
oršeniu stavu, a malo by byť teda podávané s opatrnosťou.
Ortostatická hypotenzia
V krátkodobých klinických štúdiach s teriparatidom sa pozorovali izolované epizódy prechodnej
ortostatickej hypotenzie. K tejto príhode typicky došlo v priebehu 4 hodín po podaní a príznaky spontánne odzneli v priebehu niekoľkých minút až niekoľkých hodín. Ak sa táto prechodná
ortostatická hypotenzia objavila, vyskytla sa v priebehu prvých niekoľkých dávok. Problémy ustúpili
po uložení pacienta do vodorovnej polohy a neviedli k prerušeniu liečby.
Poškodenie obličiek
Pacienti so stredne ťažkým poškodením obličiek by mali byť sledovaní so zvýšenou opatrnosťou.
Mladá dospelá populácia
Skúsenosti s mladou dospelou populáciou, vrátane predmenopauzálnych žien, sú obmedzené (pozri
časť 5.1). Liečba sa má začať len v prípade, že jej prínos zreteľne prevýši riziká v tejto populácii.
Ženy v reprodukčnom veku majú používať účinné metódy antikoncepcie počas užívania teriparatidu. Ak žena otehotnie, teriparatid sa má vysadiť.
Dĺžka liečby
Štúdie na potkanoch poukázali na zvýšený výskyt osteosarkómu pri dlhodobom podávaní teriparatidu
(pozri časť 5.3). Kým nebudú dostupné ďalšie klinické výsledky, nemala by byť prekročená odporučená dĺžka liečby teriparatidom 24 mesiacov.
Sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v maximálna denná dávke, t.j. v zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
V štúdii s 15 zdravými dobrovoľníkmi, ktorí denne užívali digoxín až do dosiahnutia rovnovážneho stavu, neovplyvnilo podanie jednorazovej dávky teriparatidom účinok digoxínu na srdce. Avšak zo sporadických hlásení vyplýva, že pacienti s hyperkalciémiou môžu mať sklon k digitalisovej toxicite. Pretože teriparatid prechodne zvyšuje koncentráciu vápnika v sére, malo by teda byť u pacientov užívajúcich digitalis podávané s opatrnosťou.
Teriparatid sa skúmalo vo farmakodynamických interakčných štúdiách s hydrochlórtiazidom. Nezaznamenali sa žiadne klinicky významné interakcie.
Súbežné podávanie raloxifénu alebo hormonálnej substitučnej terapie spolu s teriparatidom nemalo
vplyv na hladinu vápnika v sére alebo v moči alebo na výskyt klinických nežiaducich účinkov.
4.6 Fertilita, gravidita a laktácia
Ženy vreprodukčnomveku/antikoncepciaužien
Ženy v reprodukčnom veku majú používať účinné metódy antikoncepcie počas užívania teriparatidom.
Ak žena otehotnie, teriparatid sa má vysadiť.
Gravidita
Teriparatide SUN je kontraindikované na používanie počas gravidity (pozri časť 4.3).
DojčenieTeriparatide SUN je kontraindikované na používanie počas dojčenia. Nie je známe, či sa teriparatid
vylučuje do ľudského mlieka.
FertilitaŠtúdie na zajacoch preukázali reprodukčnú toxicitu (pozri časť 5.3). Neuskutočnili sa štúdie o vplyve
teriparatidu na vývoj ľudského plodu. Potencionálne riziko pre ľudí je neznáme.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeTeriparatide SUN nemá, alebo má zanedbateľné účinky na schopnosť viesť vozidlá, alebo obsluhovať stroje. U niektorých pacientov bola pozorovaná prechodná ortostatická hypotenzia alebo závraty. Títo pacienti by sa do ústupu príznakov mali zdržať vedenia vozidiel alebo obsluhovania strojov.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofiluMedzi najčastejšie hlásené nežiaduce reakcie u pacientov liečených teriparatidom patrili nevoľnosť,
bolesť v končatinách, bolesť hlavy a závraty.
Tabuľkový prehľad nežiaducichúčinkovAspoň jeden nežiaduci účinok bol hlásený v štúdiách s teriparatidom u 82,8 % pacientov užívajúcich
teriparatidom a u 84,5 % pacientov užívajúcich placebo.
Nežiaduce reakcie, ktoré súviseli s používaním teriparatidu pri osteoporóze v klinických štúdiách a v priebehu postmarketingového pôsobenia, sú zahrnuté nižšie v tabuľke. Nežiaduce reakcie boli klasifikované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až
< 1/100), zriedkavé (≥ 1/10 000 až <1/1 000), veľmi zriedkavé (< 1/10 000).
Tabuľka 1. Nežiaduce reakcie na liekTrieda orgánov systému
MedDRA
| Nežiaduce reakcie na liek
| Frekvencia
|
Poruchy krvi a
lymfatického systému
| Anémia
| Časté
|
Poruchy imunitného
systému
| Anafylaxia
| Zriedkavé
|
Poruchy metabolizmu a
výživy
| Hypercholesterolémia
| Časté
|
Hyperkalciémia vyššia ako 2,76 mmol/l,
hyperurikémia
| Menej časté
|
Hyperkalciémia vyššia ako 3,25 mmol/l
| Zriedkavé
|
Psychické poruchy
| Depresia
| Časté
|
Poruchy nervového
systému
| Závrat, bolesť hlavy, ischias, synkopa
| Časté
|
Poruchy ucha a labyrintu
| Vertigo
| Časté
|
Poruchy srdca a srdcovej
činnosti
| Palpitácie
| Časté
|
Tachykardia
| Menej časté
|
Poruchy ciev
| Hypotenzia
| Časté
|
| Dyspnoe
| Časté
|
Poruchy dýchacej sústavy,
hrudníka a mediastína
|
Emfyzém
|
Menej časté
|
Poruchy
gastrointestinálneho traktu
|
Nauzea, vracanie, hiátová hernia,
gastroezofageálna refluxná choroba
|
Časté
|
Hemoroidy
|
Menej časté
|
Poruchy kože a
podkožného tkaniva
|
Zvýšené potenie
|
Časté
|
Poruchy kostrovej a
svalovej sústavy a spojivového tkaniva
|
Bolesť končatín
|
veľmi časté
|
Svalové kŕče
|
Časté
|
Myalgia, artralgia, kŕč chrbtového
svalstva/bolesť*
|
Menej časté
|
Poruchy obličiek a
močových ciest
|
Inkontinencia moču, polyúria, nutkanie na
močenie, nefrolitiáza
|
Menej časté
|
Zlyhanie/poškodenie obličiek
|
Zriedkavé
|
Celkové poruchy a reakcie
v mieste podania
|
Únava, bolesť na hrudníku, asténia, mierne a
prechodné reakcie v mieste podania injekcie, vrátane bolesti, opuchu, erytému, modrín, pruritu a slabého krvácania v mieste podania injekcie
|
Časté
|
Erytém v mieste podania injekcie, reakcia v
mieste podania injekcie
|
Menej časté
|
Možné alergické udalosti ihneď po injekcii:
akútna dýchavičnosť, orofaciálny edém, generalizovaná urtikária, bolesť na hrudníku, edém (najmä periférny)
|
Zriedkavé
|
Laboratórne a funkčné
vyšetrenia
|
Zvýšenie hmotnosti, srdcový šelest, zvýšenie
hladiny alkalickej fosfatázy
|
Menej časté
|
* Závažné prípady kŕčov chrbtového svalstva alebo bolesti boli hlásené v priebehu niekoľkých minút
po injekcii.
Popis vybraných nežiaducichreakciíV klinických štúdiách boli hlásené nasledovné reakcie s ≥ 1 %-ným rozdielom vo frekvencii
v porovnaní s placebom: vertigo, nauzea, bolesť v končatinách, závrat, depresia, dýchavičnosť.
Teriparatid zvyšuje koncentráciu kyseliny močovej v sére. Zvýšenie koncentrácie kyseliny močovej v sére nad hornú hranicu normy sa v rámci klinických štúdií vyskytlo u 2,8 % pacientov užívajúcich teriparatidom v porovnaní s 0,7 % pacientami užívajúcimi placebo. Hyperurikémia však nespôsobila zvýšený výskyt dny, bolesti kĺbov alebo urolitiázy.
Protilátky vykazujúce skríženú reaktivitu s teriparatidom boli vo veľkej klinickej štúdii preukázané u 2,8 % žien užívajúcich teriparatidom. Vo všeobecnosti protilátky boli prvýkrát preukázané po
12 mesiacoch liečby a po vysadení liečby dochádzalo k ich poklesu. V tejto súvislosti neboli preukázané žiadne reakcie precitlivenosti, alergickej reakcie, zmeny koncentrácie vápnika v sére alebo
vplyv na vývoj minerálovej denzity kostí (Bone Mineral Density, BMD).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePríznakyasymptómy
Teriparatid bolo podávané v jednotlivých dávkach až do množstva 100 mikrogramov a v opakovaných dávkach až do množstva 60 mikrogramov/deň po dobu 6 týždňov.
Príznaky, ktoré možno očakávať pri predávkovaní, zahŕňajú oneskorenú hyperkalcémiu a riziko ortostatickej hypotenzie. Ďalej sa môžu vyskytnúť nauzea, vracanie, závraty a bolesť hlavy.
Poznatky o predávkovanízaloženénaspontánnychpostmarketingovýchhláseniach
V spontánnych postmarketingových hláseniach boli popísané omyly v dávkovaní, kedy bol celý obsah
(až 800 µg) teriparatidového pera podaný ako jedna dávka. Hlásené prechodné účinky zahŕňali nevoľnosť, slabosť/letargiu a hypotenziu. V niektorých prípadoch nevznikli následkom predávkovania
žiadne nežiaduce účinky. V súvislosti s predávkovaním neboli hlásené žiadne úmrtia.
Liečba predávkovania
Žiadne špecifické antidotum pre teriparatid neexistuje. Pri podozrení na predávkovanie by malo byť
teriparatidu dočasne vysadené, mala by byť sledovaná koncentrácia vápnika v sére a mala by byť začatá príslušná liečba, napr. hydratácia.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: homeostáza vápnika, hormóny prištítnych teliesok a analógy, ATC kód:
H05AA02
Mechanizmus účinku
Endogénny parathormón (PTH) obsahujúci 84 aminokyselín je hlavným regulátorom metabolizmu
vápnika a fosforu v kosti a v obličkách. Teriparatid (rhPTH(1-34)) je aktívny fragment (1-34) endogénneho ľudského parathormónu. Fyziologické účinky PTH zahŕňajú stimuláciu kostnej novotvorby priamym pôsobením na bunky tvoriace kostnú hmotu (osteoblasty), a tým nepriame zvýšenie črevnej absorpcie vápnika, zvýšenie tubulárnej reabsorpcie vápnika a vylučovanie fosfátov obličkami.
Farmakodynamické účinky
Teriparatid je látka podporujúca tvorbu kostí určená na liečbu osteoporózy. Účinky teriparatidu na
skelet závisia od charakteru systémovej expozície. Podávanie teriparatidu jedenkrát denne zvyšuje apozíciu novej kosti na povrchoch trámcovej a kortikálnej kosti preferenčnou stimuláciou aktivity
osteoblastov, ktorá prevláda nad aktivitou osteoklastov.
Klinická účinnosť
Rizikové faktory
Je potrebné vziať do úvahy nezávislé rizikové faktory, napríklad nízku BMD, vek, výskyt zlomeniny v minulosti, rodinnú anamnézu zlomeniny krčka stehnovej kosti, vysoký kostný obrat a nízky body mass index pri identifikácii žien a mužov so zvýšeným rizikom osteoporotických zlomenín, u ktorých môže byť liečba prínosom.
U predmenopauzálnych žien s osteoporózou indukovanou glukokortikoidmi je potrebné vziať do úvahy vysoké riziko zlomenín, ak majú bežnú zlomeninu alebo kombináciu rizikových faktorov, ktoré ich zaraďujú do skupiny s vysokým rizikom zlomenín (napr. nízka kostná denzita [napr. T skóre ≤−2], pretrvávajúca liečba vysokými dávkami glukokortikoidov [napr. ≥7,5 mg/deň počas minimálne
6 mesiacov], vysoká aktivita základného ochorenia, nízka hladina pohlavných steroidných hormónov).
Postmenopauzálna osteoporóza
Do hlavnej klinickej štúdie bolo zaradených 1 637 postmenopauzálnych žien (priemerný vek
69,5 roka). Pri vstupe do štúdie malo deväťdesiat percent pacientov jednu alebo viac zlomenín stavcov a v priemere vertebrálna BMD bola 0,82 g/cm2 (čo zodpovedá T skóre = - 2,6 SD). Všetky pacientky denne dostávali 1 000 mg vápnika a aspoň 400 IU vitamínu D. Výsledky liečby teriparatidom trvajúcej až 24 mesiacov (priemerná doba: 19 mesiacov) preukázali štatisticky významné zníženie rizika zlomenín (Tabuľka 1). Aby sa zabránilo jednej alebo viacerým zlomeninám stavcov je potrebné liečiť
11 žien počas priemerne 19 mesiacov.
Tabuľka 2. Výskyt zlomenín u postmenopauzálnych žien
|
| Placebo
(N = 544) (%)
| Teriparatid
| Relatívne riziko (95 % CI) oproti placebu
|
Nová zlomenina stavcov (> 1) a
| 14,3
| 5,
0 b
| 0,35
(0,22; 0,55)
|
Mnohopočetné zlomeniny
stavcov (> 2) a
| 4,9
| 1,
1 b
| 0,23
(0,09; 0,60)
|
Nevertebrálne osteoporotické
zlomeniny c
| 5,5 %
| 2,6 % d
| 0,47
|
Závažné nevertebrálne
osteoporotické zlomeniny c
(krčka stehnovej kosti, vretennej,
ramennej kosti, rebier a panvy)
| 3,9 %
| 1,5 % d
| 0,38
(0,17; 0,86)
|
Skratky: N = počet pacientov náhodne pridelených do každej liečenej skupiny: CI = interval spoľahlivosti

a Výskyt zlomenín stavcov bol hodnotený u 448 pacientov dostávajúcich placebo a 444 pacientov dostávajúcich teriparatid,
ktorí mali vstupné i kontrolné snímky chrbtice.

b p≤ 0,001 v porovnaní s placebom

c Signifikantné zníženie výskytu zlomeniny krčka stehnovej kosti nebolo preukázané
d p≤ 0,025 v porovnaní s placebom.
Po 19 mesiacoch liečby (priemerná doba) sa v porovnaní s placebom zvýšila (BMD) v bedrovej
chrbtici o 9 % a v celkovom krčku stehnovej kosti o 4 % (p< 0,001).
Sledovanie po ukončení liečby: Po ukončení liečby teriparatidom bolo 1 262 postmenopauzálnych žien z hlavnej klinickej štúdie zaradených do nasledujúcej štúdie. Primárnym cieľom tejto fázy štúdie bolo zhromaždiť viac informácií o bezpečnosti teriparatidu. V priebehu tejto observačnej fázy bola povolená iná liečba osteoporózy a bolo vykonané ďalšie zhodnotenie výskytu zlomenín stavcov.
V priebehu priemerného obdobia 18 mesiacov po vysadení teriparatidom bol počet pacientov s aspoň jednou novou zlomeninou stavca o 41 % nižší v porovnaní s placebom (p = 0,004).
V otvorenej štúdii sa liečilo teriparatidom 503 postmenopauzálnych žien so závažnou osteoporózou
a zlomeninou v predošlých 3 rokoch (83 % z nich už predtým dostávalo osteoporotickú liečbu) počas až 24 mesiacov. Po 24 mesiacoch liečby bol priemerný nárast BMD (minerálnej denzity kostí)
v porovnaní s východiskovými hodnotami 10,5 % v bedrovej chrbtici, 2,6 % v celom bedrovom kĺbe
a 3,9 % v krčku stehnovej kosti. Priemerný nárast BMD medzi 18 a 24 mesiacmi bol 1,4 % v bedrovej chrbtici, 1,2 % v celom bedrovom kĺbe a 1,6 % v krčku stehnovej kosti.
Do 24-mesačnej randomizovanej, dvojito zaslepenej, komparátorom kontrolovanej štúdie fázy, 4 bolo zahrnutých 1 360 postmenopauzálnych žien s rozvinutou osteoporózou. 680 pacientok bolo randomizovaných na teriparatid a 680 pacientok bolo randomizovaných na 35 mg/týždeň rizendronátu, perorálne. Na začiatku mali ženy vek v priemere 72,1 rokov a medián 2 bežných vertebrálnych zlomenín; 57,9 % pacientok užívalo pôvodnú bisfosfonátovú liečbu a počas štúdie 18,8 % užívalo súbežne glukokortikoidy. 1 013 (74,5 %) pacientok ukončilo 24-mesačné pokračovanie štúdie.
Priemerná kumulatívna dávka (medián) glukokortikoidov bola 474,3 (66,2) mg v teriparatidovej
skupine a 898,0 (100,0) mg v rizendronátovej skupine. Priemerný príjem vitamínu D (medián)
v teriparatidovej skupine bol 1433 IU/deň (1400 IU/deň) a v rizendronátovej skupine 1 191 IU/deň
(900 IU/deň). Výskyt nových vertebrálnych zlomenín u pacientok, ktorým vyhotovili vstupné aj následné rádiogramy chrbtice, bol 28/516 (5,4 %) u pacientok liečených teriparatidom a 64/533 (12,0 %) u pacientok liečených rizendronátom, relatívne riziko (95 % CI) = 0,44 (0,29-0,68),
P< 0,0001. Kumulatívny výskyt združených klinických zlomenín (klinické vertebrálne a nevertebrálne zlomeniny) bol 4,8 % u pacientok liečených teriparatidom a 9,8 % u pacientok liečených
rizedronátom, pomer rizika (95 % CI) = 0,48 (0,32-0,74), P = 0,0009.
Osteoporóza u mužov
Do klinickej štúdie u mužov bolo zaradených 437 pacientov (priemerný vek 58,7 roka)
s hypogonadálnou osteoporózou (definovanou ako nízky ranný voľný testosterón alebo zvýšený FSH alebo LH) alebo idiopatickou osteoporózou. Vstupná minerálna denzita kostí chrbtice a krčka stehnovej kosti, priemerné T skóre boli -2,2 resp. -2,1. Pri vstupe do štúdie 35 % pacientov malo zlomeninu stavcov a 59 % malo nevertebrálnu zlomeninu.
Všetci pacienti denne dostávali 1 000 mg vápnika a aspoň 400 IU vitamínu D. Po 3 mesiacoch došlo k významnému zvýšeniu BMD v bedrovej časti chrbtice. Za 12 mesiacov sa BMD bedrovej časti chrbtice zvýšila v porovnaní s placebom o 5 %, celkového krčka stehnovej kosti o 1 %. Nebol však preukázaný významný účinok na výskyt zlomenín.
Osteoporóza indukovaná glukokortikoidmi
Účinnosť teriparatidu u mužov a žien (N = 428), ktorí dostávajú pretrvávajúcu systémovú liečbu glukokortikoidmi (ekvivalentnú 5 mg alebo viac prednizónu počas minimálne 3 mesiacov) bola preukázaná v 18-mesačnej primárnej fáze randomizovanej, dvojito zaslepenej, 36-mesačnej komparátorom kontrolovanej štúdii (alendronát 10 mg/deň). Dvadsaťosem percent pacientov malo na začiatku jednu alebo viac rádiograficky potvrdených vertebrálnych zlomenín. Všetci pacienti dostávali
1 000 mg vápnika denne a 800 IU vitamínu D denne.
Táto štúdia zahŕňala postmenopauzálne ženy (N = 277), predmenopauzálne ženy (N = 67) a mužov (N
= 83). Na začiatku mali postmenopauzálne ženy vekový priemer 61 rokov, stredné BMD T skóre
v bedrovej časti chrbtice -2,7, medián dávky glukokortikoidu zodpovedajúci dávke prednizónu
7,5 mg/deň a 34 % pacientov malo jednu alebo viac rádiograficky potvrdených vertebrálnych zlomenín; predmenopauzálne ženy mali vekový priemer 37 rokov, stredné BMD T skóre v bedrovej časti chrbtice -2,5, medián dávky glukokortikoidu zodpovedajúci dávke prednizónu 10 mg/deň a 9 % pacientov malo jednu alebo viac rádiograficky potvrdených vertebrálnych zlomenín; a muži mali vekový priemer 57 rokov, stredné BMD T skóre v bedrovej časti chrbtice -2,2, medián dávky glukokortikoidu zodpovedajúci dávke prednizónu 10 mg/deň a 24 % pacientov malo jednu alebo viac rádiograficky potvrdených vertebrálnych zlomenín.
Šesťdesiatdeväť percent pacientov dokončilo 18-mesačnú primárnu fázu štúdie. V koncovom hodnotení po 18 mesiacoch, teriparatid signifikantne zvýšilo BMD v bedrovej časti chrbtice (7,2 %)
v porovnaní s alendronátom (3,4 %) (p< 0,001). Teriparatid zvýšilo BMD v oblasti celého bedrového kĺbu (3,6 %) v porovnaní s alendronátom (2,2 %) (p<0,01), takisto ako krčka stehnovej kosti (3,7 %)
v porovnaní s alendronátom (2,1 %) (p< 0,05).
U pacientov liečených teriparatidom vzrástla hodnota BMD v období medzi 18 a 24 mesiacmi liečby
o ďalších 1,7 % v bedrovej chrbtici, 0,9 % v celom bedrovom kĺbe a 0,4 % v krčku stehennej kosti.
Analýza röntgenových snímok chrbtice po 36 mesiacoch liečby 169 pacientov alendronátom a 173 pacientov teriparatidom preukázala, že u 13 pacientov v skupine liečenej alendronátom (7,7 %) sa vyskytla nová vertebrálna zlomenina v porovnaní s 3 pacientmi v skupine užívajúcich teriparatidom (1,7 %) (p = 0,01). Okrem toho u 15 z 214 pacientov v skupine liečenej alendronátom (7,0 %) sa vyskytla nevertebrálna zlomenina v porovnaní so 16 pacientami z 214 v skupine
užívajúcich teriparatidom (7,5 %) (p = 0,84).
U predmenopauzálnych žien bolo zvýšenie BMD z východiskovej hodnoty na hodnotu koncového bodu po 18 mesiacoch signifikantne vyššie v skupine s teriparatidom v porovnaní s alendronátovou skupinou v bedrovej časti chrbtice (4,2 % oproti −1,9%; p< 0,001) a v oblasti celého bedrového kĺbu (3,8 % oproti 0,9 %; p = 0,005). Žiadny signifikantný účinok na výskyt zlomeniny sa však nedokázal.
5.2 Farmakokinetické vlastnosti
Distribúcia
Distribučný objem je približne 1,7 l/kg. Po subkutánnom podaní je polčas teriparatidu približne
1 hodina, čo odráža čas potrebný na vstrebanie z miesta vpichu.
Biotransformácia
S teriparatidom neboli vykonané žiadne štúdie metabolizmu alebo exkrécie, zdá sa však, že periférny
metabolizmus parathormónu prebieha prevažne v pečeni a obličkách.
Eliminácia
Teriparatid sa vylučuje hepatálnym a extrahepatálnym klírensom (približne 62 l/hod u žien a 94 l/hod
u mužov)
Starší pacienti
Vo farmakokinetike teriparatidu neboli zaznamenané žiadne rozdiely s ohľadom na vek (rozpätie
od 31 do 85 rokov). Prispôsobenie dávky veku sa nevyžaduje.
5.3 Predklinické údaje o bezpečnosti
Teriparatid nebol genotoxický v žiadnom zo štandardne vykonávaných testov. Pri podávaní potkanom, myšiam alebo králikom neboli preukázané žiadne teratogénne účinky. Nepozorovali sa žiadne významné účinky u gravidných potkanov alebo myší, ktoré dostávali teriparatid v denných dávkach 30 až 1 000 µg/kg. Objavila sa však fetálna resorpcia a znížená veľkosť vrhu u gravidných králikov, ktoré dostávali teriparatid v denných dávkach 3 až 100 µg/kg. Embryotoxicita pozorovaná u králikov môže súvisieť s oveľa väčšou citlivosťou králikov na účinky PTH na ionizovaný vápnik v krvi v porovnaní
s hlodavcami.
U potkanov, liečených takmer celoživotne dennou injekciou teriparatidu, bolo preukázané dávkovo závislé zvýšenie kostnej novotvorby a zvýšený výskyt osteosarkómu, veľmi pravdepodobne epigenetickým mechanizmom. Podávanie teriparatidu potkanom nezvýšilo incidenciu žiadneho iného typu nádorového ochorenia. Klinický význam týchto nálezov je vďaka odlišnej fyziológii kosti
u potkanov a u ľudí pravdepodobne zanedbateľný. Žiadne kostné tumory neboli zaznamenané u opíc po ovarektómii liečených počas 18 mesiacov ani počas ďalších 3 rokov po ukončení liečby. Navyše
v klinických štúdiách alebo štúdiách nasledujúcich po ukončení liečby nebol žiadny osteosarkóm
preukázaný.
Štúdie na zvieratách preukázali, že významne znížený prietok krvi pečeňou znižuje expozíciu PTH
hlavnému štiepnemu systému (Kupfferove bunky) a následne klírens PTH(1-84).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
ľadová kyselina octová (E260)
bezvodý octan sodný (E262)
manitol (E421)
metakrezol
kyselina chlorovodíková (na úpravu pH) (E507)
hydroxid sodný (na úpravu pH) (E524)
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
Po prvom otvorení
Chemická, fyzikálna a mikrobiologická stabilita v priebehu užívania lieku bola preukázaná počas
28 dní pri 2°C – 8°C. Po otvorení sa môže liek uchovávať pri 2°C – 8°C maximálne 28 dní.
Za nedodržanie odporučených podmienok a doby uchovávania je zodpovedný užívateľ.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke. Podmienky na uchovávanie po prvom otvorení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
2,4 ml roztoku v náplni (silikónové sklo) s piestom (guma z halobutylu), tesniaci disk (gumový laminát z polyizoprén/brómbutylu)/hliník vložené do jednorazového pera.
Teriparatide SUN sa dodáva v balení obsahujúcom 1 naplnené pero alebo 3 naplnené perá. Každé pero obsahuje 28 dávok s obsahom 20 mikrogramov (na 80 mikrolitrov).
Nie všetky veľkosti balenia musia byť uvedené na trh.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Zaobchádzanie
Teriparatide SUN je dostupné ako naplnené pero. Každé pero má používať len jeden pacient. Na každú injekciu sa musí použiť nová sterilná ihla s priemerom 31 a dĺžkou 5 mm. Balenie neobsahuje ihly. Po každej aplikácii musí byť naplnené pero s Teriparatide SUN vrátené do chladničky bezprostredne po použití.
Naplnené pero neskladujte s nasadenou ihlou.
Teriparatide SUN by nemalo byť použité, ak je roztok zakalený, sfarbený alebo obsahuje častice. Likvidácia
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s miestnymi požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Sun Pharmaceutical Industries
Europe B.V. Polarisavenue 87
2132 JH Hoofddorp
Holandsko
8. REGISTRAČNÉ ČÍSLAEU/1/22/1697/001
EU/1/22/1697/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
Dátum posledného predĺženia registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.'
NÁVOD NA POUŽITIE PERA
Terparatide SUN 20 mikrogramov/80 mikrolitrov, injekčný roztok naplnený v pere
Návod na použitie
Skôr ako začnete používať nové pero, prečítajte si celý Návod na použitie. Pri používaní pera
postupujte presne podľa pokynov. Taktiež si prečítajte písomnú informáciu pre používateľa.
Nepožičiavajte si s nikým svoje pero ani ihly, aby ste sa vyhli nebezpečenstvu prenosu infekcie. Vaše pero obsahuje liek na 28 dní.
Časti Teriparatide SUN*

Čierne injekčné tlačidlo
Žltý valec
Červený pásik Zelené telo Náplň s liekom Biely uzáver
Papierová fólia Ihla Veľký kryt ihly
Malý ochranný kryt ihly
* Ihly nie sú súčasťou pera. Možno použiť ihlu s priemerom 31 a dĺžkou 5 mm. Spýtajte sa svojho lekára, ktorá veľkosť
a dĺžka ihly je pre vás najvhodnejšia.
Pred každou aplikáciou si umyte ruky. Miesto podania si pripravte podľa pokynov lekára alebo
lekárnika.
Krok 1 Stiahnite biely uzáver
Stiahnite biely uzáver Odstráňte biely uzáver tak, že ho rovno stiahnete z pera
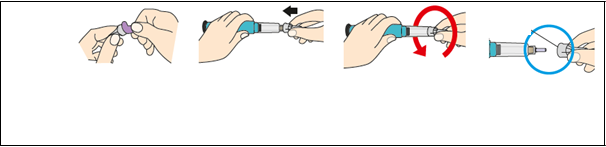
 K
rok 2
N
asaďt e novú
i
hlu
K
rok 2
N
asaďt e novú
i
hlu
a) Odstráňte papierovú fóliu.
b)
Zatlačte ihlu
priamona náplň s liekom.
c)
Otáčajte ihlou až kým nie je pevne
nasadená.
Veľký kryt ihly
d)
Odstráňte veľký kryt ihly a
odložtesi ho.
K
rok 3
N
astavte dávku
e)
Vytiahnite čierne injekčné tlačidlo
až kým sa nezastaví.Ak nemôžete vytiahnuť čierne injekčné tlačidlo, prečítajte si
Riešenie problémov, problém E.
Červený pásik

f)
Skontrolujte, či je vidieť červený
pásik.
Malý ochranný kryt ihly
g)
Odstráňte malý ochranný kryt ihly a zahoďte ho.
Poznámka: Po odstránení vnútorného chrániča ihly, môžete vidieť kvapky lieku vychádzajúce z ihly.

Je to normálne a neovplyvní to vašu dávku.
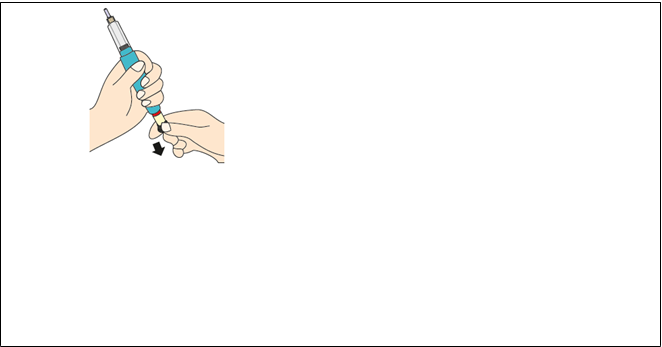
K
rok 4
A
plikácia dávky
|
|
|
|
h)
Držte jemne kožnú riasu na stehne
alebo bruchu a vpichnite ihlu priamo do kože.
|
i)
Zatlačte čierne injekčné tlačidlo, až kým sa nezastaví. Držte ho zatlačené a
p-o-m-a-l-y počítajte do 5. Potom vytiahnite
ihlu z kože.
|
K
rok 5
P
o
t
vrďte
dávku
D
Ô
LE
Ž
I
TÉ
j)
P
o skončení injekčnej
aplikácie:
Po vytiahnutí ihly
z kože skontrolujte, či je čierne injekčné
tlačidlo úplne zatlačené. Ak sa
neukáže žltý valec, dokončili ste injekčnú
aplikáciu správne.
k)
NEMALI by ste vidieť ani kúsok žltého valca. Ak ho vidíte a už ste si podali dávku, neaplikujte si ju znova
v ten istý deň. Namiesto toho, si musíte znovu nastaviť Teriparatide SUN (pozri Riešenie problémov, problém A).
K
rok 6
O
dstráňte
i
hlu
Veľký
kryt ihly

l)
Nasaďte veľký kryt ihly na ihlu.
m)
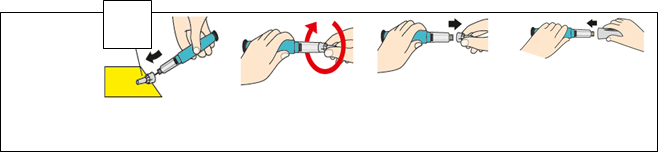
Odskrutkujte ihlu
a to tak, že otočíte
n)
Vytiahnite ihlu
a zlikvidujte ju podľa
o)
Nasaďte späť biely uzáver. Ihneď po
veľký kryt ihly o 3

až 5 celých otáčok.
pokynov lekára alebo
lekárnika.
použití odložte Teriparatide SUN do chladničky.

Tieto usmernenia týkajúce sa manipulácie s ihlou nenahrádzajú národné pokyny ani pokyny lekára či ústavné predpisy.
Riešenie problémov
|
Problém Riešenie
A. Žltý valec je stále Pri opätovnom nastavovaní Teriparatide SUN
vidieť po stlačení postupujte podľa týchto pokynov.
čierneho injekčného 1) Odporúčaná dávka je 20 mikrogramov tlačidla. Ako mám podávaných raz denne. Ak ste si už podali znovu nastaviť moje dávku, neaplikujte si znova ďalšiu dávku Teriparatide SUN? v ten istý deň.
2) Odstráňte ihlu.
3) Nasaďte novú ihlu, odstráňte veľký kryt ihly a odložte si ho.
4) Vytiahnite čierne injekčné tlačidlo, až kým
sa nezastaví. Skontrolujte, či je vidieť červený pásik. (Pozri krok 3)
5) Odstráňte malý ochranný kryt ihly
a zahoďte ho.
6) Nasmerujte ihlu nadol do prázdnej nádoby.
Zatlačte čierne injekčné tlačidlo, až kým sa nezastaví. Držte ho stlačené a p-o-m-a-l-y počítajte do 5. Môžete uvidieť malý prúd alebo kvapku tekutiny. Ak ste skončili, čierne injekčné tlačidlo by malo byť úplne zatlačené.
7) Ak stále vidíte žltý valec, obráťte sa prosím
na svojho lekára alebo lekárnika.
8) Nasaďte veľký kryt ihly na ihlu. Úplne odskrutkujte ihlu a to tak, že otočíte kryt
ihly o 3 až 5 celých otáčok. Stiahnite kryt
z ihly a zlikvidujte ho podľa pokynov lekára alebo lekárnika. Nasaďte späť biely
uzáver a odložte Teriparatide SUN do
chladničky. (Pozri krok 6)
|
| Tomuto problému môžete predchádzať, ak vždy
použijete NOVÚ ihlu na každú injekciu a ak úplne zatlačíte čierne injekčné tlačidlo a budete p-o-m-a-l-y počítať do 5.
|
 B
. Ako zistím, či môj
T
eriparatid SUN
účinkuje?
B
. Ako zistím, či môj
T
eriparatid SUN
účinkuje?
Teriparatide SUN je určené na injekčné podanie
celej dávky vždy, keď sa používa podľa pokynov

v časti
Návod na použitie. Keď je čierne injekčné tlačidlo úplne zatlačené, znamená to, že bola podaná celá dávka Teriparatide SUN.
Nezabudnite používať pri každej aplikácii novú ihlu, aby ste zabezpečili, že Teriparatide SUN bude fungovať správne.
C
. Vidím vzduchovú
bublinku v mojom
T
eriparatide
SUN.
Malá vzduchová bublinka neovplyvní vašu dávku,
ani vám neuškodí. Môžete pokračovať
v podávaní vašej dávky ako zvyčajne.
D
. Nemôžem odstrániť
i
hlu.
1) Nasaďte veľký kryt ihly na ihlu. (Pozri krok 6)
2) Použite veľký kryt ihly na odskrutkovanie ihly.
3) Úplne odskrutkujte ihlu a to tak, že otočíte kryt ihly o 3 až 5 celých otáčok.
4) Ak stále nemôžete odstrániť ihlu, požiadajte
niekoho o pomoc.
E
. Čo mám robiť, ak
nemôžem vytiahnuť čierne injekčné
tl
ačidlo?
Z
oberte si nové Teriparatide SUN na podanie
s
vojej dávky podľa pokynov vášho lekára alebo lekárnika.
Naznačuje to, že ste už použili všetok liek, ktorý môže byť presne aplikovaný, dokonca i vtedy, keď stále vidíte nejaký liek v náplni.
Č
i
stenie vášho Teriparatide SUN
Č
i
stenie a uchovávanie





• Očistite vonkajšiu časť Teriparatide SUN vlhkou utierkou.
• Neponárajte Teriparatide SUN do vody, neumývajte ho ani nečistite tekutinou.
Uchovávanie vášho Teriparatide SUN
• Ihneď po každom použití uložte Teriparatide SUN do chladničky. Prečítajte si informácie v
písomnej informácii pre používateľa o uchovávaní lieku a postupujte podľa týchto pokynov.
• Neuchovávajte Teriparatide SUN s nasadenou ihlou, pretože to môže spôsobiť tvorbu vzduchových bubliniek v náplni.
• Uchovávajte Teriparatide SUN s nasadeným bielym uzáverom.
• Ak bol liek zmrazený, zahoďte ho a použite nové Teriparatide SUN.
• Ak ste Teriparatide SUN nechali vonku z chladničky, pero nezahadzujte. Uložte pero naspäť do chladničky a kontaktujte svojho lekára alebo lekárnika.
Likvidácia ihiel a peraLikvidácia ihiel a naplneného pera Teriparatide SUN• Pred likvidáciou naplneného pera Teriparatide SUN sa uistite, že je odstránená ihla.
• Použité ihly dajte do nádoby odolnej voči prepichnutiu alebo do uzatvárateľnej nádoby z tvrdého plastu s bezpečným uzáverom. Nehádžte ihly priamo do domového odpadu.
• Naplnenú nádobu odolnú voči prepichnutiu opakovane nepoužívajte.
• Opýtajte sa vášho lekára na možnosti správnej likvidácie pera a nádoby odolnej voči
prepichnutiu.
• Pokyny týkajúce sa zaobchádzania s ihlami nemajú nahradiť miestne zdravotnícke alebo inštitucionálne predpisy.
• Zlikvidujte pero 28 dní po prvom použití.
 I
né dôležité poznámky
I
né dôležité poznámky
• Teriparatide SUN obsahuje liek na 28 dní.
• Neprenášajte liek do injekčnej striekačky.
• Zapíšte si dátum vašej prvej injekčnej aplikácie do kalendára.
• Skontrolujte etiketu Teriparatide SUN a uistite sa, že máte správny liek a že jeho použiteľnosť ešte neuplynula.
• Počas aplikácie môžete počuť jedno alebo viac kliknutí – to je bežná činnosť naplneného
pera.
• Teriparatide SUN sa neodporúča používať u nevidiacich osôb alebo osôb s poruchou zraku bez pomoci osoby, ktorá je poučená o správnom používaní pera.
• Uchovávajte Teriparatide SUN mimo dohľadu a dosahu detí.