/>
-druhý výskyt
na 0. alebo I. stupeň
50 %
-tretí výskyt Liečbu ukončiť natrvalo Neaplikovateľné
• Stupeň 4
-prvý výskyt Liečbu vysadiť natrvalo
alebo
prerušiť liečbu až do úpravy toxicity na 0. alebo I. stupeň, ak si lekár myslí, že je to v najlepšom záujme pacienta
50 %
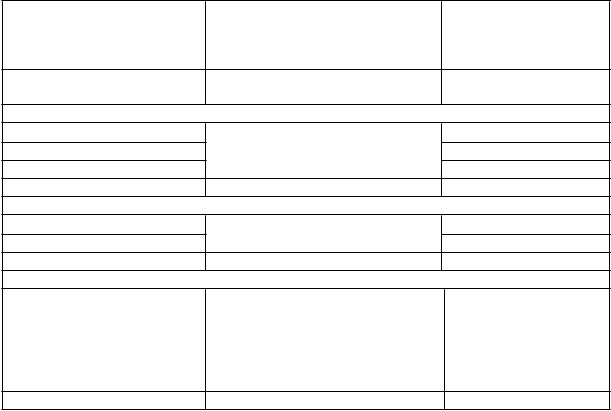
druhý výskyt Liečbu vysadiť natrvalo Neaplikovateľné
*Podľa všeobecne používaných kritérií toxicity (verzia 1) Skupiny pre klinické štúdie Národného onkologického ústavu v Kanade (NCIC CTG) alebo všeobecne používaných terminologických kritérií pre nežiaduce udalosti (CTCAE) Programu hodnotiaceho liečbu rakoviny, Národného onkologického ústavu v USA, verzia 4.0. Syndróm „ruka-noha“ a hyperbilirubinémia, pozri časť 4.4.
HematológiaPacienti s východiskovým počtom neutrofilov < 1,5 x 109/l a/alebo počtom trombocytov < 100 x 109/l pred liečbou sa nemajú liečiť kapecitabínom. Ak neplánované laboratórne vyšetrenia počas liečebného
cyklu ukazujú, že počet neutrofilov klesol pod 1,0 x 109/l alebo že počet trombocytov klesol pod
75 x 109/l, liečba kapecitabínom sa má prerušiť.
Úpravy dávkovania kvôli prejavom toxicity, keď sa Capecitabine Teva používa v 3-týždňovom liečebnom cykle v kombinácii s inými liekmiKeď sa kapecitabín používa v 3-týždňovom liečebnom cykle v kombinácii s inými liekmi, úpravy
dávkovania kvôli prejavom toxicity sa majú robiť podľa vyššie uvedenej tabuľky 3 pre kapecitabine a podľa príslušného súhrnu charakteristických vlastností.
Ak sa v čase začatia ďalšieho liečebného cyklu musí odložiť podanie buď kapecitabínu, alebo iného lieku (liekov), má sa prerušiť podávanie celej liečby až do času, kedy sa môže obnoviť liečba všetkými liekmi.
Pri prejavoch toxicity počas liečebného cyklu, ktoré podľa lekára nesúvisia s kapecitabínom, má liečba kapecitabínom pokračovať a dávka druhého lieku sa má upraviť podľa príslušného súhrnu charakteristických vlastností.
Ak sa podávanie ďalšieho/ďalších liekov musí ukončiť natrvalo, liečba kapecitabínom môže pokračovať, ak sa dodržia požiadavky pre znovu začatie liečby.
Toto odporúčanie platí pre všetky indikácie a pre všetky osobitné skupiny pacientov.
Ú
pravy dávkovania kvôli prejavom toxicity, keď sa kapecitabín používa kontinuálne v kombinácii s inými látkami
Keď sa kapecitabín používa kontinuálne v kombinácii s inými látkami, úpravy dávkovania kvôli
prejavom toxicity sa majú robiť podľa vyššie uvedenej tabuľky 3 pre kapecitabín a podľa príslušného súhrnu charakteristických vlastností iného lieku (liekov).
Úprava dávkovaniauosobitnýchskupínpacientov
Porucha funkcie pečene
Vzhľadom na nedostatočné údaje o bezpečnosti a účinnosti lieku u pacientov s poškodením funkcie pečene nie je možné odporučiť úpravu dávkovania v tejto populácii pacientov. Podobne nie sú k
dispozícii žiadne údaje u pacientov s ochorením pečene ako je cirhóza alebo hepatitída.
Porucha funkcie obličiek
Kapecitabín je kontraindikovaný u pacientov so závažnou poruchou funkcie obličiek (východisková hodnota klírensu kreatinínu < 30 ml/min [Cockroft a Gault]). Výskyt nežiaducich reakcií 3. alebo 4.
stupňa u pacientov so stredne závažnou poruchou funkcie obličiek (východisková hodnota klírensu
kreatinínu 30 – 50 ml/min) je vyšší ako v celkovej populácii. U pacientov so stredne závažnou poruchou funkcie obličiek na začiatku liečby sa odporúča znížiť úvodnú dávku na 75 % úvodnej dávky 1250 mg/m2. U pacientov so stredne závažnou poruchou funkcie obličiek na začiatku liečby sa nevyžaduje zníženie dávky, ak úvodná dávka bola 1000 mg/m2. Začiatočná dávka sa nemá meniť u pacientov s miernou poruchou funkcie obličiek (východisková hodnota klírensu kreatinínu
51 - 80 ml/min). Ak sa u pacienta počas liečby vyskytne nežiaduci účinok 2., 3. alebo 4. stupňa, je potrebné ho starostlivo sledovať, odporúča sa urýchlene prerušiť liečbu a ďalšiu dávku upraviť podľa tabuľky 3 uvedenej vyššie. Ak počas liečby vypočítaná hodnota klírensu kreatinínu klesne pod hodnotu 30 ml/min, liečba Capecitabine Teva sa má ukončiť. Odporúčania k úprave dávkovania pri poruche funkcie obličiek platia pre monoterapiu aj pre kombinovanú liečbu kapecitabín/docetaxel (pozri tiež nižšie uvedenú časť „Starší pacienti“).
Starší pacienti:
Pri monoterapii kapecitabínom nie je potrebná úprava úvodnej dávky. Na druhej strane výskyt nežiaducich reakcií 3. alebo 4. stupňa bol častejší v skupine pacientov ≥ 60 rokov v porovnaní s mladšími pacientmi.
Keď sa kapecitabín používal v kombinácii s inými liekmi, u starších pacientov (≥ 65 rokov) sa v porovnaní s mladšími pacientmi vyskytlo viac nežiaducich reakcií na liek 3. stupňa a 4. stupňa,
vrátane nežiaducich reakcií na liek, ktoré viedli k ukončeniu liečby. U pacientov ≥ 60 rokov sa odporúča starostlivé sledovanie.
- V kombinácii s docetaxelom: U pacientov vo veku ≥ 60 rokov sa pozoroval vyšší výskyt nežiaducich reakcií a závažných nežiaducich reakcií 3. alebo 4. stupňa spojených s liečbou (pozri časť 5.1). U pacientov vo veku 60 rokov alebo starších sa odporúča znížiť úvodnú dávku kapecitabínu na 75 % (950 mg/m2 dvakrát denne). Ak sa u pacientov vo veku ≥ 60 rokov
liečených zníženou dávkou kapecitabínu v kombinácii s docetaxelom neprejaví toxicita, môže sa dávka kapecitabínu opatrne zvýšiť na 1 250 mg/m2 dvakrát denne.
Pediatrická populácia
Použitie kapecitabínu sanetýkapediatrickejpopulácie pre indikácie karcinóm hrubého čreva, konečníka, žalúdka a prsníka.
Spôsobpodávania
Capecitabine Teva filmom obalené tablety sa prehĺtajú a zapíjajú vodou do 30 minút po jedle.
4.3 Kontraindikácie
• Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1 alebo fluorouracil.
• Anamnéza ťažkých a neočakávaných reakcií na liečbu fluórpyrimidínmi.
• • Pacienti so známou úplnou absenciou aktivity dihydropyrimidíndehydrogenázy (DPD) (pozri časť 4.4).
• Počas gravidity a dojčenia.
• Pacienti s ťažkou leukopéniou, neutropéniou alebo trombocytopéniou.
• Pacienti so závažným poškodením pečene.
• Pacienti so závažným poškodením funkcie obličiek (klírens kreatinínu pod 30 ml/min).
• Liečba sorivudínom alebo jeho chemicky príbuznými analógmi, napr. brivudínom (pozri časť 4.5).
• Ak je kontraindikácia na niektorý z liekov používaných v kombinovanom režime, tento liek sa nemá používať.
4.4 Osobitné upozornenia a opatrenia pri používaní
Toxicképrejavy,ktoréobmedzujúdávkovanie
Medzi toxické prejavy, ktoré limitujú dávkovanie lieku, patrí hnačka, bolesť brucha, nevoľnosť, stomatitída a syndróm „ruka-noha“ (kožná reakcia na ruke a nohe, palmárno-plantárna
erytrodyzestézia). Väčšina nežiaducich reakcií má reverzibilný charakter a nevyžaduje si trvalé
ukončenie liečby, hoci môže byť potrebné vysadiť niektoré dávky alebo znížiť dávkovanie.
Hnačka
Pacienti so závažnou hnačkou sa majú starostlivo sledovať a v prípade rozvoja dehydratácie sa im majú doplniť tekutiny a ióny. Okrem toho sa môžu použiť bežné lieky proti hnačke (napr. loperamid). Hnačka 2. stupňa podľa NCIC CTC sa definuje ako 4 - 6 stolíc denne alebo v noci, hnačka 3. stupňa ako 7 – 9 stolíc denne alebo inkontinencia a malabsorpcia a hnačka 4. stupňa ako 10 stolíc denne a viac alebo hnačka s prímesou krvi, príp. nutnosť parenterálnej podpornej výživy. V prípade potreby sa má uplatniť zníženie dávky (pozri časť 4.2).
Dehydratácia
Pred liečbou sa má predísť dehydratácii alebo ju korigovať. Pacienti s anorexiou, asténiou, nauzeou, vracaním alebo hnačkou sa môžu rýchlo
dehydratovať. Dehydratácia môže spôsobiť akútne zlyhanie obličiek, najmä u
pacientov s už existujúcou zníženou funkciou obličiek alebo ak sa kapecitabín podáva súbežne so známymi nefrotoxickými liekmi. Akútne zlyhanie obličiek sekundárne
po dehydratácii môže byť potenciálne smrteľné. Ak dôjde k 2. (alebo vyššiemu) stupňu dehydratácie,
musí sa ihneď prerušiť liečba Capecitabine Teva a upraviť dehydratácia. V liečbe sa nesmie pokračovať, pokiaľ nie je pacient rehydratovaný a pokiaľ sa príčiny dehydratácie neodstránia alebo nie
sú pod kontrolou. V prípade potreby sa má uplatniť úprava dávky kvôli príčine dehydratácie (pozri
časť 4.2).
Syndróm„ruka-noha“
Syndróm „ruka-noha“ (známy tiež ako kožná reakcia na ruke a nohe alebo palmárno-plantárna erytrodyzestézia alebo akrálny erytém navodený chemoterapiou). 1. stupeň syndrómu „ruka-noha“ sa definuje ako necitlivosť, dyzestézia/parestézia, mravenčenie, nebolestivý opuch alebo erytém
na rukách a/alebo nohách, prípadne nepríjemný pocit, ktorý nenarúša každodenné aktivity pacienta.
2. stupeň syndrómu „ruka-noha“ je bolestivý erytém a opuch rúk a/alebo nôh, prípadne nepríjemný pocit, ktorý ovplyvňuje každodenné aktivity pacienta.
3. stupeň syndrómu „ruka-noha“ je vlhké olupovanie, tvorba vriedkov a pľuzgierov a veľká bolesť rúk
a/alebo nôh a/alebo veľmi nepríjemný pocit, ktorý znemožňuje pacientovi pracovať alebo vykonávať každodenné aktivity. Pretrvávajúci alebo závažný syndróm „ruka-noha“ (2. stupeň a vyšší) môže eventuálne viesť k vymiznutiu odtlačkov prstov, čo môže znemožniť identifikáciu pacienta. Ak dôjde
k rozvoju 2. alebo 3. stupňa syndrómu „ruka-noha“, podávanie kapecitabínu sa musí okamžite prerušiť až do ústupu tohto nežiaduceho účinku alebo zníženia jeho závažnosti na 1. stupeň. Po 3. stupni
syndrómu „ruka-noha“ sa majú znížiť nasledujúce dávky kapecitabínu. Keď sa kapecitabín a cisplatina používajú v kombinácii, použitie vitamínu B6 (pyridoxín) na symptomatickú alebo sekundárnu
profylaktickú liečbu syndrómu „ruka-noha“ sa neodporúča, pretože boli publikované správy o tom, že môže znížiť účinnosť cisplatiny. K dispozícii sú údaje, ktoré naznačujú, že dexpantenol je účinný v profylaxii syndrómu „ruka-noha“ u pacientov liečených Capecitabinom Teva.
