
2" src="PublicData/Liekinfo/Spc/13638_files/image001.png" />a Odporúčania týkajúce sa antiagregačných prípravkov/NSAID/antikoagulancií nájdete v časti 4.4.
b Pre zváženie pomeru prínosu a rizika prípadnej ďalšej liečby volanesorsenom je potrebná konzultácia s hematológom.
Osobitné skupiny pacientov
Starší ľudia
U starších pacientov nie je potrebná úprava úvodnej dávky. Existujú obmedzené klinické údaje pre
pacientov vo veku 65 a viac rokov (pozri časti 5.1 a 5.2).
Pacienti s poruchou funkcie obličiek
U pacientov s miernou až stredne závažnou poruchou funkcie obličiek nie je potrebné upravovať úvodné dávkovanie. Bezpečnosť a účinnosť u pacientov so závažnou poruchou funkcie obličiek nebola stanovená a títo pacienti by mali byť pozorne sledovaní.
Pacienti s poruchou funkcie pečene
Tento liek nebol skúmaný u pacientov s poruchou funkcie pečene. Tento liek nie je metabolizovaný prostredníctvom enzýmového systému cytochrómu P450 v pečeni, a preto nie je pravdepodobné, že by bola potrebná úprava dávky u pacientov s poruchou funkcie pečene.
Pediatrická populácia
Bezpečnosť a účinnosť tohto lieku u detí a dospievajúcich mladších ako 18 rokov nebola doposiaľ
stanovená. Nie sú k dispozícii žiadne údaje.
Spôsob podávania
Tento liek je určený len pre subkutánnu aplikáciu. Nepodávajte vo forme intramuskulárnej alebo
intravenóznej injekcie.
Každá naplnená injekčná striekačka je určená len na jednorazové použitie.
Waylivra sa musí pred podaním vizuálne skontrolovať. Roztok musí byť číry a bezfarebný až nažltlý. Ak je roztok zakalený alebo obsahuje viditeľné čiastočky, obsah sa nesmie podať a liek sa musí vrátiť do lekárne.
Prvá injekcia, ktorú si podá pacient alebo ošetrovateľ, sa musí vykonať pod vedením primerane kvalifikovaného zdravotníckeho personálu. Pacienti a/alebo ošetrovatelia majú byť vyškolení v podávaní tohto lieku v súlade s písomnou informáciou pre používateľa.
Naplnenú injekčnú striekačku pred podaním injekcie nechajte ohriať na izbovú teplotu. Z chladničky (2 ° až 8 ° C) by sa mala vybrať najneskôr 30 minút pred použitím. Nemali by sa používať iné metódy ohrievania. Bežne je viditeľná veľká vzduchová bublina. Túto vzduchovú bublinu sa nepokúšajte odstrániť.
Je dôležité striedať miesta vpichu injekcie. Medzi miesta vpichu injekcie patrí brucho, oblasť hornej časti stehna alebo vonkajšia časť horného ramena. Pri injekcii do hornej časti ramena injekciu musí podať iná osoba. Treba sa vyhnúť podaniu injekcie v mieste obvodu pása a iných miest, kde odev môže spôsobiť otlak alebo oder. Tento liek sa nesmie injikovať do tetovania, znamienok, materských znamienok, modrín, vyrážok alebo oblastí, kde je koža jemná, červená, tvrdá, pomliaždená, poškodená, popálená alebo zapálená.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Chronická alebo nevysvetliteľná trombocytopénia. Liečba sa nemá začať u pacientov s
trombocytopéniou (počet krvných doštičiek <140 x 109/l).
4.4 Osobitné upozornenia a opatrenia pri používaní
Trombocytopénia
Waylivrasa veľmi často spája so znížením počtu krvných doštičiek u pacientov s FCS, čo môže viesť k trombocytopénii (pozri časť 4.8). Pacienti s nižšou telesnou hmotnosťou (menej ako 70 kg) môžu byť počas liečby týmto liekom viac náchylní na trombocytopéniu. Počas liečby týmto liekom je u
pacientov s FCS dôležité pozorné sledovanie trombocytopénie (pozri časť 4.2). Odporúčania na
úpravu frekvencie monitorovania a dávkovania sú špecifikované v tabuľke 1 (pozri časť 4.2).
Pri hladinách krvných doštičiek < 75 x 109/l sa musí zvážiť prerušenie liečby antiagregačnými prípravkami/NSAID/antikoagulanciami. Liečba s týmito liekmi sa musí prerušiť pri hladinách krvných doštičiek < 50 x 109/l (pozri časť 4.5).
Pacientov treba poučiť, aby okamžite hlásili svojmu lekárovi, ak sa objavia príznaky krvácania, ktoré môžu zahŕňať petéchie, spontánne podliatiny, subkonjunktiválne krvácanie alebo iné neobvyklé krvácanie (vrátane krvácania z nosa, krvácania z ďasien, do stolice alebo neobvykle ťažkého menštruačného krvácania), stuhnutosť krku, atypická ťažká bolesť hlavy alebo akékoľvek dlhodobé krvácanie.
Hladiny LDL-C
Pri liečbe Waylivrou sa hladiny LDL-C môžu zvyšovať, ale zvyčajne zostanú v normálnom rozmedzí.
Obličkovátoxicita
Renálna toxicita bola pozorovaná po podaní volanesorsenu a iných subkutánne a intravenózne aplikovaných antisense oligonukleotidov. Sledovanie výskytu nefrotoxicity prostredníctvom prúžkových testov z moču sa odporúča štvrťročne. V prípade pozitívneho výsledku je potrebné vykonať rozsiahlejšie hodnotenie funkcie obličiek, vrátane sérového kreatinínu a 24-hodinového zberu na kvantifikáciu proteinúrie a hodnotenie klírensu kreatinínu. Liečbu je potrebné ukončiť, ak došlo
k proteinúrii ≥500 mg/24 hodín alebo ak došlo k zvýšeniu kreatinínu v sére ≥0,3 mg/dl (26,5 μmol/l),
ktoré je >horný limit normálu, alebo ak klírens kreatinínu odhadovaný podľa rovnice CKD-EPI dosahuje ≤30 ml/min/1,73 m2. Liečbu je potrebné ukončiť aj v prípade výskytu akýchkoľvek klinických prejavov alebo príznakov poruchy funkcie obličiek, ktoré čakajú na potvrdzujúce hodnotenie.
