
iečriev
Pri užívaní bimekizumabu boli hlásené prípady nových prípadov alebo exacerbácií zápalového
ochorenia čriev (pozri časť 4.8). Bimekizumab sa neodporúča podávať pacientom so zápalovým ochorením čriev. Ak sa u pacienta vyskytnú prejavy a príznaky zápalového ochorenia čriev alebo
dôjde k exacerbácii už existujúceho zápalového ochorenia čriev, podávanie bimekizumabu sa má
ukončiť a má sa začať príslušná lekárska starostlivosť.
Precitlivenosť
Pri inhibítoroch IL-17 boli pozorované závažné hypersenzitívne reakcie vrátane anafylaktických
reakcií. Ak sa vyskytne závažná hypersenzitívna reakcia, podávanie bimekizumabu sa má ihneď ukončiť a má sa začať náležitá liečba.
Očkovania
Pred začatím liečby bimekizumabom sa má zvážiť absolvovanie všetkých príslušných imunizácií vo
vekových skupinách podľa aktuálnych pokynov pre imunizáciu.
Živé vakcíny sa nemajú podávať pacientom liečeným bimekizumabom.
Pacientom liečeným bimekizumabom sa môžu podať inaktivované alebo neživé vakcíny. Zdraví jedinci, ktorí užili jednu 320 mg dávku bimekizumabu dva týždne pred očkovaním inaktivovanou vakcínou proti sezónnej chrípke, mali podobné odpovede na protilátky v porovnaní s jedincami, ktorí pred očkovaním neužili bimekizumab.
Pomocné látky
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej dávke, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Neexistujú žiadne priame dôkazy o úlohe IL-17A alebo IL-
17F pri expresii enzýmov CYP450. Tvorba niektorých enzýmov CYP450 je potlačená zvýšenými hladinami cytokínov počas chronického zápalu. Protizápalové liečby, napríklad bimekizumabom ako
inhibítorom IL-17A a IL-17F, môžu viesť k normalizácii hladín CYP450 so sprievodnou nižšou expozíciou liekom metabolizovaným CYP450. Preto nie je možné vylúčiť klinicky významný účinok
na substráty CYP450 s úzkym terapeutickým indexom, pri ktorých je dávka individuálne upravená (napr. warfarín). Na začiatku liečby bimekizumabom u pacientov liečených týmito druhmi liekov sa má zvážiť monitorovanie liečby.
Živé vakcíny sa nemajú podávať súbežne s bimekizumabom (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Ž
eny vofertilnomveku
Ženy vo fertilnom veku majú používať účinnú antikoncepciu počas liečby a po dobu
najmenej 17 týždňov po liečbe.
Gravidita
Je iba obmedzené množstvo údajov o použití bimekizumabu u gravidných žien. Štúdie na zvieratách
nepreukázali priame alebo nepriame účinky z hľadiska gravidity, embryonálneho/fetálneho vývinu, pôrodu alebo postnatálneho vývinu (pozri časť 5.3). Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu Bimzelxu počas gravidity.
Laktácia
Nie je známe, či sa bimekizumab vylučuje do materského mlieka. Riziko u novorodencov/dojčiat
nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Bimzelxom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Účinok bimekizumabu na fertilitu u ľudí nebol hodnotený. Štúdie na zvieratách nepreukázali priame
alebo nepriame škodlivé účinky z hľadiska fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Bimzelx nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn profilubezpečnosti
Najčastejšie hlásenými nežiaducimi reakciami boli infekcie horných dýchacích ciest (14,5 %)
(najčastejšie nazofaryngitída) a orálna kandidóza (7,3 %).
Súhrnný zoznam nežiaducich reakciíNežiaduce reakcie z klinických štúdií (Tabuľka č. 1) sú klasifikované podľa triedy orgánových
systémov podľa MedDRA a frekvencie podľa nasledovnej konvencie: veľmi časté (≥ 1/10), časté
(≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Tabuľka č. 1: Zoznam nežiaducich reakciíTrieda orgánových systémov
| Frekvencia
| Nežiaduca reakcia
|
Infekcie a nákazy
| Veľmi časté
| infekcia horných dýchacích ciest
|
Časté
| orálna kandidóza,
infekcie tinea, infekcie ucha,
infekcie herpes simplex, orofaryngeálna kandidóza,
gastroenteritída, folikulitída
|
Menej časté
| kandidóza sliznice a kože (vrátane
kandidózy pažeráka), konjunktivitída
|
Poruchy krvi a lymfatického
systému
| Menej časté
| neutropénia
|
Poruchy nervového systému
| Časté
| bolesť hlavy
|
Poruchy gastrointestinálneho
traktu
| Menej časté
| zápalové ochorenie čriev
|
Poruchy kože a podkožného
tkaniva
| Časté
| dermatitída a ekzém,
akné
|
Celkové poruchy a reakcie
v mieste podania
| Časté
| reakcie v mieste podania injekciea,
únava
|
a) Patria sem: erytém, reakcia, edém, bolesť, opuch v mieste podania injekcie.
|
PopisvybranýchnežiaducichreakciíInfekcieV placebom kontrolovanom období klinických štúdií v III. fáze zameraných na ložiskovú psoriázu boli infekcie hlásené u 36,0 % pacientov liečených bimekizumabom po dobu najviac 16 týždňov v porovnaní s 22,5 % pacientov liečených placebom. Závažné infekcie sa vyskytli u 0,3 % pacientov liečených bimekizumabom a u 0 % pacientov liečených placebom.
Väčšina infekcií pozostávala z nezávažných miernych až stredne závažných infekcií horných dýchacích ciest, ako je nazofaryngitída. U pacientov liečených bimekizumabom bola vyššia miera výskytu orálnej a orofaryngálnej kandidózy v súlade s mechanizmom účinku (7,3 % a 1,2 % v porovnaní s 0 % u pacientov liečených placebom v uvedenom poradí). Viac ako 98 % prípadov bolo nezávažných, miernych alebo stredne závažných, a nevyžadovalo sa ukončenie liečby. Mierne vyšší výskyt orálnej kandidózy bol hlásený u pacientov s telesnou hmotnosťou < 70 kg (8,5 % oproti 7,0 % u pacientov s telesnou hmotnosťou ≥ 70 kg).