K
a
r
diotoxicita
Liečbu fluórpyrimidínmi sprevádzali kardiotoxické prejavy vrátane infarktu myokardu, anginy pectoris, arytmií, kardiogénneho šoku, náhlej smrti a zmien na EKG (vrátane veľmi zriedkavých
prípadov predĺženia QT intervalu). Tieto nežiaduce účinky sa môžu častejšie vyskytnúť u pacientov s
anamnézou ischemickej choroby srdca. U pacientov, ktorí užívajú kapecitabín, sa hlásili srdcové arytmie (vrátane fibrilácie komôr, „torsade de pointes“ a bradykardie), angina pectoris, infarkt myokardu, srdcové zlyhanie a kardiomyopatia. U pacientov s anamnézou závažného ochorenia srdca, arytmií a anginy pectoris sa vyžaduje opatrnosť (pozri časť 4.8).
Hypo-alebohyperkalciémia
Počas liečby Capecitabine Teva sa vyskytli prípady hypokalciémie alebo hyperkalciémie. U pacientov s existujúcou hypokalciémiou alebo hyperkalciémiou sa vyžaduje opatrnosť (pozri časť 4.8).
Ochoreniacentrálnehoaleboperiférnehonervovéhosystému
Zvýšená opatrnosť je potrebná u pacientov s ochoreniami centrálneho alebo periférneho nervového systému, napr. metastázami v mozgu alebo neuropatiou (pozri časť 4.8).
Diabetesmellitusaleboporuchyelektrolytov
U diabetikov alebo u pacientov s poruchami iónov sa vyžaduje zvýšená opatrnosť pre možné zhoršenie týchto stavov počas liečby kapecitabínom.
Antikoaguláciakumarínovýmiderivátmi
V interakčnej štúdii s jednorazovými dávkami warfarínu došlo k signifikantnému zvýšeniu (+ 57 %) priemerných hodnôt AUC S-warfarínu. Tieto výsledky naznačujú interakciu, pravdepodobne vďaka inhibícii izoenzymového systému cytochrómu P450 2C9 kapecitabínom. Pacienti liečení súčasne kapecitabínom a perorálnymi antikoagulanciami kumarínového typu sa musia pozorne sledovať (INR alebo protrombínový čas) a dávka antikoagulancia sa má podľa výsledkov adekvátne upraviť (pozri časť 4.5 ).
Poruchafunkciepečene
Vzhľadom na chýbajúce údaje o bezpečnosti a účinnosti kapecitabínu u pacientov s poškodením funkcie pečene sa majú pacienti s miernou až stredne závažnou dysfunkciou pečene starostlivo
sledovať, bez ohľadu na prítomnosť alebo chýbanie metastáz v pečeni. Podávanie kapecitabínu sa má
prerušiť, ak sa v dôsledku liečby zvýši hladina bilirubínu nad 3-násobok hornej hranice normy (ULN)
alebo ak sa hodnoty transamináz (AST, ALT) zvýšia > 2,5 x ULN. Monoterapia kapecitabínom môže pokračovať, keď hodnoty bilirubínu klesnú na ≤ 3 x ULN alebo pečeňové aminotransferázy klesnú na
≤ 2,5 x ULN.
Poruchafunkcieobličiek
Výskyt nežiaducich reakcií 3. alebo 4 stupňa u pacientov so stredne závažnou poruchou funkcie obličiek (klírens kreatinínu 30 – 50 ml/min) je vyšší ako v celkovej populácii (pozri časti 4.2 a 4.3).
Deficienciadihydropyrimidíndehydrogenázy(DPD)
Zriedkavo bola neočakávaná, závažná toxicita (napr. stomatitída, hnačka, zápal slizníc, neutropénia a neurotoxicita) spojená s 5-FU pripisovaná deficiencii aktivity DPD.
Pacienti s nízkou alebo absentujúcou aktivitou DPD, enzýmom, ktorý ovplyvňuje odbúravanie
fluóruracilu, sú vystavení zvýšenému riziku závažných, život ohrozujúcich alebo fatálnych nežiaducich reakcií spôsobených fluóruracilom. Hoci deficiencia DPD nemusí byť presne stanovená, je známe, že pacienti s istými homozygotnými alebo s istými zlúčeninami heterozygotných mutácií v géne DPYD, ktoré môžu spôsobiť úplnú alebo takmer úplnú absenciu enzymatickej aktivity DPD (stanovené laboratórnymi vyšetreniami), sú vystavení najvyššiemu riziku život ohrozujúcej alebo fatálnej toxicity a nemajú byť liečení Capecitabinom Teva (pozri časť 4.3). U pacientov s úplnou absenciou aktivity DPD sa nepreukázala žiadna dávka ako bezpečná.
Pacienti s čiastočnou deficienciou DPD (napríklad pacienti s heterozygotnými mutáciami v géne
DPYD) a tí, u ktorých sa zváži, že prínos Capecitabinu Teva prevažuje nad rizikom (musí sa zvážiť aj
vhodnosť alternatívnej chemoterapeutickej liečby bez fluórpyrimidínu), musia byť liečení
s maximálnou opatrnosťou a častým následným sledovaním s úpravou dávky podľa stupňa toxicity. Pre odporúčanie špecifickej dávky u pacientov s čiastočnou aktivitou DPD nie sú dostupné dostatočné údaje, ako sa zistilo špecifickým testovaním.
U pacientov s neznámou deficienciou DPD liečených kapecitabínom, sa môže objaviť život ohrozujúca toxicita, prejavujúca sa ako akútne predávkovanie (pozri časť 4.9). V prípade 2.-4. stupňa akútnej toxicity sa liečba musí okamžite prerušiť. Trvalé ukončenie liečby sa má zvážiť na základe klinického zhodnotenia nástupu, trvania a závažnosti pozorovanej toxicity.
Oftalmologickékomplikácie
Pacienti sa majú starostlivo sledovať pre oftalmologické komplikácie, ako je keratitída a poruchy rohovky, najmä ak majú v anamnéze ochorenie očí. Liečba porúch očí sa má začať podľa klinickej
potreby.
Závažnékožnéreakcie
Capecitabine Teva môže vyvolať závažné kožné reakcie, napr. Stevensov-Johnsonov syndróm a toxickú epidermálnu nekrolýzu. Liečba Capecitabinom Teva sa musí natrvalo ukončiť u pacientov, u
ktorých počas liečby vznikne závažná kožná reakcia.
Pomocnélátky
Pretože tento liek obsahuje laktózu ako pomocnú látku, pacienti so zriedkavými dedičnými poruchami intolerancie galaktózy, s laponskou laktázovou deficienciou alebo poruchou absorpcie glukózy a
galaktózy nemajú užívať tento liek.
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých
Interakcie s inými liekmi:
CytochrómP-4502C9substráty
Okrem warfarínu, žiadne formálne interakčné štúdie medzi kapecitabínom a inými CYP2C9 substrátmi neboli vykonané. Starostlivo sa má postupovať, ak sa kapecitabín podáva s 2C9 substrátmi (napr. fenytoín). Pozri tiež interakcie s kumarínovými antikoagulanciami nižšie a časť 4.4.
Kumarínovéantikoagulanciá
U pacientov, ktorí užívajú kapecitabín súčasne s antikoagulanciami kumarínového typu, napr. warfarínom a fenprokumonom, sa opísali odchýlky koagulačných parametrov a/alebo krvácanie. K týmto nežiaducim reakciám došlo v období niekoľkých dní až mesiacov od začiatku liečby kapecitabínom, v niektorých prípadoch aj v priebehu jedného mesiaca od ukončenia liečby kapecitabínom. V klinickej farmakokinetickej interakčnej štúdii zvýšila liečba kapecitabínom AUC S- warfarínu o 57 % po jednotlivej dávke 20 mg warfarínu a hodnotu INR o 91 %. Keďže metabolizmus R-warfarínu nebol ovplyvnený, výsledky naznačujú, že kapecitabín negatívne ovplyvňuje izoenzým
2C9, ale nemá vplyv na izoenzýmy 1A2 a 3A4. Pacienti, ktorí užívajú kapecitabín súčasne s antikoagulanciami kumarínového typu sa musia pravidelne vyšetrovať z hľadiska odchýlok
koagulačných parametrov (PT alebo INR) a podľa toho upraviť dávky antikoagulancia.
Fenytoín
Počas súbežnej liečby kapecitabínom a fenytoínom sa v jednotlivých prípadoch opísali zvýšené plazmatické koncentrácie fenytoínu aj s následnými príznakmi intoxikácie fenytoínom. Pacienti
užívajúci kapecitabín v kombinácii s fenytoínom sa musia pravidelne sledovať z hľadiska zvýšených
hladín fenytoínu v plazme.
Kyselinalistová/kyselinafolinová
V štúdii kombinovanej liečby kapecitabínom a kyselinou listovou sa zistilo, že kyselina listová významnejšie neovplyvňuje farmakokinetiku kapecitabínu a jeho metabolitov. Na druhej strane má
kyselina listová vplyv na farmakodynamiku kapecitabínu a kyselina listová môže zvýšiť toxicitu kapecitabínu: maximálna tolerovaná dávka (MTD) kapecitabín podávaný samostatne v rámci intermitentnej liečby je 3 000 mg/m2 denne, kým v kombinácii s kyselinou listovou (2 x 30 mg p.o. denne) je táto hodnota iba 2 000 mg/m2 denne. Zvýšená toxicita môže súvisieť s prechodom z režimu
5-FU/LV na režim s kapecitabínom. Môže to tiež súvisieť so suplementáciou kyseliny listovej pri deficite folátov v dôsledku podobnosti medzi kyselinou folinovou a kyselinou listovou.
Sorivudínaanalógy
Pozorovala sa klinicky významná lieková interakcia medzi sorivudínom a 5-FU vyplývajúca z inhibície dihydropyrimidíndehydrogenázy navodenej sorivudínom. Táto interakcia vedie k vyššej toxicite fluórpyrimidínov a môže byť fatálna. Z toho dôvodu sa kapecitabín nesmie podávať súbežne
so sorivudínom alebo s jeho chemicky príbuznými analógmi, napr. brivudínom (pozri časť 4.3). Medzi ukončením liečby sorivudínom alebo jeho chemicky príbuznými analógmi, napr. brivudínom, a
začiatkom liečby kapecitabínom musia uplynúť minimálne 4 týždne.
Antacidá
Skúmal sa vplyv antacíd, ktoré obsahujú hydroxid hlinitý a hydroxid horečnatý na farmakokinetiku kapecitabínu. Plazmatické koncentrácie kapecitabínu a jedného metabolitu (5'-DFCR) sa mierne
zvýšili, avšak nezistil sa žiadny vplyv na 3 hlavné metabolity kapecitabínu (5'-DFUR, 5-FU a FBAL).
Alopurinol
Pozorovali sa interakcie 5-FU s alopurinolom, ktoré viedli k možnej nižšej účinnosti 5-FU. Alopurinol sa nemá podávať súčasne s kapecitabínom.
Interferón alfa
Hodnota MTD kapecitabínu podávaného v kombinácii s interferónom alfa-2a (3 MIU/m2 denne) bola
2 000 mg/m2, kým hodnota MTD kapecitabínu v monoterapii bola 3 000 mg/m2 denne.
Rádioterapia
MTD kapecitabín v monoterapii pri prerušovanom režime je 3 000 mg/m2 denne, zatiaľ čo v kombinácii s rádioterapiou rektálneho karcinómu je MTD kapecitabín 2 000 mg/m2 denne pri
kontinuálnom režime alebo pri dennom podávaní od pondelka do piatku počas 6-týždňovej liečby
rádioterapiou.
Oxaliplatina
Keď sa kapecitabín podával v kombinácii s oxaliplatinou alebo v kombinácii s oxaliplatinou a bevacizumabom, nedošlo k žiadnym klinicky významným rozdielom v expozícii kapecitabínu alebo jeho metabolitov, voľnej (neviazanej) platine alebo celkovej platine.
Bevacizumab
Za prítomnosti oxaliplatiny nemal bevacizumab klinicky významný vplyv na farmakokinetické parametre kapecitabínu alebo jeho metabolitov.
Interakciesjedlom
Vo všetkých klinických štúdiách dostávali pacienti kapecitabín do 30 minút po jedle. Keďže súčasné údaje o bezpečnosti a účinnosti sú založené na podávaní kapecitabínu s jedlom, odporúča sa užívať
tento liek spolu s jedlom. Podávanie lieku s jedlom znižuje rýchlosť vstrebávania kapecitabínu (pozri
časť 5.2).
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku/Antikoncepciaumužovažien
Ženy vo fertilnom veku by mali byť poučené, aby sa zabránilo otehotneniu počas
liečby kapecitabínom. Ak pacientka otehotnie počas liečby kapecitabínom, musí jej byť potenciálne nebezpečenstvo pre plod vysvetlené. Počas liečby by mala byť použitá efektívna metóda
antikoncepcie.
