůPokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo

zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného Dodatku V. Uveďte přitom číslo šarže, je-li
k dispozici. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak vakcínu Comirnaty uchovávatUchovávejte tento přípravek mimo dohled a dosah dětí.
Následující informace o uchovávání, době použitelnosti a použití a manipulaci jsou určeny pro
zdravotnické pracovníky.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C. Neotevřené injekční lahvičky mohou být uchovávány a transportovány při teplotě -25 °C až -15 °C po jednotlivou dobu až 2 týdny a mohou být vráceny do prostoru pro uchovávání při teplotě -90 °C až -60 °C, nesmí být překročeno vytištěné datum použitelnosti (EXP).
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
195 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 3 hodin nebo mohou být jednotlivé
injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Přenesení zmrazených injekčních lahviček uchovávaných při velmi nízké teplotě (< -60 °C)
• Zásobníkyinjekčníchlahvičekuzavřenévíkem obsahující 195 injekčních lahviček, které byly vyjmuty z prostoru pro uchovávání při velmi nízké teplotě ve zmrazeném stavu (< -60 °C), smí být uchovávány maximálně 5 minut při teplotě až 25 °C.
• Zásobníkyinjekčníchlahvičeksotevřenýmvíkem nebo zásobníky injekčních lahviček obsahující méně než 195 injekčních lahviček, které byly vyjmuty z prostoru pro uchovávání při velmi nízké teplotě ve zmrazeném stavu (< -60 °C), smí být uchovávány maximálně 3 minuty při teplotě až 25 °C.
• Poté, co jsou zásobníky s injekčními lahvičkami vráceny do prostoru pro uchovávání ve zmrazeném stavu po vystavení teplotě až 25 °C, musí zůstat ve zmrazeném stavu po dobu nejméně 2 hodin než je lze znovu vyjmout.
Přenesení zmrazených injekčních lahviček uchovávaných při teplotě -25 °C až -15 °C
• Zásobníkyinjekčníchlahvičekuzavřené víkem obsahující 195 injekčních lahviček, které byly vyjmuty z prostoru pro uchovávání ve zmrazeném stavu (-25 °C až -15 °C), smí být uchovávány maximálně 3 minuty při teplotě až 25 °C.
• Zásobníkyinjekčníchlahvičeksotevřenýmvíkem nebo zásobníky injekčních lahviček obsahující méně než 195 injekčních lahviček, které byly vyjmuty z prostoru pro uchovávání ve zmrazeném stavu (-25 °C až -15 °C), smí být uchovávány maximálně 1 minutu při teplotě až
25 °C.
Jakmile je injekční lahvička vyjmuta ze zásobníku na injekční lahvičky, má být rozmrazena pro
použití.
Po rozmrazení se má vakcína naředit a okamžitě použít. Údaje o stabilitě v době používání prokázaly, že po vyjmutí z mrazáku lze neředěnou vakcínu před použitím uchovávat po dobu až 1 měsíce při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP). Během doby použitelnosti trvající 1 měsíc při teplotě 2 °C až 8 °C lze přípravek transportovat až po dobu 48 hodin. Neotevřenou vakcínu lze před použitím uchovávat po dobu až 2 hodin při teplotách do 30 °C.
S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Po naředění vakcínu uchovávejte a transportujte při teplotě 2 °C až 30 °C a použijte ji do 6 hodin.
Případnou nepoužitou vakcínu zlikvidujte.
Po jednorázovém vyjmutí z mrazničky a naředění se mají injekční lahvičky označit novým datem
a časem pro likvidaci. Po jednorázovém rozmrazení se vakcína nesmí znovu zmrazit.
Vakcínu nepoužívejte, pokud si všimnete, že jsou v naředěném roztoku částice nebo že roztok změnil
barvu.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co vakcína Comirnaty obsahuje
• Léčivou látkou je mRNA vakcína proti onemocnění COVID-19 označená tozinameran. Po naředění injekční lahvička obsahuje 6 dávek, jedna dávka 0,3 ml obsahuje 30 mikrogramů tozinameranu.
• Dalšími složkami jsou:
- ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
- 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
- kolfosceryl-stearát
- cholesterol
- chlorid draselný
- dihydrogenfosforečnan draselný
- chlorid sodný
- dihydrát hydrogenfosforečnanu sodného
- sacharosa
- voda pro injekci
- hydroxid sodný (pro úpravu pH)
- kyselina chlorovodíková (pro úpravu pH)
Jak vakcína Comirnaty vypadá a co obsahuje toto baleníVakcína je bílá až téměř bílá disperze (pH: 6,9–7,9), která se dodává ve vícedávkové čiré injekční
lahvičce (sklo třídy I) o objemu 2 ml obsahující 6 dávek. Injekční lahvička je uzavřena pryžovou
zátkou a odtrhovacím fialovým plastovým víčkem s hliníkovým krytem.
Velikost balení: 195 injekčních lahviček
Držitel rozhodnutí o registraci BioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.deVýrobciBioNTech Manufacturing GmbH Kupferbergterrasse 17-19
55116 Mainz
Německo
Pfizer Manufacturing Belgium NV Rijksweg 12
2870 Puurs
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
B
e
l
gië/Belgique/Belgien
L
uxembourg/Luxemburg
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +370 52 51 4000
Б
ълг
ария
Пфайзер Люксембург САРЛ, Клон
България
Teл: +359 2 970 4333
Magyarország
Pfizer Kft
Tel: +36 1 488 3700
Č
eská republika
Pfizer, spol. s r.o.
Tel: +420 283 004 111
Malta
Vivian Corporation Ltd.
Tel: +35621 344610
D
anmark
Pfizer ApS
Tlf: +45 44 201 100
Norge
Pfizer AS
Tlf: +47 67 526 100
D
eutschland
BioNTech Manufacturing GmbH Tel: +49 6131 90840
Nederland
Pfizer BV
Tel: +31 (0)10 406 43 01
E
esti
Pfizer Luxembourg SARL Eesti filiaal
Tel: +372 666 7500
Österreich
Pfizer Corporation Austria Ges.m.b.H
Tel: +43 (0)1 521 15-0
Ε
λλάδα
Pfizer Ελλάς A.E.
Τηλ.: +30 210 6785 800
Polska
Pfizer Polska Sp. z o.o. Tel.: +48 22 335 61 00
E
s
paña
Pfizer, S.L.
Tél: +34914909900
Portugal
Laboratórios Pfizer, Lda.
Tel: +351 21 423 5500
F
rance
Pfizer
Tél +33 1 58 07 34 40
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
H
rvatska
Pfizer Croatia d.o.o. Tel: +385 1 3908 777
Slovenija
Pfizer Luxembourg SARL
Pfizer, podružnica za svetovanje s področja
farmacevtske dejavnosti, Ljubljana
Tel.: +386 (0) 1 52 11 400
Ireland
Pfizer Healthcare Ireland
Tel: 1800 633 363 (toll free)
+44 (0)1304 616161
Slovenská republika
Pfizer Luxembourg SARL,
organizačná zložka
Tel: +421 2 3355 5500
Ísland
Icepharma hf
Simi: +354 540 8000
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
It
alia
Pfizer S.r.l.
Tel: +39 06 33 18 21
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
Κύ
πρ
ος
Pfizer Ελλάς Α.Ε. (Cyprus Branch) Tηλ: +357 22 817690
United Kingdom (Northern Ireland)
Pfizer Limited
Tel: +44 (0) 1304 616161
L
atvija
Pfizer Luxembourg SARL filiāle Latvijā
Tel.: +371 670 35 775
Tato příbalová informace byla naposledy revidována
Pro zobrazení příbalové informace v různých jazycích sejměte následující kód pomocí chytrého zařízení.

URL:
www.comirnatyglobal.comPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
------------------------------------------------------------------------------------------------------------------------
Následující informace jsou určeny pouze pro zdravotnické pracovníky:Podávejte vakcínu Comirnaty intramuskulárně po naředění jako základní očkování 2 dávkami (každá po 0,3 ml) s odstupem 3 týdnů.
Pokud má pacient oslabenou imunitu, může dostat třetí dávku vakcíny Comirnaty nejméně 28 dní po druhé dávce.
Posilovací dávka vakcíny Comirnaty (0,3 ml) smí být podána nejdříve 3 měsíce po poslední podané
dávce vakcíny proti onemocnění COVID-19 u osob ve věku 12 let a starších.
SledovatelnostAby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Instrukce pro zacházení s vakcínouVakcína Comirnaty má být připravována zdravotnickým pracovníkem pomocí aseptické techniky, aby
byla zajištěna sterilita připravené disperze.
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY
30 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (12 LET A STARŠÍ)
 F
i
alové víčko
P
o naředění
F
i
alové víčko
P
o naředění
• Zkontrolujte, zda má injekční lahvička fialové plastové víčko.
• Pokud má injekční lahvička šedé
plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku injekční disperze,
Comirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční disperze nebo Comirnaty Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze.
• Pokud má injekční lahvička oranžové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi nebo Comirnaty
Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi.

R
O
Z
MRAZENÍ VAKCÍNY COMIRNATY 30 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (12 LET A STARŠÍ) PŘED ŘEDĚNÍM
|
Ne déle než
2 hodiny při pokojové teplotě
(
d
o 30 °C).
|
• Vícedávková injekční lahvička se uchovává zmrazená a před ředěním se musí rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se rozmrazily. Rozmrazení balení 195 injekčních lahviček může trvat 3 hodiny. Alternativně lze zmrazené injekční lahvičky rozmrazovat po dobu 30 minut při teplotě do 30 °C pro okamžité použití.
• Neotevřenou injekční lahvičku lze
uchovávat až 1 měsíc při teplotě 2 °C až
8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP). Během doby použitelnosti trvající 1 měsíc při teplotě
2 °C až 8 °C lze přípravek transportovat
až po dobu 48 hodin.
• Rozmrazenou injekční lahvičku nechejte dosáhnout pokojové teploty. Neotevřenou injekční lahvičku lze před použitím uchovávat až 2 hodiny při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
• Před ředěním injekční lahvičku 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Před naředěním může rozmrazená disperze obsahovat bílé až téměř bílé neprůhledné amorfní částice.
|
ŘEDĚN
Í VAKCÍNY COMIRNATY 30 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (12 LET A STARŠÍ)
|
1,8 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9 %).
|
• Rozmrazená vakcína se musí naředit v původní injekční lahvičce 1,8 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%) za použití jehly
o velikosti 21 gauge (21G) nebo užší a uplatnění aseptických metod.
|
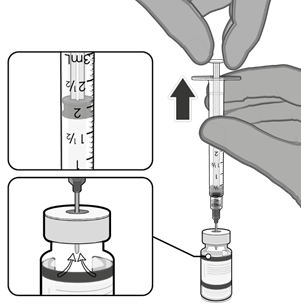
V
ytáhněte píst na značku 1,8 ml pro
odstranění vzduchu z injekční lahvičky.
|
• Nasátím 1,8 ml vzduchu do prázdné stříkačky na rozpouštědlo vyrovnejte před vyjmutím jehly z injekční lahvičky tlak v injekční lahvičce.
|
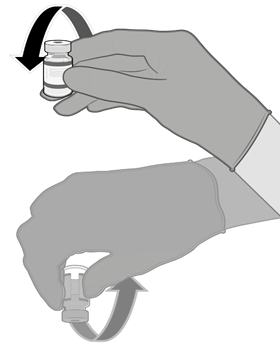
Jemně x 10
|
• Disperzi 10krát jemně převraťte.
Lahvičkou netřepejte.
• Naředěná vakcína se má jevit jako téměř bílá disperze bez viditelných částic. Jestliže jsou v naředěné vakcíně patrné částice nebo vakcína změnila barvu, nepoužívejte ji.
|
Č
as likvidace
Z
a
z
namenejte příslušné datum a čas.
P
oužijte do 6 hodin po naředění.
|
• Injekční lahvičky s naředěnou vakcínou je třeba označit příslušným datem
a časem.
• Po naředění uchovávejte vakcínu při teplotě 2 °C až 30 °C a použijte ji během
6 hodin, včetně doby transportu.
• Naředěnou disperzi nezmrazujte ani s ní netřepejte. Pokud je v chladu, nechte naředěnou disperzi před použitím dosáhnout pokojové teploty.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,3 ml VAKCÍNY COMIRNATY
30 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (12 LET A STARŠÍ)
|
N
aředěná vakcína o objemu 0,3 ml
|
• Po naředění obsahuje injekční lahvička
2,25 ml, ze kterých lze získat 6 dávek po
0,3 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím
antiseptického tampónu.
• Natáhněte 0,3 ml vakcíny Comirnaty.
K získání šesti dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční stříkačky
a jehly s malým mrtvým prostorem má
mít mrtvý objem maximálně
35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání šesté dávky z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Veškerou vakcínu, jež nebyla použita do
6 hodin po naředění, zlikvidujte.
|
L
i
kvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
P
ř
í
balová informace: informace pro uživatele
C
omirnaty 30 mikrogramů/dávku injekční disperze
D
ospělí a dospívající od 12 let
m
RN
A vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou.
Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tato vakcína podána,protože obsahuje pro Vás důležité údaje.• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci1. Co je vakcína Comirnaty a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude vakcína Comirnaty podána
3. Jak se vakcína Comirnaty podává
4. Možné nežádoucí účinky
5. Jak vakcínu Comirnaty uchovávat
6. Obsah balení a další informace
1. Co je vakcína Comirnaty a k čemu se používáComirnaty je vakcína používaná pro prevenci onemocnění COVID-19, které způsobuje SARS-CoV-2. Vakcína Comirnaty 30 mikrogramů/dávku injekční disperze se podává dospělým a dospívajícím ve
věku 12 let a starším.
Vakcína způsobuje, že imunitní systém (přirozená obrana těla) vytváří protilátky a krevní buňky, které působí proti viru, a tím chrání před onemocněním COVID-19.
Jelikož vakcína Comirnaty neobsahuje virus k vyvolání imunity, nemůže u Vás vyvolat onemocnění
COVID-19.
2. Čemu musíte věnovat pozornost, než Vám bude vakcína Comirnaty podánaVakcína Comirnaty Vám nesmí být podána• jestliže jste alergický(á) na léčivou látku nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Upozornění a opatřeníPřed podáním vakcíny se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou, jestliže:
• jste někdy měl(a) závažnou alergickou reakci nebo potíže s dýcháním po jakékoli jiné vakcíně
podané v injekci nebo poté, co Vám byla v minulosti podána vakcína Comirnaty,
• jste v souvislosti s očkováním nervózní nebo jste někdy po jakékoli injekci omdlel(a),
• máte závažné onemocnění nebo infekci s vysokou horečkou. Avšak očkování můžete podstoupit, pokud máte mírně zvýšenou teplotu nebo lehkou infekci horních cest dýchacích, jako je nachlazení,
• máte krvácivé potíže, lehce se Vám tvoří modřiny nebo užíváte lék, který zabraňuje srážení
krve,
• máte oslabený imunitní systém, kvůli onemocnění, jako je infekce virem HIV, nebo užíváte lék, jako je kortikosteroid, který imunitní systém ovlivňuje.
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy (zánětu srdečního svalu) a perikarditidy (zánětu osrdečníku (perikardu)) (viz bod 4). Tato onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhém očkování a častěji u mladších mužů a chlapců. Riziko myokarditidy a perikarditidy je pravděpodobně nižší u dětí ve věku od 5 do 11 let v porovnání s dospívajícími ve věku od 12 do
17 let. Po očkování u Vašeho dítěte pozorně sledujte příznaky myokarditidy a perikarditidy, například dušnost, silný tlukot srdce a bolest na hrudi, a pokud se u Vás vyskytnou, ihned vyhledejte lékařskou pomoc.
Podobně jako u jiných vakcín je možné, že vakcína Comirnaty nebude plně chránit všechny osoby, kterým bude vakcína podána a není známo, jak dlouho budou chráněny.
Můžete dostat posilovací dávku vakcíny Comirnaty. I po posilovací dávce může být účinnost vakcíny Comirnaty nižší u osob s oslabenou imunitou. V těchto případech byste měl(a) i nadále dodržovat preventivní opatření, která pomáhají předcházet onemocnění COVID-19. Kromě toho by měly být podle potřeby očkovány i Vaše blízké kontakty. Proberte příslušná individuální doporučení se svým lékařem.
