
ený k použitiu a je určený len na jednorazové použitie.
Vizuálne skontrolujte roztok na prítomnosť častíc a zmenu farby. Nepoužívajte pri zmene farby alebo prítomnosti častíc.
Ak sa naplnená injekčná striekačka skladuje v chlade, pred podaním sa má nechať zohriať tak, že sa balenie nechá približne 30 minút pri izbovej teplote.
• Subkutánna injekcia sa má podať do jedného z nasledujúcich miest: brucho, stehná alebo horná časť ramena. Amvuttra sa nesmie podávať do zjazveného tkaniva ani do oblastí, ktoré sú začervenané, zapálené alebo opuchnuté.
• Ak sa injekcia podáva do brucha, treba sa vyhnúť oblasti okolo pupka.
4.3 Kontraindikácie
Závažná precitlivenosť (napr. anafylaxia) na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Nedostatok vitamínu A
Zníženie sérového proteínu transtyretínu (TTR) pri liečbe Amvuttrou spôsobuje zníženie sérovej
hladiny vitamínu A (retinolu) (pozri časť 5.1). Pred začatím liečby Amvuttrou musia byť upravené sérové hladiny vitamínu A, ak sú pod dolnou hranicou normy, a musia byť vyhodnotené akékoľvek očné symptómy alebo prejavy spôsobené nedostatkom vitamínu A.
Pacienti užívajúci Amvuttru musia užívať perorálny doplnok obsahujúci vitamín A v dennej dávke približne 2 500 IU až 3 000 IU, ktorá sa však nemá prekročiť, na zníženie potenciálneho rizika očných príznakov v dôsledku nedostatku vitamínu A. Ak sa u pacientov objavia očné príznaky naznačujúce nedostatok vitamínu A, ako je zhoršené nočné videnie alebo nočná slepota, dlhodobo suché oči, zápal očí, zápal alebo ulcerácie rohovky, zhrubnutie rohovky alebo perforácia rohovky, odporúča sa poslať ich na oftalmologické vyšetrenie.
Počas prvých 60 dní tehotenstva môžu byť príliš vysoké aj príliš nízke hladiny vitamínu A asociované so zvýšeným rizikom malformácie plodu. Z tohto dôvodu je potrebné pred začatím liečby Amvuttrou
vylúčiť tehotenstvo a ženy vo fertilnom veku musia používať účinnú antikoncepciu (pozri časť 4.6). Ak žena plánuje otehotnieť, predtým, ako sa o to pokúsi, musí byť ukončené podávanie Amvuttry
a doplnku obsahujúceho vitamín A, sérové hladiny vitamínu A sa musia monitorovať a musia sa vrátiť
k normálu. Sérové hladiny vitamínu A môžu zostať znížené viac ako 12 mesiacov po poslednej dávke
Amvuttry.
V prípade neplánovaného tehotenstva musí byť liečba Amvuttrou ukončená (pozri časť 4.6). Počas prvého trimestra neplánovaného tehotenstva nie je možné odporučiť, či sa má pokračovať v podávaní doplnku obsahujúceho vitamín A, alebo sa má jeho podávanie ukončiť. Pokiaľ sa v podávaní doplnku obsahujúceho vitamín A pokračuje, denná dávka nesmie prekročiť 3 000 IU denne, pretože nie sú dostatočné údaje podporujúce užívanie vyšších dávok. Pokiaľ sa sérové hladiny vitamínu A ešte nevrátia k normálnym hodnotám, je následne potrebné kvôli zvýšenému riziku nedostatku vitamínu A v treťom trimestri obnoviť užívanie doplnku obsahujúceho vitamín A v dennej dávke 2 500 IU až
3 000 IU v druhom a treťom trimestri.
