
(hlavne pľúcne udalosti, srdcové udalosti alebo
hypotenzia) vrátane udalostí po predchádzajúcich chemoterapiách.
• Aktívne infekcie alebo zápalové ochorenia (vrátane pneumonitídy, myokarditídy alebo hepatitídy).
• Aktívna reakcia štepu proti príjemcovi (GVHD, graft-versus-host disease).
Autol
ó
gne použitie
Abecma je určená výlučne na autológne použitie a nemá sa za žiadnych podmienok podávať iným
pacientom. Pred podaním infúzie sa totožnosť pacienta musí zhodovať s identifikačnými údajmi
pacienta na infúznom vaku, kazete a certifikáte o uvoľnení infúzie (RfIC) Abecmy. Abecma sa nesmie
podávať, ak sa špecifické informácie pacienta uvedené na štítku nezhodujú s daným pacientom.
Súbežné ochorenie
Pacienti s aktívnym ochorením centrálneho nervového systému (CNS) alebo s neadekvátnou funkciou obličiek, pečene, pľúc alebo srdca budú pravdepodobne vystavení väčšiemu riziku následných nežiaducich reakcií opísaných nižšie a vyžaduje sa u nich osobitná pozornosť.
Patológia centrálneho nervového systému
K dispozícii nie sú žiadne skúsenosti s používaním Abecmy u pacientov s postihnutím CNS
myelómom alebo s inými už existujúcimi klinicky relevantnými patológiami CNS.
Alogénna transplantácia kmeňových buniek v minulosti
Neodporúča sa podávať Abecmu pacientom v priebehu 4 mesiacov po alogénnej transplantácii
kmeňových buniek (SCT, stem cell transplant) z dôvodu možného rizika, že Abecma zhorší GVHD.
Leukaferéza na výrobu Abecmy sa má vykonať minimálne 12 týždňov po alogénnej SCT.
Predchádzajúce ošetrenie liečbou anti-BCMA
K dispozícii je obmedzené množstvo skúseností s Abecmou u pacientov v minulosti vystavených
liečbe zamerané proti BCMA.
K dispozícii sú obmedzené skúsenosti s preliečením pacientov druhou dávkou Abecmy. Odpovede po opakovanej liečbe Abecmou neboli časté a mali kratšie trvanie v porovnaní s úvodnou liečbou. Navyše sa u opakovane liečených pacientov pozorovali úmrtia.
Syndróm uvoľňovania cytokínov
Po infúznom podaní Abecmy sa vyskytol syndróm uvoľňovania cytokínov (CRS, cytokine release
syndrome) vrátane fatálnych alebo život ohrozujúcich reakcií. Takmer u všetkých pacientov sa
vyskytol určitý stupeň CRS. Medián času nástupu CRS bol 1 deň (rozsah: 1 až 12) (pozri časť 4.8).
Monitorovanie a liečba CRS
CRS je potrebné identifikovať na základe klinických prejavov. U pacientov sa majú vyšetriť a liečiť iné príčiny horúčky, hypoxie a hypotenzie. CRS sa hlásil v súvislosti s nálezmi hemofagocytovej
lymfohistiocytózy/syndrómu aktivácie makrofágov (HLH, haemophagocytic lymphohistiocytosis/MAS, macrophage activation syndrome) a fyziológia syndrómov sa môže prekrývať. MAS je potenciálne život ohrozujúce ochorenie a u pacientov je potrebné dôkladne monitorovať výskyt MAS. Liečba MAS sa má vykonávať podľa smerníc zdravotníckeho zariadenia.
Pred infúznym podaním Abecmy musí byť na mieste k dispozícii jedna dávka tocilizumabu na pacienta pripravená na podanie. Liečebné centrum musí mať k dispozícii ďalšiu dávku tocilizumabu vždy do 8 hodín po podaní predchádzajúcej dávky. Pacienti majú byť v kvalifikovanom liečebnom centre monitorovaní na prípadný výskyt prejavov a symptómov CRS počas prvých 10 dní po infúznom podaní Abecmy. Po prvých 10 dňoch po podaní infúzie má byť pacient monitorovaný podľa uváženia lekára. Pacienti majú byť poučení, aby minimálne 4 týždne po podaní infúzie zotrvali v blízkosti kvalifikovaného liečebného centra (do 2 hodín cesty) a kedykoľvek v prípade výskytu prejavov alebo symptómov CRS ihneď vyhľadali lekársku pomoc.
Pri prvom prejave CRS sa má začať liečba podpornou starostlivosťou, tocilizumabom alebo tocilizumabom a kortikosteroidmi, ako je uvedené v tabuľke 1. Abecma sa môže naďalej šíriť a pretrvávať po podaní tocilizumabu a kortikosteroidov (pozri časť 4.5).
Pacienti, u ktorých sa vyskytne CRS, musia byť dôkladne monitorovaný z dôvodu funkcie srdca
a orgánov až do ustúpenia symptómov. V prípade závažného alebo život ohrozujúceho CRS sa má
zvážiť monitorovanie na jednotke intenzívnej starostlivosti a podporná liečba.
Ak existuje podozrenie na súbežnú neurologickú toxicitu počas CRS, liečte neurologickú toxicitu podľa odporúčaní v tabuľke 2 a z dvoch reakcií uvedených v tabuľke 1 a 2 použite tú intervenciu, ktorá je agresívnejšia.
U pacientov s refraktérnym CRS charakterizovaným pretrvávajúcou horúčkou, toxicitou v prípade koncového orgánu (napr. hypoxia, hypotenzia) a/alebo HLH/MAS bez zmiernenia stupňa do 12 hodín od intervencií prvej línie sa odporúča skoršie eskalovanie (t. j. vyššia dávka kortikosteroidov, alternatívne anticytokíny, anti-T-bunkové liečby) do 72 hodín po infúznom podaní Abecmy.
Tabuľka 1. Stupne CRS a odporúčania liečbyStupeň CRSa
|
Tocilizumab
|
Kortikosteroidy
|
1. stupeň
Symptómy si vyžadujú iba symptomatickú liečbu (napr. horúčka, nevoľnosť, únava, bolesť hlavy, myalgia, malátnosť).
|
V prípade nástupu
po 72 hodinách alebo neskôr po podaní infúzie liečte
symptomaticky.
Ak je nástup kratší ako
do 72 hodín po podaní infúzie
a symptómy nie sú kontrolovateľné samotnou podpornou starostlivosťou, zvážte podanie tocilizumabu v
i.v. dávke 8 mg/kg
počas1 hodiny (neprekračujte
dávku 800 mg).
|
─
|
2. stupeň
Symptómy si vyžadujú
stredne závažný zásah a
reagujú na tento zásah.
Potreba kyslíka nižšia
ako 40 % FiO2 alebo
hypotenzia reagujúca na tekutiny alebo nízku dávku jedného vazopresora alebo
orgánová toxicita 2. stupňa.
|
Podajte tocilizumab v i.v. dávke 8 mg/kg počas1 hodiny (neprekračujte dávku 800 mg).
|
Zvážte dexametazón v i.v.
dávke 10 mg
každých 12 až 24 hodín.
|
Stupeň CRS
a
|
Tocilizumab
|
Kortikosteroidy
|
3
. stupeň
Symptómy si vyžadujú
agresívny zásah a reagujú na
tento zásah.
Horúčka, potreba kyslíka vyššia ako alebo
zodpovedajúca 40 %
FiO2 alebo hypotenzia
vyžadujúca si vysokú dávku
alebo opakované podávanie
vazopresoru alebo orgánová toxicita 3. stupňa alebo transaminitída 4. stupňa.
|
Podajte tocilizumab v i.v. dávke 8 mg/kg počas1 hodiny (neprekračujte dávku 800 mg).
|
Podávajte dexametazón
(napr. 10 mg i.v.
každých 12 hodín).
|
Pri 2. a 3. stupni:
Ak nedôjde k zmierneniu do 24 hodín alebo ak dôjde k rýchlej progresii, zopakujte tocilizumab a
zvýšte dávku a frekvenciu dexametazónu (20 mg i.v. každých 6 až 12 hodín).
