
strointestinálneho traktu (pozri časť 4.3). U pacientov s akýmikoľvek chorobami, ktoré by mohli viesť k narušeniu integrity steny gastrointestinálneho traktu (napr. peptickou vredovou chorobou, Ogilvieho syndrómom, malignitou gastrointestinálneho traktu,
Crohnovou chorobou) sa má použitie naledemedínu zvažovať s opatrnosťou. U každého pacienta sa má zohľadniť celkový pomer prínosu a rizika. Pacienti majú byť sledovaní kvôli rozvoju závažnej, pretrvávajúcej alebo zhoršujúcej sa bolesti brucha. Ak je podozrenie na obštrukciu alebo perforáciu, podávanie naldemedínu sa musí ukončiť (pozri časť 4.3).
Gastrointestinálnenežiaducereakcie
Pri použití Rizmoicu boli hlásené abdominálne nežiaduce reakcie (napr. bolesť brucha, vracanie
a hnačka). Pacienti majú byť upozonení, aby hlásili závažné, pretrvávajúce alebo zhoršujúce sa príznaky svojmu lekárovi. V prípade závažnej hnačky alebo bolesti brucha má byť pacient sledovaný a liečený na dehydratáciu s použitím rehydratačnej a inej vhodnej liečby podľa potreby (pozri
časť 4.8).
Syndrómzvysadeniaopioidov
Syndróm z vysadenia opioidov je skupinou troch alebo viacerých z nasledujúcich prejavov alebo
príznakov: dysforická nálada, nauzea alebo vracanie, bolesti svalov, slzenie alebo rinorea, pupilárna dilatácia alebo piloerekcia alebo potenie, hnačka, zívanie, horúčka alebo nespavosť. Syndróm
z vysadenia opioidov sa typicky rozvíja v priebehu niekoľkých minút až niekoľkých dní po podaní
antagonistu opioidov. S ohľadom na syndróm z vysadenia opioidov je potrebná opatrnosť. Pacienti majú byť upozornení, aby ukončili liečbu naldemedínom a kontaktovali svojho lekára, ak dôjde
k rozvinutiu príznakov syndrómu z vysadenia opioidov. V klinickom programe naldemedínu boli hlásené prípady možného syndrómu z vysadenia opioidov (pozri časť 4.8).
U pacientov s narušenou hematoencefalickou bariérou (napr. pri primárnych mozgových malignitách, metastázách centrálneho nervového systému (CNS) alebo iných zápalových stavoch, aktívnej skleróze multiplex a pokročilej Alzheimerovej chorobe) môže byť zvýšené riziko syndrómu z vysadenia opioidov alebo zníženej analgézie. Celkový pomer prínosu a rizika naldemedínu sa má u týchto pacientov zvážiť starostlivým sledovaním príznakov z vysadenia opioidov.
Pacientiskardiovaskulárnymiochoreniami
Naldemedín nebol študovaný v programe klinických štúdií u pacientov, ktorí nedávno prekonali infarkt myokardu, cievnu mozgovú príhodu alebo tranzitórny ischemický atak počas 3 mesiacov pred skríningom. Títo pacienti majú byť počas užívania Rizmoicu klinicky sledovaní.
Štúdia QTc vykonaná s naldemedínom u zdravých dobrovoľníkov nepreukázala žiadne predĺženie QT
intervalu. Pacienti s rizikovými faktormi kardiovaskulárnych ochorení neboli vylúčení z programu klinických štúdií s naldemedínom, pričom BMI ≥ 30 kg/m2 a anamnéza hypertenzie a/alebo dyslipidémie sú najčastejšie hlásenými rizikovými faktormi.
Ťažkáporuchafunkciepečene
Naldemedín nebol skúmaný u pacientov s ťažkou poruchou funkciou pečene. Použitie naldemedínu sa
u týchto pacientov neodporúča (pozri časť 4.2).
Súbežnépoužitiesosilnýmiinhibítormia induktormiCYP3A
Súbežné použitie naldemedínu so silnými inhibítormi CYP3A (napr. grapefruitovým džúsom,
itrakonazolom, ketokonazolom, ritonavirom, indinavirom, sakvinavirom, telitromycínom
a klaritromycínom) vedie k zvýšeniu expozície naldemedínu a môže zvýšiť riziko nežiaducich reakcií. Je potrebné vyhnúť sa súbežnému použitiu so silnými inhibítormi CYP3A.
Súbežné použitie naldemedínu so silnými induktormi CYP3A (napr. ľubovníkom bodkovaným
(Hypericum perforatum), rifampicínom, karbamazepínom, fenobarbitalom a fenytoínom) vedie
k zníženiu expozície naldemedínu a môže znížiť účinnosť naldemedínu. Súbežné použitie so silnými
induktormi CYP3A sa neodporúča (pozri časť 4.5). Súbežné použitie naldemedínu so stredne silnými CYP3A induktormi (napr. efavirenzom) sa nestanovilo a preto sa majú používať s opatrnosťou (pozri časť 4.5).
Sodík
Tento liek obsahuje menej než 1 mmol sodíka (23 mg) v tablete, t.j. v podstate zanedbateľné
množstvo sodíka.