Nie je známe, či podávanie doplnku obsahujúceho vitamín A v tehotenstve bude stačiť na prevenciu nedostatku vitamínu A, ak tehotná žena naďalej dostáva Amvuttru. Zvýšenie podávania doplnku obsahujúceho vitamín A na viac ako 3 000 IU denne počas tehotenstva však pravdepodobne neprinesie úpravu plazmatickej hladiny retinolu vzhľadom na mechanizmus účinku Amvuttry a môže byť
škodlivé pre matku a plod.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v ml, t. j. v podstate zanedbateľné množstvo
sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne klinické interakčné štúdie. Nepredpokladá sa, že vutrisiran bude spôsobovať interakcie alebo bude ovplyvňovaný inhibítormi alebo induktormi enzýmov cytochrómu P450 alebo že bude modulovať aktivitu transportérov. Preto, sa nepredpokladajú klinicky významné interakcie vutrisiranu s inými liekmi.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku
Liečba Amvuttrou znižuje sérové hladiny vitamínu A. Príliš vysoké aj príliš nízke hladiny
vitamínu A môžu byť asociované so zvýšeným rizikom malformácie plodu. Preto musí byť pred začatím liečby vylúčené tehotenstvo a ženy vo fertilnom veku musia používať účinnú antikoncepciu.
Ak žena plánuje otehotnieť, predtým ako sa o to pokúsi, liečba Amvuttrou a doplnkovým
vitamínom A sa má ukončiť a je potrebné sledovať sérové hladiny vitamínu A, ktoré sa musia vrátiť
k normálu (pozri časť 4.4). Hladiny vitamínu A v sére môžu zostať znížené viac ako 12 mesiacov po poslednej dávke Amvuttry.
Gravidita
Nie sú k dispozícii údaje o použití Amvuttry u gravidných žien. Štúdie na zvieratách sú z hľadiska
reprodukčnej toxicity nedostatočné (pozri časť 5.3). Z dôvodu potenciálneho teratogénneho rizika vyplývajúceho z nevyvážených hladín vitamínu A sa Amvuttra nemá používať počas tehotenstva. Ako
preventívne opatrenie je potrebné vo včasnom štádiu tehotenstva stanoviť hladinu vitamínu A (pozri časť 4.4) a hormónu stimulujúceho štítnu žľazu (thyroid stimulating hormone, TSH). Najmä počas
prvého trimestra je potrebné podrobne sledovať plod.
D
ojčenie
Nie je známe, či sa vutrisiran vylučuje do ľudského mlieka. Nie sú dostatočné informácie o vylučovaní
vutrisiranu do mlieka u zvierat (pozri časť 5.3).
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Amvuttrou, sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby týmto liekom pre ženu.
FertilitaNie sú k dispozícii žiadne údaje o účinkoch Amvuttry na fertilitu u ľudí. Štúdie na zvieratách
nepreukázali žiaden vplyv na fertilitu u samcov alebo samíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeAmvuttra nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofiluPočas 18-mesačného obdobia liečby v klinickej štúdii HELIOS-A boli najčastejšie hlásenými
nežiaducimi reakciami u pacientov liečených Amvuttrou bolesť v končatinách (15 %) a artralgia
(11 %).
Tabuľkový zoznamnežiaducichreakciíNežiaduce reakcie sú zoradené podľa konvencií MedDRA do tried orgánových systémov (System
Organ Class, SOC). Frekvencia nežiaducich reakcií je uvedená podľa nasledujúcich kategórií:
• Veľmi časté (≥ 1/10)
• Časté (≥ 1/100 až < 1/10)
• Menej časté (≥ 1/1000 až < 1/100)
Tabuľka 1: Nežiaduce reakcie hlásené pri AmvuttreTrieda orgánových systémov
| Nežiaduca reakcia
| Frekvencia
|
Poruchy dýchacej sústavy, hrudníka
a mediastína
| Dyspnoea
| Časté
|
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
| Artralgia
| Veľmi časté
|
Bolesť v končatinách
| Veľmi časté
|
Celkové poruchy a reakcie v mieste
podania
| Reakcie v mieste podania injekcieb
| Časté
|
Laboratórne a funkčné vyšetrenia
| Zvýšená hladina alkalickej fosfatázy
v krvi
| Časté
|
a Patria sem dyspnoe, námahové dyspnoe a paroxyzmálne nočné dyspnoe.
b Medzi hlásené príznaky patrili modriny, erytém, bolesť, pruritus a teplo. Reakcie v mieste podania injekcie boli mierne, prechodné a neviedli k ukončeniu liečby.
|
PopisvybranýchnežiaducichreakciíImunogenitaPočas 18-mesačného obdobia liečby v klinickej štúdii HELIOS-A sa u 4 (3,3 %) pacientov liečených
Amvuttrou objavili protilátky proti lieku (anti-drug antibodies, ADA). Titre ADA boli nízke a prechodné a nepreukázal sa žiaden vplyv na klinickú účinnosť, bezpečnosť alebo profil farmakokinetiky či farmakodynamiky vutrisiranu.
