QINLOCK 50 MG TABLETY tbl 90x50 mg (fľ.HDPE)

valign="top"> 3. alebo
4. stupeň
Natrvalo ukončite liečbu.
| Artralgia alebo myalgia (pozri časť 4.8)
| 2. stupeň
| • Prerušte liečbu kým sa nedosiahne stav ≤ 1. stupňu alebo
východiskový stav. Ak sa zotaví do 7 dní, pokračujte v liečbe
s rovnakou dávkou. V opačnom prípade pokračujte v liečbe so zníženou dávkou.
• Ak sa udrží na stupni ≤ 1 alebo vo východiskovom stave najmenej 28 dní, zvážte opätovné zvýšenie dávky.
• Ak sa artralgia alebo myalgia znova objaví, liečbu prerušte až do dosiahnutia stavu ≤ 1. stupňu alebo východiskového stavu, a potom pokračujte so zníženou dávkou bez ohľadu na čas do zlepšenia.
|
3. stupeň
| • Prerušte liečbu aspoň na 7 dní alebo kým sa nedosiahne stav
≤ 1. stupňu alebo východiskový stav (maximálne 28 dní). Pokračujte v liečbe so zníženou dávkou.
• Ak sa udrží na stupni ≤ 1 alebo vo východiskovom stave najmenej 28 dní, zvážte opätovné zvýšenie dávky.
|
Iné nežiaduce reakcie (pozri časť 4.8)
| 3. alebo
4. stupeň
| • Prerušte liečbu, kým sa nedosiahne stav ≤ 1. stupňu alebo východiskový stav (maximálne 28 dní), a potom v liečbe
pokračujte so zníženou dávkou. V opačnom prípade liečbu natrvalo ukončite.
• Ak sa nežiaduca reakcia nezopakuje najmenej 28 dní, zvážte opätovné zvýšenie dávky.
• Ak sa 3. alebo 4. stupeň znova objaví, liečbu natrvalo ukončite.
|
a Odstupňovaná podľa spoločných terminologických kritérií pre nežiaduce udalosti Národného inštitútu pre rakovinu verzia 4.03 (National Cancer Institute Common Terminology Criteria for Adverse Events, NCI CTCAE v4.03).
S
úbežne podávané lieky
Má sa predchádzať súbežnému podávaniu liekov, ktoré sú silnými alebo stredne silnými induktormi
enzýmu CYP3A (pozri časti 4.4 a 4.5). Ak sa musí súbežne podávať silný alebo stredne silný induktor enzýmu CYP3A, frekvencia dávkovania lieku QINLOCK sa môže počas obdobia súbežného podávania zvýšiť. V prípade silných induktorov enzýmu sa dávka môže zvýšiť zo 150 mg raz denne
na 150 mg dvakrát denne. V prípade, že pacient užíva liek QINLOCK dvakrát denne a vynechá dávku
do 4 hodín od času, kedy liek zvyčajne užíva, má byť poučený o tom, aby vynechanú dávku užil čo najskôr a ďalšiu dávku potom užil v pravidelnom naplánovanom čase. Ak pacient vynechá dávku
o viac ako 4 hodiny po čase, kedy liek zvyčajne užíva, má byť poučený o tom, aby vynechanú dávku neužil a jednoducho pokračoval vo zvyčajnom dávkovacom režime. U týchto pacientov sa odporúča dôkladné monitorovanie celkovej účinnosti a bezpečnosti.
Osobitné populácie
Porucha funkcie obličiek
V prípade pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek sa úprava dávky
neodporúča (pozri časť 5.2). V prípade pacientov so závažnou poruchou funkcie obličiek [klírens kreatinínu (CLcr) < 30 ml/min] sú k dispozícii len obmedzené klinické údaje. V prípade pacientov so závažnou poruchou funkcie obličiek nebola odporúčaná dávka lieku QINLOCK stanovená (pozri
časť 5.2).
Porucha funkcie pečene
U pacientov s miernou poruchou funkcie pečene sa úprava dávky neodporúča. V prípade pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene nebola odporúčaná dávka lieku QINLOCK stanovená. U týchto pacientov sa odporúča dôkladné monitorovanie celkovej bezpečnosti (pozri
časť 5.2).
Starší pacienti
V klinických štúdiách neboli pozorované žiadne klinicky významné rozdiely medzi staršími (vo veku
> 65 rokov) a mladšími pacientmi (vo veku ≤ 65 a ≥ 18 rokov) (pozri časť 5.1).
Pediatrická populácia
Bezpečnosť a účinnosť lieku QINLOCK u detí mladších ako 18 rokov nebola stanovená (pozri časť 5.1). K dispozícii nie sú žiadne údaje.
Spôsob podávania
Liek QINLOCK je určený na perorálne použitie.
Tablety sa majú užívať každý deň v rovnakom čase s jedlom alebo bez jedla (pozri časť 5.2).
Lekári predpisujúci liek majú pacientov poučiť, aby tablety užili celé a aby ich nežuvali, nelámali ani nedrvili. Pacienti nemajú užívať tablety, ktoré sú zlomené, prasknuté alebo inak porušené, pretože
v prípade takýchto zmien neboli vyhodnotené potenciálne účinky.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Syndrómpalmárno-plantárnejerytrodyzestézie(syndrómPPE)
U pacientov liečených ripretinibom sa vyskytol syndróm PPE (pozri časť 4.8). Na základe závažnosti
sa má podávanie ripretinibu prerušiť, a potom sa má v ňom pokračovať v rovnakej alebo zníženej dávke (pozri časť 4.2).
H
ypertenzia
Pri liečbe ripretinibom bola pozorovaná hypertenzia (pozri časť 4.8). Ripretinib sa nesmie začať podávať, pokiaľ krvný tlak nie je pod primeranou kontrolou. Krvný tlak sa má monitorovať podľa klinických indikácií. Na základe závažnosti sa má podávanie ripretinibu prerušiť, a potom sa má v ňom pokračovať v rovnakej alebo zníženej dávke, alebo sa má jeho podávanie natrvalo ukončiť (pozri časť 4.2).
