
votnícky pracovník vymieňať infúzny vak najmenej každých 96 hodín.
4.3 Kontraindikácie
- Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
- Dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaní
Neurologické udalosti
Pozorovali sa neurologické udalosti vrátane udalostí s fatálnym následkom. Medzi neurologické
udalosti 3. stupňa (CTCAE verzia 4.0) alebo vyššieho (závažné alebo život ohrozujúce) po začatí podávania blinatumomabu patrili encefalopatia, záchvaty, poruchy reči, poruchy vedomia, zmätenosť a dezorientácia a poruchy koordinácie a rovnováhy.
Medián času od začiatku liečby blinatumomabom po výskyt neurologickej udalosti bol 9 dní. Väčšina
udalostí po prerušení liečby vymizla.
U starších pacientov sa pozoroval vyšší výskyt neurologických toxicít vrátane kognitívnych porúch, encefalopatie a zmätenosti.
Pacienti s neurologickými prejavmi a príznakmi v anamnéze (ako sú závrat, hypoestézia, hyporeflexia, tremor, dysestézia, parestézia, poruchy pamäti) vykazovali vyšší výskyt neurologických udalostí (ako
sú tremor, závraty, stavy zmätenosti, encefalopatia a ataxia). Medián času do výskytu neurologickej udalosti bol u týchto pacientov 12 dní.
U pacientov s klinicky relevantnou patológiou centrálneho nervového systému (CNS) (napr. epilepsia, záchvat, paréza, afázia, mozgová príhoda, závažné poškodenia mozgu, demencia, Parkinsonova choroba, cerebrálne ochorenie, organický mozgový syndróm, psychóza) alebo u tých, ktorí majú túto patológiu v anamnéze, sú obmedzené skúsenosti, pretože z klinických skúšaní boli vylúčení. V tejto populácii je možnosť vyššieho rizika neurologických udalostí. Potenciálne prínosy liečby sa majú dôkladne zvážiť oproti riziku neurologických udalostí a pri podávaní BLINCYTA týmto pacientom treba postupovať so zvýšenou opatrnosťou.
S použitím blinatumomabu u pacientov s doloženou aktívnou ALL v CNS alebo cerebrospinálnej tekutine (cerebrospinal fluid, CSF) sú obmedzené skúsenosti. V klinických štúdiách však boli pacienti liečení blinatumomabom po odstránení blastov z CSF liečbou cielenou na CNS (ako je intratekálna chemoterapia). Keď sa teda CSF vyčistí, liečbu BLINCYTOM možno začať.
Odporúča sa, aby sa u týchto pacientov pred začatím liečby BLINCYTOM vykonalo neurologické vyšetrenie a aby boli títo pacienti klinicky sledovaní na prejavy a príznaky neurologických udalostí (napr. test písania). Manažment týchto prejavov a príznakov do ich vymiznutia môže vyžadovať buď dočasné prerušenie, alebo trvalé ukončenie liečby BLINCYTOM (pozri časť 4.2). V prípade záchvatu sa odporúča sekundárna profylaxia vhodnými antikonvulzívami (napr. levetiracetamom).
Infekcie
U pacientov dostávajúcich blinatumomab sa pozorovali ťažké infekcie vrátane sepsy, pneumónie,
bakteriémie, oportúnnych infekcií a infekcie v mieste katétra, z ktorých niektoré boli život ohrozujúce alebo fatálne. U pacientov s výkonnostným stavom 2 podľa Eastern Cooperative Oncology Group
(ECOG) sa vyskytlo na začiatku liečby viac závažných infekcií v porovnaní s pacientmi
s výkonnostným stavom ECOG < 2. U pacientov s aktívnou nekontrolovanou infekciou sú s liečbou
BLINCYTOM obmedzené skúsenosti.
Pacientov dostávajúcich BLINCYTO treba klinicky sledovať na prejavy a príznaky infekcie a vhodne liečiť. Manažment infekcií môže vyžadovať buď dočasné prerušenie, alebo ukončenie liečby BLINCYTOM (pozri časť 4.2).
Syndróm uvoľňovania cytokínov a reakcie na infúziu
U pacientov dostávajúcich BLINCYTO bol hlásený syndróm uvoľňovania cytokínov (cytokine release
syndrome, CRS), ktorý môže byť život ohrozujúci alebo fatálny (stupeň ≥ 4) (pozri časť 4.8).
Medzi závažné nežiaduce udalosti, ktoré môžu byť prejavmi a príznakmi CRS, patrili horúčka, asténia,
bolesť hlavy, hypotenzia, zvýšený celkový bilirubín a nauzea; tieto udalosti menej často viedli
k ukončeniu liečby BLINCYTOM. Medián času do výskytu príhody CRS bol 2 dni. Pacientov treba
pozorne sledovať na prejavy a príznaky týchto udalostí.
S CRS často súvisela diseminovaná intravaskulárna koagulácia (disseminated intravascular coagulation, DIC) a syndróm kapilárneho presakovania (capillary leak syndrome, CLS, napr. hypotenzia, hypoalbuminémia, edém a hemokoncentrácia) (pozri časť 4.8). Pacientov, u ktorých sa vyskytne syndróm kapilárneho presakovania, treba okamžite liečiť.
V kontexte CRS bola menej často hlásená hemofagocytová lymfohistiocytóza/syndróm aktivácie makrofágov (haemophagocytic lymphohistiocytosis/macrophage activation syndrome, HLH/MAS).
Reakcie na infúziu môžu byť klinicky nerozoznateľné od prejavov CRS (pozri časť 4.8). Reakcie na infúziu boli zvyčajne rýchle, pričom sa vyskytli do 48 hodín po začatí infúzie. U niektorých pacientov sa však zaznamenal nástup reakcií na infúziu oneskorene alebo v neskorších cykloch. Pacientov treba na reakcie na infúziu dôkladne sledovať, predovšetkým počas iniciácie prvého a druhého liečebného
cyklu, a vhodne liečiť. Na zníženie pyrexie počas prvých 48 hodín každého cyklu sa odporúča použitie antipyretík (napr. paracetamolu). Manažment týchto príhod môže vyžadovať buď dočasné prerušenie, alebo ukončenie liečby BLINCYTOM (pozri časť 4.2).
Syndróm z rozpadu nádoru
U pacientov dostávajúcich BLINCYTO sa pozoroval syndróm z rozpadu nádoru (tumour lysis
syndrome, TLS), ktorý môže byť život ohrozujúci alebo fatálny (stupeň ≥ 4).
Na prevenciu a liečbu TLS počas terapie BLINCYTOM sa majú použiť vhodné profylaktické opatrenia vrátane agresívnej hydratácie a antihyperurikemickej terapie (ako je alopurinol alebo rasburikáza), predovšetkým u pacientov s vyššou leukocytózou alebo vysokým nádorovým zaťažením. Počas prvých 48 hodín po prvej infúzii treba pacientov dôkladne sledovať na prejavy a príznaky TLS vrátane funkcie obličiek a rovnováhy tekutín v tele. V klinických štúdiách pacienti so stredne závažným poškodením funkcie obličiek vykazovali zvýšený výskyt TLS v porovnaní s pacientmi
s miernym poškodením funkcie obličiek alebo normálnou funkciou obličiek. Manažment týchto udalostí môže vyžadovať buď dočasné prerušenie, alebo ukončenie liečby BLINCYTOM (pozri časť
4.2).
Neutropénia a febrilná neutropénia
U pacientov dostávajúcich BLINCYTO sa pozorovala neutropénia a febrilná neutropénia vrátane život
ohrozujúcich prípadov. Laboratórne parametre (vrátane, ale nie výlučne počtu bielych krviniek a absolútneho počtu neutrofilov) sa majú počas infúzie BLINCYTA pravidelne sledovať, predovšetkým
počas prvých 9 dní prvého cyklu, a vhodne liečiť.
Zvýšená hladina pečeňových enzýmov
Liečba BLINCYTOM sa spájala s prechodným zvýšením hladiny pečeňových enzýmov. Väčšina
týchto udalostí sa pozorovala v priebehu prvého týždňa od začatia liečby a nevyžadovala prerušenie
ani ukončenie liečby BLINCYTOM (pozri časť 4.8).
Pred začiatkom a počas liečby BLINCYTOM, predovšetkým počas prvých 48 hodín prvých 2 cyklov, treba sledovať hladiny alanínaminotransferázy (ALT), aspartátaminotransferázy (AST), gamaglutamyltransferázy (GGT) a celkový bilirubín v krvi. Manažment týchto udalostí môže vyžadovať buď dočasné prerušenie, alebo ukončenie liečby BLINCYTOM (pozri časť 4.2).