H
l
ásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania sa odporúča monitorovať pacienta podľa lekárskych pokynov vzhľadom na prejavy a príznaky nežiaducich reakcií a podávať príslušnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Ostatné lieky na nervový systém, ATC kód: N07XX18
Mechanizmus účinkuAmvuttra obsahuje vutrisiran, chemicky stabilizovanú dvojvláknovú malú interferujúcu ribonukleovú
kyselinu (siRNA), ktorá sa špecificky zameriava na variantný a divý typ transtyretínovej (
TTR)
messengerovej RNA (mRNA) a je kovalentne spojená s ligandom obsahujúcim tri
N-acetylgalaktozamínové (GalNAc) skupiny, ktoré umožňujú doručenie siRNA do hepatocytov.
Prostredníctvom prirodzeného procesu nazývaného interferencia RNA (RNA interference, RNAi) vutrisiran spôsobuje katalytickú degradáciu
TTR mRNA v pečeni, čo vedie k zníženiu sérových hladín variantného a divého typu proteínu TTR.
Farmakodynamické účinkyPriemerná hodnota sérovej hladiny TTR sa znížila už na 22. deň, pričom do 6. týždňa bola priemerná
hodnota TTR takmer v ustálenom stave znížená o 73 %. Pri opakovanom dávkovaní 25 mg jedenkrát za 3 mesiace bolo priemerné zníženie sérovej hladiny TTR po 9 mesiacoch liečby 83 % a po 18 mesiacoch 88 %. Podobné zníženie hladiny TTR sa pozorovalo bez ohľadu na genotyp (V30M alebo iný ako V30M), predchádzajúce užívanie stabilizátora TTR, telesnú hmotnosť, pohlavie, vek alebo rasu.
Sérový TTR je nosičom retinol viažuceho proteínu 4, ktorý je hlavným nosičom vitamínu A v krvi. Amvuttra znížila hladiny vitamínu A s priemerným znížením maxima o 70 % a minima o 63 %
v ustálenom stave (pozri časti 4.4 a 4.5).
Klinická účinnosť a bezpečnosťÚčinnosť Amvuttry sa skúmala v globálnej, randomizovanej, otvorenej klinickej štúdii (HELIOS-A)
u dospelých pacientov s hATTR amyloidózou s polyneuropatiou. Pacienti boli randomizovaní
v pomere 3 : 1 na podávanie 25 mg Amvuttry (N = 122) subkutánne jedenkrát za 3 mesiace alebo
0,3 mg/kg patisiranu (N = 42) intravenózne jedenkrát za 3 týždne. Obdobie liečby v rámci štúdie
trvalo 18 mesiacov s dvoma analýzami v 9. a 18. mesiaci. Deväťdesiatsedem percent (97 %) pacientov liečených Amvuttrou dokončilo najmenej 18 mesiacov pridelených liečob (vutrisiran alebo patisiran).
Hodnotenie účinnosti vychádzalo z porovnania ramena s vutrisiranom s externou skupinou s placebom
(rameno s placebom štúdie fázy 3 APOLLO), ktorú tvorila podobná populácia pacientov s hATTR
amyloidózou s polyneuropatiou. Hodnotenie neinferiority zníženia sérovej hladiny TTR bolo založené na porovnaní ramena s vutrisiranom s ramenom s patisiranom v rámci štúdie.
