
ia pri hematologických toxicitách na začiatku liečebného cyklu
(1. deň)
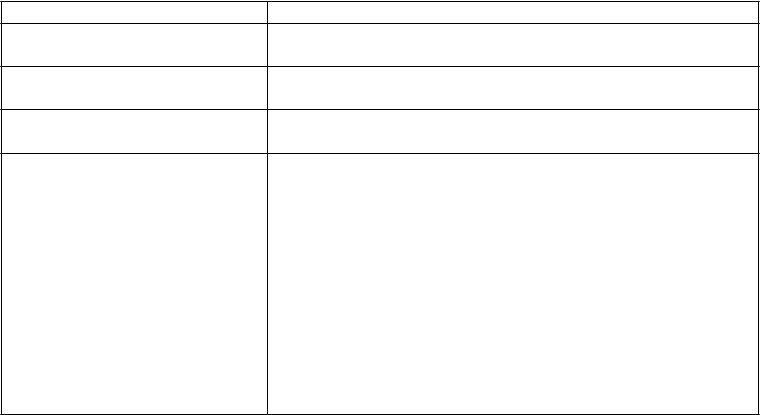
Hematologická toxicita Toxicita a úprava dávkovania
Hladiny pred liečbou
BESPONSOU:
ANC bol ≥ 1 × 109/l Ak sa ANC zníži, prerušte ďalší liečebný cyklus, kým sa ANC
nevráti na ≥ 1 × 109/l.
Počet krvných doštičiek bol ≥ 50 × 109/la
ANC bol < 1 × 109/l a/alebo počet krvných doštičiek
bol < 50 × 109/la
Ak sa počet krvných doštičiek zníži, prerušte ďalší liečebný cyklus, kým sa počet krvných doštičiek nevráti na ≥ 50 × 109/la. Ak sa ANC a/alebo počet krvných doštičiek zníži, prerušte
ďalší liečebný cyklus, kým nedôjde aspoň k jednému z nasledujúceho:
- ANC a počet krvných doštičiek sa vrátia aspoň na
východiskové hladiny z predchádzajúceho cyklu, alebo
- ANC sa vráti na ≥ 1 × 109/l a počet krvných doštičiek sa vráti na ≥ 50 × 109/la, alebo
- stabilné alebo zlepšené ochorenie (na základe najaktuálnejšieho hodnotenia kostnej drene) a hodnoty ANC a počtu krvných doštičiek sú považované za následok primárneho ochorenia (nepovažujú sa za toxicitu súvisiacu
s BESPONSOU).
Skratka: ANC = absolútny počet neutrofilov.
a Počet krvných doštičiek použitý pri dávkovaní musí byť nezávislý od transfúzie krvi.
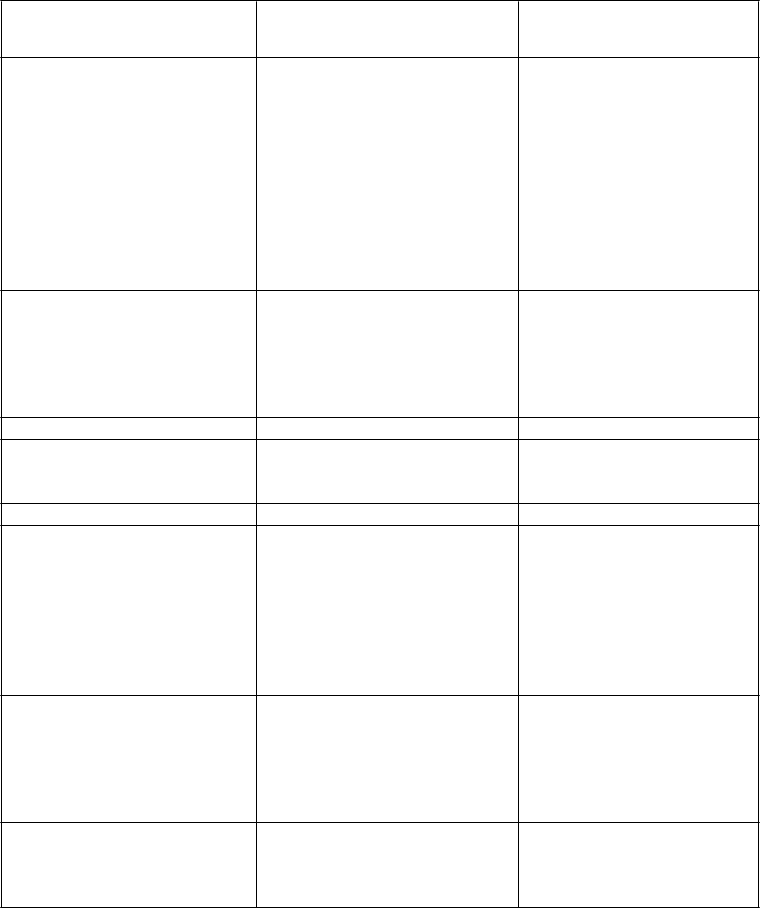
Tabuľka 3. Úprava dávkovania pri nehematologických toxicitách kedykoľvek počas liečby
Nehematologická toxicita Úprava dávkovania
VOD/SOS alebo iná závažná hepatálna toxicita
Hladina celkového bilirubínu > 1,5 × ULN
a AST/ALT > 2,5 × ULN
Natrvalo ukončite liečbu (pozri časť 4.4).
Prerušte podávanie, kým sa hladina celkového bilirubínu nevráti na ≤ 1,5 × ULN a AST/ALT na ≤ 2,5 × ULN pred každou dávkou, pokiaľ zvýšená hladina nesúvisí s Gilbertovým ochorením alebo hemolýzou. Ak sa hladina celkového bilirubínu nevráti na hodnotu ≤ 1,5 × ULN alebo sa AST/ALT nevráti na hodnotu ≤ 2,5 × ULN, natrvalo ukončite liečbu
(pozri časť 4.4).

Reakcia súvisiaca s infúziou Prerušte infúziu a zahájte odpovedajúcu farmakologickú liečbu.. V závislosti od závažnosti reakcie súvisiacej s infúziou zvážte ukončenie infúzie alebo podávanie steroidov
a antihistaminík. Pri závažných alebo život ohrozujúcich reakciách na infúziu natrvalo ukončite liečbu (pozri časť 4.4).
Tabuľka 3. Úprava dávkovania pri nehematologických toxicitách kedykoľvek počas liečby
Nehematologická toxicita Úprava dávkovania

Nehematologická toxicita ≥ 2. stupňaa (súvisiaca
s BESPONSOU)
Prerušte liečbu, kým nedôjde k zlepšeniu na 1. stupeň alebo stupeň toxicity aký bol pred podaním liečby pred každou dávkou.
Skratky: ALT = alanínaminotransferáza; AST = aspartátaminotransferáza; ULN = horný limit normálu; VOD/SOS = venookluzívna choroba/sínusoidný obštrukčný syndróm.
a Stupeň závažnosti podľa Všeobecných kritérií pre terminológiu nežiaducich udalostí podľa Národného inštitútu pre výskum rakoviny (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE) verzia 3.0.
Tabuľka 4 zobrazuje návod na úpravu dávkovania v závislosti od trvania prerušení dávkovania kvôli
toxicite.
Tabuľka 4. Úprava dávkovania v závislosti od trvania prerušenia podávania kvôli toxicite
Trvanie prerušenia podávania
kvôli toxicite
Úprava dávkovania
< 7 dní (v rámci cyklu) Nepodajte nasledujúcu dávku (zachovajte interval minimálne

6 dní medzi dávkami).
≥ 7 dní Vynechajte ďalšiu dávku v rámci cyklu.
≥ 14 dní Keď sa dosiahne adekvátne zotavenie, znížte celkovú dávku pre nasledujúci cyklus o 25 %. Ak sa vyžaduje ďalšia úprava dávkovania, znížte počet dávok pre nasledujúce cykly na 2 na cyklus. Ak nie je tolerované zníženie celkovej dávky o 25 %
a následné zníženie počtu dávok na 2 na cyklus, natrvalo ukončite liečbu.
> 28 dní Zvážte trvalé ukončenie podávania BESPONSY.
Špeciálne populácieStaršie osobyNa základe veku nie je potrebná žiadna úprava počiatočnej dávky (pozri časť 5.2).
Porucha funkcie pečeneU pacientov s poruchou funkcie pečene definovanou hladinou celkového bilirubínu ≤ 1,5 × horný limit normálu (ULN) a hladinami aspartátaminotransferázy (AST)/alanínaminotransferázy
(ALT) ≤ 2,5 × ULN sa nevyžaduje žiadna úprava počiatočnej dávky (pozri časť 5.2). O bezpečnosti
u pacientov s hladinou celkového bilirubínu > 1,5 × ULN a AST/ALT > 2,5 × ULN pred podaním sú k dispozícii len obmedzené údaje. Prerušte podávanie, kým sa hladina celkového bilirubínu nevráti
na ≤ 1,5 × ULN a AST/ALT na ≤ 2,5 × ULN pred každým podaním, pokiaľ hladina nesúvisí s Gilbertovým syndrómom alebo hemolýzou. Ak sa hladina celkového bilirubínu nevráti na
hodnotu ≤ 1,5 × ULN alebo sa AST/ALT nevráti na hodnotu ≤ 2,5 × ULN, natrvalo ukončite liečbu
(pozri tabuľku 3 a časť 4.4).
Porucha funkcie obličiekU pacientov s miernou, strednou alebo závažnou poruchou funkcie obličiek (klírens kreatinínu [CLcr]
60 – 89 ml/min, 30 – 59 ml/min, resp. 15 – 29 ml/min) sa nevyžaduje žiadna úprava počiatočnej dávky
(pozri časť 5.2). Bezpečnosť a účinnosť BESPONSY sa neštudovali u pacientov v terminálnom štádiu ochorenia obličiek.