G
r
avidita
U gravidných žien sa nevykonali žiadne klinické štúdie s kapecitabine; je však možné, že ak sa kapecitabín podá gravidným ženám, môže zapríčiniť poškodenie plodu. V štúdiách reprodukčnej toxicity na zvieratách sa ukázalo, že kapecitabine viedol k embryotoxicite a teratogenicite. Tieto poznatky patria medzi očakávané účinky derivátov fluórpyrimidínu. Kapecitabín je kontraindikovaný počas gravidity.
Dojčenie
Nie je známe, či sa kapecitabín vylučuje do materského mlieka u ľudí. U dojčiacich myší sa v mlieku zistili vysoké hladiny kapecitabínu a jeho metabolitov. Počas liečby kapecitabine sa musí prerušiť dojčenie.
Fertilita
Nie sú k dispozícii žiadne údaje o kapecitabínu a jeho vplyvu na plodnosť. V pivotnej štúdii s kapecitabínom
boli zahrnuté ženy vo fertilnom veku a muži len vtedy, ak súhlasili s používaním prijateľnej metódy an tikoncepcie na zabránenie otehotneniu počas doby trvania štúdie a po primeranej dobe potom.
V štúdiách na zvieratách účinky na plodnosť boli pozorované (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Kapecitabín má malý alebo mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Kapecitabín môže vyvolať závrat, únavu a nevoľnosť.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Celkový bezpečnostný profil kapecitabínu vychádza z údajov od viac ako 3 000 pacientov liečených
kapecitabínom v monoterapii alebo kapecitabínom v kombinácii s rôznymi chemoterapeutickými režimami pri viacerých indikáciách. Kapecitabín má porovnateľný bezpečnostný profil, keď sa
používa v monoterapii metastatického karcinómu prsníka, metastatického kolorektálneho karcinómu a
v adjuvantnej liečbe pacientov s karcinómom hrubého čreva. Podrobnosti o najdôležitejších štúdiách, vrátane dizajnu štúdií a hlavných výsledkov účinnosti - pozri časť 5.1.
K najčastejšie hláseným a/alebo klinicky významným nežiaducim reakciám na liek (adverse drug reactions, ADR) v súvislosti s liečbou patrili gastrointestinálne poruchy (najmä hnačka, nevoľnosť, vracanie, abdominálna bolesť, stomatitída), syndróm ruka-noha (palmárno-plantárna erytrodyzestézia), únava, asténia, anorexia, kardiotoxicita, zvýšená dysfunkcia obličiek u pacientov s už existujúcou poškodenou funkciou obličiek a trombóza/embólia.
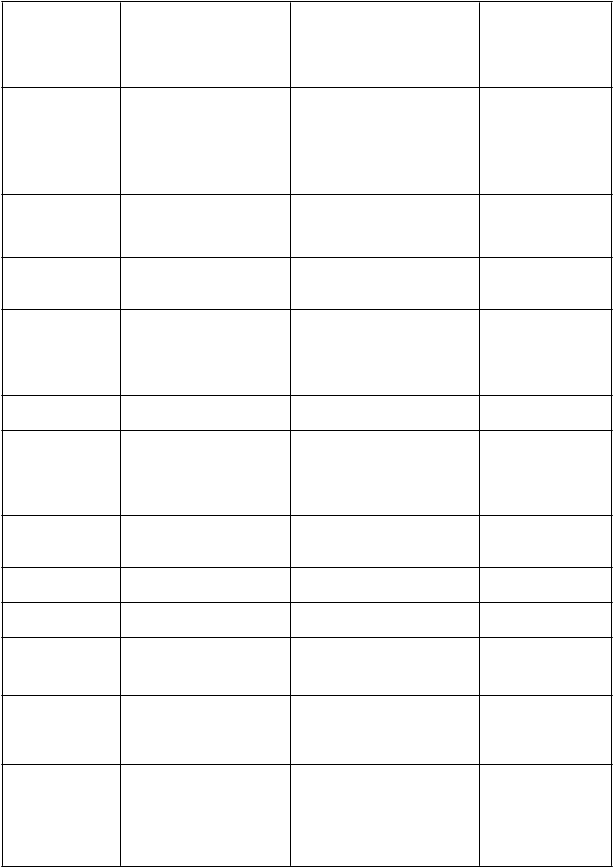
Tabuľkový zoznamnežiaducichreakcií
ADR, ktoré skúšajúci lekári dávali do možnej, pravdepodobnej alebo nepriamej súvislosti s
podávaním kapecitabínu, sú uvedené v tabuľke 4 pre kapecitabín podávaný v monoterapii a
v tabuľke 5 pre kapecitabín podávaný v kombinácii s rôznymi chemoterapeutickými režimami pri viacerých indikáciách. Na označenie výskytu ADR sa používajú nasledovné kategórie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až
< 1/1 000), veľmi zriedkavé (< 1/10 000). Výskyt ADR je v každej skupine usporiadaný v poradí klesajúcej závažnosti.
Monoterapiakapecitabínom
Tabuľka 4 uvádza ADR súvisiace s použitím kapecitabínu v monoterapii na základe súhrnnej analýzy údajov o bezpečnosti z troch najdôležitejších štúdií zahŕňajúcich viac ako 1 900 pacientov (štúdie M66001, SO14695 a SO14796). ADR sa doplnili do príslušnej skupiny výskytu podľa celkovej incidencie z tejto súhrnnej analýzy.
Tabuľka 4 Súhrn ADR súvisiacich s liečbou, ktoré sa hlásili u pacientov liečených kapecitabínom v monoterapii
T
elový systém Veľmi časté
V
šetky stupne
Č
asté
V
šetky stupne
Menej časté
Z
ávažné a/alebo život ohrozujúce
(
3.-4. stupňa) alebo považované za
dôležité z medicínskeho hľadiska
Z
riedkavé/veľmi
z
riedkavé
(
skúsenosti po uvedení lieku na trh)
I
nfekcie a nákazy
- Infekcia herpesovým vírusom, nazofaryngitída, infekcia dolných dýchacích ciest
Sepsa, infekcia močových ciest, celulitída, tonzilitída, faryngitída, ústna kandidóza, chrípka, gastroenteritída, plesňová infekcia, infekcia, absces zubov
B
enígne a malígne nádory, vrátane nešpecifikovanýc h novotvarov
- - Lipóm
P
oruchy krvi a lymfatického systému
- Neutropénia, anémia
Febrilná neutropénia, pancytopénia, granulocytopénia, trombocytopénia, leukopénia, hemolytická anémia, zvýšený International Normalised Ratio (INR)/predĺžený protrombínový čas
P
oruchy imunitného systému
- - Precitlivenosť
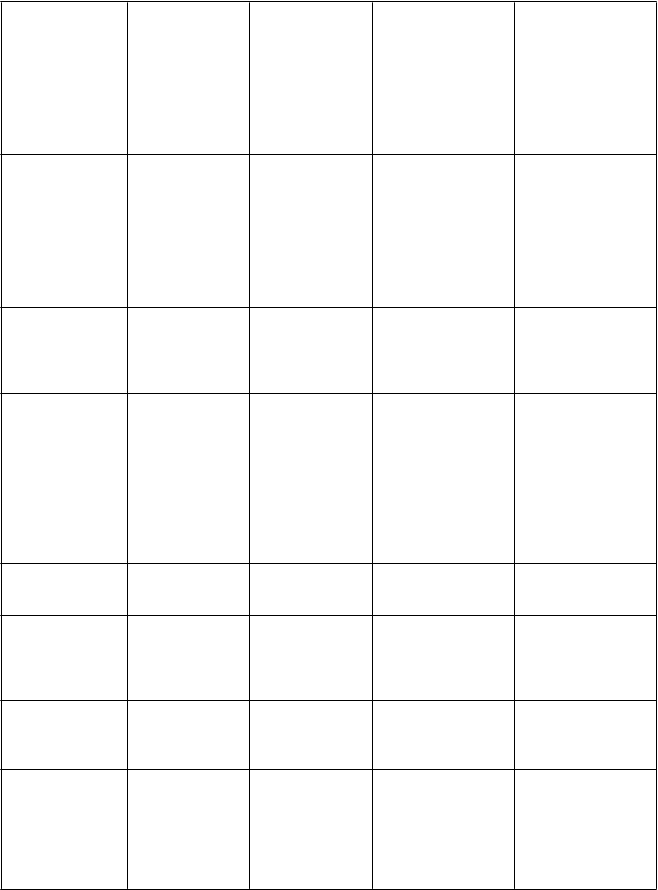 P
oruchy metabolizmu a výživy
P
sychické poruchy
P
oruchy nervového systému
P
oruchy metabolizmu a výživy
P
sychické poruchy
P
oruchy nervového systému
Anorexia Dehydratácia, úbytok telesnej hmotnosti
- Nespavosť, depresia
- Bolesť hlavy, letargia, závrat, parastézia, dysgeúzia
Diabetes mellitus, hypokaliémia, porucha chuti do jedla, malnutrícia, hypertriglyceridémia Stavy zmätenosti, záchvaty paniky, depresívna nálada, znížené libido Afázia, poruchy pamäti, ataxia, synkopa, porucha rovnováhy,
ochorenie
zmyslových orgánov, periférna neuropatia
Toxická leukoencefalopatia (veľmi zriedkavé)
T
elový systém Veľmi časté
V
šetky stupne
Č
asté
V
šetky stupne
Menej časté
Z
ávažné a/alebo život ohrozujúce
(
3.-4. stupňa) alebo
považované za dôležité z medicínskeho hľadiska
Z
riedkavé/veľmi zriedkavé (skúsenosti po uvedení lieku na trh)
P
oruchy oka - Zvýšené slzenie, konjunktivitída, podráždenie očí
Znížená zraková ostrosť, diplopia
Stenóza slzného kanálika (zriedkavé), chorobné zmeny rohovky (zriedkavé), keratitída
(zriedkavé), bodkovaná keratitída (zriedkavé)
P
oruchy ucha a labyrintu
P
oruchy srdca a srdcovej činnosti
- - Vertigo, bolesť uší
- - Nestabilná angína, angína pectoris, ischémia myokardu, fibrilácia predsiení, arytmia, tachykardia, sínusová tachykardia, palpitácie
Fibrilácia komôr (zriedkavé), predĺženie QT intervalu (zriedkavé),
„Torsade de pointes“ (zriedkavé), bradykardia (zriedkavé), vazospazmus (zriedkavé)
P
oruchy ciev - Tromboflebitída Trombóza hĺbkových žíl, hypertenzia, petechia, hypotenzia, návaly horúčavy, chlad v periférnych častiach tela
P
oruchy dýchacej sústavy, hrudníka a mediastína
- Dyspnoe, epistaxa, kašeľ, nádcha
Pľúcna embólia, pneumotorax, hemoptýza, astma, dyspnoe pri námahe
P
oruchy gastrointestináln eho traktu
Hnačka, vracanie, nevoľnosť, stomatitída, abdominálna bolesť
Gastrointestinálne krvácanie,
zápcha, bolesť v
hornej časti brucha, dyspepsia,
flatulencia, sucho v ústach
Črevná obštrukcia, ascites, enteritída, gastritída, dysfágia, bolesť v spodnej časti brucha,
ezofagitída, zažívacie ťažkosti, gastroezofageálny reflux, kolitída, krv v stolici
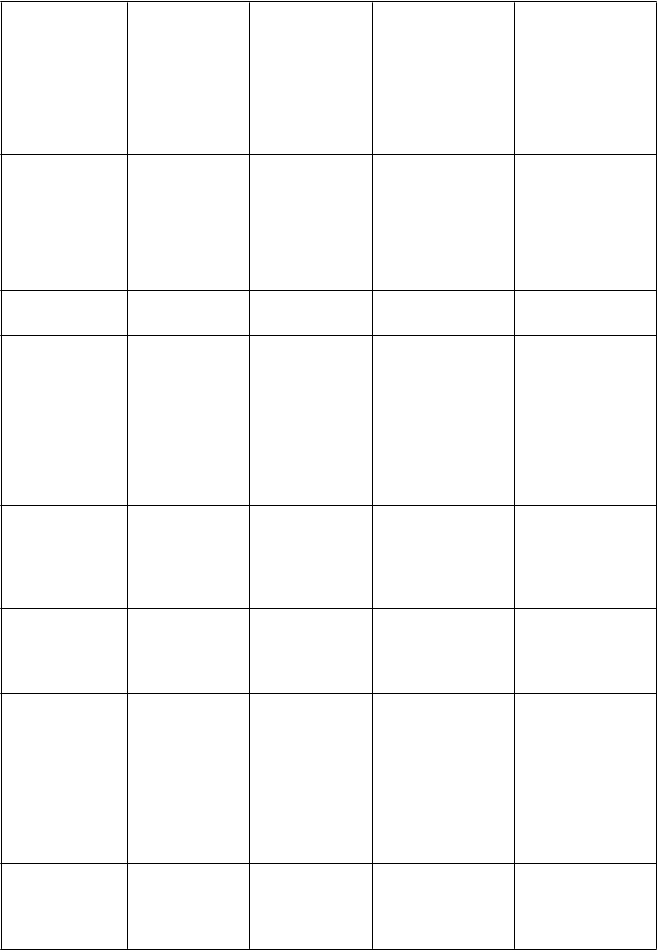 P
oruchy pečene a žlčových ciest
P
oruchy pečene a žlčových ciest
- Hyperbilirubiném ia, abnormality funkčných pečeňových
testov
Žltačka Zlyhanie pečene (zriedkavé), cholestatická hepatitída (zriedkavé)
T
elový systém Veľmi časté
V
šetky stupne
Č
asté
V
šetky stupne
Menej časté
Z
ávažné a/alebo život ohrozujúce
(
3.-4. stupňa) alebo
považované za dôležité z medicínskeho hľadiska
Z
riedkavé/veľmi zriedkavé (skúsenosti po uvedení lieku na trh)
P
oruchy kože a podkožného tkaniva
Syndróm palmárno- plantárnej erytrodyzestézie
**
Vyrážka, alopécia, erytém, suchá koža, svrbenie, hyperpigmentácia kože, makulárna vyrážka, odlupovanie kože, dermatitída, poruchy pigmentácie, poruchy nechtov
Pľuzgiere, kožný vred, vyrážka, urtikária, fotosenzitívna reakcia, palmárny erytém, opuch tváre, purpura, radiačný recall syndróm
Kožný lupus erythematosus (zriedkavé), závažné kožné reakcie ako Stevens-Johnsonov syndróm a toxická epidermálna nekrolýza (veľmi zriedkavé) (pozri časť 4.4.)
