
40 % oproti východiskovej hodnote
a
• Potvrdené zníženie KKr > 40 % oproti východiskovej hodnote; vypočítajte pomocou Cockcroftovho-Gaultovho vzorca pre aktuálnu telesnú hmotnosť
Vysaďte Pluvicto až do zlepšenia stavu alebo návratu na východiskovú hodnotu. Znížte dávku Pluvicta o 20 %.
|
Rekurentná nefrotoxicita
(stupeň ≥ 3)
|
Trvalo ukončite liečbu Pluvictom.
|
Kompresia miechy
|
Akákoľvek
|
Vysaďte Pluvicto až do podania patričnej liečby kompresie miechy a stabilizovania
akýchkoľvek neurologických následkov a stabilizovania stavu výkonnosti podľa ECOG.
|
Zlomenina nosných kostí
|
Akákoľvek
|
Vysaďte Pluvicto až do primeraného
stabilizovania/liečby zlomeniny
a stabilizovania stavu výkonnosti podľa
ECOG.
|
Únava
|
Stupeň ≥ 3
|
Vysaďte Pluvicto až do zlepšenia na stupeň 2 alebo na východiskovú hodnotu.
|
Elektrolytové alebo metabolické abnormality
|
Stupeň ≥ 2
|
Vysaďte Pluvicto až do zlepšenia na stupeň 1 alebo na východiskovú hodnotu.
|
Nehematologická toxicita (klinicky
významná, inak neuvedená)
|
Stupeň ≥ 2
|
Vysaďte Pluvicto až do zlepšenia na stupeň 1 alebo na východiskovú hodnotu.
|
Zvýšenie AST alebo
ALT
|
AST alebo ALT > 5-násobok
ULN bez metastáz v pečeni
|
Trvalo ukončite liečbu Pluvictom.
|
Skratky: KKr, klírens kreatinínu; ECOG, Eastern Cooperative Oncology Group; AST, aspartátaminotransferáza; ALT, alanínaminotransferáza; ULN, horná hranica normy.
Hodnotenie podľa najnovšej verzie Common Terminology Criteria for Adverse Events (CTCAE).
a Rovnaké prahové hodnoty sa vzťahujú aj na východiskové hodnoty v čase začatia liečby Pluvictom.
|
O
sobitné skupiny pacientov
Starší ľudia
Úprava dávkovania sa neodporúča u pacientov vo veku 65 rokov a starších.
Porucha funkcie obličiekÚprava dávky sa neodporúča u pacientov s ľahkou až stredne ťažkou poruchou funkcie obličiek (s východiskovým KKr ≥ 50 ml/min podľa Cockcroftovho-Gaultovho vzorca). Liečba Pluvictom sa neodporúča u pacientov so stredne ťažkou až ťažkou poruchou funkcie obličiek (s východiskovým KKr < 50 ml/min) alebo chorobou obličiek v terminálnom štádiu, pretože farmakokinetický profil
a bezpečnosť Pluvicta sa u týchto pacientov neskúmali (pozri časti 4.4 a 5.2).
Porucha funkcie pečeneÚprava dávkovania sa neodporúča u pacientov s poruchou funkcie pečene. Pluvicto sa neskúmalo u
pacientov so stredne ťažkou až ťažkou poruchou funkcie pečene (pozri časť 5.2).
Pediatrická populáciaPoužitie Pluvicta sa netýka pediatrickej populácie pre indikáciu liečby karcinómu prostaty s expresiou
PSMA.
Spôsob podávaniaPluvicto je injekčný/infúzny roztok pripravený na použitie a určený len na jednorazové použitie.
Pokyny na podávanieOdporúčaná dávka Pluvicta sa môže podať intravenózne ako injekcia pomocou jednorazovej injekčnej striekačky vybavenej tieniacim krytom (s injekčnou pumpou alebo bez nej), ako infúzia gravitačnou
metódou (s infúznou pumpou alebo bez nej), alebo ako infúzia z injekčnej liekovky (s peristaltickou
infúznou pumpou).
Znížená dávka Pluvicta sa má podávať metódou injekčnej striekačky (s injekčnou pumpou alebo
bez nej) alebo injekčnej liekovky (s peristaltickou infúznou pumpou). Použitie gravitačnej metódy
na podanie zníženej dávky Pluvicta sa neodporúča, pretože môže dôjsť k podaniu nesprávneho objemu
Pluvicta, ak sa dávka pred podaním neupraví.
Pred podaním prepláchnite intravenózny katéter použitý výlučne na podanie Pluvicta s ≥ 10 ml
sterilného injekčného roztoku chloridu sodného 9 mg/ml (0,9 %), aby sa zaistila priechodnosť
a minimalizovalo sa riziko extravazácie. V prípadoch extravazácie sa má postupovať podľa smerníc
zdravotníckeho zariadenia. Pacientom sa má odporučiť, aby pred a po podaní Pluvicta boli dostatočne
hydratovaní a často močili (pozri časť 4.4).
Pokyny na spôsob prípravy a metódy intravenózneho podania, pozri časť 12.
Príprava pacienta, pozri časť 4.4.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Individuálne odôvodnenie prínosu/rizika
Expozícia žiareniu u každého pacienta musí byť odôvodnená pravdepodobným prínosom. Podaná
rádioaktivita má byť v každom prípade taká nízka, aká sa dá racionálne dosiahnuť pri získaní požadovaného terapeutického účinku.
Riziko expozíciežiareniu
Pluvicto prispieva k celkovej dlhodobej kumulatívnej expozícii pacienta žiareniu. Dlhodobá
kumulatívna expozícia žiareniu je spojená so zvýšeným rizikom karcinómu.
Expozícia žiareniu u pacientov, zdravotníckych pracovníkov a členov spoločnej domácnosti sa má minimalizovať počas a po liečbe Pluvictom v súlade s predpismi správnej radiačnej bezpečnostnej praxe zdravotníckeho zariadenia, postupmi pri starostlivosti o pacienta a pokynmi pacientovi
pre následnú ochranu pred žiarením doma.
Príprava pacienta
Pacientom je potrebné odporučiť, aby pili viac tekutín, a zdôrazniť, aby močili tak často, ako je to možné, čím sa zníži žiarenie v močovom mechúre najmä po vysokých dávkach rádioaktivity, napr. Pri rádionuklidovej liečbe.
Po ukončení procedúry
Pred prepustením pacienta má lekár alebo zdravotnícky pracovník špecializujúci sa na nukleárnu medicínu vysvetliť pacientovi potrebné bezpečnostné opatrenia na ochranu pred žiarením, ktoré má pacient dodržiavať, aby u iných minimalizoval expozíciu žiareniu.
Po každom podaní Pluvicta sa môžu zvážiť nasledujúce všeobecné odporúčania pre pacientov spolu
s národnými, miestnymi a inštitucionálnymi postupmi a predpismi.
• Obmedziť blízky kontakt (menej ako 1 meter) s členmi domácnosti počas 2 dní alebo s deťmi
a gravidnými ženami počas 7 dní.
• Zdržať sa sexuálnej aktivity počas 7 dní.
• Spať sám v spálni oddelene od iných členov domácnosti počas 3 dní, oddelene od detí počas
7 dní a od gravidných žien počas 15 dní.
Myelosupresia
V štúdii VISION sa myelosupresia, vrátane smrteľných prípadov, vyskytla častejšie u pacientov, ktorí
dostali Pluvicto spolu s najlepšou štandardnou liečbou (best standard of care, BSoC) v porovnaní s pacientmi, ktorí dostali len BSoC (pozri časť 4.8).