Leukoencefalopatia vrátane progresívnej multifokálnej leukoencefalopatie
U pacientov dostávajúcich BLINCYTO, predovšetkým u pacientov s predchádzajúcou liečbou
kraniálnym ožarovaním a chemoterapiou na liečbu leukémie (vrátane systémového vysokodávkového metotrexátu alebo intratekálne podaného cytarabínu), sa pozorovali zmeny v kraniálnej magnetickej rezonancii (MRI) svedčiace o leukoencefalopatii. Klinický význam týchto zmien v zobrazení nie je známy.
Vzhľadom na možnosť progresívnej multifokálnej leukoencefalopatie (PML) sa pacienti majú sledovať na jej prejavy a príznaky. Pri podozrení na tieto udalosti zvážte konzultáciu s neurológom, MRI mozgu a vyšetrenie cerebrospinálnej tekutiny (CSF), pozri časť 4.8.
Imunizácie
Bezpečnosť imunizácie vakcínami so živým vírusom počas terapie BLINCYTOM alebo po nej sa
neskúmala. Očkovanie vakcínami so živým vírusom sa neodporúča najmenej 2 týždne pred začatím liečby BLINCYTOM, počas tejto liečby a do úpravy počtu B-lymfocytov na normálne rozsahy po poslednom liečebnom cykle.
Vzhľadom na možnú depléciu B-lymfocytov u novorodencov po expozícii blinatumomabu počas gravidity sa novorodenci majú sledovať na depléciu B-lymfocytov a očkovanie vakcínami so živým vírusom treba odložiť, kým sa počet B-lymfocytov dojčaťa neupraví (pozri časť 4.6).
Antikoncepcia
Ženy, ktoré môžu otehotnieť, musia používať účinnú antikoncepciu počas liečby BLINCYTOM a
najmenej 48 hodín po nej (pozri časť 4.6).
Chyby v predpisovaní liečby
Pri liečbe BLINCYTOM sa pozorovali chyby v predpisovaní liečby. Je veľmi dôležité, aby sa na
minimalizovanie chýb v predpisovaní liečby (vrátane poddávkovania a predávkovania) dôsledne dodržiavali pokyny na prípravu (vrátane rekonštitúcie a riedenia) a podávanie (pozri časť 4.2).
Pomocné látky so známymúčinkom
Tento liek obsahuje menej ako 1 mmol (23 mg) sodíka počas 24-hodinovej infúzie, t. j.
v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne formálne interakčné štúdie. Výsledky z testu in vitro s ľudskými hepatocytmi
svedčia o tom, že blinatumomab neovplyvnil aktivity enzýmov CYP450.
Začiatok liečby BLINCYTOM spôsobuje prechodné uvoľňovanie cytokínov počas prvých dní liečby, čo môže potlačiť enzýmy CYP450. Pacientov dostávajúcich lieky, ktoré sú substráty pre CYP450 a transportéry s úzkym terapeutickým indexom, treba počas tohto obdobia sledovať z hľadiska výskytu nežiaducich účinkov (napr. warfarín) alebo koncentrácií liečiva (napr. cyklosporín). Dávka súbežne užívaného lieku sa má podľa potreby upraviť.
4.6 Fertilita, gravidita a laktácia
Gravidita
Štúdie s blinatumomabom zamerané na reprodukčnú toxicitu sa nevykonali. V embryonálno-fetálnej
vývojovej štúdii toxicity vykonanej na myšiach náhradná myšia molekula prenikla cez placentu a nevyvolala embryotoxicitu ani teratogenitu (pozri časť 5.3). Predpokladané deplécie B- a T- lymfocytov sa pri gravidných myšiach zaznamenali, ale hematologické účinky na plody neboli hodnotené.
Nie sú k dispozícii údaje o použití blinatumomabu u gravidných žien.
Blinatumomab sa počas gravidity nemá užívať, pokiaľ potenciálny prínos nepreváži potenciálne riziko pre plod.
Ženy vo fertilnom veku musia počas liečby blinatumomabom a najmenej 48 hodín po nej používať účinnú antikoncepciu (pozri časť 4.4).
V prípade expozície počas gravidity možno vzhľadom na farmakologické vlastnosti lieku očakávať depléciu B-lymfocytov u novorodencov. V dôsledku toho treba novorodencov sledovať na depléciu B- lymfocytov a očkovanie vakcínami obsahujúcimi živý vírus sa má odložiť, kým sa počet B- lymfocytov dojčaťa neupraví (pozri časť 4.4).
D
ojčenie
Nie je známe, či sa blinatumomab alebo metabolity vylučujú do materského mlieka. Vzhľadom na
jeho farmakologické vlastnosti nemožno vylúčiť riziko pre dojčené dieťa. V dôsledku toho je v rámci bezpečnostného opatrenia dojčenie počas liečby blinatumomabom a najmenej 48 hodín po nej kontraindikované.
Fertilita
Nevykonali sa žiadne štúdie na vyhodnotenie účinkov blinatumomabu na fertilitu. V 13-týždenných
štúdiách toxicity s náhradnou myšou molekulou neboli žiadne nežiaduce účinky na reprodukčné orgány myších samčekov alebo samičiek (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Blinatumomab má veľký vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Môže sa vyskytnúť zmätenosť a dezorientácia, poruchy koordinácie a rovnováhy, riziko záchvatov a poruchy vedomia (pozri časť 4.4). Kým sa podáva blinatumomab, vzhľadom na možnosť neurologických udalostí pacienti užívajúci blinatumomab nemajú viesť vozidlá, zamestnávať sa v rizikových povolaniach alebo vykonávať nebezpečné činnosti, ako je vedenie alebo obsluhovanie ťažkých alebo potenciálne nebezpečných strojov. Pacientov treba upozorniť, že sa u nich môžu vyskytnúť neurologické udalosti.
4.8 Nežiaduce účinky
Súhrn profilubezpečnosti
Nežiaduce reakcie uvedené v tejto časti sa zistili v pivotnej klinickej štúdii (N = 189).
Medzi najzávažnejšie nežiaduce reakcie, ktoré sa môžu vyskytnúť počas liečby blinatumomabom, patria: infekcie (31,7 %), neurologické udalosti (16,4 %), neutropénia/febrilná neutropénia (15,3 %), syndróm uvoľňovania cytokínov (0,5 %) a syndróm z rozpadu nádoru (0,5 %).
Najčastejšie nežiaduce reakcie boli: reakcie súvisiace s infúziou (67,2 %), infekcie (63,0 %), horúčka
(59,8 %), bolesť hlavy (34,4 %), febrilná neutropénia (28 %), periférny edém (25,9 %), nauzea
(24,3 %), hypokaliémia (23,8 %), zápcha (20,6 %), anémia (20,1 %), kašeľ (18,5 %), diarea (18,0 %), tremor (17,5 %), neutropénia (17,5 %), bolesť brucha (16,9 %), insomnia (15,3 %), únava (15,3 %) a
triaška (15,3 %).
Zoznam nežiaducich reakcií zoradených dotabuľky
Nežiaduce reakcie sú uvedené nižšie podľa triedy orgánových systémov a kategórie frekvencií.
Kategórie frekvencií boli stanovené na základe približnej miery výskytu hlásenej pri každej nežiaducej reakcii v pivotnej klinickej štúdii (N = 189). V rámci každej triedy orgánových systémov sú nežiaduce
reakcie usporiadané v poradí klesajúcej závažnosti.
T
rieda orgánových
systémov MedDRA
V
eľmi časté
(
≥ 1/10)
Č
asté
(
≥ 1/100 až < 1/10)
Menej časté
(
≥ 1/1 000 až
< 1/100)
Infekcie a nákazy Bakteriálne infekciea, b Fungálne infekciea, b Vírusové infekciea, b
Iné patogénne infekcieb
Sepsa
Pneumónia
Poruchy krvi
a lymfatického systému
Febrilná neutropénia
Anémia Neutropénia Trombocytopénia Leukopénia
Leukocytóza
Lymfopénia
T
rieda orgánových systémov MedDRA
Poruchy imunitného systému
Poruchy metabolizmu a výživy
Veľmi časté
(≥ 1/10)
Syndróm uvoľňovania cytokínova Hypokaliémia Hypomagneziémia Hyperglykémia Znížená chuť do jedla
Časté
(≥ 1/100 až < 1/10)
Cytokínová ,,búrka“ Hypersenzitivita Hypofosfatémia Hypoalbuminémia Syndróm z rozpadu nádoru
Menej časté
(≥ 1/1 000 až
< 1/100)
Psychické poruchy Insomnia Stavy zmätenostia
Dezorientácia
Poruchy nervového systému
Poruchy srdca
a srdcovej činnosti
Bolesť hlavy Tremora Závrat
Encefalopatiaa
Afázia
Parestézia
Kŕče
Kognitívne poruchy
Poruchy pamäti
Tachykardia
Poruchy ciev Hypotenzia Syndróm kapilárneho presakovania
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Poruchy kože a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Celkové poruchy
a reakcie v mieste podania
Laboratórne
a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
Kašeľ
Nauzea Konstipácia Diarea
Bolesť brucha
Vracanie
Vyrážka
Bolesť chrbta Bolesť končatiny Artralgia
Bolesť kostí
Horúčka Periférny edém Triaška
Únava
Bolesť na hrudi
Zvýšená alanínaminotransferázaa Zvýšená aspartátaminotransferázaa
Reakcie súvisiace s infúziou (a súvisiace príznaky vrátane sipotu, sčervenania, opuchu tváre, dyspnoe,
hypotenzie a hypertenzie)
Edém
Znížená hladina imunoglobulínov
Zvýšená hladina bilirubínu v krvi
Zvýšená hladina
pečeňových enzýmov
(gamaglutamyltransferázy)
a Ďalšie informácie sú uvedené v „Opise vybraných nežiaducich reakcií“
b Skupinové termíny MedDRA vysokej úrovne (MedDRA, verzia 16.1)
O
pis vybraných nežiaducich reakcií
N
eurologické udalosti
V pivotnej klinickej štúdii (N = 189) sa u 51,9 % pacientov vyskytla jedna alebo viac neurologických nežiaducich reakcií (vrátane psychických porúch), pričom sa týkali najmä centrálneho nervového systému. Závažné neurologické nežiaduce reakcie a nežiaduce reakcie stupňa ≥ 3 sa pozorovali u
16,4 % a 12,7 % pacientov, v danom poradí, pričom najčastejšie boli encefalopatia, tremor a stav zmätenosti. Hlásená bola fatálna encefalopatia, väčšina neurologických udalostí (74,5 %) však bola
klinicky reverzibilná a odznela po prerušení liečby BLINCYTOM. Medián času do výskytu neurologických udalostí bol 9 dní. Klinický manažment neurologických udalostí, pozri časť 4.4.
Infekcie
U pacientov liečených BLINCYTOM boli hlásené život ohrozujúce alebo fatálne (stupeň ≥ 4) vírusové, bakteriálne a fungálne infekcie. Okrem toho sa pozorovali reaktivácie vírusovej infekcie (napr. Polyoma (BK) vírus). U pacientov s výkonnostným stavom podľa ECOG 2 na začiatku liečby sa zaznamenal vyšší výskyt závažných infekcií v porovnaní s pacientmi s výkonnostným stavom podľa ECOG < 2. Klinický manažment infekcií, pozri časť 4.4.
Syndróm uvoľňovania cytokínov (CRS)
V pivotnej klinickej štúdii (N = 189) boli závažné reakcie CRS hlásené u 0,5 % pacientov s mediánom
času do výskytu 2 dni. Klinický manažment CRS, pozri časť 4.4.
Zvýšená hladina pečeňových enzýmov
V pivotnej klinickej štúdii (N = 189) bola približne u 27,5 % pacientov hlásená zvýšená hladina pečeňových enzýmov. Závažné nežiaduce reakcie a nežiaduce reakcie stupňa ≥ 3 (ako je zvýšená ALT, zvýšená AST a zvýšený bilirubín v krvi) sa zaznamenali u 2,1 % a 15,3 % pacientov, v danom poradí. Medián času do výskytu prvej udalosti bol 3 dni od iniciácie liečby BLINCYTOM. Trvanie hepatálnych nežiaducich reakcií bolo celkovo krátke a tieto reakcie rýchlo odozneli, často pri pokračujúcej nepretržitej liečbe BLINCYTOM. Klinický manažment zvýšenej hladiny pečeňových enzýmov, pozri časť 4.4.
Leukoencefalopatia vrátane progresívnej multifokálnej leukoencefalopatie
Hlásená bola leukoencefalopatia. U pacientov s nálezmi MRI/CT mozgu zodpovedajúcimi leukoencefalopatii sa zároveň vyskytli závažné nežiaduce udalosti vrátane stavu zmätenosti, tremoru, kognitívnych porúch, encefalopatie a kŕčov. Hoci možnosť rozvoja progresívnej multifokálnej leukoencefalopatie (PML) existuje, v pivotnej štúdii nebol hlásený žiadny prípad PML.
Pediatrická populácia
U pediatrických pacientov sú obmedzené skúsenosti. Liek BLINCYTO bol hodnotený u pediatrických
pacientov s relapsujúcou alebo refraktérnou B-prekurzorovou ALL v štúdii fázy I/II zameranej na zvyšovanie/hodnotenie dávky. V dávke vyššej ako odporúčaná dávka pre dospelých pacientov sa
vyskytol prípad fatálneho zlyhania srdca v kontexte život ohrozujúceho syndrómu uvoľňovania
cytokínov (CRS) a syndrómu z rozpadu nádoru (TLS), pozri časť 4.4.
Iné osobitné populácie
U pacientov vo veku ≥ 75 rokov sú obmedzené skúsenosti s BLINCYTOM. Celkovo bola bezpečnosť
podobná medzi staršími pacientmi (vo veku ≥ 65 rokov) a pacientmi vo veku menej ako 65 rokov liečených BLINCYTOM. Starší pacienti však môžu byť väčšmi náchylní na závažné neurologické udalosti, ako sú kognitívne poruchy, encefalopatia a zmätenosť.
U pacientov so závažným poškodením funkcie obličiek sa bezpečnosť BLINCYTA neskúmala.
ImunogenicitaV pivotnej klinickej štúdii (N = 189) malo menej ako 1,4 % pacientov liečených blinatumomabom
pozitívny test na väzbové a neutralizačné protilátky proti blinatumomabu. Všetci pacienti
s pozitívnymi výsledkami testov na väzbové protilátky mali aj pozitívne výsledky testov na
neutralizačné protilátky proti blinatumomabu. Tvorba protilátok proti blinatumomabu by mohla
ovplyvniť farmakokinetiku blinatumomabu.
Ak je podozrenie na tvorbu protilátok proti blinatumomabu s klinicky významným účinkom, obráťte sa na držiteľa rozhodnutia o registrácii ohľadne vyšetrenia protilátok. Kontaktné údaje sú uvedené v časti 6 písomnej informácie pre používateľa.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili

akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného v
Prílohe V.4.9 PredávkovanieZaznamenali sa prípady predávkovania vrátane jedného pacienta, ktorý dostal 133-násobok odporúčanej terapeutickej dávky BLINCYTA podanej za krátke obdobie. Predávkovania mali za následok nežiaduce reakcie, ktoré boli zhodné s reakciami pozorovanými pri odporúčanej terapeutickej dávke, a zahŕňali horúčku, tremor a bolesť hlavy. V prípade predávkovania treba infúziu dočasne prerušiť a pacienti sa majú sledovať. O obnovení liečby BLINCYTOM v správnej
terapeutickej dávke sa má uvažovať po ústupe všetkých toxicít a najskôr 12 hodín po prerušení infúzie
(pozri časť 4.2).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Cytostatiká – iné cytostatiká, ATC kód: L01XC19.
Mechanizmus účinkuBlinatumomab je bišpecifický T-lymfocyty angažujúci protilátkový konštrukt vo formáte BiTE (z
anglického bispecific T-cell engager antibody construct), ktorý sa špecificky viaže na CD19
s expresiou na povrchu buniek z B línie a na CD3 s expresiou na povrchu T-lymfocytov. Aktivuje endogénne T-lymfocyty spojením CD3 s antigénovým receptorom T-lymfocytov (TCR) v komplexe s
CD19 na benígnych a malígnych B-lymfocytoch. Protinádorový účinok imunoterapie
blinatumomabom nezávisí od T-lymfocytov nesúcich špecifický TCR alebo od peptidových antigénov prezentovaných nádorovými bunkami, ale je polyklonálnej povahy a nezávislý od molekúl ľudského
leukocytového antigénu (HLA) na cieľových bunkách. Blinatumomab sprostredkúva tvorbu
cytolytickej synapsy medzi T-lymfocytom a nádorovou bunkou, pričom uvoľňuje proteolytické
enzýmy, aby usmrtil tak proliferujúce, ako aj pokojové cieľové bunky. Blinatumomab sa spája
s prechodnou zvýšenou reguláciou molekúl bunkovej adhézie, produkciou cytolytických proteínov,
uvoľňovaním zápalových cytokínov a proliferáciou T-lymfocytov a vedie k eliminácii buniek CD19+.
Farmakodynamické účinky
U skúmaných pacientov sa pozorovali konzistentné farmakodynamické odpovede v imunite. Počas
kontinuálnej intravenóznej infúzie počas 4 týždňoch bola farmakodynamická odpoveď
charakteristická aktiváciou a iniciálnou redistribúciou T-lymfocytov, rýchlou depléciou periférnych B- lymfocytov a prechodným zvýšením hladiny cytokínov.
Redistribúcia periférnych T-lymfocytov (t. j. adhézia T-lymfocytov k endotelu krvných ciev a/alebo transmigrácia do tkaniva) nastala po začatí infúzie blinatumomabu alebo zvýšení dávky. Počet T- lymfocytov sprvu v priebehu 1 až 2 dní klesol a potom sa vrátil na východiskové hodnoty do 7 až
14 dní u väčšiny pacientov. U niekoľkých pacientov sa pozoroval nárast počtu T-lymfocytov nad východiskové hodnoty (expanzia T-lymfocytov).
Počet periférnych B-lymfocytov rýchlo klesol na nezistiteľnú úroveň počas liečby v dávkach
≥ 5 µg/m2/deň alebo ≥ 9 µg/deň u väčšiny pacientov. Počas 2-týždňového obdobia bez liečby medzi liečebnými cyklami sa nepozorovala normalizácia počtu periférnych B-lymfocytov. Neúplná deplécia
B-lymfocytov nastala v dávkach 0,5 µg/m2/deň a 1,5 µg/m2/deň a u niekoľkých pacientov
neodpovedajúcich na liečbu pri vyšších dávkach.
Merali sa hladiny cytokínov IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, TNF-α a IFN-γ a najviac zvýšené boli hladiny IL-6, IL-10 a IFN-γ. Prechodné zvýšenie hladín cytokínov sa pozorovalo v prvých dvoch dňoch po začatí infúzie blinatumomabu. Zvýšené hladiny cytokínov sa vrátili na východiskové hodnoty do 24 až 48 hodín počas infúzie. V ďalších liečebných cykloch sa zvýšenie hladín cytokínov vyskytlo u menšieho počtu pacientov s nižšou intenzitou v porovnaní s prvými 48 hodinami prvého liečebného cyklu.
Klinická účinnosť abezpečnosť
Počas klinických skúšaní bolo vystavených BLINCYTU spolu 225 pacientov vo veku ≥ 18 rokov
s relapsujúcou alebo refraktérnou B-prekurzorovou ALL.
Liek BLINCYTO sa hodnotil v otvorenej multicentrickej štúdii fázy II s jedným ramenom so
189 pacientmi. Vhodní pacienti boli vo veku ≥ 18 rokov s relapsujúcou alebo refraktérnou B- prekurzorovou ALL s negatívnym chromozómom Philadelphia (relapsujúcou s trvaním prvej remisie Ł 12 mesiacov v prvej záchrannej liečbe, relapsujúcou alebo refraktérnou po prvej záchrannej liečbe alebo relapsujúcou počas 12 mesiacov od alogénnej transplantácie krvotvorných kmeňových buniek (HSCT) a s ł 10 % blastov v kostnej dreni).
Pacienti boli premedikovaní povinnou profylaktickou liečbou cerebrospinálnej tekutiny skladajúcou sa z intratekálneho režimu počas 1 týždňa pred začiatkom liečby blinatumomabom podľa ústavných
alebo národných smerníc. Liek BLINCYTO bol podávaný vo forme kontinuálnej intravenóznej infúzie. V prvom cykle bola začiatočná dávka 9 µg/deň 1. týždeň, potom 28 µg/deň zvyšné 3 týždne. Cieľová dávka 28 µg/deň bola podaná v 2. cykle a v ďalších cykloch od 1. dňa každého cyklu.
V prípade nežiaducich udalostí bola možná úprava dávky. Liečená populácia zahŕňala 189 pacientov, ktorí dostali najmenej 1 infúziu BLINCYTA; priemerný počet cyklov na pacienta bol 1,6. Pacienti,
ktorí na BLINCYTO odpovedali, ale neskôr sa u nich vyskytol relaps, mali možnosť opakovanej liečby BLINCYTO. Medzi liečenými pacientmi bol medián veku 39 rokov (rozpätie: 18 až 79 rokov vrátane 25 pacientov vo veku ≥ 65 rokov), 64 zo 189 (33,9 %) podstúpili transplantácie krvotvorných
kmeňových buniek (HSCT) pred používaním BLINCYTA a 32 zo 189 (16,9 %) absolvovali viac ako
2 predchádzajúce záchranné liečby.
Primárnym koncovým ukazovateľom bola miera úplnej remisie/úplnej remisie s čiastočnou obnovou krvotvorby (CR/CRh*) počas 2 cyklov liečby BLINCYTOM. Osemdesiatjeden zo 189 (42,9 %) pacientov dosiahlo CR/CRh* v priebehu prvých 2 liečebných cyklov, pričom väčšina odpovedí (64 z
81) sa vyskytla v priebehu 1 cyklu liečby. U staršej populácie (vo veku ≥ 65 rokov) 11 z 25 pacientov
(44,0 %) dosiahlo CR/CRh* v priebehu prvých 2 liečebných cyklov (bezpečnosť u starších pacientov,
pozri časť 4.8). Štyria pacienti dosiahli CR počas konsolidačných cyklov, čo viedlo ku kumulatívnej
miere CR 35,4 % (67/189; 95 % CI: 28,6 % – 42,7 %). Tridsaťdva zo 189 (17 %) pacientov podstúpilo alogénnu HSCT počas CR/CRh* indukovanej BLINCYTOM (pozri tabuľku 1).
Tabuľka 1. Výsledky účinnosti u pacientov vo veku ≥ 18 rokov s relapsujúcou alebo refraktérnou B-prekurzorovou akútnou lymfoblastovou leukémiou (ALL) s negatívnym chromozómom Philadelphia
 n ( %)
n = 189
95 % CI
n ( %)
n = 189
95 % CI
Úplná remisia (CR)1/Úplná remisia s čiastočnou
obnovou krvotvorby (CRh*)2
81 (42,9 %) [35,7 % – 50,2 %]
CR 63 (33,3 %) [26,7 % – 40,5 %]
CRh* 18 (9,5 %) [5,7 % – 14,6 %]
Hypoplastická alebo aplastická kostná dreň bez blastov3
17 (9 %) [5,3 % – 14,0 %]
Čiastočná remisia4 5 (2,6 %) [0,9 % – 6,1 %] Prežívanie bez relapsu (RFS) pri CR/CRh* 5,9 mesiaca [4,8 až 8,3 mesiaca]
Celkové prežívanie 6,1 mesiaca [4,2 až 7,5 mesiaca]
1. CR bola definovaná ako Ł 5 % blastov v kostnej dreni, žiadny dôkaz o ochorení a úplná obnova počtu
krviniek v periférnej krvi (trombocyty > 100 000/mikroliter a absolútny počet neutrofilov [ANC]
> 1 000/mikroliter).
2. CRh* bola definovaná ako Ł 5 % blastov v kostnej dreni, žiadny dôkaz o ochorení, čiastočná obnova počtu
krviniek v periférnej krvi (trombocyty > 50 000/mikroliter a ANC > 500/mikroliter).
3. Hypoplastická alebo aplastická kostná dreň bez blastov bola definovaná ako blasty v kostnej dreni Ł 5 %, žiadny dôkaz o ochorení, nedostatočná obnova počtu krviniek v periférnej krvi: trombocyty
≤ 50 000/mikroliter a/alebo ANC ≤ 500/mikroliter
4. Čiastočná remisia bola definovaná ako blasty v kostnej dreni 6 % až 25 % najmenej s 50 % znížením od
začiatku liečby
Vo vopred špecifikovanej exploratórnej analýze malo 60 zo 73 pacientov hodnotiteľných na MRD s
CR/CRh* (82,2 %) aj odpoveď MRD (definovanú ako minimálne reziduálne ochorenie (MRD) na základe PCR < 1 x 10-4).
Pacienti s predchádzajúcou alogénnou HSCT mali podobné miery odpovedí ako pacienti bez predchádzajúcej HSCT, starší pacienti mali podobné miery odpovedí ako mladší pacienti a na základe počtu línií predchádzajúcej záchrannej liečby sa v mierach remisie nepozoroval podstatný rozdiel.