U pacientov, ktorí dostávali Amvuttru, bol medián veku pacientov na začiatku liečby 60 rokov
(rozpätie 34 až 80 rokov), 38 % pacientov malo ≥ 65 rokov a 65 % pacientov boli muži. Zastúpených
bolo dvadsaťdva (22) rôznych variantov TTR: V30M (44 %), T60A (13 %), E89Q (8 %), A97S (6 %), S50R (4 %), V122I (3 %), L58H (3 %) a iné (18 %). Dvadsať percent (20 %) pacientov malo genotyp V30M a skorý nástup príznakov (< 50 rokov). Vo východiskovom stave malo 69 % pacientov
1. štádium ochorenia (nenarušená pohyblivosť; mierna senzorická, motorická a autonómna neuropatia v dolných končatinách) a 31 % pacientov malo 2. štádium ochorenia (potrebná pomoc pri chôdzi; stredná progresia poruchy dolných končatín, horných končatín a trupu). Štúdie sa nezúčastnili žiadni pacienti s ochorením v 3. štádiu. Šesťdesiatjeden percent (61 %) pacientov bolo predtým liečených stabilizátormi tetraméru TTR. Podľa klasifikácie srdcového zlyhávania Newyorskej srdcovej asociácie (New York Heart Association, NYHA) bolo 9 % pacientov v triede I a 35 % v triede II. Tridsaťtri percent (33 %) pacientov spĺňalo vopred definované kritériá pre postihnutie srdca (východisková hrúbka steny ĽK ≥ 13 mm bez anamnézy hypertenzie alebo ochorenia aortálnej chlopne).
Primárnym ukazovateľom účinnosti bola zmena v modifikovanom skóre neuropatického postihnutia
+7 (modified Neuropathy Impairment Score, mNIS+7) oproti východiskovému stavu za 18 mesiacov . Tento ukazovateľ je kompozitným meradlom motorickej, senzorickej a autonómnej neuropatie vrátane hodnotenia motorickej sily, reflexov, kvantitatívneho senzorického testovania, vyšetrenia nervového vedenia a posturálneho krvného tlaku, pričom skóre sa môže pohybovať v rozmedzí od 0 do 304 bodov, kde vyššie skóre indikuje zhoršujúce sa postihnutie.
Ako sekundárny ukazovateľ sa hodnotila zmena v celkovom skóre dosiahnutom v škále Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) oproti východiskovému stavu za 18 mesiacov. Dotazník Norfolk QoL-DN (podľa hlásenia pacientom) zahŕňal funkcie súvisiace s tenkými, veľkými a autonómnymi nervovými vláknami, príznakmi polyneuropatie a činnosťami každodenného života,
s celkovým skóre v rozsahu od –4 do 136, pričom vyššie skóre indikuje zhoršujúcu sa kvalitu života.
Ďalšie sekundárne ukazovatele zahŕňali rýchlosť chôdze (test chôdze na 10 metrov), nutričný stav (mBMI) a pacientom udávanú schopnosť vykonávať činnosti každodenného života a zapojenie sa do spoločenského života (Rasch-Built Overall Disability Scale [R-ODS]).
Liečba Amvuttrou v štúdii HELIOS-A preukázala štatisticky významné zlepšenie všetkých ukazovateľov (tabuľka 2 a obrázok 1) meraných od východiskového stavu do 9. a 18. mesiaca v porovnaní s externou skupinou s placebom štúdie APOLLO (všetky p < 0,0001).
Časovo spriemerované percentuálne zníženie minimálnej hladiny TTR do 18. mesiaca bolo 84,7 % pre vutrisiran a 80,6 % pre patisiran. Percentuálne zníženie sérových hladín TTR v ramene s vutrisiranom bolo neinferiórne (podľa vpred stanovených kritérií) v porovnaní s ramenom s patisiranom v rámci štúdie do 18. mesiaca s mediánom rozdielu 5,3 % (95 % CI 1,2 %, 9,3 %).
