
boli pozorované žiadne bezpečnostné riziká. Neprejavil sa žiadny aditívny účinok
brivaracetamu a valproátu na AUC pri epoxide karbamazepínu.
Perorálne kontraceptíva
Súčasné podávanie brivaracetamu (100 mg/deň) spolu s perorálnym kontraceptívom obsahujúcim
etinylestradiol (0,03 mg) a levonorgestrel (0,15 mg) neovplyvňovalo farmakokinetiku žiadnej látky. Keď bol brivaracetam v dávke 400 mg/deň (dvojnásobok odporúčanej najvyššej dennej dávky) súčasne podávaný s perorálnym kontraceptívom obsahujúcim etinylestradiol (0,03 mg) a levonorgestrel (0,15 mg), bolo pozorované zníženie AUC estrogénu o 27 % a zníženie AUC progestínu o 23 %, a to bez dopadu na potlačenie ovulácie. Všeobecne nenastala žiadna zmena
v profiloch koncentrácií v čase u endogénnych markerov estradiolu, progesterónu, luteinizačného
hormónu (LH), folikuly stimulujúceho hormónu a globulínu viažúceho pohlavné hormóny (SHBG).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Lekári by mali so ženami vo fertilnom veku, ktoré užívajú brivaracetam (pozri Gravidita)
prediskutovať plánované rodičovstvo a antikoncepciu.
Ak sa žena rozhodne otehotnieť, užívanie brivaracetamu sa má dôkladne prehodnotiť.
Gravidita
Riziko spojené s epilepsiou a s antiepileptikami všeobecne
Pre všetky antiepileptiká bolo preukázané, že u potomkov liečených žien s epilepsiou je prevalencia malformácií dvakrát až trikrát vyššia, ako je približne 3% výskyt malformácií v bežnej populácii.
V liečenej populácii bol pozorovaný nárast malformácií pri polyterapii, ale rozsah, za ktorý zodpovedá liečba a/alebo základné ochorenie, nebol objasnený. Prerušenie antiepileptickej liečby
môže viesť k exacerbácii ochorenia, ktoré môže poškodiť matku a plod.
Riziko spojené s brivaracetamom
K dispozícii je iba obmedzené množstvo údajov o použití brivaracetamu u tehotných žien. Nie sú k dispozícii žiadne údaje o placentárnom transfere u ľudí, ale brivaracetam rýchlo prechádza
placentou u potkanov (pozri časť 5.3). Potenciálne riziko u ľudí nie je známe. Štúdie na zvieratách nepreukázali žiadny teratogénny potenciál brivaracetamu (pozri časť 5.3).
Brivaracetam bol použitý v klinických štúdiách ako prídavná liečba a keď bol používaný spoločne s karbamazepínom, viedol k nárastu koncentrácie aktívneho metabolitu karbamazepín-epoxidu závislého na dávke (pozri časť 4.5). Nie sú k dispozícii dostatočné údaje na určenie klinického významu tohto účinku v tehotenstve.
Z preventívnych dôvodov sa brivaracetam nemá užívať počas tehotenstva, pokiaľ to nie je klinicky nevyhnutné (v prípade, ak prínos pre matku jednoznačne prevyšuje potenciálne riziko pre plod).
Dojčenie
Nie je známe, či sa u ľudí brivaracetam vylučuje do materského mlieka. Štúdie na potkanoch preukázali vylučovanie brivaracetamu do materského mlieka (pozri časť 5.3). Je potrebné sa rozhodnúť, či prerušiť dojčenie alebo prerušiť podávanie brivaracetamu, pričom je potrebné zhodnotiť prínos lieku pre matku. V prípade súčasného podávania brivaracetamu a karbamazepínu by sa mohlo množstvo karbamazepín-epoxidu vylučovaného do materského mlieka zvýšiť. Nie sú k dispozícii dostatočné údaje na stanovenie klinického významu.
Fertilita
Nie sú k dispozícii žiadne údaje o účinku brivaracetamu na fertilitu u ľudí. U potkanov nebol pri liečbe brivaracetamom pozorovaný žiadny účinok na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Brivaracetam má zanedbateľný alebo malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Vzhľadom na možnú rozdielnu individuálnu citlivosť môžu niektorí pacienti pociťovať somnolenciu, závrat alebo iné príznaky súvisiace s centrálnym nervovým systémom (CNS). Pacienti majú byť poučení, aby neviedli vozidlá a neobsluhovali iné potenciálne nebezpečné stroje, kým sa neoboznámia s účinkami brivaracetamu na svoju schopnosťvykonávať tieto činnosti.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Vo všetkých kontrolovaných a nekontrolovaných štúdiách u pacientov s epilepsiou dostávalo
brivaracetam 2 388 subjektov, z ktorých 1 740 bolo liečených ≥6 mesiacov, 1 363 ≥12 mesiacov, 923
≥24 mesiacov a 569 ≥60 mesiacov (5 rokov).
Najčastejšie hlásené nežiaduce účinky (>10%) pri liečbe brivaracetamom boli somnolencia (14,3 %) a závrat (11,0 %). Boli miernej až strednej intenzity. Somnolencia a únava (8,2 %) boli hlásené vo vyššej miere pri zvyšujúcej sa dávke. Typy nežiaducich účinkov hlásených v priebehu prvých 7 dní liečby sa podobali tým, ktoré boli hlásené počas celkovej doby liečby.
Frekvencia ukončenia liečby z dôvodu nežiaducich reakcií bola 3,5 %, 3,4 % a 4,0 % u pacientov randomizovaných na užívanie brivaracetamu v príslušnej dávke 50 mg/deň, 100 mg/deň a
200 mg/deň, a 1,7 % u pacientov randomizovaných na užívanie placeba. Nežiaducim účinkom, ktorý viedol najčastejšie k ukončeniu liečby brivaracetamom, boli závrat (0,8 %) a kŕč (0,8 %).
Zoznam nežiaducich účinkov zoradených do tabuľky
V nasledujúcej tabuľke sú uvedené nežiaduce účinky, ktoré boli identifikované na základe prehľadu
úplnej bezpečnostnej databázy klinických štúdií brivaracetamu, podľa tried orgánových systémov a
frekvencie.
Frekvencie sú definované nasledovne: veľmi časté: (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100). V každej skupine frekvencií sú nežiaduce účinky zoradené v poradí podľa klesajúcej závažnosti.
Trieda orgánových systémov Frekvencia Nežiaduce účinky z klinických štúdií
Infekcie a nákazy časté chrípka
Poruchy krvi a lymfatického systému
menej časté neutropénia
Poruchy metabolizmu a výživy časté znížená chuť do jedla
Poruchy imunitného systému menej časté hypersenzitívne reakcie typu I
Psychické poruchy časté depresia, anxieta, insomnia, iritabilita
menej časté samovražedné myšlienky, psychotická
porucha, agresivita, agitovanosť
Poruchy nervového systému veľmi časté závrat, somnolencia
časté kŕč, vertigo
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinálneho traktu
Celkové poruchy a reakcie v mieste podania
časté infekcia horných ciest dýchacích, kašeľ časté nauzea, vracanie, zápcha
časté únava
Popis vybraných nežiaducich reakcií
Neutropénia bola hlásená u 0,5 % (6/1 099) pacientov s brivaracetamom a u 0 % (0/459) pacientov
s placebom. Štyria z týchto subjektov mali znížený počet neutrofilov vo východiskovom stave a po zahájení liečby brivaracetamom došlo k ďalšiemu zníženiu počtu neutrofilov. Žiadny z týchto
6 prípadov neutropénie nebol závažný, nevyžadoval špecifickú liečbu, ani neviedol k ukončeniu liečby brivaracetamom a žiadny nemal pridružené infekcie.
Samovražedné myšlienky boli hlásené u 0,3 % (3/1 099) pacientov s brivaracetamom a u 0,7 % (3/459) pacientov s placebom. V krátkodobých klinických štúdiách s brivaracetamom u pacientov s epilepsiou nedošlo k žiadnemu prípadu dokonanej samovraždy a k pokusu o samovraždu, avšak obidve boli hlásené v otvorených predĺžených štúdiách (pozri časť 4.4).
V priebehu klinického vývoja boli u malého počtu pacientov s brivaracetamom (9/3022) hlásené reakcie pripomínajúce hypersenzitívne reakcie typu I.
Otvorené predĺžené štúdie
U pacientov, ktorí boli sledovaní v otvorených predĺžených štúdiách po dobu až 8 rokov, bol
bezpečnostný profil podobný profilu pozorovanému v krátkodobých placebom kontrolovaných štúdiách.
Pediatrická populácia
K dispozícii sú obmedzené bezpečnostné údaje z otvorených štúdií u detí vo veku od 1 mesiaca do
<16 rokov. Celkovo 152 detí (1 mesiac až <16 rokov) bolo liečených brivaracetamom vo
farmakokinetickej štúdii a v súvisiacej nadväzujúcej štúdii. Z obmedzených dostupných údajov
najčastejšie hlásenými TEAEs (nežiaduce účinky spojené s liečbou) považovanými za súvisiace so skúšaným liekom boli somnolencia (10 %), znížená chuť do jedla (8 %), únava (5 %) a zníženie telesnej hmotnosti (5 %). Bezpečnostný profil sa zdá byť v súlade s bezpečnostným profilom známym u dospelých. V súčasnosti nie sú k dispozícii žiadne klinické údaje u novorodencov.
Starší pacientiZo 130 starších subjektov zahrnutých do fázy 2/3 vývojového programu brivaracetamu (44
s epilepsiou) bolo 100 vo veku 65-74 rokov a 30 vo veku 75-84 rokov. Bezpečnostný profil u starších pacientov sa zdá byť podobný ako je bezpečnostný profil pozorovaný u mladších dospelých pacientov.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSymptómyK dispozícii sú obmedzené klinické skúsenosti s predávkovaním brivaracetamu u ľudí. U zdravých
subjektov, ktorí užili jednotlivú dávku 1 400 mg brivaracetamu, bola hlásená somnolencia a závrat.
Liečba predávkovaniaNie je k dispozícii žiadne špecifické antidotum na predávkovanie brivaracetamom. Liečba
predávkovania zahŕňa všeobecné podporné opatrenia. Keďže sa močom vylučuje menej ako 10 %
brivaracetamu, neočakáva sa, že by hemodialýza významne zvýšila klírens brivaracetamu (pozri časť
5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antiepileptiká, iné antiepileptiká, ATC kód: N03AX23
Mechanizmus účinkuBrivaracetam vykazuje vysokú a selektívnu afinitu k 2A proteínu synaptických vezikúl (SV2A),
transmembránový glykoproteín bol nájdený na presynaptickej úrovni v neurónoch a v endokrinných bunkách. Hoci presnú úlohu tohto proteínu je potrebné ešte objasniť, bolo preukázané, že moduluje exocytózu neurotransmiterov. Predpokladá sa, že väzba na SV2A predstavuje primárny mechanizmus antikonvulzívnej aktivity brivaracetamu.
Klinická účinnosť a bezpečnosťÚčinnosť brivaracetamu v prídavnej terapii parciálnych záchvatov (partial onset seisures-POS) bola
stanovená v 3 randomizovaných dvojito zaslepených, placebom kontrolovaných multicentrických štúdiách s fixnou dávkou u subjektov vo veku 16 rokov a starších. Denná dávka brivaracetamu sa
v týchto štúdiách pohybovala v rozmedzí 5 až 200 mg/deň. Všetky štúdie začínali základnou periódou trvajúcou 8 týždňov, nasledovanou 12 týždňov trvajúcou liečebnou periódou bez titrácie v zmysle zvyšovania dávky. 1 558 pacientov dostávalo liek zo štúdie, z toho 1 099 dostávalo brivaracetam. Kritériá pre zaradenie do štúdie vyžadovali, aby mali pacienti nekontrolované parciálne záchvaty
napriek liečbe buď 1 alebo 2 súčasne podávanými antiepileptikami. Podmienkou bolo, aby pacienti prekonali najmenej 8 parciálnych záchvatov počas základnej periódy. Primárnymi koncovými ukazovateľmi pri štúdii fázy 3 bolo percento zníženia frekvencie POS oproti placebu a pomer respondérov s dosiahnutou 50 % odpoveďou založenou na 50 % znížení frekvencie POS od východiskového stavu.
Najčastejšie užívanými antiepileptikami na začiatku štúdie boli karbamazepín (40,6 %), lamotrigín (25,2 %), valproát (20,5 %), oxkarbazepín (16,0 %), topiramát (13,5 %), fenytoín (10,2 %) a levetiracetam (9,8 %). Medián východiskovej frekvencie záchvatov vo všetkých 3 štúdiách bol
9 záchvatov v priebehu 28 dní. Pacienti mali epilepsiu v priemere približne 23 rokov.
Výsledky účinnosti sú zhrnuté v tabuľke 2. Celkovo bol brivaracetam účinný pri prídavnej terapii
parciálnych záchvatov u pacientov vo veku 16 rokov a starších v dávke medzi 50 mg/deň a
200 mg/deň.
Tabuľka 2: Kľúčové výsledky účinnosti pre frekvenciu parciálnych záchvatov v priebehu 28 dní
Štúdia Placebo Brivaracetam
*štatisticky významné (hodnota p)
50 mg/deň 100 mg/deň 200 mg/deň
Štúdia N01253(1)
n = 96 n = 101
Dosiahnutie 50% odpovede respondéra 16,7 32,7* ~ ~
(p=0,008)
Percentuálne zníženie voči placebu (%) NA 22,0* ~ ~
(p=0,0040)
Štúdia N01252(1)
n = 100 n = 99 n = 100
Dosiahnutie 50% odpovede respondéra 20,0 27,3
(p=0,372)
Percentuálne zníženie voči placebu (%) NA 9,2
(p=0,0274)
36,0(2) ~
(p=0,023)
20,5(2) ~
(p=0,0097)
Štúdia N01358
n = 259 n = 252 n = 249
Dosiahnutie 50% odpovede respondéra 21,6 ~ 38,9
(p<0,001)
37,8
(p<0,001)
Percentuálne zníženie voči placebu (%) Medián percentuálneho zníženia oproti východiskovému stavu
NA ~ 22,8* (p<0,001)
23,2* (p<0,001)
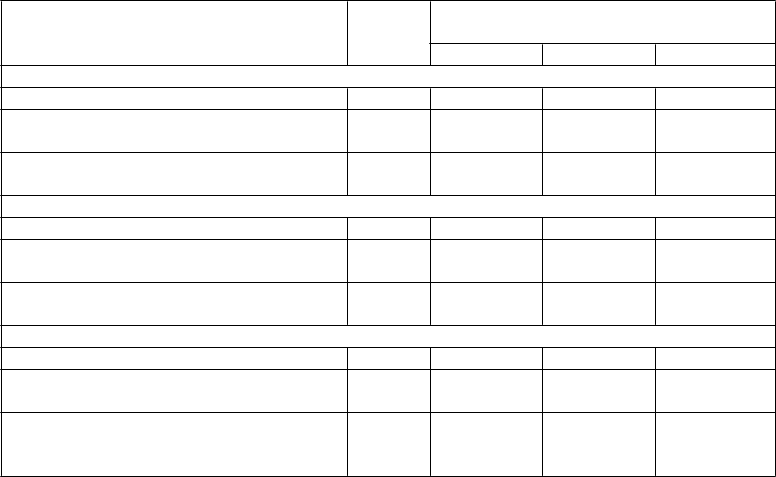
n = randomizovaní pacienti, ktorí dostali najmenej 1 dávku liečiva v štúdii
~ dávka nebola študovaná
* štatisticky významné
(1) Približne 20 % pacientov dostávalo súčasne levetiracetam
(2) Primárny výsledok pre N01252 nedosiahol štatistickú významnosť na základe sekvenčného
skúšania. Dávka 100 mg/deň bola nominálne významná.
V klinických štúdiách bolo zníženie frekvencie záchvatov vyššie oproti placebu pri dávke 100 mg/deň ako pri dávke 50 mg/deň. Na rozdiel od zvýšenia výskytu somnolencie a únavy v závislosti na dávke, mal brivaracetam pri dávke 50 mg/deň a 100 mg/deň podobný bezpečnostný profil vrátane
nežiaducich účinkov so vzťahom k CNS a pri dlhodobom užívaní.
