
> 1 až ≤ 1,5 x ULN
a aspartátaminotransferáza [AST] ≤ 10 x ULN) nie je potrebná úprava dávky, bez ohľadu na indikáciu. Lieči sa rovnakými dávkami ako u pacientov s normálnou funkciou pečene.
U pacientov s metastatickým karcinómom prsníka a u pacientov s nemalobunkovým karcinómom pľúc
so stredne závažným až závažným poškodením pečene (celkový bilirubín > 1,5 až ≤ 5 x ULN a AST
≤ 10 x ULN) je odporúčané zníženie dávky o 20 %. Znížená dávka môže byť zvýšená až na dávku
rovnakú ako pre pacientov s normálnou funkciou pečene, ak pacient toleruje liečbu po dobu aspoň
dvoch cyklov (pozri časti 4.4 a 5.2).
U pacientov s celkovým bilirubínom > 5 x ULN alebo AST > 10 x ULN nie sú dostupné dostatočné údaje, ktoré by umožnili odporúčanie na dávkovanie, bez ohľadu na indikáciu (pozri časti 4.4 a 5.2).
Pacienti s poškodením obličiek
Úprava začiatočnej dávky Pazeniru nie je potrebná u pacientov s miernym až stredne závažným
poškodením obličiek (odhadovaný klírens kreatinínu ≥ 30 až < 90 ml/min). U pacientov so závažným poškodením obličiek alebo u pacientov v terminálnom štádiu ochorenia obličiek nie sú dostupné dostatočné údaje o odporúčaniach na zmeny dávkovania Pazeniru (odhadovaný klírens kreatinínu
< 30 ml/min) (pozri časť 5.2).
Starší pacienti
Pre pacientov vo veku 65 rokov a viac sa neodporúča žiadne ďalšie znižovanie dávkovania, okrem
zmien odporúčaných pre všetkých pacientov.
Z 229 pacientov v randomizovanej štúdii, ktorí dostávali nanočastice paklitaxelu-ľudského sérového albumínu v monoterapii pri liečbe karcinómu prsníka, malo 13 % minimálne 65 rokov a < 2 % pacientov malo 75 rokov a viac. Neobjavili sa žiadne toxicity, ktoré by sa vyskytovali oveľa častejšie u pacientov vo veku minimálne 65 rokov, ktorí dostávali nanočastice paklitaxelu-ľudského sérového albumínu. Avšak následná analýza 981 pacientov, ktorí dostávali nanočastice paklitaxelu-ľudského
sérového albumínu v monoterapii na liečbu metastatického karcinómu prsníka, z ktorých 15 % bolo vo veku ≥ 65 rokov a 2 % boli vo veku ≥ 75 rokov, ukázala vyšší výskyt epistaxy, hnačky, dehydratácie, únavy a periférneho edému u pacientov vo veku ≥ 65 rokov.
Z 514 pacientov s nemalobunkovým karcinómom pľúc v randomizovanej štúdii, ktorí dostávali
nanočastice paklitaxelu-ľudského sérového albumínu v kombinácii s karboplatinou, malo 31 %
65 rokov alebo viac a 3,5 % bolo vo veku 75 rokov alebo starších. Prípady myelosupresie, periférnej neuropatie a artralgie boli častejšie u pacientov vo veku 65 rokov a starších v porovnaní s pacientmi
mladšími ako 65 rokov. K dispozícii sú len obmedzené skúsenosti s použitím nanočastíc paklitaxelu-
ľudského sérového albumínu/karboplatiny u pacientov vo veku 75 rokov alebo starších.
Farmakokinetický/farmakodynamický model využívajúci údaje od 125 pacientov s pokročilými solídnymi tumormi naznačuje, že pacienti vo veku ≥ 65 rokov môžu byť náchylnejší k rozvoju neutropénie počas prvého liečebného cyklu.
Pediatrická populácia
Bezpečnosť a účinnosť nanočastíc paklitaxelu-ľudského sérového albumínu u detí a dospievajúcich vo veku 0 - 17 rokov neboli stanovené. Použitie nanočastíc paklitaxelu-ľudského sérového albumínu sa
netýka pediatrickej populácie pre indikáciu metastatického karcinómu prsníka alebo
nemalobunkového karcinómu pľúc.
Spôsob podávania
Pazenir je určený na intravenózne použitie. Rekonštituovaná disperzia Pazeniru sa má podať intravenózne pomocou infúznej súpravy obsahujúcej 15 µm filter. Po podaní infúzie sa odporúča prepláchnuť intravenóznu linku injekčným roztokom chloridu sodného 9 mg/ml (0,9 %), aby sa
zabezpečilo podanie úplnej dávky.
Pokyny na rekonštitúciu lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Dojčenie (pozri časť 4.6).
Pacienti s východiskovým počtom neutrofilov < 1 500 buniek/mm3.
4.4 Osobitné upozornenia a opatrenia pri používaní
Pazenir obsahuje paklitaxel vo forme nanočastíc viazaných na albumín, ktorá môže mať podstatne odlišné farmakologické vlastnosti v porovnaní s inými formami paklitaxelu (pozri časti 5.1 a 5.2). Liek nemá byť náhradou iných liekov obsahujúcich paklitaxel, ani sa nemá nimi nahrádzať.
Precitlivenosť
Hlásený bol zriedkavý výskyt závažných reakcií precitlivenosti, vrátane veľmi zriedkavých prípadov anafylaktických reakcií so smrteľným koncom. Ak sa vyskytne reakcia z precitlivenosti, liek sa má
okamžite vysadiť, má sa začať symptomatická liečba a pacientovi sa nemá znovu podať paklitaxel.
Hematológia
Pri liečbe nanočasticami paklitaxelu-ľudského sérového albumínu často dochádza k potlačeniu
krvotvorby v kostnej dreni (najmä k neutropénii). Neutropénia je toxicita závislá od dávky
a obmedzujúca dávku. Počas liečby Pazenirom sa má vykonávať časté monitorovanie krvného obrazu. Pacienti nemajú pokračovať v liečbe následnými cyklami Pazeniru, kým sa hladina neutrofilov nevráti na > 1 500 buniek/mm3 a kým sa trombocyty nevrátia na > 100 000 buniek/mm3 (pozri časť 4.2).
Neuropatia
Pri liečbe nanočasticami paklitaxelu-ľudského sérového albumínu sa často vyskytuje senzorická neuropatia, hoci vývoj závažných príznakov je menej častý. Výskyt senzorickej neuropatie 1.
a 2. stupňa si zvyčajne nevyžaduje zníženie dávky. Ak sa pri podávaní Pazeniru v monoterapii vyvinie
senzorická neuropatia 3. stupňa, liečba sa má pozastaviť dovtedy, kým sa nezlepší na 1. alebo
2. stupeň a pre všetky nasledujúce cykly Pazeniru sa odporúča zníženie dávky (pozri časť 4.2).
V prípade kombinovaného použitia Pazeniru s karboplatinou, keď sa vyvinie periférna neuropatia
3. stupňa alebo vyššia, sa má liečba prerušiť, až kým sa neuropatia nezlepší na 0. alebo 1. stupeň, a má
nasledovať zníženie dávky pre všetky nasledujúce cykly Pazeniru a karboplatiny (pozri časť 4.2).
Pneumonitída
Pneumonitída sa objavila u 1 % pacientov, keď sa nanočastice paklitaxelu-ľudského sérového
albumínu podávali v monoterapii. Všetci pacienti majú byť pozorne sledovaní ohľadne prejavov
a príznakov pneumonitídy. Po vylúčení infekčnej etiológie a po určení diagnózy pneumonitídy sa má
pri výskyte pneumonitídy liečba Pazenirom trvalo vysadiť a okamžite začať s primeranou liečbou
a podpornými opatreniami (pozri časť 4.2).
Poškodeniepečene
Keďže toxicita paklitaxelu môže byť zvýšená pri poškodení pečene, má sa Pazenir pacientom
s poškodením pečene podávať s opatrnosťou. Pacienti s poškodením pečene môžu mať zvýšené riziko toxicity, najmä kvôli myelosupresii, a títo pacienti sa majú pozorne monitorovať, či sa u nich nevyvíja ťažká myelosupresia.