T
abuľka č. 2: Súhrn výsledkov klinickej účinnosti v štúdii HELIOS-A
Ko
ncový ukazovateľ
a
|
P
riemerná východisková hodnota (SD)
|
Z
m
ena oproti východiskovému stavu, priemerná hodnota LS (SEM)
|
Rozdiel
v liečbe medzi
Amvuttrou
a placebom
b
, priemerná
hodnota LS
(9
5 % CI)
|
p-hodnota
|
Amvuttra
N = 122
|
P
lacebo
b
N = 77
|
Amvuttra
|
P
lacebo
b
|
9
. mesiac
|
mNIS+7c
|
60,6 (36,0)
|
74,6 (37,0)
|
–2,2 (1,4)
|
14,8 (2,0)
|
–17,0 (–21,8, –12,2)
|
p < 0,0001
|
Norfolk
QoL-DNc
|
47,1 (26,3)
|
55,5 (24,3)
|
–3,3 (1,7)
|
12,9 (2,2)
|
–16,2 (–21,7, –10,8)
|
p < 0,0001
|
Test chôdze na
10 metrov
(m/s)d
|
1,01 (0,39)
|
0,79 (0,32)
|
0 (0,02)
|
–0,13 (0,03)
|
0,13 (0,07, 0,19)
|
p < 0,0001
|
18
. mesiac
|
mNIS+7c
|
60,6 (36,0)
|
74,6 (37,0)
|
–0,5 (1,6)
|
28,1 (2,3)
|
–28,5 (–34,0, –23,1)
|
p < 0,0001
|
Norfolk
QoL-DNc
|
47,1 (26,3)
|
55,5 (24,3)
|
–1,2 (1,8)
|
19,8 (2,6)
|
–21,0 (–27,1, –14,9)
|
p < 0,0001
|
Test chôdze na
10 metrov
(m/s)d
|
1,01 (0,39)
|
0,79 (0,32)
|
–0,02 (0,03)
|
–0,26 (0,04)
|
0,24 (0,15, 0,33)
|
p < 0,0001
|
mBMIe
|
1 057,5 (233,8)
|
989,9 (214,2)
|
25,0 (9,5)
|
–115,7 (13,4)
|
140,7 (108,4, 172,9)
|
p < 0,0001
|
R-ODSf
|
34,1 (11,0)
|
29,8 (10,8)
|
–1,5 (0,6)
|
–9,9 (0,8)
|
8,4 (6,5, 10,4)
|
p < 0,0001
|
Skratky: CI = confidence interval – interval spoľahlivosti; LS mean = least squares mean – priemer najmenších štvorcov; mBMI = modified body mass index – modifikovaný index telesnej hmotnosti; mNIS = modified Neuropathy Impairment Score – modifikované skóre neuropatického poškodenia; QoL-DN = Quality of Life - Diabetic Neuropathy – kvalita života pri diabetickej neuropatii; SD = standard deviation – štandardná odchýlka; SEM = standard error of the mean – štandardná
chyba priemeru
a Všetky koncové ukazovatele v 9. mesiaci analyzované pomocou analýzy kovariancie (ANCOVA) s metódou viacnásobnej imputácie (MI) a všetky koncové ukazovatele v 18. mesiaci analyzované metódou opakovaných meraní modelu zmiešaného účinku (mixed-effect model repeated measures, MMRM).
b Externá skupina s placebom z randomizovanej kontrolovanej klinickej štúdie APOLLO.
c Nižšie číslo indikuje menšiu poruchu/menej príznakov.
d Vyššie číslo indikuje menšie postihnutie/menšiu poruchu.
e mBMI: index telesnej hmotnosti (BMI; kg/m2) vynásobený sérovou hladinou albumínu (g/l); vyššie číslo indikuje lepší nutričný stav.
f Vyššie číslo indikuje menšie postihnutie/menšiu poruchu.
 O
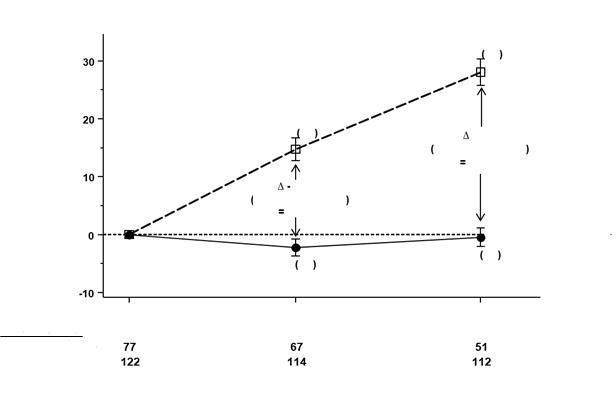
brázok 1: Zmena oproti východiskovej hodnote v mNIS+7 (9. a 18. mesiac)
28
,1 2,
P
la
c
e
bo
a
O
brázok 1: Zmena oproti východiskovej hodnote v mNIS+7 (9. a 18. mesiac)
28
,1 2,
P
la
c
e
bo
a
14
,8 2,
-
9
5 % IS : -34,0, -23,1
 17
,0
9
5 % IS : -21,8, -12,2
p
6
,5x10
-
20
17
,0
9
5 % IS : -21,8, -12,2
p
6
,5x10
-
20
p
3
,5x10
-
12
-
2
,2 1,4
'
A
m
vu
ttr
a
-
0
,5
1,
Vyhodnotiteľné N Placebo
a
A
m
vuttr
Východiskový stav
9
. mesiac 18. mesiac
Pokles mNIS+7 znamená zlepšenie.
