
j randomizovanej aktívnej kontrolovanej štúdii zameranej na inhibítor JAK u pacientov s reumatoidnou artritídou (RA) vo veku 50 rokov a starších s aspoň jedným ďalším rizikovým faktorom kardiovaskulárnych ochorení sa v prípade inhibítora JAK pozorovala
vyššia miera výskytu malignít, najmä rakoviny pľúc, lymfómu a NMSC, v porovnaní s inhibítormi tumor nekrotizujúceho faktora (tumour necrosis factor, TNF).
Na posúdenie potenciálnej súvislosti medzi expozíciou deukravacitinibu a vznikom malignít sú dostupné len obmedzené klinické údaje. Prebieha hodnotenie dlhobobej bezpečnosti. Pred začatím liečby sa u pacientov majú zvážiť riziká a prínosy liečby deukravacitinibom.
Závažnénežiaducekardiovaskulárneudalosti(major adverse cardiovascular events,MACE),hlbokážilovátrombóza(deep venous thrombosis,DVT)apľúcnaembólia(pulmonary embolism,PE)
Nie je známe, či môže inhibícia TYK2 súvisieť s nežiaducimi reakciami inhibície JAK. Vo veľkej
randomizovanej aktívnej kontrolovanej štúdii zameranej na inhibítor JAK sa v prípade inhibítora JAK
v porovnaní s inhibítormi TNF u pacientov s RA vo veku 50 rokov a starších s aspoň jedným ďalším rizikovým faktorom kardiovaskulárnych ochorení pozorovala vyššia miera výskytu MACE,
definovaná ako kardiovaskulárna smrť, nefatálny infarkt myokardu a nefatálna cievna mozgová
príhoda, a vyššia miera výskytu venózneho tromboembolizmu závislého od dávky vrátane DVT a PE .
V klinických skúšaniach s deukravacitinibom sa nepozorovalo zvýšené riziko MACE, DVT ani PE. Prebieha hodnotenie dlhobobej bezpečnosti deukravacitinibu. Pred začatím liečby sa u pacientov majú zohľadniť riziká a prínosy liečby deukravacitinibom.
Imunizácie
Pred začatím liečby deukravacitinibom zvážte absolvovanie všetkých imunizácií primeraných veku
podľa súčasných smerníc na imunizáciu. U pacientov liečených deukravacitinibom sa treba vyhnúť použitiu živých vakcín. Odpoveď na živé alebo neživé vakcíny sa nehodnotila.
Pomocné látky
Laktóza
Tento liek obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v tablete, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Klinické štúdie naznačujú, že pri súbežnom podávaní s nasledovnými liekmi nemá deukravacitinib klinicky významné liekové interakcie, a preto nie sú potrebné úpravy dávky.
Účinok deukravacitinibunainélieky
Deukravacitinib významne neovplyvňuje plazmatické expozície rosuvastatínu (substrát BCRP
a OATP), metotrexátu (substrát BCRP a renálnych transportérov), mofetil-mykofenolátu (MMF) (substrát CES1 a CES2) alebo perorálnych kontraceptív (noretindrón acetát a etinylestradiol).
Účinok iných liekovnadeukravacitinib
Lieky, ktoré sú ihnibítormi alebo induktormi enzýmov CYP alebo transportérov, ako sú cyklosporín
(dvojitý inhibítor P-gp/inhibítor proteínu rezistencie rakoviny prsníka [breast cancer resistance protein, BCRP]), fluvoxamín (silný inhibítor CYP 1A2), ritonavir (stredne silný induktor CYP 1A2), diflunizal (inhibítor UGT 1A9), pyrimetamín (inhibítor OCT1), famotidín (antagonista H2 receptora) alebo rabeprazol (inhibítor protónovej pumpy) významne neovplyvňujú plazmatické expozície deukravacitinibu (pozri časť 5.2).
4.6 Fertilita, gravidita a laktácia
G
r
avidita
Je iba obmedzené množstvo údajov o použití deukravacitinibu u gravidných žien. Štúdie na zvieratách
nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu deukravacitinibu počas gravidity.
Dojčenie
Nie je známe, či sa deukravacitinib/metabolity vylučujú do ľudského mlieka.
Dostupné údaje u zvierat preukázali vylučovanie deukravacitinibu do mlieka (pozri časť 5.3).
Riziko u novorodencov/dojčiat nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu deukravacitinibom sa má urobiť po zvážení prínosu dojčenia pre dieťa
a prínosu liečby pre ženu.