Hematologické laboratórne testy, vrátane hemoglobínu, počtu bielych krviniek, absolútneho počtu neutrofilov a počtu trombocytov, sa majú vykonať pred a počas liečby Pluvictom. Podávanie Pluvicta sa má prerušiť, dávka sa má znížiť alebo sa liečba má trvalo ukončiť a pacienti majú dostať klinickú starostlivosť, ktorá sa považuje za primeranú vzhľadom na závažnosť myelosupresie (pozri časť 4.2).
N
efrotoxicita
V štúdii VISION sa nefrotoxicita vyskytla častejšie u pacientov, ktorí dostali Pluvicto spolu s BSoC
v porovnaní s pacientmi, ktorí dostali len BSoC (pozri časť 4.8).
Pacientom je potrebné odporučiť, aby pred a po podaní Pluvicta pili viac tekutín, a zdôrazniť, aby močili tak často, ako je to možné, najmä pri vysokých dávkach rádioaktivity, napr. pri rádionuklidovej liečbe. Laboratórne testy funkcie obličiek, vrátane sérového kreatinínu a vypočítaného KKr, sa majú vykonať pred a počas liečby Pluvictom. Podávanie Pluvicta sa má prerušiť, dávka sa má znížiť alebo sa liečba má trvalo ukončiť na základe závažnosti nefrotoxicity (pozri časť 4.2).
Porucha funkcieobličiek/pečene
U týchto pacientov sa vyžaduje starostlivé zváženie pomeru prínosu a rizika, keďže je možná zvýšená
expozícia žiareniu. Očakáva sa zvyšovanie expozície (AUC) lutécium (177Lu) vipivotid tetraxetanu so stupňom poruchy funkcie obličiek (pozri časť 5.2). U pacientov s ľahkou alebo stredne ťažkou
poruchou funkcie obličiek môže byť vyššie riziko toxických účinkov. Funkcia obličiek a nežiaduce reakcie sa majú často kontrolovať u pacientov s ľahkou až stredne ťažkou poruchou funkcie obličiek
(pozri časť 4.2). Liečba Pluvictom sa neodporúča u pacientov so stredne ťažkou až ťažkou poruchou funkcie obličiek s východiskovým KKr < 50 ml/min alebo chorobou obličiek v terminálnom štádiu.
Fertilita
Žiarenie lutécium (177Lu) vipivotid tetraxetanu môže mať potenciálne toxické účinky na mužské
gonády a spermatogenézu. Odporúčaná kumulatívna dávka Pluvicta 44 400 MBq má za následok
dávku žiarenia absorbovanú semenníkmi v rozmedzí, kde Pluvicto môže spôsobiť neplodnosť. Odporúča sa genetická konzultácia, ak si pacient želá mať po liečbe deti. U pacientov pred liečbou sa môže ako možnosť prediskutovať kryoprezervácia spermií (pozri časť 4.6).
Antikoncepcia u mužov
Pacientom sa odporúča nesplodiť dieťa a používať kondóm pri pohlavnom styku počas liečby
Pluvictom a počas 14 týždňov od poslednej dávky (pozri časť 4.6).
Osobitné upozornenia
Obsah sodíka
Tento liek obsahuje 3,9 mmol (88,75 mg) sodíka v každej liekovke, čo zodpovedá 4,4 % WHO
odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
Bezpečnostné opatrenia v súvislosti s ohrozením životného prostredia, pozri časť 6.6.
4.5 Liekové a iné interakcie
Nevykonali sa klinické štúdie liekových interakcií.
4.6 Fertilita, gravidita a laktácia
A
ntikoncepcia u mužov
Vzhľadom na možné účinky na spermatogenézu spojené so žiarením lutécium (177Lu) vipivotid
tetraxetanu sa pacientom odporúča nesplodiť dieťa a používať kondóm pri pohlavnom styku počas liečby Pluvictom a počas 14 týždňov od poslednej dávky (pozri časť 4.4).
Gravidita
Pluvicto nie je indikované na použitie u žien. S lutécium (177Lu) vipivotid tetraxetanom sa nevykonali
žiadne štúdie na zvieratách na vyhodnotenie jeho účinku na reprodukčnú schopnosť žien a vývin embrya a fétu. Všetky rádiofarmaká, vrátane Pluvicta, však majú potenciál spôsobiť poškodenie plodu,
keď sa podajú gravidnej žene.
Dojčenie
Pluvicto nie je indikované na použitie u žien. Nie sú žiadne údaje o prítomnosti lutécium (177Lu)
vipivotid tetraxetanu v ľudskom mlieku alebo o jeho účinkoch na dojčeného novorodenca/dieťa alebo
na tvorbu mlieka.
Fertilita
Nevykonali sa žiadne štúdie na stanovenie účinkov lutécium (177Lu) vipivotid tetraxetanu na fertilitu.
Žiarenie lutécium (177Lu) vipivotid tetraxetanu môže mať potenciálne toxické účinky na mužské
gonády a spermatogenézu. Odporúčaná kumulatívna dávka Pluvicta 44 400 MBq má za následok dávku žiarenia absorbovanú semenníkmi v rozmedzí, kde Pluvicto môže spôsobiť neplodnosť. Odporúča sa genetická konzultácia, ak si pacient želá mať po liečbe deti. U pacientov pred liečbou sa môže ako možnosť prediskutovať kryoprezervácia spermií (pozri časť 4.4).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Pluvicto môže mať malý vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn profilubezpečnosti
Pokiaľ nie je uvedené inak, frekvencie nežiaducich účinkov v zozname vychádzajú z údajov zo štúdie
VISION, v ktorej 529 pacientov dostalo aspoň jednu dávku 7 400 MBq (medián počtu dávok bol päť).
K najčastejším nežiaducim reakciám patria: únava (43,1 %), suchosť v ústach (39,3 %), nauzea (35,3 %), anémia (31,8 %), znížená chuť do jedenia (21,2 %) a zápcha (20,2 %). Najčastejšie nežiaduce reakcie stupňov 3 až 4 zahŕňajú: anémia (12,9 %), trombocytopénia (7,9 %), lymfopénia (7,8 %) a únava (5,9 %).
Tabuľkový zoznamnežiaducichreakcií
Nežiaduce reakcie (Tabuľka 2) sú uvedené podľa triedy orgánových systémov MedDRA. V každej
triede orgánových systémov sú nežiaduce reakcie zoradené podľa frekvencie, s najčastejšími
reakciami ako prvými. Okrem toho je zodpovedajúca kategória frekvencie pre každú nežiaducu reakciu založená na nasledujúcej konvencii (CIOMS III): veľmi časté (≥ 1/10); časté (≥ 1/100 až
< 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé
(< 1/10 000).