Pediatrická populácia
Bezpečnosť a účinnosť BESPONSY u detí vo veku od 0 do <18 rokov neboli stanovené. Nie sú dostupné žiadne údaje.
Spôsob podávania
BESPONSA je určená na intravenózne použitie. Infúzia sa musí podávať počas 1 hodiny.
BESPONSA sa nemá podávať ako intravenózny bolus.
BESPONSA sa musí pred podaním rekonštituovať a nariediť. Pre pokyny na rekonštitúciu a riedenie
BESPONSY pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
- Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
- Pacienti, u ktorých sa v minulosti potvrdila závažná alebo pretrvávajúca venookluzívna choroba pečene/sinusoidný obštrukčný syndróm (VOD/SOS).
- Pacienti so závažným pretrvávajúcim ochorením pečene (napríklad cirhóza, nodulárna
regeneratívna hyperplázia, aktívna hepatitída).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila sledovateľnosť biologických liekov, do záznamu pacienta by sa mali prehľadne
zaznamenať obchodný názov a číslo šarže podávaného lieku.
Hepatotoxicitavrátanevenookluzívnejchorobypečene/sínusoidnéhoobštrukčnéhosyndrómu
(VOD/SOS)
U pacientov s relapsujúcou alebo refraktérnou ALL, ktorým sa podávala BESPONSA bola hlásená
hepatotoxicita vrátane závažnej, život ohrozujúcej a niekedy smrteľnej hepatálnej VOD/SOS (pozri časť 4.8). BESPONSA v tejto populácii pacientov významne zvýšila riziko VOD/SOS oproti
štandardným chemoterapeutickým režimom. Toto riziko bolo najvýraznejšie u pacientov, ktorí podstúpili následnú HSCT.
V nasledujúcich podskupinách bola hlásená frekvencia VOD/SOS po HSCT ≥ 50 %:
- pacienti s prípravnými režimami HSCT s obsahom 2 alkylačných látok,
- pacienti vo veku ≥ 65 rokov, a
- pacienti s hladinou bilirubínu v sére ≥ ULN pred HSCT.
Používaniu prípravných režimov pred HSCT s obsahom 2 alkylačných látok sa má vyhnúť. Prínos/riziko sa má dôkladne zvážiť pred podaním BESPONSY pacientom, u ktorých sa veľmi pravdepodobne nebude dať vyhnúť budúcemu použitiu prípravných režimov HSCT s obsahom
2 alkylačných látok.
U pacientov, ktorí majú pred HSCT hladinu bilirubínu v sére ≥ ULN, HSCT po liečbe BESPONSOU má byť vykonaná len po dôkladnom zvážení prínosu/rizika. Ak budú títo pacienti pokračovať HSCT, prejavy a príznaky VOD/SOS majú byť pozorne sledované (pozri časť 4.2).
Ďalšie faktory na strane pacienta, ktoré sa zdajú byť spojené so zvýšeným rizikom VOD/SOS po HSCT, zahŕňajú predchádzajúcu HSCT, vek ≥ 55 rokov, anamnézu ochorenia pečene a/alebo hepatitídu pred liečbou, neskoršie záchranné línie a vyšší počet liečebných cyklov.
Pred podaním BESPONSY pacientom, ktorí podstúpili predchádzajúcu HSCT, sa vyžaduje dôkladné zváženie liečby. Ani jeden z pacientov s relapsujúcou alebo refraktérnou ALL, ktorí boli liečení BESPONSOU, v klinických štúdiách nepodstúpil HSCT v predchádzajúcich 4 mesiacoch.
Pacientov s anamnézou ochorenia pečene je pred liečbou BESPONSOU potrebné dôkladne vyšetriť (napríklad ultrazvukové vyšetrenie, testovanie na prítomnosť vírusu hepatitídy), aby sa vylúčilo závažné prebiehajúce ochorenie pečene (pozri časť 4.3).
Aby sa znížilo riziko VOD/SOS u pacientov pokračujúcich HSCT je odporúčané trvanie liečby
2 cykly, s maximom 3 cykly (pozri časť 4.2).
U všetkých pacientov majú byť prejavy a príznaky VOD/SOS dôkladne sledované, hlavne u tých po HSCT. Prejavy môžu zahŕňať zvýšené hladiny celkového bilirubínu, hepatomegáliu (ktorá môže byť bolestivá), rýchly prírastok hmotnosti a ascites. Len sledovanie hladiny celkového bilirubínu nemusí identifikovať všetkých pacientov s rizikom VOD/SOS. U všetkých pacientov pred každou dávkou BESPONSY a po nej hodnoty pečeňových testov vrátane ALT, AST, celkového bilirubínu a alkalickej fosfatázy majú byť sledované. U pacientov, u ktorých boli zistené nezvyčajné hodnoty pečeňových testov, sa tieto testy a klinické prejavy a príznaky hepatotoxicity majú sledovať častejšie. U pacientov pokračujúcich HSCT majú byť hodnoty pečeňových testov dôkladne sledované počas prvého mesiaca po HSCT a následne menej často podľa štandardnej klinickej praxe. Zvýšenie hodnôt pečeňových testov môže vyžadovať prerušenie podávania, zníženie dávky alebo trvalé ukončenie liečby BESPONSOU (pozri časť 4.2).
Ak dôjde k VOD/SOS, liečba by mala byť natrvalo ukončená (pozri časť 4.2). Ak dôjde k závažnej
VOD/SOS, pacient by mal byť liečený podľa štandardnej klinickej praxe.
Myelosupresia/cytopénie
U pacientov dostávajúcich inotuzumab ozogamicín bola hlásená neutropénia, trombocytopénia,
anémia, leukopénia, febrilná neutropénia, lymfopénia a pancytopénia, pričom niektoré stavy boli život ohrozujúce (pozri časť 4.8).
Spomedzi pacientov, ktorým sa podáva inotuzumab ozogamicín boli u niektorých hlásené komplikácie spojené s neutropéniou a trombocytopéniou (vrátane infekcií, resp. krvácavých/hemoragických
epizód) (pozri časť 4.8).
Pred každou dávkou BESPONSY má byť úplný krvný obraz sledovaný, prejavy a príznaky infekcie, krvácania/hemorágie a iných následkov myelosupresie majú byť sledované počas liečby. Podľa potreby majú byť podané profylaktické antiinfektíva a počas liečby a po nej má byť zaradené dozorné testovanie.
Manažment závažnej infekcie, krvácania/hemorágie a iných následkov myelosupresie vrátane závažnej neutropénie alebo trombocytopénie môže vyžadovať prerušenie podávania, zníženie dávky alebo ukončenie liečby (pozri časť 4.2).
Reakcie súvisiacesinfúziou
U pacientov dostávajúcich inotuzumab ozogamicín boli hlásené reakcie súvisiace s infúziou (pozri
časť 4.8).
Pred podaním dávky sa odporúča premedikácia kortikosteroidmi, antipyretikami a antihistaminikami
(pozri časť 4.2).
Počas infúzie a aspoň 1 hodinu po dokončení infúzie majú byť pacienti dôkladne sledovaní ohľadom možného nástupu reakcií súvisiacich s infúziou vrátane príznakov ako hypotenzia, návaly tepla alebo problémy s dýchaním. Ak dôjde k reakcii súvisiacej s infúziou, infúzia sa má prerušiť a majú sa vykonať príslušné klinické opatrenia. V závislosti od závažnosti reakcie súvisiacej s infúziou sa má zvážiť prerušenie infúzie alebo podávanie steroidov a antihistaminík (pozri časť 4.2).
Pri závažných alebo život ohrozujúcich reakciách na infúziu sa má liečba natrvalo ukončiť (pozri časť 4.2).
Syndróm nádorovéhorozpadu(TLS,tumorlysissyndrome)
U pacientov dostávajúcich inotuzumab ozogamicín bol hlásený TLS, ktorý môže byť život ohrozujúci
alebo smrteľný (pozri časť 4.8).
U pacientov s veľkou nádorovou záťažou sa pred podaním dávky odporúča premedikácia na zníženie hladín kyseliny močovej a hydratácia (pozri časť 4.2).
Pacienti majú byť ohľadom prejavov a príznakov TLS sledovaní a liečení podľa štandardnej klinickej praxe.
Predĺženie QTintervalu
U pacientov dostávajúcich inotuzumab ozogamicín bolo pozorované predĺženie QT intervalu (pozri
časti 4.8 a 5.2).
BESPONSA sa musí podávať s opatrnosťou u pacientov s anamnézou alebo predispozíciou ku predĺženiu QT intervalu, u pacientov ktorí užívajú lieky predlžujúce QT interval (pozri časť 4.5), a u pacientov s nerovnováhou elektrolytov. Pred začiatkom liečby sa musí urobiť EKG a stanoviť hladina elektrolytov a musia sa pravidelne sledovať počas liečby (pozri časti 4.8 a 5.2).
Zvýšené hladinyamylázya lipázy
U pacientov dostávajúcich inotuzumab ozogamicín boli hlásené zvýšené hladiny amylázy a lipázy
(pozri časť 4.8).
U pacientov sa má sledovať zvýšenie hladín amylázy a lipázy. Možné hepatobiliárne ochorenie má vyhodnotiť a liečiť podľa bežnej klinickej praxe.
Imunizácia
Bezpečnosť imunizácie živými vírusovými očkovacími látkami počas alebo následne po liečbe
BESPONSOU nebola študovaná. Vakcinácia živými vírusovými očkovacími látkami sa neodporúča aspoň
2 týždne pred začiatkom liečby BESPONSOU, počas liečby a kým nepríde k zotaveniu B lymfocytov po poslednom liečebnom cykle.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne formálne klinické štúdie liekovej interakcie (pozri časť 5.2).
