
Starší pacienti (≥ 60 rokov)
U starších pacientov budú zvyčajne potrebné nižšie dávky somapacitanu. Ďalšie informácie sú uvedené v časti 5.2.
Pohlavie
Muži vykazujú v priebehu času zvyšujúcu sa citlivosť na IGF-I. To znamená, že existuje riziko predávkovania u mužov. Ženy, najmä tie, ktoré užívajú perorálny estrogén, môžu vyžadovať vyššie dávky a dlhšie obdobie titrácie ako muži, pozri časti 5.1 a 5.2. U žien užívajúcich perorálny estrogén sa má zvážiť zmena spôsobu podávania estrogénu (napr. transdermálne, vaginálne), pozri časť 4.4.
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek nie je potrebná úprava úvodnej dávky. Pacienti s poruchou funkcie obličiek budú možno potrebovať nižšie dávky somapacitanu, ale keďže dávka somapacitanu
sa upravuje individuálne podľa potrieb každého pacienta, ďalšia úprava dávky nie je potrebná, pozri
časť 5.2.
Porucha funkcie pečene
U pacientov s poruchou funkcie pečene nie je potrebná úprava úvodnej dávky. Pacienti so stredne závažnou poruchou funkcie pečene budú možno potrebovať vyššie dávky somapacitanu, ale keďže dávka somapacitanu sa upravuje individuálne podľa potrieb každého pacienta, ďalšia úprava dávky nie je potrebná. K dispozícii nie sú žiadne informácie týkajúce sa použitia somapacitanu u pacientov so závažnou poruchou funkcie pečene. Pri liečbe týchto pacientov somapacitanom je potrebná opatrnosť, pozri časť 5.2.
Pediatrická populácia
Bezpečnosť a účinnosť somapacitanu u detí a dospievajúcich mladších ako 18 rokov neboli doteraz stanovené. Nie sú k dispozícii žiadne údaje.
Spôsob podávania
Somapacitan sa má podávať jedenkrát týždenne kedykoľvek počas dňa.
Somapacitan sa podáva subkutánne injekciou do brucha alebo stehna. Miesto podávania injekcie sa môže meniť bez úpravy dávky. Miesto podávania injekcie sa má obmieňať každý týždeň.
Naplnené pero poskytuje dávky od 0,05 mg do 4 mg s pridávaním po 0,05 mg (0,075 ml). Pokyny pre liek pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Somapacitan sa nesmie používať, ak existuje akýkoľvek dôkaz o nádorovej aktivite. Intrakraniálne nádory musia byť inaktívne a pred začatím liečby somapacitanom musí byť dokončená protinádorová liečba. Ak sú k dispozícii dôkazy o raste nádoru, liečbu je potrebné prerušiť, pozri časť 4.4.
Pacienti s akútnym kritickým ochorením s komplikáciami po operácii na otvorenom srdci, abdominálnej operácii, polytraume spôsobenej nehodou, akútnom zlyhaní dýchania alebo podobných stavoch nesmú byť liečení somapacitanom (informácie o pacientoch podstupujúcich substitučnú liečbu, pozri časť 4.4).
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Adrenokortikálna insuficiencia
Zavedenie liečby rastovým hormónom môže viesť k inhibícii 11βHSD-1 a zníženým koncentráciám
kortizolu v sére. U pacientov liečených rastovým hormónom môže byť odhalený v minulosti nediagnostikovaný centrálny (sekundárny) hypoadrenalizmus a možno bude potrebná substitučná liečba glukokortikoidmi. Okrem toho, pacienti, ktorí dostávajú substitučnú liečbu glukokortikoidmi na hypoadrenalizmus, ktorý im bol v minulosti diagnostikovaný, budú možno potrebovať po začatí liečby rastovým hormónom zvýšenie udržiavacej dávky alebo stresových dávok. Pacientov so známym hypoadrenalizmom je potrebné monitorovať kvôli zníženej hladine kortizolu v sére a/alebo kvôli potrebe zvýšenia dávky glukokortikoidov, pozri časť 4.5.
Porucha metabolizmu glukózy
Liečba rastovým hormónom môže znížiť citlivosť na inzulín, najmä pri vyšších dávkach u citlivých
pacientov a následne sa môže vyskytnúť hyperglykémia u osôb s nedostatočnou schopnosťou
vylučovania inzulínu. Počas liečby rastovým hormónom sa preto môže odhaliť porucha tolerancie glukózy, ktorá nebola v minulosti diagnostikovaná, a manifestný diabetes mellitus. Preto je potrebné pravidelne sledovať hladinu glukózy u všetkých pacientov liečených rastovým hormónom, najmä
u pacientov s rizikovými faktormi pre diabetes mellitus, ako je obezita alebo diabetes mellitus
v rodinnej anamnéze. Pacientov s už existujúcim ochorením diabetes mellitus 1. alebo 2. typu alebo s poruchou tolerancie glukózy je potrebné pozorne monitorovať počas liečby rastovým hormónom.