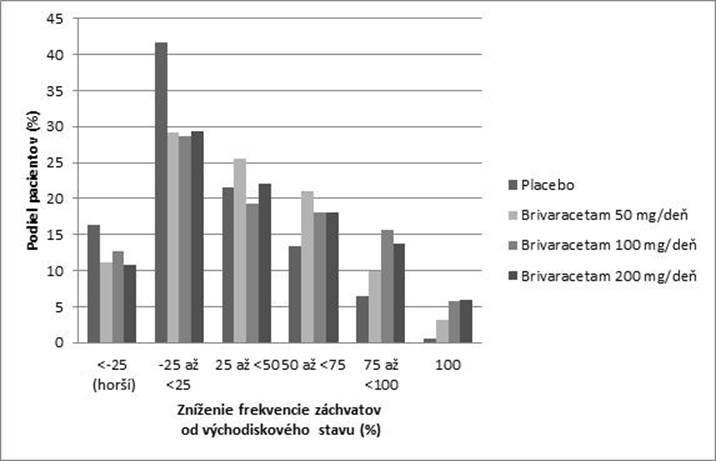
Obrázok 1 ukazuje percento pacientov (s výnimkou pacientov súčasne užívajúcich levetiracetam)
podľa kategórie zníženia frekvencie POS v priebehu 28 dní od východiskového stavu vo všetkých
3 štúdiách. Pacienti s viac ako 25 % zvýšením parciálnych záchvatov sú uvedení úplne naľavo ako
„horší“. Pacienti so zlepšením percentuálneho zníženia frekvencie POS od východiskového stavu sú uvedení v 4 kategóriách napravo. Percento pacientov s najmenej 50 % znížením frekvencie záchvatov bolo 20,3 %, 34,2 %, 39,5 %, a 37,8 % pre placebo, zodpovedajúce 50 mg/deň, 100 mg/deň a
200 mg/deň.
Obrázok 1: Podiel pacientov s brivaracetamom a placebom podľa kategórie odpovedi záchvatov
p
o dobu 12 týždňov vo všetkých troch dvojito zaslepených pivotných štúdiách
V súhrnnej analýze troch pivotných štúdií neboli pozorované žiadne rozdiely v účinnosti (merané ako
50 % odpoveď respondérov) v rozmedzí dávok 50 mg/deň až 200 mg/deň, keď je brivaracetam
kombinovaný s antiepileptikami vyvolávajúcimi alebo nevyvolávajúcimi indukciu enzýmov.
V klinických štúdiách dosiahlo stav bez záchvatov 2,5 % (4/161), 5,1 % (17/332) a 4,0 % (10/249)
pacientov s brivaracetamom v dávke zodpovedajúcej 50 mg/deň, 100 mg/deň a 200 mg/deň a to
v priebehu liečebnej periódy v trvaní 12 týždňov v porovnaní s 0,5 % (2/418) pacientov s placebom. Zlepšenie mediánu percentuálneho zníženia frekvencie záchvatov od začiatku liečby za 28 dní bolo pozorované u pacientov s typom záchvatov IC (sekundárne generalizované tonicko-klonické
záchvaty) vo východiskovom stave, liečených brivaracetamom (66,6 % (n=62), 61,2% (n=100) a 82,1
% (n=75) z pacientov s brivaracetamom v zodpovedajúcej dávke 50 mg/deň, 100 mg/deň a
200 mg/deň v porovnaní s placebom 33,3 % (n=115)).
Účinnosť brivaracetamu v monoterapii nebola ešte stanovená. Použitie brivaracetamu v monoterapii
sa neodporúča.
Liečba levetiracetamomV 2 randomizovaných placebom kontrolovaných štúdiách fázy 3 sa levetiracetam podával ako
súbežné antiepileptikum u asi 20 % pacientov. Aj keď je počet subjektov limitovaný, nebol
u pacientov, ktorí súčasne užívali levetiracetam, pozorovaný žiadny prínos brivaracetamu oproti placebu, ktorý by reflektoval kompetíciu vo väzbovom mieste SVA2. Neboli zistené žiadne ďalšie okolnosti týkajúce sa bezpečnosti a znášanlivosti.
V 3. štúdii vopred špecifikovaná analýza preukázala účinnosť oproti placebu pre dávky 100 mg/deň a
200 mg/deň u pacientov predtým užívajúcich levetiracetam. Nižšia účinnosť pozorovaná u týchto pacientov v porovnaní s pacientmi neužívajúcimi levetiracetam bola pravdepodobne dôsledkom užívania vyššieho počtu predchádzajúcich antiepileptík a vyššej východiskovej hodnoty frekvencie záchvatov.
Starší pacienti (65 rokov a starší)Tri pivotné, dvojito zaslepené, placebom kontrolované štúdie zahŕňali 38 pacientov vo veku 65 až
80 rokov. Aj keď sú údaje obmedzené, účinnosť bola porovnateľná s účinnosťou u mladších
subjektov.
Otvorené predĺžené štúdie
Vo všetkých štúdiách bolo zaradených do dlhodobých, otvorených, predĺžených štúdií 81,7 %
pacientov, ktorí dokončili randomizované štúdie. Od vstupu do randomizovaných štúdií bolo 5,3 % subjektov s brivaracetamom po dobu 6 mesiacov (n=1500) bez záchvatov v porovnaní s 4,6 % a 3,7 % u subjektov exponovaných po dobu 12 mesiacov (n=1188) a 24 mesiacov (n=847). Nakoľko však vysoké percento subjektov (26 %) prerušilo liečbu v otvorených štúdiách z dôvodu nedostatočnej účinnosti, mohlo dôjsť ku skresleniu, nakoľko subjekty, ktoré zostali v štúdii, reagovali lepšie ako tí, ktorí ju predčasne ukončili.
Pediatrická populácia
Účinnosť a znášanlivosť brivaracetamu nebola u pediatrických pacientov stanovená (pozri časť 4.2).
Brivaracetam bol hodnotený u týchto pacientov v krátkodobej otvorenej farmakokinetickej štúdií a prebiehajúcej otvorenej, predĺženej štúdií u 152 subjektov vo veku od 1 mesiaca do 16 rokov (pozri časť 5.2).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s brivaracetamom v jednej alebo vo viacerých podskupinách pediatrickej populácie s epilepsiou s parciálnymi záchvatmi.
5.2 Farmakokinetické vlastnosti
Brivaracetam filmom obalené tablety, perorálny roztok a roztok na intravenóznu injekciu vykazujú identickú AUC, zatiaľ čo maximálna plazmatická koncentrácia je mierne vyššia po intravenóznom podaní. Brivaracetam vykazuje lineárnu a na čase nezávislú farmakokinetiku s nízkou intra- a interindividuálnou variabilitou a ďalej úplnou absorpciou, nízku väzbu na proteíny, renálnu exkréciu po rozsiahlej biotransformácii a farmakologicky inaktívne metabolity.
Absorpcia
Brivaracetam sa rýchlo a úplne absorbuje po perorálnom podaní a absolútna biologická dostupnosť je
približne 100 %. Medián tmax pre tablety užité bez jedla je 1 hodina (rozsah tmax je 0,25 až 3 hod.).
Súčasné podávanie s jedlom s vysokým obsahom tuku spomalilo rýchlosť absorpcie (medián tmax 3 h) a znížilo maximálnu plazmatickú koncentráciu (37 % pokles) brivaracetamu, pričom rozsah absorpcie zostal nezmenený.
Distribúcia
Brivaracetam sa slabo viaže (≤ 20 %) na plazmatické proteíny. Distribučný objem je 0,5 l/kg, čo je
hodnota blízka celkovému množstvu telesnej vody.
Bunkové membrány sú pre brivaracetam vysoko permeabilné z dôvodu jeho lipofílie (log P).
Biotransformácia
Brivaracetam je primárne metabolizovaný hydrolýzou svojej amidovej časti za vzniku zodpovedajúcej
karboxylovej kyseliny (približne 60 % eliminácie) a sekundárne hydroxyláciou propylového vedľajšieho reťazca (približne 30 % eliminácie). Hydrolýza amidovej časti , ktorá vedie k vzniku metabolitu povahy karboxylovej kyseliny (34 % dávky v moči) je podporovaná pečeňovou a mimopečeňovou amidázou. In vitro je hydroxylácia brivaracetamu sprostredkovaná v prvom rade CYP2C19. Obidva metabolity sú ďalej metabolizované za vzniku bežnej hydroxylovanej kyseliny, ktorá vzniká prevažne hydroxyláciou postranného propylového reťazca metabolitu kyseliny
karboxylovej (hlavne prostredníctvom CYP2C9). In vivo u ľudí s neúčinnou mutáciou CYP2C19, sa
tvorba hydroxymetabolitu znižuje 10x, zatiaľ čo samotný brivaracetam sa zvyšuje o 22 % alebo 42 %
u jedincov s jednou alebo s obidvomi mutovanými alelami. Tri metabolity nie sú farmakologicky
aktívne.
Eliminácia
Brivaracetam je primárne eliminovaný metabolizáciou a vylučovaním močom. Viac ako 95 % dávky,
vrátane metabolitov, sa vylučuje močom v priebehu 72 hodín po užití. Menej ako 1 % dávky sa vylučuje stolicou a menej ako 10 % brivaracetamu sa vylučuje bez zmeny močom. Terminálny plazmatický polčas (t1/2) je približne 9 hodín. Celkový plazmatický klírens bol u pacientov odhadnutý na 3,6 l/hod.
Linearita
Farmakokinetika je úmerná dávke od 10 mg do najmenej 600 mg.
Interakcie s liekmi
Brivaracetam je eliminovaný viacerými cestami, vrátane vylučovania obličkami, na CYP nezávislou
hydrolýzou a CYP sprostredkovanou oxidáciou. In vitro, brivaracetam nie je substrátom ľudského P- glykoproteínu (P-gp), proteínom rezistentným na viacpočetné lieky (MRP – multidrug resistance proteins) 1 a 2 a pravdepodobne ani organickým transportérom aniónu polypeptidu 1B1 (OATP1B1) a OATP1B3. Testy in vitro ukázali, že metabolizmus brivaracetamu by nemal byť významne ovplyvnený CYP inhibítormi (napr. CYP1A, 2C8, 2C9, 2C19, 2D6 a 3A4).
In vitro, brivaracetam nebol inhibítorom CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 3A4 ani transportérom P-gp, BCRP, BSEP MRP2, MATE-K, MATE-1, OATP1B1, OATP1B3, OAT1 a OCT1 v klinicky relevantných koncentráciách. In vitro, brivaracetam neindukoval CYP1A2.
Farmakokinetika u osobitných skupín pacientov
Starší pacienti (65 rokov a starší)
V štúdii u starších pacientov (vo veku 65 až 79 rokov; s klírensom kreatinínu 53 až 98 ml/min/1,73 m2), ktorí užívali brivaracetam v dávke 400 mg/deň s podávaním 2x denne, bol plazmatický polčas brivaracetamu 7,9 hodiny u skupiny vo veku 65 až 75 rokov a 9,3 hodiny u skupiny >75 rokov. Plazmatický klírens rovnovážneho stavu brivaracetamu bol podobný (0,76 ml/min/kg) ako u mladých zdravých mužov (0,83 ml/min/kg) (pozri časť 4.2).
Porucha funkcie obličiek
Štúdia u subjektov so závažnou poruchou funkcie obličiek (klírens kreatinínu <30 ml/min/1,73 m2 bez nutnosti dialýzy) odhalila, že plazmatická AUC brivaracetamu bola stredne zvýšená (+21%) voči zdravým subjektom, zatiaľ čo AUC kyseliny, hydroxymetabolitu a metabolitu hydroxykyseliny boli zvýšené 3x, 4x, a 21x (v uvedenom poradí). Renálny klírens týchto neaktívnych metabolitov bol znížený 10x. Metabolit hydroxykyseliny v predklinických štúdiách nevyvolal žiadne obavy zo strany bezpečnosti. Brivaracetam nebol študovaný u pacientov liečených hemodialýzou (pozri časť 4.2).
Porucha funkcie pečene
Farmakokinetická štúdia u subjektov s cirhózou pečene (Child-Pugh triedy A, B, a C) preukázala podobné zvýšenie pri expozícii brivaracetamu bez ohľadu na závažnosť ochorenia (50 %, 57 %
a 59 %) v pomere k zodpovedajúcim zdravým subjektom (pozri časť 4.2).
Pediatrická populácia
Vo farmakokinetickej štúdii u 99 subjektov vo veku 1 mesiac až <16 rokov, ktorí dostávali brivaracetam perorálny roztok bolo preukázané, že plazmatické koncentrácie sú úmerné dávke vo všetkých vekových skupinách. Farmakokinetické populačné modelovanie ukázalo, že dávka
2,0 mg/kg dvakrát denne vedie
k rovnakej priemernej plazmatickej koncentrácii rovnovážneho stavu ako u dospelých užívajúcich
100 mg dvakrát denne.
Telesná hmotnosť
Bol odhadnutý 40 % pokles plazmatickej koncentrácie rovnovážneho stavu v rozsahu telesnej hmotnosti od 46 kg do 115 kg. To však nie je považované za klinicky významný rozdiel vo farmakokinetike brivaracetamu.
Pohlavie
Nie sú žiadne klinicky významné rozdiely vo farmakokinetike brivaracetamu medzi pohlaviami.
Rasa
Farmakokinetika brivaracetamu nebola významne ovplyvnená rasou (kaukazská, ázijská) pri
farmakokinetickom populačnom modelovaní u pacientov s epilepsiou. Počet pacientov s iným etnickým pôvodom bol obmedzený.
Farmakokinetický/farmakodynamický vzťah
EC50 (plazmatická koncentrácia brivaracetamu zodpovedajúca 50 % maximálneho účinku) bola
odhadnutá na 0,57 mg/l. Táto plazmatická koncentrácia je mierne nad mediánom expozície po podávaní brivaracetamu v dávkach 50 mg/deň. Ďalšie zníženie frekvencie záchvatov sa dostavuje pri zvýšení dávky na 100 mg/deň a dosahuje stabilizovaný stav pri dávke 200 mg/deň.
5.3 Predklinické údaje o bezpečnosti
Vo farmakologických štúdiách bezpečnosti mali prevládajúce účinky súvislosť s CNS (najmä prechodná depresia CNS a zníženie spontánnej pohybovej aktivity) a boli pozorované pri násobkoch (vyšších než 50x) farmakologicky účinnej dávky brivaracetamu 2 mg/kg. Brivaracetam neovplyvnil učenie a funkciu pamäte.
Nálezy, ktoré neboli pozorované v klinických štúdiách, ale boli pozorované v toxikologických štúdiách s opakovaným podávaním u psov pri expozícii podobnej ako pri klinickej plazmatickej AUC, boli hepatotoxické účinky (hlavne porfýria). Toxikologické údaje zhromaždené o brivaracetame a
o štrukturálne príbuzných látkach ale ukazujú, že sa pečeňové zmeny u psov vyvinuli prostredníctvom mechanizmov nerelevantných pre ľudí. Žiadne nežiaduce zmeny na pečeni neboli pozorované
u potkanov a opíc po dlhodobom podávaní brivaracetamu s expozíciou zreteľne prevyšujúcu AUC expozíciu 5 až 42x. CNS príznaky u opíc (vyčerpanosť, strata rovnováhy, nemotorné pohyby) sa vyskytli pri 64-násobkoch klinickej Cmax. Tieto účinky boli menej zrejmé v priebehu času.
Štúdie genotoxicity nepreukázali žiadnu mutagénnu alebo klastogénnu aktivitu. Štúdie kancerogenity u potkanov nepreukázali žiadny onkogénny potenciál, zatiaľ čo zvýšený výskyt hepatocelulárnych nádorov u samcov myší je považovaný za dôsledok známeho negenotoxického fenoménu, známeho
u hlodavcov, ktorého mechanizmus účinku sa vzťahuje k indukcii pečeňových enzýmov, podobnej ako po fenobarbitále.