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
- Bolesť končatín, bolesť chrbta, artralgia
Opuch kĺbov, bolesť kostí, bolesť tváre, stuhnutosť kostrového svalstva, svalová slabosť
P
oruchy obličiek a močových ciest
P
oruchy reprodukčného systému a prsníkov
- - Hydronefróza, inkontinencia moču, hematúria, noktúria, zvýšená hladina kreatinínu v krvi
- - Vaginálne krvácanie
C
elkové poruchy a reakcie v mieste podania
Únava, asténia Horúčka, letargia, periférny edém, malátnosť, bolesť na hrudi
Edém, triaška, ochorenie podobné chrípke, stuhnutosť, zvýšená telesná teplota
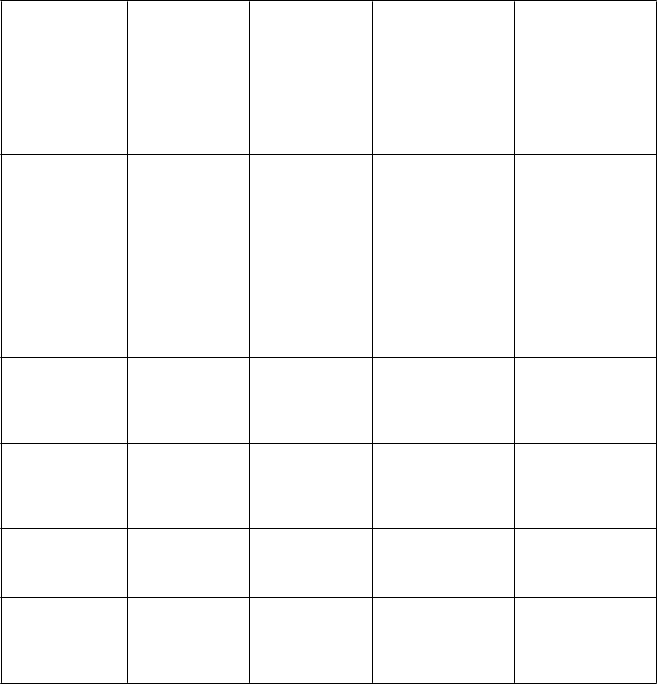
** Vychádzajúc zo skúseností po uvedení lieku na trh, môže pretrvávajúci alebo závažný syndróm palmárno- plantárnej erytrodyzestézie eventuálne viesť k vymiznutiu odtlačkov prstov (pozri časť 4.4).
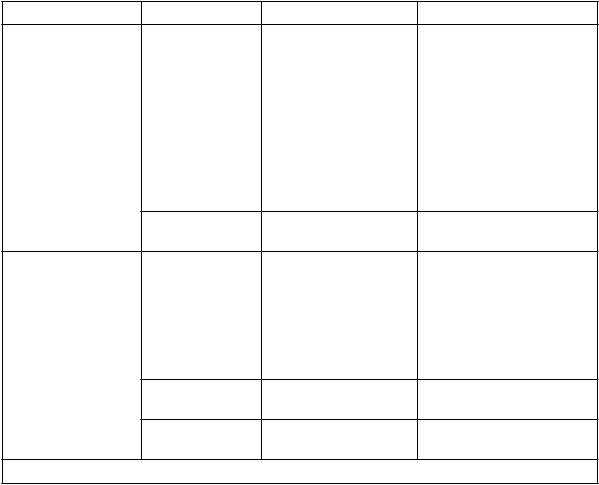
KapecitabínvkombinovanejterapiiTabuľka 5 uvádza ADR súvisiace s použitím kapecitabínu v kombinácii s rôznymi chemoterapeutickými režimami pri viacerých indikáciách na základe údajov bezpečnosti od viac ako
3 000 pacientov. ADR sa doplnili do príslušnej skupiny výskytu (veľmi časté alebo časté) podľa
najvyššej incidencie pozorovanej v ktorejkoľvek z najdôležitejších klinických štúdií a doplnili sa iba vtedy, keď boli pozorované
naviac k nežiaducim reakciám na liek pozorovaným pri kapecitabíne v monoterapii, alebo keď boli pozorované
s vyšším výskytom v porovnaní s kapecitabínom v monoterapii (pozri tabuľku 4). Menej časté ADR hlásené pri kapecitabíne v kombinovanej liečbe sa zhodujú s ARD hlásenými pri kapecitabíne v monoterapii alebo hlásenými pri kombinovanom lieku v monoterapii (v literatúre a/alebo príslušnom súhrne charakteristických vlastností lieku).
Niektoré z ADR sú reakcie často pozorované pri podávaní liekov, ktoré sa kombinujú s kapecitabínom (napr. periférna senzorická neuropatia pri docetaxele alebo oxaliplatine, hypertenzia pozorovaná pri bevacizumabe); ich zhoršenie spôsobené liečbou kapecitabínom však nie je možné vylúčiť.
Tabuľka 5 Súhrn ADR súvisiacich s liečbou, ktoré boli hlásené u pacientov liečených kapecitabín v kombinovanej liečbe naviac k nežiaducim reakciám na liek pozorovaným pri kapecitabíne v monoterapii, alebo ktoré boli pozorované s vyšším výskytom v porovnaní s kapecitabínom v monoterapii
T
elový systém Veľmi časté
V
šetky stupne
Č
asté
V
šetky stupne
Z
riedkavé/ veľmi
z
riedkavé (skúsenosti po uvedení lieku na trh)
I
nfekcie a nákazy - Herpes zoster, infekcia močových ciest, ústna kandidóza, infekcia horných dýchacích ciest, rinitída,
+
chrípka,
herpes
infekcia, orálny
P
oruchy krvi a
+ +
Neutropénia, leukopénia
+
Útlm kostnej drene, febrilná
l
ymfatického
systému
+
, anémia,
+
neutropenická
neutropénia
P
oruchy imunitného systému Poruchy metabolizmu a výživy
P
sychické poruchy
horúčka, trombocytopénia
- Precitlivenosť
Znížená chuť do jedla Hypokaliémia, hyponatriémia, hypomagneziémia, hypokalciémia, hyperglykémia
- Porucha spánku, úzkosť
Poruchy nervového systému
Parestézia, dyzestézia, periférna neuropatia, periférna senzorická neuropatia, dysgeúzia, bolesť hlavy
Neurotoxicita, tremor, neuralgia, reakcia z precitlivenosti, hypoestézia
P
oruchy oka Zvýšené slzenie Poruchy videnia, suché oko, bolesť oka, zhoršenie videnia, rozmazané videnie
P
oruchy ucha a labyrintu Poruchy srdca a srdcovej činnosti
- Tinitus, hypoakúzia
- Fibrilácia predsiení, srdcová ischémia/srdcový infarkt
P
oruchy ciev Opuch dolných končatín,
+
Sčervenanie, hypotenzia,
hypertenzia, trombóza
embólia a
hypotenzná kríza, nával
horúčavy, flebitída
P
oruchy
dýchacej sústavy, hrudníka a mediastína
Bolesť v hrdle, faryngeálna dyzestézia
Čkanie, faryngolaryngeálna bolesť, dysfónia
 P
oruchy gastrointestináln eho traktu
P
oruchy gastrointestináln eho traktu
Zápcha, dyspepsia Krvácanie z hornej časti gastrointestinálneho traktu, vriedky v ústnej dutine, gastritída, abdominálna distenzia, gastroezofageálna refluxná choroba, bolesť v
T
elový systém Veľmi časté
V
šetky stupne
Č
asté
V
šetky stupne
ústnej dutine, dysfágia, krvácanie z konečníka, bolesť v dolnej časti brucha, orálna dyzestézia, orálna parestézia, orálna hypoestézia, abdominálny diskomfort
Zriedkavé/ veľmi zriedkavé (skúsenosti po uvedení lieku na trh)
P
oruchy pečene a žlčových ciest
- Abnormálna funkcia pečene
P
oruchy kože a podkožného tkaniva Poruchy kostrovej a
svalovej sústavy
a spojivového tkaniva
Alopécia, poruchy nechtov
Myalgia, artralgia, bolesť končatín
Hyperhidróza, erytematózna vyrážka, urtikária, nočné potenie
Bolesť v čeľusti, svalové spazmy, trizmus, svalová slabosť
P
oruchy obličiek a močových ciest
- Hematúria, proteinúria, znížený renálny klírens kreatinínu, dyzúria
Akútne zlyhanie obličiek následkom dehydratácie (zriedkavé)
C
elkové poruchy a reakcie v mieste podania
Pyrexia, slabosť,
+letargia, teplotná intolerancia,
Zápal slizníc, bolesť končatín, bolesť, zimnica, bolesť na hrudníku, ochorenie podobné chrípke,
+horúčka, reakcia súvisiaca s podaním infúzie, reakcia v
mieste podania injekcie, bolesť súvisiaca s podaním
infúzie, bolesť v mieste podania injekcie
Ú
razy, otravy a komplikácie liečebného postupu
+
- Kontúzia

Pri každom výraze sa výpočet výskytu zakladal na ADR všetkých stupňov. Pri výrazoch označených “+” sa
výpočet výskytu zakladal na ADR 3.-4. stupňa. ADR sa doplnili podľa najvyššej incidencie pozorovanej v ktorejkoľvek z najdôležitejších štúdií kombinovanej liečby.
Popis vybraných nežiaducichreakciíSyndrómruka-noha(pozričasť4.4)V štúdiách kapecitabínu v monoterapii (pozostávajúcich zo štúdií overujúcich adjuvantnú liečbu karcinómu hrubého čreva, liečbu metastatického kolorektálneho karcinómu a liečbu karcinómu prsníka) sa pri podávaní kapecitabínu v dávke 1 250 mg/m2 dvakrát denne v 1. až 14. deň raz za
3 týždne pozoroval 53 % až 60 % výskyt syndrómu ruka-noha (hand-foot syndrome, HFS) všetkých stupňov a v skupine užívajúcej kapecitabín/docetaxel na liečbu metastatického karcinómu prsníka sa pozoroval 63 % výskyt. V štúdiách kapecitabínu v kombinovanej liečbe sa pri podávaní kapecitabínu v dávke 1 000 mg/m2 dvakrát denne v 1. až 14. deň raz za 3 týždne pozoroval 22 % až 30 % výskyt HFS všetkých stupňov.
Metaanalýza 14 klinických štúdií s údajmi od viac ako 4 700 pacientov liečených kapecitabínom v monoterapii alebo v kombinácii s rôznymi režimami chemoterapie vo viacerých indikáciách (karcinóm hrubého čreva, kolorektálny karcinóm, karcinóm žalúdka a karcinóm prsníka) ukázala, že HFS (všetky stupne) sa vyskytol u 2 066 (43 %) pacientov po mediáne 239 dní (95 % interval spoľahlivosti 201, 288) od začiatku liečby kapecitabínom. Nasledujúce premenné boli vo všetkých štúdiách štatisticky významne spojené so zvýšeným rizikom objavenia sa HFS: stúpajúca začiatočná dávka kapecitabínu (v gramoch), klesajúca kumulatívna dávka kapecitabínu (0,1*kg), stúpajúca relatívna dávková intenzita v prvých 6 týždňoch, stúpajúce trvanie liečby v rámci klinickej štúdie (v týždňoch), stúpajúci vek (v prírastkoch 10 rokov), ženské pohlavie, a dobrý ECOG výkonnostný stav na začiatku (0 verzus ≥1).