T
abuľka 2 Nežiaduce reakcie vyskytujúce sa vyššou incidenciou u pacientov, ktorí dostali
Pluvicto spolu s BSoC v porovnaní so samotnou BSoC v štúdii VISION
a
T
rieda orgánových systémov
N
ežiaduca reakcia
|
K
ategória frekvencie
|
V
šetky
stupne n (%)
|
Stupne 3 až
4
b
n (%)
|
Poruchy krvi a lymfatického systému
|
Anémia
|
Veľmi časté
|
168 (31,8)
|
68 (12,9)
|
Trombocytopénia
|
Veľmi časté
|
91 (17,2)
|
42 (7,9)
|
Leukopéniac
|
Veľmi časté
|
83 (15,7)
|
22 (4,2)
|
Lymfopénia
|
Veľmi časté
|
75 (14,2)
|
41 (7,8)
|
Pancytopéniad
|
Časté
|
9 (1,7)
|
7 (1,3)b
|
Poruchy nervového systému
|
Závraty
|
Časté
|
44 (8,3)
|
5 (0,9)
|
Bolesť hlavy
|
Časté
|
37 (7,0)
|
4 (0,8)
|
Dysgeúziae
|
Časté
|
37 (7,0)
|
0 (0,0)
|
Poruchy oka
|
Suchosť očí
|
Časté
|
16 (3,0)
|
0 (0,0)
|
Poruchy ucha a labyrintu
|
Vertigo
|
Časté
|
11 (2,1)
|
0 (0,0)
|
Poruchy gastrointestinálneho traktu
|
Suchosť v ústachf
|
Veľmi časté
|
208 (39,3)
|
0 (0,0)
|
Nauzea
|
Veľmi časté
|
187 (35,3)
|
7 (1,3)
|
Zápcha
|
Veľmi časté
|
107 (20,2)
|
6 (1,1)
|
Vracanieg
|
Veľmi časté
|
101 (19,1)
|
5 (0,9)
|
Hnačka
|
Veľmi časté
|
100 (18,9)
|
4 (0,8)
|
Bolesť bruchah
|
Veľmi časté
|
59 (11,2)
|
6 (1,1)
|
Poruchy obličiek a močových ciest
|
Infekcia močových ciesti
|
Veľmi časté
|
61 (11,5)
|
20 (3,8)
|
Akútne poškodenie obličiekj
|
Časté
|
45 (8,5)
|
17 (3,2)
|
C
elkové poruchy a reakcie v mieste podania
|
Únava
|
Veľmi časté
|
228 (43,1)
|
31 (5,9)
|
Znížená chuť do jedenia
|
Veľmi časté
|
112 (21,2)
|
10 (1,9)
|
Pokles telesnej hmotnosti
|
Veľmi časté
|
57 (10,8)
|
2 (0,4)
|
Periférny edémk
|
Časté
|
52 (9,8)
|
2 (0,4)
|
Pyrexia
|
Časté
|
36 (6,8)
|
2 (0,4)
|
Skratka: BSoC, najlepšia štandardná liečba.
a National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE)
verzia 5.0.
b Zahŕňa len nežiaduce reakcie stupňov 3 až 4, s výnimkou pancytopénie. Pancytopénia stupňa 5 (fatálna) bola hlásená u 2 pacientov, ktorí dostali Pluvicto spolu s BSoC.
c Leukopénia zahŕňa leukopéniu a neutropéniu.
d Pancytopénia zahŕňa pancytopéniu a bicytopéniu.
e Dysgeúzia zahŕňa dysgeúziu a poruchu chuti.
f Suchosť v ústach zahŕňa suchosť v ústach, aptyalizmus a suchosť hrdla.
g Vracanie zahŕňa vracanie a napínanie na vracanie.
h Bolesť brucha zahŕňa bolesť brucha, bolesť v hornej časti brucha, nepríjemný pocit v bruchu,
bolesť v dolnej časti brucha, citlivosť brucha na tlak a gastrointestinálnu bolesť.
i Infekcia močových ciest zahŕňa infekciu močových ciest, cystitídu a bakteriálnu cystitídu.
j Akútne poškodenie obličiek zahŕňa zvýšenie kreatinínu v krvi, akútne poškodenie obličiek, zlyhanie obličiek a zvýšenie močoviny v krvi.
k Periférny edém zahŕňa periférny edém, retenciu tekutiny a hyperhydratáciu.
|
O
pis vybranýchnežiaducichreakciíMyelosupresiaV štúdii VISION sa myelosupresia vyskytovala častejšie u pacientov, ktorí dostali Pluvicto spolu s BSoC v porovnaní s pacientmi, ktorí dostali samotnú BSoC (všetky stupne/stupeň ≥ 3): anémia (31,8 %/12,9 %) oproti (13,2 %/4,9 %); trombocytopénia (17,2 %/7,9 %) oproti (4,4 %/1,0 %); leukopénia (12,5 %/2,5 %) oproti (2,0 %/0,5 %); lymfopénia (14,2 %/7,8 %) oproti (3,9 %/0,5 %);
neutropénia (8,5 %/3,4 %) oproti (1,5 %/0,5 %); pancytopénia (1,5 %/1,1 %) oproti (0 %/0 %) vrátane dvoch fatálnych udalostí pancytopénie u pacientov, ktorí dostali Pluvicto spolu s BSoC; a bicytopénia
(0,2 %/0,2 %) oproti (0 %/0 %).
Nežiaduce reakcie myelosupresie, ktoré mali za následok trvalé ukončenie účasti v štúdii u ≥ 0,5 % pacientov, ktorí dostali Pluvicto spolu s BSoC, zahŕňali: anémia (2,8 %), trombocytopénia (2,8 %), leukopénia (1,3 %), neutropénia (0,8 %) a pancytopénia (0,6 %). Nežiaduce reakcie myelosupresie, ktoré mali za následok prerušenie podávania/zníženie dávky u ≥ 0,5 % pacientov, ktorí dostali Pluvicto spolu s BSoC, zahŕňali: anémia (5,1 %/1,3 %), trombocytopénia (3,6 %/1,9 %), leukopénia (1,5 %/0,6 %) a neutropénia (0,8 %/0,6 %).
NefrotoxicitaV štúdii VISION sa nefrotoxicita vyskytovala častejšie u pacientov, ktorí dostali Pluvicto spolu
s BSoC v porovnaní s pacientmi, ktorí dostali samotnú BSoC (všetky stupne/stupne 3 až 4): zvýšenie kreatinínu v krvi (5,3 %/0,2 %) oproti (2,4 %/0,5 %); akútne poškodenie obličiek (3,6 %/3,0 %) oproti (3,9 %/2,4 %); zlyhanie obličiek (0,2 %/0 %) oproti (0 %/0 %); a zvýšenie močoviny v krvi
(0,2 %/0 %) oproti (0 %/0 %).
Nežiaduce reakcie týkajúce sa obličiek, ktoré mali za následok trvalé ukončenie účasti v štúdii
u ≥ 0,2 % pacientov, ktorí dostali Pluvicto spolu s BSoC, zahŕňali: zvýšenie kreatinínu v krvi (0,2 %).
Nežiaduce reakcie týkajúce sa obličiek, ktoré mali za následok prerušenie podávania/zníženie dávky u
≥ 0,2 % pacientov, ktorí dostali Pluvicto spolu s BSoC, zahŕňali: zvýšenie kreatinínu v krvi
(0,2 %/0,4 %) a akútne poškodenie obličiek (0,2 %/0 %).
Druhé primárne malignityExpozícia ionizujúcemu žiareniu je spojená s indukciou zhubných nádorov a potenciálom pre vznik dedičných chýb. Dávka žiarenia pri terapeutickej expozícii môže mať za následok vyššiu incidenciu zhubných nádorov a mutácií. Vo všetkých prípadoch je potrebné zabezpečiť, aby riziká žiarenia boli menšie ako riziká samotnej choroby. Keďže Pluvicto prispieva k celkovej dlhodobej expozícii pacienta žiareniu, ktorá môže byť spojená so zvýšeným rizikom rakoviny (pozri časť 4.4), nemožno vylúčiť potenciálne riziko druhých primárnych malignít pre rádiofarmaká ako je Pluvicto. V čase primárnej analýzy štúdie VISION (k dátumu ukončenia 27-Jan-2021) boli hlásené prípady
skvamocelulárneho karcinómu (4 pacienti; 0,8 %) a bazocelulárneho karcinómu, malígneho melanómu a skvamocelulárneho karcinómu kože (po 1 pacientovi; po 0,2 %) u pacientov, ktorí dostali Pluvicto
spolu s BSoC.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade podania príliš vysokej dávky žiarenia s Pluvictom sa má absorbovaná dávka pacientom znížiť, pokiaľ je to možné, zvýšením vylučovania rádionuklidu z tela častým močením alebo nútenou diurézou a častým vyprázdňovaním močového mechúra. Môže byť užitočné odhadnúť skutočnú dávku, ktorá bola podaná.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Terapeutické rádiofarmaká, iné terapeutické rádiofarmaká, ATC kód: V10XX05
Mechanizmus účinku
Aktívna zložka Pluvicta je rádionuklid lutécium-177 naviazaný na malomolekulový ligand, ktorý sa
zameriava a viaže s vysokou afinitou na PSMA, transmembránový proteín s vysokou expresiou
pri karcinóme prostaty, vrátane mCRPC. Po naviazaní sa Pluvicta na nádorové bunky exprimujúce
PSMA je emisia beta-mínus z lutécia-177 zdrojom terapeutického žiarenia na zacielenú bunku, ako aj na okolité bunky, a vyvoláva poškodenie DNA, ktoré môže viesť k bunkovej smrti.