Zlyhaniesrdca
Pri liečbe ripretinibom bolo pozorované zlyhanie srdca (vrátane zlyhania srdca, akútneho zlyhania srdca, akútneho zlyhania ľavej komory a diastolickej dysfunkcie) (pozri časť 4.8). Pred začatím podávania ripretinibu a počas liečby sa podľa klinickej indikácie má ejekčná frakcia posúdiť pomocou echokardiogramu alebo skenovania MUGA (multiple-gated acquisition) Pri systolickej dysfunkcii ľavej komory 3. alebo 4. stupňa je potrebné podávanie ripretinibu natrvalo ukončiť (pozri časť 4.2). Bezpečnosť ripretinibu nebola hodnotená u pacientov s východiskovou ejekčnou frakciou ľavej komory nižšou ako 50 %.
Skvamocelulárnykarcinómkože(CuSCC)
U pacientov užívajúcich ripretinib bol hlásený CuSCC (pozri časť 4.8). Na začiatku podávania ripretinibu a pravidelne počas liečby sa majú vykonať dermatologické hodnotenia. Podozrivé kožné lézie sa majú riešiť excíziou a dermatopatologickým hodnotením. Ripretinib sa má naďalej podávať v rovnakej dávke.
Komplikácieprihojenírán
Nevykonali sa žiadne formálne štúdie na vyhodnotenie účinku ripretinibu na hojenie rán. U pacientov, ktorí užívajú lieky inhibujúce signálnu dráhu vaskulárneho endotelového rastového faktora (VEGF) sa môžu vyskytnúť komplikácie pri hojení rán. Ripretinib má preto potenciál nepriaznivo vplývať na hojenie rán.
Liečba ripretinibom sa má prerušiť najmenej na 3 dni pred menšou operáciou a po nej a najmenej
5 dní pred veľkou operáciou a po nej. Na základe klinického posúdenia primeraného hojenia rán možno potom po operácii v liečbe ripretinibom pokračovať.
Embryofetálnatoxicita
Na základe zistení zo štúdií na zvieratách môže ripretinib spôsobiť poškodenie plodu, ak sa podáva tehotným ženám (pozri časti 4.6 a 5.3). Odporúča sa ženy poučiť, aby počas užívania ripretinibu predchádzali otehotneniu. Pred začatím užívania ripretinibu a počas liečby sa musí u žien s reprodukčným potenciálom overiť, či nie sú tehotné. Ženy s reprodukčným potenciálom a muži
s partnerkami s reprodukčným potenciálom musia počas liečby a aspoň 1 týždeň po poslednej dávke ripretinibu používať účinnú antikoncepciu (pozri časti 4.6 a 5.3). Účinky ripretinibu na antikoncepčné steroidy neboli skúmané. Ak sa používajú systémové antikoncepčné steroidy, má sa pridať bariérová metóda antikoncepcie.
Fototoxicita
Ripretinib vykazuje potenciál fototoxicity (pozri časť 5.3). Odporúča sa poučiť pacientov, aby sa
vzhľadom na riziko fototoxicity súvisiace s ripretinibom vyhli vystaveniu priamemu slnečnému svetlu, solárnym svietidlám a iným zdrojom ultrafialového žiarenia alebo aby takéto vystavenie minimalizovali. Pacienti majú byť poučení o opatreniach, ako je používanie ochranného odevu (dlhé rukávy a pokrývka hlavy) a opaľovacích krémov s vysokým slnečným ochranným faktorom (SPF).
InhibítoryainduktoryenzýmuCYP3A
Ripretinib je substrát enzýmu CYP3A. Súbežné podávanie ripretinibu so silným inhibítorom enzýmu CYP3A a P-glykoproteínu (P-gp) itrakonazolom viedlo k zvýšeniu plazmatickej expozície ripretinibu (pozri časť 4.5). Pri podávaní ripretinibu s látkami, ktoré sú silnými inhibítormi enzýmu CYP3A a P- gp, sa vyžaduje opatrnost.
Súbežné podávanie ripretinibu so silným induktorom enzýmu CYP3A rifampicínom viedlo k zníženiu plazmatickej expozície ripretinibu. Má sa preto predchádzať chronickému podávaniu silných alebo stredne silných induktorov enzýmu CYP3A s ripretinibom (pozri časti 4.2 a 4.5).
Dôležitéinformácieoniektorýchpomocnýchlátkach
Liek QINLOCK obsahuje laktózu.
Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nemajú tento liek užívať.
4.5 Liekové a iné interakcie
Ripretinib aj jeho aktívny metabolit DP-5439 sa eliminujú najmä prostredníctvom enzýmu CYP3A4/5
a sú substrátmi P-gp a proteínu rezistencie voči rakovine prsníka (BCRP).
Účinok iných liekov na ripretinib
Účinok silných inhibítorov enzýmu CYP3A/P-gp
Súbežné podávanie itrakonazolu (silného inhibítora enzýmu CYP3A) a tiež inhibítora P-gp zvýšilo hodnoty ripretinibu Cmax o 36 % a AUC0-∞ o 99 %. Hodnota DP-5439 Cmax sa nezmenila; hodnota AUC0-∞ sa zvýšila o 99 %. Silné inhibítory enzýmu CYP3A/P-gp (napr. ketokonazol, erytromycín, klaritromycín, itrakonazol, ritonavir, posakonazol a vorikonazol) sa majú používať opatrne a pacienti sa majú monitorovať. Neodporúča sa podávanie grapefruitovej šťavy.
Účinok induktorov enzýmu CYP3A
Súbežné podávanie lieku QINLOCK so silným induktorom enzýmu CYP3A rifampicínom znížilo hodnoty ripretinibu Cmax o 18 % a AUC0-∞ o 61 %, znížilo hodnotu DP-5439 AUC0-∞ o 57 % a zvýšilo hodnotu DP-5439 Cmax o 37 %.