Na základe in vitro údajov nie je pravdepodobné, že by súčasné podávanie inotuzumab ozogamicínu s inhibítormi alebo induktormi cytochrómu P450 (CYP) alebo ridíndifosfátglukuronozyltransferázových (UGT) liek metabolizujúcich enzýmov menilo expozíciu
N-acetyl-gama-kalicheamicíndimetylhydrazidu. Okrem toho nie je pravdepodobné, že by inotuzumab
ozogamicín a N-acetyl-gama-kalicheamicíndimetylhydrazid menili expozíciu substrátov CYP
enzýmov, a že by N-acetyl-gama-kalicheamicíndimetylhydrazid menil expozíciu substrátov enzýmov
UGT alebo hlavných transportérov lieku.
U pacientov dostávajúcich inotuzumab ozogamicín bolo pozorované predĺženie QT intervalu (pozri časť 4.4). Preto je potrebné dôkladne zvážiť súčasné používanie inotuzumab ozogamicínu s liekmi, ktoré predlžujú QT interval alebo indukujú torsades de pointes. V prípade kombinácií týchto liekov sa má QT interval sledovať (pozri časti 4.4, 4.8 a 5.2).
4.6 Fertilita, gravidita a laktácia
Ženy
vo
fertilnom
veku/antikoncepcia
u
mužo
v
ažien
Ženy vo fertilnom veku počas používania BESPONSY sa majú vyhnúť otehotneniu.
Ženy majú používať účinnú antikoncepciu počas liečby BESPONSOU a aspoň 8 mesiacov po poslednej dávke. Muži s partnerkami vo fertilnom veku musia používať účinnú antikoncepciu počas liečby BESPONSOU a aspoň 5 mesiacov po poslednej dávke.
Gravidita
Nie sú k dispozícii žiadne údaje o používaní inotuzumab ozogamicínu u gravidných žien. Na základe
neklinických bezpečnostných nálezov môže inotuzumab ozogamicín pri podávaní gravidnej žene spôsobiť poškodenie embrya a plodu. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
BESPONSA sa nesmie používať počas gravidity pokiaľ možný prínos pre matku neprevyšuje možné riziká pre plod. Gravidné ženy alebo pacientky, ktoré otehotneli počas liečby inotuzumab ozogamicínom, či liečení mužskí pacienti, ktorí sú partnermi gravidných žien, musia byť oboznámení s možnými rizikami pre plod.
Dojčenie
Nie sú k dispozícii žiadne údaje o prítomnosti inotuzumab ozogamicínu alebo jeho metabolitov
v ľudskom mlieku, účinkoch na dojčené dieťa ani účinkoch na tvorbu mlieka. Kvôli možným nežiaducim reakciám u dojčených detí nesmú ženy počas liečby BESPONSOU a aspoň 2 mesiace po poslednej dávke dojčiť (pozri časť 5.3).
Fertilita
Na základe neklinických nálezov môže byť liečbou inotuzumab ozogamicínom narušená plodnosť
u mužov a žien (pozri časť 5.3). O plodnosti u pacientov nie sú žiadne informácie. Muži aj ženy musia pred liečbou vyhľadať poradenstvo ohľadom zachovania plodnosti.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
BESPONSA môže mať stredne závažný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti môžu počas liečby BESPONSOU pociťovať únavu (pozri časť 4.8). Preto sa pri vedení vozidiel alebo obsluhe strojov odporúča opatrnosť.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Najčastejšie (≥ 20 %) nežiaduce reakcie boli trombocytopénia (51 %), neutropénia (49 %), infekcia
(48 %), anémia (36 %), leukopénia (35 %), únava (35 %), krvácanie (33 %), pyrexia (32 %), nevoľnosť (31 %), bolesť hlavy (28 %), febrilná neutropénia (26 %), zvýšená hladina transamináz
(26 %), bolesť brucha (23 %), zvýšená hladina gama-glutamyltransferázy (21 %) a hyperbilirubinémia
(21 %).
U pacientov, ktorí dostali BESPONSU boli najčastejšie (≥ 2 %) závažné nežiaduce reakcie infekcia (23 %), febrilná neutropénia (11 %), krvácanie (5 %), bolesť brucha (3 %), pyrexia (3 %), VOD/SOS (2 %) a únava (2 %).
Tabuľkový zoznamnežiaducichreakcií
Tabuľka 5 zobrazuje nežiaduce reakcie hlásené u pacientov s relapsujúcou alebo refraktérnou ALL,
ktorí dostávali BESPONSU.
Nežiaduce reakcie sú uvedené podľa triedy orgánových systémov (SOC) a kategórií frekvencie definovaných pomocou nasledujúceho dohovoru: veľmi časté (³ 1/10), časté (³ 1/100 až < 1/10), menej časté (³ 1/1 000 až < 1/100), zriedkavé (³ 1/10 000 až < 1/1 000), veľmi zriedkavé
(< 1/10 000), neznáme (nemôžu byť určené z dostupných údajov). V každej skupine frekvencie sú
nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 5. Nežiaduce reakcie hlásené u pacientov s relapsujúcou alebo refraktérnou ALL
z prekurzorov B-buniek, ktorí dostávali BESPONSU
Trieda orgánových systémov
podľa databázy MedDRA
Veľmi časté Časté
Infekcie a nákazy Infekcia (48 %)a (vrátane sepsy a bakteriémie [16 %],
hubovej infekcie [9 %],
infekcie dolných dýchacích ciest
[12 %], infekcie horných dýchacích ciest [12 %], bakteriálnej infekcie [1 %], vírusovej infekcie [8 %], infekcie gastrointestinálneho traktu [4 %], kožnej infekcie
[4 %])
Poruchy krvi a lymfatického systému
Febrilná neutropénia (26 %) Neutropénia (49 %) Trombocytopénia (51 %) Leukopénia (35 %) Lymfopénia (18 %)
Anémia (36 %)
Pancytopéniab (2 %)
Poruchy imunitného systému Precitlivenosť (1 %)
Poruchy metabolizmu a výživy Znížená chuť do jedla (12 %) Syndróm nádorového rozpadu
(2 %)
Hyperurikémia (4 %)
Poruchy nervového systému Bolesť hlavy (28 %) Poruchy ciev Krvácanie c (33 %) (vrátane
krvácania v centrálnom
nervovom systéme [1 %], krvácania v hornej časti gastrointestinálneho traktu [5 %], krvácania v dolnej časti gastrointestinálneho traktu [4 %], epistaxy [15 %])
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových ciest
Bolesť brucha (23 %) Vracanie (15 %) Hnačka (17 %) Nevoľnosť (31 %) Stomatitída (13 %) Zápcha (17 %)
Hyperbilirubinémia (21 %) Zvýšené hladiny transamináz (26 %)
Zvýšená hladina GMT (21 %)
Ascites (4 %)
Abdominálna distenzia (6 %)
Venookluzívna choroba pečene (sínusoidný obštrukčný syndróm) (3 %
[pred HSCT]d)
Trieda orgánových systémov podľa databázy MedDRA
Celkové poruchy a reakcie v mieste podania
Veľmi časté Časté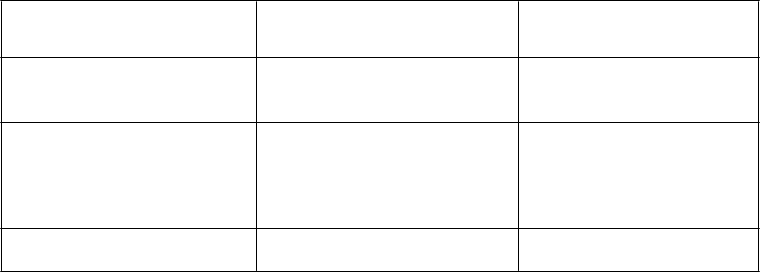
Pyrexia (32 %) Únava (35 %) Zimnica (11 %)
Laboratórne a funkčné vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
Zvýšená hladina alkalickej fosfatázy (13 %)
Reakcia súvisiaca s infúziou
(10 %)
Predĺženie QT intervalu na
EKG (1 %)
Zvýšená hladina amylázy
(5 %)
Zvýšená hladina lipázy (9 %)
Nežiaduce reakcie zahŕňali udalosti z akejkoľvej príčiny, ktoré vznikli počas liečby, počnúc 1. dňom 1. cyklu alebo po ňom, končiac 42 dní od poslednej dávky BESPONSY, avšak pred začiatkom novej protinádorovej liečby (vrátane HSCT).
Preferované termíny sa získali použitím slovníka medicínskej terminológie pre regulačné činnosti (Medical
Dictionary for Regulatory Activities, MedDRA) verzie 18.1.
Skratky: ALL = akútna lymfoblastová leukémia; EKG = elektrokardiogram; GMT = gamaglutamyltransferáza; HSCT = transplantácia hematopoetických kmeňových buniek.
a Infekcia tiež zahŕňa iné typy infekcie (11 %). Poznámka: pacienti mohli mať > 1 typ infekcie.
b Pancytopénia zahŕňa nasledujúce hlásené preferované termíny: zlyhanie kostnej drene, febrilná aplázia kostnej drene a pancytopénia.
c Krvácanie tiež zahŕňa iné typy krvácania (16 %). Poznámka: pacienti mohli mať > 1 typ krvácania.
d VOD zahŕňa 1 ďalšieho pacienta s venookluzívnou chorobou pečene, ktorá sa objavila 56. deň bez zásahu
HSCT. VOD/SOS boli hlásené aj u 17 pacientov po nasledujúcej HSCT.