Fertilita
Účinok deukravacitinibu na fertilitu u ľudí sa nehodnotil. Štúdie na zvieratách nepreukázali priame
alebo nepriame škodlivé účinky z hľadiska fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Deukravacitinib nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
Najčastejšie hlásenou nežiaducou reakciou sú infekcie horných dýchacích ciest (18,9 %), najčastejšie
nazofaryngitída. Dlhodobý bezpečnostný profil deukravacitinibu bol podobný a v súlade s predchádzajúcimi skúsenosťami.
Tabuľkový zoznamnežiaducichreakcií
Nasledovný zoznam nežiaducich reakcií deukravacitinibu je z klinických skúšaní s ložiskovou
psoriázou (tabuľka 1). Tieto reakcie sú uvedené podľa triedy orgánových systémov podľa MedDRA
a podľa frekvencie.
Frekvencie sú definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej
časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000);
neznáme (z dostupných údajov).
T
abuľka 1: Zoznam nežiaducich reakcií
T
rieda orgánových systémov
|
F
rekvencia
|
Nežiaduca reakcia
|
I
nfekcie a nákazy
|
Veľmi časté
|
Infekcie horných dýchacích ciesta
|
Časté
|
Infekcie herpes simplexb
|
Menej časté
|
Herpes zoster
|
Po
ruchy gastrointestinálneho traktu
|
Časté
|
Vredy v ústachc
|
Po
ruchy kože a podkožného tkaniva
|
Časté
|
Akneiformná vyrážkad
Folikulitída
|
L
a
boratórne a funkčné vyšetrenia
|
Časté
|
Zvýšené hodnoty kreatínfosfokinázy v krvi
|
a Infekcie horných dýchacích ciest zahŕňajú nazofaryngitídu, infekciu horných dýchacích ciest, vírusovú infekciu horných dýchacích ciest, faryngitídu, sinusitídu, akútnu sinusitídu, rinitídu, tonzilitídu, peritonzilárny absces, laryngitídu, tracheitídu a rinotracheitídu.
b Infekcie herpes simplex zahŕňajú orálny herpes, herpes simplex, genitálny herpes a herpetickú vírusovú
infekciu.
c Vredy v ústach zahŕňajú afty, ulceráciu v ústach, ulceráciu jazyka a stomatitídu.
d Akneiformná vyrážka zahŕňa akné, akneiformnú dermatitídu, vyrážku, rosaceu, pustuly, pustulárnu vyrážku a papuly.
|
O
pis
vybraných
nežiaducich
reakcií
I
nfekcie
V štúdiách POETYK PSO-1 a POETYK PSO-2 (pozri časť 5.1) sa infekcie vyskytli
u 29,1 % pacientov v skupine s deukravacitinibom (116,0 udalostí na 100 osoborokov) v porovnaní
s 21,5 % pacientov v skupine s placebom (83,7 udalostí na 100 osoborokov) počas prvých 16 týždňov. Väčšina infekcií bola nezávažná a mierna až stredne závažná a neviedla k prerušeniu liečby
deukravacitinibom. Incidencia závažných infekcií v skupine s deukravacitinibom bola 0,6 %
(2,0 udalosti na 100 osoborokov) a v skupine s placebom bola 0,5 % (1,6 udalostí na 100 osoborokov).
Miera infekcií v skupine s deukravacitinibom sa do 52. týždňa nezvýšila (95,4 udalostí na
100 osoborokov). Miera závažných infekcií v skupine s deukravacitinibom sa do 52. týždňa nezvýšila
(1,7 udalostí na 100 osoborokov).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieDeukravacitinib sa podával zdravým osobám v jednorazových dávkach až do 40 mg (> 6-násobok odporúčanej dávky u ľudí 6 mg/deň) a v opakovane podávaných dávkach až do 24 mg/deň (12 mg dvakrát denne) počas 14 dní bez toxicity obmedzujúcej dávku.
V prípade predávkovania sa odporúča sledovať u pacienta akékoľvek prejavy alebo symptómy nežiaducich reakcií a okamžite začať primeranú symptomatickú liečbu. Dialýzou sa deukravacitinib zo systémovej cirkulácie podstatne neodstráni (pozri časť 5.2).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, selektívne imunosupresíva, ATC kód: L04AA56
Mechanizmus účinku
Deukravacitinib selektívne inhibuje enzým TYK2 (TYK2 patrí do skupiny JAK). Deukravacitinib sa
viaže na regulačnú doménu TYK2 čím stabilizuje interakciu inhibície medzi regulačnou a katalytickou doménou enzýmu. To vedie k alosterickej inhibícii receptorom sprostredkovanej aktivácie TYK2 a jej následných funkcií v bunkách. TYK2 sprostredkúva signalizáciu interleukínu-23 (IL-23),
interleukínu-12 (IL-12) a interferónov typu I (I interferons, IFN), čo sú prirodzene sa vyskytujúce cytokíny zapojené do zápalových a imunitných reakcií. Deukravacitinib inhibuje uvoľňovanie
prozápalových cytokínov a chemokínov.