Děti
Vakcína Comirnaty 30 mikrogramů/dávku injekční disperze není určena pro děti ve věku do 12 let.
Pro kojence a děti ve věku 6 měsíců až 4 roky je k dispozici léková forma pro děti. Podrobnosti naleznete v příbalové informaci vakcíny Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi.
Pro děti ve věku 5 až 11 let (tj. 5 až méně než 12 let) je k dispozici léková forma pro děti. Podrobnosti naleznete v příbalové informaci vakcíny Comirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi.
Vakcína Comirnaty není určena pro kojence mladší 6 měsíců.
Další léčivé přípravky a vakcína Comirnaty
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době
užíval(a) nebo které možná budete užívat, a o tom, zda Vám byla v nedávné době podána jiná vakcína.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte
se se svým lékařem, zdravotní sestrou nebo lékárníkem dříve, než Vám bude tato vakcína podána.
Vakcínu Comirnaty lze podávat během těhotenství. Velké množství informací o těhotných ženách očkovaných vakcínou Comirnaty během druhého a třetího trimestru neprokázalo negativní účinky na těhotenství nebo novorozence. Informace o účincích na těhotenství nebo novorozence po očkování během prvního trimestru jsou sice omezené, ale nebylo pozorováno zvýšení rizika potratu.
Vakcínu Comirnaty lze podávat během kojení.
Řízení dopravních prostředků a obsluha strojů
Některé účinky očkování uvedené v bodě 4 (Možné nežádoucí účinky) mohou dočasně ovlivnit Vaši schopnost řídit nebo obsluhovat stroje. Před řízením nebo obsluhou strojů počkejte, dokud tyto účinky
neodezní.
3. Jak se vakcína Comirnaty podává
Vakcína Comirnaty se podává jako injekce o objemu 0,3 ml do svalu v horní části paže.
Budou Vám podány 2 injekce.
Doporučuje se, abyste druhou dávku téže vakcíny dostal(a) 3 týdny po první dávce pro dokončení základního očkování.
Pokud máte oslabenou imunitu, můžete dostat třetí dávku vakcíny Comirnaty nejméně 28 dní po druhé dávce.
Posilovací dávka vakcíny Comirnaty smí být podána nejdříve 3 měsíce po poslední podané dávce
vakcíny proti onemocnění COVID-19 u osob ve věku 12 let a starších.
O způsobilosti a načasování posilovací dávky se prosím poraďte se svým zdravotnickým pracovníkem.
Máte-li jakékoli další otázky týkající se používání vakcíny Comirnaty, zeptejte se svého lékaře,
lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny vakcíny může mít i vakcína Comirnaty nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Nežádoucí účinky se mohou vyskytnout s následující četností:
Velmi časté nežádoucí účinky: mohou postihnout více než 1 z 10 lidí
• v místě injekce: bolest, zduření
• únava
• bolest hlavy
• bolest svalů
• zimnice
• bolest kloubů
• průjem
• horečka
Některé z těchto nežádoucích účinků se o něco častěji vyskytovaly u dospívajících ve věku 12 až
15 let než u dospělých.
Časté nežádoucí účinky: mohou postihnout až 1 z 10 lidí
• zarudnutí v místě injekce
• pocit na zvracení
• zvracení
Méně časté nežádoucí účinky: mohou postihnout až 1 ze 100 lidí
• zvětšené mízní uzliny (častěji pozorované po posilovací dávce)
• malátnost
• bolest paže
• nespavost
• svědění v místě injekce
• alergické reakce, jako je vyrážka nebo svědění
• pocit slabosti nebo nedostatku energie/ospalost
• snížená chuť k jídlu
• nadměrné pocení
• noční pocení
Vzácné nežádoucí účinky: mohou postihnout až 1 z 1 000 lidí
• dočasná jednostranná obrna lícního nervu
• alergické reakce, jako je kopřivka nebo otok obličeje
Velmi vzácné nežádoucí účinky: mohou postihnout až 1 z 10 000 lidí
• zánět srdečního svalu (myokarditida) nebo zánět osrdečníku (perikarditida), který může vést
k dušnosti, silnému tlukotu srdce nebo bolesti na hrudi
Není známo (z dostupných údajů nelze určit)
• závažná alergická reakce
• rozsáhlý otok končetiny, do které byla vakcína podána
• otok obličeje (otok obličeje se může objevit u pacientů, kterým byly do obličeje aplikovány
injekce kožních výplní)
• kožní reakce způsobující červené skvrny nebo fleky na kůži, které mohou vypadat jako terč (nebo střed terče) s tmavě červeným středem, který je obklopen kruhy světlejší červené barvy (erythema multiforme)
• neobvyklý pocit na kůži, jako je brnění nebo pocit šimrání (parestezie)
• snížený pocit nebo citlivost, zejména v kůži (hypestezie)
• silné menstruační krvácení (většina případů se zdála být nezávažné a dočasné povahy)
Hlášení nežádoucích účinkůPokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo
zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného Dodatku V. Uveďte přitom číslo šarže, je-li
k dispozici. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti
tohoto přípravku.
5. Jak vakcínu Comirnaty uchovávatUchovávejte tento přípravek mimo dohled a dosah dětí.
Následující informace o uchovávání, době použitelnosti a použití a manipulaci jsou určeny pro
zdravotnické pracovníky.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána buď při teplotě -90 °C až -60 °C a nebo 2 °C až 8 °C.
Jednodávkové injekční lahvičky: Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C
až -60 °C, lze balení obsahující 10 jednodávkových injekčních lahviček rozmrazit při teplotě 2 °C
až 8 °C po dobu 2 hodin nebo mohou být jednotlivé injekční lahvičky rozmrazeny při pokojové teplotě
(do 30 °C) po dobu 30 minut.
Vícedávkové injekční lahvičky: Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C
až -60 °C, lze balení obsahující 10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu
6 hodin nebo mohou být jednotlivé injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Rozmrazené injekční lahvičky: Po vyjmutí z prostoru pro uchovávání ve zmrazeném stavu může být neotevřená injekční lahvička uchovávána a transportována chlazená při teplotě 2 °C až 8 °C po dobu až 10 týdnů; nesmí být překročeno vytištěné datum použitelnosti (EXP). Vnější obal má být označen novým datem likvidace při teplotě 2 °C až 8 °C. Po rozmrazení nemůže být vakcína znovu zmrazena.
Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Otevřené injekční lahvičky: Po prvním vpichu vakcínu uchovávejte při teplotě 2 °C až 30 °C
a použijte ji do 12 hodin, která zahrnuje až 6 hodin transportu. Případnou nepoužitou vakcínu
zlikvidujte.
Vakcínu nepoužívejte, pokud si všimnete částic nebo změny barvy.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co vakcína Comirnaty obsahuje
• Léčivou látkou je mRNA vakcína proti onemocnění COVID-19 označená tozinameran.
– Jednodávková injekční lahvička obsahuje 1 dávku, jedna dávka 0,3 ml obsahuje
30 mikrogramů tozinameranu.
– Vícedávková injekční lahvička obsahuje 6 dávek, jedna dávka 0,3 ml obsahuje 30 mikrogramů tozinameranu.
• Dalšími složkami jsou:
- ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
- 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
- kolfosceryl-stearát
- cholesterol
- trometamol
- trometamol-hydrochlorid
- sacharosa
- voda pro injekci
Jak vakcína Comirnaty vypadá a co obsahuje toto balení
Vakcína je bílá až téměř bílá disperze (pH: 6,9–7,9), která se dodává buď:
• v jednodávkové čiré injekční lahvičce (sklo třídy I) o objemu 2 ml obsahující 1 dávku.
Injekční lahvička je uzavřena pryžovou zátkou a šedým odtrhovacím plastovým víčkem
s hliníkovým krytem; nebo
• ve vícedávkové čiré injekční lahvičce (sklo třídy I) o objemu 2 ml obsahující 6 dávek. Injekční lahvička je uzavřena pryžovou zátkou a šedým odtrhovacím plastovým víčkem s hliníkovým krytem.
Velikost balení jednodávkových injekčních lahviček: 10 injekčních lahviček.
Velikost balení vícedávkových injekčních lahviček: 10 nebo 195 injekčních lahviček. Na trhu nemusí být všechny velikosti balení.
D
ržitel rozhodnutí o registraci BioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.deVýrobciBioNTech Manufacturing GmbH Kupferbergterrasse 17-19
55116 Mainz
Německo
Pfizer Manufacturing Belgium NV Rijksweg 12
2870 Puurs
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
B
e
l
gië/Belgique/Belgien
L
uxembourg/Luxemburg
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +370 52 51 4000
Б
ълг
ария
Пфайзер Люксембург САРЛ, Клон
България
Teл: +359 2 970 4333
Magyarország
Pfizer Kft
Tel: +36 1 488 3700
Č
eská republika
Pfizer, spol. s r.o.
Tel: +420 283 004 111
Malta
Vivian Corporation Ltd.
Tel: +35621 344610
D
anmark
Pfizer ApS
Tlf: +45 44 201 100
Norge
Pfizer AS
Tlf: +47 67 526 100
D
eutschland
BioNTech Manufacturing GmbH
Tel: +49 6131 90840
Nederland
Pfizer BV
Tel: +31 (0)10 406 43 01
E
esti
Pfizer Luxembourg SARL Eesti filiaal
Tel: +372 666 7500
Österreich
Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1 521 15-0
Ε
λλάδα
Pfizer Ελλάς A.E.
Τηλ.: +30 210 6785 800
Polska
Pfizer Polska Sp. z o.o.
Tel.: +48 22 335 61 00
E
s
paña
Pfizer, S.L.
Tél: +34914909900
Portugal
Laboratórios Pfizer, Lda.
Tel: +351 21 423 5500
F
rance
Pfizer
Tél +33 1 58 07 34 40
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
H
rvatska
Pfizer Croatia d.o.o. Tel: +385 1 3908 777
Slovenija
Pfizer Luxembourg SARL
Pfizer, podružnica za svetovanje s področja
farmacevtske dejavnosti, Ljubljana
Tel.: +386 (0) 1 52 11 400
Ireland
Pfizer Healthcare Ireland
Tel: 1800 633 363 (toll free)
+44 (0)1304 616161
Slovenská republika
Pfizer Luxembourg SARL,
organizačná zložka
Tel: +421 2 3355 5500
Ísland
Icepharma hf
Simi: +354 540 8000
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
It
alia
Pfizer S.r.l.
Tel: +39 06 33 18 21
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
Κύ
πρ
ος
Pfizer Ελλάς Α.Ε. (Cyprus Branch) Tηλ: +357 22 817690
United Kingdom (Northern Ireland)
Pfizer Limited
Tel: +44 (0) 1304 616161
L
atvija
Pfizer Luxembourg SARL filiāle Latvijā
Tel.: +371 670 35 775
Tato příbalová informace byla naposledy revidovánaPro zobrazení příbalové informace v různých jazycích sejměte následující kód pomocí chytrého zařízení.

URL:
www.comirnatyglobal.comPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
------------------------------------------------------------------------------------------------------------------------
N
ásledující informace jsou určeny pouze pro zdravotnické pracovníky:
Podávejte vakcínu Comirnaty intramuskulárně jako základní očkování 2 dávkami (každá po 0,3 ml)
s odstupem 3 týdnů.
Pokud má pacient oslabenou imunitu, může dostat třetí dávku vakcíny Comirnaty nejméně 28 dní po druhé dávce.
Posilovací dávka vakcíny Comirnaty (0,3 ml) smí být podána nejdříve 3 měsíce po poslední podané
dávce vakcíny proti onemocnění COVID-19 u osob ve věku 12 let a starších.
Sledovatelnost
Aby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Instrukce pro zacházení s vakcínou
Vakcína Comirnaty má být připravována zdravotnickým pracovníkem pomocí aseptické techniky, aby
byla zajištěna sterilita připravené disperze.
NÁV
O
D SE VZTAHUJE NA JEDNODÁVKOVÉ I VÍCEDÁVKOVÉ INJEKČNÍ LAHVIČKY
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY
30 MIKROGRAMŮ/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
• Zkontrolujte, zda má injekční lahvička
Š
e
d
é víčko
Neřeďte
šedé plastové víčko a šedý okraj kolem štítku a název přípravku je Comirnaty
30 mikrogramů/dávku injekční
disperze.
• Zkontrolujte, zda se jedná
o jednodávkovou nebo vícedávkovou injekční lahvičku, a řiďte se příslušnými pokyny pro zacházení uvedenými níže.
• Pokud má injekční lahvička šedé plastové víčko a šedý okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.1
(15/15 mikrogramů)/dávku injekční
disperze nebo Comirnaty
Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze, přečtěte si souhrn údajů
o přípravku pro příslušné složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro
injekční disperzi nebo Comirnaty
Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte
si souhrn údajů o přípravku pro vakcínu Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi.

Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY 30 MIKROGRAMŮ/DÁVKU INJEKČNÍ
D
I
SPERZE (12 LET A STARŠÍ) PŘED POUŽITÍM
|
U
chovávejte po dobu až 10 týdnů při teplotě 2 °C
až 8 °C, aktualizujte datum použitelnosti na krabičce.
|
• Pokud se jednodávková nebo vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se
rozmrazily. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
o Jednodávkové injekční lahvičky:
Rozmrazení balení
10 jednodávkových injekčních lahviček může trvat 2 hodiny.
o Vícedávkové injekční lahvičky:
Rozmrazení balení
10 vícedávkových injekčních lahviček může trvat 6 hodin.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
Jemně
× 10
|
• Injekční lahvičky před použitím 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Před smícháním může rozmrazená disperze obsahovat bílé až téměř bílé, matné amorfní částice.
• Po smíchání se má vakcína jevit jako bílá až téměř bílá disperze bez viditelných částic. Jestliže jsou
v naředěné vakcíně patrné částice nebo vakcína změnila barvu, nepoužívejte ji.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,3 ml VAKCÍNY COMIRNATY
30 MIKROGRAMŮ/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
J
ednodávkové
i
n
j
ekční
l
ahvičky
• Natáhněte jednu 0,3ml dávku vakcíny.
• Injekční lahvičku a veškerý přebytečný
objem zlikvidujte.
Vícedávkovéinjekčnílahvičky• Vícedávkové injekční lahvičky obsahují
6 dávek vakcíny po 0,3 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím antiseptického tampónu.
• Natáhněte 0,3 ml vakcíny Comirnaty.
V
akcína o objemu 0,3 ml
K získání šesti dávek z jedné injekční
lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční
stříkačky a jehly s malým mrtvým
prostorem má mít mrtvý objem
maximálně 35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání šesté dávky z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Zaznamenejte příslušné datum/čas na injekční lahvičku. Veškerou vakcínu, jež nebyla použita do 12 hodin po prvním vpichu, zlikvidujte.
L
i
kvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
P
ř
í
balová informace: informace pro uživatele
C
omirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi
D
ěti ve věku 5 až 11 let
m
RN
A vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vašeho dítěte vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tato vakcína podána, protože obsahuje pro Vás důležité údaje.• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci1. Co je vakcína Comirnaty a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vašemu dítěti bude vakcína Comirnaty podána
3. Jak se vakcína Comirnaty podává
4. Možné nežádoucí účinky
5. Jak vakcínu Comirnaty uchovávat
6. Obsah balení a další informace
1. Co je vakcína Comirnaty a k čemu se používáComirnaty je vakcína používaná pro prevenci onemocnění COVID-19, které způsobuje SARS-CoV-2. Vakcína Comirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi se podává dětem ve věku 5
až 11 let.
Vakcína způsobuje, že imunitní systém (přirozená obrana těla) vytváří protilátky a krevní buňky, které působí proti viru, a tím chrání před onemocněním COVID-19.
Jelikož vakcína Comirnaty neobsahuje virus k vyvolání imunity, nemůže u Vašeho dítěte vyvolat onemocnění COVID-19.
