
. Vo fáze po uvedení na trh bol hlásený opuch tváre u očkovaných osôb s anamnézou podania injekcie s dermatologickým výplňovým materiálom.
Opis vybraných nežiaducich reakciíMyokarditídaZvýšené riziko myokarditídy po očkovaní očkovacou látkou Comirnaty je najvyššie u mladších mužov
(pozri časť 4.4).
V dvoch veľkých, európskych, farmako-epidemiologických štúdiách sa určilo zvýšené riziko u mladších mužov po druhej dávke očkovacej látky Comirnaty.
Z jednej štúdie vyplynulo, že v období 7 dní po podaní druhej dávky sa u mužov vo veku 12–29 rokov
vyskytlo približne o 0,265 (95 % IS 0,255–0,275) prípadov myokarditídy na 10 000 osôb viac ako u
neexponovaných osôb. V ďalšej štúdii sa v období 28 dní po podaní druhej dávky u mužov vo veku
16–24 rokov vyskytlo o 0,57 (95 % IS 0,39–0,75) prípadov myokarditídy na 10 000 osôb viac ako u neexponovaných osôb.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V
a uviedli číslo šarže, ak je k dispozícii.
4.9 PredávkovanieÚdaje o predávkovaní sú dostupné od 52 účastníkov zahrnutých do klinického skúšania, ktorí dostali
z dôvodu chybného nariedenia 58 mikrogramov očkovacej látky Comirnaty. Očkované osoby nehlásili
zvýšenie reaktogenity ani výskytu nežiaducich reakcií.
V prípade predávkovania sa odporúča sledovanie životných funkcií a prípadne symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: očkovacie látky, iné vírusové očkovacie látky, ATC kód: J07BX03
Mechanizmus účinkuMediátorová RNA s modifikovaným nukleozidom v očkovacej látke Comirnaty (tozinameran) je
zapuzdrená v lipidových nanočasticiach, ktoré umožňujú dopravenie nereplikujúcej sa RNA do hostiteľských buniek na priamu dočasnú expresiu S antigénu vírusu SARS-CoV-2. mRNA kóduje
S proteín plnej dĺžky, ukotvený v membráne, s dvoma bodovými mutáciami v rámci centrálnej špirály. Mutácia týchto dvoch aminokyselín na prolín uzamyká S proteín v antigénne uprednostňovanej pred-
fúznej konformácii. Očkovacia látka vyvoláva odpoveď vo forme tvorby neutralizujúcich protilátok ako aj bunkovú imunitnú odpoveď na „spike“ (S) antigén, čo môže prispievať k ochrane pred
ochorením COVID-19.
Ú
činnosť
Štúdia 2 je multicentrová, medzinárodná, randomizovaná, placebom kontrolovaná, pre pozorovateľov
zaslepená štúdia fázy 1/2/3 na stanovenie dávky, výber kandidátov na očkovaciu látku a stanovenie
účinnosti u účastníkov vo veku 12 rokov a starších. Randomizácia bola stratifikovaná podľa veku: vek
12 až 15 rokov, vek 16 až 55 rokov alebo vek 56 rokov a viac, pričom minimálne 40 % účastníkov bolo vo vekovej skupine ≥ 56 rokov. Z účasti na štúdii boli vylúčení imunokompromitovaní účastníci a tí, ktorí mali v minulosti klinicky alebo mikrobiologicky diagnostikovaný COVID-19. Účastníci
s prebiehajúcim stabilným ochorením definovaným ako ochorenie, ktoré si nevyžaduje významnú
zmenu liečby ani hospitalizáciu z dôvodu zhoršenia ochorenia počas 6 týždňov pred zaradením, boli
do tejto štúdie zahrnutí, ako aj účastníci so známou stabilnou infekciou vírusom ľudskej
imunodeficiencie (HIV), vírusom hepatitídy C (HCV) alebo vírusom hepatitídy B (HBV).
Účinnosť u účastníkov vo veku 16 rokov a starších - po 2 dávkachV časti fázy 2/3 štúdie 2 bolo na základe údajov zozbieraných do 14. novembra 2020 približne
44 000 účastníkov rovnomerne randomizovaných do skupín, ktoré dostali 2 dávky mRNA očkovacej látky proti COVID-19 alebo placebo. Analýzy účinnosti zahŕňali účastníkov, ktorí dostali druhú dávku očkovacej látky v intervale 19 až 42 dní po ich prvom očkovaní. Väčšina (93,1 %) očkovaných osôb dostala druhú dávku 19 až 23 dní po 1. dávke. Je plánované sledovanie účastníkov po dobu až
24 mesiacov po 2. dávke na účely hodnotení bezpečnosti a účinnosti proti ochoreniu COVID-19.
V klinickej štúdii sa vyžadovalo, aby sa u účastníkov pri podaní buď placeba alebo mRNA očkovacej látky proti COVID-19 dodržal interval minimálne 14 dní pred a po podaní očkovacej látky proti chrípke. V klinickej štúdii sa vyžadovalo, aby sa u účastníkov pri podaní buď placeba alebo mRNA očkovacej látky proti COVID-19 dodržal interval minimálne 60 dní pred alebo po podaní krvných/plazmových produktov alebo imunoglobulínov v rámci obdobia do ukončenia štúdie.
Populácia na analýzu primárneho cieľového ukazovateľa účinnosti zahŕňala 36 621 účastníkov vo
veku 12 rokov a starších (18 242 v skupine s mRNA očkovacou látkou proti COVID-19
a 18 379 v skupine s placebom), u ktorých nebol do doby 7 dní po druhej dávke zistený dôkaz predchádzajúcej infekcie vírusom SARS-CoV-2. Ďalej, 134 účastníkov bolo vo veku medzi 16 až
17 rokov (66 v skupine s mRNA očkovacou látkou proti COVID-19 a 68 v skupine s placebom)
a 1 616 účastníkov bolo vo veku 75 rokov a starších (804 v skupine s mRNA očkovacou látkou proti
COVID-19 a 812 v skupine s placebom).
V čase primárnej analýzy účinnosti boli účastníci sledovaní z hľadiska symptomatického COVID-19 po dobu spolu 2 214 osoborokov v skupine s mRNA očkovacou látkou proti COVID-19 a po dobu spolu 2 222 osoborokov v skupine s placebom.
U účastníkov ohrozených závažným priebehom COVID-19 vrátane tých s 1 alebo viacerými komorbiditami, ktoré zvyšujú riziko závažného priebehu COVID-19 (napr. astma, index telesnej hmotnosti (BMI) ≥ 30 kg/m2, chronické ochorenie pľúc, diabetes mellitus, hypertenzia), sa nevyskytli žiadne významné klinické rozdiely v celkovej účinnosti očkovacej látky.
Informácie o účinnosti očkovacej látky sú uvedené v tabuľke 2.
Prvý výskyt COVID-19 od 7 dní po 2. dávke u účastníkov bez dôkazu predchádzajúcej
infekcie SARS-CoV-2*
|
Podskupina
| mRNA očkovacia látka
proti COVID-19
na = 18 198
Prípady n1b
Čas sledovaniac (n2d)
|
Placebo na = 18,325
Prípady n1b
Čas sledovaniac (n2d)
|
Účinnosť očkovacej
látky % (95 % IS)e
| Všetci účastníci
| 8
| 162
| 95,0
|
|
|
Tabuľka 2: Účinnosť očkovacej látky - prvý výskyt COVID-
19 od 7 dní po 2. dávke podľa vekových podskupín - účastníci bez dôkazu infekcie do 7 dní po 2. dávke - populácia hodnotiteľná z hľadiska účinnosti (7 dní)
|
2,214 (17 411)
|
2,222 (17 511)
|
(90,0; 97,9)
|
16 až 64 rokov
|
7
1,706 (13 549)
|
143
1,710 (13 618)
|
95,1
(89,6; 98,1)
|
65 rokov a starší
|
1
0,508 (3 848)
|
19
0,511 (3 880)
|
94,7
(66,7; 99,9)
|
65 až 74 rokov
|
1
0,406 (3 074)
|
14
0,406 (3 095)
|
92,9
(53,1; 99,8)
|
75 rokov a starší
|
0
0,102 (774)
|
5
0,106 (785)
|
100,0
(-13,1; 100,0)
|
Poznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou
(RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 [*Definícia prípadu: (aspoň 1 z) horúčka, nový alebo zhoršený kašeľ, nová alebo zhoršená dýchavičnosť, triaška, nová alebo zhoršená bolesť svalov, nová strata chuti alebo čuchu, bolesť hrdla, hnačka alebo vracanie.]
* Do analýzy boli zahrnutí účastníci, ktorí nemali sérologický ani virologický dôkaz (do 7 dní po podaní druhej dávky) predchádzajúcej infekcie SARS-CoV-2 (t.j. negatívni na N-viažuce protilátky [sérum] pri
1. kontrole a nezistený SARS-CoV-2 použitím testu amplifikácie nukleových kyselín (NAAT) [výter
z nosa] pri 1. a 2. kontrole) a mali negatívny NAAT (výter z nosa) pri akejkoľvek neplánovanej kontrole do
7 dní po podaní 2. dávky.
a. n = počet účastníkov v špecifikovanej skupine.
b. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
c. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po podaní 2. dávky po koniec obdobia sledovania.
d. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
e. Dvojstranný interval spoľahlivosti (IS) pre účinnosť očkovacej látky je odvodený na základe Clopperovej a
Pearsonovej metódy upravenej podľa doby sledovania. IS nie je upravený pre multiplicitu.
Účinnosť mRNA očkovacej látky proti COVID-19 v prevencii prvého výskytu COVID-19 od 7. dňa
po 2. dávke v porovnaní s placebom u účastníkov vo veku 16 rokov a starších s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 bola 94,6 % (95 % interval spoľahlivosti 89,6 % až
97,6 %).
Okrem toho, analýzy primárneho cieľového ukazovateľa účinnosti v rámci jednotlivých podskupín ukázali podobné bodové odhady medzi jednotlivými pohlaviami, etnickými skupinami a účastníkmi so zdravotnými komorbiditami spojenými s vysokým rizikom závažného priebehu COVID-19.
Vykonali sa aktualizované analýzy účinnosti s ďalšími potvrdenými prípadmi COVID-19, ktoré sa objavili počas zaslepeného, placebom kontrolovaného sledovania, čo v populácii skúmanej z hľadiska účinnosti predstavuje až 6 mesiacov po 2. dávke.
Aktualizované informácie o účinnosti očkovacej látky sú uvedené v tabuľke 3.
Podskupina
| mRNA očkovacia látka
proti COVID-19
na = 20 998
Prípady n1b
Čas sledovaniac (n2d)
|
Placebo na = 21 096
Prípady
n1b
Čas sledovaniac (n2d)
|
Účinnosť očkovacej
látky % (95 % IS)e
| Všetci účastnícif
| 77
6,247 (20 712)
| 850
6,003 (20 713)
| 91,3
(89,0; 93,2)
| 16 až 64 rokov
| 70
4,859 (15 519)
| 710
4,654 (15 515)
| 90.6
(87,9; 92,7)
| 65 rokov a starší
| 7
1,233 (4 192)
| 124
1,202 (4 226)
| 94.5
(88,3; 97,8)
|
|
|
Tabuľka 3: Účinnosť očkovacej látky - prvý výskyt COVID-
19 od 7 dní po 2. dávke podľa vekových podskupín - účastníci bez dôkazu predchádzajúcej infekcie SARS-CoV-2* do 7 dní po 2. dávke – populácia hodnotiteľná z hľadiska účinnosti (7 dní) počas placebom kontrolovaného obdobia sledovania
65 až 74 rokov
|
6
0,994 (3 350)
|
98
0,966 (3 379)
|
94.1
(86,6; 97,9)
|
75 rokov a starší
|
1
0,239 (842)
|
26
0,237 (847)
|
96.2
(76,9; 99.9)
|
Poznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou (RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 (príznaky zahŕňali horúčku, nový alebo zhoršený kašeľ, novú alebo zhoršenú dýchavičnosť, triašku, novú alebo zhoršenú bolesť svalov, novú stratu chuti alebo čuchu, bolesť hrdla, hnačku, vracanie).
* Do analýzy boli zahrnutí účastníci, ktorí nemali dôkaz predchádzajúcej infekcie SARS-CoV-2 (t.j.
negatívni na N-viažuce protilátky [sérum] pri 1. kontrole a nezistený SARS-CoV-2 použitím NAAT [výter z nosa] pri 1. a 2. kontrole) a mali negatívny NAAT (výter z nosa) pri akejkoľvek nenaplánovanej kontrole
do 7 dní po podaní 2. dávky.
a. n = počet účastníkov v špecifikovanej skupine.
b. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
c. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po podaní 2. dávky po koniec obdobia sledovania.
d. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
e. Dvojstranný 95 % interval spoľahlivosti (IS) pre účinnosť očkovacej látky je odvodený na základe
Clopperovej a Pearsonovej metódy upravenej podľa doby sledovania.
f. Zahrnuté sú potvrdené prípady u účastníkov vo veku 12 až 15 rokov: 0 v skupine s mRNA očkovacou
látkou proti COVID-19, 16 v skupine s placebom.
V aktualizovanej analýze účinnosti bola účinnosť mRNA očkovacej látky proti COVID-19 v prevencii
prvého výskytu COVID-19 od 7. dňa po 2. dávke v porovnaní s placebom 91,1 % (95 % IS 88,8 % až
93,0 %) u účastníkov v populácii hodnotiteľnej z hľadiska účinnosti s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2.
Okrem toho, aktualizované analýzy účinnosti podľa podskupín ukázali podobné bodové odhady účinnosti medzi pohlaviami, etnickými skupinami, geografickými oblasťami a účastníkmi so zdravotnými komorbiditami a obezitou spojenou s vysokým rizikom závažného priebehu COVID-19.
Účinnosť voči závažnému priebehu COVID-19Aktualizované analýzy účinnosti sekundárnych cieľových ukazovateľov účinnosti podporovali prínos mRNA očkovacej látky proti COVID-19 v prevencii závažného priebehu COVID-19.
K 13. marcu 2021 je účinnosť očkovacej látky voči závažnému priebehu COVID-19 uvádzaná len pre účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 (tabuľka 4), pretože počty prípadov COVID-19 u účastníkov bez predchádzajúcej infekcie SARS-CoV-2 boli rovnaké ako u účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 v skupine s mRNA očkovacou látkou proti COVID-19 aj v skupine s placebom.
Tabuľka 4: Účinnosť očkovacej látky - prvý výskyt závažného priebehu COVID-19u účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 na základe údajov Úradu pre potraviny a liečivá (Food and Drug Administration,
Podskupina
| mRNA očkovacia látka
proti COVID-19
Prípady n1a
Čas sledovania (n2b)
|
Placebo Prípady n1a
Čas sledovania (n2b)
|
Účinnosť očkovacej
látky % (95 % ISc)
| Po 1. dávked
| 1
8,439e (22 505)
| 30
8,288e (22 435)
| 96,7
(80,3; 99,9)
| 7 dní po 2. dávkef
| 1
6,522g (21 649)
| 21
6,404g (21 730)
| 95,3
(70,9; 99,9)
|
|
|
FDA)* po 1. dávke alebo od 7 dní po 2. dávke v placebom kontrolovanom období sledovania

Poznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou (RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 (príznaky zahŕňali horúčku, nový alebo zhoršený kašeľ, novú alebo zhoršenú dýchavičnosť, triašku, novú alebo zhoršenú bolesť svalov, novú stratu chuti alebo čuchu, bolesť hrdla, hnačku, vracanie).
* Závažné ochorenie spôsobené COVID-19 definované podľa FDA je potvrdený COVID-19 a prítomnosť
aspoň 1 z nasledujúcich:
• klinické prejavy v pokoji naznačujúce závažné systémové ochorenie (dychová frekvencia ≥ 30 dychov za minútu, srdcová frekvencia ≥ 125 úderov za minútu, saturácia kyslíkom ≤ 93 % pri izbovom vzduchu vo výške hladiny mora alebo pomer parciálneho artériového tlaku kyslíka
k frakčnému inspirovanému kyslíku < 300 mm Hg),
• zlyhanie dýchania [definované ako potreba vysokého prietoku kyslíka, neinvazívnej ventilácie, mechanickej ventilácie alebo mimotelovej membránovej oxygenácie (
extracorporeal membrane oxygenation, ECMO)],
• dôkaz šoku (systolický krvný tlak < 90 mm Hg, diastolický krvný tlak < 60 mm Hg alebo potreba použitia vazopresív),
• významná akútna obličková, pečeňová alebo neurologická dysfunkcia,
• prijatie na jednotku intenzívnej starostlivosti,
• úmrtie.
a. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
b. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
c. Dvojstranný interval spoľahlivosti (IS) pre účinnosť očkovacej látky je odvodený na základe Clopperovej
a Pearsonovej metódy upravenej podľa doby sledovania.
d. Účinnosť hodnotená na základe celej dostupnej populácie (modifikovaná „
intention-to-treat“) účinnosti po
1. dávke zahŕňajúca všetkých randomizovaných účastníkov, ktorí dostali aspoň 1 dávku skúmanej liečby.
e. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 1. dávky po koniec obdobia sledovania.
f. Účinnosť hodnotená na základe populácie hodnotiteľnej z hľadiska účinnosti (7 dní), ktorá zahŕňala všetkých spôsobilých randomizovaných účastníkov, ktorí dostali všetky dávky skúmanej liečby podľa randomizácie v rámci preddefinovaného časového obdobia a nemali žiadne iné dôležité odchýlky od
protokolu stanovené lekárom.
g. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ pre všetkých účastníkov v každej skupine s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po 2. dávke po koniec obdobia sledovania.
Účinnosť a imunogenita u dospievajúcich vo veku 12 až 15 rokov - po 2 dávkachV analýze štúdie 2 u dospievajúcich vo veku 12 až 15 rokov bez dôkazu predchádzajúcej infekcie sa nevyskytli žiadne prípady u 1 005 účastníkov, ktorí dostali očkovaciu látku a vyskytlo sa 16 prípadov z 978 u účastníkov, ktorí dostali placebo. Bodový odhad účinnosti je 100 % (95 % interval spoľahlivosti 75,3; 100,0). U účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie sa vyskytlo 0 prípadov u 1 119 účastníkov, ktorí dostali očkovaciu látku a 18 prípadov
u 1 110 účastníkov, ktorí dostali placebo. To tiež ukazuje bodový odhad účinnosti 100 % (95 %
interval spoľahlivosti 78,1; 100,0).
V štúdii 2 sa vykonala analýza neutralizačných titrov SARS-CoV-2 1 mesiac po 2. dávke v náhodne vybranej podskupine účastníkov, ktorí nemali žiadny sérologický ani virologický dôkaz predchádzajúcej infekcie SARS-CoV-2 až do 1 mesiaca po 2. dávke porovnávajúca odpoveď
u dospievajúcich vo veku 12 až 15 rokov (n = 190) s účastníkmi vo veku 16 až 25 rokov (n = 170).
Pomer geometrických priemerných titrov (
Geometric Mean Titres, GMT) v skupine 12 až 15 rokov ku skupine vo veku 16 až 25 rokov bol 1,76 s 2-stranným 95 % IS 1,47 až 2,10. Keďže dolná hranica
2-stranného 95 % IS pre pomer geometrického priemeru [
Geometric Mean Ratio, GMR] bola > 0,67, bolo splnené kritérium 1,5-násobnej non-inferiority.
Imunogenita u účastníkov vo veku 18 rokov a starších – po posilňovacej dávke (tretia dávka) Účinnosť posilňovacej dávky Comirnaty je založená na vyhodnotení 50 % titrov neutralizačných protilátok (NT50) proti SARS-CoV-2 (USA_WA1/2020). V štúdii 2 preukázali analýzy porovnávajúce NT50 1 mesiac po posilňovacej dávke s 1 mesiacom po primárnom cykle u osôb vo veku 18 až 55 rokov, ktoré nemali sérologický ani virologický dôkaz predchádzajúcej infekcie
SARS-CoV-2 až do 1 mesiaca po posilňovacom očkovaní non-inferioritu pre pomer geometrického priemeru (GMR) aj rozdiel v miere sérologickej odpovede. Sérologická odpoveď sa definovala ako dosiahnutie ≥ 4-násobného zvýšenia NT50 oproti východiskovému stavu (pred primárnym cyklom). Tieto analýzy sú zhrnuté v tabuľke 5.