Popisvybranýchnežiaducichreakcií
Hepatotoxicita vrátane venookluzívnej choroby pečene/sínusoidného obštrukčného syndrómu
(VOD/SOS)
V pivotnej klinickej štúdii (N = 164) boli VOD/SOS hlásené u 22 (13 %) pacientov vrátane 5 (3 %)
pacientov počas podávania skúšanej liečby alebo počas sledovania bez intervencie v zmysle HSCT. Zo
77 pacientov, ktorí pokračovali následnou HSCT (z ktorých 6 dostalo ďalšiu záchrannú liečbu po liečbe BESPONSOU pred pokračovaním HSCT), boli VOD/SOS hlásené u 17 (22 %) pacientov. Päť
zo 17 prípadov VOD/SOS, ktoré nastali po HSCT, bolo smrteľných.
VOD/SOS boli hlásené až do 56 dní po poslednej dávke inotuzumab ozogamicínu bez intervencie v zmysle HSCT. Medián času od HSCT do nástupu VOD/SOS bol 15 dní (rozsah: 3 – 57 dní).
Z 5 pacientov, u ktorých došlo k VOD/SOS počas liečby inotuzumab ozogamicínom, ale bez intervencie v zmysle HSCT, 2 pacienti podstúpili HSCT aj pred liečbou BESPONSOU.
U pacientov, ktorí po liečbe BESPONSOU pokračovali HSCT, boli VOD/SOS hlásené u 5/11 (46 %) pacientov, ktorí podstúpili HSCT pred aj po liečbe BESPONSOU a u 12/66 (18 %) pacientov, ktorí podstúpili HSCT len po liečbe BESPONSOU.
Pokiaľ ide o iné rizikové faktory, boli VOD/SOS hlásené u 6/11 (55 %) pacientov, ktorí dostali prípravný režim pred HSCT obsahujúci 2 alkylačné látky, a u 8/52 (15 %) pacientov, ktorí dostali prípravný režim pred HSCT obsahujúci 1 alkylačnú látku, u 7/17 (41 %) pacientov vo veku ≥ 55 rokov a u 10/60 (17 %) pacientov vo veku < 55 rokov, u 7/12 (58 %) pacientov s hladinou bilirubínu
v sére ≥ ULN pred HSCT a u 10/65 (15 %) pacientov s hladinou bilirubínu v sére < ULN pred HSCT.
V pivotnej štúdii (N = 164) bola hlásená hyperbilirubinémia u 35 (21 %) pacientov a zvýšená hladina transamináz u 43 (26 %) pacientov. Hyperbilirubinémia stupňa ≥ 3 a zvýšené hladiny transamináz boli hlásené u 9 (6 %), resp. 11 (7 %) pacientov. Medián času do nástupu hyperbilirubinémie a zvýšenej hladiny transamináz bol 73 dní, resp. 29 dní.
Klinické opatrenia v prípade hepatotoxicity vrátane VOD/SOS, pozri časť 4.4.
Myelosupresia/cytopénie
V pivotnej štúdii (N = 164) boli hlásené trombocytopénia a neutropénia u 83 (51 %), resp. 81 (49 %) pacientov. Trombocytopénia a neutropénia stupňa 3 boli hlásené u 23 (14 %), resp. u 33 (20 %) pacientov. Trombocytopénia a neutropénia stupňa 4 boli hlásené u 46 (28 %), resp. u 45 (27 %) pacientov. Febrilná neutropénia, ktorá môže byť život ohrozujúca, bola hlásená u 43 (26 %) pacientov.
Klinické opatrenia v prípade myelosupresie/cytopénií, pozri časť 4.4.
Infekcie
V pivotnej štúdii (N = 164) boli infekcie vrátane závažných infekcií, z ktorých niektoré boli život ohrozujúce alebo smrteľné, hlásené u 79 (48 %) pacientov. Frekvencie konkrétnych infekcií boli:
sepsa a bakteriémia (16 %), infekcia dolných dýchacích ciest (12 %), infekcia horných dýchacích ciest (12 %), mykotická infekcia (9 %), vírusová infekcia (8 %), infekcia gastrointestinálneho traktu (4 %), kožná infekcia (4 %) a bakteriálna infekcia (1 %). Smrteľné infekcie vrátane pneumónie,
neutropenickej sepsy, sepsy, septického šoku a pseudomonádovej sepsy boli hlásené u 8 (5 %) pacientov.
Klinické opatrenia v prípade infekcií, pozri časť 4.4.
Krvácanie/hemorágia
V pivotnej klinickej štúdii (N = 164) boli krvácavé/hemoragické príhody, väčšinou miernej závažnosti, hlásené u 54 (33 %) pacientov. Frekvencie konkrétnych krvácavých/hemoragických príhod takéto: epistaxa (15 %), krvácanie v hornej časti gastrointestinálneho traktu (5 %), krvácanie v dolnej časti gastrointestinálneho traktu (4 %) a krvácanie v centrálnej nervovej sústave (CNS) (1 %). Krvácavé/hemoragické príhody stupňa 3/4 boli hlásené u 8/164 (5 %) pacientov. Bola hlásená jedna krvácavá/hemoragická príhoda stupňa 5 (vnútrobrušná hemorágia).
Klinické opatrenia v prípade krvácavých/hemoragických príhod, pozri časť 4.4.
Reakcie súvisiace s infúziou
V pivotnej štúdii (N = 164) boli reakcie súvisiace s infúziou hlásené u 17 (10 %) pacientov. Všetky udalosti mali závažnosť stupňa ≤ 2. Reakcie súvisiace s infúziou všeobecne vznikli počas 1. cyklu
a krátko po konci infúzie inotuzumab ozogamicínu a odzneli spontánne alebo pomocou klinických opatrení.
Klinické opatrenia v prípade reakcií súvisiacich s infúziou, pozri časť 4.4.
Syndróm nádorového rozpadu (TLS)
V pivotnej štúdii (N = 164) bol TLS, ktorý môže byť život ohrozujúci alebo smrteľný, hlásený
u 4/164 (2 %) pacientov. TLS stupňa 3/4 bol hlásený u 3 (2 %) pacientov. TLS vznikol krátko po skončení infúzie inotuzumab ozogamicínu a odznel po klinických opatreniach.
Klinické opatrenia v prípade TLS, pozri časť 4.4.
Predĺženie QT intervalu
V pivotnej štúdii (N = 164) boli predĺženia QT intervalu upravené podľa frekvencie srdca pomocou Fridericiovho vzorca (QTcF) ≥ 60 ms od počiatočnej hodnoty namerané u 4/162 (3 %) pacientov. Žiadni pacienti nemali hodnoty QTcF > 500 ms. Predĺženie QT intervalu stupňa 2 bolo hlásené
u 2/164 (1 %) pacientov. Neboli hlásené žiadne predĺženia intervalu QT stupňa ≥ 3 ani príhody torsades de pointes.
Pravidelné sledovanie EKG a hladiny elektrolytov, pozri časť 4.4.
Zvýšené hladiny amylázy a lipázyV pivotnej štúdii (N = 164) boli zvýšenia hladín amylázy a lipázy hlásené u 8 (5 %), resp. 15 (9 %) pacientov. Zvýšenia hladín amylázy a lipázy stupňa ≥ 3 boli hlásené u 3 (2 %), resp. 7 (4 %) pacientov.
Pravidelné sledovanie zvýšených hladín amylázy a lipázy, pozri časť 4.4.
ImunogenicitaV klinických štúdiách BESPONSY u pacientov s relapsujúcou alebo refraktérnou ALL malo 7/236
(3 %) pacientov pozitívny test na protilátky proti inotuzumab ozogamicínu. Žiadni pacienti nemali pozitívny test na neutralizačné protilátky proti inotuzumab ozogamicínu. U pacientov s pozitívnym
testom na protilátky proti inotuzumab ozogamicínu nebol na základe populačnej farmakokinetickej analýzy po liečbe BESPONSOU pozorovaný žiadny účinok na klírens BESPONSY. Počet pacientov
bol príliš nízky, aby bolo možné hodnotiť vplyv protilátok proti inotuzumab ozogamicínu na účinnosť a bezpečnosť.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV klinických štúdiách u pacientov s relapsujúcou alebo refraktérnou ALL boli maximálne jednorazové a viacnásobné dávky inotuzumab ozogamicínu 0,8 mg/m2, resp. 1,8 mg/m2 na cyklus, podané ako
3 rozdelené dávky v 1. deň (0,8 mg/m2), 8. deň (0,5 mg/m2) a 15. deň (0,5 mg/m2) (pozri časť 4.2).
Predávkovania môžu viesť k nežiaducim reakciám, ktoré sú rovnaké ako reakcie pozorované pri odporúčanej liečebnej dávke (pozri časť 4.8).
V prípade predávkovania sa musí infúzia dočasne prerušiť a pacientov je potrebné sledovať ohľadom pečeňovej a hematologickej toxicity (pozri časť 4.2). Keď všetky toxicity odznejú, je potrebné zvážiť opätovné začatie podávania BESPONSY v správnej liečebnej dávke.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastické látky, iné antineoplastické látky, monoklonálne protilátky, ATC kód: L01XC26.
Mechanizmus účinkuInotuzumab ozogamicín je ADC zložený z monoklonálnej protilátky cielenej proti CD22, ktorá je
kovalentne viazaná na N-acetyl-gama-kalicheamicíndimetylhydrazid. Inotuzumab je humanizovaná protilátka imunoglobulínu triedy G podtypu 4 (IgG4), ktorá špecificky rozpoznáva ľudský antigén CD22. Malá molekula, N-acetyl-gama-kalicheamicín, je cytotoxický produkt.