Za celé liečebné obdobie klinických štúdií v III. fáze zameraných na ložiskovú psoriázu boli infekcie hlásené u 63,2 % pacientov liečených bimekizumabom (120,4 na 100 pacientorokov). Závažné infekcie boli hlásené u 1,5 % pacientov liečených bimekizumabom (1,6 na 100 pacientorokov) (pozri časť 4.4).
NeutropéniaNeutropénia bola pozorovaná pri bimekizumabe v klinických štúdiách v III. fáze zameraných na
ložiskovú psoriázu. Za celé liečebné obdobie klinických štúdií v III. fáze bola neutropénia 3./4. stupňa pozorovaná u 1 % pacientov liečených bimekizumabom. Väčšina prípadov bola prechodná a nevyžadovalo sa ukončenie liečby. S neutropéniou nesúviseli žiadne závažné infekcie.
HypersenzitivitaPri inhibítoroch IL-17 boli pozorované závažné reakcie z precitlivenosti vrátane anafylaktických reakcií.
ImunogenicitaPribližne u 45 % pacientov s ložiskovou psoriázou liečených bimekizumabom po dobu
najviac 56 týždňov s odporúčanou dávkovacou schémou (320 mg každé 4 týždne po 16. týždeň a následne 320 mg každých 8 týždňov) sa vytvorili protilátky proti lieku. Spomedzi pacientov, u ktorých
sa vytvorili protilátky proti lieku, malo približne 34 % (16 % zo všetkých pacientov liečených
bimekizumabom) protilátky, ktoré boli klasifikované ako neutralizujúce. S tvorbou protilátok proti bimekizumabu nesúviseli žiadne dôkazy o zmene v klinickej odpovedi ani o výraznej zmene v profile bezpečnosti.
Staršie osoby (vo veku ≥ 65 rokov)U starších pacientov môže byť pri užívaní bimekizumabu vyššia pravdepodobnosť výskytu určitých nežiaducich reakcií, ako je orálna kandidóza, dermatitída a ekzém. V placebom kontrolovanom období klinických štúdií v III. fáze zameraných na ložiskovú psoriázu bola pozorovaná orálna kandidóza u
18,2 % pacientov vo veku ≥ 65 rokov oproti 6,3 % vo veku < 65 rokoch, dermatitída a ekzém u 7,3 %
pacientov vo veku ≥ 65 rokov oproti 2,8 % vo veku < 65 rokoch.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieJednorazové 640 mg intravenózne dávky alebo 640 mg subkutánne dávky, po ktorých
nasledovala 320 mg subkutánna dávka každé dva týždne po podanie piatich dávok, boli podávané v klinických štúdiách bez toxicity obmedzujúcej dávku. V prípade predávkovania sa odporúča sledovať
u pacienta prípadný výskyt prejavov alebo príznakov nežiaducich reakcií a v prípade ich výskytu sa
má ihneď zaviesť náležitá symptomatická liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, inhibítory interleukínov, ATC kód: L04AC21
Mechanizmus účinkuBimekizumab je humanizovaná IgG1/κ monoklonálna protilátka, ktorá sa selektívne viaže s vysokou
afinitou na cytokíny IL-17A, IL-17F a IL-17AF, čím blokuje ich interakciu s receptorovým komplexom IL-17RA/IL-17RC. Zvýšené koncentrácie IL-17A a IL-17F sa podieľajú na patogenéze
niekoľkých imunitne sprostredkovaných zápalových ochorení vrátane ložiskovej psoriázy.
Bimekizumab inhibuje tieto prozápalové cytokíny, čo vedie k normalizácii zápalu kože a následne k zmierneniu klinických príznakov súvisiacich so psoriázou. Z in vitro modelov sa preukázalo, že bimekizumab inhibuje expresiu génov a tvorbu cytokínov v súvislosti so psoriázou, a to vo väčšom
rozsahu, než v prípade inhibície samotného IL-17A.
Klinická účinnosťabezpečnosť
Bezpečnosť a účinnosť bimekizumabu sa hodnotila u 1480 pacientov so stredne závažnou až závažnou
ložiskovou psoriázou v troch multicentrických, randomizovaných, placebom a/alebo aktívnou referenčnou vzorkou kontrolovaných štúdiách v 3. fáze. Pacienti boli vo veku najmenej 18 rokov, mali skóre indexu plochy a závažnosti psoriázy (PASI) ≥ 12, skóre plochy telesného povrchu (BSA) postihnutého psoriázou (PSO) ≥ 10 % a skóre celkového hodnotenia skúšajúcim (IGA) ≥ 3 dosiahnuté na 5-bodovej stupnici a boli kandidátmi na systémovú liečbu psoriázy a/alebo fototerapiu. Účinnosť a bezpečnosť bimekizumabu sa hodnotili v porovnaní s placebom a ustekinumabom (BE VIVID – PS0009), v porovnaní s placebom (BE READY – PS0013) a v porovnaní s adalimumabom (BE SURE
– PS0008).
Štúdia BE VIVID hodnotila 567 pacientov po dobu 52 týždňov, v ktorej boli pacienti randomizovaní pre užívanie 320 mg bimekizumabu každé 4 týždne, ustekinumabu (45 mg alebo 90 mg, v závislosti od telesnej hmotnosti pacienta, na začiatku liečby a v 4. týždni a potom každých 12 týždňov) alebo placeba počas počiatočných 16 týždňov, po ktorých užívali 320 mg bimekizumab každé 4 týždne.