Brivaracetam neovplyvnil fertilitu samíc ani samcov, nepreukázal sa žiadny teratogénny potenciál u potkanov alebo králikov. Embryotoxicita bola pozorovaná u králikov pri dávke brivaracetamu toxickej pre matky s expozíciou 8x vyššou než klinická AUC expozícia pri maximálnej odporúčanej dávke. U potkanov bolo preukázané, že brivaracetam ľahko prestupuje placentou a je vylučovaný do materského mlieka u dojčiacich samíc potkanov v koncentráciách podobných plazmatickým koncentráciám u matiek.
Brivaracetam u potkanov nepreukázal žiadny potenciál na závislosť. Štúdie u nedospelých zvierat
U nedospelých potkanov expozičné hladiny 6 -15 násobnej klinickej AUC expozície brivaracetamu
pri maximálnej odporúčanej dávke vyvolávali vývojové nežiaduce účinky (napr. mortalitu, klinické príznaky, zníženie telesnej hmotnosti a nižšiu hmotnosť mozgu). Neboli zaznamenané žiadne nežiaduce účinky na funkciu CNS, žiadne neuropatologické a histopatologické vyšetrenia mozgu.
U nedospelých psov boli brivaracetamom indukované zmeny pri dávke 100 mg/kg/deň spojené s 6- násobným zvýšením hladín AUC, podobným zmenám pozorovaným u dospelých zvierat. Neboli pozorované žiadne nežiaduce účinky v štandardných ukazovateľoch vývoja alebo maturácie.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
sodná soľ kroskarmelózy monohydrát laktózy betadex
bezvodá laktóza magnéziumstearát.
Filmotvorná vrstva
Briviact 10 mg filmom obalené tablety
polyvinylalkohol
oxid titaničitý (E171) makrogol 3350 mastenec.
Briviact 25 mg filmom obalené tablety polyvinylalkohol
oxid titaničitý (E171) makrogol 3350 mastenec
žltý oxid železitý (E172)
čierny oxid železitý (E172).
Briviact 50 mg filmom obalené tablety polyvinylalkohol
oxid titaničitý (E171) makrogol 3350 mastenec
žltý oxid železitý(E172)
červený oxid železitý(E172).
Briviact 75 mg filmom obalené tablety polyvinylalkohol
oxid titaničitý (E171) makrogol 3350 mastenec
žltý oxid železitý (E172) červený oxid železitý (E172) čierny oxid železitý (E172).
Briviact 100 mg filmom obalené tablety polyvinylalkohol
oxid titaničitý (E171) makrogol 3350 mastenec
žltý oxid železitý (E172)
čierny oxid železitý (E172).
6
.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
4 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Briviact 10 mg filmom obalené tablety
• Balenie obsahujúce 14, 56 filmom obalených tabliet a multibalenie obsahujúce 168 (3 balenie po 56) filmom obalených tabliet v PVC/PCTFE/Al blistroch
• Balenie 14 x 1 a 100 x 1 filmom obalená tableta v PVC/PCTFE/Al blistroch
Briviact 25 mg filmom obalené tablety
• Balenie obsahujúce 14, 56 filmom obalených tabliet a multibalenie obsahujúce 168 (3 balenia po 56) filmom obalených tabliet v PVC/PCTFE/Al blistroch
• Balenie 14 x 1 a 100 x 1 filmom obalená tableta v PVC/PCTFE/Al blistroch
Briviact 50 mg filmom obalené tablety
• Balenie obsahujúce 14, 56 filmom obalených tabliet a multibalenie obsahujúce 168 (3 balenia po 56) filmom obalených tabliet v PVC/PCTFE/Al blistroch
• Balenie 14 x 1 a 100 x 1 filmom obalená tableta v PVC/PCTFE/Al blistroch
Briviact 75 mg filmom obalené tablety
• Balenie obsahujúce 14, 56 filmom obalených tabliet a multibalenie obsahujúce 168 (3 balenia po 56) filmom obalených tabliet v PVC/PCTFE/Al blistroch
• Balenie 14 x 1 a 100 x 1 filmom obalená tableta v PVC/PCTFE/Al blistroch
Briviact 100 mg filmom obalené tablety
• Balenie obsahujúce 14, 56 filmom obalených tabliet a multibalenie obsahujúce 168 (3 balenia po 56) filmom obalených tabliet v PVC/PCTFE/Al blistroch
• Balenie 14 x 1 a 100 x 1 filmom obalená tableta v PVC/PCTFE/Al blistroch
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
UCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8
. REGISTRAČNÉ ČÍSLO(A)
EU/1/15/1073/001
EU/1/15/1073/002
EU/1/15/1073/003
EU/1/15/1073/004
EU/1/15/1073/005
EU/1/15/1073/006
EU/1/15/1073/007
EU/1/15/1073/008
EU/1/15/1073/009
EU/1/15/1073/010
EU/1/15/1073/011
EU/1/15/1073/012
EU/1/15/1073/013
EU/1/15/1073/014
EU/1/15/1073/015
EU/1/15/1073/016
EU/1/15/1073/017
EU/1/15/1073/018
EU/1/15/1073/019
EU/1/15/1073/020
EU/1/15/1073/023
EU/1/15/1073/024
EU/1/15/1073/025
EU/1/15/1073/026
EU/1/15/1073/027
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 14. január 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUBriviact 10 mg/ml perorálny roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždý ml obsahuje 10 mg brivaracetamu.
Pomocnélátkysoznámymúčinkom:
Každý ml perorálneho roztoku obsahuje 239,8 mg sorbitolu (E420), 1 mg metylparabénu (E218) a
1,16 mg sodíka.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAPerorálny roztok
Mierne viskózna, číra, bezfarebná až nažltlá tekutina.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieBriviact je indikovaný ako prídavná terapia pri liečbe parciálnych záchvatov s alebo bez sekundárnej
generalizácie u dospelých a dospievajúcich pacientov s epilepsiou vo veku od 16 rokov.
4.2 Dávkovanie a spôsob podávaniaDávkovanieOdporúčaná začiatočná dávka potrebná na zníženie počtu záchvatov je buď 50 mg/deň alebo 100
mg/deň, na základe posúdenia potenciálnych nežiaducich účinkov lekárom. Dávka sa má podávať v dvoch rovnakých rozdelených dávkach, jedenkrát ráno a jedenkrát večer. Na základe individuálnej odpovede
a znášanlivosti pacienta sa môže dávka upraviť v rozmedzí 50 mg/deň až 200 mg/deň.
Zabudnutá dávkaAk pacienti zabudnú užiť jednu alebo viac dávok, odporúča sa užiť jednu dávku ihneď, ako si
spomenú
a nasledujúcu dávku užiť vo zvyčajnom čase ráno alebo večer. To môže zabrániť poklesu plazmatickej koncentrácie brivaracetamu pod účinnú hladinu, a tým opätovnému prepuknutiu záchvatov.
Ukončenie liečbyAk sa má podávanie brivaracetamu ukončiť, odporúča sa postupné znižovanie o 50 mg/deň
v týždennom intervale. Po týždni liečby dávkou 50 mg/deň sa odporúča v poslednom týždni liečby
dávka 20 mg/deň.
Osobitné skupiny pacientovStarší pacienti (65 rokov a starší)Nie je potrebná žiadna úprava dávky u starších pacientov (pozri časť 5.2). Klinická skúsenosť u pacientov ≥65 rokov je obmedzená.
Porucha funkcie obličiekU pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávky (pozri časť 5.2). Vzhľadom na nedostatok údajov, pacientom v konečnom štádiu ochorenia obličiek, ktorí sú liečení
dialýzou, sa podávanie brivaracetamu neodporúča.
Porucha funkcie pečeneExpozícia brivaracetamu bola zvýšená u pacientov s chronickým ochorením pečene. Začiatočná
dávka 50 mg/deň sa má zvážiť. Vo všetkých štádiách poruchy funkcie pečene sa odporúča maximálna
denná dávka 150 mg, podávaná v 2 rozdelených dávkach (pozri časti 4.4 a 5.2).
Pediatrická populáciaBezpečnosť a účinnosť brivaracetamu u novorodencov a detí vo veku menej ako 16 rokov neboli doteraz stanovené.
V súčasnosti dostupné údaje sú uvedené v časti 4.8, 5.1, a 5.2, ale na ich základe sa nedajú urobiť žiadne odporúčania ohľadne dávkovania.
Spôsob podávaniaPerorálny roztok brivaracetamu sa môže krátko pred užitím riediť vodou alebo džúsom a môže sa
užívať s jedlom alebo bez jedla (pozri časť 5.2). Perorálny roztok brivaracetamu sa môže podávať
nazogastrickou alebo gastrostomickou sondou.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na iné deriváty pyrolidónu alebo na ktorúkoľvek z pomocných látok
uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaníSamovražedné myšlienky a správanieU pacientov liečených antiepileptikami (AEDs), vrátane brivaracetamu, boli pri niekoľkých
indikáciách hlásené samovražedné myšlienky a správanie. Meta-analýza randomizovaných, placebom kontrolovaných štúdií s antiepileptikami preukázala malé zvýšenie rizika samovražedných myšlienok a správania. Mechanizmus vzniku tohto rizika nie je známy a dostupné údaje nevylučujú možnosť zvýšeného rizika pre brivaracetam.
U pacientov sa majú sledovať príznaky samovražedných myšlienok a správania a má sa zvážiť
vhodná liečba. Pacientom (a ich opatrovateľom) sa má odporučiť, aby v prípade výskytu akýchkoľvek príznakov samovražedných myšlienok a správania okamžite vyhľadali lekársku pomoc.
Porucha funkcie pečeneK dispozícii sú obmedzené klinické údaje o použití brivaracetamu u pacientov s už existujúcou
poruchou funkcie pečene. U pacientov s poruchou funkcie pečene sa odporúča úprava dávky (pozri časť 4.2).
Pomocné látkyObsah sodíkaPerorálny roztok brivaracetamu obsahuje sodík. Toto je potrebné vziať do úvahy u pacientov
nastavených na diétny režim s kontrolovaným obsahom sodíka.
Intolerancia fruktózyPerorálny roztok obsahuje sorbitol (E420). Pacienti so zriedkavými dedičnými
problémami intolerancie fruktózy nemajú užívať tento liek.
Pomocné látky, ktoré môžu spôsobiť intoleranciuPerorálny roztok obsahuje metylparabén (E218), ktorý môže spôsobiť alergické reakcie
(pravdepodobne oneskorené).
4.5 Liekové a iné interakcieFormálne štúdie interakcie sa uskutočnili iba u dospelých.
Farmakodynamické interakcieSúčasná liečba s levetiracetamomV klinických štúdiách, aj keď ich počet bol obmedzený, nebol pozorovaný žiadny prínos brivaracetamu oproti placebu u pacientov súčasne užívajúcich levetiracetam. Neboli pozorované žiadne ďalšie obavy ohľadne bezpečnosti a znášanlivosti (pozri časť 5.1).
Interakcie s alkoholomV štúdii farmakokinetickej a farmakodynamickej interakcie medzi brivaracetamom v jednorazovej dávke 200 mg a etanolom v kontinuálnej infúzii 0,6 g/l u zdravých subjektov nenastala žiadna farmakokinetická interakcia ale brivaracetam približne zdvojnásobil účinky alkoholu na psychomotorické funkcie, pozornosť a pamäť. Podávanie brivaracetamu s alkoholom sa neodporúča.
Farmakokinetické interakcieÚčinky iných látok na farmakokinetiku brivaracetamuIn vitro údaje naznačujú, že brivaracetam má nízky interakčný potenciál. Hlavnou metabolickou cestou brivaracetamu je na CYP nezávislá hydrolýza. Druhá cesta zahŕňa hydroxyláciu, ktorá je
sprostredkovaná CYP2C19 (pozri časť 5.2).
Plazmatické koncentrácie brivaracetamu sa môžu zvýšiť, ak je súčasne podávaný so silnými inhibítormi CYP2C19 (ako flukonazol, fluvoxamín), ale riziko klinicky významnej interakcie sprostredkované CYP2C19 je považované za nízke.
RifampicínU zdravých dobrovoľníkov súčasné podávanie silného induktora enzýmov rifampicínu (600 mg/deň po dobu 5 dní) znížilo plochu brivaracetamu pod krivkou (AUC) o 45 %. Predpisujúci lekári musia zvážiť úpravu dávky brivaracetamu u pacientov, u ktorých sa zahajuje alebo ukončuje liečba rifampicínom.
Antiepileptiká so silnou indukciou enzýmovPlazmatické koncentrácie brivaracetamu klesajú pri súčasnom podávaní s antiepileptikami silne indukujúcimi enzýmy (karbamazepín, fenobarbital, fenytoín), nie je však potrebná žiadna úprava dávky (pozri tabuľka 1).
Iné induktory enzýmovOčakáva sa, že iné silné induktory enzýmov (ako ľubovník bodkovaný (Hypericum perforatum)), môžu tiež znížiť systémovú expozíciu brivaracetamu. Preto zahájenie alebo ukončenie liečby ľubovníkom bodkovaným sa má uskutočniť opatrne.
Účinok brivaracetamu na iné liekyBrivaracetam podávaný v dávkach 50 mg alebo 150 mg/deň neovplyvňoval AUC midazolamu
(metabolizovaný CYP3A4). Riziko klinicky relevantných CYP3A4 interakcií je považované za nízke.
Štúdie
in vitro preukázali, že brivaracetam vykazuje malú alebo žiadnu inhibíciu izoforiem CYP450, s
výnimkou CYP2C19. Brivaracetam môže zvyšovať plazmatické koncentrácie liečiv metabolizovaných CYP2C19 (napr. lanzoprazol, omeprazol, diazepam). Pri skúšaní
in vitro brivaracetam neindukoval CYP1A1/2, ale indukoval CYP3A4 a CYP2B6. Žiadna CYP3A4 indukcia nebola zistená
in vivo (pozri midazolam vyššie). CYP2B6 indukcia nebola skúmaná
in vivo a brivaracetam môže znižovať plazmatické koncentrácie liečiv metabolizovaných CYP2B6 (napr. efavirenz).
In vitro interakčné štúdie na určenie potenciálnych inhibičných účinkov na transportéry viedli k záveru, že nedochádza k žiadnym klinicky významným účinkom, s výnimkou OAT3.
In vitro brivaracetam inhibuje OAT3 s polovičnou maximálnou inhibičnou koncentráciou 42-krát vyššou ako Cmax pri najvyšších klinických dávkach. Brivaracetam v dávke 200 mg/deň môže zvýšiť plazmatické koncentrácie liekov transportovaných AOT3.
AntiepileptikáPotenciálne interakcie medzi brivaracetamom (50 mg/deň až 200 mg/deň) a inými antiepileptikami
boli skúmané v súhrnnej analýze plazmatických liekových koncentrácií zo všetkých štúdií fázy 2-3 a v populačnej farmakokinetickej analýze placebom kontrolovaných štúdií fázy 2-3 a vo vyhradených štúdiách liekových interakcií (pre nasledujúce antiepileptiká: karbamazepín, lamotrigín, fenytoín a topiramát). Účinok vzájomných interakcií na plazmatickú koncentráciu je zhrnutý v tabuľke 1 (zvýšenie je označené ako “↑” a zníženie ako “↓”, plocha pod krivkou plazmatickej koncentrácie
versus čas ako “AUC“, maximálna pozorovaná koncentrácia ako “Cmax“).