Preto sa má predchádzať súbežnému užívaniu lieku QINLOCK so silnými induktormi enzýmu
CYP3A (ako sú napr. karbamazepín, fenytoín, rifampicín, fenobarbital a ľubovník bodkovaný)
a stredne silnými induktormi enzýmu CYP3A (ako sú napr. efavirenz a etravirín). Ak sa musí súbežne podávať silný alebo stredne silný induktor enzýmu CYP3A, frekvencia dávkovania lieku QINLOCK sa môže počas obdobia súbežného podávania zvýšiť. V prípade silných induktorov enzýmu sa dávka môže zvýšiť zo 150 mg raz denne na 150 mg dvakrát denne. V prípade, že pacient užíva liek QINLOCK dvakrát denne a vynechá dávku do 4 hodín od času, kedy liek zvyčajne užíva, má byť poučený o tom, aby vynechanú dávku užil čo najskôr a ďalšiu dávku potom užil v pravidelnom naplánovanom čase. Ak pacient vynechá dávku o viac ako 4 hodiny od času, kedy liek zvyčajne užíva, pacienta je potrebné poučiť, aby vynechanú dávku neužil a jednoducho pokračoval vo zvyčajnom dávkovacom režime. Monitorujte klinickú odpoveď a znášanlivosť.
Účinok látok znižujúcich kyslosť
Pri súbežnom podávaní lieku QINLOCK s pantoprazolom (inhibítorom protónovej pumpy) neboli pozorované žiadne klinicky významné rozdiely v plazmatickej expozícii ripretinibu a DP-5439.
Systémy transportérov liekov
Na základe údajov in vitro sa lieky, ktoré sú inhibítormi BCRP (napr. cyklosporín A, eltrombopag), majú v kombinácii s liekom QINLOCK používať opatrne, pretože môže dôjsť k zvýšeným plazmatickým koncentráciám ripretinibu alebo DP-5439.
Účinok ripretinibu na iné lieky
Substráty selektívne pre izoformu CYP
Zo štúdií in vitro vyplynulo, že ripretinib môže inhibovať CYP2C8. QINLOCK sa má používať opatrne v kombinácii so substrátmi CYP2C8 (ako sú napr. repaglinid, paklitaxel), pretože súbežné podávanie môže viesť k zvýšenej expozícii substrátov CYP2C8.
Čistý účinok in vivo inhibície CYP3A4 v čreve a systémovej indukcie CYP3A4 nie je známy. Pri súbežnom podávaní ripretinibu s citlivými substrátmi CYP3A4 s úzkym terapeutickým oknom (ako sú napr. cyklosporín, takrolimus) alebo s takými, ktoré sa metabolizujú zväčša v čreve (napr. Midazolam) sa odporúča opatrnosť.
Ripretinib a DP-5439 indukovali CYP2B6 in vitro. Súbežné podávanie ripretinibu so substrátmi CYP2B6 s úzkym terapeutickým indexom (napr. efavirenzom) môže viesť k strate ich účinnosti. Ripretinib a DP-5439 znížili reguláciu CYP1A2 in vitro. Súbežné podávanie ripretinibu so substrátmi CYP1A2 s úzkym terapeutickým indexom (napr. tizanidínom) môže viesť k zvýšeniu koncentrácií
a odporúča sa monitorovanie.
Nie je známe, či ripretinib može znížiť účinnosť systémovo pôsobiacej hormonálnej antikoncepcie. Ženy, ktoré užívajú hormonálnu antikoncepciu so systémovým pôsobením, majú preto zároveň používať aj bariérovú metódu antikoncepcie.
Systémy transportérov liekov
Zo štúdií in vitro vyplýva, že ripretinib je inhibítor P-gp a BCRP. DP-5439 je substrátom pre P-gp a BCRP. DP-5439 je inhibítorom BCRP a multiliekového a toxínového extrúzneho proteínu 1 (MATE-1).
Lieky, ktoré sú substrátmi P-gp s úzkym terapeutickým indexom (napr. digoxín, dabigatran etexilát), sa majú v kombinácii s liekom QINLOCK používať opatrne vzhľadom na pravdepodobnosť zvýšenia plazmatických koncentrácií týchto substrátov.
Liek QINLOCK sa má v kombinácii so substrátmi BCRP (ako sú napr. rosuvastatín, sulfasalazín a irinotekan) a substrátmi MATE-1 (ako je napr. metformín) používať opatrne, pretože súbežné podávanie lieku QINLOCK so substrátmi BCRP a MATE-1 môže viesť k zvýšeniu ich expozície. Klinické štúdie so substrátmi BCRP alebo MATE-1 neboli vykonané.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku/antikoncepciaumužovažien
Ženy vo fertilnom veku a muži s partnerkami s reprodukčným potenciálom musia byť informovaní, že QINLOCK môže poškodiť plod, a počas liečby a aspoň 1 týždeň po poslednej dávke lieku QINLOCK (pozri časť 4.4) musia používať účinnú antikoncepciu.
Pred začatím užívania lieku QINLOCK a počas liečby sa musí u žien s reprodukčným potenciálom overiť, či nie sú tehotné.
Účinky lieku QINLOCK na antikoncepčné steroidy neboli skúmané. Ak sa ako antikoncepcia používajú systémové steroidy, má sa používať aj bariérová metóda.
Gravidita
Nie sú k dispozícii žiadne údaje o použití ripretinibu u gravidných žien.
Na základe mechanizmu účinku existuje podozrenie, že ripretinib spôsobuje poškodenie plodu, ak sa podáva počas gravidity, a v štúdiách na zvieratách sa preukázala reprodukčná toxicita (pozri časti 4.4 a 5.3). Liek QINLOCK sa nemá počas gravidity používať, pokiaľ si klinický stav ženy nevyžaduje liečbu ripretinibom.
Dojčenie
Nie je známe, či sa ripretinib/metabolity vylučujú do ľudského mlieka. Riziko pre dojčené deti nie je
možné vylúčiť. Počas liečby liekom QINLOCK a aspoň jeden týždeň po poslednej dávke sa má dojčenie prerušiť.
Fertilita
K dispozícii nie sú žiadne údaje o vplyve ripretinibu na fertilitu u ľudí. Na základe zistení zo štúdií na
zvieratách môže liečba liekom QINLOCK zhoršiť fertilitu samcov a samíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liek QINLOCK nemá žiadny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U niektorých pacientov bola po podaní lieku QINLOCK hlásená únava. Ak pacient pociťuje únavu, môže to mať vplyv na jeho schopnosť viesť vozidlá alebo obsluhovať stroje.
4.8 Nežiaduce účinky
Zhrnutiebezpečnostnéhoprofilu
V dvojito zaslepenej, randomizovanej (2:1), placebom kontrolovanej štúdii fázy 3 (INVICTUS) bolo
129 účastníkov s diagnózou pokročilého GIST, u ktorých zlyhali aspoň 3 schválené predchádzajúce línie liečby, randomizovaných do skupiny s liekom QINLOCK (n = 85) alebo do skupiny s placebom (n = 44) (pozri časť 5.1). Do štúdie fázy 1 DCC-2618-01-001 bolo zaradených celkovo 277 pacientov s pokročilými malignitami a 218 pacientov bolo liečených odporúčanou dávkou vo fáze 2, a síce
150 mg lieku QINLOCK raz denne.
Medián trvania liečby s liekom QINLOCK v dvojito zaslepenej fáze štúdie INVICTUS bol
5,49 mesiaca.
Najčastejšie pozorované nežiaduce reakcie (≥ 25 %) u pacientov liečených liekom QINLOCK v súhrnnej populácii na hodnotenie bezpečnosti (n = 392) boli únava (51,0 %), alopécia (50,8 %), nauzea (39,8 %), myalgia (37,8 %), zápcha (37,2 %), hnačka (32,7 %), PPES (29,8 %), zníženie hmotnosti (26,5 %) a vracanie (25,8 %).
Nežiaduce reakcie (≥ 10 až < 25 %) pozorované u pacientov liečených liekom QINLOCK v súhrnnej populácii na hodnotenie bezpečnosti (n = 392) boli zvýšená lipáza (23,7 %), svalové kŕče (23,7 %), artralgia (21,2 %), bolesť hlavy (20,7 %), dyspnoe (20,2 %), hypertenzia (19,4 %), suchá koža
(17,6 %), bolesť chrbta (15,6 %), kašeľ (15,6 %), zvýšená hladina bilirubínu v krvi (14,0 %), periférny edém (13,8 %), hypofosfatémia (12,2 %), bolesť končatín (12,0 %), pruritus (11,0 %) a seboroická keratóza (11,0 %).
Nežiaduce reakcie 3./4. stupňa (≥ 2 %) pozorované u pacientov liečených liekom QINLOCK v súhrnnej populácii na hodnotenie bezpečnosti (n = 392) boli zvýšená lipáza (14,8 %), anémia
(14,0 %), bolesť brucha (8,2 %), hypertenzia (6,9 %), únava (4,1 %), hypofosfatémia (4,1 %), vracanie
(2,6 %), dyspnoe (2,0 %), hnačka (2,0 %) a zvýšená hladina bilirubínu v krvi (2,0 %). Závažné nežiaduce reakcie (≥ 1 %) pozorované u pacientov liečených liekom QINLOCK boli anémia (3,8 %), dyspnoe (2,3 %), vracanie (2,0 %), nauzea (1,8 %), únava (1,5 %), zvýšená hladina bilirubínu v krvi (1,3 %), zápcha (1,0 %) a svalová slabosť (1,0 %).
Tabuľkovýzoznamnežiaducichreakcií
Celkový bezpečnostný profil lieku QINLOCK je založený na súhrnných údajoch zhromaždených od
392 pacientov (súhrnná populácia na hodnotenie bezpečnosti), ktorí dostali najmenej 1 dávku lieku QINLOCK. Vykonali sa dve klinické štúdie s liekom QINLOCK u dospelých pacientov s pokročilými malignitami, ktoré tvoria primárny základ celkového hodnotenia bezpečnosti: hlavná štúdia fázy 3
u dospelých pacientov s diagnózou GIST, štúdia DCC-2618-03-001 (INVICTUS) (pozri časť 5.1)
a otvorená štúdia, ktorá sa ako prvá vykonala na ľuďoch, u dospelých pacientov s pokročilými malignitami (štúdia DCC-2618-01-001).
Dvojito zaslepená fáza štúdie INVICTUS vytvorila primárny základ pre stanovenie nežiaducich reakcií. Za nežiaduce reakcie na liek sa považovali nežiaduce udalosti, ktoré sa objavili počas liečby a ktoré mali aspoň o 5 % vyššie hodnoty v skupine s liekom QINLOCK v porovnaní so skupinou
s placebom, a nežiaduce udalosti, ktoré mali aspoň 1,5-krát vyššie hodnoty v skupine s liekom QINLOCK v porovnaní so skupinou s placebom v rámci štúdie INVICTUS. Nežiaduce udalosti, ktoré sa objavili počas liečby a ktoré boli identifikované v rámci štúdie INVICTUS, sa hodnotili aj v rámci súhrnnej populácie na hodnotenie bezpečnosti (n = 392). Podľa posúdenia zadávateľa boli tieto
udalosti považované za nežiaduce reakcie na liek. Sú klasifikované podľa triedy orgánových systémov
a na opis určitej reakcie, jej synoným a súvisiacich stavov sa používa najvhodnejší pojem podľa databázy MedDRA.
Závažnosť nežiaducich reakcií na liek bola hodnotená na základe spoločných terminologických kritérií pre nežiaduce udalosti (Common Terminology Criteria for Adverse Events, CTCAE), ktoré definujú
1. stupeň = mierny, 2. stupeň = stredne závažný, 3. stupeň = závažný, 4. stupeň = život ohrozujúci a 5. stupeň = úmrtie.
Frekvencie sú definované ako veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000
až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000) a neznáme
(z dostupných údajov). Sú uvedené v tabuľke 2. V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie uvedené v poradí podľa klesajúcej závažnosti.
Tabuľka 2: Nežiaduce reakcie na liek hlásené v štúdii INVICTUS a DCC-2618-01-001Benígne a malígne nádory vrátane nešpecifikovaných novotvarov (vrátane cýst a polypov)
| Veľmi časté
| Seboroická keratóza
| Časté
| Melanocytický naevus, kožný papilóm, skvamocelulárny karcinóm kožea,
fibrózny histiocytóm
| Poruchy endokrinného systému
| Časté
| Hypotyreóza
| Poruchy metabolizmu a výživy
| Veľmi časté
| Hypofosfatémia
| Psychické poruchy
| Časté
| Depresia
| Poruchy nervového systému
| Veľmi časté
| Bolesť hlavy
| Časté
| Periférna senzorická neuropatia
| Poruchy srdca a srdcovej činnosti
| Časté
| Zlyhanie srdcab, tachykardia
| Poruchy ciev
| Veľmi časté
| Hypertenziac
| Poruchy dýchacej sústavy, hrudníka a mediastína
| Veľmi časté
| Dyspnoe, kašeľ
| Poruchy gastrointestinálneho traktu
| Veľmi časté
| Nauzea, zápcha, hnačka, vracanie
| Časté
| Stomatitída, bolesť v hornej časti brucha
| Poruchy kože a podkožného tkaniva
| Veľmi časté
| Alopécia, syndróm PPE, suchá koža, pruritus
| Časté
| Hyperkeratóza, makulopapulárna vyrážka, generalizovaný pruritus,
akneiformná dermatitída
| Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| Veľmi časté
| Myalgia, svalové kŕče, artralgia, bolesť chrbta, bolesť v končatinách
| Časté
| Svalová slabosť, muskuloskeletálna bolesť v hrudníku
| Celkové poruchy a reakcie v mieste podania
| Veľmi časté
| Únava, periférny edém
| Laboratórne a funkčné vyšetrenia
| Veľmi časté
| Zníženie telesnej hmotnosti, zvýšenie lipázy, zvýšenie hladiny bilirubínu v krvi
| Časté
| Zvýšená hladina alanínaminotransferázy
|
|
|
aSkvamocelulárny karcinóm kože (Skvamocelulárny karcinóm kože, keratoakantóm, skvamocelulárny karcinóm hla vy a krku).
bZlyha nie srdca (zlyhanie srdca, a kútne zlyhanie ľa vej komory, a kútne zlyhanie srdca, diastolická dysfunkcia).
cHypertenzia (hypertenzia, zvýšený krvný tlak).
O
pis vybraných nežiaducich reakcií na liekSyndrómpalmárno-plantárnejerytrodyzestézie(syndrómPPE)V dvojito zaslepenej fáze štúdie INVICTUS bol syndróm PPE hlásený u 19 z 85 (22,4 %) pacientov v skupine s liekom QINLOCK a v skupine s placebom nebol hlásený u žiadneho pacienta. Syndróm PPE viedol k ukončeniu podávania dávok u 1,2 % pacientov, prerušeniu podávania dávok
u 3,5 % pacientov a zníženiu dávky u 2,4 % pacientov. Všetky udalosti boli mierne alebo stredne závažné (58 % udalostí bolo 1. stupňa a 42 % udalostí bolo 2. stupňa).
V súhrnnej populácii na hodnotenie bezpečnosti sa syndróm PPE vyskytol u 29,8 % z 392 pacientov vrátane nežiaducich reakcií 3. stupňa u 0,5 %. Medián času do nástupu a trvanie prvej udalosti bol
8,1 týždňa (rozsah: 0,3 týždňa až 112,1 týždňa) a 24,3 týždňa (rozsah: 0,9 týždňa až 191,7 týždňa)
v uvedenom poradí. Ďalšie informácie nájdete v častiach 4.2 a 4.4.
HypertenziaV dvojito zaslepenej fáze štúdie INVICTUS sa u pacientov liečených pomocou lieku QINLOCK (15,3 %) zistil vyšší výskyt hypertenzie (všetky udalosti bez ohľadu na príčinnú súvislosť)
v porovnaní so 4,7 % z pacientov, ktorí dostávali placebo.
V súhrnnej populácii na hodnotenie bezpečnosti sa hypertenzia vyskytla u 19,4 % z 392 pacientov vrátane nežiaducich reakcií 3. stupňa u 6,9 %. Ďalšie informácie nájdete v častiach 4.2 a 4.4.
ZlyhaniesrdcaV dvojito zaslepenej fáze štúdie INVICTUS sa vyskytlo zlyhanie srdca (všetky udalosti bez ohľadu na
príčinnú súvislosť) u 1,2 % z 85 pacientov, ktorí dostávali liek QINLOCK. Zlyhanie srdca viedlo k ukončeniu podávania dávok u 1,2 % z 85 pacientov, ktorí dostávali liek QINLOCK.
V súhrnnej populácii na hodnotenie bezpečnosti sa srdcové zlyhanie vyskytlo u 1,5 % z 392 pacientov vrátane nežiaducich reakcií 3. stupňa u 1,0 %.
V súhrnnej populácii na hodnotenie bezpečnosti malo 299 z 392 pacientov k dispozícii echokardiogram vo východiskovom stave a aspoň jeden echokardiogram po východiskovom stave. Znížená ejekčná frakcia ľavej komory 3. stupňa sa vyskytla u 4,0 % z 299 pacientov.
Ďalšie informácie sú uvedené v časti 4.4.
KožnémalignityV dvojito zaslepenej fáze štúdie INVICTUS bol CuSCC (všetky udalosti bez ohľadu na príčinnú súvislosť) hlásený u 5,9 % z 85 pacientov, ktorí dostávali liek QINLOCK. CuSCC kože nebol hlásený u pacientov liečených placebom. Ďalšie informácie nájdete v častiach 4.2 a 4.4.
V spoločnej bezpečnostnej populácii sa CuSCC vyskytol u 8,7 % z 392 pacientov vrátane nežiaducich reakcií 3. stupňa u 0,5 %. Melanóm (všetky udalosti bez ohľadu na príčinnú súvislosť) sa vyskytol
u 0,3 % z 392 pacientov.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNa predávkovanie liekom QINLOCK nie je známe žiadne konkrétne antidotum.
V prípade podozrenia na predávkovanie sa liečba liekom QINLOCK musí okamžite zastaviť
a zdravotnícky pracovník má začať poskytovať najlepšiu podpornú starostlivosť, pričom pacient musí
byť pozorovaný až do klinickej stabilizácie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: antineoplastické látky, iné inhibítory proteínkinázy, ATC kód: L01EX19
Mechanizmusúčinku
Ripretinib je nový inhibítor tyrozínkinázy, ktorý inhibuje KIT proto-onkogénový receptor tyrozínkinázu a PDGFRA kinázu vrátane mutácií divokého typu, primárnych a sekundárnych mutácií. Ripretinib tiež inhibuje ďalšie kinázy in vitro, ako sú PDFRB, TIE2, VEGFR2 a BRAF.
Klinickáúčinnosťabezpečnosť
INVICTUS (štúdia DCC-2618-03-001)
Účinnosť a bezpečnosť lieku QINLOCK boli hodnotené v randomizovanej (2:1) dvojito zaslepenej, placebom kontrolovanej štúdii (štúdia INVICTUS) u pacientov s neresekovateľným, lokálne pokročilým alebo metastatickým GIST, ktorí boli v minulosti liečení najmenej tromi protirakovinovými terapiami vrátane liečby imatinibom, sunitinibom a regorafenibom, alebo ich netolerovali. V rámci randomizácie boli pacienti rozdelení podľa predchádzajúcich línií liečby (3 proti
≥ 4) a podľa výkonnostného stavu skupiny Eastern Cooperative Oncology Group (ECOG) (0 proti 1
alebo 2).
Primárnym meradlom výslednej účinnosti bolo prežitie bez progresie (PFS) na základe posúdenia ochorenia podľa zaslepeného nezávislého centrálneho preskúmania (BICR) s použitím upravených kritérií RECIST 1.1, podľa ktorých lymfatické uzliny a kostné lézie neboli cieľové lézie a postupne rastúca nová uzlina nádoru v rámci už existujúcej nádorovej hmoty musí spĺňať osobitné kritériá, aby sa mohla považovať za jednoznačný dôkaz progresie. Sekundárne koncové ukazovatele účinnosti zahŕňali mieru objektívnej odpovede (ORR) podľa BICR, celkové prežitie (OS) a zdravotný stav hlásený pacientom, fyzickú funkciu (PF) a rolovú funkciu (RF).
Účastníci boli randomizovaní do skupín a podávalo sa im 150 mg lieku QINLOCK (n = 85) alebo placebo (n = 44), a to perorálne raz denne v kontinuálnych 28-dňových cykloch. Liečba pokračovala, kým sa nezaznamenala progresia ochorenia alebo neakceptovateľná toxicita. Jednotlivé liečebné skupiny prestali byť zaslepené v čase progresie ochorenia podľa hodnotenia BICR a všetkým pacientom v skupine s placebom sa ponúkol prechod na užívanie lieku QINLOCK.
Demografické charakteristiky boli medián veku 60 rokov (29 až 83 rokov), so 79 (61,2 %) pacientmi vo veku 18 až 64 rokov, s 32 (24,8 %) pacientmi vo veku 65 až 74 rokov a s 18 (13,9 %) pacientmi vo veku ≥ 75 rokov (neboli randomizovaní žiadni pacienti vo veku ≥ 85 rokov); muži (56,6 %); belosi (75,2 %); a výkonnostný stav skupiny ECOG sa rovnal 0 (41,9 %), 1 (49,6 %) alebo 2 (8,5 %). Šesťdesiattri percent (63 %) pacientov absolvovalo 3 predchádzajúce liečby a približne 37 % absolvovalo 4 alebo viaceré predchádzajúce liečby. Šesťdesiatšesť percent (66 %) pacientov randomizovaných do skupiny s placebom prešlo počas otvorenej fázy na užívanie lieku QINLOCK.
V primárnej analýze (uzávierka údajov 31. mája 2019) sa liek QINLOCK porovnával s placebom v štúdii INVICTUS. Liek QINLOCK preukázal z hľadiska prežitia bez progresie (PFS) prínos vo všetkých hodnotených podskupinách pacientov. Medián PFS určený podľa BICR (v mesiacoch) (95 % CI) bol 6,3 (4,6; 6,9) v prípade lieku QINLOCK oproti 1,0 (0,9; 1,7) v prípade placeba, HR'
(95 % CI) 0,15 (0,09; 0,25), hodnota p < 0,0001. Sekundárny koncový ukazovateľ ORR (%) bol 9,4 (4,2; 18) v prípade lieku QINLOCK oproti 0 (0; 8) v prípade placeba, pričom hodnota p predstavovala
0,0504 a nebola štatisticky významná. Medián OS (v mesiacoch) (95 % CI) bol 15,1 (12,3; 15,1) v prípade lieku QINLOCK oproti 6,6 (4,1; 11,6) v prípade placeba, pričom nominálna hodnota p
< 0,0004. OS sa nehodnotilo z hľadiska štatistickej významnosti v dôsledku postupu sekvenčného testovania pre sekundárne koncové ukazovatele ORR a OS.
Výsledky PFS, ORR a OS z novšej uzávierky údajov (10. augusta 2020) sú uvedené v tabuľke 3 a na obrázkoch 1 a 2. Výsledky PFS boli v jednotlivých podskupinách podobné na základe veku, pohlavia, regiónu, stavu ECOG a počtu predchádzajúcich línií liečby.