Ak nedôjde k zmierneniu do 24 hodín alebo ak pretrváva rýchla progresia, prejdite na metylprednizolón v dávke 2 mg/kg a potom v dávke 2 mg/kg rozdelenej na 4 podania počas dňa. Ak sa začnú podávať steroidy, pokračujte v podávaní steroidov minimálne v 3 dávkach, a znižujte počas maximálne 7 dní.
Po 2 dávkach tocilizumabu zvážte alternatívne anticytokíny. Neprekračujte 3 dávky tocilizumabu za 24 hodín alebo celkovo 4 dávky.
|
4
. stupeň
Život ohrozujúce symptómy.
Potreba podpornej ventilácie,
kontinuálna venovenózna hemodialýza (CVVHD) alebo
orgánová toxicita 4. stupňa
(okrem transaminitídy).
|
Podajte tocilizumab v i.v. dávke 8 mg/kg počas1 hodiny (neprekračujte dávku 800 mg).
|
Podávajte 20 mg dexametazónu
i.v. každých 6 hodín.
|
Pri 4. stupni:
Po 2 dávkach tocilizumabu zvážte alternatívne anticytokíny. Neprekračujte 3 dávky tocilizumabu
za 24 hodín alebo celkovo 4 dávky.
Ak nedôjde k zmierneniu do 24 hodín, zvážte metylprednizolón (1 až 2 g, v prípade potreby zopakujte každých 24 hodín, podľa klinickej indikácie znižujte) alebo anti-T-bunkové liečby, ako
je cyklofosfamid v dávke 1,5 g/m2 alebo iné.
|
a Lee a kol., 2014.
Neurologické nežiaduce reakciePo liečbe Abecmou sa vyskytli neurologické toxicity, ako je afázia a encefalopatia, ktoré môžu byť
závažné alebo život ohrozujúce. Medián času nástupu prvej udalosti neurotoxicity bol 2 dni
(rozsah: 1 až 10 dní). Neurologická toxicita sa môže vyskytnúť súbežne so CRS, po ustúpení CRS
alebo bez výskytu CRS (pozri časť 4.8).
Monitorovanie a liečba neurologických toxicítPacienti majú byť v kvalifikovanom liečebnom centre monitorovaní na prípadný výskyt prejavov a symptómov neurologických toxicít počas prvých 10 dní po infúznom podaní Abecmy. Po prvých
10 dňoch po podaní infúzie má byť pacient monitorovaný podľa uváženia lekára. Pacientov je potrebné poučiť, aby minimálne 4 týždne po podaní infúzie zotrvali v blízkosti kvalifikovaného liečebného centra (do 2 hodín cesty) a kedykoľvek v prípade výskytu prejavov a symptómov neurologických toxicít ihneď vyhľadali lekársku pomoc.
Ak je podozrenie na neurologickú toxicitu, liečte podľa odporúčaní v tabuľke 2. Je potrebné vylúčiť iné príčiny neurologických symptómov. V prípade závažných alebo život ohrozujúcich neurologických toxicít sa má poskytnúť podporná liečba v rámci intenzívnej starostlivosti.
Ak je podozrenie na súbežný CRS počas reakcie neurologickej toxicity, má sa liečiť podľa odporúčaní v tabuľke 1, a z dvoch reakcií uvedených v tabuľke 1 a 2 sa má použiť intervencia, ktorá je agresívnejšia.
Tabuľka 2. Stupne neurologickej toxicity a odporúčania liečbyStupeň neurologickej toxicitya
|
Kortikosteroidy a lieky proti záchvatom
|
1. stupeň Mierna alebo asymptomatická.
|
Začnite podávať lieky proti záchvatom bez sedatívneho účinku (napr.
levetiracetam) na profylaxiu záchvatov.
Ak uplynulo 72 hodín alebo viac po podaní infúzie, pozorujte pacienta. Ak uplynulo menej ako 72 hodín po podaní infúzie a symptómy nie sú
kontrolované samotnou podpornou starostlivosťou, zvážte podávanie
dexametazónu v i.v. dávke 10 mg/kg každých 12 až 24 hodín počas 2 až 3 dní.
|
2. stupeň
Stredne závažná.
|
Začnite podávať lieky proti záchvatom bez sedatívneho účinku (napr.
levetiracetam) na profylaxiu záchvatov.
Začnite podávať dexametazónu v i.v. dávke 10 mg každých 12 hodín počas 2 až 3 dní alebo dlhšie pri pretrvávajúcich symptómoch. Zvážte znižovanie dávky pri celkovej expozícii steroidu dlhšej ako 3 dni. Steroidy sa neodporúčajú pri ojedinelých bolestiach hlavy 2. stupňa.
Ak nedôjde k zmierneniu po 24 hodinách alebo ak dôjde k zhoršeniu
neurologickej toxicity, zvýšte dávku dexametazónu a/alebo frekvenciu
podávania až na maximálne 20 mg i.v. každých 6 hodín.
|
3. stupeň
Závažná alebo
medicínsky
významná, no nie bezprostredne
život
ohrozujúca;
hospitalizácia alebo jej
predĺženie;
postihnutie.
|
Začnite podávať lieky proti záchvatom bez sedatívneho účinku (napr.
levetiracetam) na profylaxiu záchvatov.
Začnite podávať dexametazón v i.v. dávkach 10 až 20 mg
každých 8 až 12 hodín. Steroidy sa neodporúčajú pri ojedinelých bolestiach
hlavy 3. stupňa.
Ak nedôjde k zmierneniu po 24 hodinách alebo ak dôjde k zhoršeniu
neurologickej toxicity, zvýšte dávku metylprednizolónu (2 mg/kg nasycovacia
dávka, po ktorej nasleduje dávka 2 mg/kg rozdelená na 4 podania počas dňa;
znižujte v priebehu 7 dní).
Pri podozrení na edém mozgu zvážte hyperventiláciu a hyperosmolárnu liečbu.
Podajte vysokú dávku metylprednizolónu (1 až 2 g, v prípade potreby
zopakujte každých 24 hodín; znižujte podľa klinickej indikácie) a
cyklofosfamid v dávke 1,5 g/m2.
|
Stupeň neurologickej toxicity
a
|
Kortikosteroidy a lieky proti záchvatom
|
4
. stupeň
Život
ohrozujúca.
|
Začnite podávať lieky proti záchvatom bez sedatívneho účinku (napr.
levetiracetam) na profylaxiu záchvatov.
Začnite podávať dexametazónu v i.v. dávke 20 mg každých 6 hodín. Ak nedôjde k zmierneniu po 24 hodinách alebo ak dôjde k zhoršeniu neurologickej toxicity, zvýšte dávku na vysokú dávku metylprednizolónu (1 až 2 g, v prípade potreby zopakujte každých 24 hodín; znižujte podľa klinickej indikácie). Zvážte podanie cyklofosfamidu v dávke 1,5 g/m2.
Pri podozrení na edém mozgu zvážte hyperventiláciu a hyperosmolárnu liečbu.
Podajte vysokú dávku metylprednizolónu (1 až 2 g, v prípade potreby
zopakujte každých 24 hodín; znižujte podľa klinickej indikácie) a
cyklofosfamid v dávke 1,5 g/m2.
|
a NCI CTCAE v. 4 – kritériá na určovanie závažnosti neurologických toxicít.
Dlhodobé cytopénieU pacientov sa môžu prejavovať dlhodobé cytopénie počas niekoľkých týždňov po lymfodeplečnej
chemoterapii a infúznom podaní Abecmy (pozri časť 4.8). Pred infúznym podaním Abecmy a po
podaní sa má monitorovať krvný obraz. Cytopénie sa majú liečiť myeloidným rastovým faktorom
a podpornou transfúziou krvi podľa smerníc zdravotníckeho zariadenia.
Infekcie a febrilná neutropéniaAbecma sa nemá podávať pacientom s aktívnymi infekciami alebo zápalovými ochoreniami. Po
podaní Abecmy sa u pacientov vyskytli závažné infekcie vrátane život ohrozujúcich alebo smrteľných
infekcií (pozri časť 4.8). Pred infúznym podaním Abecmy a po jej podaní majú byť pacienti
monitorovaní na prípadný výskyt prejavov a symptómov infekcie, a podľa toho ich treba liečiť.
Profylaktické, preemptívne a/alebo terapeutické bakteriostatiká sa majú podávať v súlade so
smernicami zdravotníckeho zariadenia.