Tabuľka 5: Neutralizačný test SARS-CoV-2 - NT50 (titer)† (SARS-CoV-2 USA_WA1/2020) –porovnanie GMT a miery sérologickej odpovede 1 mesiac po posilňovacej dávkes 1 mesiacom po primárnom cykle – účastníci vo veku 18 až 55 rokov bez dôkazu infekcie až do 1 mesiaca po posilňovacej dávke* – populácia s posilňovacou dávkou hodnotiteľná z hľadiska imunogenity±
|
n
|
1 mesiac po posilňovacej dávke
(95 % IS)
|
1 mesiac po primárnom cykle
(95 % IS)
| 1 mesiac po
posilňovacej
dávke/-
1 mesiac po primárnom
cykle
(97,5 % IS)
|
Dosiahnutý cieľ non- inferiority (Á/N)
|
Geometrický priemer 50 %
neutralizačného
titra (GMTb)
|
212a
|
2 466,0b
(2 202,6; 2 760,8)
|
750,6b
(656,2; 858,6)
|
3,29c
(2,77; 3,90)
|
Ád
|
Miera sérologickej odpovede (%) pre
50 % neutralizačný
titer†
|
200e
|
199f
99,5 %
(97,2 %, 100,0 %)
|
196f
98,0%
(95,0 %, 99,5 %)
|
1,5 %g
(-0,7 %, 3,7 %h)
|
Ái
|
Skratky: IS = interval spoľahlivosti, GMR = pomer geometrického priemeru, GMT = geometrický priemerný
titer, LLOQ = dolná hranica kvantifikácie (
lower limit of quantitation), N-väzba = SARS-CoV-2 nukleoproteínová väzba, NAAT = test amplifikácie nukleových kyselín, NT50 = 50 % neutralizačný titer, SARS-CoV-2 = závažný akútny respiračný syndróm vyvolaný koronavírusom 2, Á/N = áno/nie.
† NT50 SARS-CoV-2 sa stanovili použitím mikroneutralizačného testu SARS-CoV-2 mNeonGreen. Test používa fluorescenčný reportérový vírus odvodený z kmeňa USA_WA1/2020 a neutralizácia vírusu sa odčíta na monovrstvách buniek Vero. Vzorka NT50 je definovaná ako vzájomné riedenie séra, pri ktorom je neutralizovaných 50 % vírusu.
* Do analýzy boli zahrnutí účastníci, ktorí nemali sérologický ani virologický dôkaz (až do 1 mesiaca po podaní posilňovacej dávky Comirnaty) predchádzajúcej infekcie SARS-CV-2 (t.j. negatívni na N-viažuce protilátky [sérum] a nezistený SARS-CoV-2 použitím NAAT [výter z nosa]) a mali negatívny NAAT (výter z nosa) pri akejkoľvek neplánovanej kontrole až do 1 mesiaca po posilňovacej dávke.
± Všetci spôsobilí účastníci, ktorí podľa pôvodnej randomizácie dostali 2 dávky Comirnaty, s 2. dávkou podanou v rámci preddefinovaného časového obdobia (počas 19 až 42 dní po 1. dávke), dostali posilňovaciu dávku Comirnaty, mali aspoň 1 platný a určený výsledok imunogenity po posilňovacej dávke z odberu krvi v rámci príslušného časového obdobia (počas 28 až 42 dní po posilňovacej dávke) a nemali žiadne iné dôležité odchýlky od protokolu stanovené lekárom.
a. n = počet účastníkov s platnými a určenými výsledkami testu v oboch časových bodoch odberu vzoriek v rámci stanoveného časového obdobia.
b. Hodnoty GMT a 2-stranného 95 % IS sa vypočítali exponenciálnym vyjadrením priemerného logaritmu titrov a príslušných hodnôt IS (na základe Studentovho t rozdelenia). Výsledky testu pod hodnotou LLOQ boli stanovené ako 0,5-násobok LLOQ.
c. Hodnoty GMR a 2-stranného 97,5 % IS sa vypočítali umocnením priemerných rozdielov logaritmov testu a zodpovedajúcich hodnôt IS (na základe Studentovho t rozdelenia).
d. Non-inferiorita sa deklarovala, ak bola dolná hranica 2-stranného 97,5 % IS pre GMR > 0,67 a bodový
odhad GMR ≥ 0,80.
e. n = počet účastníkov s platnými a určenými výsledkami špecifikovaného testu na začiatku štúdie, 1 mesiac po 2. dávke a 1 mesiac po posilňovacej dávke v rámci stanoveného časového obdobia. Tieto hodnoty sú
menovateľmi vo výpočte percentuálnych hodnôt.
f. Počet účastníkov so sérologickou odpoveďou na daný test v danom časovom bode podania dávky/odberu vzorky. Presný 2-stranný IS je odvodený na základe Clopperovej a Pearsonovej metódy.
g. Rozdiel v podieloch vyjadrený v percentách (1 mesiac po posilňovacej dávke – 1 mesiac po 2. dávke). h. Upravený Waldov 2-stranný IS pre rozdiel v podieloch, vyjadrený v percentách.
i. Non-inferiorita sa deklarovala, ak bola dolná hranica 2-stranného 97,5 % IS pre percentuálny rozdiel
> -10 %.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s očkovacou látkou
Comirnaty u pediatrickej populácie na prevenciu ochorenia COVID-19 (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku. Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Neaplikovateľné.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní a reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Celková toxicita
U potkanov, ktorým sa intramuskulárne podávala očkovacia látka Comirnaty (dostávajúc 3 plné
ľudské dávky jedenkrát týždenne, ktoré viedli u potkanov k celkovo vyšším hladinám z dôvodu rozdielu v telesnej hmotnosti) sa pozoroval mierny edém a erytém v mieste vpichu a zvýšenie počtu
leukocytov (vrátane bazofilov a eozinofilov), čo je konzistentné so zápalovou odpoveďou, ako aj
vakuolizácia portálnych hepatocytov bez dôkazu poškodenia pečene. Všetky účinky boli reverzibilné.
Genotoxicita/karcinogenita
Nevykonali sa štúdie genotoxicity ani karcinogenity. Pre komponenty očkovacej látky (lipidy
a mRNA) sa neočakáva genotoxický potenciál.
Reprodukčná toxicita
Reprodukčná a vývinová toxicita sa skúmala na potkanoch v kombinovanej štúdii fertility a vývinovej
toxicity, pri ktorej sa samiciam potkanov intramuskulárne podávala očkovacia látka Comirnaty pred párením a počas gravidity (dostali 4 plné ľudské dávky, ktoré viedli u potkanov k celkovo vyšším hladinám z dôvodu rozdielu v telesnej hmotnosti, a to v období 21 dní pred párením do 20. dňa gravidity). U samíc bola prítomná imunitná odpoveď prostredníctvom neutralizačných protilátok proti SARS-CoV-2 počas obdobia pred párením až po koniec štúdie v 21. postnatálny deň, pričom neutralizujúce protilátky boli prítomné aj u plodov a mláďat. Nevyskytli sa žiadne účinky na samičiu fertilitu, graviditu alebo embryofetálny vývin alebo vývin mláďat súvisiace s očkovacou látkou. Nie sú dostupné žiadne údaje pre očkovaciu látku Comirnaty týkajúce sa prechodu očkovacej látky cez placentu alebo jej vylučovania do mlieka.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
((4-hydroxybutyl)azándiyl)bis(hexán-6,1-diyl)bis(2-hexyldekanoát) (ALC-0315)
2-[(polyetylénglykol)-2000]-N,N-ditetradecylacetamid (ALC-0159)
1,2-distearoyl-sn-glycero-3-fosfocholín (DSPC)
cholesterol chlorid draselný
dihydrogenfosforečnan draselný
chlorid sodný
dihydrát hydrogenfosforečnanu sodného
sacharóza
voda na injekcie
hydroxid sodný (na úpravu pH)
kyselina chlorovodíková (na úpravu pH)
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
Zmrazená injekčná liekovka
9 mesiacov, ak sa uchováva pri teplote -90 °C až -60 °C.
V rámci 9-mesačného času použiteľnosti sa môžu neotvorené injekčné liekovky uchovávať
a prepravovať pri teplote -25 °C až -15 °C jednorazovo po dobu maximálne 2 týždne a môžu sa vrátiť
do podmienok -90 °C až -60 °C.
Ak sa uchovávajú zmrazené pri teplote -90 °C až -60 °C, môžu sa balenia po 195 injekčných liekoviek očkovacej látky rozmraziť pri teplote 2 °C až 8 °C po dobu 3 hodín alebo jednotlivé injekčné liekovky sa môžu rozmraziť pri izbovej teplote (maximálne 30 °C) po dobu 30 minút.
Rozmrazená injekčná liekovka
1 mesiac pri teplote 2 °C až 8 °C v rámci 9-mesačného času použiteľnosti.
V rámci 1-mesačného času použiteľnosti pri teplote 2 °C až 8 °C sa môže injekčná liekovka prepravovať až po dobu 12 hodín.
Pred použitím sa môže neotvorená injekčná liekovka uchovávať maximálne 2 hodiny pri teplotách do
30 °C.
S rozmrazenými injekčnými liekovkami je možné manipulovať v podmienkach s umelým osvetlením miestnosti.
Po rozmrazení sa očkovacia látka nesmie opakovane zmraziť.
Manipulácia pri teplotných výkyvoch po vybratí z mrazničky
Údaje o stabilite naznačujú, že neotvorená injekčná liekovka je stabilná až do:
• 24 hodín, ak sa uchováva pri teplotách od -3 °C do 2 °C,
• spolu 4 hodiny, ak sa uchováva pri teplotách od 8 °C do 30 °C, vrátane 2 hodín pri teplotách až do 30 °C, ako je opísané vyššie.
Tieto informácie sú určené na pomoc zdravotníckym pracovníkom len v prípade dočasných teplotných
výkyvov.
Preprava zmrazených injekčných liekoviek pri ultra nízkej teplote (< -60 °C)
• Škatule na liekovky s uzatvoreným vekom obsahujúce 195 injekčných liekoviek, ktoré sa vybrali z mraziaceho uchovávacieho priestoru s ultra nízkou teplotou (< -60 °C), môžu byť vystavené teplotám až do 25 °C maximálne 5 minút.
• Škatule na liekovky s otvoreným vekom alebo škatule na liekovky obsahujúce menej ako
195 injekčných liekoviek, ktoré sa vybrali z mraziaceho uchovávacieho priestoru s ultra nízkou teplotou (< -60 °C), môžu byť vystavené teplotám až do 25 °C maximálne 3 minúty.
• Po vystavení teplote do 25 °C sa škatule s injekčnými liekovkami vrátia do mraziaceho
uchovávacieho priestoru, kde musia zostať aspoň 2 hodiny pred ďalším vybratím.
Preprava zmrazených injekčných liekoviek uchovávaných pri teplote -25 °C až -15 °C
• Škatule na liekovky s uzatvoreným vekom obsahujúce 195 injekčných liekoviek, ktoré sa
vybrali z mraziaceho uchovávacieho priestoru (-25 °C až -15 °C), môžu byť vystavené teplotám
až do 25 °C maximálne 3 minúty.
• Škatule na liekovky s otvoreným vekom alebo škatule na liekovky obsahujúce menej ako
195 injekčných liekoviek, ktoré sa vybrali z mraziaceho uchovávacieho priestoru (-25 °C
až -15 °C), môžu byť vystavené teplotám až do 25 °C maximálne 1 minútu.
Po vybratí injekčnej liekovky zo škatule na liekovky sa má injekčná liekovka rozmraziť na použitie.
Nariedený liek
Chemická a fyzikálna stabilita počas používania, vrátane prepravy, sa po nariedení s injekčným
roztokom chloridu sodného 9 mg/ml (0,9 %) preukázala počas 6 hodín pri teplote 2 °C až 30 °C.
Z mikrobiologického hľadiska, ak metóda riedenia nevylučuje riziko mikrobiálnej kontaminácie, sa má liek použiť okamžite. Ak sa liek nepoužije okamžite, za čas použiteľnosti a podmienky uchovávania počas používania zodpovedá používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v mrazničke pri teplote -90 °C až -60 °C. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Počas uchovávania minimalizujte pôsobenie svetla v miestnosti na liek a nevystavujte priamemu
slnečnému svetlu ani ultrafialovému žiareniu.
Podmienky na uchovávanie po rozmrazení a riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
0,45 ml koncentrátu v 2 ml čírej, viacdávkovej, injekčnej liekovke (sklo typu I) so zátkou (syntetická brómbutylová guma) a fialovým odklápacím plastovým viečkom s hliníkovým tesnením. Jedna injekčná liekovka obsahuje 6 dávok, pozri časť 6.6.
Veľkosť balenia: 195 injekčných liekoviek
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Pokyny na zaobchádzanie s liekom
Comirnaty má pripravovať zdravotnícky pracovník pomocou aseptického postupu na zabezpečenie
sterility pripravenej disperzie.


KO
NT
R
O
L
A DÁVKY OČKOVACEJ LÁTKY COMIRNATY
30 MIKROGRAMOV/DÁVKA KONCENTRÁT NA INJEKČNÚ DISPERZIU (12 ROKOV A STARŠÍ)
|
F
ialové viečko
|
• Skontrolujte, či má injekčná liekovka
fialové plastové viečko.
• Ak má injekčná liekovka sivé plastové viečko, prečítajte si súhrn charakteristických vlastností lieku očkovacej látky Comirnaty
30 mikrogramov/dávka injekčná
disperzia.
• Ak má injekčná liekovka oranžové plastové viečko, prečítajte si súhrn charakteristických vlastností lieku očkovacej látky Comirnaty
10 mikrogramov/dávka koncentrát na
injekčnú disperziu.
|
R
O
Z
MRAZENIE PRED NARIEDENÍM OČKOVACEJ LÁTKY COMIRNATY
30 MIKROGRAMOV/DÁVKA KONCENTRÁT NA INJEKČNÚ DISPERZIU (12 ROKOV A STARŠÍ)
|
Nie dlhšie ako
2 hodiny
p
r
i izbovej teplote
(
m
aximálne 30 °C)
|
• Viacdávková injekčná liekovka sa uchováva zmrazená a pred nariedením sa musí rozmraziť. Zmrazené injekčné liekovky sa majú premiestniť do prostredia s teplotou od 2 °C do 8 °C, aby sa rozmrazili; rozmrazenie balenia
195 injekčných liekoviek môže trvať až
3 hodiny. Zmrazené injekčné liekovky je možné alternatívne tiež nechať
rozmraziť po dobu 30 minút pri teplotách do 30 °C na okamžité
použitie.
• Neotvorená injekčná liekovka sa môže
uchovávať po dobu maximálne
1 mesiaca pri teplote 2 °C až 8 °C
v rámci 9-mesačného času použiteľnosti. V rámci 1-mesačného času použiteľnosti pri teplote 2 °C až
8 °C sa môže injekčná liekovka prepravovať až po dobu 12 hodín.
• Rozmrazenú injekčnú liekovku nechajte
dosiahnuť izbovú teplotu. Pred použitím sa môže neotvorená injekčná liekovka uchovávať maximálne
2 hodiny pri teplotách do 30 °C.
S rozmrazenými injekčnými liekovkami je možné manipulovať v podmienkach
s umelým osvetlením miestnosti.
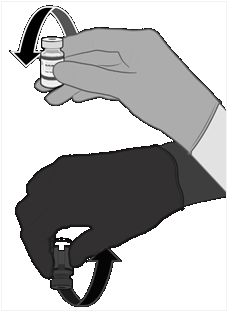
• Injekčnú liekovku pred nariedením
10-krát opatrne prevráťte.
Nepretrepávajte.
• Pred nariedením môže rozmrazená disperzia obsahovať biele až sivobiele nepriehľadné amorfné častice.
|
R
I
EDEN
I
E OČKOVACEJ LÁTKY COMIRNATY 30 MIKROGRAMOV/DÁVKA
KO
NCENTRÁ
T NA INJEKČNÚ DISPERZIU (12 ROKOV A STARŠÍ)
|
1,8 ml 0,9 % injekčného roztoku
c
h
loridu sodného
|
• Rozmrazená očkovacia látka musí byť nariedená v jej pôvodnej injekčnej liekovke s 1,8 ml injekčného roztoku chloridu sodného s koncentráciou
9 mg/ml (0,9 %) pomocou ihly veľkosti
21 G alebo tenšej a pomocou aseptického postupu.
|
Z
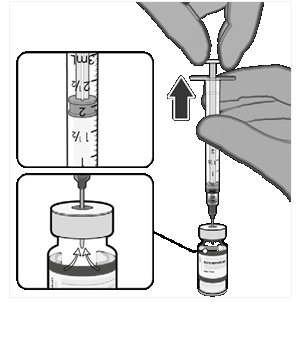
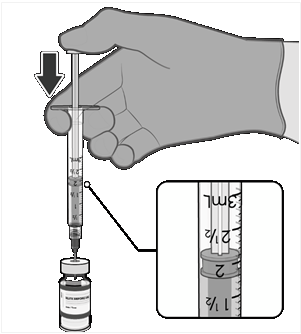
atiahnite piest na 1,8 ml na odstránenie vzduchu z injekčnej liekovky.
|
• Pred vytiahnutím ihly zo zátky
injekčnej liekovky vyrovnajte tlak
v injekčnej liekovke odobratím 1,8 ml vzduchu do prázdnej injekčnej striekačky, ktorou sa vstreklo rozpúšťadlo.
|
Jemne 10x
|
• Nariedenú disperziu 10-krát jemne
prevráťte. Nepretrepávajte.
• Nariedená očkovacia látka má byť vo forme sivobielej disperzie bez viditeľných častíc. Ak nariedená očkovacia látka obsahuje častice, alebo ak došlo k zmene jej sfarbenia, nepoužívajte ju.
|
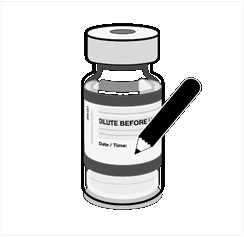
Z
aznamenajte príslušný dátum a čas.
P
oužite do 6 hodín po nariedení.
|
• Po nariedení treba injekčné liekovky označiť príslušným dátumom a časom.
• Po nariedení uchovávajte pri teplote
2 °C až 30 °C a použite do 6 hodín,
vrátane času prepravy.
• Nariedenú disperziu nezmrazujte ani
ňou netraste. Pri uchovávaní
v chladničke nechajte pred použitím nariedenú disperziu dosiahnuť izbovú
teplotu.
|
P
R
Í
P
RAV
A JEDNOTLIVÝCH 0,3 ml DÁVOK OČKOVACEJ LÁTKY COMIRNATY
30 MIKROGRAMOV/DÁVKA KONCENTRÁT NA INJEKČNÚ DISPERZIU (12 ROKOV A STARŠÍ)

• Injekčná liekovka obsahuje po nariedení 2,25 ml, z ktorých je možné získať 6 dávok po 0,3 ml
• Zátku injekčnej liekovky očistite aseptickým postupom pomocou jednorazového antiseptického tampónu.
• Odoberte dávku 0,3 ml očkovacej látky
Comirnaty.
Na získanie 6 dávok z jednej injekčnej liekovky sa majú používať injekčné striekačky a/alebo ihly s malým mŕtvym priestorom. Kombinácia injekčnej striekačky s malým mŕtvym priestorom a ihly nemá mať mŕtvy priestor väčší ako 35 mikrolitrov.
0,3 ml nariedenej očkovacej látky
Pri použití štandardných injekčných
striekačiek a ihiel nemusí byť objem dostatočný na získanie šiestej dávky z jednej injekčnej liekovky.
• Každá dávka musí obsahovať 0,3 ml
očkovacej látky.