Štúdia BE READY hodnotila 435 pacientov po dobu 56 týždňov. Pacienti boli randomizovaní pre užívanie 320 mg bimekizumabu každé 4 týždne alebo placeba. V 16. týždni pacienti, ktorí dosiahli odpoveď PASI 90, vstúpili do 40-týždňového randomizovaného obdobia s vysadzovaním. Pacienti pôvodne randomizovaní pre užívanie 320 mg bimekizumabu každé 4 týždne boli opätovne randomizovaní pre užívanie 320 mg bimekizumabu každé 4 týždne alebo 320 mg bimekizumabu každých 8 týždňov alebo pre užívanie placeba (t. j. vysadenie bimekizumabu). Pacienti pôvodne randomizovaní pre užívanie placeba pokračovali v užívaní placeba za predpokladu, že dosiahli odpoveď PASI 90. Pacienti, ktorí nedosiahli odpoveď PASI 90 v 16. týždni, vstúpili do nezaslepenej únikovej skupiny a užívali 320 mg bimekizumabu každé 4 týždne po dobu 12 týždňov. Pacienti s recidívou (nedosiahli odpoveď PASI 75) počas randomizovaného obdobia s vysadzovaním takisto vstúpili do 12- týždňovej únikovej skupiny.
Štúdia BE SURE hodnotila 478 pacientov počas 56 týždňov. Pacienti boli randomizovaní pre užívanie 320 mg bimekizumabu každé 4 týždne po 56. týždeň, pre užívanie 320 mg bimekizumabu každé 4 týždne po 16. týždeň, po ktorom užívali 320 mg bimekizumabu každých 8 týždňov po 56. týždeň alebo adalimumab podľa odporúčaní v označení po 24. týždeň, po ktorom užívali 320 mg bimekizumabu každé 4 týždne po 56. týždeň.
Východiskové charakteristiky boli konzistentné v rámci všetkých 3 štúdií: pacientmi boli prevažne muži (70,7 %) a belosi (84,1 %), pričom priemerný vek bol 45,2 rokov (18 až 83 rokov) a 8,9 % bolo vo veku ≥65 rokov. Medián východiskovej hodnoty BSA bol 20 %, medián východiskového skóre PASI bol 18 a východiskové skóre IGA predstavovalo „závažné ochorenie“ u 33 % pacientov. Medián východiskových skóre dosiahnutých v položkách bolesť, svrbenie a tvorba šupín v pacientovom denníku na zaznamenávanie príznakov (PSD) bol v rozmedzí od 6 do 7 na stupnici od 0-10 bodov a medián celkového východiskového skóre dosiahnutého v indexe dermatologickej kvality života
(DLQI) bol 9.
Vo všetkých 3 štúdiách 38 % pacientov predtým užívalo liečbu biologikom, 23 % predtým užívalo najmenej jednu anti-IL17 látku (primárne neúspešné anti-IL17 boli vylúčené) a 13 % predtým užívalo najmenej jedného antagonistu TNF. Dvadsaťdva percent dovtedy neužívalo žiadnu systémovú liečbu (vrátane nebiologických a biologických látok) a 39 % pacientov predtým užívalo fototerapiu alebo fotochemoterapiu.
Účinnosť bimekizumabu bola hodnotená z hľadiska celkového kožného ochorenia, špecifického miesta na tele (koža hlavy, nechty, dlane a spodok chodidiel), príznakov hlásených pacientmi a dopadu na kvalitu života. Dvoma súbežne primárnymi ukazovateľmi vo všetkých 3 štúdiách boli podiel pacientov, ktorí dosiahli 1) odpoveď PASI 90 a 2) odpoveď IGA klasifikovanú ako „bez nálezov alebo takmer bez nálezov“ (IGA 0/1 najmenej s dvojbodovým zmiernením ochorenia oproti
východiskovému stavu) v 16. týždni. Odpoveď PASI 100, IGA 0 v 16. týždni a odpoveď
PASI 75 vo 4. týždni boli sekundárnymi ukazovateľmi vo všetkých 3 štúdiách.
Celkové kožnéochorenieLiečba bimekizumabom viedla k výraznému zlepšeniu v rámci ukazovateľov účinnosti v porovnaní s
placebom, ustekinumabom alebo adalimumabom v 16. týždni. Hlavné výsledky účinnosti sú uvedené v Tabuľke č. 2.