Febrilná neutropénia sa pozorovala u pacientov po infúznom podaní Abecmy (pozri časť 4.8) a môže byť súbežná so CRS. V prípade výskytu febrilnej neutropénie sa má posúdiť infekcia a liečiť širokospektrálnymi antibiotikami, tekutinami a inou podpornou starostlivosťou podľa zdravotnej
indikácie.
Reaktivácia vírusuPo podaní Abecmy sa vyskytla infekcia vyvolaná cytomegalovírusom (CMV), ktorá spôsobila pneumóniu a úmrtie (pozri časť 4.8). Pacienti majú byť monitorovaní a liečení na prípadný výskyt prejavov a symptómov infekcie vyvolanej CMV podľa klinických pokynov.
Reaktivácia HBV, v niektorých prípadoch vedúca k fulminantnej hepatitíde, zlyhaniu pečene a úmrtiu, sa môže vyskytnúť u pacientov liečených liekmi zacielenými proti plazmatickým bunkám (pozri
časť 4.8).
Skríning zameraný na CMV, HBV, aktívnu infekciu HIV a aktívnu infekciu HCV sa musí vykonať pred odberom buniek na výrobu (pozri časť 4.2).
HypogamaglobulinémiaU pacientov liečených Abecmou sa môže vyskytnúť aplázia plazmatických buniek a
hypogamaglobulinémia (pozri časť 4.8). Po liečbe Abecmou sa majú monitorovať hladiny
imunoglobulínu a majú sa liečiť podľa smerníc zdravotníckeho zariadenia vrátane preventívnych
opatrení proti infekciám, profylaxie antibiotikami alebo antivirotikami a náhrady imunoglobulínu.
Sekundárne malignity
U pacientov liečených Abecmou sa môžu vyskytnúť sekundárne malignity. Pacienti majú byť po celý
život monitorovaní na prípadný výskyt sekundárnych malignít. V prípade výskytu sekundárnej
malignity T-buniek je potrebnékontaktovať spoločnosť a získať pokyny týkajúce sa odberu vzoriek
pacienta na testovanie.
Reakcie z precitlivenosti
Pri infúznom podávaní Abecmy sa môžu vyskytnúť alergické reakcie. Závažné reakcie z
precitlivenosti vrátane anafylaxie môžu byť spôsobené dimetylsulfoxidom (DMSO), pomocnou látkou
v Abecme. Pacienti, ktorí neboli v minulosti exponovaní DMSO, majú byť dôkladne monitorovaní.
Pred začatím podávania infúzie, približne každých desať minút počas podávania infúzie a každú
hodinu počas 3 hodín po podaní infúzie sa majú monitorovať vitálne funkcie (krvný tlak, tep srdca
a saturácia kyslíka) a výskyt akýchkoľvek symptómov.
Interferencia so sérologickým testovaním
HIV a lentivírus používaný pri výrobe Abecmy majú obmedzené, krátke rozpätie identického
genetického materiálu (RNA). Preto niektoré komerčné testy zamerané na nukleové kyseliny HIV
môžu viesť k falošne pozitívnym výsledkom u pacientov, ktorí dostali Abecmu.
Darovanie krvi, orgánov, tkanív a buniek
Pacienti liečení Abecmou nesmú darovať krv, orgány, tkanivá alebo bunky na transplantáciu.
Dlhodobé následné sledovanie
Od pacientov sa očakáva, že sa zaradia do registra a že budú v rámci registra sledovaní s cieľom lepšie
porozumieť dlhodobej bezpečnosti a účinnosti Abecmy.
Pomocné látky
Tento liek obsahuje až 33 mmol (752 mg) sodíka v každej dávke, čo zodpovedá 37,6 % WHO
odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
Tento liek obsahuje až 7 mmol (274 mg) draslíka v každej dávke. Musí sa vziať do úvahy u pacientov so zníženou funkciou obličiek alebo u pacientov na diéte s kontrolovaným obsahom draslíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Súbežné podávanie látok, o ktorých je známe, že inhibujú funkciu T-buniek, sa oficiálne neskúmalo. Súbežné podávanie látok, o ktorých je známe, že stimulujú funkciu T-buniek, sa neskúmalo a ich účinky nie sú známe.
Tocilizumab a použitie kortikosteroidov
Niektorí pacienti potrebovali tocilizumab a/alebo kortikosteroid na liečbu CRS (pozri časť 4.8).
Použitie tocilizumabu a/alebo steroidov na liečbu CRS bolo častejšie u pacientov s vyššou expanziou
buniek.
Pacienti s CRS liečení tocilizumabom mali vyššie bunkové expanzie Abecmy podľa
nameraného 1,4-násobne a 1,6-násobne vyššieho mediánu Cmax (N = 66) a AUC0–28 dní (N = 65), v
uvedenom poradí, v porovnaní s pacientmi, ktorí neužívali tocilizumab (N = 61 pri Cmax a N = 60 pri
AUC0–28 dní). Podobne pacienti s CRS liečení kortikosteroidmi mali vyššie bunkové expanzie Abecmy
podľa nameraného 1,7-násobne a 2,2-násobne vyššieho mediánu Cmax (N = 18) a AUC0–28 dní (N = 18),
v uvedenom poradí, v porovnaní s pacientmi, ktorí neužívali kortikosteroidy (N = 109 pri Cmax a
N = 107 pri AUC0‐-28 dní).
Živé vakcíny
Bezpečnosť imunizácie živými vírusovými očkovacími látkami počas liečby Abecmou alebo po nej sa
neskúmala. Vakcinácia živými vírusovými očkovacími látkami sa neodporúča počas minimálne
6 týždňov pred začatím lymfodeplečnej chemoterapie, počas liečby Abecmou a pred dosiahnutím
imunitného zotavenia po liečbe Abecmou.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku/antikoncepcia u mužov a žien
Prípadná gravidita u žien vo fertilnom veku sa má preveriť tehotenským testom pred začatím liečby
Abecmou.
Informácie o potrebe účinnej antikoncepcie u pacientov, ktorí postávajú lymfodeplečnú chemoterapiu,
sú uvedené v informáciách o lieku fludarabínu a cyklofosfamidu.
K dispozícii nie sú dostatočné údaje o expozícii, ktoré by poskytli odporúčanie týkajúce sa dĺžky používania antikoncepcie po liečbe Abecmou.
Gravidita
Nie sú k dispozícii údaje o použití idekabtagén-vikleucelu u gravidných žien. S Abecmou sa na
zvieratách nevykonali žiadne reprodukčné štúdie ani štúdie vývinovej toxicity s cieľom zistiť, či
idekabtagén-vikleucel môže poškodiť plod pri podávaní gravidným ženám (pozri časť 5.3).
Nie je známe, či idekabtagén-vikleucel môže prechádzať do plodu. Na základe mechanizmu účinku, ak transdukované bunky prechádzajú placentou, môžu spôsobiť u plodu toxicitu vrátane aplázie plazmatických buniek alebo hypogamaglobulinémie. Preto sa Abecma neodporúča používať počas gravidity a u žien vo fertilnom veku nepoužívajúcich antikoncepciu. Gravidné ženy majú byť poučené o možných rizikách pre plod. Gravidita po liečbe Abecmou sa má prekonzultovať s ošetrujúcim
lekárom.
Je potrebné zvážiť zhodnotenie hladín imunoglobulínu u novorodencov matiek liečených Abecmou. Dojčenie
Nie je známe, či sa bunky idekabtagén-vikleucelu vylučujú do ľudského mlieka alebo či prechádzajú
do dojčaťa. Riziko u dojčaťa nemôže byť vylúčené. Ženy, ktoré dojčia, majú byť poučené o
potenciálnom riziku pre dojča.
Fertilita
K dispozícii nie sú žiadne údaje o účinku idekabtagén-vikleucelu na fertilitu. Účinky idekabtagén-
vikleucelu na fertilitu mužov a žien sa v štúdiách na zvieratách nehodnotili.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Abecma môže mať veľký vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
Vzhľadom na možný výskyt neurologických nežiaducich reakcií vrátane zmeneného duševného stavu alebo záchvatov pri podávaní Abecmy sa pacienti, ktorí dostávajú Abecmu, majú vyhýbať vedeniu vozidiel alebo obsluhovaniu ťažkých alebo potenciálne nebezpečných strojov počas minimálne
8 týždňov po infúznom podaní Abecmy alebo do úpravy neurologických nežiaducich reakcií.