Tabuľka 1: Farmakokinetické interakcie medzi brivaracetamom a inými antiepileptikami
Súčasne podávané
a
ntiepiletikum
Vplyv antiepiletika na koncentráciu brivaracetamu v plazme
Vplyv brivaracetamu na koncentráciu antiepiletika v plazme
Karbamazepín AUC 29 % ↓
Cmax 13 % ↓
nie je potrebná žiadna úprava dávky
karbamazepín - žiadny
epoxid karbamazepínu ↑ (pozri
nižšie)
nie je potrebná žiadna úprava dávky
Klobazam žiadne údaje žiadny Klonazepam žiadne údaje žiadny Lakosamid žiadne údaje žiadny Lamotrigín žiadny žiadny Levetiracetam žiadny žiadny
Oxkarbazepín žiadny žiadny (monohydroxyderivát, MHD)
Fenobarbital AUC 19 % ↓
nie je potrebná žiadna úprava dávky
Fenytoín AUC 21 % ↓
nie je potrebná žiadna úprava dávky
žiadny
žiadny
a AUC 20 % ↑
a Cmax 20 % ↑

Pregabalín žiadne údaje žiadny Topiramát žiadny žiadny Kyselina valproová žiadny žiadny Zonisamid žiadne údaje žiadny
a na základe štúdie zahŕňajúcej podávanie superterapeutických dávok brivaracetamu 400 mg/deň
KarbamazepínBrivaracetam je stredne účinný reverzibilný inhibítor epoxidovej hydrolázy vyvolávajúci zvýšenú koncentráciu karbamazepín-epoxidu, aktívneho metabolitu karbamazepínu. V kontrolovaných štúdiách sa plazmatická koncentrácia karbamazepín-epoxidu zvýšila v priemere o 37 %, 62 % a 98 % s malou variabilitou pri dávkach brivaracetamu zodpovedajúcich 50 mg/deň, 100 mg/ deň a
200 mg/deň. Neboli pozorované žiadne bezpečnostné riziká. Neprejavil sa žiadny aditívny účinok
brivaracetamu a valproátu na AUC pri epoxide karbamazepínu.
Perorálne kontraceptíva
Súčasné podávanie brivaracetamu (100 mg/deň) spolu s perorálnym kontraceptívom obsahujúcim
etinylestradiol (0,03 mg) a levonorgestrel (0,15 mg) neovplyvňovalo farmakokinetiku žiadnej látky. Keď bol brivaracetam v dávke 400 mg/deň (dvojnásobok odporúčanej najvyššej dennej dávky) súčasne podávaný s perorálnym kontraceptívom obsahujúcim etinylestradiol (0,03 mg) a levonorgestrel (0,15 mg), bolo pozorované zníženie AUC estrogénu o 27 % a zníženie AUC progestínu o 23 %, a to
bez dopadu na potlačenie ovulácie. Všeobecne nenastala žiadna zmena v profiloch koncentrácií v čase u endogénnych markerov estradiolu, progesterónu, luteinizačného hormónu (LH), folikuly stimulujúceho hormónu a globulínu viažúceho pohlavné hormóny (SHBG).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Lekári by mali so ženami vo fertilnom veku, ktoré užívajú brivaracetam (pozri Gravidita)
prediskutovať plánované rodičovstvo a antikoncepciu.
Ak sa žena rozhodne otehotnieť, užívanie brivaracetamu sa má dôkladne prehodnotiť.
Gravidita
Riziko spojené s epilepsiou a s antiepileptikami všeobecne
Pre všetky antiepileptiká bolo preukázané, že u potomkov liečených žien s epilepsiou je prevalencia malformácií dvakrát až trikrát vyššia, ako je približne 3% výskyt malformácií v bežnej populácii.
V liečenej populácii bol pozorovaný nárast malformácií pri polyterapii, ale rozsah, za ktorý zodpovedá liečba a/alebo základné ochorenie, nebol objasnený. Prerušenie antiepileptickej liečby
môže viesť k exacerbácii ochorenia, ktoré môže poškodiť matku a plod.
Riziko spojené s brivaracetamom
K dispozícii je iba obmedzené množstvo údajov o použití brivaracetamu u tehotných žien. Nie sú k dispozícii žiadne údaje o placentárnom transfere u ľudí, ale brivaracetam rýchlo prechádza
placentou u potkanov (pozri časť 5.3). Potenciálne riziko u ľudí nie je známe. Štúdie na zvieratách
nepreukázali žiadny teratogénny potenciál brivaracetamu (pozri časť 5.3).
Brivaracetam bol použitý v klinických štúdiách ako prídavná liečba a keď bol používaný spoločne s karbamazepínom, viedol k nárastu koncentrácie aktívneho metabolitu karbamazepín-epoxidu závislého na dávke (pozri časť 4.5). Nie sú k dispozícii dostatočné údaje na určenie klinického významu tohto účinku v tehotenstve.
Z preventívnych dôvodov sa brivaracetam nemá užívať počas tehotenstva, pokiaľ to nie je klinicky nevyhnutné (v prípade, ak prínos pre matku jednoznačne prevyšuje potenciálne riziko pre plod).
Dojčenie
Nie je známe, či sa u ľudí brivaracetam vylučuje do materského mlieka. Štúdie na potkanoch preukázali vylučovanie brivaracetamu do materského mlieka (pozri časť 5.3). Je potrebné sa rozhodnúť, či prerušiť dojčenie alebo prerušiť podávanie brivaracetamu, pričom je potrebné zhodnotiť prínos lieku pre matku. V prípade súčasného podávania brivaracetamu a karbamazepínu by sa mohlo množstvo karbamazepín-epoxidu vylučovaného do materského mlieka zvýšiť. Nie sú k dispozícii dostatočné údaje na stanovenie klinického významu.
Fertilita
Nie sú k dispozícii žiadne údaje o účinku brivaracetamu na fertilitu u ľudí. U potkanov nebol pri liečbe brivaracetamom pozorovaný žiadny účinok na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Brivaracetam má zanedbateľný alebo malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Vzhľadom na možnú rozdielnu individuálnu citlivosť môžu niektorí pacienti pociťovať somnolenciu, závrat alebo iné príznaky súvisiace s centrálnym nervovým systémom (CNS). Pacienti majú byť poučení, aby neviedli vozidlá a neobsluhovali iné potenciálne nebezpečné stroje, kým sa neoboznámia s účinkami brivaracetamu na svoju schopnosť vykonávať tieto činnosti.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Vo všetkých kontrolovaných a nekontrolovaných štúdiách u pacientov s epilepsiou dostávalo
brivaracetam 2 388 subjektov, z ktorých 1 740 bolo liečených ≥6 mesiacov, 1 363 ≥12 mesiacov, 923
≥24 mesiacov a 569 ≥60 mesiacov (5 rokov).
Najčastejšie hlásené nežiaduce účinky (>10%) pri liečbe brivaracetamom boli somnolencia (14,3 %) a závrat (11,0 %). Boli miernej až strednej intenzity. Somnolencia a únava (8,2 %) boli hlásené vo vyššej miere pri zvyšujúcej sa dávke. Typy nežiaducich účinkov hlásených v priebehu prvých 7 dní liečby sa podobali tým, ktoré boli hlásené počas celkovej doby liečby.
Frekvencia ukončenia liečby z dôvodu nežiaducich reakcií bola 3,5 %, 3,4 % a 4,0 % u pacientov randomizovaných na užívanie brivaracetamu v príslušnej dávke 50 mg/deň, 100 mg/deň a
200 mg/deň, a 1,7 % u pacientov randomizovaných na užívanie placeba. Nežiaducim účinkom, ktorý viedol najčastejšie k ukončeniu liečby brivaracetamom, boli závrat (0,8 %) a kŕč (0,8 %).
Zoznam nežiaducich účinkov zoradených do tabuľky
V nasledujúcej tabuľke sú uvedené nežiaduce účinky, ktoré boli identifikované na základe prehľadu
úplnej bezpečnostnej databázy klinických štúdií brivaracetamu, podľa tried orgánových systémov a
frekvencie.
Frekvencie sú definované nasledovne: veľmi časté: (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100). V každej skupine frekvencií sú nežiaduce účinky zoradené v poradí podľa klesajúcej závažnosti.
Trieda orgánových systémov Frekvencia Nežiaduce účinky z klinických štúdií
Infekcie a nákazy časté chrípka
Poruchy krvi a lymfatického systému
menej časté neutropénia
Poruchy metabolizmu a výživy časté znížená chuť do jedla
Poruchy imunitného systému menej časté hypersenzitívne reakcie typu I
Psychické poruchy časté depresia, anxieta, insomnia, iritabilita
menej časté samovražedné myšlienky, psychotická
porucha, agresivita, agitovanosť
Poruchy nervového systému veľmi časté závrat, somnolencia
časté kŕč, vertigo
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinálneho traktu
Celkové poruchy a reakcie v mieste podania
časté infekcia horných ciest dýchacích, kašeľ časté nauzea, vracanie, zápcha
časté únava
Popis vybraných nežiaducich reakcií
Neutropénia bola hlásená u 0,5 % (6/1 099) pacientov s brivaracetamom a u 0 % (0/459) pacientov
s placebom. Štyria z týchto subjektov mali znížený počet neutrofilov vo východiskovom stave a po zahájení liečby brivaracetamom došlo k ďalšiemu zníženiu počtu neutrofilov. Žiadny z týchto
6 prípadov neutropénie nebol závažný, nevyžadoval špecifickú liečbu, ani neviedol k ukončeniu liečby brivaracetamom a žiadny nemal pridružené infekcie.
Samovražedné myšlienky boli hlásené u 0,3 % (3/1 099) pacientov s brivaracetamom a u 0,7 % (3/459) pacientov s placebom. V krátkodobých klinických štúdiách s brivaracetamom u pacientov s epilepsiou nedošlo k žiadnemu prípadu dokonanej samovraždy a k pokusu o samovraždu, avšak obidve boli hlásené v otvorených predĺžených štúdiách (pozri časť 4.4).
V priebehu klinického vývoja boli u malého počtu pacientov s brivaracetamom (9/3022) hlásené reakcie pripomínajúce hypersenzitívne reakcie typu I.
Otvorené predĺžené štúdieU pacientov, ktorí boli sledovaní v otvorených predĺžených štúdiách po dobu až 8 rokov, bol
bezpečnostný profil podobný profilu pozorovanému v krátkodobých placebom kontrolovaných štúdiách.
Pediatrická populáciaK dispozícii sú obmedzené bezpečnostné údaje z otvorených štúdií u detí vo veku od 1 mesiaca do
<16 rokov. Celkovo 152 detí (1 mesiac až <16 rokov) bolo liečených brivaracetamom vo farmakokinetickej štúdii a v súvisiacej nadväzujúcej štúdii. Z obmedzených dostupných údajov najčastejšie hlásenými TEAEs (nežiaduce účinky spojené s liečbou) považovanými za súvisiace so skúšaným liekom boli somnolencia (10 %), znížená chuť do jedla (8 %), únava (5 %) a zníženie telesnej hmotnosti (5 %). Bezpečnostný profil sa zdá byť v súlade s bezpečnostným profilom známym u dospelých. V súčasnosti nie sú k dispozícii žiadne klinické údaje u novorodencov.
Starší pacientiZo 130 starších subjektov zahrnutých do fázy 2/3 vývojového programu brivaracetamu (44
s epilepsiou) bolo 100 vo veku 65-74 rokov a 30 vo veku 75-84 rokov. Bezpečnostný profil u starších pacientov sa zdá byť podobný ako je bezpečnostný profil pozorovaný u mladších dospelých pacientov.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSymptómyK dispozícii sú obmedzené klinické skúsenosti s predávkovaním brivaracetamu u ľudí. U zdravých
subjektov, ktorí užili jednotlivú dávku 1 400 mg brivaracetamu, bola hlásená somnolencia a závrat.
Liečba predávkovaniaNie je k dispozícii žiadne špecifické antidotum na predávkovanie brivaracetamom. Liečba
predávkovania zahŕňa všeobecné podporné opatrenia. Keďže sa močom vylučuje menej ako 10 %
brivaracetamu, neočakáva sa, že by hemodialýza významne zvýšila klírens brivaracetamu (pozri časť
5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI
5
.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptiká, iné antiepileptiká, ATC kód: N03AX23
Mechanizmus účinku
Brivaracetam vykazuje vysokú a selektívnu afinitu k 2A proteínu synaptických vezikúl (SV2A),
transmembránový glykoproteín bol nájdený na presynaptickej úrovni v neurónoch a v endokrinných bunkách. Hoci presnú úlohu tohto proteínu je potrebné ešte objasniť, bolo preukázané, že moduluje exocytózu neurotransmiterov. Predpokladá sa, že väzba na SV2A predstavuje primárny mechanizmus antikonvulzívnej aktivity brivaracetamu.
Klinická účinnosť a bezpečnosť
Účinnosť brivaracetamu v prídavnej terapii parciálnych záchvatov (partial onset seisures-POS) bola
stanovená v 3 randomizovaných dvojito zaslepených, placebom kontrolovaných multicentrických štúdiách s fixnou dávkou u subjektov vo veku 16 rokov a starších. Denná dávka brivaracetamu sa
v týchto štúdiách pohybovala v rozmedzí 5 až 200 mg/deň. Všetky štúdie začínali základnou periódou trvajúcou 8 týždňov, nasledovanou 12 týždňov trvajúcou liečebnou periódou bez titrácie v zmysle zvyšovania dávky. 1 558 pacientov dostávalo liek zo štúdie, z toho 1 099 dostávalo brivaracetam. Kritériá pre zaradenie do štúdie vyžadovali, aby mali pacienti nekontrolované parciálne záchvaty
napriek liečbe buď 1 alebo 2 súčasne podávanými antiepileptikami. Podmienkou bolo, aby pacienti
prekonali najmenej 8 parciálnych záchvatov počas základnej periódy. Primárnymi koncovými ukazovateľmi pri štúdii fázy 3 bolo percento zníženia frekvencie POS oproti placebu a pomer respondérov s dosiahnutou 50 % odpoveďou založenou na 50 % znížení frekvencie POS od východiskového stavu.
Najčastejšie užívanými antiepileptikami na začiatku štúdie boli karbamazepín (40,6 %), lamotrigín (25,2 %), valproát (20,5 %), oxkarbazepín (16,0 %), topiramát (13,5 %), fenytoín (10,2 %) a levetiracetam (9,8 %). Medián východiskovej frekvencie záchvatov vo všetkých 3 štúdiách bol
9 záchvatov v priebehu 28 dní. Pacienti mali epilepsiu v priemere približne 23 rokov.
Výsledky účinnosti sú zhrnuté v tabuľke 2. Celkovo bol brivaracetam účinný pri prídavnej terapii
parciálnych záchvatov u pacientov vo veku 16 rokov a starších v dávke medzi 50 mg/deň a
200 mg/deň.
Tabuľka 2: Kľúčové výsledky účinnosti pre frekvenciu parciálnych záchvatov v priebehu 28 dní
Štúdia Placebo Brivaracetam
*štatisticky významné (hodnota p)
50 mg/deň 100 mg/deň 200 mg/deň
Štúdia N01253(1)
n = 96 n = 101
Dosiahnutie 50% odpovede respondéra 16,73 32,7* ~ ~
(p=0,008)
Percentuálne zníženie voči placebu (%) NA 22,0* ~ ~
(p=0,0040)
Štúdia N01252(1)
n = 100 n = 99 n = 100
Dosiahnutie 50% odpovede respondéra 20,0 27,3
(p=0,372)
Percentuálne zníženie voči placebu (%) NA 9,2
(p=0,0274)
36,0(2) ~
(p=0,023)
20,5(2) ~
(p=0,0097)
Štúdia N01358
n = 259 n = 252 n = 249

Dosiahnutie 50% odpovede respondéra 21,6 ~ 38,9
(p<0,001)
37,8
(p<0,001)
Percentuálne zníženie voči placebu (%) Medián percentuálneho zníženia oproti východiskovému stavu
NA ~ 22,8* (p<0,001)
23,2* (p<0,001)

n = randomizovaní pacienti, ktorí dostali najmenej 1 dávku liečiva v štúdii
~ dávka nebola študovaná
* štatisticky významné
(1) Približne 20 % pacientov dostávalo súčasne levetiracetam
(2) Primárny výsledok pre N01252 nedosiahol štatistickú významnosť na základe sekvenčného
skúšania. Dávka 100 mg/deň bola nominálne významná.
V klinických štúdiách bolo zníženie frekvencie záchvatov vyššie oproti placebu pri dávke 100 mg/deň ako pri dávke 50 mg/deň. Na rozdiel od zvýšenia výskytu somnolencie a únavy v závislosti na dávke, mal brivaracetam pri dávke 50 mg/deň a 100 mg/deň podobný bezpečnostný profil vrátane
nežiaducich účinkov so vzťahom k CNS a pri dlhodobom užívaní.
