
ene
K dispozícii sú len obmedzené údaje u pacientov so stredne ťažkou poruchou funkcie pečene,
darolutamid sa neskúmal u pacientov s ťažkou poruchou funkcie pečene.
Keďže expozícia môže byť zvýšená, u týchto pacientov sa majú dôkladne sledovať nežiaduce reakcie
(pozri časti 4.2 a 5.2).
Nedávne kardiovaskulárne ochorenie
Pacienti s klinicky významným kardiovaskulárnym ochorením v posledných 6 mesiacoch vrátane
cievnej mozgovej príhody, infarktu myokardu, závažnej/nestabilnej angíny pektoris, koronárneho/periférneho arteriálneho bypassu a symptomatického kongestívneho zlyhania srdca boli vylúčení z klinických štúdií. Z tohto dôvodu nebola bezpečnosť darolutamidu u týchto pacientov preukázaná.
Ak je predpísaný liek NUBEQA, pacienti s klinicky významným kardiovaskulárnym ochorením majú byť liečení podľa stanovených liečebných postupov.
Súbežné užívanie s inými liekmi
Používanie silných induktorov CYP3A4 a P-gp počas liečby darolutamidom môže znížiť plazmatické
koncentrácie darolutamidu a neodporúča sa, pokiaľ existuje terapeutická alternatíva. Má sa zvážiť výber alternatívneho súbežne podávaného lieku, ktorý má menší potenciál indukovať CYP3A4 alebo P-gp (pozri časť 4.5).
U pacientov sa majú sledovať nežiaduce reakcie spôsobené substrátmi BCRP, OATP1B1 a OATP1B3, pretože súbežné podávanie s darolutamidom môže zvýšiť plazmatické koncentrácie týchto substrátov. Treba sa vyhnúť súbežnému podávaniu s rosuvastatínom, pokiaľ existuje terapeutická alternatíva
(pozri časť 4.5).
Androgénna deprivačná terapia môže predlžovať QT interval
U pacientov s rizikovými faktormi predĺženia QT v anamnéze a u pacientov užívajúcich súbežne
lieky, ktoré môžu predĺžiť QT interval (pozri časť 4.5), majú lekári zvážiť pomer prínosu a rizika vrátane potenciálneho rizika Torsade de pointes pred začatím liečby liekom NUBEQA.
Informácie o pomocných látkach
Liek NUBEQA obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej
intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
4.5 Liekové a iné interakcie
Účinky iných liekov na darolutamid
Induktory CYP3A4 a P-gp
Darolutamid je substrátom CYP3A4 a P-glykoproteínu (P-gp).
Užívanie silných a stredne silných induktorov CYP3A4 a induktorov P-gp (napr. karbamazepín, fenobarbital, ľubovník bodkovaný, fenytoín a rifampicín) počas liečby darolutamidom sa neodporúča, pokiaľ existuje terapeutická alternatíva. Musí sa uvážiť výber alternatívneho súbežného lieku, ktorý nemá žiadny alebo má iba slabý potenciál indukovať CYP3A4 alebo P-gp.
Opakované podávanie rifampicínu (600 mg), silného induktora CYP3A4 a P-gp, s jednorazovou dávkou darolutamidu (600 mg) spolu s jedlom viedlo ku zníženiu priemernej expozície o 72 %
(AUC0 - 72) a zníženiu hodnoty Cmax darolutamidu o 52 %.
Inhibítory CYP3A4, P-gp a BCRP
Darolutamid je substrátom CYP3A4, P-gp a proteínu rezistencie karcinómu prsníka (BCRP).
V prípade podania inhibítorov CYP3A4, P-gp alebo BCRP sa neočakáva žiadna klinicky významná lieková interakcia. Darolutamid sa môže podávať súbežne s inhibítormi CYP3A4, P-gp alebo BCRP. Súbežné užívanie darolutamidu s kombináciou P-gp a silného inhibítora CYP3A4 zvyšuje expozíciu darolutamidu, čo môže zvýšiť riziko nežiaducich reakcií v súvislosti s darolutamidom. Odporúča sa častejšie monitorovanie pacientov na nežiaduce reakcie v súvislosti s darolutamidom a, ak je to potrebné, úprava dávky darolutamidu.
