)
Nie je potrebná úprava dávkovania (pozri časť 5.2).
Porucha funkcie obličiekNie je potrebná úprava dávkovania (pozri časť 5.2).
Porucha funkcie pečene- Mierna porucha funkcie pečene (trieda A podľa Childa-Pugha) – odporúčaná dávka je 7,5 mg denne.
- Stredne závažná porucha funkcie pečene (trieda B podľa Childa-Pugha) – odporúčaná dávka je 5 mg denne.
- Závažná porucha funkcie pečene (trieda C podľa Childa-Pugha) – Verimmus sa odporúča len vtedy, ak želaný prínos preváži riziko. V takom prípade sa nesmie prekročiť dávka 2,5 mg denne.
Dávka sa má upravovať, ak sa počas liečby mení stav pečene pacienta (podľa Childa-Pugha) (pozri tiež časti 4.4 a 5.2).
Pediatrická populáciaBezpečnosť a účinnosť Verimmu u detí vo veku 0 až 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávaniaVerimmus sa má podávať perorálne raz denne v rovnakom čase každý deň, vždy buď s jedlom, alebo bez jedla (pozri časť 5.2). Tablety Verimmu sa majú prehĺtať celé a zapíjať pohárom vody. Tablety sa nemajú hrýzť ani drviť.
4.3 KontraindikáciePrecitlivenosť na liečivo, na iné deriváty rapamycínu alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaníNeinfekčná pneumonitídaNeinfekčná pneumonitída je účinok triedy derivátov rapamycínu vrátane everolimu. Neinfekčná pneumonitída (vrátane intersticiálnej choroby pľúc) bola často hlásená u pacientov užívajúcich Verimmus (pozri časť 4.8). Niektoré prípady boli závažné a zriedkavo sa pozorovalo, že sa skončili fatálne. Diagnóza neinfekčnej pneumonitídy sa má vziať do úvahy u pacientov, u ktorých sa objavia nešpecifické respiračné prejavy a príznaky, napr. hypoxia, pleurálny výpotok, kašeľ alebo dyspnoe a u ktorých sa príslušnými vyšetreniami vylúčili infekčné, neoplastické alebo iné nemedicínske príčiny. Pri diferenciálnej diagnostike neinfekčnej pneumonitídy sa majú vylúčiť oportúnne infekcie ako pneumónia spôsobená
Pneumocystis jirovecii (
Pneumocystis carinii) (PJP, PCP) (pozri „Infekcie“ nižšie). Pacientov je potrebné upozorniť, aby ihneď hlásili akékoľvek nové alebo zhoršujúce sa respiračné príznaky.
Pacienti, u ktorých nastanú rádiologické zmeny poukazujúce na neinfekčnú pneumonitídu a majú len málo príznakov alebo nemajú žiadne príznaky, môžu naďalej dostávať liečbu Verimmom bez úpravy dávkovania. Ak sú príznaky stredne závažné (stupeň 2) alebo závažné (stupeň 3), môže byť indikované podávanie kortikosteroidov až do ústupu klinických príznakov.
U pacientov s neinfekčnou pneumonitídou, ktorých je potrebné liečiť kortikosteroidmi, je vhodné zvážiť profylaxiu pneumónie spôsobenej
Pneumocystis jirovecii (
Pneumocystis carinii) (PJP, PCP).
InfekcieEverolimus má imunosupresívne vlastnosti a môže u pacientov vyvolať predispozíciu na bakteriálne, hubové, vírusové alebo protozoálne infekcie vrátane infekcií oportúnnymi patogénmi (pozri časť 4.8). U pacientov užívajúcich everolimus boli opísané lokalizované a systémové infekcie vrátane pneumónie, iné bakteriálne infekcie, invazívne hubové infekcie, napr. aspergilóza, kandidóza alebo pneumónia spôsobená
Pneumocystis jirovecii (
Pneumocystis carinii) (PJP, PCP) a vírusové infekcie vrátane reaktivácie vírusu hepatitídy B. Niektoré z týchto infekčných ochorení boli závažné (napr. viedli k sepse, zlyhaniu dýchania alebo pečene) a ojedinele sa skončili fatálne.
Lekári aj pacienti si majú byť vedomí zvýšeného rizika infekcie pri užívaní Verimmu. Už existujúce infekčné ochorenia sa majú primerane liečiť a majú úplne vymiznúť pred začatím liečby Verimmom. Počas užívania Verimmu je potrebné dávať pozor na príznaky a prejavy infekcie; ak sa diagnostikuje infekcia, má sa ihneď začať náležitá liečba a má sa zvážiť prerušenie alebo ukončenie liečby Verimmom.
Ak sa diagnostikuje invazívna systémová hubová infekcia, liečba Verimmom sa má ihneď a natrvalo ukončiť a pacient má dostať primeranú antimykotickú liečbu.
U pacientov užívajúcich everolimus sa zaznamenali prípady pneumónie spôsobenej
Pneumocystis jirovecii (
Pneumocystis carinii) (PJP, PCP), niektoré s fatálnym koncom. PJP/PCP môže súvisieť so súbežnou liečbou kortikosteroidmi alebo inými imunosupresívami. Ak je potrebná súbežná liečba kortikosteroidmi alebo inými imunosupresívami, má sa zvážiť profylaxia PJP/PCP.
Reakcie z precitlivenostiPri everolime sa pozorovali reakcie z precitlivenosti prejavujúce sa príznakmi, ktoré zahŕňali, ale neboli obmedzené len na anafylaxiu, dyspnoe, návaly horúčavy, bolesť na hrudi alebo angioedém (napr. edém dýchacích ciest alebo jazyka, s poruchou dýchania alebo bez nej) (pozri časť 4.3).
Súbežná liečba s inhibítormi enzýmu konvertujúceho angiotenzín (ACE)Pacienti užívajúci súbežne liečbu ACE inhibítormi (napr. ramipril) môžu mať zvýšené riziko angioedému (napr. opuch dýchacích ciest alebo jazyka, s poruchou dýchania alebo bez nej) (pozri časť 4.5).
StomatitídaStomatitída, vrátane ulcerácií ústnej dutiny a orálnej mukozitídy, je najčastejšie hlásenou nežiaducou reakciou u pacientov liečených everolimom (pozri časť 4.8). Stomatitída sa väčšinou vyskytuje počas prvých 8 týždňov liečby. Štúdia s jednou skupinou postmenopauzálnych pacientok s karcinómom prsníka liečených everolimom a exemestánom ukázala, že orálny roztok kortikosteroidu neobsahujúci alkohol, ktorý sa používa na vyplachovanie ústnej dutiny počas prvých 8 týždňov liečby, môže znížiť incidenciu a závažnosť stomatitídy (pozri časť 5.1). Zvládnutie stomatitídy preto môže zahŕňať profylaktické a/alebo terapeutické použitie topických liekov, ako je orálny roztok kortikosteroidu neobsahujúci alkohol na vyplachovanie ústnej dutiny. Je však potrebné vyhnúť sa produktom obsahujúcim alkohol, peroxid vodíka, jód a deriváty tymiánu, pretože môžu stav zhoršiť. Odporúča sa monitorovanie a liečba hubovej infekcie, hlavne u pacientov liečených liekmi steroidného charakteru. Antimykotiká sa nemajú používať, pokiaľ sa nediagnostikovala hubová infekcia (pozri časť 4.5).
Prípady zlyhania obličiekU pacientov liečených everolimom sa pozorovali prípady zlyhania obličiek (vrátane akútneho zlyhania obličiek), ktoré sa niekedy skončili fatálne (pozri časť 4.8). Funkcia obličiek sa má monitorovať najmä u pacientov s ďalšími rizikovými faktormi, ktoré môžu funkciu obličiek ďalej zhoršovať.
Laboratórne testy a monitorovanieFunkcia obličiekBolo hlásené zvýšenie sérového kreatinínu, zvyčajne mierne a proteinúria (pozri časť 4.8). Monitorovanie funkcie obličiek, vrátane stanovenia dusíka močoviny v krvi (z angl. blood urea nitrogen, BUN), bielkovín v moči alebo sérového kreatinínu, sa odporúča pred začatím liečby Verimmom a následne v pravidelných intervaloch.
Glukóza v krviBola hlásená hyperglykémia (pozri časť 4.8). Monitorovanie glukózy v sére nalačno sa odporúča pred začatím liečby Verimmom a následne v pravidelných intervaloch. Ak sa Verimmus podáva spolu s inými liekmi, ktoré môžu indukovať hyperglykémiu, odporúča sa častejšie monitorovanie. Ak je to možné, optimálna úprava glykémie sa má u pacienta dosiahnuť pred začiatkom liečby Verimmom.
Lipidy v krviBola hlásená dyslipidémia (vrátane hypercholesterolémie a hypertriglyceridémia). Monitorovanie cholesterolu a triacylglycerolov v krvi sa odporúča pred začatím liečby Verimmom a následne v pravidelných intervaloch ako aj ich kontrola vhodnou liečbou.