Farmakodynamické účinky
Neoznačený vipivotid tetraxetan nemá žiadne farmakodynamické účinky.
Klinická účinnosť abezpečnosť
VISION
Účinnosť Pluvicta u pacientov s progresívnym mCRPC pozitívnym na PSMA sa vyhodnotila vo
VISION, randomizovanej, multicentrickej, otvorenej štúdii fázy III. Osemstotridsaťjeden (N=831)
dospelých pacientov bolo randomizovaných (2:1) na podanie buď Pluvicta 7 400 MBq každých
6 týždňov až do celkovo 6 dávok spolu s najlepšou štandardnou liečbou (BSoC) (N=551), alebo len
na samotnú BSoC (N=280). U pacientov, ktorí dostali 4 dávky Pluvicta, sa opätovne vyhodnotili údaje o odpovedi, prejavoch reziduálnej choroby a znášanlivosti, a títo pacienti mohli podľa uváženia lekára
dostať až 2 ďalšie dávky.
Na udržiavanie stavu kastrácie všetci pacienti naďalej dostávali analóg GnRH alebo podstúpili predtým bilaterálnu orchietómiu. Pacienti vhodní na zaradenie mali mať progresívny mCRPC pozitívny na PSMA, stav výkonnosti (PS) podľa Eastern Cooperative Oncology Group (ECOG) 0 až
2, najmenej jednu metastatickú léziu prítomnú na zobrazení počítačovou tomografiou (CT),
magnetickou rezonanciou (MRI) alebo pri skenovaní kostí a primeranú renálnu, hepatálnu a hematologickú funkciu.
U pacientov vhodných na zaradenie sa tiež požadovalo, aby dostali najmenej jeden inhibítor dráhy
AR, napr. abiraterón-acetát alebo enzalutamid, a 1 alebo 2 režimy chemoterapie na báze taxánov
(s režimom definovaným ako podanie minimálne 2 cyklov taxánu). Pacienti liečení predtým len
1 režimom chemoterapie na báze taxánov boli vhodní na zaradenie, ak pacient nebol ochotný alebo lekár nepovažoval pacienta za schopného dostať druhý režim. Pacienti s nestabilnými symptomatickými metastázami v centrálnom nervovom systéme alebo symptomatickou alebo klinicky/rádiologicky potvrdenou hroziacou kompresiou miechy neboli vhodní na zaradenie do štúdie. U pacientov sa vykonalo zobrazovacie vyšetrenie pozitrónovou emisnou tomografiou (PET) s gálium (68Ga) gozetotidom na vyhodnotenie expresie PSMA v léziách, ktoré sa riadilo kritériami centrálneho hodnotenia. U pacientov vhodných na zaradenie sa požadovalo, aby mali mCRPC pozitívny na PSMA definovanú ako prítomnosť najmenej jednej nádorovej lézie s absorpciou gálium (68Ga) gozetotidu väčšou ako v normálnej pečeni. Pacienti boli vylúčení, ak ktorékoľvek z lézií presahujúcich kritériá
pre veľkosť krátkej osi (orgány ≥ 1 cm, lymfatické uzliny ≥ 2,5 cm, kosti [časť tvorená mäkkým tkanivom] ≥ 1 cm) mali absorpciu nižšiu alebo rovnakú, ako bola absorpcia v normálnej pečeni.
BSoC podávaná na základe rozhodnutia lekára zahŕňala: podporné opatrenia vrátane liekov
proti bolesti, hydratácia, transfúzie krvi, atď.; ketokonazol; rádioterapia (vrátane zrnovej formy alebo akejkoľvek vonkajšej lúčovej rádioterapie [vrátane stereotaktickej telovej rádioterapie a paliatívneho externého lúča]) na lokalizovanie cieľov pri karcinóme prostaty; látky cieliace na kosti vrátane kyseliny zoledrónovej, denosumabu a akýchkoľvek bisfosfonátov; látky znižujúce hladinu androgénov vrátane analógov GnRH, akýchkoľvek kortikosteroidov a 5-alfa reduktáz; inhibítory dráhy AR.
Z BSoC boli vylúčené skúšané lieky, cytotoxická chemoterapia, imunoterapia, iné systémové rádioizotopy a rádioterapia polovice tela.
Pacienti pokračovali v randomizovanej liečbe až do preukázania progresie nádoru (na základe
hodnotenia skúšajúceho lekára podľa kritérií Prostate Cancer Working Group 3 [PCWG3]),
do výskytu neprijateľnej toxicity, použitia zakázanej liečby, nedodržania požiadaviek alebo vystúpenia zo štúdie, alebo do nedostatočného klinického prínosu.
Primárne koncové ukazovatele boli celkové prežívanie (OS) a prežívanie bez rádiografickej progresie (rPFS) určené zaslepeným nezávislým centrálnym hodnotením (BICR) v súlade s kritériami PCWG3. K sekundárnym cieľom účinnosti patrili celková miera odpovedí (ORR) určená BICR v súlade
s Response Evaluation Criteria in Solid Tumors (RECIST) v1.1 a čas do prvej symptomatickej udalosti týkajúcej sa skeletu (SSE), ktorá bola definovaná ako prvá nová symptomatická patologická
zlomenina kosti, kompresia miechy, ortopedický chirurgický zákrok súvisiaci s nádorom, potreba
radiačnej liečby na zmiernenie bolesti kostí, alebo smrť z akejkoľvek príčiny, čokoľvek nastalo ako prvé. Rádiografické zobrazovacie vyšetrenie na hodnotenie nádoru (CT s kontrastným MRI/zobrazovacie vyšetrenie a skenovanie kostí) sa vykonalo každých 8 týždňov (±4 dni) po prvej dávke počas prvých 24 týždňov (bez ohľadu na oneskorenia dávky), potom každých 12 týždňov
(±4 dni).
Demografické znaky a východiskový stav choroby boli vyvážené medzi obidvomi skupinami liečby. Medián veku bol 71 rokov (rozmedzie: 40 až 94 rokov); 86,8 % belosi; 6,6 % černosi alebo Afroameričania; 2,4 % Ázijci; 92,4 % malo PS 0-1 podľa ECOG; 7,6 % malo PS 2 podľa ECOG. Randomizácia bola stratifikovaná podľa východiskovej hodnoty laktátdehydrogenázy (LDH
≤ 260 IU/l oproti >260 IU/l), prítomnosti metastáz v pečeni (áno oproti nie), skóre PS podľa ECOG (0 alebo 1 oproti 2) a použitie inhibítora dráhy AR ako súčasť BSoC v čase randomizácie (áno oproti nie). V čase do randomizácie všetci pacienti (100,0 %) dostali najmenej jeden režim chemoterapie založenej na taxánoch, 41,2 % pacientov dostalo dva; 97,1 % pacientov dostalo docetaxel a 38,0 % pacientov dostalo kabazitaxel. V čase do randomizácie dostalo 51,3 % pacientov jeden inhibítor dráhy AR, 41,0 % pacientov dostalo 2 a 7,7 % pacientov dostalo 3 alebo viac. Počas obdobia randomizovanej liečby dostalo 52,6 % pacientov v skupine Pluvicta spolu s BSoC a 67,8 % pacientov v skupine samotnej BSoC najmenej jeden inhibítor dráhy AR.