2. Čemu musíte věnovat pozornost, než Vašemu dítěti bude vakcína Comirnaty podánaVakcína Comirnaty Vám nesmí být podána• jestliže je Vaše dítě alergické na léčivou látku nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Upozornění a opatřeníPřed podáním vakcíny Vašemu dítěti se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou,
jestliže Vaše dítě:
• někdy mělo závažnou alergickou reakci nebo potíže s dýcháním po jakékoli jiné vakcíně podané
v injekci nebo poté, co mu byla v minulosti podána vakcína Comirnaty,
• je v souvislosti s očkováním nervózní nebo někdy po jakékoli injekci omdlelo,
• má závažné onemocnění nebo infekci s vysokou horečkou. Avšak očkování může Vaše dítě podstoupit, pokud má mírně zvýšenou teplotu nebo lehkou infekci horních cest dýchacích, jako
je nachlazení,
• má krvácivé potíže, lehce se mu tvoří modřiny nebo užívá lék, který zabraňuje srážení krve,
• má oslabený imunitní systém, kvůli onemocnění, jako je infekce virem HIV, nebo užívá lék,
jako je kortikosteroid, který imunitní systém ovlivňuje.
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy (zánětu srdečního svalu) a perikarditidy (zánětu osrdečníku (perikardu)) (viz bod 4). Tato onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhém očkování a častěji u mladších mužů a chlapců. Riziko myokarditidy a perikarditidy je pravděpodobně nižší u dětí ve věku od 5 do 11 let v porovnání s dospívajícími ve věku od 12 do
17 let. Po očkování u Vašeho dítěte pozorně sledujte příznaky myokarditidy a perikarditidy, například dušnost, silný tlukot srdce a bolest na hrudi, a pokud se u něj vyskytnou, ihned vyhledejte lékařskou pomoc.
Podobně jako u jiných vakcín je možné, že vakcína Comirnaty nebude plně chránit všechny osoby,
kterým bude vakcína podána a není známo, jak dlouho budou chráněny.
Vaše dítě může dostat třetí dávku vakcíny Comirnaty. Účinnost vakcíny Comirnaty může být i po třetí dávce nižší u osob s oslabenou imunitou. V těchto případech byste měli i nadále dodržovat preventivní opatření, která pomáhají předcházet onemocnění COVID-19. Kromě toho by měly být podle potřeby očkovány i Vaše blízké kontakty. Proberte příslušná individuální doporučení s lékařem.
Děti
Pro kojence a děti ve věku 6 měsíců až 4 roky je k dispozici léková forma pro děti. Podrobnosti
naleznete v příbalové informaci vakcíny Comirnaty 3 mikrogramy/dávku koncentrát pro injekční
disperzi.
Vakcína Comirnaty není určena pro kojence mladší 6 měsíců.
Další léčivé přípravky a vakcína Comirnaty
Informujte svého lékaře nebo lékárníka o všech lécích, které Vaše dítě užívá, které v nedávné době
užívalo nebo které možná bude užívat, a o tom, zda mu byla v nedávné době podána jiná vakcína.
Těhotenství a kojení
Pokud je Vaše dcera těhotná nebo kojí, poraďte s lékařem, zdravotní sestrou nebo lékárníkem dříve,
než jí bude tato vakcína podána.
Vakcínu Comirnaty lze podávat během těhotenství. Velké množství informací o těhotných ženách očkovaných vakcínou Comirnaty během druhého a třetího trimestru neprokázalo negativní účinky na těhotenství nebo novorozence. Informace o účincích na těhotenství nebo novorozence po očkování během prvního trimestru jsou sice omezené, ale nebylo pozorováno zvýšení rizika potratu.
Vakcínu Comirnaty lze podávat během kojení.
Řízení dopravních prostředků a obsluha strojů
Některé účinky očkování uvedené v bodě 4 (Možné nežádoucí účinky) mohou dočasně ovlivnit
schopnost řídit nebo obsluhovat stroje nebo provádět aktivity, jako je jízda na kole. Před znovuzahájením aktivit, které vyžadují plnou pozornost počkejte, dokud tyto účinky neodezní.
3. Jak se vakcína Comirnaty podává
Vakcína Comirnaty se podává po naředění jako injekce o objemu 0,2 ml do svalu v horní části paže. Vašemu dítěti budou podány 2 injekce.
Doporučuje se, aby druhou dávku téže vakcíny dostalo 3 týdny po první dávce pro dokončení základního očkování.
Pokud má Vaše dítě oslabenou imunitu, může dostat třetí dávku vakcíny Comirnaty nejméně 28 dní po druhé dávce.
Pokud dítě mezi jednotlivými dávkami základního očkování dosáhne věku 12 let, má sérii dokončit se
stejnou dávkou 10 mikrogramů.
Posilovací dávka vakcíny Comirnaty může být u dětí ve věku 5 až 11 let podána za nejméně 6 měsíců po základním očkování.
Máte-li jakékoli další otázky týkající se používání vakcíny Comirnaty, zeptejte se svého lékaře,
lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny vakcíny může mít i vakcína Comirnaty nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Nežádoucí účinky se mohou vyskytnout s následující četností:
Velmi časté nežádoucí účinky: mohou postihnout více než 1 z 10 lidí
• v místě injekce: bolest, zduření
• únava
• bolest hlavy
• bolest svalů
• zimnice
• bolest kloubů
• průjem
• horečka
Časté nežádoucí účinky: mohou postihnout až 1 z 10 lidí
• pocit na zvracení
• zvracení
• zarudnutí v místě injekce (‘velmi časté’ ve věku od 5 do 11 let)
Méně časté nežádoucí účinky: mohou postihnout až 1 ze 100 lidí
• zvětšené mízní uzliny (častěji pozorované po posilovací dávce)
• malátnost
• bolest paže
• nespavost
• svědění v místě injekce
• alergické reakce, jako je vyrážka nebo svědění
• pocit slabosti nebo nedostatku energie/ospalost
• snížená chuť k jídlu
• nadměrné pocení
• noční pocení
Vzácné nežádoucí účinky: mohou postihnout až 1 z 1 000 lidí
• dočasná jednostranná obrna lícního nervu
• alergické reakce, jako je kopřivka nebo otok obličeje
Velmi vzácné nežádoucí účinky: mohou postihnout až 1 z 10 000 lidí
• zánět srdečního svalu (myokarditida) nebo zánět osrdečníku (perikarditida), který může vést
k dušnosti, silnému tlukotu srdce nebo bolesti na hrudi
Není známo (z dostupných údajů nelze určit)
• závažná alergická reakce
• rozsáhlý otok končetiny, do které byla vakcína podána
• otok obličeje (otok obličeje se může objevit u pacientů, kterým byly do obličeje aplikovány
injekce kožních výplní)
• kožní reakce způsobující červené skvrny nebo fleky na kůži, které mohou vypadat jako terč (nebo střed terče) s tmavě červeným středem, který je obklopen kruhy světlejší červené barvy (erythema multiforme)
• neobvyklý pocit na kůži, jako je brnění nebo pocit šimrání (parestezie)
• snížený pocit nebo citlivost, zejména v kůži (hypestezie)
• silné menstruační krvácení (většina případů se zdála být nezávažné a dočasné povahy)
Hlášení nežádoucích účinků
Pokud se u Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím
národního systému hlášení nežádoucích účinků uvedeného Dodatku V. Uveďte přitom číslo šarže, je-li k dispozici. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti
tohoto přípravku.
5. Jak vakcínu Comirnaty uchovávatUchovávejte tento přípravek mimo dohled a dosah dětí.
Následující informace o uchovávání, době použitelnosti a použití a manipulaci jsou určeny pro
zdravotnické pracovníky.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána buď při teplotě -90 °C až -60 °C a nebo 2 °C až 8 °C.
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 4 hodin nebo mohou být jednotlivé
injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Po jednorázovém vyjmutí z mrazničky může být neotevřená injekční lahvička uchovávána a transportována chlazená při teplotě 2 °C až 8 °C po dobu až 10 týdnů; nesmí být překročeno vytištěné datum použitelnosti (EXP). Vnější obal má být označen novým datem likvidace při teplotě 2 °C až
8 °C. Po jednorázovém rozmrazení se vakcína nesmí znovu zmrazit.
Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Po naředění vakcínu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin, která zahrnuje až
6 hodin transportu. Případnou nepoužitou vakcínu zlikvidujte.
Vakcínu nepoužívejte, pokud si všimnete, že jsou v naředěném roztoku částice nebo že roztok změnil
barvu.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informaceCo vakcína Comirnaty obsahuje• Léčivou látkou je mRNA vakcína proti onemocnění COVID-19 označená tozinameran. Po naředění injekční lahvička obsahuje 10 dávek, jedna dávka 0,2 ml obsahuje 10 mikrogramů tozinameranu.
• Dalšími složkami jsou:
- ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
- 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
- kolfosceryl-stearát
- cholesterol
- trometamol
- trometamol-hydrochlorid
- sacharosa
- voda pro injekci
Jak vakcína Comirnaty vypadá a co obsahuje toto baleníVakcína je bílá až téměř bílá disperze (pH: 6,9–7,9), která se dodává ve vícedávkové čiré injekční lahvičce (sklo třídy I) o objemu 2 ml obsahující 10 dávek. Injekční lahvička je uzavřena pryžovou
zátkou a odtrhovacím oranžovým plastovým víčkem s hliníkovým krytem.
Velikosti balení: 10 nebo 195 injekčních lahviček
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci BioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.deVýrobciBioNTech Manufacturing GmbH
Kupferbergterrasse 17-19
55116 Mainz
Německo
Pfizer Manufacturing Belgium NV Rijksweg 12
2870 Puurs
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
B
e
l
gië/Belgique/Belgien Luxembourg/Luxemburg Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +370 52 51 4000
Б
ълг
ария
Пфайзер Люксембург САРЛ, Клон
България
Teл: +359 2 970 4333
Magyarország
Pfizer Kft
Tel: +36 1 488 3700
Č
eská republika
Pfizer, spol. s r.o.
Tel: +420 283 004 111
Malta
Vivian Corporation Ltd.
Tel: +35621 344610
D
anmark
Pfizer ApS
Tlf: +45 44 201 100
Norge
Pfizer AS
Tlf: +47 67 526 100
D
eutschland
BioNTech Manufacturing GmbH
Tel: +49 6131 90840
Nederland
Pfizer BV
Tel: +31 (0)10 406 43 01
E
esti
Pfizer Luxembourg SARL Eesti filiaal
Tel: +372 666 7500
Österreich
Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1 521 15-0
Ε
λλάδα
Pfizer Ελλάς A.E.
Τηλ.: +30 210 6785 800
Polska
Pfizer Polska Sp. z o.o. Tel.: +48 22 335 61 00
E
s
paña
Pfizer, S.L.
Tél: +34914909900
Portugal
Laboratórios Pfizer, Lda.
Tel: +351 21 423 5500
F
rance
Pfizer
Tél +33 1 58 07 34 40
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
H
rvatska
Pfizer Croatia d.o.o.
Tel: +385 1 3908 777
Slovenija
Pfizer Luxembourg SARL
Pfizer, podružnica za svetovanje s področja
farmacevtske dejavnosti, Ljubljana
Tel.: +386 (0) 1 52 11 400
Ireland
Pfizer Healthcare Ireland
Tel: 1800 633 363 (toll free)
+44 (0)1304 616161
Slovenská republika
Pfizer Luxembourg SARL,
organizačná zložka
Tel: +421 2 3355 5500
Ísland
Icepharma hf
Simi: +354 540 8000
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
It
alia
Pfizer S.r.l.
Tel: +39 06 33 18 21
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
Κύ
πρ
ος
Pfizer Ελλάς Α.Ε. (Cyprus Branch)
Tηλ: +357 22 817690
United Kingdom (Northern Ireland)
Pfizer Limited
Tel: +44 (0) 1304 616161
L
atvija
Pfizer Luxembourg SARL filiāle Latvijā
Tel.: +371 670 35 775
Tato příbalová informace byla naposledy revidovánaPro zobrazení příbalové informace v různých jazycích sejměte následující kód pomocí chytrého zařízení.

URL:
www.comirnatyglobal.comPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
------------------------------------------------------------------------------------------------------------------------
Následující informace jsou určeny pouze pro zdravotnické pracovníky:Podávejte vakcínu Comirnaty intramuskulárně po naředění jako cyklus 2 dávek (každá po 0,2 ml)
s odstupem 3 týdnů.
Posilovací dávka vakcíny Comirnaty může být u dětí ve věku 5 až 11 let podána za nejméně 6 měsíců po základním očkování.
Třetí dávka může být podána nejméně 28 dní po druhé dávce jedincům s těžkou poruchou imunity.
SledovatelnostAby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Instrukce pro zacházení s vakcínouVakcína Comirnaty 10 mikrogramů/dávku má být připravována zdravotnickým pracovníkem pomocí
aseptické techniky, aby byla zajištěna sterilita připravené disperze.

KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY
10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
|
O
ranžové
víčko
P
o naředění
10 mikrogramů
|
• Zkontrolujte, zda má injekční lahvička oranžové plastové víčko a oranžový okraj kolem štítku a název přípravku je Comirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko a oranžový okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro injekční disperzi, přečtěte si souhrn údajů o přípravku pro toto složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička šedé
plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku injekční disperze Comirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční
disperze nebo Comirnaty
Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi.
|
Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY 10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET) PŘED POUŽITÍM
|
U
chovávejte až
10 týdnů při teplotě 2 °C až
8 °C.
|
• Pokud se vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se rozmrazily; rozmrazení balení 10 injekčních lahviček může trvat 4 hodiny. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené
injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
MÍCHÁNÍ VAKCÍNY COMIRNATY 10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO
I
N
JEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET) PŘED ŘEDĚNÍM
|

Jemně × 10
|
• Rozmrazenou injekční lahvičku nechte ohřát na pokojovou teplotu a před ředěním ji 10krát jemně převraťte. Netřepejte.
• Před ředěním může rozmrazená disperze obsahovat bílé až téměř bílé matné amorfní částice.
|
ŘEDĚN
Í VAKCÍNY COMIRNATY 10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
|
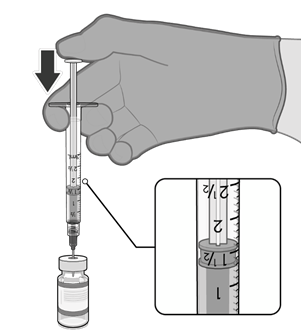
1,3 ml injekčního roztoku chloridu
sodného 9 mg/ml (0,9 %)
|
• Rozmrazená vakcína se musí naředit v původní injekční lahvičce 1,3 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%) za použití jehly
o velikosti 21 gauge (21G) nebo užší a uplatnění aseptických metod.
|

V
ytáhněte píst na značku 1,3 ml pro
odstranění vzduchu z injekční lahvičky.
|
• Nasátím 1,3 ml vzduchu do prázdné stříkačky na rozpouštědlo vyrovnejte před vyjmutím jehly z injekční lahvičky tlak v injekční lahvičce.
|
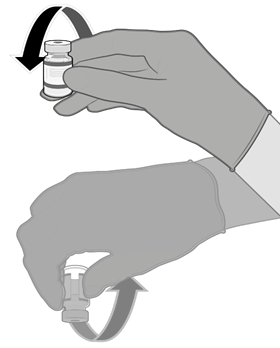
Jemně x 10
|
• Disperzi 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Naředěná vakcína se má jevit jako bílá až téměř bílá disperze bez viditelných
částic. Jestliže jsou v naředěné vakcíně
patrné částice nebo vakcína změnila
barvu, nepoužívejte ji.
|
Č
as likvidace
Z
a
z
namenejte příslušné datum a čas. Použijte během 12 hodin po naředění.
|
• Injekční lahvičky s naředěnou vakcínou je třeba označit příslušným datem
a časem.
• Po naředění uchovávejte vakcínu při teplotě 2 °C až 30 °C a použijte ji během
12 hodin.