∆ označuje rozdiel v liečbe medzi skupinami, zobrazený ako priemerný rozdiel LS (95 % IS) pre porovnanie AMVUTTRA –
externé placebo.
Všetky koncové ukazovatele v 9. mesiaci analyzované pomocou analýzy kovariancie (ANCOVA) s metódou viacnásobnej imputácie (MI) a všetky koncové ukazovatele v 18. mesiaci analyzované metódou opakovaných meraní modelu zmiešaného
účinku (mixed-effect model repeated measures, MMRM).
a Externá skupina s placebom z randomizovanej kontrolovanej klinickej štúdie APOLLO.
U pacientov užívajúcich Amvuttru došlo oproti placebu k podobným zlepšeniam v celkovom skóre mNIS+7 a Norfolk QoL-DN v 9. a 18. mesiaci vo všetkých podskupinách vrátane veku, pohlavia, rasy, regiónu, skóre NIS, stavu genotypu V30M (V30M alebo iný ako V30M), predchádzajúceho užívania stabilizátora TTR, štádia ochorenia a pacientov s vopred definovanými kritériami pre postihnutie srdca alebo bez nich.
N-terminálny fragment prohormónu natriuretického peptidu typu B (NT-proBNP) je prognostický biomarker srdcovej dysfunkcie. Východiskové hodnoty hladiny NT-proBNP (geometrický priemer) boli 273 ng/l u pacientov liečených Amvuttrou a 531 ng/l u pacientov liečených placebom.
V 18. mesiaci sa geometrický priemer hladín NT-proBNP u pacientov s Amvuttrou znížil o 6 %, zatiaľ čo u pacientov s placebom sa zvýšil o 96 %.
Centrálne hodnotené echokardiogramy preukázali zmeny hrúbky steny ĽK (priemerná hodnota LS rozdielu: –0,18 mm [95 % IS –0,74, 0,38]) a pozdĺžnej deformácie (longitudinal strain, priemerná hodnota LS rozdielu: –0,4 % [95 % IS –1,2, 0,4]) pri liečbe Amvuttrou oproti placebu.
Napriek pozorovaným hodnotám NT proBNP a hrúbky steny ĽK je potrené klinický prínos s ohľadom na kardiomiopatiu ešte potvrdiť.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s vutrisiranom
vo všetkých podskupinách pediatrickej populácie pri hATTR amyloidóze (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetické vlastnosti Amvuttry boli charakterizované meraním plazmatických a močových koncentrácií vutrisiranu.
Absorpcia
Po subkutánnom podaní sa vutrisiran rýchlo absorbuje s časom do maximálnej plazmatickej
koncentrácie (tmax) 3,0 hodiny (rozsah: 2,0 až 6,5). Pri odporúčanom dávkovacom režime 25 mg jedenkrát za 3 mesiace subkutánne bola priemerná (% variačný koeficient [%CV]) maximálna koncentrácia v ustálenom stave (Cmax) 0,12 μg/ml (64,3 %) a plocha pod krivkou koncentrácie v čase od 0 do 24 hodín (AUC0-24) 0,80 μg-h/ml (35,0 %). Po opakovanom štvrťročnom dávkovaní nedošlo ku kumulácii vutrisiranu v plazme.
Distribúcia
Vutrisiran sa viaže na plazmatické bielkoviny z viac ako 80 % v rozsahu koncentrácií pozorovaných
u ľudí pri dávke 25 mg jedenkrát za 3 mesiace subkutánne. Väzba vutrisiranu na plazmatické bielkoviny bola závislá od koncentrácie a klesala so zvyšujúcou sa koncentráciou vutrisiranu (zo 78 %
pri 0,5 µg/ml na 19 % pri 50 µg/ml). Populačný odhad zjavného distribučného objemu centrálneho
kompartmentu (Vd/F) vutrisiranu u ľudí bol 10,2 l (% relatívna štandardná odchýlka [RSE] = 5,71 %). Vutrisiran sa po subkutánnom podaní distribuuje primárne do pečene.