Podávanie itrakonazolu (200 mg dvakrát denne v 1. deň a jedenkrát denne nasledujúcich 7 dní), silného inhibítora CYP3A4, P-gp a BCRP, s jednorazovou dávkou darolutamidu (600 mg v 5. deň spolu s jedlom) viedlo k 1,7-násobnému zvýšeniu priemernej expozície (AUC0 - 72] a 1,4-násobnému zvýšeniu hodnoty Cmax darolutamidu.
Inhibítory UGT1A9
Darolutamid je substrátom UGT1A9.
Neočakáva sa žiadna klinicky významná interakcia typu liek-liek v prípade podávania inhibítora
UGT1A9.
Darolutamid môže byť podávaný súbežne s inhibítormi UGT1A9.
Populačná farmakokinetická analýza preukázala, že súbežné podávanie inhibítorov UGT1A9
s darolutamidom malo za následok 1,2-násobné zvýšenie expozície (AUC0-72) darolutamidu.
Účinky darolutamidu na iné lieky
Substráty BCRP, OATP1B1 a OATP1B3
Darolutamid je inhibítorom proteínu rezistencie karcinómu prsníka (BCRP) a polypeptidov transportujúcich organické anióny (OATP) 1B1 a 1B3.
Treba sa vyhnúť súbežnému podávaniu s rosuvastatínom, pokiaľ existuje terapeutická alternatíva. Má sa zvážiť výber alternatívneho súbežne podávaného lieku, ktorý má menší potenciál inhibovať BCRP, OATP1B1 a OATP1B3.
Podávanie darolutamidu (600 mg dvakrát denne po dobu 5 dní) pred súbežným podaním jednorazovej dávky rosuvastatínu (5 mg) spolu s potravou viedlo k 5-násobnému zvýšeniu priemernej expozície (AUC) a hodnoty Cmax rosuvastatínu.
Ak je to možné, má sa vyhnúť súbežnému podávaniu darolutamidu s inými substrátmi BCRP. Súbežné
podávanie darolutamidu môže zvýšiť plazmatické koncentrácie iných súbežne podávaných substrátov BCRP, OATP1B1 a OATP1B3 (napr. metotrexát, sulfasalazín, fluvastatín, atorvastatín, pitavastatín). Preto sa odporúča u pacientov sledovať nežiaduce reakcie na substráty BCRP, OATP1B1
a OATP1B3. Okrem toho sa majú dodržiavať príslušné odporúčania v informáciách o lieku týchto substrátov, ak sa podávajú súbežne s darolutamidom.
Substráty P-gp
V prípade podania substrátu P-gp sa neočakáva žiadna klinicky významná lieková interakcia. Darolutamid sa môže podávať súbežne so substrátmi P-gp (napr. digoxín, verapamil alebo nifedipín). Súbežné podávanie darolutamidu spolu so substrátom citlivým na P-gp, dabigatran etexilátom, neodhalilo žiadne zvýšenie expozície (AUC a Cmax) dabigatranu.
Substráty CYP3A4
Darolutamid je miernym induktorom CYP3A4.
V prípade podania substrátu CYP sa neočakáva žiadna klinicky významná lieková interakcia. Darolutamid sa môže podávať súbežne so substrátmi CYP (napr. warfarín, L-tyroxín, omeprazol). Podávanie darolutamidu (600 mg dvakrát denne po dobu 9 dní) pred súbežným podaním jednorazovej dávky substrátu citlivého na CYP3A4, midazolamu (1 mg), spolu s jedlom znížilo priemernú expozíciu (AUC) a Cmax midazolamu o 29 % a 32 %, v uvedenom poradí.