Výsledky účinnosti vo VISION sú uvedené v Tabuľke 3 a na Obrázkoch 1 a 2. Konečné analýzy OS
a rPFS záviseli od výskytu udalostí a vykonali sa, až keď došlo k 530 úmrtiam a vyskytlo sa
347 udalostí.
T
abuľka 3 Výsledky účinnosti vo VISION

C
elkové prežívanie (OS)a
|
N=551
|
N=280
|
Úmrtia, n (%)
|
343 (62,3 %)
|
187 (66,8 %)
|
Medián, mesiace (95 % IS)b
|
15,3 (14,2; 16,9)
|
11,3 (9,8; 13,5)
|
Pomer rizík (95 % IS)c
|
0,62 (0,52; 0,74)
|
Hodnota pd
|
< 0,001
|
Prežívanie bez rádiografickej progresie (rPFS)
e
,f
|
N=385
|
N=196
|
Udalosti (progresia alebo úmrtie), n (%)
|
254 (66,0 %)
|
93 (47,4 %)
|
Rádiografické progresie, n (%)
|
171 (44,4 %)
|
59 (30,1 %)
|
Úmrtia, n (%)
|
83 (21,6 %)
|
34 (17,3 %)
|
Medián, mesiace (99,2 % IS)b
|
8,7 (7,9; 10,8)
|
3,4 (2,4; 4,0)
|
Pomer rizík (99,2 % IS)c
|
0,40 (0,29; 0,57)
|
Hodnota pd
|
< 0,001
|
|
|
Parametre účinnosti Pluvicto spolu s BSoC BSoC Alternatívne primárne koncové ukazovatele
Sekundárne ciele účinnosti
Č
as do prvej symptomatickej udalosti týkajúcej sa skeletu (SSE)
f
|
N=385
|
N=196
|
Udalosti (SSE alebo úmrtie), n (%)
|
256 (66,5 %)
|
137 (69,9 %)
|
SSE, n (%)
|
60 (15,6 %)
|
34 (17,3 %)
|
Úmrtia, n (%)
|
196 (50,9 %)
|
103 (52,6 %)
|
Medián, mesiace (95 % IS)b
|
11,5 (10,3; 13,2)
|
6,8 (5,2; 8,5)
|
Pomer rizík (95 % IS)c
|
0,50 (0,40; 0,62)
|
Hodnota pg
|
< 0,001
|
N
ajlepšia celková odpoveď (BOR)
|
|
|
Pacienti s vyhodnotiteľným východiskovým
stavom choroby
|
N=319
|
N=120
|
Kompletná odpoveď (CR), n (%)
|
18 (5,6 %)
|
0 (0 %)
|
Čiastočná odpoveď (PR), n (%)
|
77 (24,1 %)
|
2 (1,7 %)
|
C
elková miera odpovede (ORR)
h
,
i
|
95 (29,8 %)
|
2 (1,7 %)
|
Hodnota pj
|
< 0,001
|
T
rvanie odpovede (DOR)
h
|
|
|
Medián, mesiace (95 % IS)b
|
9,8 (9,1; 11,7)
|
10,6 (NE; NE)k
|
BSoC: Najlepšia štandardná liečba; IS: Interval spoľahlivosti; NE: Nevyhodnotiteľné; BICR: Zaslepené nezávislé centrálne hodnotenie; PCWG3: Prostate Cancer Working Group 3; RECIST: Kritériá hodnotenia odpovede pri solídnych nádoroch.
a Analyzované podľa liečebného zámeru (ITT) u všetkých randomizovaných pacientov.
b Na základe odhadu podľa Kaplana-Meiera.
c Pomer rizík na základe stratifikovaného modelu Cox PH. Pomer rizík < 1 svedčí v prospech Pluvicta spolu s BSoC.
d Hodnota p jednostranného stratifikovaného log-rank testu.
e BICR podľa kritérií PCWG3. Primárna analýza rPFS zahŕňala cenzurovanie pacientov, u ktorých došlo
k vynechaniu ≥ 2 po sebe nasledujúcich hodnotení nádoru bezprostredne pred progresiou alebo úmrtím. Výsledky rPFS s cenzurovaním pre vynechané hodnotenia alebo bez neho boli zhodné.
f Analyzované na základe ITT u všetkých pacientov randomizovaných 05. marca 2019 alebo neskôr, keď
boli zavedené opatrenia na zníženie predčasného ukončenia účasti v skupine BSoC.
g Hodnota p dvojstranného stratifikovaného log-rank testu.
h BICR podľa RECIST v1.1.
i ORR: CR+PR. Potvrdená odpoveď CR a PR.
j Hodnota p dvojstranného stratifikovaného Waldovho chí-kvadrát testu.
k Medián DOR v skupine samotnej BSoC nebol spoľahlivý, pretože len u 1 z 2 pacientov s odpoveďou došlo
k rádiografickej progresii podľaRECISTv1.1alebok úmrtiu.
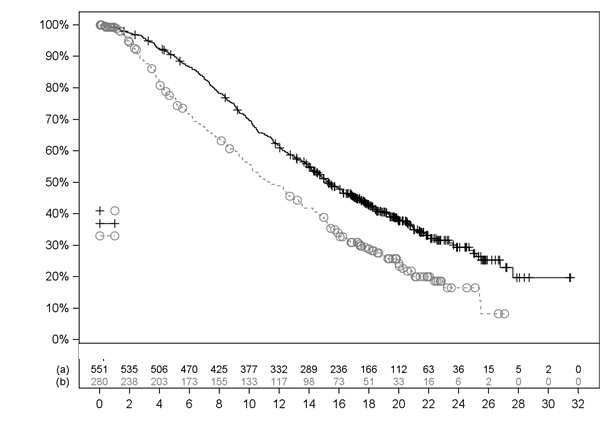 O
brázok 1 Krivka podľa Kaplana-Meiera pre OS vo VISION
O
brázok 1 Krivka podľa Kaplana-Meiera pre OS vo VISION
Časy cenzurovania
(a) Lu-PSMA-617+BSoC (b) Len BSoC
Počet pacientov s pretrvávajúcim rizikom
Čas od randomizácie (mesiace)
Stratifikovaný log-rank test a Coxov model používajúci stratifikáciu podľa Interaktívnej technológie
odozvy(IRT) definovanú hladinou LDH, prítomnosťou metastáz v pečeni, skóre podľa ECOG a zahrnutím inhibítora dráhy AR do BSoC v čase randomizácie.
n/N: Počet udalostí/počet pacientov v skupine liečby.
