
mi na zvrátenie poklesu počtu krvných doštičiek. U pacientov, u ktorých bola liečba inotersenom prerušená pre pokles počtu krvných doštičiek pod 25 x 109/l , sa liečba nemá znovu začínať.
Vynechané dávky
Ak sa vynechá dávka inotersenu, nasledovná dávka sa má podať čo najskôr, pokiaľ nie je podanie
nasledovnej dávky plánované do dvoch dní; v takom prípade sa vynechaná dávka má preskočiť a
nasledovná dávka sa má podať podľa rozvrhu.
Osobitné populácie
Staršie osoby
U pacientov vo veku 65 rokov a starších nie je potrebná žiadna úprava dávkovania (pozri časť 5.2).
Porucha funkcie obličiek
U pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek nie je potrebná žiadna
úprava dávky (pozri časť 5.2). Inotersen sa nemá používať u pacientov, ktorí majú pomer bielkovín ku kreatinínu v moči (urine protein to creatinine ratio, UPCR) ≥ 113 mg/mmol (1 g/g) alebo odhadovanú rýchlosť glomerulárnej filtrácie (estimated glomerular filtration rate, eGFR) < 45 ml/min/1,73m2
(pozri časť 4.3).
Kvôli riziku glomerulonefritídy a možného poklesu funkcie obličiek sa počas liečby inotersenom majú sledovať UPCR a eGFR (pozri časť 4.4). Ak sa potvrdí akútna glomerulonefritída, má sa zvážiť trvalé prerušenie liečby.
Porucha funkcie pečene
U pacientov s miernou alebo stredne závažnou poruchou funkcie pečene nie je potrebná žiadna úprava dávky (pozri časť 5.2). U pacientov so závažnou poruchou funkcie pečene sa inotersen nesmie používať (pozri časť 4.3).
Pacienti podstupujúci transplantáciu pečene
Inotersen nebol hodnotený u pacientov podstupujúcich transplantáciu pečene. Preto sa odporúča, aby
bola liečba inotersenom prerušená u osôb podstupujúcich transplantáciu pečene.
Pediatrická populácia
Bezpečnosť a účinnosť inotersenu u detí a dospievajúcich vo veku do 18 rokov neboli stanovené.
K dispozícii nie sú žiadne údaje.
Spôsob podávania
Iba na subkutánne použitie.
Prvá injekcia, podaná pacientom alebo opatrovateľom, sa má podať za prítomnosti príslušne kvalifikovaného zdravotníckeho pracovníka. Pacienti a/alebo opatrovatelia majú byť zaškolení v subkutánnom podávaní Tegsedi.
Vhodné miesta na injekciu sú brucho, oblasť horného stehna alebo vonkajšia plocha ramena. Je dôležité obmieňať miesta na injekciu. Ak sa injekcia podáva do ramena, má ju podať iná osoba. Je potrebné vyhnúť sa podávaniu injekcie do oblasti pásu a iných miest, kde môže dôjsť k tlaku alebo treniu oblečenia. Tegsedi sa nemá podávať do oblastí postihnutých kožným ochorením alebo do poranených oblastí. Tiež je potrebné vyhnúť sa tetovaniam a jazvám.
Naplnená injekčná striekačka by mala dosiahnuť izbovú teplotu pred podaním injekcie. Je potrebné
vybrať ju z chladničky aspoň 30 minút pred použitím. Nemajú sa používať iné metódy zohrievania.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Počet krvných doštičiek pred liečbou < 100 x 109/l.
Pomer bielkovín ku kreatinínu v moči (urine protein to creatinine ratio, UPCR) pred liečbou
≥ 113 mg/mmol (1 g/g).
Odhadovaná rýchlosť glomerulárnej filtrácie (estimated glomerular filtration rate, eGFR)
< 45 ml/min/1,73 m2.
Závažná porucha funkcie pečene.
4.4 Osobitné upozornenia a opatrenia pri používaní
Trombocytopénia
Inotersen sa spája so znižovaním počtu krvných doštičiek, čo môže viesť k trombocytopénii (pozri
časť 4.8). Počet krvných doštičiek sa má monitorovať každé 2 týždne počas liečby inotersenom
a počas 8 týždňov po ukončení liečby. Odporúčania na úpravu frekvencie monitorovania a dávkovania
inotersenu sú upresnené v Tabuľke 1 (pozri časť 4.2).
Pacientov je potrebné poučiť o tom, aby ihneď oznámili svojmu lekárovi, ak sa u nich vyskytnú prejavy neobvyklého alebo predĺženého krvácania (napr. petechie, spontánne modriny, subkonjunktiválne krvácanie, krvácanie z nosa), stuhnutosť krku alebo nezvyčajne silná bolesť hlavy.
Osobitnú pozornosť treba venovať starším pacientom, pacientom užívajúcim antitrombotické lieky, protidoštičkové lieky alebo lieky, ktoré môžu znížiť počet krvných doštičiek (pozri časť 4.5),
a pacientom s príhodami veľkého krvácania v anamnéze.