Tabuľka 3: Výsledky účinnosti štúdie INVICTUS (10. augusta 2020)
| QINLOCK (n = 85)
| Placebo (n = 44)
|
PFSa
|
Počet udalostí (%)
| 68 (80)
| 37 (84)
|
Progresívne ochorenie
| 62 (73)
| 32 (73)
|
Úmrtia
| 6 (7)
| 5 (11)
|
Medián PFS (v mesiacoch) (95 % IS)
| 6,3 (4,6; 8,1)
| 1,0 (0,9; 1,7)
|
HR (95 % IS)b
| 0,16 (0,10; 0,27)
|
ORRa
|
ORR (%)
| 11,8
| 0
|
(95 % IS)
| (5,8; 20,6)
| (0; 8)
|
OS
|
Počet úmrtí (%)
| 44 (52)
| 35 (80)
|
Medián OS (v mesiacoch) (95 % IS)
| 18,2 (13,1, NE)
| 6,3 (4,1; 10,0)
|
HR (95 % IS)b
| 0,42 (0,27; 0,67)
|
BICR = zas lepené nezávis lé centrálne pres kúmanie; CI = interval s poľahlivos ti (IS); HR = pomer rizika; ORR = miera objektívnej odpovede; NE = neodhadnuteľné; PFS = prežitie bez progres ie; OS = celkové prežitie
a Hodnotené podľa BICR.
b Pomer rizika je založený na Coxovom modeli proporcionálnej regres ie. Tento model zahŕňa liečbu a faktory randomizácie s tratifikácie ako fixné faktory.
 Obrázok 1: INVICTUS – Kaplan-Meierova krivka prežitia bez progresiea
Obrázok 1: INVICTUS – Kaplan-Meierova krivka prežitia bez progresieaMedián PFS (v mesiacoch) 95 % IS
Ripretinib 150 mg
raz denne: 6,3 (4,6; 8,1) Placebo: 1,0 (0,9; 1,7)
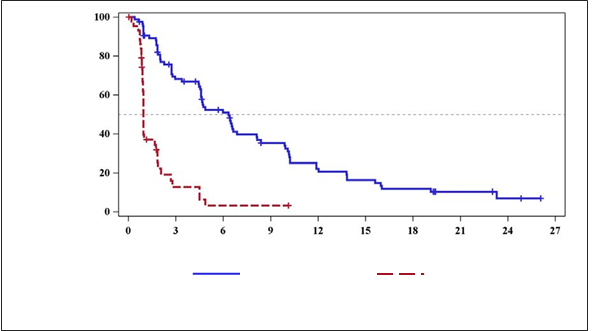 + cenzurovanéPrežitie bez progresie (v mesiacoch)
+ cenzurovanéPrežitie bez progresie (v mesiacoch)Počet rizikových pacientov
|
|
|
| i
| ti
|
| r
|
|
| l
|
|
|
|
Ripretinib 150 mg
|
|
|
|
|
|
|
|
|
|
|
|
|
|
raz denne
| 85
| 54
| 37
|
| 24
| 15
|
| 11
| 8
|
| 4
| 2
| 0
|
Placebo
| 44
| 4
| 1
|
| 1
| 0
|
|
|
|
|
|
|
|
R pre nib 150 mg az denne P acebo
|
|
a Ukončenie zberu údajov: 10. a ugust 2020
 O
b
r
ázok 2: INVICTUS – Kaplan-Meierova krivka celkového prežitia
a
O
b
r
ázok 2: INVICTUS – Kaplan-Meierova krivka celkového prežitia
a
Medián OS (v mesiacoch) 95 % IS
Ripretinib: 18,2 (13,1, NE) Placebo: 6,3 (4,1; 10,0)
+ cenzurovanéCelkové prežitie (v mesiacoch)
Ri
zikoví pacienti:

Ripretinib
Placebo
Ripretinib 85 76 59 49 39 32 12 2 0
Placebo 44 29 17 12 12 12 4 1 0
a Ukončenie zberu údajov: 10. a ugust 2020
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom QINLOCK vo všetkých podskupinách pediatrickej populácie pri liečbe GIST (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Absorpcia
Ripretinib dosahuje maximálne plazmatické koncentrácie v mediáne 4 hodiny po perorálnom podaní jednej dávky 150 mg ripretinibu (za predpokladu, že každá z troch tabliet obsahuje 50 mg). Priemerná hodnota (variačný koeficient v %) AUC0-∞ po jednej dávke 150 mg ripretinibu bola
9 856 (39 %) ng•h/ml pre ripretinib a 8 146 (56 %) ng•h/ml pre DP-5439.
Pri podávaní s jedlom s vysokým obsahom tuku sa pre ripretinib zvýšili hodnoty AUC0-24 o 30 %, a Cmax o 22 %. Pre DP-5439 sa zvýšili hodnoty AUC0-24 o 47 % a Cmax o 66 %.
Distribúcia
Ripretinib a jeho aktívny metabolit DP-5439 sa viažu na plazmatické proteíny s koncentráciou ≥ 99 %. Priemerný (variačný koeficient v %) zdanlivý objem distribúcie (Vss/F) je približne 302 (35 %) l pre ripretinib a 491 (38 %) l pre DP-5439.
Biotransformácia
CYP3A4/5 je hlavným metabolizátorom ripretinibu a jeho aktívneho metabolitu DP-5439, zatiaľ čo
CYP2C8 a CYP2D6 sú slabými metabolizátormi.
Eliminácia
Po perorálnom podaní jednej dávky 150 mg ripretinibu u ľudí bol priemerný (variačný koeficient v %) zdanlivý perorálny klírens (CL/F) 15,2 (39 %) l/h pre ripretinib a 17,9 (56 %) l/h pre DP-5439. Priemerný (variačný koeficient v %) polčas rozpadu (t½) bol 12,6 (17 %) hodiny pre ripretinib
a 15,6 (23 %) hodiny pre DP-5439.
Systémová eliminácia ripretinibu nebola primárne pripísaná obličke, pričom 0,02 % dávky ripretinibu sa vylúčilo v moči ako ripretinib a 0,1 % dávky ripretinibu ako DP-5439 a 34 % dávky ripretinibu sa vylúčilo v stolici ako ripretinib a 6 % dávky ripretinibu ako DP-5439.
P
r
oporcionalita
dávky
V dávkovacom rozsahu 20 až 250 mg sa ripretinib a DP-5439 PK javili byť menej ako úmerné dávke, najmä pri dávkach ripretinibu vyšších ako 150 mg.
Časovázávislosť
Podmienky ustáleného stavu sa dosiahnu do 14 dní.
Osobitnéskupiypacientov
Vo farmakokinetike lieku QINLOCK neboli pozorované žiadne klinicky významné rozdiely na základe veku (19 až 87 rokov), pohlavia, rasy (biela, čierna a ázijská), telesnej hmotnosti (39 až
138 kg) a nádoru (GIST alebo iné solídne nádory).