• Ak zvyšné množstvo očkovacej látky
v injekčnej liekovke nie je dostatočné
na podanie plnej dávky 0,3 ml, injekčnú liekovku a všetok zvyšný objem zlikvidujte.
• Zlikvidujte všetku očkovaciu látku, ktorú nespotrebujete do 6 hodín po nariedení.
 L
i
kv
i
dácia
L
i
kv
i
dácia
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Nemecko
Telefón: +49 6131 9084-0
Fax: +49 6131 9084-2121
service@biontech.de8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/20/1528/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 21. decembra 2020
Dátum posledného predĺženia registrácie: 03. novembra 2021
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu. Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUComirnaty 30 mikrogramov/dávka injekčná disperzia
mRNA očkovacia látka proti COVID-19 (modifikovaný nukleozid)
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEToto je viacdávková injekčná liekovka. Pred použitím nerieďte.
Jedna injekčná liekovka (2,25 ml) obsahuje 6 dávok po 0,3 ml, pozri časti 4.2 a 6.6.
Jedna dávka (0,3 ml) obsahuje 30 mikrogramov tozinameranu, mRNA očkovacej látky proti
COVID-19 (zapuzdrenej do lipidových nanočastíc).
Tozinameran je jednovláknová mediátorová RNA (mRNA) s čiapočkou na 5’ konci produkovaná pomocou bezbunkovej transkripcie
in vitro z príslušných matríc DNA, kódujúca „spike“ (S) proteín vírusu SARS-CoV-2.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInjekčná disperzia.
Očkovacia látka je vo forme bielej až sivobielej zmrazenej disperzie (pH 6,9 - 7,9).
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieOčkovacia látka Comirnaty 30 mikrogramov/dávka injekčná disperzia je indikovaná na aktívnu imunizáciu na prevenciu ochorenia COVID-19 spôsobeného vírusom SARS-CoV-2 u osôb vo veku
12 rokov a starších.
Použitie tejto očkovacej látky má byť v súlade s oficiálnymi odporúčaniami.
4.2 Dávkovanie a spôsob podávaniaDávkovanieOsoby vo veku 12 rokov a staršieComirnaty sa podáva intramuskulárne ako primárny cyklus 2 dávok (každá má 0,3 ml). Druhú dávku
sa odporúča podať 3 týždne po prvej dávke (pozri časti 4.4 a 5.1).
Osobám vo veku 18 rokov a starším sa môže minimálne 6 mesiacov po druhej dávke intramuskulárne podať posilňovacia dávka (tretia dávka) Comirnaty. Rozhodnutie, kedy a komu podať tretiu dávku Comirnaty, sa má urobiť na základe dostupných údajov o účinnosti očkovacej látky, pričom sa majú vziať do úvahy limitované údaje o bezpečnosti (pozri časti 4.4 a 5.1).
Zameniteľnosť očkovacej látky Comirnaty s očkovacími látkami proti COVID-19 iných výrobcov na účely dokončenia primárneho očkovacieho cyklu alebo ako posilňovacej dávky (tretia dávka) nebola stanovená. Osoby, ktoré dostali 1. dávku očkovacej látky Comirnaty, majú na dokončenie primárneho očkovacieho cyklu dostať aj druhú dávku Comirnaty a aj v prípade ďalších dávok. Dávky Comirnaty
30 mikrogramov/dávka koncentrát na injekčnú disperziu po nariedení a Comirnaty
30 mikrogramov/dávka injekčná disperzia sa považujú za zameniteľné.
Ťažko imunokompromitované osoby vo veku 12 rokov a staršieŤažko imunokompromitovaným osobám sa môže minimálne 28 dní po druhej dávke podať tretia dávka (pozri časť 4.4).
Pediatrická populáciaK dispozícii je pediatrická lieková forma pre deti vo veku 5 až 11 rokov (t.j. vo veku 5 až menej ako
12 rokov). Ďalšie informácie nájdete v súhrne charakteristických vlastností lieku očkovacej látky
Comirnaty 10 mikrogramov/dávka koncentrát na injekčnú disperziu.
Populácia starších osôbU starších osôb vo veku ≥ 65 rokov nie je potrebná žiadna úprava dávky. Bezpečnosť a imunogenita
posilňovacej dávky (tretia dávka) Comirnaty u osôb vo veku 65 rokov a starších je založená na údajoch o bezpečnosti a imunogenite u dospelých vo veku 18 až 55 rokov.
Spôsob podávaniaComirnaty 30 mikrogramov/dávka injekčná disperzia sa má podávať intramuskulárne (pozri časť 6.6).
Pred použitím nerieďte.
Injekčné liekovky Comirnaty obsahujú 6 dávok po 0,3 ml očkovacej látky. Aby bolo možné získať
6 dávok z jednej injekčnej liekovky, majú sa používať injekčné striekačky a/alebo ihly s malým mŕtvym priestorom. Kombinácia injekčnej striekačky s malým mŕtvym priestorom a ihly nemá mať mŕtvy priestor väčší ako 35 mikrolitrov. Pri použití štandardných injekčných striekačiek a ihiel nemusí byť objem dostatočný na získanie šiestej dávky z jednej injekčnej liekovky. Nezávisle od typu injekčnej striekačky a ihly:
• Každá dávka musí obsahovať 0,3 ml očkovacej látky.
• Ak zvyšné množstvo očkovacej látky v injekčnej liekovke nie je dostatočné na podanie plnej
dávky 0,3 ml, injekčnú liekovku a všetok zvyšný objem zlikvidujte.
• Nezlievajte zvyšné množstvo očkovacej látky z viacerých injekčných liekoviek. Uprednostňované miesto podania je deltový sval ramena.
Očkovaciu látku nepodávajte injekčne intravaskulárne, subkutánne ani intradermálne.
Očkovacia látka sa nemá miešať v rovnakej injekčnej striekačke so žiadnymi inými očkovacími
látkami ani liekmi.
Opatrenia, ktoré sa majú urobiť pred podaním očkovacej látky, pozri časť 4.4.
Pokyny týkajúce sa rozmrazenia, zaobchádzania a likvidácie očkovacej látky, pozri časť 6.6.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Všeobecné odporúčania
Precitlivenosť a anafylaxia
Boli hlásené prípady anafylaxie. Pre prípad vzniku anafylaktickej reakcie po podaní očkovacej látky má byť vždy okamžite dostupné vhodné lekárske ošetrenie a dohľad.
Po očkovaní sa odporúča dôkladné sledovanie po dobu aspoň 15 minút. Osobám, u ktorých po prvej dávke očkovacej látky Comirnaty vznikla anafylaxia, sa nemá podať druhá dávka očkovacej látky.
Myokarditída a perikarditída
Existuje zvýšené riziko myokarditídy a perikarditídy po očkovaní očkovacou látkou Comirnaty. Tieto ochorenia sa môžu objaviť už do niekoľkých dní po očkovaní a vyskytli sa najmä počas prvých 14 dní.
Boli pozorované častejšie po druhom očkovaní a častejšie u mladších mužov (pozri časť 4.8). Z
dostupných údajov vyplýva, že priebeh myokarditídy a perikarditídy po očkovaní sa nelíši od
myokarditídy alebo perikarditídy vo všeobecnosti.
Zdravotnícki pracovníci majú venovať zvýšenú pozornosť prejavom a príznakom myokarditídy a perikarditídy. Očkované osoby (vrátane rodičov alebo opatrovateľov) majú byť poučené, aby okamžite vyhľadali lekársku pomoc, ak sa u nich po očkovaní objavia príznaky naznačujúce myokarditídu alebo perikarditídu, napríklad (akútna a pretrvávajúca) bolesť v hrudníku, dýchavičnosť alebo palpitácie.
Zdravotnícki pracovníci si majú pri diagnostike a liečbe tohto ochorenia prečítať usmernenia a/alebo sa poradiť so špecialistami.
Riziko myokarditídy po tretej dávke Comirnaty ešte nebolo zistené.
Reakcie spojené s úzkosťou
Pri očkovaní sa môžu v spojení so samotným procesom očkovania vyskytnúť reakcie spojené
s úzkosťou vrátane vazovagálnych reakcií (synkopa), hyperventilácie alebo reakcií spojených so
stresom (napr. závrat, palpitácie, zvýšený tep srdca, zmeny krvného tlaku, parestézia, hypestézia
a potenie). Reakcie spojené so stresom sú dočasné a vymiznú samé. Osoby je potrebné poučiť, aby oznámili príznaky poskytovateľovi očkovania za účelom vyhodnotenia. Je dôležité urobiť opatrenia na zabránenie poranenia spôsobeného omdlievaním.
Súbežné ochorenie
Očkovanie sa má odložiť u osôb so závažným akútnym ochorením s horúčkou alebo akútnou infekciou. Kvôli miernej infekcii a/alebo nízkej horúčke sa očkovanie nemá odkladať.
Trombocytopénia a poruchy koagulácie
Tak ako pri iných intramuskulárnych injekciách sa má očkovacia látka podávať s opatrnosťou u osôb
liečených antikoagulanciami alebo u osôb s trombocytopéniou alebo akoukoľvek poruchou koagulácie (ako je napríklad hemofília), pretože u týchto osôb sa môže po intramuskulárnom podaní vyskytnúť krvácanie alebo tvorba podliatin.
Imunokompromitované osoby
Účinnosť a bezpečnosť očkovacej látky sa nehodnotila u imunokompromitovaných osôb vrátane osôb
liečených imunosupresívami. Účinnosť očkovacej látky Comirnaty môže byť
u imunokompromitovaných osôb nižšia.
Odporúčanie zvážiť podanie tretej dávky u ťažko imunokopromitovaných osôb je založené na limitovaných sérologických dôkazoch z prípadových štúdií v literatúre týkajúcich sa klinickej liečby pacientov s iatrogénnou imunosupresiou po transplantácii solídnych orgánov (pozri časť 4.2).
Trvanie ochrany
Trvanie ochrany vyvolanej očkovacou látkou nie je známe, keďže sa ešte stále stanovuje
v prebiehajúcich klinických skúšaniach.
Obmedzenia účinnosti očkovacej látky
Tak ako každá očkovacia látka, ani očkovanie očkovacou látkou Comirnaty nemusí chrániť každého, kto ju dostane. Jednotlivci nemusia byť chránení, kým neuplynie minimálne 7 dní po podaní druhej
dávky očkovacej látky.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Súbežné podávanie očkovacej látky Comirnaty s inými očkovacími látkami sa neskúmalo.
4.6 Fertilita, gravidita a laktácia
Gravidita
Sú k dispozícii iba obmedzené skúsenosti s použitím očkovacej látky Comirnaty u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska gravidity, embryofetálneho vývinu, pôrodu alebo postnatálneho vývinu (pozri časť 5.3). Podanie očkovacej látky Comirnaty počas gravidity sa má zvažovať len v prípade, ak možné prínosy prevažujú nad akýmikoľvek možnými rizikami pre matku a plod.
Dojčenie
Nie je známe, či sa Comirnaty vylučuje do ľudského mlieka.
Fertilita
Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej
toxicity (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Comirnaty nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Niektoré z účinkov uvedených v časti 4.8 však môžu dočasne ovplyvniť schopnosť viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Bezpečnosť očkovacej látky Comirnaty bola vyhodnocovaná u účastníkov vo veku 12 rokov a starších
v 2 klinických štúdiách zahŕňajúcich 23 205 účastníkov (pozostávajúcich z 22 074 účastníkov vo veku
16 rokov a starších a 1 131 dospievajúcich vo veku 12 až 15 rokov), ktorí dostali aspoň jednu dávku očkovacej látky Comirnaty.
Celkový bezpečnostný profil očkovacej látky Comirnaty u dospievajúcich vo veku 12 až 15 rokov bol
podobný ako bezpečnostný profil pozorovaný u účastníkov vo veku 16 rokov a starších.
306 účastníkov vo veku 18 až 55 rokov dostalo dodatočne v prebiehajúcej fáze 3 posilňovaciu dávku (tretia dávka) Comirnaty približne 6 mesiacov po druhej dávke. Celkový bezpečnostný profil posilňovacej dávky (tretia dávka) bol podobný ako bezpečnostný profil pozorovaný po 2 dávkach.
Účastníci vo veku 16 rokov a starší - po 2 dávkach
V štúdii 2 dostalo spolu 22 026 účastníkov vo veku 16 rokov alebo starších najmenej 1 dávku očkovacej látky Comirnaty a spolu 22 021 účastníkov vo veku 16 rokov alebo starších dostalo placebo (vrátane dospievajúcich vo veku 16 a 17 rokov, v počte 138 v skupine s očkovacou látkou a 145
v skupine s placebom). Spolu 20 519 účastníkov vo veku 16 rokov alebo starších dostalo 2 dávky
očkovacej látky Comirnaty.
V čase analýzy štúdie 2 s dátumom ukončenia zberu údajov 13. marca 2021 pre placebom kontrolované, zaslepené obdobie sledovania až do dátumov odslepenia účastníkov bolo spolu 25 651 (58,2 %) účastníkov (13 031 dostalo očkovaciu látku Comirnaty a 12 620 dostalo placebo) vo veku
16 rokov a starších sledovaných po dobu ≥ 4 mesiace po druhej dávke. To zahŕňalo spolu
15 111 účastníkov (7 704 dostalo očkovaciu látku Comirnaty a 7 407 dostalo placebo) vo veku 16 až
55 rokov a spolu 10 540 účastníkov (5 327 dostalo očkovaciu látku Comirnaty a 5 213 dostalo placebo) vo veku 56 rokov a starších.
Najčastejšími nežiaducimi reakciami u účastníkov vo veku 16 rokov a starších, ktorí dostali 2 dávky, boli bolesť v mieste vpichu (> 80 %), únava (> 60 %), bolesť hlavy (> 50 %), myalgia (> 40 %), triaška (> 30 %), artralgia (> 20 %), pyrexia a opuch v mieste vpichu (> 10 %) a mali zvyčajne miernu alebo strednú intenzitu a do niekoľkých dní po podaní očkovacej látky ustúpili. Mierne nižšia frekvencia príhod reaktogenity súvisela s vyšším vekom.
Bezpečnostný profil u 545 účastníkov vo veku 16 rokov a starších dostávajúcich očkovaciu látku Comirnaty, ktorí boli na začiatku štúdie séropozitívni na SARS-CoV-2, bol podobný ako bezpečnostný profil pozorovaný v celkovej populácii.
Dospievajúci vo veku 12 až 15 rokov - po 2 dávkach
V analýze štúdie 2 zakladajúcej sa na údajoch získaných až do dátumu ukončenia sledovania (cut-off)
13. marca 2021 bolo zahrnutých 2 260 dospievajúcich (1 131 dostalo očkovaciu látku Comirnaty a 1 129 dostalo placebo) vo veku 12 až 15 rokov. Z nich sa 1 308 dospievajúcich (660 dostalo
očkovaciu látku Comirnaty a 648 dostalo placebo) sledovalo aspoň 2 mesiace po druhej dávke
očkovacej látky Comirnaty. Vyhodnocovanie bezpečnosti v štúdii 2 ešte prebieha.
Najčastejšími nežiaducimi reakciami u dospievajúcich vo veku 12 až 15 rokov, ktorí dostali 2 dávky, boli bolesť v mieste vpichu (> 90 %), únava a bolesť hlavy (> 70 %), myalgia a triaška (> 40 %), artralgia a pyrexia (> 20 %).
Účastníci vo veku 18 rokov a starší - po posilňovacej dávke (tretia dávka)
Podskupina účastníkov v štúdii 2 fázy 2/3 pozostávajúca z dospelých vo veku 18 až 55 rokov, ktorí
ukončili pôvodný 2-dávkový cyklus Comirnaty, dostali posilňovaciu dávku (tretia dávka) Comirnaty
približne 6 mesiacov (rozsah 4,8 až 8,0 mesiacov) po podaní 2. dávky.
Najčastejšími nežiaducimi reakciami u účastníkov vo veku 18 až 55 rokov boli bolesť v mieste vpichu
(> 80 %), únava (> 60 %), bolesť hlavy (> 40 %), myalgia (> 30 %), triaška a artralgia (> 20 %).
Tabuľkovýzoznamnežiaducichreakciívklinických štúdiách a zo skúseností po uvedení lieku na trhu osôb vo veku 12 rokov a starších
Nežiaduce reakcie pozorované počas klinických štúdií sú uvedené nižšie podľa nasledujúcich kategórií
frekvencie:
veľmi časté (≥ 1/10),
časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100),
zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Trieda orgánových systémov
| Veľmi časté
(≥ 1/10)
| Časté
(≥ 1/100 až
< 1/10)
| Menej časté
(≥ 1/1 000 až
< 1/100)
| Zriedkavé (≥ 1/10 000 až
< 1/1 000)
| Veľmi
zriedkavé
(< 1/10 000)
| Neznáme
(z dostupných údajov)
| Poruchy krvi
a lymfatického systému
|
|
| lymfadenopatiaa
|
|
|
| Poruchy imunitného systému
|
|
| reakcie
z precitlivenosti (napr. vyrážka, pruritus, urtikáriab, angioedémb)
|
|
| anafylaxia
| Poruchy metabolizmu a výživy
|
|
| znížená chuť do
jedla
|
|
|
| Psychické poruchy
|
|
| nespavosť
|
|
|
| Poruchy nervového systému
| bolesť hlavy
|
| letargia
| akútna periférna paralýza tvárec
|
| parestéziad, hypestéziad
| Poruchy srdca
|
|
|
|
| myokarditídad, perikarditídad
|
| Poruchy
gastrointestinálneho traktu
| hnačkad
| nevoľnosť,
vracanied
|
|
|
|
| Poruchy kože
a podkožného tkaniva
|
|
| hyperhidróza,
nočné potenie
|
|
| multiformný erytémd
| Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
| artralgia, myalgia
|
| bolesť
v končatinee
|
|
|
| Celkové poruchy
a reakcie v mieste podania
| bolesť v mieste vpichu, únava, triaška, pyrexiaf, opuch v mieste
vpichu
| začervenanie v mieste vpichu
| asténia, malátnosť, pruritus
v mieste vpichu
|
|
| rozsiahly opuch očkovanej končatinyd, opuch tváreg
|
|
|
Tabuľka 1: Nežiaduce reakcie z klinických skúšaní a zo skúseností po uvedení lieku na trh s očkovacou látkou Comirnaty u osôb vo veku 12 rokov a staršícha. U účastníkov, ktorí dostali posilňovaciu dávku (tretia dávka), sa v porovnaní s účastníkmi, ktorí dostali
2 dávky, pozorovala vyššia frekvencia výskytu lymfadenopatie (5,2 % oproti 0,4 %). b. Kategória frekvencie pre urtikáriu a angioedém bola zriedkavé.
c. Počas obdobia sledovania bezpečnosti v rámci klinického skúšania až do 14. novembra 2020 bola hlásená akútna periférna paralýza (alebo ochrnutie) tváre u štyroch účastníkov v skupine s mRNA očkovacou látkou proti COVID-19. Nástup reakcie bol 37. deň po 1. dávke (účastník nedostal 2. dávku) a 3., 9. a 48. deň po
2. dávke. V skupine s placebom sa nehlásili žiadne prípady akútnej periférnej paralýzy (alebo ochrnutia)
tváre.
d. Nežiaduca reakcia zistená po uvedení lieku na trh.
e. Týka sa ruky, do ktorej bola podaná očkovacia látka.
f. Po druhej dávke sa pozorovala vyššia frekvencia pyrexie v porovnaní s prvou dávkou.
g. Vo fáze po uvedení na trh bol hlásený opuch tváre u očkovaných osôb s anamnézou podania injekcie s dermatologickým výplňovým materiálom.
O
pis vybraných nežiaducich reakcií
M
yokarditída
Zvýšené riziko myokarditídy po očkovaní očkovacou látkou Comirnaty je najvyššie u mladších mužov
(pozri časť 4.4).
V dvoch veľkých, európskych, farmako-epidemiologických štúdiách sa určilo zvýšené riziko u mladších mužov po druhej dávke očkovacej látky Comirnaty.