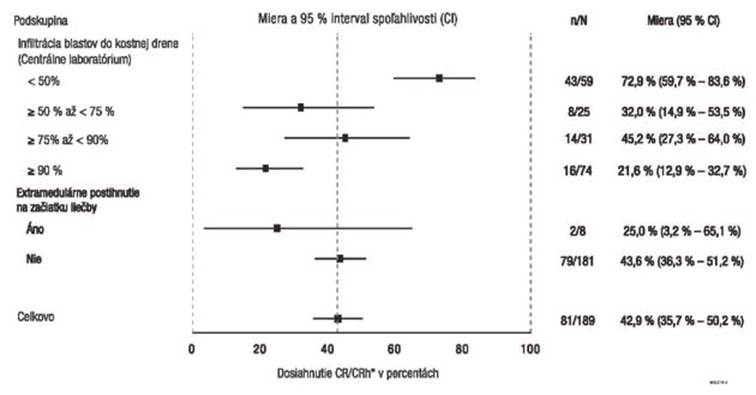
U pacientov s extramedulárnym ochorením bez postihnutia CNS/semenníkov (definovaným ako najmenej 1 lézia ≥ 1,5 cm) pri skríningu (N = 8/189) boli miery klinickej odpovede (25 % [95 % CI
3,2 – 65,1] nižšie v porovnaní s pacientmi bez dôkazu o extramedulárnom postihnutí (N = 181, 43,6 %
[95 % CI 36,3 – 51,2]) (pozri obrázok 1).
Pacienti s najvyšším nádorovým zaťažením stanoveným na základe percentuálneho podielu blastov v kostnej dreni na začiatku liečby (≥ 90 %) ešte stále mali klinicky významnú odpoveď s mierou CR/CRh* 21,6 % (CI 12,9 – 32,7) (pozri obrázok 1). Pacienti s nízkym nádorovým zaťažením
(< 50 %) odpovedali na liečbu blinatumomabom najlepšie s mierou CR/CRh* 72,9 % (CI 59,7 – 83,6).
O
brázok 1. Znázornenie „Forest plot“ miery CR/CRh* počas prvých dvoch cyklov v štúdii
MT103-211
(
súbor na primárnu analýzu)
Bezpečnosť a účinnosť blinatumomabu sa hodnotila v otvorenej multicentrickej štúdii fázy II so stupňovaním dávky s 36 pacientmi (vo veku ≥ 18 rokov s relapsom B-prekurzorovej ALL aspoň po indukcii a konsolidácii alebo s refraktérnym ochorením s > 5 % blastov v kostnej dreni, výkonnostným stavom ECOG ≤ 2, predpokladanou dĺžkou života ≥ 12 týždňov, ktorí nepodstúpili autológnu transplantáciu krvotvorných kmeňových buniek (HSCT) v priebehu 6 týždňov pred začatím liečby blinatumomabom, alogénnu HSCT v priebehu 3 mesiacov pred začatím liečby blinatumomabom alebo predchádzajúcu liečbu blinatumomabom). Pätnásť z 36 (41,7 %) pacientov absolvovalo alogénnu HSCT pred podávaním BLINCYTA. Miera CR/CRh* bola 69,4 % (25 z 36 pacientov: 15 [41,7 %;
95 % CI: 25,5 % – 59,2 %] CR; 10 [27,8 %; 95 % CI: 14,2 % – 45,2 %] CRh*). U staršej populácie (vo veku ≥ 65 rokov) 4 z 5 pacientov (80,0 %) dosiahli CR/CRh* v priebehu 2 liečebných cyklov (bezpečnosť u starších pacientov, pozri časť 4.8). Dvadsaťdva z 25 (88 %) pacientov s kompletnou hematologickou remisiou malo aj odpovede minimálneho reziduálneho ochorenia (MRD) (definované ako MRD na základe PCR < 1 x 10-4). Medián trvania remisie bol 8,9 mesiaca a medián prežívania
bez relapsu (RFS) bol 7,6 mesiaca. Medián celkového prežívania (OS) bol 9,8 mesiaca.
Obmedzené údaje sú u pacientov s oneskoreným prvým relapsom B-prekurzorovej ALL definovaným ako relaps, ktorý sa vyskytne viac ako 12 mesiacov po prvej remisii alebo viac ako 12 mesiacov po HSCT v prvej remisii. V klinických štúdiách 88,9 % (8/9) pacientov s oneskoreným prvým relapsom, ako sa uvádza v jednotlivých štúdiách, dosiahlo CR/CRh* v priebehu prvých 2 liečebných cyklov, pričom 62,5 % (6/9) dosiahlo odpoveď MRD a 37,5 % (3/9) podstúpilo alogénnu HSCT po liečbe blinatumomabom. Medián celkového prežívania (OS) bol 17,7 mesiaca (CI 3,1 – nehodnotiteľné).
Pediatrická populáciaU pediatrických pacientov sú obmedzené skúsenosti, pozri časť 4.8.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s BLINCYTOM u detí vo veku od 1 mesiaca do menej ako 18 rokov s akútnou lymfoblastovou leukémiou (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku. Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Zdá sa, že farmakokinetika blinatumomabu je u dospelých pacientov lineárna v rozpätí dávok od 5 do
90 µg/m2/deň (približne ekvivalentná s dávkou 9 – 162 µg/deň). Po kontinuálnej intravenóznej infúzii sa rovnovážna koncentrácia v sére (Css) dosiahla v priebehu jedného dňa a postupom času ostala
stabilná. Nárast priemerných hodnôt Css bol približne úmerný dávke v skúmanom rozsahu.
V klinických dávkach 9 µg/deň a 28 µg/deň na liečbu relapsujúcej/refraktérnej ALL bola priemerná
(SD) Css 211 (258) pg/ml a 621 (502) pg/ml, v príslušnom poradí.
Distribúcia
Odhadovaný priemerný (SD) distribučný objem na základe terminálnej fázy (Vz) pri kontinuálnej
intravenóznej infúzii blinatumomabu bol 4,52 (2,89) l.
Biotransformácia
Metabolická cesta blinatumomabu nebola charakterizovaná. Tak ako pri iných proteínových
terapeutikách, aj pri blinatumomabe sa predpokladá, že sa katabolickými cestami bude degradovať na
malé peptidy a aminokyseliny.
Eliminácia
Odhadovaný priemerný (SD) systémový klírens pri kontinuálnej intravenóznej infúzii u pacientov
dostávajúcich blinatumomab v klinických štúdiách bol 2,92 (2,83) l/hodina. Priemerný (SD) polčas bol 2,11 (1,42) hodiny. Zanedbateľné množstvá blinatumomabu sa vylučovali močom pri skúšaných klinických dávkach.
Telesná hmotnosť, plocha povrchu tela, pohlavie a vek
Vykonala sa populačná farmakokinetická analýza na vyhodnotenie vplyvu demografických
charakteristík na farmakokinetiku blinatumomabu. Výsledky naznačujú, že vek (18 až 80 rokov), pohlavie, telesná hmotnosť (44 až 134 kg) a plocha povrchu tela (1,39 až 2,57) neovplyvňujú farmakokinetiku blinatumomabu. S používaním blinatumomabu u dospelých vážiacich menej ako
45 kg sú veľmi obmedzené skúsenosti.
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa nevykonali žiadne formálne farmakokinetické štúdie s
blinatumomabom.
Farmakokinetické analýzy preukázali približne 2-násobný rozdiel v priemerných hodnotách klírensu blinatumomabu medzi pacientmi so stredne závažnou renálnou dysfunkciou a normálnou funkciou obličiek. Zaznamenala sa však vysoká variabilita medzi pacientmi (CV % až 95,6 %) a hodnoty klírensu u pacientov s poruchou funkcie obličiek sa v podstate pohybovali v rámci rozpätia pozorovaného u pacientov s normálnou funkciou obličiek, a tak sa neočakáva klinicky významný vplyv funkcie obličiek na klinické výsledky.
Porucha funkcie pečene
U pacientov s poruchou funkcie pečene sa nevykonali žiadne formálne farmakokinetické štúdie s
blinatumomabom. Na posúdenie vplyvu poruchy funkcie pečene na klírens blinatumomabu sa použili východiskové hladiny ALT a AST. Populačná farmakokinetická analýza naznačila, že medzi hladinami ALT alebo AST a klírensom blinatumomabu nie je súvislosť.
Pediatrická populácia
U pediatrických pacientov sú obmedzené skúsenosti.
5.3 Predklinické údaje o bezpečnosti
Štúdie toxicity s opakovanými dávkami vykonané s blinatumomabom a myšou náhradou odhalili očakávané farmakologické účinky (vrátane uvoľnenia cytokínov, zníženia počtu leukocytov, deplécie B-lymfocytov, zníženia počtu T-lymfocytov, zníženej celularity v lymfoidných tkanivách). Tieto zmeny boli po ukončení liečby reverzibilné.
S blinatumomabom sa neuskutočnili štúdie reprodukčnej toxicity. Vo vývojovej štúdii embryonálnej/fetálnej toxicity vykonanej na myšiach, myšia náhrada prenikla cez placentu v obmedzenom rozsahu (pomer koncentrácie v sére plodu a matky < 1 %) a nevyvolala
embryonálnu/fetálnu toxicitu ani teratogenitu. Pri gravidných myšiach sa pozorovala predpokladaná deplécia B-lymfocytov a T-lymfocytov, ale pri plodoch sa hematologické účinky nehodnotili.