Hepatotoxicita
Zvýšenie pečeňových enzýmov bolo pozorované po podaní iných subkutánne a intravenózne aplikovaných antisense oligonukleotidov. Sledovanie výskytu hepatotoxicity prostredníctvom pečeňových enzýmov a bilirubínu v sére sa odporúča štvrťročne. Liečbu je potrebné ukončiť, ak došlo k zvýšeniu hladiny ALT alebo AST >8-násobok horného limitu normálu alebo k zvýšeniu >5-násobok horného limitu normálu, ktoré pretrváva ≥2 týždne alebo menšie zvýšenia hladiny ALT alebo AST, ktoré súvisia s hladinou celkového bilirubínu >2-násobok horného limitu normálu alebo INR >1,5. Liečbu je potrebné ukončiť aj v prípade výskytu akýchkoľvek klinických prejavov alebo príznakov poruchy funkcie pečene alebo hepatitídy.
Imunogenita a zápal
S prítomnosťou protilátok na liek sa nespája žiadny dôkaz úpravy bezpečnostného profilu ani žiadna klinická odpoveď. Ak sa očakáva tvorba protilátok proti lieku s klinicky významným účinkom,
kontaktujte držiteľa rozhodnutia o registrácii a poraďte sa s ním o testovaní protilátok.
Sledovanie zápalu je potrebné hodnotiť prostredníctvom štvrťročného hodnotenia sedimentácie
erytrocytov (ESR).
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) na dávku 285 mg, t.j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
Neočakávajú sa klinicky významné farmakokinetické interakcie medzi volanesorsenom a substrátmi, induktormi alebo inhibítormi enzýmov cytochrómu P450 (CYP) a transportérmi liekov. Nie je známe, či zníženie hladiny triglyceridov volanesorsenom a potenciálne nasledujúce zníženie zápalu vedie k normalizácii expresie enzýmu CYP.
V klinických štúdiách sa tento liek používal v kombinácii s fibrátmi a rybacími olejmi bez toho, aby sa to prejavilo na farmakodynamike alebo farmakokinetike tohto liečiva. Počas klinického programu neboli hlásené žiadne nežiaduce príhody, ktoré by súviseli s interakciami liekov, avšak tento záver sa zakladá len na obmedzených údajoch.
Účinok súbežného podávania tohto lieku s alkoholom alebo s liekmi, o ktorých je známe, že sú potenciálne hepatotoxické (napr. paracetamol), nie je známy. Ak sa objavia znaky a príznaky hepatotoxicity, liečba hepatotoxickým liekom sa musí prerušiť.
Antitrombotickéčinidláalieky,ktorémôžuznížiťpočetkrvnýchdoštičiek
Nie je známe, či sa riziko krvácania zvyšuje súbežným používaním volanesorsenových a antitrombotických liekov alebo liekov, ktoré môžu znížiť počet krvných doštičiek alebo ovplyvniť funkciu krvných doštičiek.
Pri hladinách krvných doštičiek <75 x 109/l sa musí zvážiť prerušenie antiagregačnej
liečby/NSAID/antikoagulanciami a liečba s týmito liekmi sa má zastaviť pri hladinách krvných
doštičiek <50 x 109/l (pozri časť 4.4).
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii žiadne údaje o použití volanesorsenu u gravidných žien.
Pokusy na zvieratách nepreukázali priamy alebo nepriamy škodlivý účinok pokiaľ ide o reprodukčnú toxicitu (pozri časť 5.3).
Ako preventívne opatrenie sa odporúča vyhnúť sa užívaniu tohto lieku počas tehotenstva. Dojčenie
V predklinických štúdiách boli u laktujúcich myší hladiny volanesorsenu v mlieku veľmi nízke.
Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie veľmi nízkych množstiev volanesorsenu v mlieku (pozri časť 5.3). Vzhľadom na slabú perorálnu biologickú dostupnosť tohto lieku sa nepovažuje za pravdepodobné, že by tieto nízke koncentrácie mlieka mali za následok systémovú expozíciu v dôsledku kojenia.
Nie je známe, či sa volanesorsen alebo jeho metabolity vylučujú do materského mlieka. Nemôže sa vylúčiť riziko pre novorodenca.
Musí sa prijať rozhodnutie, či má matka prerušiť dojčenie alebo prerušiť/zdržať sa liečby, pričom sa
berie do úvahy prínos dojčenia pre dieťa a prínos liečby pre ženu.
Fertilita
Nie sú k dispozícii žiadne klinické údaje o účinku tohto lieku na ľudskú fertilitu. Volanesorsen nemal
žiadny vplyv na plodnosť myší.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Volanesorsen nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať
stroje.
4.8 Nežiaduce účinky
Súhrnný bezpečnostný profil
V klinických štúdiách u pacientov s FCS boli počas liečby najčastejšie hlásenými nežiaducimi účinkami: znížený počet krvných doštičiek (pozri časť 4.4), ktorý sa počas kľúčových štúdií vyskytol u 40 % pacientov a reakcie v mieste vpichu, ktoré sa vyskytli u 82 % pacientov.
Tabuľkovýzoznamnežiaducichúčinkov
V tabuľke 2 sú uvedené nežiaduce účinky zo štúdií fázy 3 u pacientov s FCS, ktorým sa subkutánne podával volanesorsen.
Frekvencia nežiaducich reakcií je definovaná pomocou nasledujúcej konvencie: veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté (≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1 000); veľmi zriedkavé (<1/10 000) a neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencie výskytu sú nežiaduce reakcie usporiadané do poradia podľa klesajúcej frekvencie.