Z jednej štúdie vyplynulo, že v období 7 dní po podaní druhej dávky sa u mužov vo veku 12–29 rokov vyskytlo približne o 0,265 (95 % IS 0,255–0,275) prípadov myokarditídy na 10 000 osôb viac ako u
neexponovaných osôb. V ďalšej štúdii sa v období 28 dní po podaní druhej dávky u mužov vo veku
16–24 rokov vyskytlo o 0,57 (95 % IS 0,39–0,75) prípadov myokarditídy na 10 000 osôb viac ako u neexponovaných osôb.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V
a uviedli číslo šarže, ak je k dispozícii.
4.9 PredávkovanieÚdaje o predávkovaní sú dostupné od 52 účastníkov zahrnutých do klinického skúšania, ktorí dostali
z dôvodu chybného nariedenia 58 mikrogramov očkovacej látky Comirnaty. Očkované osoby nehlásili zvýšenie reaktogenity ani výskytu nežiaducich reakcií.
V prípade predávkovania sa odporúča sledovanie životných funkcií a prípadne symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: očkovacie látky, iné vírusové očkovacie látky, ATC kód: J07BX03
Mechanizmus účinkuMediátorová RNA s modifikovaným nukleozidom v očkovacej látke Comirnaty (tozinameran) je
zapuzdrená v lipidových nanočasticiach, ktoré umožňujú dopravenie nereplikujúcej sa RNA do
hostiteľských buniek na priamu dočasnú expresiu S antigénu vírusu SARS-CoV-2. mRNA kóduje
S proteín plnej dĺžky, ukotvený v membráne, s dvoma bodovými mutáciami v rámci centrálnej špirály. Mutácia týchto dvoch aminokyselín na prolín uzamyká S proteín v antigénne uprednostňovanej pred-
fúznej konformácii. Očkovacia látka vyvoláva odpoveď vo forme tvorby neutralizujúcich protilátok
ako aj bunkovú imunitnú odpoveď na „spike“ (S) antigén, čo môže prispievať k ochrane pred ochorením COVID-19.
ÚčinnosťŠtúdia 2 je multicentrová, medzinárodná, randomizovaná, placebom kontrolovaná, pre pozorovateľov
zaslepená štúdia fázy 1/2/3 na stanovenie dávky, výber kandidátov na očkovaciu látku a stanovenie
účinnosti u účastníkov vo veku 12 rokov a starších. Randomizácia bola stratifikovaná podľa veku: vek
12 až 15 rokov, vek 16 až 55 rokov alebo vek 56 rokov a viac, pričom minimálne 40 % účastníkov bolo vo vekovej skupine ≥ 56 rokov. Z účasti na štúdii boli vylúčení imunokompromitovaní účastníci a tí, ktorí mali v minulosti klinicky alebo mikrobiologicky diagnostikovaný COVID-19. Účastníci
s prebiehajúcim stabilným ochorením definovaným ako ochorenie, ktoré si nevyžaduje významnú
zmenu liečby ani hospitalizáciu z dôvodu zhoršenia ochorenia počas 6 týždňov pred zaradením, boli
do tejto štúdie zahrnutí, ako aj účastníci so známou stabilnou infekciou vírusom ľudskej
imunodeficiencie (HIV), vírusom hepatitídy C (HCV) alebo vírusom hepatitídy B (HBV).
Účinnosť u účastníkov vo veku 16 rokov a starších - po 2 dávkachV časti fázy 2/3 štúdie 2 bolo na základe údajov zozbieraných do 14. novembra 2020 približne
44 000 účastníkov rovnomerne randomizovaných do skupín, ktoré dostali 2 dávky mRNA očkovacej látky proti COVID-19 alebo placebo. Analýzy účinnosti zahŕňali účastníkov, ktorí dostali druhú dávku očkovacej látky v intervale 19 až 42 dní po ich prvom očkovaní. Väčšina (93,1 %) očkovaných osôb dostala druhú dávku 19 až 23 dní po 1. dávke. Je plánované sledovanie účastníkov po dobu až
24 mesiacov po 2. dávke na účely hodnotení bezpečnosti a účinnosti proti ochoreniu COVID-19.
V klinickej štúdii sa vyžadovalo, aby sa u účastníkov pri podaní buď placeba alebo mRNA očkovacej látky proti COVID-19 dodržal interval minimálne 14 dní pred a po podaní očkovacej látky proti chrípke. V klinickej štúdii sa vyžadovalo, aby sa u účastníkov pri podaní buď placeba alebo mRNA očkovacej látky proti COVID-19 dodržal interval minimálne 60 dní pred alebo po podaní krvných/plazmových produktov alebo imunoglobulínov v rámci obdobia do ukončenia štúdie.
Populácia na analýzu primárneho cieľového ukazovateľa účinnosti zahŕňala 36 621 účastníkov vo
veku 12 rokov a starších (18 242 v skupine s mRNA očkovacou látkou proti COVID-19
a 18 379 v skupine s placebom), u ktorých nebol do doby 7 dní po druhej dávke zistený dôkaz predchádzajúcej infekcie vírusom SARS-CoV-2. Ďalej, 134 účastníkov bolo vo veku medzi 16 až
17 rokov (66 v skupine s mRNA očkovacou látkou proti COVID-19 a 68 v skupine s placebom)
a 1 616 účastníkov bolo vo veku 75 rokov a starších (804 v skupine s mRNA očkovacou látkou proti
COVID-19 a 812 v skupine s placebom).
V čase primárnej analýzy účinnosti boli účastníci sledovaní z hľadiska symptomatického COVID-19 po dobu spolu 2 214 osoborokov v skupine s mRNA očkovacou látkou proti COVID-19 a po dobu spolu 2 222 osoborokov v skupine s placebom.
U účastníkov ohrozených závažným priebehom COVID-19 vrátane tých s 1 alebo viacerými komorbiditami, ktoré zvyšujú riziko závažného priebehu COVID-19 (napr. astma, index telesnej hmotnosti (BMI) ≥ 30 kg/m2, chronické ochorenie pľúc, diabetes mellitus, hypertenzia), sa nevyskytli žiadne významné klinické rozdiely v celkovej účinnosti očkovacej látky.
Informácie o účinnosti očkovacej látky sú uvedené v tabuľke 2.
Prvý výskyt COVID-19 od 7 dní po 2. dávke u účastníkov bez dôkazu predchádzajúcej infekcie SARS-CoV-2*
|
Podskupina
| mRNA očkovacia látka
proti COVID-19
na = 18 198
Prípady n1b
Čas sledovaniac (n2d)
|
Placebo na = 18,325
Prípady n1b
Čas sledovaniac (n2d)
|
Účinnosť očkovacej
látky % (95 % IS)e
| Všetci účastníci
| 8
2,214 (17 411)
| 162
2,222 (17 511)
| 95,0
(90,0; 97,9)
| 16 až 64 rokov
| 7
1,706 (13 549)
| 143
1,710 (13 618)
| 95,1
(89,6; 98,1)
| 65 rokov a starší
| 1
0,508 (3 848)
| 19
0,511 (3 880)
| 94,7
(66,7; 99,9)
| 65 až 74 rokov
| 1
0,406 (3 074)
| 14
0,406 (3 095)
| 92,9
(53,1; 99,8)
| 75 rokov a starší
| 0
0,102 (774)
| 5
0,106 (785)
| 100,0
(-13,1; 100,0)
|
|
|
Tabuľka 2: Účinnosť očkovacej látky - prvý výskyt COVID-
19 od 7 dní po 2. dávke podľa vekových podskupín - účastníci bez dôkazu infekcie do 7 dní po 2. dávke - populácia hodnotiteľná z hľadiska účinnosti (7 dní)
Poznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou
(RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 [*Definícia prípadu: (aspoň 1 z) horúčka, nový alebo zhoršený kašeľ, nová alebo zhoršená dýchavičnosť, triaška, nová alebo zhoršená bolesť svalov, nová strata chuti alebo čuchu, bolesť hrdla, hnačka alebo vracanie.]
* Do analýzy boli zahrnutí účastníci, ktorí nemali sérologický ani virologický dôkaz (do 7 dní po podaní druhej dávky) predchádzajúcej infekcie SARS-CoV-2 (t.j. negatívni na N-viažuce protilátky [sérum] pri
1. kontrole a nezistený SARS-CoV-2 použitím testu amplifikácie nukleových kyselín (NAAT) [výter
z nosa] pri 1. a 2. kontrole) a mali negatívny NAAT (výter z nosa) pri akejkoľvek neplánovanej kontrole do
7 dní po podaní 2. dávky.
a. n = počet účastníkov v špecifikovanej skupine.
b. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
c. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po podaní 2. dávky po koniec obdobia sledovania.
d. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
e. Dvojstranný interval spoľahlivosti (IS) pre účinnosť očkovacej látky je odvodený na základe Clopperovej a
Pearsonovej metódy upravenej podľa doby sledovania. IS nie je upravený pre multiplicitu.
Účinnosť mRNA očkovacej látky proti COVID-19 v prevencii prvého výskytu COVID-19 od 7. dňa
po 2. dávke v porovnaní s placebom u účastníkov vo veku 16 rokov a starších s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 bola 94,6 % (95 % interval spoľahlivosti 89,6 % až
97,6 %).
Okrem toho, analýzy primárneho cieľového ukazovateľa účinnosti v rámci jednotlivých podskupín ukázali podobné bodové odhady medzi jednotlivými pohlaviami, etnickými skupinami a účastníkmi so zdravotnými komorbiditami spojenými s vysokým rizikom závažného priebehu COVID-19.
Vykonali sa aktualizované analýzy účinnosti s ďalšími potvrdenými prípadmi COVID-19, ktoré sa objavili počas zaslepeného, placebom kontrolovaného sledovania, čo v populácii skúmanej z hľadiska účinnosti predstavuje až 6 mesiacov po 2. dávke.
Aktualizované informácie o účinnosti očkovacej látky sú uvedené v tabuľke 3.
Podskupina
| mRNA očkovacia látka proti COVID-19
na = 20 998
Prípady n1b
Čas sledovaniac (n2d)
|
Placebo na = 21 096
Prípady n1b
Čas sledovaniac (n2d)
|
Účinnosť očkovacej
látky % (95 % IS)e
| Všetci účastnícif
| 77
6,247 (20 712)
| 850
6,003 (20 713)
| 91,3
(89,0; 93,2)
| 16 až 64 rokov
| 70
4,859 (15 519)
| 710
4,654 (15 515)
| 90.6
(87,9; 92,7)
| 65 rokov a starší
| 7
1,233 (4 192)
| 124
1,202 (4 226)
| 94.5
(88,3; 97,8)
| 65 až 74 rokov
| 6
0,994 (3 350)
| 98
0,966 (3 379)
| 94.1
(86,6; 97,9)
| 75 rokov a starší
| 1
0,239 (842)
| 26
0,237 (847)
| 96.2
(76,9; 99.9)
|
|
|
Tabuľka 3: Účinnosť očkovacej látky - prvý výskyt COVID-
19 od 7 dní po 2. dávke podľa vekových podskupín - účastníci bez dôkazu predchádzajúcej infekcie SARS-CoV-2* do 7 dní po 2. dávke – populácia hodnotiteľná z hľadiska účinnosti (7 dní) počas placebom kontrolovaného obdobia sledovaniaPoznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou
(RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 (príznaky zahŕňali horúčku, nový alebo zhoršený kašeľ, novú alebo zhoršenú dýchavičnosť, triašku, novú alebo zhoršenú bolesť svalov, novú stratu chuti alebo čuchu, bolesť hrdla, hnačku, vracanie).

* Do analýzy boli zahrnutí účastníci, ktorí nemali dôkaz predchádzajúcej infekcie SARS-CoV-2 (t.j. negatívni na N-viažuce protilátky [sérum] pri 1. kontrole a nezistený SARS-CoV-2 použitím NAAT [výter z nosa] pri 1. a 2. kontrole) a mali negatívny NAAT (výter z nosa) pri akejkoľvek nenaplánovanej kontrole
do 7 dní po podaní 2. dávky.
a. n = počet účastníkov v špecifikovanej skupine.
b. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
c. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po podaní 2. dávky po koniec obdobia sledovania.
d. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
e. Dvojstranný 95 % interval spoľahlivosti (IS) pre účinnosť očkovacej látky je odvodený na základe
Clopperovej a Pearsonovej metódy upravenej podľa doby sledovania.
f. Zahrnuté sú potvrdené prípady u účastníkov vo veku 12 až 15 rokov: 0 v skupine s mRNA očkovacou
látkou proti COVID-19, 16 v skupine s placebom.
V aktualizovanej analýze účinnosti bola účinnosť mRNA očkovacej látky proti COVID-19 v prevencii
prvého výskytu COVID-19 od 7. dňa po 2. dávke v porovnaní s placebom 91,1 % (95 % IS 88,8 % až
93,0 %) u účastníkov v populácii hodnotiteľnej z hľadiska účinnosti s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2.
Okrem toho, aktualizované analýzy účinnosti podľa podskupín ukázali podobné bodové odhady účinnosti medzi pohlaviami, etnickými skupinami, geografickými oblasťami a účastníkmi so zdravotnými komorbiditami a obezitou spojenou s vysokým rizikom závažného priebehu COVID-19.
Účinnosť voči závažnému priebehu COVID-19Aktualizované analýzy účinnosti sekundárnych cieľových ukazovateľov účinnosti podporovali prínos mRNA očkovacej látky proti COVID-19 v prevencii závažného priebehu COVID-19.
K 13. marcu 2021 je účinnosť očkovacej látky voči závažnému priebehu COVID-19 uvádzaná len pre účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 (tabuľka 4), pretože počty prípadov COVID-19 u účastníkov bez predchádzajúcej infekcie SARS-CoV-2 boli rovnaké ako u účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 v skupine s mRNA očkovacou látkou proti COVID-19 aj v skupine s placebom.
Tabuľka 4: Účinnosť očkovacej látky - prvý výskyt závažného priebehu COVID-19u účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 na základe údajov Úradu pre potraviny a liečivá (Food and Drug Administration,
Podskupina
| mRNA očkovacia látka
proti COVID-19
Prípady n1a
Čas sledovania (n2b)
|
Placebo Prípady n1a
Čas sledovania (n2b)
|
Účinnosť očkovacej
látky % (95 % ISc)
| Po 1. dávked
| 1
8,439e (22 505)
| 30
8,288e (22 435)
| 96,7
(80,3; 99,9)
| 7 dní po 2. dávkef
| 1
6,522g (21 649)
| 21
6,404g (21 730)
| 95,3
(70,9; 99,9)
|
|
|
FDA)* po 1. dávke alebo od 7 dní po 2. dávke v placebom kontrolovanom období sledovaniaPoznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou
(RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 (príznaky zahŕňali horúčku, nový alebo zhoršený kašeľ, novú alebo zhoršenú dýchavičnosť, triašku, novú alebo zhoršenú bolesť svalov, novú stratu chuti alebo čuchu, bolesť hrdla, hnačku, vracanie).
* Závažné ochorenie spôsobené COVID-19 definované podľa FDA je potvrdený COVID-19 a prítomnosť
aspoň 1 z nasledujúcich:
• klinické prejavy v pokoji naznačujúce závažné systémové ochorenie (dychová frekvencia ≥ 30 dychov za minútu, srdcová frekvencia ≥ 125 úderov za minútu, saturácia kyslíkom ≤ 93 % pri izbovom vzduchu vo výške hladiny mora alebo pomer parciálneho artériového tlaku kyslíka
k frakčnému inspirovanému kyslíku < 300 mm Hg),
• zlyhanie dýchania [definované ako potreba vysokého prietoku kyslíka, neinvazívnej ventilácie, mechanickej ventilácie alebo mimotelovej membránovej oxygenácie (
extracorporeal membrane oxygenation, ECMO)],
• dôkaz šoku (systolický krvný tlak < 90 mm Hg, diastolický krvný tlak < 60 mm Hg alebo potreba použitia vazopresív),
• významná akútna obličková, pečeňová alebo neurologická dysfunkcia,
• prijatie na jednotku intenzívnej starostlivosti,
• úmrtie.
a. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
b. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
c. Dvojstranný interval spoľahlivosti (IS) pre účinnosť očkovacej látky je odvodený na základe Clopperovej
a Pearsonovej metódy upravenej podľa doby sledovania.
d. Účinnosť hodnotená na základe celej dostupnej populácie (modifikovaná „
intention-to-treat“) účinnosti po
1. dávke zahŕňajúca všetkých randomizovaných účastníkov, ktorí dostali aspoň 1 dávku skúmanej liečby.
e. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 1. dávky po koniec obdobia sledovania.
f. Účinnosť hodnotená na základe populácie hodnotiteľnej z hľadiska účinnosti (7 dní), ktorá zahŕňala všetkých spôsobilých randomizovaných účastníkov, ktorí dostali všetky dávky skúmanej liečby podľa randomizácie v rámci preddefinovaného časového obdobia a nemali žiadne iné dôležité odchýlky od protokolu stanovené lekárom.
g. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ pre všetkých účastníkov v každej skupine s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po 2. dávke po koniec obdobia sledovania.
Účinnosť a imunogenita u dospievajúcich vo veku 12 až 15 rokov - po 2 dávkachV analýze štúdie 2 u dospievajúcich vo veku 12 až 15 rokov bez dôkazu predchádzajúcej infekcie sa nevyskytli žiadne prípady u 1 005 účastníkov, ktorí dostali očkovaciu látku a vyskytlo sa 16 prípadov z 978 u účastníkov, ktorí dostali placebo. Bodový odhad účinnosti je 100 % (95 % interval spoľahlivosti 75,3; 100,0). U účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie sa vyskytlo 0 prípadov u 1 119 účastníkov, ktorí dostali očkovaciu látku a 18 prípadov
u 1 110 účastníkov, ktorí dostali placebo. To tiež ukazuje bodový odhad účinnosti 100 % (95 %
interval spoľahlivosti 78,1; 100,0).
V štúdii 2 sa vykonala analýza neutralizačných titrov SARS-CoV-2 1 mesiac po 2. dávke v náhodne vybranej podskupine účastníkov, ktorí nemali žiadny sérologický ani virologický dôkaz predchádzajúcej infekcie SARS-CoV-2 až do 1 mesiaca po 2. dávke porovnávajúca odpoveď
u dospievajúcich vo veku 12 až 15 rokov (n = 190) s účastníkmi vo veku 16 až 25 rokov (n = 170).
Pomer geometrických priemerných titrov (
Geometric Mean Titres, GMT) v skupine 12 až 15 rokov ku skupine vo veku 16 až 25 rokov bol 1,76 s 2-stranným 95 % IS 1,47 až 2,10. Keďže dolná hranica
2-stranného 95 % IS pre pomer geometrického priemeru [
Geometric Mean Ratio, GMR] bola > 0,67,
bolo splnené kritérium 1,5-násobnej non-inferiority.
Imunogenita u účastníkov vo veku 18 rokov a starších – po posilňovacej dávke (tretia dávka) Účinnosť posilňovacej dávky Comirnaty je založená na vyhodnotení 50 % titrov neutralizačných protilátok (NT50) proti SARS-CoV-2 (USA_WA1/2020). V štúdii 2 preukázali analýzy porovnávajúce NT50 1 mesiac po posilňovacej dávke s 1 mesiacom po primárnom cykle u osôb vo veku 18 až 55 rokov, ktoré nemali sérologický ani virologický dôkaz predchádzajúcej infekcie SARS-CoV-2 až do 1 mesiaca po posilňovacom očkovaní non-inferioritu pre pomer geometrického priemeru (GMR) aj rozdiel v miere sérologickej odpovede. Sérologická odpoveď sa definovala ako dosiahnutie ≥ 4-násobného zvýšenia NT50 oproti východiskovému stavu (pred primárnym cyklom). Tieto analýzy sú zhrnuté v tabuľke 5.