4.8 Nežiaduce účinky
Súhrn profilu bezpečnosti
Údaje o bezpečnosti opísané v tejto časti uvádzajú expozíciu Abecme v štúdiách KarMMa a CRB-401,
v ktorých Abecmu dostalo 184 pacientov s recidivovaným a refraktérnym mnohopočetným
myelómom. Medián trvania ďalšieho sledovania bol 15,5 mesiaca. K najčastejším nežiaducim
reakciám patrili neutropénia (91,3 %), CRS (81,0 %), anémia (70,7 %), trombocytopénia (66,8 %), infekcie – nešpecifikovaný patogén (53,8 %), leukopénia (48,4 %), únava (39,1 %), hnačka (36,4 %), hypokaliémia (34,2 %), hypofosfatémia (32,6 %), nevoľnosť (32,6 %), lymfopénia (31,5 %), pyrexia (28,8 %), kašeľ (27,2 %), hypokalciémia (26,6 %), infekcie – vírusové (26,1 %), bolesť hlavy
(23,9 %), hypomagneziémia (22,3 %), infekcia horných dýchacích ciest (21,7 %), artralgia (20,7 %), periférny edém (20,1 %), znížená chuť na jedlo (19,6 %), hypogamaglobulinémia (19,6 %) a febrilná neutropénia (16,3 %); k iným častým nežiaducim udalostiam vyskytujúcim sa s nižšou frekvenciou a považovaným za klinicky významné patrili pneumónia (10,3 %), tras (8,2 %), somnolencia (5,4 %),
afázia (4,3 %), encefalopatia (4,3 %) a synkopa (4,3 %).
Závažné nežiaduce reakcie sa vyskytli u 70,1 % pacientov. K najčastejším závažným nežiaducim
reakciám patrili CRS (17,4 %), pneumónia (7,1 %), febrilná neutropénia (6,0 %) a pyrexia (6,0 %); k
iným závažným nežiaducim udalostiam vyskytujúcim sa s nižšou frekvenciou a považovaným za
klinicky významné patrili neutropénia (4,3 %), sepsa (3,8 %), trombocytopénia (3,8 %), stav
zmätenosti (2,2 %), dyspnoe (2,2 %), hypoxia (1,6 %), zmeny duševného stavu (1,6 %) a encefalopatia
(1,6 %).
K najčastejším nežiaducim reakciám 3. alebo 4. stupňa patrili neutropénia (88,6 %), anémia (58,2 %), trombocytopénia (53,5 %), leukopénia (45,1 %), lymfopénia (30,4 %), infekcie – nešpecifikovaný
patogén (17,9 %), hypofosfatémia (17,4 %), febrilná neutropénia (14,7 %), hypokalciémia (7,1 %), infekcia – vírusová (7,1 %), pneumónia (6,0 %), CRS (5,4 %), hypertenzia (5,4 %) a hyponatriémia
(5,4 %).
Nežiaduce reakcie 3. alebo 4. stupňa sa častejšie pozorovali v priebehu začiatočných 8 týždňov po podaní infúzie (97,8 %) v porovnaní s 8 týždňami po podaní infúzie (60,8 %). K najčastejšie hláseným nežiaducim reakciám 3. alebo 4. stupňa v priebehu prvých 8 týždňov po podaní infúzie patrili
neutropénia (87,0 %), anémia (56,0 %), trombocytopénia (48,4 %), leukopénia (44,0 %), lymfopénia
(27,7 %) a hypofosfatémia (16,3 %).
Zoznam nežiaducich reakcií v tabuľke
V tabuľke 3 sa uvádza súhrn nežiaducich reakcií pozorovaných u 128 a 56 pacientov liečených
Abecmou v rámci hladín cieľových dávok 150 až 450 x 106 CAR-pozitívnych T-buniek (pozri
tabuľku 4 v časti 5.1 ohľadom zodpovedajúceho rozmedzia dávky životaschopných CAR-pozitívnych
T-buniek) v štúdiách KarMMa a CRB-401, v uvedenom poradí. Nežiaduce reakcie sú zoradené podľa
tried orgánových systémov podľa databázy MedDRA a podľa frekvencie. Frekvencie sú definované
ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé
(≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov). V rámci
jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 3. Nežiaduce reakcie pozorované u pacientov liečených Abecmou
Trieda orgánového systému
|
Nežiaduca reakcia
|
Všetky stupne frekvencií
|
Infekcie a nákazya
|
Infekcie – bakteriálne
Infekcie – vírusové
Infekcie – nešpecifikovaný patogén
Infekcie – plesňové
|
Veľmi časté Veľmi časté Veľmi časté Časté
|
Poruchy krvi
a lymfatického systému
|
Neutropénia Leukopénia Trombocytopénia Febrilná neutropénia
Lymfopénia
Anémia
Diseminovaná intravaskulárna koagulopatia
|
Veľmi časté Veľmi časté Veľmi časté Veľmi časté Veľmi časté Veľmi časté Časté
|
Poruchy imunitného systému
|
Syndróm uvoľňovania cytokínov
Hypogamaglobulinémia
Hemofagocytová lymfohistiocytóza*
|
Veľmi časté Veľmi časté Časté
|
Poruchy metabolizmu a výživy
|
Hypofosfatémia
Hypokaliémia Hyponatriémia Hypokalciémia Hypoalbuminémia
Znížená chuť na jedlo
Hypomagneziémia
|
Veľmi časté Veľmi časté Veľmi časté Veľmi časté Veľmi časté Veľmi časté Veľmi časté
|
Psychické poruchy
|
Delíriumb
Insomnia
|
Časté
Časté
|
Poruchy nervového systému
|
Encefalopatiac
Bolesť hlavy*
Závratd
Záchvat Hemiparéza Afáziae Ataxiaf
Motorická dysfunkciag
Tras
|
Veľmi časté Veľmi časté Veľmi časté Časté
Časté
Časté
Časté
Časté
Časté
|
Poruchy srdca a
srdcovej činnosti
|
Tachykardia* Fibrilácia predsiene*
|
Veľmi časté
Časté
|
Poruchy ciev
|
Hypertenzia
Hypotenzia*h
|
Veľmi časté
Veľmi časté
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Dyspnoe
Kašeľ
Pľúcny edém
Hypoxia*
|
Veľmi časté Veľmi časté Časté
Časté
|
Poruchy gastrointestinálneho
traktu
|
Vracanie Hnačka Nevoľnosť
Zápcha
Gastrointestinálna hemorágiai
|
Veľmi časté Veľmi časté Veľmi časté Veľmi časté Časté
|
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
|
Artralgia
Myalgia
|
Veľmi časté
Časté
|
Trieda orgánového systému
|
Nežiaduca reakcia
|
Všetky stupne frekvencií
|
Celkové poruchy
a reakcie v mieste podania
|
Pyrexia* Únava*j Asténia Edémk Zimnica*
|
Veľmi časté Veľmi časté Veľmi časté Veľmi časté Veľmi časté
|
Laboratórne a funkčné vyšetrenia
|
Zvýšená hladina
alanínaminotransferázy
Zvýšená hladina
aspartátaminotransferázy
Zvýšená hladina alkalickej fosfatázy
v krvi
Zvýšená hladina C-reaktívneho
proteínu*
|
Veľmi časté
Veľmi časté Veľmi časté Časté
|
* Udalosť, ktorá sa hlásila ako prejav CRS.
a Nežiaduce udalosti triedy orgánového systému Infekcie a nákazy sú zoskupené podľa typu patogénu.
b Delírium zahŕňa delírium, dezorientáciu, halucináciu.
c Encefalopatia zahŕňa amnéziu, bradyfréniu, poruchu kognitívnej funkcie, stav zmätenosti, poruchu pozornosti, dyskalciu,
dysgrafiu, encefalopatiu, letargiu, poruchu pamäti, zmeny duševného stavu, metabolickú encefalopatiu, somnolenciu,
toxickú encefalopatiu.
d K závratom patria závraty, presynkopa, synkopa, vertigo.
e Afázia zahŕňa afáziu, dysartriu.
f Ataxia zahŕňa ataxiu, poruchu chôdze.
g Motorická dysfunkcia zahŕňa motorickú dysfunkciu, svalové kŕče, svalovú slabosť.
h Hypotenzia zahŕňa hypotenziu, ortostatickú hypotenziu.
i Gastrointestinálna hemorágia zahŕňa gastrointestinálnu hemorágiu, hemoroidnú hemorágiu, melénu, krvácanie v ústach.
j Únava zahŕňa únavu, malátnosť.
k Edém zahŕňa edém, edém tváre, generalizovaný edém, periférny edém, periférny opuch.