Pacienti s poruchou funkcie obličiek
V klinických štúdiách neboli pozorované žiadne relevantné rozdiely v expozícii medzi pacientmi
s miernou a stredne závažnou poruchou funkcie obličiek (CLcr 30 až 89 ml/min. podľa odhadu na základe Cockcroft-Gaultovho vzorca) a pacientmi s normálnou funkciou obličiek. Na základe populačnej farmakokinetickej analýzy sa neodporúča úprava dávky u pacientov s miernou a stredne závažnou poruchou funkcie obličiek. Farmakokinetika a bezpečnosť lieku QINLOCK u pacientov
so závažnou poruchou funkcie obličiek (CLcr 15 až 29 ml/min podľa odhadu na základe Cockcroft-
Gaultovho vzorca) je obmedzená. U pacientov so závažnou poruchou funkcie obličiek nie je možné stanoviť žiadne odporúčanie týkajúce sa dávkovania (pozri časť 4.2).
Pacienti s poruchou funkcie pečene
V klinických štúdiách neboli pozorované žiadne relevantné rozdiely v expozícii medzi pacientmi
s miernou (celkový bilirubín ≤ horný limit normálnej hodnoty (ULN) a AST > ULN alebo celkový bilirubín > ULN až ≤ 1,5 × ULN a akákoľvek hodnota AST) poruchou funkcie pečene a normálnou funkciou pečene. Na základe populačnej farmakokinetickej analýzy sa neodporúča úprava dávky
u pacientov s miernou poruchou funkcie pečene. Farmakokinetika a bezpečnosť lieku QINLOCK
u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene nebola skúmaná. V tejto podskupine nie je možné stanoviť žiadne odporúčanie týkajúce sa dávkovania (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Predklinický bezpečnostný profil ripretinibu bol hodnotený v prípade potkanov a psov až 13 týždňov. V prípade potkanov sa zaznamenali odpovede vo forme zápalu korelujúce so zmenami kože (zmena farby, lézie) (približne 1,12-násobok expozície u ľudí pri dávke 150 mg raz denne). V prípade oboch druhov bola hlásená zvýšená aktivita pečeňových enzýmov (v prípade potkanov približne 1,12- násobok a v prípade psov 1,3-násobok expozície u ľudí pri dávke 150 mg raz denne). V prípade psov sa prejavili gastrointestinálne účinky (vracanie a/alebo abnormálna stolica) (približne 1,3-násobok expozície u ľudí pri dávke 150 mg raz denne) a odpovede vo forme zápalu prejavujúce sa nežiaducimi kožnými léziami (približne 0,14-násobok expozície ľudí pri dávke 150 mg raz denne).
Karcinogenita
S ripretinibom sa neuskutočnili štúdie karcinogenity.
Genotoxicita
V mikronukleovej skúške in vitro bol ripretinib hodnotený ako pozitívny. Ripretinib nebol mutagénny v skúške bakteriálnej reverznej mutácie (Ames) in vitro ani v skúške kostnej drene potkanov in vivo,
čo dokazuje neprítomnosť významného genotoxického rizika.
Reprodukčnáavývojovátoxicita
Špecializované štúdie fertility v prípade samcov a samíc zvierat sa s ripretinibom neuskutočnili. V 13- týždňovej štúdii toxicity po opakovaných dávkach sa však v prípade samcov potkanov pozorovala degenerácia semenotvorného epitelu semenníkov a bunkové zvyšky nadsemenníkov v
prípade samcov, ktorým sa podávala dávka 30 alebo 300 mg/kg/deň, tieto nálezy sa ale považovali za
dostatočne závažné, aby ovplyvnili reprodukciu len pri dávke 300 mg/kg/deň (približne 1,4-násobok expozície u ľudí pri dávke 150 mg raz denne).
V hlavnej štúdii embryofetálneho vývoja bol ripretinib teratogénny v prípade potkanov, pričom vyvolal malformácie súvisiace s dávkou primárne spojené s viscerálnymi a skeletálnymi systémami pri materskej dávke 20 mg/kg/deň (približne 1,0-násobok expozície u ľudí pri dávke 150 mg raz denne). Okrem toho sa už pri dávke 5 mg/kg/deň pozorovali zmeny na skelete. Vývojová úroveň NOAEL pre ripretinib sa preto stanovila na dávke 1 mg/kg/deň (približne 0,02-násobok expozície u ľudí pri dávke
150 mg raz denne).
Štúdia skúmajúca účinky ripretinibu na prenatálny/postnatálny vývoj sa neuskutočnila. Fototoxicita
Ripretinib indikuje potenciál fotopodráždenia/fototoxicity na základe absorpcie vo viditeľnom rozsahu UV žiarenia (nad 290 nm). Z hodnotenia fototoxicity in vitro v myších fibroblastových bunkách 3T3 vyplýva, že ripretinib vykazuje potenciál fototoxicity v klinicky relevantných koncentráciách po vystavení UVA a UVB žiareniu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
krospovidón (E1202),
acetát-sukcinát hypromelózy, monohydrát laktózy,
stearát horečnatý (E470b), mikrokryštalická celulóza (E460),
koloidný hydratovaný oxid kremičitý (E551).
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek si nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie.
Uchovávajte v pôvodnom obale a udržiavajte fľašu tesne uzavretú na ochranu pred svetlom a vlhkosťou.
6.5 Druh obalu a obsah balenia
Biela fľaša z polyetylénu s vysokou hustotou (HDPE) s poistným krúžkom z hliníkovej fólie/polyetylénu (PE) a bielym polypropylénovým (PP) detským bezpečnostným uzáverom spolu s jednou nádobkou s desikantom PE obsahujúcou silikagél. Každá fľaša obsahuje 30 alebo 90 tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Deciphera Pharmaceuticals (Netherlands) B.V. Atrium Building 4th Floor
Strawinskylaan 3051
1077ZX, Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/21/1569/001
EU/1/21/1569/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 18. november 2021
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.