Biotransformácia
Vutrisiran sa v pečeni metabolizuje endo- a exonukleázami na krátke nukleotidové fragmenty rôznej
veľkosti. U ľudí neboli zistené žiadne významné cirkulujúce metabolity. Štúdie in vitro naznačujú, že vutrisiran nepodlieha metabolizmu enzýmami CYP450.
Eliminácia
Po jednorazovej subkutánnej dávke 25 mg bol medián zjavného plazmatického klírensu 21,4 l/h
(rozsah: 19,8, 30,0). Medián terminálneho polčasu eliminácie (t1/2) vutrisiranu bol 5,23 hodiny (rozsah:
2,24, 6,36). Po jednorazovej subkutánnej dávke 5 až 300 mg sa priemerná frakcia nezmenenej účinnej látky vylúčená močom pohybovala od 15,4 do 25,4 % a priemerný renálny klírens vutrisiranu sa
pohyboval od 4,45 do 5,74 l/h.
Linearita/nelinearita
Po podaní jednorazových subkutánnych dávok v rozsahu 5 až 300 mg sa ukázalo, že hodnota Cmax
vutrisiranu je úmerná dávke, zatiaľ čo plocha pod krivkou koncentrácie v čase od okamihu podania dávky extrapolovanej do nekonečna (AUCinf) a plocha pod krivkou koncentrácie v čase od okamihu podania dávky do poslednej merateľnej koncentrácie (AUClast) boli mierne zvýšené oproti proporcionálnej dávke.
Farmakokinetický/farmakodynamický vzťah
Populačné farmakokinetické/farmakodynamické analýzy u zdravých osôb a pacientov s hATTR
amyloidózou (n = 202) ukázali vzťah závislý od dávky medzi predpokladanými koncentráciami vutrisiranu v pečeni a znížením sérovej hladiny TTR. Modelom predpovedaný medián maximálneho
zníženia TTR bol 88 %, minimálneho zníženia 86 % a priemerného zníženia 87 % v ustálenom stave, čo potvrdzuje minimálnu variabilitu medzi maximálnou a minimálnou hodnotou počas 3-mesačného
dávkovacieho intervalu. Analýza kovariátov ukázala podobné zníženie TTR u pacientov s miernou až stredne závažnou poruchou funkcie obličiek alebo miernou poruchou funkcie pečene, ako aj
pri porovnaní podľa pohlavia, rasy, predchádzajúceho užívania stabilizátorov TTR, genotypu (V30M
alebo iný ako V30M), veku a telesnej hmotnosti.
Špeciálne populácie
Pohlavie a rasa
V klinických štúdiách sa nezistili významné rozdiely vo farmakokinetických parametroch v ustálenom stave ani v redukcii hladiny TTR podľa pohlavia a rasy.
Starší pacienti
V štúdii HELIOS-A malo 46 (38 %) pacientov liečených vutrisiranom ≥ 65 rokov a z nich 7 (5,7 %)
pacientov malo ≥ 75 rokov. Nezistili sa žiadne významné rozdiely vo farmakokinetických parametroch v ustálenom stave ani v redukcii hladiny TTR pri porovnaní pacientov vo veku < 65 rokov a vo veku
≥ 65 rokov.
Porucha funkcie pečene
Populačné analýzy farmakokinetiky a farmakodynamiky nepreukázali žiaden vplyv miernej poruchy funkcie pečene (celkový bilirubín ≤ 1 x HHN a AST > 1 x HHN alebo celkový bilirubín > 1,0 až 1,5 x HHN a akákoľvek hodnota AST) na expozíciu vutrisiranu ani na redukciu hladiny TTR pri porovnaní
s pacientmi s normálnou funkciou pečene. Vutrisiran nebol skúmaný u pacientov so stredne závažnou až závažnou poruchou funkcie pečene.
Porucha funkcie obličiek
Populačné analýzy farmakokinetiky a farmakodynamiky nepreukázali žiaden vplyv miernej alebo stredne závažnej poruchy funkcie obličiek (eGFR ≥ 30 až < 90 ml/min/1,73 m2) na expozíciu vutrisiranu ani na redukciu hladiny TTR pri porovnaní s pacientmi s normálnou funkciou obličiek. Vutrisiran nebol skúmaný u pacientov so závažnou poruchou funkcie obličiek alebo v poslednom štádiu ochorenia obličiek.