Farmakodynamické účinky
U pacientov so psoriázou deukravacitinib znížil expresiu génov súvisiacu s psoriázou v psoriatickej
pokožke zahŕňajúc najmä utlmenia génov regulovaných dráhou IL-23 a dráhou IFN typu I. Deukravacitinib po 16 týždňoch liečby jedenkrát denne znížil množstvo IL-17A o 47-50 %, IL-19 o 72 % a β-defenzín o 81-84 %.
Klinická účinnosť abezpečnosť
Účinnosť a bezpečnosť deukravacitinibu sa hodnotili v dvoch multicentrických, randomizovaných,
dvojito zaslepených, placebom a apremilastom kontrolovaných klinických štúdiách (POETYK PSO-1 a POETYK PSO-2) s pacientmi vo veku 18 rokov a vyššom so stredne ťažkou až ťažkou ložiskovou psoriázou, ktorí boli vhodní na systémovú liečbu alebo fototerapiu. Pacienti mali postihnutých ≥ 10 % plochy povrchu tela (body surface area, BSA), skóre plochy a indexu závažnosti psoriázy (Psoriasis Area and Severity Index, PASI) ≥ 12 a statické celkové hodnotenie lekára (static Physician’s Global Assessment, sPGA) ≥ 3 (stredne závažné alebo závažné) na 5-bodovej škále celkovej závažnosti ochorenia.
V štúdiách POETYK PSO-1 a POETYK PSO-2 sa hodnotilo celkovo 1 686 pacientov, z nich 843 bolo randomizovaných na deukravacitinib v dávke 6 mg jedenkrát denne, 422 na apremilast v dávke 30 mg dvakrát denne a 421 na placebo.
V oboch štúdiách boli pacienti, ktorí dostávali placebo, prestavení na deukravacitinib v 16. týždni, a v liečbe pokračovali až do 52. týždňa. Pacienti randomizovaní na užívanie apremilastu, ktorí nedosiahli odpoveď na liečbu PASI 50 (POETYK PSO-1) alebo PASI 75 (POETYK PSO-2)
v 24. týždni, boli prestavení na deukravacitinib a pokračovali v liečbe až do 52. týždňa. Pacienti
v štúdii POETYK PSO-1, ktorí boli randomizovaní na liečbu deukravacitinibom, pokračovali v liečbe až do 52. týždňa. V štúdii POETYK PSO-2 boli pacienti liečení deukravacitinibom, ktorí dosiahli
PASI 75 v 24. týždni, opätovne randomizovaní v pomere 1: 1 buď na pokračovanie liečby
deukravacitinibom (udržiavacia liečba), alebo boli prestavení na placebo (ukončenie liečby).
V oboch štúdiách boli základné charakteristiky ochorenia v skúmanej populácii zhodné: väčšina pacientov boli muži (67 %), priemerný vek bol približne 47 rokov, pričom väčšina pacientov bola vo veku medzi 40 a 64 rokov. 10 % pacientov bolo vo veku ≥ 65 rokov. Celkový medián skóre PASI
bol 18,7 a medián BSA bol 20 %. Východiskové skóre sPGA bolo 3 (stredné) u 79,8 % pacientov a 4
(závažné) u 20,2 %. Medián skóre dermatologického indexu kvality života pacienta (Dermatology Life
Quality Index, DLQI) bol 11. Celkovo 18,4 % pacientov v štúdii malo psoriatickú artritídu v anamnéze.
V oboch štúdiách 40 % pacientov dostalo predtým fototerapiu, 42,4 % pacientov predtým nedostalo žiadnu systémovú liečbu (vrátane biologickej a/alebo nebiologickej liečby), 41 % pacientov už predtým dostalo nebiologickú systémovú liečbu a 34,8 % dostalo predtým biologickú liečbu (16,1 % inhibítory TNF, 4,9 % IL-12/23, 16,6 % IL-17 a 4,4 % IL-23).
Koprimárnymi koncovými ukazovateľmi v týchto dvoch štúdiách boli podiely pacientov, ktorí dosiahli
1) minimálne 75 % zlepšenie skóre PASI (PASI 75) oproti východiskovej hodnote a 2) jasné alebo takmer jasné (0 alebo 1) skóre sPGA v 16. týždni oproti placebu.