• Naředěnou disperzi nezmrazujte ani s ní netřepejte. Pokud je v chladu, nechte naředěnou disperzi před použitím dosáhnout pokojové teploty.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,2 ml VAKCÍNY COMIRNATY
10 MIKROGRAMŮ/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
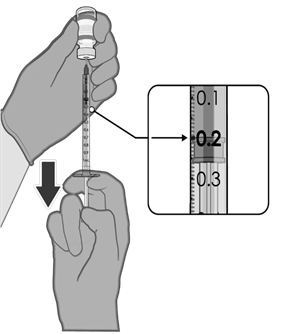 N
aředěná vakcína o objemu 0,2 ml
N
aředěná vakcína o objemu 0,2 ml
• Po naředění obsahuje injekční lahvička
2,6 ml, ze kterých lze získat 10 dávek po
0,2 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím
antiseptického tampónu.
• Natáhněte 0,2 ml vakcíny Comirnaty pro
děti ve věku 5 až 11 let.
K získání 10 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně
35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání deseti dávek z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,2 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,2 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Veškerou vakcínu, jež nebyla použita do
12 hodin po naředění, zlikvidujte.
L
i
kvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
P
ř
í
balová informace: informace pro uživatele
C
omirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi
K
ojenci a děti ve věku 6 měsíců až 4 roky
m
RN
A vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vašeho dítěte vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tato vakcína podána, protože obsahuje pro Vás důležité údaje.• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci1. Co je vakcína Comirnaty a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vašemu dítěti bude vakcína Comirnaty podána
3. Jak se vakcína Comirnaty podává
4. Možné nežádoucí účinky
5. Jak vakcínu Comirnaty uchovávat
6. Obsah balení a další informace
1. Co je vakcína Comirnaty a k čemu se používáComirnaty je vakcína používaná pro prevenci onemocnění COVID-19, které způsobuje SARS-CoV-2. Vakcína Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi se podává kojencům a
dětem ve věku 6 měsíců až 4 roky.
Vakcína způsobuje, že imunitní systém (přirozená obrana těla) vytváří protilátky a krevní buňky, které působí proti viru, a tím chrání před onemocněním COVID-19.
Jelikož vakcína Comirnaty neobsahuje virus k vyvolání imunity, nemůže u Vašeho dítěte vyvolat onemocnění COVID-19.
2. Čemu musíte věnovat pozornost, než Vašemu dítěti bude vakcína Comirnaty podánaVakcína Comirnaty Vám nesmí být podána• jestliže je Vaše dítě alergické na léčivou látku nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Upozornění a opatřeníPřed podáním vakcíny Vašemu dítěti se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou, jestliže Vaše dítě:
• někdy mělo závažnou alergickou reakci nebo potíže s dýcháním po jakékoli jiné vakcíně podané
v injekci nebo poté, co mu byla v minulosti podána vakcína Comirnaty,
• je v souvislosti s očkováním nervózní nebo někdy po jakékoli injekci omdlelo,
• má závažné onemocnění nebo infekci s vysokou horečkou. Avšak očkování může Vaše dítě podstoupit, pokud má mírně zvýšenou teplotu nebo lehkou infekci horních cest dýchacích, jako
je nachlazení,
• má krvácivé potíže, lehce se mu tvoří modřiny nebo užívá lék, který zabraňuje srážení krve,
• má oslabený imunitní systém, kvůli onemocnění, jako je infekce virem HIV, nebo užívá lék,
jako je kortikosteroid, který imunitní systém ovlivňuje.
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy (zánětu srdečního svalu) a perikarditidy (zánětu osrdečníku (perikardu)) (viz bod 4). Tato onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhém očkování a častěji u mladších mužů a chlapců. Riziko myokarditidy a perikarditidy je pravděpodobně nižší u dětí ve věku od 5 do 11 let v porovnání s dospívajícími ve věku od 12 do
17 let. Po očkování u Vašeho dítěte pozorně sledujte příznaky myokarditidy a perikarditidy, například dušnost, silný tlukot srdce a bolest na hrudi, a pokud se u něj vyskytnou, ihned vyhledejte lékařskou pomoc.
Podobně jako u jiných vakcín je možné, že vakcína Comirnaty nebude plně chránit všechny osoby, kterým bude vakcína podána a není známo, jak dlouho budou chráněny.
Účinnost vakcíny Comirnaty může být i po třetí dávce nižší u osob s oslabenou imunitou. V těchto případech byste měli i nadále dodržovat preventivní opatření, která pomáhají předcházet onemocnění COVID-19. Kromě toho by měly být podle potřeby očkovány i Vaše blízké kontakty. Proberte příslušná individuální doporučení s lékařem.
Děti
Vakcína Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi není určena pro děti ve věku
od 5 do 11 let. Pro děti ve věku od 5 do 11 let je k dispozici léková forma pro děti. Podrobnosti
naleznete v příbalové informaci vakcíny Comirnaty 10 mikrogramů/dávku koncentrát pro injekční
disperzi.
Vakcína Comirnaty není určena pro kojence mladší 6 měsíců.
Další léčivé přípravky a vakcína Comirnaty
Informujte svého lékaře nebo lékárníka o všech lécích, které Vaše dítě užívá, které v nedávné době
užívalo nebo které možná bude užívat, a o tom, zda mu byla v nedávné době podána jiná vakcína.
Těhotenství a kojení
Vakcína Comirnaty 3 mikrogramy/dávku injekční disperze není určena pro osoby starší 5 let.
Podrobnosti o použití u osob starších 5 let naleznete v příbalové informaci vakcíny Comirnaty
30 mikrogramů/dávku koncentrát pro injekční disperzi, Comirnaty 30 mikrogramů/dávku injekční
disperze nebo Comirnaty 10 mikrogramů/dávku koncentrát pro injekční disperzi.
Řízení dopravních prostředků a obsluha strojů
Některé účinky očkování uvedené v bodě 4 (Možné nežádoucí účinky) mohou dočasně ovlivnit schopnost řídit nebo obsluhovat stroje nebo provádět aktivity, jako je jízda na kole. Před
znovuzahájením aktivit, které vyžadují plnou pozornost počkejte, dokud tyto účinky neodezní.
3. Jak se vakcína Comirnaty podává
Vakcína Comirnaty se podává po naředění jako injekce o objemu 0,2 ml do stehenního svalu
u kojenců od 6 do méně než 12 měsíců věku. U kojenců a dětí od 1 roku věku se vakcína Comirnaty
podává po naředění jako injekce o objemu 0,2 ml do stehenního svalu nebo do svalu v horní části
paže.
Vašemu dítěti budou podány 3 injekce.
Doporučuje se, aby druhou dávku téže vakcíny dostalo 3 týdny po první dávce a následně třetí dávku alespoň 8 týdnů po druhé dávce pro dokončení základního očkování.
Pokud dítě mezi jednotlivými dávkami základního očkování dosáhne věku 5 let, má sérii dokončit se
stejnou dávkou 3 mikrogramů.
Máte-li jakékoli další otázky týkající se používání vakcíny Comirnaty, zeptejte se svého lékaře,
lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny vakcíny může mít i vakcína Comirnaty nežádoucí účinky, které se ale nemusí
vyskytnout u každého.
Nežádoucí účinky se mohou vyskytnout s následující četností:
Velmi časté nežádoucí účinky: mohou postihnout více než 1 z 10 lidí
• podrážděnost (od 6 měsíců do < 2 let)
• v místě injekce: bolest/citlivost, zduření
• únava
• bolest hlavy
• ospalost (od 6 měsíců do < 2 let)
• bolest svalů
• zimnice
• bolest kloubů
• průjem
• horečka
Časté nežádoucí účinky: mohou postihnout až 1 z 10 lidí
• pocit na zvracení
• zvracení
• zarudnutí v místě injekce (‘velmi časté’ ve věku od 6 měsíců do 11 let)
Méně časté nežádoucí účinky: mohou postihnout až 1 ze 100 lidí
• zvětšené mízní uzliny (častěji pozorované po posilovací dávce)
• malátnost
• bolest paže
• nespavost
• svědění v místě injekce
• alergické reakce, jako je vyrážka (‘časté’ ve věku od 6 měsíců do < 2 let) nebo svědění
• pocit slabosti nebo nedostatku energie/ospalost
• snížená chuť k jídlu (‘velmi časté’ ve věku od 6 měsíců do < 2 let)
• nadměrné pocení
• noční pocení
Vzácné nežádoucí účinky: mohou postihnout až 1 z 1 000 lidí
• dočasná jednostranná obrna lícního nervu
• alergické reakce, jako je kopřivka nebo otok obličeje
Velmi vzácné nežádoucí účinky: mohou postihnout až 1 z 10 000 lidí
• zánět srdečního svalu (myokarditida) nebo zánět osrdečníku (perikarditida), který může vést
k dušnosti, silnému tlukotu srdce nebo bolesti na hrudi
N
ení známo (z dostupných údajů nelze určit)
• závažná alergická reakce
• rozsáhlý otok končetiny, do které byla vakcína podána
• otok obličeje (otok obličeje se může objevit u pacientů, kterým byly do obličeje aplikovány
injekce kožních výplní)
• kožní reakce způsobující červené skvrny nebo fleky na kůži, které mohou vypadat jako terč (nebo střed terče) s tmavě červeným středem, který je obklopen kruhy světlejší červené barvy (erythema multiforme)
• neobvyklý pocit na kůži, jako je brnění nebo pocit šimrání (parestezie)
• snížený pocit nebo citlivost, zejména v kůži (hypestezie)
• silné menstruační krvácení (většina případů se zdála být nezávažné a dočasné povahy)
Hlášení nežádoucích účinkůPokud se u Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou
uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného Dodatku V. Uveďte přitom číslo šarže, je-li k dispozici. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti
tohoto přípravku.
5. Jak vakcínu Comirnaty uchovávatUchovávejte tento přípravek mimo dohled a dosah dětí.
Následující informace o uchovávání, době použitelnosti a použití a manipulaci jsou určeny pro
zdravotnické pracovníky.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána buď při teplotě -90 °C až -60 °C a nebo 2 °C až 8 °C.
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 2 hodin nebo mohou být jednotlivé
injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Po jednorázovém vyjmutí z mrazničky může být neotevřená injekční lahvička uchovávána a transportována chlazená při teplotě 2 °C až 8 °C po dobu až 10 týdnů; nesmí být překročeno vytištěné datum použitelnosti (EXP). Vnější obal má být označen novým datem likvidace při teplotě 2 °C až
8 °C. Po jednorázovém rozmrazení se vakcína nesmí znovu zmrazit.
Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Po naředění vakcínu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin, která zahrnuje až
6 hodin transportu. Případnou nepoužitou vakcínu zlikvidujte.
Vakcínu nepoužívejte, pokud si všimnete, že jsou v naředěném roztoku částice nebo že roztok změnil
barvu.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informaceCo vakcína Comirnaty obsahuje• Léčivou látkou je mRNA vakcína proti onemocnění COVID-19 označená tozinameran. Po naředění injekční lahvička obsahuje 10 dávek, jedna dávka 0,2 ml obsahuje 3 mikrogramy tozinameranu.
• Dalšími složkami jsou:
- ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
- 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
- kolfosceryl-stearát
- cholesterol
- trometamol
- trometamol-hydrochlorid
- sacharosa
- voda pro injekci
Jak vakcína Comirnaty vypadá a co obsahuje toto baleníVakcína je bílá až téměř bílá disperze (pH: 6,9–7,9), která se dodává ve vícedávkové čiré injekční lahvičce (sklo třídy I) o objemu 2 ml obsahující 10 dávek. Injekční lahvička je uzavřena pryžovou zátkou a odtrhovacím hnědočerveným plastovým víčkem s hliníkovým krytem.
Velikosti balení: 10 injekčních lahviček
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci BioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.deVýrobciBioNTech Manufacturing GmbH
Kupferbergterrasse 17-19
55116 Mainz
Německo
Pfizer Manufacturing Belgium NV Rijksweg 12
2870 Puurs
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
B
e
l
gië/Belgique/Belgien
L
uxembourg/Luxemburg
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +370 52 51 4000
Б
ълг
ария
Пфайзер Люксембург САРЛ, Клон
България
Teл: +359 2 970 4333
Magyarország
Pfizer Kft
Tel: +36 1 488 3700
Č
eská republika
Pfizer, spol. s r.o.
Tel: +420 283 004 111
Malta
Vivian Corporation Ltd.
Tel: +35621 344610
D
anmark
Pfizer ApS
Tlf: +45 44 201 100
Norge
Pfizer AS
Tlf: +47 67 526 100
D
eutschland
BioNTech Manufacturing GmbH
Tel: +49 6131 90840
Nederland
Pfizer BV
Tel: +31 (0)10 406 43 01
E
esti
Pfizer Luxembourg SARL Eesti filiaal
Tel: +372 666 7500
Österreich
Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1 521 15-0
Ε
λλάδα
Pfizer Ελλάς A.E.
Τηλ.: +30 210 6785 800
Polska
Pfizer Polska Sp. z o.o.
Tel.: +48 22 335 61 00
E
s
paña
Pfizer, S.L.
Tél: +34914909900
Portugal
Laboratórios Pfizer, Lda.
Tel: +351 21 423 5500
F
rance
Pfizer
Tél +33 1 58 07 34 40
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
H
rvatska
Pfizer Croatia d.o.o.
Tel: +385 1 3908 777
Slovenija
Pfizer Luxembourg SARL
Pfizer, podružnica za svetovanje s področja
farmacevtske dejavnosti, Ljubljana
Tel.: +386 (0) 1 52 11 400
Ireland
Pfizer Healthcare Ireland
Tel: 1800 633 363 (toll free)
+44 (0)1304 616161
Slovenská republika
Pfizer Luxembourg SARL,
organizačná zložka
Tel: +421 2 3355 5500
Ísland
Icepharma hf
Simi: +354 540 8000
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
It
alia
Pfizer S.r.l.
Tel: +39 06 33 18 21
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
Κύ
πρ
ος
Pfizer Ελλάς Α.Ε. (Cyprus Branch) Tηλ: +357 22 817690
United Kingdom (Northern Ireland)
Pfizer Limited
Tel: +44 (0) 1304 616161
L
atvija
Pfizer Luxembourg SARL filiāle Latvijā
Tel.: +371 670 35 775
Tato příbalová informace byla naposledy revidovánaPro zobrazení příbalové informace v různých jazycích sejměte následující kód pomocí chytrého zařízení.

URL:
www.comirnatyglobal.comPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
------------------------------------------------------------------------------------------------------------------------
Následující informace jsou určeny pouze pro zdravotnické pracovníky:Podávejte vakcínu Comirnaty intramuskulárně po naředění jako cyklus 3 dávek (každá po 0,2 ml); druhá dávka téže vakcíny má být podána 3 týdny po první dávce, a následně má být podána třetí dávka nejdříve 8 týdnů po druhé dávce k dokončení očkovacího schématu.
SledovatelnostAby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Instrukce pro zacházení s vakcínouVakcína Comirnaty 3 mikrogramy/dávku má být připravována zdravotnickým pracovníkem pomocí
aseptické techniky, aby byla zajištěna sterilita připravené disperze.
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY
3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (KOJENCI A
DĚT
I VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY)
H
nědočervené víčko
P
o naředění
3 mikrogramy
• Zkontrolujte, zda má injekční lahvička hnědočervené plastové víčko.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička šedé
plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku injekční disperze, Comirnaty Original/Omicron BA.1
(15/15 mikrogramů)/dávku injekční
disperze nebo Comirnaty
Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze.
• Pokud má injekční lahvička oranžové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi nebo Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro injekční disperzi.
Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY 3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (KOJENCI A DĚTI VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY) PŘED POUŽITÍM
|
U
chovávejte až
10 týdnů při teplotě 2 °C až
8 °C.
|
• Pokud se vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se rozmrazily;
rozmrazení balení 10 injekčních lahviček může trvat 2 hodiny. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
MÍCHÁNÍ VAKCÍNY COMIRNATY 3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO
I
N
JEKČNÍ DISPERZI (KOJENCI A DĚTI VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY) PŘED ŘEDĚNÍM
|

Jemně × 10
|
• Rozmrazenou injekční lahvičku nechte ohřát na pokojovou teplotu a před ředěním ji 10krát jemně převraťte. Netřepejte.