Obrázok 1 ukazuje percento pacientov (s výnimkou pacientov súčasne užívajúcich levetiracetam)
podľa kategórie zníženia frekvencie POS v priebehu 28 dní od východiskového stavu vo všetkých
3 štúdiách. Pacienti s viac ako 25 % zvýšením parciálnych záchvatov sú uvedení úplne naľavo ako
„horší“. Pacienti so zlepšením percentuálneho zníženia frekvencie POS od východiskového stavu sú uvedení v 4 kategóriách napravo. Percento pacientov s najmenej 50 % znížením frekvencie záchvatov bolo 20,3 %, 34,2 %, 39,5 %, a 37,8 % pre placebo, zodpovedajúce 50 mg/deň, 100 mg/deň a
200 mg/deň.
Obrázok 1: Podiel pacientov s brivaracetamom a placebom podľa kategórie odpovedi záchvatovpo dobu 12 týždňov vo všetkých troch dvojito zaslepených pivotných štúdiáchV súhrnnej analýze troch pivotných štúdií neboli pozorované žiadne rozdiely v účinnosti (merané ako
50 % odpoveď respondérov) v rozmedzí dávok 50 mg/deň až 200 mg/deň, keď je brivaracetam
kombinovaný s antiepileptikami vyvolávajúcimi alebo nevyvolávajúcimi indukciu enzýmov.
V klinických štúdiách dosiahlo stav bez záchvatov 2,5 % (4/161), 5,1 % (17/332) a 4,0 % (10/249)
pacientov s brivaracetamom v dávke zodpovedajúcej 50 mg/deň, 100 mg/deň a 200 mg/deň a to
v priebehu liečebnej periódy v trvaní 12 týždňov v porovnaní s 0,5 % (2/418) pacientov s placebom.
Zlepšenie mediánu percentuálneho zníženia frekvencie záchvatov od začiatku liečby za 28 dní bolo pozorované u pacientov s typom záchvatov IC (sekundárne generalizované tonicko-klonické záchvaty)
vo východiskovom stave, liečených brivaracetamom (66,6 % (n=62), 61,2% (n=100) a 82,1 % (n=75) z pacientov s brivaracetamom v zodpovedajúcej dávke 50 mg/deň, 100 mg/deň a 200 mg/deň v porovnaní s placebom 33,3 % (n=115)).
Účinnosť brivaracetamu v monoterapii nebola ešte stanovená. Použitie brivaracetamu v monoterapii
sa neodporúča.
Liečba levetiracetamom
V 2 randomizovaných placebom kontrolovaných štúdiách fázy 3 sa levetiracetam podával ako
súbežné antiepileptikum u asi 20 % pacientov. Aj keď je počet subjektov limitovaný, nebol
u pacientov, ktorí súčasne užívali levetiracetam, pozorovaný žiadny prínos brivaracetamu oproti placebu, ktorý by reflektoval kompetíciu vo väzbovom mieste SVA2. Neboli zistené žiadne ďalšie okolnosti týkajúce sa bezpečnosti a znášanlivosti.
V 3. štúdii vopred špecifikovaná analýza preukázala účinnosťoproti placebu pre dávky 100 mg/deň a
200 mg/deň u pacientov predtým užívajúcich levetiracetam. Nižšia účinnosť pozorovaná u týchto pacientov v porovnaní s pacientmi neužívajúcimi levetiracetam bola pravdepodobne dôsledkom užívania vyššieho počtu predchádzajúcich antiepileptík a vyššej východiskovej hodnoty frekvencie záchvatov.
Starší pacienti (65 rokov a starší)
Tri pivotné, dvojito zaslepené, placebom kontrolované štúdie zahŕňali 38 pacientov vo veku 65 až
80 rokov. Aj keď sú údaje obmedzené, účinnosť bola porovnateľná s účinnosťou u mladších subjektov.
Otvorené predĺžené štúdie
Vo všetkých štúdiách bolo zaradených do dlhodobých, otvorených, predĺžených štúdií 81,7 %
pacientov, ktorí dokončili randomizované štúdie. Od vstupu do randomizovaných štúdií bolo 5,3 % subjektov s brivaracetamom po dobu 6 mesiacov (n=1500) bez záchvatov v porovnaní s 4,6 % a 3,7 % u subjektov exponovaných po dobu 12 mesiacov (n=1188) a 24 mesiacov (n=847). Nakoľko však vysoké percento subjektov (26 %) prerušilo liečbu v otvorených štúdiách z dôvodu nedostatočnej účinnosti, mohlo dôjsť ku skresleniu, nakoľko subjekty, ktoré zostali v štúdii, reagovali lepšie ako tí, ktorí ju predčasne ukončili.
Pediatrická populácia
Účinnosť a znášanlivosť brivaracetamu nebola u pediatrických pacientov stanovená (pozri časť 4.2).
Brivaracetam bol hodnotený u týchto pacientov v krátkodobej otvorenej farmakokinetickej štúdií a prebiehajúcej otvorenej, predĺženej štúdií u 152 subjektov vo veku od 1 mesiaca do 16 rokov (pozri časť 5.2).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s brivaracetamom v jednej alebo vo viacerých podskupinách pediatrickej populácie s epilepsiou s parciálnymi záchvatmi.
5.2 Farmakokinetické vlastnosti
Brivaracetam filmom obalené tablety, perorálny roztok a roztok na intravenóznu injekciu vykazujú identickú AUC, zatiaľ čo maximálna plazmatická koncentrácia je mierne vyššia po intravenóznom podaní. Brivaracetam vykazuje lineárnu a na čase nezávislú farmakokinetiku s nízkou intra- a interindividuálnou variabilitou a ďalej úplnou absorpciou, nízku väzbu na proteíny, renálnu exkréciu po rozsiahlej biotransformácii a farmakologicky inaktívne metabolity.
Absorpcia
Brivaracetam sa rýchlo a úplne absorbuje po perorálnom podaní a absolútna biologická dostupnosť je
približne 100 %. Medián tmax pre tablety užité bez jedla je 1 hodina (rozsah tmax je 0,25 až 3 hod.).
Súčasné podávanie s jedlom s vysokým obsahom tuku spomalilo rýchlosť absorpcie (medián tmax 3 h) a znížilo maximálnu plazmatickú koncentráciu (37 % pokles) brivaracetamu, pričom rozsah absorpcie zostal nezmenený.
Distribúcia
Brivaracetam sa slabo viaže (≤ 20 %) na plazmatické proteíny. Distribučný objem je 0,5 l/kg, čo je
hodnota blízka celkovému množstvu telesnej vody.
Bunkové membrány sú pre brivaracetam vysoko permeabilné z dôvodu jeho lipofílie (log P).
Biotransformácia
Brivaracetam je primárne metabolizovaný hydrolýzou svojej amidovej časti za vzniku zodpovedajúcej
karboxylovej kyseliny (približne 60 % eliminácie) a sekundárne hydroxyláciou propylového vedľajšieho reťazca (približne 30 % eliminácie). Hydrolýza amidovej časti, ktorá vedie k vzniku metabolitu povahy karboxylovej kyseliny (34 % dávky v moči) je podporovaná pečeňovou a mimopečeňovou amidázou. In vitro je hydroxylácia brivaracetamu sprostredkovaná v prvom rade CYP2C19. Obidva metabolity sú ďalej metabolizované za vzniku bežnej hydroxylovanej kyseliny, ktorá vzniká prevažne hydroxyláciou postranného propylového reťazca metabolitu kyseliny karboxylovej (hlavne prostredníctvom CYP2C9). In vivo u ľudí s neúčinnou mutáciou CYP2C19, sa tvorba hydroxymetabolitu znižuje 10x, zatiaľ čo samotný brivaracetam sa zvyšuje o 22 % alebo 42 % u jedincov s jednou alebo s obidvomi mutovanými alelami. Tri metabolity nie sú farmakologicky aktívne.
Eliminácia
Brivaracetam je primárne eliminovaný metabolizáciou a vylučovaním močom. Viac ako 95 % dávky,
vrátane metabolitov, sa vylučuje močom v priebehu 72 hodín po užití. Menej ako 1 % dávky sa vylučuje stolicou a menej ako 10 % brivaracetamu sa vylučuje bez zmeny močom. Terminálny plazmatický polčas (t1/2) je približne 9 hodín. Celkový plazmatický klírens bol u pacientov odhadnutý na 3,6 l/hod.
Linearita
Farmakokinetika je úmerná dávke od 10 mg do najmenej 600 mg.
Interakcie s liekmi
Brivaracetam je eliminovaný viacerými cestami, vrátane vylučovania obličkami, na CYP nezávislou
hydrolýzou a CYP sprostredkovanou oxidáciou. In vitro, brivaracetam nie je substrátom ľudského P-glykoproteínu (P-gp), proteínom rezistentným na viacpočetné lieky (MRP – multidrug resistance proteins) 1 a 2 a pravdepodobne ani organickým transportérom aniónu polypeptidu 1B1 (OATP1B1) a OATP1B3. Testy in vitro ukázali, že metabolizmus brivaracetamu by nemal byť významne ovplyvnený CYP inhibítormi (napr. CYP1A, 2C8, 2C9, 2C19, 2D6 a 3A4).
In vitro, brivaracetam nebol inhibítorom CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 3A4 ani transportérom P-gp, BCRP, BSEP MRP2, MATE-K, MATE-1, OATP1B1, OATP1B3, OAT1 a OCT1 v klinicky relevantných koncentráciách. In vitro, brivaracetam neindukoval CYP1A2.
Farmakokinetika u osobitných skupín pacientov
Starší pacienti (65 rokov a starší)
V štúdii u starších pacientov (vo veku 65 až 79 rokov; s klírensom kreatinínu 53 až 98 ml/min/1,73 m2), ktorí užívali brivaracetam v dávke 400 mg/deň s podávaním 2x denne, bol plazmatický polčas brivaracetamu 7,9 hodiny u skupiny vo veku 65 až 75 rokov a 9,3 hodiny u skupiny >75 rokov. Plazmatický klírens rovnovážneho stavu brivaracetamu bol podobný (0,76 ml/min/kg) ako u mladých zdravých mužov (0,83 ml/min/kg) (pozri časť 4.2).
Porucha funkcie obličiek
Štúdia u subjektov so závažnou poruchou funkcie obličiek (klírens kreatinínu <30 ml/min/1,73 m2 bez nutnosti dialýzy) odhalila, že plazmatická AUC brivaracetamu bola stredne zvýšená (+21%) voči zdravým subjektom zatiaľ čo AUC kyseliny, hydroxymetabolitu a metabolitu hydroxykyseliny boli
zvýšené 3x, 4x, a 21x (v uvedenom poradí). Renálny klírens týchto neaktívnych metabolitov bol
znížený 10x. Metabolit hydroxykyseliny v predklinických štúdiách nevyvolal žiadne obavy zo strany
bezpečnosti. Brivaracetam nebol študovaný u pacientov liečených hemodialýzou (pozri časť 4.2).
Porucha funkcie pečene
Farmakokinetická štúdia u subjektov s cirhózou pečene (Child-Pugh triedy A, B, a C) preukázala
podobné zvýšenie pre expozíciu brivaracetamu bez ohľadu na závažnosť ochorenia (50 %, 57 % a 59
%) v pomere k zodpovedajúcim zdravým subjektom (pozri časť 4.2).
Pediatrická populácia
Vo farmakokinetickej štúdii u 99 subjektov vo veku 1 mesiac až <16 rokov, ktorí dostávali brivaracetam perorálny roztok bolo preukázané, že plazmatické koncentrácie sú úmerné dávke vo
všetkých vekových skupinách. Farmakokinetické populačné modelovanie ukázalo, že dávka
2,0 mg/kg dvakrát denne vedie
k rovnakej priemernej plazmatickej koncentrácii rovnovážneho stavu ako u dospelých užívajúcich
100 mg dvakrát denne.
Telesná hmotnosť
Bol odhadnutý 40 % pokles plazmatickej koncentrácie rovnovážneho stavu v rozsahu telesnej hmotnosti od 46 kg do 115 kg. To však nie je považované za klinicky významný rozdiel vo farmakokinetike brivaracetamu.
Pohlavie
Nie sú žiadne klinicky významné rozdiely vo farmakokinetike brivaracetamu medzi pohlaviami.
Rasa
Farmakokinetika brivaracetamu nebola významne ovplyvnená rasou (kaukazská, ázijská) pri
farmakokinetickom populačnom modelovaní u pacientov s epilepsiou. Počet pacientov s iným etnickým pôvodom bol obmedzený.
Farmakokinetický/farmakodynamický vzťah
EC50 (plazmatická koncentrácia brivaracetamu zodpovedajúca 50 % maximálneho účinku) bola
odhadnutá na 0,57 mg/l. Táto plazmatická koncentrácia je mierne nad mediánom expozície po podávaní brivaracetamu v dávkach 50 mg/deň. Ďalšie zníženie frekvencie záchvatov sa dostavuje pri zvýšení dávky na 100 mg/deň a dosahuje stabilizovaný stav pri dávke 200 mg/deň.
5.3 Predklinické údaje o bezpečnosti
Vo farmakologických štúdiách bezpečnosti mali prevládajúce účinky súvislosť s CNS (najmä prechodná depresia CNS a zníženie spontánnej pohybovej aktivity) a boli pozorované pri násobkoch (vyšších než 50x) farmakologicky účinnej dávky brivaracetamu 2 mg/kg. Brivaracetam neovplyvnil učenie a funkciu pamäte.
Nálezy, ktoré neboli pozorované v klinických štúdiách, ale boli pozorované v toxikologických
štúdiách s opakovaným podávaním u psov pri expozícii podobnej ako pri klinickej plazmatickej AUC, boli hepatotoxické účinky (hlavne porfýria). Toxikologické údaje zhromaždené o brivaracetame a
o štrukturálne príbuzných látkach ale ukazujú, že sa pečeňové zmeny u psov vyvinuli prostredníctvom mechanizmov nerelevantných pre ľudí. Žiadne nežiaduce zmeny na pečeni neboli pozorované
u potkanov a opíc po dlhodobom podávaní brivaracetamu s expozíciou zreteľne prevyšujúcu AUC expozíciu 5 až 42x. CNS príznaky u opíc (vyčerpanosť, strata rovnováhy, nemotorné pohyby) sa vyskytli pri 64-násobkoch klinickej Cmax. Tieto účinky boli menej zrejmé v priebehu času.
Štúdie genotoxicity nepreukázali žiadnu mutagénnu alebo klastogénnu aktivitu. Štúdie kancerogenity u potkanov nepreukázali žiadny onkogénny potenciál, zatiaľ čo zvýšený výskyt hepatocelulárnych nádorov u samcov myší je považovaný za dôsledok známeho negenotoxického fenoménu, známeho
u hlodavcov, ktorého mechanizmus účinku sa vzťahuje k indukcii pečeňových enzýmov, podobnej ako po fenobarbitále.
Brivaracetam neovplyvnil fertilitu samíc ani samcov, nepreukázal sa žiadny teratogénny potenciál u potkanov alebo králikov. Embryotoxicita bola pozorovaná u králikov pri dávke brivaracetamu toxickej pre matky s expozíciou 8x vyššou než klinická AUC expozícia pri maximálnej odporúčanej dávke. U potkanov bolo preukázané, že brivaracetam ľahko prestupuje placentou a je vylučovaný do materského mlieka u dojčiacich samíc potkanov v koncentráciách podobných plazmatickým koncentráciám u matiek.
Brivaracetam u potkanov nepreukázal žiadny potenciál na závislosť. Štúdie u nedospelých zvierat
U nedospelých potkanov expozičné hladiny 6 -15 násobnej klinickej AUC expozície brivaracetamu
pri maximálnej odporúčanej dávke vyvolávali vývojové nežiaduce účinky (napr. mortalitu, klinické príznaky, zníženie telesnej hmotnosti a nižšiu hmotnosť mozgu). Neboli zaznamenané žiadne nežiaduce účinky na funkciu CNS, žiadne neuropatologické a histopatologické vyšetrenia mozgu.