Opis vybraných nežiaducich reakciíSyndróm uvoľňovania cytokínovPodľa súhrnu zo štúdií (KarMMa a CRB‐401) sa CRS vyskytol u 81,0 % pacientov liečených Abecmou. 3. alebo vyšší stupeň CRS (Lee a kol., 2014) sa vyskytol u 5,4 % pacientov s fatálnym (5. stupňom) CRS hláseným u 0,5 % pacientov. Medián času do nástupu pri akomkoľvek stupni bol 1 deň (rozsah: 1 až 17) a medián trvania CRS bol 5 dní (rozsah: 1 až 63).
K najčastejším prejavom CRS patrili pyrexia (78,3 %), hypotenzia (32,1 %), tachykardia (25,5 %), zimnica (23,4 %), hypoxia (16,3 %), zvýšená hladina C-reaktívneho proteínu (16,3 %), bolesť hlavy (14,7 %) a únava (10,9 %). K udalostiam 3. alebo vyššieho stupňa, ktoré možno pozorovať v súvislosti
s CRS, patrili fibrilácia predsiene, syndróm kapilárneho úniku, hypotenzia, hypoxia a HLH/MAS.
Zo 184 pacientov dostalo 45,1 % pacientov tocilizumab, pričom 32,6 % dostalo jednu dávku a 12,5 % dostalo viac ako 1 dávku tocilizumabu na liečbu CRS. Celkovo v rámci hladín cieľovej dávky 15,8 % pacientov dostalo minimálne 1 dávku kortikosteroidov na liečbu CRS. Z 92 pacientov pri cieľovej
dávke 450 x 106 CAR-pozitívnych T-buniek dostalo 54,3 % pacientov tocilizumab a 22,8 % dostalo minimálne 1 dávku kortikosteroidov na liečbu CRS. Pokyny týkajúce sa monitorovania a liečby, pozri časť 4.4.
Neurologické nežiaduce reakciePodľa súhrnu zo štúdií u 184 pacientov, nezávisle od prisúdenia neurotoxicity skúšajúcim, k najčastejším neurologickým alebo psychiatrickým nežiaducim reakciám patrili bolesť hlavy (28,8 %), závrat (15,2 %), stav zmätenosti (13,0 %), nespavosť (9,8 %), anxieta (8,2 %), tras (8,2 %) a somnolencia (6,5 %). K iným neurologickým nežiaducim reakciám vyskytujúcim sa s nižšou frekvenciou a považovaným za klinicky významné patrili afázia (4,3 %) a encefalopatia (4,3 %).
Neurotoxicita zistená skúšajúcimi, ktorá bola primárnou metódou hodnotenia neurotoxicity súvisiacej
s CAR T-bunkami iba v štúdii KarMMa, sa vyskytla u 18,0 % zo 128 pacientov liečených Abecmou
vrátane 3. stupňa u 3,1 % pacientov (bez udalostí 4. alebo 5. stupňa). Medián času do nástupu prvej udalosti bol 2 dni (rozsah: 1 až 10). Medián trvania bol 3 dni (rozsah: 1 až 26). Celkovo 7,8 % pacientov dostalo minimálne 1 dávku kortikosteroidu na liečbu neurotoxicity súvisiacej s CAR
T-bunkami, zatiaľ čo pri cieľovej dávke 450 x 106 CAR-pozitívnych T-buniek dostalo 14,8 % pacientovminimálne 1 dávku kortikosteroidov. K najčastejším prejavom neurotoxicity zisteným skúšajúcim patrili stav zmätenosti (9,4 %), encefalopatia (5,5 %), afázia (4,7 %), halucinácia (3,1 %) a zmeny duševného stavu (3,1 %). Pokyny týkajúce sa monitorovania a liečby, pozri časť 4.4.
Febrilná neutropénia a infekciePodľa súhrnu zo štúdií sa infekcie vyskytli u 71,2 % pacientov. Infekcie 3. alebo 4. stupňa sa vyskytli u 23,4 % pacientov. Infekcie 3. alebo 4. stupňa s nešpecifikovaným patogénom sa vyskytli u 17,9 %, vírusové infekcie u 7,1 %, bakteriálne infekcie u 3,8 % a plesňové infekcie u 0,5 % pacientov. Fatálne infekcie s nešpecifikovaným patogénom sa hlásili u 1,6 % pacientov a 0,5 % pacientov malo fatálnu plesňovú alebo vírusovú infekciu. Pokyny týkajúce sa monitorovania a liečby, pozri časť 4.4.
Febrilná neutropénia (3. alebo 4. stupňa) sa pozorovala u 14,7 % pacientov po infúznom podaní Abecmy. Febrilná neutropénia môže byť súbežná so CRS. Pokyny týkajúce sa monitorovania a liečby, pozri časť 4.4.
Dlhodobá cytopéniaU pacientov sa môžu prejavovať dlhodobé cytopénie po lymfodeplečnej chemoterapii a infúznom podaní Abecmy. Podľa súhrnu zo štúdií u 34,8 % zo 178 pacientov, ktorí mali neutropéniu 3.
alebo 4. stupňa, a u 72,7 % zo 110 pacientov, ktorí mali trombocytopéniu 3. alebo 4. stupňa počas
prvého mesiaca po infúznom podaní Abecmy, tieto reakcie neustúpili do posledného hodnotenia počas
prvého mesiaca. U 62 pacientov s neutropéniou, ktorá neustúpila do 1. mesiaca, sa 82,3 % zotavilo z neutropénie 3. alebo 4. stupňa s mediánom času do zotavenia od infúzneho podania Abecmy
1,9 mesiacov. U 80 pacientov s trombocytopéniou, ktorá neustúpila do 1. mesiaca, sa 71,3 % zotavilo
z trombocytopénie 3. alebo 4. stupňa s mediánom času do zotavenie 2,2 mesiacov. Pokyny týkajúce sa monitorovania a liečby, pozri časť 4.4.
HypogamaglobulinémiaPodľa súhrnu zo štúdií sa hypogamaglobulinémia hlásila u 19,6 % pacientov liečených Abecmou
s mediánom času do nástupu 100 dní (rozmedzie 15 až 326). Pokyny týkajúce sa monitorovania a
liečby, pozri časť 4.4.
ImunogenicitaAbecma má potenciál indukovať tvorbu protilátok proti CAR. V klinických štúdiách sa humorálna
imunogenicita Abecmy merala stanovením protilátok proti CAR v sére pred podaním a po podaní.
Podľa súhrnu zo štúdií 4,3 % pacientov malo pozitívny výsledok testu na protilátky proti CAR pred
podaním infúzie a protilátky proti CAR po podaní infúzie boli zistené u 50,5 % pacientov. Nie je k dispozícii žiaden dôkaz, že prítomnosť existujúcich protilátok proti CAR alebo protilátok po podaní infúzie ovplyvňuje bunkovú expanziu, bezpečnosť alebo účinnosť Abecmy.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieK dispozícii sú obmedzené údaje o predávkovaní Abecmou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: zatiaľ nepridelená, ATC kód: zatiaľ nepridelený
Mechanizmus účinku
Abecma je liečba T-bunkami pozitívnymi na chimérický antigénový receptor (CAR) zacielenými proti
antigénu zrenia B-buniek (BCMA, B-cell maturation antigen), ktorý sa exprimuje na povrchu
normálnych a malígnych plazmatických buniek. CAR je zostavený z scFv anti-BCMA domény pre špecifickosť antigénu, transmembránovej domény, CD3-zeta T-bunkovej aktivačnej domény a kostimulačnej domény 4-1BB. Aktivácia Abecmy špecifickým antigénom vedie k proliferácii
CAR-pozitívnych T-buniek, k sekrécii cytokínov a k následnému cytolytickému usmrcovaniu buniek
exprimujúcich BCMA.