N-acetyl-gama-kalicheamicín je kovalentne viazaný na protilátku pomocou linkera štiepiteľného kyselinou. Neklinické údaje ukazujú, že protinádorová aktivita BESPONSY je spôsobená väzbou ADC na nádorové bunky exprimujúce CD22 s následnou internalizáciou komplexu ADC-CD22
a uvoľnením N-acetyl-gama-kalicheamicíndimetylhydrazidu vo vnútri bunky hydrolytickým štiepením linkera. Aktivácia N-acetyl-gama-kalicheamicíndimetylhydrazidu indukuje zlomy v dvojvláknovej DNA a následne indukciu zastavenia bunkového cyklu a apoptotickú bunkovú smrť.
Klinická účinnosťabezpečnosť
Pacienti s relapsujúcou alebo refraktérnou ALL, ktorí predtým mali podané 1 alebo 2 liečebné režimy
na liečbu ALL – Štúdia 1
Bezpečnosť a účinnosť BESPONSY u pacientov s relapsujúcou alebo refraktérnou CD22-pozitívnou ALL sa hodnotili v otvorenej medzinárodnej multicentrickej štúdii fázy 3 (Štúdia 1), v ktorej boli pacienti randomizovaní tak, že dostávali BESPONSU (N = 164 [164 bolo liečených]) alebo chemoterapiu podľa voľby skúšajúceho lekára (N=162 [143 bolo liečených]), konkrétne fludarabín plus cytarabín plus faktor stimulujúci kolónie granulocytov (FLAG) (N = 102 [93 bolo liečených]), mitoxantrón/cytarabín (MXN/Ara-C) (N = 38 [33 bolo liečených]) alebo vysokú dávku cytarabínu (HIDAC) (N = 22 [17 bolo liečených]).
Vhodní pacienti boli vo veku ≥ 18 rokov s Philadelphia chromozómom negatívnym (Ph-) alebo pozitívnym (Ph+) relapsujúcou alebo refraktérnou CD22-pozitívnou ALL z prekurzorov B-buniek.
Expresia CD22 bola hodnotená prietokovou cytometriou z aspirátov kostnej drene. U pacientov
s nedostatočnou vzorkou z aspirátu kostnej drene sa hodnotila vzorka periférnej krvi. Alternatívne sa
u pacientov s nevhodným aspirátom kostnej drene a nedostatkom cirkulujúcich blastov expresia CD22
hodnotila imunohistochemickými metódami.
V klinickej štúdii bola citlivosť niektorých lokálne použitých testov nižšia ako citlivosť testu použitého v centrálnom laboratóriu. Preto sa majú používať iba validované testy s preukázanou vysokou citlivosťou.
Všetci pacienti museli mať ≥ 5 % blastov v kostnej dreni a predtým podstúpiť 1 alebo 2 indukčné chemoterapeutické režimy na liečbu ALL. Pacienti s Ph+ ALL z prekurzorov B-buniek museli predtým zlyhať na liečbe aspoň jedným druho- alebo treťogeneračným TKI a štandardnou chemoterapiou. Tabuľka 1 (pozri časť 4.2) zobrazuje režimy dávkovania použité na liečbu pacientov.
Primárne ukazovatele boli CR/CRi, hodnotené zaslepeným spôsobom nezávislou posudkovou komisiou (EAC), a celkové prežívanie (OS). Sekundárne ukazovatele zahŕňali negativitu MRD (pozri tabuľku 1 v časti 4.2), trvanie remisie (DoR), frekvenciu HSCT a prežívanie bez progresie ochorenia (PFS). Primárna analýza CR/CRi a negativity MRD sa vykonala u počiatočných
218 randomizovaných pacientov a analýza OS, PFS, DoR a frekvencie HSCT sa vykonala u všetkých
326 randomizovaných pacientov.
Spomedzi všetkých 326 randomizovaných pacientov (ITT populácia) absolvovalo 215 (66%) pacientov 1 predchádzajúci liečebný režim a 108 (33%) pacientov 2 predchádzajúce liečebné režimy v liečbe ALL. Medián veku bol 47 rokov (rozsah: 18 - 79 rokov), u 206 (63%) pacientov trvala prvá remisia < 12 mesiacov a 55 (17%) pacientov podstúpilo HSCT predtým, ako dostali BESPONSU alebo chemoterapiu podľa výberu skúšajúceho lekára. Celkovo malo 276 (85%) pacientov Ph- ALL. Zo 49 (15%) pacientov s Ph+ ALL nedostali 4 pacienti predchádzajúci TKI, 28 pacientov dostalo 1 predchádzajúci TKI a 17 pacientov dostalo 2 predchádzajúce TKI. Dasatinib bol najčastejšie podávaným TKI (42 pacientov), po ňom nasledoval imatinib (24 pacientov).
Východiskové charakteristiky u 218 iniciálne randomizovaných pacientov boli podobné.
Z 326 pacientov (ITT populácia) boli u 253 pacientov vzorky hodnotiteľné na CD22 lokálnym aj centrálnym laboratórnym testovaním. Na začiatku liečby malo 231/253 (91,3%) pacientov na základe
centrálneho testovania a 130/253 (51,4%) pacientov na základe lokálneho testovania ≥ 70% CD22- pozitívnych leukemických blastov.
Tabuľka 6 zobrazuje výsledky účinnosti z tejto štúdie.
Tabuľka 6. Štúdia 1: Výsledky účinnosti u pacientov vo veku ≥ 18 rokov s relapsujúcou alebo refraktérnou ALL z prekurzorov B-buniek, ktorí podstúpili 1
alebo 2 liečebné režimy v liečbe ALL
BESPONSA
(
N = 109)
CRa/ CRib; n (%) [95 % CI] 88 (80,7 %)
[72,1 % – 87,7 %]
HIDAC, FLAG alebo
MXN/Ara-C (N = 109)
32 (29,4 %)
[21,0 % – 38,8 %]
2-stranná p-hodnota < 0,0001
CRa; n (%) [95 % CI] 39 (35,8 %)
[26,8 % – 45,5 %]
19 (17,4 %)
[10,8 % – 25,9 %]
2-stranná p-hodnota = 0,0022
CRib; n (%) [95 % CI] 49 (45,0 %)
[35,4 % – 54,8 %]
13 (11,9 %)
[6,5 % – 19,5 %]
2-stranná p-hodnota < 0,0001
Negativita MRDc u pacientov, ktorí dosiahli CR/CRi; frekvenciad (%)
69/88 (78,4 %)
[68,4 % – 86,5 %]
9/32 (28,1 %)
[13,7 % – 46,7 %]
[95 % CI]
2-stranná p-hodnota < 0,0001
BESPONSA (N = 164)
Medián OS; mesiace [95 % CI] 7,7
[6,0 až 9,2]
HIDAC, FLAG alebo
MXN/Ara-C (N = 162)
6,7
[4,9 až 8,3]
Miera rizika [95 % CI] = 0,770 [0,599 – 0,990]
2-stranná p-hodnota = 0,0407
Medián PFSe,f; mesiace [95 % CI] 5,0
[3,7 až 5,6]
1,8
[1,5 až 2,2]
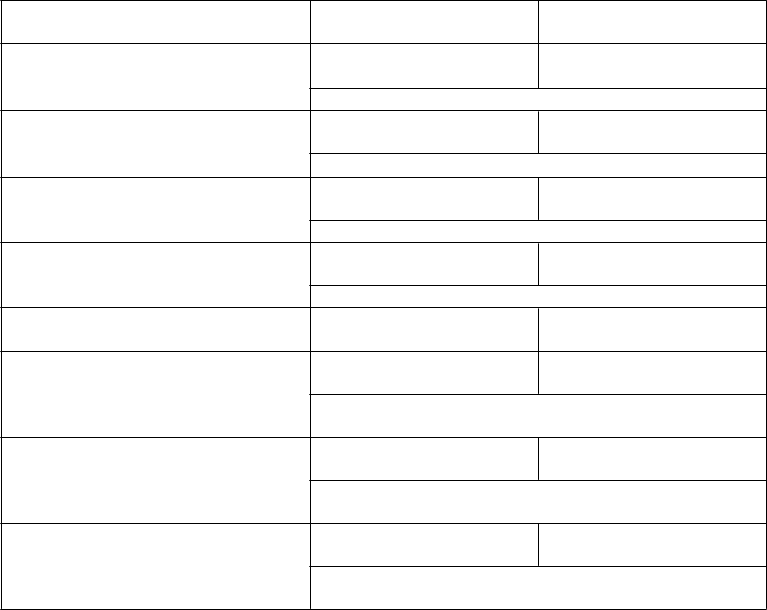
Miera rizika [95 % CI] = 0,452 [0,349 – 0,586]
2-stranná p-hodnota < 0,0001
Medián DoRg; mesiace [95 % CI] 3,7[2,8 až 4,3] 0,0
[–,–]
Miera rizika [95 % CI] = 0,468 [0,363 – 0,603]
2-stranná p-hodnota < 0,0001
Skratky: ALL = akútna lymfoblastová leukémia; ANC = absolútny počet neutrofilov; Ara-C = cytarabín; CI = interval spoľahlivosti; CR = úplná remisia; CRi = úplná remisia s neúplnou obnovou krvného obrazu; DoR = trvanie remisie; EAC = komisia pre posúdenie ukazovateľov; FLAG = fludarabín + cytarabín + faktor stimulujúci kolónie granulocytov; HIDAC = vysoká dávka cytarabínu; HSCT = transplantácia hematopoetických kmeňových buniek; ITT = snaha liečiť; MRD = minimálne reziduálne ochorenie;
MXN = mitoxantrón; N/n = počet pacientov; OS = celkové prežívanie; PFS = prežívanie bez progresie.
a CR, podľa EAC, bola definovaná ako < 5 % blastov v kostnej dreni a neprítomnosťou leukemických blastov v periférnej krvi, s úplným obnovením periférneho krvného obrazu (krvné doštičky ≥ 100 × 109/l
a ANC ≥ 1 × 109/l) a vyliečením akéhokoľvek extramedulárneho ochorenia.