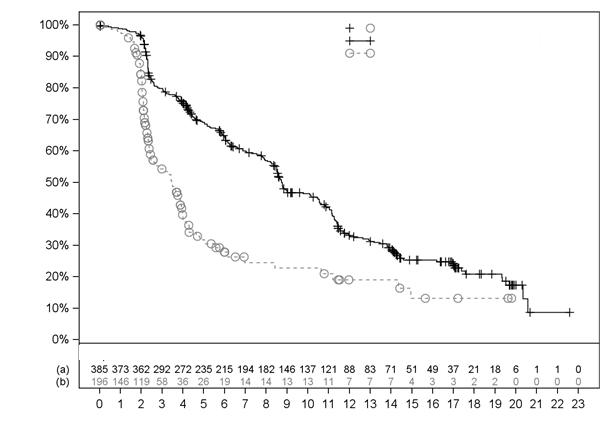
 O
brázok 2 Krivka podľa Kaplana-Meiera pre rPFS hodnotenej BICR vo VISION
O
brázok 2 Krivka podľa Kaplana-Meiera pre rPFS hodnotenej BICR vo VISION
Časy cenzurovania
(a) Lu-PSMA-617+BSoC (b) Len BSoC
Počet pacientov s pretrvávajúcim rizikom
Čas od randomizácie (mesiace)
Stratifikovaný log-rank test a Coxov model používajúci stratifikáciu podľa IRT definovanú hladinou LDH,
prítomnosťou metastáz v pečeni, skóre podľa ECOG a zahrnutím inhibítora dráhy AR do BSoC v čase
randomizácie.
n/N: Počet udalostí/počet pacientov v skupine liečby.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Pluvictom vo
všetkých podskupinách pediatrickej populácie v liečbe karcinómu prostaty s expresiou PSMA (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika lutécium (177Lu) vipivotid tetraxetanu sa skúmala u 30 pacientov v podštúdii fázy III VISION.
Absorpcia
Pluvicto sa podáva intravenózne a je okamžite a úplne biologicky dostupné.
Geometrický priemer expozície v krvi (plocha pod krivkou [AUCinf]) lutécium (177Lu) vipivotid tetraxetanu pri odporúčanej dávke je 52,3 ng.h/ml (geometrický priemer variačného koeficientu [CV]
31,4 %). Geometrický priemer maximálnej koncentrácie v krvi (Cmax) lutécium (177Lu) vipivotid tetraxetanu je 6,58 ng/ml (CV 43,5 %).
Distribúcia
Geometrický priemer distribučného objemu (Vz) lutécium (177Lu) vipivotid tetraxetanu je 123 l (CV
78,1 %).
Na bielkoviny ľudskej plazmy sa viaže 60 % až 70 % neoznačeného vipivotid tetraxetanu aj nerádioaktívneho lutécium (175Lu) vipivotid tetraxetanu.
Absorpcia orgánmi
Pri biodistribúcii lutécium (177Lu) vipivotid tetraxetanu sa primárna absorpcia pozoruje v slzných
žľazách, slinných žľazách, obličkách, stenách močového mechúra, pečeni, tenkom čreve a hrubom
čreve (pravá a ľavá časť hrubého čreva).
Eliminácia
Geometrický priemer klírensu (CL) lutécium (177Lu) vipivotid tetraxetanu je 2,04 l/h (CV 31,5 %).
Lutécium (177Lu) vipivotid tetraxetan sa vylučuje primárne obličkami. Polčas
Pluvicto vykazuje biexponenciálnu elimináciu s geometrickým priemerom terminálneho polčasu
eliminácie (T½) 41,6 hodín (CV 68,8 %).
Biotransformácia
Lutécium (177Lu) vipivotid tetraxetan nie je metabolizovaný v pečeni ani v obličkách.
Hodnotenie potenciálu iekových interakcií in vitro
Enzýmy CYP450
Vipivotid tetraxetan nie je substrát enzýmov cytochrómu P450 (CYP450). Neindukuje cytochróm
P450 (CYP) 1A2, 2B6 alebo 3A4 a neinhibuje cytochróm P450 (CYP) 1A2, 2B6, 2C8, 2C9, 2C19,
2D6 alebo 3A4/5 in vitro.
Transportéry
Vipivotid tetraxetan nie je substrát BCRP, P-gp, MATE1, MATE2-K, OAT1, OAT3 alebo OCT2 a nie je inhibítor BCRP, P-gp, BSEP, MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, OCT1 alebo OCT2 in vitro.
Osobitné skupiny pacientov
Vplyv veku a telesnej hmotnosti
Žiadne klinicky významné účinky na farmakokinetické parametre lutécium (177Lu) vipivotid tetraxetanu sa nezistili pri nasledujúcich kovariantách vyhodnotených u 30 pacientov v podštúdii
fázy III VISION: vek (medián: 67 rokov; rozmedzie: 52 až 80 rokov) a telesná hmotnosť (medián:
88,8 kg; rozmedzie: 63,8 až 143,0 kg).
Porucha f unkc ie obl i či ek
Expozícia (AUC) lutécium (177Lu) vipivotid tetraxetanu sa zvýšila o 20% u pacientov s ľahkou poruchou funkcie obličiek v porovnaní s normálnou funkciou obličiek. Polčas dozimetrie sa tiež zvýšil
u pacientov s ľahkou poruchou fukncie obličiek v porovnaní s normálnou funkciou obličiek, 51 hodín verzus 37 hodín, v uvedenom poradí. Pacienti so stredne ťažkou až ťažkou pourchou funkcie obličiek
môžu mať väčšie riziko toxicity (pozri časť 4.4). Nie sú k dispozícii žiadne farmakokinetické údaje u pacientov so stredne ťažkou až ťažkou poruchou funkcie obličiek s vychodikovým KKr < 50 ml/min
alebo chorobou obličiek v terminálnom štádiu.
5.3 Predklinické údaje o bezpečnosti
Žiadne toxikologické účinky sa nepozorovali v štúdiách farmakologickej bezpečnosti alebo v štúdiách toxicity po jednorazovom podaní na potkanoch a miniprasiatkach, ktorým sa podal nerádioaktívny prípravok obsahujúci neoznačený vipivotid tetraxetan a lutécium (175Lu) vipivotid tetraxetan, alebo
v štúdiách toxicity po opakovanom podávaní na potkanoch, ktorým sa podával neoznačený vipivotid tetraxetan.
Karcinogenita a mutagenita'
Štúdie mutagenity a dlhodobé štúdie karcinogenity sa s lutécium (177Lu) vipivotid tetraxetanom
nevykonali; žiarenie je však karcinogénne a mutagénne.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
kyselina octová octan sodný kyselina gentisová askorbát sodný kyselina pentetová voda na injekcie
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v častiach 4.2 a 12.
6.3 Čas použiteľnosti
120 hodín (5 dní) od dátumu a času kalibrácie.
6.4 Špeciálne upozornenia na uchovávanie
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred ionizujúcim žiarením (tienenie olovom).
Uchovávanie rádiofarmák má byť v súlade s národnými predpismi pre rádioaktívne materiály.
6.5 Druh obalu a obsah balenia
Injekčná liekovka z číreho, bezfarebného skla typu I, uzavretá brómbutylovou gumovou zátkou a hliníkovým tesniacim viečkom.
Každá injekčná liekovka obsahuje objem roztoku, ktorý môže byť v rozmedzí od 7,5 ml do 12,5 ml, zodpovedajúci rádioaktivite 7 400 MBq ±10 % v deň a čase podania.
Injekčná liekovka je uzavretá v olovenej nádobe použitej na ochranné tienenie.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všeobecné upozornenie
Rádiofarmaká majú preberať, používať a podávať len oprávnené osoby v určených klinických
zariadeniach. Ich prevzatie, uchovávanie, používanie, presun a likvidácia podliehajú predpisom
a/alebo príslušným povoleniam kompetentnej oficiálnej organizácie.
Rádiofarmaká sa majú pripravovať spôsobom, ktorý zodpovedá požiadavkám na radiačnú bezpečnosť
a kvalitu lieku. Majú sa dodržiavať príslušné aseptické opatrenia.
Pokyny na prípravu lieku pred podaním, pozri časť 12.
Ak sa kedykoľvek počas prípravy tohto lieku poruší celistvosť olovenej nádoby alebo injekčnej liekovky, liek sa nemá použiť.
Pri podaní lieku sa má postupovať tak, aby sa minimalizovalo riziko kontaminácie lieku a ožiarenia
pracovníkov. Povinné je patričné tienenie žiarenia.