U nedospelých psov boli brivaracetamom indukované zmeny pri dávke 100 mg/kg/deň spojené s 6- násobným zvýšením hladín AUC, podobným zmenám pozorovaným u dospelých zvierat. Neboli pozorované žiadne nežiaduce účinky v štandardných ukazovateľoch vývoja alebo maturácie.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
nátriumcitrát
bezvodá kyselina citrónová (na úpravu pH)
metylparabén (E218) sodná soľ karmelózy sukralóza
roztok sorbitolu glycerol (E422)
malinová aróma (propylénglykol 90 % - 98 %)
čistená voda
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
Po prvom otvorení: 5 mesiacov
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie. Podmienky na uchovávanie po prvom otvorení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia300 ml jantárovo hnedá fľaška (triedy III) s bielym detským bezpečnostným uzáverom
(z polypropylénu) v papierovej škatuľke s 10 ml kalibrovanou striekačkou na perorálne podanie
(polypropylén, polyetylén) a adaptérom na striekačku (polyetylén).
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky.
Všetok nepoužitý liek (riedený aj neriedený) alebo odpad vzniknutý z lieku sa má zlikvidovať
v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIUCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8. REGISTRAČNÉ ČÍSLO(A)EU/1/15/1073/021
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 14. január 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií
o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na
nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUBriviact 10 mg/ml injekčný/infúzny roztok
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždý ml obsahuje 10 mg brivaracetamu.
Jedna 5 ml liekovka obsahuje brivaracetamum 50 mg.
Pomocnélátkysoznámymúčinkom:
Každý ml injekčného/infúzneho roztoku obsahuje 3,8 mg sodíka.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAInjekčný/infúzny roztok (injekcia/infúzia)
Číry, bezfarebný roztok.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieBriviact je indikovaný ako prídavná terapia pri liečbe parciálnych záchvatov s alebo bez sekundárnej
generalizácie u dospelých a dospievajúcich pacientov s epilepsiou vo veku od 16 rokov.
4.2 Dávkovanie a spôsob podávaniaDávkovanieLiečbu brivaracetamom možno zahájiť vnútrožilovou alebo perorálnou aplikáciou. Keď sa prechádza
z perorálneho na intravenózne podávanie alebo naopak, celková denná dávka a frekvencia podávania zostávajú rovnaké. Injekčný/infúzny roztok brivaracetamu je alternatívou pre pacientov, u ktorých nie je dočasne možné perorálne podávanie.
Odporúčaná začiatočná dávka potrebná na zníženie počtu záchvatov je buď 50 mg/deň alebo 100 mg/deň, na základe posúdenia potenciálnych nežiaducich účinkov lekárom. Dávka sa má podávať v dvoch rovnakých rozdelených dávkach, jedenkrát ráno a jedenkrát večer. Na základe individuálnej odpovede a znášanlivosti pacienta sa môže dávka upraviť v rozmedzí 50 mg/deň až 200 mg/deň.
Nie sú žiadne skúsenosti s intravenóznym podávaním brivaracetamu trvajúce dlhšie ako 4 dni.
Zabudnutá dávkaAk pacienti vynechali jednu alebo viac dávok, odporúča sa podať jednu dávku ihneď, ako si spomenú a nasledujúcu dávku podať vo zvyčajnom čase ráno alebo večer. To môže zabrániť poklesu plazmatickej koncentrácie brivaracetamu pod účinnú hladinu, a tým opätovnému prepuknutiu
záchvatov.
Ukončenie liečby
Ak sa má podávanie brivaracetamu ukončiť, odporúča sa postupné znižovanie o 50 mg/deň
v týždennom intervale. Po týždni liečby dávkou 50 mg/deň sa odporúča v poslednom týždni liečby
dávka 20 mg/deň.
Osobitné skupiny pacientov
Starší pacienti (65 rokov a starší)
Nie je potrebná žiadna úprava dávky u starších pacientov (pozri časť 5.2). Klinická skúsenosť u pacientov ≥65 rokov je obmedzená.
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná žiadna úprava dávky (pozri časť 5.2). Vzhľadom na nedostatok údajov, pacientom v konečnom štádiu ochorenia obličiek, ktorí sú liečení dialýzou, sa podávanie brivaracetamu neodporúča.
Porucha funkcie pečene
Expozícia brivaracetamu bola zvýšená u pacientov s chronickým ochorením pečene. Začiatočná
dávka 50 mg/deň sa má zvážiť. Vo všetkých štádiách poruchy funkcie pečene sa odporúča maximálna
denná dávka 150 mg, podávaná v 2 rozdelených dávkach (pozri časti 4.4 a 5.2).
Pediatrická populácia
Bezpečnosť a účinnosť brivaracetamu u novorodencov a detí vo veku menej ako 16 rokov neboli doteraz stanovené.
V súčasnosti dostupné údaje sú uvedené v časti 4.8, 5.1, a 5.2, ale na ich základe sa nedajú urobiť žiadne odporúčania ohľadne dávkovania.
Spôsob podávania
• Intravenózny bolus: brivaracetam sa môže podávať bez riedenia ako intravenózny bolus.'
• Intravenózna infúzia: brivaracetam sa môže nariediť kompatibilným riedidlom a podávať 15
minútovou intravenóznou infúziou (pozri časť 6.6). Tento liek sa nesmie miešať s inými liekmi.
Podávanie brivaracetamu bolusovou injekciou alebo intravenóznou infúziou nebolo skúmané pri akútnych stavoch; napr. status epilepticus, a preto sa pri takýchto stavoch neodporúča.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na iné deriváty pyrolidónu alebo na ktorúkoľvek z pomocných látok
uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Samovražedné myšlienky a správanie
U pacientov liečených antiepileptikami (AEDs), vrátane brivaracetamu, boli pri niekoľkých
indikáciách hlásené samovražedné myšlienky a správanie. Meta-analýza randomizovaných, placebom kontrolovaných štúdií s antiepileptikami preukázala malé zvýšenie rizika samovražedných myšlienok a správania. Mechanizmus vzniku tohto rizika nie je známy a dostupné údaje nevylučujú možnosť zvýšeného rizika pre brivaracetam.
U pacientov sa majú sledovať príznaky samovražedných myšlienok a správania a má sa zvážiť
vhodná liečba. Pacientom (a ich opatrovateľom) sa má odporučiť, aby v prípade výskytu akýchkoľvek príznakov samovražedných myšlienok a správania okamžite vyhľadali lekársku pomoc.
Porucha funkcie pečene
K dispozícii sú obmedzené klinické údaje o použití brivaracetamu u pacientov s už existujúcou
poruchou funkcie pečene. U pacientov s poruchou funkcie pečene sa odporúča úprava dávky (pozri časť 4.2).
Obsah sodíka
Injekčný/infúzny roztok obsahuje 0,83 mmol (zodp. 19,14 mg) sodíka v injekčnej liekovke. Toto je
potrebné vziať do úvahy u pacientov nastavených na diétny režim s kontrolovaným obsahom sodíka.
4.5 Liekové a iné interakcie
Formálne štúdie interakcie sa uskutočnili iba u dospelých.
Farmakodynamické interakcie
Súčasná liečba s levetiracetamom
V klinických štúdiách, aj keď ich počet bol obmedzený, nebol pozorovaný žiadny prínos brivaracetamu oproti placebu u pacientov súčasne užívajúcich levetiracetam. Neboli pozorované žiadne ďalšie obavy ohľadne bezpečnosti a znášanlivosti (pozri časť 5.1).
Interakcie s alkoholom
V štúdii farmakokinetickej a farmakodynamickej interakcie medzi brivaracetamom v jednorazovej dávke 200 mg a etanolom v kontinuálnej infúzii 0,6 g/l u zdravých subjektov nenastala žiadna farmakokinetická interakcia ale brivaracetam približne zdvojnásobil účinky alkoholu na psychomotorické funkcie, pozornosť a pamäť. Podávanie brivaracetamu s alkoholom sa neodporúča.
Farmakokinetické interakcie
Účinky iných látok na farmakokinetiku brivaracetamu
In vitro údaje naznačujú, že brivaracetam má nízky interakčný potenciál. Hlavnou metabolickou cestou brivaracetamu je na CYP nezávislá hydrolýza. Druhá cesta zahŕňa hydroxyláciu, ktorá je
sprostredkovaná CYP2C19 (pozri časť 5.2).
Plazmatické koncentrácie brivaracetamu sa môžu zvýšiť, ak je súčasne podávaný so silnými inhibítormi CYP2C19 (ako flukonazol, fluvoxamín), ale riziko klinicky významnej interakcie sprostredkované CYP2C19 je považované za nízke.
Rifampicín
U zdravých dobrovoľníkov súčasné podávanie silného induktora enzýmov rifampicínu (600 mg/deň po dobu 5 dní) znížilo plochu brivaracetamu pod krivkou (AUC) o 45 %. Predpisujúci lekári musia zvážiť úpravu dávky brivaracetamu u pacientov, u ktorých sa zahajuje alebo ukončuje liečba
rifampicínom.
Antiepileptiká so silnou indukciou enzýmov
Plazmatické koncentrácie brivaracetamu klesajú pri súčasnom podávaní s antiepileptikami silne indukujúcimi enzýmy (karbamazepín, fenobarbital, fenytoín), nie je však potrebná žiadna úprava
dávky (pozri tabuľka 1).
Iné induktory enzýmov
Očakáva sa, že iné silné induktory enzýmov (ako ľubovník bodkovaný (Hypericum perforatum)), môžu tiež znížiť systémovú expozíciu brivaracetamu. Preto zahájenie alebo ukončenie liečby ľubovníkom bodkovaným sa má uskutočniť opatrne.
Účinok brivaracetamu na iné lieky
Brivaracetam podávaný v dávkach 50 mg alebo 150 mg/deň neovplyvňoval AUC midazolamu
(metabolizovaný CYP3A4). Riziko klinicky relevantných CYP3A4 interakcií je považované za nízke.
Štúdie in vitro preukázali, že brivaracetam vykazuje malú alebo žiadnu inhibíciu izoforom CYP450, s
výnimkou CYP2C19. Brivaracetam môže zvyšovať plazmatické koncentrácie liečiv metabolizovaných CYP2C19 (napr. lanzoprazol, omeprazol, diazepam). Pri skúšaní in vitro brivaracetam neindukoval CYP1A1/2, ale indukoval CYP3A4 a CYP2B6. Žiadna CYP3A4 indukcia nebola nájdená in vivo (pozri midazolam vyššie). CYP2B6 indukcia nebola skúmaná in vivo a brivaracetam môže znižovať plazmatické koncentrácie liečiv metabolizovaných CYP2B6 (napr.
efavirenz). In vitro interakčné štúdie na určenie potenciálnych inhibičných účinkov na transportéry
viedli k záveru, že nedochádza k žiadnym klinicky významným účinkom, s výnimkou OAT3. In vitro brivaracetam inhibuje OAT3 s polovičnou maximálnou inhibičnou koncentráciou 42-krát vyššou ako Cmax pri najvyšších klinických dávkach. Brivaracetam v dávke 200 mg/deň môže zvýšiť plazmatické koncentrácie liekov transportovaných AOT3.
Antiepileptiká
Potenciálne interakcie medzi brivaracetamom (50 mg/deň až 200 mg/deň) a inými antiepileptikami
boli skúmané v súhrnnej analýze plazmatických liekových koncentrácií zo všetkých štúdií fázy 2-3 a v populačnej farmakokinetickej analýze placebom kontrolovaných štúdií fázy 2-3 a vo vyhradených štúdiách liekových interakcií (pre nasledujúce antiepileptiká: karbamazepín, lamotrigín, fenytoín a topiramát). Účinok vzájomných interakcií na plazmatickú koncentráciu je zhrnutý v tabuľke 1 (zvýšenie je označené ako “↑” a zníženie ako “↓”, plocha pod krivkou plazmatickej koncentrácie versus čas ako “AUC“, maximálna pozorovaná koncentrácia ako “Cmax“).
Tabuľka 1: Farmakokinetické interakcie medzi brivaracetamom a inými antiepileptikami
Súčasne podávané
a
ntiepiletikum
Vplyv antiepiletika na koncentráciu brivaracetamu v plazme
Vplyv brivaracetamu na koncentráciu antiepiletika v plazme
Karbamazepín AUC 29 % ↓
Cmax 13 % ↓
nie je potrebná žiadna úprava dávky
karbamazepín - žiadny
epoxid karbamazepínu ↑ (pozri
nižšie)
nie je potrebná žiadna úprava dávky
Klobazam žiadne údaje žiadny Klonazepam žiadne údaje žiadny Lakosamid žiadne údaje žiadny Lamotrigín žiadny žiadny Levetiracetam žiadny žiadny
Oxkarbazepín žiadny žiadny (monohydroxyderivát, MHD)
Fenobarbital AUC 19 % ↓
nie je potrebná žiadna úprava dávky
Fenytoín AUC 21 % ↓
nie je potrebná žiadna úprava dávky
žiadny
žiadny
a AUC 20 % ↑
a Cmax 20 % ↑
Pregabalín žiadne údaje žiadny Topiramát žiadny žiadny Kyselina valproová žiadny žiadny Zonisamid žiadne údaje žiadny
a na základe štúdie zahŕňajúcej podávanie superterapeutických dávok brivaracetamu 400 mg/deň
KarbamazepínBrivaracetam je stredne účinný reverzibilný inhibítor epoxidovej hydrolázy vyvolávajúci zvýšenú
koncentráciu karbamazepín-epoxidu, aktívneho metabolitu karbamazepínu. V kontrolovaných štúdiách sa plazmatická koncentrácia karbamazepín-epoxidu zvýšila v priemere o 37 %, 62 % a 98 % s malou variabilitou pri dávkach brivaracetamu zodpovedajúcich 50 mg/deň, 100 mg/ deň a
200 mg/deň. Neboli pozorované žiadne bezpečnostné riziká. Neprejavil sa žiadny aditívny účinok
brivaracetamu a valproátu na AUC pri epoxide karbamazepínu.
Perorálne kontraceptíva
Súčasné podávanie brivaracetamu (100 mg/deň) spolu s perorálnym kontraceptívom obsahujúcim
etinylestradiol (0,03 mg) a levonorgestrel (0,15 mg) neovplyvňovalo farmakokinetiku žiadnej látky. Keď bol brivaracetam v dávke 400 mg/deň (dvojnásobok odporúčanej najvyššej dennej dávky) súčasne podávaný s perorálnym kontraceptívom obsahujúcim etinylestradiol (0,03 mg) a levonorgestrel (0,15 mg), bolo pozorované zníženie AUC estrogénu o 27 % a zníženie AUC progestínu o 23 %, a to
bez dopadu na potlačenie ovulácie. Všeobecne nenastala žiadna zmena v profiloch koncentrácií v čase u endogénnych markerov estradiolu, progesterónu, luteinizačného hormónu (LH), folikuly stimulujúceho hormónu a globulínu viažúceho pohlavné hormóny (SHBG).
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Lekári by mali so ženami vo fertilnom veku, ktoré užívajú brivaracetam (pozri Gravidita)
prediskutovať plánované rodičovstvo a antikoncepciu.
Ak sa žena rozhodne otehotnieť, užívanie brivaracetamu sa má dôkladne prehodnotiť.
Gravidita
Riziko spojené s epilepsiou a s antiepileptikami všeobecne
Pre všetky antiepileptiká bolo preukázané, že u potomkov liečených žien s epilepsiou je prevalencia malformácií dvakrát až trikrát vyššia, ako je približne 3% výskyt malformácií v bežnej populácii.
V liečenej populácii bol pozorovaný nárast malformácií pri polyterapii, ale rozsah, za ktorý zodpovedá liečba a/alebo základné ochorenie, nebol objasnený. Prerušenie antiepileptickej liečby môže viesť k exacerbácii ochorenia, ktoré môže poškodiť matku a plod.
Riziko spojené s brivaracetamom
K dispozícii je iba obmedzené množstvo údajov o použití brivaracetamu u tehotných žien. Nie sú k dispozícii žiadne údaje o placentárnom transfere u ľudí, ale brivaracetam rýchlo prechádza placentou u potkanov (pozri časť 5.3). Potenciálne riziko u ľudí nie je známe. Štúdie na zvieratách nepreukázali žiadny teratogénny potenciál brivaracetamu (pozri časť 5.3).
Brivaracetam bol použitý v klinických štúdiách ako prídavná liečba a keď bol používaný spoločne s karbamazepínom, viedol k nárastu koncentrácie aktívneho metabolitu karbamazepín-epoxidu závislého na dávke (pozri časť 4.5). Nie sú k dispozícii dostatočné údaje na určenie klinického významu tohto účinku v tehotenstve.