Keď sa u týchto pacientov zavedie liečba rastovým hormónom, dávky antihyperglykemických liekov
bude možno potrebné upraviť.
Novotvary
K dispozícii nie sú dôkazy o zvýšenom riziku nových primárnych druhov rakoviny u dospelých
liečených rastovým hormónom.
U pacientov s úplnou remisiou malígnych ochorení alebo u pacientov liečených na benígne nádory liečba rastovým hormónom nesúvisela so zvýšenou mierou relapsu.
Pacientov, ktorí dosiahli úplnú remisiu malígnych ochorení alebo boli liečení na benígne nádory, je
potrebné po začatí liečby rastovým hormónom pozorne sledovať kvôli relapsu. Liečbu rastovým hormónom je potrebné prerušiť v prípade vzniku alebo opätovného výskytu malígneho alebo benígneho nádoru.
Celkovo bol pozorovaný mierny nárast sekundárnych novotvarov u pacientov, ktorí v detstve prekonali rakovinu, a ktorí boli liečení rastovým hormónom, pričom najčastejšími boli intrakraniálne
nádory. Dominantným rizikovým faktorom senkundárnych novotvarov sa zdá byť predchádzajúce vystavenie sa pôsobeniu radiácie.
Benígna intrakraniálna hypertenzia
V prípade závažnej alebo rekurentnej bolesti hlavy, vizuálnych symptómov, nauzey a/alebo vracania
sa odporúča fundoskopia na zistenie papiloedému. Ak sa papiloedém potvrdí, je potrebné uvažovať o diagnóze benígnej intrakraniálnej hypertenzie a v prípade potreby sa má liečba rastovým hormónom
prerušiť. V súčasnosti nie sú k dispozícii dostatočné dôkazy na odporučenie klinického rozhodnutia v prípade pacientov s vyliečenou intrakraniálnou hypertenziou. Ak sa znova začne liečba rastovým
hormónom, je potrebné pozorné monitorovanie symptómov intrakraniálnej hypertenzie.
Funkcia štítnej žľazy
Rastový hormón zvyšuje extratyreoidálnu konverziu T4 na T3, a preto môže odhaliť incipientný
hypotyreoidizmus. Keďže hypotyreóza interferuje s odpoveďou na liečbu rastovým hormónom,
u pacientov je potrebné pravidelne vyšetrovať funkciu štítnej žľazy a mali by dostávať substitučnú liečbu hormónom štítnej žľazy, keď je to indikované, pozri časti 4.5 a 4.8.
Použitie s perorálnym estrogénom
Perorálny estrogén ovplyvňuje odpoveď IGF-I na rastový hormón vrátane somapacitanu.
Ženy používajúce akúkoľvek formu perorálneho estrogénu (hormonálna liečba alebo antikoncepcia) majú zvážiť zmenu spôsobu podávania estrogénu (napr. transdermálne, vaginálne hormonálne lieky) alebo užívať inú formu antikoncepcie. Ak žena používajúca perorálny estrogén začína s liečbou somapacitanom, môžu byť potrebné vyššie úvodné dávky a dlhší čas titrácie (pozri časť 4.2).
Ak žena používajúca somapacitan začína perorálnu liečbu estrogénom, dávka somapacitanu sa môžno bude musieť zvýšiť, aby sa zachovala sérová hladina IGF-I v normálnom rozsahu úmerne k veku. Ak
však žena používajúca somapacitan preruší perorálnu liečbu estrogénom, dávka somapacitanu sa
možno bude musieť znížiť, aby sa zabránilo nadbytku somapacitanu a/alebo nežiaducim účinkom, pozri časti 4.2 a 4.5.
Lipohypertrofia
Keď sa somapacitan dlhodobo podáva do rovnakého miesta, môže dôjsť k lipohypertrofii. Na zníženie
tohto rizika treba obmieňať miesto podávania injekcie, pozri časti 4.2 a 4.8.
Protilátky
Hoci sa po liečbe somapacitanom nepozorovali žiadne protilátky, mohli by sa očakávať, ako sa
pozorovalo v prípade iných terapeutických proteínov. Testovanie na prítomnosť protilátok proti somapacitanu je potrebné vykonať u pacientov, ktorí nereagujú na liečbu.