b CRi, podľa EAC, bola definovaná ako < 5 % blastov v kostnej dreni a neprítomnosťou leukemických blastov v periférnej krvi, s čiastočným obnovením krvného obrazu (krvné doštičky < 100 × 109/l a/alebo ANC < 1 × 109/l) a vyliečením akéhokoľvek extramedulárneho ochorenia.
c Negativita MRD bola definovaná prietokovou cytometriou ako leukemické bunky pozostávajúce z < 1 × 10-
4 (< 0,01 %) jadrových buniek kostnej drene.
d Frekvencia bola definovaná ako počet pacientov, ktorí dosiahli negativitu MRD vydelenú celkovým počtom pacientov, ktorí dosiahli CR/CRi podľa EAC.
e PFS bolo definované ako čas od dátumu randomizácie do najskoršieho dátumu nasledujúcich udalostí:
úmrtie, progresia ochorenia (vrátanie objektívnej progresie, relapsu z CR/CRi, ukončenia liečby kvôli celkovému zhoršeniu zdravotného stavu) a začiatok novej indukčnej liečby alebo poliečebnej HSCT bez dosiahnutia CR/CRi.
f V štandardnej definícii PFS, definovanej ako čas od dátumu randomizácie do najskoršieho dátumu
nasledujúcich udalostí: úmrtie, progresia ochorenia (vrátane objektívnej progresie a relapsu z CR/CRi), bolo
HR 0,535 (2-stranná p-hodnota < 0,0001) a medián PFS bol 5,6 mesiacov pri BESPONSE a 3,6 mesiacov pri chemoterapií podľa výberu skúšajúceho lekára.
g Trvanie remisie bolo definované ako čas od prvej odpovede CRa alebo CRib podľa hodnotenia skúšajúceho lekára do dátumu udalosti PFS alebo dátumu cenzúry, ak nebola zdokumentovaná žiadna udalosť PFS. Analýza bola založená na populácii ITT, pričom bolo pacientom bez remisie pripísané trvanie nula a bolo
považované za udalosť.
Z 218 počiatočne randomizovaných pacientov 64/88 (73 %) a 21/88 (24 %) odpovedajúcich pacientov podľa EAC dosiahlo CR/CRi v 1., resp. 2. cykle v ramene s BESPONSOU. Žiadni ďalší pacienti nedosiahli CR/CRi po 3. cykle v ramene BESPONSY.
Výsledky CR/CRi a negativity MRD u 218 počiatočne randomizovaných pacientov boli v súlade s výsledkami pozorovanými u všetkých 326 randomizovaných pacientov.
U všetkých 326 randomizovaných pacientov bola pravdepodobnosť prežívania po 24 mesiacoch
22,6 % v ramene BESPONSY a 9,6 % v ramene chemoterapie podľa výberu skúšajúceho lekára.
Následnú HSCT malo celkovo 77/164 (47,0%) pacientov v ramene s BESPONSOU a 33/162 (20,4%) pacientov v ramene s chemoterapiou podľa výberu skúšajúceho lekára, vrátane 71 pacientov v ramene s BESPONSOU a 18 pacientov v ramene s chemoterapiou podľa výberu skúšajúceho lekára, ktorí priamo pokračovali HSCT. U pacientov, ktorí priamo pokračovali HSCT, bol medián obdobia medzi poslednou dávkou inotuzumab ozogamicínu a HSCT 4,9 týždňov (rozsah: 1 - 19 týždňov). Zlepšenie OS pri BESPONSE oproti chemoterapií podľa výberu skúšajúceho lekára bolo pozorované u pacientov, ktorí podstúpili HSCT. Hoci došlo k vyššej frekvencii skorých úmrtí po HSCT (až do 100. dňa ) v ramene s BESPONSOU, bol evidentný prínos BESPONSY pre neskoré prežívanie. U pacientov, ktorí podstúpili následnú HSCT, bol medián OS 11,9 mesiacov (95% CI: 8,6; 20,6) v porovnaní s 16,7 mesiacov (95% CI: 14,6; 27,8) a pravdepodobnosť prežitia v 24. mesiaci bola 38,9% (95% CI: 27,6; 50,0) oproti 35,7% (95% CI: 16,3; 55,8) pre BESPONSU oproti chemoterapie podľa výberu skúšajúceho lekára, v danom poradí.
Na základe výskumných analýz mali pacienti s priaznivejšími prognostickými faktormi (trvanie prvej remisie ≥ 12 mesiacov, 1 záchranná liečba, vek < 55 rokov, Ph-, bez predchádzajúcej HSCT, ³ 90 % CD22-pozitívnych leukemických blastov na počiatku, žiadne periférne blasty na počiatku a počiatočná hladina hemoglobínu ≥ 10 g/dl) lepšie výsledky OS. Pacienti s prestavbami génu leukémie zmiešaných línií (MLL) vrátane t (4;11), ktorí majú pred liečbou vo všeobecnosti nižšiu expresiu CD22, mali po liečbe BESPONSOU alebo chemoterapiou podľa výberu skúšajúceho lekára horšie výsledky OS.
U výsledkov hlásených pacientmi boli v prospech BESPONSY väčšina skóre funkčnosti a skóre príznakov, v porovnaní s chemoterapiou podľa výberu skúšajúceho lekára. U výsledkov hlásených pacientmi hodnotených pomocou základného dotazníka kvality života Európskej organizácie pre výskum a liečbu rakoviny (European Organisation for Research and Treatment of Cancer Quality of Life Core Questionnaire, EORTC QLQ-C30), mala BESPONSA významne lepšie odhadované priemerné skóre po počiatku (BESPONSA, resp. chemoterapia podľa výberu skúšajúceho lekára)'
v plnení role (64,7 oproti 53,4; p = 0,0065), fyzickej funkčnosti (75,0 oproti 68,1; p = 0,0139), sociálnom fungovaní (68,1 oproti 59,8; p = 0,0336) a strate chuti do jedla (17,6 oproti 26,3;
p = 0,0193) v porovnaní s chemoterapiou podľa výberu skúšajúceho lekára. Aj keď sa nedosiahla štatistická významnosť, BESPONSA viedla k zlepšeniu odhadovaných priemerných skóre po počiatku (BESPONSA, resp. chemoterapia podľa výberu skúšajúceho lekára) v celkovom zdravotnom
stave/kvalite života (QoL) (62,1 oproti 57,8, p = 0,1572), kognitívnej funkčnosti (85,3 oproti 82,5,
p = 0,1904), dýchavičnosti (14,7 oproti 19,4, p = 0,1281), hnačke (5,9 oproti 8,9, p = 0,1534), únave
(35,0 oproti 39,4, p = 0,1789), nevoľnosti a vracaní (8,7 oproti 10,4, p = 0,4578), finančných ťažkostiach (29,5 oproti 32,0, p = 0,4915), nespavosti (25,4 oproti 27,1, p = 0,6207) a bolesti (21,3 oproti 22,0, p = 0,8428). Aj keď sa nedosiahla štatistická významnosť, BESPONSA viedla k zhoršeniu odhadovaných priemerných skóre po počiatku (BESPONSA, resp. chemoterapia podľa výberu skúšajúceho lekára) v emočnej funkčnosti (77,4 oproti 79,6, p = 0,3307) a zápche (12,1 oproti 10,7,
p = 0,6249). U výsledkov hlásených pacientami meraných pomocou dotazníka EuroQoL 5 Dimension
(EQ-5D) viedla BESPONSA k lepším odhadovaným priemerným skóre po počiatku (BESPONSA,
resp. chemoterapia podľa výberu skúšajúceho lekára) v indexe EQ-5D (0,80 oproti 0,76, p = 0,1710) a vo vizuálnej analógovej škále EQ (EQ-VAS) (67,1 oproti 62,5, p = 0,1172), aj keď sa nedosiahla štatistická významnosť.
Pacienti s relapsujúcou alebo refraktérnou ALL, ktorí predtým mali 2 alebo viac liečebných režimov v liečbe ALL – Štúdia 2Bezpečnosť a účinnosť BESPONSY sa hodnotili v jednoramennej otvorenej multicentrickej štúdii vo fáze 1/2 (Štúdia 2). Vhodní pacienti mali ≥ 18 rokov a relapsujúcu alebo refraktérnu ALL
z prekurzorov B-buniek.
Z 93 vyšetrených pacientov bol 72 pacientom priradený skúšaný liek a boli liečení BESPONSOU. Medián veku bol 45 rokov (rozsah 20 - 79); 76,4% bola podávaná ≥ 2 záchranná liečba; 31,9% podstúpilo predchádzajúcu HSCT a 22,2% bolo Ph+. Najčastejšie dôvody pre prerušenie liečby boli: progresia/relaps ochorenia (30 [41,7%)], rezistencia ochorenia (4 [5,6%]); HSCT (18 [25,0%]) a nežiaduce udalosti (13 [18,1%]).
V časti fázy 1 štúdie dostalo 37 pacientov BESPONSU v celkovej dávke 1,2 mg/m2 (n = 3), 1,6 mg/m2
(n = 12) alebo 1,8 mg/m2 (n = 22). Odporúčaná dávka BESPONSY bola stanovená na
1,8 mg/m2/cyklus podaná v dávke 0,8 mg/m2 v 1. deň a 0,5 mg/m2 v 8. deň a 15. deň pri 28-dňovom cykle s redukciou dávky po dosiahnutí CR/CRi.
V časti fázy 2 štúdie mali mať pacienti predtým najmenej 2 liečebné režimy v liečbe ALL a u pacientov s Ph+ ALL z B-buniek musela zlyhať liečba aspoň 1 TKI. Z 9 pacientov s Ph+ ALL z B-buniek dostával predtým 1 pacient 1 TKI a 1 pacient nedostával predtým žiaden TKI.
Tabuľka 7 zobrazuje výsledky účinnosti z tejto štúdie.