Neuskutočnili sa žiadne štúdie na hodnotenie účinkov súvisiacich s liečbou na fertilitu. V štúdiách
toxicity s myšou náhradou sa účinky na samčie alebo samičie reprodukčné orgány nepozorovali.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok
monohydrát kyseliny citrónovej (E330)
dihydrát trehalózy lyzín hydrochlorid polysorbát 80
hydroxid sodný (na úpravu pH)
Roztok (stabilizátor)
monohydrát kyseliny citrónovej (E330)
lyzín hydrochlorid polysorbát 80
hydroxid sodný (na úpravu pH)
voda na injekciu
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorené injekčné liekovky
3 roky
Rekonštituovaný roztok
Bola preukázaná chemická a fyzikálna stabilita po rekonštituovaní po dobu 24 hodín pri teplote 2°C –
8°C alebo 4 hodiny pri teplote 27°C alebo nižšej.
Z mikrobiologického hľadiska sa má rekonštituovaný roztok nariediť okamžite, pokiaľ metóda rekonštituovania nevylúči riziko mikrobiálnej kontaminácie. Ak sa nenariedi okamžite, za dobu a podmienky uchovávania pred použitím zodpovedá používateľ.
Nariedený roztok (pripravený infúzny vak)
Bola preukázaná chemická a fyzikálna stabilita po nariedení po dobu 10 dní pri teplote 2°C – 8°C
alebo 96 hodín pri teplote 27°C alebo nižšej.
Z mikrobiologického hľadiska sa majú pripravené infúzne vaky použiť okamžite.
Ak sa nepoužijú okamžite, za dobu a podmienky uchovávania pred použitím zodpovedá používateľ a za normálnych okolností by to nemalo byť dlhšie ako 24 hodín pri teplote 2°C – 8°C, pokiaľ sa nariedenie neuskutočnilo v kontrolovaných a validovaných aseptických podmienkach.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte a prepravujte v chlade (2°C – 8°C).
Neuchovávajte v mrazničke.
Injekčné liekovky uchovávajte v pôvodnom obale na ochranu pred svetlom. Podmienky na uchovávanie lieku po rekonštitúcii a nariedení, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Každé balenie BLINCYTA obsahuje 1 injekčnú liekovku s práškom na prípravu koncentrátu na infúzny roztok a 1 injekčnú liekovku s roztokom (stabilizátorom):
· 38,5 mikrogramov prášku blinatumomab v injekčnej liekovke (zo skla typu I) so zátkou (z elastomérovej gumy), tesnením (z hliníka) a odklápacím viečkom a
· 10 ml roztoku v injekčnej liekovke (zo skla typu I) so zátkou (z elastomérovej gumy), tesnením
(z hliníka) a odklápacím viečkom.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Aseptická príprava
Pri príprave infúzie sa musí zabezpečiť aseptické zaobchádzanie. Príprava BLINCYTA sa má
vykonávať:
- za aseptických podmienok školeným personálom v súlade s pravidlami správnej praxe, predovšetkým pokiaľ ide o aseptickú prípravu parenterálnych produktov.
- v boxe alebo biologicky bezpečnej miestnosti s laminárnym prúdením vzduchu s použitím
štandardných opatrení na bezpečné zaobchádzanie s intravenóznymi látkami.
Na minimalizovanie chýb v predpisovaní liečby (vrátane poddávkovania a predávkovania) je veľmi
dôležité, aby sa dôsledne dodržiavali pokyny na prípravu a podávanie uvedené v tejto časti.
Osobitné pokyny na podporu správnej prípravy
· Roztok (stabilizátor) sa dodáva v balení s BLINCYTOM a používa sa na potiahnutie
naplneného infúzneho vaku pred pridaním rekonštituovaného BLINCYTA. Nepoužívajte tento roztok (stabilizátor) na rekonštitúciu prášku na koncentrát BLINCYTO.
· Celý objem rekonštituovaného a nariedeného BLINCYTA bude väčší ako objem, ktorý sa podá pacientovi (240 ml). Dôvodom sú straty v intravenóznej infúznej súprave a potreba zabezpečiť, aby pacient dostal plnú dávku BLINCYTA.
· Pri príprave infúzneho vaku, odstráňte z neho všetok vzduch. Obzvlášť dôležité je to pri
používaní ambulantnej infúznej pumpy.
· Na minimalizovanie chýb vo výpočte použite špecifické objemy uvedené ďalej v pokynoch na rekonštitúciu a riedenie.
Iné pokyny
· BLINCYTO je kompatibilný s infúznymi vakmi/kazetami pumpy z polyolefínu, PVC bez
obsahu dietylhexylftalátu (non-DEHP) alebo z etylvinylacetátu (EVA).
· Špecifikácie pumpy: Infúzna pumpa na podanie infúzneho roztoku BLINCYTO má byť programovateľná, uzamykateľná a má mať alarm. Elastomérové pumpy sa nemajú používať.
· Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
Príprava infúzneho roztoku
Pre každú dávku a čas podávania infúzie sú uvedené osobitné pokyny na rekonštitúciu a riedenie.
Overte predpísanú dávku a čas podávania infúzie BLINCYTA a nájdite príslušnú časť o príprave dávok uvedenú nižšie. Postupujte podľa krokov na rekonštituovanie BLINCYTA a prípravu infúzneho
vaku.
a) pre 9 µg/deň podávaných infúziou počas 24 hodín rýchlosťou 10 ml/h b) pre 9 µg/deň podávaných infúziou počas 48 hodín rýchlosťou 5 ml/h
c) pre 9 µg/deň podávaných infúziou počas 72 hodín rýchlosťou 3,3 ml/h
d) pre 9 µg/deň podávaných infúziou počas 96 hodín rýchlosťou 2,5 ml/h e) pre 28 µg/deň podávaných infúziou počas 24 hodín rýchlosťou 10 ml/h f) pre 28 µg/deň podávaných infúziou počas 48 hodín rýchlosťou 5 ml/h
g) pre 28 µg/deň podávaných infúziou počas 72 hodín rýchlosťou 3,3 ml/h h) pre 28 µg/deň podávaných infúziou počas 96 hodín rýchlosťou 2,5 ml/h
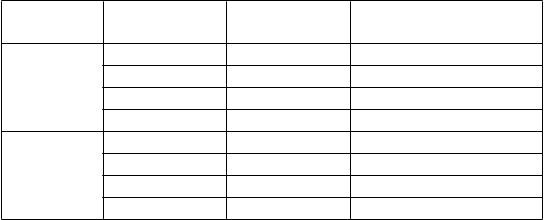
Zabezpečte, aby ste pred prípravou mali pripravený tento materiál:
D
ávka Trvanie
i
n
f
úzie (h)
R
ýchlosť
 i
n
f
úzie (ml/h)
P
očet balení s
B
L
INCYTOM
i
n
f
úzie (ml/h)
P
očet balení s
B
L
INCYTOM
9 µg/deň 24 10 1
48 5 1
72 3,3 1
96 2,5 2
28 µg/ deň 24 10 1
48 5 2
72 3,3 3
96 2,5 4
Potrebné sú aj nasledujúce pomôcky, nie sú však súčasťou balenia
· Sterilné jednorazové injekčné striekačky
· Ihla (ihly) s hrúbkou 21 – 23 G (odporúčané)
· Voda na injekciu
· Infúzny vak s 250 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %);
o Na minimalizovanie počtu aseptických transferov použite 250 ml naplnený infúzny vak.
Vypočítanie dávky BLINCYTA vychádza zo zvyčajného celkového objemu 265 až
275 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %).
o Používajte iba infúzne vaky/kazety pumpy z polyolefínu, PVC bez obsahu dietylhexylftalátu (non-DEHP) alebo z etylvinylacetátu (EVA).
· Intravenózne hadičky vyrobené z polyolefínu, PVC bez DEHP alebo EVA so sterilným, nepyrogénnym in-line filtrom s nízkou afinitou k bielkovinám s veľkosťou pórov 0,2 μm o Zabezpečte, aby boli hadičky kompatibilné s infúznou pumpou.
 a) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 24 hodín rýchlosťou 10 ml/h
a) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 24 hodín rýchlosťou 10 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Jednu injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste
predišli nadmernému speneniu.
Netraste.
·
Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom(stabilizátorom).· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte
na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne
opaleskujúci, bezfarebný až žltkastý.
Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 0,83 ml rekonštituovaného
BLINCYTA do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte
ibapripraveným infúznym roztokom.