Pri podávaní rádiofarmák vzniká riziko vonkajšieho ožiarenia ďalších osôb alebo kontaminácie
z vyliateho moču, zvratkov, atď. Je preto potrebné dodržiavať bezpečnostné opatrenia na ochranu pred žiarením v súlade s národnými smernicami.
Tento liek môže mať za následok pomerne vysokú dávku žiarenia u väčšiny pacientov. Podanie Pluvicta môže predstavovať významné ohrozenie životného prostredia. Môže to byť problém pre rodinných príslušníkov osôb, ktoré podstupujú liečbu, alebo pre širšiu verejnosť v závislosti od dávky podanej aktivity. V súlade s národnými smernicami je potrebné prijať primerané
bezpečnostné opatrenia týkajúce sa aktivity vylučovanej pacientmi, aby sa zabránilo akejkoľvek
kontaminácii.
Na prípravu lutécia-177 pre Pluvicto sa môžu použiť dva rôzne zdroje stabilných izotopov (buď lutécium-176, alebo yterbium-176). Lutécium-177 pripravené pre Pluvicto použitím stabilného izotopu lutécium-176 (“pridaný nosič”) vyžaduje osobitnú pozornosť s ohľadom na zaobchádzanie s odpadnom kvôli prítomnosti dlho žijúcej metastabilnej nečistoty lutécium-177 (177mLu) s polčasom rozpadu 160,4 dní. Lutécium-177 pre Pluvicto sa pripravuje z yterbia-176 („bez pridaného nosiča“), pokiaľ v certifikáte na uvoľnenie šarže produktu nie je uvedené inak. Používateľ musí pred použitím Pluvicta konzultovať poskytnuté osvedčenie o uvoľnení šarže, aby sa zabezpečilo správne zaobchádzanie s odpadom.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Novartis Europharm Limited
Vista Building
Elm Park, Merrion Road
Dublin 4
Írsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/22/1703/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
09. december 2022
10. DÁTUM REVÍZIE TEXTU
11. DOZIMETRIA
Dávka žiarenia absorbovaná jednotlivými orgánmi, ktoré nemusia byť cieľovým orgánom liečby, môže byť významne ovplyvnená patofyziologickými zmenami vyvolanými chorobným procesom. Je potrebné to zohľadniť pri použití nasledujúcich informácií.
Dozimetrické údaje pre lutécium (177Lu) vipivotid tetraxetan sa získali u 29 pacientov v podštúdii fázy III VISION, aby sa vypočítala dozimetria žiarenia pre celé telo a orgány. Priemer a smerodajná odchýlka (SD) odhadovaných absorbovaných dávok jednotlivými orgánmi dospelých pacientov, ktorým sa podá Pluvicto, sú uvedené v Tabuľke 4. Orgány s najvyššími absorbovanými dávkami sú slzné žľazy a slinné žľazy.
Maximálny prienik lutécia-177 do tkaniva je približne 2 mm a priemerný prienik je 0,67 mm.
T
abuľka 4 Odhadovaná absorbovaná dávka pri Pluvicte v podštúdii VISION
|
A
bsorbovaná dávka
na jednotku rádioaktivity (mGy/MBq)
a
(
N=
29)
|
V
ypočítaná
absorbovaná dávka pri podaní
7 400 MBq
(G
y
)
a
|
V
ypočítaná
absorbovaná dávka pri 6 x 7 400 MBq (kumulatívna rádioaktivita
44 400 MBq) (Gy)
a
|
O
rgán
|
Priemer
|
SD
|
Priemer
|
SD
|
Priemer
|
SD
|
Nadobličky
|
0,033
|
0,025
|
0,24
|
0,19
|
1,5
|
1,1
|
Mozog
|
0,007
|
0,005
|
0,049
|
0,035
|
0,30
|
0,22
|
Oči
|
0,022
|
0,024
|
0,16
|
0,18
|
0,99
|
1,1
|
Stena žlčníka
|
0,028
|
0,026
|
0,20
|
0,19
|
1,2
|
1,1
|
Stena srdca
|
0,17
|
0,12
|
1,2
|
0,83
|
7,8
|
5,2
|
Obličky
|
0,43
|
0,16
|
3,1
|
1,2
|
19
|
7,3
|
Slzné žľazy
|
2,1
|
0,47
|
15
|
3,4
|
92
|
21
|
Ľavá časť hrubého
čreva
|
0,58
|
0,14
|
4,1
|
1,0
|
26
|
6,0
|
Pečeň
|
0,090
|
0,044
|
0,64
|
0,32
|
4,0
|
2,0
|
Pľúca
|
0,11
|
0,11
|
0,76
|
0,81
|
4,7
|
4,9
|
Pažerák
|
0,025
|
0,026
|
0,18
|
0,19
|
1,1
|
1,1
|
Osteogénne bunky
|
0,036
|
0,028
|
0,26
|
0,21
|
1,6
|
1,3
|
Pankreas
|
0,027
|
0,026
|
0,19
|
0,19
|
1,2
|
1,1
|
Prostata
|
0,027
|
0,026
|
0,19
|
0,19
|
1,2
|
1,1
|
Červená kostná dreň
|
0,035
|
0,020
|
0,25
|
0,15
|
1,5
|
0,90
|
Rektum
|
0,56
|
0,14
|
4,0
|
1,1
|
25
|
6,2
|
Pravá časť hrubého čreva
|
0,32
|
0,078
|
2,3
|
0,58
|
14
|
3,4
|
Slinné žľazy
|
0,63
|
0,36
|
4,5
|
2,6
|
28
|
16
|
Tenké črevo
|
0,071
|
0,031
|
0,50
|
0,23
|
3,1
|
1,4
|
Slezina
|
0,067
|
0,027
|
0,48
|
0,20
|
3,0
|
1,2
|
Stena žalúdka
|
0,025
|
0,026
|
0,18
|
0,19
|
1,1
|
1,1
|
Semenníky
|
0,023
|
0,025
|
0,16
|
0,18
|
1,0
|
1,1
|
Týmus
|
0,025
|
0,026
|
0,18
|
0,19
|
1,1
|
1,1
|
Štítna žľaza
|
0,26
|
0,37
|
1,8
|
2,7
|
11
|
16
|
Celé telo
|
0,037
|
0,027
|
0,27
|
0,20
|
1,6
|
1,2
|
Stena močového
mechúra
|
0,32
|
0,025
|
2,3
|
0,19
|
14
|
1,1
|
Účinná dávkab
|
0,120
mSv/MBq
|
0,043
mSv/MBq
|
0,886
Sv
|
0,315
Sv
|
5,319
Sv
|
1,892
Sv
|
a Odhady absorbovanej dávky boli odvodené s použitím OLINDA v2.2. Hodnoty boli vypočítané s úplnou
presnosťou na základe dozimetrických odhadov a zaokrúhlené na patričné číslice.
b Odvodené podľa publikácie ICRP 103.
|
12. POKYNY NA PRÍPRAVU RÁDIOFARMÁK
Používateľ musí pred použitím Pluvicta konzultovať poskytnuté osvedčenie o uvoľnení šarže, aby sa
zabezpečilo správne zaobchádzanie s odpadom (pozri časť 6.6).
Odoberania roztoku sa majú vykonať za aseptických podmienok. Injekčné liekovky sa nesmú otvoriť pred dezinfikovaním zátky, roztok sa má odobrať cez zátku pomocou jednodávkovej injekčnej striekačky vybavenej vhodným ochranným krytom a jednorazovou sterilnou ihlou alebo pomocou schváleného automatizovaného aplikačného systému.