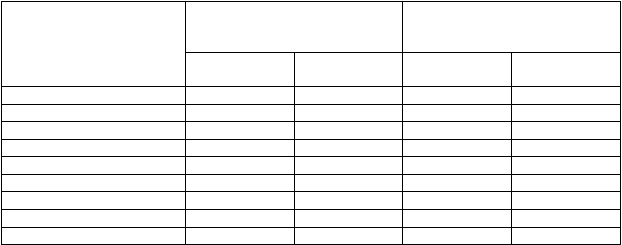
Tabuľka 2: Súhrn nežiaducich účinkov v klinických štúdiách u pacientov s FCS (N=86)
Trieda orgánových
systémov Veľmi časté (N, %) Časté (N, %)
Poruchy krvi a
lymfatického systému
Poruchy imunitného systému
Poruchy metabolizmu a výživy
Trombocytopénia (10, 12 %) Leukopénia (2, 2 %)
Eozinofília (1, 1 %)
Imúnna trombocytopenická purpura (1, 1 %)
Spontánny hematóm (1, 1 %)
Imunizačná reakcia (3, 3 %)
Precitlivelosť (1, 1 %)
Reakcia podobná sérovej chorobe
(1, 1 %)
Diabetes mellitus (1, 1 %)
Psychické poruchy Nespavosť (1, 1 %) Poruchy nervového systému Bolesť hlavy (8, 9 %)
Hypoestézia (1, 1 %)
Presynkopa (1, 1 %) Retinálna migréna (1, 1 %) Synkopa (2, 2 %)
Točenie hlavy (1, 1 %) Trasenie (1, 1 %)
Poruchy oka Konjuktiválna hemorágia (1, 1 %) Rozmazané videnie (1, 1 %)
Poruchy ciev Hematóm (3, 3 %) Hypertenzia (1, 1 %) Hemorágia (1, 1 %)
Nával horúčavy (1, 1 %)
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Epistaxa (3, 3 %) Kašeľ (1, 1 %) Dyspnoe (2, 2 %)
Kongescia nosa (1, 1 %) Faryngeálny edém (1, 1 %) Ťažkosti s dýchaním (1, 1 %)
Nevoľnosť (8, 9 %) Diarea (4, 5 %)
Sucho v ústach (1, 1 %) Gingiválne krvácanie (1, 1 %) Krvácanie z úst (1, 1 %)
T
rieda orgánových
systémov
V
eľmi časté (N, %) Časté (N, %)
Zväčšenie príušnej žľazy (1, 1 %) Zvracanie (4, 5 %)
Bolesť brucha (4, 5 %) Abdominálna distenzia (1, 1 %) Dyspepsia (1, 1 %)
Gingiválny opuch (1, 1 %)

Poruchy kože a podkožného tkaniva
Poruchy kostrovej
a svalovej sústavy a
spojivového tkaniva
Poruchy obličiek a močových ciest
Celkové poruchy a reakcie v mieste podania
Erytém v mieste vpichu (67, 78 %) Bolesť miesta vpichu (38, 44 %) Zblednutie miesta vpichu (37,
43 %)
Opuch miesta vpichu (25, 29 %) Pruritus miesta vpichu (22, 26 %) Sfarbenie miesta vpichu (19,
22 %)
Stvrdnutie miesta vpichu (17,
20 %)
Podliatina v mieste vpichu (10,
12 %)
Erytém (4, 5 %) Pruritus (4, 5 %) Urtikária (3, 3 %) Hyperhidróza (2, 2 %) Vyrážka (3, 3 %) Petechia (1, 1 %) Ekchymóza (1, 1 %) Nočné potenie (1, 1 %) Vezikuly (1, 1 %)
Hypertrofia kože (1, 1 %) Opuch tváre (1, 1 %)
Bolesť svalov (8, 9 %)
Bolesť kĺbov (6, 7 %)
Bolesť v končatinách (5, 6 %) Artritída (2, 2 %)
Bolesť chrbtice (2, 2 %) Muskuloskeletová bolesť (2, 2 %) Bolesť krku (2, 2 %)
Svalový spazmus (1, 1 %) Stuhnutosť kĺbov (1, 1 %) Myozitída (1, 1 %)
Bolesť v čeľusti (1, 1 %) Reumatická polymyalgia (1, 1 %)
Hematúria (1, 1 %) Proteinúria (1, 1 %)
Asténia (8, 9 %) Únava (8, 9 %)
Hematóm v mieste vpichu (7,
8 %)
Reakcia v mieste vpichu (6, 7 %) Urtikária v mieste vpichu (5, 6 %) Teplo v mieste vpichu (5, 6 %) Zimnica (5, 6 %)
Pyrexia (4, 5 %)
Sucho v mieste vpichu (4, 5 %) Krvácanie miesta vpichu (4, 5 %) Hypoestézia miesta vpichu (4,
T
rieda orgánových
systémov
V
eľmi časté (N, %) Časté (N, %)
Edém miesta vpichu (10, 12 %) 5 %)
Vezikuly v mieste vpichu (3, 3 %) Celkový pocit nepohodlia (2, 2 %) Pocit tepla (2, 2 %)
Choroba podobná chrípke (2,
2 %)
Diskomfort v mieste vpichu (2,
2 %)
Zápal miesta vpichu (2, 2 %) Zdurenie miesta vpichu (2, 2 %) Bolesť (2, 2 %)
Parestézia miesta vpichu (1, 1 %) Chrasta v mieste vpichu (1, 1 %) Pľuzgier v mieste vpichu (1, 1 %) Edém (1, 1 %)
Nonkardiálna bolesť v hrudi (1,
1 %)
Krvácanie cievy v mieste vpichu
(1, 1 %)
Laboratórne a funkčné
vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
Znížený počet krvných doštičiek
(34, 40 %)
Kreatinín v krvi zvýšený (1, 1 %)
Močovina v krvi zvýšená (1, 1 %)
Obličkový klírens kreatinínu
znížený (1, 1 %)
Transaminázy zvýšené (1, 1 %)
Počet bielych krviniek znížený (1,
1 %)
Hemoglobín znížený (1, 1 %)
Hladiny pečeňových enzýmov
zvýšené (1, 1 %) Medzinárodný normalizovaný
pomer (INR) zvýšený (1, 1 %)
Kontúzia (3, 3 %)
 Popis
vy
braných
nežiaducich
účinkov
Trombocytopénia
Popis
vy
braných
nežiaducich
účinkov
Trombocytopénia
V kľúčovej štúdii fázy 3 u pacientov s FCS (štúdia APPROACH) sa potvrdilo zníženie počtu krvných
doštičiek pod normálnu hladinu (140 x 109/l) u 75 % pacientov s FCS liečených s volanesorsenom a
24 % pacientov užívajúcich placebo; potvrdilo sa zníženie pod 100 x 109/l u 47 % pacientov liečených volanesorsenom v porovnaní s pacientmi bez placeba. V štúdii APPROACH a jej otvorenom rozšírení (CS7) pacienti, ktorí prerušili liečbu kvôli hladinám krvných doštičiek, zahŕňali 3 pacientov s počtom krvných doštičiek <25 x 109/l, 2 s počtom krvných doštičiek medzi 25 x 109/l a 50 x 109/l a 5 s počtom krvných doštičiek medzi 50 x 109/l a 75 x 109/l. Žiadny z týchto pacientov nemal nijaké závažné krvácavé príhody a všetci sa regenerovali na normálny počet krvných doštičiek po vysadení lieku u
všetkých pacientov a pri podávaní glukokortikosteroidov pacientom, u ktorých to bolo medicínsky indikované.
ImunogenitaV klinických štúdiách fázy 3 (CS16 a APPROACH) 16 % a 30 % pacientov liečených volanesorsenom malo počas 6-mesačnej a 12-mesačnej liečby pozitívne testy na protilátky proti volanesorsenu. Prítomnosť protilátok proti lieku nebola spojená so žiadnymi dôkazmi o zmenenom bezpečnostnom profile alebo klinickej odpovedi, avšak toto pozorovanie je založené na obmedzených dlhodobých údajoch (pozri časť 4.4).
Reakcie v mieste vpichu injekcieV štúdii APPROACH na pacientoch s FCS, kde reakcie v mieste vpichu boli definované ako akákoľvek lokálna kožná reakcia v mieste vpichu pretrvávajúca dlhšie ako 2 dni, sa vyskytli u 82 % pacientov liečených volanesorsenom v priebehu štúdie APPROACH a jej otvoreného rozšírenia (CS7). Tieto lokálne reakcie boli väčšinou mierne a typicky pozostávali z jedného alebo viacerých príznakov ako: erytém, bolesť, svrbenie alebo lokálne opuchnutie. Reakcie v mieste vpichu sa nevyskytli pri všetkých injekciách a v štúdii APPROACH si vyžiadali prerušenie liečby u 1 pacienta.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce účinky na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeexistujú žiadne klinické skúsenosti s predávkovaním tohto lieku. V prípade predávkovania je potrebné starostlivo sledovať pacientov a podľa potreby poskytovať podpornú starostlivosť. Predpokladá sa, že príznaky predávkovania budú obmedzené na konštitučné symptómy a reakcie v mieste vpichu.
Je nepravdepodobné, že by hemodialýza bola prospešná, nakoľko volanesorsen sa rýchlo distribuuje do buniek.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina:{zatiaľ nepridelená}, ATC kód: {zatiaľ nepridelený}
Mechanizmus účinkuVolanesorsen je antisense oligonukleotid zameraný na inhibíciu tvorby apoC-III, proteínu, ktorý je
rozpoznaný ako proteín regulujúci ako metabolizmus triglyceridov, tak aj pečeňový klírens chylomikrónov a iných lipoproteínov bohatých na triglyceridy. Selektívna väzba volanesorsenu na apoC-III mediátor ribonukleovej kyseliny (mRNA) v 3'netranslatovanej oblasti v bázickej pozícii 489-
508 spôsobuje degradáciu mRNA. Táto väzba zabraňuje translácii proteínu apoC-III, čím sa odstráni
inhibítor klírensu triglyceridov a umožní sa metabolizmus prostredníctvom LPL-nezávislej dráhy.
Farmakodynamické účinkyÚčinky Waylivry na lipidové parametreV štúdii APPROACH klinická štúdia fázy 3 u pacientov s FCS, Waylivraznížila hladiny triglyceridov nalačno, celkového cholesterolu, non-HDL cholesterolu, hladiny apoC-III, apoB-48 a triglyceridov chylomikrónu a zvýšil hladiny LDL-C, HDL-C a apoB (pozri Tabuľku 3).
T
abuľka 3: Priemerná východisková a percentuálna zmena parametrov lipidov od východiskového stavu do tretieho mesiaca
L
i
pidový parameter (g/l pre apoC-III, apoB, apoB-
48, mmol/l pre cholesterol,
t
riglyceridy)
Placebo
(
N=
33)
Východiskový stav
Volanesorsen285 mg(N=33)Východiskový
stav % zmena
Triglyceridy 24,3 +24 % 25,6 -72 % Celkový cholesterol 7,3 +13 % 7,6 -39 % LDL-C 0,72 +7 % 0,73 +139 % HDL-C 0,43 +5 % 0,44 +45 % Non-HDL-C 6,9 +14 % 7,1 -45 % ApoC-III 0,29 +6 % 0,31 -84 % ApoB 0,69 +2 % 0,65 +20 % ApoB-48 0,09 +16 % 0,11 -75 % Triglyceridy chylomikrónu 20 +38 % 22 -77 %
Kardiálna elektrofyziológiaPri koncentrácii lieku 4,1-násobku maximálnych plazmatických koncentrácií lieku (Cmax) maximálnej
odporúčanej dávky (285 mg subkutánnej injekcie), volanesorsen nepredĺžil korigovaný interval QT
(QTc) na srdcovú frekvenciu.
Klinická účinnosť abezpečnosťŠtúdia APPROACH u pacientov s FCSŠtúdia APPROACH je randomizovaná, dvojito zaslepená placebom kontrolovaná 52-týždňová
multicentrická klinická štúdia u 66 pacientov s FCS, hodnotiaca 285 mg volanesorsenu podaného ako subkutánna injekcia (33 pacientov liečených volanesorsenom, 33 s placebom). Hlavné kritériá zaradenia boli: diagnóza FCS (hyperlipoproteinémia typu 1) v kombinácii s anamnézou chylomikronémie preukázanej dokumentáciou chylózneho séra alebo dokumentáciou merania TG nalačno ≥880 mg/dl.
Diagnóza FCS s požadovanou dokumentáciou aspoň jednej z nasledujúcich:
a) potvrdený homozygot, zmes heterozygotov alebo dvojitý heterozygot pre známe stratové funkcie mutácií v génoch spôsobujúcich typ 1 (ako je LPL, APOC2, GPIHBP1 alebo LMF1),
b) aktivita LPL v postheparínovej plazme ≤20 % normálnej hodnoty.
Pacienti užívajúci liek Glybera v období 2 rokov pred skríningom boli vylúčení zo štúdie.
Devätnásť z 33 pacientov vo volanesorsenovej skupine absolvovalo 12-mesačnú štúdiovú liečbu. Trinásti z týchto pacientov mali počas štúdie upravenú dávku/prestávku. Z týchto 13 pacientov 5 mali prestávku v dávkovaní, 5 mali upravenú dávku a 3 mali pozastavenú aj upravenú dávku.
Priemerný vek bol 46 rokov (rozpätie 20-75 rokov, 5 pacienti ≥65 rokov); 45 % boli muži; 80 % belosi, 17 % ázijskej rasy a 3 % boli inej rasy. Priemerný index telesnej hmotnosti bol 25 kg/m2. Anamnéza preukázanej akútnej pankreatitídy bola hlásená u 76 % pacientov a anamnéza diabetu
u 15 % pacientov; 21 % pacientov malo zaznamenanú anamnézu lipémie retinalis a 23 % pacientov malo zaznamenanú anamnézu eruptívnych xantómov. Stredný vek pri diagnostike bol 27 rokov, u
23 % sa preukázalo, že nemajú známu genetickú mutáciu FCS.
Pri zaradení do štúdie, 55 % pacientov absolvovalo terapiu zníženia hladiny lipidov (48 % bolo na fibrátoch, 29 % na rybích olejoch, 20 % na inhibítoroch HMG-CoA reduktázy), 27 % na liekoch proti bolesti, 20 % na inhibítoroch agregácie krvných doštičiek a 14 % užívalo výživové doplnky. Súčasne prebiehajúce terapie zamerané na zníženie hladiny lipidov zostali počas celej štúdie konzistentné. V priebehu 4 týždňov pred skríningom alebo počas štúdie pacienti nesmeli dostávať aferézu plazmy;
11 % pacientov predtým dostalo génovú terapiu nedostatku lipoproteínovej lipázy (t. j. alipogén tiparvovec); bolo to v priemere 8 rokov pred začiatkom tejto štúdie. Po 6-týždňovej dobe trvania diéty bola priemerná hladina triglyceridov nalačno na začiatku 2 209 mg/dl (25,0 mmol/l). Dodržiavanie
diéty a obmedzenie alkoholu bolo v priebehu štúdie podporované pravidelnými poradenskými stretnutiami.
Waylivra viedla k štatisticky významnému zníženiu hladín triglyceridov v porovnaní s placebom pri primárnej cieľovej hodnote účinnosti, ktorá bola definovaná ako percentuálna zmena v porovnaní východiskového stavu s 3. mesiacom triglyceridov nalačno, okrem zníženia výskytu pankreatitídy počas 52-týždňového liečebného obdobia v následnej analýze (Tabuľka 4).
Pri primárnej cieľovej hodnote účinnosti bol rozdiel medzi liečebným postupom volanesorsenom a placebom v percentách zmeny priemerného triglyceridu nalačno, a to: -94 % (95 % CI: -122 %, -67 %; p˂0,0001), s poklesom o -77 % oproti východiskovej hodnote (95 % CI: -97, -56) u pacientov užívajúcich volanesorsen a zvýšenie o 18 % oproti východiskovej hodnote (95 % CI: -4, 39) u pacientov, ktorí dostávali placebo (Tabuľka 4).
Tabuľka4: Priemerná zmena z východiskovej hodnoty u triglyceridov nalačno vo fáze 3
placebom kontrolovanej štúdie u pacientov s FCS v mesiaci 3 (APPROACH)
Stredná zmena
Placebo
(N=33)
Volanesorsen 285 mg
(N=33)
Relatívny rozdiel v
zmene vs placebo
percenta (95 % CI) +18 % (-4, 39) -77 % (-97, -56) -94 %* (-122, -67)
Priemerná
absolútna zmena
LS (95 % CI)
mg/dl alebo mmol/l
+92 (-301, +486) mg/dl
+1 (-3, +5) mmol/l
-1 712 (-2 094, -
1 330) mg/dl
-19 (-24, -15) mmol/l
-1 804 (-2 306, -
1 302) mg/dl
-20 (-26, -15) mmol/l

*hodnota p <0,0001 (primárny koncový ukazovateľ účinnosti)
Rozdiel = LS stredný priemer [volanesorsen % zmena - placebo % zmena] (model ANCOVA)
Nástup zníženia bol rýchly s odstupom od placeba pozorovaným takmer 4 týždne a maximálna odpoveď pozorovaná po 12 týždňoch s klinicky a štatisticky významnou redukciou triglyceridov v priebehu 52 týždňov (Obrázok 1). Stredná zmena percenta triglyceridov nalačno bola významne rozdielna medzi ramenami s volanesorsenom a placebom po 3, 6 a 12 mesiacoch; rameno s volanesorsenom zahŕňalo pacientov, ktorí celkom nedokončili dávkovanie, ale ktorí sa vrátili pre hodnotenia počas 52-týždňovej štúdie. Neexistujú žiadne významné rozdiely v účinku liečby naprieč stratifikačnými faktormi prítomnosti alebo neprítomnosti súbežných omega-3 mastných kyselín alebo fibrátov.
Obrázok 1: Stredná percentuálna zmena hladiny triglyceridov nalačno v štúdii fázy 3 u pacientov s FCS (APPROACH)
Percentuálna zmena oproti východiskovej hodnote u triglyceridov nalačno:
Podsúbor pacientov s nechýbajúcimi koncovými hodnotami, úplná analytická sada

20%
18%
10%
0%
-10%
-20%
Placebo Waylivra
-30%
-40%
-50%
-60%
-70%
-80%
-77%
0 2 4 6 8 10 12
Mesiac
LS znamená priemernú percentuálnu zmenu triglyceridov nalačno na základe pozorovaných údajov oproti
východiskovej hodnote.
Rozdiel = stredný priemer [volanesorsen % zmena - placebo % zmena] (model ANCOVA)
p-hodnota modelu ANCOVA <0,0001 v 3. mesiaci (primárny cieľový ukazovateľ účinnosti), 6. mesiaci a 12. mesiaci
Ďalšie výsledky účinnosti pre zmeny triglyceridov sú uvedené v tabuľke 5. Väčšina pacientov, ktorí
dostávali liek volanesorsen, zaznamenala klinicky významné zníženie triglyceridov.
Tabuľka 5: Ďalšie výsledky pre zmeny triglyceridov v štúdii APPROACH (primárny koncový
ukazovateľ v 3. mesiaci)
V
ýskyt v 3. mesiacia
Placebo
(N=31)
Volanesorsen 285 mg
(N=30)
Percento pacientovb s triglyceridom
plazmy nalačno <750 mg/dl
(8,5 mmol/l)*
10 % 77 %
Percento pacientovc s ≥40 %
znížením triglyceridov nalačno**
9 % 88 %


a Koncový bod 3. mesiaca bol definovaný ako priemer hodnôt nalačno v 12. týždni (78. dni) a 13. týždni
(85. dni). Ak jedna návšteva chýbala, druhá návšteva bola použitá ako koncový bod.
b Menovateľom percentuálneho výpočtu bol celkový počet pacientov v systéme FAS s triglyceridom nalačno na začiatku dávky ≥750 mg/dl (alebo 8,5 mmol/l) v každej liečebnej skupine.
c Menovateľom percentuálneho výpočtu bol celkový počet pacientov v každej liečebnej skupine.
* Hodnota p = 0,0001
**Hodnota p <0,0001
Hodnoty P z logistického regresného modelu s liečbou, prítomnosťou pankreatitídy a prítomnosťou súbežných omega-3 mastných kyselín a/alebo fibrátov ako faktorov a logaritmicky transformované základné triglyceridy nalačno ako kovariát.'
V štúdii APPROACH bola numerická incidencia pankreatitídy u pacientov liečených volanesorsenom
nižšia v porovnaní s placebom (3 pacienti 4 udalosti v skupine 33 pacientov s placebom v porovnaní s
1 pacientom 1 udalosťou v skupine 33 pacientov s volanesorsenom).
Analýza pacientov s príhodami recidivujúcej pankreatitídy v anamnéze (≥2 prípady v priebehu 5 rokov pred 1. dňom štúdie) ukázala významné zníženie záchvatov pankreatitídy u pacientov liečených volanesorsenom v porovnaní s pacientmi liečenými placebom (p = 0,0242). Vo volanesorsenovej skupine pacientov zo 7 pacientov, u ktorých sa v predchádzajúcich 5 rokoch vyskytlo 24 atakov pankreatitídy, sa u žiadneho pacienta nedostavil počas 52 týždňov trvajúceho liečebného obdobia žiaden atak pankreatitídy. V placebovej skupine pacientov, zo 4 pacientov, u ktorých sa v predchádzajúcich 5 rokoch vyskytlo 17 atakov pankreatitídy, 3 pacienti podstúpili 4 ataky
pankreatitídy počas 52-týždňového liečebného obdobia.
Otvorená rozšírená štúdia u pacientov s FCS
Štúdia CS7 je pretrvávajúca multicentrická, otvorená rozšírená štúdia fázy 3 určená na hodnotenie bezpečnosti a účinnosti dávkovania a rozšíreného dávkovania volanesorsenu u pacientov s FCS. Všetci zapísaní pacienti buď sa zúčastnili štúdie APPROACH, v štúdii CS16, alebo boli noví pacienti
s FCS a dokončili kvalifikačné hodnotenia predtým, ako dostávali liek volanesorsen 285 mg jedenkrát
týždenne alebo znížili frekvenciu z dôvodov bezpečnosti alebo znášanlivosti v ich indexovej štúdii. Celkovo bolo liečených 67 pacientov a 50 (74 %) pacientov zotrvalo v liečbe; z nich 38 (76 %) pacientov tvorilo skupinu bez predchádzajúcej liečby, 9 (18 %) skupinu APPROACH, ktorá dostávala volanesorsen a 3 (6 %) tvorili skupinu CS16, liečenú volanesorsenom. Z 50 pacientov, ktorí boli ešte stále na liečbe, malo 8 pacientov pozastavené dávky, 8 pacientom upravili dávkovanie a 29 pacientov malo pozastavené dávky, ako aj upravené dávkovanie.
Najnovšie údaje prebiehajúcej štúdie CS7 sú uvedené v tabuľke 6. Percentuálna zmena hladiny TG nalačno z východiskového indexu štúdie na otvorený mesiac 3 u pacientov s APPROACH- a CS16- volanesorsenom bola -49,2 %, resp. -64,9 %. Percentuálna zmena hladiny TG od východiskového indexu štúdie na otvorený mesiac 6 a mesiac 12 u APPROACH-volanesorsenových pacientov bola -
54,8 %, resp. -35,1 %.
Tabuľka 6: Súhrn triglyceridov nalačno (priemerný (SD, SEM), mg/dl) počas štúdie CS7
Časový
bod
S
k
upina bez predchádzajúcej liečby (Otvorená štúdia Baseline
a
, N=51)
% zmeny oproti východisk
APPROACH-volanesorsen
(
I
ndex Study Baseline
a
, N=14)
% zmeny oproti východisk ovej
CS16-volanesorsen
(
I
ndex Study Baseline
a
, N=3)
% zmeny oproti východisk
Východisko
Pozorova ná
n hodnota
2341
ovej hodnote v
CS7 n
Pozorova ná hodnota
2641
hodnote v
APPROA
CH n
Pozorova ná hodnota
2288
ovej hodnote v CS16
vá hodnotaa 51
3. mesiac
(1193;
167)
- 14
(1228;
328)
1266
- 3 (1524; -
880)
6. mesiac
12. mesiac
47 804 (564;
82)
49 1032 (695; 99)
1345
-59,8 (37,0;
5,4) 14
-45,5 (42,9;
6,1) 13
-31,6 (44,6;
(812;
217)
1248 (927;
257)
1670
-49,2
(34,8; 9,3) 3
-54,8
(23,8; 6,6) 3
-35,1
855 (651;
376)
1215 (610;
352)
1369
-64,9 (9,1;
5,3)
-43,0 (19,7;
11,4)
-39,9
39
15. mesiac
(959;
154)
1374
7,1)
12 (1198;
346)
1886
(45,6;
13,2)
-26,5
3 (897;
518)
(34,2;
19,7)

22 (1090;
232)
-36,4 (41,0;
8,7) 10
(1219;
386)
(57,4;
18,1)
0 NV NV
18. mesiac
9
1139 (690;
230)
-38,7 (42,1;
14,0) 7
1713 (1122;
424)
-38,4 (32,2;
12,2)
0 NV NV

a Východiskové hodnoty pre skupinu pacientov bez predchádzajúcej liečby boli prevzaté z otvorenej štúdie CS7
a východisková hodnota pre skupiny APPROACH-volanesorsen a CS16-volanesorsen bola prevzatá z príslušnej indexovej štúdie.
NC = not calculated (nevypočítané)
Starší ľudiaKlinické štúdie zahŕňali 4 pacientov s FCS vo veku 65 rokov liečených s volanesorsenom v
randomizovaných kontrolných štúdiách (štúdia fázy II CS2, 1 pacient; APPROACH 3 pacienti)
a 6 pacientov vo veku 65 rokov a viac v otvorenej rozšírenej štúdii (CS7). Medzi týmito pacientmi a mladšími pacientmi neboli pozorované žiadne celkové rozdiely v bezpečnosti alebo účinnosti, avšak údaje o tejto subpopulácii sú obmedzené.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s volanesorsenom vo všetkých podskupinách pediatrickej populácie {v súlade so schváleným výskumným pediatrickým plánom (PIP) pre schválenú indikáciu} (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPo subkutánnej injekcii sa maximálne plazmatické koncentrácie volanesorsenu obvykle dosiahnu za 2 až 4 hodiny. Absolútna biologická dostupnosť volanesorsenu po jednorazovom podkožnom podaní je približne 80 % (s najväčšou pravdepodobnosťou vyššia, pretože sa použila AUC od 0 do 24 hodín a polčas rozpadu volanesorsenu bol >2 týždne).
Po podaní dávky 285 mg jedenkrát týždenne u pacientov s FCS je odhadovaný geometrický priemer Cmax v rovnovážnom stave (koeficientu variácie % geometrického priemeru) je 8,92 μg/ml (35 %), AUC0-168h je 136 μg*h/ml (38 %) a Ctrough je 127 ng/ml (58 %) u pacientov, ktorí zostávajú negatívni
na protilátky proti lieku. Alternatívny dávkovací režim 285 mg volanesorsenu každé dva týždne má za
následok Ctrough,ss približne 58,0 ng/ml s Cmax a AUC podobnými v porovnaní s dávkovacím režimom raz týždenne.
DistribúciaVšetky hodnotené vzorky volanesorsenu boli po subkutánnom alebo intravenóznom podaní rýchlo a široko distribuované do tkanív. Odhadovaný distribučný objem v rovnovážnom stave (Vss) u pacientov s FCS je 330 l. Volanesorsen je vysoko viazaný na proteíny ľudskej plazmy (>98 %) a táto väzba nezávisí od koncentrácie.
In vitro štúdie ukazujú, že volanesorsen nie je substrátom alebo inhibítorom P-glykoproteínu (P-gp), proteín rezistencie voči rakovine prsníka (breast cancer resistance protein - BCRP), polypeptidov transportujúcich organické anióny (organic anion transporting polypeptides - OATP1B1, OATP1B3), pumpy na báze žlčových solí (bile salt export pump - BSEP), transportérov organických katiónov (organic cation transporters - OCT1, OCT2) alebo transportérov organických katiónov (organic anion transporters - OAT1, OAT3).
BiotransformáciaVolanesorsen nie je substrátom metabolizmu CYP a metabolizuje sa v tkanivách endonukleázami za vzniku kratších oligonukleotidov, ktoré sú potom substrátmi pre ďalší metabolizmus pomocou exonukleáz. Nezmenený volanesorsen je prevládajúca cirkulujúca zložka.
Štúdie in vitro naznačujú, že volanesorsen nie je inhibítorom CYP1A2, CYP2B6, CYP2C8, CYP2C9,
CYP2C19, CYP2D6, CYP2E1 alebo CYP3A4 alebo induktorom CYP1A2, CYP2B6 alebo CYP3A4.
E
li
m
i
nácia
Eliminácia zahŕňa metabolizmus v tkanivách a vylučovanie močom. Opätovný zisk materského liečiva v moči bol u ľudí obmedzený <3 % subkutánne podanej dávky získanej do 24 hodín po podaní dávky. Materské liečivo a 5- až 7-merové metabolity so skráteným reťazcom predstavovali približne 26 % a
55 % oligonukleotidov získaných z moču. Po subkutánnom podaní je terminálny polčas eliminácie približne 2 až 5 týždňov.
U zvierat bola eliminácia volanesorsenu pomalá a vyskytuje sa hlavne vylučovaním močom, čo odráža rýchly plazmatický klírens hlavne v tkanivách. V ľudskom moči bol zistený ako volanesorsen, tak aj kratší oligonukleotidový metabolit (prevažne 7-merné metabolity (generované buď z 3'-delécií alebo
5'-delécií)).
Linearita/nelinearita
Farmakokinetika jednorazovej a viacnásobnej dávky volanesorsenu u zdravých dobrovoľníkov a
pacientov s hypertriglyceridémiou ukázala, že Cmax volanesorsenu je úmerná dávke v rozsahu dávok
100 až 400 mg a AUC je o niečo viac ako proporcionálna dávka v tom istom rozpätí dávok. Ustálený stav bol dosiahnutý približne 3 mesiace po začatí liečby volanesorsenom. Bola pozorovaná akumulácia v Ctrough (7- až 14-krát) a po týždennom SC podávaní v dávke 200 až 400 mg bol pozorovaný len malý alebo žiadny nárast Cmax alebo AUC. Pri dávke 50 až 100 mg bola pozorovaná určitá akumulácia v AUC a Cmax. Keďže každé dva týždne bude podaná dávka 285 mg alebo každý týždeň 142,5 mg, očakáva sa len malý nárast Cmax alebo AUC po opakovanom podávaní v klinickom nastavení.
Osobitné skupiny pacientov
Porucha funkcie obličiek
Populačná farmakokinetická analýza naznačuje, že mierna a stredne závažná porucha funkcie obličiek nemá klinicky významný vplyv na systémovú expozíciu volanesorsenu. Nie sú dostupné žiadne údaje u pacientov so závažnou poruchou funkcie obličiek.
Porucha funkcie pečene
Farmakokinetika volanesorsenu u pacientov s poruchou funkcie pečene nie je známa.
Vek, pohlavie, váha a rasa
Na základe populačnej farmakokinetickej analýzy nemá vek, telesná hmotnosť, pohlavie alebo rasa žiadny klinicky relevantný účinok na expozíciu volanesorsenu. Pre osoby staršie ako 75 rokov sú k dispozícii obmedzené údaje.
Tvorba anti-volanesorsenovýchprotilátokovplyvňujúcichfarmakokinetiku
Zdá sa, že tvorba väzobných protilátok proti volanesorsenu zvyšuje celkový Ctrough 2- až 19-násobne.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých štúdií farmakologickej bezpečnosti, genotoxicity, karcinogenicity alebo reprodukčnej a vývinovej toxicity neodhalili žiadne osobitné riziko pre ľudí.
Dávkovo a časovo podmienené zníženie počtu krvných doštičiek bolo pozorované v štúdiách s opakovanými dávkami u opice Cynomolgus. Pokles bol postupný, udržateľný a neklesol na nepriaznivé úrovne. U jednotlivých opíc bola zaznamenaná ťažká trombocytopénia v 9-mesačnej štúdii liečebných skupín pri klinicky významných expozíciách a bola tiež pozorovaná aj v klinických štúdiách. Pokles počtu doštičiek nebol akútny a znížil sa pod 50 000 buniek/μl. Počet trombocytov sa vrátil späť po zastavení liečby, ale po obnovení liečby u niektorých opíc opäť klesol pod
50 000 buniek/μl. Znížený počet krvných doštičiek bol pozorovaný aj v štúdiách hlodavcov s
opakovanými dávkami. Mechanizmus účinku na pozorovanú trombocytopéniu nie je v súčasnosti
známy.
V neklinických štúdiách laktujúcich myší boli hladiny volanesorsenu v mlieku veľmi nízke. Koncentrácie v materskom mlieku myší boli >800-krát nižšie ako účinné koncentrácie v tkanivách materskej pečene. Vzhľadom na slabú perorálnu biologickú dostupnosť volanesorsenu sa nepovažuje za pravdepodobné, že tieto nízke koncentrácie mlieka budú mať za následok systémovú expozíciu v dôsledku ošetrovania (pozri časť 4.6).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
hydroxid sodný (na úpravu pH)
kyselina chlorovodíková (na úpravu pH)
voda na injekcie.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
Tento liek možno vybrať z chladničky a skladovať v pôvodnom obale pri izbovej teplote (do 30 °C) až
6 týždňov. Počas tohto šesťtýždňového obdobia sa môže podľa potreby uchovávať medzi chladiacou a izbovou teplotou (do 30 °C). Tento liek sa musí okamžite zlikvidovať, ak sa nepoužije v priebehu 6 týždňov po jeho prvom vybratí z chladeného skladu.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Nezmrazujte.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Jednorazová naplnená injekčná striekačka zo skla typu I so silikonizovanou chlórbutylovou gumovou zátkou a nasadenou ihlou s krytom, naplnená pre podanie 1,5 ml roztoku.
Veľkosti balenia jednej naplnenej striekačky alebo štvorbalení obsahujúcich 4 (4 balenia po 1)
naplnené injekčné striekačky.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Waylivra sa musí pred podaním vizuálne skontrolovať. Roztok musí byť číry a bezfarebný až nažltlý. Ak je roztok zakalený alebo obsahuje viditeľné čiastočky, obsah sa nesmie podať a liek sa musí vrátiť do lekárne.
Každú naplnenú injekčnú striekačku použite iba raz a potom ju vložte do nádoby na likvidáciu ostrých
predmetov podľa miestnych nariadení.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Akcea Therapeutics Ireland Ltd. Regus House, Harcourt Centre, Harcourt Road,
Dublin 2
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/19/1360/001
EU/1/19/1360/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 03. máj 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.