Nenaplňte injekčným roztokom chloridu sodného9 mg/ml (0,9 %).8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C
– 8°C.
b) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 48 hodín rýchlosťou 5 ml/h1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Jednu injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte
tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie
nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu.
Netraste.
·
Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom(stabilizátorom).· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte
na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý.
Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 1,7 ml rekonštituovaného BLINCYTA
do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte
ibapripraveným infúznym roztokom.
Nenaplňte injekčným roztokom chloridu sodného9 mg/ml (0,9 %).8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C
– 8°C.
c) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 72 hodín rýchlosťou 3,3 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku (stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Jednu injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte
tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie
nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte
na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 2,5 ml rekonštituovaného BLINCYTA
do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
d) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 96 hodín rýchlosťou 2,5 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku (stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Použite dve injekčné liekovky s práškom na prípravu koncentrátu BLINCYTO. Každú
injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte
na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne
opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 3,5 ml rekonštituovaného BLINCYTA do infúzneho vaku (2,0 ml z jednej injekčnej liekovky a zvyšných 1,5 ml z druhej injekčnej liekovky). Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok
zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
e) Príprava BLINCYTA 28 µg/deň podaného infúziou počas 24 hodín rýchlosťou 10 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Jednu injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte
tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 2,6 ml rekonštituovaného BLINCYTA
do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
f) Príprava BLINCYTA 28 µg/deň podaného infúziou počas 48 hodín rýchlosťou 5 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Použite dve injekčné liekovky s práškom na prípravu koncentrátu BLINCYTO. Každú injekčnú liekovku s práškom na koncentrát BLINCYTO rekonštituujte tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte
vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte
na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 5,2 ml rekonštituovaného BLINCYTA
do infúzneho vaku (2,7 ml z jednej injekčnej liekovky a zvyšných 2,5 ml z druhej
injekčnej liekovky). Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného'
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
g) Príprava BLINCYTA 28 µg/deň podaného infúziou počas 72 hodín rýchlosťou 3,3 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Použite tri injekčné liekovky s práškom na prípravu koncentrátu BLINCYTO. Každú
injekčnú liekovku s BLINCYTOM rekonštituujte tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte
na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 8 ml rekonštituovaného BLINCYTA
do infúzneho vaku (2,8 ml z každej z prvých dvoch injekčných liekoviek a zvyšných
2,4 ml z tretej injekčnej liekovky). Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
h) Príprava BLINCYTA 28 µg/deň podaného infúziou počas 96 hodín rýchlosťou 2,5 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Použite štyri injekčné liekovky s práškom na prípravu koncentrátu BLINCYTO. Každú
injekčnú liekovku s BLINCYTOM rekonštituujte tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte
na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 10,7 ml rekonštituovaného
BLINCYTA do infúzneho vaku (2,8 ml z každej z troch injekčných liekoviek a zvyšných
2,3 ml zo štvrtej injekčnej liekovky). Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte
ibapripraveným infúznym roztokom.
Nenaplňte injekčným roztokom chloridu sodného9 mg/ml (0,9 %).8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C
– 8°C.
Pokyny na podávanie, pozri časť 4.2.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAmgen Europe B.V. Minervum 7061
4817 ZK Breda
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/15/1047/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.Nasledujúca informácia je určená len pre zdravotníckych pracovníkov:BLINCYTO infúzny roztok sa podáva vo forme kontinuálnej intravenóznej infúzie konštantnou
rýchlosťou pomocou infúznej pumpy počas až 96 hodín.
Odporúčaná začiatočná dávka BLINCYTA v prvom cykle je 9 µg/deň prvý týždeň (prvých 7 dní)
liečby.
Dávka sa má zvýšiť na 28 µg/deň od 2. týždňa po 4. týždeň prvého cyklu. Všetky ďalšie cykly sa má liek podávať v dávke 28 µg/deň po celé 4-týždňové liečebné obdobie.
Terapeutická dávka 9 µg/deň alebo 28 µg/deň sa má pacientovi podať infúziou v celkovom objeme
240 ml infúzneho roztoku BLINCYTO jednou zo 4 konštantných rýchlostí infúzie s priradeným trvaním infúzie:
· Rýchlosť infúzie 10 ml/h v trvaní 24 hodín
· Rýchlosť infúzie 5 ml/h v trvaní 48 hodín
· Rýchlosť infúzie 3,3 ml/h v trvaní 72 hodín
· Rýchlosť infúzie 2,5 ml/h v trvaní 96 hodín
Dĺžku trvania infúzie má zvoliť ošetrujúci lekár a vziať pritom do úvahy frekvenciu výmeny infúznych
vakov. Cieľová podaná terapeutická dávka BLINCYTA sa nemení.
Aseptická prípravaPri príprave infúzie sa musí zabezpečiť aseptické zaobchádzanie. Príprava BLINCYTA sa má
vykonávať:
- za aseptických podmienok školeným personálom v súlade s pravidlami správnej praxe,
predovšetkým pokiaľ ide o aseptickú prípravu parenterálnych produktov.
- v boxe alebo biologicky bezpečnej miestnosti s laminárnym prúdením vzduchu s použitím štandardných opatrení na bezpečné zaobchádzanie s intravenóznymi látkami.
Na minimalizovanie chýb v predpisovaní liečby (vrátane poddávkovania a predávkovania) je veľmi
dôležité, aby sa dôsledne dodržiavali pokyny na prípravu a podávanie uvedené v tejto časti.
Osobitné pokyny na podporu správnej prípravy· Roztok (stabilizátor) sa dodáva v balení s BLINCYTOM a používa sa na potiahnutie
naplneného infúzneho vaku pred pridaním rekonštituovaného BLINCYTA.
Nepoužívajte tento roztok (stabilizátor) na rekonštitúciu prášku na koncentrát BLINCYTO.· Celý objem rekonštituovaného a nariedeného BLINCYTA bude väčší ako objem, ktorý sa podá pacientovi (240 ml). Dôvodom sú straty v intravenóznej infúznej súprave a potreba zabezpečiť, aby pacient dostal plnú dávku BLINCYTA.
· Pri príprave infúzneho vaku, odstráňte z neho všetok vzduch. Obzvlášť dôležité je to pri
používaní ambulantnej infúznej pumpy.
· Na minimalizovanie chýb vo výpočte použite špecifické objemy uvedené ďalej v pokynoch na rekonštitúciu a riedenie.
Iné pokyny· BLINCYTO je kompatibilný s infúznymi vakmi/kazetami pumpy z polyolefínu, PVC bez
obsahu dietylhexylftalátu (non-DEHP) alebo z etylvinylacetátu (EVA).
· Špecifikácie pumpy: Infúzna pumpa na podanie infúzneho roztoku BLINCYTO má byť programovateľná, uzamykateľná a má mať alarm. Elastomérové pumpy sa nemajú používať.
· Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
Príprava infúzneho roztokuPre každú dávku a čas podávania infúzie sú uvedené osobitné pokyny na rekonštitúciu a riedenie.
Overte predpísanú dávku a čas podávania infúzie BLINCYTA a nájdite príslušnú časť o príprave dávok uvedenú nižšie. Postupujte podľa krokov na rekonštituovanie BLINCYTA a prípravu infúzneho vaku.
a) pre 9 µg/deň podávaných infúziou počas 24 hodín rýchlosťou 10 ml/h b) pre 9 µg/deň podávaných infúziou počas 48 hodín rýchlosťou 5 ml/h
c) pre 9 µg/deň podávaných infúziou počas 72 hodín rýchlosťou 3,3 ml/h d) pre 9 µg/deň podávaných infúziou počas 96 hodín rýchlosťou 2,5 ml/h
e) pre 28 µg/deň podávaných infúziou počas 24 hodín rýchlosťou 10 ml/h f) pre 28 µg/deň podávaných infúziou počas 48 hodín rýchlosťou 5 ml/h
g) pre 28 µg/deň podávaných infúziou počas 72 hodín rýchlosťou 3,3 ml/h
h) pre 28 µg/deň podávaných infúziou počas 96 hodín rýchlosťou 2,5 ml/h
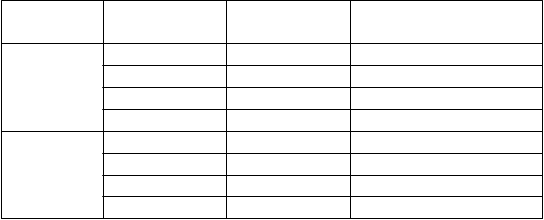
Zabezpečte, aby ste pred prípravou mali pripravený tento materiál:
D
ávka Trvanie
i
n
f
úzie (h)
R
ýchlosť
 i
n
f
úzie (ml/h)
P
očet balení s
B
L
INCYTOM
i
n
f
úzie (ml/h)
P
očet balení s
B
L
INCYTOM
9 µg/deň 24 10 1
48 5 1
72 3,3 1
96 2,5 2
28 µg/ deň 24 10 1
48 5 2
72 3,3 3
96 2,5 4
Potrebné sú aj nasledujúce pomôcky,
nie sú však súčasťou balenia
· Sterilné jednorazové injekčné striekačky
· Ihla (ihly) s hrúbkou 21 – 23 G (odporúčané)
· Voda na injekciu
· Infúzny vak s 250 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %);
o Na minimalizovanie počtu aseptických transferov použite 250 ml naplnený infúzny vak.