Pazenir nie je odporúčaný u pacientov, ktorí majú celkový bilirubín > 5 x ULN alebo
AST > 10 x ULN (pozri časť 5.2).
Kardiotoxicita
U jedincov liečených nanočasticami paklitaxelu-ľudského sérového albumínu sa pozorovali zriedkavé
prípady kongestívneho srdcového zlyhania a dysfunkcie ľavej komory. Väčšina jedincov bola predtým
vystavená kardiotoxickým liekom, ako sú antracyklíny, alebo mali srdcové ochorenia v anamnéze. Preto majú lekári u pacientov dostávajúcich Pazenir dôsledne monitorovať výskyt srdcových príhod.
Metastázy v centrálnom nervovom systéme
Účinnosť a bezpečnosť nanočastíc paklitaxelu-ľudského sérového albumínu u pacientov s metastázami v centrálnom nervovom systéme (CNS) nebola stanovená. Metastázy v CNS zvyčajne nie sú dobre
kontrolované systémovou chemoterapiou.
Gastrointestinálne príznaky
Ak pacienti po podaní Pazeniru cítia nevoľnosť, vracajú alebo majú hnačku, možno ich liečiť bežne
používanými antiemetikami a konstipačnými látkami.
Pomocné látky
Po rekonštitúcii obsahuje každý ml koncentrátu Pazeniru 4,2 mg sodíka. Musí sa vziať do úvahy
u pacientov na diéte s kontrolovaným obsahom sodíka.
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v injekčnej liekovke, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcie
Metabolizmus paklitaxelu je čiastočne katalyzovaný izoenzýmami cytochrómu P450, CYP2C8
a CYP3A4 (pozri časť 5.2). Preto pri chýbajúcich farmakokinetických štúdiách liekových interakcií, je pri podávaní paklitaxelu súbežne s liekmi, ktoré sú známe ako inhibítory buď CYP2C8 alebo CYP3A4
(napr. ketokonazol a iné imidazolové antimykotiká, erytromycín, fluoxetín, gemfibrozil, klopidogrel,
cimetidín, ritonavir, saquinavir, indinavir a nelfinavir) potrebná opatrnosť z dôvodu možného zvýšenia toxicity paklitaxelu spôsobeného vyššou expozíciou paklitaxelu. Podávanie paklitaxelu súbežne
s liekmi, o ktorých je známe, že indukujú buď CYP2C8 alebo CYP3A4 (napr. rifampicín, karbamazepín, fenytoín, efavirenz, nevirapín) sa neodporúča, pretože účinnosť by mohla byť znížená kvôli nižšej expozícii paklitaxelu.
Klírens paklitaxelu primárne určuje metabolizmus sprostredkovaný CYP2C8 a CYP3A4, potom
nasleduje exkrécia žlčou.
Farmakokinetická štúdia bola vykonaná s nanočasticami paklitaxelu-ľudského sérového albumínu a karboplatinou u pacientov s nemalobunkovým karcinómom pľúc. Neboli zistené žiadne klinicky významné farmakokinetické interakcie medzi nanočasticami paklitaxelu-ľudského sérového albumínu a karboplatinou.
Pazenir je indikovaný v monoterapii na liečbu karcinómu prsníka alebo v kombinácii s karboplatinou na liečbu nemalobunkového karcinómu pľúc (pozri časť 4.1). Pazenir sa nemá používať v kombinácii s inými protinádorovými liekmi.
4.6 Fertilita, gravidita a laktácia
Antikoncepcia u mužov a žien
Ženy vo fertilnom veku majú používať účinnú antikoncepciu počas liečby až do 1 mesiaca po
ukončení liečby Pazenirom. Mužským pacientom liečeným Pazenirom sa odporúča nesplodiť dieťa počas liečby a do šiestich mesiacov po liečbe.
Gravidita
Existujú len veľmi obmedzené údaje o používaní paklitaxelu počas gravidity u ľudí. Predpokladá sa, že paklitaxel spôsobuje závažné vrodené defekty, keď sa podáva počas gravidity. Štúdie na zvieratách
preukázali reprodukčnú toxicitu (pozri časť 5.3). U žien vo fertilnom veku sa má pred začatím liečby
Pazenirom previesť tehotenský test. Pazenir sa nemá používať počas gravidity a ani u žien vo
fertilnom veku, ktoré nepoužívajú účinnú antikoncepciu, pokiaľ klinický stav matky nevyžaduje liečbu
paklitaxelom.
D
ojčenie
Paklitaxel a/alebo jeho metabolity sa vylučovali do mlieka dojčiacich potkanov (pozri časť 5.3). Nie je
známe, či sa paklitaxel vylučuje do ľudského mlieka. Vzhľadom k možnému výskytu závažných nežiaducich reakcií u dojčených detí je Pazenir počas dojčenia kontraindikovaný. Dojčenie musí byť počas liečby ukončené.
FertilitaNanočastice paklitaxelu-ľudského sérového albumínu spôsobili neplodnosť u samcov potkanov (pozri časť 5.3). Na základe zistení u zvierat sa môže znížiť plodnosť mužov a žien. Mužskí pacienti sa majú pred liečbou poradiť o konzervácii spermií, pretože existuje možnosť trvalej neplodnosti spôsobenej liečbou Pazenirom.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojePaklitaxel má malý alebo mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Paklitaxel môže spôsobovať nežiaduce reakcie, ako napríklad únavu (veľmi často) a závrat (často), ktoré môžu ovplyvniť schopnosť viesť vozidlá a obsluhovať stroje. Pacienti majú byť upozornení, aby neviedli vozidlá a neobsluhovali stroje, keď pociťujú únavu alebo závrat.
4.8 Nežiaduce účinkySúhrnbezpečnostnéhoprofiluNajčastejšie klinicky signifikantné nežiaduce reakcie spojené s podávaním nanočastíc paklitaxelu-
ľudského sérového albumínu boli neutropénia, periférna neuropatia, artralgia/myalgia a gastrointestinálne poruchy.
Frekvencie výskytu nežiaducich reakcií spojené s podávaním Pazeniru sú uvedené v tabuľke 3 (nanočastice paklitaxelu-ľudského sérového albumínu v monoterapii) a v tabuľke 4 (nanočastice paklitaxelu-ľudského sérového albumínu v kombinácii s karboplatinou).
Frekvencie výskytu sú definované ako: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Karcinóm prsníka (nanočastice paklitaxelu-ľudského sérovéhoalbumínupodávanévmonoterapii)TabuľkovýzoznamnežiaducichreakciíV tabuľke 3 sú uvedené nežiaduce reakcie spojené s podávaním nanočastíc paklitaxelu-ľudského
sérového albumínu u pacientov zo štúdií, v ktorých sa nanočastice paklitaxelu-ľudského sérového
albumínu podávali v monoterapii v akejkoľvek dávke pre akúkoľvek indikáciu (n = 789).