V štúdii POETYK PSO-1 sa v 16. týždni dosiahlo skóre PASI 75 s deukravacitinibom u 58,4 % pacientov, s apremilastom u 35,1 % pacientov a s placebom u 12,7 % pacientov. Statické celkové hodnotenie lekára (
Static Physician’s Global Assessment, sPGA) jasné alebo celkom jasné v 16. týždni sa dosiahlo u 53,6 % pacientov v skupine s deukravacitinibom, 32,1 % v skupine s apremilastom
a 7,2 % v skupine s placebom. V prípade týchto koprimárnych koncových ukazovateľov sa preukázala superiorita deukravacitinibu oproti placebu. V štúdii POETYK PSO-2 sa pozorovali konzistentné výsledky.
V tabuľke 2 sú uvedené hlavné výsledky účinnosti koprimárnych a ďalších koncových ukazovateľov.
Tabuľka 2: Hlavné výsledky účinnosti u dospelých s ložiskovou psoriázou
| POETYK PSO-1
| POETYK PSO-2
|
Koncový ukazova- teľ
| Deukravacitinib
(N = 332)
n (%)
| Apremilast
(N = 168)
n (%)
| Placebo (N = 166) n (%)
| Deukravacitinib
(N = 511)
n (%)
| Apremilast
(N = 254)
n (%)
| Placebo (N = 255) n (%)
|
sPGA 0/1
|
16. týždeň
| 178 (53,6)
| 54 (32,1)d
| 12 (7,2)a,d
| 253 (49,5)
| 86 (33,9)d
| 22 (8,6)a,d
|
24. týždeň
| 195 (58,7)
| 52 (31,0)d
| -
| 251 (49,8)b
| 75 (29,5)d
| -
|
sPGA 0
|
16. týždeň
| 58 (17,5)
| 8 (4,8)d
| 1 (0,6)d
| 80 (15,7)
| 16 (6,3)e
| 3 (1,2)d
|
PASI 75
|
16. týždeň
| 194 (58,4)
| 59 (35,1)d
| 21 (12,7)a,d
| 271 (53,0)
| 101 (39,8)e
| 24 (9,4)a,d
|
24. týždeň
| 230 (69,3)
| 64 (38,1)d
| -
| 296 (58,7)b
| 96 (37,8)d
| -
|
PASI 90
|
16. týždeň
| 118 (35,5)
| 33 (19,6)e
| 7 (4,2)d
| 138 (27,0)
| 46 (18,1)f
| 7 (2,7)d
|
24. týždeň
| 140 (42,2)
| 37 (22,0)d
| -
| 164 (32,5)b
| 50 (19,7)d
| -
|
PASI 100
|
16. týždeň
| 47 (14,2)
| 5 (3,0)d
| 1 (0,6)d
| 52 (10,2)
| 11 (4,3)f
| 3 (1,2)d
|
PGA 0/1 špecifické pre pokožku hlavyc
|
(N = 209)
|
(N = 110)
|
(N = 121)
|
(N = 305)
|
(N = 166)
|
(N = 173)
|
16. týždeň
| 147 (70,3)
| 43 (39,1)d
| 21 (17,4)d
| 182 (59,7)
| 61 (36,7)d
| 30 (17,3)d
|
Použila sa imputácia nereagujúceho na liečbu (Non-responder imputation, NRI). Pacienti, ktorí ukončili liečbu
alebo štúdiu pred koncovým ukazovateľom alebo im chýbali údaje, boli počítaní ako nereagujúci na liečbu.
a Koprimárny koncový ukazovateľ porovnávajúci deukravacitinib s placebom
b N = 504 predstavuje neuskutočnené hodnotenia v dôsledku pandémie ochorenia COVID-19
c Zahŕňa pacientov s východiskovým skóre PGA špecifickým pre pokožku hlavy ≥ 3
d p ≤ 0,0001 na porovnanie medzi deukravacitinibom a placebom alebo deukravacitinibom a apremilastom
e p < 0,001 na porovnanie medzi deukravacitinibom a apremilastom
f p < 0,01 na porovnanie medzi deukravacitinibom a apremilastom
|
Vyšetrením veku, pohlavia, rasy, telesnej hmotnosti, dĺžky ochorenia, závažnosti ochorenia na
začiatku štúdie a predchádzajúcej liečby biologickými alebo nebiologickými liečivami sa neidentifikovali rozdiely v odpovedi na liečbu deukravacitinibom medzi týmito podskupinami.
Odpoveď v priebehu časuDeukravacitinib vykazoval rýchly nástup účinnosti s maximálnou odpoveďou PASI 75 dosiahnutou do
24. týždňa (POETYK PSO-1 a PSO-2), ktorá sa udržala až do 52. týždňa (POETYK PSO-1) (pozri obrázok 1).