Hnačka(pozričasť4.4)
Capecitabine Teva môže vyvolať hnačku, ktorej výskyt sa pozoroval až u 50 % pacientov. Metaanalýza 14 klinických štúdií s údajmi od viac ako 4 700 pacientov liečených kapecitabínom
ukázala, že vo všetkých štúdiách boli nasledujúce premenné štatisticky významne spojené so
zvýšeným rizikom objavenia sa hnačky: stúpajúca počiatočná dávka kapecitabínu (v gramoch), stúpajúce trvanie liečby v rámci klinickej štúdie (v týždňoch), stúpajúci vek (v násobkoch 10 rokov) a ženské pohlavie. Nasledujúce premenné boli štatisticky významne spojené so zníženým rizikom objavenia sa hnačky: stúpajúca kumulatívna dávka kapecitabínu (0,1*kg) a stúpajúca relatívna dávková intenzita v prvých 6 týždňoch.
Kardiotoxicita(pozričasť4.4)
Navyše k ADR popísaným v tabuľkách 4 a 5 súviseli s použitím kapecitabínu v monoterapii nasledujúce ADR s incidenciou nižšou ako 0,1 %, a to na základe súhrnnej analýzy klinických údajov
o bezpečnosti zo 7 klinických štúdií zahŕňajúcich 949 pacientov (2 klinické štúdie fázy III a 5
klinických štúdií fázy II u pacientov s metastatickým kolorektálnym karcinómom a u pacientov s metastatickým karcinómom prsníka): kardiomyopatia, srdcové zlyhanie, náhla smrť a ventrikulárne extrasystoly.
Encefalopatia
Navyše k ADR popísaným v tabuľkách 4 a 5 a na základe vyššie uvedenej súhrnnej analýzy
klinických údajov o bezpečnosti zo 7 klinických štúdií súvisela s použitím kapecitabínu v monoterapii aj encefalopatia s incidenciou nižšou ako 0,1 %.
Osobitné skupiny pacientov
Starší pacienti (pozri časť 4.2)
Analýza údajov o bezpečnosti u pacientov vo veku ≥ 60 rokov liečených kapecitabínom v monoterapii a analýza pacientov liečených kapecitabínom v kombinovanej liečbe s docetaxelom preukázala zvýšený výskyt nežiaducich reakcií 3. a 4. stupňa súvisiacich s liečbou a závažných nežiaducich
reakcií súvisiacich s liečbou v porovnaní s pacientmi vo veku < 60 rokov. U pacientov vo veku
≥ 60 rokov liečených kapecitabínom a docetaxelom sa zistil aj zvýšený výskyt predčasného ukončenia liečby kvôli nežiaducim reakciám v porovnaní s pacientmi vo veku < 60 rokov.
Výsledky metaanalýzy 14 klinických štúdií s údajmi od viac ako 4 700 pacientov liečených kapecitabínom ukázali, že vo všetkých štúdiách bol stúpajúci vek (v prírastkoch 10 rokov) štatisticky významne spojený so zvýšeným rizikom vzniku HFS a hnačky a so zníženým rizikom vzniku neutropénie.
Pohlavie
Výsledky metaanalýzy 14 klinických štúdií s údajmi od viac ako 4 700 pacientov liečených kapecitabínom ukázali, že vo všetkých štúdiách bolo ženské pohlavie štatisticky významne spojené so zvýšeným rizikom vzniku HFS a hnačky a so zníženým rizikom vzniku neutropénie.
Pacienti s poruchou funkcie obličiek (pozri časti 4.2, 4.4 a 5.2):
Analýza údajov o bezpečnosti u pacientov liečených kapecitabínom v monoterapii (kolorektálny karcinóm), ktorí už pred začiatkom liečby mali poruchu funkcie obličiek, preukázala zvýšený výskyt
nežiaducich reakcií 3. a 4. stupňa súvisiacich s liečbou v porovnaní s pacientmi s normálnou funkciou obličiek (36 % u pacientov bez poruchy funkcie obličiek n=268 oproti 41 % u pacientov s miernou poruchou funkcie obličiek n=257 a 54 % u pacientov so stredne ťažkou poruchou funkcie obličiek n=59) (pozri časť 5.2). U pacientov so stredne ťažkou poruchou funkcie obličiek sa zistil zvýšený výskyt potreby zníženia dávky (44 %) oproti 33 % u pacientov bez poruchy funkcie obličiek a 32 % u pacientov s miernou poruchou funkcie a zvýšený výskyt predčasného ukončenia liečby (predčasné ukončenie liečby u 21 % pacientov počas prvých dvoch cyklov) oproti 5 % u pacientov bez poruchy funkcie obličiek a 8 % u pacientov s miernou poruchou funkcie obličiek.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieAkútne predávkovanie sa prejavuje nevoľnosťou, vracaním, hnačkou, mukozitídou, podráždením gastrointestinálneho traktu, krvácaním do zažívacieho traktu a útlmom kostnej drene. Liečba predávkovania zahŕňa zvyčajné liečebné a podporné zákroky zamerané na úpravu klinických príznakov a prevenciu možných komplikácií.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antineoplastiká, antimetabolity, ATC kód: L01BC06
Kapecitabín je necytotoxický karbamát fluórpyrimidínu, ktorý účinkuje ako perorálne podávaný prekurzor cytotoxickej funkčnej skupiny 5-fluóruracilu (5-FU). K aktivácii kapecitabínu dochádza v priebehu niekoľkých enzymatických krokov (pozri časť 5.2). Enzým, ktorý riadi záverečnú premenu na 5-FU, tymidínfosforyláza (ThyPase) sa nachádza v nádorovom tkanive, ale taktiež v normálnych tkanivách, hoci obyčajne v nízkych koncentráciách. V štúdiách s ľudskými karcinómami vykázal kapecitabín synergický efekt v kombinácii s docetaxelom, čo môže byť následkom ovplyvnenia tymidínfosforylázy docetaxelom.
Zistilo sa, že metabolizmus 5-FU v anabolickej dráhe blokuje metylačnú premenu kyseliny deoxyuridylovej na kyselinu tymidylovú, čím dochádza k poruche syntézy kyseliny deoxyribonukleovej (DNA). Inkorporácia 5-FU taktiež vedie k inhibícii syntézy kyseliny ribonukleovej (RNA) a syntézy proteínov. Vzhľadom na zásadný význam DNA a RNA pre delenie a rast buniek, 5-FU môže spôsobiť deficit tymidínu, ktorý vedie k nevyváženému rastu buniek a ich následnému odumretiu. Nedostatok DNA a RNA najvýraznejšie postihuje bunky s rýchlou proliferáciou a rýchlym metabolizmom 5-FU.
Karcinóm hrubého čreva a kolorektálny karcinómMonoterapiakapecitabínomprikarcinómehrubéhočrevaÚdaje z jednej multicentrickej, randomizovanej, kontrolovanej klinickej štúdie v III. fáze, skúmajúcej pacientov s karcinómom hrubého čreva štádia III (podľa Dukesa stupeň C), podporujú používanie kapecitabínu pri adjuvantnej liečbe pacientov s karcinómom hrubého čreva (štúdia XACT; M66001). V tejto štúdii bolo 1987 pacientov randomizovaných na liečbu kapecitabínom (1250 mg/m2 dvakrát denne počas 2 týždňov, potom nasledovala týždňová prestávka; liek sa podával v trojtýždňových cykloch počas 24 týždňov) alebo 5-FU plus leukovorín (režim kliniky Mayo: 20 mg/m2 i.v. leukovorín, potom 425 mg/m2 intrvenózne bolus 5-FU, od 1. do 5.dňa, každých 28 dní počas
24 týždňov). Kapecitabín bol prinajmenšom ekvivalentný i.v. 5-FU/LV, čo sa týka prežitia bez ochorenia u populácie podľa protokolu (miera rizika 0,92; 95 % IS 0,80 – 1,06). Testy rozdielnosti kapecitabínu a 5-FU/LV vzhľadom na prežitie bez ochorenia a celkové prežitie v celej
randomizovanej populácii dokázali mieru rizika 0,88 (95 % IS 0,77 – 1,01; p = 0,068) a 0,86 (95 % IS
0,74 – 1,01; p = 0,060). Stredný čas sledovania v čase analýzy bol 6,9 rokov. V multivariantnej Coxovej analýze, ktorá bola vopred plánovaná, sa dokázala superiorita kapecitabínu v porovnaní s bolusovým 5-FU/LV. Pre zaradenie do modelu boli v štatistickom pláne vopred špecifikované nasledovné faktory: vek, čas od chirurgického výkonu do randomizácie, pohlavie, hladiny CEA a počet lymfatických uzlín pri zaradení do klinickej štúdie, krajina. V populácii všetkých randomizovaných pacientov bola dokázaná superiorita kapecitabínu oproti 5-FU/LV z hľadiska prežívania bez choroby (miera rizika 0,849; 95 % IS 0,739 - 0,976; p = 0,0212) a aj z hľadiska celkového prežívania (miera rizika 0,828; 95 % IS 0,705 - 0,971; p = 0,0203).
Kombinovanáterapiaprikarcinómehrubéhočreva
Údaje z jednej multicentrickej, randomizovanej, kontrolovanej klinickej štúdie fázy III, ktorá skúmala pacientov s karcinómom hrubého čreva v III. štádiu (podľa Dukesa stupeň C), podporujú používanie
Capecitabine Teva v kombinácii s oxaliplatinou (XELOX) pri adjuvantnej liečbe pacientov s karcinómom hrubého čreva (štúdia NO16968). V tejto klinickej štúdii bolo 944 pacientov
randomizovaných na liečbu v 3-týždňových cykloch počas 24 týždňov kapecitabínom
(1 000 mg/m2 dvakrát denne počas 2 týždňov, potom nasledovala týždňová prestávka) v kombinácii s oxaliplatinou (130 mg/m2 intravenózna infúzia podávaná počas 2 hodín v 1. deň každé 3 týždne);
942 pacientov bolo randomizovaných na bolus 5-FU a leukovorín. V primárnej analýze prežívania bez choroby (DFS) v populácii ITT (intent-to-treat, „s úmyslom liečiť”) sa dokázala signifikantná
superiorita XELOXu v porovnaní s 5-FU/LV (miera rizika HR 0,80, 95 % IS [0,69; 0,93]; p = 0,0045). Miera 3-ročného DFS bola pri XELOXe 71 % oproti 67 % pri 5-FU/LV. Analýza sekundárneho cieľa RFS podporuje tieto výsledky s HR 0,78 (95 % IS = [0,67; 0,92]; p = 0,0024) pri XELOXe oproti 5-
FU/LV. XELOX vykazoval tendenciu k lepšiemu celkovému prežívaniu (OS) s HR 0,87 (95 %
CI = [0,72; 1,05]; p = 0,1486), čo sa premieta do 13 %-ného zníženia rizika úmrtia. Miera 5-ročného
OS bola 78 % pri XELOXe oproti 74 % pri 5-FU/LV. Údaje o účinnosti vychádzajú zo stredného času pozorovania 59 mesiacov so zreteľom na OS a 57 mesiacov so zreteľom na DFS. V ITT populácii bol počet pacientov, ktorí ukončili účasť v štúdii kvôli nežiaducim účinkom vyšší v skupine s kombinovanou liečbou XELOX (21 %) v porovnaní so skupinou liečenou monoterapiou 5-FU/LV
(9 %).
Monoterapiakapecitabínomprimetastatickomkolorektálnomkarcinóme
Údaje získané v dvoch multicentrických, randomizovaných, kontrolovaných klinických štúdiách III. fázy (SO14695; SO14796) s rovnakým dizajnom podporujú podávanie kapecitabínu v liečbe prvej línie u pacientov s metastázujúcim kolorektálnym karcinómom. V rámci týchto štúdií sa
603 pacientom náhodne pridelila liečba kapecitabínom (1 250 mg/m2 dvakrát denne počas 2 týždňov, po ktorých nasledoval 1 týždeň bez liečby; podávaný v 3-týždňových cykloch). A 604 pacientom sa
náhodne pridelila liečba 5-FU a leukovorínom (Mayo režim: leukovorín v dávke
20 mg/m2 intravenózne, po ktorom nasleduje bolus intravenózne 5-FU v dávke 425 mg/m2 na 1. a
5. deň; každých 28 dní). V celej randomizovanej populácii sa celková objektívna odpoveď na liečbu
(podľa hodnotenia skúšajúceho) zaznamenala u 25,7 % (kapecitabín) v porovnaní s 16,7 % (Mayo režim); p < 0,0002. Stredná doba do progresie ochorenia bola 140 dní (kapecitabín), v porovnaní so
144 dňami (Mayo režim). Stredná doba prežívania bola 392 dní (kapecitabín), v porovnaní
s 391 dňami (Mayo režim). V súčasnosti nie sú dostupné žiadne údaje vyplývajúce z porovnania monoterapie kapecitabínom a kombinovaných režimov prvej línie pri liečbe kolorektálneho
karcinómu.