Glomerulonefritída/pokles funkcie obličiek
U pacientov liečených inotersenom došlo ku glomerulonefritíde (pozri časť 4.8). Pokles funkcie
obličiek bol tiež pozorovaný u veľkého počtu pacientov bez prejavov glomerulonefritídy (pozri časť
4.8).
UPCR a eGFR sa majú monitorovať každé 3 mesiace alebo častejšie, ako je klinicky indikované na základe prítomnosti chronického ochorenia obličiek a/alebo obličkovej amyloidózy v anamnéze. UPCR a eGFR sa majú monitorovať 8 týždňov po ukončení liečby. Pacienti s UPCR rovnakým alebo vyšším ako dvojnásobok hornej hranice normálnych hodnôt, alebo s eGFR < 60 ml/min, ktorá je potvrdená pri opakovanom testovaní, a v neprítomnosti alternatívneho vysvetlenia, sa majú sledovať každé 4 týždne.
V prípade poklesu eGFR >30 %, v neprítomnosti alternatívneho vysvetlenia, má sa zvážiť pozastavenie liečby inotersenom, kým sa ďalším skúmaním nezistí príčina.
V prípade, že UPCR je ≥ 2 g/g (226 mg/mmol), čo sa potvrdí opakovaným testom, liečba inotersenom sa má pozastaviť, kým prebieha ďalšie skúmanie akútnej glomerulonefritídy. Liečba inotersenom sa má trvalo prerušiť, ak sa potvrdí akútna glomerulonefritída. Ak sa nepotvrdí glomerulonefritída, v liečbe možno pokračovať, ak je to klinicky indikované a po zlepšení funkcie obličiek (pozri časť 4.3).
Ak sa potvrdí diagnóza glomerulonefritídy, má sa zvážiť včasné začatie imunosupresívnej liečby. Odporúča sa opatrnosť s nefrotoxickými liekmi a inými liekmi, ktoré môžu poškodiť funkciu obličiek
(pozri časť 4.5).
Nedostatok vitamínu A
Na základe mechanizmu účinku sa očakáva, že inotersen bude znižovať hladinu vitamínu A
(retinolu) v plazme pod normálnu hladinu (pozri časť 5.1).
Hladina vitamínu A (retinolu) v plazme nižšia ako dolná hranica normálneho rozsahu sa má upraviť a očné príznaky alebo prejavy nedostatku vitamínu A majú ustúpiť pred začiatkom liečby inotersenom.
Pacienti, ktorí dostávajú inotersen majú perorálne užívať suplementáciu približne 3 000 IU vitamínu A denne, za účelom zníženia potenciálneho rizika očnej toxicity zapríčinenej nedostatkom vitamínu A. Odporúča sa vyslanie pacientov na oftalmologické vyšetrenie, ak sa vyskytnú očné príznaky zodpovedajúce nedostatku vitamínu A vrátane zníženej schopnosti nočného videnia alebo nočnej slepoty, pretrvávajúcej suchosti očí, zápalov očí, zápalov rohovky alebo vredov na rohovke,
zhrubnutia rohovky, perforácie rohovky.
Počas prvých 60 dní tehotenstva príliš vysoké aj príliš nízke hladiny vitamínu A môžu byť spojené so
zvýšeným rizikom fetálnych malformácií. Preto sa pred začiatkom liečby musí vylúčiť tehotenstvo a ženy v plodnom veku musia užívať účinnú antikoncepciu (pozri časť 4.6). Ak má žena v úmysle otehotnieť, liečba inotersenom a suplementácia vitamínom A sa majú prerušiť a majú sa sledovať hladiny vitamínu A v plazme a vrátiť sa na normálne hodnoty pred pokusom o počatie.
V prípade neplánovaného tehotenstva sa má prerušiť podávanie inotersenu. Kvôli dlhému polčasu inotersenu (časť 5.2) môže dôjsť k nedostatku vitamínu A aj po ukončení liečby. Nemožno poskytnúť žiadne odporúčanie, či pokračovať alebo prerušiť suplementáciu vitamínu A počas prvého trimestra neplánovaného tehotenstva. Ak sa pokračuje so suplementáciou vitamínu A, denná dávka nemá presiahnuť 3 000 IU pre nedostatok údajov podporujúcich vyššie dávky. Potom sa má pokračovať so
suplementáciou vitamínu A v dávke 3 000 IU denne v druhom a treťom trimestri, ak sa hladina retinolu v plazme ešte nevrátila na normálnu hodnotu, kvôli zvýšenému riziku nedostatku vitamínu A v treťom trimestri.
Nie je známe, či suplementácia vitamínu A v tehotenstve bude dostatočná na zabránenie nedostatku vitamínu A ak tehotná žena bude pokračovať v užívaní inotersenu. Avšak zvýšenie suplementácie vitamínu A na viac ako 3 000 IU denne počas tehotenstva pravdepodobne neupraví hladinu retinolu v plazme kvôli mechanizmu účinku inotersenu, a môže uškodiť matke a plodu.
Monitorovanie pečene
Pečeňové enzýmy sa majú vyšetriť 4 mesiace po začatí liečby inotersenom a potom každoročne alebo
častejšie, ak je to klinicky indikované, aby sa odhalili prípady poruchy funkcie pečene (pozri časť 4.8).
Opatrenia pred začatím liečby inotersenom
Pred začatím liečby s Tegsedi je potrebné zistiť počet krvných doštičiek a hodnoty odhadovanej
rýchlosti glomerulárnej filtrácie (eGFR), pomeru bielkovín ku kreatinínu v moči (UPCR) a pečeňových enzýmov.
Po začatí liečby s inotersenom môže u niektorých pacientov dôjsť k prechodným zvýšeniam hladín
CRP a krvných doštičiek. Táto reakcia obyčajne spontánne ustúpi po niekoľkých dňoch liečby.
4.5 Liekové a iné interakcie
Odporúča sa opatrnosť pri používaní antitrombotických liekov, protidoštičkových liekov a liekov, ktoré môžu znižovať počet krvných doštičiek, napríklad kyselina acetylsalicylová, klopidogrel, warfarín, heparín, nízkomolekulové heparíny, inhibítory faktoru Xa ako sú rivaroxabán a apixabán, a trombínové inhibítory ako dabigatran (pozri časť 4.4).
Odporúča sa opatrnosť pri súbežnom používaní nefrotoxických liekov a iných liekov, ktoré môžu poškodiť funkciu pečene, ako sú sulfónamidy, antagonisty aldosterónu, anilidy, prirodzené ópiové alkaloidy a iné ópioidy (pozri časť 4.4). I keď populačná farmakokinetická analýza neidentifikovala klinicky významné účinky niektorých nefrotoxických liekov na klírens inotersenu alebo na potenciál ovplyvnenia funkcie obličiek, nebolo vykonané systematické hodnotenie súbežného podávania inotersenu a potenciálne nefrotoxických liekov.
4.6 Fertilita, gravidita a laktácia
Ženy v plodnom veku
Inotersen zníži plazmatickú hladinu vitamínu A, ktorý je zásadný pre normálny vývoj plodu. Nie je
známe, či suplementácia vitamínu A bude dostatočná na zníženie rizika pre plod (pozri časť 4.4).
Z tohto dôvodu je potrebné vylúčiť tehotenstvo pred začiatkom liečby inotersenom a ženy v plodnom
veku by mali užívať účinnú antikoncepciu.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití inotersenu u gravidných žien.
Štúdie na zvieratách sú nedostatočné z hľadiska reprodukčnej toxicity (pozri časť 5.3). Z dôvodu
potenciálneho teratogénneho rizika vyplývajúceho z nevyvážených hladín vitamínu A, inotersen sa nemá používať počas tehotenstva pokiaľ klinický stav ženy nevyžaduje liečbu inotersenom. Ženy v plodnom veku musia používať účinnú antikoncepciu počas liečby inotersenom.
Dojčenie
Nie je známe, či sa inotersen/metabolity vylučujú do ľudského mlieka. Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie metabolitov inotersenu do mlieka (pozri časť 5.3). Riziko u dojčených novorodencov/dojčiat nemôže byť vylúčené.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu liekom Tegsedi sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú žiadne údaje o účinkoch inotersenu na fertilitu u ľudí. Štúdie na zvieratách
nepreukázali žiadny účinok inotersenu na fertilitu samcov alebo samíc.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Tegsedi nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najčastejšie pozorovanými nežiaducimi reakciami počas liečby inotersenom boli príhody spojené s
reakciami v mieste vpichu* (50,9 %). Iné najčastejšie hlásené nežiaduce reakcie v súvislosti s inotersenom boli nauzea (31,3 %), anémia (27,7 %), bolesť hlavy (23,2 %), pyrexia (19,6 %), periférny opuch (18,8 %), triaška (17,9 %), vracanie (15,2 %), trombocytopénia (13,4 %) a zníženie počtu krvných doštičiek (10,7 %).
Tabuľkový súhrn nežiaducich reakcií
Tabuľka 2 obsahuje zoznam nežiaducich reakcií (NR) podľa triedy orgánových systémov databázy
MedDRA. V rámci každej triedy orgánových systémov sú NR zoradené podľa frekvencie,
s najčastejšie vyskytujúcimi sa reakciami uvedenými ako prvými. V rámci každej kategórie frekvencie sú nežiaduce reakcie na liečivo uvedené v poradí klesajúcej závažnosti. Okrem toho, kategória frekvencie pre každú NR vychádza z nasledovnej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až
< 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé
(< 1/10 000).
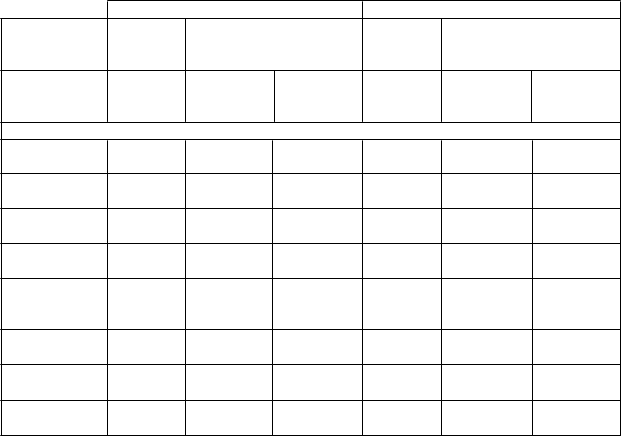
Tabuľka 2. Zoznam nežiaducich reakcií v klinických štúdiách
Trieda orgánových systémov Veľmi časté Časté
Poruchy krvi a lymfatického
systému
Trombocytopénia
Anémia
Znížený počet krvných doštičiek
Eozinofília
Poruchy metabolizmu a výživy Znížená chuť do jedla
Poruchy nervového systému Bolesť hlavy
Poruchy ciev Ortostatická hypotenzia
Hypotenzia
Hematóm
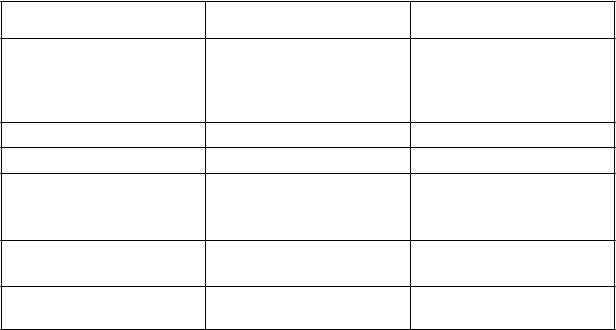
Poruchy gastrointestinálneho traktu
Poruchy pečene a žlčových
ciest
Vracanie
Nevoľnosť
Zvýšená hladina transamináz
T
r
i
e
d
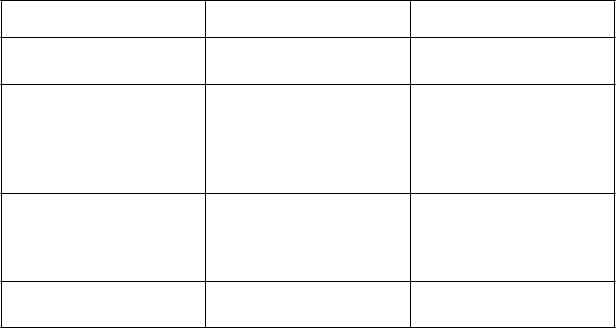
a orgánových systémov Veľmi časté Časté
Poruchy kože a podkožného
tkaniva
Poruchy obličiek a močových
ciest
Celkové poruchy a reakcie v mieste podania
Úrazy, otravy a komplikácie
liečebného postupu
Horúčka
Triaška
Reakcie v mieste vpichu
Periférny edém
Svrbenie
Vyrážky
Glomerulonefritída Proteinúria Zlyhanie obličiek
Akútne poškodenie obličiek
Porucha funkcie obličiek
Ochorenie podobné chrípke
Periférne opuchy
Zmena farby v mieste vpichu
Pomliaždenina
 P
o
pis
vybraných
nežiaducich
r
e
a
kcií
Reakcie v mieste vpichu
P
o
pis
vybraných
nežiaducich
r
e
a
kcií
Reakcie v mieste vpichu
Najčastejšie pozorované príhody zahŕňali príhody spojené s reakciami v mieste vpichu (vrátane bolesti v mieste vpichu, sčervenania, svrbenia, opuchu, vyrážky, tvrdnutia, modriny alebo krvácania). Tieto príhody boli zvyčajne buď obmedzené alebo ich bolo možné liečiť symptomatickou liečbou.
TrombocytopéniaInotersen sa spája so znižovaním počtu krvných doštičiek, čo môže viesť k trombocytopénii. V štúdii
3. fázy NEURO-TTR boli pozorované zníženia počtu krvných doštičiek pod normálne hodnoty (140 x 109/l ) u 54 % pacientov liečených inotersenom a u 13 % pacientov, ktorí dostali placebo; zníženia pod 100 x 109/l boli pozorované u 23 % pacientov liečených inotersenom a u 2 % pacientov, ktorí dostali placebo; potvrdené počty krvných doštičiek < 75 x 109/l boli pozorované u 10,7 % pacientov liečených inotersenom. Traja pacienti (3 %) mali počet krvných doštičiek < 25 x 109/l;
u jedného z týchto pacientov došlo k smrteľnému intrakraniálnemu krvácaniu. Počas liečby inotersenom je potrebné monitorovať pacientov, či u nich nedošlo k trombocytopénii (pozri časť 4.4).
Glomerulonefritída/pokles funkcie obličiekPočas liečby inotersenom je potrebné monitorovať pacientov, aby sa zistili prejavy zvýšenej
proteinúrie a zníženia eGFR (pozri časť 4.4).
ImunogenicitaV hlavnej štúdii 2./3. fázy bolo po 15 mesiacoch liečby 30,4 % pacientov liečených inotersenom
pozitívnych na protilátky proti liečivu. Tvorba protilátok proti inotersenu bola charakterizovaná
oneskoreným nástupom (medián nástupu bol > 200 dní) a nízkym titrom (medián maximálneho titru bol 284 v hlavnej štúdii). Prítomnosť protilátok proti liečivu nemala žiadny vplyv na farmakokinetické parametre (Cmax, AUC alebo polčas) a účinnosť inotersenu, ale pacienti s protilátkami proti liečivu
mali viac reakcií v mieste vpichu.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 Predávkovanie
V prípade predávkovania sa má poskytnúť podporná lekárska starostlivosť vrátane konzultácie so
zdravotníckym pracovníkom a pozorného sledovania klinického stavu pacienta.
Potrebné je pravidelne sledovať vyšetrenia krvných doštičiek a funkcie obličiek.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: zatiaľ nepridelená, ATC kód: zatiaľ nepridelený
Mechanizmus účinkuInotersen je 2′-
O-2-metoxyetyl (2′-MOE) fosforotioát antisense oligonukleotid (ASO), ktorý účinkuje
ako inhibítor produkcie transtyretínu (TTR) u ľudí. Selektívna väzba inotersenu na mediátorovú RNA
(mRNA) pre TTR spôsobuje degradáciu mutantného a divokého (normálneho) typu mRNA pre TTR. Toto bráni syntéze bielkoviny TTR v pečeni, čo vedie k významnému zníženiu hladín mutovanej a normálnej bielkoviny TTR vylučovanej pečeňou do krvného obehu.
TTR je nosnou bielkovinou pre retinol-viažuci proteín 4 (retinol binding protein 4, RBP4), ktorý je hlavným nosičom vitamínu A (retinol). Preto sa očakáva, že zníženie hladiny TTR v plazme bude viesť k zníženiu hladiny retinolu v plazme na hladinu nižšiu ako je dolná hranica normálneho rozsahu.
Farmakodynamické účinkyV hlavnej štúdii NEURO-TTR, v skupine liečenej inotersenom bolo pozorované výrazné zníženie
hladiny cirkulujúceho TTR počas liečby trvajúcej 15 mesiacov, pričom priemerné percentuálne zmeny
oproti východiskovej hodnote TTR v sére boli v rozsahu 68,41 % až 74,03 % (medián rozsahu:
74,64 % až 78,98 %) od 13. do 65. týždňa (Obrázok 1). V skupine s placebom skupine klesla priemerná koncentrácia TTR v sére o 8,50 % v 3.týždni a potom zostala pomerne konštantná počas zostatku liečebného obdobia.
e n
i
l
e s
a
)
B E S
R m
o
T r (
f
T
e M
g S n L a
h
C
%
|
|
 0
0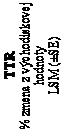 - 1 0- 2 0- 3 0
- 1 0- 2 0- 3 0
- 4 0

P l a c e b o
I n o t e r s e n
- 5 0
- 6 0
- 7 0
- 8 0
 0 1 0 2 0 3 0 4 0 5 0 6 0 7 0
0 1 0 2 0 3 0 4 0 5 0 6 0 7 0
Týždeň štúdie
O
b
r
ázok 1 Percentuálna zmena hladiny TTR v sére v čase oproti východiskovej hodnote
K
l
i
nická
účinnosť
a
bezpečnosť
Multicentrická, dvojito zaslepená, placebom kontrolovaná štúdia NEURO-TTR obsahovala 172 liečených pacientov s dedičnou transtyretínovou amyloidózou s polyneuropatiou (hATTR-PN). Choroba hATTR-PN je rozdelená do 3 štádií, kde i) pacienti v 1. štádiu nepotrebujú pomoc s chôdzou, ii) pacienti v 2. štádiu potrebujú pomoc s chôdzou, a iii) pacienti v 3. štádiu sú odkázaní na invalidný vozík. Osoby s hATTR-PN v 1. a 2. štádiu a NIS ≥10 a ≤130 boli zaradené do hlavnej štúdie NEURO- TTR. Štúdia hodnotila 284 mg inotersenu, podaného ako jedna subkutánna injekcia raz týždenne,
počas 65 týždňov liečby. Pacienti boli randomizovaní v pomere 2:1 na liečbu buď inotersenom alebo placebom. Primárnymi koncovými ukazovateľmi účinnosti boli zmena z východiskovej hodnoty po
66. týždeň v kompozitnom skóre Modifikovaného skóre neuropatického poškodenia + 7 testov (modified Neuropathy Impairment Score + 7 tests, mNIS+7) a v celkovom skóre dotazníka Kvalita života Norfolk - diabetická neuropatia (Norfolk Quality of Life – Diabetic Neuropathy, QoL-DN). Pacienti boli stratifikovaní podľa štádia choroby (1. štádium oproti 2. štádiu), mutácie TTR (V30M oproti non-V30M) a predošlej liečby s buď tafamidisom alebo diflunisalom (áno - nie). Východiskové demografické údaje a charakteristiky choroby sú prezentované v Tabuľke 3.
Tabuľka 3. Východiskové demografické údaje
Placebo
(N=60)
Inotersen
(N=112)
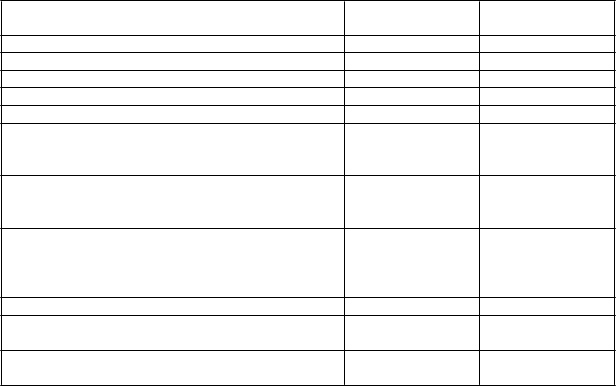
Vek (roky), priemer (SD – smerodajná odchýlka) 59,5 (14,05) 59,0 (12,53) Vek 65 rokov a starší, n (%) 26 (43,3) 48 (42,9) Muži, n (%) 41 (68,3) 77 (68,8) mNIS+7, priemer (SD) 74,75 (39,003) 79,16 (36,958) Norfolk QoL-DN, priemer (SD) 48,68 (26,746) 48,22 (27,503) Štádium choroby, n (%)
1. štádium 42 (70,0) 74 (66,1)
2. štádium 18 (30,0) 38 (33,9) TTR, mutácia V30M1, n (%)
Áno 33 (55,0) 56 (50,0) Nie 27 (45,0) 56 (50,0)
Predchádzajúca liečba tafamidisom alebo
diflusinalom1, n (%)
Áno 36 (60,0) 63 (56,3) Nie 24 (40,0) 49 (43,8) hATTR-CM2, n (%) 33 (55,0) 75 (66,4)
hATTR-PN, trvanie choroby3 (mesiace)
priemer (SD) 64,0 (52,34) 63,9 (53,16)
hATTR-CM, trvanie choroby3 (mesiace)
priemer (SD) 34,1 (29,33) 44,7 (58,00)
1 Na základe klinickej databázy
2 Definované ako všetci pacienti s diagnózou dedičnej transtyretínovej amyloidózy s
kardiomyopatiou (hereditary transthyretin amyloidosis with cardiomyopathy, hATTR-CM) na začiatku štúdie, alebo s hrúbkou steny ľavej komory >1,3 cm na echokardiograme, bez prítomnosti pretrvávajúcej hypertenzie v anamnéze.
3 Trvanie od nástupu príznakov po dátum informovaného súhlasu
Zmeny z východiskovej hodnoty pre obidva primárne koncové ukazovatele (mNIS+7 a Norfolk QoL- DN) preukázali štatisticky významný prínos v prospech liečby inotersenom v 66. týždni (Tabuľka 4). Výsledky pre viaceré charakteristiky choroby [mutácia TTR (V30M, non-V30M)], štádium choroby (1. štádium, 2. štádium), predchádzajúca liečba tafamidisom alebo diflunisalom (áno, nie), prítomnosť hATTR-CM (áno, nie) v 66. týždni preukázali štatisticky významný prínos vo všetkých podskupinách na základe kompozitného skóre mNIS+7 a tiež vo všetkých podskupinách okrem jednej (súbor CM- Echo; p=0,067) na základe celkového skóre Norfolk QoL-DN (Tabuľka 5). Okrem toho, výsledky u zložiek mNIS+7 a domén kompozitných skóre Norfolk QoL-DN boli v zhode s analýzou primárnych'
koncových ukazovateľov, preukazujúc prínos u motorických, senzorických a autonómnych neuropatií
(Obrázok 2).
Tabuľka 4. Analýza primárneho koncového ukazovateľa mNIS+7 a Norfolk QoL-DN
mNIS+7 Norfolk-QOL-DN
Východisková hodnota n
Priemer (SD)
Placebo
(N=60)
60
74,75 (39,003)
Inotersen
(N=112)
112
79,16 (36,958)
Placebo
(N=60)
59
48,68 (26,746)
Inotersen
(N=112)
111
48,22 (27,503)
Zmena v 66. týždni
n
LSM (SE)
95% IS
Rozdiel v hodnote
LSM
(Tegsedi – Placebo)
95% IS
Hodnota p
60
25,43 (3,225)
19,11, 31,75
112
10,54 (2,397)
5,85, 15,24
-14,89
-22,55, -7,22
<0,001
59
12,94 (2,840)
7,38, 18,51
111
4,38 (2,175)
0,11, 8,64
-8,56
-15,42, -1,71
0,015
LSM - ,,priemerná hodnota najmenších štvorcov“
SE - „štandardná chyba
Tabuľka 5. Podskupinová analýza mNIS+7 a Norfolk QoL-DN
mNIS+7 Norfolk QoL-DN
Zmena oproti východiskovej hodnote (inotersen – placebo)
Zmena oproti východiskovej hodnote (inotersen – placebo)
Podskupina n (placebo, inotersen)
Rozdiel v LSM (SE)
Hodnota p n (placebo, inotersen)
66. týždeň
Rozdiel v LSM (SE)
Hodnota P
V30M 32, 58 -13,52 (3,795)
Non-V30 28, 54 -19,06 (5,334)
p<0,001 32, 58 -8,14 (3,998)
p<0,001 27, 53 -9,87 (4,666)
p=0,042
p=0,034
I. štádium
choroby
II. štádium choroby Predchádzajúce použitie stabilizátorov Predtým neliečení
Súbor CM- Echo
Súbor Non- CM-Echo
39, 74 -12,13 (3,838)
21, 38 -24,79 (5,601)
33, 61 -18,04 (4,591)
27, 51 -14,87 (4,377)
33, 75 -14,94 (4,083)
27, 37 -18,79 (5,197)
p=0,002 38, 73 -8,44 (3,706)
p<0,001 21, 38 -11,23 (5,271)
p<0,001 32, 60 -9,26 (4,060)
p<0,001 27, 51 -10,21 (4,659)
p<0,001 33, 75 -7,47 (4,075)
p<0,001 26, 36 -11,67 (4,213)
p=0,023 p=0,033 p=0,022
p=0,028 p=0,067 p=0,006
 m
N
I
S
+7 (p<0,001) NIS (p<0,001)
M
o
d
i
f
i
k
o
v
a
n
ý
+7
(
p
=0
,
020
)
N
I
S
-
W (p<0,001)
N
I
S
-
R (p=0,177) NIS-S (p<0,001) HRDB (p=0,704)
N
er
v
o
v
é vedenie (p=0,051) Dotyk-tlak (p=0,101)
T
ep
l
o
-
b
o
l
es
ť (p=0,007)
v prospech inotersenu v prospech placeba
LSM rozdiel v zmene oproti východiskovej hodnote v 66. týždni
(inotersen – placebo veľkosť účinku, 95 % IS)
O
b
r
ázok 2 Rozdiel v zmene priemernej hodnoty najmenších štvorcov (Least Squares Mean,
L
S
M
) oproti východiskovej hodnote medzi liečebnými skupinami v mNIS+7 a zložkami
m
N
I
S
+7 (p<0,001) NIS (p<0,001)
M
o
d
i
f
i
k
o
v
a
n
ý
+7
(
p
=0
,
020
)
N
I
S
-
W (p<0,001)
N
I
S
-
R (p=0,177) NIS-S (p<0,001) HRDB (p=0,704)
N
er
v
o
v
é vedenie (p=0,051) Dotyk-tlak (p=0,101)
T
ep
l
o
-
b
o
l
es
ť (p=0,007)
v prospech inotersenu v prospech placeba
LSM rozdiel v zmene oproti východiskovej hodnote v 66. týždni
(inotersen – placebo veľkosť účinku, 95 % IS)
O
b
r
ázok 2 Rozdiel v zmene priemernej hodnoty najmenších štvorcov (Least Squares Mean,
L
S
M
) oproti východiskovej hodnote medzi liečebnými skupinami v mNIS+7 a zložkami
Analýza mNIS+7 z hľadiska pacientov reagujúcich na liečbu, s použitím prahov v rozsahu od 0- po
30-bodové zvýšenie oproti východiskovej hodnote (s použitím bezpečnostného súboru), ukázala, že skupina s inotersenom mala približne 2-krát vyššiu mieru odpovede ako skupina s placebom pri každom testovanom prahu, dokazujúc tak konzistentnosť odpovede. Pacient reagujúci na liečbu bol definovaný ako osoba, u ktorej zmena oproti východiskovej hodnote bola menšia ako alebo rovnaká ako prahová hodnota. Osoby, ktoré predčasne ukončia liečbu bez ohľadu na dôvod alebo chýbajú údaje pre 66. týždeň sú považované za nereagujúce na liečbu. Štatistický význam v prospech inotersenu bol preukázaný pre všetky prahové hodnoty vyššie ako 0-bodová zmena.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Tegsedi vo
všetkých podskupinách pediatrickej populácie pre transtyretínovú amyloidózu (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaPo subkutánnom podaní sa inotersen rýchlo absorbuje do systémového obehu závisle od dávky,
s mediánom času dosiahnutia maximálnej koncentrácie v plazme (Cmax) inotersenu typicky v rozsahu 2
až 4 hodiny.
DistribúciaInotersen sa vo veľkej miere viaže na bielkoviny ľudskej plazmy (> 94 %) a viazaný podiel je
nezávislý od koncentrácie liečiva. U pacientov s hATTR je zdanlivý distribučný objem inotersenu v rovnovážnom stave 293 l. Vysoká hodnota distribučného objemu nasvedčuje, že inotersen sa po subkutánnom podaní v značnej miere distribuuje do tkanív.
Biotransformácia
Inotersen nie je substrátom pre metabolizmus CYP450 a v tkanivách je metabolizovaný endonukleázami na kratšie inaktívne oligonukleotidy, ktoré sú substrátmi pre ďalšiu metabolizáciu exonukleázami. Prevládajúcou zložkou v obehu je nezmenený inotersen.
Eliminácia
Eliminácia inotersenu zahŕňa metabolizmus v tkanivách aj vylučovanie močom. Inotersen aj jeho
kratšie oligonukleotidové metabolity sa vylučujú ľudským močom. Prítomnosť pôvodného lieku
v moči je obmedzená na menej ako 1 % do 24 hodín po podaní dávky. Po subkutánnom podaní je
polčas eliminácie inotersenu približne 1 mesiac.
Osobitné populácie
Na základe populačnej farmakokinetickej analýzy vyplýva, že vek, telesná hmotnosť, pohlavie alebo
rasa nemajú klinicky významný vplyv na expozíciu inotersenu. Definitívne vyhodnotenia boli v určitých prípadoch obmedzené, lebo boli obmedzené i premenné celkovo nízkym počtom pozorovaní.
Staršie osoby
Neboli pozorované celkové rozdiely vo farmakokinetike medzi inými dospelými pacientmi a staršími
pacientmi.
Porucha funkcie obličiek
Populačná farmakokinetická analýza naznačuje, že mierna a stredne závažná porucha funkcie obličiek nemá klinicky významný vplyv na systémovú expozíciu inotersenu. U pacientov so závažnou poruchou funkcie obličiek nie sú dostupné žiadne údaje.
Porucha funkcie pečene
Farmakokinetika inotersenu u pacientov s poruchou funkcie pečene nebola skúmaná. Primárna metabolizácia inotersenu neprebieha v pečeni, inotersen nie je substrátom pre oxidáciu pomocou CYP450 a je extenzívne metabolizovaný nukleázami vo všetkých tkanivách, do ktorých sa distribuuje. Preto by nemalo dôjsť k zmene farmakokinetiky pri miernej alebo stredne závažnej poruche funkcie pečene.
5.3 Predklinické údaje o bezpečnosti
Toxikológia
Znížený počet krvných doštičiek bol pozorovaný v štúdiách chronickej toxicity u myší, potkanov
a opíc s 1,4 až 2-násobkom ľudskej AUC pri odporúčanej terapeutickej dávke inotersenu.
U jednotlivých opíc boli pozorované výrazné poklesy v počte krvných doštičiek spojené so zvýšeným krvácaním alebo modrinami. Počty krvných doštičiek sa vrátili na normálne hodnoty po zastavení liečby, ale klesli na ešte nižšie hladiny po obnovení podávania inotersenu. Toto nasvedčuje, že ide
o imunologicky spätý mechanizmus.
Bolo pozorované extenzívne a pretrvávajúce vychytávanie inotersenu viacerými druhmi buniek vo viacerých orgánoch u všetkých skúmaných zvieracích druhov, vrátane monocytov/makrofágov, epitelu proximálnych tubulov obličiek, Kupfferových buniek pečene a infiltrátov histiocytických buniek
v lymfatických uzlinách a miestach vpichu. Akumulácia inotersenu v obličkách bola spojená
s proteinúriou u potkanov pri 13,4-násobku ľudskej AUC pri odporúčanej terapeutickej dávke inotersenu. Okrem toho bola u myší a potkanov pozorovaná znížená hmotnosť týmusu spôsobená depléciou lymfocytov. U opíc bola vo viacerých orgánoch zistená infiltrácia lymfohistiocytických buniek medzi perivaskulárne bunky. Tieto prozápalové zmeny orgánov boli pozorované pri 1,4 až 6,6- násobku ľudskej AUC pri odporúčanej terapeutickej dávke u všetkých skúmaných druhov zvierat a
boli sprevádzané zvýšením hladín rôznych plazmatických cytokínov/chemokínov.
G
e
notoxicita/karcinogenita
Inotersen nepreukázal genotoxický potenciál v podmienkach in vitro a in vivo a nebol karcinogénny u
kmeňa rasH2 transgénnych myší.
Reprodukčná toxikológia
Inotersen nemal žiadne účinky na plodnosť, embryofetálny alebo postnatálny vývin u myší a králikov
v dávke ekvivalentnej približne 3-násobku maximálnej odporúčanej dávky u ľudí. Prestup inotersenu do mlieka bol u myší nízky. Inotersen je však farmakologicky neúčinný u myší a králikov. Z toho dôvodu bolo v týchto pokusoch možné zachytiť iba účinky spojené s chemickými vlastnosťami inotersenu. Aj napriek tomu nebol zistený žiadny účinok na embryofetálny vývoj u myší po podaní analógu inotersenu špecifického pre myši, čo bolo spojené s ~60 % inhibíciou (individuálny rozsah až po 90 % zníženie) expresie mRNA pre TTR.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Voda na injekcie
Kyselina chlorovodíková (na úpravu pH) Hydroxid sodný (na úpravu pH)
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
18 mesiacov.
Tegsedi možno uchovávať nechladené do 6 týždňov pri teplotách do 30 °C. Ak sa nepoužije do
6 týždňov, je potrebné ho zlikvidovať.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke (2 °C - 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
1,5 ml roztoku v naplnenej injekčnej striekačke z číreho skla typu 1. Zásobník s odtrhávacím viečkom.
Veľkosti balenia: 1 naplnená injekčná striekačka alebo 4 naplnené injekčné striekačky. Na trh nemusia
byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Tegsedi sa má pred podaním vizuálne skontrolovať. Roztok má byť číry a bezfarebný až bledožltý. Ak
je roztok zakalený alebo obsahuje viditeľné častice, nesmie sa podať.
Každá naplnená injekčná striekačka sa má použiť iba raz a potom sa má vyhodiť do nádoby na ostré
predmety na likvidáciu.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIIONIS USA Limited Tower 42, Level 30, International Finance Centre,
25 Old Broad Street
Londýn,
EC2N 1HQ
Spojené kráľovstvo.
8. REGISTRAČNÉ ČÍSLAEU/1/18/1296/001
EU/1/18/1296/002
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.