
na čas potrebný na zotavenie zo závažnej neutropénie u pacientov s akútnou myeloidnou leukémiou (acute myeloid leukaemia, AML) de novo (pozri časť 5.1). Avšak dlhodobé účinky Pelmegu pri AML sa neskúmali; preto je potrebné u tejto populácie pacientov tento liek používať
s opatrnosťou.
Faktor stimulujúci kolónie granulocytov (granulocyte-colony stimulating factor, G-CSF) môže urýchľovať rast myeloidných buniek in vitro a podobné účinky možno pozorovať aj u niektorých iných ako myeloidných buniek in vitro.
Bezpečnosť a účinnosť Pelmegu sa neskúmala u pacientov s myelodysplastickým syndrómom, chronickou myelogénnou leukémiou a u pacientov so sekundárnou AML; preto sa u týchto pacientov nemá používať. Zvláštnu pozornosť je potrebné venovať odlíšeniu diagnózy nárazovej transformácie chronickej myeloidnej leukémie od AML.
Bezpečnosť a účinnosť Pelmegu podávaného pacientom s AML de novo vo veku < 55 rokov s cytogenetickým nálezom t (15; 17) neboli stanovené.
Bezpečnosť a účinnosť Pelmegu sa neskúmali u pacientov užívajúcich vysoké dávky chemoterapie. Tento liek sa nemá používať na zvýšenie dávok cytotoxickej chemoterapie nad stanovené dávkovacie režimy.
Pľúcne nežiaduce udalosti
Po podaní G-CSF boli hlásené pľúcne nežiaduce reakcie, osobitne intersticiálna pneumónia. Zvýšené
riziko je u pacientov, ktorí majú v nedávnej anamnéze pulmonálne infiltráty alebo pneumóniu (pozri časť 4.8). Výskyt pulmonálnych prejavov, ako sú kašeľ, horúčka a dyspnoe, v spojení
s rádiologickými dôkazmi pulmonálnych infiltrátov a zhoršenie pulmonálnych funkcií spolu so zvýšeným počtom neutrofilov môže predstavovať začiatočné prejavy syndrómu akútnej respiračnej
tiesne (acute respiratory distress syndrome, ARDS). Za takýchto okolností sa má podávanie Pelmegu podľa uváženia lekára prerušiť a má sa začať vhodná liečba (pozri časť 4.8).
Glomerulonefritída
U pacientov užívajúcich filgrastim a pegfilgrastim bola hlásená glomerulonefritída. Vo všeobecnosti
sa po znížení dávky alebo po vysadení filgrastimu a pegfilgrastimu prípady glomerulonefritídy upravili. Odporúča sa sledovať rozbor moču.
Syndróm kapilárneho presakovania
Po podaní faktora stimulujúceho kolónie granulocytov sa zaznamenal syndróm kapilárneho
presakovania, ktorý je charakterizovaný hypotenziou, hypoalbuminémiou, edémom
a hemokoncentráciou. Pacienti, u ktorých sa vyvinú príznaky syndrómu kapilárneho presakovania, sa majú starostlivo sledovať a majú dostať štandardnú symptomatickú liečbu, ktorá môže zahŕňať potrebu intenzívnej starostlivosti (pozri časť 4.8).
Splenomegália a ruptúrasleziny
Po podaní pegfilgrastimu boli hlásené zvyčajne asymptomatické prípady splenomegálie a prípady
ruptúry sleziny, vrátane niekoľkých fatálnych prípadov (pozri časť 4.8). Z toho dôvodu je potrebné starostlivo sledovať veľkosť sleziny (napr. fyzikálnym vyšetrením, ultrazvukom). Diagnóza ruptúry sleziny sa má vziať do úvahy u pacientov s bolesťami v oblasti brušnej dutiny vľavo hore alebo
s bolesťami hornej časti ramena.