• Před ředěním může rozmrazená disperze obsahovat bílé až téměř bílé matné amorfní částice.
|
ŘEDĚN
Í VAKCÍNY COMIRNATY 3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO
I
N
JEKČNÍ DISPERZI (KOJENCI A DĚTI VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY)
|
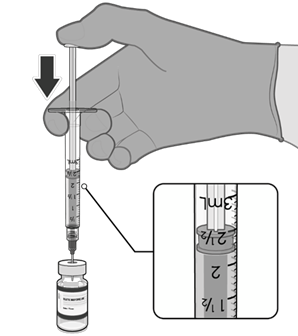
2,2 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9 %)
|
• Rozmrazená vakcína se musí naředit v původní injekční lahvičce 2,2 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%) za použití jehly
o velikosti 21 gauge (21G) nebo užší a uplatnění aseptických metod.
|
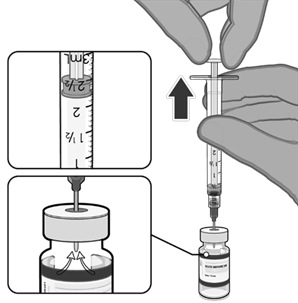
V
ytáhněte píst na značku 2,2 ml pro
odstranění vzduchu z injekční lahvičky.
|
• Nasátím 2,2 ml vzduchu do prázdné stříkačky na rozpouštědlo vyrovnejte před vyjmutím jehly z injekční lahvičky tlak v injekční lahvičce.
|
Jemně x 10
|
• Disperzi 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Naředěná vakcína se má jevit jako bílá až téměř bílá disperze bez viditelných
částic. Jestliže jsou v naředěné vakcíně
patrné částice nebo vakcína změnila
barvu, nepoužívejte ji.
|
Č
as likvidace
Z
a
z
namenejte příslušné datum a čas. Použijte během 12 hodin po naředění.
|
• Injekční lahvičky s naředěnou vakcínou je třeba označit příslušným datem
a časem.
• Po naředění uchovávejte vakcínu při teplotě 2 °C až 30 °C a použijte ji během
12 hodin.
• Naředěnou disperzi nezmrazujte ani s ní netřepejte. Pokud je v chladu, nechte naředěnou disperzi před použitím dosáhnout pokojové teploty.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,2 ml VAKCÍNY COMIRNATY
3 MIKROGRAMY/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (KOJENCI A
DĚT
I VE VĚKU 6 MĚSÍCŮ AŽ 4 ROKY)
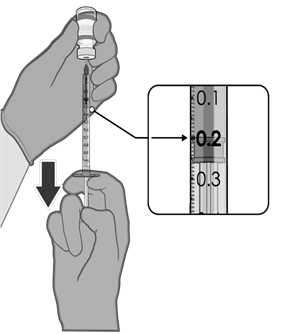 N
aředěná vakcína o objemu 0,2 ml
N
aředěná vakcína o objemu 0,2 ml
• Po naředění obsahuje injekční lahvička
2,6 ml, ze kterých lze získat 10 dávek po
0,2 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím
antiseptického tampónu.
• Natáhněte 0,2 ml vakcíny Comirnaty pro kojence a děti ve věku 6 měsíců až
4 roky.
K získání 10 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční stříkačky
a jehly s malým mrtvým prostorem má
mít mrtvý objem maximálně
35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání deseti dávek z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,2 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,2 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Veškerou vakcínu, jež nebyla použita do
12 hodin po naředění, zlikvidujte.
L
i
kvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními
požadavky.
P
ř
í
balová informace: informace pro uživatele
C
omirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční disperze
D
ospělí a dospívající od 12 let
m
RN
A vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran/riltozinameran
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou.
Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tato vakcína podána, protože obsahuje pro Vás důležité údaje.• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci1. Co je vakcína Comirnaty Original/Omicron BA.1 a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude vakcína Comirnaty Original/Omicron BA.1
podána
3. Jak se vakcína Comirnaty Original/Omicron BA.1 podává
4. Možné nežádoucí účinky
5. Jak vakcínu Comirnaty Original/Omicron BA.1 uchovávat
6. Obsah balení a další informace
1. Co je vakcína Comirnaty Original/Omicron BA.1 a k čemu se používáComirnaty Original/Omicron BA.1 je vakcína používaná pro prevenci onemocnění COVID-19, které
způsobuje SARS-CoV-2.
Podává se dospělým a dospívajícím ve věku 12 let a starším.
Vakcína Comirnaty Original/Omicron BA.1 je určena pouze pro jednotlivce, kteří již předtím
absolvovali alespoň základní očkování proti onemocnění COVID-19.
Vakcína způsobuje, že imunitní systém (přirozená obrana těla) vytváří protilátky a krevní buňky, které
působí proti viru, a tím chrání před onemocněním COVID-19.
Jelikož vakcína Comirnaty Original/Omicron BA.1 neobsahuje virus k vyvolání imunity, nemůže u
Vás vyvolat onemocnění COVID-19.
2. Čemu musíte věnovat pozornost, než Vám bude vakcína Comirnaty Original/OmicronBA.1 podánaVakcína Comirnaty Original/Omicron BA.1 Vám nesmí být podána• jestliže jste alergický(á) na léčivou látku nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Upozornění a opatřeníPřed podáním vakcíny se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou, jestliže:
• jste někdy měl(a) závažnou alergickou reakci nebo potíže s dýcháním po jakékoli jiné vakcíně
podané v injekci nebo poté, co Vám byla v minulosti podána vakcína Comirnaty nebo
Comirnaty Original/Omicron BA.1,
• jste v souvislosti s očkováním nervózní nebo jste někdy po jakékoli injekci omdlel(a),
• máte závažné onemocnění nebo infekci s vysokou horečkou. Avšak očkování můžete podstoupit, pokud máte mírně zvýšenou teplotu nebo lehkou infekci horních cest dýchacích, jako je nachlazení,
• máte krvácivé potíže, lehce se Vám tvoří modřiny nebo užíváte lék, který zabraňuje srážení
krve,
• máte oslabený imunitní systém, kvůli onemocnění, jako je infekce virem HIV, nebo užíváte lék, jako je kortikosteroid, který imunitní systém ovlivňuje.
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy (zánětu srdečního svalu) a perikarditidy (zánětu osrdečníku (perikardu)) (viz bod 4). Tato onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhém očkování a častěji u mladších mužů a chlapců. Riziko myokarditidy a perikarditidy je pravděpodobně nižší u dětí ve věku od 5 do 11 let v porovnání s dospívajícími ve věku od 12 do
17 let. Po očkování u Vašeho dítěte pozorně sledujte příznaky myokarditidy a perikarditidy, například dušnost, silný tlukot srdce a bolest na hrudi, a pokud se u Vás vyskytnou, ihned vyhledejte lékařskou
pomoc.
Podobně jako u jiných vakcín je možné, že vakcína Comirnaty Original/Omicron BA.1 nebude plně chránit všechny osoby, kterým bude vakcína podána a není známo, jak dlouho budou chráněny.
Účinnost vakcíny Comirnaty Original/Omicron BA.1 nižší u osob s oslabenou imunitou. V těchto případech byste měl(a) i nadále dodržovat preventivní opatření, která pomáhají předcházet onemocnění COVID-19. Kromě toho by měly být podle potřeby očkovány i Vaše blízké kontakty. Proberte příslušná individuální doporučení se svým lékařem.
Děti
Vakcína Comirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční disperze není určena
pro děti ve věku do 12 let.
Další léčivé přípravky a vakcína Comirnaty Original/Omicron BA.1
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době
užíval(a) nebo které možná budete užívat, a o tom, zda Vám byla v nedávné době podána jiná vakcína.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte
se se svým lékařem, zdravotní sestrou nebo lékárníkem dříve, než Vám bude tato vakcína podána.
Údaje o použití vakcíny Comirnaty Original/Omicron BA.1 během těhotenství nejsou k dispozici. Ovšem velké množství informací o těhotných ženách očkovaných původně schválenou vakcínou Comirnaty během druhého a třetího trimestru neprokázalo negativní účinky na těhotenství nebo novorozence. Informace o účincích na těhotenství nebo novorozence po očkování během prvního trimestru jsou sice omezené, ale nebylo pozorováno zvýšení rizika potratu. Vakcínu Comirnaty Original/Omicron BA.1 lze podávat během těhotenství.
Údaje o použití vakcíny Comirnaty Original/Omicron BA.1 během kojení nejsou k dispozici. Nicméně se neočekávají žádné účinky na kojeného novorozence/kojence. Údaje od žen, které kojily po
očkování původně schválenou vakcínou Comirnaty, neudávaly riziko nežádoucích účinků kojených
novorozenců/kojenců. Vakcínu Comirnaty Original/Omicron BA.1 lze podávat během kojení.
Řízení dopravních prostředků a obsluha strojů
Některé účinky očkování uvedené v bodě 4 (Možné nežádoucí účinky) mohou dočasně ovlivnit Vaši
schopnost řídit nebo obsluhovat stroje. Před řízením nebo obsluhou strojů počkejte, dokud tyto účinky
neodezní.
3. Jak se vakcína Comirnaty Original/Omicron BA.1 podává
Vakcína Comirnaty Original/Omicron BA.1 se podává jako injekce o objemu 0,3 ml do svalu v horní
části paže.
Vakcínu Comirnaty Original/Omicron BA.1 lze podávat nejméně 3 měsíce po poslední minulé dávce vakcíny proti onemocnění COVID-19.
Vakcína Comirnaty Original/Omicron BA.1 je indikována pouze u jedinců, kteří již dříve absolvovali alespoň základní očkování proti onemocnění COVID-19.
O způsobilosti a načasování posilovací dávky se prosím poraďte se svým zdravotnickým pracovníkem.
Podrobnosti o základním očkování u jedinců ve věku 12 let a starších naleznete v příbalové informaci vakcíny Comirnaty 30 mikrogramů/dávku injekční disperze nebo Comirnaty 30 mikrogramů/dávku koncentrátu injekční disperze.
Máte-li jakékoli další otázky týkající se používání vakcíny Comirnaty Original/Omicron BA.1,
zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny vakcíny může mít i vakcína Comirnaty Original/Omicron BA.1 nežádoucí
účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky se mohou vyskytnout s následující četností:
Velmi časté nežádoucí účinky: mohou postihnout více než 1 z 10 lidí
• v místě injekce: bolest, zduření
• únava
• bolest hlavy
• bolest svalů
• zimnice
• bolest kloubů
• průjem
• horečka
Některé z těchto nežádoucích účinků se o něco častěji vyskytovaly u dospívajících ve věku 12 až
15 let než u dospělých.
Časté nežádoucí účinky: mohou postihnout až 1 z 10 lidí
• zarudnutí v místě injekce
• pocit na zvracení
• zvracení
Méně časté nežádoucí účinky: mohou postihnout až 1 ze 100 lidí
• zvětšené mízní uzliny (častěji pozorované po posilovací dávce)
• malátnost
• bolest paže
• nespavost
• svědění v místě injekce
• alergické reakce, jako je vyrážka nebo svědění
• pocit slabosti nebo nedostatku energie/ospalost
• snížená chuť k jídlu
• nadměrné pocení
• noční pocení
Vzácné nežádoucí účinky: mohou postihnout až 1 z 1 000 lidí
• dočasná jednostranná obrna lícního nervu
• alergické reakce, jako je kopřivka nebo otok obličeje
Velmi vzácné nežádoucí účinky: mohou postihnout až 1 z 10 000 lidí
• zánět srdečního svalu (myokarditida) nebo zánět osrdečníku (perikarditida), který může vést
k dušnosti, silnému tlukotu srdce nebo bolesti na hrudi
Není známo (z dostupných údajů nelze určit)
• závažná alergická reakce
• rozsáhlý otok končetiny, do které byla vakcína podána
• otok obličeje (otok obličeje se může objevit u pacientů, kterým byly do obličeje aplikovány
injekce kožních výplní)
• kožní reakce způsobující červené skvrny nebo fleky na kůži, které mohou vypadat jako terč (nebo střed terče) s tmavě červeným středem, který je obklopen kruhy světlejší červené barvy (erythema multiforme)
• neobvyklý pocit na kůži, jako je brnění nebo pocit šimrání (parestezie)
• snížený pocit nebo citlivost, zejména v kůži (hypestezie)
• silné menstruační krvácení (většina případů se zdála být nezávažné a dočasné povahy)
Hlášení nežádoucích účinkůPokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo
zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného Dodatku V. Uveďte přitom číslo šarže, je-li
k dispozici. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti
tohoto přípravku.
5. Jak vakcínu Comirnaty Original/Omicron BA.1 uchovávatUchovávejte tento přípravek mimo dohled a dosah dětí.
Následující informace o uchovávání, době použitelnosti a použití a manipulaci jsou určeny pro
zdravotnické pracovníky.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána buď při teplotě -90 °C až -60 °C a nebo 2 °C až 8 °C.
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 6 hodin nebo mohou být jednotlivé
injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Po vyjmutí z prostoru pro uchovávání ve zmrazeném stavu může být neotevřená injekční lahvička uchovávána a transportována chlazená při teplotě 2 °C až 8 °C po dobu až 10 týdnů; nesmí být překročeno vytištěné datum použitelnosti (EXP). Vnější obal má být označen novým datem likvidace při teplotě 2 °C až 8 °C. Po rozmrazení nemůže být vakcína znovu zmrazena.
Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Po prvním vpichu vakcínu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin, která zahrnuje až 6 hodin transportu. Případnou nepoužitou vakcínu zlikvidujte.
Vakcínu nepoužívejte, pokud si všimnete částic nebo změny barvy.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informaceCo vakcína Comirnaty Original/Omicron BA.1 obsahuje• Léčivými látkami mRNA vakcíny proti onemocnění COVID-19 jsou tozinameran a riltozinameran. Injekční lahvička obsahuje 6 dávek, jedna dávka 0,3 ml
obsahuje 15 mikrogramů tozinameranu (Original) a 15 mikrogramů riltozinameranu (Omikron
BA.1).
• Dalšími složkami jsou:
- ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
- 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
- kolfosceryl-stearát
- cholesterol
- trometamol
- trometamol-hydrochlorid
- sacharosa
- voda pro injekci
Jak vakcína Comirnaty Original/Omicron BA.1 vypadá a co obsahuje toto baleníVakcína je bílá až téměř bílá disperze (pH: 6,9–7,9), která se dodává ve vícedávkové čiré injekční
lahvičce (sklo třídy I) o objemu 2 ml obsahující 6 dávek. Injekční lahvička je uzavřena pryžovou
zátkou a šedým odtrhovacím plastovým víčkem s hliníkovým krytem.
Velikost balení: 10 nebo 195 injekčních lahviček
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci BioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.deVýrobciBioNTech Manufacturing GmbH
Kupferbergterrasse 17-19
55116 Mainz
Německo
Pfizer Manufacturing Belgium NV
Rijksweg 12
2870 Puurs
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
B
e
l
gië/Belgique/Belgien
L
uxembourg/Luxemburg
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +370 52 51 4000
Б
ълг
ария
Пфайзер Люксембург САРЛ, Клон
България
Teл: +359 2 970 4333
Magyarország
Pfizer Kft
Tel: +36 1 488 3700
Č
eská republika
Pfizer, spol. s r.o.
Tel: +420 283 004 111
Malta
Vivian Corporation Ltd.
Tel: +35621 344610
D
anmark
Pfizer ApS
Tlf: +45 44 201 100
Norge
Pfizer AS
Tlf: +47 67 526 100
D
eutschland
BioNTech Manufacturing GmbH Tel: +49 6131 90840
Nederland
Pfizer BV
Tel: +31 (0)10 406 43 01
E
esti
Pfizer Luxembourg SARL Eesti filiaal
Tel: +372 666 7500
Österreich
Pfizer Corporation Austria Ges.m.b.H
Tel: +43 (0)1 521 15-0
Ε
λλάδα
Pfizer Ελλάς A.E.
Τηλ.: +30 210 6785 800
Polska
Pfizer Polska Sp. z o.o.
Tel.: +48 22 335 61 00
E
s
paña
Pfizer, S.L.