T
abuľka 5: Neutralizačný test SARS-CoV-2 - NT50 (titer)
†
(
SARS-CoV-2 USA_WA1/2020) –
porovnanie GMT a miery sérologickej odpovede 1 mesiac po posilňovacej dávke
s 1 mesiacom po primárnom cykle – účastníci vo veku 18 až 55 rokov bez dôkazu infekcie až do 1 mesiaca po posilňovacej dávke* – populácia s posilňovacou dávkou hodnotiteľná z hľadiska imunogenity
±
|
n
|
1 mesiac po posilňovacej dávke
(
95 % IS)
|
1 mesiac po primárnom cykle
(
95 % IS)
|
1 mesiac po
posilňovacej
dávke/-
1 mesiac po primárnom
cykle
(
97,5 % IS)
|
D
osiahnutý cieľ non- inferiority (Á/N)
|
G
eometrický priemer 50 %
neutralizačného
ti
t
ra (GMT
b
)
|
212a
|
2 466,0b
(2 202,6; 2 760,8)
|
750,6b
(656,2; 858,6)
|
3,29c
(2,77; 3,90)
|
Ád
|
Miera sérologickej odpovede (%) pre
50 % neutralizačný
ti
t
er
†
|
200e
|
199f
99,5 %
(97,2 %, 100,0 %)
|
196f
98,0%
(95,0 %, 99,5 %)
|
1,5 %g
(-0,7 %, 3,7 %h)
|
Ái
|
Skratky: IS = interval spoľahlivosti, GMR = pomer geometrického priemeru, GMT = geometrický priemerný
titer, LLOQ = dolná hranica kvantifikácie (
lower limit of quantitation), N-väzba = SARS-CoV-2 nukleoproteínová väzba, NAAT = test amplifikácie nukleových kyselín, NT50 = 50 % neutralizačný titer, SARS-CoV-2 = závažný akútny respiračný syndróm vyvolaný koronavírusom 2, Á/N = áno/nie.
† NT50 SARS-CoV-2 sa stanovili použitím mikroneutralizačného testu SARS-CoV-2 mNeonGreen. Test používa fluorescenčný reportérový vírus odvodený z kmeňa USA_WA1/2020 a neutralizácia vírusu sa odčíta na monovrstvách buniek Vero. Vzorka NT50 je definovaná ako vzájomné riedenie séra, pri ktorom je neutralizovaných 50 % vírusu.
* Do analýzy boli zahrnutí účastníci, ktorí nemali sérologický ani virologický dôkaz (až do 1 mesiaca po podaní posilňovacej dávky Comirnaty) predchádzajúcej infekcie SARS-CV-2 (t.j. negatívni na N-viažuce protilátky [sérum] a nezistený SARS-CoV-2 použitím NAAT [výter z nosa]) a mali negatívny NAAT (výter z nosa) pri akejkoľvek neplánovanej kontrole až do 1 mesiaca po posilňovacej dávke.
± Všetci spôsobilí účastníci, ktorí podľa pôvodnej randomizácie dostali 2 dávky Comirnaty, s 2. dávkou podanou v rámci preddefinovaného časového obdobia (počas 19 až 42 dní po 1. dávke), dostali posilňovaciu dávku Comirnaty, mali aspoň 1 platný a určený výsledok imunogenity po posilňovacej dávke z odberu krvi v rámci príslušného časového obdobia (počas 28 až 42 dní po posilňovacej dávke) a nemali žiadne iné dôležité odchýlky od protokolu stanovené lekárom.
a. n = počet účastníkov s platnými a určenými výsledkami testu v oboch časových bodoch odberu vzoriek
v rámci stanoveného časového obdobia.
b. Hodnoty GMT a 2-stranného 95 % IS sa vypočítali exponenciálnym vyjadrením priemerného logaritmu titrov a príslušných hodnôt IS (na základe Studentovho t rozdelenia). Výsledky testu pod hodnotou LLOQ boli stanovené ako 0,5-násobok LLOQ.
c. Hodnoty GMR a 2-stranného 97,5 % IS sa vypočítali umocnením priemerných rozdielov logaritmov testu a zodpovedajúcich hodnôt IS (na základe Studentovho t rozdelenia).
d. Non-inferiorita sa deklarovala, ak bola dolná hranica 2-stranného 97,5 % IS pre GMR > 0,67 a bodový
odhad GMR ≥ 0,80.
e. n = počet účastníkov s platnými a určenými výsledkami špecifikovaného testu na začiatku štúdie, 1 mesiac po 2. dávke a 1 mesiac po posilňovacej dávke v rámci stanoveného časového obdobia. Tieto hodnoty sú
menovateľmi vo výpočte percentuálnych hodnôt.
f. Počet účastníkov so sérologickou odpoveďou na daný test v danom časovom bode podania dávky/odberu
vzorky. Presný 2-stranný IS je odvodený na základe Clopperovej a Pearsonovej metódy.
g. Rozdiel v podieloch vyjadrený v percentách (1 mesiac po posilňovacej dávke – 1 mesiac po 2. dávke). h. Upravený Waldov 2-stranný IS pre rozdiel v podieloch, vyjadrený v percentách.
i. Non-inferiorita sa deklarovala, ak bola dolná hranica 2-stranného 97,5 % IS pre percentuálny rozdiel
> -10 %.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s očkovacou látkou
Comirnaty u pediatrickej populácie na prevenciu ochorenia COVID-19 (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku. Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Neaplikovateľné.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní a reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Celková toxicita
U potkanov, ktorým sa intramuskulárne podávala očkovacia látka Comirnaty (dostávajúc 3 plné
ľudské dávky jedenkrát týždenne, ktoré viedli u potkanov k celkovo vyšším hladinám z dôvodu rozdielu v telesnej hmotnosti) sa pozoroval mierny edém a erytém v mieste vpichu a zvýšenie počtu
leukocytov (vrátane bazofilov a eozinofilov), čo je konzistentné so zápalovou odpoveďou, ako aj
vakuolizácia portálnych hepatocytov bez dôkazu poškodenia pečene. Všetky účinky boli reverzibilné.
Genotoxicita/karcinogenita
Nevykonali sa štúdie genotoxicity ani karcinogenity. Pre komponenty očkovacej látky (lipidy
a mRNA) sa neočakáva genotoxický potenciál.
Reprodukčná toxicita
Reprodukčná a vývinová toxicita sa skúmala na potkanoch v kombinovanej štúdii fertility a vývinovej
toxicity, pri ktorej sa samiciam potkanov intramuskulárne podávala očkovacia látka Comirnaty pred párením a počas gravidity (dostali 4 plné ľudské dávky, ktoré viedli u potkanov k celkovo vyšším hladinám z dôvodu rozdielu v telesnej hmotnosti, a to v období 21 dní pred párením do 20. dňa gravidity). U samíc bola prítomná imunitná odpoveď prostredníctvom neutralizačných protilátok proti SARS-CoV-2 počas obdobia pred párením až po koniec štúdie v 21. postnatálny deň, pričom neutralizujúce protilátky boli prítomné aj u plodov a mláďat. Nevyskytli sa žiadne účinky na samičiu fertilitu, graviditu alebo embryofetálny vývin alebo vývin mláďat súvisiace s očkovacou látkou. Nie sú dostupné žiadne údaje pre očkovaciu látku Comirnaty týkajúce sa prechodu očkovacej látky cez placentu alebo jej vylučovania do mlieka.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
((4-hydroxybutyl)azándiyl)bis(hexán-6,1-diyl)bis(2-hexyldekanoát) (ALC-0315)
2-[(polyetylénglykol)-2000]-N,N-ditetradecylacetamid (ALC-0159)
1,2-distearoyl-sn-glycero-3-fosfocholín (DSPC)
cholesterol trometamol
trometamólium-chlorid
sacharóza
voda na injekcie
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
Zmrazená injekčná liekovka
9 mesiacov, ak sa uchováva pri teplote -90 °C až -60 °C.
Očkovacia látka sa môže dodávať zmrazená pri teplote -90 °C až -60 °C alebo -25 °C až -15 °C.
Zmrazená očkovacia látka sa môže po prijatí uchovávať buď pri teplote -90 °C až -60 °C alebo 2 °C až
8 °C.
Ak sa uchovávajú zmrazené pri teplote -90 °C až -60 °C, balenia po 10 injekčných liekoviek očkovacej látky sa môžu rozmraziť pri teplote 2 °C až 8 °C po dobu 6 hodín alebo jednotlivé injekčné liekovky sa môžu rozmraziť pri izbovej teplote (maximálne 30 °C) po dobu 30 minút.
Rozmrazená injekčná liekovka
10 týždňov uchovávania a prepravy pri teplote 2 °C až 8 °C v rámci 9-mesačného času použiteľnosti.
• Po premiestnení do podmienok uchovávania pri teplote 2 °C až 8 °C sa musí aktualizovaný dátum exspirácie napísať na vonkajšiu škatuľku a očkovacia látka sa má použiť alebo zlikvidovať do aktualizovaného dátumu exspirácie. Pôvodný dátum exspirácie sa má preškrtnúť.
• Ak sa očkovacia látka dodá pri teplote 2 °C až 8 °C, má sa uchovávať pri teplote 2 °C až 8 °C.
Skontrolujte, či bol dátum exspirácie na vonkajšej škatuli aktualizovaný tak, aby zobrazoval dátum exspirácie očkovacej látky uchovávanej v chladničke a či bol pôvodný dátum exspirácie preškrtnutý.
Pred použitím sa môžu neotvorené injekčné liekovky uchovávať maximálne 12 hodín pri teplotách medzi 8 °C a 30 °C.
S rozmrazenými injekčnými liekovkami je možné manipulovať v podmienkach s umelým osvetlením miestnosti.
Po rozmrazení sa očkovacia látka nesmie opakovane zmraziť.
Manipulácia pri teplotných výkyvoch počas uchovávania v chladničke
• Údaje o stabilite naznačujú, že neotvorená injekčná liekovka je stabilná až do 10 týždňov, ak sa uchováva pri teplotách od -2 °C do 2 °C, v rámci 10-týždňového obdobia uchovávania pri teplote medzi 2 °C a 8 °C.
• Údaje o stabilite naznačujú, že injekčná liekovka sa môže uchovávať maximálne 24 hodín pri teplotách 8 °C až 30 °C, vrátane maximálne 12 hodín po prvom prepichnutí zátky.
Tieto informácie sú určené na pomoc zdravotníckym pracovníkom len v prípade dočasných teplotných výkyvov.
Otvorená injekčná liekovka
Chemická a fyzikálna stabilita sa preukázala počas 12 hodín pri teplote 2 °C až 30 °C.
Z mikrobiologického hľadiska, ak metóda otvárania nevylučuje riziká mikrobiálnej kontaminácie, sa má liek použiť okamžite. Ak sa liek nepoužije okamžite, za čas použiteľnosti a podmienky
uchovávania počas používania zodpovedá používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v mrazničke pri teplote -90 °C až -60 °C. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Počas uchovávania minimalizujte pôsobenie svetla v miestnosti na liek a nevystavujte priamemu
slnečnému svetlu ani ultrafialovému žiareniu.
Podmienky na uchovávanie po prvom otvorení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
2,25 ml roztoku v 2 ml čírej, viacdávkovej, injekčnej liekovke (sklo typu I) so zátkou (syntetická brómbutylová guma) a sivým odklápacím plastovým viečkom s hliníkovým tesnením. Jedna injekčná liekovka obsahuje 6 dávok, pozri časť 6.6.
Veľkosti balenia: 195 injekčných liekoviek alebo 10 injekčných liekoviek
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Pokyny na zaobchádzanie s liekom
Comirnaty má pripravovať zdravotnícky pracovník pomocou aseptického postupu na zabezpečenie
sterility pripravenej disperzie.
KONTROLA DÁVKY OČKOVACEJ LÁTKY COMIRNATY
30 MIKROGRAMOV/DÁVKA INJEKČNÁ DISPERZIA (12 ROKOV A STARŠÍ)
• Skontrolujte, či má injekčná liekovka sivé plastové viečko.
• Ak má injekčná liekovka fialové


 S
ivé viečko
S
ivé viečko
plastové viečko, prečítajte si súhrn charakteristických vlastností lieku očkovacej látky Comirnaty
30 mikrogramov/dávka koncentrát na
injekčnú disperziu.
• Ak má injekčná liekovka oranžové plastové viečko, prečítajte si súhrn charakteristických vlastností lieku očkovacej látky Comirnaty
10 mikrogramov/dávka koncentrát na
injekčnú disperziu.
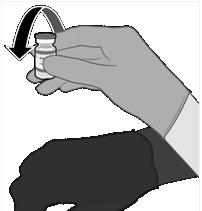
MANIPULÁCIA PRED POUŽITÍM OČKOVACEJ LÁTKY COMIRNATY
30 MIKROGRAMOV/DÁVKA INJEKČNÁ DISPERZIA (12 ROKOV A STARŠÍ)
|
U
chovávajte maximálne
10 týždňov pri
t
eplote 2 °C až
8 °C, aktualizujte dátum exspirácie na škatuli
|
• Ak sa viacdávková injekčná liekovka uchováva zmrazená, pred použitím sa musí rozmraziť. Zmrazené injekčné liekovky sa majú premiestniť do prostredia s teplotou od 2 °C do 8 °C, aby sa rozmrazili; rozmrazenie balenia
10 injekčných liekoviek môže trvať až
6 hodín. Zaistite, aby boli pred
použitím injekčné liekovky úplne
rozmrazené.
• Po premiestnení injekčných liekoviek
do podmienok uchovávania pri teplote
2 °C až 8 °C aktualizujte dátum
exspirácie na škatuľke.
• Neotvorené injekčné liekovky sa môžu uchovávať maximálne 10 týždňov pri teplote 2 °C až 8 °C v rámci
9-mesačného času použiteľnosti.
• Jednotlivé zmrazené injekčné liekovky je možné alternatívne nechať rozmraziť po dobu 30 minút pri teplotách do
30 °C.
• Pred použitím sa môže neotvorená injekčná liekovka uchovávať maximálne 12 hodín pri teplotách do
30 °C. S rozmrazenými injekčnými liekovkami je možné manipulovať
v podmienkach s umelým osvetlením miestnosti.
|
Jemne 10x
|
• Pred použitím jemne premiešajte prevrátením injekčnej liekovky 10-krát. Nepretrepávajte.
• Pred premiešaním môže rozmrazená injekčná disperzia obsahovať biele až sivobiele, nepriehľadné, amorfné častice.
• Po premiešaní má byť očkovacia látka vo forme bielej až sivobielej disperzie bez viditeľných častíc. Ak očkovacia látka obsahuje častice, alebo ak došlo
k zmene jej sfarbenia, nepoužívajte ju.
|
P
R
Í
P
RAV
A JEDNOTLIVÝCH 0,3 ml DÁVOK OČKOVACEJ LÁTKY COMIRNATY OČKOVACEJ LÁTKY COMIRNATY 30 MIKROGRAMOV/DÁVKA INJEKČNÁ DISPERZIA (12 ROKOV A STARŠÍ)

• Zátku injekčnej liekovky očistite aseptickým postupom pomocou jednorazového antiseptického tampónu.
• Odoberte dávku 0,3 ml očkovacej látky
Comirnaty.
Na získanie 6 dávok z jednej injekčnej liekovky sa majú používať injekčné striekačky a/alebo ihly s malým mŕtvym priestorom. Kombinácia injekčnej striekačky s malým mŕtvym priestorom a ihly nemá mať mŕtvy priestor väčší ako 35 mikrolitrov.
0,3 ml očkovacej látky
Pri použití štandardných injekčných
striekačiek a ihiel nemusí byť objem dostatočný na získanie šiestej dávky z jednej injekčnej liekovky.
• Každá dávka musí obsahovať 0,3 ml
očkovacej látky.
• Ak zvyšné množstvo očkovacej látky v injekčnej liekovke nie je dostatočné na podanie plnej dávky 0,3 ml, injekčnú liekovku a všetok zvyšný objem zlikvidujte.
• Zlikvidujte všetku očkovaciu látku, ktorú nespotrebujete do 12 hodín po prvom prepichnutí zátky. Napíšte správny dátum/čas na injekčnú liekovku.
 L
i
kv
i
dácia
L
i
kv
i
dácia
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Nemecko
Telefón: +49 6131 9084-0
Fax: +49 6131 9084-2121
service@biontech.de8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/20/1528/003
EU/1/20/1528/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 21. decembra 2020
Dátum posledného predĺženia registrácie: 03. novembra 2021
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu. Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUComirnaty 10 mikrogramov/dávka koncentrát na injekčnú disperziu
mRNA očkovacia látka proti COVID-19 (modifikovaný nukleozid)
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEToto je viacdávková injekčná liekovka a pred použitím sa musí zriediť.
Jedna injekčná liekovka (1,3 ml) obsahuje po nariedení 10 dávok po 0,2 ml, pozri časti 4.2 a 6.6. Jedna dávka (0,2 ml) obsahuje 10 mikrogramov tozinameranu, mRNA očkovacej látky proti
COVID-19 (zapuzdrenej do lipidových nanočastíc).
Tozinameran je jednovláknová mediátorová RNA (mRNA) s čiapočkou na 5’ konci produkovaná pomocou bezbunkovej transkripcie
in vitro z príslušných matríc DNA, kódujúca „spike“ (S) proteín vírusu SARS-CoV-2.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAKoncentrát na injekčnú disperziu (sterilný koncentrát).
Očkovacia látka je vo forme bielej až sivobielej zmrazenej disperzie (pH 6,9 - 7,9).
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieOčkovacia látka Comirnaty 10 mikrogramov/dávka koncentrát na injekčnú disperziu je indikovaná na aktívnu imunizáciu na prevenciu ochorenia COVID-19 spôsobeného vírusom SARS-CoV-2 u detí vo veku 5 až 11 rokov.
Použitie tejto očkovacej látky má byť v súlade s oficiálnymi odporúčaniami.
4.2 Dávkovanie a spôsob podávaniaDávkovanieDeti vo veku 5 až 11 rokov (t.j. 5 až menej ako 12 rokov)Comirnaty 10 mikrogramov/dávka sa podáva intramuskulárne po nariedení ako cyklus 2 dávok (každá má 0,2 ml). Druhú dávku sa odporúča podať 3 týždne po prvej dávke (pozri časti 4.4 a 5.1).
Zameniteľnosť očkovacej látky Comirnaty s očkovacími látkami proti COVID-19 iných výrobcov na účely dokončenia očkovacieho cyklu nebola stanovená. Osoby, ktoré dostali 1. dávku očkovacej látky Comirnaty, majú na dokončenie očkovacieho cyklu dostať aj druhú dávku Comirnaty.
Comirnaty 10 mikrogramov/dávka sa má používať len u detí vo veku 5 až 11 rokov.
Ťažko imunokompromitované osoby vo veku 5 rokov a staršieŤažko imunokompromitovaným osobám sa môže minimálne 28 dní po druhej dávke podať tretia dávka (pozri časť 4.4).
Pediatrická populáciaBezpečnosť a účinnosť Comirnaty u detí vo veku menej ako 5 rokov neboli doteraz stanovené.
Spôsob podávaniaComirnaty 10 mikrogramov/dávka koncentrát na injekčnú disperziu sa má podávať intramuskulárne
po
nariedení (pozri časť 6.6).
Po nariedení obsahujú injekčné liekovky Comirnaty 10 dávok po 0,2 ml očkovacej látky. Aby bolo
možné získať 10 dávok z jednej injekčnej liekovky, majú sa používať injekčné striekačky a/alebo ihly s malým mŕtvym priestorom. Kombinácia injekčnej striekačky s malým mŕtvym priestorom a ihly nemá mať mŕtvy priestor väčší ako 35 mikrolitrov. Pri použití štandardných injekčných striekačiek
a ihiel nemusí byť objem dostatočný na získanie 10 dávok z jednej injekčnej liekovky. Nezávisle od typu injekčnej striekačky a ihly:
• Každá dávka musí obsahovať 0,2 ml očkovacej látky.
• Ak zvyšné množstvo očkovacej látky v injekčnej liekovke nie je dostatočné na podanie plnej
dávky 0,2 ml, injekčnú liekovku a všetok zvyšný objem zlikvidujte.
• Nezlievajte zvyšné množstvo očkovacej látky z viacerých injekčných liekoviek. Uprednostňované miesto podania je deltový sval ramena.
Očkovaciu látku nepodávajte injekčne intravaskulárne, subkutánne ani intradermálne.
Očkovacia látka sa nemá miešať v rovnakej injekčnej striekačke so žiadnymi inými očkovacími
látkami ani liekmi.