Klinická účinnosť a bezpečnosť
KarMMa bola nezaslepená, multicentrická klinická štúdia s jednou skupinou, ktorá hodnotila účinnosť
a bezpečnosť Abecmy u dospelých pacientov s recidivujúcim a refraktérnym mnohopočetným
myelómom, ktorí dostali minimálne 3 predchádzajúce liečby proti myelómu vrátane
imunomodulátora, inhibítora proteazómu a protilátky proti CD38, a ktorí boli refraktérni na posledný
liečebný režim. Pacienti s myelómom postihnutím CNS, s anamnézou iných terapií zameraných proti
BCMA, alogénnej SCT alebo s predchádzajúcou génovou terapiou alebo s inou geneticky modifikovanou T-bunkovou terapiou boli vylúčení. Pacienti s ochoreniami CNS v anamnéze (ako sú záchvaty), s nedostatočnou funkciou pečene, obličiek, kostnej drene, srdca, pľúc alebo s prebiehajúcou liečbou imunosupresívami boli vylúčení.
Štúdia pozostávala z predbežnej liečby (skríning, leukaferéza a z preklenovacej liečby [v prípade potreby]); z liečby (lymfodeplečná chemoterapia a infúzne podanie Abecmy); a z obdobia po liečbe
(prebiehajúceho) trvajúceho minimálne 24 mesiacov po infúznom podaní Abecmy alebo do zdokumentovanej progresie ochorenia, podľa toho, čo trvalo dlhšie. Obdobie lymfodeplečnej chemoterapie predstavovalo jeden 3-dňový cyklus podávania cyklofosfamidu (300 mg/m2 vo forme i.v. infúzie denne počas 3 dní) a fludarabínu (30 mg/m2 vo forme i.v. infúzie denne počas 3 dní) ktorý začínal 5 dní pred cieľovým dátum infúzneho podania Abecmy. Pacienti boli hospitalizovaní počas
14 dní po infúznom podaní Abecmy s cieľom monitorovať a liečiť potenciálny CRS a neurotoxicitu.
Zo 140 zaradených pacientov (t.j. ktorí podstúpili leukaferézu) dostalo 128 pacientov infúziu Abecmy. Iba jednému zo 140 pacientov nebol liek podaný z dôvodu neúspešnej výroby. Jedenásť ďalších pacientov nebolo liečených Abecmou z dôvodu rozhodnutia lekára (n = 3), vylúčenia pacienta (n = 4), nežiaducich udalostí (n = 1), progresie ochorenia (n = 1) alebo úmrtia (n = 2) pred podaním Abecmy.
Medzi aferézou a lymfodepléciou bola povolená protinádorová liečba na kontrolu ochorenia (preklenutie), posledná dávka sa podala minimálne 14 dní pred začatím lymfodeplečnej chemoterapie. Zo 128 pacientov liečených Abecmou väčšina pacientov (87,5 %) dostala protinádorovú liečbu na kontrolu ochorenia podľa uváženia skúšajúceho.
Cieľovými dávkami v klinickej štúdii bolo 150, 300 alebo 450 x 106 CAR-pozitívnych T-buniek
v podanej infúzii. Povolený rozsah dávok bol 150 až 540 x 106 CAR-pozitívnych T-buniek. Nižšie
v tabuľke 4 sú uvedené cieľové hladiny dávky použité v klinickej štúdii na základe celkových
CAR-pozitívnych T-buniek a zodpovedajúceho rozmedzia aktuálne podanej dávky definované ako
životaschopné CAR-pozitívne T-bunky.
Tabuľka 4. Celková dávka CAR-pozitívnych T-buniek so zodpovedajúcim rozsahom dávky
životaschopných CAR-pozitívnych T-buniek (x10
6
)
Cieľová dávka podľa celkových CAR-pozitívnych T-buniek, vrátane životaschopných a neživotaschopných buniek (x10
6
)
|
Životaschopné CAR pozitívne T-bunky
(x10
6
) (min,
max)
|
150
|
133 až 181
|
300
|
254 až 299
|
450
|
307 až 485
|
V tabuľke 5 je uvedený súhrn východiskových charakteristík pacientov a ochorenia v zaradenej a
liečenej populácii v štúdii.
Tabuľka 5. Východiskové demografické údaje/charakteristiky ochorenia v štúdiovej populácii
Charakteristika
| Zaradených celkovo
(N = 140)
| Liečených celkovo
(N = 128)
|
Vek (roky)
|
|
|
Medián (min., max.)
| 60,5 (33; 78)
| 60,5 (33; 78)
|
≥ 65 rokov, n (%)
| 48 (34,3)
| 45 (35,2)
|
≥ 75 rokov, n (%)
| 5 (3,6)
| 4 (3,1)
|
Pohlavie, mužské, n (%)
| 82 (58,6)
| 76 (59,4)
|
Rasa, n (%)
|
|
|
Ázijská
| 3 (2,1)
| 3 (2,3)
|
Černošská
| 8 (5,7)
| 6 (4,7)
|
Belošská
| 113 (80,7)
| 103 (80,5)
|
Stav výkonnosti podľa ECOG, n (%)
|
|
|
0
| 60 (42,9)
| 57 (44,5)
|
1
| 77 (55,0)
| 68 (53,1)
|
2a
| 3 (2,1)
| 3 (2,3)
|
Pacienti s extramedulárnym plazmocytómom, n (%)
|
52 (37,1)
|
50 (39,1)
|
Čas, ktorý uplynul od začiatku diagnózy (roky), medián (min., max.)
|
6 (1,0; 17,9)
|
6 (1,0; 17,9)
|
Transplantácia kmeňových buniek v
minulosti, n (%)
|
131 (93,6)
|
120 (93,8)
|
Východiskové vysoké cytogenetické rizikob,c
|
46 (32,9)
|
45 (35,2)
|
Východiskové revidované ISS
štádium (derivované)d, n (%)
|
|
|
I. štádium
| 14 (10,0)
| 14 (10,9)
|
II. štádium
| 97 (69,3)
| 90 (70,3)
|
III. štádium
| 26 (18,6)
| 21 (16,4)
|
Nie je známe
| 3 (2,1)
| 3 (2,3)
|
Počet predchádzajúcich terapií proti myelómue, medián (min., max.)
|
6 (3;17)
|
6 (3;16)
|
Trojito refraktérnif, n (%)
| 117 (83,6)
| 108 (84,4)
|
Charakteristika
|
Zaradených celkovo
(N = 140)
|
Liečených celkovo
(N = 128)
|
Klírens kreatinínu (ml/min), n (%)
|
|
|
< 30
|
3 (2,1)'
|
1 (0,8)
|
30 až < 45
|
9 (6,4)
|
8 (6,3)
|
45 až < 60
|
13 (9,3)
|
10 (7,8)
|
60 až < 80
|
38 (27,1)
|
36 (28,1)
|
≥ 80
|
77 (55,0)
|
73 (57,0)
|
max. = maximum; min. = minimum
a Títo pacienti mali pri skríningu skóre podľa ECOG < 2, aby splnili podmienky, ale následne sa im zhoršilo na skóre podľa
ECOG ≥ 2 vo východiskovom stave pred začatím LD chemoterapie.
b Východisková cytogenetická abnormalita vychádzala z východiskovej cytogenetiky z centrálneho laboratória, ak bola k dispozícii. Ak centrálne laboratórium nebolo k dispozícii alebo ak nebolo známe, použila sa cytogenetika pred skríningom.
c Vysoké riziko definované ako delécia v chromozóme 17p (del[17p]), translokácia zahŕňajúca chromozómy 4 a 14 (t[4;14])
alebo translokácia zahŕňajúca chromozómy 14 a 16 (t[14;16]).
d Revidované štádium podľa ISS bolo odvodené od východiskového štádia podľa ISS, cytogénnej abnormality a sérovej
laktátdehydrogenázy.
e Indukcia s transplantáciou hematopoetických kmeňových buniek alebo bez nej a s udržiavacou liečbou alebo bez nej bola považovaná za jedinú liečbu.
f Trojito refraktérni sú definovaní ako refraktérni proti imunomodulátoru, inhibítoru proteazómu a protilátke proti CD38.
Medián času od leukaferézy po dostupnosť lieku bol 32 dní (rozsah: 24 až 55 dní) a medián času od leukaferézy po podanie infúzie bol 40 dní (rozsah: 33 až 79 dní). Medián skutočnej dávky podanej v prípade všetkých cieľových dávok v klinickej štúdii bol 315,3 x 106 CAR-pozitívnych T-buniek (rozsah 150,5 až 518,4).
Účinnosť sa hodnotila na základe celkovej miery odpovede (ORR, overall response rate), miery kompletnej odpovede (CR, complete response) a dĺžky trvania odpovede (DOR, duration of response) na základe stanovenia nezávislým kontrolným výborom. Ďalšie koncové ukazovatele účinnosti zahŕňali minimálne reziduálne ochorenia (MRD, minimal residual disease) použitím sekvenovania
novej generácie (NGS, next-generation sequencing).
Výsledky účinnosti v prípade cieľových dávok v klinickej štúdii (150 až 450 x 106 CAR-pozitívnych
T-buniek) sú uvedené v tabuľke 6. Medián následného sledovania bol u všetkých pacientov liečených
Abecmou 15,4 mesiacov.
Tabuľka 6. Súhrn účinnosti v klinickej štúdii KarMMa
|
Zaradení
a
(N = 140)
|
Liečená populácia
Cieľová dávka Abecmy (CAR-pozitívne T-bunky)
|
15
0 x 10
6b
(N = 4)
|
30
0 x 10
6
(N = 70)
|
45
0 x 10
6
(N = 54)
|
Celkovo
15
0 až 450 x 10
6
(N = 128)
|
Celková miera odpovede
(sCR+CR+VGPR+PR), n (%)
|
9
4 (67,1)
|
2 (50,0)
|
4
8 (68,6)
|
4
4 (81,5)
|
9
4 (73,4)
|
95 % CIc
|
59,4; 74,9
|
6,8; 93,2
|
56,4; 79,1
|
68,6; 90,7
|
65,8; 81,1
|
CR alebo lepšie, n (%)
|
4
2 (30,0)
|
1 (25,0)
|
2
0 (28,6)
|
2
1 (38,9)
|
4
2 (32,8)
|
95 % CIc
|
22,4; 37,6
|
0,6; 80,6
|
18,4; 40,6
|
25,9; 53,1
|
24,7; 40,9
|
VGPR alebo lepšie, n(%)
|
6
8 (48,6)
|
2 (50,0)
|
3
1 (44,3)
|
3
5 (64,8)
|
6
8 (53,1)
|
95 % CIc
|
40,3; 56,9
|
6,8; 93,2
|
32,4; 56,7
|
50,6; 77,3
|
44,5; 61,8
|
MRD-negatívny stav
d
a ³ CR
|
|
|
|
|
|
Na základe liečených
pacientov
|
–
|
4
|
70
|
54
|
128
|
n (%)
|
–
|
1 (25,0)
|
17 (24,3)
|
15 (27,8)
|
33 (25,8)
|
95% CIb
|
–
|
0,6; 80,6
|
14,8; 36,0
|
16,5; 41,6
|
18,5; 34,3
|
Čas do odpovede, n
|
94
|
2
|
48
|
44
|
94
|
Medián (mesiace)
|
1,0
|
1,0
|
1,0
|
1,0
|
1,0
|
Min., max.
|
0,5; 8,8
|
1,0; 1,0
|
0,5; 8,8
|
0,9; 2,0
|
0,5; 8,8
|
Dĺžka trvania odpovede
(PR alebo lepšie)
e
, n
|
94
|
2
|
48
|
44
|
94
|
Medián (mesiace)
|
10,6
|
13,0
|
8,5
|
11,3
|
10,6
|
95 % CI
|
8,0; 11,4
|
2,8; 23,3
|
5,4; 10,9
|
10,3, NE
|
8,0; 11,4
|
CAR = chimérický antigénový receptor; CI = interval spoľahlivosti; CR = complete response = kompletná odpoveď; MRD = minimal residual disease = minimálne reziduálne ochorenie; NE = nevypočítateľné; PR = partial response = parciálne odpoveď; sCR = stringent complete response = striktná kompletná odpoveď; VGPR = very good partial response = veľmi dobrá parciálne odpoveď.
a Všetci pacienti, ktorí podstúpili leukaferézu.
b Dávka 150 x 106 CAR-pozitívnych T-buniek nepatrí do schváleného rozmedzia dávky.
c Pri „Celkovo ( liečenej populácii“ a „zaradenej populácii“): Waldov CI; pri jednotlivých cieľových dávkach:
Clopperov-Pearsonov presný CI.
d Na základe prahovej hodnoty 10-5 pomocou analýzy sekvenovania novej generácie. 95 % CI pre percento negativity MRD
používa Clopperov-Pearsonov presný CI pre jednotlivé cieľové dávky, a rovnako pre liečenú populáciu.
e Medián a 95 % CI sú založené na Kaplan-Meierovom prístupe.
Poznámka: Cieľová dávka je 450 x 106 CAR-pozitívnych T-buniek v rozsahu 150 až 540 × 106 CAR-pozitívnych T-buniek.
Dávka 150 x 106 CAR-pozitívnych T-buniek nepatrí do schváleného rozmedzia dávky.
Kaplan-Meierova krivka trvania odpovede podľa najlepšej celkovej odpovede je zobrazená na
Obrázku 1.
Obrázok 1. Kaplan-Meierova krivka trvania odpovede na základe posúdenia odpovede nezávislým kontrolným výborom podľa kritérií IMWG – podľa najlepšej celkovej odpovede (populácia liečená Abecmou)
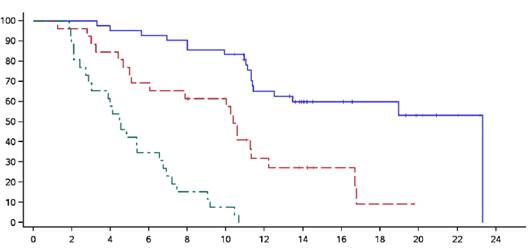
Čas, (mesiace)
CR alebo
lepšie
| 42
| 42
| 40
| 39
| 36
| 35
| 25
| 19
| 12
| 9
| 5
| 3
| 0
|
VGPR
| 26
| 25
| 22
| 18
| 16
| 15
| 7
| 5
| 3
| 1
| 0
| 0
|
|
PR
| 26
| 23
| 15
| 9
| 4
| 2
| 0
| 0
| 0
| 0
| 0
| 0
|
|
CR alebo lepšie: jedinec: 42; udalosti: 18; medián: 23,29 (95 % CI: 11,43; 23,29)
VGPR: jedinec: 26; udalosti: 20; medián: 10,38 (95 % CI: 5,09; 12,22)
PR: jedinec: 26; udalosti: 26; medián: 4,50 (95 % CI: 2,86; 6,54)
CI = interval spoľahlivosti; IMWG = medzinárodné zoskupenie International Myeloma Working Group; NE =
nevypočítateľné. Dvaja pacienti s dávkou 150 x 106 CAR-pozitívnych T-buniek, ktorá nepatrí do schváleného rozmedzia
dávok, sú tiež zahrnutí v Obrázku 1.
Špeciálne populácieStarší pacientiV klinickom skúšaní s Abecmou bolo 48 (34,3 %) pacientov v štúdii KarMMa vo veku od 65 rokov alebo starší a 5 (3,6 %) bolo vo veku od 75 rokov alebo starší (pozri tabuľku 5). Medzi týmito pacientmi a pacientmi mladšími ako 65 rokov sa nepozorovali žiadne klinicky významné rozdiely v bezpečnosti alebo účinnosti Abecmy.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Abecmou vo všetkých podskupinách pediatrickej populácie pri liečbe neoplaziem zrelých B-buniek (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku.
Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn
charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Po infúznom podaní Abecmy sa CAR-pozitívne T-bunky proliferujú a podstupujú rýchlu
multi-logarimickú expanziu, po ktorej nasleduje pomalší biexponenciálny pokles. Medián času
maximálnej expanzie v periférnej krvi (Tmax) nastal 11 dní po podaní infúzie.
Abecma môže pretrvávať v periférnej krvi až do 1 roka po podaní infúzie.
Hladiny transgénov Abecmy sa pozitívne spájali s objektívnou odpoveďou nádoru (parciálne odpoveď alebo lepšia). Medián hodnôt Cmax u pacientov odpovedajúcich na liečbu (N = 93) bol
približne 4,5-násobne vyšší v porovnaní s príslušnými hodnotami u pacientov neodpovedajúcich na liečbu (N = 34). Medián hodnôt AUC0-28 dní u pacientov odpovedajúcich na liečbu (N = 93) bol približne 5,5-násobne vyšší v porovnaní s hodnotami u pacientov neodpovedajúcich na liečbu
(N = 32).
Špeciálne populácie
Porucha funkcie obličiek a pečene
Neboli vykonané štúdie Abecmy zamerané na poruchy funkcie pečene a obličiek.
Vplyvy veku, telesnej hmotnosti, pohlavia alebo rasy
Vek (rozsah: 33 až 78 rokov) nemal žiadny vplyv na parametre expanzie Abecmy.Farmakokinetika
Abecmy u pacientov mladších ako 18 rokov nebola hodnotená.
Pacienti s nižšou telesnou hmotnosťou mali vyššiu expanziu buniek. Vzhľadom na vysokú variabilitu
vo farmakokinetickej expanzii buniek sa celkový vplyv telesnej hmotnosti na parametre expanzie
Abecmy nepovažuje za klinicky významný.
Pohlavie nemalo žiadny vplyv na parametre expanzie Abecmy.
Rasa a etnická príslušnosť nemali žiadny významný vplyv na parametre expanzie Abecmy.
5.3 Predklinické údaje o bezpečnosti
Abecma obsahuje upravené ľudské T-bunky, preto neexistujú žiadne reprezentatívne in vitro analýzy, ex vivo modely ani in vivo modely, ktoré by mohli presne vyjadrovať toxikologické charakteristiky lieku u ľudí. Z tohto dôvodu neboli vykonané typické toxikologické štúdie vykonávané pri vývoji
liekov.
Neboli vykonané analýzy genotoxicity a štúdie zamerané na karcinogenitu.
Štúdie in vitro zamerané na expanziu u zdravých darcov a pacientov nepreukázali žiadne dôkazy transformácie a/alebo imortalizácie ani žiadnu preferenčnú integráciu problémových blízkych génov
T-buniek Abecmy.
Vzhľadom na charakter tohto lieku sa nevykonali predklinické štúdie zamerané na fertilitu,
reprodukciu a vývin.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
CryoStor CS10 chlorid sodný glukónát sodný
octan sodný, trihydrát chlorid draselný
chlorid horečnatý
voda na injekcie
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Abecma je stabilná 1 rok, ak sa uchováva v plynnej fáze tekutého dusíka (≤ -130 °C).
Každý vak sa musí infúzne podať do 1 hodiny od začatia rozmrazovania. Po rozmrazení sa objem lieku určený na infúzne podanie musí uchovávať pri izbovej teplote (20 – 25 °C).
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte a prepravujte v mraze v plynnej fáze tekutého dusíka (≤ –130 °C) a musí sa ponechať zmrazené, až kým je pacient pripravený na liečbu, čím sa zabezpečí, že na podanie pacientovi sú
k dispozícii životaschopné autológne bunky. Po rozmrazení sa liek nesmie znova zmrazovať.
Podmienky na uchovávanie po rozmrazení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Kryoprezervačný vak (vaky) z etylénvinylacetátu s uzavretou prídavnou hadičkou obsahujúci
10 – 30 ml (50 ml vak), 30 – 70 ml (250 ml vak) alebo 55 – 100 ml (500 ml vak) bunkovej disperzie.
Každý kryoprezervačný vak je samostatne balený v kovovej kazete.
Jedna individuálna terapeutická dávka sa skladá z jedného alebo viacerých infúznych vakov s
rovnakou veľkosťou a objemom náplne.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Opatrenia pred zaobchádzaním alebo podaním lieku
Abecma sa má v rámci liečebného centra prepravovať v uzavretých, nerozbitných a nepriepustných
nádobách.
Tento liek obsahuje geneticky modifikované ľudské krvinky. Zdravotnícki pracovníci musia pri zaobchádzaní s Abecmou dodržiavať príslušné opatrenia (použitie rukavíc a okuliarov), aby sa zabránilo prípadnému prenosu infekčných ochorení.
Príprava pred podávaním
Pred infúznym podaním Abecmy sa musí potvrdiť, či sa totožnosť pacienta zhoduje s identifikačnými
údajmi pacienta uvedenými na kazete (kazetách), na infúznom vaku (infúznych vakoch) a na certifikáte RfIC Abecmy. Infúzny vak Abecmy sa nesmie vyberať z kazety, ak sa špecifické informácie pacienta uvedené na štítku nezhodujú s daným pacientom. V prípade akýchkoľvek nezrovnalostí medzi štítkami a identifikačnými údajmi pacienta sa musí okamžite kontaktovať príslušná spoločnosť.
Ak ste na liečbu dostali viac ako jeden infúzny vak, rozmrazujte ich po jednom. Načasovanie rozmrazenia Abecmy a podania infúzie sa majú skoordinovať. Čas začatia podávania infúzie sa má potvrdiť vopred a upraviť podľa neho rozmrazenie, aby bola Abecma k dispozícii na infúzne podávanie, keď bude pacient pripravený.
Rozmrazovanie
• Pred rozmrazením vyberte infúzny vak Abecmy z kazety a skontrolujte, či infúzny vak nemá
nejaké porušenia obalu, ako sú trhliny alebo praskliny. Ak sa zdá, že je infúzny vak poškodený
alebo nie je tesný, infúzia sa nemá podať a vak sa má zlikvidovať v súlade s miestnymi
predpismi na zaobchádzanie s odpadovým materiálom z ľudských tkanív.
• Infúzny vak vložte do druhého sterilného vaku.
• Abecmu rozmrazujte približne pri teplote 37 °C v schválenom rozmrazovacom zariadení alebo
vo vodnom kúpeli tak, aby v infúznom vaku nebol žiaden viditeľný ľad. Jemne premiešajte
obsah vaku, aby sa rozptýlili zhluky bunkového materiálu. Ak zostanú viditeľné zhluky buniek,
pokračujte v jemnom premiešavaní obsahu vaku. Malé zhluky bunkového materiálu sa musia
rozptýliť jemným manuálnym premiešaním. Abecmu pred infúznym podávaním nepremývajte,
neodstreďujte a/ani neresuspendujte v nových médiách.
Podávanie Abecmy
• Pred podaním infúzie prepláchnite hadičky infúznej súpravy 9 mg/ml (0,9 %) injekčným
roztokom chloridu sodného.
• Abecmu podajte do 1 hodiny od začatia rozmrazovania rýchlosťou, ktorá umožňuje prietok
samospádom.
• Po infúznom podaní celého obsahu infúzneho vaku prepláchnite hadičky 9 mg/ml (0,9 %)
injekčným roztokom chloridu sodného s rovnakou rýchlosťou infúzie, aby sa zabezpečilo
podanie celého lieku.
• Pri všetkých nasledujúcich infúznych vakoch pre daného pacienta postupujte rovnako.
Opatrenia vzťahujúce sa na likvidáciu lieku
S nepoužitým liekom a s každým materiálom, ktorý bol v kontakte s Abecmou (pevný a kvapalný
odpad), sa má zaobchádzať ako s potenciálne infekčným odpadom a má sa zlikvidovať v súlade
s miestnymi predpismi na zaobchádzanie s odpadovým materiálom z ľudských tkanív.
Náhodná expozícia
V prípade náhodnej expozície Abecmy sa majú dodržiavať miestne predpisy na zaobchádzanie
s materiálom z ľudských tkanív. Pracovné povrchy a materiály, ktoré mohli byť potenciálne v kontakte
s Abecmou, sa musia dekontaminovať vhodným dezinfekčným prostriedkom.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Celgene Europe B.V. Winthontlaan 6 N
3526 KV Utrecht
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/21/1539/001
9
. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUMM/RRRR
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.