Tabuľka 3: Nežiaduce reakcie hlásené pri podávaní nanočastíc paklitaxelu-ľudského sérového albumínu v monoterapii pri akejkoľvek dávke v klinických štúdiáchČasté
albumínu v monoterapii pri akejkoľvek dávke v klinických štúdiáchČasté: Infekcia, infekcia močových ciest, folikulitída, infekcia horných dýchacích ciest, kandidóza, sinusitída
Infekcie a nákazy
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
Menej časté: Orálna kandidóza, nazofaryngitída, celulitída, herpes
simplex, vírusová infekcia, pneumónia, infekcia spojená so zavedením katétra, plesňová infekcia, herpes zoster, infekcia v mieste vpichu, sepsa2, neutropenická sepsa2
Menej časté: Bolesť súvisiaca s metastázami, nekróza tumoru
Poruchy krvi a lymfatického systému
Veľmi časté: Neutropénia, anémia, leukopénia, trombocytopénia,
lymfopénia, potlačenie krvotvorby v kostnej dreni
Časté: Febrilná neutropénia
Zriedkavé: Pancytopénia
Poruchy imunitného systému
Menej časté1: Precitlivenosť
Zriedkavé: Závažná precitlivenosť
V
eľmi časté: Anorexia
Poruchy metabolizmu a
výživy
Časté: Dehydratácia, znížená chuť do jedla, hypokaliémia
Menej časté: Hypofosfatémia, zadržiavanie tekutín, hypoalbuminémia, polydipsia, hyperglykémia, hypokalciémia, hypoglykémia, hyponatriémia
Psychické poruchy
Poruchy nervového systému
Poruchy oka
Poruchy ucha a labyrintu
Časté: Nespavosť, depresia, úzkosť
Menej časté: Nepokoj
Veľmi časté: Periférna neuropatia, neuropatia, hypoestézia, parestézia
Časté: Periférna senzorická neuropatia, bolesť hlavy, dysgeúzia, závrat,
periférna motorická neuropatia, ataxia, zmyslové poruchy, somnolencia
Menej časté: Polyneuropatia, areflexia, dyskinéza, hyporeflexia, neuralgia, strata zmyslov, synkopa, závrat v stoji, neuropatická bolesť, tremor
Časté: Zvýšené slzenie, rozmazané videnie, suché oči,
keratoconjunctivitis sicca, madaróza
Menej časté: Podráždenie očí, bolesť očí, abnormálne videnie, znížená ostrosť videnia, konjunktivitída, poruchy videnia, svrbenie očí, keratitída
Zriedkavé: Cystoidný edém makuly2
Časté: Vertigo (závrat)
Menej časté: Bolesť ucha, tinitus (hučanie v ušiach)
Poruchy srdca
a srdcovej činnosti
Časté: Tachykardia, arytmia, supraventrikulárna tachykardia
Zriedkavé: Bradykardia, zástava srdca, dysfunkcia ľavej komory,
kongestívne srdcové zlyhanie, atrioventrikulárna blokáda2
Č
asté: Sčervenanie pokožky, návaly horúčavy, hypertenzia, lymfedém
Poruchy ciev
Poruchy dýchacej sústavy, hrudníka a mediastína
Menej časté: Hypotenzia, periférny chlad, ortostatická hypotenzia
Zriedkavé: Trombóza
Časté: Intersticiálna pneumonitída3, dýchavičnosť, epistaxa, bolesť hltana
a hrtana, kašeľ, nádcha, výtok z nosa
Menej časté: Produktívny kašeľ, námahová dýchavičnosť, upchanie dutín, znížené dychové ozvy, pleurálny výpotok, alergická nádcha, chrapot, upchanie nosa, suchý nos, sipot, pľúcna embólia, pľúcna tromboembólia
Poruchy gastrointestinálneho traktu
Veľmi časté: Nevoľnosť, hnačka, vracanie, zápcha, stomatitída
Časté: Bolesť brucha, nadúvanie, bolesť hornej časti brucha, dyspepsia,
gastroezofageálna refluxná choroba, orálna hypoestézia
Menej časté: Dysfágia, plynatosť, glosodýnia, sucho v ústach, bolesť ďasien, riedka stolica, ezofagitída, bolesť v podbrušku, vredy v ústach, bolesť úst, krvácanie z rekta
Poruchy pečene
a žlčových ciest Menej časté: Hepatomegália
Veľmi časté: Alopécia, vyrážka
Časté: Porucha nechtov, svrbenie, suchá koža, erytém, pigmentácia/zmena sfarbenia nechtov, hyperpigmentácia kože, onycholýza, zmeny nechtov
Poruchy kože
a podkožného tkaniva
Menej časté: Citlivosť v nechtovom lôžku, urtikária, bolesť kože,
precitlivenosť na svetlo, porucha pigmentácie, svrbivá vyrážka, porucha kože, hyperhidróza, onychomadéza, erytematózna vyrážka, generalizovaná vyrážka, dermatitída, nočné potenie, makulopapulárna vyrážka, vitiligo, hypotrichóza, nepríjemné pocity v nechtoch, generalizovaný pruritus, makulárna vyrážka, papulárna vyrážka, kožné lézie, opuch tváre
Veľmi zriedkavé: Stevensov-Johnsonov syndróm2, toxická epidermálna nekrolýza2
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Veľmi časté: Artralgia, myalgia
Časté: Bolesť v končatinách, bolesť kostí, bolesť chrbta, svalové kŕče, bolesti končatín
Menej časté: Bolesť hrudnej steny, svalová slabosť, bolesť krku, bolesť v slabinách, svalové kŕče, muskuloskeletálna bolesť, bolesť v boku, nepríjemné pocity v končatinách, svalová slabosť
Poruchy obličiek
a močových ciest
Menej časté: Dyzúria, polakizúria, hematúria, noktúria, polyúria, močová
inkontinencia
Poruchy reprodukčného
systému a prsníkov
Menej časté: Bolesť prsníkov
V
eľmi časté: Únava, asténia, pyrexia
Celkové poruchy
a reakcie v mieste podania
Laboratórne a funkčné
vyšetrenia
Úrazy, otravy a komplikácie liečebného postupu
Časté: Periférny edém, zápal slizníc, bolesť, zimnica, edém, slabosť,
znížená výkonnosť, bolesť hrudníka, ochorenie podobné chrípke, nevoľnosť, letargia, hyperpyrexia
Menej časté: Nepríjemné pocity v hrudníku, abnormálna chôdza, opuch, reakcia v mieste vpichu
Zriedkavé: Extravazácia
Časté: Pokles telesnej hmotnosti, zvýšená hodnota alanínaminotransferázy, zvýšená hodnota aspartátaminotransferázy, znížený hematokrit, znížený počet červených krviniek, zvýšená telesná teplota, zvýšená hodnota gamaglutamyltransferáza, zvýšená hladina alkalická fosfatáza v krvi
Menej časté: Zvýšený krvný tlak, zvýšenie telesnej hmotnosti, zvýšená hladina laktátdehydrogenázy v krvi, zvýšená hladina kreatinínu v krvi, zvýšená hladina glukózy v krvi, zvýšená hladina fosforu v krvi, znížená hladina draslíka v krvi, zvýšená hodnota bilirubínu
Menej časté: Kontúzia
Zriedkavé: radiačný fenomén (radiation recall phenomenon), radiačná
pneumonitída
MedDRA (z angl.
Medical Dictionary for Regulatory Activities) = Slovník medicínskej terminológie pre regulačné činnosti. SMQ (z angl.
Standardized MedDRA Query) = štandardizovaný dotazník MedDRA; SMQ je zoskupenie niekoľkých preferovaných termínov MedDRA pre vystihnutie medicínskeho pojmu.
1 Frekvencia reakcií z precitlivenosti sa vypočítala na základe jedného určite súvisiaceho prípadu v skupine 789 pacientov.
2 Ako sa zaznamenalo podľa hlásení vychádzajúcich zo sledovaní nanočastíc paklitaxelu-ľudského sérového albumínu po
uvedení lieku na trh.
3 Frekvencia výskytu pneumonitídy sa vypočítala na základe združených údajov od 1 310 pacientov z klinických štúdií, ktorí dostávali nanočastice paklitaxelu-ľudského sérového albumínu v monoterapii na liečbu karcinómu prsníka a v ďalších indikáciách zahrnutých MedDRA SMQ pod pojmom intersticiálne ochorenie pľúc. Pozri časť 4.4.
Popis vybraných nežiaducich reakciíĎalej nasledujú najčastejšie a klinicky relevantné nežiaduce reakcie od 229 pacientov s metastatickým
karcinómom prsníka, ktorí boli liečení nanočasticami paklitaxelu-ľudského sérového albumínu
v dávke 260 mg/m2 jedenkrát za tri týždne v pivotnej klinickej štúdii fázy III.
Poruchy krvi a lymfatického systémuNajvýznamnejšia dôležitá hematologická toxicita bola neutropénia (hlásená u 79 % pacientov), ktorá bola rýchlo reverzibilná a závisela od dávky; leukopénia bola hlásená u 71 % pacientov. Neutropénia
4. stupňa (< 500 buniek/mm3) sa vyskytla u 9 % pacientov liečených nanočasticami paklitaxelu-
ľudského sérového albumínu. Febrilná neutropénia sa vyskytla u štyroch pacientov liečených nanočasticami paklitaxelu-ľudského sérového albumínu. Anémia (Hb < 10 g/dl) sa pozorovala u 46 % pacientov liečených nanočasticami paklitaxelu-ľudského sérového albumínu a bola závažná
(Hb < 8 g/dl) v troch prípadoch. Lymfopénia sa pozorovala u 45 % pacientov.
Poruchy nervového systémuFrekvencia a závažnosť neurotoxicity u pacientov dostávajúcich nanočastice paklitaxelu-ľudského
sérového albumínu bola vo všeobecnosti závislá od dávky. Periférna neuropatia (najmä senzorická neuropatia 1. alebo 2. stupňa) sa pozorovala u 68 % pacientov liečených nanočasticami paklitaxelu- ľudského sérového albumínu, pričom 10 % malo 3. stupeň a 4. stupeň nemal žiadny pacient.
Poruchy gastrointestinálneho traktuU 29 % pacientov sa vyskytla nevoľnosť a u 25 % pacientov hnačka.
P
oruchy kože a podkožného tkaniva
Alopécia sa pozorovala u > 80 % pacientov liečených nanočasticami paklitaxelu-ľudského sérového albumínu. Väčšina prípadov alopécie sa vyskytla v období kratšom ako jeden mesiac od začiatku podávania nanočastíc paklitaxelu-ľudského sérového albumínu. U väčšiny pacientov, u ktorých sa vyskytla alopécia, sa očakáva výrazný úbytok vlasov ≥ 50 %.
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Artralgia sa vyskytla u 32 % pacientov liečených nanočasticami paklitaxelu-ľudského sérového
albumínu a v 6 % prípadov bola závažná. Myalgia sa vyskytla u 24 % pacientov liečených nanočasticami paklitaxelu-ľudského sérového albumínu a v 7 % prípadov bola závažná. Príznaky boli zvyčajne prechodné, obvykle sa vyskytli tri dni po podaní nanočastíc paklitaxelu-ľudského sérového albumínu a vymizli do jedného týždňa.
Celkové poruchy a reakcie v mieste podania
Asténia/únava sa hlásili u 40 % pacientov.
Nemalobunkovýkarcinómpľúc(nanočasticamipaklitaxelu-ľudskéhosérovéhoalbumínupodávaný
v kombinácii s karboplatinou)
Tabuľkovýzoznamnežiaducichreakcií
Tabuľka 4 uvádza nežiaduce reakcie spojené s podávaním nanočastíc paklitaxelu-ľudského sérového
albumínu v kombinácii s karboplatinou.
Tabuľka 4: Nežiaduce reakcie hlásené pri liečbe nanočasticami paklitaxelu-ľudského sérového
albumínu v kombinácii s karboplatinou (n = 514)
Infekcie a nákazy Časté: Pneumónia, bronchitída, infekcia horných dýchacích ciest, infekcie
močových ciest
Poruchy krvi
a lymfatického systému1
Poruchy imunitného systému
Poruchy metabolizmu a výživy
Menej časté: Sepsa, orálna kandidóza
Veľmi časté: Neutropénia1, trombocytopénia1, anémia1, leukopénia1
Časté: Febrilná neutropénia, lymfopénia
Menej časté: Pancytopénia
Menej časté: Precitlivenosť na liek, precitlivenosť
Veľmi časté: Znížená chuť do jedla
Časté: Dehydratácia
Psychické poruchy Časté: Nespavosť
Poruchy nervového systému
Veľmi časté: Periférna neuropatia2
Časté: Poruchy chuti, bolesť hlavy, závraty
Poruchy oka Časté: Rozmazané videnie
Poruchy ciev Časté: Hypotenzia, hypertenzia
Poruchy dýchacej sústavy, hrudníka a mediastína
Poruchy gastrointestinálneho traktu
Poruchy pečene
a žlčových ciest
Menej časté: Návaly horúčavy
Veľmi časté: Dyspnoe
Časté: Hemoptýza, epistaxa, kašeľ
Menej časté: Pneumonitída3
Veľmi časté: Hnačka, vracanie, nevoľnosť, zápcha
Časté: Stomatitída, dyspepsia, abdominálna bolesť, dysfágia
Časté: Hyperbilirubinémia
Poruchy kože
a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
Celkové poruchy
a reakcie v mieste podania
Laboratórne
a funkčné vyšetrenia
Veľmi časté: Vyrážka, alopécia
Časté: Pruritus, poruchy nechtov
Menej časté: Kožná exfoliácia, alergická dermatitída, urtikária
Veľmi časté: Artralgia, myalgia
Časté: Bolesť chrbta, bolesť v končatinách, muskuloskeletálna bolesť
 Veľmi časté:
Veľmi časté: Únava, asténia, periférny edém
Časté: Pyrexia, bolesť na hrudníku
Menej časté: Sliznicové zápaly, extravazácia v mieste podania infúzie, zápal v mieste podania infúzie, vyrážka v mieste podania infúzie
Časté: Zvýšená hodnota alanínaminotransferázy, zvýšená hodnota aspartátaminotransferázy, zvýšená hladina alkalickej fosfatázy v krvi, úbytok telesnej hmotnosti
MedDRA (z angl. Medical Dictionary for Regulatory Activities) = Slovník medicínskej terminológie pre regulačné činnosti:
SMQ (z angl. Standardized MedDRA Query) = štandardizovaný dotazník MedDRA.
1 Na základe laboratórnych posúdení: maximálny stupeň myelosupresie (liečenej populácie).
2 Periférna neuropatia je hodnotená pomocou SMQ neuropatie (broad scope).
3 Pneumonitída je hodnotená pomocou SMQ intersticiálneho pľúcneho ochorenia (broad scope).
U pacientov s nemalobunkovým karcinómom pľúc liečených paklitaxelom a karboplatinou bol medián času do prvého výskytu 3. stupňa s liečbou súvisiacej periférnej neuropatie 121 dní a medián času do zlepšenia z 3. stupňa s liečbou súvisiacej periférnej neuropatie na 1. stupeň bol 38 dní. U žiadneho
z pacientov liečených nanočasticami paklitaxelu-ľudského sérového albumínu a karboplatinou sa neprejavila periférna neuropatia 4. stupňa.
Anémia a trombocytopénia boli častejšie hlásené v ramene nanočastíc paklitaxelu-ľudského sérového albumínu ako v ramene Taxolu (54 % v porovnaní s 28 % a 45 % v porovnaní s 27 %, v uvedenom poradí).
Pacientom hlásená toxicita taxánu bola hodnotená pomocou 4 podstupníc dotazníka funkčného posudku nádorovej liečby (Functional Assessment of Cancer Therapy, FACT) pre taxánového dotazníka. Použitie opakovanej analýzy mier, 3 zo 4 podstupníc (periférna neuropatia, bolesti rúk/nôh a sluch), zvýhodňovalo nanočastice paklitaxelu-ľudského sérového albumínu s karboplatinou
(p ≤ 0,002). Pre inú podstupnicu (edém) nebol žiadny rozdiel v liečebných ramenách.
Skúsenosti po uvedení lieku na trh
Hlásili sa ochrnutie hlavových nervov, paréza hlasiviek a zriedkavo prípady závažných reakcií
z precitlivenosti počas sledovania nanočastíc paklitaxelu-ľudského sérového albumínu po uvedení na
trh.
Počas liečby nanočasticami paklitaxelu-ľudského sérového albumínu boli zriedkavo hlásené prípady zníženej zrakovej ostrosti zapríčinenej cystoidným edémom makuly. Po diagnostike cystoidného edému makuly sa má liečba Pazenirom ukončiť.
Počas liečby nanočasticami paklitaxelu-ľudského sérového albumínu sa hlásili prípady syndrómu
nádorového rozpadu.
U niektorých pacientov už predtým vystavených kapecitabinu sa hlásili prípady palmárno-plantárnej erytrodyzestézie v rámci pokračujúceho sledovania nanočastíc paklitaxelu-ľudského sérového albumínu. Pretože tieto príhody sa hlásili dobrovoľne počas klinickej praxe, nemožno urobiť skutočné odhady frekvencie výskytu a kauzálny vzťah k týmto príhodám sa nezistil.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNa predávkovanie paklitaxelom nie je známe žiadne antidotum. V prípade predávkovania sa má pacient pozorne monitorovať. Liečba sa má zamerať na hlavné očakávané toxicity, čo je potlačenie krvotvorby v kostnej dreni, mukozitída a periférna neuropatia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Antineoplastické látky, rastlinné alkaloidy a iné prírodné liečivá,
taxány, ATC kód: L01CD01
MechanizmusúčinkuPaklitaxel je antimikrotubulárna látka, ktorá podporuje zoskupenie mikrotubulov z tubulínových dimérov a mikrotubuly stabilizuje tým, že bráni ich depolymerizácii. Dôsledkom tejto stabilizácie je inhibícia normálnej dynamickej reorganizácie mikrotubulárnej siete, ktorá je nevyhnutná pre vitálne interfázové a mitotické bunkové funkcie. Okrem toho paklitaxel indukuje abnormálne usporiadanie alebo „zväzky“ mikrotubulov v celom bunkovom cykle a viacpočetné mikrotubulárne astery počas mitózy.
Pazenir obsahuje nanočastice o veľkosti približne 130 nm zložené z ľudského sérového albumínu
a paklitaxelu, v ktorých sa paklitaxel nachádza v nekryštalickom, amorfnom stave. Po intravenóznom
podaní sa nanočastice rýchlo disociujú na rozpustné komplexy paklitaxelu viazané na albumín
o veľkosti približne 10 nm. Albumín je známy ako sprostredkovateľ endotelovej dutinovej transcytózy
zložiek plazmy a štúdie
in vitro preukázali, že prítomnosť albumínu zlepšuje transport paklitaxelu cez
endotelové bunky. Existuje hypotéza, že tento zlepšený transendoteliálny dutinový transport je sprostredkovaný albumínovým receptorom gp-60, a že v oblasti tumoru dochádza k zvýšenému nahromadeniu paklitaxelu následkom proteínu viazaného na albumín, tzv. vylučovaný kyslý proteín bohatý na cysteín (
Secreted Protein Acidic Rich in Cysteine, SPARC).
Klinická účinnosť abezpečnosťKarcinóm prsníkaNa podporu používania nanočastíc paklitaxelu-ľudského sérového albumínu pri metastatickom
karcinóme prsníka sú k dispozícii údaje od 106 pacientov zhromaždené v dvoch otvorených jednoramenných štúdiách a od 454 pacientov liečených v randomizovanej komparatívnej štúdii
fázy III. Tieto informácie sú uvedené nižšie.
Otvorené jednoramenné štúdieV jednej štúdii sa nanočastice paklitaxelu-ľudského sérového albumínu podávali ako 30-minútová infúzia v dávke 175 mg/m2 43 pacientom s metastatickým karcinómom prsníka. V druhom klinickom skúšaní sa používala dávka 300 mg/m2 ako 30 minútová infúzia u 63 pacientov s metastatickým karcinómom prsníka. Pacienti sa liečili bez premedikácie steroidmi alebo plánovanej podpory G-CSF. Cykly sa podávali v 3-týždňových intervaloch. Miera odpovede u všetkých pacientov bola 39,5 %
v jednej štúdii (95 % CI: 24,9 % - 54,2 %) a 47,6 % v druhej štúdii (95 % CI: 35,3 % - 60,0 %).
Medián času do progresie ochorenia bol 5,3 mesiaca (175 mg/m2; 95 % CI: 4,6 - 6,2 mesiaca)
a 6,1 mesiaca (300 mg/m2; 95 % CI: 4,2 - 9,8 mesiaca).
Randomizovaná komparatívna štúdiaToto multicentrické klinické skúšanie sa uskutočnilo u pacientov s metastatickým karcinómom prsníka, ktorí sa liečili každé 3 týždne paklitaxelom ako jedinou látkou, buď ako paklitaxel na báze rozpúšťadla 175 mg/m2, podávaný ako 3-hodinová infúzia s premedikáciou na zabránenie vzniku
precitlivenosti (n = 225), alebo ako nanočastice paklitaxelu-ľudského sérového albumínu 260 mg/m2, podávaný ako 30-minútová infúzia bez premedikácie (n = 229).
64 % pacientov malo oslabený stav výkonnosti (ECOG 1 alebo 2) pri nástupe do štúdie, 79 % malo viscerálne metastázy a 76 % malo > 3 miesta metastáz. 14 % pacientov nedostávalo predtým chemoterapiu; 27 % dostávalo len podpornú chemoterapiu, 40 % len na liečbu metastáz a 19 % aj na liečbu metastáz a aj ako podpornú liečbu. 59 % dostávalo skúšaný liek ako druhú alebo vyššiu ako druhú líniu liečby. 77 % pacientov už bolo skôr vystavených účinku antracyklínov.
Výsledky miery celkovej odpovede a čas do progresie ochorenia, čas prežívania bez progresie
ochorenia a čas prežívania u pacientov, ktorí dostávali > ako liečbu 1. línie, sú uvedené nižšie.
Tabuľka 5: Výsledky miery celkovej odpovede, medián času do progresie ochorenia a čas prežívania bez progresie ochorenia, podľa vyhodnotenia skúšajúceho
Premenná účinnosti
Nanočastice paklitaxelu- ľudského sérového albumínu (260 mg/m2)
Paklitaxel na báze
rozpúšťadla
(175 mg/m2)
p-hodnota
 Mi
e
r
a odpovede [95 % CI] (%)
Mi
e
r
a odpovede [95 % CI] (%)
> liečba 1. línie 26,5 [18,98; 34,05] (n = 132) 13,2 [7,54; 18,93] (n = 136) 0,006a
* Medián času do progresie ochorenia [95 % CI] (týždne)> liečba 1. línie 20,9 [15,7; 25,9] (n = 131) 16,1 [15,0; 19,3] (n = 135) 0,011b
* Medián prežívania bez progresie ochorenia [95 % CI] (týždne)> liečba 1. línie 20,6 [15,6; 25,9] (n = 131) 16,1 [15,0; 18,3] (n = 135) 0,010b
*Prežívanie [95 % CI] (týždne)> liečba 1. línie 56,4 [45,1; 76,9] (n = 131) 46,7 [39,0; 55,3] (n = 136) 0,020b
*Tieto údaje sa zakladajú na správe z klinickej štúdie: CA012-0 dodatok datovaný ako finálny (23. marca 2005)
a Chi-kvadrát test
b Log-rank test
U 229 pacientov liečených nanočasticami paklitaxelu-ľudského sérového albumínu
v randomizovanom, kontrolovanom klinickom skúšaní sa hodnotila bezpečnosť. Neurotoxicita
paklitaxelu sa vyhodnotila prostredníctvom zlepšenia o jeden stupeň u pacientov, ktorí mali periférnu neuropatiu 3. stupňa kedykoľvek v priebehu liečby. Prirodzený priebeh periférnej neuropatie do vyriešenia na východiskový stav sa vzhľadom na kumulatívnu toxicitu nanočastíc paklitaxelu- ľudského sérového albumínu po > 6 cykloch liečby nehodnotil a ostáva neznámy.
NemalobunkovýkarcinómpľúcMulticentrická, randomizovaná, otvorená klinická štúdia bola vykonaná u 1 052 chemoterapiou
neliečených pacientov s nemalobunkovým karcinómom pľúc v štádiu IIIb/IV. Štúdia porovnávala nanočastice paklitaxelu-ľudského sérového albumínu v kombinácii s karboplatinou s paklitaxelom na báze rozpúšťadla v kombinácii s karboplatinou v prvej línii liečby u pacientov s pokročilým nemalobunkovým karcinómom pľúc. Viac ako 99 % pacientov malo stav výkonnosti ECOG (
Eastern Cooperative Oncology Group) 0 alebo 1. Pacienti s už existujúcou neuropatiou ≥ 2. stupňa alebo so závažnými zdravotnými rizikovými faktormi ktoréhokoľvek z hlavných orgánových systémov, boli vylúčení. Nanočastice paklitaxelu-ľudského sérového albumínu sa podávali pacientom (n = 521) ako intravenózna infúzia po dobu 30 minút v dávke 100 mg/m2 v 1., 8. a 15 deň každého 21-dňového cyklu bez akejkoľvek steroidovej premedikácie a bez profylaxie faktorom stimulujúcim kolónie granulocytov. Karboplatina sa podávala intravenózne len v 1. deň každého 21-dňového cyklu v dávke AUC = 6 mg•min/ml ihneď po skončení podávania nanočastíc paklitaxelu-ľudského sérového albumínu. Paklitaxel na báze rozpúšťadla sa podával pacientom (n = 531) v dávke 200 mg/m2 formou intravenóznej infúzie po dobu 3 hodín so štandardnou premedikáciou, po ktorej ihneď nasledovala
karboplatina intravenóznym podávaním v dávke AUC = 6 mg•min/ml. Každý liek sa podával v 1. deň každého 21-dňového cyklu. V oboch liečebných ramenách štúdie sa liečba podávala až do progresie alebo rozvoja neprijateľnej toxicity. Pacienti dostávali medián 6 cyklov liečby v oboch ramenách štúdie.
Primárnym cieľovým ukazovateľom účinnosti bola miera celkovej odpovede definovaná ako percento pacientov, ktorí dosiahli objektívne potvrdenú úplnú odpoveď alebo čiastočnú odpoveď na základe nezávislého, centrálneho, zaslepeného rádiologického vyšetrenia s použitím kritérií RECIST
(verzia 1.0). Pacienti v ramene s nanočasticami paklitaxelu-ľudského sérového albumínu/karboplatinou mali významne vyššiu mieru celkovej odpovede v porovnaní s pacientmi'
v kontrolnom ramene: 33 % v porovnaní s 25 %, p = 0,005 (tabuľka 6). V miere celkovej odpovede existuje významný rozdiel v ramene s nanočasticami paklitaxelu-ľudského sérového albumínu/karboplatinou v porovnaní s kontrolným ramenom u pacientov s nemalobunkovým
karcinómom pľúc skvamózneho histologického typu (n = 450, 41 % oproti 24 %, p ˂ 0,001), avšak tento rozdiel sa nepreniesol do rozdielu v PFS alebo OS. Nebol žiadny rozdiel v ORR medzi
liečebnými ramenami u pacientov s neskvamóznou histológiou (n = 602, 26 % oproti 25 %, p = 0,808).
Tabuľka 6: Miera celkovej odpovede v randomizovanej štúdii nemalobunkového karcinómu
pľúc („intent-to-treat“ populácia).
N
anočastice
paklitaxelu-ľudského
P
aklitaxel na báze
rozpúšťadla
2
P
arameter účinnosti
s
érového albumínu
(
200 mg/m
každé
Miera celkovej odpovede (nezávislý prieskum)
(
100 mg/m
2
/
t
ýždeň)
+ karboplatina
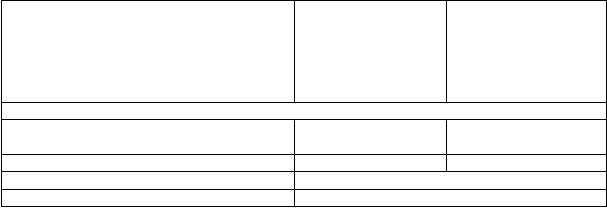 (
n = 521)
3 týždne)
+ karboplatina
(
n = 531)
(
n = 521)
3 týždne)
+ karboplatina
(
n = 531)
Potvrdená úplná alebo čiastočná celková odpoveď, n (%)
170 (33 %) 132 (25 %)
95 % CI (%) 28,6; 36,7 21,2; 28,5
pA/pT (95,1 % CI) 1,313 (1,082; 1,593) P-hodnotaa 0,005
CI = interval spoľahlivosti; HRA/T = pomer rizika nanočastice paklitaxelu-ľudského sérového albumínu/karboplatina oproti paklitaxelu na báze rozpúšťadla/karboplatine; pA/pT = pomer miery odpovede nanočastice paklitaxelu-ľudského sérového albumínu/karboplatina oproti paklitaxelu na báze rozpúšťadla/karboplatine.
a P-hodnota je založená na chi-square teste.
Nezaznamenalo sa štatisticky významné zlepšenie v celkovom prežívaní bez progresie (podľa zaslepeného rádiologického hodnotenia) ani v celkovom prežívaní medzi oboma liečebnými ramenami. Analýza porovnateľnosti (non-inferiority) bola vykonaná pre PFS a OS, s vopred
špecifikovanou hranicou porovnateľnosti vo výške 15 %. Kritérium porovnateľnosti spĺňali obe PFS aj
OS s hornou hranicou intervalu spoľahlivosti 95 % pre súvisiace pomery rizík, ktoré boli menšie ako
1,176 (tabuľka 7).
T
abuľka 7: Analýzy porovnateľnosti prežívania bez progresie a celkového prežívania
v randomizovanej štúdii nemalobunkového karcinómu pľúc (
„
i
n
t
ent-to-treat
“
populácia).
P
arameter účinnosti
P
r
ežívanie bez progresie
a
(
nezávislý prieskum)
N
anočastice paklitaxelu- ľudského sérového albumínu
(
100 mg/m
2
/
t
ýždeň)
+ karboplatina
 (
n = 521)
P
aklitaxel na báze
rozpúšťadla
(
200 mg/m
2
každé
3 týždne)
+ karboplatina
(
n = 531)
(
n = 521)
P
aklitaxel na báze
rozpúšťadla
(
200 mg/m
2
každé
3 týždne)
+ karboplatina
(
n = 531)
Smrť alebo progresia, n (%) 429 (82 %) 442 (83 %) Medián PFS (95 % CI) (mesiace) 6,8 (5,7; 7,7) 6,5 (5,7; 6,9) HRA/T (95 % CI) 0,949 (0,830; 1,086)
Celkové prežívanie
Počet úmrtí, n (%) 360 (69 %) 384 (72 %) Medián OS (95 % CI) (mesiace) 12,1 (10,8; 12,9) 11,2 (10,3; 12,6) HRA/T (95,1 % CI) 0,922 (0,797; 1,066)
CI = interval spoľahlivosti; HRA/T = pomer rizika nanočastice paklitaxelu-ľudského sérového albumínu/karboplatina oproti
paklitaxelu na báze rozpúšťadla/karboplatine; pA/pT = pomer miery odpovede nanočastice paklitaxelu-ľudského sérového
albumínu/karboplatina oproti paklitaxelu na báze rozpúšťadla/karboplatine.
a Na EMA metodické úvahy pre PFS koncový bod, chýbajúce pozorovania alebo začatie následnej novej terapie neboli
použité pre cenzúru.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s nanočasticami
paklitaxelu-ľudského sérového albumínu vo všetkých podskupinách pediatrickej populácie v liečbe
metastatického karcinómu prsníka a nemalobunkového karcinómu pľúc (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
V klinických štúdiách sa stanovili farmakokinetické vlastnosti celkového podaného paklitaxelu po
30 a 180-minútových infúziách nanočastíc paklitaxelu-ľudského sérového albumínu pri hladinách dávok 80 až 375 mg/m2. Expozícia paklitaxelu (AUC) sa zvyšovala lineárne od 2 653 do
16 736 ng.hod/ml po podaní dávok od 80 do 300 mg/m2.
V štúdii s pacientmi s pokročilými solídnymi tumormi sa farmakokinetické parametre paklitaxelu po
intravenóznom podaní nanočastíc paklitaxelu-ľudského sérového albumínu v dávke 260 mg/m2 počas
30 minút porovnávali s dávkami 175 mg/m2 paklitaxelu na báze rozpúšťadla podávaného infúzne počas 3 hodín. Na základe nekompartmentovej farmakokinetickej analýzy vyplýva, že plazmatický
klírens paklitaxelu v nanočasticach paklitaxelu-ľudského sérového albumínu bol väčší (43 %) ako po
infúzii paklitaxelu na báze rozpúšťadla a aj jeho distribučný objem bol vyšší (53 %). V terminálnom
polčase neboli žiadne rozdiely.
V štúdii s opakovaným podávaním uskutočnenej na 12 pacientoch, ktorí dostávali nanočastice paklitaxelu-ľudského sérového albumínu intravenózne v dávke 260 mg/m2, bola intraindividuálna variabilita hodnôt AUC 19 % (rozsah = 3,21 % - 37,70 %). Pri viacnásobných liečebných cykloch sa nepozoroval žiadny dôkaz kumulácie paklitaxelu.
Distribúcia
Po podaní nanočastíc paklitaxelu-ľudského sérového albumínu pacientom so solídnymi tumormi je
paklitaxel rovnomerne distribuovaný do krvných buniek a plazmy a je vo vysokej miere viazaný na plazmatické proteíny (94 %).
Množstvo proteínu, na ktorý sa viaže paklitaxel po podaní nanočastíc paklitaxelu-ľudského sérového albumínu, bolo vyhodnotené ultrafiltráciou v rámci porovnávacej štúdie pacienta. Frakcia voľného paklitaxelu bola významne vyššia pri nanočasticiach paklitaxelu-ľudského sérového albumínu (6,2 %) v porovnaní s paklitaxelom na báze rozpúšťadla (2,3 %). To malo za následok významne vyššiu
expozíciu neviazaného paklitaxelu pri nanočasticiach paklitaxelu-ľudského sérového albumínu
v porovnaní s paklitaxelom na báze rozpúšťadla, hoci celková expozícia bola porovnateľná. Je to pravdepodobne z dôvodu nezachytenia paklitaxelu v micelách Cremophor EL, ako je to v prípade paklitaxelu na báze rozpúšťadla. Z publikácií v literatúre vyplýva, že in vitro štúdie väzby na ľudské proteíny v sére (použitím paklitaxelu v koncentráciách v rozmedzí od 0,1 do 50 µg/ml) naznačujú, že prítomnosť cimetidínu, ranitidínu, dexametazónu ani difenhydramínu neovplyvnila väzbu paklitaxelu na proteíny.
Na základe populačnej farmakokinetickej analýzy je celkový distribučný objem približne 1 741 l; veľký distribučný objem naznačuje rozsiahlu extravaskulárnu distribúciu a/alebo väzbu paklitaxelu v tkanivách.
Biotransformácia a eliminácia
Z publikácií v literatúre vyplýva, že in vitro štúdie s mikrozómami z ľudskej pečene a rezmi tkaniva ukazujú, že paklitaxel sa metabolizuje primárne na 6α-hydroxypaklitaxel a na dva menej významné metabolity, 3’-p-hydroxypaklitaxel a 6α-3’-p-dihydroxypaklitaxel. Vznik týchto hydroxylovaných metabolitov je katalyzovaný prostredníctvom CYP2C8 v prípade 6α -hydroxypaklitaxelu, CYP3A4 v prípade 3’-p-hydroxypaklitaxelu, a oboma typmi izoenzýmov CYP2C8 aj CYP3A4 v prípade
6α-3’-p-dihydroxypaklitaxelu.
U pacientov s metastatickým karcinómom prsníka bola po 30-minútovej infúzii nanočastíc paklitaxelu-ľudského sérového albumínu v dávke 260 mg/m2 priemerná hodnota kumulatívneho vylučovania nezmeneného liečiva močom 4 % z celkovej podanej dávky, menej ako 1 % bolo vylúčené vo forme metabolitov 6α-hydroxypaklitaxelu a 3’-p-hydroxypaklitaxelu, čo naznačuje rozsiahly klírens mimo obličiek. Paklitaxel sa primárne eliminuje metabolizáciou v pečeni
a vylučovaním žlčou.
V rozmedzí klinických dávok 80 až 300 mg/m2 sa priemerný plazmatický klírens paklitaxelu pohybuje v rozmedzí od 13 do 30 l/h/m2 a priemerný terminálny polčas sa pohybuje v rozmedzí od 13 do
27 hodín.
Poškodeniepečene
Vplyv poškodenia pečene na populačné farmakokinetické vlastnosti nanočastíc paklitaxelu-ľudského sérového albumínu bol skúmaný u pacientov s pokročilými solídnymi nádormi. Táto analýza zahŕňala pacientov s normálnou funkciou pečene (n = 130) a už existujúcim miernym (n = 8), stredne závažným (n = 7) alebo závažným (n = 5) poškodením pečene (podľa kritérií pracovnej skupiny Organ Dysfunction Working Group NCI). Výsledky ukazujú, že mierne poškodenie pečene (celkový bilirubín > 1 až ≤ 1,5 x ULN) nemá žiadny klinicky významný vplyv na farmakokinetiku paklitaxelu. U pacientov so stredne závažným (celkový bilirubín > 1,5 až ≤ 3 x ULN) alebo závažným (celkový bilirubín > 3 to ≤ 5 x ULN) poškodením pečene dochádza k poklesu maximálnej miery eliminácie paklitaxelu o 22 % až 26 % a k zvýšeniu priemernej hodnoty AUC paklitaxelu približne o 20 %
v porovnaní s pacientmi s normálnou funkciou pečene. Poškodenie pečene nemá žiadny vplyv na
priemernú hodnotu Cmax paklitaxelu. Okrem toho eliminácia paklitaxelu inverzne koreluje s množstvom celkového bilirubínu a pozitívne koreluje s množstvom sérového albumínu.
Farmakokinetický/farmakodynamický model naznačuje, že neexistuje žiadna korelácia medzi funkciou pečene (stanovenou počiatočnou hladinou albumínu alebo hladinou celkového bilirubínu) a neutropéniou po úprave expozície nanočasticiam paklitaxelu-ľudského sérového albumínu.
Farmakokinetické údaje nie sú k dispozícii pre pacientov s celkovým bilirubínom > 5 x ULN (pozri
časť 4.2).
Poškodenieobličiek
Farmakokinetická populačná analýza zahŕňala pacientov s normálnou funkciou obličiek (n = 65) a s už existujúcim miernym (n = 61), stredne závažným (n = 23) alebo závažným (n = 1) poškodením
obličiek (podľa kritérií FDA 2010 - draft FDA guidance criteria 2010). Mierne až stredne ťažké
poškodenie obličiek (klírens kreatinínu ≥ 30 až < 90 ml/min) nemá žiadny klinicky významný vplyv
na maximálnu mieru eliminácie a systémovú expozíciu paklitaxelu (AUC a Cmax). Farmakokinetické údaje sú nedostatočné pre pacientov so závažným poškodením obličiek a nie sú k dispozícii pre pacientov v terminálnom štádiu ochorenia obličiek.
Starší pacienti
Farmakokinetická populačná analýza nanočastíc paklitaxelu-ľudského sérového albumínu zahŕňala pacientov vo vekovom rozmedzí 24 až 85 rokov a ukazuje, že vek nemá významný vplyv na maximálnu mieru eliminácie a systémovú expozíciu (AUC a Cmax) paklitaxelu. Farmakokinetický/farmakodynamický model využívajúci údaje od 125 pacientov s pokročilými solídnymi tumormi naznačuje, že pacienti vo veku ≥ 65 rokov môžu byť náchylnejší k rozvoju neutropénie počas prvého liečebného cyklu, hoci expozícia paklitaxelu v plazme nie je ovplyvnená vekom.
Ostatné vnútorné faktory
Farmakokinetické populačné analýzy nanočastíc paklitaxelu-ľudského sérového albumínu naznačujú,
že pohlavie, rasa (ázijská oproti bielej) a typ solídnych nádorov nemajú klinicky významný vplyv na systémovú expozíciu (AUC a Cmax) paklitaxelu. Pacienti s hmotnosťou 50 kg mali hodnotu AUC paklitaxelu približne o 25 % nižšiu ako tí, ktorí vážili 75 kg. Klinický význam týchto zistení je neistý.
5.3 Predklinické údaje o bezpečnosti
Karcinogénny potenciál paklitaxelu sa neskúmal. Na základe publikácií v literatúre je však paklitaxel potenciálne karcinogénnou a genotoxickou látkou pri klinických dávkach na základe jeho farmakodynamického mechanizmu účinku. Ukázalo sa, že paklitaxel je klastogénny in vitro (chromozómové aberácie v ľudských lymfocytoch) a in vivo (mikronukleárny test u myší). Ukázalo sa, že paklitaxel je genotoxický in vivo (mikronukleárny test u myší), no nevyvolával mutagenitu
v Amesovom teste ani na ováriách čínskeho škrečka/hypoxantín-guanín fosforibozyltransferáza
(CHO/HGPRT).
Paklitaxel pri nižších dávkach ako je terapeutická dávka pre ľudí sa spájal s nízkou plodnosťou
a fetálnou toxicitou u potkanov. Štúdie na zvieratách s nanočasticami paklitaxelu-ľudského sérového albumínu preukázali ireverzibilné toxické účinky na mužské reprodukčné orgány v klinicky relevantných hladinách expozície.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
albumín (ľudský)
kaprylát sodný
N-acetyl-DL-tryptofán chlorid sodný
kyselina chlorovodíková
hydroxid sodný
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorenéinjekčnéliekovky
2 roky
Stabilita rekonštituovanej disperzie v injekčnejliekovke
Chemická a fyzikálna stabilita použiteľnosti bola preukázaná počas 8 hodín pri teplote 2 - 8 °C, ak je
injekčná liekovka v pôvodnom obale a chránená pred silným svetlom. Alternatívne sa môže použiť ochrana pred svetlom na čistom mieste. Z mikrobiologického hľadiska sa má liek naplniť do infúzneho vaku ihneď, ak nebola použitá metóda otvorenia/rekonštitúcie/zriedenia vylučujúca riziko mikrobiologickej kontaminácie. Ak sa liek nepoužije ihneď, za časy a podmienky uchovávania počas použitia je zodpovedný používateľ.
Stabilita rekonštituovanej disperzie v infúznom vaku
Chemická a fyzikálna stabilita použiteľnosti bola preukázaná počas 8 hodín pri teplote neprevyšujúcej
25 °C. Z mikrobiologického hľadiska sa má liek použiť ihneď, ak nebola použitá metóda otvorenia/rekonštitúcie/zriedenia vylučujúca riziko mikrobiologickej kontaminácie. Ak sa liek nepoužije ihneď, za časy a podmienky uchovávania počas použitia je zodpovedný používateľ.
6.4 Špeciálne upozornenia na uchovávanie
Neotvorenéinjekčnéliekovky
Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie. Injekčné liekovky uchovávajte vo vonkajšom obale na ochranu pred svetlom. Zmrazenie ani chladenie nemajú na stabilitu tohto lieku nežiaduce účinky.
Rekonštituovaná disperzia
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
50 ml injekčná liekovka (sklo typu I) so zátkou (butylkaučuk), s vrchným uzáverom (hliník), obsahujúca 100 mg paklitaxelu vo forme nanočastíc viazaných na albumín.
Veľkosť balenia je jedna injekčná liekovka.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Bezpečnostnéopatreniapriprípravea podávaní
Paklitaxel je cytotoxický protinádorový liek a preto, tak ako pri iných potenciálne toxických zlúčeninách, je pri manipulácii s Pazenirom potrebné postupovať opatrne. Odporúča sa použitie rukavíc, ochranných okuliarov a ochranného oblečenia. Ak sa disperzia dostane do kontaktu
s pokožkou, pokožka sa musí okamžite a dôkladne umyť mydlom a vodou. Ak sa dostane do kontaktu so sliznicami, musia sa okamžite a dôkladne opláchnuť vodou. Pazenir majú pripravovať a podávať
len pracovníci primerane vyškolení na manipuláciu s cytotoxickými látkami. Tehotné pracovníčky nemajú manipulovať s Pazenirom.
Vzhľadom na možnosť extravazácie sa odporúča starostlivo sledovať miesto infúzie z dôvodu možnej infiltrácie počas podávania lieku. Obmedzenie infúzie Pazeniru podľa odporúčania na 30 minút znižuje pravdepodobnosť reakcií súvisiacich s infúziou.
Rekonštitúcia a podávanie lieku
Pazenir sa dodáva ako sterilný lyofilizovaný prášok na rekonštitúciu pred použitím. Po rekonštitúcii obsahuje každý ml disperzie 5 mg paklitaxelu vo forme nanočastíc viazaných na albumín.
Pomocou sterilnej striekačky pomaly, minimálne počas 1 minúty, vstrekujte 20 ml infúzneho roztoku chloridu sodného 9 mg/ml (0,9 %) do injekčnej liekovky s Pazenirom.
Roztok sa má nasmerovať navnútornústenuinjekčnejliekovky. Roztok sa nemá vstrekovať priamo
do prášku, pretože to spôsobí spenenie.
Po ukončení vstrekovania roztoku nechajte injekčnú liekovku stáť minimálne 5 minút, aby sa tuhé častice dôkladne navlhčili. Potom je potrebné injekčnú liekovku jemne a pomaly miešať a/alebo preklápať najmenej 2 minúty, kým sa prášok úplne nerozpustí. Musí sa vyhnúť vytvoreniu peny. Ak
dôjde k vytvoreniu peny alebo zhlukov, roztok sa má nechať stáť najmenej 15 minút, kým pena neopadne.
Rekonštituovaná disperzia má byť mliečna a homogénna bez viditeľných zrazenín. Môže dôjsť
k drobnému usadeniu rekonštituovanej disperzie. Ak sú viditeľné zrazeniny alebo usadeniny, injekčnú liekovku treba znova jemne preklopiť, aby sa pred použitím zabezpečila úplná redisperzia. Disperzia
v injekčnej liekovke sa má skontrolovať, či neobsahuje pevné častice. Rekonštituovaná disperzia sa
nesmie podať, ak sa v injekčnej liekovke spozorujú pevné častice.
Je potrebné vypočítať presný objem celkovej dávky na 5 mg/ml disperzie, potrebný pre pacienta,
a vstreknúť príslušné množstvo rekonštituovaného Pazeniru do prázdneho, sterilného intravenózneho vaku z PVC alebo iného materiálu.
Používanie zdravotníckych pomôcok obsahujúcich silikónový olej ako lubrikant (t.j. striekačky a i.v. vaky) na rekonštitúciu alebo podávanie Pazeniru môže mať za následok tvorbu bielkovinových vláken. Pazenir sa má podávať pomocou infúznej súpravy obsahujúcej 15 µm filter, aby sa vyhlo podaniu týchto vlákien. Použitie 15 µm filtra odstraňuje vlákna a nemení fyzikálne ani chemické vlastnosti rekonštituovaného lieku.
Používanie filtrov s veľkosťou pórov menšou ako 15 µm môže mať za následok upchatie filtra.
Na prípravu ani na podávanie infúzií Pazeniru nie je nutné použitie špecializovaných nádob na roztoky alebo infúznych súprav bez obsahu di(2-etylhexyl)ftalátu (DEHP).
Po podaní infúzie sa odporúča prepláchnuť intravenóznu linku injekčným roztokom chloridu sodného
9 mg/ml (0,9 %), aby sa zabezpečilo podanie úplnej dávky.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIratiopharm GmbH Graf-Arco-Straße 3
89079 Ulm
Nemecko
8. REGISTRAČNÉ ČÍSLOEU/1/18/1317/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.