O
brázok 1: Odpoveď PASI 75 (NRI) do 52. týždňa podľa kontroly v rámci POETYK PSO-1

deukravacitinib, n = 332
apremilast, n = 168
placebo, n = 166

Týždeň
Udržanie a trvanie odpovedeV štúdii POETYK PSO-2 na hodnotenie udržania a trvania odpovede boli pacienti, ktorí boli pôvodne randomizovaní na liečbu deukravacitinibom a dosiahli odpoveď PASI 75 v 24. týždni, opätovne randomizovaní na pokračovanie liečby deukravacitinibom alebo na placebo. U odpovedajúcich na liečbu v 24. týždni, ktorí boli opätovne randomizovaní na placebo, bol medián času do straty odpovede PASI 75 približne 12 týždňov. Na obrázku 2 sú uvedené odpovede PASI 75 v dvoch skupinách
z 24-52. týždňa.
Obrázok 2: Odpoveď PASI 75 (NRI) po opätovnej randomizácii v 24. týždni v rámciPOETYK PSO-2deukravacitinib, n = 148
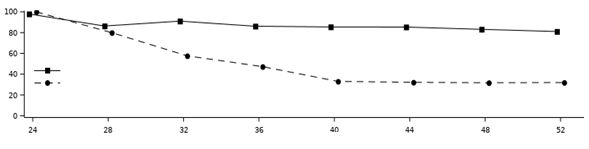
placebo, n = 150
Výsledky hlásené od pacientov
Týždeň
Výrazne väčšie zlepšenie kvality života súvisiacej so zdravím merané pomocou dermatologického indexu kvality života pacienta (
Dermatology Life Quality Index, DLQI) a symptómov (svrbenie, bolesť, pálenie, štípanie a napätá pokožka) a prejavov (suchá pokožka, praskanie, šupinatenie, vypadávanie alebo odlupovanie, sčervenenie a krvácanie) psoriázy merané pomocou denníka symptómov a prejavov psoriázy (
Psoriasis Symptoms and Signs Diary, PSSD), sa pozorovali
u pacientov liečených deukravacitinibom v porovnaní s placebom v 16. týždni a s apremilastom v 16. a 24. týždni. Zlepšenie týchto odpovedí u pacientov, ktorí dostávali kontinuálnu liečbu deukravacitinibom, sa v štúdii POETYK PSO-1 udržalo až do 52. týždňa.
Tabuľka 3: Výsledky hlásené od pacientov v štúdii POETYK PSO-1 a POETYK PSO-2
| POETYK PSO-1
| POETYK PSO-2
|
Deukravacitinib
| Apremilast
| Placebo
| Deukravacitinib
| Apremilast
| Placebo
|
DLQI
Pacienti dosahujúci
0 alebo 1 (NRI)*
|
N = 322
|
N = 161
|
N = 160
|
N = 495
|
N = 247
|
N = 246
|
16. týždeň, n (%)
| 132 (41,0)
| 46 (28,6)a
| 17 (10,6)b
| 186 (37,6)
| 57 (23,1)b
| 24 (9,8)b
|
24. týždeň, n (%)
| 155 (48,1)
| 39 (24,2)b
| -
| 205 (41,4)
| 53 (21,5)b
| -
|
|
PO
ET
YK PSO-1
|
PO
ET
YK PSO-2
|
Deukravacitinib
|
Apremilast
|
Placebo
|
Deukravacitinib
|
Apremilast
|
Placebo
|
Skóre symptómu PSSD Zmena oproti východiskovej hodnote (mBOCF)**
|
N = 306
|
N = 158
|
N = 151
|
N = 466
|
N = 233
|
N = 239
|
16. týždeň, priemer
(SE)
|
-26,7 (1,8)
|
-17,8 (2,2)b
|
-3,6 (2,1)b
|
-28,3 (1,1)
|
-21,1 (1,4)b
|
-4,7 (1,4)b
|
24. týždeň, priemer
(SE)
|
-31,9 (2,0)
|
-20,7 (2,4)b
|
-
|
-29,1 (1,1)
|
-21,4 (1,5)b
|
-
|
Skóre prejavu PSSD Zmena oproti východiskovej hodnote
(mBOCF)*
|
N = 306
|
N = 158
|
N = 151
|
N = 466
|
N = 233
|
N = 239
|
16. týždeň, priemer'
(SE)
|
-28,9 (1,8)
|
-20,0 (2,2)b
|
-5,3 (2,1)a
|
-31,9 (1)
|
-23,8 (1,4)b
|
-7,1 (1,4)b
|
24. týždeň, priemer
(SE)
|
-33,8 (2,0)
|
-22,5 (2,4)b
|
-
|
-32,4 (1,1)
|
-24,2 (1,5)b
|
-
|
* Pacienti s východiskovým skóre ≥ 2
** Upravená priemerná zmena; mBOCF – upravené základné prenesené pozorovanie; štandardná chyba (SE)
a p < 0,01 na porovnanie medzi deukravacitinibom a placebom alebo deukravacitinibom a apremilastom
b p < 0,0001 na porovnanie medzi deukravacitinibom a placebom alebo deukravacitinibom a apremilastom
|
Populácia
s
t
arších
ľudí
Z 1 519 pacientov s ložiskovou psoriázou liečených deukravacitinibom v klinických štúdiách bolo
152 pacientov vo veku 65 rokov alebo starších vrátane 21 pacientov vo veku 75 rokov alebo starších
(pozri časť 4.2). V expozícii, bezpečnosti alebo účinnosti medzi staršími a mladšími pacientmi, ktorí dostávali deukravacitinib, sa nepozorovali žiadne celkové rozdiely.