Trombocytopénia a anémia
Liečba samotným pegfilgrastimom nezabraňuje trombocytopénii a anémii, keďže myelosupresívna
chemoterapia sa podľa predpísaného režimu udržiava v plných dávkach. Odporúča sa pravidelné sledovanie počtu krvných doštičiek a hematokritu. Špeciálna opatrnosť je potrebná pri podávaní jednej chemoterapie alebo kombinácie chemoterapií, o ktorých je známe, že spôsobujú závažnú trombocytopéniu.
Kosáčikovitá anémia
Pri podaní pegfilgrastimu prenášačom kosáčikovitej anémie alebo pacientom s kosáčikovitou anémiou
sa vyskytli krízy kosáčikovitej anémie (pozri časť 4.8). Preto majú lekári pri predpisovaní Pelmegu prenášačom kosáčikovitej anémie alebo pacientom s kosáčikovitou anémiou postupovať opatrne, monitorovať príslušné klinické parametre a laboratórne funkcie a venovať pozornosť možnej spojitosti medzi týmto liekom a zväčšením sleziny a vazooklúznou krízou.
Leukocytóza
Menej ako 1 % pacientov liečených pegfilgrastimom vykazovalo počet bielych krviniek (white blood
cell, WBC) 100 x 109/l alebo vyšší. Neboli hlásené nežiaduce udalosti, ktoré by sa dali priamo pripísať tomuto stupňu leukocytózy. Takéto zvýšenie počtu bielych krviniek je prechodné, zvyčajne sa
objavuje 24 až 48 hodín po podaní a je v súlade s farmakodynamickými účinkami tohto lieku.
V súlade s klinickými účinkami a potenciálom pre leukocytózu sa má počet WBC počas liečby kontrolovať v pravidelných intervaloch. Ak počet leukocytov po očakávanom minime prevýši
50 x 109/l , podávanie tohto lieku sa má okamžite ukončiť.
Precitlivenosť
U pacientov liečených pegfilgrastimom sa pri úvodnej alebo následnej liečbe zaznamenala
precitlivenosť, vrátane anafylaktických reakcií. U pacientov s klinicky významnou precitlivenosťou vysaďte Pelmeg natrvalo. Nepodávajte Pelmeg pacientom s precitlivenosťou na pegfilgrastim alebo
filgrastim v anamnéze. Ak sa vyskytne závažná alergická reakcia, je potrebné podať vhodnú liečbu a po dobu niekoľkých dní pacienta starostlivo sledovať.
Imunogenicita
Rovnako ako u všetkých terapeutických proteínov existuje potenciál pre imunogenicitu. Výskyt tvorby
protilátok proti pegfilgrastimu je vo všeobecnosti nízky. Naviazané protilátky sa vyskytujú v takej miere, ako sa očakáva u všetkých biologických liekov; avšak doteraz neboli spojené s neutralizačnou aktivitou.
Aortitída
Aortitída bola hlásená po podaní G-CSF zdravým pacientom a pacientom s malígnym nádorovým
ochorením. Medzi hlásené príznaky patrili horúčka, abdominálna bolesť, malátnosť, bolesť chrbta
a zvýšená hladina zápalových markerov (napr. C-reaktívneho proteínu a počtu bielych krviniek). Vo väčšine prípadov bola aortitída diagnostikovaná pomocou snímky počítačovej tomografie (computed tomography, CT) a vo všeobecnosti ustúpila po vysadení G-CSF (pozri časť 4.8).
Iné upozornenia
Bezpečnosť a účinnosť Pelmegu na mobilizáciu krvných kmeňových buniek u pacientov alebo
zdravých darcov sa primerane nehodnila.
Zvýšenie hematopoetickej aktivity kostnej drene ako odpoveď na liečbu rastovým faktorom sa spája s prechodnými pozitívnymi nálezmi na kostných snímkach. Túto skutočnosť je potrebné zvážiť pri interpretácii výsledkov kostných snímok.
Tento liek obsahuje 30 mg sorbitolu v jednej naplnenej injekčnej striekačke, čo zodpovedá 50 mg/ml. Musí sa vziať do úvahy aditívny účinok súbežne podávaných liekov obsahujúcich sorbitol (alebo fruktózu) a príjem sorbitolu (alebo fruktózy) v strave.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v jednej 6 mg dávke, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Vzhľadom na potenciálnu senzitivitu rýchlo sa deliacich myeloidných buniek na cytotoxickú chemoterapiu sa má Pelmeg podávať minimálne 24 hodín po podaní cytotoxickej chemoterapie. V klinických skúšaniach sa pegfilgrastim bezpečne podával 14 dní pred chemoterapiou. Súbežné použitie Pelmegu s niektorým chemoterapeutikom sa u pacientov nehodnotilo. U zvierat viedlo
súčasné podanie pegfilgrastimu a 5-fluorouracilu (5-FU) alebo iných antimetabolitov k potencovaniu myelosupresie.
Možné interakcie s inými hematopoetickými rastovými faktormi a cytokínmi sa v klinických skúšaniach špeciálne nehodnotili.
Možnosť interakcií s lítiom, ktoré taktiež podporuje uvoľňovanie neutrofilov, sa špeciálne neskúmala. Nie sú k dispozícii dôkazy, že by takéto interakcie boli škodlivé.
Bezpečnosť a účinnosť Pelmegu sa nehodnotili u pacientov liečených chemoterapiou spojenou s oneskorenou myelosupresiou, napr. derivátmi nitrózomočoviny.
Špecifické interakčné alebo metabolické štúdie sa neuskutočnili, avšak klinické skúšania neindikovali interakcie pegfilgrastimu s inými liekmi.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití pegfilgrastimu u gravidných
žien. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3). Pelmeg sa neodporúča používať počas gravidity a u žien vo fertilnom veku nepoužívajúcich antikoncepciu.
Dojčenie
Nie sú dostatočné informácie o vylučovaní pegfilgrastimu/metabolitov do ľudského mlieka; riziko
u novorodencov/dojčiat nemôže byť vylúčené. Rozhodnutie či ukončiť dojčenie alebo či ukončiť/prerušiť liečbu Pelmegom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
Pegfilgrastim neovplyvnil reprodukčnú schopnosť ani fertilitu samcov alebo samíc pri podávaní
kumulatívnych týždenných dávok približne 6 až 9-krát vyšších, ako je odporúčaná dávka u ľudí (na základe plochy povrchu tela) (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Pelmeg nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najčastejšie hlásenými nežiaducimi reakciami boli bolesť v kostiach (veľmi častá [≥ 1/10])
a muskuloskeletálna bolesť (častá). Bolesť v kostiach bola väčšinou miernej až strednej intenzity, prechodnej povahy a u väčšiny pacientov bola kontrolovateľná štandardnými analgetikami.
Reakcie hypersenzitívneho typu vrátane kožnej vyrážky, žihľavky, angioedému, dyspnoe, erytému, sčervenania a hypotenzie sa vyskytli pri začiatočnej alebo následnej liečbe pegfilgrastimom (menej časté [≥ 1/1 000 až < 1/100]). U pacientov liečených pegfilgrastimom sa môžu menej často vyskytnúť závažné alergické reakcie vrátane anafylaxie (pozri časť 4.4).
Syndróm kapilárneho presakovania, ktorý môže ohroziť život, ak sa liečba oneskorí, sa zaznamenal ako menej častý (≥ 1/1 000 až < 1/100) po podaní faktorov stimulujúcich kolónie granulocytov pacientom s malígnym ochorením, ktorí podstupujú chemoterapiu; pozri časť 4.4 a časť “Opis vybraných nežiaducich reakcií“ nižšie.
Splenomegália, zvyčajne asymptomatická, je menej častá.
Ruptúra sleziny, vrátane fatálnych prípadov, je po podaní pegfilgrastimu hlásená menej často (pozri časť 4.4). Zaznamenali sa menej časté pľúcne nežiaduce reakcie vrátane intersticiálnej pneumónie, pľúcneho edému, pľúcnych infiltrátov a pľúcnej fibrózy. Menej často tieto prípady prerástli do respiračného zlyhania alebo syndrómu akútnej respiračnej tiesne (ARDS), ktoré môžu byť fatálne (pozri časť 4.4).