Hematologické parametreBol hlásený pokles hemoglobínu, lymfocytov, neutrofilov a trombocytov (pozri časť 4.8). Monitorovanie kompletného krvného obrazu sa odporúča pred začatím liečby Verimmom a následne v pravidelných intervaloch.
Funkčné karcinoidné nádoryV randomizovanom, dvojito zaslepenom, multicentrickom klinickom skúšaní s pacientmi s funkčnými karcinoidnými nádormi sa everolimus a depotný oktreotid porovnali s placebom a depotným oktreotidom. V štúdii sa nedosiahol primárny koncový ukazovateľ účinnosti (prežívanie bez progresie – z angl. progression-free-survival, PFS) a predbežná analýza celkového prežívania (overall survival, OS) ukázala numericky priaznivejšie výsledky v skupine s placebom a depotným oktreotidom. Bezpečnosť a účinnosť everolimu u pacientov s funkčnými karcinoidnými nádormi sa preto nepreukázali.
Prognostické faktory u neuroendokrinných nádorov gastrointestinálneho alebo pľúcneho pôvoduU pacientov s nefunkčnými neuroendokrinnými gastrointestinálnymi alebo pľúcnymi nádormi a dobrými prognostickými faktormi pri vstupnom vyšetrení, napr. ileum ako primárny pôvod nádoru a normálne hodnoty chromogranínu A alebo bez postihnutia kostí, sa má individuálne zhodnotenie pomeru prínosu a rizika vykonať pred začatím liečby Verimmom. Obmedzené dôkazy o prínose pre PFS sa zaznamenali v podskupine pacientov s ileom ako primárnym pôvodom nádoru (pozri časť 5.1).
InterakcieSúbežnému podávaniu s inhibítormi a induktormi CYP3A4 a/alebo nešpecifickej efluxnej pumpy glykoproteínu P (PgP) je potrebné sa vyhnúť. Ak sa nemožno vyhnúť súbežnému podávaniu
strednesilného inhibítora alebo induktora CYP3A4 a/alebo PgP, na základe predpokladanej AUC sa môže zvážiť úprava dávky Verimmu (pozri časť 4.5).
Súbežná liečba
silnými inibítormi CYP3A4 má za následok dramaticky zvýšené plazmatické koncentrácie everolimu (pozri časť 4.5). V súčasnosti nie sú dostatočné údaje, ktoré by umožnili odporučiť dávkovanie v tejto situácii. Preto sa súbežná liečba Verimmom a
silnými inhibítormi neodporúča.
Vzhľadom na možnosť liekových interakcií je potrebná opatrnosť pri užívaní Verimmu v kombinácii s perorálne podávanými substrátmi CYP3A4 s úzkym terapeutickým indexom. Ak sa Verimmus užíva s perorálne podávanými substrátmi CYP3A4 s úzkym terapeutickým indexom (napr. pimozidom, terfenadínom, astemizolom, cisapridom, chinidínom alebo derivátmi námeľových alkaloidov), pacienta je potrebné sledovať pre nežiaduce účinky opísané v informácii o lieku perorálne podávaného substrátu CYP3A4 (pozri časť 4.5).
Porucha funkcie pečeneExpozícia everolimu sa zvýšila u pacientov s miernou (trieda A podľa Childa-Pugha), stredne závažnou (trieda B podľa Childa-Pugha) a závažnou (trieda C podľa Childa-Pugha) poruchou funkcie pečene (pozri časť 5.2).
Použitie Verimmu sa odporúča u pacientov so závažnou poruchou funkcie pečene (trieda C podľa Childa-Pugha), len ak možný prínos preváži riziko (pozri časti 4.2 a 5.2).
V súčasnosti nie sú k dispozícii klinické údaje o bezpečnosti a účinnosti, na základe ktorých by bolo možné odporučiť úpravu dávky pri zvládaní nežiaducich reakcií u pacientov s poruchou funkcie pečene.
VakcináciePoužitiu živých vakcín počas liečby Verimmom je potrebné sa vyhnúť (pozri časť 4.5).
Komplikácie hojenia ránZhoršené hojenie rán je účinok triedy derivátov rapamycínu, vrátane everolimu. Preto je potrebná opatrnosť pri použití Verimmu v perioperačnom období.
Upozornenia súvisiace s pomocnou látkouPacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
4.5 Liekové a iné interakcieEverolimus je substrát CYP3A4 a tiež substrát a stredne silný inhibítor PgP. Preto môže byť absorpcia a následná eliminácia everolimu ovplyvnená liekmi, ktoré pôsobia na CYP3A4 a/alebo PgP. Everolimus je
in vitro kompetitívny inhibítor CYP3A4 a zmiešaný inhibítor CYP2D6.
Známe a teoretické interakcie s vybranými inhibítormi a induktormi CYP3A4 a PgP sú uvedené nižšie v tabuľke 2.
Inhibítory CYP3A4 a PgP zvyšujúce koncentrácie everolimuLátky, ktoré sú inhibítormi CYP3A4 alebo PgP, môžu zvýšiť koncentrácie everolimu v krvi znížením metabolizmu alebo efluxu everolimu z črevných buniek.
Induktory CYP3A4 a PgP znižujúce koncentrácie everolimuLátky, ktoré sú induktormi CYP3A4 alebo PgP, môžu znížiť koncentrácie everolimu v krvi zvyšovaním metabolizmu alebo efluxu everolimu z črevných buniek.
Tabuľka 2 Účinky iných liečiv na everolimus
Liečivo podľa interakcie
| Interakcia - zmena AUC/Cmax
everolimu
Pomer geometrických priemerov (pozorované rozmedzie)
| Odporúčania pre súbežné podávanie
|
|
Silné inhibítoryCYP3A4/PgP
|
Ketokonazol
| AUC ↑ 15,3-násobné
(rozmedzie 11,2 – 22,5)
Cmax ↑ 4,1-násobné
(rozmedzie 2,6 – 7,0)
| Súbežná liečba Verimmom a silnými inhibítormi sa neodporúča.
|
Itrakonazol,
posakonazol,
vorikonazol
| Nesledovalo sa. Očakáva sa výrazné zvýšenie koncentrácie everolimu.
|
Telitromycín,
klaritromycín
|
Nefazodón
|
Ritonavir, atazanavir, sakvinavir, darunavir, indinavir, nelfinavir
|
|
Stredne silné inhibítory CYP3A4/PgP
|
Erytromycín
| AUC ↑ 4,4-násobné
(rozmedzie 2,0 – 12,6)
Cmax ↑ 2,0-násobné
(rozmedzie 0,9 – 3,5)
| Postupujte opatrne, keď sa nemožno vyhnúť súbežnému podávaniu so stredne silnými inhibítormi CYP3A4 alebo inhibítormi PgP. Ak sa u pacientov vyžaduje súbežné podávanie so stredne silným inhibítorom CYP3A4 alebo PgP, možno zvážiť zníženie dávky na 5 mg denne alebo 2,5 mg denne. Nie sú však klinické údaje pri tejto úprave dávky. Vzhľadom na variabilitu medzi jedincami nemusia byť odporúčané úpravy dávky optimálne pre všetkých jedincov, preto sa odporúča dôsledné sledovanie vedľajších účinkov. Po ukončení podávania stredne silného inhibítora je potrebné zvážiť dobu potrebnú na elimináciu v dĺžke aspoň 2 až 3 dní (priemerný eliminačný polčas pre väčšinu bežne používaných stredne silných inhibítorov) predtým, ako sa dávka Verimmu vráti na dávku používanú pred začatím súbežného podávania.
|
Imatinib
| AUC ↑ 3,7-násobné
Cmax 2,2-násobné
|
Verapamil
| AUC ↑ 3,5-násobné
(rozmedzie 2,2 – 6,3)
Cmax ↑ 2,3-násobné
(rozmedzie 1,3 – 3,8)
|
Cyklosporín perorálne
| AUC↑ 2,7-násobné
(rozmedzie 1,5 – 4,7)
Cmax ↑ 1,8-násobné
(rozmedzie 1,3 – 2,6)
|
Flukonazol
| Nesledovalo sa. Očakáva sa zvýšená expozícia.
|
Diltiazem
|
Dronedarón
| Nesledovalo sa. Očakáva sa zvýšená expozícia.
|
Amprenavir,
fosamprenavir
| Nesledovalo sa. Očakáva sa zvýšená expozícia.
|
Grapefruitová šťava alebo iné jedlo ovplyvňujúce
CYP3A4/PgP
| Nesledovalo sa. Očakáva sa zvýšená expozícia (účinok sa značne líši).
| Kombinácii je potrebné sa vyhnúť.
|
|
Silné a stredne silné induktory CYP3A4
|
Rifampicín
| AUC ↓ 63 %
(rozmedzie 0 – 80 %)
Cmax↓ 58 %
(rozmedzie 10 – 70 %)
| Vyhýbajte sa súbežnému podávaniu so silnými induktormi CYP3A4. Ak sa u pacientov vyžaduje súbežné podávanie so silným induktorom CYP3A4, má sa zvážiť zvýšenie dávky Verimmu z 10 mg denne až na 20 mg denne, so zvyšovaním po 5 mg alebo menej uskutočneným na 4. a 8. deň po začatí liečby induktorom. Pri tejto dávke Verimmu sa predpokladá, že sa AUC upraví do rozmedzia pozorovaného bez induktorov. Nie sú však klinické údaje pri tejto úprave dávky. Ak sa liečba induktorom ukončí, je potrebné zvážiť dobu potrebnú na elimináciu v dĺžke aspoň 3 až 5 dní (dostatočný čas na značnú enzýmovú deindukciu) predtým, ako sa dávka Verimmu vráti na dávku používanú pred začatím súbežného podávania.
|
Dexametazón
| Nesledovalo sa. Očakáva sa znížená expozícia.
|
Karbamazepín, fenobarbital, fenytoín
| Nesledovalo sa. Očakáva sa znížená expozícia.
|
Efavirenz, nevirapín
| Nesledovalo sa. Očakáva sa znížená expozícia.
|
Ľubovník bodkovaný
(Hypericum perforatum)
| Nesledovalo sa. Očakáva sa výrazne znížená expozícia.
| Lieky obsahujúce ľubovník bodkovaný sa počas liečby everolimom nemajú používať.
|
Vzhľadom na výsledky
in vitroje nepravdepodobná inhibícia PgP, CYP3A4 a CYP2D6 systémovými koncentráciami, ktoré sa dosiahnu po perorálnych denných dávkach 10 mg. Inhibíciu CYP3A4 a PgP v čreve však nemožno vylúčiť. Štúdia interakcií u zdravých osôb ukázala, že súbežné perorálne podanie dávky midazolamu, citlivého skúšobného substrátu CYP3A a everolimu malo za následok zvýšenie C
max midazolamu o 25 % a zvýšenie AUC
(0-inf) midazolamu o 30 %. Tento účinok pravdepodobne vyvoláva inhibícia črevného CYP3A4 everolimom. Preto everolimus môže ovplyvniť biologickú dostupnosť súbežne perorálne podávaných substrátov CYP3A4. Klinicky významný účinok na expozíciu systémovo podávaným substrátom CYP3A4 sa však neočakáva (pozri časť 4.4).
Súbežné podávanie everolimu a depotného oktreotidu zvýšilo C
min oktreotidu pričom pomer geometrických priemerov (everolimus/placebo) bol 1,47. Klinicky významné ovplyvnenie účinnosti odpovede na everolimus u pacientov s pokročilými neuroendokrinnými nádormi sa nepreukázalo.
Súbežné podávanie everolimu a exemestánu zvýšilo C
min exemestánu o 45 % a C
2h o 64 %. Avšak v rovnovážnom stave (4 týždne) sa zodpovedajúce koncentrácie estradiolu medzi oboma skupinami liečby nelíšili. U pacientok s pokročilým karcinómom prsníka s pozitivitou hormonálnych receptorov, ktoré dostávali kombináciu, sa nepozorovalo zvýšenie nežiaducich reakcií súvisiacich s exemestánom. Nie je pravdepodobné, že zvýšenie koncentrácií exemestánu má vplyv na účinnosť alebo bezpečnosť.
Súbežná liečba s inhibítormi enzýmu konvertujúceho angiotenzín (ACE)Pacienti užívajúci súbežne liečbu ACE inhibítormi (napr. ramipril) môžu mať zvýšené riziko angioedému (pozri časť 4.4)
VakcináciePočas liečby Verimmom môže byť ovplyvnená imunitná odpoveď na vakcináciu, preto vakcinácia môže byť menej účinná. Použitiu živých vakcín počas liečby Verimmom je potrebné sa vyhnúť (pozri časť 4.4). Príkladom živých vakcín je: intranazálna vakcína proti chrípke, vakcíny proti osýpkam, mumpsu, rubeole, perorálna vakcína proti poliomyelitíde, BCG (Bacillus Calmette-Guérin), vakcíny proti žltej zimnici, varicelle a týfu TY21a.
4.6 Fertilita, gravidita a laktáciaŽeny vo fertilnom veku/Antikoncepcia u mužov a žienŽeny vo fertilnom veku musia používať vysoko účinnú metódu antikoncepcie (napr. hormonálnu antikoncepciu bez obsahu estrogénov používanú vo forme perorálnych tabliet, injekcií alebo implantátu, antikoncepciu založenú na progesteróne, hysterektómiu, tubálnu ligáciu, úplnú abstinenciu, bariérové metódy, vnútromaternicové teliesko [VMT] a/alebo ženskú/mužskú sterilizáciu) počas užívania everolimu a až do 8 týždňov po ukončení liečby. Pacientom mužského pohlavia nemá byť zakázané pokúšať sa počať deti.
GraviditaNie sú k dispozícii dostatočné údaje o použití everolimu u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu vrátane toxického účinku na embryo a plod (pozri časť 5.3). Potenciálne riziko pre ľudí nie je známe.
Everolimus sa neodporúča podávať počas gravidity a ženám vo fertilnom veku, ktoré nepoužívajú antikoncepciu.
DojčenieNie je známe, či sa everolimus vylučuje do ľudského materského mlieka. U potkanov však everolimus a/alebo jeho metabolity ľahko prechádzajú do mlieka (pozri časť 5.3). Preto ženy užívajúce everolimus nemajú dojčiť počas liečby a počas 2 týždňov od užitia poslednej dávky.
FertilitaNie je známe, či everolimus môže zapríčiniť u pacientov mužského a ženského pohlavia neplodnosť, u pacientok ženského pohlavia však bola pozorovaná amenorea (sekundárna amenorea a iné nepravidelnosti menštruačného cyklu) a s ňou spojená nerovnováha medzi luteinizačným hormónom (LH) a folikuly stimulujúcim hormónom (FSH). Podľa predklinických zistení môže liečba everolimom zhoršiť mužskú a ženskú fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeVerimmus môže mať malý alebo mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacientov je potrebné upozorniť, aby boli opatrní pri vedení vozidiel alebo obsluhe strojov, ak počas liečby Verimmom pocítia únavu.
4.8 Nežiaduce účinkyZhrnutie profilu bezpečnostiBezpečnostný profil je založený na združených údajoch od 2 672 pacientov liečených everolimom v desiatich klinických štúdiách pozostávajúcich z piatich randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdií fázy III a z piatich otvorených skúšaní fázy I a fázy II súvisiacich so schválenými indikáciami.
Najčastejšími nežiaducimi reakciami (incidencia ≥ 1/10) zo združených údajov o bezpečnosti boli (v klesajúcom poradí): stomatitída, vyrážka, únava, hnačka, infekcie, nauzea, znížená chuť do jedla, anémia, dysgeúzia, pneumonitída, periférny edém, hyperglykémia, asténia, pruritus, pokles hmotnosti, hypercholesterolémia, epistaxa, kašeľ a bolesť hlavy.
Najčastejšie nežiaduce reakcie stupňa 3 – 4 (incidencia ≥ 1/100 až < 1/10) boli stomatitída, anémia, hyperglykémia, infekcie, únava, hnačka, pneumonitída, asténia, trombocytopénia, neutropénia, dyspnoe, proteínúria, lymfopénia, krvácanie, hypofosfatémia, vyrážka, hypertenzia, pneumónia, zvýšená hladina alanínaminotransferázy (ALT), zvýšená hladina aspartátaminotransferázy (AST) a diabetes mellitus. Stupne zodpovedajú CTCAE, verzii 3.0 a 4.03.
Tabuľkový zoznam nežiaducich reakciíTabuľka 3 uvádza kategórie frekvencie nežiaducich reakcií udávané na základe analýzy združených údajov o bezpečnosti. Nežiaduce reakcie sú uvedené podľa triedy orgánových systémov a kategórie frekvencií MedDRA. Kategórie frekvencie sú definované s použitím nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až <1/1 000); veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 3 Nežiaduce reakcie hlásené v klinických skúšaniach
Infekcie a nákazy
|
Veľmi časté
| Infekcie a, *
|
Poruchy krvi a lymfatického systému
|
Veľmi časté
| Anémia
|
Časté
| Trombocytopénia, neutropénia, leukopénia, lymfopénia
|
Menej časté
| Pancytopénia
|
Zriedkavé
| Čistá aplázia erytrocytov
|
Poruchy imunitného systému
|
Menej časté
| Precitlivenosť
|
Poruchy metabolizmu a výživy
|
Veľmi časté
| Znížená chuť do jedla, hyperglykémia, hypercholesterolémia
|
Časté
| Hypertriglyceridémia, hypofosfatémia, diabetes mellitus, hyperlipidémia, hypokaliémia, dehydratácia, hypokalciémia
|
Psychické poruchy
|
Časté
| Nespavosť
|
Poruchy nervového systému
|
Veľmi časté
| Dysgeúzia, bolesť hlavy
|
Menej časté
| Ageúzia
|
Poruchy oka
|
Časté
| Edém očného viečka
|
Menej časté
| Konjuktivitída
|
Poruchy srdca a srdcovej činnosti
|
Menej časté
| Kongestívne zlyhávanie srdca
|
Poruchy ciev
|
Časté
| Krvácanie b, hypertenzia
|
Menej časté
| Návaly tepla, trombóza hlbokých žíl
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
Veľmi časté
| Pneumonitída c, epistaxa, kašeľ
|
Časté
| Dyspnoe
|
Menej časté
| Hemoptýza, pľúcna embólia
|
Zriedkavé
| Syndróm akútnej respiračnej tiesne
|
Poruchy gastrointestinálneho traktu
|
Veľmi časté
| Stomatitída d, hnačka, nauzea
|
Časté
| Vracanie, sucho v ústach, abdominálna bolesť, zápal slizníc, bolesť úst, dyspepsia, dysfágia
|
Poruchy pečene a žlčových ciest
|
Časté
| Zvýšenie hladiny aspartátaminotransferázy, zvýšenie hladiny alanínaminotransferázy
|
Poruchy kože a podkožného tkaniva
|
Veľmi časté
| Vyrážka, pruritus
|
Časté
| Suchosť kože, ochorenie nechtov, mierna alopécia, akné, erytém, onychoklázia, syndróm palmárno-plantárnej erytrodyzestézie, exfoliácia kože, kožná lézia
|
Zriedkavé
| Angioedém
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
Časté
| Artralgia
|
Poruchy obličiek a močových ciest
|
Časté
| Proteínúria*, zvýšenie kreatinínu v krvi, zlyhanie obličiek*
|
Menej časté
| Zvýšené močenie počas dňa, akútne zlyhanie obličiek*
|
Poruchy reprodukčného systému a prsníkov
|
Časté
| Nepravidelná menštruácia e
|
Menej časté
| Amenorea e
|
Celkové poruchy a reakcie v mieste podania
|
Veľmi časté
| Únava, asténia, periférny edém
|
Časté
| Pyrexia
|
Menej časté
| Bolesť na hrudi iného ako srdcového pôvodu, zhoršenie hojenia rán
|
Laboratórne a funkčné vyšetrenia
|
Veľmi časté
| Zníženie telesnej hmotnosti
|
* Pozri aj odsek „Popis vybraných nežiaducich reakcií“
a Zahŕňa všetky reakcie v triede orgánových systémov „Infekcie a nákazy“ vrátane (časté) pneumónie, infekcie močových ciest; (menej časté) bronchitídy, herpes zoster, sepsy, abscesu a ojedinelých prípadov oportúnnych infekcií (napr. aspergilóza, kandidóza, pneumónia spôsobená Pneumocystis jirovecii (Pneumocystis carinii) (PJP, PCP) a hepatitídy B (pozri aj časť 4.4)) a (zriedkavé) vírusovej myokarditídy
b Zahŕňa rôzne prípady krvácania na rôznych miestach, ktoré nie sú uvedené osobitne
c Zahŕňa (časté) pneumonitídu, intersticiálnu chorobu pľúc, pľúcnu infiltráciu a (zriedkavé) pľúcnu alveolárnu hemorágiu, pľúcnu toxicitu a alveolitídu
d Zahŕňa (veľmi časté) stomatitídu, (časté) aftóznu stomatitídu, ulceráciu úst a jazyka a (menej časté) glosodýniu, glositídu
e Frekvencia určená zo združených údajov podľa počtu žien vo veku od 10 do 55 rokov
|
Popis vybraných nežiaducich reakciíEverolimus sa v klinických štúdiách a v spontánnych hláseniach po uvedení lieku na trh dával do súvislosti so závažnými prípadmi reaktivácie hepatitídy B, vrátane smrteľných prípadov. Reaktivácia infekcie je očakávaná udalosť v obdobiach imunosupresie.
V klinických štúdiách a v spontánnych hláseniach po uvedení lieku na trh sa everolimus dával do súvislosti s prípadmi zlyhania obličiek (vrátane smrteľných prípadov) a proteinúriou. Odporúča sa monitorovanie funkcie obličiek (pozri časť 4.4).
V klinických štúdiách a v spontánnych hláseniach po uvedení lieku na trh bolo používanie everolimu spojené s prípadmi amenorey (sekundárna amenorea a iné nepravidelnosti menštruačného cyklu).
V klinických štúdiách a v spontánnych hláseniach po uvedení lieku na trh bolo používanie everolimu spojené s prípadmi pneumónie spôsobenej
Pneumocystis jirovecii (
Pneumocystiscarinii) (PJP, PCP), niektoré s fatálnym koncom (pozri časť 4.4).
V klinických skúšaniach a v spontánnych hláseniach po uvedení lieku na trh bol zaznamenaný angioedém pri súbežnom užívaní s ACE inhibítormi aj bez nich (pozri časť 4.4).
Starší pacientiV združených údajoch o bezpečnosti bolo 37 % pacientov liečených everolimom vo veku ≥ 65 rokov. Počet pacientov, u ktorých bola liečba z dôvodu nežiaducej reakcie ukončená, bol vyšší vo vekovej skupine ≥ 65 rokov (20 %
vs. 13 %). Najčastejšie nežiaduce reakcie vedúce k ukončeniu liečby boli pneumonitída (vrátane intersticiálnej choroby pľúc), stomatitída, únava a dyspnoe.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 PredávkovanieHlásené skúsenosti s predávkovaním u ľudí sú veľmi obmedzené. Jednorazové dávky až do 70 mg boli podané s prijateľnou akútnou znášanlivosťou. Vo všetkých prípadoch predávkovania je potrebné začať s celkovými podpornými opatreniami.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Cytostatiká, iné cytostatiká, inhibítory proteínkinázy, ATC kód: L01XE10
Mechanizmus účinkuEverolimus je selektívny inhibítor mTOR (cieľové miesto rapamycínu u cicavcov). mTOR je kľúčová seríntreonínkináza o aktivite ktorej je známe, že je zvýšená pri niekoľkých druhoch rakoviny u ľudí. Everolimus sa viaže na vnútrobunkovú bielkovinu FKBP-12, čím tvorí komplex, ktorý inhibuje aktivitu mTOR komplexu-1 (mTORC1). Inhibícia signálnej dráhy mTORC1 narúša transláciu a syntézu bielkovín znížením aktivity ribozómovej proteínkinázy S6 (S6K1) a väzobného proteínu 4E eukaryotického elongačného faktora (4EBP-1), ktoré regulujú bielkoviny podieľajúce sa na bunkovom cykle, angiogenéze a glykolýze. Predpokladá sa, že S6K1 fosforyluje aktivačnú funkčnú doménu 1 estrogénového receptora, ktorý zodpovedá za aktiváciu receptora nezávislú od ligandu. Everolimus znižuje hladiny rastového faktora cievneho endotelu (VEGF), ktorý potenciuje angiogénne procesy v nádore. Everolimus je účinný inhibítor rastu a proliferácie nádorových buniek, endotelových buniek, fibroblastov a buniek hladkého svalstva súvisiacich s krvnými cievami a preukázalo sa, že znižuje glykolýzu v solídnych nádoroch
in vitro a
in vivo.
Klinická účinnosť a bezpečnosťPokročilý karcinóm prsníka s pozitivitou hormonálnych receptorovBOLERO-2 (štúdia CRAD001Y2301), randomizovaná, dvojito zaslepená, multicentrická štúdia fázy III everolimu + exemestánu oproti placebu + exemestánu, sa vykonalo u postmenopauzálnych žien s pokročilým karcinómom prsníka s pozitivitou estrogénových receptorov a negativitou HER2/neu pri recidíve alebo progresii po predchádzajúcej liečbe letrozolom alebo anastrozolom. Randomizácia bola stratifikovaná podľa zdokumentovanej citlivosti na predchádzajúcu hormonálnu liečbu a podľa prítomnosti viscerálnych metastáz. Citlivosť na predchádzajúcu hormonálnu liečbu bola definovaná buď ako (1) zdokumentovaný klinický prínos (kompletná odpoveď [CR], čiastočná odpoveď [PR], stabilizovaná choroba ≥ 24 týždňov) vyvolaný najmenej jednou predchádzajúcou hormonálnou liečbou pri pokročilej chorobe, alebo (2) najmenej 24 mesiacov adjuvantnej hormonálnej liečby pred recidívou.
Primárnym koncovým ukazovateľom v štúdii bolo prežívanie bez progresie (PFS) vyhodnotené prostredníctvom RECIST (Response Evaluation Criteria in Solid Tumors, kritériá vyhodnotenia odpovede pri solídnych nádoroch), pri ktorom sa vychádzalo z hodnotenia skúšajúcim lekárom (lokálna rádiológia). Podporné analýzy PFS sa zakladali na posúdení nezávislou centrálnou rádiológiou.
Sekundárne koncové ukazovatele zahŕňali celkové prežívanie (OS), podiel objektívnych odpovedí, podiel klinického prínosu, bezpečnosť, zmenu kvality života (QoL) a čas do zhoršenia výkonnostného stavu (PS) podľa ECOG (Eastern Cooperative Oncology Group performance status).
Celkovo bolo randomizovaných 724 pacientok v pomere 2:1 buď do skupiny kombinácie everolimu (10 mg denne) + exemestánu (25 mg denne) (n = 485), alebo do skupiny placeba + exemestánu (25 mg denne) (n = 239). V čase finálnej analýzy celkového prežívania bol medián trvania liečby everolimom 24,0 týždňov (rozmedzie 1,0 – 199,1 týždňov). Medián trvania liečby exemestánom bol dlhší v skupine everolimus + exemestán v trvaní 29,5 týždňov (1,0 – 199,1) v porovnaní s 14,1 týždňami (1,0 – 156,0) v skupine placebo + exemestán.
Výsledky účinnosti z hľadiska primárneho koncového ukazovateľa sa získali z finálnej analýzy PFS (pozri tabuľku 4 a obrázok 1). Pacientky v skupine placeba + exemestánu neprešli v čase progresie na liečbu everolimom.
Tabuľka 4 BOLERO-2 – výsledky účinnosti
Analýza
| Everolimusa
n = 485
| Placeboa
n = 239
| Pomer rizika
| Hodnota p
|
Medián prežívania bez progresie (mesiace) (95 % IS)
|
Rádiologické hodnotenie skúšajúcim lekárom
| 7,8
(6,9 až 8,5)
| 3,2
(2,8 až 4,1)
| 0,45
(0,38 až 0,54)
| < 0,0001
|
Nezávislé rádiologické hodnotenie
| 11,0
(9,7 až 15,0)
| 4,1
(2,9 až 5,6)
| 0,38
(0,31 až 0,48)
| < 0,0001
|
Medián celkového prežívania (mesiace) (95 % IS)
|
Medián celkového prežívania
| 31,0
(28,0 – 34,6)
| 26,6
(22,6 – 33,1)
| 0,89
(0,73 – 1,10)
| 0,1426
|
Najlepšia celková odpoveď (%) (95 % IS)
|
Podiel objektívnych odpovedíb
| 12,6 %
(9,8 až 15,9)
| 1,7 %
(0,5 až 4,2)
| n/ad
| < 0,0001e
|
|
|
|
|
|
Podiel klinického prínosuc
| 51,3 %
(46,8 až 55,9)
| 26,4 %
(20,9 až 32,4)
| n/ad
| < 0,0001e
|
a Plus exemestán
b Podiel objektívnych odpovedí = podiel pacientok s kompletnou alebo čiastočnou odpoveďou
c Podiel klinického prínosu = podiel pacientok s kompletnou alebo čiastočnou odpoveďou alebo stabilizovanou chorobou ≥ 24 týždňov
d Neaplikovateľné
e Hodnota p získaná z exaktného testu podľa Cochrana-Mantela-Haenszela pomocou stratifikovanej verzie permutačného testu podľa Cochrana-Armitagea.
|
Obrázok 1 BOLERO-2 – krivky prežívania bez progresie podľa Kaplana-Meiera (rádiologické hodnotenie skúšajúcim lekárom) Everolimus 10 mg + exemestán (n/N = 310/485)
|
|
Placebo + exemestán (n/N = 200/239)
|
|
Pomer rizika = 0,45
95 % IS [0,38; 0,54]
p‑hodnota log-rank testu: < 0,0001
Mediány PFS podľa Kaplana‑Meiera
Everolimus 10 mg + exemestán: 7,82 mesiacov
Placebo + exemestán: 3,19 mesiacov
|
|
Pravdepodobnosť (%) udalosti (Probability (%) of event); Čas (týždne) (Time (weeks))
Počet pacientok s pretrvávajúcim rizikom
Čas (týždne)
| 0
| 6
| 12
| 18
| 24
| 30
| 36
| 42
| 48
| 54
| 60
| 66
| 72
| 78
| 84
| 90
| 96
| 102
| 108
| 114
| 120
|
Everolimus
| 485
| 436
| 366
| 304
| 257
| 221
| 185
| 158
| 124
| 91
| 66
| 50
| 35
| 24
| 22
| 13
| 10
| 8
| 2
| 1
| 0
|
Placebo
| 239
| 190
| 132
| 96
| 67
| 50
| 39
| 30
| 21
| 15
| 10
| 8
| 5
| 3
| 1
| 1
| 1
| 0
| 0
| 0
| 0
|
Odhadovaný účinok liečby z hľadiska PFS potvrdila plánovaná analýza PFS v podskupinách podľa hodnotenia skúšajúcim lekárom. Vo všetkých analyzovaných podskupinách (vek, citlivosť na predchádzajúcu hormonálnu liečbu, počet postihnutých orgánov, východiskový stav lézií len v kostiach a prítomnosť viscerálnych metastáz, ako aj naprieč hlavnými demografickými a prognostickými podskupinami) sa pozoroval pozitívny účinok liečby pri everolime + exemestáne s odhadovaným pomerom rizika (HR) oproti placebu + exemestánu v rozmedzí 0,25 až 0,60.
V oboch skupinách liečby sa nepozorovali rozdiely v čase do zhoršenia (≥ 5 %) celkového skóre a skóre vo funkčných oblastiach podľa QLQ-C30.
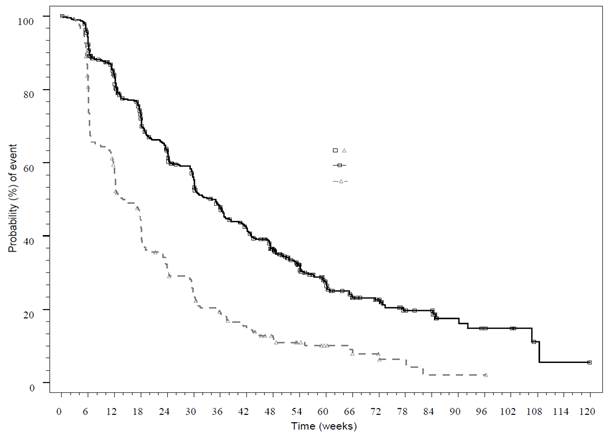
Pokročilé neuroendokrinné nádory pankreatického pôvodu (pNET)RADIANT-3 (štúdia CRAD001C2324), multicentrická, randomizovaná, dvojito zaslepená štúdia fázy III everolimu s najlepšou podpornou starostlivosťou (z angl. best supportive care, BSC) oproti placebu s BSC u pacientov s pokročilými pNET preukázala v porovnaní s placebom štatisticky významný prínos everolimu, ktorý 2,4-násobne predĺžil medián prežívania bez progresie (PFS) (11,04 mesiacov oproti 4,6 mesiacom) (HR 0,35; 95 % IS: 0,27, 0,45; p < 0,0001) (pozri tabuľku 5 a obrázok 2).
Do RADIANT-3 boli zaradení pacienti s dobre a stredne diferencovanými pokročilými pNET, ktorých choroba progredovala počas predchádzajúcich 12 mesiacov. Liečba analógmi somatostatínu bola povolená ako súčasť BSC.
Primárnym koncovým ukazovateľom v štúdii bolo PFS hodnotené prostredníctvom RECIST (kritériá vyhodnotenia odpovede pri solídnych nádoroch, z angl. Response Evaluation Criteria in Solid Tumors). Po rádiologicky zdokumentovanej progresii mohol skúšajúci lekár vykonať odslepenie liečby. Pacienti randomizovaní do skupiny s placebom mohli potom dostávať everolimus v otvorenej štúdii.
K sekundárnym koncovým ukazovateľom patrila bezpečnosť, podiel objektívnych odpovedí, trvanie odpovede a celkové prežívanie (OS).
Celkovo bolo randomizovaných 410 pacientov v pomere 1:1, aby dostávali buď everolimus 10 mg/deň (n = 207) alebo placebo (n = 203). Demografické parametre boli dobre vyvážené (medián veku 58 rokov, 55 % muži, 78,5 % belosi). Systémovú liečbu dostalo predtým 58 % pacientov v oboch skupinách. Medián trvania liečby v zaslepenej štúdii bol 37, 8 týždňov (rozmedzie 1,1 – 129,9 týždňov) u pacientov, ktorí dostávali everolimus a 16,1 týždňov (rozmedzie 0,4 – 147,0 týždňov) u pacientov, ktorí dostávali placebo.
Po progresii ochorenia alebo po odslepení štúdie prešlo 172 z 203 pacientov (84,7 %) pôvodne randomizovaných na placebo na otvorené podávanie everolimu. Medián trvania otvorenej liečby bol 47,7 týždňov spomedzi všetkých pacientov; 67,1 týždňov u 53 pacientov randomizovaných na everolimus, ktorí prestúpili na otvorenú liečbu everolimom a 44,1 týždňov u 172 pacientov randomizovaných na placebo, ktorí prestúpili na otvorenú liečbu everolimom.
Tabuľka 5 RADIANT-3 – výsledky účinnosti
Populácia
| Everolimus
n = 207
| Placebo
n = 203
| Pomer rizika (95 % IS)
| Hodnota p
|
Medián prežívania bez progresie (mesiace) (95 % IS)
|
Rádiologické hodnotenie skúšajúcim lekárom
| 11,04
(8,41; 13,86)
| 4,60
(3,06; 5,39)
| 0,35
(0,27; 0,45)
| < 0,0001
|
Nezávislé rádiologické hodnotenie
| 13,67
(11,17; 18,79)
| 5,68
(5,39; 8,31)
| 0,38
(0,28; 0,51)
| < 0,0001
|
Medián celkového prežívania (mesiace) (95 % IS)
|
Medián celkového prežívania
| 44,02
(35,61; 51,75)
| 37,68
(29,14; 45,77)
| 0,94
(0,73; 1,20)
| 0,300
|
Obrázok 2 RADIANT-3 – krivky prežívania bez progresie podľa Kaplana-Meiera (rádiologické hodnotenie skúšajúcim lekárom) Pomer rizika = 0,35
95 % IS [0,27; 0,45]
p‑hodnota log-rank testu = < 0,001
Mediány PFS podľa Kaplana‑Meiera
Everolimus: 11,04 mesiaca
Placebo: 4,60 mesiaca
|
|

Pravdepodobnosť (%) (Probability (%)); Čas (mesiace) (Time (months))
Počet pacientov s pretrvávajúcim rizikom
Everolimusus
| 207
| 189
| 153
| 126
| 114
| 80
| 49
| 36
| 28
| 21
| 10
| 6
| 2
| 0
| 0
| 0
|
Placebo
| 203
| 117
| 98
| 59
| 52
| 24
| 16
| 7
| 4
| 3
| 2
| 1
| 1
| 1
| 1
| 0'
|
Pokročilé neuroendokrinné nádory gastrointestinálneho alebo pľúcneho pôvoduRADIANT-4 (štúdia CRAD001T2302), randomizovaná, dvojito zaslepená, multicentrická štúdia fázy III everolimu s najlepšou podpornou starostlivosťou (BSC) oproti placebu s BSC bola vykonaná u pacientov s pokročilými, dobre diferencovanými (stupeň 1 alebo stupeň 2) nefunkčnými neuroendokrinnými nádormi gastrointestinálneho alebo pľúcneho pôvodu bez predošlej anamnézy a aktívnych príznakov spojených s karcinoidným syndrómom.
Primárnym koncovým ukazovateľom štúdie bolo prežívanie bez progresie (PFS) hodnotené podľa RECIST (Response Evaluation Criteria in Solid Tumors) na základe nezávislého rádiologického hodnotenia. Podporná analýza PFS bola založená na hodnotení miestneho skúšajúceho lekára. Sekundárne koncové ukazovatele zahŕňali celkové prežívanie (OS), mieru celkovej odpovede, mieru kontroly ochorenia, bezpečnosť, zmenu v kvalite života (FACT-G) a čas do zhoršenia výkonnostného stavu podľa WHO (WHO PS).
Celkovo bolo randomizovaných 302 pacientov v pomere 2:1 na everolimus (10 mg denne) (n = 205) alebo placebo (n = 97). Demografické údaje a charakteristika ochorenia boli v zásade vyvážené (medián veku pacientov 63 rokov [rozsah 22 až 86], 76 % kaukazskej rasy, predošlé užívanie somatostatínového analógu [SSA]). Medián trvania zaslepenej liečby bol 40,4 týždňov u pacientov užívajúcich everolimus a 19,6 týždňov u pacientov užívajúcich placebo. Pacienti užívajúci placebo neprestúpili v čase progresie do skupiny s everolimom.
Výsledky účinnosti z hľadiska primárneho koncového ukazovateľa boli získané zo záverečnej analýzy PFS (pozri tabuľku 6 a obrázok 3).
Tabuľka 6 RADIANT-4 – výsledky prežívania bez progresie
Populácia
| Everolimus
n = 205
| Placebo
n = 97
| Pomer rizika (95 % IS)
| Hodnota pa
|
Medián prežívania bez progresie (mesiace) (95% IS)
|
Nezávislé rádiologické hodnotenie
| 11,01
(9,2; 13,3)
| 3,91
(3,6; 7,4)
| 0,48
(0,35; 0,67)
| < 0,0001
|
Rádiologické hodnotenie skúšajúcim lekárom
| 13,96
(11,2; 17,7)
| 5,45
(3,7; 7,4)
| 0,39
(0,28; 0,54)
| < 0,0001
|
aJednostranná hodnota p podľa stratifikovaného log-rank testu
|
Obrázok 3 RADIANT-4 – krivky prežívania bez progresie podľa Kaplana-Meiera (nezávislé rádiologické hodnotenie)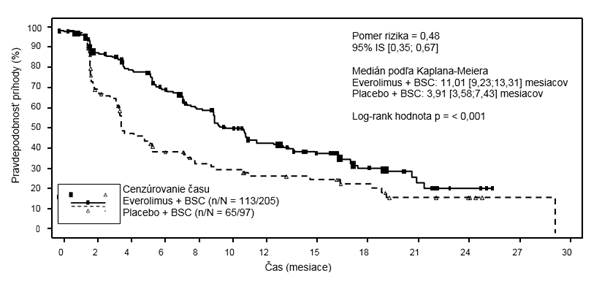
Počet pacientov s pretrvávajúcim rizikom
Čas (mesiace)
| 0
| 2
| 4
| 6
| 8
| 10
| 12
| 15
| 18
| 21
| 24
| 27
| 30
|
Everolimus
| 205
| 168
| 145
| 124
| 101
| 81
| 65
| 52
| 26
| 10
| 3
| 0
| 0
|
Placebo
| 97
| 65
| 39
| 30
| 24
| 21
| 17
| 15
| 11
| 6
| 5
| 1
| 0
|
Podľa podpornej analýzy bol pozitívny účinok liečby zaznamenaný vo všetkých podskupinách pacientov s výnimkou podskupiny pacientov, kde bolo ileum primárne miesto vzniku nádoru (ileum: HR = 1,22 [95 % IS: 0,56 až 2,65]; iné ako ileum: HR = 0,34 [95 % IS: 0,22 až 0,54]; pľúca: HR = 0,43 [95 % IS: 0,24 až 0,79]) (pozri obrázok 4).
Obrázok 4 RADIANT-4 – Výsledky prežívania bez progresie podľa vopred špecifikovaných podskupín pacientov (nezávislé rádiologické hodnotenie) Stupeň 1 (n = 194)
Stupeň 2 (n = 107)
|
|
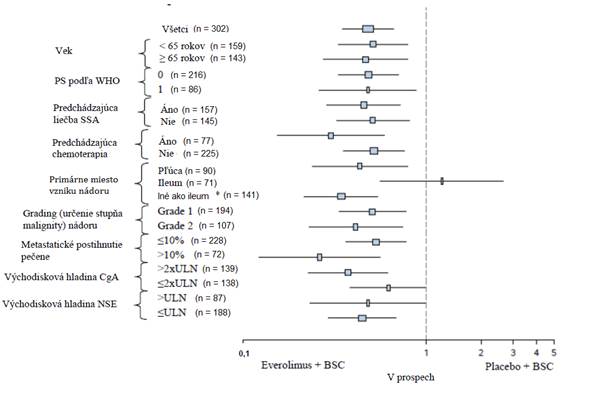
*Iné ako ileum: žalúdok, hrubé črevo, konečník, prívesok slepého čreva, slepé črevo, dvanástnik, stredná časť tenkého čreva, karcinóm s neznámym miestom primárneho vzniku a iného gastrointestinálneho pôvodu
ULN: horná hranica normálu
CgA: chromogranín A
NSE: neurónová špecifická enoláza
Pomer rizika (95 % IS) podľa stratifikovaného Coxovho modelu
Výsledky vopred plánovanej predbežnej analýzy celkového prežívania po výskyte 101 úmrtí (z celkového počtu 191 úmrtí potrebného pre záverečnú analýzu) a 33 mesačné obdobie následného sledovania boli v prospech skupiny s everolimom; štatisticky významný rozdiel v celkovom prežívaní sa však nezaznamenal (HR = 0,73 [95 % IS: 0,48 až 1,11; p = 0,071]).
Medzi oboma skupinami sa nepozoroval rozdiel v čase do konečného zhoršenia WHO PS (≥ 1 bod) a v čase do konečného zhoršenia kvality života (celkové skóre FACT-G ≥ 7 bodov).
Pokročilý karcinóm obličkových buniekRECORD-1 (štúdia CRAD001C2240), medzinárodná, multicentrická, randomizovaná, dvojito zaslepená štúdia fázy III, porovnávajúca everolimus 10 mg/deň a placebo, oboje spojené s najlepšou podpornou starostlivosťou, sa vykonalo u pacientov s metastatickým karcinómom obličkových buniek, ktorých ochorenie progredovalo počas alebo po liečbe VEGFR-TKI (inhibítormi tyrozínkinázy receptora pre rastový faktor cievneho endotelu) (sunitinibom, sorafenibom, alebo sunitinibom aj sorafenibom). Povolená bola tiež predchádzajúca liečba bevacizumabom a interferónom-α. Pacienti boli stratifikovaní podľa prognostického skóre Memorial Sloan-Kettering Cancer Center (MSKCC) (skupiny s priaznivým
vs. stredným
vs. vysokým rizikom) a predchádzajúcej protinádorovej liečby (1
vs. 2 predchádzajúce liečby VEGFR-TKI).
Prežívanie bez progresie zdokumentované pomocou RECIST (Response Evaluation Criteria in Solid Tumours) a stanovené zaslepeným, nezávislým centrálnym hodnotením, bolo primárnym koncovým ukazovateľom. Medzi sekundárne koncové ukazovatele patrila bezpečnosť, miera objektívnej odpovede nádoru, celkové prežívanie, symptómy súvisiace s ochorením a kvalita života. Po zdokumentovaní rádiologickej progresie mohol skúšajúci lekár vykonať odslepenie liečby: pacienti randomizovaní do skupiny placeba mohli potom dostávať otvorenú liečbu everolimom 10 mg/deň. Nezávislý výbor pre monitorovanie údajov (Independent Data Monitoring Committee) odporučil ukončiť toto skúšanie v čase druhej predbežnej analýzy, keďže sa dosiahol primárny koncový ukazovateľ.
Celkovo bolo randomizovaných 416 pacientov v pomere 2:1 na podávanie everolimu (n = 277) alebo placeba (n = 139). Demograficky bola štúdia dobre vyvážená (zlúčený medián veku [61 rokov; rozmedzie 27 – 85], 78 % mužov, 88 % belochov, počet podaní predchádzajúcej liečby VEGFR-TKI [1 – 74 %, 2 – 26 %]). Medián trvania liečby v zaslepenej štúdii bol 141 dní (rozmedzie 19 – 451 dní) u pacientov, ktorí dostávali everolimus, a 60 dní (rozmedzie 21 – 295 dní) u pacientov, ktorí dostávali placebo.
Everolimus bol lepší než placebo z hľadiska primárneho koncového ukazovateľa prežívania bez progresie, so štatisticky významným poklesom rizika progresie alebo úmrtia o 67 % (pozri tabuľku 7 a obrázok 5).
Tabuľka 7 RECORD-1 – výsledky prežívania bez progresie
Populácia
| n
| Everolimus
n = 277
| Placebo
n = 139
| Pomer rizika (95 % IS)
| p‑hodnota
|
|
| Medián prežívania bez progresie (mesiace) (95 % IS)
|
|
|
Primárna analýza
|
Všetci (zaslepené nezávislé centrálne hodnotenie)
| 416
| 4,9
(4,0 – 5,5)
| 1,9
(1,8 – 1,9)
| 0,33
(0,25 – 0,43)
| < 0,0001a
|
Podporné analýzy/analýzy citlivosti
|
Všetci (lokálne hodnotenie skúšajúcim lekárom)
| 416
| 5,5
(4,6 – 5,8)
| 1,9
(1,8 – 2,2)
| 0,32
(0,25 – 0,41)
| < 0,0001a
|
Prognostické skóre MSKCC (zaslepené nezávislé centrálne hodnotenie)
|
Priaznivé riziko
| 120
| 5,8
(4,0 – 7,4)
| 1,9
(1,9 – 2,8)
| 0,31
(0,19 – 0,50)
| < 0,0001
|
Stredné riziko
| 235
| 4,5
(3,8 – 5,5)
| 1,8
(1,8 – 1,9)
| 0,32
(0,22 – 0,44)
| < 0,0001
|
Vysoké riziko
| 61
| 3,6
(1,9 – 4,6)
| 1,8
(1,8 – 3,6)
| 0,44
(0,22 – 0,85)
| 0,007
|
a Stratifikovaný log-rank test
|
Obrázok 5 RECORD-1 ‑ Kaplanove‑Meierove krivky prežívania bez progresie (nezávislé centrálne hodnotenie) Pomer rizika = 0,33
95 % IS [0,25; 0,43]
Mediány PFS podľa Kaplana‑Meiera
Everolimus: 4,90 mesiacov
Placebo: 1,87 mesiacov
p-hodnota log-rank testu = < 0,0001
|
|
Everolimus (n/N = 155/277)
|
|

*Pravdepodobnosť (%) (Probability (%)); Čas (mesiace) (Time (months))
Počet pacientov s pretrvávajúcim rizikom
Čas (mesiace)
| 0
| 2
| 4
| 6
| 8
| 10
| 12
| 14
|
Everolimus
| 277
| 192
| 115
| 51
| 26
| 10
| 1
| 0
|
Placebo
| 139
| 47
| 15
| 6
| 2
| 0
| 0
| 0
|
| | | | | | | | | |
Podiely prežívania bez progresie po 6 mesiacoch boli 36 % pri liečbe everolimom v porovnaní s 9 % pri placebe.
Potvrdená objektívna odpoveď nádoru sa pozorovala u 5 pacientov (2 %), ktorí dostávali everolimus, zatiaľ čo u pacientov dostávajúcich placebo sa nepozorovala žiadna. Preto výhoda z hľadiska prežívania bez progresie primárne platí pre populáciu so stabilizáciou ochorenia (zodpovedá 67 % v skupine liečby everolimom).
Štatisticky významný rozdiel v celkovom prežívaní súvisiaci s liečbou sa nezaznamenal (pomer rizika 0,87; interval spoľahlivosti: 0,65 – 1,17; p = 0,177). Prechod na otvorenú liečbu everolimom po progresii ochorenia u pacientov zadelených do skupiny placeba znemožnil zistenie rozdielu v celkovom prežívaní súvisiacom s liečbou.
Iné štúdieStomatitída je najčastejšie hlásenou nežiaducou reakciou u pacientov liečených everolimom (pozri časti 4. 4 a 4. 8). Po uvedení lieku na trh sa v štúdii s jednou skupinou u postmenopauzálnych žien s pokročilým karcinómom prsníka (n = 92) ako topická liečba použil orálny roztok dexametazónu 0,5 mg/5 ml neobsahujúci alkohol na vyplachovanie ústnej dutiny (4-krát denne počas prvých 8 týždňov liečby), ktorý sa pacientkam podával na začiatku liečby everolimom (10 mg/deň) a exemestánom (25 mg/deň) na zníženie incidencie a závažnosti stomatitídy. Incidencia stomatitídy stupňa ≥ 2 po 8 týždňoch bola 2,4 % (n = 2/85 vyhodnotiteľných pacientok), čo bolo menej ako historicky hlásené hodnoty. Incidencia stomatitídy stupňa 1 bola 18,8 % (n = 16/85) a neboli hlásené prípady stomatitídy stupňa 3 alebo 4. Celkový profil bezpečnosti v tejto štúdii sa zhodoval s tým, ktorý sa stanovil pre everolimus pri liečbe onkologických chorôb a komplexu tuberóznej sklerózy (z angl. tumor sclerosis complex, TSC), s výnimkou mierne zvýšeného výskytu orálnej kandidózy, ktorá sa zaznamenala u 2,2 % (n = 2/92) pacientok.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s referenčným liekom obsahujúcim everolimus vo všetkých podskupinách pediatrickej populácie pri neuroendokrinných nádoroch pankreatického pôvodu, hrudníkových neuroendokrinných nádoroch a karcinóme obličkových buniek (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaU pacientov s pokročilými solídnymi nádormi sa maximálne koncentrácie everolimu (C
max) dosahujú v priemere do 1 hodiny po dennom podávaní dávky 5 a 10 mg everolimu nalačno alebo s ľahkým jedlom bez obsahu tuku. C
max je úmerná dávke v rozmedzí od 5 do 10 mg. Everolimus je substrát a stredne silný inhibítor PgP.
Vplyv jedlaU zdravých osôb jedlá s vysokým obsahom tuku znižovali systémovú expozíciu everolimu 10 mg (stanovené na základe AUC) o 22 % a maximálnu plazmatickú koncentráciu C
max o 54 %. Jedlá s nízkym obsahom tuku znižovali AUC o 32 % a C
max o 42 %. Jedlo však zjavne nemalo účinok na profil závislosti koncentrácie od času v postabsorpčnej fáze.
DistribúciaPomer everolimu v krvi a plazme, ktorý závisí od koncentrácie v rozmedzí od 5 až do 5 000 ng/ml, je 17 % až 73 %. Približne 20 % koncentrácie everolimu v celej krvi je obsiahnutých v plazme onkologických pacientov, ktorým sa podáva everolimus 10 mg/deň. Väzba na bielkoviny plazmy je približne 74 % u zdravých osôb aj u pacientov so stredne závažnou poruchou funkcie pečene. U pacientov s pokročilými solídnymi nádormi bola hodnota distribučného objemu V
d pre zdanlivý centrálny kompartment 191 l a pre zdanlivý periférny kompartment 517 l.
BiotransformáciaEverolimus je substrát CYP3A4 a PgP. Po perorálnom podaní je everolimus hlavnou cirkulujúcou zložkou v ľudskej krvi. V ľudskej krvi sa zistilo šesť hlavných metabolitov everolimu, vrátane troch monohydroxylovaných metabolitov, dvoch hydrolytických produktov s otvoreným reťazcom a fosfatidylcholínového konjugátu everolimu. Tieto metabolity sa identifikovali aj u zvieracích druhov, ktoré sa použili v štúdiách toxicity, pričom vykazovali približne 100-násobne nižšiu aktivitu než samotný everolimus. Predpokladá sa preto, že everolimus zodpovedá za väčšinu celkovej farmakologickej aktivity.
ElimináciaPriemerný perorálny klírens (CL/F) everolimu po dennej dávke 10 mg u pacientov s pokročilými solídnymi nádormi bol 24,5 l/h. Priemerný polčas eliminácie everolimu je približne 30 hodín.
U onkologických pacientov sa nevykonali žiadne špecifické štúdie zamerané na vylučovanie; dostupné sú však údaje zo štúdií s pacientmi po transplantácii. Po podaní jednorazovej dávky rádioaktívne značeného everolimu spolu s cyklosporínom sa 80 % rádioaktivity zistilo v stolici, zatiaľ čo 5 % sa vylúčilo močom. Nezmenená látka sa nezistila v moči ani v stolici.
Farmakokinetika v rovnovážnom stavePo podaní everolimu pacientom s pokročilými solídnymi nádormi bola hodnota AUC
0 - τ v rovnovážnom stave úmerná dávke v rozmedzí dennej dávky od 5 až do 10 mg. Rovnovážny stav sa dosiahol v priebehu 2 týždňov. C
max je úmerná dávke v rozsahu od 5 až do 10 mg. t
max sa dosahuje 1 až 2 hodiny po užití dávky. Medzi AUC
0 - τ a minimálnou koncentráciou pred podaním dávky v rovnovážnom stave sa zistila významná korelácia.
Osobitné skupinyPorucha funkcie pečeneBezpečnosť, znášanlivosť a farmakokinetika everolimu sa hodnotili v dvoch štúdiách s jednorazovým perorálnym podaním tabliet everolimu u 8 a 34 jedincov s poruchou funkcie pečene vo vzťahu k jedincom s normálnou funkciou pečene.
V prvej štúdii bola u 8 jedincov so stredne závažnou poruchou funkcie pečene (trieda B podľa Childa-Pugha) priemerná hodnota AUC everolimu dvojnásobne vyššia ako u 8 jedincov s normálnou funkciou pečene.
V druhej štúdii u 34 jedincov s rozdielnym stupňom poruchy funkcie pečene mali v porovnaní so zdravými osobami jedinci s miernou (trieda A podľa Childa-Pugha), stredne závažnou (trieda B podľa Childa-Pugha) a závažnou poruchou funkcie pečene (trieda C podľa Childa-Pugha) 1,6-násobne, 3,3-násobne a 3,6-násobne zvýšenú expozíciu (t.j. AUC
0-inf), v uvedenom poradí.
Odporúčania na dávkovanie u pacientov s poruchou funkcie pečene podľa tried Childa-Pugha sú podložené aj simuláciou farmakokinetiky po opakovanom podaní.
Na základe výsledkov z uvedených dvoch štúdii sa u pacientov s poruchou funkcie pečene odporúča úprava dávky (pozri časti 4.2 a 4.4).
Porucha funkcie obličiekV populačnej farmakokinetickej analýze 170 pacientov s pokročilými solídnymi nádormi sa nezistil významný vplyv klírensu kreatinínu (25 – 178 ml/min) na CL/F everolimu. Potransplantačná porucha funkcie obličiek (klírens kreatinínu v rozmedzí 11 – 107 ml/min) nemala vplyv na farmakokinetiku everolimu u pacientov po transplantácii.
Starší pacientiVo vyhodnotení populačnej farmakokinetickej analýzy u onkologických pacientov sa nezistil významný vplyv veku (27 – 85 rokov) na klírens everolimu po perorálnom podaní.
Etnická príslušnosťKlírens po perorálnom podaní (CL/F) je podobný u japonských a belošských onkologických pacientov s podobnou funkciou pečene. Na základe populačnej farmakokinetickej analýzy je CL/F v priemere o 20 % vyšší u černošských pacientov po transplantácii.
5.3 Predklinické údaje o bezpečnostiPredklinický bezpečnostný profil everolimu sa hodnotil na myšiach, potkanoch, miniatúrnych prasiatkach, opiciach a králikoch. Hlavnými cieľovými orgánmi boli samčie a samičie reprodukčné systémy (degenerácia tubulov v semenníkoch, znížené množstvo spermií v nadsemenníkoch a atrofia maternice) u viacerých druhov zvierat; pľúca (zvýšené hodnoty alveolárnych makrofágov) u potkanov a myší; pankreas (degranulácia exokrinných buniek u opíc a vakuolizácia exokrinných buniek u miniatúrnych prasiatok a degenerácia buniek Langerhansových ostrovčekov u opíc) a oči (zákaly línie šva prednej časti šošovky) len u potkanov. Malé zmeny na obličkách sa pozorovali u potkanov (exacerbácia lipofuscínu súvisiaceho s vekom v epiteli tubulov, zvýšený počet prípadov hydronefrózy) a u myší (exacerbácia už existujúcich lézií). Nič nepoukazovalo na toxicitu pre obličky u opíc a miniatúrnych prasiatok.
Everolimus zjavne spontánne zhoršoval už existujúce ochorenia (chronickú myokarditídu u potkanov, infekciu vírusom Coxsackie v plazme a srdci opíc, kokcídiovú nákazu gastrointestinálneho traktu u miniatúrnych prasiatok, kožné lézie u myší a opíc). Tieto nálezy sa spravidla pozorovali pri hladinách systémovej expozície v rozmedzí terapeutickej expozície alebo vyšších, s výnimkou nálezov u potkanov, kde sa pre vysokú distribúciu do tkanív vyskytovali pri nižšej ako terapeutickej expozícii.
V štúdii samčej fertility na potkanoch bola morfológia semenníkov ovplyvnená pri dávke 0,5 mg/kg a viac; pohyblivosť spermií, počet spermií a hladiny testosterónu v plazme boli znížené pri dávke 5 mg/kg, čo spôsobilo zníženie samčej fertility. Preukázala sa reverzibilita.
V reprodukčných štúdiách na zvieratách nebola samičia fertilita ovplyvnená. Avšak pri perorálnych dávkach everolimu ≥ 0, 1 mg/kg (približne 4 % AUC
0-24h u pacientov užívajúcich dennú dávku 10 mg) boli u samíc potkanov zvýšené predimplantačné straty.
Everolimus prestupoval cez placentu a mal toxické účinky na plod. U potkanov everolimus vyvolával embryo/fetotoxicitu pri systémovej expozícii nižšej, ako je terapeutická hladina. Prejavilo sa to ako mortalita a znížená hmotnosť plodov. Incidencia zmien a malformácií kostry (napr. rázštep hrudnej kosti) sa zvýšila pri dávkach 0,3 a 0,9 mg/kg. U králikov sa prejavila embryotoxicita zvýšením výskytu neskorých resorpcií.
Štúdie genotoxicity, ktoré skúmali významné koncové ukazovatele genotoxicity, nepreukázali klastogénnu alebo mutagénnu aktivitu. Podávanie everolimu trvajúce až 2 roky neodhalilo onkogénny potenciál u myší a potkanov ani pri najvyšších dávkach, ktoré zodpovedajú 3,9-násobku a 0,2-násobku odhadovanej klinickej expozície.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokbutylhydroxytoluén (E 321)hypromelóza (E 464)
laktóza
monohydrát laktózy
krospovidón (E 1202)
stearan horečnatý (E 470b)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnosti2 roky
6.4 Špeciálne upozornenia na uchovávanieUchovávajte v pôvodnom obale na ochranu pred svetlom.
Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaOPA/Al/PVC/Al blister
Verimmus 5 mg a Verimmus 10 mg sú dostupné v baleniach obsahujúcich 10, 30 alebo 90 tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIEgis Pharmaceuticals PLC
1106 Budapešť, Keresztúri út 30-38.
Maďarsko
8. REGISTRAČNÉ ČÍSLAVerimmus 5 mg tablety: 44/0266/19-S
Verimmus 10 mg tablety: 44/0267/19-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
Dátum posledného predĺženia registrácie:
10. DÁTUM REVÍZIE TEXTU08/2019