Opatrenia, ktoré sa majú urobiť pred podaním očkovacej látky, pozri časť 4.4.
Pokyny týkajúce sa rozmrazenia, zaobchádzania a likvidácie očkovacej látky, pozri časť 6.6.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaníSledovateľnosťAby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Všeobecné odporúčaniaPrecitlivenosť a anafylaxiaBoli hlásené prípady anafylaxie. Pre prípad vzniku anafylaktickej reakcie po podaní očkovacej látky má byť vždy okamžite dostupné vhodné lekárske ošetrenie a dohľad.
Po očkovaní sa odporúča dôkladné sledovanie po dobu aspoň 15 minút. Osobám, u ktorých po prvej
dávke očkovacej látky Comirnaty vznikla anafylaxia, sa nemá podať druhá dávka očkovacej látky.
M
yokarditída a perikarditída
Existuje zvýšené riziko myokarditídy a perikarditídy po očkovaní očkovacou látkou Comirnaty. Tieto ochorenia sa môžu objaviť už do niekoľkých dní po očkovaní a vyskytli sa najmä počas prvých 14 dní. Boli pozorované častejšie po druhom očkovaní a častejšie u mladších mužov (pozri časť 4.8). Z dostupných údajov vyplýva, že priebeh myokarditídy a perikarditídy po očkovaní sa nelíši od myokarditídy alebo perikarditídy vo všeobecnosti.
Zdravotnícki pracovníci majú venovať zvýšenú pozornosť prejavom a príznakom myokarditídy a perikarditídy. Očkované osoby (vrátane rodičov alebo opatrovateľov) majú byť poučené, aby okamžite vyhľadali lekársku pomoc, ak sa u nich po očkovaní objavia príznaky naznačujúce myokarditídu alebo perikarditídu, napríklad (akútna a pretrvávajúca) bolesť v hrudníku, dýchavičnosť alebo palpitácie.
Zdravotnícki pracovníci si majú pri diagnostike a liečbe tohto ochorenia prečítať usmernenia a/alebo sa poradiť so špecialistami.
Riziko myokarditídy po tretej dávke Comirnaty ešte nebolo zistené.
Reakcie spojené s úzkosťou
Pri očkovaní sa môžu v spojení so samotným procesom očkovania vyskytnúť reakcie spojené
s úzkosťou vrátane vazovagálnych reakcií (synkopa), hyperventilácie alebo reakcií spojených so stresom (napr. závrat, palpitácie, zvýšený tep srdca, zmeny krvného tlaku, parestézia, hypestézia
a potenie). Reakcie spojené so stresom sú dočasné a vymiznú samé. Osoby je potrebné poučiť, aby
oznámili príznaky poskytovateľovi očkovania za účelom vyhodnotenia. Je dôležité urobiť opatrenia na
zabránenie poranenia spôsobeného omdlievaním.
Súbežné ochorenie
Očkovanie sa má odložiť u osôb so závažným akútnym ochorením s horúčkou alebo akútnou infekciou. Kvôli miernej infekcii a/alebo nízkej horúčke sa očkovanie nemá odkladať.
Trombocytopénia a poruchy koagulácie
Tak ako pri iných intramuskulárnych injekciách sa má očkovacia látka podávať s opatrnosťou u osôb liečených antikoagulanciami alebo u osôb s trombocytopéniou alebo akoukoľvek poruchou koagulácie (ako je napríklad hemofília), pretože u týchto osôb sa môže po intramuskulárnom podaní vyskytnúť krvácanie alebo tvorba podliatin.
Imunokompromitované osoby
Účinnosť a bezpečnosť očkovacej látky sa nehodnotila u imunokompromitovaných osôb vrátane osôb
liečených imunosupresívami. Účinnosť očkovacej látky Comirnaty môže byť
u imunokompromitovaných osôb nižšia.
Odporúčanie zvážiť podanie tretej dávky u ťažko imunokopromitovaných osôb je založené na limitovaných sérologických dôkazoch z prípadových štúdií v literatúre týkajúcich sa klinickej liečby dospelých pacientov s iatrogénnou imunosupresiou po transplantácii solídnych orgánov (pozri
časť 4.2).
Trvanie ochrany
Trvanie ochrany vyvolanej očkovacou látkou nie je známe, keďže sa ešte stále stanovuje
v prebiehajúcich klinických skúšaniach.
Obmedzenia účinnosti očkovacej látky
Tak ako každá očkovacia látka, ani očkovanie očkovacou látkou Comirnaty nemusí chrániť každého, kto ju dostane. Jednotlivci nemusia byť chránení, kým neuplynie minimálne 7 dní po podaní druhej dávky očkovacej látky.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Súbežné podávanie očkovacej látky Comirnaty s inými očkovacími látkami sa neskúmalo.
4.6 Fertilita, gravidita a laktácia
Gravidita
Sú k dispozícii iba obmedzené skúsenosti s použitím očkovacej látky Comirnaty u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska gravidity, embryofetálneho vývinu, pôrodu alebo postnatálneho vývinu (pozri časť 5.3). Podanie očkovacej látky Comirnaty počas gravidity sa má zvažovať len v prípade, ak možné prínosy prevažujú nad akýmikoľvek možnými rizikami pre matku a plod.
Dojčenie
Nie je známe, či sa Comirnaty vylučuje do ľudského mlieka.
Fertilita
Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej
toxicity (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Comirnaty nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Niektoré z účinkov uvedených v časti 4.8 však môžu dočasne ovplyvniť schopnosť viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Bezpečnosť očkovacej látky Comirnaty bola vyhodnocovaná u účastníkov vo veku 5 rokov a starších
v 3 klinických štúdiách zahŕňajúcich 24 675 účastníkov (pozostávajúcich z 22 026 účastníkov vo veku
16 rokov a starších, 1 131 dospievajúcich vo veku 12 až 15 rokov a 3 109 detí vo veku 5 až 11 rokov),
ktorí dostali aspoň 1 dávku očkovacej látky Comirnaty.
Celkový bezpečnostný profil očkovacej látky Comirnaty u účastníkov vo veku 5 až 15 rokov bol
podobný ako bezpečnostný profil pozorovaný u účastníkov vo veku 16 rokov a starších.
306 účastníkov vo veku 18 až 55 rokov dostalo dodatočne v prebiehajúcej fáze 3 posilňovaciu dávku (tretia dávka) Comirnaty približne 6 mesiacov po druhej dávke. Celkový bezpečnostný profil posilňovacej dávky (tretia dávka) bol podobný ako bezpečnostný profil pozorovaný po 2 dávkach.
Deti vo veku 5 až 11 rokov (t.j. vo veku 5 až menej ako 12 rokov) – po 2 dávkach
V štúdii 3 dostalo spolu 1 518 detí vo veku 5 až 11 rokov najmenej 1 dávku očkovacej látky
Comirnaty 10 µg a spolu 750 detí vo veku 5 až 11 rokov dostalo placebo. V čase analýzy štúdie 3
fázy 2/3 s údajmi až do ukončenia zberu údajov 6. septembra 2021 bolo 2 158 (95,1 %) (1 444 dostalo
očkovaciu látku Comirnaty 10 µg a 714 dostalo placebo) detí sledovaných po dobu aspoň 2 mesiace po druhej dávke očkovacej látky Comirnaty 10 µg. Analýza údajov týkajúcich sa nežiaducich príhod zo štúdie 3 fázy 2/3 tiež zahŕňala ďalších 2 379 účastníkov [1 591 dostalo očkovaciu látku Comirnaty
10 µg a 788 dostalo placebo], z čoho u 71,2 % trvalo obdobie sledovania aspoň 2 týždne po 2. dávke
až do ukončenia zberu údajov 8. októbra 2021. Vyhodnocovanie bezpečnosti v štúdii 3 ešte prebieha.
Najčastejšími nežiaducimi reakciami u detí vo veku 5 až 11 rokov boli bolesť v mieste vpichu
(> 80 %), únava (> 50 %), bolesť hlavy (> 30 %), začervenanie v mieste vpichu a opuch (> 20 %), myalgia a triaška (> 10 %).
Ú
častníci vo veku 16 rokov a starší - po 2 dávkach
V štúdii 2 dostalo spolu 22 026 účastníkov vo veku 16 rokov alebo starších najmenej 1 dávku
očkovacej látky Comirnaty 30 µg a spolu 22 021 účastníkov vo veku 16 rokov alebo starších dostalo placebo (vrátane dospievajúcich vo veku 16 a 17 rokov, v počte 138 v skupine s očkovacou látkou
a 145 v skupine s placebom). Spolu 20 519 účastníkov vo veku 16 rokov alebo starších dostalo
2 dávky očkovacej látky Comirnaty.
V čase analýzy štúdie 2 s dátumom ukončenia zberu údajov 13. marca 2021 pre placebom kontrolované, zaslepené obdobie sledovania až do dátumov odslepenia účastníkov bolo spolu 25 651 (58,2 %) účastníkov (13 031 dostalo očkovaciu látku Comirnaty a 12 620 dostalo placebo) vo veku
16 rokov a starších sledovaných po dobu ≥ 4 mesiace po druhej dávke. To zahŕňalo spolu
15 111 účastníkov (7 704 dostalo očkovaciu látku Comirnaty a 7 407 dostalo placebo) vo veku 16 až
55 rokov a spolu 10 540 účastníkov (5 327 dostalo očkovaciu látku Comirnaty a 5 213 dostalo placebo) vo veku 56 rokov a starších.
Najčastejšími nežiaducimi reakciami u účastníkov vo veku 16 rokov a starších, ktorí dostali 2 dávky, boli bolesť v mieste vpichu (> 80 %), únava (> 60 %), bolesť hlavy (> 50 %), myalgia (> 40 %), triaška (> 30 %), artralgia (> 20 %), pyrexia a opuch v mieste vpichu (> 10 %) a mali zvyčajne miernu alebo strednú intenzitu a do niekoľkých dní po podaní očkovacej látky ustúpili. Mierne nižšia frekvencia príhod reaktogenity súvisela s vyšším vekom.
Bezpečnostný profil u 545 účastníkov vo veku 16 rokov a starších dostávajúcich očkovaciu látku Comirnaty, ktorí boli na začiatku štúdie séropozitívni na SARS-CoV-2, bol podobný ako bezpečnostný profil pozorovaný v celkovej populácii.
Dospievajúci vo veku 12 až 15 rokov - po 2 dávkach
V analýze štúdie 2 zakladajúcej sa na údajoch získaných až do dátumu ukončenia sledovania (cut-off)
13. marca 2021 bolo zahrnutých 2 260 dospievajúcich (1 131 dostalo očkovaciu látku Comirnaty
30 µg a 1 129 dostalo placebo) vo veku 12 až 15 rokov. Z nich sa 1 308 dospievajúcich (660 dostalo
očkovaciu látku Comirnaty a 648 dostalo placebo) sledovalo aspoň 2 mesiace po druhej dávke
očkovacej látky Comirnaty. Vyhodnocovanie bezpečnosti v štúdii 2 ešte prebieha.
Najčastejšími nežiaducimi reakciami u dospievajúcich vo veku 12 až 15 rokov, ktorí dostali 2 dávky, boli bolesť v mieste vpichu (> 90 %), únava a bolesť hlavy (> 70 %), myalgia a triaška (> 40 %), artralgia a pyrexia (> 20 %).
Účastníci vo veku 18 rokov a starší - po posilňovacej dávke (tretia dávka)
Podskupina účastníkov v štúdii 2 fázy 2/3 pozostávajúca z dospelých vo veku 18 až 55 rokov, ktorí
ukončili pôvodný 2-dávkový cyklus Comirnaty, dostali posilňovaciu dávku (tretia dávka) Comirnaty
približne 6 mesiacov (rozsah 4,8 až 8,0 mesiacov) po podaní 2. dávky.
Najčastejšími nežiaducimi reakciami u účastníkov vo veku 18 až 55 rokov boli bolesť v mieste vpichu
(> 80 %), únava (> 60 %), bolesť hlavy (> 40 %), myalgia (> 30 %), triaška a artralgia (> 20 %).
Tabuľkovýzoznamnežiaducichreakciívklinických štúdiách a zo skúseností po uvedení lieku na trhu osôb vo veku 5 rokov a starších
Nežiaduce reakcie pozorované počas klinických štúdií sú uvedené nižšie podľa nasledujúcich kategórií
frekvencie:
veľmi časté (≥ 1/10),
časté (≥ 1/100 až < 1/10),
menej časté (≥ 1/1 000 až < 1/100),
zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
T
rieda orgánových systémov
|
Veľmi časté
(
≥ 1/10)
|
Časté
(
≥ 1/100 až
< 1/10)
|
M
enej časté
(
≥ 1/1 000 až
< 1/100)
|
Z
r
iedkavé
(
≥ 1/10 000 až
< 1/1 000)
|
Veľmi
zriedkavé
(
< 1/10 000)
|
Neznáme
(
z dostupných údajov)
|
Poruchy krvi
a lymfatického systému
|
|
|
lymfadenopatiaa
|
'
|
|
|
Poruchy imunitného systému
|
|
|
reakcie
z precitlivenosti (napr. vyrážka, pruritus, urtikáriab, angioedémb)
|
|
|
anafylaxia
|
Poruchy metabolizmu a výživy
|
|
|
znížená chuť do
jedla
|
|
|
|
Psychické poruchy
|
|
|
nespavosť
|
|
|
|
Poruchy nervového systému
|
bolesť hlavy
|
|
letargia
|
akútna periférna paralýza tvárec
|
|
parestéziad, hypestéziad
|
Poruchy srdca
|
|
|
|
|
myokarditídad, perikarditídad
|
|
Poruchy
gastrointestinálneho traktu
|
hnačkad
|
nevoľnosť,
vracanied
|
|
|
|
|
Poruchy kože
a podkožného tkaniva
|
|
|
hyperhidróza,
nočné potenie
|
|
|
multiformný erytémd
|
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
|
artralgia, myalgia
|
|
bolesť
v končatinee
|
|
|
|
Celkové poruchy
a reakcie v mieste podania
|
bolesť v mieste vpichu, únava, triaška, pyrexiaf, opuch v mieste
vpichu
|
začervenanie v mieste vpichuh
|
asténia, malátnosť, pruritus
v mieste vpichu
|
|
|
rozsiahly opuch očkovanej končatinyd, opuch tváreg
|
|
|
T
abuľka 1: Nežiaduce reakcie z klinických skúšaní a zo skúseností po uvedení lieku na trh s očkovacou látkou Comirnaty u osôb vo veku 5 rokov a starších
a. U účastníkov, ktorí dostali posilňovaciu dávku (tretia dávka), sa v porovnaní s účastníkmi, ktorí dostali
2 dávky, pozorovala vyššia frekvencia výskytu lymfadenopatie (5,2 % oproti 0,4 %). b. Kategória frekvencie pre urtikáriu a angioedém bola zriedkavé.
c. Počas obdobia sledovania bezpečnosti v rámci klinického skúšania až do 14. novembra 2020 bola hlásená akútna periférna paralýza (alebo ochrnutie) tváre u štyroch účastníkov v skupine s mRNA očkovacou látkou proti COVID-19. Nástup reakcie bol 37. deň po 1. dávke (účastník nedostal 2. dávku) a 3., 9. a 48. deň po
2. dávke. V skupine s placebom sa nehlásili žiadne prípady akútnej periférnej paralýzy (alebo ochrnutia)
tváre.
d. Nežiaduca reakcia zistená po uvedení lieku na trh.
e. Týka sa ruky, do ktorej bola podaná očkovacia látka.
f. Po druhej dávke sa pozorovala vyššia frekvencia pyrexie v porovnaní s prvou dávkou.
g. Vo fáze po uvedení na trh bol hlásený opuch tváre u očkovaných osôb s anamnézou podania injekcie s dermatologickým výplňovým materiálom.
h. Začervenanie v mieste vpichu sa vyskytlo s vyššou frekvenciou (veľmi často) u detí vo veku 5 až 11 rokov.
O
pis vybraných nežiaducich reakcií
M
yokarditída
Zvýšené riziko myokarditídy po očkovaní očkovacou látkou Comirnaty je najvyššie u mladších mužov
(pozri časť 4.4).
V dvoch veľkých, európskych, farmako-epidemiologických štúdiách sa určilo zvýšené riziko u mladších mužov po druhej dávke očkovacej látky Comirnaty.
Z jednej štúdie vyplynulo, že v období 7 dní po podaní druhej dávky sa u mužov vo veku 12–29 rokov vyskytlo približne o 0,265 (95 % IS 0,255–0,275) prípadov myokarditídy na 10 000 osôb viac ako u
neexponovaných osôb. V ďalšej štúdii sa v období 28 dní po podaní druhej dávky u mužov vo veku
16–24 rokov vyskytlo o 0,57 (95 % IS 0,39–0,75) prípadov myokarditídy na 10 000 osôb viac ako u neexponovaných osôb.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V
a uviedli číslo šarže, ak je k dispozícii.
4.9 PredávkovanieÚdaje o predávkovaní sú dostupné od 52 účastníkov zahrnutých do klinického skúšania, ktorí dostali
z dôvodu chybného nariedenia 58 mikrogramov očkovacej látky Comirnaty. Očkované osoby nehlásili
zvýšenie reaktogenity ani výskytu nežiaducich reakcií.
V prípade predávkovania sa odporúča sledovanie životných funkcií a prípadne symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: očkovacie látky, iné vírusové očkovacie látky, ATC kód: J07BX03
Mechanizmus účinkuMediátorová RNA s modifikovaným nukleozidom v očkovacej látke Comirnaty je zapuzdrená
v lipidových nanočasticiach, ktoré umožňujú dopravenie nereplikujúcej sa RNA do hostiteľských buniek na priamu dočasnú expresiu S antigénu vírusu SARS-CoV-2. mRNA kóduje S proteín plnej
dĺžky, ukotvený v membráne, s dvoma bodovými mutáciami v rámci centrálnej špirály. Mutácia
týchto dvoch aminokyselín na prolín uzamyká S proteín v antigénne uprednostňovanej pred-fúznej konformácii. Očkovacia látka vyvoláva odpoveď vo forme tvorby neutralizujúcich protilátok ako aj bunkovú imunitnú odpoveď na „spike“ (S) antigén, čo môže prispievať k ochrane pred ochorením COVID-19.
ÚčinnosťŠtúdia 2 je multicentrová, medzinárodná, randomizovaná, placebom kontrolovaná, pre pozorovateľov
zaslepená štúdia fázy 1/2/3 na stanovenie dávky, výber kandidátov na očkovaciu látku a stanovenie
účinnosti u účastníkov vo veku 12 rokov a starších. Randomizácia bola stratifikovaná podľa veku: vek
12 až 15 rokov, vek 16 až 55 rokov alebo vek 56 rokov a viac, pričom minimálne 40 % účastníkov bolo vo vekovej skupine ≥ 56 rokov. Z účasti na štúdii boli vylúčení imunokompromitovaní účastníci a tí, ktorí mali v minulosti klinicky alebo mikrobiologicky diagnostikovaný COVID-19. Účastníci
s prebiehajúcim stabilným ochorením definovaným ako ochorenie, ktoré si nevyžaduje významnú
zmenu liečby ani hospitalizáciu z dôvodu zhoršenia ochorenia počas 6 týždňov pred zaradením, boli
do tejto štúdie zahrnutí, ako aj účastníci so známou stabilnou infekciou vírusom ľudskej
imunodeficiencie (HIV), vírusom hepatitídy C (HCV) alebo vírusom hepatitídy B (HBV).
Účinnosť u účastníkov vo veku 16 rokov a starších - po 2 dávkachV časti fázy 2/3 štúdie 2 bolo na základe údajov zozbieraných do 14. novembra 2020 približne
44 000 účastníkov rovnomerne randomizovaných do skupín, ktoré dostali 2 dávky mRNA očkovacej látky proti COVID-19 alebo placebo. Analýzy účinnosti zahŕňali účastníkov, ktorí dostali druhú dávku očkovacej látky v intervale 19 až 42 dní po ich prvom očkovaní. Väčšina (93,1 %) očkovaných osôb dostala druhú dávku 19 až 23 dní po 1. dávke. Je plánované sledovanie účastníkov po dobu až
24 mesiacov po 2. dávke na účely hodnotení bezpečnosti a účinnosti proti ochoreniu COVID-19.