5.3 Predklinické údaje o bezpečnosti
Všeobecná toxikológia
Opakované subkutánne podávanie vutrisiranu v dávke ≥ 30 mg/kg jedenkrát mesačne opiciam
spôsobilo očakávané trvalé zníženie cirkulujúceho TTR (až o 99 %) a vitamínu A (až o 89 %) bez zjavných toxikologických nálezov.
Po opakovanom podávaní jedenkrát mesačne počas maximálne 6 mesiacov u potkanov a 9 mesiacov u opíc boli zistené mierne a konzistentné, neadverzné, histologické zmeny v pečeni (hepatocyty, Kupfferove bunky), obličkách (renálne tubuly), lymfatických uzlinách a miestach podania injekcie (makrofágy), ktoré odzrkadľovali hlavnú distribúciu a kumuláciu vutrisiranu. Nezistila sa však žiadna toxicita pri viac ako 1 000 a 3 000-násobnom zvýšení hodnoty AUC v plazme, pokiaľ sa normalizujú na štvrťročné dávkovanie a porovnajú s predpokladanou expozíciou pri maximálnej odporúčanej dávke pre ľudí [MRHD].
Genotoxicita/karcinogenita
Vutrisiran nevykazoval žiadny genotoxický potenciál in vitro ani in vivo. Štúdie karcinogenity neboli
ukončené.
Reprodukčná toxicita
Vutrisiran nie je farmakologicky aktívny na potkanoch a králikoch, čo obmedzuje prediktívnosť týchto
sledovaní. Jednorazová dávka ortológu vutrisiranu špecifického pre potkany však nemala v kombinovanej štúdii na potkanoch vplyv na fertilitu a skorý embryonálny vývin.
Týždenné subkutánne podávanie vutrisiranu nemalo vplyv na fertilitu a skorý embryonálny vývin pri viac ako 300-násobku normalizovanej hodnoty MRHD. V embryofetálnej štúdii so subkutánnym podávaním vutrisiranu jedenkrát denne u gravidných potkanov sa pozorovali nežiaduce účinky na telesnú hmotnosť matky, konzumáciu potravy, zvýšený počet predčasných pôrodov a postimplantačné
straty s hodnotou NOAEL matky 10 mg/kg/deň, čo bol viac ako 300-násobok normalizovanej hodnoty MRHD 0,005 mg/kg/deň. Na základe nežiaduceho zníženia telesnej hmotnosti plodu a zvýšených skeletálnych variácií pri ≥ 10 mg/kg/deň bola hodnota NOAEL vutrisiranu pre plod 3 mg/kg/deň, čo je
97-násobok normalizovanej hodnoty MRHD.
V štúdii embryofetálneho vývinu na gravidných králikoch sa nepozorovali žiadne nežiaduce účinky na embryofetálny vývin pri ≤ 30 mg/kg/deň vutrisiranu, čo je viac ako 1900-násobok normalizovanej hodnoty MRHD.
V štúdii prenatálno-postnatálneho vývinu nemalo subkutánne podávanie vutrisiranu každý šiesty deň žiadny vplyv na rast a vývin mláďat s hodnotou NOAEL 20 mg/kg, čo je viac ako 90-násobok normalizovanej hodnoty MRHD.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
dihydrogenfosforečnan sodný, dihydrát hydrogenfosforečnan sodný, dihydrát chlorid sodný
voda na injekcie
hydroxid sodný (na úpravu pH)
kyselina fosforečná (na úpravu pH).
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote neprevyšujúcej 30 °C. Neuchovávajte v mrazničke.
6.5 Druh obalu a obsah balenia
Naplnená injekčná striekačka (zo skla typu I) s ihlou veľkosti 29 G z nehrdzavejúcej ocele s chráničom ihly.
Amvuttra je dostupná v baleniach obsahujúcich jednu jednorazovú naplnenú injekčnú striekačku.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Alnylam Netherlands B.V. Antonio Vivaldistraat 150
1083 HP Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/22/1681/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 15. september 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu