
ok užívajúcich lieky, ktoré môžu ovplyvniť BMD (napr. systémové kortikosteroidy, antikonvulzíva). U týchto rizikových pacientok sa odporúča pred začatím liečby liekom Yselty vykonať vyšetrenie pomocou snímok DXA.
Vyšetrenie DXA sa po jednom roku liečby ďalej odporúča u všetkých žien, aby sa overilo, že pacientka nemá nežiaduci stupeň zníženia BMD. Následne sa v závislosti od predpísanej dávky lieku Yselty každý rok odporúča vykonať hodnotenie BMD (Yselty 100 mg) alebo s frekvenciou, ktorú určí ošetrujúci lekár na základe individuálneho rizika ženy a predchádzajúceho hodnotenia BMD (Yselty
100 mg so súbežnou ABT a Yselty 200 mg so súbežnou ABT).
Ak riziká zníženia BMD presiahnu potenciálny prínos liečby liekom Yselty, liečba sa má ukončiť. Poruchafunkciepečene
Liek Yselty sa nemá používať u žien so závažnou poruchou funkcie pečene (trieda C podľa Childa-
Pugha). U žien s miernou alebo stredne závažnou poruchou funkcie pečene (trieda A alebo B podľa
Childa-Pugha) nie je potrebná úprava dávky, pozri časti 4.2 a 5.2.
Porucha funkcie obličiek
Liek Yselty sa nemá používať u žien so stredne závažnou (eGFR = 30 – 59 ml/min), závažnou
poruchou funkcie obličiek (eGFR < 30 ml/min) alebo s ochorením obličiek v konečnom štádiu (pozri časť 4.2). Predpisujúcim lekárom sa odporúča sledovať výskyt nežiaducich reakcií u žien s miernou poruchou funkcie obličiek (eGFR = 60 - 89 ml/min; pozri časť 5.2), aj keď úprava dávky nie je potrebná (pozri časť 4.2).
Kardiovaskulárne poruchy/predĺženie intervalu QT
Linzagolix len nepatrne predlžuje interval QT, nevyskytol sa však žiadny dôkaz klinicky významného
rizika predĺženia intervalu QT ani torsade de pointes (pozri časť 5.1). Má sa postupovať opatrne u pacientok so známym kardiovaskulárnym ochorením, s predĺžením QT alebo hypokaliémiou v
rodinnej anamnéze a pri súčasnom užívaní liekov, o ktorých je známe, že predlžujú QT interval. Má sa postupovať opatrne aj u pacientok so súčasne existujúcimi poruchami, ktoré vedú k zvýšeným plazmatickým hladinám linzagolixu (pozri časť 5.2).
Antikoncepcia
Nepreukázalo sa, že by linzagolix so súbežnou ABT alebo bez nej poskytoval antikoncepciu. Ženy vo
fertilnom veku, u ktorých existuje riziko otehotnenia, musia počas liečby liekom Yselty používať účinnú nehormonálnu antikoncepciu (pozri časť 4.6).
Zmena vzorca menštruačného krvácania a zníženáschopnosťrozpoznaniatehotenstva
Ženy majú byť poučené o tom, že liečba liekom Yselty zvyčajne vedie k významnému zníženiu straty
menštruačnej krvi a často vedie k amenorei, čo môže znížiť schopnosť včas rozpoznať vznik tehotenstva. V prípade podozrenia na tehotenstvo sa má vykonať tehotenský test a liečba sa má prerušiť, ak je gravidita potvrdená (pozri časti 4.3 a 4.6).
Pečeňové enzýmy
Bolo hlásené asymptomatické prechodné zvýšenie hladín pečeňových enzýmov (pozri časť 4.8).
Pacientky majú byť poučené o tom, aby v prípade príznakov alebo prejavov, ktoré môžu signalizovať poškodenie pečene, ako je žltačka, okamžite vyhľadali lekársku pomoc. Ak sa objaví žltačka, liečba sa má prerušiť. Akútne abnormality pečeňových testov si môžu vyžadovať, aby sa liečba linzagolixom prerušila, kým sa výsledky pečeňových testov nevrátia do normálu.
Ženy s abnormálnymi parametrami funkcie pečene (≥ 2 x horná hranica normálu, upper limit of normal ULN) boli zo štúdií s linzagolixom vylúčené. U žien so známou abnormálnou anamnézou pečene sa má preto určiť východisková úroveň testov funkcie pečene a má sa vykonávať ďalšie pravidelné monitorovanie. Pri liečbe týchto pacientok sa má postupovať opatrne.
Hladiny lipidov
Pri liečbe linzagolixom sa pozorovalo zvýšenie hladín lipidov (pozri časť 5.1). Toto zvýšenie nebolo
vo všeobecnosti klinicky významné. Avšak u žien s už existujúcimi zvýšenými lipidovými profilmi sa odporúča monitorovanie hladín lipidov.
Poruchy nálady
Pri liečbe antagonistami GnRH vrátane linzagolixu boli pozorované poruchy nálady vrátane depresie,
zmien nálady a emocionálnej lability (pozri časť 4.8). U žien s depresiou a/alebo samovražednými myšlienkami v anamnéze sa má postupovať opatrne. Pacientky so známou depresiou alebo depresiou v anamnéze majú byť počas liečby dôkladne sledované. Ak depresia znova dosiahne závažný stupeň, liečba sa má prerušiť.
Substráty CYP2C8
Liek Yselty sa nemá používať u pacientok užívajúcich lieky so substrátom citlivým na CYP2C8 s
úzkym terapeutickým indexom (napr. paklitaxel, sorafenib a repaglinid, pozri časť 4.5). Odporúča sa sledovať, či pri súbežnom podávaní s liekom Yselty nedochádza k zvýšeniu nežiaducich reakcií spojených s inými substrátmi CYP2C8.
Upozornenia a opatreniatýkajúcesaABT
Ak sa súbežne predpisuje ABT, majú sa zvážiť všetky upozornenia a opatrenia týkajúce sa ABT.
Laktóza
Pacientky so zriedkavými dedičnými problémami galaktózovej intolerancie, celkovým deficitom
laktázy alebo glukózo-galaktózovou malabsorpciou nemajú tento liek užívať.
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej tablete, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Lieky, ktoré sú substrátmi CYP2C8
Preukázalo sa, že linzagolix u zdravých subjektov zvyšuje priemernú expozíciu repaglinidu (substrát
citlivý na CYP2C8) menej ako dvojnásobne. Vzhľadom na riziko zvýšených plazmatických koncentrácií sa má predchádzať súbežnému podávaniu lieku Yselty a liekov, ktoré sa primárne vylučujú metabolizmom CYP2C8 a majú úzky terapeutický index, ako napr. paklitaxel, sorafenib a repaglinid (pozri časť 4.4). Predpisujúcim lekárom sa odporúča sledovať, či sa pri súbežnom podávaní s liekom Yselty nezvýšia nežiaduce reakcie spojené s inými substrátmi CYP2C8.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Nepreukázalo sa, že by linzagolix s ABT alebo bez nej poskytoval antikoncepciu. Ženy vo fertilnom
veku, u ktorých existuje riziko otehotnenia, musia počas liečby liekom Yselty používať účinnú nehormonálnu antikoncepciu.
Gravidita
O používaní linzagolixu u gravidných žien nie sú k dispozícii žiadne údaje alebo je k dispozícii iba
obmedzené množstvo údajov.
Štúdie na zvieratách preukázali, že expozícia linzagolixu na začiatku gravidity môže zvýšiť riziko skorého zániku gravidity (pozri časť 5.3). Na základe farmakologických účinkov nemožno nežiaduce účinky na graviditu vylúčiť.
Podávanie lieku Yselty počas gravidity je kontraindikované (pozri časť 4.3). Ak sa gravidita potvrdí, liečba sa má prerušiť.
Dojčenie
Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie linzagolixu do
mlieka (podrobnosti pozri v časti 5.3).
Nie je známe, či sa linzagolix/metabolity vylučujú do ľudského mlieka. Riziko u novorodencov a dojčiat nemožno vylúčiť.
Podávanie lieku Yselty dojčiacim matkám je kontraindikované (pozri časť 4.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Yselty nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4
.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najčastejšími nežiaducimi reakciami hlásenými v pivotných klinických štúdiách fázy 3 boli návaly
tepla a bolesti hlavy, ktorých frekvencia bola pri vyšších dávkach väčšia a menej častá bola, keď sa súbežne užívala ABT (uvádzané ako „s ABT“). Návaly tepla boli hlásené u 5,2 %; 9,6 %; 10,1 % a 31
% žien liečených dávkami 100 mg s ABT, 200 mg s ABT, 100 mg a 200 mg, v uvedenom poradí. Podobne boli bolesti hlavy hlásené častejšie pri vyšších dávkach a pri podávaní ABT boli hlásené menej často (1,4 %; 2,4 %; 4 % a 6,2 % pri 100 mg s ABT, 200 mg s ABT, 100 mg a 200 mg, v uvedenom poradí). Všetky ostatné nežiaduce reakcie uvedené ďalej boli hlásené u menej ako 3 % subjektov.
Tabuľkový zoznam nežiaducich reakcií
Nežiaduce reakcie spájané s linzagolixom sú uvedené na základe súhrnných údajov z dvoch pivotných
štúdií fázy 3 zahŕňajúcich 828 pacientok s myómami maternice, ktoré užívali linzagolix, a 209
pacientok, ktoré užívali placebo, až počas šiestich mesiacov. Nežiaduce reakcie sú uvedené v tabuľke
1.
Nežiaduce reakcie uvedené v tabuľke 1 sú klasifikované podľa kategórie frekvencie a triedy orgánových systémov podľa databázy MedDRA. V rámci jednotlivých skupín frekvencie sú nežiaduce reakcie uvedené v poradí podľa klesajúcej závažnosti. Frekvencie sú definované ako veľmi časté
(≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až
<1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
Tabuľka 1: Nežiaduce reakcie na liek z pivotných klinických štúdií
|
Linzagolix
10
0 mg
|
Linzagolix
10
0 mg s ABT
|
Linzagolix
20
0 mg
|
Linzagolix
20
0 mg s ABT
|
Psychické poruchy
|
Časté
|
a/
|
a/
znížené libido
|
a/
znížené libido
|
a/
znížené libido
|
Menej časté
|
znížené libido
|
|
|
|
Poruchy nervového systému
|
Časté
|
bolesť hlavy
|
bolesť hlavy
|
bolesť hlavy
|
bolesť hlavy
|
Poruchy ciev
|
Veľmi časté
|
návaly tepla
|
|
návaly tepla
|
|
Časté
|
|
návaly tepla
|
|
návaly tepla hypertenzia
|
Menej časté
|
hypertenzia
|
hypertenzia
|
hypertenzia
|
|
Poruchy gastrointestinálneho traktu
|
Časté
|
|
nauzea/vracanie bolesť v hornej časti
brucha
|
nauzea/vracanie zápcha
|
nauzea/vracanie
|
Menej časté
|
bolesť v hornej časti brucha
|
|
bolesť v hornej časti brucha
|
zápcha
|
Poruchy pečene a žlčových ciest
|
Časté
|
zvýšené hladiny pečeňových enzýmov
|
zvýšené hladiny pečeňových
enzýmov
|
zvýšené hladiny pečeňových enzýmov
|
zvýšené hladiny pečeňových enzýmov
|
Poruchy kože a podkožného tkaniva
|
Časté
|
hyperhidróza
|
|
hyperhidróza
|
|
|
|
poruchy nálady
* poruchy nálady *
poruchy nálady *
poruchy nálady *
|
|
|
nočné potenie
|
|
Menej časté
|
nočné potenie
|
|
|
nočné potenie
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
Časté
|
bolesť kĺbov
|
znížená hustota minerálov v kostiach*
|
bolesť kĺbov znížená hustota minerálov v kostiach*
|
bolesť kĺbov
|
Menej časté
|
hustota minerálov v kostiach*
|
|
|
hustota minerálov v kostiach*
|
Poruchy reprodukčného systému a prsníkov
|
Časté
|
vaginálne krvácanieb* bolesť v panve zmena vzorca menštruačného
krvácaniac/*
|
vaginálne krvácanieb/* bolesť v panve
|
vaginálne krvácanieb/* bolesť v panve vulvovaginálna suchosť
|
vaginálne krvácanieb/* bolesť v panve zmena vzorca menštruačného
krvácania c/*
|
Menej časté
|
vulvovaginálna suchosť
|
vulvovaginálna suchosť
zmena vzorca menštruačného krvácaniac/*
|
zmena vzorca menštruačného krvácaniac/*
|
|
Celkové poruchy a reakcie v mieste podania
|
Časté
|
asténia
|
|
|
|
Menej časté
|
|
|
asténia
|
asténia
|
ABT – estradiol 1 mg a noretisterón-acetát 0,5 mg tableta raz denne
*Ďalšie informácie sa nachádzajú v častiach 4.4 a/alebo 4.8 Opis vybraných nežiaducich reakcií.
aPoruchy nálady zahŕňajú hlásenia o zmenách nálady, afektívnej labilite, emočnej poruche, podráždenosti, zmenenej nálade, úzkosti, depresii, depresívnej nálade.
bVaginálne krvácanie zahŕňa hlásenia vaginálneho krvácania, metrorágie, menorágie, menometrorágie a krvácania z maternice.
cZmena vzorca menštruačného krvácania zahŕňa hlásenia oneskorenej menštruácie, nepravidelnej menštruácie a amenorey.
Opis vybraných nežiaducich reakciíPoruchy náladyMedzi najčastejšími nežiaducimi reakciami v rámci porúch nálady boli oznamované zmeny nálady, ktoré boli hlásené až u 1,5 % subjektov vo všetkých skupinách užívajúcich dávky linzagolixu. Afektívna labilita a úzkosť boli hlásené u 0,6 % subjektov užívajúcich linzagolix. Úzkosť bola hlásená len v skupinách s 200 mg linzagolixu s ABT alebo bez ABT. Hlásenia o depresii a depresívnej nálade boli zriedkavé. V klinických štúdiách fázy 2 alebo fázy 3 nehlásil depresiu alebo depresívnu náladu viac ako jeden subjekt v každej z liečených skupín s linzagolixom. Konkrétne odporúčania sú uvedené v časti 4.4.
Zvýšené hladiny pečeňových enzýmovBolo hlásené asymptomatické zvýšenie hladiny pečeňových enzýmov, najmä alanínaminotransferázy a aspartátaminotransferázy (ALT a AST). Väčšina zvýšení bola nízkeho stupňa a počas pokračujúcej liečby sa hodnoty zvyčajne vrátili na normálnu úroveň. Incidencia zvýšenia hladín ALT a/alebo AST v skupinách s linzagolixom bola nižšia ako 3 %. Približne u 1 % subjektov sa hladiny ALT/AST zvýšili najmenej na trojnásobok ULN, pričom najvyšší nárast bol hlásený pri užívaní linzagolixu v dávke 200
mg alebo linzagolixu 200 mg s ABT. Nebolo pozorované žiadne súbežné zvýšenie hladiny bilirubínu.
Konkrétne odporúčania sú uvedené v časti 4.4.
Zmeny hustoty minerálov v kostiachÚčinok linzagolixu na BMD sa posudzoval vyšetrením pomocou snímok DXA. V dvoch klinických štúdiách fázy 3 boli zaznamenané zmeny BMD závislé od dávky a času. Súbežné podávanie ABT zmiernilo zníženie BMD (pozri tabuľku 2).
Zmeny BMD boli najvýraznejšie pri dávke 200 mg; po šiestich mesiacoch liečby bol pozorovaný priemerný pokles v BMD driekovej chrbtice oproti východiskovej hodnote > 3 % a > 8 % u 55 %, resp. 4 % pacientok.
Po 12 mesiacoch liečby linzagolixom bol pozorovaný priemerný pokles v BMD driekovej chrbtice oproti východiskovej hodnote > 3 % a > 8% u 38 %, resp. 7 % pri dávke 100 mg, 16 %, resp. 0 % pri dávke 100 mg s ABT a 27 %, resp. 1 % pri dávke 200 mg s ABT pacientok.
| Linzagolix
100 mg
| Linzagolix
100 mg s ABT
| Linzagolix
200 mg
| Linzagolix 200
mg s ABT
|
24 týždňov liečby
| Percento subjektov (%) s BMD
CfB > 3 % /
> 8 %
|
36/3
|
20/0
|
55/4
|
26/1
|
52 týždňov liečby
| Percento subjektov (%)
s BMD
CfB > 3 %/> 8 %
|
38/7
|
16/0
|
–*
|
27/1
|
|
|
Tabuľka 2: Podiel pacientok so zmenou BMD driekovej chrbtice oproti východiskovej hodnote o > 3 % a > 8 % po 24 týždňoch a po 52 týždňoch liečby v štúdiách PRIMROSE 1 a 2ABT – estradiol 1 mg a noretisterón-acetát 0,5 mg tableta raz denne, CfB – zmena oproti východiskovej hodnote (change from
baseline)
* Podávanie linzagolixu 200 mg sa skúmalo najviac 6 mesiacov.
Šesť mesiacov po ukončení liečby bolo vo všetkých liečebných skupinách zaznamenané zvýšenie BMD, čo poukazuje na čiastočnú obnovu. Konkrétne odporúčania sú uvedené v častiach 4.2 a 4.4. Podrobné informácie o znížení BMD sú uvedené v časti 5.1.
Vaginálne krvácaniePočas liečby linzagolixom bolo hlásené vaginálne krvácanie (zahŕňajúce hlásenia vaginálneho krvácania, krvácania z maternice, metrorágie, menorágie a menometrorágie). Najčastejšími nežiaducimi reakciami boli vaginálne krvácanie, metrorágia a menorágia, ktoré boli hlásené u 13 (1,6
%), 11 (1,3 %) a 5 (0,6 %) subjektov liečených linzagolixom, v uvedenom poradí. Vaginálne krvácanie bolo hlásené častejšie u subjektov v skupinách s dávkami linzagolixu 100 mg a 200 mg s ABT (do 2,4 %) v porovnaní so skupinami bez ABT (1 %). Metrorágia bola hlásená u 3 (1,5 %), 3
(1,4 %), 1 (0,5 %) a 4 (1,9 %) subjektov v skupinách s dávkami 100 mg, 100 mg s ABT, 200 mg a 200 mg s ABT, v uvedenom poradí, a menorágia bola hlásená u 1 (0,5 %), 1 (0,5 %), 2 (1,0 %) a 1 (0,5 %) subjektov v skupinách s dávkami linzagolixu 100 mg, 100 mg s ABT, 200 mg a 200 mg s ABT, v uvedenom poradí.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieNebol hlásený žiadny prípad predávkovania.
V prípade predávkovania sa pacientky majú pozorne sledovať a má sa im poskytnúť symptomatická a podporná liečbu.
U žien, ktoré užívajú liečebné režimy so súbežnou ABT, môže predávkovanie estrogénom a progestínom spôsobiť príznaky súvisiace s hormónmi, okrem iného nevoľnosť, vracanie, citlivosť prsníkov, bolesť brucha, ospalosť, únavu a krvácanie z vysadenia.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antagonisty hormónov uvoľňujúcich gonadotropín, ATC kód: H01CC04.
Mechanizmus účinku
Linzagolix je selektívny nepeptidový antagonista receptora pre hormón uvoľňujúci gonadotropín
(gonadotropin-releasing hormone, GnRH), ktorý inhibuje endogénnu signalizáciu GnRH tak, že sa kompetitívne viaže na receptory GnRH v hypofýze, čím moduluje os hypotalamus – hypofýza – gonády (hypothalamic-pituitary-gonadal axis, HPG axis), .
Farmakodynamické účinky
Účinky na hypofýzu a ovariálne hormóny
Podávanie linzagolixu vedie k supresii luteinizačného hormónu a hormónu stimulujúceho folikuly, ktorá je závislá od dávky, čo vedie k zníženiu koncentrácií estradiolu a progesterónu v krvi.
V štúdiách fázy 3 sa pri dávke 200 mg linzagolixu od 4 do 24 týždňov pozorovala úplná supresia estradiolu v sére (medián < 20 pg/ml). Čiastočná supresia sa od 4 do 52 týždňov pozorovala pri dávkach linzagolixu 100 mg, 100 mg so súbežnou ABT (označované ako „s ABT“) a 200 mg s ABT, s mediánom hladiny estradiolu v sére v rozsahu 20 až 60 pg/ml. Hladiny progesterónu sa zachovali na
úrovni ≤ 3,1 ng/ml u 83 % žien užívajúcich linzagolix 200 mg počas 24 týždňov a 68 % žien
užívajúcich linzagolix 100 mg počas 52 týždňov a približne 90 % žien užívajúcich linzagolix 100 mg s
ABT alebo 200 mg s ABT počas 52 týždňov.
Elektrofyziológia srdca
V jednej randomizovanej, placebom a pozitívne kontrolovanej, otvorenej, prekríženej QTc štúdii s jedinou dávkou sa hodnotil účinok linzagolixu na QTc interval. Štyridsaťosem zdravých žien dostalo
200 mg dávku linzagolixu (terapeutická cieľová expozícia), 700 mg dávku linzagolixu (supraterapeutická cieľová expozícia), 400 mg dávku moxifloxacínu (pozitívna kontrola) alebo placebo s vhodným vymývaním (washout). Pri dávkach linzagolixu 200 mg a 700 mg sa zistil nevýznamný účinok na predĺženie intervalu QT korigovaného vzhľadom na srdcovú frekvenciu s
maximálnym pozorovaným priemerom 3 hodiny po podaní dávky 8,34 ms (90 % CI 6,44 – 10,23) a
9,92 ms (90 % CI 8,03 – 11,81), v uvedenom poradí. Na základe rozsahu predĺženia intervalu QTc, modelovania následného účinku koncentrácie a podintervalu QT (JTpeakc) sa pozorované účinky nepovažujú za klinicky významné. Najvyššia predpokladaná koncentrácia v ustálenom stave v štúdii QT bola odhadnutá u zdravých subjektov bez prihliadnutia na zvýšenie expozície neviazanému linzagolixu v dôsledku existujúcich porúch (pozri časť 5.2).
Zmeny lipidových parametrov
Hladiny lipidov (HDL, LDL a celkového cholesterolu a triglyceridov) nalačno sa hodnotili každé tri mesiace od začiatku liečby linzagolixom až do troch mesiacov po liečbe. Vo všetkých skupinách s linzagolixom došlo k zvýšeniu hladiny LDL cholesterolu, HDL cholesterolu a triglyceridov (v prípade LDL to bolo zvyčajne o menej ako 15 % a v prípade triglyceridov o menej ako 20 %) a toto zvýšenie bolo zvyčajne vyššie v prípade režimov s linzagolixom samotným. Zvýšenia boli zrejmé od 12. týždňa a lipidové parametre sa vo všeobecnosti stabilizovali po 52 týždňoch liečby. Po zastavení liečby linzagolixom vykazovali hladiny lipidov známky návratu k východiskovej hodnote do 12 týždňov po zastavení liečby, ale aj tak zostali v porovnaní s východiskovou hodnotou mierne zvýšené (pozri časť
4.4).
Klinická účinnosť a bezpečnosťÚčinnosť lieku Yselty sa hodnotila v dvoch randomizovaných, dvojito zaslepených a placebom
kontrolovaných štúdiách fázy 3, PRIMROSE 1 s 511 ženami a PRIMROSE 2 s 501 ženami. Štúdia PRIMROSE 1 sa uskutočnila v USA a štúdia PRIMROSE 2 sa uskutočnila predovšetkým v Európe, pričom asi 10 % subjektov bolo z USA. Štúdie mali v podstate rovnaký dizajn: liečba trvala 52 týždňov a sledovanie po liečbe 24 týždňov. Nie sú k dispozícii žiadne údaje o účinnosti ani bezpečnosti liečby dlhšej ako 52 týždňov.
Spôsobilé pacientky mali silné menštruačné krvácanie (heavy menstrual bleeding, HMB: menštruačná strata krvi [menstrual blood loss, MBL] > 80 ml/cyklus) a myomatózna maternica s najmenej jedným myómom ≥ 2 cm potvrdeným ultrazvukom a bez myómu > 12 cm. MBL sa merala alkalickou hematínovou metódou.
Priemerný vek žien bol 42 rokov (rozpätie od 20 do 58 rokov) a priemerný index telesnej hmotnosti bol 29,9 kg/m2 (rozpätie od 16,8 do 58,6). Približne 34,5 % žien boli černošky, 63,5 % belošky a 2 % inej rasy. Najčastejšie hlásenými príznakmi okrem HMB boli bolesť brucha (67,9 % žien), tlak v bruchu (52,5 %), menštruácia trvajúca dlhšie ako zvyčajne (50,4 %), bolesť v dolnej časti chrbta (50,2
%), zvýšená frekvencia močenia (34,5 %) a bolesť pri pohlavnom styku (27,7 %). Medián objemu maternice bol 241 cm3 (rozpätie 32 až 2 075 cm3) a medián objemu myómov bol 53 cm3 (rozpätie 0 až
1 142 cm3). Takmer všetky ženy (99,7 %) mali aspoň jeden myóm ≥ 2 cm dlhý a 97,5 % malo
klasifikáciu FIGO od 1 do 6.
Subjekty boli randomizované do jednej z piatich možností liečby: placebo, Yselty 100 mg, Yselty 200
mg, Yselty 100 mg so súbežnou ABT (estradiol 1 mg/noretisterón-acetát 0,5 mg, označované ako
„s ABT“) alebo Yselty 200 mg s ABT, pričom všetky sa užívali jedenkrát denne. Subjekty randomizované do skupiny dostávajúcej placebo alebo liek Yselty v dávke 200 mg prešli na liečbu liekom Yselty v dávke 200 mg s ABT po 24 týždňoch, s výnimkou štúdie PRIMROSE 1, v ktorej
50 % subjektov dostávajúcich placebo pokračovalo v liečbe placebom až do 52 týždňov.
Primárnym cieľovým ukazovateľom účinnosti bola odpoveď definovaná ako MBL ≤ 80 ml a ≥ 50 % zníženie oproti východiskovej hodnote počas posledných 28 dní pred 24. týždňom. Liečba liekom Yselty s ABT alebo bez nej viedla v 24. Týždni k vyššiemu podielu žien so zníženou MBL v porovnaní s placebom. Na liečbu odpovedalo 56,4 %; 66,4 %; 71,4 % a 75,5 % subjektov, ktorým sa podával liek Yselty 100 mg, 100 mg s ABT, 200 mg a 200 mg s ABT v uvedenom poradí v štúdii PRIMROSE 1 a 56,7 %; 77,2 %; 77,7 % a 93,9 % subjektov v uvedenom poradí v štúdii PRIMROSE
2 (tabuľka 3). V 52. týždni na liečbu odpovedalo 57,4 %; 79,9 % a 87,9 % subjektov, ktorým sa podával liek Yselty 100 mg, 100 mg s ABT a 200 mg s ABT v uvedenom poradí v štúdii PRIMROSE
1 a 53,2 %; 91,3 % a 91,6 % subjektov v uvedenom poradí v štúdii PRIMROSE 2.
Tabuľka 3: Subjekty, ktoré odpovedali na liečbu (ženy so zníženou menštruačnou stratou krvi)po 24 týždňoch liečby Štúdia
| PRIMROSE 1
| PRIMROSE 2
|
Liečba
|
Placebo
| Yselty
|
Placebo
| Yselty
|
100 m
g
| 100 m
g
+ ABT
|
200 m
g
| 200 m
g
+ ABT
|
100 m
g
| 100 m
g
+ ABT
|
200 m
g
| 200 m
g
+ ABT
|
N
| 103
| 94
| 107
| 105
| 102
| 102
| 97
| 101
| 103
| 98
|
Percentuálny podiel (95 % CI)
subjektov odpovedajúc
ich na liečbu1, 2
|
35,0 (25,8;
45,0)
|
56,4 (45,8;
66,6)
|
66,4 (56,6;
75,2)
|
71,4 (61,8;
79,8)
|
75,5 (66,0;
83,5)
|
29,4 (20,8;
39,3)
|
56,7 (46,3;
66,7)
|
77,2 (67,8;
85,0)
|
77,7 (68,4;
85,3)
|
93,9 (87,1;
97,7)
|
1 Subjekty odpovedajúce na liečbu boli ženy s MBL ≤ 80 ml a ≥ 50 % znížením oproti východiskovej hodnote.
2 Clopperov-Pearsonov 95 % CI, hodnoty p ≤ 0,003 pre pomer pravdepodobnosti k placebu podľa Cochranovho-Mantelovho-
Haenszelovho testu s rasou ako stratifikačným faktorom. ABT – estradiol 1 mg/noretisterón-acetát 0,5 mg
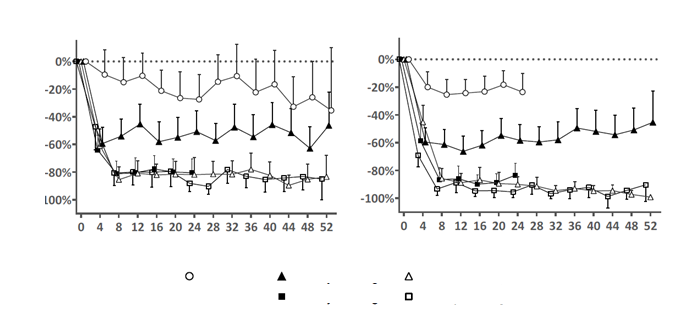
Priemerné percentuálne zníženie MBL v priebehu času je znázornené na obrázku 1. Pri liečbe liekom Yselty v dávke 100 mg sa dosiahol maximálny účinok v podobe približne 60 % zníženia MBL do 4 týždňov. Pri liečbe liekom Yselty 100 mg s ABT alebo 200 mg s ABT alebo bez ABT sa dosiahol maximálny účinok v podobe približne 80 % až 95 % zníženia MBL do 8 týždňov. Tieto zníženia sa zachovali až 52 týždňov.
 Obrázok 1: Priemerná percentuálna zmena menštruačnej straty krvi za každé 28-dňové obdobie do 52. týždňaPRIMROSE 1 PRIMROSE 2Týždne Týždne
Obrázok 1: Priemerná percentuálna zmena menštruačnej straty krvi za každé 28-dňové obdobie do 52. týždňaPRIMROSE 1 PRIMROSE 2Týždne Týždne Placebo Yselty 100 mg
Yselty 200 mg
Yselty 100 mg + ABT
Yselty 200 mg + ABT
V obidvoch pivotných štúdiách fázy 3 boli po 24 týždňoch v skupinách s dávkami lieku Yselty v
porovnaní s placebom (tabuľka 4) pozorované zlepšenia v sekundárnych cieľových ukazovateľoch vrátane zvýšeného podielu žien, ktoré dosiahli amenoreu, znížené skóre bolesti, vyššie hladiny hemoglobínu u anemických pacientok (< 12 g/dl na začiatku) a zvýšené skóre kvality života súvisiaceho so zdravím. Tieto zlepšenia boli výraznejšie pri použití lieku Yselty 200 mg (s ABT alebo bez ABT) a lieku Yselty 100 mg s ABT v porovnaní s liekom Yselty 100 mg.
Zlepšenia v sekundárnych cieľových parametroch po 24 týždňoch sa zvyčajne zachovali po 52 týždňoch v skupinách s liekom Yselty 100 mg s ABT a bez ABT a v skupine s liekom Yselty 200 mg s ABT. Objemy maternice a myómov sa po 24 týždňoch výrazne a konzistentne znížili len v skupine užívajúcej liek Yselty 200 mg bez ABT. V štúdiách PRIMROSE 1 a 2 sa objem maternice znížil o
31 % a 43 % a objem myómov sa znížil o 43 % a 49 % v uvedenom poradí. Keď bola pridaná ABT po
6 mesiacoch liečby liekom Yselty 200 mg bez ABT, priemerné objemy maternice a myómov sa zvýšili smerom k východiskovým objemom.
Tabuľka 4: Sekundárne cieľové ukazovatele po 24 týždňoch
Štúdia
|
PRIMROSE 1
|
PRIMROSE 2
|
Liečba
|
Placebo
|
Yselty
|
Placebo
|
Yselty
|
100 m
g
|
100 m
g
+
ABT
|
200 m
g
|
200 m
g
+
ABT
|
100 m
g
|
100 m
g
+
ABT
|
200 m
g
|
200 m
g
+
ABT
|
N
|
103
|
94
|
107
|
105
|
102
|
102
|
97
|
101
|
103
|
98
|
Percentuálny podiel žien s amenoreou (95 % CI)1
|
21,4 (13,9;
30,5)
|
38,3 (28,5;
48,9)
|
42,1 (32,6;
52,0)
|
60,0 (50,0;
69,4)
|
57,8 (47,7;
67,6)
|
11,8 (6,2;
19,6)
|
34,0 (24,7;
44,3)
|
63,4 (53,2;
72,7)
|
70,9 (61,1;
79,4)
|
80,6 (71,4;
87,9)
|
Priemerná zmena hladín hemoglobínu oproti východiskovej hodnote – g/dl (SD, n)2
|
0,30 (1,57;
45)
|
1,36 (1,82;
42)
|
'
1,87 (1,57;
52)
|
2,22 (1,58;
53)
|
2,00 (1,60;
50)
|
0,38 (1,69;
43)
|
1,36 (1,50;
49)
|
1,88 (1,58;
45)
|
2,10 (1,77;
46)
|
2,27 (1,43;
47)
|
Odhadovaná priemerná zmena skóre bolesti (95 % CI)3 oproti východiskovej hodnote
|
-1,06
(-1,74; -
0,37)
|
-2,70 (-
3,38; -
2,02)
|
-3,11 (-
3,81; -
2,41)
|
-3,85 (-
4,47; -
3,23)
|
-3,68 (-
4,34; -
3,01)
|
-0,44
(-1,14;
0,27)
|
-1,61 (-
2,35; -
0,88)
|
-1,91 (-
2,64; -
1,18)
|
-2,55 (-
3,25; -
1,84)
|
-2,27 (-
3,00;
-1,55)
|
Odhadovaný priemerný pomer
objemu maternice
k východiskovej hodnote(95 % CI)
|
1,02 (0,91;
1,15)
|
0,83 (0,74;
0,94)
|
1,06 (0,94;
1,20)
|
0,69 (0,62;
0,77)
|
0,92 (0,82;
1,03)
|
1,04 (0,92;
1,17)
|
0,85 (0,75;
0,96)
|
0,88 (0,77;
0,99)
|
0,57 (0,50;
0,64)
|
0,80 (0,71;
0,91)
|
Odhadovaný priemerný pomer objemu myómov k východiskovej hodnote(95 % CI)
|
0,95 (0,75;
1,19)
|
0,75 (0,60;
0,94)
|
0,98 (0,77;
1,24)
|
0,57 (0,46;
0,70)
|
0,88 (0,70;
1,09)
|
1,04 (0,84;
1,29)
|
0,85 (0,68;
1,06)
|
0,93 (0,75;
1,17)
|
0,51 (0,41;
0,63)
|
0,79 (0,63;
0,99)
|
Odhadovaná priemerná zmena
skóre HRQL (95 % CI)4 oproti východiskovej
hodnote
|
15,5 (9,4;
21,6)
|
26,1 (20,0;
32,2)
|
37,2
31,0;
43,5)
|
35,5 (29,8;
41,1)
|
34,2 (28,3;
40,1)
|
10,3 (4,0;
16,6)
|
20,6 (14,1;
27,2)
|
22,9 (16,4;
29,5)
|
30,2 (23,9;
36,5)
|
30,7 (24,2;
37,1)
|
1 Amenorea bola definovaná ako negatívny nález menštruačnej krvi alkalickou hematínovou metódou (nezahŕňalo špinenie
alebo MBL < 1 až 3 ml) počas 35 dní a do konca liečby počas až 24 týždňov.
2 U žien s východiskovou anémiou (hladina hemoglobínu < 12 g/dl) „n“ predstavuje počet žien s nechýbajúcimi údajmi po 24
týždňoch.
3 Bolesť sa posudzovala pomocou numerickej hodnotiacej stupnice (NRS) od 0 do 10.
4 Skóre kvality života súvisiacej so zdravím (Health-Related Quality of Life, HRQL) je súčasťou schváleného dotazníka
Príznaky myómu maternice – kvalita života (Uterine Fibroid Symptoms – Quality of Life, UFS-QoL). Skóre sa pohybuje od
0 do 100, pričom vyššie skóre znamená lepšiu kvalitu života súvisiacu so zdravím. Východiskové skóre bolo približne 40. ABT – estradiol 1 mg/noretisterón-acetát 0,5 mg; SD – štandardná odchýlka, CI – interval spoľahlivosti (confidence interval)
Hustota minerálov v kostiachMinerálna hustota kostí sa hodnotila vyšetrenia pomocou snímok DXA na začiatku liečby, počas liečby (24. a 52. týždeň) a 6 mesiacov po skončení liečby (76. týždeň). Subjekty s významným rizikom vzniku osteoporózy, s osteoporózou v anamnéze, s preukázanou osteoporózou alebo iným metabolickým ochorením kostí alebo známou osteoporózou alebo iným metabolickým ochorením kostí boli zo skúšaní PRIMROSE 1 a PRIMROSE 2 vylúčené.
Priemerné percentuálne zníženie BMD pozorované po 24 a 52 týždňoch bolo závislé od dávky a času a zmiernené súbežnou ABT (tabuľka 5).
Po 24 týždňoch bola zmena BMD najvýraznejšia u žien s úplnou supresiou estradiolu s liekom Yselty v dávke 200 mg (-3,70 %). V tomto režime sa nepokračovalo dlhšie ako 6 mesiacov (pozri časť 4.2). Uvedené zmeny boli menej výrazné u žien, ktoré podstúpili iné režimy: -1,99 % s liekom Yselty 100 mg, -0,96 % s liekom Yselty 100 mg s ABT a -1,13 % s liekom Yselty 200 mg s ABT.
Po 52 týždňoch priemerné percentuálne zmeny oproti východiskovej hodnote naznačovali menšiu mieru zníženia BMD: -2,36 % s liekom Yselty 100 mg, -0,93 % s liekom Yselty 100 mg s ABT a -1,61 % s liekom Yselty 200 mg s ABT. Úroveň zníženia BMD v dôsledku liečby v tejto populácii, ktorá sa považuje za klinicky významnú, nie je presne stanovená a závisí od konkrétnej ženy, avšak vo všeobecnosti zníženia BMD na úrovni približne 3 % alebo viac sa majú pozorne preskúmať a monitorovať. Pri posudzovaní zníženia BMD u konkrétnej ženy je nevyhnutné zvážiť východiskovú hodnotu BMD, vek a celkový profil rizika osteoporózy u konkrétnej ženy, ako aj pomer prínosu a rizík pokračovania v liečbe.
.
Po 24 týždňoch od ukončenia liečby došlo u väčšiny pacientok k úplnému alebo čiastočnému obnoveniu BMD driekovej chrbtice: 53 % s liekom Yselty 100 mg, 52 % s liekom Yselty 100 mg s ABT a 64 % s liekom Yselty 200 mg s ABT v štúdii PRIMROSE 1 a 59 % s liekom Yselty 100 mg, 80 % s liekom Yselty 100 mg s ABT a 67 % s liekom Yselty 200 mg s ABT v štúdii PRIMROSE 2.
Rozsah a miera zníženia BMD pri liečbe žien dlhšej ako 12 mesiacov nie je v súčasnosti známa.
| Placebo
| Yselty
100 mg
| Yselty
100 mg + ABT
| Yselty
200 mg*
| Yselty
200 mg + ABT
| 24 týždňov liečby
| Počet subjektov
| 130
| 121
| 122
| 138
| 127
| Priemerná percentuálna CfB
|
0,46
|
-1,99
|
-0,96
|
-3,70
|
-1,13
| 95 % CI
| 0,06; 0,85
| -2,47; -1,50
| -1,45; -0,48
| -4,18; -3,22
| -1,60; -0,66
| 52 týždňov liečby
| Počet subjektov
| 19
| 93
| 84
| -
| 97
| Priemerná percentuálna CfB
|
-0,83 **
|
-2,36
|
-0,93
|
-
|
-1,61
| 95 % CI
| -2,08; 0,42
| -3,10; -1,63
| -1,40; -0,47
| -
| -2,22; -0,99
|
|
|
Tabuľka 5: Priemerná percentuálna zmena BMD driekovej chrbtice oproti východiskovej hodnote (CfB) po 24 a 52 týždňoch liečby v štúdiách PRIMROSE 1 a 2* Liek Yselty 200 mg sa skúmal po dobu 6 mesiacov.
** V štúdii PRIMROSE 1 sa placebo podávalo až 12 mesiacov.
Účinky na endometriumV obidvoch štúdiách fázy 3 boli v rámci hodnotenia bezpečnosti u podskupiny pacientok vykonané biopsie endometria na začiatku liečby, v 24. a 52. týždni. Výsledky nevyvolali žiadne obavy týkajúce sa bezpečnosti.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Yselty
vo všetkých podskupinách pediatrickej populácie pri liečbe leiomyómu maternice (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5
.2 Farmakokinetické vlastnosti
Absorpcia
Po perorálnom podaní jednej dávky 100 mg alebo 200 mg sa linzagolix rýchlo absorbuje, pričom Cmax
nastáva približne 2 hodiny po podaní. Linzagolix vykazuje dávkovo lineárnu farmakokinetiku a nevykazuje žiadnu relevantnú akumuláciu v ustálenom stave.
Zdá sa, že podávanie linzagolixu (200 mg) s jedlom s vysokým obsahom tuku oddialilo a mierne znížilo maximálne koncentrácie v plazme, čo je v súlade s oneskoreným vyprázdnením žalúdka po jedle s vysokým obsahom tuku, rozsah expozície to však neovplyvnilo. Táto skutočnosť sa nepovažuje za klinicky významnú.
Distribúcia
Linzagolix sa vo veľkej miere viazal (> 99 %) na plazmatické proteíny, najmä na albumín, a
neprenikol do červených krviniek. Distribučný objem (Vd/F) po siedmich po sebe nasledujúcich dňoch perorálneho podávania linzagolixu v dávke 100 mg bol 11,067 l (CV: 20,4 %) a v dávke 200 mg 11,178 l (CV: 11,8 %).
Biotransformácia
Pri profilácii metabolitov a identifikácii linzagolixu bolo kvantifikovaných až 7 metabolitov v plazme,
moči a stolici. Prevládajúcou zložkou v profiloch ľudskej plazmy bol nezmenený linzagolix. Podobne Linzagolix prevládal v moči a bol jednou z hlavných zložiek v stolici. Všetky metabolity v plazme boli prítomné na úrovni menej ako 10 % celkovej expozície súvisiacej s linzagolixom.
Eliminácia
Po viacerých dávkach linzagolixu bol t1/2 linzagolixu približne 15 hodín. Linzagolix sa vylučoval
najmä močom a približne jedna tretina bola vylúčená stolicou. Po podaní viacerých dávok 100 mg a
200 mg linzagolixu bol geometrický priemer zjavného klírensu (CL/F) linzagolixu 0,522 l/h (CV: 20,1
%) a 0,499 l/h (CV: 15,2 %), v uvedenom poradí.
Osobitné populácie
Z farmakokinetickej analýzy populácie vyplýva, že vek nemá na expozíciu linzagolixu významný
vplyv. V analýze sa ukázalo, že u subjektov černošskej rasy došlo v porovnaní so subjektmi bielej rasy k poklesu CL/F o 22,5 %, avšak bezpečnostný profil linzagolixu u subjektov černošskej a bielej rasy bol podobný.
Na základe farmakokinetickej analýzy populácie sa zistilo, že hmotnosť ovplyvňuje farmakokinetiku linzagolixu. CL/F u pacientok s hmotnosťou 52,7 kg (5. percentil) sa predpokladal asi o 19,2 % nižší a u pacientok s hmotnosťou 112 kg (95. percentil) asi o 42 % vyšší ako u pacientok s hmotnosťou 70 kg. Analýzy údajov o podskupinách z pivotných štúdií fázy 3 však nepreukázali žiadne klinicky
významné rozdiely, pokiaľ ide o bezpečnosť a účinnosť, a úprava dávky sa neodporúča.
Porucha funkcie pečene
V klinickej štúdii vykonanej u žien s poruchou funkcie pečene (mierna: trieda A podľa Childa-Pugha, stredne závažná: trieda B podľa Childa-Pugha a závažná: trieda C podľa Childa-Pugha) sa po podaní jednej 200 mg dávky linzagolixu nezistil významný účinok na celkovú plazmatickú expozíciu linzagolixu. Neviazaná frakcia linzagolixu nebola miernou a stredne závažnou poruchou funkcie pečene ovplyvnená. U pacientok s miernou a stredne závažnou poruchou funkcie pečene sa úprava dávky lieku Yselty nevyžaduje (pozri časť 4.2). Yselty sa nemá používať u žien so závažnou poruchou funkcie pečene (trieda C podľa Childa-Pugha), keďže boli zaznamenané dvoj- až trojnásobne vyššie stredné expozície neviazanému linzagolixu (pozri časť 4.4).
Porucha funkcie obličiek
V klinickej štúdii vykonanej u žien s poruchou funkcie obličiek (mierna, stredne závažná, závažná a konečné štádium ochorenia obličiek), v ktorej sa rýchlosť glomerulárnej filtrácie (GFR) posudzovala pomocou klírensu kreatínu, nebol po podaní jednorazovej dávky 200 mg linzagolixu zistený významný účinok na celkovú plazmatickú expozíciu linzagolixu. Cmaxu, AUCu0-t a AUCu0-inf neviazaného linzagolixu v plazme sa zvýšili o 30 %, 32 % a 33 % u žien s miernou poruchou funkcie obličiek v porovnaní so zdravými účastníčkami s normálnou funkciou obličiek. Keďže nie je možné vylúčiť potenciálne obavy týkajúce sa bezpečnosti pri dlhodobom užívaní, predpisujúcim lekárom sa odporúča sledovať nežiaduce reakcie u žien s miernou poruchou funkcie obličiek (pozri časť 4.4). Úprava dávky sa však nevyžaduje (pozri časť 4.2). Liek Yselty sa nemá používať u žien so stredne závažnou alebo závažnou poruchou funkcie obličiek alebo s ochorením obličiek v konečnom štádiu, pretože sa pozorovali približne 1,5-násobne (u stredne závažnej) a dvojnásobne (u závažnej poruchy funkcie obličiek a ESRD) vyššie priemerné expozície neviazanému linzagolixu (pozri časť 4.4).
5.3 Predklinické údaje o bezpečnosti
Reprodukčná a vývojová toxicita
V štúdiách fertility potkanov linzagolix vzhľadom na svoj mechanizmus účinku zabránil počatiu, znížil počet uhniezdení a v embryofetálnych štúdiách na potkanoch a králikoch viedol
k embryofetálnej mortalite, úplnej strate vrhu alebo zrušeniu gravidity.
V štúdii na potkanoch sa nepozorovali žiadne teratogénne účinky ani nežiaduce účinky na prenatálny a postnatálny vývoj.
V hlavných štúdiách embryofetálneho vývoja u potkanov a králikov sa preukázalo, že úrovne dávok
100 mg/kg a 3 mg/kg linzagolixu, v uvedenom poradí, zodpovedajú hladine bez pozorovaného nepriaznivého účinku (NOAEL, no observed adverse effect level) na reprodukčnú funkciu a embryofetálny vývoj (čo na základe AUC zodpovedá 5,9-násobku a 0,004-násobku maximálnej odporúčanej dávky pre ľudí, v uvedenom poradí).
Laktácia
Preukázalo sa, že linzagolix sa vylučuje do mlieka potkanov. Až do 96 hodín po podaní bola koncentrácia rádioaktivity nižšia v mlieku ako v plazme (menej ako 0,3-krát).
Mutagenita
Štandardná séria testov in vitro a in vivo nepreukázala mutagénny ani klinicky relevantný genotoxický potenciál lieku.
Karcinogenita
Karcinogénne vlastnosti linzagolixu sa hodnotili v 26-týždňovej štúdii karcinogenity u transgénnych myší Tg RasH2. Nebol zistený žiadny dôkaz karcinogenity vyvolanej linzagolixom až do najvyššej dávky 500 mg/kg (čo zodpovedá 13,2-násobku maximálnej odporúčanej dávky u ľudí na základe AUC).
V dvojročnej štúdii karcinogenity u potkanov bol pozorovaný zvýšený výskyt adenokarcinómu endometria maternice v skupinách so strednou (50 mg/kg) a vysokou (500 mg/kg) dávkou (čo v uvedenom poradí zodpovedá 6,8-násobku a 9,6-násobku maximálnej odporúčanej dávky u ľudí na základe AUC) a marginálne zvýšenie frekvencie výskytu adenokarcinómu mliečnej žľazy, ktoré bolo pozorované len so strednou dávkou (50 mg/kg) (čo zodpovedá 6,8-násobku maximálnej odporúčanej dávky u ľudí na základe AUC). Klinický význam týchto zistení zostáva neznámy.
Predpokladá sa, že nekarcinogénne histopatologické nálezy vo vaječníku a maternici (myš) alebo vaječníku a samičej mliečnej žľaze (potkan) súvisia s farmakologickým účinkom linzagolixu.
6
. FARMACEUTICKÉ ÚDAJE
6
.1 Zoznam pomocných látok
Jadro tablety
Monohydrát laktózy
Mikrokryštalická celulóza
Čiastočne substituovaná hydroxypropylcelulóza
Hydroxypropylcelulóza Sodná soľ kroskaramelózy Stearát horečnatý
Obal tablety
Makrogol a polyvinylalkohol vrúbľovaný, kopolymér (E1209)
Mastenec (E553b) Oxid titaničitý (E171) Žltý oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
PVC-PVDC/hliníkový blister obsahujúci 14 filmom obalených tabliet v blistri.
Veľkosť balenia 28 filmom obalených tabliet (dva blistre so 14 filmom obalenými tabletami) alebo 84
filmom obalených tabliet (šesť blistrov so 14 filmom obalenými tabletami) v jednej kartónovej krabičke. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Thermaex Ireland Limited
3rd Floor, Kilmore House, Park Lane, Spencer Dock, Dublin 1,
D01 YE64
Irsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/21/1606/001
EU/1/21/1606/002
EU/1/21/1606/003
EU/1/21/1606/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 14. jún 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.