Z preventívnych dôvodov sa brivaracetam nemá užívať počas tehotenstva, pokiaľ to nie je klinicky nevyhnutné (v prípade, ak prínos pre matku jednoznačne prevyšuje potenciálne riziko pre plod).
Dojčenie
Nie je známe, či sa u ľudí brivaracetam vylučuje do materského mlieka. Štúdie na potkanoch preukázali vylučovanie brivaracetamu do materského mlieka (pozri časť 5.3). Je potrebné sa rozhodnúť, či prerušiť dojčenie alebo prerušiť podávanie brivaracetamu, pričom je potrebné zhodnotiť prínos lieku pre matku. V prípade súčasného podávania brivaracetamu a karbamazepínu by sa mohlo množstvo karbamazepín-epoxidu vylučovaného do materského mlieka zvýšiť. Nie sú k dispozícii dostatočné údaje na stanovenie klinického významu.
Fertilita
Nie sú k dispozícii žiadne údaje o účinku brivaracetamu na fertilitu u ľudí. U potkanov nebol pri liečbe brivaracetamom pozorovaný žiadny účinok na fertilitu (pozri časť 5.3).
4
.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Brivaracetam má zanedbateľný alebo malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Vzhľadom na možnú rozdielnu individuálnu citlivosť môžu niektorí pacienti pociťovať somnolenciu,
závrat alebo iné príznaky súvisiace s centrálnym nervovým systémom (CNS). Pacienti majú byť poučení, aby neviedli vozidlá a neobsluhovali iné potenciálne nebezpečné stroje, kým sa neoboznámia s účinkami brivaracetamu na svoju schopnosť vykonávať tieto činnosti.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Vo všetkých kontrolovaných a nekontrolovaných štúdiách u pacientov s epilepsiou dostávalo
brivaracetam 2 388 subjektov, z ktorých 1 740 bolo liečených ≥6 mesiacov, 1 363 ≥12 mesiacov, 923
≥24 mesiacov a 569 ≥60 mesiacov (5 rokov).
Najčastejšie hlásené nežiaduce účinky (>10%) pri liečbe brivaracetamom boli somnolencia (14,3 %) a závrat (11,0 %). Boli miernej až strednej intenzity. Somnolencia a únava (8,2 %) boli hlásené vo vyššej miere pri zvyšujúcej sa dávke. Typy nežiaducich účinkov hlásených v priebehu prvých 7 dní liečby sa podobali tým, ktoré boli hlásené počas celkovej doby liečby.
Frekvencia ukončenia liečby z dôvodu nežiaducich reakcií bola 3,5 %, 3,4 % a 4,0 % u pacientov randomizovaných na užívanie brivaracetamu v príslušnej dávke 50 mg/deň, 100 mg/deň a
200 mg/deň, a 1,7 % u pacientov randomizovaných na užívanie placeba. Nežiaducim účinkom, ktorý viedol najčastejšie k ukončeniu liečby brivaracetamom, boli závrat (0,8 %) a kŕč (0,8 %).
Zoznam nežiaducich účinkov zoradených do tabuľky
V nasledujúcej tabuľke sú uvedené nežiaduce účinky, ktoré boli identifikované na základe prehľadu
úplnej bezpečnostnej databázy klinických štúdií brivaracetamu, podľa tried orgánových systémov a
frekvencie.
Frekvencie sú definované nasledovne: veľmi časté: (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100). V každej skupine frekvencií sú nežiaduce účinky zoradené v poradí podľa klesajúcej závažnosti.
Trieda orgánových systémov Frekvencia Nežiaduce účinky z klinických štúdií
Infekcie a nákazy časté chrípka
Poruchy krvi a lymfatického systému
Poruchy metabolizmu a výživy
menej časté neutropénia
časté znížená chuť do jedla
Poruchy imunitného systému menej časté hypersenzitívne reakcie typu I
Psychické poruchy časté depresia, anxieta, insomnia, iritabilita
menej časté samovražedné myšlienky, psychotická
porucha, agresivita, agitovanosť
Poruchy nervového systému veľmi časté závrat, somnolencia
časté kŕč, vertigo
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinálneho traktu
Celkové poruchy a reakcie v mieste podania
časté infekcia horných ciest dýchacích, kašeľ časté nauzea, vracanie, zápcha
časté únava
Popis vybraných nežiaducich reakcií
Neutropénia bola hlásená u 0,5 % (6/1 099) pacientov s brivaracetamom a u 0 % (0/459) pacientov
s placebom. Štyria z týchto subjektov mali znížený počet neutrofilov vo východiskovom stave a po zahájení liečby brivaracetamom došlo k ďalšiemu zníženiu počtu neutrofilov. Žiadny z týchto
6 prípadov neutropénie nebol závažný, nevyžadoval špecifickú liečbu, ani neviedol k ukončeniu liečby brivaracetamom a žiadny nemal pridružené infekcie.
Samovražedné myšlienky boli hlásené u 0,3 % (3/1 099) pacientov s brivaracetamom a u 0,7 % (3/459) pacientov s placebom. V krátkodobých klinických štúdiách s brivaracetamom u pacientov s epilepsiou nedošlo k žiadnemu prípadu dokonanej samovraždy a k pokusu o samovraždu, avšak obidve boli hlásené v otvorených predĺžených štúdiách (pozri časť 4.4).
Nežiaduce reakcie spojené s vnútrožilovým podaním sa všeobecne zdajú byť podobné tým, ktoré pozorované pri perorálnom podaní. Intravenózne podávanie bolo spojené s bolesťou mieste aplikácie u 2,8 % pacientov.
V priebehu klinického vývoja boli u malého počtu pacientov s brivaracetamom (9/3022) hlásené reakcie pripomínajúce hypersenzitívne reakcie typu I.
Otvorené predĺžené štúdieU pacientov, ktorí boli sledovaní v otvorených predĺžených štúdiách po dobu až 8 rokov, bol
bezpečnostný profil podobný profilu pozorovanému v krátkodobých placebom kontrolovaných štúdiách.
Pediatrická populáciaK dispozícii sú obmedzené bezpečnostné údaje z otvorených štúdií u detí vo veku od 1 mesiaca do
<16 rokov. Celkovo 152 detí (1 mesiac až <16 rokov) bolo liečených brivaracetamom vo farmakokinetickej štúdii a v súvisiacej nadväzujúcej štúdii. Z obmedzených dostupných údajov najčastejšie hlásenými TEAEs (nežiaduce účinky spojené s liečbou) považovanými za súvisiace so skúšaným liekom boli somnolencia (10 %), znížená chuť do jedla (8 %), únava (5 %) a zníženie telesnej hmotnosti (5 %). Bezpečnostný profil sa zdá byť v súlade s bezpečnostným profilom známym u dospelých. V súčasnosti nie sú k dispozícii žiadne klinické údaje u novorodencov.
Starší pacientiZo 130 starších subjektov zahrnutých do fázy 2/3 vývojového programu brivaracetamu (44
s epilepsiou) bolo 100 vo veku 65-74 rokov a 30 vo veku 75-84 rokov. Bezpečnostný profil u starších pacientov sa zdá byť podobný ako je bezpečnostný profil pozorovaný u mladších dospelých pacientov.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSymptómyK dispozícii sú obmedzené klinické skúsenosti s predávkovaním brivaracetamu u ľudí. U zdravých
subjektov, ktorí užili jednotlivú dávku 1 400 mg brivaracetamu, bola hlásená somnolencia a závrat.
Liečba predávkovania
Nie je k dispozícii žiadne špecifické antidotum na predávkovanie brivaracetamom. Liečba
predávkovania zahŕňa všeobecné podporné opatrenia. Keďže sa močom vylučuje menej ako 10 %
brivaracetamu, neočakáva sa, že by hemodialýza významne zvýšila klírens brivaracetamu (pozri časť
5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antiepileptiká, iné antiepileptiká, ATC kód: N03AX23
Mechanizmus účinku
Brivaracetam vykazuje vysokú a selektívnu afinitu k 2A proteínu synaptických vezikúl (SV2A),
transmembránový glykoproteín bol nájdený na presynaptickej úrovni v neurónoch a v endokrinných bunkách. Hoci presnú úlohu tohto proteínu je potrebné ešte objasniť, bolo preukázané, že moduluje exocytózu neurotransmiterov. Predpokladá sa, že väzba na SV2A predstavuje primárny mechanizmus antikonvulzívnej aktivity brivaracetamu.
Klinická účinnosť a bezpečnosť
Účinnosť brivaracetamu v prídavnej terapii parciálnych záchvatov (partial onset seisures-POS) bola
stanovená v 3 randomizovaných dvojito zaslepených, placebom kontrolovaných multicentrických štúdiách s fixnou dávkou u subjektov vo veku 16 rokov a starších. Denná dávka brivaracetamu sa
v týchto štúdiách pohybovala v rozmedzí 5 až 200 mg/deň. Všetky štúdie začínali základnou periódou trvajúcou 8 týždňov, nasledovanou 12 týždňov trvajúcou liečebnou periódou bez titrácie v zmysle zvyšovania dávky. 1 558 pacientov dostávalo liek zo štúdie, z toho 1 099 dostávalo brivaracetam. Kritériá pre zaradenie do štúdie vyžadovali, aby mali pacienti nekontrolované parciálne záchvaty napriek liečbe buď 1 alebo 2 súčasne podávanými antiepileptikami. Podmienkou bolo, aby pacienti
prekonali najmenej 8 parciálnych záchvatov počas základnej periódy. Primárnymi koncovými
ukazovateľmi pri štúdii fázy 3 bolo percento zníženia frekvencie POS oproti placebu a pomer respondérov s dosiahnutou 50 % odpoveďou založenou na 50 % znížení frekvencie POS od východiskového stavu.
Najčastejšie užívanými antiepileptikami na začiatku štúdie boli karbamazepín (40,6 %), lamotrigín (25,2 %), valproát (20,5 %), oxkarbazepín (16,0 %), topiramát (13,5 %), fenytoín (10,2 %) a levetiracetam (9,8 %). Medián východiskovej frekvencie záchvatov vo všetkých 3 štúdiách bol
9 záchvatov v priebehu 28 dní. Pacienti mali epilepsiu v priemere približne 23 rokov.
Výsledky účinnosti sú zhrnuté v tabuľke 2. Celkovo bol brivaracetam účinný pri prídavnej terapii
parciálnych záchvatov u pacientov vo veku 16 rokov a starších v dávke medzi 50 mg/deň a
200 mg/deň.
Tabuľka 2: Kľúčové výsledky účinnosti pre frekvenciu parciálnych záchvatov v priebehu 28 dní
Štúdia Placebo Brivaracetam
*štatisticky významné (hodnota p)
50 mg/deň 100 mg/deň 200 mg/deň
Štúdia NO1253(1)
n = 96 n = 101
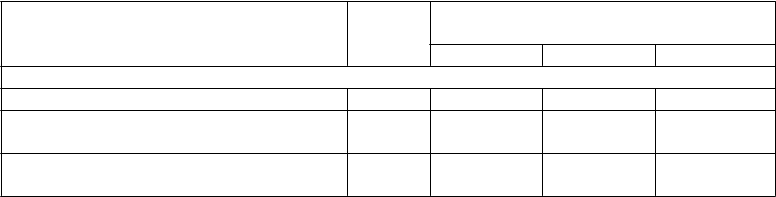
Dosiahnutie 50% odpovede respondéra 16,73 32,7* ~ ~
(p=0,008)
Percentuálne zníženie voči placebu (%) NA 22,0 ~ ~
(p=0,0040)
Štúdia NO1252(1)
n = 100 n = 99 n = 100
Dosiahnutie 50% odpovede respondéra 20,0 27,3
(p=0,372)
Percentuálne zníženie voči placebu (%) NA 9,2
(p=0,0274)
36,0(2) ~
(p=0,023)
20,5(2) ~
(p=0,0097)
Štúdia N01358
n = 259 n = 252 n = 249
Dosiahnutie 50% odpovede respondéra 21,6 ~ 38,9
(p<0,001)
37,8
(p<0,001)
Percentuálne zníženie voči placebu (%) Medián percentuálneho zníženia oproti východiskovému stavu
NA ~ 22,8* (p<0,001)
23,2* (p<0,001)

n = randomizovaní pacienti, ktorí dostali najmenej 1 dávku liečiva v štúdii
~ dávka nebola študovaná
* štatisticky významné
(1) Približne 20 % pacientov dostávalo súčasne levetiracetam
(2) Primárny výsledok pre N01252 nedosiahol štatistickú významnosť na základe sekvenčného
skúšania. Dávka 100 mg/deň bola nominálne významná.
V klinických štúdiách bolo zníženie frekvencie záchvatov vyššie oproti placebu pri dávke 100 mg/deň ako pri dávke 50 mg/deň. Na rozdiel od zvýšenia výskytu somnolencie a únavy v závislosti na dávke, mal brivaracetam pri dávke 50 mg/deň a 100 mg/deň podobný bezpečnostný profil vrátane
nežiaducich účinkov so vzťahom k CNS a pri dlhodobom užívaní.
Obrázok 1 ukazuje percento pacientov (s výnimkou pacientov súčasne užívajúcich levetiracetam)
podľa kategórie zníženia frekvencie POS v priebehu 28 dní od východiskového stavu vo všetkých
3 štúdiách. Pacienti s viac ako 25 % zvýšením parciálnych záchvatov sú uvedení úplne naľavo ako
„horší“. Pacienti so zlepšením percentuálneho zníženia frekvencie POS od východiskového stavu sú uvedení v 4 kategóriách napravo. Percento pacientov s najmenej 50 % znížením frekvencie záchvatov bolo 20,3 %, 34,2 %, 39,5 %, a 37,8 % pre placebo, zodpovedajúce 50 mg/deň, 100 mg/deň a
200 mg/deň.
Obrázok 1: Podiel pacientov s brivaracetamom a placebom podľa kategórie odpovedi záchvatovpo dobu 12 týždňov vo všetkých troch dvojito zaslepenými pivotnými štúdiami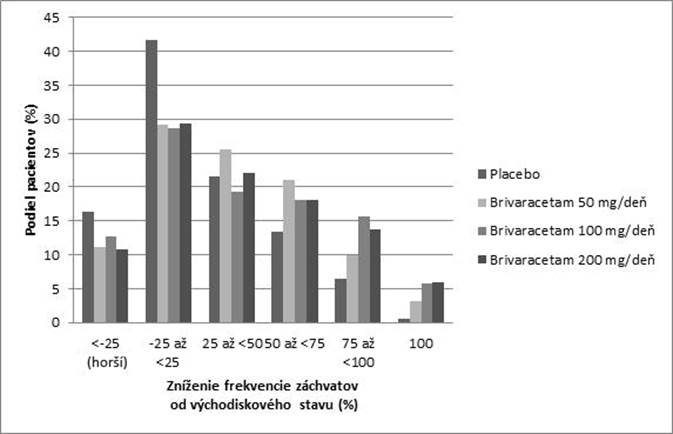
V súhrnnej analýze troch pivotných štúdií neboli pozorované žiadne rozdiely v účinnosti (merané ako
50 % odpoveď respondérov) v rozmedzí dávok 50 mg/deň až 200 mg/deň, keď je brivaracetam
kombinovaný s antiepileptikami vyvolávajúcimi alebo nevyvolávajúcimi indukciu enzýmov.
V klinických štúdiách dosiahlo stav bez záchvatov 2,5 % (4/161), 5,1 % (17/332) a 4,0 % (10/249)
pacientov s brivaracetamom v dávke zodpovedajúcej 50 mg/deň, 100 mg/deň a 200 mg/deň a to
v priebehu liečebnej periódy v trvaní 12 týždňov v porovnaní s 0,5 % (2/418) pacientov s placebom. Zlepšenie mediánu percentuálneho zníženia frekvencie záchvatov od začiatku liečby za 28 dní bolo pozorované u pacientov s typom záchvatov IC (sekundárne generalizované tonicko-klonické záchvaty)
vo východiskovom stave, liečených brivaracetamom (66,6 % (n=62), 61,2% (n=100) a 82,1 % (n=75)
z pacientov s brivaracetamom v zodpovedajúcej dávke 50 mg/deň, 100 mg/deň a 200 mg/deň v
porovnaní s placebom 33,3 % (n=115)).