U prenášačov kosáčikovitej anémie alebo u pacientov s kosáčikovitou anémiou sa zaznamenali izolované prípady kríz kosáčikovitej anémie (menej často u pacientov s kosáčikovitou anémiou) (pozri časť 4.4).
Súhrn nežiaducich reakcií zoradených do tabuľky
Údaje uvedené v tabuľke nižšie opisujú nežiaduce reakcie hlásené z klinických skúšaní a spontánnych
hlásení. V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Trieda
orgánových systémov MedDRA
Nežiaduce reakcie
Veľmi časté Časté Menej časté Zriedkavé
Veľmi
z
riedkavé
(≥ 1/10) (≥ 1/100
až < 1/10)
(≥ 1/1 000
až < 1/100)
(≥ 1/10 000
až < 1/1 000) (< 1/10 000)
Poruchy krvi
Trombocytopénia1 Kríza kosáčikovitej
a lymfatického systému
Poruchy imunitného systému Poruchy metabolizmu a výživy Poruchy nervového systému
Bolesť hlavy1
Leukocytóza 1
anémie2; Splenomegália2; Ruptúra sleziny2
Hypersenzitívne reakcie; Anafylaxia
Zvýšenie hladín kyseliny močove
Poruchy ciev Syndróm kapilárneho presakovania1
Aortitída
Poruchy dýchacej sústavy, hrudníka
 a mediastína
Poruchy gastrointestinál neho traktu Poruchy kože
a podkožného
tkaniva
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Poruchy obličiek a močových ciest
a mediastína
Poruchy gastrointestinál neho traktu Poruchy kože
a podkožného
tkaniva
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Poruchy obličiek a močových ciest
Nauzea1
Bolesť
v kostiach
Muskuloskeletálna bolesť (myalgia, artralgia, bolesť
v končatinách, bolesť chrbta, muskuloskeletálna bolesť, bolesť krku)
Syndróm akútnej respiračnej tiesne2; Pľúcne nežiaduce reakcie (intersticiálna pneumónia, pľúcny edém, pľúcne infiltráty a pľúcna fibróza)
Hemoptýza
Sweetov syndrom (akútna febrilná dermatóza) 1,2; Kožná vaskulitída1,2
Glomerulonefritída2
Pulmonálne krvácanie
Trieda orgánových systémov MedDRA
Nežiaduce reakcie
Veľmi časté Časté Menej časté Zriedkavé
Veľmi
z
riedkavé
Celkové poruchy a reakcie
v mieste podania
Laboratórne a funkčné vyšetrenia
(≥ 1/10) (≥ 1/100
až < 1/10) Bolesť v mieste vpichu,
Bolesť na hrudi,
ktorá nesúvisí so srdcom1
(≥ 1/1 000
až < 1/100) Reakcie v mieste vpichu2
Zvýšenie hladiny laktátdehydrogenáz y a alkalickej fosfatázy1; Prechodné zvýšenie LFT pre ALT alebo AST 1
(≥ 1/10 000
až < 1/1 000) (< 1/10 000)
1 Pozri časť nižšie “Opis vybraných nežiaducich reakcií”.
2 Táto nežiaduca reakcia sa zistila po uvedení lieku na trh, nepozorovala sa však v randomizovaných, kontrolovaných klinických skúšaniach u dospelých, ktoré boli podkladom pre registráciu. Kategória frekvencie bola odhadovaná zo štatistického výpočtu na základe 1 576 pacientov liečených
pegfilgrastimom v deviatich randomizovaných klinických skúšaniach.
Opis vybraných nežiaducich reakcií
Zaznamenali sa menej časté prípady Sweetovho syndrómu, hoci v niektorých prípadoch môže k ich
vzniku prispievať aj základné hematologické nádorové ochorenie.
U pacientov liečených pegfilgrastimom sa menej často zaznamenali prípady kožnej vaskulitídy. Mechanizmus vaskulitídy u pacientov liečených pegfilgrastimom nie je známy.
Pri začiatočnej alebo následnej liečbe pegfilgrastimom sa objavili reakcie v mieste vpichu, vrátane erytému v mieste vpichu (menej časté), ako aj bolesť v mieste vpichu (časté).
Často sa zaznamenali prípady leukocytózy (počet bielych krviniek [WBC] > 100 x 109/l) (pozri časť 4.4).
Vratný, mierny až stredný nárast hladiny kyseliny močovej a alkalickej fosfatázy bez pridružených klinických účinkov sa vyskytoval menej často; vratný, mierny až stredný nárast hladiny laktátdehydrogenázy bez pridružených klinických účinkov sa objavil menej často u pacientov liečených pegfilgrastimom po cytotoxickej chemoterapii.
Nevoľnosť a bolesť hlavy boli zaznamenané veľmi často u pacientov užívajúcich chemoterapiu. U pacientov po podaní pegfilgrastimu následne po cytotoxickej chemoterapii sa menej často
pozorovalo zvýšenie hodnôt funkčných pečeňových testov – ALT (alanínaminotransferázy) alebo AST (aspartátaminotransferázy). Tieto zvýšenia boli prechodné, s návratom na pôvodné hodnoty.
Zaznamenali sa časté prípady trombocytopénie.
Po uvedení faktora stimulujúceho kolónie granulocytov na trh sa zaznamenali prípady syndrómu kapilárneho presakovania. Zvyčajne sa vyskytovali u pacientov s pokročilým nádorovým ochorením, sepsou, u pacientov liečených kombinovanou chemoterapiou alebo podstupujúcich aferézu (pozri časť 4.4).
Pediatrická populáciaSkúsenosti u detí sú obmedzené. U mladších detí vo veku 0 – 5 rokov sa v porovnaní so staršími
deťmi vo veku 6 - 11 rokov (80 %) a 12 - 21 rokov (67 %) a dospelými pozoroval vyšší výskyt závažných nežiaducich reakcií (92 %). Najčastejšie hlásená nežiaduca reakcia bola bolesť kostí (pozri časti 5.1 a 5.2).
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieObmedzenému počtu zdravých dobrovoľníkov a pacientov s nemalobunkovým karcinómom pľúc sa subkutánne podávali jednorazové dávky 300 μg/kg bez závažných nežiaducich reakcií. Nežiaduce udalosti boli podobné udalostiam, ktoré sa pozorovali u jedincov, ktorým sa podávali nižšie dávky pegfilgrastimu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunostimulanciá, faktor stimulujúci kolónie; ATC kód: L03AA13
Pelmeg je biologicky podobný liek. Podrobné informácie sú dostupné na internetovej stránke
Európskej agentúry pre lieky
http://www.ema.europa.eu.
Ľudský faktor stimulujúci kolónie granulocytov (G-CSF) je glykoproteín, ktorý reguluje produkciu
a uvoľňovanie neutrofilov z kostnej drene. Pegfilgrastim je kovalentný konjugát rekombinantného ľudského G-CSF (r-metHuG-CSF) a jednej 20 kd molekuly polyetylénglykolu (PEG). Pegfilgrastim je vzhľadom na znížený renálny klírens trvácnejšou formou filgrastimu. Mechanizmus účinku pegfilgrastimu a filgrastimu sa ukázal byť identický a vedie k značnému zvýšeniu počtu periférnych krvných neutrofilov do 24 hodín, s miernym nárastom hladiny monocytov a/alebo lymfocytov. Podobne ako v prípade filgrastimu, neutrofily produkované ako odpoveď na pegfilgrastim vykazujú normálne alebo silnejšie funkcie ako sa demonštrovalo v testoch chemotaktických a fagocytárnych funkcií. Ako aj ostatné hematopoetické rastové faktory,
in vitro G-CSF vykazuje stimulujúce
vlastnosti na ľudské endoteliálne bunky. G-CSF môže
in vitro podporovať rast myeloidných buniek, vrátane malígnych. Podobné efekty možno
in vitro pozorovať na niektorých iných ako myeloidných bunkách.
V dvoch randomizovaných, dvojito zaslepených, pivotných štúdiách s pacientmi s vysoko rizikovým karcinómom prsníka II-IV štádia, ktorí podstupovali myelosupresívnu chemoterapiu pozostávajúcu'
z doxorubicínu a docetaxelu, podávanie pegfilgrastimu jedenkrát počas cyklu znížilo trvanie neutropénie a výskyt febrilnej neutropénie podobne, ako sa pozorovalo v prípade denného podávania filgrastimu (medián 11 denných podaní). Bez podpory rastového faktora viedol takýto režim k stredne dlho trvajúcej (5 až 7 dní) neutropénii stupňa 4 a 30–40 % výskytu febrilnej neutropénie. V jednej štúdii (n = 157), kde sa podávala stála dávka 6 mg pegfilgrastimu, sa stredná dĺžka trvania neutropénie stupňa 4 v skupine s pegfilgrastimom pohybovala na úrovni 1,8 dňa, v porovnaní s 1,6 dňa v skupine
s filgrastimom (rozdiel 0,23 dňa, 95 % interval spoľahlivosti −0,15; 0,63). Počas celej štúdie bol výskyt febrilnej neutropénie 13 % u pacientov liečených pegfilgrastimom, v porovnaní s 20 %
u pacientov liečených filgrastimom (rozdiel 7 %, 95 % interval spoľahlivosti −19 %; 5 %). V druhej štúdii (n = 310), kde sa podávala dávka upravená podľa hmotnosti (100 μg/kg), bola stredná dĺžka trvania neutropénie stupňa 4 1,7 dňa v skupine s pegfilgrastimom, v porovnaní s 1,8 dňa v skupine
s filgrastimom (rozdiel 0,03 dňa, 95 % interval spoľahlivosti −0,36; 0,30). Celkový výskyt febrilnej
neutropénie bol 9 % u pacientov liečených pegfilgrastimom v porovnaní s 18 % u pacientov liečených filgrastimom (rozdiel 9 %, 95 % interval spoľahlivosti −16,8 %; −1,1 %).
V placebom kontrolovanej, dvojito zaslepenej štúdii u pacientov s karcinómom prsníka sa hodnotil účinok pegfilgrastimu na ovplyvnenie incidencie febrilnej neutropénie po podávaní chemoterapeutického režimu spojeného s 10–20 % výskytom febrilnej neutropénie (docetaxel
100 mg/m2 každé 3 týždne počas 4 cyklov). Deväťstodvadsaťosem pacientov bolo randomizovaných buď do skupiny, ktorá dostávala jednorazovú dávku pegfilgrastimu alebo ktorá dostávala placebo
približne 24 hodín (deň 2) po chemoterapii v každom cykle. Incidencia febrilnej neutropénie bola
nižšia u pacientov randomizovaných do skupiny, ktorá dostávala pegfilgrastim v porovnaní so skupinou s placebom (1 % verzus 17 %, p < 0,001). Výskyt hospitalizácie a podanie i.v. antiinfektív v súvislosti s klinickou diagnózou febrilnej neutropénie bol nižší v skupine pacientov
s pegfilgrastimom v porovnaní so skupinou s placebom (1 % verzus 14 %, p < 0,001; a 2 % verzus
10 %, p < 0,001).
V malej (n = 83) randomizovanej dvojito zaslepenej štúdii fázy II u pacientov, ktorí dostávali chemoterapiu na liečbu akútnej myeloidnej leukémie de novo, sa porovnával pegfilgrastim (jednorazová dávka 6 mg) s filgrastimom s podávaním počas indukčnej chemoterapie. Medián času na zotavenie z ťažkej neutropénie bol stanovený na 22 dní v oboch liečebných skupinách. Dlhodobé skúšky sa nevykonali (pozri časť 4.4).
V multicentrickej, randomizovanej, otvorenej štúdii fázy II (n = 37) s pediatrickými pacientmi so sarkómom, ktorí dostávali 100 μg/kg pegfilgrastimu po prvom cykle chemoterapie vinkristínom, doxorubicínom a cyklofosfamidom (VAdriaC/IE), sa pozorovalo dlhšie trvanie závažnej neutropénie (neutrofily < 0,5 x 109) u mladších detí vo veku 0-5 rokov (8,9 dní), v porovnaní so staršími deťmi
vo veku 6–11 rokov (6 dní) a 12–21 rokov (3,7 dní) a dospelými. Okrem toho sa pozoroval vyšší
výskyt febrilnej neutropénie u mladších detí vo veku 0–5 rokov (75 %), v porovnaní so staršími deťmi vo veku 6–11 rokov (70 %) a 12–21 rokov (33 %) a dospelými (pozri časti 4.8 a 5.2).
5.2 Farmakokinetické vlastnosti
Po jednej subkutánnej dávke pegfilgrastimu sa maximálna sérová koncentrácia pegfilgrastimu dosiahne 16 až 120 hodín po podaní a sérové koncentrácie pegfilgrastimu sa udržujú počas obdobia neutropénie po myelosupresívnej chemoterapii. Vzhľadom na dávku je eliminácia pegfilgrastimu nelineárna; sérový klírens pegfilgrastimu klesá s narastajúcou dávkou. Zdá sa, že pegfilgrastim sa zväčša eliminuje klírensom sprostredkovaným neutrofilmi, ktoré sú pri vyšších dávkach saturované. Sérová koncentrácia pegfilgrastimu prudko klesá s nástupom obnovy neutrofilov, čo je v súlade
s mechanizmom spätnej regulácie klírensu (pozri obrázok 1).
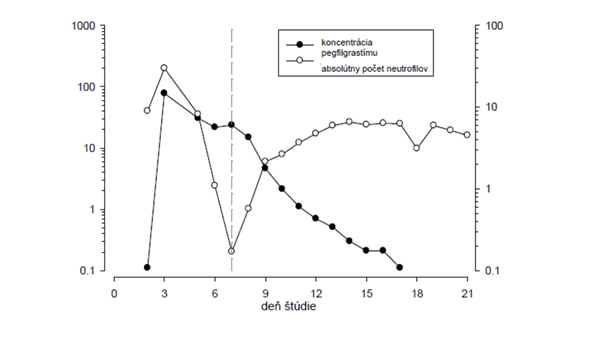
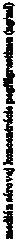
 Obrázok 1. Profil mediánov sérovej koncentrácie pegfilgrastimu a absolútneho počtu neutrofilov (absolute neutrophil count, ANC) po jednorazovom injekčnom podaní (6 mg) pacientom užívajúcim chemoterapiu
Obrázok 1. Profil mediánov sérovej koncentrácie pegfilgrastimu a absolútneho počtu neutrofilov (absolute neutrophil count, ANC) po jednorazovom injekčnom podaní (6 mg) pacientom užívajúcim chemoterapiu
Vzhľadom na neutrofilmi sprostredkovaný mechanizmus klírensu sa vplyv poruchy funkcie obličiek alebo pečene na farmakokinetiku pegfilgrastimu nepredpokladá. V otvorenej štúdii (n = 31)
s jednorazovou dávkou nemal rôzny stupeň poruchy funkcie obličiek, vrátane konečného štádia ochorenia obličiek (
end stage renal disease, ESRD), žiaden vplyv na farmakokinetiku pegfilgrastimu.
Starší pacientiObmedzené údaje poukazujú, že farmakokinetika pegfilgrastimu u starších pacientov (> 65 rokov) je
podobná ako u dospelých.
Pediatrická populáciaFarmakokinetika pegfilgrastimu sa skúmala u 37 pediatrických pacientov so sarkómom, ktorí dostávali
100 μg/kg pegfilgrastimu po skončení VAdriaC/IE chemoterapie. Najmladšia veková skupina
(0-5 rokov) mala vyššiu priemernú expozíciu pegfilgrastimu (AUC) (±SD) (47,9±22,5 μgꞏhod/ml) ako staršie deti vo veku 6–11 rokov (22,0±13,1 μg hod/ml) a 12–21 rokov (29,3±23,2 μg hod/ml) (pozri časť 5.1). S výnimkou najmladšej vekovej skupiny (0–5 rokov) sa zdalo, že priemerná hodnota AUC
u pediatrických jedincov je podobná ako u dospelých pacientov s vysoko rizikovým štádiom II - IV karcinómu prsníka, ktorí užívali 100 μg/kg pegfilgrastimu po skončení chemoterapie. doxorubicínom/docetaxelom (pozri časti 4.8 a 5.1).
5.3 Predklinické údaje o bezpečnostiPredklinické údaje z obvyklých štúdií toxicity po opakovanom podávaní odhalili očakávané farmakologické účinky vrátane zvýšenia počtu leukocytov, myeloidnej hyperplázie v kostnej dreni, extramedulárnej hematopoézy a zväčšenia sleziny.
U mláďat potkanov, ktorým sa v období gravidity subkutánne podal pegfilgrastim, sa nepozorovali nežiaduce účinky, avšak u králikov sa preukázala embryonálna/fetálna toxicita (strata embrya) spôsobená pegfilgrastimom pri kumulatívnych dávkach približne 4-krát vyšších, ako je odporúčaná dávka u ľudí. Embryonálna/fetálna toxicita (strata embrya) sa nepozorovala, keď boli gravidné králičice vystavené dávke, ktorá sa odporúča u ľudí. V štúdiách s potkanmi sa dokázalo, že
pegfilgrastim môže prenikať placentou. Štúdie u potkanov naznačili, že subkutánne podaný pegfilgrastim neovplyvňuje reprodukčnú výkonnosť, fertilitu, estrálny cyklus, dni medzi párením
a pohlavným stykom a vnútromaternicové prežívanie. Význam týchto nálezov pre ľudí nie je známy.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
octan sodný* sorbitol (E 420) polysorbát 20 voda na injekciu
kyselina chlorovodíková (na úpravu pH)
hydroxid sodný (na úpravu pH)
*Octan sodný pripravený zmiešaním trihydrátu octanu sodného a kyseliny octovej.
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi, najmä nie s roztokmi chloridu sodného.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 ºC – 8 ºC).
Pelmeg možno vystaviť izbovej teplote (nie nad 30 ºC) najviac jedenkrát na maximálne 96 hodín. Pelmeg ponechaný pri izbovej teplote po dobu dlhšiu ako 96 hodín sa musí zlikvidovať.
Neuchovávajte v mrazničke. Náhodná expozícia mrazu dvakrát, vždy na čas kratší ako 72 hodín neovplyvnila nepriaznivo stabilitu Pelmegu.
Obal uchovávajte vo vonkajšej škatuli na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
Naplnená injekčná striekačka (sklo typu I) s brómbutylovou gumovou zátkou a ihlou z nehrdzavejúcej ocele s automatickým chráničom ihly.
Každá naplnená injekčná striekačka obsahuje 0,6 ml injekčného roztoku. Veľkosť balenia je jedna naplnená injekčná striekačka v blistrovom balení.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Pred podávaním je nutné vizuálne skontrolovať, či roztok Pelmegu neobsahuje viditeľné častice. Podávať sa môžu iba číre a bezfarebné roztoky.
Nadmerné pretrepávanie môže viesť k agregácii pegfilgrastimu a tak spôsobiť inaktiváciu jeho biologických vlastností.
Pred podaním ponechajte naplnenú injekčnú striekačku, dosiahnuť izbovú teplotu.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIICinfa Biotech S.L.
Travesía de Roncesvalles, 1
31699 Olloki (Navarra), Španielsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/18/1328/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu/.