Darolutamid neinhiboval metabolizmus vybraných substrátov CYP in vitro pri klinicky významných koncentráciách.
Lieky, ktoré predlžujú interval QT
Vzhľadom na to, že androgénna deprivačná liečba môže predlžovať interval QT, súbežné podávanie
s liekmi, o ktorých je známe, že predlžujú QT interval, alebo s liekmi schopnými vyvolať Torsade de pointes, sa má dôkladne vyhodnotiť. Tieto zahŕňajú lieky ako sú antiarytmiká triedy IA (napr. chinidín, disopyramid) alebo triedy III (napr. amiodarón, sotalol, dofetilid, ibutilid), metadón, moxifloxacín a antipsychotiká (napr. haloperidol).
4
.6 Fertilita, gravidita a laktácia
Tento liek nie je indikovaný u žien vo fertilnom veku. Nemá sa používať u žien, ktoré sú alebo môžu byť tehotné alebo dojčia (pozri časti 4.1 a 4.3).
Ženy vo fertilnom veku/antikoncepcia u mužov a žien
Nie je známe, či sa darolutamid alebo jeho metabolity nachádzajú v spermiách. Ak je pacient zapojený
do sexuálnych aktivít so ženou vo fertilnom veku, musí počas liečby a po dobu 1 týždňa po ukončení liečby liekom NUBEQA používať vysoko účinný spôsob antikoncepcie (ročná miera zlyhania < 1 %), aby sa zabránilo tehotenstvu.
Gravidita
Vychádzajúc z mechanizmu účinku darolutamid môže poškodiť plod. Nevykonali sa žiadne
predklinické štúdie reprodukčnej toxicity (pozri časť 5.3).
Nie je známe, či sú darolutamid alebo jeho metabolity prítomné v spermiách. Ak je pacient sexuálne aktívny s tehotnou ženou, má sa počas liečby liekom NUBEQA a počas 1 týždňa po ukončení liečby liekom NUBEQA používať kondóm. Musí sa vyhnúť expozícii plodu inhibítoru androgénneho receptora prenosom spermiami na tehotnú ženu, pretože to môže ovplyvniť vývoj plodu.
Dojčenie
Nie je známe, či sa darolutamid alebo jeho metabolity vylučujú do ľudského mlieka. Nevykonali sa
žiadne štúdie na zvieratách na vyhodnotenie vylučovania darolutamidu alebo jeho metabolitov do mlieka (pozri časť 5.3). Riziko u dojčiat nemôže byť vylúčené.
Fertilita
Nie sú k dispozícii žiadne údaje o účinku darolutamidu na ľudskú fertilitu.
Na základe štúdií na zvieratách, NUBEQA môže zhoršiť fertilitu samcov s reprodukčným potenciálom
(pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
NUBEQA nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Najčastejšie pozorovanou nežiaducou reakciou je únava/astenické stavy (15,8 %).
Tabuľkový zoznam nežiaducich reakcií
Pozorované nežiaduce reakcie sú uvedené nižšie v tabuľke 1. Sú klasifikované podľa tried orgánových
systémov.
Nežiaduce reakcie sú zoskupené podľa ich frekvencií výskytu. Skupiny frekvencií výskytu sú definované podľa nasledujúcej konvencie: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme
(z dostupných údajov).
V rámci skupín frekvencie výskytu sú nežiaduce reakcie uvedené v poradí podľa klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie hlásené v rámci štúdie ARAMIS
Trieda systémových orgánov
(MedDRA)
|
Veľmi časté
|
Časté
|
Poruchy srdca a srdcovej činnosti
|
|
ischemická choroba srdcab, zlyhanie srdcac
|
Poruchy kože a podkožného tkaniva
|
|
vyrážka
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
|
bolesť v končatinách, bolesť svalov a kostí, zlomeniny
|
Celkové poruchy a reakcie v mieste podania
|
únava/astenické stavya
|
|
Laboratórne a funkčné vyšetreniad
|
znížený počet neutrofilov, zvýšená hladina bilirubínu,
zvýšená hladina AST
|
|
a Zahŕňa únavu a asténiu, letargiu a malátnosť.