4.5 Liekové a iné interakcie
Vplyvinýchliekovnanaldemedín
Naldemedín sa metabolizuje predovšetkým prostredníctvom CYP3A s určitým podielom UGT1A3
a je substrátom P-glykoproteínu (P-gp) (pozri časť 5.2).
Interakcie s inhibítormi CYP3A
Itrakonazol, silný inhibítor CYP3A, zvýšil expozíciu naldemedínu 2,9-násobne, čo môže viesť
k zvýšenému riziku nežiaducich reakcií.
Súbežnému použitiu so silnými inhibítormi CYP3A, ako sú grapefruitový džús, itrakonazol, ketokonazol, ritonavir, indinavir, sakvinavir, telitromycín a klaritromycín, je potrebné sa vyhnúť. Ak je použitie so silnými inhibítormi CYP3A nevyhnutné, sledujte nežiaduce reakcie (pozri časť 4.4). Súbežné použitie stredne silných inhibítorov CYP3A, ako je flukonazol, môže zvýšiť plazmatickú koncentráciu naldemedínu. Pri použití so stredne silnými inhibítormi CYP3A, sledujte nežiaduce reakcie.
Neexistuje riziko interakcie pri súbežnom použití s miernymi inhibítormi CYP3A.
Interakcie so silnými a stredne silnými induktormi CYP3A
Rifampicín, silný induktor CYP3A, významne znížil expozíciu naldemedínu o 83 %.
Súbežné použitie silných induktorov CYP3A, ako sú ľubovník bodkovaný (Hypericum perforatum), rifampín, karbamazepín, fenobarbital a fenytoín, sa neodporúča. Súbežné použitie naldemedínu
so stredne silnými induktormi (napr. efavirenzom) sa nestanovilo a preto sa majú pacienti sledovať
(pozri časť 4.4).
Interakcie so silnými inhibítormi P-gp
Súbežné použitie inhibítorov P-gp, ako je cyklosporín, môže zvýšiť plazmatickú koncentráciu naldemedínu. Ak sa naldemedín používa so silnými inhibítormi P-gp, sledujte nežiaduce reakcie.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii nie sú žiadne údaje o použití naldemedínu u gravidných žien.
Štúdie na zvieratách nepreukázali priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Použitie naldemedínu počas gravidity môže vyvolať syndróm z vysadenia opioidov u plodu
v dôsledku nezrelej hematoencefalickej bariéry.
Naldemedín sa nemá používať počas tehotenstva, pokiaľ klinický stav ženy nevyžaduje liečbu naldemedínom.
Dojčenie
Nie je známe, či sa naldemedín/metabolity vylučujú do ľudského mlieka. Dostupné údaje u potkanov preukázali vylučovanie naldemedínu do mlieka (pozri časť 5.3).
V terapeutických dávkach sa väčšina opioidov (napr. morfin, meperidín, metadón) vylučuje
v minimálnych množstvách do materského mlieka. Existuje teoretická možnosť, že naldemedín vyvoláva syndróm z vysadenia opioidov u dojčeného novorodenca, ktorého matka užíva agonistu opioidného receptora.
Riziko u dojčiat nemôže byť vylúčené. Naldemedín sa nemá používať počas dojčenia.
Fertilita
K dispozícii nie sú žiadne údaje o účinku naldemedínu na fertilitu u ľudí. Zistilo sa, že naldemedín nemá žiadne klinicky významné nežiaduce účinky na fertilitu alebo reprodukčnú schopnosť u samcov a samíc potkanov (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Naldemedín nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšie hlásené nežiaduce reakcie u pacientov s chronickou bolesťou nekancerózneho pôvodu
a OIC boli bolesť brucha (7,8 %), hnačka (5,9 %), nauzea (3,6 %) a vracanie (1,1 %). Väčšina týchto gastrointestinálnych nežiaducich reakcií bola mierna až stredne závažná a vyriešila sa bez ukončenia liečby naldemedínom. U pacientov s chronickou bolesťou nekancerózneho pôvodu a OIC bol hlásený jeden vážny prípad bolesti brucha a jeden vážny prípad nauzey.
Najčastejšie hlásené nežiaduce reakcie u pacientov s nádorovým ochorením a OIC boli hnačka
(24,5 %) a bolesť brucha (3,9 %). Väčšina týchto gastrointestinálnych nežiaducich reakcií bola mierna
až stredne závažná a vyriešila sa liečbou. U pacientov s nádorovým ochorením a OIC boli hlásené dva vážne prípady hnačky.
Tabuľkovýzoznamnežiaducichreakcií
Nežiaduce reakcie pri liečbe naldemedínom 200 mikrogramovými tabletami u pacientov s chronickou bolesťou nekancerózneho pôvodu a OIC a u pacientov s nádorovým ochorením a OIC, hlásené
v klinických štúdiách, sú uvedené v tabuľkách podľa klasifikácie orgánových systémov MedDRA. Kategórie frekvencie sú definované použitím nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (
≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov nie je možné určiť frekvenciu). V rámci každej skupiny frekvencií sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 1. Nežiaduce reakcie uvedené podľa triedy orgánových systémov
a frekvencie u pacientov s chronickou bolesťou nekancerózneho pôvodu a opioidmi indukovanou obstipáciou
T
r
i
e
d
a
orgánových
systémov Poruchy imunitného systému
Časté Menej časté Zriedkavé Neznáme
Precitlivenosťa
Poruchy gastrointestinálne ho traktu
Celkové poruchy
a reakcie v mieste podania
Hnačka
Bolesť bruchab
Nauzea
Vracanie

Syndróm
z vysadenia opioidov
Gastrointestinálna perforácia
a V klinických štúdiách s naldemedínom bol hlásený jeden vážny prípad precitlivenosti. Pacient sa zotavil po odstúpení zo štúdie
b MedDRA preferované termíny: bolesť brucha, bolesť v hornej časti brucha, bolesť v dolnej
časti brucha a brušný dyskomfort
Tabuľka 2. Nežiaduce reakcie uvedené podľa triedy orgánových systémov a frekvencie u pacientov s nádorovým ochorením a opioidmi indukovanou obstipáciou
T
r
i
e
d
a
orgánových systémov Poruchy
gastrointestinálne
ho traktu
Celkové poruchy a reakcie
v mieste podania
Veľmi časté Časté Menej časté NeznámeHnačka Bolesť bruchaa Gastrointestinálna perforácia
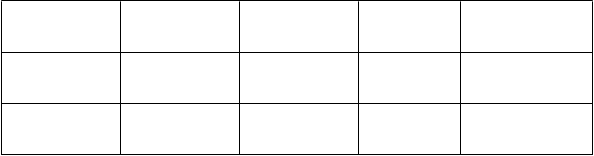
Syndróm
z vysadenia opioidov
aMedDRA preferované termíny: bolesť brucha, bolesť v hornej časti brucha, bolesť v dolnej
časti brucha a brušný dyskomfort
Popisvybranýchnežiaducichreakcií
Syndróm z vysadenia opioidov
Možný syndróm z vysadenia opioidov, definovaný ako aspoň tri nežiaduce reakcie potenciálne
súvisiace s vysadením opioidov, ktoré sa objavili v ten istý deň a nesúviseli výhradne
s gastrointestinálnym systémom, sa objavil u 0,8 % (9/1 163) pacientov s chronickou bolesťou nekancerózneho pôvodu a OIC užívajúcich naldemedín v porovnaní s 0,2 % (2/1 165) pacientov uživajúcich placebo bez ohľadu na udržiavaciu liečbu opioidmi, a u 0,6 % (1/155) pacientov
s nádorovým ochorením a OIC uživajúcich naldemedín 200 mikrogramov v porovnaní s 0 % (0/152) pacientov užívajúcich placebo. Príznaky zahŕňali napr. hyperhidrózu, zimnicu, zvýšenú lakrimáciu, nával tepla/sčervenanie, pyrexiu, kýchanie, pocit chladu, bolesť brucha, hnačku, nauzeu, vracanie, artralgiu, myalgiu a tachykardiu (pozri časť 4.4).
Poruchy gastrointestinálneho traktu
Bolesť brucha, hnačka, nauzea a vracanie boli najčastejšie hlásené nežiaduce reakcie v klinických štúdiách u pacientov s chronickou bolesťou nekancerózneho pôvodu a OIC, a u pacientov
s nádorovým ochorením a OIC. Väčšina týchto gastrointestinálnych nežiaducich reakcií bola mierna až stredne závažná a vyriešila sa liečbou. Miera ukončenia liečby v dôsledku gastrointestinálnych nežiaducich udalostí pri liečbe naldemedínom 200 mikrogramov v porovnaní s placebom bola 3,2 % a 1 % v uvedenom poradí u pacientov s chronickou bolesťou nekancerózneho pôvodu a OIC, a 4,5 % a 0 % v uvedenom poradí u pacientov s nádorovým ochorením a OIC.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieZdravídobrovoľníciV klinických štúdiách bola zdravým dobrovoľníkom podaná jednorazová dávka naldemedínu až do
100 mg a viacnásobná dávka až do 30 mg/deň počas 10 dní. Bol pozorovaný na dávke závislý nárast nežiaducich reakcií súvisiacich s gastrointestinálnym systémom, vrátane bolesti brucha, hnačky
a nauzey. Tieto boli mierne až stredne závažné a vyriešili sa.
PacientisOICV klinických štúdiách bola pacientom s OIC podávaná jednorazová dávka naldemedínu (0,01 mg až
3 mg) a viacnásobné dávky 0,4 mg/deň. U pacienta, ktorý užil jednorazovú dávku naldemedínu 1 mg, vznikol vážny syndróm z vysadenia lieku, vrátane nauzey a žalúdočných kŕčov, a bol mu podaný ezomeprazol a ondansetrón na nauzeu, a midazolam hydrochlorid na žalúdočné kŕče. Príznaky sa vyriešili. V klinických štúdiách mali pacienti s OIC, ktorým bol podávaný naldemedín 0,4 mg/deň (dvojnásobok odporúčanej dávky) počas 4 týždňov, zvýšený výskyt nežiaducich liekových reakcií súvisiacich s gastrointestinálnym traktom, vrátane hnačky a bolesti brucha zvyčajne v priebehu 1-2 dní po úvodnej dávke.
ManažmentNeexistuje špecifické antidotum pre naldemedín. Naldemedín sa neodstraňuje z tela hemodialýzou.
V prípade predávkovania sa majú pacienti starostlivo sledovať pre potenciálne prejavy a príznaky syndrómu z vysadenia opioidov (pozri časť 4.4) a je potrebné poskytnúť im vhodnú podpornú starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Liečivá na zápchu, antagonisty periférnych opioidných receptorov, ATC kód: A06AH05.
MechanizmusúčinkuNaldemedín je antagonista opioidnej väzby na mí-, delta- a kappa-opioidných receptoroch.
Naldemedín funguje ako periférne pôsobiaci antagonista mí-opioidného receptora v tkanivách ako je gastrointestinálny trakt, a tým znižuje obstipačné účinky opioidov bez reverzie opioidných účinkov
v centrálnom nervovom systéme (CNS).
Naldemedín je derivát naltrexónu, ku ktorému bol pridaný bočný reťazec, ktorý zvyšuje molekulárnu hmotnosť a plochu polárneho povrchu, a tým znižuje jeho schopnosť prechodu cez hematoencefalickú bariéru (
blood-brain barrier, BBB); predpokladá sa, že penetrácia naldemedínu do CNS je pri odporúčanej dávke zanedbateľná. Naldemedín je navyše substrátom P-glykoproteínového (P-gp) efluxového transportéra, ktorý môže byť tiež zapojený do zníženia penetrácie naldemedínu do CNS. Na základe toho sa očakáva, že naldemedín uplatňuje svoje anti-obstipačné účinky na opioidy bez reverzie ich analgetických účinkov sprostredkovaných CNS.
K
l
i
nická
účinnosť
a
bezpečnosť
Účinnosť a bezpečnosť naldemedínu bola stanovená u pacientov s chronickou bolesťou nekancerózneho pôvodu a OIC a u pacientov s nádorovým ochorením a OIC.
Klinické štúdie u pacientov s chronickou bolesťou nekancerózneho pôvodu a OIC. Bezpečnosť a účinnosť naldemedínu bola hodnotená v dvoch identických, 12-týždňových randomizovaných, dvojito zaslepených, placebom kontrolovaných štúdiách (štúdiách V9231 a V9232), v ktorých bol naldemedín použitý bez laxatív, a v tretej, dlhodobej, 52-týždňovej
randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii (štúdii V9235), v ktorej bol naldemedín použitý so stabilnými laxatívami alebo bez nich u pacientov s chronickou bolesťou nekancerózneho pôvodu a OIC.
Pacienti, ktorí dostávali stabilnú dennú dávku opioidov ekvivalentnú morfínu ≥ 30 mg aspoň 4 týždne pred zaradením do štúdie a sami hlásili OIC, boli vhodní pre účasť v štúdii.
V štúdiách V9231 a V9232 bola OIC potvrdená počas 2-týždňového prípravného obdobia a bola definovaná ako nie viac ako 4 spontánne stolice (spontaneous bowel movement, SBM) celkovo počas
14 po sebe nasledujúcich dní a < 3 SBM v danom týždni s aspoň 25 % SBM spojenými s jedným
alebo s viacerými nasledujúcimi stavmi:(1) namáhavé vyprázdňovanie; (2) tvrdá alebo hrudkovitá stolica; (3) pocit neúplného vyprázdnenia a (4) pocit anorektálnej obštrukcie/prekážky. V štúdii V9235 bola OIC potvrdená počas 2-týždňového prípravného obdobia a bola definovaná ako nie viac ako
4 SBM celkovo počas 14 po sebe nasledujúcich dní a < 3 SBM v danom týždni.
SBM bola definovaná ako stolica (bowel movement, BM) bez záchranných laxatív užitých v priebehu posledných 24 hodín.
V štúdiách V9231 a V9232 pacienti buď nesmeli používať laxatíva alebo museli byť ochotní ukončiť používanie laxatív v čase skríningu a počas obdobia skríningu a obdobia liečby používať iba poskytnuté záchranné laxatíva. Všetci účastníci štúdie užívali predtým na liečbu OIC laxatíva. V štúdii V9235 bolo pacientom so stabilným laxatívnym režimom pri skríningu (52,4 %) dovolené pokračovať v používaní rovnakého režimu bez zmeny počas trvania štúdie. Počas prípravného obdobia a obdobia liečby vo všetkých troch štúdiách sa ako záchranné laxatívum používal bisakodyl, ak pacienti nemali BM počas 72 hodín, a ak po 24 hodín od užitia bisakodylu stále nemali BM, bolo im dovolené jednorazové použitie klyzmy.
Pacienti s dôkazom významných štrukturálnych abnormalít gastrointestinálneho traktu neboli zapojení do týchto štúdií.
Celkovo bolo randomizovaných 547 pacientov v štúdii V9231, 551 pacientov v štúdii V9232
a 1 246 pacientov v štúdii V9235 v pomere 1:1 k liečbe naldemedínom 200 mikrogramov alebo placebom raz denne počas 12 týždňov v štúdiách V9231 a V9232, 52 týždňov v štúdii V9235.
Celkovo bol v štúdiách V9231, V9232 a V9235 priemerný vek pacientov 53,2 rokov; 14,8 % bolo vo veku 65 rokov alebo starších; 62,0 % boli ženy; 80,2 % boli belosi.
V štúdii V9231 boli tri najčastejšie typy bolesti bolesť chrbta (62,0 %); bolesť krku (8,3 %)
a osteoartritída (5,3 %). V štúdii V9232 to boli bolesť chrbta (53,6 %); bolesť (10,2 %) a artralgia
(7,8 %). V štúdii V9235 boli tri najčastejšie typy bolesti bolesť chrbta (58,0 %); osteoartritída (9,5 %)
a bolesť krku (8,1 %).
Pred zaradením do štúdie pacienti používali svoj aktuálny opioid v priemere 5 rokov. Pacienti, ktorí sa zúčastnili štúdií V9231, V9232 a V9235 užívali širokú škálu opioidov.
Priemerná denná dávka opioidu ekvivalentná morfínu na začiatku štúdie bola 132,42 mg, 120,93 mg
a 122,06 mg na deň v štúdiách V9231, V9232 a V9235, v uvedenom poradí. Priemerné SBM
na začiatku štúdíí V9231, V9232 a V9235 boli 1,31, 1,17 a 1,60 v tomto poradí.
Primárnym koncovým ukazovateľom pre štúdie V9231 a V9232 bol podiel SBM respondérov, definovaný ako: ≥ 3 SBM na týždeň a zmena oproti hodnote na začiatku štúdie o ≥ 1 SBM na týždeň počas aspoň 9 z 12 týždňov štúdie a 3 z posledných 4 týždňov. Primárnym koncovým ukazovateľom účinnosti pre štúdiu V9235 bola zmena frekvencie BM na týždeň oproti hodnote na začiatku štúdie vo
12., 24., 36. a 52. týždni.
V štúdiách V9231 a V9232 bol štatisticky významný rozdiel pre primárny koncový ukazovateľ v skupine liečenej naldemedínom oproti skupine s placebom (pozri tabuľku 3).
V štúdiách V9231 a V9232 boli 4 sekundárne koncové ukazovatele (pozri tabuľku 3).
Tabuľka 3. Klinické výsledky pre štúdie V9231 a V9232
V9231 V9232
Naldemedín
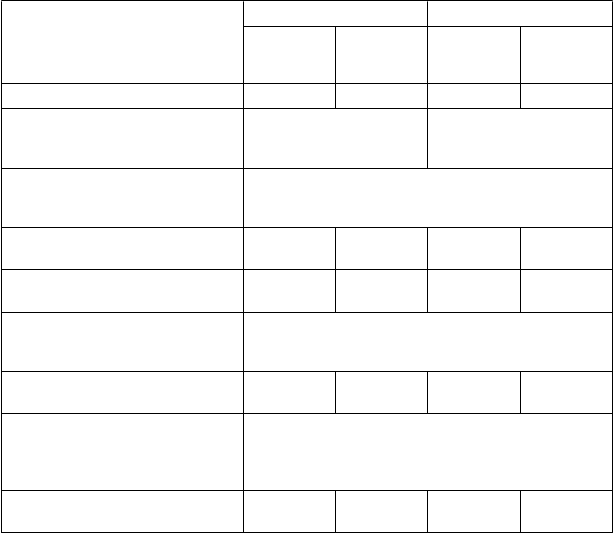
(N=273)
Placebo
(N=272)
Naldemedín
(N=276)
Placebo
(N=274)
P
odiel SBM respondérov 47,6 % 34,6 % 52,5 % 33,6 %
Rozdiel v liečbe 13,0 %
(95 % CI: 4,8 %, 21,3 %, p=0,0020*)
18,9 %
(95 % CI: 10,8 %, 27,0 %, p<0,0001*)
Z
m
e
n
y frekvencie SBM na týždeň (priemer metódou najmenších štvorcov)
V posledných 2 týždňoch liečby
oproti hodnote na začiatku štúdie** 3,42 2,12 3,56 2,16
V 1. týždni oproti hodnote na začiatku
štúdie** 3,48 1,36 3,86 1,69
Zmeny frekvencie CSBM na týždeň (priemer metódou najmenších štvorcov)
V posledných 2 týždňoch liečby
oproti hodnote na začiatku štúdie** 2,58 1,57 2,77 1,62
Zmeny frekvencie SBM bez namáhavého vyprázdnenia na týždeň (priemer metódou najmenších štvorcov)
V posledných 2 týždňoch liečby
oproti hodnote na začiatku štúdie*** 1,46 0,73 1,85 1,10
CI = Interval spoľahlivosti (Confidence Interval)
*Štatisticky významné: p-hodnoty podľa Cochranovho-Mantelovho-Haenszelovho testu.
** p<0,0001
*** p=0,0003 v štúdii V9231 a p=0,0011 v štúdii V9232
V štúdii V9235 sa ako sekundárny koncový ukazovateľ hodnotila účinnosť naldemedínu oproti placebu frekvenciou BM, ako je uvedené v tabuľke 4.
T
a
b
u
ľ
k
a 4. Zmena frekvencie BM na týždeň pri každej návšteve oproti hodnote na začiatku štúdie (priemer metódou najmenších štvorcov) ITT populácie v štúdii V9235
Naldemedín
(N=621)
Placebo
(N=620)
Priemerná frekvencia BM na začiatku štúdie
Zmena frekvencie BM na týždeň
2,02 2,02

12. týždeň* 3,70 2,42
24. týždeň* 3,77 2,77
36. týždeň* 3,88 2,88
52. týždeň* 3,92 2,92
*nominálny p≤0,0001
Účinnosť a bezpečnosť sa tiež hodnotili v podskupinách respondérov nedostatočne odpovedajúcich na laxatíva
(laxative inadequate responders, LIR) a non-LIR.
V štúdiách V9231 a V9232 sa za LIR považovali pacienti, ktorí boli na základe záznamov súbežnej medikácie pred vstupom do štúdie na liečbe laxatívami, a ktorí ukončili ich užívanie v priebehu 30 dní pred skríningom, a sami hlásili OIC.
Okrem toho sa pacienti, ktorí neužívali laxatíva v priebehu 30 dní pred skríningom a užívali iba záchranné laxatíva počas alebo po skríningu, považovali za non-LIR. Počet pacientov v podskupinách LIR a non-LIR bol 629 (naldemedín: 317 a placebo: 312) a 451 (naldemedín: 223 a placebo: 228) pre súhrnné štúdie V9231 a V9232. Všetci účastníci štúdie pred vstupom do štúdií V9231 a V9232 užívali určitý čas na liečbu OIC laxatíva.
V podskupine LIR bol pozorovaný väčší podiel respondérov s naldemedínom (46,4 %) v porovnaní s placebom (30,2 %) a rozdiel medzi skupinami (16,2 %) bol štatisticky významný (p<0,0001).
V podskupine non-LIR, v súlade s výsledkami v LIR podskupine, bol pozorovaný väčší podiel respondérov s naldemedínom (54,3 %) v porovnaní s placebom (38,9 %) a rozdiel medzi skupinami (15,6 %) bol štatisticky významný (p=0,0009).
V štúdii V9235 preukázali údaje o dlhodobej účinnosti, ktoré sa hodnotili ako sekundárny koncový ukazovateľ a boli definované ako zmena vo frekvencii BM medzi začiatkom štúdie a týždňom 52, že u pacientov v skupine na naldemedíne oproti skupine s placebom sa frekvencia BM zlepšila aj
v LIR (3,10 oproti 1,90, p=0,0210) aj v non-LIR (4,26 oproti 3,39, p=0,1349) podskupine.'
Klinické štúdie u pacientov s nádorovým ochorením a OICBezpečnosť a účinnosť naldemedínu bola tiež hodnotená v 2 randomizovaných, dvojito zaslepených
a placebom kontrolovaných štúdiách (V9222 a V9236) u pacientov s nádorovým ochorením a OIC.
Pacientov bolo potrebné liečiť opioidmi po dobu ≥ 14 dní pred skríningom a museli dostávať stabilnú dávku. Štúdie zahŕňali 2-týždňové obdobie skríningu, 2-týždňové obdobie liečby a 4-týždňové obdobie následného sledovania. U pacientov, ktorí boli liečení laxatívami pri návšteve pri skríningu,
musela liečba v stabilnej dávke pokračovať do konca obdobia liečby. Pacientom bolo dovolené užívať záchranné laxatívum (laxatíva) podľa potreby bez ohľadu na to, že boli na stabilnom laxatívnom režime na začiatku štúdie (okrem 24 hodín na začiatku obdobia liečby).
V štúdiách V9222 a V9236 bola OIC potvrdená počas 2-týždňového prípravného obdobia a bola definovaná ako ≤ 5 SBM počas 14 po sebe nasledujúcich dní pred randomizáciou
a ≥ 1 z nasledujúcich črevných príznakov u ≥ 25 % všetkých BM bez ohľadu na použitie záchranných laxatív: prítomnosť namáhania počas vyprázdňovania, pocit neúplného vyprázdnenia, prechod tvrdej stolice alebo malých hrudiek.
V štúdiách V9222 a V9236 bol priemerný vek pacientov 64,3 rokov; 51,8 % bolo vo veku 65 rokov alebo starších; 39,4 % boli ženy; 97,1 % boli Japonci.
Naldemedín 200 mikrogramov alebo placebo sa podávali pacientom s nádorovým ochorením a s OIC po dobu 2 týždňov. Primárnym koncovým ukazovateľom pre štúdiu V9236 a sekundárnym koncovým ukazovateľom, bez úpravy na multiplicitu, pre štúdiu V9222 boli podiel SBM respondérov počas
2-týždňového obdobia liečby. Respondér bol definovaný ako pacient s frekvenciou ≥ 3 SBM
na týždeň a s nárastom o ≥ 1 SBM na týždeň počas 2-týždňového obdobia liečby oproti hodnote na začiatku štúdie.
Tabuľka 5. Podiel SBM respondérov u pacientov s nádorovým ochorením a OIC počas
2-týždňového obdobia liečby (štúdie V9222 a V9236)
V9222 V9236
Pacienti odpovedajúc
Naldemedín
(N=58)
Placebo
(N=56)
Rozdiel v liečbe
[95 % Cl]
40,1 %
Naldemedín
(N=97)
Placebo
(N=96)
Rozdiel v liečbe
[95 % Cl]
36,8 %
i na liečbu, n
(%)
45 (77,6 %) 21 (37,5 %)
[23,5 %;
56,7 %]
69 (71,1 %) 33 (34,4 %)
[23,7 %;
49,9 %]

p-hodnota* <0,0001 <0,0001
*Štatisticky významné: p-hodnoty podľa chí-kvadrát testu.
PediatrickápopuláciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Rizmoicom
v jednej alebo vo viacerých podskupinách pediatrickej populácie pre opioidmi indukovanú obstipáciu
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiAbsorpciaNaldemedín sa absorbuje s časom do dosiahnutia maximálnej plazmatickej koncentrácie približne
0,75 hodiny v stave nalačno. Absolútna biologická dostupnosť naldemedínu nebola stanovená.
Absolútna biologická dostupnosť naldemedínu sa odhaduje na rozmedzie 20 % až 56 %.
Neexistuje žiadny klinicky významný účinok jedla. Maximálna plazmatická koncentrácia sa znížila o 35 % a čas dosiahnutia maximálnej plazmatickej koncentrácie sa predlžil z 0,75 hodiny v stave nalačno na 2,5 hodiny po jedle, zatiaľ čo v ploche pod krivkou závislosti plazmatickej koncentrácie na čase sa pri príjme potravy žiadny významný rozdiel nepozoroval. Na základe týchto údajov sa naldemedín môže užívať s jedlom alebo bez jedla (pozri časť 4.2).
DistribúciaNaldemedín sa s vysokou afinitou viaže na sérové proteíny, predovšetkým na ľudský sérový albumín
a v menšej miere na α1-kyslý-glykoproteín a γ-globulín s priemerným pomerom väzby na proteíny
u ľudí 93,2 %. Zdanlivý distribučný objem je približne 155 litrov.
BiotransformáciaNaldemedín sa metabolizuje predovšetkým prostredníctvom CYP3A na nor-naldemedín, s malým podielom UGT1A3 za vzniku naldemedínu 3-G.
Po perorálnom podaní [14C]-označeného naldemedínu bol primárnym metabolitom v plazme nor-
naldemedín s relatívnou expozíciou v porovnaní s naldemedínom približne 9 až 13 %. Naldemedín
3-G bol vedľajším metabolitom v plazme s relatívnou expozíciou vzhľadom k naldemedínu menej ako
3 %.
Naldemedín tiež podlieha štiepeniu v gastrointestinálnom trakte za vzniku benzamidínu a karboxylovej kyseliny naldemedínu.
Naldemedín v štúdiách in vitro pri klinicky významných koncentráciách neinhiboval hlavné CYP enzýmy (vrátane CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A alebo CYP4A11 izoenzýmy) a nebol inhibítorom transportérov OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, BCRP, P-gp, MATE1, MATE2-K alebo BSEP. Naldemedín nespôsobil významnú indukciu izoenzýmov CYP1A2, CYP2B6 alebo CYP3A4. Preto sa neočakáva, že by liečba naldemedínom zmenila farmakokinetiku súbežne podávaných liekov, ktoré sú substrátmi týchto enzýmov a transportérov.
Eliminácia
Zdanlivý terminálny polčas eliminácie naldemedínu je približne 11 hodín a zdanlivý celkový klírens (CL/F) naldemedínu je 8,4 l/h. Po perorálnom podaní rádiologicky označeného naldemedínu sa 57,3 % a 34,8 % dávky vylúčilo močom a stolicou vo forme [oxadiazol-14C]-naldemedín, a 20,4 % a 64,3 % dávky sa vylúčilo močom a stolicou ako [karbonyl-14C]-naldemedín, v uvedenom poradí. Približne
20 % dávky naldemedínu sa vylučuje močom v nezmenenej forme.
Linearita/nelinearita
Maximálna plazmatická koncentrácia a plocha pod krivkou závislosti plazmatickej koncentrácie na čase sa zvýšili spôsobom takmer proporcionálnym dávke v rozsahu dávok od 0,1 do 100 mg. Po podávaní viacnásobnej dávky raz denne nalačno po dobu 10 dní sa pozorovala mierna akumulácia (1- až 1,3-násobne) pre maximálnu plazmatickú koncentráciu a plochu pod krivkou závislosti plazmatickej koncentrácie na čase.
Farmakokinetikavsubpopuláciách
Vek, pohlavie, telesná hmotnosť a rasa
Populačná farmakokinetická analýza z klinických štúdií s naldemedínom neidentifikovala klinicky
významný účinok veku, pohlavia, telesnej hmotnosti alebo rasy na farmakokinetiku naldemedínu. Farmakokinetika naldemedínu v pediatrickej populácii sa neskúmala (pozri časť 4.2).
Porucha funkcie obličiek
Farmakokinetika naldemedínu po podaní jednorazovej dávky naldemedínu 200 mikrogramov sa
skúmala u pacientov s miernou, stredne ťažkou alebo ťažkou poruchou funkcie obličiek, alebo
u pacientov s terminálnym štádiom ochorenia obličiek (end-stage renal disease, ESRD), ktorých stav vyžadoval hemodialýzu, a porovnávala sa so zdravými jedincami s normálnou funkciou obličiek.
Farmakokinetika naldemedínu u pacientov s miernou, stredne ťažkou alebo ťažkou poruchou funkcie obličiek, alebo pacientov s ESRD, ktorých stav vyžadoval hemodialýzu, a u zdravých jedincov
s normálnou funkciou obličiek, bola podobná.
Plazmatická koncentrácia naldemedínu u pacientov s ESRD, ktorých stav vyžadoval dialýzu, bola podobná pri podávaní naldemedínu či pred alebo po hemodialýze, čo naznačuje, že naldemedín nebol odstránený z krvi hemodialýzou.
Porucha funkcie pečene
Vplyv poruchy funkcie pečene na farmakokinetiku jednorazovej dávky naldemedínu
200 mikrogramov sa skúmal u pacientov s poruchou funkcie pečene klasifikovanou ako mierna (Child-Pugh trieda A) alebo stredne ťažká (Child-Pugh trieda B), a porovnával sa so zdravými jedincami s normálnou funkciou pečene. Farmakokinetika naldemedínu u pacientov s miernou alebo stredne ťažkou poruchou funkcie pečene a zdravých jedincov s normálnou funkciou pečene bola podobná. Vplyv ťažkej poruchy funkcie pečene (Child-Pugh trieda C) na farmakokinetiku naldemedínu sa nehodnotil.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity, karcinogénneho potenciálu a embryo-fetálneho vývinu neodhalili žiadne osobitné riziko pre ľudí.
V štúdii fertility a skorého embryonálneho vývoja na potkanoch bolo pri dávke 10 mg/kg/deň a vyššej pozorované predĺženie fázy diestru, čo však nebolo pozorované pri dávke 1 mg/kg/deň (12-násobok expozície [AUC0-24h] u ľudí pri perorálnej dávke 200 mikrogramov). Vplyv na estrálny cyklus sa nepovažuje za klinicky významný pri navrhovanej terapeutickej dávke. Nepozorovali sa žiadne nežiaduce účinky na fertilitu u samcov alebo samíc a reprodukčnú schopnosť pri dávkach až
do 1 000 mg/kg/deň (presahujúcich 16 000-násobok expozície [AUC0-24h] u ľudí pri perorálnej dávke
200 mikrogramov).
V štúdii pre- a postnatálneho vývoja na potkanoch uhynula jedna samica pri pôrode pri dávke
1 000 mg/kg/deň, a pri dávkach 30 a 1 000 mg/kg/deň bolo zaznamenané slabé dojčenie, menšie prírastky telesnej hmotnosti a zníženie konzumácie potravy. Pri dávkach 30 a 1 000 mg/kg/deň bol zaznamenaný pokles indexu životaschopnosti na 4. deň po narodení a pri dávke 1 000 mg/kg/deň sa u mláďat zaznamenalo oneskorené rozloženie ušnice. Nepozorovali sa žiadne nežiaduce účinky na
pre- a postnatálny vývoj pri dávke 1 mg/kg/deň (12-násobok expozície [AUC0-24h] u ľudí pri perorálnej dávke 200 mikrogramov).
U gravidných potkanov sa pozoroval placentárny prenos rádioaktivity odvodenej od [karbonyl-14C]- naldemedínu. U dojčiacich potkanov sa rádioaktivita odvodená od [karbonyl-14C]-naldemedínu vylučovala do mlieka.
V štúdiách toxicity na juvenilných potkanoch pri rovnakých dávkach sa expozícia (PND 10)
u mladých zvierat zvýšila porovnateľne s dospelými zvieratami (2,3- až 7,4-krát). Pri všetkých
testovaných dávkach sa u samíc vo vaječníkoch pozorovali ďalšie histopatologické nálezy (terciálne folikuly/luteálne cysty) popri nepravidelných estrálnych cykloch, hyperplázii prsných žliaz a zmene vaginálneho epitelu na bunky produkujúce mukózny sekrét, ktoré sa už predtým pozorovali
u dospelých zvierat (najnižšia testovaná dávka korešpondovala s hraničnou expozíciou 6 alebo viac
v závislosti od veku mláďat). Tiež sa pozorovalo o tri dni skoršie otváranie vagíny naznačujúce včasný nástup sexuálnej zrelosti, čo sa ale vyskytlo len pri vysokých expozíciách, ktoré sa považovali za dostatočne presahujúce maximum expozície u ľudí pri perorálnej dávke 200 mikrogramov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety
Manitol
Sodná soľ kroskarmelózy
Stearan horečnatý
Obaltablety Hypromelóza Mastenec
Žltý oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie. Uchovávajte v pôvodnom obale na ochranu pred svetlom a vlhkosťou.
6.5 Druh obalu a obsah baleniaHliník/hliníkový blister obsahujúci 7, 10 alebo 14 filmom obalených tabliet. Veľkosti balenia: 7, 10, 28, 30, 84 alebo 100 filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIShionogi B.V. Kingsfordweg 151
1043GR Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/18/1291/001
EU/1/18/1291/002
EU/1/18/1291/003
EU/1/18/1291/004
EU/1/18/1291/005
EU/1/18/1291/006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 18. február 2019
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.