Akútne kritické ochorenie
Účinok rastového hormónu na uzdravenie sa skúmal v dvoch placebom kontrolovaných skúšaniach
zahŕňajúcich 522 kriticky chorých dospelých pacientov s komplikáciami po operácii na otvorenom srdci, abdominálnej operácii, polytraume v dôsledku nehody alebo akútnom zlyhaní dýchania. Mortalita bola vyššia u pacientov liečených rastovým hormónom podávaným denne v dávke 5,3 alebo
8 mg v porovnaní s pacientmi používajúcimi placebo, 42 % oproti 19 %. Na základe týchto informácií nemajú byť títo pacienti liečení somapacitanom. Keďže nie sú k dispozícii žiadne informácie o
bezpečnosti substitučnej liečby rastovým hormónom u akútne kriticky chorých pacientov, prínosy pokračovania v liečbe v tejto situácii treba zvážiť oproti potenciálnym rizikám.
Deficiencia rastového hormónu u dospelých je celoživotné ochorenie a má sa tak liečiť, avšak skúsenosti s pacientmi staršími ako 60 rokov a pacientmi, ktorí sa liečia dlhšie ako päť rokov na deficienciu rastového hormónu v dospelosti, sú stále obmedzené.
Pediatrická populácia
Somapacitan sa nemá podávať pacientom mladším ako 18 rokov, pretože bezpečnosť a účinnosť
somapacitanu u detí a dospievajúcich mladších ako 18 rokov neboli doteraz stanovené.
Sodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v dávke t. j. v podstate zanedbateľné množstvo
sodíka.
4.5 Liekové a iné interakcie
Liečivá metabolizované cytochrómom P450
Údaje z interakčnej štúdie vykonanej u dospelých s deficienciou rastového hormónu naznačujú, že
podávanie rastového hormónu môže zvýšiť klírens liečiv, o ktorých je známe, že sú metabolizované izoenzýmami cytochrómu P450. Klírens liečiv metabolizovaných cytochrómom P450 (napr. pohlavné steroidy, kortikosteroidy, antikonvulzíva a cyklosporín) môže byť mimoriadne zvýšený, čo vedie k nižším plazmatickým koncentráciám týchto liečiv. Klinický význam tohto zistenia nie je známy.
Glukokortikoidy
Rastový hormón znižuje konverziu kortizónu na kortizol a môže odhaliť centrálny hypoadrenalizmus,
ktorý nebol v minulosti zistený alebo môže spôsobiť neúčinnosť nízkych dávok glukokortikoidov v rámci substitučnej liečby, pozri časť 4.4.
Perorálne estrogény
U žien liečených perorálnymi estrogénmi bude možno potrebná vyššia dávka somapacitanu na
dosiahnutie cieľa liečby, pozri časti 4.2 a 4.4.
Antihyperglykemické lieky
Antihyperglykemická liečba vrátane inzulínu môže vyžadovať úpravu dávky v prípade súbežného
podávania somapacitanu, pretože somapacitan môže znížiť citlivosť na inzulín, pozri časti 4.4 a 4.8.
I
né
Metabolické účinky somapacitanu môžu byť ovplyvnené aj súbežnou liečbou inými hormónmi, napr.
testosterónom a hormónmi štítnej žľazy, pozri časť 4.4.
4.6 Fertilita, gravidita a laktáciaGraviditaNie sú k dispozícii žiadne údaje o použití somapacitanu u tehotných žien.
Štúdie na zvieratách preukázali reprodukčnú toxicitu, pozri časť 5.3.
Sogroya sa neodporúča počas gravidity a u žien vo fertilnom veku neužívajúcich antikoncepciu.
DojčenieNie je známe, či sa somapacitan/metabolity vylučuje/vylučujú do materského mlieka.
Dostupné farmakodynamické/toxikologické údaje u zvierat preukázali vylučovanie somapacitanu
do mlieka, pozri časť 5.3.
Riziko pre dojčených novorodencov/dojčatá sa nedá vylúčiť.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu liekom Sogroya sa má urobiť
po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
FertilitaNie sú žiadne klinické skúsenosti s používaním somapacitanu a jeho potenciálnym účinkom na
fertilitu.
Nepozorovali sa žiadne nežiaduce účinky na fertilitu samcov a samíc potkanov, pozri časť 5.3.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeSogroya nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrn bezpečnostnéhoprofiluČasto hlásené a závažné nežiaduce reakcie po liečbe somapacitanom sú bolesť hlavy (12 %), periférny
edém (4 %) a adrenokortikálna insuficiencia (3 %).
Tabuľkový zoznamnežiaducichreakciíNežiaduce reakcie na liek uvedené ďalej sú založené na súhrne údajov bezpečnosti z troch
dokončených skúšaní v tretej fáze u pacientov s AGHD.
Nežiaduce reakcie sú uvedené podľa triedy orgánových systémov na základe databázy MedDRA a kategórie frekvencie a sú definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000).