b Zahŕňa artériosklerózu koronárnej tepny, chorobu koronárnej tepny, oklúziu koronárnej tepny, stenózu koronárnej tepny, akútny koronárny syndróm, akútny infarkt myokardu, angínu pektoris, nestabilnú
angínu, infarkt myokardu, ischémiu myokardu.
c Zahŕňa zlyhanie srdca, akútne zlyhanie srdca, chronické zlyhanie srdca, kongestívne zlyhanie srdca, kardiogénny šok.
d Všeobecné terminologické kritériá pre nežiaduce udalosti (
Common Terminology Criteria for AdverseEvents, CTCAE) verzia 4.03.
Popis vybraných nežiaducich reakciíÚnavaÚnava/astenické stavy sa hlásili u 15,8 % pacientov liečených darolutamidom a u 11,4 % pacientov, ktorí dostávali placebo. Udalosti s najhorším 3. stupňom boli hlásené u 0,6 % pacientov liečených darolutamidom a u 1,1 % pacientov, ktorí dostávali placebo. Únava (okrem asténie, letargie alebo malátnosti) sa vyskytla u väčšiny pacientov (12,1 % pacientov liečených darolutamidom a 8,7 % pacientov dostávajúcich placebo).
FraktúryFraktúry sa vyskytli u 4,2 % pacientov liečených darolutamidom a u 3,6 % pacientov, ktorí dostávali placebo.
Ischemická choroba srdca a zlyhanie srdcaIschemická choroba srdca sa vyskytla u 3,2 % pacientov liečených darolutamidom a u 2,5 % pacientov, ktorí dostávali placebo. Udalosti 5. stupňa sa vyskytli u 0,3 % pacientov liečených darolutamidom a u 0,2 % pacientov, ktorí dostávali placebo. Zlyhanie srdca sa vyskytlo u 1,9 % pacientov liečených darolutamidom a u 0,9 % pacientov, ktorí dostávali placebo.
Znížený počet neutrofilovZnížený počet neutrofilov sa hlásil ako laboratórna abnormalita u 19,6 % pacientov liečených darolutamidom a u 9,4 % pacientov, ktorí dostávali placebo. Medián času do dosiahnutia minimálnych hodnôt bol 256 dní. Abnormality laboratórnych testov sa prejavovali prevažne ako účinky s intenzitou
1. alebo 2. stupňa. Zníženie počtu neutrofilov 3. a 4. stupňa sa hlásilo u 3,5 % a 0,5 % pacientov,
v uvedenom poradí. Iba jeden pacient natrvalo ukončil liečbu darolutamidom z dôvodu neutropénie. Neutropénia bola buď prechodná alebo reverzibilná (88 % pacientov) a nebola spojená so žiadnymi klinicky významnými prejavmi alebo príznakmi.
Zvýšená hladina bilirubínu
Zvýšená hladina bilirubínu sa hlásila ako laboratórna abnormalita u 16,4 % pacientov liečených darolutamidom a u 6,9 % pacientov, ktorí dostávali placebo. Tieto udalosti mali prevažne intenzitu
1. alebo 2. stupňa, neboli spojené so žiadnymi klinicky významnými prejavmi alebo príznakmi a po
ukončení liečby darolutamidom boli reverzibilné. Zvýšená hladina bilirubínu 3. stupňa sa hlásila
u 0,1 % pacientov liečených darolutamidom a u 0 % pacientov, ktorí dostávali placebo. V ramene
s darolutamidom bol priemerný čas do prvého nástupu zvýšenia bilirubínu 153 dní a priemerné trvanie prvej epizódy bolo 182 dní. U žiadneho z pacientov sa liečba neukončila kvôli zvýšenej hladine bilirubínu.