Pokyny na prípravu
• Pri manipulácii s Pluvictom alebo pri jeho podávaní použite aseptické postupy a ochranu
pred žiarením a podľa potreby sa majú používať kliešte, aby sa minimalizovala expozícia žiareniu.
• Vizuálne skontrolujte injekčnú liekovku pred podaním pod tieniacou clonou na prítomnosť viditeľných častíc a zmenu sfarbenia. Zlikvidujte injekčnú liekovku, ak sú prítomné viditeľné častice a/alebo zmena sfarbenia.
• Neinjekujte roztok Pluvicta priamo do akéhokoľvek iného intravenózneho roztoku.
• Množstvo rádioaktivity podané pacientovi potvrdťepomocou patrične kalibrovaného merača
dávky pred a po podaní Pluvicta.
Metódy intravenózneho podania
Pokyny pre met ódu i nj ekčnej st ri ek ačk y (so stri ekačkovou pumpou al ebo bez nej)
• Po dezinfikovaní zátky injekčnej liekovky odoberte príslušný objem roztoku Pluvicta
na podanie potrebnej rádioaktivity pomocou jednorazovej injekčnej striekačky vybavenej
tieniacim krytom a sterilnou jednorazovou ihlou.
• Podajte Pluvicto pacientovi intravenózne pomalým vstrekovaním trvajúcim približne 1 až
10 minút (buď striekačkovou pumpou, alebo manuálne bez striekačkovej pumpy)
cez intravenózny katéter, ktorý bol vopred naplnený sterilným injekčným roztokom chloridu
sodného 9 mg/ml (0,9 %) a ktorý sa použije výlučne na podanie Pluvicta pacientovi.
• Po podaní požadovanej rádioaktivity Pluvicta prepláchnite ≥ 10 ml sterilného injekčného roztoku chloridu sodného 9 mg/ml (0,9 %), ktoré podajte cez intravenózny katéter do žily pacienta.
Pokyny pre gravi t ačnú metódu (s infúznou pumpou alebo bez nej)
• Zaveďte 2,5 cm ihlu o veľkosti 20 G (krátku ihlu) do injekčnej liekovky Pluvicta a spojte ju katétrom s 500 ml sterilného injekčného roztoku chloridu sodného 9 mg/ml (0,9 %) (použitého na presun roztoku Pluvicta počas infúzie). Zabezpečte, aby sa krátka ihla nedotýkala roztoku Pluvicta v injekčnej liekovke a neprepojte krátku ihlu priamo s pacientom. Nedovoľte, aby sterilný injekčný roztok chloridu sodného 9 mg/ml (0,9 %) vtiekol do injekčnej liekovky Pluvicta pred začiatkom infúzie Pluvicta a neinjikujte roztok Pluvicta priamo do sterilného injekčného roztoku chloridu sodného 9 mg/ml (0,9 %).
• Zaveďte druhú 9 cm ihlu o veľkosti 18 G (dlhú ihlu) do injekčnej liekovky Pluvicta a zabezpečte, aby sa dlhá ihla dotýkala dna injekčnej liekovky Pluvicta a bola zaistená
proti posunutiu počas celej infúzie. Nasaďte dlhú ihlu pacientovi cez intravenózny katéter, ktorý
bol vopred naplnený sterilným injekčným roztokom chloridu sodného 9 mg/ml (0,9 %) a ktorý
sa použije výlučne na infúziu Pluvicta pacientovi.
• Použite svorku alebo infúznu pumpu na regulovanie prietoku sterilného injekčného roztoku chloridu sodného 9 mg/ml (0,9 %) cez krátku ihlu do injekčnej liekovky Pluvicta (sterilný injekčný roztok chloridu sodného 9 mg/ml (0,9 %), ktorý vtečie do injekčnej liekovky
cez krátku ihlu, prenesie roztok Pluvicta z injekčnej liekovky k pacientovi cez intravenózny
katéter spojený s dlhou ihlou približne do 30 minút).
• Počas infúzie dbajte o to, aby hladina roztoku v injekčnej liekovke Pluvicta zostala konštantná.
• Odpojte injekčnú liekovku od hadičky s dlhou ihlou a zasvorkujte prívod chloridu sodného, keď
hladina rádioaktivity bude stabilná najmenej päť minút.
• Po infúzii prepláchnite ≥ 10 ml sterilného injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %), ktoré podajte cez intravenózny katéter do žily pacienta.
Pokyny pre met ódu i nj ekčnej li ekov ky (s peristaltickou infúznou pumpou)
• Zaveďte 2,5 cm ihlu o veľkosti 20 G (krátku ventilačnú ihlu) do injekčnej liekovky Pluvicta.
Zabezpečte, aby sa krátka ihla nedotýkala roztoku Pluvicta v injekčnej liekovke a neprepojte krátku ihlu priamo s pacientom alebo s peristaltickou infúznou pumpou.
• Zaveďte druhú 9 cm ihlu o veľkosti 18 G (dlhú ihlu) do injekčnej liekovky Pluvicta a zabezpečte, aby sa dlhá ihla dotýkala dna injekčnej liekovky Pluvicta a bola zaistená proti posunutiu počas celej infúzie. Pripojte dlhú ihlu a sterilný injekčný roztok chloridu sodného 9 mg/ml (0,9 %) na 3-cestný uzatvárací ventil vhodnou hadičkou.
• Pripojte výstup 3-cestného uzatváracieho ventilu na hadičku inštalovanú na vstupnej strane peristaltickej infúznej pumpy podľa pokynov výrobcu pumpy.
• Vopred naplňte hadičku otvorením 3-cestného uzatváracieho ventilu a pumpovaním roztoku
Pluvicta cez hadičku, až dosiahne výstup ventilu.
• Vopred naplňte intravenózny katéter, ktorý bude nasadený pacientovi, otvorením 3-cestného uzatváracieho ventilu pre sterilný injekčný roztok chloridu sodného 9 mg/ml (0,9 %) a pumpovaním sterilného injekčného roztoku chloridu sodného 9 mg/ml (0,9 %), až kým vytečie na konci hadičky katétra.
• Nasaďte vopred naplnený intravenózny katéter pacientovi a nastavte 3-cestný uzatvárací ventil, aby bol roztok Pluvicta spojený s peristaltickou infúznou pumpou.
• Podajte infúziou zodpovedajúci objem roztoku Pluvicta s požadovanou rádioaktivitou rýchlosťou približne 25 ml/h.
• Po podaní požadovanej rádioaktivity Pluvicta zastavte peristaltickú infúznu pumpu a potom zmeňte polohu 3-cestného uzatváracieho ventilu, aby peristaltická infúzna pumpa bola spojená so sterilným injekčným roztokom chloridu sodného 9 mg/ml (0,9 %). Znovu spustite peristaltickú infúznu pumpu a prepláchnite ≥ 10 ml sterilného injekčného roztoku chloridu sodného 9 mg/ml (0,9 %), ktoré podajte pacientovi cez intravenózny katéter.
Kontrola kvalityRoztok sa má pred použitím vizuálne skontrolovať pre prípadné poškodenie a kontamináciu, a použiť
sa majú len číre roztoky bez viditeľných častíc. Vizuálna kontrola sa má vykonať pod tieniacou clonou
určenou na ochranu pred žiarením. Injekčná liekovka sa nesmie otvoriť.
Ak sa kedykoľvek počas prípravy tohto lieku poruší celistvosť olovenej nádoby alebo injekčnej liekovky, liek sa nemá použiť.
Množstvo rádioaktivity v injekčnej liekovke sa pred podaním musí odmerať pomocou vhodného kalibračného systému na meranie rádioaktivity, aby sa potvrdilo, že skutočné množstvo podanej rádioaktivity sa rovná plánovanému množstvu v čase podania.
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.