Pediatrická populáciaEurópska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií so SOTYKTUOM
v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe psoriázy (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiDeukravacitinib vykazoval takmer úplnú absorpciu po perorálnom podaní, zvýšenie expozície závislé
od dávky a žiadnu evidentnú farmakokinetiku závislú od času.
AbsorpciaPo perorálnom podaní tabliet vykazoval deukravacitinib rýchlu a takmer úplnú absorpciu. Medián Tmax
sa u zdravých dobrovoľníkov pohyboval od 2 do 3 hodín a absolútna perorálna biologická dostupnosť
bola 99 %. Mierna akumulácia (< 1,4-násobok v rovnovážnom stave) sa pozorovala po dávkovaní jedenkrát denne.
JedloDeukravacitinib sa môže podávať bez ohľadu na jedlo alebo modulátory pH žalúdka (blokátory
H2 receptorov a inhibítory protónovej pumpy). Súbežné podávanie jedla alebo modulátorov
pH žalúdka neovplyvnilo celkovú expozíciu (AUC[INF]) deukravacitinibu.
DistribúciaDistribučný objem v rovnovážnom stave (Vss) je 140 l, čo je viac ako celkový objem vody v tele
[42 l], čo naznačuje extravaskulárnu distribúciu. Deukravacitinib sa z 81,6 % viaže na ľudské plazmatické proteíny, predovšetkým na ľudský sérový albumín.
Deukravacitinib sa distribuuje podobne medzi zložkami plazmy a červených krviniek s pomerom
koncentrácie v krvi k plazme 1,26.
Biotransformácia
U ľudí sa deukravacitinib metabolizuje štyrmi primárnymi biotransformačnými cestami, ktoré
zahŕňajú N-demetyláciu na triazolovej časti cytochrómom P-450 (CYP) 1A2 za vzniku hlavného metabolitu BMT-153261, hydrolýzu cyklopropylkarboxamidu karboxylesterázou 2 (CES2) za vzniku
hlavného metabolitu BMT-158170, N-glukuronidáciu uridín-glukuronyltransferázou (UGT) za vzniku
BMT-334616 a monooxidáciu prostredníctvom CYP 2B6/2D6 na deuterovanej metylovej skupine za vzniku M11.
V rovnovážnom stave je deukravacitinib hlavnou cirkulujúcou zlúčeninou, ktorá tvorí 49 % meraných zložiek súvisiacich so zložkou. Identifikovali sa dva hlavné cirkulujúce metabolity (BMT-153261
a BMT-158170) pričom oba z nich majú polčasy porovnateľné s pôvodným deukravacitinibom.
BMT-153261 má porovnateľnú účinnosť s materskou zložkou a BMT-158170 nie je farmakologicky aktívny. Cirkulujúca expozícia BMT-153261 je oveľa nižšia ako u materskej zložky, a preto sa prevládajúca farmakologická aktivita pripisuje materskej zložke deukravacitinibu.
Navyše neboli identifikované žiadne metabolity jedinečné u ľudí ani žiadne metabolity s dlhou životnosťou v krvnom obehu.
Eliminácia
Deukravacitinib sa eliminuje viacerými cestami vrátane metabolizmu fázy I a II, s priamou
elimináciou obličkami a stolicou. Žiadny samostatný enzým už neprispel k celkovému klírensu viac ako 26 %. Deukravacitinib sa vo veľkej miere metabolizuje, pričom 59 % perorálne podanej dávky
[14C]-deukravacitinibu sa vylúči vo forme metabolitov močom (37 % dávky) a stolicou (22 % dávky). Nezmenený deukravacitinib v moči predstavoval 13 % a v stolici 26 % dávky.
Terminálny polčas eliminácie 6 mg deukravacitinibu u zdravých dospelých ľudí je 10 hodín,
s celkovým klírensom 15,3 l/h (CV 27 %). Deukravacitinib je substrátom efluxných transportérov,
P-glykoproteínu (P-gp) a proteínu rezistencie na rakovinu prsníka (breast cancer resistance protein, BCRP) a transportéra vychytávania OCT1. V dôsledku vysokej pasívnej permeability, vysokej perorálnej biologickej dostupnosti a nízkej afinity k týmto transportérom je príspevok týchto transportérov k farmakokinetike deukravacitinibu minimálny.
Deukravacitinib nie je substrátom transportérov OATP, NTCP, OAT1, OAT3, OCT2, MATE1 alebo
MATE2K.
Linearita/nelinearita
Farmakokinetika jednorazových dávok deukravacitinibu podávaných vo forme tabliet bola lineárna
v rozmedzí dávok 3 mg až 36 mg.
Interakcie
Účinok deukravacitinibu na iné lieky
Štúdie in vitro nepreukázali žiadne dôkazy, že deukravacitinib a jeho hlavné cirkulujúce metabolity pri klinicky relevantných expozíciách inhibujú hlavné CYP (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4), UGT
(1A1, 1A4, 1A6, 1A9, 2B7), CES2 a liekové transportéry (P-gp, BCRP, OATP1B1, OATP1B3, BSEP, MRP2, OAT1, OAT3, OCT1, OCT2, MATE1 a MATE2K). Navyše, deukravacitinib
neindukuje ani CYP 1A2, 2B6 a 3A4 (pozri časť 4.5).
O
s
obitné skupiny pacientov
Starší ľudia
Na základe populačnej farmakokinetickej analýzy bola priemerná expozícia deukravacitinibu v rovnovážnom stave (Cavg,ss) vyššia, 31 % u pacientov vo veku 65-74 rokov [n = 87 z 1 387 (6,3 %)] a 53 % u pacientov vo veku 75-84 rokov [n = 13 z 1 387 (0,94 %)]. Expozície u pacientov vo veku
≥ 85 rokov nie sú k dispozícii.
Pacienti s poruchou funkcie obličiek
Porucha funkcie obličiek nemá klinicky významný vplyv na expozície deukravacitinibu (pozri časť 4.2) na základe špecializovanej štúdie, kde sa odhadovaná rýchlosť glomerulárnej filtrácie
(estimated glomerular filtration rate, eGFR) stanovila pomocou rovnice úpravy stravy pri ochorení
obličiek (modification of diet in renal disease, MDRD). V porovnaní so skupinou s normálnou
funkciou obličiek sa hodnota Cmax deukravacitinibu zmenila až o 15 % a AUC[INF] sa zvýšila až
o 48 % v skupinách s poruchou funkcie obličiek (mierne (eGFR: ≥ 60 až < 90 ml/min.), stredne ťažké
(eGFR: ≥ 30 až < 60 ml/min.), závažné (eGFR: < 30 ml/min.) a ESRD (eGFR: < 15 ml/min.)).
V porovnaní so skupinou s normálnou funkciou obličiek sa hodnota Cmax BMT-153261 zvýšila až
o 34 % a AUC[INF] sa zvýšila až o 84 % v skupinách s poruchou funkcie obličiek.
Dialýzou sa deukravacitinib zo systémovej cirkulácie podstatne neodstráni (dialýzou sa odstráni 5,4 %
dávky).
Pacienti s poruchou funkcie pečene
Mierna (Childova-Pughova trieda A) a stredne ťažká (Childova-Pughova trieda B) porucha funkcie pečene nemá klinicky významný vplyv na expozíciu deukravacitinibu (pozri časť 4.2). V porovnaní so skupinou s normálnou funkciou pečene sa celková hodnota Cmax deukravacitinibu v skupine s miernou a stredne ťažkou poruchou funkcie pečene zvýšila o 10 % a AUC[INF] o 40 %, zatiaľ čo Cmax neviazaného deukravacitinibu sa zvýšila o 26 % a AUC(INF) o 60 %. U dospelých so závažnou poruchou funkcie pečene (Childova-Pughova trieda C) bola celková hodnota Cmax deukravacitinibu porovnateľná a celková AUC o 43 % vyššia v porovnaní so zodpovedajúcimi zdravými dospelými.
U týchto dospelých sa neviazaná Cmax zvýšila o 62 % a AUC(INF) o 131 %. Deukravacitinib sa neodporúča používať u pacientov s ťažkou poruchou funkcie pečene (pozri časť 4.2).