Trieda orgánových systémov podľa databázy MedDRA
| Veľmi časté
| Časté
| Menej časté
|
|
|
Tabuľka 2: Nežiaduce reakcie
Poruchy endokrinného systému
|
|
Adrenokortikálna insuficiencia Hypotyreoidizmus
|
|
Poruchy
metabolizmu a výživy
|
|
Hyperglykémia*
|
|
Poruchy nervového
systému
|
Bolesť hlavy
|
Parestézie
|
Syndróm karpálneho
tunela
|
Poruchy kože
a podkožného tkaniva
|
|
Vyrážka*
Urtikária*
|
Lipohypertrofia*
Pruritus*
|
Poruchy kostrovej
a svalovej
sústavy
a spojivového
tkaniva
|
|
Artralgia
Myalgia
Stuhnutosť svalov*
|
Stuhnutosť kĺbov
|
Celkové poruchy
a reakcie
v mieste podania
|
|
Periférny edém
Únava
Asténia
Reakcie v mieste
podania injekcie*
|
|
*Vo všeobecnosti boli tieto nežiaduce reakcie zvyčajne nezávažné, mierne alebo stredne závažné a
prechodné
Opis vybraných nežiaducichreakciíPeriférny edémČasto sa pozoroval periférny edém (4 %). Pacienti s deficienciou rastového hormónu sa vyznačujú deficitom extracelulárneho objemu. Keď sa začne liečba liekmi obsahujúcimi rastový hormón, tento deficit sa napraví. Môže sa vyskytnúť zadržiavanie tekutín s periférnym edémom. Symptómy sú zvyčajne prechodné, závislé od dávky a môžu vyžadovať dočasné zníženie dávky.
Adrenokortikálna insuficienciaČasto sa pozorovala adrenokortikálna insuficiencia (3 %) (pozri časť 4.4).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieLiečba rastovým hormónom môže viesť k akútnemu predávkovaniu, najskôr s nízkou hladinou glukózy v krvi a následne s vysokou hladinou glukózy v krvi. Táto znížená hladina glukózy sa zistila biochemicky, ale bez klinických prejavov hypoglykémie.
Dlhodobé predávkovanie môže mať za následok prejavy a príznaky zodpovedajúce známym
účinkom nadbytku rastového hormónu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: hormóny hypofýzy a hypotalamu a analógy, somatropín a agonisty somatropínu, ATC kód: H01AC07.
Mechanizmus účinkuSomapacitan je derivát rekombinantného ľudského rastového hormónu s dlhodobým účinkom. Skladá
sa zo 191 aminokyselín podobných endogénnemu ľudskému rastovému hormónu s jednou substitúciou v aminokyselinovej kostre (L101C), na ktorú je naviazané väzbové miesto albumínu. Väzbové miesto
albumínu (bočný reťazec) sa skladá z mastnej kyseliny a hydrofilného nadstavca (spacera)
naviazaného na polohu 101 v proteíne.
Mechanizmus účinku somapacitanu je buď priamo prostredníctvom GH-receptora a/alebo nepriamo prostredníctvom IGF-I vytváraného v tkanivách celého tela, ale prevažne v pečeni.
Keď sa deficiencia rastového hormónu lieči somapacitanom, dosiahne sa normalizácia stavby tela (t. j. znížená hmotnosť telesného tuku, zvýšená svalová hmota) a metabolizmu.
Farmakodynamické účinkyIGF-
IIGF-I je všeobecne akceptovaný biomarker účinnosti pri AGHD.
Po podaní somapacitanu u pacientov s AGHD je indukovaná odpoveď IGF-I v závislosti od dávky. Rovnovážny stav IGF-I sa dosiahne po 1 − 2 týždenných dávkach.

Hladiny IGF-I počas týždňa kolíšu. Maximálna odpoveď IGF-I sa dosiahne po 2 až 4 dňoch. V
porovnaní s dennou liečbou pomocou GH profil IGF-I je odlišný, pozri obrázok 1.
somapacitan
somatropín
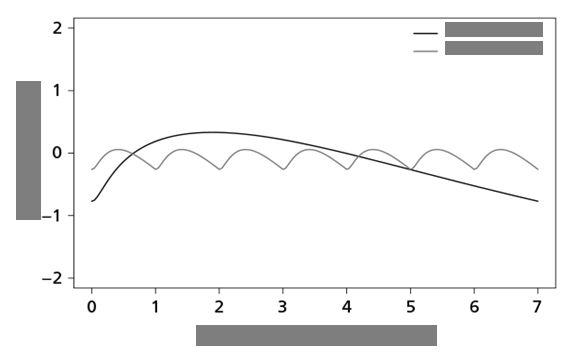
Dni v ustálenom stave
Obrázok 1: Profily IGF-
I odvodené z modelu počas rovnovážneho stavu somapacitanua somatropínuKlinickáúčinnosťabezpečnosť V 34-týždňovom, placebom kontrolovanom (dvojito zaslepenom) a aktívne kontrolovanom (otvorenom) skúšaní bolo 301 predtým neliečených dospelých pacientov s GHD randomizovaných (v pomere 2:1:2) a exponovaných jedenkrát týždenne somapacitanom alebo placebom alebo somatropínom denne počas obdobia liečby trvajúceho 34 týždňov (hlavná fáza skúšania). Populácia pacientov mala priemerný vek 45,1 roka (rozsah 23 − 77 rokov; 41 pacientov malo 65 rokov alebo viac), 51,7 % boli ženy a 69,7 % malo GHD s nástupom v dospelosti.
Celkovo 272 pacientov s AGHD, ktorí dokončili 34-týždňovú hlavnú fázu, pokračovalo v 53-
týždňovom predĺženom otvorenom období. Účastníci, ktorí dostávali placebo, prešli na somapacitan a pacienti, ktorí dostávali somatropín, boli opätovne randomizovaní (v pomere 1:1) buď na somapacitan
alebo somatropín.
Ďalej sú uvedené pozorované klinické účinky pre hlavné ukazovatele vo fáze hlavnej liečby (tabuľka
3) a vo fáze predĺženej liečby (tabuľka 4).
Tabuľka 3: Výsledky v 34. týždni
zmena v 34. týždni oproti východiskovému stavua
|
somapacitan
|
somatropín
|
placebo
| rozdiel
somapacitan- placebo
[95 % IS]
p-hodnota
| rozdiel somapacitan- somatropín
[95 % IS]
p-hodnota
|
počet účastníkov (n)
|
120
|
119
|
61
|
|
|
trunkálny tuk % (primárny cieľový ukazovateľ)
|
-1,06
|
-2,23
|
0,47
|
-1,53
[-2,68; -0,38]
0,0090b
| 1,17
[0,23; 2,11]
|
viscerálne adipózne tkanivo (cm2)
|
-10
|
-9
|
3
|
-14
[-21; -7]
|
-1
[-7; 4]
|
hmotnosť apendikulárneho kostrového svalstva (g)
|
558
|
462
|
-121
| 679
[340; 1 019]
| 96
[-182; 374]
|
svalová hmota (g)
|
1 394
|
1 345
|
250
| 1 144
[459; 1 829]
| 49
[-513; 610]
|
hladiny IGF-I SDS
|
2,40
|
2,37
|
-0.01
|
2,40
[2,09; 2,72]
|
0,02
[-0,23; 0,28]
|
Skratky: n = počet účastníkov v úplnom analyzovanom súbore, IS = interval spoľahlivosti, DM =
diabetesmellitus. SDS IGF-I: Skóre štandardnej odchýlky pre rastový faktor podobný inzulínu-I.
a Parametre stavby tela sú založené na vyšetrení pomocou duálnej RTG absorpciometrie (DXA).
b Primárnu analýzu predstavovalo porovnanie zmien v % trunkálneho tuku pri použití somapacitanu a placeba v porovnaní s východiskovými hodnotami. Zmeny v percentuálnom obsahu trunkálneho tuku z východiskovej
hodnoty do merania v 34. týždni boli analyzované s použitím analýzy kovariančného modelu s liečbou, typom nástupu deficitu rastového hormónu (GHD), pohlavím, regiónom,
diabetom mellitus (DM) a pohlavia podľa oblasti interakcií
diabetu mellitus ako faktorov a východiskovej hodnoty ako kovariátu so začlenením viacnásobnej imputačnej techniky, kde chýbajúce hodnoty v týždni 34 boli imputované na základe údajov zo skupiny s placebom.
Post-hoc analýza podskupiny zmien oproti východiskovej hodnote percenta trunkálneho tuku (%)
porovnané s placebom v 34. týždni ukázala odhadovaný rozdiel (somapacitan-placebo) -2,49% [-4,19;
-0,79] u mužov, -0,80% [-2,99; 1,39] u žien neliečených perorálnym estrogénom, -1,44% [-3,97; 1,09]
u žien na perorálnej liečbe estrogénom.
Tabuľka 4: Výsledky v 87. týždni
zmena
v 87. týždni oproti východiskovému stavu
a
|
s
o
m
a
pacitan/
s
o
m
a
pacitan
|
s
o
m
at
ropín/
s
o
m
at
ropín
|
placebo/
s
o
m
a
pacitan
|
s
o
m
at
ropín/
s
o
m
a
pacitan
|
rozdiel somapacitan/ somapacitan oproti somatropín/somatropín
[9
5 % IS]
p-hodnota
|
počet účastníkov
(n)
|
114
|
52
|
54
|
51
|
|
trunkálny tuk %
|
-1,51
|
-2,67
|
-2,28
|
-1,35
|
1,15
[-0,10; 2,40]
|
viscerálne adipózne tkanivo (cm2)
|
-6,64
|
-6,85
|
-10,21
|
-8,77
|
0,22
[-10; 10]
|
hmotnosť apendikulárneho
kostrového
svalstva (g)
|
546,11
|
449,09
|
411,05
|
575,80
|
97,02
[-362; 556]
0,3247b
|
svalová hmota hmota (g)
|
1 739,05
|
1 305,73
|
1 660,56
|
1 707,82'
|
433,32
[-404; 1 271]
|
a Parametre stavby tela sú založené na vyšetrení DXA.
Pozorované a simulované hladiny IGF-I SDS v klinickom skúšaníV hlavnej fáze klinického skúšania boli hodnoty SDF IGF-I 0 a vyššie celkovo dosiahnuté u 53%
pacientov v skúšaní AGHD liečených somapacitanom po 8 týždňovej titrácii dávky. Tento podiel bol však nižší, najmä v podskupinách, ako sú ženy užívajúce perorálny estrogén (32%) a pacienti s nástupom do detstva (39 %) (tabuľka 5).
Post-hoc simulačné analýzy naznačili, že podiely pacientov s AGHD, ktorí dosahujú hladiny IGF-I SDS nad 0, budú vyššie v prípade, že by bola povolená titrácia dávky somapacitanu dlhšia ako 8 týždňov. V tejto simulačnej analýze sa predpokladalo, že titrácia dávky somapacitanu bola u všetkých pacientov dobre tolerovaná, až kým nebolo dosiahnuté cieľové rozpätie IGF-I SDS alebo dávka somapacitanu 8 mg týždenne.
podskupiny
|
muži
| ženy bez perorálneho estrogénu
| ženy s perorálnym estrogénom
| nástup AGHD v detstve
| nástup AGHD v dospelosti
|
všetci
| pozorovanáa
| 71%
| 46%
| 32%
| 39%
| 60%
| 53%
| post-hoc
simulácia
|
100%
|
96%
|
70%
|
84%
|
92%
|
90%
|
|
|
Tabuľka5 PodielpacientovsAGHDliečenýchsomapacitanomshladinamiIGF-I SDS nad 0 3Skúšanie bolo dizajnované na titráciu smerom k hladine IGF-I SDS nad -0,5
Udržiavacia dávkaUdržiavacia dávka kolíše medzi pacientami a medzi mužmi a ženami. Priemerná udržiavacia dávka somapacitanu pozorovaná v klinických skúšaniach v 3. fáze bola 2,4 mg/týždeň.
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Sogroya vo všetkých podskupinách pediatrickej populácie s deficienciou rastového hormónu (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiSomapacitan má farmakokinetické vlastnosti kompatibilné s podávaním lieku jedenkrát týždenne. Reverzibilné naviazanie na endogénny albumín oddiaľuje elimináciu somapacitanu, a tým predlžuje
in vivo polčas a trvanie účinku.
Farmakokinetika somapacitanu po subkutánnom podaní sa skúmala v dávkových hladinách od 0,01 do
0,32 mg/kg u zdravých dospelých a v dávkach do 0,12 mg/kg u dospelých s GHD.
Somapacitan celkovo vykazuje nelineárnu farmakokinetiku, ale v klinicky relevantnom rozsahu dávok
somapacitanu u dospelých s GHD je farmakokinetika somapacitanu približne lineárna.
Absorpcia
U dospelých pacientov s GHD sa medián tmax pohyboval v rozsahu od 4 do 24 hodín v dávkach od
0,02 mg/kg/týždeň do 0,12 mg/kg/týždeň.
Expozícia v rovnovážnom stave sa dosiahla po 1-2 týždennom podávaní. Absolútna biologická dostupnosť somapacitanu u ľudí sa neskúmala.
Distribúcia
Somapacitan sa výrazne viaže (> 99 %) na plazmatické proteíny a očakáva sa, že sa bude distribuovať
ako albumín. Na základe populačných farmakokinetických analýz bol odhadovaný distribučný objem
(V/F) 14,6 l.
Eliminácia
Terminálny polčas bol odhadnutý s geometrickými priemermi v rozsahu približne 2 až 3 dni
v rovnovážnom stave u pacientov s AGHD (dávky: 0,02 až 0,12 mg/kg).
Somapacitan bude prítomný v krvnom obehu približne 2 týždne po poslednej dávke. Pozorovala sa mierna až nulová akumulácia (priemerný pomer akumulácie: 1-2) somapacitanu po opakovaných dávkach u pacientov s AGHD.
Biotransformácia
Somapacitan sa extenzívne metabolizuje proteolytickým rozkladom a štiepením spájajúcej sekvencie
medzi peptidom a väzbovým miestom albumínu.
Somapacitan sa extenzívne metabolizoval pred exkréciou a intaktný somapacitan sa nezistil v moči, ktorý bol hlavnou cestou exkrécie (81 %), ani v stolici, v ktorej sa našlo 13 % častí
molekuly somapacitanu, z čoho vyplýva úplná biotransformácia pred exkréciou.
Osobitné populácie
Vek
Osoby staršie ako 60 rokov majú vyššiu expozíciu (29 %) ako mladšie osoby pri rovnakej dávke somapacitanu. Nižšia úvodná dávka pre osoby staršie ako 60 rokov je opísaná v časti 4.2.
Pohlavie
Ženy, najmä tie, ktoré užívajú perorálne estrogén, majú nižšiu expozíciu (53 % pre ženy užívajúce perorálny estrogén a 30 % pre ženy, ktoré neužívajú perorálny estrogén) ako muži používajúci rovnakú dávku somapacitanu. Vyššia úvodná dávka pre ženy užívajúce perorálny estrogén je opísaná v časti 4.2.
Rasa
Nepozoroval sa rozdiel v expozícii somapacitanu a v odpovedi na IGF-I medzi Japoncami a pacientmi bielej rasy. Napriek vyššej expozícii u Ázijčanov (okrem Japoncov), v porovnaní s pacientmi bielej
rasy pri rovnakej dávke somapacitanu, potrebovali pacienti bielej rasy, Japonci a Ázijčania (okrem
Japoncov) rovnakú dávku na dosiahnutie podobnej hladiny IGF-I. Preto nie je k dispozícii žiadne odporúčanie týkajúce sa úpravy dávky na základe rasy.
Etnicita
Etnicita [hispánska alebo latinskoamerická 4,5 % (15 osôb dostávalo somapacitan)] sa neskúmala vzhľadom na malú veľkosť vzorky vo vývojovom programe.
Telesná hmotnosť
Napriek vyššej expozícii u osôb s nízkou telesnou hmotnosťou v porovnaní s osobami s vysokou telesnou hmotnosťou pri rovnakej dávke somapacitanu, účastníci potrebovali rovnakú dávku
na dosiahnutie podobnej hladiny IGF-I v rozsahu telesnej hmotnosti 35 kg až 150 kg. Preto nie je
k dispozícii žiadne odporúčanie týkajúce sa úpravy dávky na základe telesnej hmotnosti.
Porucha funkcie obličiek
Dávka somapacitanu 0,08 mg/kg v rovnovážnom stave mala za následok vyššie expozície u osôb
s poruchou funkcie obličiek, najvýraznejšie u osôb so závažnou poruchou funkcie obličiek a u osôb
vyžadujúcich hemodialýzu, keď pomery AUC 0-168h k normálnej funkcii obličiek boli 1,75 a 1,63. Expozícia somapacitanu mala vo všeobecnosti tendenciu zvyšovať sa s klesajúcou rýchlosťou glomerulárnej filtrácie (GFR).
Vyššie hladiny IGF-I AUC0-168h sa pozorovali u osôb so stredne závažnou a závažnou poruchou funkcie obličiek a u osôb, ktoré potrebovali hemodialýzu, s pomerom k normálnej funkcii obličiek 1,35; 1,40 a
1,24 v uvedenom poradí.
Vzhľadom na pozorované mierne zvýšenie hladiny IGF-I spolu s nízkymi odporúčanými úvodnými dávkami a individuálnou titráciou dávky somapacitanu nie je k dispozícii žiadne odporúčanie týkajúce
sa úpravy dávky pre pacientov s poruchou funkcie obličiek.
Porucha funkcie pečene
Dávka somapacitanu 0,08 mg/kg v rovnovážnom stave viedla k vyššej expozícii u osôb so stredne
závažnou poruchou funkcie pečene s pomerom k normálnej funkcii pečene 4,69 pre AUC0-168h a 3,52
pre Cmax.
Nižšie hladiny IGF-I stimulované somapacitanom sa pozorovali u osôb s miernou a stredne závažnou
poruchou funkcie pečene v porovnaní s osobami s normálnou funkciou pečene (pomer k normálu bol
0,85 pre miernu poruchu a 0,75 pre stredne závažnú poruchu).
Vzhľadom na pozorované mierne zníženie hladiny IGF-I spolu s individuálnou titráciou dávky somapacitanu nie je k dispozícii žiadne odporúčanie týkajúce sa úpravy dávky pre pacientov
s poruchou funkcie pečene.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity
po opakovanom podávaní, genotoxicity alebo prenatálneho/postnatálneho vývinu, neodhalili žiadne osobitné riziko pre ľudí.
So somapacitanom sa neuskutočnili žiadne štúdie karcinogenicity.
Nepozorovali sa nežiaduce účinky na fertilitu samcov a samíc potkanov pri dávke vedúcej k expozícii, ktorá je vyššia aspoň 13-krát u mužov a 15-krát u žien ako očakávaná maximálna klinická expozícia pri dávke 8 mg/týždeň. Pri všetkých použitých dávkach sa však pozoroval nepravidelný estrálny
cyklus u samíc.