Tél: +34914909900
Portugal
Laboratórios Pfizer, Lda. Tel: +351 21 423 5500
F
rance
Pfizer
Tél +33 1 58 07 34 40
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
H
rvatska
Pfizer Croatia d.o.o. Tel: +385 1 3908 777
Slovenija
Pfizer Luxembourg SARL
Pfizer, podružnica za svetovanje s področja
farmacevtske dejavnosti, Ljubljana
Tel.: +386 (0) 1 52 11 400
Ireland
Pfizer Healthcare Ireland
Tel: 1800 633 363 (toll free)
+44 (0)1304 616161
Slovenská republika
Pfizer Luxembourg SARL,
organizačná zložka
Tel: +421 2 3355 5500
Ísland
Icepharma hf
Simi: +354 540 8000
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
It
alia
Pfizer S.r.l.
Tel: +39 06 33 18 21
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
Κύ
πρ
ος
Pfizer Ελλάς Α.Ε. (Cyprus Branch) Tηλ: +357 22 817690
United Kingdom (Northern Ireland)
Pfizer Limited
Tel: +44 (0) 1304 616161
L
atvija
Pfizer Luxembourg SARL filiāle Latvijā
Tel.: +371 670 35 775
Tato příbalová informace byla naposledy revidována {MM/RRRR}Pro zobrazení příbalové informace v různých jazycích sejměte následující kód pomocí chytrého zařízení.

URL:
www.comirnatyglobal.comPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
------------------------------------------------------------------------------------------------------------------------
Následující informace jsou určeny pouze pro zdravotnické pracovníky:Dávka vakcíny Comirnaty Original/Omicron BA.1 je 0,3 ml podávaná intramuskulárně.
Mezi podáním vakcíny Comirnaty Original/Omicron BA.1 a poslední předchozí dávkou vakcíny proti
COVID-19 má uplynout interval nejméně 3 měsíce.
Vakcína Comirnaty Original/Omicron BA.1 je indikována pouze u jedinců, kteří již dříve podstoupili alespoň základní očkování proti onemocnění COVID-19.
SledovatelnostAby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Instrukce pro zacházení s vakcínou
Vakcína Comirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku má být připravována zdravotnickým pracovníkem pomocí aseptické techniky, aby byla zajištěna sterilita připravené
disperze.
KONTROLA INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.1 (15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
Š
e
d
é víčko
C
omirnaty Original/ Omicron BA.1
N
eřeďte
• Zkontrolujte, zda má injekční lahvička šedé plastové víčko a šedý okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.1
(15/15 mikrogramů)/dávku injekční
disperze.
• Pokud má injekční lahvička šedé plastové víčko a šedý okraj a název přípravku je Comirnaty
30 mikrogramů/dávku injekční disperze
nebo Comirnaty Original/Omicron
BA.4-5 (15/15 mikrogramů)/dávku injekční disperze, přečtěte si, prosím, souhrn údajů o přípravku pro příslušné složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi nebo Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte
si souhrn údajů o přípravku pro vakcínu
Comirnaty 3 mikrogramy/dávku
koncentrát pro injekční disperzi.
Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.1
(
15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ) PŘED
P
O
U
Ž
ITÍM
|
U
chovávejte po dobu až 10 týdnů při teplotě 2 °C
až 8 °C,
aktualizujte datum použitelnosti na krabičce.
|
• Pokud se vícedávková injekční lahvička uchovává zmrazená, musí
se před použitím rozmrazit. Zmrazené
injekční lahvičky je třeba přenést do prostředí o teplotě 2 °C až 8 °C, aby se rozmrazily; rozmrazení balení
10 injekčních lahviček může trvat
6 hodin. Ujistěte se, že jsou injekční lahvičky před použitím úplně
rozmrazené.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
Jemně
× 10
|
• Injekční lahvičky před použitím 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Před smícháním může rozmrazená disperze obsahovat bílé až téměř bílé, matné amorfní částice.
• Po smíchání se má vakcína jevit jako bílá až téměř bílá disperze bez viditelných částic. Jestliže jsou
v naředěné vakcíně patrné částice nebo vakcína změnila barvu, nepoužívejte ji.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,3 ml VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.1 (15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
 V
akcína o objemu 0,3 ml
V
akcína o objemu 0,3 ml
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím antiseptického tampónu.
• Natáhněte 0,3 ml vakcíny Comirnaty
Original/Omicron BA.1.
K získání šesti dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání šesté dávky z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Zaznamenejte příslušné datum/čas na injekční lahvičku. Veškerou vakcínu, jež nebyla použita do 12 hodin po prvním vpichu, zlikvidujte.
L
i
kvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
P
ř
í
balová informace: informace pro uživatele
C
omirnaty Original/Omicron BA.4-5 (15/15 mikrogramů)/dávku injekční disperze
D
ospělí a dospívající od 12 let
m
RN
A vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran/famtozinameran
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou.
Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tato vakcína podána, protože obsahuje pro Vás důležité údaje.• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci1. Co je vakcína Comirnaty Original/Omicron BA.4-5 a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude vakcína Comirnaty Original/Omicron BA.4-5
podána
3. Jak se vakcína Comirnaty Original/Omicron BA.4-5 podává
4. Možné nežádoucí účinky
5. Jak vakcínu Comirnaty Original/Omicron BA.4-5 uchovávat
6. Obsah balení a další informace
1. Co je vakcína Comirnaty Original/Omicron BA.4-5 a k čemu se používáComirnaty Original/Omicron BA.4-5 je vakcína používaná pro prevenci onemocnění COVID-19,
které způsobuje SARS-CoV-2.
Podává se dospělým a dospívajícím ve věku 12 let a starším.
Vakcína Comirnaty Original/Omicron BA.4-5 je určena pouze pro jednotlivce, kteří již předtím
absolvovali alespoň základní očkování proti onemocnění COVID-19.
Vakcína způsobuje, že imunitní systém (přirozená obrana těla) vytváří protilátky a krevní buňky, které
působí proti viru, a tím chrání před onemocněním COVID-19.
Jelikož vakcína Comirnaty Original/Omicron BA.4-5 neobsahuje virus k vyvolání imunity, nemůže u
Vás vyvolat onemocnění COVID-19.
2. Čemu musíte věnovat pozornost, než Vám bude vakcína Comirnaty Original/OmicronBA.4-5 podánaVakcína Comirnaty Original/Omicron BA.4-5 Vám nesmí být podána• jestliže jste alergický(á) na léčivou látku nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Upozornění a opatřeníPřed podáním vakcíny se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou, jestliže:
• jste někdy měl(a) závažnou alergickou reakci nebo potíže s dýcháním po jakékoli jiné vakcíně
podané v injekci nebo poté, co Vám byla v minulosti podána vakcína Comirnaty nebo
Comirnaty Original/Omicron BA.4-5,
• jste v souvislosti s očkováním nervózní nebo jste někdy po jakékoli injekci omdlel(a),
• máte závažné onemocnění nebo infekci s vysokou horečkou. Avšak očkování můžete podstoupit, pokud máte mírně zvýšenou teplotu nebo lehkou infekci horních cest dýchacích, jako je nachlazení,
• máte krvácivé potíže, lehce se Vám tvoří modřiny nebo užíváte lék, který zabraňuje srážení
krve,
• máte oslabený imunitní systém, kvůli onemocnění, jako je infekce virem HIV, nebo užíváte lék, jako je kortikosteroid, který imunitní systém ovlivňuje.
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy (zánětu srdečního svalu) a perikarditidy (zánětu osrdečníku (perikardu)) (viz bod 4). Tato onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhém očkování a častěji u mladších mužů a chlapců. Riziko myokarditidy a perikarditidy je pravděpodobně nižší u dětí ve věku od 5 do 11 let v porovnání s dospívajícími ve věku od 12 do
17 let. Po očkování u Vašeho dítěte pozorně sledujte příznaky myokarditidy a perikarditidy, například dušnost, silný tlukot srdce a bolest na hrudi, a pokud se u Vás vyskytnou, ihned vyhledejte lékařskou
pomoc.
Podobně jako u jiných vakcín je možné, že vakcína Comirnaty Original/Omicron BA.4-5 nebude
plně chránit všechny osoby, kterým bude vakcína podána a není známo, jak dlouho budou chráněny.
Účinnost vakcíny Comirnaty Original/Omicron BA.4-5 nižší u osob s oslabenou imunitou. V těchto případech byste měl(a) i nadále dodržovat preventivní opatření, která pomáhají předcházet onemocnění COVID-19. Kromě toho by měly být podle potřeby očkovány i Vaše blízké kontakty. Proberte příslušná individuální doporučení se svým lékařem.
Děti
Vakcína Comirnaty Original/Omicron BA.4-5 (15/15 mikrogramů)/dávku injekční disperze není
určena pro děti ve věku do 12 let.
Pro děti ve věku 5 až 11 let je k dispozici léková forma pro děti. Podrobnosti naleznete v příbalové informaci vakcíny Comirnaty Original/Omicron BA.4-5 (5/5 mikrogramů)/dávku koncentrát pro injekční disperzi.
Další léčivé přípravky a vakcína Comirnaty Original/Omicron BA.4-5
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době
užíval(a) nebo které možná budete užívat, a o tom, zda Vám byla v nedávné době podána jiná vakcína.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte
se se svým lékařem, zdravotní sestrou nebo lékárníkem dříve, než Vám bude tato vakcína podána.
Údaje o použití vakcíny Comirnaty Original/Omicron BA.4-5 během těhotenství nejsou k dispozici. Ovšem velké množství informací o těhotných ženách očkovaných původně schválenou vakcínou Comirnaty během druhého a třetího trimestru neprokázalo negativní účinky na těhotenství nebo novorozence. Informace o účincích na těhotenství nebo novorozence po očkování během prvního trimestru jsou sice omezené, ale nebylo pozorováno zvýšení rizika potratu. Vakcínu Comirnaty Original/Omicron BA.4-5 lze podávat během těhotenství.
Údaje o použití vakcíny Comirnaty Original/Omicron BA.4-5 během kojení nejsou k dispozici.
Nicméně se neočekávají žádné účinky na kojeného novorozence/kojence. Údaje od žen, které kojily
po očkování původně schválenou vakcínou Comirnaty, neudávaly riziko nežádoucích účinků kojených novorozenců/kojenců. Vakcínu Comirnaty Original/Omicron BA.4-5 lze podávat během kojení.
Řízení dopravních prostředků a obsluha strojů
Některé účinky očkování uvedené v bodě 4 (Možné nežádoucí účinky) mohou dočasně ovlivnit Vaši
schopnost řídit nebo obsluhovat stroje. Před řízením nebo obsluhou strojů počkejte, dokud tyto účinky
neodezní.
3. Jak se vakcína Comirnaty Original/Omicron BA.4-5 podává
Vakcína Comirnaty Original/Omicron BA.4-5 se podává jako injekce o objemu 0,3 ml do svalu v horní části paže.
Vakcínu Comirnaty Original/Omicron BA.4-5 lze podávat nejméně 3 měsíce po poslední minulé dávce vakcíny proti onemocnění COVID-19.
Vakcína Comirnaty Original/Omicron BA.4-5 je indikována pouze u jedinců, kteří již dříve
absolvovali alespoň základní očkování proti onemocnění COVID-19.
O způsobilosti a načasování posilovací dávky se prosím poraďte se svým zdravotnickým pracovníkem.
Podrobnosti o základním očkování u jedinců ve věku 12 let a starších naleznete v příbalové informaci vakcíny Comirnaty 30 mikrogramů/dávku injekční disperze nebo Comirnaty 30 mikrogramů/dávku koncentrátu injekční disperze.
Máte-li jakékoli další otázky týkající se používání vakcíny Comirnaty Original/Omicron BA.4-5,
zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny vakcíny může mít i vakcína Comirnaty Original/Omicron BA.4-5 nežádoucí
účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky se mohou vyskytnout s následující četností:
Velmi časté nežádoucí účinky: mohou postihnout více než 1 z 10 lidí
• v místě injekce: bolest, zduření
• únava
• bolest hlavy
• bolest svalů
• zimnice
• bolest kloubů
• průjem
• horečka
Některé z těchto nežádoucích účinků se o něco častěji vyskytovaly u dospívajících ve věku 12 až
15 let než u dospělých.
Časté nežádoucí účinky: mohou postihnout až 1 z 10 lidí
• zarudnutí v místě injekce
• pocit na zvracení
• zvracení
Méně časté nežádoucí účinky: mohou postihnout až 1 ze 100 lidí
• zvětšené mízní uzliny (častěji pozorované po posilovací dávce)
• malátnost
• bolest paže
• nespavost
• svědění v místě injekce
• alergické reakce, jako je vyrážka nebo svědění
• pocit slabosti nebo nedostatku energie/ospalost
• snížená chuť k jídlu
• nadměrné pocení
• noční pocení
Vzácné nežádoucí účinky: mohou postihnout až 1 z 1 000 lidí
• dočasná jednostranná obrna lícního nervu
• alergické reakce, jako je kopřivka nebo otok obličeje
Velmi vzácné nežádoucí účinky: mohou postihnout až 1 z 10 000 lidí
• zánět srdečního svalu (myokarditida) nebo zánět osrdečníku (perikarditida), který může vést
k dušnosti, silnému tlukotu srdce nebo bolesti na hrudi
Není známo (z dostupných údajů nelze určit)
• závažná alergická reakce
• rozsáhlý otok končetiny, do které byla vakcína podána
• otok obličeje (otok obličeje se může objevit u pacientů, kterým byly do obličeje aplikovány
injekce kožních výplní)
• kožní reakce způsobující červené skvrny nebo fleky na kůži, které mohou vypadat jako terč (nebo střed terče) s tmavě červeným středem, který je obklopen kruhy světlejší červené barvy (erythema multiforme)
• neobvyklý pocit na kůži, jako je brnění nebo pocit šimrání (parestezie)
• snížený pocit nebo citlivost, zejména v kůži (hypestezie)
• silné menstruační krvácení (většina případů se zdála být nezávažné a dočasné povahy)
Hlášení nežádoucích účinkůPokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního
systému hlášení nežádoucích účinků uvedeného Dodatku V. Uveďte přitom číslo šarže, je-li
k dispozici. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak vakcínu Comirnaty Original/Omicron BA.4-5 uchovávatUchovávejte tento přípravek mimo dohled a dosah dětí.
Následující informace o uchovávání, době použitelnosti a použití a manipulaci jsou určeny pro
zdravotnické pracovníky.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána buď při teplotě -90 °C až -60 °C a nebo 2 °C až 8 °C.
Jednodávkové injekční lahvičky: Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C
až -60 °C, lze balení 10 jednodávkových injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu
2 hodin nebo mohou být jednotlivé injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Vícedávkové injekční lahvičky: Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C
až -60 °C, lze balení obsahující 10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu
6 hodin nebo mohou být jednotlivé injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Rozmrazené injekční lahvičky: Po vyjmutí z prostoru pro uchovávání ve zmrazeném stavu může být neotevřená injekční lahvička uchovávána a transportována chlazená při teplotě 2 °C až 8 °C po dobu až 10 týdnů; nesmí být překročeno vytištěné datum použitelnosti (EXP). Vnější obal má být označen novým datem likvidace při teplotě 2 °C až 8 °C. Po rozmrazení nemůže být vakcína znovu zmrazena.
Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Otevřené injekční lahvičky: Po prvním vpichu vakcínu uchovávejte při teplotě 2 °C až 30 °C
a použijte ji do 12 hodin, která zahrnuje až 6 hodin transportu. Případnou nepoužitou vakcínu
zlikvidujte.
Vakcínu nepoužívejte, pokud si všimnete částic nebo změny barvy.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co vakcína Comirnaty Original/Omicron BA.4-5 obsahuje
• Léčivými látkami mRNA vakcíny proti onemocnění COVID-19 jsou tozinameran a famtozinameran.
– Jednodávková injekční lahvička obsahuje 1 dávku, jedna dávka 0,3 ml obsahuje
15 mikrogramů tozinameranu a 15 mikrogramů famtozinameranu (Omikron BA.4-5).
– Vícedávková injekční lahvička obsahuje 6 dávek, jedna dávka 0,3 ml
obsahuje 15 mikrogramů tozinameranu (Original) a 15 mikrogramů famtozinameranu
(Omikron BA.4-5).
• Dalšími složkami jsou:
- ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
- 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
- kolfosceryl-stearát
- cholesterol
- trometamol
- trometamol-hydrochlorid
- sacharosa
- voda pro injekci
Jak vakcína Comirnaty Original/Omicron BA.4-5 vypadá a co obsahuje toto balení
Vakcína je bílá až téměř bílá disperze (pH: 6,9–7,9), která se dodává:
• V jednodávkové čiré injekční lahvičce (sklo třídy I) o objemu 2 ml obsahující 1 dávku.
Injekční lahvička je uzavřena pryžovou zátkou a šedým odtrhovacím plastovým víčkem
s hliníkovým krytem.
• Ve vícedávkové čiré injekční lahvičce (sklo třídy I) o objemu 2 ml obsahující 6 dávek.
Injekční lahvička je uzavřena pryžovou zátkou a šedým odtrhovacím plastovým víčkem
s hliníkovým krytem.
Velikosti balení jednodávkových injekčních lahviček: 10 injekčních lahviček.
Velikosti balení vícedávkových injekčních lahviček: 10 nebo 195 injekčních lahviček. Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci BioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.deVýrobciBioNTech Manufacturing GmbH
Kupferbergterrasse 17-19
55116 Mainz
Německo
Pfizer Manufacturing Belgium NV Rijksweg 12
2870 Puurs
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
B
e
l
gië/Belgique/Belgien
L
uxembourg/Luxemburg
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +370 52 51 4000
Б
ълг
ария
Пфайзер Люксембург САРЛ, Клон
България
Teл: +359 2 970 4333
Magyarország
Pfizer Kft
Tel: +36 1 488 3700
Č
eská republika
Pfizer, spol. s r.o.
Tel: +420 283 004 111
Malta
Vivian Corporation Ltd. Tel: +35621 344610
D
anmark
Pfizer ApS
Tlf: +45 44 201 100
Norge
Pfizer AS
Tlf: +47 67 526 100
D
eutschland
BioNTech Manufacturing GmbH Tel: +49 6131 90840
Nederland
Pfizer BV
Tel: +31 (0)10 406 43 01
E
esti
Pfizer Luxembourg SARL Eesti filiaal
Tel: +372 666 7500
Österreich
Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1 521 15-0
Ε
λλάδα
Pfizer Ελλάς A.E.
Τηλ.: +30 210 6785 800
Polska
Pfizer Polska Sp. z o.o. Tel.: +48 22 335 61 00
E
s
paña
Pfizer, S.L.
Tél: +34914909900
Portugal
Laboratórios Pfizer, Lda.
Tel: +351 21 423 5500
F
rance
Pfizer
Tél +33 1 58 07 34 40
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
H
rvatska
Pfizer Croatia d.o.o.
Tel: +385 1 3908 777
Slovenija
Pfizer Luxembourg SARL
Pfizer, podružnica za svetovanje s področja
farmacevtske dejavnosti, Ljubljana
Tel.: +386 (0) 1 52 11 400
Ireland
Pfizer Healthcare Ireland
Tel: 1800 633 363 (toll free)
+44 (0)1304 616161
Slovenská republika
Pfizer Luxembourg SARL,
organizačná zložka
Tel: +421 2 3355 5500
Ísland
Icepharma hf
Simi: +354 540 8000
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
It
alia
Pfizer S.r.l.
Tel: +39 06 33 18 21
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
Κύ
πρ
ος
Pfizer Ελλάς Α.Ε. (Cyprus Branch)
Tηλ: +357 22 817690
United Kingdom (Northern Ireland)
Pfizer Limited
Tel: +44 (0) 1304 616161
L
atvija
Pfizer Luxembourg SARL filiāle Latvijā
Tel.: +371 670 35 775
Tato příbalová informace byla naposledy revidována {MM/RRRR}Pro zobrazení příbalové informace v různých jazycích sejměte následující kód pomocí chytrého zařízení.
URL:
www.comirnatyglobal.com
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
------------------------------------------------------------------------------------------------------------------------
Následující informace jsou určeny pouze pro zdravotnické pracovníky:Dávka vakcíny Comirnaty Original/Omicron BA.4-5 je 0,3 ml podávaná intramuskulárně.
Mezi podáním vakcíny Comirnaty Original/Omicron BA.4-5 a poslední předchozí dávkou vakcíny
proti COVID-19 má uplynout interval nejméně 3 měsíce.
Vakcína Comirnaty Original/Omicron BA.4-5 je indikována pouze u jedinců, kteří již dříve
podstoupili alespoň základní očkování proti onemocnění COVID-19.
SledovatelnostAby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Instrukce pro zacházení s vakcínouVakcína Comirnaty Original/Omicron BA.4-5 (15/15 mikrogramů)/dávku má být připravována zdravotnickým pracovníkem pomocí aseptické techniky, aby byla zajištěna sterilita připravené
disperze.
NÁV
O
D SE VZTAHUJE NA JEDNODÁVKOVÉ I VÍCEDÁVKOVÉ INJEKČNÍ LAHVIČKY
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.4-5 (15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
Š
e
d
é víčko
C
omirnaty Original/ Omicron BA.4-
5
N
eřeďte
• Zkontrolujte, zda má injekční lahvička šedé plastové víčko a šedý okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze.
• Zkontrolujte, zda se jedná
o jednodávkovou nebo vícedávkovou injekční lahvičku, a řiďte se příslušnými pokyny pro zacházení uvedenými níže.
• Pokud má injekční lahvička šedé plastové víčko a šedý okraj a název přípravku je Comirnaty
30 mikrogramů/dávku injekční disperze
nebo Comirnaty Original/Omicron
BA.1 (15/15 mikrogramů)/dávku injekční disperze, přečtěte si, prosím, souhrn údajů o přípravku pro příslušné složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi nebo Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte
si souhrn údajů o přípravku pro vakcínu
Comirnaty 3 mikrogramy/dávku
koncentrát pro injekční disperzi.
Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.4-5
(
15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ) PŘED
P
O
U
Ž
ITÍM
|
U
chovávejte po dobu až 10 týdnů při teplotě 2 °C
až 8 °C,
aktualizujte datum použitelnosti na krabičce.
|
• Pokud se jednodávková nebo vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se rozmrazily. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
o Jednodávkové injekční lahvičky:
Rozmrazení balení
10 jednodávkových injekčních lahviček může trvat 2 hodiny.
o Vícedávkové injekční lahvičky:
Rozmrazení balení
10 vícedávkových injekčních lahviček může trvat 6 hodin.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
Jemně
× 10
|
• Injekční lahvičky před použitím 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Před smícháním může rozmrazená disperze obsahovat bílé až téměř bílé, matné amorfní částice.
• Po smíchání se má vakcína jevit jako bílá až téměř bílá disperze bez viditelných částic. Jestliže jsou
v naředěné vakcíně patrné částice nebo vakcína změnila barvu, nepoužívejte ji.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,3 ml VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.4-5 (15/15 MIKROGRAMŮ)/DÁVKU INJEKČNÍ DISPERZE (12 LET A STARŠÍ)
 V
akcína o objemu 0,3 ml
J
ednodávkové
i
n
j
ekční
l
ahvičky
V
akcína o objemu 0,3 ml
J
ednodávkové
i
n
j
ekční
l
ahvičky
• Natáhněte jednu 0,3ml dávku vakcíny.
• Injekční lahvičku a veškerý přebytečný
objem zlikvidujte.
Vícedávkovéinjekčnílahvičky• Vícedávkové injekční lahvičky obsahují
6 dávek po 0,3 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím antiseptického tampónu.
• Natáhněte 0,3 ml vakcíny Comirnaty
Original/Omicron BA.4-5.
K získání šesti dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně 35 mikrolitrů.
Pokud se používají standardní injekční stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání šesté dávky z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,3 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,3 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Zaznamenejte příslušné datum/čas na injekční lahvičku. Veškerou vakcínu, jež nebyla použita do 12 hodin po prvním vpichu, zlikvidujte.
L
i
kvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
P
ř
í
balová informace: informace pro uživatele
C
omirnaty Original/Omicron BA.4-5 (5/5 mikrogramů)/dávku koncentrát pro injekční disperzi
D
ěti ve věku 5 až 11 let
m
RN
A vakcína proti onemocnění COVID-19 (modifikovaný nukleosid)
tozinameran/famtozinameran
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací
o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vašeho dítěte
vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tato vakcína podána, protože obsahuje pro Vás důležité údaje.• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Pokud se u Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci1. Co je vakcína Comirnaty Original/Omicron BA.4-5 a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vašemu dítěti bude vakcína Comirnaty Original/Omicron
BA.4-5 podána
3. Jak se vakcína Comirnaty Original/Omicron BA.4-5 podává
4. Možné nežádoucí účinky
5. Jak vakcínu Comirnaty Original/Omicron BA.4-5 uchovávat
6. Obsah balení a další informace
1. Co je vakcína Comirnaty Original/Omicron BA.4-5 a k čemu se používáComirnaty Original/Omicron BA.4-5 je vakcína používaná pro prevenci onemocnění COVID-19,
které způsobuje SARS-CoV-2. Podává se dětem ve věku 5 až 11 let.
Vakcína Comirnaty Original/Omicron BA.4-5 je určena pouze pro jednotlivce, kteří již předtím
absolvovali alespoň základní očkování proti onemocnění COVID-19.
Vakcína způsobuje, že imunitní systém (přirozená obrana těla) vytváří protilátky a krevní buňky, které
působí proti viru, a tím chrání před onemocněním COVID-19.
Jelikož vakcína Comirnaty Original/Omicron BA.4-5 neobsahuje virus k vyvolání imunity, nemůže u
Vašeho dítěte vyvolat onemocnění COVID-19.
2. Čemu musíte věnovat pozornost, než Vašemu dítěti bude vakcína ComirnatyOriginal/Omicron BA.4-5 podánaVakcína Comirnaty Original/Omicron BA.4-5 Vám nesmí být podána• jestliže je Vaše dítě alergické na léčivou látku nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Upozornění a opatřeníPřed podáním vakcíny Vašemu dítěti se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou,
jestliže Vaše dítě:
• někdy mělo závažnou alergickou reakci nebo potíže s dýcháním po jakékoli jiné vakcíně podané v injekci nebo poté, co mu byla v minulosti podána vakcína Comirnaty nebo Comirnaty Original/Omicron BA.4-5,
• je v souvislosti s očkováním nervózní nebo někdy po jakékoli injekci omdlelo,
• má závažné onemocnění nebo infekci s vysokou horečkou. Avšak očkování může Vaše dítě podstoupit, pokud má mírně zvýšenou teplotu nebo lehkou infekci horních cest dýchacích, jako je nachlazení,
• má krvácivé potíže, lehce se mu tvoří modřiny nebo užívá lék, který zabraňuje srážení krve,
• má oslabený imunitní systém, kvůli onemocnění, jako je infekce virem HIV, nebo užívá lék,
jako je kortikosteroid, který imunitní systém ovlivňuje.
Po očkování vakcínou Comirnaty existuje zvýšené riziko myokarditidy (zánětu srdečního svalu) a perikarditidy (zánětu osrdečníku (perikardu)) (viz bod 4). Tato onemocnění se mohou objevit během několika málo dnů po očkování a vyskytla se zejména v průběhu prvních 14 dnů. Byla pozorována častěji po druhém očkování a častěji u mladších mužů a chlapců. Riziko myokarditidy a perikarditidy je pravděpodobně nižší u dětí ve věku od 5 do 11 let v porovnání s dospívajícími ve věku od 12 do
17 let. Po očkování u Vašeho dítěte pozorně sledujte příznaky myokarditidy a perikarditidy, například
dušnost, silný tlukot srdce a bolest na hrudi, a pokud se u něj vyskytnou, ihned vyhledejte lékařskou
pomoc.
Podobně jako u jiných vakcín je možné, že vakcína Comirnaty Original/Omicron BA.4-5 nebude
plně chránit všechny osoby, kterým bude vakcína podána a není známo, jak dlouho budou chráněny.
Účinnost vakcíny Comirnaty Original/Omicron BA.4-5 může být nižší u osob s oslabenou imunitou. V těchto případech byste měli i nadále dodržovat preventivní opatření, která pomáhají předcházet onemocnění COVID-19. Kromě toho by měly být podle potřeby očkovány i Vaše blízké kontakty. Proberte příslušná individuální doporučení s lékařem.
Děti
Vakcína Comirnaty Original/Omicron BA.4-5 (5/5 mikrogramů)/dávku koncentrát pro injekční
disperzi není určena pro děti mladší 5 let.
Další léčivé přípravky a vakcína Comirnaty Original/Omicron BA.4-5
Informujte svého lékaře nebo lékárníka o všech lécích, které Vaše dítě užívá, které v nedávné době
užívalo nebo které možná bude užívat, a o tom, zda mu byla v nedávné době podána jiná vakcína.
Těhotenství a kojení
Pokud je Vaše dcera těhotná nebo kojí, poraďte s lékařem, zdravotní sestrou nebo lékárníkem dříve,
než jí bude tato vakcína podána.
Údaje o použití vakcíny Comirnaty Original/Omicron BA.4-5 během těhotenství nejsou k dispozici. Ovšem velké množství informací o těhotných ženách očkovaných původně schválenou vakcínou Comirnaty během druhého a třetího trimestru neprokázalo negativní účinky na těhotenství nebo novorozence. Informace o účincích na těhotenství nebo novorozence po očkování během prvního trimestru jsou sice omezené, ale nebylo pozorováno zvýšení rizika potratu. Vakcínu Comirnaty Original/Omicron BA.4-5 lze podávat během těhotenství.
Údaje o použití vakcíny Comirnaty Original/Omicron BA.4-5 během kojení nejsou k dispozici.
Nicméně se neočekávají žádné účinky na kojeného novorozence/kojence. Údaje od žen, které kojily
po očkování původně schválenou vakcínou Comirnaty, neudávaly riziko nežádoucích účinků kojených novorozenců/kojenců. Vakcínu Comirnaty Original/Omicron BA.4-5 lze podávat během kojení.
Řízení dopravních prostředků a obsluha strojů
Některé účinky očkování uvedené v bodě 4 (Možné nežádoucí účinky) mohou dočasně ovlivnit
schopnost řídit nebo obsluhovat stroje nebo provádět aktivity, jako je jízda na kole. Před znovuzahájením aktivit, které vyžadují plnou pozornost počkejte, dokud tyto účinky neodezní.
3. Jak se vakcína Comirnaty Original/Omicron BA.4-5 podává
Vakcína Comirnaty Original/Omicron BA.4-5 se podává po naředění jako injekce o objemu 0,2 ml do svalu v horní části paže.
Vakcínu Comirnaty Original/Omicron BA.4-5 lze podávat nejméně 3 měsíce po poslední minulé
dávce vakcíny proti onemocnění COVID-19.
Vakcína Comirnaty Original/Omicron BA.4-5 je indikována pouze u jedinců, kteří již dříve
absolvovali alespoň základní očkování proti onemocnění COVID-19.
O způsobilosti a načasování posilovací dávky se prosím poraďte se svým zdravotnickým pracovníkem. Podrobnosti o základním očkování u dětí ve věku od 5 do 11 let naleznete v příbalové informaci
vakcíny Comirnaty 10 mikrogramů/dávku koncentrátu injekční disperze.
Máte-li jakékoli další otázky týkající se používání vakcíny Comirnaty Original/Omicron BA.4-5,
zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny vakcíny může mít i vakcína Comirnaty Original/Omicron BA.4-5 nežádoucí
účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky se mohou vyskytnout s následující četností:
Velmi časté nežádoucí účinky: mohou postihnout více než 1 z 10 lidí
• v místě injekce: bolest, zduření
• únava
• bolest hlavy
• bolest svalů
• zimnice
• bolest kloubů
• průjem
• horečka
Časté nežádoucí účinky: mohou postihnout až 1 z 10 lidí
• pocit na zvracení
• zvracení
• zarudnutí v místě injekce (‘velmi časté’ ve věku od 5 do 11 let)
Méně časté nežádoucí účinky: mohou postihnout až 1 ze 100 lidí
• zvětšené mízní uzliny (častěji pozorované po posilovací dávce)
• malátnost
• bolest paže
• nespavost
• svědění v místě injekce
• alergické reakce, jako je vyrážka nebo svědění
• pocit slabosti nebo nedostatku energie/ospalost
• snížená chuť k jídlu
• nadměrné pocení
• noční pocení
Vzácné nežádoucí účinky: mohou postihnout až 1 z 1 000 lidí
• dočasná jednostranná obrna lícního nervu
• alergické reakce, jako je kopřivka nebo otok obličeje
Velmi vzácné nežádoucí účinky: mohou postihnout až 1 z 10 000 lidí
• zánět srdečního svalu (myokarditida) nebo zánět osrdečníku (perikarditida), který může vést
k dušnosti, silnému tlukotu srdce nebo bolesti na hrudi
Není známo (z dostupných údajů nelze určit)
• závažná alergická reakce
• rozsáhlý otok končetiny, do které byla vakcína podána
• otok obličeje (otok obličeje se může objevit u pacientů, kterým byly do obličeje aplikovány
injekce kožních výplní)
• kožní reakce způsobující červené skvrny nebo fleky na kůži, které mohou vypadat jako terč (nebo střed terče) s tmavě červeným středem, který je obklopen kruhy světlejší červené barvy (erythema multiforme)
• neobvyklý pocit na kůži, jako je brnění nebo pocit šimrání (parestezie)
• snížený pocit nebo citlivost, zejména v kůži (hypestezie)
• silné menstruační krvácení (většina případů se zdála být nezávažné a dočasné povahy)
Hlášení nežádoucích účinkůPokud se u Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi
nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného Dodatku V. Uveďte přitom číslo šarže, je-li k dispozici. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak vakcínu Comirnaty Original/Omicron BA.4-5 uchovávatUchovávejte tento přípravek mimo dohled a dosah dětí.
Následující informace o uchovávání, době použitelnosti a použití a manipulaci jsou určeny pro
zdravotnické pracovníky.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v mrazničce při teplotě -90 °C až -60 °C.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Vakcína bude obdržena ve zmrazeném stavu při teplotě -90 °C až -60 °C. Zmrazená vakcína může být po přijetí uchovávána buď při teplotě -90 °C až -60 °C a nebo 2 °C až 8 °C.
Pokud je vakcína uchovávána ve zmrazeném stavu při teplotě -90 °C až -60 °C, lze balení obsahující
10 injekčních lahviček rozmrazit při teplotě 2 °C až 8 °C po dobu 4 hodin nebo mohou být jednotlivé
injekční lahvičky rozmrazeny při pokojové teplotě (do 30 °C) po dobu 30 minut.
Po jednorázovém vyjmutí z mrazničky může být neotevřená injekční lahvička uchovávána a transportována chlazená při teplotě 2 °C až 8 °C po dobu až 10 týdnů; nesmí být překročeno vytištěné datum použitelnosti (EXP). Vnější obal má být označen novým datem likvidace při teplotě 2 °C až
8 °C. Po jednorázovém rozmrazení se vakcína nesmí znovu zmrazit.
Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě 8 °C až 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
Po naředění vakcínu uchovávejte při teplotě 2 °C až 30 °C a použijte ji do 12 hodin, která zahrnuje až
6 hodin transportu. Případnou nepoužitou vakcínu zlikvidujte.
Vakcínu nepoužívejte, pokud si všimnete, že jsou v naředěném roztoku částice nebo že roztok změnil
barvu.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informaceCo vakcína Comirnaty Original/Omicron BA.4-5 obsahuje• Léčivými látkami mRNA vakcíny proti onemocnění COVID-19 jsou tozinameran
a famtozinameran. Po naředění injekční lahvička obsahuje 10 dávek, jedna dávka 0,2 ml obsahuje 5 mikrogramů tozinameranu (Original) a 5 mikrogramů famtozinameranu (Omikron BA.4-5).
• Dalšími složkami jsou:
- ((4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate) (ALC-0315)
- 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide (ALC-0159)
- kolfosceryl-stearát
- cholesterol
- trometamol
- trometamol-hydrochlorid
- sacharosa
- voda pro injekci
Jak vakcína Comirnaty Original/Omicron BA.4-5 vypadá a co obsahuje toto baleníVakcína je bílá až téměř bílá disperze (pH: 6,9–7,9), která se dodává ve vícedávkové čiré injekční
lahvičce (sklo třídy I) o objemu 2 ml obsahující 10 dávek. Injekční lahvička je uzavřena pryžovou
zátkou a odtrhovacím oranžovým plastovým víčkem s hliníkovým krytem.
Velikosti balení: 10 nebo 195 injekčních lahviček
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci BioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Německo
tel.: +49 6131 9084-0
fax: +49 6131 9084-2121
service@biontech.deVýrobciBioNTech Manufacturing GmbH Kupferbergterrasse 17-19
55116 Mainz
Německo
Pfizer Manufacturing Belgium NV Rijksweg 12
2870 Puurs
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
B
e
l
gië/Belgique/Belgien
L
uxembourg/Luxemburg
Pfizer S.A./N.V.
Tél/Tel: +32 (0)2 554 62 11
Lietuva
Pfizer Luxembourg SARL filialas Lietuvoje
Tel. +370 52 51 4000
Б
ълг
ария
Пфайзер Люксембург САРЛ, Клон
България
Teл: +359 2 970 4333
Magyarország
Pfizer Kft
Tel: +36 1 488 3700
Č
eská republika
Pfizer, spol. s r.o.
Tel: +420 283 004 111
Malta
Vivian Corporation Ltd. Tel: +35621 344610
D
anmark
Pfizer ApS
Tlf: +45 44 201 100
Norge
Pfizer AS
Tlf: +47 67 526 100
D
eutschland
BioNTech Manufacturing GmbH
Tel: +49 6131 90840
Nederland
Pfizer BV
Tel: +31 (0)10 406 43 01
E
esti
Pfizer Luxembourg SARL Eesti filiaal
Tel: +372 666 7500
Österreich
Pfizer Corporation Austria Ges.m.b.H Tel: +43 (0)1 521 15-0
Ε
λλάδα
Pfizer Ελλάς A.E.
Τηλ.: +30 210 6785 800
Polska
Pfizer Polska Sp. z o.o.
Tel.: +48 22 335 61 00
E
s
paña
Pfizer, S.L.
Tél: +34914909900
Portugal
Laboratórios Pfizer, Lda.
Tel: +351 21 423 5500
F
rance
Pfizer
Tél +33 1 58 07 34 40
România
Pfizer Romania S.R.L
Tel: +40 (0) 21 207 28 00
H
rvatska
Pfizer Croatia d.o.o.
Tel: +385 1 3908 777
Slovenija
Pfizer Luxembourg SARL
Pfizer, podružnica za svetovanje s področja
farmacevtske dejavnosti, Ljubljana
Tel.: +386 (0) 1 52 11 400
Ireland
Pfizer Healthcare Ireland
Tel: 1800 633 363 (toll free)
+44 (0)1304 616161
Slovenská republika
Pfizer Luxembourg SARL,
organizačná zložka
Tel: +421 2 3355 5500
Ísland
Icepharma hf
Simi: +354 540 8000
Suomi/Finland
Pfizer Oy
Puh/Tel: +358 (0)9 430 040
It
alia
Pfizer S.r.l.
Tel: +39 06 33 18 21
Sverige
Pfizer AB
Tel: +46 (0)8 550 520 00
Κύ
πρ
ος
Pfizer Ελλάς Α.Ε. (Cyprus Branch)
Tηλ: +357 22 817690
United Kingdom (Northern Ireland)
Pfizer Limited
Tel: +44 (0) 1304 616161
L
atvija
Pfizer Luxembourg SARL filiāle Latvijā
Tel.: +371 670 35 775
Tato příbalová informace byla naposledy revidována {MM/RRRR}Pro zobrazení příbalové informace v různých jazycích sejměte následující kód pomocí chytrého
zařízení.

URL:
www.comirnatyglobal.comPodrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské
agentury pro léčivé přípravky
http://www.ema.europa.eu/Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
------------------------------------------------------------------------------------------------------------------------
Následující informace jsou určeny pouze pro zdravotnické pracovníky:Dávka vakcíny Comirnaty Original/Omicron BA.4-5 je 0,2 ml podávaná intramuskulárně.
Mezi podáním vakcíny Comirnaty Original/Omicron BA.4-5 a poslední předchozí dávkou vakcíny
proti COVID-19 má uplynout interval nejméně 3 měsíce.
Vakcína Comirnaty Original/Omicron BA.4-5 je indikována pouze u jedinců, kteří již dříve
podstoupili alespoň základní očkování proti onemocnění COVID-19.
SledovatelnostAby se zlepšila sledovatelnost biologických léčivých přípravků, má se přehledně zaznamenat název
podaného přípravku a číslo šarže.
Instrukce pro zacházení s vakcínouVakcína Comirnaty Original/Omicron BA.4-5 (5/5 mikrogramů)/dávku má být připravována
zdravotnickým pracovníkem pomocí aseptické techniky, aby byla zajištěna sterilita připravené
disperze.
KO
NT
R
O
L
A INJEKČNÍ LAHVIČKY S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.4-5 (5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
• Zkontrolujte, zda má injekční lahvička
O
ranžové
víčko
C
omirnaty Original/ Omicron BA.4-5
P
o naředění
5/5 mikrogramů
oranžové plastové víčko a oranžový okraj kolem štítku a název přípravku je Comirnaty Original/Omicron BA.4-5
(5/5 mikrogramů)/dávku koncentrát pro
injekční disperzi.
• Pokud má injekční lahvička oranžové plastové víčko a oranžový okraj kolem štítku a název přípravku je Comirnaty
10 mikrogramů/dávku koncentrát pro injekční disperzi, přečtěte si souhrn údajů o přípravku pro toto složení.
• Pokud má injekční lahvička fialové plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku koncentrát pro injekční disperzi.
• Pokud má injekční lahvička šedé
plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty
30 mikrogramů/dávku injekční disperze, Comirnaty Original/Omicron BA.1 (15/15 mikrogramů)/dávku injekční
disperze nebo Comirnaty
Original/Omicron BA.4-5
(15/15 mikrogramů)/dávku injekční
disperze.
• Pokud má injekční lahvička hnědočervené plastové víčko, přečtěte si souhrn údajů o přípravku pro vakcínu Comirnaty 3 mikrogramy/dávku koncentrát pro injekční disperzi.
Z
A
C
H
Á
Z
E
N
Í S VAKCÍNOU COMIRNATY ORIGINAL/OMICRON BA.4-5
(
5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET) PŘED POUŽITÍM
|
U
chovávejte až
10 týdnů při teplotě 2 °C až
8 °C.
|
• Pokud se vícedávková injekční lahvička uchovává zmrazená, musí se před použitím rozmrazit. Zmrazené injekční lahvičky je třeba přenést do prostředí
o teplotě 2 °C až 8 °C, aby se rozmrazily;
rozmrazení balení 10 injekčních lahviček může trvat 4 hodiny. Ujistěte se, že jsou injekční lahvičky před použitím úplně rozmrazené.
• Po přenesení injekčních lahviček pro uchovávání při teplotě 2 °C až 8 °C aktualizujte datum použitelnosti na krabičce.
• Neotevřené injekční lahvičky mohou být uchovávány po dobu až 10 týdnů při teplotě 2 °C až 8 °C; nesmí být překročeno vytištěné datum použitelnosti (EXP).
• Alternativně lze jednotlivé zmrazené injekční lahvičky rozmrazovat po dobu
30 minut při teplotě do 30 °C.
• Neotevřenou injekční lahvičku lze před použitím uchovávat až 12 hodin při teplotě do 30 °C. S rozmrazenými injekčními lahvičkami lze manipulovat za podmínek osvětlení místnosti.
|
MÍCHÁNÍ VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.4-5
(
5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET) PŘED ŘEDĚNÍM
|
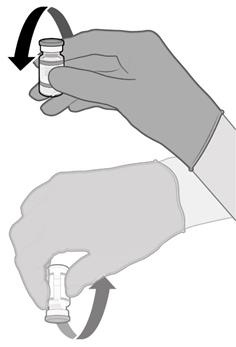
Jemně × 10
|
• Rozmrazenou injekční lahvičku nechte ohřát na pokojovou teplotu a před ředěním ji 10krát jemně převraťte. Netřepejte.
• Před ředěním může rozmrazená disperze obsahovat bílé až téměř bílé matné amorfní částice.
|
ŘEDĚN
Í VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.4-5
(
5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
|
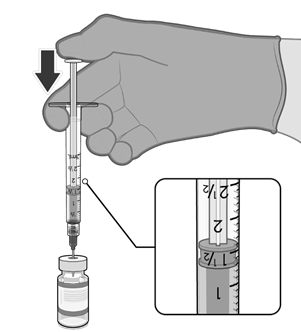
1,3 ml injekčního roztoku chloridu
sodného 9 mg/ml (0,9 %)
|
• Rozmrazená vakcína se musí naředit v původní injekční lahvičce 1,3 ml injekčního roztoku chloridu sodného
9 mg/ml (0,9%) za použití jehly
o velikosti 21 gauge (21G) nebo užší a uplatnění aseptických metod.
|
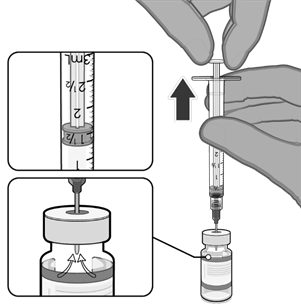
V
ytáhněte píst na značku 1,3 ml pro
odstranění vzduchu z injekční lahvičky.
|
• Nasátím 1,3 ml vzduchu do prázdné stříkačky na rozpouštědlo vyrovnejte před vyjmutím jehly z injekční lahvičky tlak v injekční lahvičce.
|

Jemně x 10
|
• Disperzi 10krát jemně převraťte. Injekční lahvičkou netřepejte.
• Naředěná vakcína se má jevit jako bílá až téměř bílá disperze bez viditelných
částic. Jestliže jsou v naředěné vakcíně
patrné částice nebo vakcína změnila
barvu, nepoužívejte ji.
|
Č
as likvidace
Z
a
z
namenejte příslušné datum a čas. Použijte během 12 hodin po naředění.
|
• Injekční lahvičky s naředěnou vakcínou je třeba označit příslušným datem
a časem.
• Po naředění uchovávejte vakcínu při teplotě 2 °C až 30 °C a použijte ji během
12 hodin.
• Naředěnou disperzi nezmrazujte ani s ní netřepejte. Pokud je v chladu, nechte naředěnou disperzi před použitím dosáhnout pokojové teploty.
|
P
Ř
Í
P
RAV
A JEDNOTLIVÝCH DÁVEK O OBJEMU 0,2 ml VAKCÍNY COMIRNATY ORIGINAL/OMICRON BA.4-5 (5/5 MIKROGRAMŮ)/DÁVKU KONCENTRÁT PRO INJEKČNÍ DISPERZI (DĚTI VE VĚKU 5 AŽ 11 LET)
• Po naředění obsahuje injekční lahvička
2,6 ml, ze kterých lze získat 10 dávek po
0,2 ml.
• Pomocí aseptické techniky očistěte zátku injekční lahvičky použitím
antiseptického tampónu.
• Natáhněte 0,2 ml vakcíny Comirnaty pro
děti ve věku 5 až 11 let.
K získání 10 dávek z jedné injekční lahvičky je třeba použít injekční stříkačky a/nebo jehly s malým mrtvým
prostorem. Kombinace injekční stříkačky a jehly s malým mrtvým prostorem má mít mrtvý objem maximálně
35 mikrolitrů.
N
aředěná vakcína o objemu 0,2 ml
Pokud se používají standardní injekční
stříkačky a jehly, nemusí být zajištěn dostatečný objem k získání deseti dávek
z jedné injekční lahvičky.
• Každá dávka musí obsahovat 0,2 ml vakcíny.
• Pokud množství vakcíny zbývající v injekční lahvičce nemůže poskytnout plnou dávku 0,2 ml, injekční lahvičku a veškerý přebytečný objem zlikvidujte.
• Veškerou vakcínu, jež nebyla použita do
12 hodin po naředění, zlikvidujte.
L
i
kvidace
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.