Vypočítanie dávky
BLINCYTA vychádza zo zvyčajného celkového objemu 265 až275 ml injekčného roztoku chloridu sodného 9 mg/ml (0,9 %).o Používajte iba infúzne vaky/kazety pumpy z polyolefínu, PVC bez obsahu dietylhexylftalátu (non-DEHP) alebo z etylvinylacetátu (EVA).
· Intravenózne hadičky vyrobené z polyolefínu, PVC bez DEHP alebo EVA so sterilným, nepyrogénnym in-line filtrom s nízkou afinitou k bielkovinám s veľkosťou pórov 0,2 μm o Zabezpečte, aby boli hadičky kompatibilné s infúznou pumpou.
 a) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 24 hodín rýchlosťou 10 ml/h
a) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 24 hodín rýchlosťou 10 ml/h1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli
speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Jednu injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte
tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu.
Netraste.
·
Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom(stabilizátorom).· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý.
Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 0,83 ml rekonštituovaného
BLINCYTA do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte
ibapripraveným infúznym roztokom.
Nenaplňte injekčným roztokom chloridu sodného9 mg/ml (0,9 %).8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C
– 8°C.
b) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 48 hodín rýchlosťou 5 ml/h1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli
speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Jednu injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte
tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu.
Netraste.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 1,7 ml rekonštituovaného BLINCYTA
do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
c) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 72 hodín rýchlosťou 3,3 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli
speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Jednu injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte
tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 2,5 ml rekonštituovaného BLINCYTA
do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
d) Príprava BLINCYTA 9 µg/deň podaného infúziou počas 96 hodín rýchlosťou 2,5 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli
speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Použite dve injekčné liekovky s práškom na prípravu koncentrátu BLINCYTO. Každú
injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 3,5 ml rekonštituovaného BLINCYTA
do infúzneho vaku (2,0 ml z jednej injekčnej liekovky a zvyšných 1,5 ml z druhej injekčnej liekovky). Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
e) Príprava BLINCYTA 28 µg/deň podaného infúziou počas 24 hodín rýchlosťou 10 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku
(stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli
speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Jednu injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte tak, že do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte
na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne
opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 2,6 ml rekonštituovaného BLINCYTA
do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
f) Príprava BLINCYTA 28 µg/deň podaného infúziou počas 48 hodín rýchlosťou 5 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku (stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Použite dve injekčné liekovky s práškom na prípravu koncentrátu BLINCYTO. Každú
injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte tak, že
do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 5,2 ml rekonštituovaného BLINCYTA do infúzneho vaku (2,7 ml z jednej injekčnej liekovky a zvyšných 2,5 ml z druhej injekčnej liekovky). Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
g) Príprava BLINCYTA 28 µg/deň podaného infúziou počas 72 hodín rýchlosťou 3,3 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku (stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Použite tri injekčné liekovky s práškom na prípravu koncentrátu BLINCYTO. Každú
injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte tak, že
do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 8 ml rekonštituovaného BLINCYTA
do infúzneho vaku (2,8 ml z každej z prvých dvoch injekčných liekoviek a zvyšných
2,4 ml z tretej injekčnej liekovky). Obsah vaku jemne premiešajte, aby ste predišli speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
h) Príprava BLINCYTA 28 µg/deň podaného infúziou počas 96 hodín rýchlosťou 2,5 ml/h
1. Použite infúzny vak naplnený 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktorý zvyčajne obsahuje celkový objem 265 až 275 ml.
2. Infúzny vak potiahnete tak, že injekčnou striekačkou asepticky prenesiete 5,5 ml roztoku (stabilizátora) do infúzneho vaku. Obsah vaku jemne premiešajte, aby ste predišli speneniu. Injekčnú liekovku so zvyšným roztokom (stabilizátorom) zlikvidujte.
3. Použite štyri injekčné liekovky s práškom na prípravu koncentrátu BLINCYTO. Každú
injekčnú liekovku s práškom na prípravu koncentrátu BLINCYTO rekonštituujte tak, že
do injekčnej striekačky natiahnete 3 ml vody na injekciu. Počas rekonštitúcie nasmerujte
vodu na injekciu na stenu injekčnej liekovky. Obsahom jemne krúžte, aby ste predišli nadmernému speneniu. Netraste.
· Prášok na prípravu koncentrátu BLINCYTO nerekonštituujte roztokom
(stabilizátorom).
· Pridaním vody na injekciu k prášku na prípravu koncentrátu dostanete celkový objem 3,08 ml pri konečnej koncentrácii BLINCYTA 12,5 µg/ml.
4. Rekonštituovaný roztok počas rekonštitúcie a pred podaním infúzie vizuálne skontrolujte
na prítomnosť častíc a zmenu sfarbenia. Výsledný roztok má byť číry až mierne opaleskujúci, bezfarebný až žltkastý. Nepoužite, ak je roztok zakalený alebo sa vyzrážal.
5. S použitím injekčnej striekačky asepticky preneste 10,7 ml rekonštituovaného
BLINCYTA do infúzneho vaku (2,8 ml z každej z troch injekčných liekoviek a zvyšných
2,3 ml zo štvrtej injekčnej liekovky). Obsah vaku jemne premiešajte, aby ste predišli
speneniu. Všetok zvyšný rekonštituovaný roztok BLINCYTA zlikvidujte.
6. Za aseptických podmienok pripojte intravenóznu hadičku k infúznemu vaku so sterilným
0,2 mikrónovým in-line filtrom.
7. Z infúzneho vaku odstráňte vzduch a intravenóznu infúznu súpravu naplňte iba
pripraveným infúznym roztokom. Nenaplňte injekčným roztokom chloridu sodného
9 mg/ml (0,9 %).
8. Ak sa pripravený infúzny roztok nepoužije okamžite, uchovávajte pri teplote 2°C – 8°C.
Pokyny na podávanie, pozri časť 4.2 súhrnu charakteristických vlastností lieku. Spôsob podávania
Dôležitá poznámka: Nepreplachujte infúznu súpravu, ktorá je zavedená v žile pacienta, pretože
to môže zapríčiniť náhodné podanie bolusu BLINCYTA. BLINCYTO sa má infúziou podávať cez určený lúmen.
BLINCYTO infúzny roztok sa podáva vo forme kontinuálnej intravenóznej infúzie konštantnou
rýchlosťou pomocou infúznej pumpy počas až 96 hodín.
BLINCYTO infúzny roztok sa musí podať cez intravenóznu hadičku, ktorá obsahuje in-line, sterilný, nepyrogénny filter s nízkou afinitou k bielkovinám s veľkosťou pórov 0,2 mikrometra.
Z dôvodov sterility musí zdravotnícky pracovník vymieňať infúzny vak najmenej každých 96 hodín. Podmienky uchovávania a časpoužiteľnosti
Neotvorené injekčné liekovky:
3 roky (2°C – 8°C)
Rekonštituovaný roztok:
Bola preukázaná chemická a fyzikálna stabilita po rekonštituovaní po dobu 24 hodín pri teplote 2°C –
8°C alebo 4 hodiny pri teplote 27°C alebo nižšej.
Z mikrobiologického hľadiska sa má rekonštituovaný roztok nariediť okamžite, pokiaľ metóda rekonštituovania nevylúči riziko mikrobiálnej kontaminácie. Ak sa nenariedi okamžite, za dobu a podmienky uchovávania pred použitím zodpovedá používateľ.
N
ariedený roztok (pripravený infúzny vak)
Bola preukázaná chemická a fyzikálna stabilita po nariedení po dobu 10 dní pri teplote 2 C – 8°C
alebo 96 hodín pri teplote 27°C alebo nižšej.
Z mikrobiologického hľadiska sa majú pripravené infúzne vaky použiť okamžite.
Ak sa nepoužijú okamžite, za dobu a podmienky uchovávania pred použitím zodpovedá používateľ a za normálnych okolností by to nemalo byť dlhšie ako 24 hodín pri teplote 2°C – 8°C, pokiaľ sa nariedenie neuskutočnilo v kontrolovaných a validovaných aseptických podmienkach.