Nebol identifikovaný žiadny dôkaz o poškodení plodu, keď sa brezivým potkanom a králikom podával subkutánne somapacitan počas organogenézy v dávkach vedúcich k expozíciám, ktoré boli výrazne vyššie ako očakávaná expozícia pri maximálnej klinickej dávke 8 mg/týždeň (aspoň 18-násobne). Pri vysokých dávkach vedúcich k expozícii aspoň 130-násobne vyššej ako očakávaná maximálna klinická expozícia pri dávke 8 mg/týždeň sa u mláďat samíc potkanov, ktoré dostávali somapacitan, zistili krátke/ohnuté/zhrubnuté dlhé kosti. Je známe, že takéto nálezy u potkanov sa vyriešia po narodení a majú sa považovať za menšie malformácie a nie za trvalé poruchy.
Keď sa brezivým králikom podávala subkutánna dávka somapacitanu pri expozíciách aspoň 9-násobne vyšších ako očakávaná expozícia pri maximálnej klinickej dávke 8 mg/týždeň, znížil sa rast plodu.
U laktujúcich potkanov sa časti molekuly somapacitanu vylučovali do mlieka, ale s nižšou hladinou ako sa pozorovalo v plazme (do 50 % hladiny v plazme).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
histidín manitol poloxamér 188 fenol
voda na injekcie
kyselina chlorovodíková (na úpravu pH)
hydroxid sodný (na úpravu pH)
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky.
Po prvom otvorení
6 týždňov. Uchovávajte v chladničke (2 °C – 8 °C).
Neuchovávajte v mrazničke. Neuchovávajte v blízkosti mraziacej jednotky. Uchovávajte vo vonkajšom obale s krytom na pere na ochranu pred svetlom.
Pred a po prvom otvorení
Ak chladenie nie je možné (napr. počas transportu), Sogroya sa môže uchovávať dočasne pri teplote do 30 °C maximálne 72 hodín (3 dni). Po uchovávaní pri tejto teplote je potrebné umiestniť liek
Sogroya znova do chladničky. Ak sa uchováva mimo chladničky, a potom sa vráti späť do chladničky,
celkový kombinovaný čas mimo chladničky nemá presiahnuť 3 dni, starostlivo sa to má sledovať. Pero Sogroya sa má zlikvidovať, ak bolo uchovávané pri teplote do 30 °C dlhšie ako 72 hodín (3 dni)
alebo akúkoľvek dobu pri teplote nad 30 °C.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke. Neuchovávajte v blízkosti mraziacej jednotky.
Uchovávajte liek Sogroya vo vonkajšom obale s krytom na pere na ochranu pred svetlom.
Podmienky na uchovávanie po prvom otvorení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
1,5 ml roztoku v sklenenej náplni (bezfarebné sklo typu I) s piestom vyrobeným z chlórbutylovej gumy a zátkou vyrobenou z brómbutylu/izoprénu utesnenej hliníkovým viečkom.
Náplň sa nachádza v jednorazovom viacdávkovom pere vyrobenom z polypropylénu, polyacetálu, polykarbonátu a akrylonitril-butadién-styrénu, ktoré obsahuje dve kovové pružiny.
Náplň je permanentne uzavretá v injekčnom pere.
Veľkosti balenia sú 1 naplnené pero a multibalenie obsahujúce 5 (5 balení po 1) naplnených pier. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Pero je určené na použitie len pre jednu osobu.
Sogroya sa nemá používať, ak roztok nie je číry až mierne opaleskujúci, bezfarebný až svetložltý a bez viditeľných častíc.
Liek Sogroya sa nesmie používať, ak bol zmrazená.
Náplň sa nesmie vyberať z naplneného pera a znova napĺňať.
Pred použitím sa vždy musí nasadiť ihla. Ihly sa nesmú používať opakovane. Injekčná ihla sa má po každej injekcii odstrániť a pero sa má uchovávať bez nasadenej ihly. Môže to zabrániť upchatiu ihly, kontaminácii, infekcii, vytekaniu roztoku a nepresnému dávkovaniu.
V prípade upchatia ihly pacienti musia dodržiavať pokyny uvedené v návode na použitie, ktorý je súčasťou písomnej informácie pre používateľa.
Ihly nie sú súčasťou balenia. Naplnené pero Sogroya bolo testované s jednorazovými ihlami 31 Gx6
mm a 32 Gx5 mm. Sogroya sa môže podávať pomocou ihiel s dĺžkou do 8 mm a s hrúbkou do 32G.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIINovo Nordisk A/S Novo Allé
DK-2880 Bagsværd
Dánsko
8. REGISTRAČNÉ ČÍSLAEU/1/20/1501/001
EU/1/20/1501/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.