Tabuľka 7. Štúdia 2: Výsledky účinnosti u pacientov vo veku ≥ 18 rokov s relapsujúcou alebo refraktérnou ALL z prekurzorov B-buniek, ktorí podstúpili 2alebo viaceré liečebné režimy v liečbe ALLBESPONSA(N = 35)CRa/CRib; n (%) [95 % CI] 24 (68,6 %)
[50,7 % – 83,2 %] CRa; n (%) [95 % CI] 10 (28,6 %)

[14,6 % – 46,3 %] CRib; n (%) [95 % CI] 14 (40,0 %)
[23,9 % – 57,9 %] Medián DoRf; mesiace [95 % CI] 2,2
[1,0 až 3,8]
Negativita MRDc u pacientov dosahujúcich CR/CRi;
frekvenciad (%) [95 % CI]
18/24 (75 %)
[53,3 % – 90,2 %]
Medián PFSe; mesiace [95 % CI] 3,7
[2,6 až 4,7]
Medián OS; mesiace [95 % CI] 6,4
[4,5 až 7,9]
Skratky: ALL = akútna lymfoblastová leukémia; ANC = absolútny počet neutrofilov; CI = interval spoľahlivosti; CR = úplná remisia; CRi = úplná remisia s neúplnou obnovou krvného obrazu; DoR = trvanie remisie; HSCT = transplantácia hematopoetických kmeňových buniek; MRD = minimálne reziduálne ochorenie; N/n = počet pacientov; OS = celkové prežívanie; PFS = prežívanie bez progresie.
a , b, c, d, e, f Definícia, pozri Tabuľku 6 (s výnimkou toho, že CR/CRi nebola hodnotená EAC v Štúdii 2)
V časti fázy 2 štúdie podstúpilo 8/35 (22, 9%) pacientov následnú HSCT.
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s BESPONSOU v 1
alebo vo viacerých podskupinách pediatrickej populácie pri liečbe relapsujúcej alebo refraktérnej ALL (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
U pacientov s relapsujúcou alebo refraktérnou ALL liečených inotuzumab ozogamicínom
v odporúčanej počiatočnej dávke 1,8 mg/m2/cyklus (pozri časť 4.2), bola ustálená expozícia dosiahnutá do 4. cyklu. Priemerná (SD) maximálna koncentrácia (Cmax) inotuzumab ozogamicínu
v sére bola 308 ng/ml (362). Priemerná (SD) simulovaná celková plocha pod krivkou koncentrácia-čas
(AUC) na cyklus bola v ustálenom stave 100 μgh/ml (32,9).
Distribúcia
In vitro sa približne 97% N-acetyl-gama-kalicheamicíndimetylhydrazidu naviaže na proteíny ľudskej
plazmy . In vitro je N-acetyl-gama-kalicheamicíndimetylhydrazid substrátom P-glykoproteínu (P-gp). U ľudí bol celkový distribučný objem inotuzumab ozogamicínu približne 12 l.
Biotransformácia
In vitro bol N-acetyl-gama-kalicheamicíndimetylhydrazid primárne metabolizovaný neenzymatickou
redukciou. U ľudí boli hladiny N-acetyl-gama-kalicheamicíndimetylhydrazidu v sére typicky pod limitom kvantifikácie (50 pg/ml).
Eliminácia
Farmakokinetika inotuzumab ozogamicínu bola dobre charakterizovaná pomocou 2-
kompartmentového modelu s lineárnymi a časovo závislými komponentmi klírensu. U 234 pacientov s relapsujúcou alebo refraktérnou ALL bol klírens inotuzumab ozogamicínu v ustálenom stave
0,0333 l/h a polčas terminálnej eliminácie (t½) na konci 4. cyklu bol približne 12,3 dňa. Po podaní viacerých dávok sa medzi 1. a 4. cyklom pozorovala 5,3-násobná akumulácia inotuzumab ozogamicínu.
Na základe populačnej farmakokinetickej analýzy u 765 pacientov sa zistilo, že plocha telesného povrchu významne ovplyvňuje dispozíciu inotuzumab ozogamicínu. Dávka inotuzumab ozogamicínu sa podáva na základe plochy telesného povrchu (pozri časť 4.2).
Vek, rasaapohlavie
Na základe populačnej farmakokinetickej analýzy vek, rasa ani pohlavie významne neovplyvňujú
dispozíciu inotuzumab ozogamicínu.
Porucha funkciepečene
U pacientov s poruchou funkcie pečene sa neuskutočnili žiadne formálne farmakokinetické štúdie
s inotuzumab ozogamicínom.
Na základe populačnej farmakokinetickej analýzy u 765 pacientov bol klírens inotuzumab ozogamicínu u pacientov s poruchou funkcie pečene definovanej pracovnou skupinou Národného onkologického ústavu zaoberajúcou sa orgánovými dysfunkciami (National Cancer Institute Organ Dysfunction Working Group, NCI ODWG) ako kategória B1 (celkový bilirubín ≤ ULN
a AST > ULN; n = 133) alebo B2 (celkový bilirubín > 1,0 – 1,5 × ULN a akákoľvek hladina AST;
n = 17) podobný ako u pacientov s normálnou funkciou pečene (celkový bilirubín/AST ≤ ULN;
n = 611) (pozri časť 4.2). U 3 pacientov s poruchou funkcie pečene kategórie C podľa NCI ODWG (celkový bilirubín > 1,5 – 3 × ULN a akákoľvek hladina AST) a 1 pacienta s poruchou funkcie pečene
kategórie D podľa NCI ODWG (celkový bilirubín > 3 × ULN a akákoľvek hladina AST) sa nejavilo, že by bol klírens inotuzumab ozogamicínu znížený.
Porucha funkcieobličiek
U pacientov s poruchou funkcie obličiek sa neuskutočnili žiadne formálne farmakokinetické štúdie
s inotuzumab ozogamicínom.
Na základe populačnej farmakokinetickej analýzy u 765 pacientov bol klírens inotuzumab ozogamicínu u pacientov s miernou poruchou funkcie obličiek (CLcr 60 – 89 ml/min; n = 237), so strednou poruchou funkcie obličiek (CLcr 30 – 59 ml/min; n = 122) alebo závažnou poruchou funkcie obličiek (CLcr 15 – 29 ml/min; n = 4) podobný ako u pacientov s normálnou funkciou obličiek (CLcr
≥ 90 ml/min; n = 402) (pozri časť 4.2). Inotuzumab ozogamicín nebol študovaný u pacientov
v konečnom štádiu ochorenia obličiek (pozri časť 4.2).
Elektrofyziológia srdca
Na základe farmakokinetickej analýzy odpovede na expozíciu u 250 pacientov s relapsujúcou alebo
refraktérnou ALL či inou hematologickou malignitou dostávajúcich 1,8 mg/m2/cyklus inotuzumab ozogamicínu podávaného ako 3 rozdelené dávky v 1. deň (0,8 mg/m2), 8. deň (0,5 mg/m2) a 15. deň (0,5 mg/m2) 21 až 28-dňového cyklu, resp. 1,8 mg/m2/cyklus podávaného každé 4 týždne sa medián
QTcF zvýšil o o 2,53 milisekúnd (ms) oproti počiatočnej hodnote (97,5. percentil: 4,92 ms) pri priemernej Cmax odhadnutej pre pacientov s relapsujúcou alebo refraktérnou ALL (371 ng/ml)
a o 3,87 ms od počiatočnej hodnoty (97,5. percentil: 7,54 ms) pri 1,5-krát vyššej priemernej Cmax
(569 ng/ml).
V randomizovanej klinickej štúdii u pacientov s relapsujúcou alebo refraktérnou ALL (Štúdia 1) boli zvýšenia QTcF o ≥ 60 ms od počiatočnej hodnoty namerané u 4/162 (3 %) pacientov v ramene inotuzumab ozogamicínu a 3/124 (2 %) v ramene chemoterapie podľa výberu skúšajúceho lekára. Zvýšenia QTcF na > 500 ms neboli pozorované u žiadneho pacienta v ramene inotuzumab ozogamicínu a u 1/124 (1 %) pacientov v ramene chemoterapie podľa výberu skúšajúceho lekára (pozri časť 4.8). Priemerné (90 % CI) maximálne zmeny QTcF od počiatočnej hodnoty boli
16,5 ms (14,3 – 18,7) v ramene inotuzumab ozogamicínu a 10,8 ms (8,0 – 13,6) v ramene chemoterapie podľa výberu skúšajúceho lekára. Centrálna analýza trendu zmien intervalu QTcF od počiatočnej hodnoty ukázala, že najvyššia horná hranica 2-stranného 90 % CI u QTcF bola 21,1 ms (pozorovaná v 4. cykle/1. deň /1 hodina) v ramene inotuzumab ozogamicínu a 21,2 ms (pozorovaná v 2. cykle/1. deň /1 hodina) v ramene chemoterapie podľa výberu skúšajúceho lekára.
5.3 Predklinické údaje o bezpečnosti
Toxicita poopakovanompodaní
U zvierat zahŕňali primárne cieľové orgány pečeň, kostnú dreň a lymfatické orgány s pridruženými
hematologickými zmenami, obličky a nervový systém. Ostatné pozorované zmeny zahŕňali účinky na samčie a samičie reprodukčné orgány (pozri nižšie) a preneoplastické a neoplastické lézie v pečeni (pozri nižšie). Väčšina účinkov bola reverzibilná až čiastočne reverzibilná okrem účinkov na pečeň
a nervový systém. Relevantnosť ireverzibilných nálezov u zvierat pre ľudí je nejasná.