V porovnaní s osobami s normálnou funkciou pečene sa u osôb s miernou poruchou funkcie pečene hodnota AUC(0-T) BMT-153261 znížila o 19 % a Cmax BMT-153261 o 25 %. U osôb so stredne ťažkou poruchou funkcie pečene sa hodnota AUC(0-T) BMT-153261 znížila o 53 % a Cmax BMT-153261
o 59 % a u osôb s ťažkou poruchou funkcie pečene sa hodnota AUC(0-T) BMT-153261 znížila o 76 %
a Cmax BMT-153261 o 79 % v porovnaní s osobami s normálnou funkciou pečene.
Pohlavie
Na základe farmakokinetického modelovania a simulácie populácie sa u žien očakáva asi o 30 %
vyššia priemerná expozícia deukravacitinibu v rovnovážnom stave (Cmax,ss a Cavg,ss) v porovnaní s mužmi.
Telesná hmotnosť
Na základe farmakokinetického modelovania a simulácie populácie sa u pacientov s nižšou telesnou hmotnosťou (< 60 kg) očakáva vyššia geometrická priemerná expozícia deukravacitinibu
v rovnovážnom stave o 37,4 % (Cmaxss) a 24,8 % (Cavgss). U pacientov s vyššou telesnou hmotnosťou (> 90 kg) sa očakáva nižšia geometrická priemerná expozícia deukravacitinibu v rovnovážnom stave o 24,8 % (Cmaxss) a 19,6 % (Cavgss) (v porovnaní s pacientmi s telesnou hmotnosťou 60-90 kg).
Inherentné faktory
Rasa a etnická príslušnosť nemali klinicky významný vplyv na expozíciu deukravacitinibu.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, karcinogénneho potenciálu, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Toxicita po opakovanompodávanídávok
V štúdii chronickej toxicity na potkanoch sa pozoroval pokles počtu lymfocytov, celularity kostnej
drene a celularity lymfatických uzlín v tkanivách imunitného systému pri expozícii (Area Under
Curve, AUC) na najnižšej hladine dávky, pri ktorej sa pozoroval účinok (lowest observed effect level, LOEL) približne 9-násobku odporúčanej dávky u ľudí (recommended human dose, RHD). Tieto účinky nesúviseli s klinickými prejavmi imunosupresie (napr. infekcie). Zníženie počtu krvných doštičiek a parametrov masy červených krviniek (red blood cell, RBC) sa pozorovalo pri expozícii (AUC) na úrovni LOEL približne 42-násobku RHD. V štúdii chronickej toxicity na opiciach sa pozorovali klinické a mikroskopické kožné zmeny a znížené parametre masy červených krviniek pri expozícii (AUC) na úrovni LOEL približne 7-násobku RHD.
Vývojová a reprodukčnátoxicita
Deukravacitinib nemal žiadne účinky na fertilitu alebo skorý embryonálny vývoj u samcov a samíc
potkanov pri expozíciách (AUC) do približne 247- a 171-násobku RHD.
Deukravacitinib nebol ani embryoletálny, ani teratogénny pri expozíciách (AUC) matiek až do približne 266-násobku RHD u potkanov alebo 91-/20-násobku (celkový/voľný) RHD u králikov.
V štúdii prenatálneho a postnatálneho vývoja u potkanov sa počas obdobia pred odstavením pri expozícii (AUC) matky približne 110-násobku RHD zaznamenali prechodne nižšie telesné hmotnosti mláďat. Počas obdobia po odstavení sa tento účinok sa úplne zregeneroval.
Po podaní rádioaktívne označeného deukravacitinibu laktujúcim potkanom sa deukravacitinib a/alebo jeho metabolity nachádzali v mlieku s pomermi koncentrácie v mlieku a plazme 2,7 až 30,9.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
Acetát-sukcinát hypromelózy
Bezvodá laktóza Mikrokryštalická celulóza Sodná soľ kroskarmelózy
Koloidný hydratovaný oxid kremičitý
Stearát horečnatý
Filmový obal
Polyvinylalkohol
Oxid titaničitý (E171) Makrogol
Mastenec
Červený oxid železitý (E172) Žltý oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Priehľadný blister z polyvinylchloridu/polychlórtrifluóretylénu (PVC/PCTFE) s pretlačovacou hliníkovou fóliou obsahujúci 7 alebo 14 filmom obalených tabliet v blistri (kalendárové alebo nekalendárové blistre).
Veľkosti balenia: 7, 14, 28 a 84 filmom obalených tabliet. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Bristol-Myers Squibb Pharma EEIG Plaza 254
Blanchardstown Corporate Park 2
Dublin 15, D15 T867
Írsko
8. REGISTRAČNÉ ČÍSLA
EU/1/23/1718/001
EU/1/23/1718/002
EU/1/23/1718/003
EU/1/23/1718/004
EU/1/23/1718/005
EU/1/23/1718/006
EU/1/23/1718/007
EU/1/23/1718/008
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.