V klinickej štúdii sa vyžadovalo, aby sa u účastníkov pri podaní buď placeba alebo mRNA očkovacej látky proti COVID-19 dodržal interval minimálne 14 dní pred a po podaní očkovacej látky proti chrípke. V klinickej štúdii sa vyžadovalo, aby sa u účastníkov pri podaní buď placeba alebo mRNA očkovacej látky proti COVID-19 dodržal interval minimálne 60 dní pred alebo po podaní krvných/plazmových produktov alebo imunoglobulínov v rámci obdobia do ukončenia štúdie.
Populácia na analýzu primárneho cieľového ukazovateľa účinnosti zahŕňala 36 621 účastníkov vo
veku 12 rokov a starších (18 242 v skupine s mRNA očkovacou látkou proti COVID-19
a 18 379 v skupine s placebom), u ktorých nebol do doby 7 dní po druhej dávke zistený dôkaz predchádzajúcej infekcie vírusom SARS-CoV-2. Ďalej, 134 účastníkov bolo vo veku medzi 16 až
17 rokov (66 v skupine s mRNA očkovacou látkou proti COVID-19 a 68 v skupine s placebom)
a 1 616 účastníkov bolo vo veku 75 rokov a starších (804 v skupine s mRNA očkovacou látkou proti
COVID-19 a 812 v skupine s placebom).
V čase primárnej analýzy účinnosti boli účastníci sledovaní z hľadiska symptomatického COVID-19 po dobu spolu 2 214 osoborokov v skupine s mRNA očkovacou látkou proti COVID-19 a po dobu spolu 2 222 osoborokov v skupine s placebom.
U účastníkov ohrozených závažným priebehom COVID-19 vrátane tých s 1 alebo viacerými komorbiditami, ktoré zvyšujú riziko závažného priebehu COVID-19 (napr. astma, index telesnej hmotnosti (BMI) ≥ 30 kg/m2, chronické ochorenie pľúc, diabetes mellitus, hypertenzia), sa nevyskytli žiadne významné klinické rozdiely v celkovej účinnosti očkovacej látky.
Informácie o účinnosti očkovacej látky sú uvedené v tabuľke 2.
Prvý výskyt COVID-19 od 7 dní po 2. dávke u účastníkov bez dôkazu predchádzajúcej
infekcie SARS-CoV-2*
|
Podskupina
| mRNA očkovacia látka
proti COVID-19
na = 18 198
Prípady n1b
Čas sledovaniac (n2d)
|
Placebo na = 18,325
Prípady n1b
Čas sledovaniac (n2d)
|
Účinnosť očkovacej
látky % (95 % IS)e
| Všetci účastníci
| 8
2,214 (17 411)
| 162
2,222 (17 511)
| 95,0
(90,0; 97,9)
| 16 až 64 rokov
| 7
1,706 (13 549)
| 143
1,710 (13 618)
| 95,1
(89,6; 98,1)
| 65 rokov a starší
| 1
0,508 (3 848)
| 19
0,511 (3 880)
| 94,7
(66,7; 99,9)
| 65 až 74 rokov
| 1
0,406 (3 074)
| 14
0,406 (3 095)
| 92,9
(53,1; 99,8)
| 75 rokov a starší
| 0
0,102 (774)
| 5
0,106 (785)
| 100,0
(-13,1; 100,0)
|
|
|
Tabuľka 2: Účinnosť očkovacej látky - prvý výskyt COVID-
19 od 7 dní po 2. dávke podľa vekových podskupín - účastníci bez dôkazu infekcie do 7 dní po 2. dávke - populácia hodnotiteľná z hľadiska účinnosti (7 dní)
Poznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou
(RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 [*Definícia prípadu: (aspoň 1 z) horúčka, nový alebo zhoršený kašeľ, nová alebo zhoršená dýchavičnosť, triaška, nová alebo zhoršená bolesť svalov, nová strata chuti alebo čuchu, bolesť hrdla, hnačka alebo vracanie.]
* Do analýzy boli zahrnutí účastníci, ktorí nemali sérologický ani virologický dôkaz (do 7 dní po podaní druhej dávky) predchádzajúcej infekcie SARS-CoV-2 (t.j. negatívni na N-viažuce protilátky [sérum] pri
1. kontrole a nezistený SARS-CoV-2 použitím testu amplifikácie nukleových kyselín (NAAT) [výter
z nosa] pri 1. a 2. kontrole) a mali negatívny NAAT (výter z nosa) pri akejkoľvek neplánovanej kontrole do
7 dní po podaní 2. dávky.
a. n = počet účastníkov v špecifikovanej skupine.
b. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
c. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po podaní 2. dávky po koniec obdobia sledovania.
d. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
e. Dvojstranný interval spoľahlivosti (IS) pre účinnosť očkovacej látky je odvodený na základe Clopperovej a
Pearsonovej metódy upravenej podľa doby sledovania. IS nie je upravený pre multiplicitu.
Účinnosť mRNA očkovacej látky proti COVID-19 v prevencii prvého výskytu COVID-19 od 7. dňa
po 2. dávke v porovnaní s placebom u účastníkov vo veku 16 rokov a starších s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 bola 94,6 % (95 % interval spoľahlivosti 89,6 % až
97,6 %).
Okrem toho, analýzy primárneho cieľového ukazovateľa účinnosti v rámci jednotlivých podskupín ukázali podobné bodové odhady medzi jednotlivými pohlaviami, etnickými skupinami a účastníkmi so zdravotnými komorbiditami spojenými s vysokým rizikom závažného priebehu COVID-19.
Vykonali sa aktualizované analýzy účinnosti s ďalšími potvrdenými prípadmi COVID-19, ktoré sa objavili počas zaslepeného, placebom kontrolovaného sledovania, čo v populácii skúmanej z hľadiska účinnosti predstavuje až 6 mesiacov po 2. dávke.
Aktualizované informácie o účinnosti očkovacej látky sú uvedené v tabuľke 3.
Podskupina
| mRNA očkovacia látka
proti COVID-19
na = 20 998
Prípady n1b
Čas sledovaniac (n2d)
|
Placebo na = 21 096
Prípady n1b
Čas sledovaniac (n2d)
|
Účinnosť očkovacej
látky % (95 % IS)e
| Všetci účastnícif
| 77
6,247 (20 712)
| 850
6,003 (20 713)
| 91,3
(89,0; 93,2)
| 16 až 64 rokov
| 70
4,859 (15 519)
| 710
4,654 (15 515)
| 90.6
(87,9; 92,7)
| 65 rokov a starší
| 7
1,233 (4 192)
| 124
1,202 (4 226)
| 94.5
(88,3; 97,8)
| 65 až 74 rokov
| 6
0,994 (3 350)
| 98
0,966 (3 379)
| 94.1
(86,6; 97,9)
| 75 rokov a starší
| 1
0,239 (842)
| 26
0,237 (847)
| 96.2
(76,9; 99.9)
|
|
|
Tabuľka 3: Účinnosť očkovacej látky - prvý výskyt COVID-
19 od 7 dní po 2. dávke podľa vekových podskupín - účastníci bez dôkazu predchádzajúcej infekcie SARS-CoV-2* do 7 dní po 2. dávke – populácia hodnotiteľná z hľadiska účinnosti (7 dní) počas placebom kontrolovaného obdobia sledovaniaPoznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou
(RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 (príznaky zahŕňali horúčku, nový alebo zhoršený kašeľ, novú alebo zhoršenú dýchavičnosť, triašku, novú alebo zhoršenú bolesť svalov, novú stratu chuti alebo čuchu, bolesť hrdla, hnačku, vracanie).
* Do analýzy boli zahrnutí účastníci, ktorí nemali dôkaz predchádzajúcej infekcie SARS-CoV-2 (t.j. negatívni na N-viažuce protilátky [sérum] pri 1. kontrole a nezistený SARS-CoV-2 použitím NAAT [výter z nosa] pri 1. a 2. kontrole) a mali negatívny NAAT (výter z nosa) pri akejkoľvek nenaplánovanej kontrole
do 7 dní po podaní 2. dávky.
a. n = počet účastníkov v špecifikovanej skupine.
b. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
c. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po podaní 2. dávky po koniec obdobia sledovania.
d. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
e. Dvojstranný 95 % interval spoľahlivosti (IS) pre účinnosť očkovacej látky je odvodený na základe
Clopperovej a Pearsonovej metódy upravenej podľa doby sledovania.
f. Zahrnuté sú potvrdené prípady u účastníkov vo veku 12 až 15 rokov: 0 v skupine s mRNA očkovacou
látkou proti COVID-19, 16 v skupine s placebom.
V aktualizovanej analýze účinnosti bola účinnosť mRNA očkovacej látky proti COVID-19 v prevencii
prvého výskytu COVID-19 od 7. dňa po 2. dávke v porovnaní s placebom 91,1 % (95 % IS 88,8 % až
93,0 %) u účastníkov v populácii hodnotiteľnej z hľadiska účinnosti s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2.
Okrem toho, aktualizované analýzy účinnosti podľa podskupín ukázali podobné bodové odhady účinnosti medzi pohlaviami, etnickými skupinami, geografickými oblasťami a účastníkmi so zdravotnými komorbiditami a obezitou spojenou s vysokým rizikom závažného priebehu COVID-19.
Účinnosť voči závažnému priebehu COVID-19Aktualizované analýzy účinnosti sekundárnych cieľových ukazovateľov účinnosti podporovali prínos mRNA očkovacej látky proti COVID-19 v prevencii závažného priebehu COVID-19.
K 13. marcu 2021 je účinnosť očkovacej látky voči závažnému priebehu COVID-19 uvádzaná len pre účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 (tabuľka 4), pretože počty prípadov COVID-19 u účastníkov bez predchádzajúcej infekcie SARS-CoV-2 boli rovnaké ako u účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 v skupine s mRNA očkovacou látkou proti COVID-19 aj v skupine s placebom.
Tabuľka 4: Účinnosť očkovacej látky - prvý výskyt závažného priebehu COVID-19u účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie SARS-CoV-2 na základe údajov Úradu pre potraviny a liečivá (Food and Drug Administration,
Podskupina
| mRNA očkovacia látka
proti COVID-19
Prípady n1a
Čas sledovania (n2b)
|
Placebo Prípady n1a
Čas sledovania (n2b)
|
Účinnosť očkovacej
látky % (95 % ISc)
| Po 1. dávked
| 1
8,439e (22 505)
| 30
8,288e (22 435)
| 96,7
(80,3; 99,9)
| 7 dní po 2. dávkef
| 1
6,522g (21 649)
| 21
6,404g (21 730)
| 95,3
(70,9; 99,9)
|
|
|
FDA)* po 1. dávke alebo od 7 dní po 2. dávke v placebom kontrolovanom období sledovaniaPoznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou
(RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 (príznaky zahŕňali horúčku, nový alebo zhoršený kašeľ, novú alebo zhoršenú dýchavičnosť, triašku, novú alebo zhoršenú bolesť svalov, novú stratu chuti alebo čuchu, bolesť hrdla, hnačku, vracanie).
* Závažné ochorenie spôsobené COVID-19 definované podľa FDA je potvrdený COVID-19 a prítomnosť
aspoň 1 z nasledujúcich:
• klinické prejavy v pokoji naznačujúce závažné systémové ochorenie (dychová frekvencia ≥ 30 dychov za minútu, srdcová frekvencia ≥ 125 úderov za minútu, saturácia kyslíkom ≤ 93 % pri izbovom vzduchu vo výške hladiny mora alebo pomer parciálneho artériového tlaku kyslíka
k frakčnému inspirovanému kyslíku < 300 mm Hg),
• zlyhanie dýchania [definované ako potreba vysokého prietoku kyslíka, neinvazívnej ventilácie, mechanickej ventilácie alebo mimotelovej membránovej oxygenácie (
extracorporeal membrane oxygenation, ECMO)],
• dôkaz šoku (systolický krvný tlak < 90 mm Hg, diastolický krvný tlak < 60 mm Hg alebo potreba použitia vazopresív),
• významná akútna obličková, pečeňová alebo neurologická dysfunkcia,
• prijatie na jednotku intenzívnej starostlivosti,
• úmrtie.
a. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
b. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
c. Dvojstranný interval spoľahlivosti (IS) pre účinnosť očkovacej látky je odvodený na základe Clopperovej
a Pearsonovej metódy upravenej podľa doby sledovania.
d. Účinnosť hodnotená na základe celej dostupnej populácie (modifikovaná „
intention-to-treat“) účinnosti po
1. dávke zahŕňajúca všetkých randomizovaných účastníkov, ktorí dostali aspoň 1 dávku skúmanej liečby.
e. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 1. dávky po koniec obdobia sledovania.
f. Účinnosť hodnotená na základe populácie hodnotiteľnej z hľadiska účinnosti (7 dní), ktorá zahŕňala všetkých spôsobilých randomizovaných účastníkov, ktorí dostali všetky dávky skúmanej liečby podľa randomizácie v rámci preddefinovaného časového obdobia a nemali žiadne iné dôležité odchýlky od protokolu stanovené lekárom.
g. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ pre všetkých účastníkov v každej skupine s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po 2. dávke po koniec obdobia sledovania.
Účinnosť a imunogenita u dospievajúcich vo veku 12 až 15 rokov - po 2 dávkachV analýze štúdie 2 u dospievajúcich vo veku 12 až 15 rokov bez dôkazu predchádzajúcej infekcie sa nevyskytli žiadne prípady u 1 005 účastníkov, ktorí dostali očkovaciu látku a vyskytlo sa 16 prípadov z 978 u účastníkov, ktorí dostali placebo. Bodový odhad účinnosti je 100 % (95 % interval spoľahlivosti 75,3; 100,0). U účastníkov s dôkazom alebo bez dôkazu predchádzajúcej infekcie sa vyskytlo 0 prípadov u 1 119 účastníkov, ktorí dostali očkovaciu látku a 18 prípadov
u 1 110 účastníkov, ktorí dostali placebo. To tiež ukazuje bodový odhad účinnosti 100 % (95 %
interval spoľahlivosti 78,1; 100,0).
V štúdii 2 sa vykonala analýza neutralizačných titrov SARS-CoV-2 1 mesiac po 2. dávke v náhodne vybranej podskupine účastníkov, ktorí nemali žiadny sérologický ani virologický dôkaz predchádzajúcej infekcie SARS-CoV-2 až do 1 mesiaca po 2. dávke porovnávajúca odpoveď
u dospievajúcich vo veku 12 až 15 rokov (n = 190) s účastníkmi vo veku 16 až 25 rokov (n = 170).
Pomer geometrických priemerných titrov (
Geometric Mean Titres, GMT) v skupine 12 až 15 rokov ku skupine vo veku 16 až 25 rokov bol 1,76 s 2-stranným 95 % IS 1,47 až 2,10. Keďže dolná hranica
2-stranného 95 % IS pre pomer geometrického priemeru [
Geometric Mean Ratio, GMR] bola > 0,67,
bolo splnené kritérium 1,5-násobnej non-inferiority.
Účinnosť a imunogenita u detí vo veku 5 až 11 rokov (t.j. vo veku 5 až menej ako 12 rokov) – po2 dávkachŠtúdia 3 je štúdiou fázy 1/2/3 pozostávajúcou z otvorenej časti zameranej na stanovenie dávky
očkovacej látky (fáza 1) a multicentrovej, medzinárodnej, randomizovanej, fyziologickým roztokom ako placebom kontrolovanej, pre pozorovateľa zaslepenej časti zameranej na účinnosť (fáza 2/3), do ktorej boli zaradení účastníci vo veku 5 až 11 rokov. Väčšina (94,4 %) randomizovaných účastníkov, ktorí dostali očkovaciu látku, dostalo druhú dávku 19 až 23 dní po 1. dávke.
Popisné výsledky účinnosti očkovacej látky u detí vo veku 5 až 11 rokov bez dôkazu predchádzajúcej infekcie SARS-CoV-2 sú uvedené v tabuľke 5. U účastníkov s predchádzajúcou infekciou
SARS-CoV-2 sa v skupine s očkovacou látkou ani v skupine s placebom nepozorovali žiadne prípady
COVID-19.
P
r
vý výskyt COVID-19 od 7 dní po 2. dávke u detí vo veku 5 až 11 rokov bez dôkazu predchádzajúcej infekcie SARS-CoV-2*
|
|
m
R
N
A očkovacia
l
átka proti COVID-19
10 µg/dávka n
a
= 1 305
Prípady n1
b
Č
a
s sledovania
c
(
n
2
d
)
|
Placebo n
a
= 663
Prípady n1
b
Č
a
s sledovania
c
(
n
2
d
)
|
Ú
č
i
n
nosť očkovacej látky
%
(
9
5 % IS)
|
Deti vo veku 5 až
11 rokov
|
3
0,322 (1 273)
|
16
0,159 (637)
|
90,7
(67,7; 98,3)
|
|
|
T
abuľka 5: Účinnosť očkovacej látky - prvý výskyt COVID-19 od 7 dní po 2. dávke: bez dôkazu infekcie do 7 dní po 2. dávke – fáza 2/3 – populácia detí vo veku 5 až 11 rokov hodnotiteľná z hľadiska účinnosti
Poznámka: Potvrdené prípady sa stanovili polymerázovou reťazovou reakciou s reverznou transkripciou
(RT-PCR) a aspoň 1 príznakom zhodným s COVID-19 (príznaky zahŕňali: horúčku; nový alebo zhoršený kašeľ, novú alebo zhoršenú dýchavičnosť, triašku, novú alebo zhoršenú bolesť svalov, novú stratu chuti alebo čuchu, bolesť hrdla, hnačku alebo vracanie.
* Do analýzy boli zahrnutí účastníci, ktorí nemali dôkaz predchádzajúcej infekcie SARS-CoV-2 (t.j. negatívni na N-viažuce protilátky [sérum] pri 1. kontrole a nezistený SARS-CoV-2 použitím testu NAAT
(výter z nosa) pri 1. a 2. kontrole) a mali negatívny NAAT (výter z nosa) pri akejkoľvek neplánovanej
kontrole do 7 dní po podaní 2. dávky.
a. n = počet účastníkov v špecifikovanej skupine.
b. n1 = počet účastníkov spĺňajúcich definíciu cieľového ukazovateľa.
c. Celkový čas sledovania vyjadrený v 1 000 osoborokoch pre daný cieľový ukazovateľ u všetkých účastníkov v rámci každej skupiny s rizikom pre cieľový ukazovateľ. Časový interval na rozlíšenie prípadov COVID-19 je od 7 dní po podaní 2. dávky po koniec obdobia sledovania.
d. n2 = počet účastníkov s rizikom pre cieľový ukazovateľ.
V štúdii 3 preukázala analýza 50 % neutralizačných titrov SARS-CoV-2 (NT50) 1 mesiac po podaní
2. dávky u náhodne vybranej podskupiny účastníkov účinnosť prostredníctvom tzv.
immunobridginguimunitnej odpovede porovnávajúceho deti vo veku 5 až 11 rokov (t.j. vo veku 5 až menej ako
12 rokov) v časti fázy 2/3 štúdie 3 s účastníkmi vo veku 16 až 25 rokov v časti fázy 2/3 štúdie 2, ktorí nemali žiadny sérologický ani virologický dôkaz predchádzajúcej infekcie SARS-CoV-2 až do
1 mesiaca po 2. dávke, spĺňajúcich predšpecifikované kritériá
immunobridgingu pre pomer geometrického priemeru (GMR) aj rozdiel v miere sérologickej odpovede definovaný ako dosiahnutie najmenej 4-násobného zvýšenia SARS-CoV-2 NT50 oproti východiskovému stavu (pred 1. dávkou).