Genotoxicita
Inotuzumab ozogamicín bol in vivo v kostnej dreni u samcov myší klastogénny. Je to v súlade so
známou indukciou DNA zlomov kalicheamicínom a inými enediynovými protinádorovými antibiotikami. Inotuzumab ozogamicín nebol mutagénny v teste bakteriálnych reverzných mutácií (Ames) in vitro pri testovaní až do maximálnej prípustnej dávky.
Karcinogenicita
Formálne štúdie karcinogenicity s inotuzumab ozogamicínom sa neuskutočnili. V štúdiách toxicity sa
u potkanov vyvinula hyperplázia oválnych buniek, zmena hepatocelulárnych ložísk a hepatocelulárne adenómy v pečeni pri približne 0,3-násobku klinickej expozície u ľudí na základe AUC. U 1 opice sa zistilo ložisko hepatocelulárnych zmien pri približne 3,1-násobku klinickej expozície u ľudí na základe AUC na konci 26-týždňového obdobia dávkovania. Relevancia týchto nálezov u zvierat pre ľudí je nejasná.
Reprodukčná toxicita
Podávanie inotuzumab ozogamicínu samiciam potkanov v dávke toxickej pre matku (približne 2,3-
násobok klinickej expozície u ľudí na základe AUC) pred párením a počas prvého týždňa brezivosti viedlo k toxicite pre embryo a plod vrátane zvýšených resorpcií a zníženia počtu životaschopných
embryí. Dávka toxická pre matku (približne 2,3-násobok klinickej expozície u ľudí na základe AUC)
tiež viedla k retardácii rastu plodu vrátane zníženej hmotnosti plodu a oneskorenej osifikácie kostí. Mierna retardácia rastu plodu u potkanov sa vyskytla aj pri dávkach približne 0,4-násobných ako je klinická expozícia u ľudí na základe AUC (pozri časť 4.6).
Na základe neklinických nálezov sa u inotuzumab ozogamicínu predpokladá, že môže potenciálne poškodiť reprodukčné funkcie a plodnosť u mužov a žien (pozri časť 4.6). V štúdiách toxicity opakovanej dávky u potkanov a opíc reprodukčné nálezy u samíc zahŕňali atrofiu vaječníkov, maternice, vagíny a mliečnej žľazy. Hladina bez pozorovaných nežiaducich účinkov (NOAEL)
u účinkov na samičie reprodukčné orgány u potkanov a opíc bola približne 2,2-, resp. 3,1-násobok klinickej expozície u ľudí na základe AUC. V štúdiách toxicity opakovanej dávky u potkanov reprodukčné nálezy u samcov zahŕňali degeneráciu semenníkov spojenú s hypospermiou a atrofiou prostaty a semenných vačkov. Pre účinky na samčie reprodukčné orgány nebola stanovená NOAEL. Tieto boli pozorované pri približne 0,3-násobku klinického vystavenia u ľudí na základe AUC.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Sacharóza
Polysorbát 80
Chlorid sodný
Trometamín
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka
2 roky
Rekonštituovaný roztok
BESPONSA neobsahuje žiadne bakteriostatické konzervačné látky. Rekonštituovaný roztok sa musí
použiť okamžite. Ak sa rekonštituovaný roztok nemôže použiť okamžite, môže sa až 4 hodiny uchovávať v chladničke (2 °C – 8 °C). Chráňte pred svetlom a neuchovávajte v mrazničke.
Zriedený roztok
Zriedený roztok sa musí použiť okamžite alebo sa môže uchovávať pri izbovej teplote (20 °C – 25 °C)
či v chladničke (2 °C – 8 °C). Maximálny čas od rekonštitúcie do podania musí byť ≤ 8 hodín, pričom čas medzi rekonštitúciou a riedením musí byť ≤ 4 hodiny. Chráňte pred svetlom a neuchovávajte
v mrazničke.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii a riedení, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Injekčná liekovka z jantárového skla typu I s chlórbutylovou gumovou zátkou, zapečatením po okrajoch a ľahko otvárateľným viečkom obsahujúca 1 mg prášku.
Každé balenie obsahuje 1 injekčnú liekovku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Návod narekonštitúciu,riedenieapodávanie
Pri procesoch rekonštitúcie a riedenia použite vhodnú aseptickú techniku. Inotuzumab ozogamicín je
citlivý na svetlo a musí sa počas rekonštitúcie, riedenia a podávania chrániť pred ultrafialovým svetlom.
Maximálny čas od rekonštitúcie do skončenia podávania musí byť ≤ 8 hodín, pričom čas medzi rekonštitúciou a riedením musí byť ≤ 4 hodiny.
Rekonštitúcia
· Vypočítajte dávku (mg) a počet potrebných injekčných liekoviek BESPONSY.
· Každú injekčnú liekovku s obsahom 1 mg rekonštituujte so 4 ml vody na injekciu, aby ste získali roztok BESPONSY s koncentráciou 0,25 mg/ml na jednorazové použitie.
· Injekčnú liekovku jemne otáčajte, aby sa uľahčilo rozpúšťanie. Nepretrepávajte.
· Skontrolujte, či rekonštituovaný roztok neobsahuje častice a či nezmenil farbu.
Rekonštituovaný roztok musí byť priehľadný až mierne zakalený, bezfarebný a hlavne nesmie obsahovať žiadne viditeľné cudzorodé častice. Ak pozorujete častice alebo zmenu farby, nepoužívajte.
· BESPONSA neobsahuje žiadne bakteriostatické konzervačné látky. Rekonštituovaný roztok sa musí použiť okamžite. Ak sa rekonštituovaný roztok nemôže použiť okamžite, môže sa až
4 hodiny uchovávať v chladničke (2 °C – 8 °C). Chráňte pred svetlom a neuchovávajte v mrazničke.
Riedenie
· Vypočítajte požadovaný objem rekonštituovaného roztoku potrebný na dosiahnutie príslušnej dávky na základe plochy telesného povrchu pacienta. Toto množstvo natiahnite z injekčnej liekovky pomocou injekčnej striekačky. Chráňte pred svetlom. Akýkoľvek nepoužitý rekonštituovaný roztok v injekčnej liekovke zlikvidujte.
· Rekonštituovaný roztok pridajte do infúzneho vaku spolu s injekčným roztokom chloridu sodného s koncentráciou 9 mg/ml (0,9 %) do celkového nominálneho objemu 50 ml. Chráňte
pred svetlom. Odporúča sa infúzny vak vyrobený z polyvinylchloridu (PVC) (di(2-
etylhexyl)ftalátu [DEHP] alebo bez obsahu-DEHP-), polyolefínu (polypropylén a/alebo polyetylén) alebo etylénvinylacetátu (EVA).
· Jemne prevráťte infúzny vak, aby ste premiešali zriedený roztok. Nepretrepávajte.
· Zriedený roztok sa musí použiť okamžite alebo sa môže uchovávať pri izbovej teplote
(20 °C – 25 °C) či v chladničke (2 °C – 8 °C). Maximálny čas od rekonštitúcie do skončenia podávania musí byť ≤ 8 hodín, pričom čas medzi rekonštitúciou a riedením musí byť ≤ 4 hodiny. Chráňte pred svetlom a neuchovávajte v mrazničke.
Podávanie
· Ak je zriedený roztok uchovávaný v chladničke (2 °C – 8 °C), musíte ho nechať približne
1 hodinu pred podávaním temperovať na izbovú teplotu (20 °C – 25 °C).
· Filtrácia zriedeného roztoku sa nevyžaduje. Ak však zriedený roztok filtrujete, odporúčajú sa filtre na báze polyétersulfónu (PES), polyvinylidénfluoridu (PVDF) alebo hydrofilného polysulfónu (HPS). Nepoužívajte filtre vyrobené z nylonu alebo zmesných esterov celulózy (MCE).
· Zriedený roztok podávajte infúziou počas 1 hodiny rýchlosťou 50 ml/h pri izbovej teplote
(20 °C – 25 °C). Chráňte pred svetlom. Odporúčajú sa infúzne sety vyrobené z PVC (s DEHP
alebo bez obsahu DEHP), polyolefínu (polypropylén a/alebo polyetylén) či polybutadiénu.
BESPONSA sa nesmie miešať ani podávať ako infúzia s inými liekmi.
Tabuľka 8 zobrazuje časy a podmienky uchovávania pre rekonštitúciu, riedenie a podávanie
BESPONSY.
Tabuľka 8. Časy a podmienky uchovávania pre rekonštituovaný a zriedený roztok
BESPONSY
Rekonštituovaný roztok
Maximálny čas od rekonštitúcie do podania ≤ 8 hodín
a
Zriedený roztok
Po začiatku riedenia Podávanie
Rekonštituovaný roztok použite okamžite alebo po uchovávaní
v chladničke (2 °C –
8 °C) do 4 hodín. Chráňte pred
svetlom.
Neuchovávajte v mrazničke.
Zriedený roztok použite okamžite alebo po uchovávaní pri izbovej teplote (20 °C – 25 °C) či
v chladničke (2 °C – 8 °C). Maximálny čas od rekonštitúcie do skončenia podávania musí
byť ≤ 8 hodín, pričom čas medzi rekonštitúciou a riedením musí
byť ≤ 4 hodiny. Chráňte pred svetlom. Neuchovávajte v mrazničke.
Ak sa zriedený roztok uchováva v chladničke (2 °C – 8 °C), pred podávaním ho nechajte približne
1 hodinu nadobudnúť izbovú teplotu (20 °C – 25 °C). Zriedený roztok podávajte infúziou počas
1 hodiny rýchlosťou 50 ml/h pri izbovej teplote (20 °C – 25 °C).
Chráňte pred svetlom.
a ≤ 4 hodiny medzi rekonštitúciou a riedením.
LikvidáciaBESPONSA je určená len na jednorazové použite.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Pfizer Limited
Ramsgate Road
Sandwich, Kent CT13 9NJ Spojené kráľovstvo
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/17/1200/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.