Účinnosť brivaracetamu v monoterapii nebola ešte stanovená. Použitie brivaracetamu v monoterapii
sa neodporúča.
Liečba levetiracetamom
V 2 randomizovaných placebom kontrolovaných štúdiách fázy 3 sa levetiracetam podával ako
súbežné antiepileptikum u asi 20 % pacientov. Aj keď je počet subjektov limitovaný, nebol
u pacientov, ktorí súčasne užívali levetiracetam, pozorovaný žiadny prínos brivaracetamu oproti placebu, ktorý by reflektoval kompetíciu vo väzbovom mieste SVA2. Neboli zistené žiadne ďalšie okolnosti týkajúce sa bezpečnosti a znášanlivosti.
V 3. štúdii vopred špecifikovaná analýza preukázala účinnosť oproti placebu pre dávky 100 mg/deň a
200 mg/deň u pacientov predtým užívajúcich levetiracetam. Nižšia účinnosť pozorovaná u týchto pacientov v porovnaní s pacientmi neužívajúcimi levetiracetam bola pravdepodobne dôsledkom užívania vyššieho počtu predchádzajúcich antiepileptík a vyššej východiskovej hodnoty frekvencie záchvatov.
Starší pacienti (65 rokov a starší)
Tri pivotné, dvojito zaslepené, placebom kontrolované štúdie zahŕňali 38 pacientov vo veku 65 až
80 rokov. Aj keď sú údaje obmedzené, účinnosť bola porovnateľná s účinnosťou u mladších subjektov.
Otvorené predĺžené štúdie
Vo všetkých štúdiách bolo zaradených do dlhodobých, otvorených, predĺžených štúdií 81,7 %
pacientov, ktorí dokončili randomizované štúdie.. Od vstupu do randomizovaných štúdií bolo 5,3 % subjektov s brivaracetamom po dobu 6 mesiacov (n=1500) bez záchvatov v porovnaní s 4,6 % a 3,7 % u subjektov exponovaných po dobu 12 mesiacov (n=1188) a 24 mesiacov (n=847). Nakoľko však vysoké percento subjektov (26 %) prerušilo liečbu v otvorených štúdiách z dôvodu nedostatočnej účinnosti, mohlo dôjsť ku skresleniu, nakoľko subjekty, ktoré zostali v štúdii, reagovali lepšie ako tí, ktorí ju predčasne ukončili.
Pediatrická populácia
Účinnosť a znášanlivosť brivaracetamu nebola u pediatrických pacientov stanovená (pozri časť 4.2).
Brivaracetam bol hodnotený u týchto pacientov v krátkodobej otvorenej farmakokinetickej štúdií a prebiehajúcej otvorenej, predĺženej štúdií u 152 subjektov vo veku od 1 mesiaca do 16 rokov (pozri časť 5.2).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s brivaracetamom v jednej alebo vo viacerých podskupinách pediatrickej populácie s epilepsiou s parciálnymi záchvatmi.
5.2 Farmakokinetické vlastnosti
Brivaracetam filmom obalené tablety, injekčný/infúzny roztok a roztok na intravenóznu injekciu vykazujú identickú AUC, zatiaľ čo maximálna plazmatická koncentrácia je mierne vyššia po intravenóznom podaní. Brivaracetam vykazuje lineárnu a na čase nezávislú farmakokinetiku s nízkou intra- a interindividuálnou variabilitou a ďalej úplnou absorpciou, nízku väzbu na proteíny, renálnu exkréciu po rozsiahlej biotransformácii a farmakologicky inaktívne metabolity.
Absorpcia
Brivaracetam sa rýchlo a úplne absorbuje po perorálnom podaní a absolútna biologická dostupnosť je
približne 100 %. Medián tmax pre tablety užité bez jedla je 1 hodina (rozsah tmax je 0,25 až 3 hod.).
Súčasné podávanie s jedlom s vysokým obsahom tuku spomalilo rýchlosť absorpcie (medián tmax 3 h) a znížilo maximálnu plazmatickú koncentráciu (37 % pokles) brivaracetamu, pričom rozsah absorpcie zostal nezmenený.
Distribúcia
Brivaracetam sa slabo viaže (≤ 20 %) na plazmatické proteíny. Distribučný objem je 0,5 l/kg, čo je
hodnota blízka celkovému množstvu telesnej vody.
Bunkové membrány sú pre brivaracetam vysoko permeabilné z dôvodu jeho lipofílie (log P).
Biotransformácia
Brivaracetam je primárne metabolizovaný hydrolýzou svojej amidovej časti za vzniku zodpovedajúcej
karboxylovej kyseliny (približne 60 % eliminácie) a sekundárne hydroxyláciou propylového vedľajšieho reťazca (približne 30 % eliminácie). Hydrolýza amidovej časti , ktorá vedie k vzniku metabolitu povahy karboxylovej kyseliny (34 % dávky v moči) je podporovaná pečeňovou a mimopečeňovou amidázou. In vitro je hydroxylácia brivaracetamu sprostredkovaná v prvom rade CYP2C19. Obidva metabolity sú ďalej metabolizované za vzniku bežnej hydroxylovanej kyseliny, ktorá vzniká prevažne hydroxyláciou postranného propylového reťazca metabolitu kyseliny karboxylovej (hlavne prostredníctvom CYP2C9). In vivo u ľudí s neúčinnou mutáciou CYP2C19, sa tvorba hydroxymetabolitu znižuje 10x, zatiaľ čo samotný brivaracetam sa zvyšuje o 22 % alebo 42 % u jedincov s jednou alebo s obidvomi mutovanými alelami. Tri metabolity nie sú farmakologicky aktívne.
Eliminácia
Brivaracetam je primárne eliminovaný metabolizáciou a vylučovaním močom. Viac ako 95 % dávky,
vrátane metabolitov, sa vylučuje močom v priebehu 72 hodín po užití. Menej ako 1 % dávky sa vylučuje stolicou a menej ako 10 % brivaracetamu sa vylučuje bez zmeny močom. Terminálny plazmatický polčas (t1/2) je približne 9 hodín. Celkový plazmatický klírens bol u pacientov odhadnutý na 3,6 l/hod.
Linearita
Farmakokinetika je úmerná dávke od 10 mg do najmenej 600 mg.
Interakcia s liekmi
Brivaracetam je eliminovaný viacerými cestami, vrátane vylučovania obličkami, na CYP nezávislou
hydrolýzou a CYP sprostredkovanou oxidáciou. In vitro, brivaracetam nie je substrátom ľudského P- glykoproteínu (P-gp), proteínom rezistentným na viacpočetné lieky (MRP – multidrug resistance proteins)
1 a 2 a pravdepodobne ani organickým transportérom aniónu polypeptidu 1B1 (OATP1B1) a OATP1B3. Testy in vitro ukázali, že metabolizmus brivaracetamu by nemal byť významne ovplyvnený CYP inhibítormi (napr. CYP1A, 2C8, 2C9, 2C19, 2D6 a 3A4).
In vitro, brivaracetam nebol inhibítorom CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6, 3A4 ani transportérom P-gp, BCRP, BSEP MRP2, MATE-K, MATE-1, OATP1B1, OATP1B3, OAT1 a OCT1 v klinicky relevantných koncentráciách. In vitro, brivaracetam neindukoval CYP1A2.
Farmakokinetika u osobitných skupín pacientov
Starší pacienti (65 rokov a starší)
V štúdii u starších pacientov (vo veku 65 až 79 rokov; s klírensom kreatinínu 53 až 98 ml/min/1,73 m2), ktorí užívali brivaracetam v dávke 400 mg/deň s podávaním 2x denne, bol plazmatický polčas brivaracetamu 7,9 hodiny u skupiny vo veku 65 až 75 rokov a 9,3 hodiny u skupiny >75 rokov. Plazmatický klírens rovnovážneho stavu brivaracetamu bol podobný (0,76 ml/min/kg) ako u mladých zdravých mužov (0,83 ml/min/kg) (pozri časť 4.2).
Porucha funkcie obličiek
Štúdia u subjektov so závažnou poruchou funkcie obličiek (klírens kreatinínu <30 ml/min/1,73 m2 bez
nutnosti dialýzy) odhalila, že plazmatická AUC brivaracetamu bola stredne zvýšená (+21%) voči zdravým subjektom zatiaľ čo AUC kyseliny, hydroxymetabolitu a metabolitu hydroxykyseliny boli zvýšené 3x, 4x, a 21x (v uvedenom poradí). Renálny klírens týchto neaktívnych metabolitov bol znížený 10x. Metabolit hydroxykyseliny v predklinických štúdiách nevyvolal žiadne obavy zo strany bezpečnosti. Brivaracetam nebol študovaný u pacientov liečených hemodialýzou (pozri časť 4.2).
Porucha funkcie pečene
Farmakokinetická štúdia u subjektov s cirhózou pečene (Child-Pugh triedy A, B, a C) preukázala podobné zvýšenie pre expozíciu brivaracetamu bez ohľadu na závažnosť ochorenia (50 %, 57 % a 59
%) v pomere k zodpovedajúcim zdravým subjektom (pozri časť 4.2).
Pediatrická populácia
Vo farmakokinetickej štúdii u 99 subjektov vo veku 1 mesiac až <16 rokov, ktorí dostávali brivaracetam injekčný/infúzny roztok bolo preukázané, že plazmatické koncentrácie sú úmerné dávke vo všetkých vekových skupinách. Farmakokinetické populačné modelovanie ukázalo, že dávka
2,0 mg/kg dvakrát denne vedie
k rovnakej priemernej plazmatickej koncentrácii rovnovážneho stavu ako u dospelých užívajúcich
100 mg dvakrát denne.
Telesná hmotnosť
Bol odhadnutý 40 % pokles plazmatickej koncentrácie rovnovážneho stavu v rozsahu telesnej hmotnosti od 46 kg do 115 kg. To však nie je považované za klinicky významný rozdiel vo
farmakokinetike brivaracetamu.
Pohlavie
Nie sú žiadne klinicky významné rozdiely vo farmakokinetike brivaracetamu medzi pohlaviami.
Rasa
Farmakokinetika brivaracetamu nebola významne ovplyvnená rasou (kaukazská, ázijská) pri farmakokinetickom populačnom modelovaní u pacientov s epilepsiou. Počet pacientov s iným etnickým pôvodom bol obmedzený.
Farmakokinetický/farmakodynamický vzťah
EC50 (plazmatická koncentrácia brivaracetamu zodpovedajúca 50 % maximálneho účinku) bola
odhadnutá na 0,57 mg/l. Táto plazmatická koncentrácia je mierne nad mediánom expozície po podávaní brivaracetamu v dávkach 50 mg/deň. Ďalšie zníženie frekvencie záchvatov sa dostavuje pri zvýšení dávky na 100 mg/deň a dosahuje stabilizovaný stav pri dávke 200 mg/deň.
5.3 Predklinické údaje o bezpečnosti
Vo farmakologických štúdiách bezpečnosti mali prevládajúce účinky súvislosť s CNS (najmä
prechodná depresia CNS a zníženie spontánnej pohybovej aktivity) a boli pozorované pri násobkoch (vyšších než 50x) farmakologicky účinnej dávky brivaracetamu 2 mg/kg. Brivaracetam neovplyvnil učenie a funkciu pamäte.
Nálezy, ktoré neboli pozorované v klinických štúdiách, ale boli pozorované v toxikologických
štúdiách s opakovaným podávaním u psov pri expozícii podobnej ako pri klinickej plazmatickej AUC, boli hepatotoxické účinky (hlavne porfýria). Toxikologické údaje zhromaždené o brivaracetame a
o štrukturálne príbuzných látkach ale ukazujú, že sa pečeňové zmeny u psov vyvinuli prostredníctvom mechanizmov nerelevantných pre ľudí. Žiadne nežiaduce zmeny na pečeni neboli pozorované
u potkanov a opíc po dlhodobom podávaní brivaracetamu s expozíciou zreteľne prevyšujúcu AUC
expozíciu 5 až 42x. CNS príznaky u opíc (vyčerpanosť, strata rovnováhy, nemotorné pohyby) sa
vyskytli pri 64-násobkoch klinickej Cmax. Tieto účinky boli menej zrejmé v priebehu času.
Štúdie genotoxicity nepreukázali žiadnu mutagénnu alebo klastogénnu aktivitu. Štúdie kancerogenity u potkanov nepreukázali žiadny onkogénny potenciál, zatiaľ čo zvýšený výskyt hepatocelulárnych nádorov u samcov myší je považovaný za dôsledok známeho negenotoxického fenoménu, známeho
u hlodavcov, ktorého mechanizmus účinku sa vzťahuje k indukcii pečeňových enzýmov, podobnej ako po fenobarbitále.
Brivaracetam neovplyvnil fertilitu samíc ani samcov, nepreukázal sa žiadny teratogénny potenciál u potkanov alebo králikov. Embryotoxicita bola pozorovaná u králikov pri dávke brivaracetamu toxickej pre matky s expozíciou 8x vyššou než klinická AUC expozícia pri maximálnej odporúčanej dávke. U potkanov bolo preukázané, že brivaracetam ľahko prestupuje placentou a je vylučovaný do materského mlieka u dojčiacich samíc potkanov v koncentráciách podobných plazmatickým koncentráciám u matiek.
Brivaracetam u potkanov nepreukázal žiadny potenciál na závislosť. Štúdie u nedospelých zvierat
U nedospelých potkanov expozičné hladiny 6 -15násobnej klinickej AUC expozície brivaracetamu pri
maximálnej odporúčanej dávke vyvolali vývojové nežiaduce účinky (napr. mortalitu, klinické príznaky, zníženie telesnej hmotnosti a nižšiu hmotnosť mozgu). Neboli zaznamenané žiadne nežiaduce účinky na funkciu CNS, žiadne neuropatologické a histopatologické vyšetrenia mozgu.
U nedospelých psov boli brivaracetamom indukované zmeny pri dávke 100 mg/kg/deň spojené s 6- násobným zvýšením hladín AUC, podobným zmenám pozorovaným u dospelých zvierat. Neboli pozorované žiadne nežiaduce účinky v štandardných ukazovateľoch vývoja alebo maturácie.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
trihydrát octanu sodného
kyselina octová 99 % (na úpravu pH)
chlorid sodný voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
Zistilo sa, že po zriedení je injekčný/infúzny roztok brivaracetamu fyzikálne kompatibilný a chemicky
stabilný pri miešaní s rozpúšťadlami uvedenými v časti 6.6 po dobu 24 hodín a pri uchovávaní v PVC
alebo polyolefínových vakoch pri teplote až do 25 ºC. Z mikrobiologického hľadiska sa má liek použiť okamžite. Pokiaľ sa nepoužije okamžite, čas a podmienky uchovávania po otvorení pred použitím sú zodpovednosťou používateľa.
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
Podmienky na uchovávanie lieku po jeho nariedení, pozri časť 6.3.
6.5 Druh obalu a obsah baleniaInjekčný/infúzny roztok 10 mg/ml je balený v 6 ml sklenených injekčných liekovkách (typu I) so silikónovou brómbutylovou gumenou zátkou a Al/polypropylénovým odtrhávacím viečkom. Každá injekčná liekovka na jednorazové použitie obsahuje extrahovateľný objem minimálne 5 ml injekčného/infúzneho roztoku.
Jedna škatuľka obsahuje 10 injekčných liekoviek.
6.6 Špeciálne opatrenia na likvidáciuTento liek je iba na jednorazové použitie, všetok nepoužitý roztok sa má zlikvidovať.
Liek s výskytom častíc alebo zmenou farby sa nesmie použiť.
Injekčný/infúzny roztok briavaracetamu je fyzikálne kompatibilný a chemicky stabilný pri miešaní s nasledujúcimi rozpúšťadlami:
Rozpúšťadlá:
- roztok chloridu sodného 9 mg/ml (0,9%) na injekciu
- roztok glukózy 50 mg/ml (5%) na injekciu
- Ringerov roztok s laktátom na injekciu.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIUCB Pharma S.A.
Allée de la Recherche 60
B-1070 Brusel
Belgicko
8. REGISTRAČNÉ ČÍSLO(A)EU/1/15/1073/022
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 14. január 2016
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu