
tenzia, vrátane závažnej hypertenzie (systolický tlak > 200 mmHg alebo diastolický tlak >110 mmHg). Pacienti majú byť preventívne vyšetrení na hypertenziu a v prípade potreby primerane liečení. Pacientom so závažnou a nedostatočne medikamentózne kontrolovanou hypertenziou sa odporúča dočasné prerušenie liečby. V liečbe je možné pokračovať, akonáhle je hypertenzia primerane kontrolovaná (pozri časť 4.8).
Hematologicképoruchy
V súvislosti so sunitinibom bol hlásený pokles absolútneho počtu neutrofilov a pokles počtu trombocytov (pozri časť 4.8). Vyššie uvedené účinky neboli kumulatívne, zvyčajne boli reverzibilné a vo všeobecnosti neviedli k prerušeniu liečby. Žiadna z týchto príhod v štúdiách fázy 3 nebola smrteľná, avšak zriedkavé smrteľné hematologické príhody, vrátane hemorágie spojenej s trombocytopéniou a neutropenickými infekciami, boli hlásené počas sledovania po uvedení lieku na trh.
Počas liečby sunitinibom bol pozorovaný včasný, ale aj neskorý výskyt anémie.
Na začiatku každého liečebného cyklu sunitinibom sa musí pacientom vyšetriť celkový krvný obraz (pozri časť 4.8).
Poruchysrdcaasrdcovejčinnosti
Kardiovaskulárne príhody, vrátane zlyhania srdca, kardiomyopatie, zníženia ejekčnej frakcie ľavej komory pod dolnú hranicu normy, myokarditídy, ischémie myokardu a infarktu myokardu, z ktorých niektoré boli smrteľné, boli hlásené u pacientov liečených sunitinibom. Tieto údaje naznačujú, že sunitinib zvyšuje riziko kardiomyopatie. U liečených pacientov neboli okrem účinku špecifického pre liek identifikované žiadne špecifické dodatočné rizikové faktory pre kardiomyopatiu indukovanú sunitinibom. Sunitinib používajte s opatrnosťou u pacientov, u ktorých sa vyskytuje riziko týchto udalostí alebo ktorí majú tieto udalosti v anamnéze (pozri časť 4.8).
Zo všetkých klinických štúdií so sunitinibom boli vylúčení pacienti s prítomnosťou závažnej srdcovej príhody do 12 mesiacov pred podaním sunitinibu, ako je infarkt myokardu (vrátane ťažkej/nestabilnej angíny), koronárny/periférny arteriálny bypass, symptomatické kongestívne zlyhávanie srdca (congestive heart failure,CHF), cievna mozgová príhoda alebo tranzitórny ischemický atak či pľúcna embólia. Nie je známe, či pacienti s týmito konkomitantnými stavmi môžu mať zvýšené riziko rozvoja dysfunkcie ľavej komory súvisiacej so sunitinibom.
Odporúča sa, aby lekár zvážil toto riziko v porovnaní s možným prínosom liečby sunitinibom.
U pacientov sa majú počas podávania sunitinibu starostlivo monitorovať klinické prejavy a príznaky CHF,
zvlášť u pacientov s kardiologickými rizikovými faktormi a/alebo ochoreniami koronárnych artérií v anamnéze. Na začiatku liečby a potom v pravidelných intervaloch počas liečby sunitinibom sa má zvážiť
vyšetrenie LVEF. U pacientov bez rizikových kardiálnych faktorov sa má zvážiť vyšetrenie ejekčnej
frakcie pred začiatkom liečby.
Ak sú prítomné klinické prejavy kongestívneho zlyhávania srdca, odporúča sa liečbu sunitinibom ukončiť. Pacientom bez klinických prejavov CHF, avšak s ejekčnou frakciou < 50 % a > 20 % poklesom oproti východiskovej hodnote, sa musí podávanie sunitinibu prerušiť a/alebo znížiť dávka.
PredĺženieQT-intervalu
U pacientov vystavených sunitinibu sa pozorovalo predĺženie QT-intervalu a torsade de pointes. Predĺženie
QT intervalu môže viesť k zvýšenému riziku ventrikulárnej arytmie vrátane torsade de pointes.
Sunitinib sa má s opatrnosťou používať u pacientov so známou anamnézou predĺženia QT-intervalu, u pacientov, ktorí užívajú antiarytmiká alebo lieky, ktoré môžu predlžovať QT-interval alebo u pacientov s už existujúcim závažným ochorením srdca, bradykardiou alebo s poruchou elektrolytovej rovnováhy. Súčasné podávanie so silnými inhibítormi CYP3A4 sa musí obmedziť kvôli možnému zvýšeniu koncentrácie sunitinibu v plazme (pozri časti 4.2, 4.5 a 4.8).
Venóznetrombembolicképríhody
U pacientov, ktorí dostávali sunitinib, boli hlásené venózne trombembolické príhody, ktoré súviseli s liečbou – vrátane hlbokej žilovej trombózy a pľúcnej embólie (pozri časť 4.8). V rámci dohľadu po uvedení
lieku na trh boli hlásené prípady pľúcnej embólie so smrteľným následkom.
Artériovétrombembolicképríhody
U pacientov liečených sunitinibom boli hlásené artériové trombembolické príhody (ATP), v niektorých prípadoch smrteľné. Najčastejšie príhody zahŕňali cerebrovaskulárnu príhodu, tranzitórny ischemický atak a mozgový infarkt. Rizikové faktory spojené s ATP, okrem základného malígneho ochorenia a veku ≥ 65 rokov, zahŕňali hypertenziu, diabetes mellitus a predchádzajúce trombembolické ochorenie.
Aneuryzmy aarteriálnedisekcie
Používanie inhibítorov dráhy vaskulárneho endotelového rastového faktora (vascular endothelial growth factor, VEGF) u pacientov s hypertenziou alebo bez hypertenzie môže podporovať tvorbu aneuryziem
a/alebo arteriálnych disekcií. Pred podaním sunitinibu sa toto riziko musí dôkladne zvážiť u pacientov s rizikovými faktormi, ako je napríklad hypertenzia alebo aneuryzma v anamnéze.
Trombotickámikroangiopatia(TMA)
Ak sa vyskytne hemolytická anémia, trombocytopénia, únava, kolísavá neurologická manifestácia, porucha funkcie obličiek a horúčka, je potrebné zobrať do úvahy diagnózu TMA, vrátane trombotickej
trombocytopenickej purpury (TTP) a hemolytického uremického syndrómu (HUS), ktoré v niektorých
prípadoch môžu viesť ku zlyhaniu obličiek alebo smrteľným následkom. U pacientov, u ktorých sa vyvinula TMA, sa musí liečba sunitinibom prerušiť a je nevyhnutné okamžite začať liečbu TMA. Po prerušení liečby sa pozorovalo vymiznutie príznakov TMA (pozri časť 4.8).
Dysfunkciaštítnejžľazy
U všetkých pacientov sa odporúča vykonať základné laboratórne vyšetrenia funkcie štítnej žľazy. Pacienti s už prítomnou hypotyreózou alebo hypertyreózou majú byť liečení podľa štandardných klinických postupov pred začiatkom liečby sunitinibom. Počas liečby sunitinibom sa má každé 3 mesiace vykonávať rutinné
monitorovanie funkcie štítnej žľazy. Okrem toho sa u pacientov počas liečby sunitinibom majú dôsledne sledovať prejavy a príznaky dysfunkcie štítnej žľazy a pacientom, u ktorých sa objavia akékoľvek prejavy a/alebo príznaky poukazujúce na dysfunkciu štítnej žľazy, sa má urobiť laboratórne vyšetrenie funkcie štítnej žľazy, ak je klinicky indikované. Pacientov, u ktorých sa vyvinie dysfunkcia štítnej žľazy, treba liečiť podľa zásad platných v medicínskej praxi.
Výskyt hypotyreózy sa pozoroval na začiatku liečby sunitinibom, ale aj neskôr počas liečby (pozri časť
4.8).
Pankreatitída
U pacientov s rôznymi nádormi, ktorí dostávali sunitinib, sa pozorovalo zvýšenie aktivity sérovej lipázy a amylázy. Zvýšenie aktivity lipázy u pacientov s rôznymi nádormi bolo prechodné a tento nález vo všeobecnosti nesprevádzali prejavy či príznaky pankreatitídy (pozri časť 4.8).
Boli hlásené závažné pankreatické príhody, niektoré so smrteľným koncom. Ak sú prítomné príznaky
pankreatitídy, sunitinib sa má pacientom vysadiť a má im byť poskytnutá primeraná podporná starostlivosť.
Hepatotoxicita
U pacientov liečených sunitinibom bola pozorovaná hepatotoxicita. Prípady zlyhania pečene, niektoré so smrteľným koncom, sa pozorovali u < 1 % pacientov s nádormi, ktorí boli liečení sunitinibom. Pred
začatím liečby, počas každého cyklu a vždy, keď je to z klinického hľadiska indikované, monitorujte testy pečeňových funkcií (alanínaminotransferázu [ALT], aspartátaminotransferázu [AST], hladiny bilirubínu).
Ak sú prítomné prejavy a príznaky zlyhávania pečene, liečba sunitinibom sa musí ukončiť a má sa poskytnúť vhodná podporná liečba (pozri časť 4.8).
Funkciaobličiek
Boli hlásené prípady poruchy funkcie obličiek, obličkového zlyhania a/alebo akútneho obličkového zlyhania, v niektorých prípadoch so smrteľným koncom (pozri časť 4.8).
Rizikové faktory spojené s poruchou funkcie/zlyhaním obličiek u pacientov užívajúcich sunitinib zahŕňali okrem prítomného RCC, vyšší vek, diabetes mellitus, prítomnosť poruchy funkcie obličiek, zlyhávanie srdca, hypertenziu, sepsu, dehydratáciu/hypovolémiu a rabdomyolýzu.
Bezpečnosť pokračujúcej liečby sunitinibom u pacientov so stredne závažnou až závažnou proteinúriou sa
systematicky nehodnotila.
Boli hlásené prípady proteinúrie a zriedkavé prípady nefrotického syndrómu. Odporúča sa vstupné vyšetrenie moču a pacienti majú byť monitorovaní na rozvoj alebo zhoršenie proteinúrie. U pacientov s nefrotickým syndrómom ukončite podávanie sunitinibu.
Fistula
Ak dôjde k vytvoreniu fistuly, liečba sunitinibom sa má prerušiť. O pokračovaní v liečbe sunitinibom u pacientov s fistulou sú dostupné obmedzené informácie (pozri časť 4.8).
Zhoršenéhojenierán
Počas liečby sunitinibom boli hlásené prípady zhoršeného hojenia rán.
Nevykonali sa žiadne formálne klinické štúdie zamerané na účinok sunitinibu na hojenie rán. U pacientov podstupujúcich veľký chirurgický zákrok sa z preventívnych dôvodov odporúča dočasné prerušenie liečby sunitinibom. Existujú iba limitované klinické skúsenosti týkajúce sa načasovania opätovného začatia liečby následne po veľkom chirurgickom zákroku. Preto rozhodnutie pokračovať v liečbe sunitinibom následne po veľkom chirurgickom zákroku má byť založené na klinickom zhodnotení zotavovania sa po zákroku.
Osteonekrózačeľuste
U pacientov liečených sunitinibom boli hlásené prípady osteonekrózy čeľuste. Väčšina prípadov bola
je osteonekróza čeľuste identifikovaným rizikom. Preto je potrebná zvýšená opatrnosť, keď sa sunitinib
používa s intravenóznymi bisfosfonátmi buď súbežne alebo následne.
Invazívne stomatologické zákroky sú tiež identifikovaným rizikovým faktorom. Pred liečbou sunitinibom treba zvážiť vyšetrenie zubov a náležité preventívne ošetrenie zubov. U pacientov, ktorí predtým dostávali alebo dostávajú intravenózne bisfosfonáty, sa treba, ak je to možné, vyhnúť invazívnym stomatologickým zákrokom (pozri časť 4.8).
Hypersenzitivita/angioedém
Ak sa v dôsledku hypersenzitivity vyskytne angioedém, má sa liečba sunitinibom prerušiť a poskytnúť štandardná lekárska starostlivosť (pozri časť 4.8).
Záchvaty
V klinických štúdiách so sunitinibom a počas dohľadu po uvedení lieku na trh sa hlásili kŕče. Pacientov s
kŕčmi a prejavmi/príznakmi, ktoré poukazujú na syndróm posteriórnej reverzibilnej leukoencefalopatie
(reversible posterior leukoencephalopathy syndrome, RPLS), ako je hypertenzia, bolesť hlavy, zníženie bdelosti, zmenené mentálne funkcie a strata zraku, vrátane kortikálnej slepoty, treba kontrolovať a liečiť vrátane liečby hypertenzie. Odporúča sa dočasne prerušiť liečbu sunitinibom; po úprave stavu sa môže liečba obnoviť podľa uváženia ošetrujúceho lekára (pozri časť 4.8).
Syndrómzrozpadunádoru(tumorlysissyndrome,TLS)
Prípady TLS, niektoré smrteľné, boli zriedkavo pozorované v klinických skúšaniach a boli hlásené aj u pacientov liečených sunitinibom v rámci sledovania po uvedení lieku na trh. Rizikové faktory pre TLS
zahŕňajú veľkú nádorovú masu už existujúcu chronickú renálnu insuficienciu, oligúriu, dehydratáciu,
hypotenziu a kyslý moč. Títo pacienti majú byť prísne monitorovaní a liečení podľa klinických indikácií a má sa zvážiť profylaktická hydratácia.
Infekcie
Boli hlásené závažné infekcie, s neutropéniou alebo bez nej, vrátane niektorých so smrteľnými následkami. Boli hlásené menej časté prípady nekrotizujúcej fasciitídy vrátane perinea, niektoré smrteľné (pozri časť
4.8).
U pacientov, u ktorých sa rozvinie nekrotizujúca fasciitída, sa má liečba sunitinibom ukončiť a okamžite sa má začať vhodná liečba.
Hypoglykémia
Počas liečby sunitinibom boli zaznamenané poklesy v hladine glukózy v krvi, ktoré boli v niektorých prípadoch klinicky symptomatické a vyžiadali si hospitalizáciu z dôvodu straty vedomia. V prípade
symptomatickej hypoglykémie sa má podávanie sunitinibu dočasne prerušiť. U pacientov s diabetes
mellitus sa majú pravidelne kontrolovať hladiny glukózy v krvi, aby sa posúdilo, či je na minimalizáciu rizika hypoglykémie potrebné upraviť dávkovanie antidiabetického lieku (pozri časť 4.8).
Sunitinib MSN obsahuje sodík
Tento liek obsahuje menej ako 1 mmol (23 mg) sodíka v jednej kapsule, t. j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Interakčné štúdie sa uskutočnili len u dospelých.
Lieky,ktorémôžuzvýšiťplazmatickékoncentráciesunitinibu
Účinok inhibítorov CYP3A4
Spoločné podávanie jednorazovej dávky sunitinibu zdravým dobrovoľníkom so silným inhibítorom CYP3A4, ketokonazolom, viedlo ku 49 % zvýšeniu maximálnej koncentrácie (Cmax) komplexu [sunitinib + primárny metabolit] a k 51 % zvýšeniu plochy pod krivkou (AUC0-∞) tohto komplexu.
Podávanie sunitinibu so silnými inhibítormi CYP3A4 (napr. ritonavirom, itrakonazolom, erytromycínom, klaritromycínom, grapefruitovou šťavou) môže zvýšiť koncentrácie sunitinibu.
Preto sa má kombináciám s CYP3A4 inhibítormi vyhnúť alebo zvážiť výber alternatívneho súčasne
podávaného lieku so žiadnou alebo minimálnou schopnosťou inhibovať CYP3A4.
Ak to nie je možné, dávku sunitinibu bude možno potrebné znížiť na minimálnu dennú dávku 37,5 mg pre GIST a MRCC alebo 25 mg denne pre pNET na základe starostlivého monitorovania znášanlivosti (pozri časť 4.2).
Účinok inhibítorov proteínu rezistencie rakoviny prsníka (Breast Cancer Resistance Protein, BCRP)
O interakcii medzi sunitinibom a inhibítormi BCRP je k dispozícii len obmedzené množstvo klinických údajov a nedá sa vylúčiť možnosť interakcie medzi sunitinibom a inými inhibítormi BCRP (pozri časť 5.2).
Lieky,ktorémôžuznížiťplazmatickékoncentráciesunitinibu
Účinok induktorov CYP3A4
Spoločné podávanie jednorazovej dávky sunitinibu zdravým dobrovoľníkom s induktorom CYP3A4, rifampicínom, viedlo k 23 % zníženiu Cmax komplexu [sunitinib + primárny metabolit] a k 46 % zníženiu AUC0-∞ tohto komplexu.
Podávanie sunitinibu so silnými induktormi CYP3A4 (napr. dexametazonom, fenytoínom, karbamazepínom, rifampicínom, fenobarbitalom alebo fytofarmakami obsahujúcimi ľubovník bodkovaný/Hypericum perforatum) môže znížiť koncentrácie sunitinibu. Preto sa má kombináciám s CYP3A4 induktormi vyhnúť, prípadne zvážiť výber alternatívneho súčasne podávaného lieku so žiadnou alebo minimálnou schopnosťou indukovať CYP3A4. Ak to nie je možné, dávku sunitinibu bude možno potrebné zvýšiť po 12,5 mg prídavkoch (až na 87,5 mg/deň pre GIST a MRCC alebo 62,5 mg pre pNET) na základe starostlivého monitorovania znášanlivosti (pozri časť 4.2).
4.6 Fertilita, gravidita a laktácia
Antikoncepciaumužovažien
Ženy vo fertilnom veku majú byť poučené, aby počas liečby sunitinibom používali účinnú antikoncepciu a
vyhli sa tak otehotneniu.
Gravidita
Nie sú k dispozícii žiadne štúdie o použití sunitinibu u gravidných žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu vrátane malformácií plodu (pozri časť 5.3). Sunitinib sa má používať počas gravidity alebo u žien, ktoré nepoužívajú účinnú antikoncepciu iba v prípade, že potenciálny prínos prevyšuje potenciálne riziko pre plod. Ak sa sunitinib používa počas gravidity, alebo ak pacientka otehotnie počas liečby sunitinibom, musí byť oboznámená s možným rizikom pre plod.
Dojčenie
Sunitinib a/alebo jeho metabolity sa u potkanov vylučujú do materského mlieka. Nie je známe, či sa sunitinib alebo jeho primárny aktívny metabolit vylučujú do materského mlieka u človeka. Vzhľadom na
to, že u človeka sa liečivá obvykle vylučujú do materského mlieka ako aj kvôli potenciálnemu riziku
závažných nežiaducich reakcií u dojčených detí, ženy nesmú dojčiť počas užívania sunitinibu.
Fertilita
Podľa výsledkov predklinických skúmaní môže liečba sunitinibom nepriaznivo vplývať na mužskú a ženskú fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Sunitinib má malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pacienti majú byť poučení, že sa u nich počas liečby sunitinibom môžu vyskytnúť závraty.
4.8 Nežiaduce účinky
Súhrn
b
ezpečnostného
profilu
Najzávažnejšie nežiaduce reakcie spájané so sunitinibom
, niekedy fatálne, sú renálne zlyhanie, srdcové zlyhanie, pľúcna embólia, gastrointestinálna perforácia a hemorágie (napr. krvácanie z dýchacej sústavy,
gastrointestinálneho traktu, močových ciest, krvácanie do nádoru a do mozgu). Najčastejšie nežiaduce reakcie akéhokoľvek stupňa závažnosti (s výskytom u pacientov v registračných klinických skúšaniach s
RCC, GIST a pNET) zahŕňali: zníženú chuť do jedla, poruchu chuti, hypertenziu, únavu, gastrointestinálne poruchy (napr. hnačku, nevoľnosť, stomatitídu, dyspepsiu a vracanie), zmeny sfarbenia kože, syndróm
palmárno-plantárnej erytrodyzestézie. Tieto príznaky môžu slabnúť pri pokračovaní liečby. Počas liečby sa môže vyvinúť hypotyreóza. Hematologické poruchy (napr. neutropénia, trombocytopénia a anémia) patria medzi najčastejšie sa vyskytujúce nežiaduce reakcie.
Smrteľné príhody, iné ako sú uvedené v časti 4.4 vyššie alebo v časti 4.8 nižšie, ktoré sa považovali za pravdepodobne súvisiace so sunitinibom, zahŕňali multiorgánové zlyhanie, rozptýlenú intravaskulárnu koaguláciu, peritoneálne krvácanie, insuficienciu nadobličiek, pneumotorax, šok a náhlu smrť.
TabuľkovýzoznamnežiaducichreakciíNežiaduce reakcie, ktoré boli hlásené u pacientov s GIST, MRCC a pNET v spoločnom súbore údajov o 7
115 pacientoch, sú uvedené nižšie a zoradené podľa tried orgánových systémov a frekvencie a stupňa závažnosti NCI-CTCAE). Uvedené sú aj nežiaduce reakcie identifikované v klinických štúdiách po uvedení lieku na trh. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Frekvencie sú definované ako: veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až
<1/100), zriedkavé (≥1/10 000 až <1/1 000), veľmi zriedkavé (<1/10 000) neznáme (z dostupných údajov).
Tabuľka1.Nežiaducereakciehlásenévklinickýchskúšaniach Trieda orgánových systémov
| Veľmi časté
| Časté
| Menej časté
| Zriedkavé
| Neznáme
|
Infekcie a nákazy
|
| vírusové infekciea, infekcie
dýchacích ciestb,*, abscesc,*, mykotické infekcied infekcia močového ústrojenstva, kožné infekciee, sepsaf,*
| nekrotizujúca fasciitída*, bakteriálne infekcieg
|
|
|
Poruchy krvi a lymfatického systému
| neutropénia, trombocytopénia, anémia, leukopénia
| lymfopénia
| pancytopénia
| trombotická mikroangiopatia h,*
|
|
Poruchy imunitného systému
|
|
| hypersenzitivita
| angioedém
|
|
Poruchy endokrinného systému
| hypotyreóza
|
| hypertyreóza
| tyreoitída
|
|
Poruchy metabolizmu a výživy
| znížená chuť do
jedlai
| dehydratácia, hypoglykémia
|
| syndróm nádorového rozpadu*
|
|
P
sychické
poruchy
|
insomnia
|
depresia
|
|
|
|
P
oruchy nervového systému
|
závraty, bolesť hlavy, poruchy chutij
|
periférna neuropatia, parestézia, hypestézia, hyperestézia
|
cerebrálne krvácanie*, cerebrovaskulár ne príhody*, tranzientný ischemický atak
|
syndróm posteriórnej reverzibilnej encefalopatie*
|
|
P
oruchy oka
|
|
opuch okolo očí, opuch očných viečok, zvýšené slzenie
|
|
|
|
P
oruchy srdca a
srdcovej činnosti
|
|
ischémia myokarduk,*, pokles ejekčnej frakciel
|
kongestívne srdcové zlyhávanie, infarkt myokardum,*, srdcové zlyhávanie*, kardiomyopatia*, perikardiálny výpotok, predĺženie QT
na elektrokardiogra
me
|
zlyhávanie ľavej srdcovej komory*, torsade de pointes
|
|
P
oruchy ciev
|
hypertenzia
|
hlboká žilová trombóza, návaly tepla, sčervenanie kože
|
krvácanie nádoru*
|
|
aneuryzmy a arteriálne disekcie*
|
P
oruchy
dýchacej sústavy, hrudníka a
m
ediastína
|
dyspnoe,
epistaxa, kašeľ
|
pľúcna embólia*, pleurálny výpotok*, hemoptýza, ponámahové dyspnoe, orofaryngeálna bolesťn, nazálna kongescia, suchosť nosa
|
pľúcne krvácanie*, respiračné zlyhanie*
|
|
|
P
oruchy gastrointestináln eho traktu
|
stomatitídao, bolesť bruchap, vracanie, hnačka, dyspepsia, nauzea, zápcha
|
gastroezofágová refluxná choroba, dysfágia, gastrointestinálne krvácanie*, ezofagitída*, abdominálna distenzia, abdominálny diskomfort, krvácanie z konečníka, krvácanie z
|
perforácia gastrointestináln eho
traktuq,*,
pankreatitída,
fistula v konečníku, kolitídar
|
|
|
|
|
ďasien, ulcerácie v ústach, proktalgia, cheilitída, hemoroidy, glosodýnia, bolesť v ústach, sucho v ústach,
flatulencia, diskomfort v ústach, eruktácia
|
|
|
|
P
oruchy pečene a žlčových ciest
|
|
|
zlyhanie pečene*, cholecystitídas,*, porucha funkcie pečene
|
hepatitída
|
|
P
oruchy kože a podkožného tkaniva
|
zmena sfarbenia pokožkyt, syndróm palmárnoplantárn ej erytrodyzestézie
,
vyrážkau, zmeny sfarbenia vlasov,
suchosť kože
|
exfoliácia kože, kožná reakciav, ekzém, pľuzgier, erytém, alopécia, akné, pruritus, hyperpigmentácia kože, kožná lézia, hyperkeratóza, dermatitída, poruchy nechtovw
|
|
multiformný erytém*, StevensovJohns onov syndróm*, pyoderma gangrenosum, toxická epidermálna nekrolýza*
|
|
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
bolesť
v končatinách, artralgia, bolesť chrbta
|
muskuloskeletálne bolesti, svalové kŕče, myalgia, svalová slabosť
|
osteonekróza čeľuste, fistula*
|
rabdomyolýza*, myopatia
|
|
P
oruchy obličiek a močových
ciest
|
|
zlyhanie obličiek*, akútne zlyhanie obličiek*, chromatúria, proteinúria
|
krvácanie z močových ciest
|
nefrotický syndróm
|
|
C
elkové poruchy a reakcie v mieste podania
|
zápal slizníc, únavax, edémy, pyrexia
|
bolesť hrudníka, bolesť, ochorenie podobné
chrípke, zimnica
|
zhoršené hojenie
|
|
|
L
aboratórne a funkčné vyšetrenia
|
|
pokles hmotnosti, znížený počet bielych krviniek, zvýšená lipáza, pokles počtu trombocytov, znížený hemoglobín, zvýšená amylázaz, zvýšená hladina aspartátaminotran sferázy, zvýšená hladina alanínaminotransf
|
zvýšená hladina kreatínfosfokiná zy v
krvi, zvýšená hladina tyreostimulačné ho hormónu v krvi
|
|
|
|
|
erázy, zvýšený kreatinín v krvi, zvýšený tlak krvi, zvýšená hladina kyseliny močovej v krvi
|
|
|
|
* Vrátane smrteľných udalostí.
Nasledujúce výrazy boli zlúčené:
a Zápal nosohltanu a ústny opar
b Zápal priedušiek, infekcia dolných dýchacích ciest, zápal pľúc a infekcia dýchacích ciest
c Absces, absces končatiny, análny absces, absces ďasien, absces pečene, absces pankreasu, perineálny absces, perirektálny absces, absces konečníka, podkožný absces a zubný absces
d Kvasinková infekcia pažeráka a úst
e Celulitída a infekcia kože
f Sepsa a septický šok
g Brušný absces, brušná sepsa, divertikulitída a osteomyelitída
h Trombotická mikroangiopatia, trombotická trombocytopenická purpura, hemolyticko-uremický syndróm
i Znížená chuť do jedla a anorexia
j Dysgeúzia, ageúzia a poruchy chuti
k Akútny koronárny syndróm, angína pektoris, nestabilná angína, oklúzia koronárnej artérie, ischémia
myokardu
l Pokles ejekčnej frakcie/abnormálna ejekčná frakcia
m Akútny infarkt myokardu, infarkt myokardu, latentný infarkt myokardu
n Orofaryngálna a faryngolaryngálna bolesť
o Stomatitída a aftózna stomatitída
p Abdominálna bolesť, bolesť v dolnej časti brucha, bolesť v hornej časti brucha
q Perforácia gastrointestinálneho traktu a perforácia čreva
r Kolitída a ischemická kolitída s Cholecystitída a akalkulózna cholecystitída
t Žlté sfarbenie kože, zmeny sfarbenia kože a porucha pigmentácie
u Psoriasiformná dermatitída, exfoliatívna vyrážka, vyrážka, erytémová vyrážka, folikulárna vyrážka, generalizovaná vyrážka, makulárna vyrážka, makulo-papulózna vyrážka, papulózna vyrážka a pruritická vyrážka
v Kožná reakcia a porucha kože
w Poškodenie a zmena sfarbenia nechtov
x Únava a asténia
y Opuch tváre, edém a periférny edém
z Amyláza a zvýšená amyláza
Opis vybranýchnežiaducichreakciíInfekcie a nákazyBoli hlásené prípady závažných infekcií (s neutropéniou alebo bez neutropénie), vrátane prípadov so smrteľným koncom. Boli hlásené prípady nekrotizujúcej fascititídy, vrátane perinea, niekedy smrteľné (pozri tiež časť 4.4).
Poruchy krvi a lymfatického systémuPokles absolútneho počtu neutrofilov 3. a 4. stupňa závažnosti bol v uvedenom poradí hlásený: u 10 % a
1,7 % pacientov v štúdii fázy 3 s GIST, u 16 % a 1,6 % pacientov v štúdii fázy 3 s MRCC a u 13 % a 2,4 %
pacientov v štúdii fázy 3 s pNET. Pokles počtu trombocytov 3. a 4. stupňa závažnosti bol v uvedenom poradí hlásený: u 3,7 % a 0,4 % pacientov v štúdii fázy 3 s GIST, u 8,2 % a 1,1 % pacientov v štúdii fázy 3 s MRCC a u 3,7 % a 1,2 % pacientov v štúdii fázy 3 s pNET (pozri časť 4.4).
Prípady krvácania boli hlásené u 18 % pacientov užívajúcich sunitinib v štúdii fázy 3 s GIST v porovnaní so 17 % pacientov užívajúcich placebo. U pacientov užívajúcich sunitinib pre predtým neliečený MRCC,
sa krvácanie vyskytlo v 39 % v porovnaní s 11 % pacientov užívajúcich interferónα (IFN-α). U
sedemnástich (4,5 %) pacientov liečených sunitinibom sa vyskytlo krvácanie 3.
alebo vyššieho stupňa v porovnaní s 5 (1,7 %) pacientami užívajúcimi IFN-α. U pacientov užívajúcich sunitinib pre MRCC rezistentný na cytokíny sa u 26 % objavilo krvácanie. Prípady krvácania, okrem
epistaxy, sa vyskytli u 21,7 % pacientov užívajúcich sunitinib v štúdii fázy 3 s pNET v porovnaní s 9,85 %
pacientov, ktorí dostávali placebo (pozri časť 4.4).
V klinických štúdiách bolo krvácanie do nádoru hlásené približne u 2 % pacientov s GIST.
Poruchy imunitného systému
Boli hlásené hypersenzitívne reakcie vrátane angioedému (pozri časť 4.4).
Endokrinné poruchy
Hypotyreóza bola hlásená ako nežiaduca reakcia u 7 pacientov (4 %), ktorí dostávali sunitinib v 2 štúdiách
s MRCC rezistentným na cytokíny; u 61 pacientov (16 %), ktorí dostávali sunitinib a u 3 pacientov (< 1 %)
v skupine s IFN-α v štúdii s predtým neliečeným MRCC.
Okrem toho bolo hlásené zvýšenie hormónu stimulujúceho štítnu žľazu (thyroid-stimulating hormone, TSH) u 4 pacientov (2 %) s MRCC rezistentným na cytokíny. Celkovo malo 7 % pacientov z MRCC populácie buď klinické, alebo laboratórne príznaky hypotyreózy, ktoré sa objavili počas liečby. Získaná hypotyreóza bola zaznamenaná u 6,2 % pacientov s GIST liečených sunitinibom v porovnaní s 1 % pacientov užívajúcich placebo. V štúdii fázy 3 s pNET sa u 6 pacientov (7,2 %) liečených sunitinibom a u jedného pacienta (1,2 %) užívajúceho placebo hlásila hypotyreóza.
Funkcia štítnej žľazy bola prospektívne monitorovaná v 2 štúdiách u pacientov s karcinómom prsníka;
sunitinib nie je schválený na použitie pri karcinóme prsníka. V 1 štúdii bola hypotyreóza hlásená u 15 (13,6
%) pacientov liečených sunitinibom a u 3 (2,9 %) pacientov so štandardnou liečbou. Zvýšenie hladín TSH
v krvi bolo hlásené u 1 (0,9 %) pacienta liečeného sunitinibom a nebolo hlásené u žiadneho pacienta so štandardnou liečbou. Hypertyreóza nebola hlásená u žiadneho pacienta liečeného sunitinibom, bola však hlásená u 1 (1,0 %) pacienta so tandardnou liečbou. V druhej štúdii bola hypotyreóza hlásená celkovo u 31 (13 %) pacientov liečených sunitinibom a u 2 (0,8 %) pacientov liečených kapecitabínom. Zvýšenie hladín TSH v krvi bolo hlásené u 12 (5,0 %) pacientov liečených sunitinibom a nebolo hlásené u žiadneho pacienta liečeného kapecitabínom. Hypertyreóza bola hlásená u 4 (1,7 %) pacientov liečených sunitinibom a nebola hlásená u žiadneho pacienta liečeného kapecitabínom. Zníženie hladín TSH v krvi bolo hlásené u
3 (1,3 %) pacientov liečených sunitinibom a nebolo hlásené u žiadneho pacienta liečeného kapecitabínom. Zvýšenie hladín T4 bolo hlásené u 2 (0,8 %) pacientov liečených sunitinibom a u 1 (0,4 %) pacienta liečeného kapecitabínom. Zvýšenie hladín T3 bolo hlásené u 1 (0,8 %) pacienta liečeného sunitinibom a nebolo hlásené u žiadneho pacienta liečeného kapecitabínom. Všetky hlásené príhody súvisiace so štítnou žľazou boli 1. – 2. stupňa (pozri časť 4.4).
Poruchy metabolizmu a výživy
U pacientov s pNET bola hlásená vyššia miera incidencie hypoglykemických udalostí v porovnaní s pacientami s MRCC a GIST. Väčšina týchto nežiaducich udalostí pozorovaných v klinických štúdiách sa
však nepovažuje za súvisiacu s liečbou v rámci štúdie (pozri časť 4.4).
Poruchy nervového systému
V klinických štúdiách so sunitinibom a v rámci dohľadu po uvedení lieku na trh bolo u pacientov hlásených niekoľko prípadov (< 1 %), niektoré z nich smrteľné, v ktorých sa udávali záchvaty a prítomnosť
rádiologicky potvrdeného RPLS. Záchvaty sa pozorovali u pacientov s rádiologicky potvrdenými
metastázami do mozgu alebo bez nich (pozri časť 4.4).
Poruchy srdca a srdcovej činnosti
V klinických štúdiách boli poklesy ejekčnej frakcie ľavej komory (left ventricular ejection fraction, LVEF)
o ≥ 20 % a pod dolnú hranicu normy hlásené u približne 2 % pacientov s GIST liečených sunitinibom, u 4
% pacientov s MRCC rezistentným na cytokíny a u 2 % pacientov s GIST, ktorí užívali placebo. Tieto poklesy LVEF nejavili známky progresie a často sa upravili pri pokračovaní liečby. V štúdii s predtým neliečeným MRCC malo 27 % pacientov liečených sunitinibom a 15 % pacientov liečených INF-α hodnotu
LVEF pod dolnou hranicou normy. Dvom pacientom (< 1 %), ktorí dostávali sunitinib, bolo diagnostikované CHF.
U pacientov s GIST boli hlásené: „zlyhávanie srdca“, „kongestívne zlyhávanie srdca“ alebo „zlyhanie ľavej komory“ – konkrétne u 1,2 % pacientov liečených sunitinibom a u 1 % pacientov, ktorí užívali placebo. V pivotnej štúdii fázy 3 u pacientov s GIST (n = 312) boli smrteľné srdcové reakcie súvisiace s liečbou hlásené u 1 % pacientov v každej skupine štúdie (t. j. v skupine so sunitinibom a v skupine s placebom). V štúdii fázy 2 u pacientov s MRCC rezistentným na cytokíny sa u 0,9 % pacientov vyskytol s liečbou súvisiaci smrteľný infarkt myokardu a v štúdii fázy 3 u predtým neliečených pacientov s MRCC sa
smrteľné srdcové udalosti vyskytli u 0,6 % pacientov v skupine s IFN-α a u 0 % pacientov v skupine so sunitinibom. V štúdii fázy 3 u pacientov s pNET sa u jedného (1 %) pacienta, ktorý dostával sunitinib,
vyskytlo smrteľné zlyhanie srdca súvisiace s liečbou.
Poruchy ciev
Hypertenzia
V klinických skúšaniach bola hypertenzia veľmi častou nežiaducou reakciou. Dávka sunitinibu bola znížená alebo jeho podávanie dočasne prerušené približne u 2,7 % pacientov, ku ktorých sa vyskytla hypertenzia. U žiadneho z týchto pacientov nebola liečba sunitinibom natrvalo ukončená. U 4,7 % pacientov s nádormi sa vyskytla závažná hypertenzia (> 200 mmHg systolického alebo 110 mmHg diastolického tlaku krvi). Hypertenzia bola hlásená približne u 33,9 % pacientov, ktorí dostávali sunitinib na liečbu predtým neliečeného MRCC, v porovnaní s 3,6 % pacientov liečených IFN-α. Závažná hypertenzia bola hlásená u 12 % predtým neliečených pacientov užívajúcich sunitinib a u < 1 % pacientov
liečených IFN-α. Hypertenzia bola hlásená u 26,5 % pacientov užívajúcich sunitinib v štúdii fázy 3 s pNET v porovnaní so 4,9 % pacientov užívajúcich placebo. Závažná hypertenzia bola hlásená u 10 % pacientov s pNET užívajúcich sunitinib a u 3 % pacientov užívajúcich placebo.
Venóznetrombembolicképríhody
Venózne trombembolické príhody súvisiace s liečbou boli hlásené u približne 1,0 % pacientov s nádormi, ktorí dostávali sunitinib v rámci klinických skúšaní, vrátane GIST a RCC.
U siedmich pacientov (3 %) užívajúcich sunitinib a u žiadneho pacienta užívajúceho placebo sa v štúdii fázy 3 s GIST vyskytli venózne trombembolické príhody; u 5 zo 7 išlo o hlbokú venóznu trombózu (deep venous thrombosis, DVT) 3. stupňa a u 2 išlo o 1. alebo 2. stupeň. Štyria z týchto 7 pacientov s GIST ukončili liečbu po prvom spozorovaní DVT.
U trinástich pacientov (3 %) užívajúcich sunitinib v štúdii fázy 3 na predtým neliečený MRCC a u 4 pacientov (2 %) v 2 štúdiách s MRCC rezistentným na cytokíny boli hlásené venózne trombembolické príhody. U deviatich z týchto pacientov sa vyskytla pľúcna embólia; 1 bola 2. stupňa a 8 bolo 4. stupňa. Osem z týchto pacientov malo DVT; jeden 1. stupňa, dvaja 2. stupňa, štyria 3. stupňa a jeden 4. stupňa. U jedného pacienta s pľúcnou embóliou v štúdii s MRCC rezistentným na cytokíny bolo prerušené podávanie lieku.
U pacientov s predtým neliečeným MRCC užívajúcich IFN-α bolo hlásených 6 (2 %) venóznych trombembolických príhod; 1 pacient (< 1 %) mal DVT 3. stupňa a 5 pacientov (1 %) malo pľúcnu embóliu, všetci 4. stupňa.
V štúdii fázy 3 u pacientov s pNET boli venózne trombembolické príhody hlásené u 1 (1,2 %) pacienta v skupine so sunitinibom a u 5 (6,1 %) pacientov v skupine s placebom. U dvoch z týchto pacientov užívajúcich placebo išlo o DVT, pričom u 1 pacienta bola 2. stupňa a u 1 bola 3. stupňa.
V registračných štúdiách u pacientov s GIST, MRCC a pNET neboli hlásené žiadne prípady so smrteľným koncom. Prípady so smrteľným koncom sa pozorovali po uvedení lieku na trh.
V štúdiách fázy 3 u pacientov, ktorí dostávali sunitinib, sa prípady pľúcnej embólie pozorovali približne u
3,1 % pacientov s GIST a približne u 1,2 % pacientov s MRCC. V štúdii fázy 3 u pacientov
s pNET, ktorí dostávali sunitinib, nebola hlásená žiadna pľúcna embólia. Zriedkavé prípady so smrteľným koncom sa pozorovali po uvedení lieku na trh.
Pacienti, u ktorých sa vyskytla pľúcna embólia v predchádzajúcich 12 mesiacoch, boli vylúčení z klinických štúdií so sunitinibom.
U pacientov, ktorí dostávali sunitinib v registračných štúdiách fázy 3, boli pľúcne príhody (t. j.
dyspnoe, pleurálny výpotok, pľúcna embólia alebo pľúcny edém) hlásené približne u 17,8 % pacientov s
GIST, približne u 26,7 % pacientov s MRCC a u 12 % pacientov s pNET.
Približne 22,2 % pacientov s nádormi, vrátane GIST a MRCC, ktorí v klinických skúšaniach dostávali sunitinib, malo pľúcne príhody.
Poruchy gastrointestinálneho traktu
U pacientov liečených sunitinibom pre GIST alebo MRCC sa pankreatitída pozorovala menej často (< 1
%). V štúdii fázy 3 u pacientov s pNET nebola hlásená žiadna pankreatitída súvisiaca s liečbou (pozri časť
4.4).
Smrteľné gastrointestinálne krvácanie bolo hlásené u 0,98 % pacientov, ktorí dostávali placebo v štúdii
fázy 3 s GIST.
Poruchy pečene a žlčových ciest
Bola hlásená hepatálna dysfunkcia, ktorá môže zahŕňať odchýlky testov pečeňových funkcií, hepatitídu alebo zlyhanie pečene (pozri časť 4.4).
Poruchy kože a podkožného tkaniva
Boli hlásené prípady pyoderma gangrenosum, vo všeobecnosti reverzibilné po prerušení liečby sunitinibom
(pozri časť 4.4).
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Boli hlásené prípady myopatie a/alebo rabdomyolýzy, niektoré s akútnym renálnym zlyhaním. Pacienti s prejavmi alebo príznakmi svalovej toxicity majú byť liečení štandardnými lekárskymi postupmi (pozri časť
4.4).
Boli hlásené prípady vytvorenia fistuly, niekedy spojené s nekrózou nádoru a regresiou, v niektorých prípadoch so smrteľným koncom (pozri časť 4.4).
U pacientov liečených sunitinibom boli hlásené prípady osteonekrózy čeľuste, z ktorých sa väčšina vyskytla u pacientov, ktorí mali identifikované rizikové faktory pre osteonekrózu čeľuste, zvlášť expozíciu intravenóznym bifosfonátom a/alebo stomatologické ochorenie v anamnéze vyžadujúce invazívny stomatologický zákrok (pozri tiež časť 4.4).
Laboratórne a funkčné vyšetrenia
Údaje z predklinických (in vitro a in vivo) štúdií pri dávkach vyšších, ako je odporúčaná dávka pre ľudí, ukazujú, že sunitinib má potenciál inhibovať repolarizačný proces srdcového akčného potenciálu (napr.
predĺženie QT-intervalu).
Predĺženie QTc-intervalu na viac ako 500 ms bolo hlásené u 0,5 % a zmeny o viac ako 60 ms oproti vstupnej hodnote boli hlásené u 1,1 % zo 450 pacientov s nádorom; oba z týchto parametrov sú uznané ako potenciálne signifikantné zmeny. Pri približne dvojnásobných terapeutických koncentráciách sa ukázalo, že sunitinib predlžuje QTcF-interval (korekcia QT-intervalu podľa Fridericia).
Predĺženie QTc-intervalu sa skúmalo v skúšaní s 24 pacientmi vo veku 20 – 87 rokov s pokročilými malignitami. Výsledky tejto štúdie ukázali, že sunitinib mal vplyv na QTc-interval (definovaný ako priemerná zmena upravená vzhľadom k placebu o > 10 ms s 90 % horným limitom intervalu spoľahlivosti (confidence interval, CI) > 15 ms) pri terapeutickej koncentrácii (3. deň) pri použití korekčnej metódy oproti vstupnej hodnote v rámci dňa a pri koncentrácii väčšej, ako je terapeutická (9. deň) pri použití oboch korekčných metód oproti vstupnej hodnote. Žiaden pacient nemal hodnotu QTc-intervalu > 500 ms. Hoci
sa vplyv na QTcF-interval pozoroval na 3. deň 24 hodín po podaní dávky (t. j. pri terapeutickej
metódy oproti vstupnej hodnote v rámci dňa, klinický význam tohto nálezu nie je jasný.
Pri použití rozsiahlych sériových vyšetrení EKG v časoch korešpondujúcich buď s terapeutickou, alebo vyššou ako terapeutickou expozíciou sa u žiadneho z pacientov v hodnotiteľnej alebo
ITT-populácii nepozoroval výskyt predĺženia QTc-intervalu, ktorý by sa považoval za „závažný“ (t. j. rovný alebo väčší ako 3. stupeň podľa všeobecných terminologických kritérií pre nežiaduce účinky
[common terminology criteria for adverse events, CTCAE] verzia 3.0).
Pri terapeutických koncentráciách v plazme bola maximálna priemerná zmena QTcF-intervalu (korekcia podľa Fridericia) oproti vstupnej hodnote 9 ms (90 % CI: 15,1 ms). Pri približne dvojnásobných terapeutických koncentráciách bola maximálna zmena QTcF-intervalu oproti vstupnej hodnote 15,4 ms (90
% CI: 22,4 ms). Moxifloxacín (400 mg), ktorý sa používal ako pozitívna kontrola, vykazoval maximálnu priemernú zmenu QTcF-intervalu 5,6 ms oproti vstupnej hodnote. Ani u jedného účastníka nebol účinok na QTc-interval vyšší ako 2. stupeň (CTCAE, verzia 3.0) (pozri časť 4.4).
DlhodobábezpečnosťpriMRCCDlhodobá bezpečnosť sunitinibu u pacientov s MRCC sa analyzovala v 9 ukončených klinických štúdiách, realizovaných v prvej línii liečby u pacientov refraktérnych na bevacizumab a cytokíny. Analýza zahŕňala 5
739 pacientov, z ktorých sa 807 (14 %) liečilo 2 roky až 6 rokov. U tých 807 pacientov, ktorí sa
dlhodobo liečili sunitinibom, sa väčšina nežiaducich reakcií súvisiacich s liečbou (treatment-related adverse
events, TRAE) po prvýkrát zaznamenala v rámci prvých 6 mesiacov až 1 roka a potom boli stabilné alebo sa ich frekvencia časom znižovala. Výnimkou bola hypotyreóza, ktorej výskyt časom postupne narastal,
pričom sa počas 6-ročného obdobia zaznamenávali nové prípady. Neukázalo sa, že by sa predĺžená liečba
sunitinibom spájala s novými typmi TRAE.
PediatrickápopuláciaBezpečnostný profil sunitinibu bol odvodený zo štúdie fázy 1 so zvyšujúcou sa dávkou, otvorenej štúdie
fázy 2, jednoramennej štúdie fázy 1/2 a z publikácií, ako je uvedené nižšie.
Štúdia fázy 1 so zvyšujúcou sa dávkou perorálneho sunitinibu sa uskutočnila u 35 pacientov, pričom 30 z nich bolo pediatrických pacientov (vo veku 3 až 17 rokov) a 5 mladých dospelých pacientov (vo veku 18 až
21 rokov), s refraktérnymi nádormi, pričom u väčšiny z nich sa primárne diagnostikoval mozgový nádor. U
všetkých účastníkov štúdie došlo k nežiaducim reakciám na liek. Väčšina z týchto reakcií bola závažná
(stupeň toxicity
≥ 3) a zahŕňala aj srdcovú toxicitu. Najbežnejšími nežiaducimi reakciami na liek boli gastrointestinálna (GI) toxicita, neutropénia, únava a zvýšenie ALT. Riziko srdcových nežiaducich reakcií na liek sa ukázalo byť vyššie u tých pediatrických pacientov, ktorí boli predtým vystavení ožarovaniu srdca alebo antracyklínu, v porovnaní s pediatrickými pacientmi bez predchádzajúcej expozície. U týchto pediatrických pacientov, ktorí predtým neboli vystavení antracyklínom alebo ožarovaniu srdca, bola identifikovaná maximálne tolerovaná dávka (MTD) (pozri časť 5.1).
Otvorená štúdia fázy 2 sa uskutočnila u 29 pacientov, z ktorých 27 bolo pediatrických pacientov (vo veku 3 až 16 rokov) a 2 boli mladí dospelí pacienti (vo veku 18 až 19 rokov), s rekurentným/progresívnym/refraktérnym gliómom vysokého stupňa (HGG) alebo ependymómom. V žiadnej zo skupín sa nevyskytli nežiaduce reakcie 5. stupňa. Najbežnejšími (≥ 10 %) nežiaducimi udalosťami súvisiacimi s liečbou boli pokles počtu neutrofilov (6 [20,7 %] pacientov) a vnútrolebečné krvácanie (3 [10,3 %] pacientov).
Jednoramenná štúdia fázy 1/2 sa uskutočnila u 6 pediatrických pacientov (vo veku 13 až 16 rokov) s pokročilým neresektovateľným GIST. Najčastejšími nežiaducimi reakciami na liek boli hnačka, nevoľnosť, pokles počtu bielych krviniek, neutropénia a bolesť hlavy, každá u 3 (50 %) pacientov, primárne 1. alebo 2. stupňa závažnosti. U štyroch zo 6 (66,7 %) pacientov sa vyskytli nežiaduce udalosti súvisiace s liečbou 3. –
4. stupňa (3. stupňa boli hypofosfatémia, neutropénia a trombocytopénia, každá u 1 pacienta, a 4. stupňa bola neutropénia u 1 pacienta). V tejto štúdii neboli hlásené žiadne závažné nežiaduce udalosti (SAE) ani nežiaduce reakcie na liek 5. stupňa. V klinickej štúdii aj publikáciách bol bezpečnostný profil konzistentný so známym bezpečnostným profilom u dospelých.
H
l
ásenie
podozrení
na
nežiaduce
r
eakcie

Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNeexistuje žiadne špecifické antidotum na predávkovanie sunitinibom a liečba predávkovania má spočívať vo všeobecných podporných opatreniach. V indikovaných prípadoch sa môže odstránenie neabsorbovaného liečiva dosiahnuť vracaním alebo výplachom žalúdka. Boli hlásené prípady predávkovania; niektoré
prípady boli spojené s nežiaducimi reakciami v súlade so známym bezpečnostným profilom sunitinibu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: cytostatiká, inhibítory proteínkinázy; ATC kód: L01EX01
MechanizmusúčinkuSunitinib inhibuje početné RTK, ktoré sa podieľajú na raste nádoru, neoangiogenéze a metastatickom šírení nádoru. Sunitinib bol identifikovaný ako inhibítor receptorov pre doštičkový rastový faktor (PDGFRα a PDGFRβ), VEGF receptorov (VEGFR1, VEGFR2 a VEGFR3), receptoru pre faktor kmeňových buniek (KIT), tyrozínkinázy 3 podobnej Fms (FLT3), receptoru pre faktor stimulujúci kolónie (CSF - 1R) a receptoru pre neurotrofický faktor odvodený od gliálnej línie buniek (RET).
V biochemických a bunkových testoch vykazuje primárny metabolit podobnú účinnosť ako sunitinib.
KlinickáúčinnosťabezpečnosťKlinická bezpečnosť a účinnosť sunitinibu bola skúmaná v liečbe pacientov s GIST, ktorí boli rezistentní na imatinib (t.j. počas alebo po liečbe imatinibom u nich došlo k progresii ochorenia), alebo neznášali
imatinib (t.j. počas liečby imatinibom u nich vznikli prejavy závažnej toxicity, ktoré znemožnili pokračovať v liečbe), v liečbe pacientov s MRCC a liečbe pacientov s neresekovateľným pNET.
Účinnosť je pri GIST založená na čase do progresie nádoru (time to tumour progression, TTP) a zlepšení prežívania, u pacientov s predtým neliečeným MRCC na prežívaní bez progresie
(progression-free survival, PFS), resp. na miere objektívnej odpovede (objective response rates, ORR) pri
MRCC rezistentnom na cytokíny a na PFS u pacientov s pNET.
Gastrointestinálne stromálne tumoryU pacientov s GIST po zlyhaní liečby imatinibom (medián maximálnej dennej dávky 800 mg) v dôsledku rezistencie alebo intolerancie bola vykonaná počiatočná otvorená štúdia so stúpajúcimi dávkami sunitinibu. Deväťdesiatsedem pacientov bolo zaradených do štúdie pri rôznom dávkovaní a schémach podávania; 55 pacientov dostávalo 50 mg sunitinibu v odporúčanej liečebnej Schéme 4 týždne liečba /2 týždne prestávka („Schéma 4/2“).
Medián TTP bol v tejto štúdii 34 týždňov (95 % CI: 22,0, 46,0).
U pacientov s GIST, ktorí neznášali imatinib alebo u nich počas alebo po tejto liečbe ochorenie progredovalo (medián maximálnej dennej dávky imatinibu - 800 mg), bola vykonaná randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia 3. fázy so sunitinibom. V tejto štúdii bolo randomizovaných 312 pacientov (2 : 1) na perorálne podávanie 50 mg sunitinibu alebo placeba raz za deň v schéme 4/2 až do progresie ochorenia alebo do vyradenia zo štúdie kvôli inej príčine (207 pacientov dostávalo sunitinib, 105 pacientov placebo). Primárny cieľový ukazovateľ v rámci hodnotenia účinnosti v tejto štúdii bol TTP definovaný ako čas od randomizácie po prvý objektívny dôkaz progresie nádoru. V
čase vopred stanovenej predbežnej analýzy bol medián TTP pri liečbe sunitinibom 28,9 týždňa (95 % CI:
21,3, 34,1) podľa hodnotenia investigátorov a 27,3 týždňa (95 % CI: 16,0, 32,1) podľa hodnotenia nezávislej komisie a zároveň bol štatisticky signifikantne dlhší ako TTP pri liečbe placebom 5,1 týždňa (95
% CI: 4,4, 10,1) podľa hodnotenia investigátorov a 6,4 týždňa (95 % CI: 4,4, 10,0) podľa hodnotenia nezávislej komisie. Rozdiel v celkovom prežívaní (overall survival, OS) vychádzal štatisticky v prospech sunitinibu [miera rizika [hazard ratio (HR): 0,491; (95 % CI: 0,290, 0,831)]; riziko úmrtia bolo v placebovom ramene dvakrát vyššie v porovnaní so sunitinibovým ramenom.
Po predbežnej analýze účinnosti a bezpečnosti na základe odporúčania nezávislej komisie na monitorovanie dát a bezpečnosti (data and safety monitoring board, DSMB) bola štúdia odslepená a pacientom v ramene s placebom bola ponúknutá liečba sunitinibom v otvorenej fáze štúdie.
V otvorenej fáze štúdie dostávalo sunitinib celkovo 255 pacientov vrátane 99 pacientov, ktorí boli pôvodne liečení placebom.
Analýza primárnych a sekundárnych cieľových ukazovateľov v otvorenej fáze štúdie opakovane potvrdila výsledky získané v čase predbežnej analýzy, ako je uvedené v Tabuľke 2:
Tabuľka2. Súhrncieľovýchukazovateľovúčinnosti(ITTpopulácia)preGIST
| Dvojito zaslepená liečbaa
|
|
| Medián (95 % CI)
| Miera rizika
| Liečebná skupina s
placebom
s prestupom na
aktívnu liečbub
|
Cieľový ukazovateľ
|
Sunitinib
|
Placebo
|
(95 % CI)
|
p-hodnota
|
Primárny:
|
|
TTP (týždne)
|
|
predbežná analýza
| 27,3 (16,0, 32,1)
| 6,4 (4,4, 10,0)
| 0,329 (0,233,
0,466)
| < 0,001
| -
|
finálna analýza
| 26,6 (16,0, 32,1)
| 6,4 (4,4, 10,0 )
| 0,339 (0,244,
0,472)
| < 0,001
| 10,4 (4,3, 22,0)
|
Sekundárny
|
|
PFS (týždne) c
|
|
predbežná analýza
| 24,1 (11,1, 28,3)
| 6,0 (4,4, 9,9)
| 0,333 (0,238,
0,467)
| < 0,001
| -
|
finálna analýza
| 22,9 (10,9, 28,0)
| 6,0 (4,4, 9,7)
| 0,347 (0,253,
0,475)
| < 0,001
| -
|
ORR (%)d
|
|
predbežná analýza
| 6,8 (3,7, 11,1)
| 0 (-)
| NA
| 0,006
| -
|
finálna analýza
| 6,6 (3,8, 10,5)
| 0 (-)
| NA
| 0,004
| 10,1 (5,0, 17,8)
|
OS (týždne)e
|
|
predbežná analýza
| -
| -
| 0,491 (0,290,
0,831)
| 0,007
| -
|
finálna analýza
| 72,7 (61,3, 83,0)
| 64,9 (45,7, 96,0)
| 0,876 (0,679,
1,129)
| 0,306
| -
|
Skratky: CI = interval spoľahlivosti; ITT = intent-to-treat; NA = neaplikovateľné; ORR = miera objektívnej
odpovede; OS = celkové prežívanie; PFS = prežívanie bez progresie; TTP = čas do progresie nádoru.
a Výsledky dvojito zaslepenej liečby pochádzajú z ITT populácie s použitím merania centrálneho rádiológa tam, kde to bolo vhodné.
b Výsledky účinnosti pre 99 subjektov, ktorým bola zmenená liečba z placeba na sunitinib po odslepení. Vstupné hodnoty boli vymazané v čase zmeny liečby a analýza účinnosti je založená na hodnotení investigátorov.
c Predbežné hodnoty PFS boli aktualizované na základe prepočítania pôvodných údajov.
d Výsledky pre ORR sú udávané ako percento pacientov, u ktorých bola potvrdená odpoveď v rámci
95 % CI.
e Medián nebol dosiahnutý, pretože údaje ešte neboli zrelé.
Medián OS v ITT populácii bol 72,7 týždňa v skupine pacientov liečených sunitinibom a 64,9 týždňa v skupine pacientov na placebe (HR: 0,876; 95 % CI: 0,679, 1,129; p = 0,306). V tejto analýze boli do liečebného ramena s placebom zaradení aj pacienti pôvodne randomizovaní na placebo, ktorí následne boli liečení sunitinibom v otvorenej fáze štúdie.
Doteraz neliečený metastatický karcinóm z obličkových buniekRandomizovaná multicentrická medzinárodná štúdia 3. fázy hodnotiaca účinnosť a bezpečnosť sunitinibu v porovnaní s interferónom IFN-α bola vykonaná u pacientov s doteraz neliečeným karcinómom z obličkových buniek MRCC. Sedemstopäťdesiat pacientov bolo randomizovaných do liečebných ramien
1:1; pacienti boli liečení buď sunitinibom v opakovaných 6-týždňových cykloch pozostávajúcich zo 4
týždňov perorálneho podávania 50 mg denne, po ktorých nasledovali 2 týždne bez liečby (Schéma 4/2),
alebo IFN-α podávaným ako subkutánna injekcia s 3 miliónmi jednotiek (MU) prvý týždeň, 6 MU druhý týždeň a 9 MU tretí týždeň a potom 3-krát týždenne obdeň.
Medián trvania liečby sunitinibom bol 11,1 mesiacov (rozsah: 0,4 - 46,1) a 4,1 mesiacov (rozsah 0,1 - 45,6) pri liečbe IFN-α. S liečbou súvisiace závažné nežiaduce účinky (treatment related serious adverse events, TRSAEs) boli hlásené u 23,7 % pacientov liečených sunitinibom a u 6,9 % pacientov liečených IFN-α. Avšak miera prerušenia z dôvodu nežiaducich účinkov bola 20 % pri sunitinibe a 23 % pri IFN-α. Prerušenie podávania sa vyskytlo u 202 pacientov (54 %) na sunitinibe a 141 pacientov (39 %) na IFN-α. Redukcia dávky sa vyskytla u 194 pacientov (52 %) na sunitinibe a 98 pacientov (27 %) na IFN-α. Pacienti boli liečení do progresie ochorenia alebo do odstúpenia zo štúdie. Primárnym cieľovým ukazovateľom hodnotenia účinnosti bolo PFS. Plánovaná priebežná analýza ukázala štatisticky signifikantnú výhodu pre sunitinib oproti IFN-α, v tejto štúdii medián PFS pre sunitinibom liečenú skupinu bol 47,3 týždňa v porovnaní s 22,0 týždňami pre skupinu liečenú IFN-α; HR bolo 0,415 (95 % CI = 0,320, 0,539, p-hodnota
< 0,001). Ostatné ciele zahŕňali ORR, OS a bezpečnosť. Centrálne rádiologické vyšetrenia boli pozastavené po dosiahnutí primárneho cieľového ukazovateľa. V čase finálnej analýzy bola ORR stanovená na
podklade vyšetrení investigátormi 46 % (95 % CI: 41%, 51%) pre rameno so sunitinibom a 12,0 % (95 %
CI: 9%, 16%) pre rameno s IFN-α (p< 0,001).
Liečba sunitinibom bola spojená s dlhším prežívaním v porovnaní s IFN-α. Medián OS bol 114,6 týždňa pre rameno so sunitinibom (95 % CI: 100,1, 142,9) a 94,9 týždňov pre rameno s IFN-α (95 % CI: 77,7,
117,0) pri HR 0,821 (95 % CI: 0,673, 1,001; p = 0,0510 podľa nestratifikovaného “log-rank“ testu).
Celkové PFS a OS pozorované v ITT populácii, tak ako boli stanovené vyšetrením v centrálnom rádiologickom laboratóriu, sú zhrnuté v Tabuľke 3.
Tabuľka3.Súhrncieľovýchukazovateľovúčinnosti(ITTpopulácia)upredtýmneliečenéhomRCC
Súhrn výsledkov prežívania bez progresie
| Sunitinib
(N = 375)
| IFN-α
(N = 375)
|
Pacienti, u ktorých ochorenie neprogredovalo, alebo ktorí nezomreli [n (%)]
| 161 (42,9)
| 176 (46,9)
|
Pacienti, u ktorých bola pozorovaná progresia, alebo ktorí zomreli [n (%)]
| 214 (57,1)
| 199 (53,1)
|
PFS (týždne)
|
Kvartil (95 % CI)
|
25 %
| 22,7 (18,0, 34,0)
| 10,0 (7,3, 10,3)
|
50 %
| 48,3 (46,4, 58,3)
| 22,1 (17,1, 24,0)
|
75 %
| 84,3 (72,9, 95,1)
| 58,1 (45,6, 82,1)
|
Nestratifikovaná analýza
|
Miera rizika (sunitinib oproti IFN-α)
| 0,5268
|
95 % CI pre mieru rizika
|
(0,4316, 0,6430)
|
hodnota pa
|
< 0,0001
|
Súhrn výsledkov celkového prežívania
|
Sunitinib
(
n = 375)
|
IFN-α
(n = 375)
|
Z
hrnutie celkového prežitia
|
Pacienti, o ktorých nie je známe, že zomreli [n
(%)]
|
185 (49,3)
|
175 (46,7)
|
Pacienti, u ktorých bolo zistené úmrtie [n (%)]
|
190 (50,7)
|
200 (53,3)
|
OS (týždne)
|
Kvartil (95 % CI)
|
25 %
|
56,6 (48,7, 68,4)
|
41,7 (32,6, 51,6)
|
50 %
|
114,6 (100,1, 142,9)
|
94,9 (77,7, 117,0)
|
75 %
|
NA (NA, NA)
|
NA (NA, NA)
|
Nestratifikovaná analýza'
|
|
Miera rizika (sunitinib versus IFN-α)
|
0,8209
|
95 % CI pre mieru rizika
|
(0,6730, 1,0013)
|
hodnota pa
|
0,0510
|
Skratky: CI = interval spoľahlivosti; INF-α = interferón-alfa; ITT = intent-to-treat; n = počet pacientov; NA
= neaplikovateľné; OS = celkové prežívanie; PFS = prežívanie bez progresie.
a Podľa dvojstranného log-rank testu
Metastatický karcinóm z obličkových buniek rezistentný na cytokínyU pacientov refraktérnych na predchádzajúcu cytokínovú liečbu interleukínom 2 alebo IFN-α bola vykonaná klinická štúdia 2. fázy so sunitinibom. Šesťdesiatim trom pacientom sa podávala úvodná dávka
sunitinibu 50 mg perorálne raz denne počas 4 po sebe nasledujúcich týždňov, po ktorých nasledovali 2
týždne bez liečby, aby sa zavŕšil kompletný 6-týždňový cyklus (Schéma 4/2). Primárnym cieľovým ukazovateľom v rámci hodnotenia účinnosti bola ORR hodnotená na podklade kritérií pre odpoveď na
liečbu u tumorov (Response Evaluation Criteria in Solid Tumours, RECIST).
U tejto štúdii bola miera objektívnej odpovede 36,5 % (95 % CI: 24,7 %, 49,6 %) a medián TTP bol 37,7
týždňa (95 % CI: 24,0, 46,4).
U pacientov s MRCC refraktérnych na predchádzajúcu cytokínovú liečbu bola vykonaná podporná, otvorená, multicentrická štúdia s jedným liečebným ramenom, ktorá hodnotila účinnosť a bezpečnosť sunitinibu. Sunitinib sa podával 106 pacientom v dávke minimálne 50 mg denne podľa Schémy 4/2.
Primárnym cieľovým ukazovateľopm v rámci hodnotenia účinnosti bola v tejto štúdii ORR. Sekundárne
ciele boli TTP, trvanie odpovede (duration of response, DR) a OS.
V tejto štúdii bola ORR 35,8 % (95 % CI: 26,8%, 47,5 %). Medián pre DR a OS sa doteraz nedosiahol.
Pankreatické neuroendokrinné nádoryPodporná otvorená, multicentrická štúdia 2. fázy hodnotila účinnosť a bezpečnosť monoterapie sunitinibom v dávke 50 mg denne v Schéme 4/2 u pacientov s neresekovateľným pNET. V kohorte 66 pacientov s
nádorom z buniek pankreatických ostrovčekov bola primárnym cieľovým ukazovateľom miera odpovede
17 %.
U pacientov s neresekovateľným pNET sa vykonala pivotná multicentrická, medzinárodná, randomizovaná, dvojito zaslepená, placebom kontrolovaná štúdia 3. fázy s monoterapiou sunitinibom.
Pacienti, u ktorých sa vyžadovalo, aby mali potvrdenú progresiu za základe RECIST kritérií v rámci predchádzajúcich 12 mesiacov, boli randomizovaní (1:1) na liečbu buď sunitinibom v dávke 37,5 mg raz denne bez plánovanej prestávky v liečbe (N = 86) alebo placebom (N = 85).
Primárnym cieľovým ukazovateľom bolo porovnanie PFS u pacientov užívajúcich sunitinib a u pacientov užívajúcich placebo. Ostatné cieľové ukazovatelezahŕňali OS, ORR, PRO a bezpečnosť.
Demografické charakteristiky skupín liečených sunitinibom a placebom boli porovnateľné. Navyše malo
49 % pacientov liečených sunitinibom a 52 % pacientov s placebom nefunkčné nádory a 92 % pacientov v oboch ramenách malo metastázy v pečeni.
Použitie somatostatínových analógov bolo v štúdii povolené.
Celkovo 66 % pacientov užívajúcich sunitinib v porovnaní so 72 % pacientov s placebom dostávalo predchádzajúcu systémovú liečbu. Navyše 24 % pacientov užívajúcich sunitinib v porovnaní s 22 % pacientov s placebom dostávalo analógy somatostatínu.
Pri PFS hodnotenom skúšajúcimi sa pozorovala klinicky signifikantná výhoda sunitinibu oproti placebu. Medián PFS bol 11,4 mesiacov pre rameno so sunitinibom v porovnaní s 5,5 mesiacmi pre rameno s placebom [HR: 0,418 (95 % CI: 0,263 - 0,662), p = 0,0001]; podobné výsledky sa pozorovali, ak sa na stanovenie progresie ochorenia použili odvodené vyšetrenia odpovede nádorov založené na aplikácii RECIST kritérií na merania nádorov skúšajúcimi, ako je uvedené v tabuľke 4. HR v prospech sunitinibu sa pozorovalo vo všetkých podskupinách pacientov odvodených od hodnotených vstupných charakteristík vrátane analýzy podľa počtu predchádzajúcich systémových terapií. Celkovo 29 pacientov v ramene so sunitinibom a 24 pacientov v ramene s placebom neužívalo predtým žiadnu systémovú terapiu; u týchto pacientov bolo HR pre PFS 0,365 (95 % CI: 0,156 - 0,857), p = 0,0156. Podobne u 57 pacientov v ramene so sunitinibom (vrátane 28 pacientov s 1 predchádzajúcou systémovou terapiou a 29 pacientov s 2 a viac predchádzajúcimi systémovými terapiami) a u 61 pacientov v ramene s placebom (vrátane 25 pacientov s 1 predchádzajúcou systémovou terapiou a 36 pacientov s 2 a viac predchádzajúcimi systémovými terapiami), bolo HR pre PFS 0,456 (95 % CI: 0,264 - 0,787), p = 0,0036.
Tam, kde bola progresia založená na meraní nádorov udávanom skúšajúcimi a kde všetci pacienti cenzurovaní pre iné príčiny ako ukončenie štúdie boli považovaní za PFS príhody, sa vykonala analýza senzitivity PFS. Táto analýza poskytla konzervatívny odhad liečebného efektu sunitinibu a podporila primárnu analýzu tým, že demonštrovala HR 0,507 (95% CI 0,350 - 0,733), p = 0,000193. Pivotná štúdia s pankreatickým NET bola predčasne ukončená na odporúčanie nezávislého Výboru pre monitorovanie liekov (Drug Monitoring Committee) a primárny cielový ukazovateľ sa založil na hodnotení skúšajúcich, pričom obe skutočnosti mohli ovplyvniť odhad efektu liečby.
S cieľom vylúčiť skreslenia (bias) v hodnotení PFS založenom na vyšetreniach investigátorov sa vykonalo
BICR skenov; toto hodnotenie potvrdilo hodnotenie investigátorov, ako je uvedené v tabuľke 4.
Tabuľka4.VýsledkyúčinnostipNETzoštúdie3.fázy Parametre účinnosti
| Sunitinib
(N = 86)
| Placebo
(N = 85)
| Miera rizika
(95 % CI)
| Hodnota-p
|
Prežívanie bez progresie
[medián, mesiace (95 % CI)] podľa
hodnotenia skúšajúcich
|
11,4 (7,4; 19,8)
|
5,5 (3,6; 7,4)
| 0,418 (0,263; 0,662)
|
0,0001a
|
Prežívanie bez progresie
[medián, mesiace (95 % CI)] podľa odvodených vyšetrení odpovede nádorov založených na aplikácii RECIST kritérií na merania nádorov skúšajúcimi
|
12,6 (7,4; 16,9)
|
5,4 (3,5; 6,0)
|
0,401 (0,252; 0,640)
|
0,000066a
|
Prežívanie bez progresie
[medián, mesiace (95 % CI)] podľa zaslepeného nezávislého centrálneho
prehodnotenia vyšetrení nádorov
|
12,6 (11,1; 20.6)
|
5,8 (3,8; 7,2)
|
0,315 (0,181; 0,546)
|
0,000015a
|
Celkové prežívanie [sledovanie počas
5 rokov]
[medián, mesiace (95 % CI)]
|
38,6 (25,6, 56,4)
|
29,1 (16,4, 36,8)
|
0,730 (0,504, 1,057)
|
0,0940a
|
Miera objektívnej odpovede [%, (95
% CI)]
|
9,3 (3,2; 15,4)
|
0
|
NA
|
0,0066b
|
Skratky: CI = interval spoľahlivosti, N = počet pacientov; NA = neaplikovateľné, pNET = pankreatické
neuroendokrinné nádory, RECIST = kritériá na hodnotenie odpovede u nádorov.
a 2-stranný nestratifikovaný log-rank test
b Fisherov exaktný test
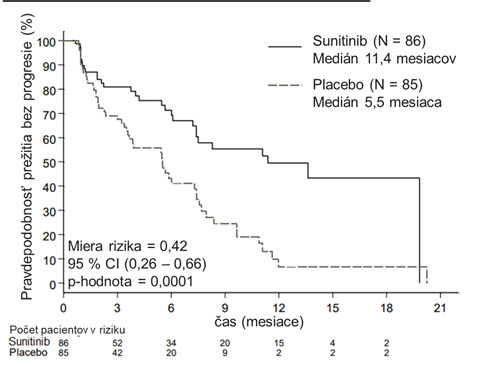 Obr. 1 Kaplanov-Meierov graf PFS v štúdii 3. fázy s pNET
Obr. 1 Kaplanov-Meierov graf PFS v štúdii 3. fázy s pNETSkratky: CI = interval spoľahlivosti; N = počet pacientov; PFS = prežívanie bez progresie; pNET =
pankreatické neuroendokrinné nádory.
Údaje o OS neboli zrelé v čase ukončenia štúdie [20,6 mesiacov (95% Cl: 20,6; NR) pre rameno so sunitinibom v porovnaní s NR (95% Cl: 15,5; NR) pre rameno s placebom, HR: 0,409 (95% Cl: 0,187;
0,894), p = 0,204]. V ramene so sunitinibom sa vyskytlo 9 úmrtí a v ramene s placebom 21 úmrtí.
Po progresii ochorenia boli pacienti odslepení a pacientom, ktorí užívali placebo, bola ponúknutá nezaslepená liečba sunitinibom v samostatnej pokračovacej štúdii. V dôsledku predčasného ukončenia štúdie bola liečba ostávajúcich pacientov odslepená a bola im ponúknutá nezaslepená liečba sunitinibom v pokračovacej štúdii. Celkovo 59 z 85 pacientov (69,4%) z ramena s placebom prešlo na nezaslepenú liečbu sunitinibom po progresii ochorenia alebo pri odslepení v čase predčasného ukončenia. OS pozorované po 5 rokoch sledovania v pokračujúcej štúdii preukázalo HR 0,730 (95% Cl: 0,504; 1,057).
Výsledky dotazníka kvality života od Európskej organizácie pre výskum a liečbu rakoviny (European Organization for Research and Treatment of Cancer Quality of Life Questionnaire, EORTC QLQ-C30) ukázali, že celková všeobecná so zdravím súvisiaca kvalita života a 5 funkčných domén (fyzická, funkčná, kognitívna, emočná a sociálna) sa zachovali u pacientov liečených sunitinibom v porovnaní s placebom s obmedzenými nežiaducimi symptomatickými prejavmi.
Vykonala sa medzinárodná, multicentrická, jednoramenná, otvorená štúdia fázy 4 hodnotiaca účinnosť a bezpečnosť sunitinibu u pacientov s progredujúcim, pokročilým/metastatickým, dobre diferencovaným, neresekovateľným pNET.
Stošesť pacientov (61 pacientov v kohorte bez predchádzajúcej liečby a 45 pacientov v kohorte neskoršej línie) dostalo perorálne liečbu sunitinibom s dávkou 37,5 mg jedenkrát denne v režime kontinuálneho denného dávkovania (CDD – continuous daily dosing).
Medián PFS hodnotený skúšajúcim lekárom bol 13,2 mesiaca v celkovej populácii (95 % CI: 10,9; 16,7) aj
v kohorte bez predchádzajúcej liečby (95 % CI: 7,4; 16,8).
Pediatrickápopulácia
Skúsenosti s používaním sunitinibu u pediatrických pacientov sú obmedzené (pozri časť 4.2).
Štúdia fázy I so zvyšujúcou sa dávkou perorálneho sunitinibu sa uskutočňila u 35 pacientov, pričom
30 bolo pediatrických pacientov (vo veku 3 až 17 rokov) a 5 mladých dospelých pacientov (vo veku 18 až
21 rokov), s refraktérnymi nádormi, pričom väčšina z nich mala pri zaradení do štúdie primárne diagnostikovaný mozgový nádor. V prvej časti štúdie sa pozorovala dávku obmedzujúca kardiotoxicita, a
preto sa štúdia zmenila tak, aby sa vylúčili pacienti, ktorí boli predtým vystavení potenciálne kardiotoxickým terapiám (vrátane antracyklínov) alebo ožarovaniu srdca. V druhej časti štúdie, do ktorej
boli zahrnutí pacienti s predchádzajúcou protinádorovou liečbou, ale bez rizikových faktorov srdcovej toxicity, bol sunitinib vo všeobecnosti tolerovateľný a klinicky manažovateľný v dávke 15 mg/m2 denne (MTD) v Schéme 4/2. U žiadneho zo subjektov sa nedosiahla kompletná odpoveď alebo čiastočná odpoveď. Stabilizované ochorenie sa pozorovalo u 6 pacientov (17 %). Jeden pacient s GIST sa zapojil do štúdie na dávkovej úrovni 15 mg/m2, pričom sa nedokázal žiadny prínos terapie. Celkovo sa pozorovali podobné nežiaduce reakcie na liek ako u dospelých (pozri časť 4.8).
Otvorená štúdia fázy 2 sa uskutočnila u 29 pacientov, pričom 27 bolo pediatrických pacientov (vo veku 3
až 16 rokov) a 2 boli mladí dospelí pacienti (vo veku 18 až 19 rokov), s HGG alebo ependymómom. Štúdia bola uzatvorená v čase plánovanej predbežnej analýzy kvôli nedostatočnej kontrole ochorenia. Medián PFS bol 2,3 mesiaca v skupine HGG a 2,7 mesiaca v skupine ependymómu. Medián celkového OS bol 5,1 mesiaca v skupine HGG a 12,3 mesiaca v skupine ependymómu. Najbežnejšími (≥ 10 %) hlásenými nežiaducimi udalosťami súvisiacimi s liečbou u pacientov v oboch skupinách dohromady boli pokles počtu neutrofilov (6 pacientov [20,7 %]) a vnútrolebečné krvácanie (3 pacienti [10,3 %]) (pozri časť 4.8).
Z údajov zo štúdie fázy 1/2 s perorálnym sunitinibom uskutočnenej u 6 pediatrických pacientov s GIST vo veku 13 rokov až 16 rokov, ktorí dostávali sunitinib v schéme 4/2 v dávkach medzi 15 mg/m2 denne a 30 mg/m2 denne, a z dostupných publikovaných údajov (20 pediatrických pacientov alebo mladých dospelých pacientov s GIST) vyplynulo, že liečba sunitinibom viedla k stabilizácii ochorenia u 18 z 26 (69,2 %) pacientov buď po zlyhaní imatinibu či jeho neznášanlivosti (16 pacientov so stabilným ochorením z 21), alebo de novo/po operácii (2 pacienti so stabilným ochorením z 5). V štúdii fázy 1/2 sa stabilné ochorenie a progresia ochorenia pozorovali každé u 3 zo 6 pacientov (1 pacient dostával imatinib ako neoadjuvantnú liečbu a 1 pacient dostával imatinib ako adjuvantnú liečbu). V tejto štúdii sa u 4 zo 6 pacientov (66,7 %) vyskytli nežiaduce udalosti súvisiace s liečbou 3. – 4. stupňa (3. stupňa boli hypofosfatémia, neutropénia a trombocytopénia, každá u 1 pacienta, a 4. stupňa bola neutropénia u 1 pacienta). Okrem toho boli v publikáciách hlásené nasledujúce nežiaduce reakcie na liek 3. stupňa u 5 pacientov: únava (2), gastrointestinálne nežiaduce reakcie na liek (vrátane hnačky) (2), hematologické nežiaduce reakcie na liek (vrátane anémie) (2), cholecystitída (1), hypertyroidizmus (1) a mukozitída (1).
Uskutočnila sa populačná farmakokinetická (PK) a farmakokineticko-farmakodynamická (PK/PD) analýza za účelom extrapolovať PK a kľúčové ukazovatele bezpečnosti a účinnosti sunitinibu u pediatrických pacientov s GIST (vo veku 6 až 17 rokov). Táto analýza bola založená na údajoch získaných od dospelých pacientov s GIST alebo nádormi a od pediatrických pacientov s nádormi. Na základe modelových analýz sa ukázalo, že nižší vek a menšia veľkosť tela nemajú negatívny vplyv na bezpečnosť a účinnosť vo vzťahu k plazmatickej expozícii sunitinibu. Neukázalo sa, že by bol pomer prínosu a rizika pre sunitinib negatívne ovplyvnený nižším vekom a menšou veľkosťou tela, a na tento pomer mala hlavný vplyv jeho plazmatická expozícia.
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s referenčným liekom obsahujúcim sunitinib vo všetkých podskupinách pediatrickej populácie na liečbu karcinómu z obličkových
sarkómu, mezoblastického nefrómu, renálneho medulárneho karcinómu a rabdoidného tumoru obličky) (pozri časť 4.2).
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s referenčným liekom obsahujúcim sunitinib vo všetkých podskupinách pediatrickej populácie na liečbu
gastroenteropankreatických neuroendokrinných tumorov (okrem neuroblastómu, neuroganglioblastómu a feochromocytómu) (pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
PK sunitinibu bola hodnotená u 135 zdravých dobrovoľníkov a 266 pacientov s nádormi. PK bola podobná u celej sledovanej populácie s nádormi a u zdravých dobrovoľníkov.
Pri dávkovaní od 25 mg do 100 mg sa proporcionálne k dávke zvyšuje plazmatická koncentrácia pod krivkou (AUC = area under curve) a Cmax. Pri opakovanom podávaní denne sa sunitinib kumuluje 3- až 4- násobne, jeho primárny metabolit sa kumuluje 7- až 10-násobne. Ustálené koncentrácie sunitinibu a jeho primárneho metabolitu sa dosiahnu do 10 až 14 dní. Na 14. deň sú kombinované plazmatické koncentrácie sunitinibu a jeho aktívneho metabolitu 62,9 - 101 ng/ml, čo predstavujú cieľové koncentrácie predpokladané z predklinických údajov na inhibíciu receptorovej fosforylácie in vitro, ktorá vedie in vivo k zastaveniu/redukcii rastu nádorov. Primárny aktívny metabolit tvorí 23 % až 37 % celkovej expozície. Pri opakovanom podávaní denne alebo opakovaných liečebných cykloch v testovaných dávkových režimoch
sa nepozorovali žiadne signifikantné zmeny PK sunitinibu alebo jeho primárneho aktívneho metabolitu.
Absorpcia
Po perorálnom podaní sunitinibu sa cmax obvykle pozoruje po 6 - 12 hodinách času do maximálnej koncentrácie (tmax) po podaní.
Potrava neovplyvňuje biologickú dostupnosť sunitinibu.
Distribúcia
V in vitro testoch sa sunitinib, resp. jeho primárny aktívny metabolit viazal na bielkoviny ľudskej plazmy v
95 %, resp. 90 % bez evidentnej závislosti od koncentrácie. Zdanlivý distribučný objem sunitinibu (Vd) bol
veľký – 2230 l, čo svedčí o distribúcii do tkanív.
Metabolickéinterakcie
In vitro kalkulované hodnoty Ki pre všetky cytochrómové testované izoformy P450 (CYP) (CYP1A2,
CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 a CYP4A9/11)
ukazujú, že je nepravdepodobné, aby sunitinib a jeho primárny aktívny metabolit indukovali, v akomkoľvek klinicky významnom rozsahu, metabolizmus iných liečiv, ktoré by mohli byť metabolizované týmito enzýmami.
Biotransformácia
Sunitinib je primárne metabolizovaný prostredníctvom CYP3A4, izoformy CYP, pričom vzniká jeho primárny aktívny metabolit, desetylsunitinib, ktorý sa takisto ďalej metabolizuje prostredníctvom toho istého izoenzýmu.
Treba sa vyhnúť súčasnému podávaniu sunitinibu so silným induktormi CYP3A4 alebo inhibítormi, lebo môžu byť zmenené plazmatické hladiny sunitinibu (pozri časť 4.4 a 4.5).
Eliminácia
Vylučovanie sa deje predovšetkým prostredníctvom stolice (61 %), vylučovanie nezmeneného liečiva a
jeho metabolitov obličkami dosahuje 16 % podanej dávky. Sunitinib a jeho primárny aktívny metabolit boli hlavnými zlúčeninami, ktoré boli identifikované v plazme, moči a stolici, čo predstavovalo 91,5 %, 86,4 %,
resp. 73,8 % rádioaktivity v odobraných vzorkách. Menej významné metabolity boli identifikované v moči
a stolici, ale spravidla sa nezistili v plazme. Celkový klírens perorálnej dávky (CL/F) je 34 – 62 l/h. Po perorálnom podaní zdravým dobrovoľníkom boli eliminačné polčasy sunitinibu a jeho primárneho aktívneho desetylmetabolitu približne 40 - 60 hodín a 80 - 110 hodín.
Súbežné
podávanie
s
liekmi,
ktoré
sú
inhibítormi
BCRP
I
n vitro je sunitinib substrátom pre efluxný transportér BCRP. V štúdii A6181038 súbežné podávanie s
gefitinibom, inhibítorom BCRP, nevyústilo do klinicky relevantného účinku na Cmax a AUC sunitinibu alebo celkového lieku (sunitinib + metabolit) (pozri časť 4.5). Táto štúdia bola multicentrická, otvorená štúdia fázy 1/2 skúmajúca bezpečnosť/tolerovateľnosť, maximálnu tolerovanú dávku a protinádorovú aktivitu sunitinibu v kombinácii s gefitinibom u pacientov s MRCC.
Ako sekundárny cieľový ukazovateľ štúdie sa vyhodnocovala PK gefitinibu (250 mg denne) a sunitinibu (37,5 mg [1. kohorta, n = 4] alebo 50 mg [2. kohorta, n = 7] denne s režimom 4 týždne liečba, po ktorej nasledovali 2 týždne bez liečby), keď sa podávali súbežne. Zmeny v PK parametroch sunitinibu nemali žiadny klinický význam a nepoukazovali na žiadne interakcie medzi liekmi. Avšak vzhľadom na relatívne nízky počet pacientov (tzn. N = 7 + 4) a strednú až veľkú variabilitu medzi pacientmi v ich farmakokinetických parametroch, sa PK zistenia z tejto štúdie, týkajúce sa interakcie medzi liekmi, musia interpretovať opatrne.
Osobitné skupinypacientov
Porucha funkcie pečene
Sunitinib a jeho primárny metabolit sa metabolizujú prevažne v pečeni. Systémové expozície po jednorazovej dávke sunitinibu boli u jedincov s miernym alebo stredne závažným (trieda A a B klasifikácie
podľa Childa-Pugha) poškodením pečene podobné v porovnaní s jedincami s normálnou funkciou pečene.
Sunitinib sa neskúmal u jedincov so závažným (trieda C klasifikácie podľa Childa-Pugha) poškodením pečene.
Zo štúdií u pacientov s nádorovým ochorením boli vylúčení pacienti s hodnotou ALT alebo AST > 2,5 x ULN (upper limit of normal = horný limit normy) alebo s hodnotou >5,0 x ULN, ak bolo zvýšenie spôsobené metastázami do pečene.
Porucha funkcie obličiek
Populačné PK analýzy ukázali, že zdanlivý klírens (CL/F) sunitinibu nebol ovplyvnený klírensom kreatinínu (CLcr) v rámci meraného rozmedzia (42 - 347 ml/min). Systémové expozície po podaní
jednotlivej dávky sunitinibu boli podobné u pacientov so závažnou poruchou funkcie obličiek (klírens
kreatinínu CLcr < 30 ml/min) v porovnaní s pacientmi s normálnou funkciou obličiek
(CLcr > 80 ml/min). Aj keď sa sunitinib a jeho primárny metabolit neeliminovali prostredníctvom hemodialýzy u pacientov s ESRD, celkové systémové expozície boli nižšie o 47 % pre sunitinib a 31 % pre
jeho primárny metabolit v porovnaní s pacientmi s normálnou funkciou obličiek.
Hmotnosť, skóre
Populačné PK analýzy hodnotiace demografické údaje svedčia, že nie sú potrebné úpravy dávky vzhľadom na hmotnosť alebo skóre ECOG (Eastern Cooperative Oncology Group).
Pohlavie
Dostupné údaje ukazujú, že ženy môžu mať asi o 30 % nižší zdanlivý klírens (CL/F) sunitinibu ako muži, tento rozdiel však nevyžaduje úpravu dávky.
Pediatrická populácia
Skúsenosti s používaním sunitinibu u pediatrických pacientov sú obmedzené (pozri časť 4.2). Realizovali sa populačné PK analýzy spojených dátových súborov od dospelých pacientov s GIST a nádormi a
pediatrických pacientov s nádormi. Uskutočnili sa postupné kovariančné modelové analýzy na
vyhodnotenie účinku veku a veľkosti tela (telesnej hmotnosti alebo plochy povrchu tela), ako aj iných spoločných premenných na dôležité PK parametre pre sunitinib a jeho aktívny metabolit. Pokiaľ ide o testované spoločné premenné súvisiace s vekom a veľkosťou tela bol vek signifikantnou spoločnou premennou pre zdanlivý klírens sunitinibu (čím nižší vek pediatrického pacienta, tým nižší zdanlivý klírens). Podobne signifikantnou spoločnou premennou zdanlivého klírensu aktívneho metabolitu bola plocha povrchu tela (čím menšia povrchová plocha tela, tým nižší zdanlivý klírens).
Ďalej, na základe integrovanej populačnej PK analýzy združených údajov z 3 pediatrických štúdií
bola významnou spoločnou premennou zdanlivého klírensu sunitinibu a jeho aktívneho metabolitu počiatočná plocha povrchu tela (BSA). Na základe tejto analýzy sa predpokladá, že dávka približne 20 mg/m2 denne (rozsah BSA: 1,10 – 1,87 m2) u pediatrických pacientov poskytne plazmatické expozície sunitinibu a jeho aktívneho metabolitu porovnateľné (75 až 125 % AUC) s tými, ktoré sa vyskytujú u dospelých s GIST, ktorým sa podáva sunitinib 50 mg denne v schéme 4/2 (AUC 1 233 ng.hod./ml). V pediatrických štúdiách bola východisková dávka sunitinibu 15 mg/m2 (na základe maximálne tolerovanej dávky (MTD) identifikovanej v štúdii fázy I so zvyšujúcou sa dávkou, pozri časť 5.1), ktorá sa u pediatrických pacientov s GIST zvyšovala na 22,5 mg/m2 a následne na 30 mg/m2 (tak, aby nepresiahla celkovú dávku 50 mg/deň) na základe individuálnej bezpečnosti/znášanlivosti pacienta. Okrem toho bola v súlade so zverejnenou literatúrou o pediatrických pacientoch s GIST vypočítaná východisková dávka v rozsahu od 16,6 mg/m2 až 36 m/m2, pričom sa dávky zvýšili až na 40,4 mg/m2 (nepresahujúc celkovú dávku
50 mg/deň).
5.3 Predklinické údaje o bezpečnosti
V štúdiách toxicity na potkanoch a opiciach boli pri opakovanom podávaní v trvaní do 9 mesiacov primárne účinky na cieľové orgány zistené v tráviacom trakte (vracanie a hnačka u opíc); v nadobličkách (kortikálna kongescia a/alebo hemorágia u potkanov a opíc, s nekrózou a následnou fibrózou u potkanov); v hemolymfopoetickom systéme (hypocelularita kostnej drene a lymfoidná deplécia týmu, sleziny a
lymfatických uzlín); v exokrinnej časti pankreasu (degranulácia acinárnych buniek s nekrózou jednotlivých buniek); v slinných žľazách (acinárna hypertrofia); v kostných spojeniach (zhrubnutie rastových
platničiek); v maternici (atrofia); a vo vaječníkoch (spomalený vývoj folikulov). Všetky tieto nálezy sa vyskytli pri klinicky relevantných expozičných plazmatických hladinách sunitinibu. Ďalšie účinky pozorované v iných štúdiách zahŕňali: predĺženie QTc intervalu, pokles LVEF a atrofiu semenníkových
tubulov, zmnoženie mezangia v obličke, hemorágie v tráviacom trakte a na sliznici úst a hypertrofiu buniek prednej hypofýzy. Predpokladá sa, že zmeny na maternici (atrofia endometria) a rastovej platničke kostí
(zahustenie epifyzárnej chrupavky alebo dysplázia chrupavky) súvisia s farmakologickým účinkom sunitinibu. Väčšina týchto prejavov bola reverzibilná po 2 až 6 týždňoch bez liečby.
Genotoxicita
Genotoxický potenciál sunitinibu bol hodnotený in vitro a in vivo. Sunitinib nebol mutagénny pre baktérie pri metabolickej aktivácii v pečeni potkanov. In vitro nevyvolal sunitinib štrukturálne chromozómové
aberácie v lymfocytoch z periférnej krvi človeka. In vitro sa pozorovala na lymfocytoch z periférnej krvi
človeka polyploidia (numerické chromozómové aberácie)
v prítomnosti aj neprítomnosti metabolickej aktivácie. U potkanov nebol sunitinib in vivo klastogénny v kostnej dreni. Hlavný aktívny metabolit sa nehodnotil z hľadiska genotoxického potenciálu.
Karcinogenicita
V 1-mesačnej štúdii s perorálnym plnením žalúdka sondou zameranej na zistenie rozsahu dávky s CDD (v dávkach 0, 10, 25, 75 alebo 200 mg/kg/deň) u rasH2 transgénnych myší sa pri najvyššej testovanej dávke (200 mg/kg/deň) pozoroval karcinóm a hyperplázia Brunnerovych žliaz duodena.
U rasH2 transgénnych myší bola uskutočnená 6-mesačná štúdia s perorálnym plnením žalúdka sondou zameraná na karcinogenicitu s denným podávaním (v dávkach 0, 8, 25, 75 [znížená na 50] mg/kg/deň). Gastroduodenálne karcinómy, zvýšený výskyt sprievodných hemangiosarkómov a/alebo hyperplázia sliznice žalúdka boli pozorované pri dávkach ≥ 25 mg/kg/deň následne po 1- až 6-mesačnej dobe podávania (≥ 7,3-násobok AUC u pacientov, ktorým sa podávala odporúčaná denná dávka).
V 2-ročnej štúdii zameranej na karcinogenicitu u potkanov (v dávkach 0, 0,33, 1 alebo 3 mg/kg/deň) malo podávanie sunitinibu počas 28-dňového cyklu s následnou 7-dňovou prestávkou v liečbe za následok zvýšenie incidencie feochromocytómu a hyperpláziu drene nadobličky potkanov mužského pohlavia pri dávke 3 mg/kg/deň po > 1-ročnom podávaní (≥ 7,8-násobok AUC u pacientov liečených odporúčanou dennou dávkou). Nádor Brunnerových žliaz duodéna sa objavil u samíc pri dávke ≥ 1 mg/kg/deň a u samcov pri dávke 3 mg/kg/deň a hyperplázia buniek sliznice v žľazovom tkanive žalúdka bola evidentná u samcov pri dávke 3 mg/kg/deň, čo predstavuje ≥ 0,9-, 7,8- a 7,8-násobok AUC v uvedenom poradí u pacientov, ktorým sa podávala odporúčaná denná dávka. Význam malígnych nálezov pozorovaných u
(rasH2 transgénnych) myší a potkanov v rámci štúdií karcinogenicity počas liečby sunitinibom pre človeka nie je známy.
ReprodukčnáavývojovágenotoxicitaV štúdiách reprodukčnej toxicity sa nepozorovalo žiadne ovplyvnenie fertility samcov alebo samíc. Avšak v štúdiách toxicity na potkanoch a opiciach sa pri opakovanom podávaní pri dosiahnutí klinicky
významných systémových expozičných hladín pozorovalo ovplyvnenie fertility samíc vo forme
folikulárnej atrézie, degenerácie žltých teliesok, zmien na endometriu v maternici a poklese hmotnosti maternice a vaječníkov. Pri plazmatických expozičných hladinách predstavujúcich 25-násobok systémovej expozície u ľudí sa pozorovalo ovplyvnenie fertility potkaních samcov vo forme tubulárnej atrofie v semenníkoch, zníženia počtu spermatozoí v nadsemenníkoch a koloidnej deplécie v prostate a semenných vačkoch.
U potkanov bola zjavná embryonálno-fetálna mortalita vo forme signifikantného poklesu živých plodov, zvýšeného počtu rezorpcií, zvýšenia postimplantačných strát a celkovej straty vrhu u 8 z 28 gravidných samíc pri plazmatických expozičných hladinách predstavujúcich 5,5-násobok systémovej expozície u ľudí. U králikov došlo pri plazmatických expozičných hladinách predstavujúcich 3-násobok systémovej expozície u ľudí k poklesu hmotnosti gravidnej maternice a počtu živých plodov v dôsledku zvýšeného počtu resorpcií, zvýšených postimplantačných strát, ako aj celkovej straty vrhu u 4 zo 6 gravidných samíc. Liečba sunitinibom počas organogenézy viedla u potkanov k vývojovým chybám pozostávajúcim zo zvýšenej incidencie malformácií kostry plodu, charakterizovaných predovšetkým ako spomalená osifikácia hrudných/bedrových stavcov, ktoré sa vyskytli pri plazmatických expozičných hladinách predstavujúcich
5,5-násobok systémovej expozície u ľudí. U králikov predstavovali vývojové chyby častejší výskyt rázštepu pery pri plazmatických expozičných hladinách približne rovnakých ako hladiny pozorované v klinickej praxi; a rázštep pery a podnebia sa vyskytol častejšie pri plazmatických expozičných hladinách predstavujúcich 2,7-násobok systémovej expozície u ľudí.
Sunitinib (v dávkach 0,3; 1,0; 3,0 mg/kg/deň) bol hodnotený v štúdii zameranej na pre- a postnatálny vývoj u gravidných potkanov. Počas gestácie a laktácie sa znížili prírastky telesnej hmotnosti matky pri dávke > 1 mg/kg/deň, ale nepozorovala sa žiadna toxicita na reprodukciu u matky až do
3 mg/kg/deň (odhadovaná expozícia > 2,3-násobok AUC u pacientov, ktorým sa podávala odporúčaná denná dávka). Pri dávke 3 mg/kg/deň sa u mláďat pozoroval pokles telesnej hmotnosti počas obdobia pred
aj po odstavení. Žiadna vývojová toxicita sa nepozorovala pri dávke 1 mg/kg/deň (približná expozícia >
0,9-násobok AUC u pacientov, ktorým sa podávala odporúčaná denná dávka).
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokObsah kapsuly Manitol (E421) Kroskarmelóza, sodná soľ
Škorb, predželatinovaný (kukuričný) Stearát horečnatý
Obal kapsuly Želatína Čistená voda
Oxid titaničitý (E171)

Červený oxid železitý (E172) [pre 12.5 mg, 25 mg and 50 mg]
Čierny oxid železitý (E172) [pre 25mg a 50 mg]
Žltý oxid železitý (E172) [pre 25 mg, 37.5 mg a 50 mg]
Potlačový atramentŠelak (E904)
Propylénglykol (E1520)

Hydroxid draselný (E525)
Oxid titaničitý (E171) [pre 12.5 mg, 25 mg and 50 mg]
Čierny oxid železitý (E172) [pre 37.5 mg]
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnostiBlistrové balenie:
2 roky

Balenie fliaš:
18 mesiacov [pre 25 mg]
2 roky [pre 12.5 mg, 37.5 mg a 50 mg]
6.4 Špeciálne upozornenia na uchovávanieBlistrové balenie
Uchovávajte pri teplote do 30 °C.
Balenie fliaš
Uchovávajte pri teplote do 25 °C.
6.5 Druh obalu a obsah baleniaFľaška z polyetylénu s vysokou hustotou (HDPE) a polypropylénovým uzáverom obsahujúca 28 alebo 30
tvrdých kapsúl.
PVC/PCTFE - Alu perforovaný blister s jednotlivými dávkami obsahujúci 14 x 1, 28 x 1 alebo 30 x 1
kapsulu.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIMSN LABS EUROPE Limited
KW20A, Corradino Park
Paola, PLA 3000
Malta
8. REGISTRAČNÉ ČÍSLASunitinib MSN 12,5 mg tvrdé kapsuly: 44/0075/22-S Sunitinib MSN 25 mg tvrdé kapsuly: 44/0076/22-S Sunitinib MSN 37,5 mg tvrdé kapsuly: 44/0077/22-S
Sunitinib MSN 50 mg tvrdé kapsuly: 44/0078/22-S
9. DÁTUM PRVEJ REGISTRÁCIE
Dátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTU
08/2022