Hodnota GMR SARS-CoV-2 NT50 1 mesiac po 2. dávke u detí vo veku 5 až 11 rokov (t.j. vo veku 5
až menej ako 12 rokov) v porovnaní s hodnotou u mladých dospelých vo veku 16 až 25 rokov bola
1,04 (2-stranný 95 % IS: 0,93; 1,18). U účastníkov bez dôkazu predchádzajúcej infekcie SARS-CoV-2 až do 1 mesiaca po 2. dávke malo 99,2 % detí vo veku 5 až 11 rokov a 99,2 % účastníkov vo veku 16 až 25 rokov sérologickú odpoveď 1 mesiac po 2. dávke. Rozdiel v pomere účastníkov so sérologickou odpoveďou medzi týmito 2 vekovými skupinami (deti – mladí dospelí) bol 0,0 % (2-stranný 95 %
IS: -2,0 %, 2,2 %). Tieto informácie sú uvedené v tabuľke 6.
T
abuľka 6: Súhrn pomeru geometrického priemeru 50 % neutralizačných titrov a rozdiel
|
m
R
N
A očkovacia látka proti
COV
I
D
-19
|
5 až 11 rokov/
16 až 25 rokov
|
10 µg/dávka
5 až 11 rokov n
a
= 264
|
30 µg/dávka
16 až 25 rokov n
a
= 253
|
|
|
Č
asový
bod
b
|
G
MT
c
(
95 % IS
c
)
|
G
MT
c
(
95 % IS
c
)
|
G
MR
d
(
95 % IS
d
)
|
Splnili cieľ
i
mm
unobridgingu
e
(
Á
/
N)
|
G
eometrický priemer 50 %
neutralizačného
ti
t
ra
f
(
G
MT
c
)
|
1 mesiac po
2. dávke
|
1 197,6
(1 106,1; 1 296,6)
|
1 146,5
(1 045,5; 1 257,2)
|
1,04 (0,93; 1,18)
|
Á
|
|
Č
asový
bod
b
|
n
g
(
%)
(
95 % IS
h
)
|
n
g
(
%)
(
95 % IS
h
)
|
R
ozdiel %
i
(
95 % IS
j
)
|
Splnili cieľ immunobridgingu
k
(
Á
/
N)
|
Miera sérologickej
odpovede (%)
pre 50 % neutralizačný titer
f
|
1 mesiac po
2. dávke
|
262 (99,2) (97,3; 99,9)
|
251 (99,2) (97,2; 99,9)
|
0,0
(-2,0; 2,2)
|
Á
|
|
|
v percentuálnych počtoch účastníkov so sérologickou odpoveďou – porovnanie detí vo veku 5 až 11 rokov (štúdia 3) s účastníkmi vo veku 16 až 25 rokov (štúdia 2) - účastníci bez dôkazu predchádzajúcej infekcie až do 1 mesiaca po 2. dávke – podskupina immunobridgingu – fáza 2/3 – populácia hodnotiteľná z hľadiska účinnosti
Skratky: IS = interval spoľahlivosti, GMR = pomer geometrického priemeru, GMT = geometrický
priemerný titer, LLOQ = dolná hranica kvantifikácie (
lower limit of quantitation), NAAT = test amplifikácie nukleových kyselín, NT50 = 50 % neutralizačný titer, SARS-CoV-2 = závažný akútny
respiračný syndróm vyvolaný koronavírusom 2.
Poznámka: Do analýzy boli zahrnutí účastníci, ktorí nemali sérologický ani virologický dôkaz (vzorka krvi odobratá až do 1 mesiaca po podaní 2. dávky) predchádzajúcej infekcie SARS-CoV-2 (t.j. negatívni na
N-viažuce protilátky [sérum] pri kontrole pri 1. dávke a 1 mesiac po 2. dávke, nezistený SARS-CoV-2
použitím testu NAAT [výter z nosa] pri kontrolách 1. dávke a 2 dávke a negatívny NAAT (výter z nosa) pri akejkoľvek neplánovanej kontrole až do 1 mesiaca po podaní 2. dávky zo vzorky krvi) a ktorí nemali COVID-19 v anamnéze.
Poznámka: Sérologická odpoveď sa definovala ako dosiahnutie ≥ 4-násobného zvýšenia oproti východiskovému stavu (pred 1. dávkou). Ak je meraná hodnota vo východiskovom stave pod hodnotou
LLOQ, výsledok testu po očkovaní ≥4 × LLOQ sa považuje za sérologickú odpoveď.
a. n = počet účastníkov s platnými a určenými výsledkami testu pred očkovaním a 1 mesiac po 2. dávke.
Tieto hodnoty sú tiež menovateľmi vo výpočte percentuálnych hodnôt miery sérologickej odpovede.
b. Protokolom špecifikované načasovanie odberu krvných vzoriek.
c. Hodnoty GMT a 2-stranného 95 % IS sa vypočítali umocnením priemerných logaritmických hodnôt titrov a príslušných IS (na základe Studentovho t rozdelenia). Výsledky testu pod hodnotou LLOQ boli
stanovené ako 0,5-násobok LLOQ.
d. Hodnoty GMR a 2-stranného 95 % IS sa vypočítali umocnením priemerného rozdielu logaritmických hodnôt titrov (vek 5 až 11 rokov mínus vek 16 až 25 rokov) a príslušných IS (na základe Studentovho t
rozdelenia).
e.
Immunobridging sa deklaroval na základe hodnoty GMT, ak bola dolná hranica 2-stranného 95 % IS
pre GMR vyššia ako 0,67 a bodový odhad GMR ≥ 0,8.
f. NT50 SARS-CoV-2 sa stanovili použitím mikroneutralizačného testu SARS-CoV-2 mNeonGreen.
Test používa fluorescenčný reportérový vírus odvodený z kmeňa USA_WA1/2020 a neutralizácia vírusu sa odčíta na monovrstvách buniek Vero. Vzorka NT50 je definovaná ako vzájomné riedenie séra, pri ktorom je neutralizovaných 50 % vírusu.
g. n = počet účastníkov so sérologickou odpoveďou na základe NT50 1 mesiac po 2. dávke. h. Presný 2-stranný IS je odvodený na základe Clopperovej a Pearsonovej metódy.

i. Rozdiel v podieloch vyjadrený v percentách (vek 5 až 11 rokov mínus vek 16 až 25 rokov).
j. 2-stranný IS na základe Miettinenovej a Nurminenovej metódy pre rozdiel v podieloch vyjadrený v percentách.
k.
Immunobridging sa deklaroval na základe miery sérologickej odpovede, ak bola dolná hranica
2-stranného 95 % IS pre sérologickú odpoveď vyššia ako -10,0 %.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s očkovacou látkou
Comirnaty u pediatrickej populácie na prevenciu ochorenia COVID-19 (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku. Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnostiNeaplikovateľné.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých štúdií toxicity po opakovanom podávaní a reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Celková toxicitaU potkanov, ktorým sa intramuskulárne podávala očkovacia látka Comirnaty (dostávajúc 3 plné
ľudské dávky jedenkrát týždenne, ktoré viedli u potkanov k celkovo vyšším hladinám z dôvodu rozdielu v telesnej hmotnosti) sa pozoroval mierny edém a erytém v mieste vpichu a zvýšenie počtu leukocytov (vrátane bazofilov a eozinofilov), čo je konzistentné so zápalovou odpoveďou, ako aj vakuolizácia portálnych hepatocytov bez dôkazu poškodenia pečene. Všetky účinky boli reverzibilné.
Genotoxicita/karcinogenitaNevykonali sa štúdie genotoxicity ani karcinogenity. Pre komponenty očkovacej látky (lipidy
a mRNA) sa neočakáva genotoxický potenciál.
Reprodukčná toxicitaReprodukčná a vývinová toxicita sa skúmala na potkanoch v kombinovanej štúdii fertility a vývinovej
toxicity, pri ktorej sa samiciam potkanov intramuskulárne podávala očkovacia látka Comirnaty pred párením a počas gravidity (dostali 4 plné ľudské dávky, ktoré viedli u potkanov k celkovo vyšším hladinám z dôvodu rozdielu v telesnej hmotnosti, a to v období 21 dní pred párením do 20. dňa gravidity). U samíc bola prítomná imunitná odpoveď prostredníctvom neutralizačných protilátok proti SARS-CoV-2 počas obdobia pred párením až po koniec štúdie v 21. postnatálny deň, pričom neutralizujúce protilátky boli prítomné aj u plodov a mláďat. Nevyskytli sa žiadne účinky na samičiu fertilitu, graviditu alebo embryofetálny vývin alebo vývin mláďat súvisiace s očkovacou látkou. Nie sú dostupné žiadne údaje pre očkovaciu látku Comirnaty týkajúce sa prechodu očkovacej látky cez placentu alebo jej vylučovania do mlieka.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
((4-hydroxybutyl)azándiyl)bis(hexán-6,1-diyl)bis(2-hexyldekanoát) (ALC-0315)
2-[(polyetylénglykol)-2000]-N,N-ditetradecylacetamid (ALC-0159)
1,2-distearoyl-sn-glycero-3-fosfocholín (DSPC)
cholesterol trometamol
trometamólium-chlorid
sacharóza
voda na injekcie
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
Zmrazená injekčná liekovka
9 mesiacov, ak sa uchováva pri teplote -90 °C až -60 °C.
Očkovacia látka sa môže dodávať zmrazená pri teplote -90 °C až -60 °C alebo -25 °C až -15 °C. Zmrazená očkovacia látka sa môže po prijatí uchovávať buď pri teplote -90 °C až -60 °C alebo pri teplote 2 °C až 8 °C.
Ak sa uchovávajú zmrazené pri teplote -90 °C až -60 °C, môžu sa balenia po 10 injekčných liekoviek očkovacej látky rozmraziť pri teplote 2 °C až 8 °C po dobu 4 hodín alebo jednotlivé injekčné liekovky sa môžu rozmraziť pri izbovej teplote (maximálne 30 °C) po dobu 30 minút.
Rozmrazená injekčná liekovka
10-týždňové uchovávanie a preprava pri teplote 2 °C až 8 °C v rámci 9-mesačného času použiteľnosti.
• Po premiestnení očkovacej látky do podmienok uchovávania pri teplote 2 °C až 8 °C sa musí aktualizovaný dátum exspirácie napísať na vonkajšiu škatuľku a očkovacia látka sa má použiť alebo zlikvidovať do aktualizovaného dátumu exspirácie. Pôvodný dátum exspirácie sa má preškrtnúť.
• Ak sa očkovacia látka dodá pri teplote 2 °C až 8 °C, má sa uchovávať pri teplote 2 °C až 8 °C.
Dátum exspirácie na vonkajšej škatuli mal byť aktualizovaný tak, aby zobrazoval dátum exspirácie očkovacej látky uchovávanej v chladničke a pôvodný dátum exspirácie sa má preškrtnúť.
Pred použitím sa môžu neotvorené injekčné liekovky uchovávať maximálne 12 hodín pri teplotách medzi 8 °C a 30 °C.
S rozmrazenými injekčnými liekovkami je možné manipulovať v podmienkach s umelým osvetlením miestnosti.
Po rozmrazení sa očkovacia látka nesmie opakovane zmraziť.
Manipulácia pri teplotných výkyvoch počas uchovávania v chladničke
• Údaje o stabilite naznačujú, že neotvorená injekčná liekovka je stabilná až do
10 týždňov, ak sa uchováva pri teplotách od -2 °C do 2 °C a v rámci 10-týždňového obdobia
uchovávania pri teplotách medzi 2 °C a 8 °C.
• Údaje o stabilite naznačujú, že injekčná liekovka sa môže uchovávať maximálne 24 hodín pri
teplotách 8 °C až 30 °C, vrátane maximálne 12 hodín po prvom prepichnutí zátky.
Tieto informácie sú určené na pomoc zdravotníckym pracovníkom len v prípade dočasných teplotných
výkyvov.
Nariedený liek
Chemická a fyzikálna stabilita počas používania, sa po nariedení s injekčným roztokom chloridu
sodného 9 mg/ml (0,9 %) preukázala počas 12 hodín pri teplote 2 °C až 30 °C. Z mikrobiologického hľadiska, ak metóda riedenia nevylučuje riziko mikrobiálnej kontaminácie, sa má liek použiť okamžite. Ak sa liek nepoužije okamžite, za čas použiteľnosti a podmienky uchovávania počas používania zodpovedá používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v mrazničke pri teplote -90 °C až -60 °C. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Počas uchovávania minimalizujte pôsobenie svetla v miestnosti na liek a nevystavujte priamemu
slnečnému svetlu ani ultrafialovému žiareniu.
Podmienky na uchovávanie po rozmrazení a riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
1,3 ml koncentrátu na injekčnú disperziu v 2 ml čírej, viacdávkovej, injekčnej liekovke (sklo typu I) so zátkou (syntetická brómbutylová guma) a oranžovým odklápacím plastovým viečkom s hliníkovým tesnením. Jedna injekčná liekovka obsahuje 10 dávok, pozri časť 6.6.
Veľkosti balenia: 10 injekčných liekoviek alebo 195 injekčných liekoviek
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Pokyny na zaobchádzanie s liekom
Comirnaty 10 mikrogramov/dávka má pripravovať zdravotnícky pracovník pomocou aseptického
postupu na zabezpečenie sterility pripravenej disperzie.
KONTROLA DÁVKY OČKOVACEJ LÁTKY COMIRNATY 10 MIKROGRAMOV/DÁVKA
KONCENTRÁT NA INJEKČNÚ DISPERZIU (DETI VO VEKU 5 AŽ 11 ROKOV)
• Skontrolujte, či má injekčná liekovka
oranžové plastové viečko.
• Ak má injekčná liekovka fialové plastové
 O
ranžové viečko
10 µg
O
ranžové viečko
10 µg
viečko, prečítajte si súhrn charakteristických vlastností lieku očkovacej látky Comirnaty
30 mikrogramov/dávka koncentrát na
injekčnú disperziu.
• Ak má injekčná liekovka sivé plastové viečko, prečítajte si súhrn charakteristických vlastností lieku očkovacej látky Comirnaty
30 mikrogramov/dávka injekčná disperzia.

MANIPULÁCIA PRED POUŽITÍM OČKOVACEJ LÁTKY COMIRNATY
10 MIKROGRAMOV/DÁVKA KONCENTRÁT NA INJEKČNÚ DISPERZIU (DETI VO VEKU 5 AŽ 11 ROKOV)
|
U
chovávajte maximálne
10 týždňov pri
t
eplote 2 °C až
8 °C
|
• Ak je viacdávková injekčná liekovka zmrazená, musí sa pred nariedením. Zmrazené injekčné liekovky sa majú premiestniť do prostredia s teplotou od
2 °C do 8 °C, aby sa rozmrazili;
rozmrazenie balenia 10 injekčných liekoviek môže trvať až 4 hodiny. Zaistite, aby boli pred použitím injekčné liekovky úplne rozmrazené.
• Po premiestnení injekčných liekoviek do podmienok uchovávania pri teplote 2 °C až
8 °C aktualizujte dátum exspirácie na
škatuľke.
• Neotvorené injekčné liekovky sa môžu uchovávať maximálne 10 týždňov pri teplote 2 °C až 8 °C v rámci 9-mesačného času použiteľnosti.
• Zmrazené jednotlivé injekčné liekovky je možné alternatívne nechať rozmraziť po dobu 30 minút pri teplotách do 30 °C.
• Pred použitím sa môže neotvorená
injekčná liekovka uchovávať maximálne
12 hodín pri teplotách do 30 °C.
S rozmrazenými injekčnými liekovkami je možné manipulovať v podmienkach
s umelým osvetlením miestnosti.
|
Z
MIEŠANIE PRED RIEDENÍM OČKOVACEJ LÁTKY COMIRNATY
10 MIKROGRAMOV/DÁVKA KONCENTRÁT NA INJEKČNÚ DISPERZIU (DETI VO VEKU 5 AŽ 11 ROKOV)
|
Jemne 10x
|
• Rozmrazenú injekčnú liekovku nechajte dosiahnuť izbovú teplotu a pred nariedením ju 10-krát opatrne prevráťte. Nepretrepávajte.
• Pred nariedením môže rozmrazená disperzia obsahovať biele až sivobiele nepriehľadné amorfné častice.
|
R
I
EDEN
I
E OČKOVACEJ LÁTKY COMIRNATY 10 MIKROGRAMOV/DÁVKA
KO
NCENTRÁ
T NA INJEKČNÚ DISPERZIU (DETI VO VEKU 5 AŽ 11 ROKOV)
• Rozmrazená očkovacia látka musí byť nariedená v jej pôvodnej injekčnej liekovke s 1,3 ml injekčného roztoku
chloridu sodného s koncentráciou 9 mg/ml
(0,9 %) pomocou ihly veľkosti 21 G alebo tenšej a pomocou aseptického postupu.
 1,3 ml 0,9 % injekčného roztoku chloridu
s
odného
1,3 ml 0,9 % injekčného roztoku chloridu
s
odného
• Pred vytiahnutím ihly zo zátky injekčnej liekovky vyrovnajte tlak v injekčnej liekovke odobratím 1,3 ml vzduchu do prázdnej injekčnej striekačky, ktorou sa vstreklo rozpúšťadlo.
 Z
atiahnite piest na 1,3 ml na
odstránenie vzduchu z injekčnej
li
ekovky.
Z
atiahnite piest na 1,3 ml na
odstránenie vzduchu z injekčnej
li
ekovky.


Jemne 10x
|
• Nariedenú disperziu 10-krát jemne
prevráťte. Nepretrepávajte.
• Nariedená očkovacia látka má byť
vo forme bielej až sivobielej disperzie bez viditeľných častíc. Ak nariedená očkovacia látka obsahuje častice, alebo ak došlo
k zmene jej sfarbenia, nepoužívajte ju.
|
Z
a
z
namenajte príslušný dátum a čas.
P
oužite do 12 hodín po nariedení.
|
• Po nariedení treba injekčné liekovky označiť príslušným dátumom a časom.
• Po nariedení uchovávajte pri teplote 2 °C
až 30 °C a použite do 12hodín.
• Nariedenú disperziu nezmrazujte ani ňou netraste. Pri uchovávaní v chladničke nechajte pred použitím nariedenú disperziu dosiahnuť izbovú teplotu.
|
P
R
Í
P
RAV
A JEDNOTLIVÝCH 0,2 ml DÁVOK OČKOVACEJ LÁTKY COMIRNATY
10 MIKROGRAMOV/DÁVKA KONCENTRÁT NA INJEKČNÚ DISPERZIU (DETI VO VEKU 5 AŽ 11 ROKOV)
• Injekčná liekovka obsahuje po nariedení
2,6 ml, z ktorých je možné získať 10 dávok po 0,2 ml
• Zátku injekčnej liekovky očistite aseptickým postupom pomocou jednorazového antiseptického tampónu.
• Odoberte dávku 0,2 ml očkovacej látky
Comirnaty pre deti vo veku 5 až 11 rokov.
 0,2 ml nariedenej očkovacej látky
0,2 ml nariedenej očkovacej látky
Na získanie 10 dávok z jednej injekčnej
liekovky sa majú používať injekčné striekačky a/alebo ihly s malým mŕtvym
priestorom. Kombinácia injekčnej
striekačky s malým mŕtvym priestorom
a ihly nemá mať mŕtvy priestor väčší ako
35 mikrolitrov.
Pri použití štandardných injekčných striekačiek a ihiel nemusí byť objem dostatočný na získanie desiatich dávok z jednej injekčnej liekovky.
• Každá dávka musí obsahovať 0,2 ml
očkovacej látky.
• Ak zvyšné množstvo očkovacej látky
v injekčnej liekovke nie je dostatočné na podanie plnej dávky 0,2 ml, injekčnú liekovku a všetok zvyšný objem zlikvidujte.
• Zlikvidujte všetku očkovaciu látku, ktorú
nespotrebujete do 12 hodín po nariedení.
 L
i
kv
i
dácia
L
i
kv
i
dácia
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBioNTech Manufacturing GmbH An der Goldgrube 12
55131 Mainz
Nemecko
Telefón: +49 6131 9084-0
Fax: +49 6131 9084-2121
service@biontech.de8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/20/1528/004
EU/1/20/1528/005
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 21. decembra 2020
Dátum posledného predĺženia registrácie: 03. novembra 2021
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.