Tabuľka č. 2: Súhrn klinických odpovedí v štúdiách BE VIVID, BE READY a BE SURE
| BE VIVID
| BE READY
| BE SURE
|
| Placebo
(N = 83)
n (%)
| Bimekizumab
320 mg Q4W (N = 321)
n (%)
| Ustekinumab
(N = 163)
n (%)
| Placebo
(N = 86)
n (%)
| Bimekizumab
320
mg Q4W (N = 349)
n (%)
| Bimekizumab
320
mg Q4W (N = 319)
n (%)
| Adalimumab
(N = 159)
n (%)
|
PASI 100
16. týždeň
|
0 (0,0)
|
188 (58,6)a
|
34 (20,9)
|
1 (1,2)
|
238 (68,2)a
|
194 (60,8)a
|
38 (23,9)
|
PASI 90
16. týždeň
|
4 (4,8)
|
273 (85,0)a, b
|
81 (49,7)
|
1 (1,2)
|
317 (90,8)a
|
275 (86,2)a
|
75 (47,2)
|
PASI 75
4. týždeň
16. týždeň
|
2 (2,4)
6 (7,2)
|
247 (76,9)a, b
296 (92,2)
|
25 (15,3)
119 (73,0)
|
1 (1,2)
2 (2,3)
|
265 (75,9)a
333 (95,4)
|
244 (76,5)a
295 (92,5)
|
50 (31,4)
110 (69,2)
|
IGA 0
16. týždeň
|
0 (0,0)
|
188 (58,6)a
|
36 (22,1)
|
1 (1,2)
|
243 (69,6)a
|
197 (61,8)
|
39 (24,5)
|
IGA 0/1
16. týždeň
|
4 (4,8)
|
270 (84,1)a, b
|
87 (53,4)
|
1 (1,2)
|
323 (92,6)a
|
272 (85,3)a
|
91 (57,2)
|
Absolútna hodnota
PASI ≤ 2
16. týždeň
|
3 (3,6)
|
273 (85,0)
|
84 (51,5)
|
1 (1,2)
|
315 (90,3)
|
280 (87,8)
|
86 (54,1)
|
Zmiernenie bolesti podľa PSD
≥4 (N)
16. týždeň
| (N = 48)
5 (10,4)
| (N = 190)
140 (73,7)
| (N = 90)
54 (60,0)
| (N = 49)
0 (0,0)
| (N = 209)
148 (70,8)
| (N = 222)
143 (64,4)
| (N = 92)
43 (46,7)
|
Zmiernenie svrbenia podľa PSD
≥4 (N)
16. týždeň
| (N = 53)
6 (11,3)
| (N = 222)
151 (68,0)
| (N = 104)
57 (54,8)
| (N = 60)
0 (0,0)
| (N = 244)
161 (66,0)
| (N = 248)
153 (61,7)
| (N = 107)
42 (39,3)
|
Zmiernenie tvorby
šupín podľa
PSD ≥4 (N)
16. týždeň
| (N = 56)
6 (10,7)
| (N = 225)
171 (76,0)
| (N = 104)
59 (56,7)
| (N = 65)
1 (1,5)
| (N = 262)
198 (75,6)
| (N = 251)
170 (67,7)
| (N = 109)
42 (38,5)
|
Bimekizumab 320 mg Q4W = bimekizumab každé 4 týždne. Používa sa imputácia účastníkov bez odpovede (NRI). Odpoveď IGA 0/1 bola definovaná ako „bez nálezov“ (0) alebo „takmer bez nálezov“ (1) so zlepšením oproti východiskovému stavu v 16. týždni najmenej o 2 kategórie. Odpoveď IGA 0 bola definovaná ako „bez nálezov“ (0) so zlepšením oproti východiskovému stavu v 16. týždni najmenej o 2 kategórie.
PSD je pacientov denník na zaznamenávanie príznakov, takisto označovaný ako meradlo príznakov a vplyvov psoriázy (Psoriasis Symptoms and Impacts Measure, P-SIM), na meranie závažnosti príznakov psoriázy na stupnici od 0 (žiadne príznaky) do 10 (veľmi závažné príznaky). Odpoveď je definovaná ako pokles oproti východiskovému stavu v 16. týždni o
≥ 4 pri bolesti, svrbení a tvorbe šupín na stupnici od 0 do 10.
a) p < 0,001 v porovnaní s placebom (BE VIVID a BE READY), v porovnaní s adalimumabom (BE SURE), s úpravou pre multiplicitu.
b) p < 0,001 v porovnaní s ustekinumabom (BE VIVID), s úpravou pre multiplicitu.
Bimekizumab súvisel s rýchlym nástupom účinnosti. V prípade štúdie BE VIVID boli miery odpovede
PASI 90 v 2. a 4. týždni výrazne vyššie u pacientov liečených bimekizumabom
(12,1 % a 43,6 % v uvedenom poradí) v porovnaní s placebom (1,2 % a 2,4 % v uvedenom poradí) a ustekinumabom (1,2 % a 3,1 % v uvedenom poradí).
V štúdii BE VIVID pacienti liečení bimekizumabom dosiahli v 52. týždni (každé 4 týždne) výrazne
vyššie miery odpovede, než pacienti liečení ustekinumabom v ukazovateľoch PASI 90 (81,9 % pri bimekizumabe v porovnaní s 55,8 % pri ustekinumabe, p < 0,001), IGA 0/1 (78,2 % pri bimekizumabe v porovnaní so 60,7 % pri ustekinumabe, p < 0,001) a PASI 100 (64,5 % pri bimekizumabe v porovnaní s 38,0 % pri ustekinumabe).
 Graf č. 1: Podiel účastníkov s odpoveďou PASI 90 na liečbu v priebehu času v štúdii BE VIVID
Graf č. 1: Podiel účastníkov s odpoveďou PASI 90 na liečbu v priebehu času v štúdii BE VIVID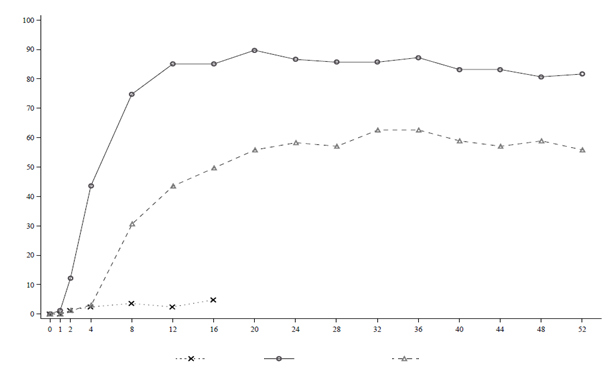
Týždeň
Placebo (N = 83) BKZ 320 mg Q4W (N = 321) Uste (N = 163)
BKZ 320 mg Q4W = bimekizumab každé 4 týždne; Uste = ustekinumab. Používa sa NRI.
V štúdii BE SURE v 24. týždni výrazne vyššie percento pacientov liečených bimekizumabom
(skupiny s kombinovanou schémou dávkovania Q4W/Q4W a Q4W/Q8W) dosiahlo odpovede
PASI 90 a IGA 0/1 v porovnaní s adalimumabom (85,6 % a 86,5 % v uvedenom poradí v porovnaní s 51,6 % a 57,9 % v uvedenom poradí, p < 0,001). V 56. týždni 70,2 % pacientov liečených
bimekizumabom Q8W dosiahlo odpoveď PASI 100. Spomedzi 65 účastníkov bez odpovede na adalimumab v 24. týždni (< PASI 90) 78,5 % dosiahlo odpoveď PASI 90 po 16 týždňoch liečby
bimekizumabom. Profil bezpečnosti pozorovaný u pacientov, ktorí prešli z adalimumabu na bimekizumab bez obdobia bez liečby („wash-out“ obdobie), bol podobný profilu bezpečnosti u pacientov, ktorí začali užívať bimekizumab po období bez liečby predchádzajúcimi systémovými
liečbami.
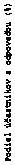 G
raf č. 2: Podiel účastníkov s odpoveďou PASI 90 na liečbu v priebehu času v štúdii BE SURE
G
raf č. 2: Podiel účastníkov s odpoveďou PASI 90 na liečbu v priebehu času v štúdii BE SURE
Týždeň
BKZ 320 mg Q4W (N = 158) ADA (N = 159)
BKZ 320 mg Q4W/Q8W (N = 161) ADA->BKZ 320 mg Q4W (N = 159)
BKZ 320 mg Q4W = bimekizumab každé 4 týždne; BKZ 320 mg Q8W = bimekizumab každých 8 týždňov; ADA =
adalimumab.
Pacienti v skupine užívajúcej BKZ Q4W/Q8W prešli z dávkovania Q4W na Q8W v 16. týždni. Pacienti v skupine užívajúcej 320 mg ADA/BKZ Q4W prešli z ADA na BKZ Q4W v 24. týždni. Používa sa NRI.
Účinnosť bimekizumabu bola preukázaná bez ohľadu na vek, pohlavie, rasu, trvanie ochorenia, telesnú hmotnosť, východiskovú závažnosť podľa PASI a predchádzajúcu liečbu biologikom. Bimekizumab bol účinný u pacientov, ktorí predtým užívali biologikum vrátane anti-TNF/anti-IL-17, a u pacientov bez systémovej liečby v minulosti. Účinnosť u pacientov s primárnou neúspešnou liečbou anti-IL17 nebola skúmaná.
Na základe populačnej analýzy FK/FD doloženej klinickými údajmi pacienti s vyššou telesnou hmotnosťou (≥ 120 kg), ktorí nedosiahli úplné vyčistenie kože v 16. týždni, vykazovali prínos z pokračovania v liečbe 320 mg bimekizumabom každé štyri týždne (Q4W) po prvých 16 týždňoch liečby. V štúdii BE SURE pacienti užívali 320 mg bimekizumab Q4W po 16. týždeň, po ktorom nasledovalo dávkovanie Q4W alebo každých osem týždňov (Q8W) po 56. týždeň bez ohľadu na stav odpovede v 16. týždni. Pacienti v skupine s telesnou hmotnosťou ≥ 120 kg (N = 37) s udržiavacou dávkovacou schémou Q4W preukázali väčšie zlepšenie v PASI 100 medzi 16. týždňom (23,5 %)
a 56. týždňom (70,6 %) v porovnaní s pacientmi s udržiavacou dávkovacou schémou Q8W (16. týždeň: 45,0 % v porovnaní s 56. týždňom: 60,0 %).
U pacientov liečených bimekizumabom bolo v 16. týždni pozorované zlepšenie psoriázy postihujúcej kožu hlavy, nechty, dlane a spodná časť chodidiel (pozri Tabuľku č. 3).
| BE VIVID
| BE READY
| BE SURE
|
| Placebo
| Bimekizumab
320
mg Q4W
| Ustekinumab
| Placebo
| Bimekizumab
320
mg Q4W
| Bimekizumab
320
mg Q4W
| Adalimumab
| Koža hlavy
– IGA (N)a
Koža hlavy
– IGA 0/1, n (%)
|
(72)
11 (15,3)
|
(285)
240 (84,2)b
|
(146)
103 (70,5)
|
(74)
5 (6,8)
|
(310)
286 (92,3)b
|
(296)
256 (86,5)
|
(138)
93 (67,4)
| pp-IGA
(N)a
pp-IGA 0/1, n (%)
|
(29)
7 (24,1)
|
(105)
85 (81,0)
|
(47)
39 (83,0)
|
(31)
10 (32,3)
|
(97)
91 (93,8)
|
(90)
75 (83,3)
|
(34)
24 (70,6)
| mNAPSI
100 (N)a
|
(51)
|
(194)
|
(109)
|
(50)
|
(210)
|
(181)
|
(95)
|
|
|
Tabuľka č. 3: Odpovede na liečbu na koži hlavy, v palmoplantárnej oblasti a na nechtoch v štúdii BE VIVID, BE READY a BE SURE v 16. týždni
mNAPSI
100, n (%)
|
4 (7,8)
|
57 (29,4)
|
15 (13,8)
|
3 (6,0)
|
73 (34,8)
|
54 (29,8)
|
21 (22,1)
|
Bimekizumab 320 mg Q4W = bimekizumab každé 4 týždne. Používa sa imputácia účastníkov bez odpovede (NRI).
Odpovede na liečbu na koži hlavy podľa IGA 0/1 a pp-IGA 0/1 boli definované ako „bez nálezov“ (0) alebo „takmer bez nálezov“ (1) so zlepšením oproti východiskovej hodnote o ≥ 2 kategórie.
a) Zahŕňa iba pacientov s celkovým hodnotením kože hlavy skúšajúcim (IGA) s hodnotou najmenej 2, s palmoplantárnou hodnotou podľa IGA najmenej 2 a so skóre podľa modifikovaného indexu závažnosti psoriázy nechtov (mNAPSI) > 0 pri
východiskovom stave.
b) p < 0,001 v porovnaní s placebom, s úpravou pre multiplicitu
Odpovede na liečbu na koži hlavy podľa IGA a palmoplantárne odpovede podľa IGA u pacientov liečených bimekizumabom sa zachovali po 52. týždeň/56. týždeň. Psoriáza na nechtoch sa naďalej zmierňovala aj po 16. týždni. V štúdii BE VIVID v 52. týždni 60,3 % pacientov liečených bimekizumabom 320 mg každé 4 týždne dosiahol úplné vyliečenie nechtov (mNAPSI 100). V štúdii BE READY v 56. týždni 67,7 % a 69,8 % pacientov s odpoveďou PASI 90 v 16. týždni dosiahlo úplné vyliečenie nechtov pri užívaní 320 mg bimekizumabu každých 8 týždňov a 320 mg bimekizumabu každé 4 týždne v uvedenom poradí.
Udržiavanie odpovedeTabuľka č. 4: Udržiavanie odpovede PASI100, PASI90, IGA 0/1 v 52. týždni a absolútnejhodnoty PASI ≤ 2 u účastníkov s odpoveďou v 16. týždni pri bimekizumabe *PASI 100
| PASI 90
| IGA 0/1
| Absolútna hodnota
PASI ≤ 2
|
320
mg Q4W (N = 355)
n (%)
| 320
mg Q8W (N = 182)
n (%)
| 320
mg Q4W (N = 516)
n (%)
| 320
mg Q8W (N = 237)
n (%)
| 320
mg Q4W (N = 511)
n (%)
| 320
mg Q8W (N = 234)
n (%)'
| 320
mg Q4W (N = 511)
n (%)
| 320
mg Q8W (N = 238)
n (%)
|
295 (83,1)
| 161 (88,5)
| 464 (89,9)
| 214 (90,3)
| 447 (87,5)
| 214 (91,5)
| 460 (90,0)
| 215 (90,3)
|
* Integrovaná analýza štúdií BE VIVID, BE READY a BE SURE. Používa sa NRI.
320 mg Q4W: 320 mg bimekizumabu každé 4 týždne, po ktorom nasledovalo 320 mg bimekizumabu každé 4 týždne od 16. týždňa.
320 mg Q8W: 320 mg bimekizumabu každé 4 týždne, po ktorom nasledovalo 320 mg bimekizumabu každých 8 týždňov od 16. týždňa.
Trvanlivosť odpovede(poprerušeníliečbybimekizumabom)Graf č. 3: Podiely účastníkov s odpoveďou PASI 90 na liečbu v priebehu času v 16. týždni – Randomizované obdobie s vysadzovaním v štúdii BE READY
Randomizované obdobie s vysadzovaním v štúdii BE READY
Používa sa NRI.
Týždeň
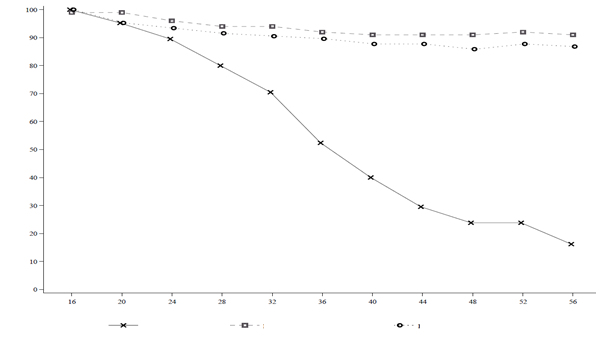
BKZ 320 mg Q4W/Placebo BKZ 320 mg Q4W/BKZ 320 mg Q8W
BKZ 320 mg Q4W/BKZ 320 mg Q4W
V 16. týždni 105 účastníkov štúdie začalo randomizované obdobie s vysadzovaním v skupine s 320 mg bimekizumabom Q4W/placebom, 100 v skupine s 320 mg bimekizumabom Q4W/Q8W a 106 v skupine s 320 mg bimekizumabom Q4W/Q4W.
V štúdii BE READY u účastníkov s odpoveďou PASI 90 v 16. týždni, ktorí boli opätovne randomizovaní pre užívanie placeba a prestali užívať bimekizumab, bol medián dĺžky času po recidívu, definovaný ako strata PASI 75, približne 28 týždňov (32 týždňov od užitia poslednej dávky bimekizumabu). Spomedzi týchto pacientov 88,1 % opäť dosiahlo odpoveď PASI 90 do 12 týždňov od opätovného začatia liečby 320 mg bimekizumabu každé 4 týždne.
Kvalita životavzhľadomnazdravotnýstav/VýsledkyhlásenépacientmiV rámci všetkých 3 štúdií u väčšieho podielu pacientov liečených bimekizumabom nedošlo k
žiadnemu vplyvu psoriázy na kvalitu ich života na základe merania indexu dermatologickej kvality života (DLQI) v porovnaní s pacientmi liečenými placebom a aktívnou referenčnou vzorkou v 16.
týždni (Tabuľka č. 5).
Tabuľka č. 5: Kvalita života v štúdiách BE VIVID, BE READY a BE SURE
| BE VIVID
| BE READY
| BE SURE
|
| Placebo
(N = 83)
n (%)
| Bimekizu
mab 320 mg Q4W (N = 321) n (%)
| Ustekinumab
(N = 163)
n (%)
| Placebo
(N = 86)
n (%)
| Bimekizumab
320 mg Q4W (N = 349)
n (%)
| Bimekizumab
320 mg Q4W (N = 319)
n (%)
| Adalimumab
(N = 159)
n (%)
|
DLQI 0/1a
Východisko
vý stav
|
3 (3,6)
|
16 (5,0)
|
5 (3,1)
|
4 (4,7)
|
11 (3,2)
|
10 (3,1)
|
13 (8,2)
|
DLQI 0/1a
16. týždeň
| 10 (12,0)
| 216 (67,3)
| 69 (42,3)
| 5 (5,8)
| 264 (75,6)
| 201 (63,0)
| 74 (46,5)
|
a) Absolútne skóre DLQI 0 alebo 1 naznačuje žiaden vplyv ochorenia na kvalitu života súvisiacu so zdravím. Používa sa NRI.
Odpovede DLQI 0/1 sa naďalej zvyšovali aj po 16. týždni a potom sa udržali po 52./56. týždeň. V štúdii BE VIVID bola miera odpovede DLQI 0/1 v 52. týždni 74,8 % u pacientov liečených bimekizumabom v dávke 320 mg každé 4 týždne. V štúdii BE SURE v 56.
týždni 78,9 % a 74,1 % pacientov malo DLQI 0/1 pri bimekizumabe v dávke 320 mg každých 8
týždňov a pri bimekizumabe v dávke 320 mg každé 4 týždne v uvedenom poradí.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Bimzelxom v
jednej alebo vo viacerých podskupinách pediatrickej populácie pri psoriáze (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaNa základe populačnej farmakokinetickej analýzy po jednorazovej subkutánnej dávke 320 mg u
pacientov s ložiskovou psoriázou dosiahol bimekizumab medián (2,5-ty a 97,5-ty percentil)
maximálnej koncentrácie v plazme 25 (12 – 50) μg/ml v rozmedzí 3 a 4 dní od podania dávky.
Populačná farmakokinetická analýza preukázala, že bimekizumab sa absorboval s vykazovaním priemernej absolútnej biodostupnosti 70,1 % u zdravých dobrovoľníkov.
Na základe simulovaných údajov medián (2,5-ty a 97,5-ty percentil) najvyššej a najnižšej koncentrácie v ustálenom stave po subkutánnom podávaní 320 mg každé 4 týždne je 43 (20 – 91) μg/ml
a 20 (7 – 50) μg/ml v uvedenom poradí a ustálený stav sa dosiahne približne po 16 týždňoch pri dávkovacej schéme každé 4 týždne. V porovnaní s expozíciou po jednorazovej dávke populačná
farmakokinetická analýza preukázala, že pacienti vykazovali 1,74-násobný nárast maximálnych
plazmatických koncentrácií a plochy pod krivkou (AUC) po opakovaných dávkach každé štyri týždne.
Po prechode z dávkovacej schémy 320 mg každé 4 týždne na dávkovaciu schému 320 mg každých 8 týždňov v 16. týždni sa ustálený stav dosiahne približne 16 týždňov po zmene. Medián (2,5-ty a 97,5-ty percentil) najvyšších a najnižších plazmatických koncentrácií je 30 (14 – 60) μg/ml a 5 (1 – 16) μg/ml v uvedenom poradí.
Distribúcia
Na základe populačných farmakokinetických analýz bol medián (koeficient variácie %) distribučného
objemu (V/F) v ustálenom stave 11,2 (30,5 %) l u pacientov s ložiskovou psoriázou.
Biotransformácia
Bimekizumab je monoklonálna protilátka a očakáva sa, že sa rozloží na malé peptidy a aminokyseliny
prostredníctvom katabolických dráh rovnakým spôsobom ako endogénne imunoglobulíny.
Eliminácia
Na základe populačných farmakokinetických analýz bol medián (koeficient variácie %) zdanlivého
klírensu (CL/F) bimekizumabu 0,337 l/deň (32,7 %) a stredná hodnota terminálneho polčasu eliminácie bimekizumabu bola 23 dní v klinických štúdiách u pacientov s ložiskovou psoriázou.
Linearita/nelinearita
Bimekizumab vykazoval farmakokinetiku úmernú dávke u pacientov s ložiskovou psoriázou pri
rozmedzí dávky od 64 mg do 480 mg po viacerých subkutánnych podaniach, pričom zdanlivý klírens
(CL/F) bol nezávislý od dávky.
Farmakokinetický/farmakodynamický vzťah
Farmakokinetický/farmakodynamický model populácie bol vyvinutý na základe všetkých dostupných
údajov u pacientov so stredne závažnou až závažnou ložiskovou psoriázou. Analýza preukázala, že vyššie koncentrácie bimekizumabu súvisia s lepším indexom plochy a závažnosti psoriázy (PASI) a
odpoveďou podľa globálneho hodnotenia skúšajúcim (IGA). Preukázalo sa, že dávka 320 mg každé 4
týždne je vhodná dávka pre počiatočné obdobie liečby a potom dávka 320 mg každých 8 týždňov je vhodná dávka pre udržiavacie obdobie pre väčšinu pacientov so stredne závažnou až závažnou
ložiskovou psoriázou (pozri časť Osobitné skupiny pacientov, telesná hmotnosť).
Osobitné skupinypacientov
Telesná hmotnosť
Populačné farmakokinetické modelovanie naznačuje, že expozícia sa znížila so zvyšovaním sa telesnej hmotnosti. Predpokladalo sa, že priemerná plazmatická koncentrácia u dospelých pacientov s telesnou
hmotnosťou ≥ 120 kg po 320 mg subkutánnej injekcii bude najmenej o 30 % nižšia ako u dospelých
pacientov s telesnou hmotnosťou 90 kg. U niektorých pacientov môže byť vhodná úprava dávky (pozri časť 4.2).
Staršie osoby
Na základe populačnej farmakokinetickej analýzy s obmedzeným počtom starších pacientov
(n = 110 vo veku ≥ 65 rokov a n = 14 vo veku ≥ 75 rokov) zdanlivý klírens (CL/F) u starších
pacientov a pacientov mladších ako 65 rokov bol podobný. Nie je potrebná žiadna úprava dávky (pozri časť 4.2).
Porucha funkcie obličiek alebo pečene
Neuskutočnili sa žiadne špecifické štúdie na stanovenie vplyvu poruchy funkcie obličiek alebo pečene na farmakokinetické vlastnosti bimekizumabu. Očakáva sa, že renálna eliminácia neporušeného bimekizumabu, monoklonálnej protilátky IgG, bude nízka a bude mať malý význam. Podobne sa IgG
eliminujú hlavne intracelulárnym katabolizmom a nepredpokladá sa, že porucha funkcie pečene ovplyvní klírens bimekizumabu. Na základe populačných farmakokinetických analýz nemali markery funkcie pečene (ALT/bilirubín) žiadny vplyv na klírens bimekizumabu u pacientov s ložiskovou psoriázou.
Rasa
V klinickej farmakokinetickej štúdii neboli pozorované žiadne klinicky významné rozdiely v expozícii bimekizumabu u japonských účastníkov v porovnaní s belošskými účastníkmi. Nie je potrebná úprava
dávky.
Pohlavie
Populačné farmakokinetické modelovanie naznačilo, že ženy môžu mať o 10 % rýchlejší zdanlivý klírens (CL/F) v porovnaní s mužmi, pričom to nie je klinicky významné. Nie je potrebná úprava
dávky.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe testovania krížovej reaktivity tkaniva, štúdií toxicity po opakovanom podávaní (vrátane farmakologických ukazovateľov bezpečnosti a hodnotenia ukazovateľov týkajúcich sa fertility) a na základe hodnotenia prenatálneho a postnatálneho vývinu u makaka dlhochvostého neodhalili žiadne osobitné riziko pre ľudí.
U makakov dlhochvostých boli účinky súvisiace s bimekizumabom obmedzené na mukokutánne zmeny zodpovedajúce farmakologickej modulácii komenzálnej mikroflóry.
Neuskutočnili sa žiadne klinické štúdie bimekizumabu zamerané na mutagenitu alebo karcinogenitu. Nepredpokladá sa však, že monoklonálne protilátky poškodzujú DNA alebo chromozómy. V 26- týždňovej klinickej štúdii zameranej na chronickú toxikológiu u makakov dlhochvostých sa nepozorovali žiadne preneoplastické ani neoplastické lézie pri dávke, ktorá viedla k 109-násobku expozície u ľudí pri dávke 320 mg každé 4 týždne.
V klinickej štúdii zameranej na perinatálny a postnatálny vývin u makakov dlhochvostých sa nepreukázali žiadne účinky na gestáciu, pôrod, prežívanie novorodených mláďat, fetálny a postnatálny vývin pri podávaní bimekizumabu počas celej organogenézy až do pôrodu v dávke, ktorá viedla k 27- násobku expozície u ľudí pri dávke 320 mg každé 4 týždne na základe AUC. Pri narodení boli koncentrácie bimekizumabu v sére u mláďat opíc porovnateľné s koncentráciami u matiek.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Glycín
Octan sodný, trihydrát
Kyselina octová, ľadová
Polysorbát 80
Voda na injekcie
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
B
i
m
z
elx 160mginjekčnýroztokvnaplnenejinjekčnejstriekačke
Uchovávajte v chladničke (2 °C – 8 °C).
Neuchovávajte v mrazničke.
Naplnenú injekčnú striekačku uchovávajte vo vonkajšom obale na ochranu pred svetlom. Naplnená injekčná striekačka sa môže uchovávať pri izbovej teplote (do 25 °C) jedenkrát po dobu
maximálne 25 dní s ochranou pred svetlom. Po vybratí z chladničky a uchovávaní v týchto
podmienkach liek zlikvidujte po 25 dňoch alebo do dátumu exspirácie vytlačeného na obale, podľa toho, ktorá lehota uplynie skôr. Na škatuli sa nachádza políčko určené pre dátum na zaznamenanie
dátumu vybratia z chladničky.
Bimzelx 160mginjekčnýroztokvnaplnenompere
Uchovávajte v chladničke (2 °C – 8 °C).
Neuchovávajte v mrazničke.
Naplnené pero uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Naplnené pero sa môže uchovávať pri izbovej teplote (do 25 °C) jedenkrát po dobu maximálne 25 dní s ochranou pred svetlom. Po vybratí z chladničky a uchovávaní v týchto podmienkach liek zlikvidujte po 25 dňoch alebo do dátumu exspirácie vytlačeného na obale, podľa toho, ktorá lehota uplynie skôr. Na škatuli sa nachádza políčko určené pre dátum na zaznamenanie dátumu vybratia z chladničky.
6.5 Druh obalu a obsah balenia
Bimzelx 160mginjekčnýroztokvnaplnenejinjekčnejstriekačke
Naplnená injekčná striekačka (sklo typu I) s objemom jeden ml s brómbutylovou gumovou zátkou
laminovanou fluoropolymérom, s nasadenou 1⁄2” tenkostennou ihlou veľkosti 27G a polypropylénovým pevným krytom ihly v pasívnom bezpečnostnom zariadení.
Veľkosť balenia – 1 naplnená injekčná striekačka. Veľkosť balenia – 2 naplnené injekčné striekačky.
Multibalenie obsahujúce 3 (3 balenia po 1) naplnené injekčné striekačky. Multibalenie obsahujúce 4 (2 balenia po 2) naplnené injekčné striekačky.
Na trh nemusia byť uvedené všetky veľkosti balenia. Bimzelx160mginjekčnýroztokvnaplnenompere
Naplnené pero s objemom jeden ml obsahujúce naplnenú injekčnú striekačku (sklo typu I) s
brómbutylovou gumovou zátkou laminovanou fluoropolymérom, s nasadenou 1⁄2” tenkostennou ihlou veľkosti 27G a polypropylénovým pevným krytom ihly.
Veľkosť balenia – 1 naplnené pero. Veľkosť balenia – 2 naplnené perá.
Multibalenie obsahujúce 3 (3 balenia po 1) naplnené perá. Multibalenie obsahujúce 4 (2 balenia po 2) naplnené perá.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIUCB Pharma S.A.
Allée de la Recherche 60
B-1070 Bruxelles
Belgicko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)Bimzelx160mginjekčnýroztokvnaplnenejinjekčnejstriekačkeEU/1/21/1575/001
EU/1/21/1575/002
EU/1/21/1575/003
EU/1/21/1575/004
Bimzelx160mginjekčnýroztokvnaplnenompereEU/1/21/1575/005
EU/1/21/1575/006
EU/1/21/1575/007
EU/1/21/1575/008
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.