Kombinovanáterapiapriliečbeprvejlíniemetastatickéhokolorektálnehokarcinómu
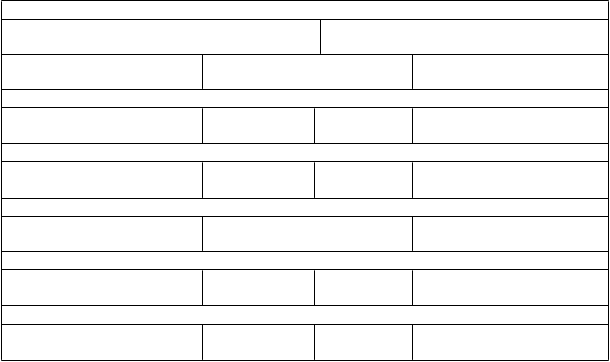
Údaje z multicentrickej, randomizovanej, kontrolovanej klinickej štúdie fázy III (NO16966) podporujú použitie kapecitabínu v kombinácii s oxaliplatinou alebo v kombinácii s oxaliplatinou a
bevacizumabom na liečbu prvej línie metastatického kolorektálneho karcinómu. Štúdia pozostávala z dvoch častí: úvodná časť s 2 skupinami, v ktorej bolo 634 pacientov randomizovaných do dvoch
rôznych liečebných skupín, zahŕňajúcich XELOX alebo FOLFOX-4, a následná 2x2 faktorová časť, v ktorej bolo 1 401 pacientov randomizovaných do štyroch rôznych liečebných skupín, zahŕňajúcich
XELOX plus placebo, FOLFOX-4 plus placebo, XELOX plus bevacizumab a FOLFOX-4 plus bevacizumab. Pozri tabuľku 6 pre liečebné režimy.
Tabuľka 6 Liečebné režimy v štúdii NO16966 (mCRC)
Liečba Počiatočná dávka Schéma
FOLFOX-4 alebo FOLFOX-4 +
bevacizumab
Oxaliplatina
Leukovorín
5-fluóruracil
Placebo alebo bevacizumab
85 mg/m2 intravenózne
2 hod
200 mg/m2
intravenózne 2 hod
400 mg/m2 intravenózn e bolus, po ktorom nasledovalo
600 mg/m2 intravenózn e 22 hod
5 mg/kg intravenózne
30-90 min.
Oxaliplatina v 1. deň, raz za
2 týždne
Leukovorín v 1. a 2. deň, raz za 2 týždne
5-fluóruracil intravenózne
bolus/infúzia, obidve v 1. a
2. deň, raz za 2 týždne
1. deň, pred FOLFOX-4, raz za 2 týždne
XELOX alebo XELOX+
bevacizumab
Oxaliplatina 130 mg/m2
intravenózne 2 hod
Kapecitabín 1000 mg/m2 perorálne dvakrát denne
Oxaliplatina v 1. deň, raz za
3 týždne
Kapecitabín perorálne dvakrát denne počas
2 týždňov (po ktorých nasledovala 1-týždňová prestávka)
Placebo alebo bevacizumab
7,5 mg/kg intravenózne
30-90 min.
1. deň, pred XELOX
5-fluóruracil: intravenózne bolus podaný ihneď po leukovoríne
Pri celkovom porovnaní sa preukázalo, že v skupinách, ktoré dostávali XELOX nebola liečba menej
účinná oproti skupinám, ktoré dostávali FOLFOX-4, a to z hľadiska prežívania bez progresie ochorenia v populácii vhodných pacientov a v populácii všetkých randomizovaných (intent-to-treat) pacientov (pozri tabuľku 7). Výsledky svedčia o tom, že XELOX je rovnako účinný ako FOLFOX-4, a to z hľadiska celkového prežívania (pozri tabuľku 7). Porovnanie XELOX plus bevacizumab oproti FOLFOX-4 plus bevacizumab bolo vopred špecifikovanou exploračnou analýzou. V tomto porovnaní
liečebných podskupín bol XELOX plus bevacizumab podobný ako FOLFOX-4 plus bevacizumab, a to z hľadiska prežívania bez progresie ochorenia (miera rizika 1,01; 97,5 % IS 0,84 - 1,22). Medián sledovania v čase primárnych analýz v populácii všetkých randomizovaných (intent-to-treat) pacientov bol 1,5 roka; údaje z analýz po ďalšom 1-ročnom sledovaní sú taktiež zhrnuté v tabuľke 7. Analýza prežívania bez progresie ochorenia (progression-free survival, PFS) počas liečby však nepotvrdila výsledky všeobecnej analýzy PFS a celkového prežívania (overall survival, OS): miera rizika pre XELOX oproti FOLFOX-4 bola 1,24 s 97,5 % IS 1,07 - 1,44. Hoci analýzy citlivosti ukazujú, že rozdiely v liečebných režimoch a v čase hodnotenia nádoru majú vplyv na analýzu PFS počas liečby, úplné vysvetlenie tohto výsledku sa nezistilo
Tabuľka 7 Hlavné výsledky účinnosti z analýzy posudzujúcej nie nižšiu účinnosť v štúdii NO16966
PRIMÁRNA ANALÝZA
XELOX/XELOX+P/ XELOX+BV (EPP*: N=967; ITT**: N=1 017)
FOLFOX-4/FOLFOX-4+P/ FOLFOX-4+BV (EPP*: N=937; ITT**: N=1 017)
Populácia Stredný čas do vzniku príhody (dni) HR (97,5 % IS)
Parameter: Prežívanie bez progresie ochorenia

EPP ITT
Parameter: Celkové prežívanie
241
244
259
259
1,05 (0,94; 1,18)
1,04 (0,93; 1,16)
EPP ITT
577
581
549
553
0,97 (0,84; 1,14)
0,96 (0,83; 1,12)
ĎALŠIE 1-ROČNÉ SLEDOVANIE
Populácia Stredný čas do vzniku príhody (dni) HR (97,5 % IS)'
Parameter: Prežívanie bez progresie ochorenia
EPP ITT
Parameter: Celkové prežívanie
EPP ITT
242
244
600
602
259
259
594
596
1,02 (0,92; 1,14)
1,01 (0,91; 1,12)
1,00 (0,88; 1,13)
0,99 (0,88; 1,12)

*EPP=populácia vhodných pacientov; **ITT=populácia všetkých randomizovaných (intent-to-treat) pacientov
V randomizovanej, kontrolovanej klinickej štúdie fázy III (CAIRO) bol študovaný účinok použitia kapecitabínu v počiatočnej dávke 1 000 mg/m2 podávanej počas 2 týždňov raz za 3 týždne v kombinácii s irinotekanom v prvej línii liečby pacientov s metastatickým kolorektálnym karcinómom.
820 pacientov bolo randomizových na sekvenčnú liečbu (n=410) alebo kombinovanú liečbu (n=410). Sekvenčná liečba pozostávala z kapecitabínu (1 250 mg/m2 dvakrát denne počas 14 dní) ako lieku prvej línie liečby, irinotekanu (350 mg/m2 v 1. deň) ako lieku druhej línie liečby a kombinácie kapecitabínu (1 000 mg/m2 dvakrát denne počas 14 dní) s oxaliplatinou (130 mg/m2 v 1. deň) ako liekov tretej línie liečby. Kombinovaná liečba pozostávala z kapecitabínu (1 000 mg/m2 dvakrát denne počas 14 dní) v kombinácii s irinotekanom (250 mg/m2 v 1. deň) ako liekov prvej línie liečby a kapecitabínu (1 000 mg/m2 dvakrát denne počas 14 dní) plus oxaliplatiny (130 mg/m2 v 1. deň) ako liekov druhej línie liečby. Všetky liečebné cykly boli podávané s časovým odstupom 3 týždňov. V prvej línii liečby bol medián prežívania bez progresie ochorenia v populácii všetkých randomizovaných (intent-to-treat) pacientov 5,8 mesiacov (95 % IS 5,1 - 6,2 mesiacov) pre
kapecitabín v monoterapii a 7,8 mesiacov (95 % IS 7,0 - 8,3 mesiacov; p=0,0002) pre XELIRI. Bolo to však spojené so zvýšeným výskytom gastrointestinálnej toxicity a neutropénie počas prvej línie liečby s XELIRI (26% pre XELIRI a 11% v prvej línii liečby kapecitabínom).
XELIRI bol porovnávaný s 5-FU + irinotekanom (FOLFIRI) v troch randomizovaných štúdiách u pacientov s metastatickým kolorektálnym karcinómom. XELIRI režimy obsahovali kapecitabín 1000 mg/m2 dvakrát denne v dňoch 1 až 14 trojtýždňového cyklu v kombinácii s irinotekanom 250
mg/m2 v deň 1. V najväčšej štúdii (BICC-C) boli pacienti randomizovaní na nezaslepené prijatie
FOLFIRI (n = 144), na bolus 5-FU (mIFL) (n = 145) alebo XELIRI (n = 141) a boli ďalej randomizovaní na prijatie buď dvojito zaslepenej liečby alebo na liečbu celekoxibom alebo placebom. Medián PFS bol 7,6 mesiacov pre FOLFIRI, 5,9 mesiaca pre mIFL (p = 0,004 pre porovnanie s FOLFIRI), a 5,8 mesiacov pre XELIRI (p = 0,015). Medián OS bol 23,1 mesiacov pre FOLFIRI, 17,6 mesiacov pre mIFL (p = 0,09) a 18,9 mesiacov pre XELIRI (p = 0,27). U pacientov liečených XELIRI došlo k excesívnej gastrointestinálnej toxicite v porovnaní s FOLFIRI (hnačka 48% pri XELIRI a 14% pri FOLFIRI).
V štúdii EORTC boli pacienti randomizovaní buď na zaslepené prijatie FOLFIRI (n = 41) alebo XELIRI (n = 44) s ďalšou randomizáciou buď do dvojito zaslepenej liečby celekoxibom alebo placebom. Medián PFS a celkové prežívanie (OS) boli kratšie pre XELIRI oproti FOLFIRI (PFS 5,9 oproti 9,6 mesiacov a OS 14,8 oproti 19,9 mesiacov), okrem toho bol hlásený výrazne excesívny výskyt hnačky u pacientov liečených režimom XELIRI (41% XELIRI, 5,1% FOLFIRI).
V štúdii, ktorú publikoval Skof a spol., boli pacienti randomizovaní na liečbu buď s FOLFIRI, alebo XELIRI. Celková miera odpovede bola 49% v skupine XELIRI a 48% v skupine FOLFIRI (p = 0,76). Na konci liečby, 37% pacientov v skupine XELIRI a 26% pacientov v skupine FOLFIRI boli bez známok ochorenia (p = 0,56). Toxicita bola podobná, s výnimkou neutropénie, ktorá bola hlásená častejšie u pacientov liečených FOLFIRI.
Montagnani a spol. použili výsledky z vyššie uvedených troch štúdií, aby poskytli celkovú analýzu randomizovaných štúdií porovnávajúcich liečebné režimy FOLFIRI a XELIRI v liečbe mCRC.
Významné zníženie rizika progresie ochorenia sa spájalo s FOLFIRI (HR, 0,76; 95 % IS, 0,62 - 0,95; P < 0,01), čo je čiastočne kvôli zlej tolerancii použitého režimu XELIRI.
Údaje z randomizovanej klinickej štúdie (Souglakos a spol., 2012) porovnávajúcej
FOLFIRI + bevacizumab s XELIRI + bevacizumab neukázali významné rozdiely v PFS alebo OS
medzi liečbami. Pacienti boli randomizovaní na liečbu buď s FOLFIRI plus bevacizumab (skupinaA n=167) alebo s XELIRI plus bevacizumab (skupinaB, n=166). Pre skupinuB v režime XELIRI boli použité dávky kapecitabínu 1000 mg/m2 dvakrát denne počas 14 dní + irinotekan 250 mg/m2 v deň 1. Medián prežívania bez progresie ochorenia (PFS) bol 10,0 a 8,9 mesiaca; p = 0,64, celkové prežívanie bolo 25,7 a 27,5 mesiaca; p = 0,55 a miera výskytu odpovede bola 45,5 a 39,8 %; p = 0,32 pri FOLFIRI-bev a pri XELIRI-bev, v uvedenom poradí. U pacientov liečených XELIRI + bevacizumab bol hlásený signifikantne vyšší výskyt hnačky, febrilnej neutropénie a syndrómu „ruka-noha“ ako u
pacientov liečených FOLFIRI + bevacizumab s výrazne zvýšeným počtom oneskorení liečby, zníženia dávky a ukončenia liečby.
Údaje z multicentrickej, randomizovanej, kontrolovanej klinickej štúdie fázy II (AIO KRK 0604) podporujú použitie kapecitabínu v počiatočnej dávke 800 mg/m2 podávanej počas 2 týždňov raz za 3 týždne v kombinácii s irinotekanom a bevacizumabom v prvej línii liečby pacientov s metastatickým kolorektálnym karcinómom. 120 pacientov bolo randomizových na liečbu modifikovaným režimom XELIRI s kapecitabínom 800 mg/m2 dvakrát denne počas dvoch týždňov, po ktorých nasleduje 7 dní bez liečby irinotekanom (200 mg/m2 formou 30 minút trvajúcej infúzie v 1. deň, raz za 3 týždne) a bevacizumabom (7,5 mg/kg formou 30 až 90 minút trvajúcej infúzie v 1. deň, raz za 3 týždne); 127 pacientov bolo randomizovaných na liečbu kapecitabínom (1 000 mg/m2 dvakrát denne počas dvoch týždňov, po ktorých nasleduje 7 dní bez liečby oxaliplatinou (130 mg/m2 formou 2 hodiny trvajúcej infúzie v 1. deň raz za 3 týždne) a bevacizumabom (7,5 mg/kg formou 30 až 90 minút trvajúcej infúzie v 1. deň raz za 3 týždne). Po priemernom sledovaní populácie pacientov v štúdii počas 26,2 mesiacov boli zaznamenané odpovede na liečbu tak ako je uvedené nižšie.
Tabuľka 8 Hlavné výsledky účinnosti v štúdii AIO KRK
XEL
O
X + bevacizumab
(I
TT
: N=127)
Celkové prežívanie po 6 mesiacoch
Modifikovaný XELIRI+ bevacizumab (ITT: N= 120)
Miera rizika
95% IS P value
ITT
95% IS
76%
69 - 84%
84%
77 - 90% -
Medián prežívania bez progresie
ITT
95% IS
10,4 mesiacov
9,0 – 12,0
12,1 mesiacov
10,8 – 13,2
0,93
0,82 – 1,07
P=0,30
Medián celkového prežívania
ITT
95% IS
24,4 mesiacov
19,3 – 30,7
25,5 mesiacov
21,0 – 31,0
0,90
0,68 – 1,19
P=0,45
 K
ombinovaná
li
ečba
v
druhej
lí
n
i
i
l
i
ečby
m
etastatického
kolorektálneho
karcinómu
K
ombinovaná
li
ečba
v
druhej
lí
n
i
i
l
i
ečby
m
etastatického
kolorektálneho
karcinómu
Údaje z multicentrickej, randomizovanej, kontrolovanej klinickej štúdie fázy III (NO16967) podporujú použitie kapecitabínu v kombinácii s oxaliplatinou v druhej línii liečby metastatického kolorektálneho
karcinómu. V tejto štúdii bolo 627 pacientov s metastatickým kolorektálnym karcinómom, ktorí
predtým dostali liečbu irinotekanom v kombinácii s fluórpyrimidínovým režimom ako liečbu prvej línie, randomizovaných do liečebnej skupiny, ktorá dostávala buď XELOX, alebo FOLFOX-4. Dávkovacia schéma pre XELOX a FOLFOX-4 (bez pridania placeba alebo bevacizumabu), pozri tabuľku 6. Preukázalo sa, že XELOX nebol menej účinný ako FOLFOX-4, a to z hľadiska prežívania bez progresie ochorenia v populácii podľa protokolu a v populácii všetkých randomizovaných (intent- to-treat) pacientov (pozri tabuľku 9). Výsledky svedčia o tom, že XELOX je rovnocenný ako FOLFOX-4, a to z hľadiska celkového prežívania (pozri tabuľku 9). Medián sledovania v čase
primárnych analýz v populácii všetkých randomizovaných (intent-to-treat) pacientov bol 2,1 rokov;
údaje z analýz po ďalšom 6-mesačnom sledovaní sú taktiež zhrnuté v tabuľke 9.
Tabuľka 9 Hlavné výsledky účinnosti z analýzy posudzujúcej nie nižšiu účinnosť v štúdii NO16967
PRIMÁRNA ANALÝZA
XELOX
(PPP*: N=251; ITT**: N=313)
FOLFOX-4
(PPP*: N=252; ITT**: N=314)
Populácia Stredný čas do vzniku príhody
(dni)
HR
(95 % IS)
Parameter: Prežívanie bez progresie ochorenia
PPP ITT
Parameter: Celkové prežívanie
PPP ITT
154
144
388
363
168
146
401
382
1,03 (0,87; 1,24)
0,97 (0,83; 1,14)
1,07 (0,88; 1,31)
1,03 (0,87; 1,23)
ĎALŠIE 6-MESAČNÉ SLEDOVANIE Populácia Stredný čas do vzniku príhody
(dni)
HR
(95 % IS)
Parameter: Prežívanie bez progresie ochorenia
PPP ITT
Parameter: Celkové prežívanie
PPP ITT
154
143
393
363
166
146
402
382
1,04 (0,87; 1,24)
0,97 (0,83; 1,14)
1,05 (0,88; 1,27)
1,02 (0,86; 1,21)
*PPP=populácia podľa protokolu; **ITT=populácia všetkých randomizovaných (intent-to-treat) pacientov
Pokročilý karcinóm žalúdkaÚdaje z multicentrickej, randomizovanej, kontrolovanej klinickej štúdie III. fázy skúmajúcej pacientov
s pokročilým karcinómom žalúdka podporujú použitie kapecitabínu ako lieku prvej línie na liečbu pokročilého karcinómu žalúdka (ML17032). V tejto štúdii bolo 160 pacientov randomizovaných na liečbu kapecitabínom (1 000 mg/m2 dvakrát denne počas 2 týždňov, po ktorých nasleduje 7 dní bez liečby) a cisplatinou (80 mg/m2 formou 2 hodiny trvajúcej infúzie, raz za 3 týždne). Celkom
156 pacientov bolo randomizovaných na liečbu 5-FU (800 mg/m2 denne, kontinuálna infúzia v 1. až
5. deň, raz za 3 týždne) a cisplatinou (80 mg/m2 formou 2 hodiny trvajúcej infúzie v 1. deň, raz za
3 týždne). Kapecitabín v kombinácii s cisplatinou nebol menej účinný ako 5-FU v kombinácii s cisplatinou z hľadiska prežívania bez progresie ochorenia v analýze podľa protokolu (miera rizika
0,81; 95 % IS 0,63 – 1,04). Medián prežívania bez progresie bol 5,6 mesiacov (kapecitabín +
cisplatina) oproti 5,0 mesiacom (5-FU + cisplatina). Miera rizika dĺžky prežívania (celkové
prežívanie) bola podobná miere rizika pre prežívanie bez progresie ochorenia (miera rizika 0,85; 95 % IS 0,64 - 1,13). Medián dĺžky prežívania bol 10,5 mesiacov (kapecitabín + cisplatina) oproti
9,3 mesiacom (5-FU + cisplatina).
Údaje z randomizovanej, multicentrickej klinickej štúdie III. fázy porovnávajúcej kapecitabín s 5-FU a oxaliplatinu s cisplatinou u pacientov s pokročilým karcinómom žalúdka podporujú použitie kapecitabínu ako lieku prvej línie na liečbu pokročilého karcinómu žalúdka (REAL-2). V tejto štúdii bolo v 2x2 faktorovom dizajne 1 002 pacientov randomizovaných do jedného z nasledujúcich
4 ramien:
- ECF: epirubicín (50 mg/m2 podávaných formou bolusu v 1. deň, raz za 3 týždne), cisplatina (60 mg/m2 podávaných formou 2 hodiny trvajúcej infúzie v 1. deň, raz za 3 týždne) a 5-FU (200 mg/m2 podávaných denne formou kontinuálnej infúzie cez centrálny venózny katéter).
- ECX: epirubicín (50 mg/m2 podávaných formou bolusu v 1. deň, raz za 3 týždne), cisplatina
(60 mg/m2 podávaných formou 2 hodiny trvajúcej infúzie v 1. deň, raz za 3 týždne) a kapecitabín (625 mg/m2 podávaných dvakrát denne kontinuálne).
- EOF: epirubicín (50 mg/m2 podávaných formou bolusu v 1. deň, raz za 3 týždne), oxaliplatina (130 mg/m2 podávaných formou 2 hodiny trvajúcej infúzie v 1. deň, raz za 3 týždne) a 5-FU (200 mg/m2 podávaných denne formou kontinuálnej infúzie cez centrálny venózny katéter).
- EOX: epirubicín (50 mg/m2 podávaných formou bolusu v 1. deň, raz za 3 týždne), oxaliplatina (130 mg/m2 podávaných formou 2 hodiny trvajúcej infúzie v 1. deň, raz za 3 týždne) a kapecitabín (625 mg/m2 podávaných dvakrát denne kontinuálne).
Analýzy primárnej účinnosti u populácie liečenej podľa protokolu preukázali nie nižšiu účinnosť v celkovom prežívaní pre režim na báze kapecitabínu oproti režimu na báze 5-FU (miera rizika 0,86;
95 % IS 0,8 - 0,99) a pre režim na báze oxaliplatiny oproti režimu na báze cisplatiny (miera rizika
0,92; 95 % IS 0,80 - 1,1). Medián celkového prežívania bol 10,9 mesiacov pre režimy na báze kapecitabínu a 9,6 mesiacov pre režimy na báze 5-FU. Medián celkového prežívania bol
10,0 mesiacov pre režimy na báze cisplatiny a 10,4 mesiacov pre režimy na báze oxaliplatiny.
Kapecitabín sa tiež používal v kombinácii s oxaliplatinou na liečbu pokročilého karcinómu žalúdka. Klinické štúdie s kapecitabínom v monoterapii svedčia o tom, že kapecitabín vykazuje účinnosť pri pokročilom karcinóme žalúdka.
Karcinómhrubéhočreva,kolorektálnykarcinómapokročilýkarcinómžalúdka:metaanalýza Metaanalýza šiestich klinických štúdií (štúdií SO14695, SO14796, M66001, NO16966, NO16967, M17032) podporuje použitie kapecitabínu ako náhrady 5-FU v monoterapii a v kombinovanej liečbe gastrointestinálneho karcinómu. Súhrnná analýza zahŕňa 3 097 pacientov liečených režimami obsahujúcimi kapecitabín a 3 074 pacientov liečených režimami obsahujúcimi 5-FU. Medián celkového prežívania bol 703 dní (95 % IS: 671; 745) u pacientov liečených režimami obsahujúcimi kapecitabín a 683 dní (95 % IS: 646; 715) u pacientov liečených režimami obsahujúcimi 5-FU. Miera rizika celkového prežívania bola 0,94 (95 % IS: 0,89; 1,00; p = 0,0489), čo svedčí o tom, že režimy obsahujúce kapecitabín sú noninferiórne v porovnaní s režimami obsahujúcimi 5-FU.
Karcinóm prsníka
Kombinovanáliečbalokálnepokročiléhoalebometastatickéhokarcinómuprsníkakapecitabínoma
docetaxelom
Údaje z jednej multicentrickej, randomizovanej, kontrolovanej klinickej štúdie III. fázy podporujú použitie kapecitabínu v kombinácii s docetaxelom na liečbu pacientov s lokálne rozvinutým alebo metastatickým karcinómom prsníka po zlyhaní cytotoxickej chemoterapie zahrňujúcej antracyklín. V tejto štúdii sa randomizovalo 255 pacientov na liečbu kapecitabínom (1 250 mg/m2 dvakrát denne počas 2 týždňov, po ktorých nasledoval 1 týždeň bez liečby a docetaxelom, ktorý sa podal intravenóznou infúziou, ktorá trvala 1 hodinu v dávke 75 mg/m2 raz za 3 týždne). 256 pacientov sa liečilo samotným docetaxelom (100 mg/m2 v intravenóznej infúzii trvajúcej 1 hodinu raz za 3 týždne). Prežitie bolo vyššie v skupine kapecitabín + docetaxel (p = 0,0126). Stredná doba prežitia bola 442 dní (kapecitabín + docetaxel), verzus 352 dní (docetaxel samotný). Celková objektívna odpoveď na liečbu (podľa hodnotenia skúšajúceho) bola 41,6 % (kapecitabín + docetaxel), verzus 29,7 % (docetaxel samotný); p = 0,0058. Čas do progresie ochorenia bol dlhší v skupine kapecitabín/docetaxel
(p < 0,0001); 186 dní (kapecitabín + docetaxel) verzus 128 dní (docetaxel samotný).
Monoterapiakapecitabínompozlyhanítaxánov,chemoterapieantracyklínomaupacientov,uktorýchniejeindikovanáliečbaantracyklínom
Údaje z dvoch multicentrických klinických štúdií II. fázy podporujú použitie kapecitabínu v monoterapii na liečbu pacientov po zlyhaní taxánov a chemoterapeutických liečebných režimov, ktoré obsahujú antracyklíny, a u ktorých ďalšie použitie antracyklínov nie je indikované. V týchto štúdiách sa celkovo 236 pacientov liečilo kapecitabínom (1 250 mg/m2 denne počas 2 týždňov, po ktorých nasledoval 1 týždeň bez liečby). Celková objektívna odpoveď na liečbu (podľa hodnotenia skúšajúceho) bola 20 % (prvá štúdia) a 25 % (druhá štúdia). Stredný čas do progresie bol 93 a 98 dní. Stredná doba prežitia bola 384 a 373 dní.
Všetkyindikácie
Metaanalýza 14 klinických štúdií s údajmi od viac ako 4 700 pacientov liečených kapecitabínom v monoterapii alebo v kombinácii s rôznymi režimami chemoterapie vo viacerých indikáciách (karcinóm hrubého čreva, kolorektálny karcinóm, karcinóm žalúdka a karcinóm prsníka) ukázala, že pacienti, ktorí užívali kapecitabín a vyskytol sa u nich syndróm ruka-noha (HFS) mali dlhšie celkové prežívanie v porovnaní s pacientmi, u ktorých sa syndróm HFS nevyvinul: medián celkového prežívania 1 100 dní (95 % interval spoľahlivosti 1 007; 1 200) verzus 691 dní (95 % interval spoľahlivosti 638; 754), hazard ratio 0,61 (95 % interval spoľahlivosti 0,56; 0,66).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti kapecitabínu sa hodnotili v intervale dávok 502 - 3 514 mg/m2 denne. Parametre kapecitabínu, 5'-deoxy-5-fluórcytidínu (5'-DFCR) a 5'-deoxy-5-fluóruridínu (5'-DFUR) boli podobné pri meraniach uskutočnených v 1. a 14. dni liečby. Na 14. deň bola hodnota AUC pre 5-FU
o 30 – 35 % vyššia. Zníženie dávkovania kapecitabínu viedlo k viac ako proporčnému zníženiu systémovej expozície 5-FU v dôsledku nelineárnej farmakokinetiky aktívneho metabolitu.
Absorpcia
Kapecitabín sa rýchlo a významne absorbuje po perorálnom podaní a ďalej sa výrazne mení na metabolity 5'-DFCR a 5'-DFUR. Podanie lieku s jedlom znižuje rýchlosť vstrebávania kapecitabínu,
avšak len minimálne ovplyvňuje hodnoty AUC pre 5'-DFUR a následného metabolitu 5-FU. Po
14 dňoch podávania kapecitabínu v dennej dávke 1 250 mg/m2 po jedle boli plazmatické koncentrácie
(Cmax v μg/ml) kapecitabínu, 5'-DFCR, 5'-DFUR, 5-FU a FBAL nasledovné: 4,67; 3,05; 12,1; 0,95 a
5,46 μg/ml. Časy potrebné na dosiahnutie maximálnych koncentrácií uvedených látok v plazme (T max
v hodinách) boli nasledovné: 1,5; 2,0; 2,0; 2,0 a 3,34 hod. Hodnoty AUCo-∞ pre uvedené látky boli
nasledovné: 7,75; 7,24; 24,6; 2,03 a 36,3 μg.h/ml.
Distribúcia
V štúdiách in vitro s ľudskou plazmou sa pri kapecitabíne, 5'-DFCR, 5'-DFUR
a 5-FU zistili nasledovné hodnoty väzby na bielkoviny: 54 %, 10 %, 62 % a 10 %, prevažne na albumín.
Biotransformácia
Kapecitabín sa najskôr mení účinkom pečeňovej karboxylesterázy na 5'-DFCR, ten sa ďalej účinkom cytidíndeaminázy mení na 5'-DFUR, ktorá sa nachádza hlavne v pečeni a nádorových tkanivách. K ďalšej katalytickej aktivácii 5'-DFUR dochádza tymidínfosforylázou (ThyPase). Enzýmy, ktoré sa zúčastňujú katalytickej aktivácie sa nachádzajú v nádorových tkanivách, ale aj v normálnych tkanivách, hoci obyčajne v nízkych koncentráciách. Následná enzymatická biotransformácia kapecitabínu na 5-FU vedie k vysokým koncentráciám v nádorových tkanivách. V prípade kolorektálneho karcinómu sa zdá, že k tvorbe 5-FU z veľkej časti dochádza v bunkách strómy nádorového tkaniva. Po perorálnom podaní kapecitabínu pacientom s kolorektálnym karcinómom bol pomer koncentrácie 5-FU v nádore a okolitých tkanivách 3,2 (interval: 0,9 - 8,0). Pomer koncentrácie
5-FU v nádore a plazme bol 21,4 (interval: 3,9 - 59,9; n = 8), kým pomer jeho koncentrácie v zdravých tkanivách a plazme bol 8,9 (interval: 3 - 25,8; n = 8). Aktivita tymidínfosforylázy bola 4x vyššia v
primárnom kolorektálnom karcinóme ako v okolitom normálnom tkanive. Na základe
imunohistochemických štúdií sa zdá, že tymidínfosforyláza sa z veľkej časti nachádza v bunkách strómy nádorového tkaniva.
5-FU sa ďalej katabolizuje enzýmom dihydropyrimidíndehydrogenáza (DPD) na omnoho menej toxický dihydro-5-fluóruracil (FUH2). Dihydropyrimidináza štiepi pyrimidínový kruh na kyselinu 5- fluór-ureidopropiónovú. Konečná β-ureido-propionáza štiepi FUPA na α-fluór-β-alanín (FBAL), ktorý sa vylučuje močom. Aktivita dihydropyrimidíndehydrogenázy (DPD) je rýchlosť limitujúci krok degradácie. Nedostatok DPD môže viesť k zvýšeniu toxicity kapecitabínu (pozri časť 4.3 a 4.4).
Eliminácia
Eliminačný polčas (t1/2 ) kapecitabínu, 5'-DFCR, 5'-DFUR, 5-FU a FBAL bol 0,85; 1,11; 0,66; 0,76 a
3,23 hodín. Kapecitabín a jeho metabolity sa vylučujú prevažne do moču, pričom 95,5 % z podanej
dávky kapecitabínu sa zachytilo v moči. Vylučovanie stolicou je minimálne (2,6 %). Hlavný metabolit
vylučovaný močom je FBAL, jeho podiel predstavuje 57 % z podanej dávky. Približne 3 % z podanej dávky sa vylučujú močom v nezmenenej forme.
Kombinovanáliečba
Štúdie I. fázy hodnotiace vplyv kapecitabínu na farmakokinetiku docetaxelu a paklitaxelu a opačne
nepreukázali žiadny vplyv kapecitabínu na farmakokinetiku docetaxelu alebo paklitaxelu (Cmax a
AUC) a žiadny efekt docetaxelu a paklitaxelu na farmakokinetiku 5´-DFUR.
Farmakokinetikavosobitnýchpopuláciách
V populácii 505 pacientov s kolorektálnym karcinómom dostávajúcich kapecitabín v dávke
1 250 mg/m2 dvakrát denne sa analyzovali farmakokinetické parametre. Pohlavie, prítomnosť alebo chýbanie metastáz v pečeni v čase zaradenia do klinickej štúdie, stav výkonnosti podľa Karnofského,
celkový bilirubín, sérový albumín, ani hodnoty AST a ALT nemali štatisticky významný vplyv na farmakokinetiku 5'-DFUR, 5-FU a FBAL.
Pacientisporuchoufunkciepečenevdôsledkumetastázvpečeni.
Vo farmakokinetickej štúdii, ktorá zahŕňala onkologických pacientov s miernou až stredne závažnou poruchou funkcie pečene v dôsledku metastáz v tomto orgáne sa ukázalo, že biologická dostupnosť kapecitabínu a expozícia 5-FU sa môže zvýšiť v porovnaní s pacientmi bez poruchy funkcie pečene. O pacientoch so závažnou hepatálnou dysfunkciou nie sú k dispozícii žiadne farmakokinetické údaje.
Pacientisporuchoufunkcieobličiek
Vo farmakokinetickej štúdii, ktorá zahŕňala onkologických pacientov s miernou až závažnou poruchou funkcie obličiek sa nezistil žiadny vplyv klírensu kreatinínu na farmakokinetiku nezmeneného lieku a
5-FU. Zistilo sa, že klírens kreatinínu ovplyvňuje systémovú expozíciu 5'-DFUR (zníženie klírensu kreatinínu o 50 % viedlo k zvýšeniu AUC o 35 %) a FBAL (zníženie klírensu kreatinínu o 50 %
viedlo k zvýšeniu AUC o 114 %). FBAL je metabolit, ktorý nemá žiadnu antiproliferačnú aktivitu.
Staršíľudia
Pri analýze farmakokinetických parametrov v populácii zahŕňajúcej pacientov z rozličných vekových skupín (27 – 86 rokov), pričom počet pacientov s vekom ≥ 65 rokov bol 234 (46 %), sa ukázalo, že vek nemal žiadny vplyv na farmakokinetiku 5'-DFUR a 5-FU. Hodnota AUC pre FBAL sa zvyšovala s vekom (zvýšenie veku o 20 % viedlo k 15 % zvýšeniu hodnoty AUC pre FBAL). Toto zvýšenie je pravdepodobne dôsledkom zmeny funkcie obličiek.
Etnické faktory
Po orálnom podaní 825 mg/m2 kapecitabínu dvakrát denne počas 14 dní mali japonskí pacienti (n=18) o 36 % nižšiu Cmax a o 24 % nižšiu AUC kapecitabínu než kaukažskí pacienti (n=22). Japonskí pacienti mali tiež o 25 % nižšiu Cmax a o 34 % nižšiu AUC FBAL než kaukažskí pacienti. Klinická
relevancia týchto rozdielov je neznáma. Rozdiely v ostatných metabolitoch (5´-DFCR, 5´-DFUR a 5- FU) sa nevyskytovali.
5.3 Predklinické údaje o bezpečnosti
V štúdiách toxicity opakovanej dávky sa po dennom perorálnom podávaní kapecitabínu opiciam cynomolgus a myšiam zistili toxické účinky na gastrointestinálny, lymfatický a hemopoetický systém, ktoré sú typické pre fluórpyrimidíny. Tieto toxické prejavy boli reverzibilné. Pri liečbe kapecitabínom sa pozorovali prejavy kožnej toxicity charakterizované degeneratívnymi/regresívnymi zmenami. Kapecitabín nemal v štúdiách žiadne toxické účinky na pečeň a centrálny nervový systém. U opíc cynomolgus sa po intravenóznom podaní kapecitabínu (100 mg/kg) zistili známky kardiovaskulárnej toxicity (napr. predĺženie intervalov PR a QT), hoci pri opakovanom perorálnom podávaní kapecitabínu (1 379 mg/m2 denne) tieto známky neboli prítomné.
V 2-ročnej štúdii, ktorá na myšiach skúmala karcinogénne vlastnosti kapecitabínu, sa nepotvrdil karcinogénny účinok tejto látky.
Počas štandardných štúdií, ktoré skúmali vplyv kapecitabínu na fertilitu u samíc myší, sa pozorovala porucha plodnosti, avšak tento účinok bol reverzibilný po vysadení lieku po určitom období neužívania kapecitabínu. Okrem toho sa v 13-týždňovej štúdii zistili atrofické a degeneratívne zmeny na reprodukčných orgánoch samcov myší, avšak aj tieto zmeny ustúpili po určitom období bez liečby (pozri časť 4.6).
V štúdiách, ktoré skúmali embryotoxické a teratogénne účinky na myšiach, sa pozorovalo na dávke závislé zvýšenie počtu rezorpcií plodov a teratogenity. Po podávaní lieku vo vysokých dávkach sa u opíc pozorovali potraty a úmrtia zárodkov, avšak žiadne známky teratogenity.
Kapecitabín nemal mutagénne účinky v in vitro štúdiách s baktériami (Amesov test) alebo cicavčími bunkami (test génovej mutácie na čínskych škrečkoch V79/HPRT). Avšak podobne ako pri iných analógoch nukleozidov (t.j. 5-FU), kapecitabín mal klastogénne účinky v ľudských lymfocytoch (in vitro), pričom pozitívny trend sa taktiež zaznamenal v mikronukleus teste kostnej drene myší (in vivo).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety laktóza,
mikrokryštalická celulóza,
hypromelóza,
sodná soľ kroskarmelózy, magnéziumstearát.
Obaltablety Mastenec (400) hypromelóza,
dioxid titaničitý (E171), žltý oxid železitý (E172),
červený oxid železitý (E172).
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30°C.
Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia
CapecitabineTeva150mgfilmomobalenétablety
PVC/PE/PVDC – Alumíniový blister obsahujúci 10 filmom obalených tabliet. Každé balenie obsahuje
60 tabliet.
CapecitabineTeva500mgfilmomobalenétablety
PVC/PE/PVDC – Alumíniový blister obsahujúci 10 filmom obalených tabliet. Každé balenie obsahuje
120 tabliet.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIITeva B.V. Swensweg 5
2031 GA Haarlem
Holandsko
8. REGISTRAČNÉ ČÍSLACapecitabineTeva150mgfilmomobalenétabletyEU/1/12/761/001
CapecitabineTeva500mgfilmomobalenétabletyEU/1/12/761/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 20. apríl 2012
Dátum posledného predĺženia registrácie: 9. január 2017
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.