Zvýšená hladina ASTZvýšená hladina AST sa hlásila ako laboratórna abnormalita u 22,5 % pacientov liečených darolutamidom a u 13,6 % pacientov, ktorí dostávali placebo. Tieto udalosti mali prevažne intenzitu
1. alebo 2. stupňa, neboli spojené so žiadnymi klinicky významnými prejavmi alebo príznakmi a po ukončení liečby darolutamidom boli reverzibilné. Zvýšená hladina AST 3. stupňa sa hlásila u 0,5 % pacientov liečených darolutamidom a u 0,2 % pacientov, ktorí dostávali placebo. V ramene
s darolutamidom bol priemerný čas do prvého nástupu zvýšenia AST 258 dní a priemerné trvanie prvej epizódy bolo 118 dní. U žiadneho z pacientov sa liečba neukončila kvôli zvýšenej hladine AST.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieNajvyššia klinicky skúmaná dávka darolutamidu bola 900 mg dvakrát denne, čo zodpovedá celkovej dennej dávke 1 800 mg. Pri tejto dávke neboli pozorované žiadne toxicity obmedzujúce dávku. Vzhľadom na saturabilnú absorpciu (pozri časť 5.2) a neprítomnosť dôkazov o akútnej toxicite sa neočakáva, že bude užitie vyššej ako odporúčanej dávky darolutamidu viesť k toxicite.
V prípade užitia vyššej ako odporúčanej dávky sa môže v liečbe darolutamidom pokračovať ďalšou plánovanou dávkou.
Neexistuje žiadne špecifické antidotum pre darolutamid a príznaky predávkovania sa nestanovili.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Endokrinná liečba, antiandrogény: ATC kód: L02BB06
Mechanizmus účinkuDarolutamid je inhibítorom androgénneho receptora (AR) s flexibilnou polárne substituovanou
pyrazolovou štruktúrou, ktorá sa s vysokou afinitou priamo viaže na oblasť receptora viažucu ligand. Darolutamid kompetitívne inhibuje väzbu androgénu, nukleárnu translokáciu AR a transkripciu sprostredkovanú AR. Hlavný metabolit, keto-darolutamid, vykazoval in vitro aktivitu podobnú ako darolutamid. Liečba darolutamidom znižuje proliferáciu nádorových buniek prostaty, čo vedie k silnej protinádorovej aktivite.
Farmakodynamické účinkyPo perorálnom podaní 600 mg darolutamidu dvakrát denne sa v porovnaní s placebom nepozorovalo
predĺženie priemerného intervalu QTcF (t. j. viac ako 10 ms).
Klinická účinnosť a bezpečnosť
Účinnosť a bezpečnosť darolutamidu sa hodnotila v randomizovanej, dvojito zaslepenej, placebom
kontrolovanej multicentrickej štúdii fázy III (ARAMIS) u pacientov s nemetastatickým karcinómom prostaty (stanoveným konvenčným zobrazením CT, kostným skenovaním, NMR) rezistentným na kastráciu s časom do zdvojnásobenia prostatického špecifického antigénu (PSADT) ≤ 10 mesiacov. Pacienti boli zaradení do štúdie, ak mali 3 zvýšenia hladiny prostatického špecifického antigénu (PSA) oproti nadiru, ktoré sa namerali v minimálne 1 týždňových odstupoch počas androgén deprivačnej liečby, PSA ≥ 2 ng/ml pri skríningu a kastračnej hladine testosterónu v sére < 1,7 nmol/l.
Do štúdie mohli byť zaradení pacienti so záchvatmi v anamnéze. Do skupiny s darolutamidom bolo zaradených 12 pacientov (0,21 %) so záchvatmi v anamnéze.
Pacienti s nekontrolovanou hypertenziou alebo nedávnou (v posledných 6 mesiacoch) cievnou mozgovou príhodou, infarktom myokardu, ťažkou/nestabilnou angínou pektoris, koronárnym/periférnym arteriálnym bypassom, kongestívnym zlyhaním srdca triedy III alebo IV podľa asociácie New York Heart Association (NYHA) boli vylúčení zo štúdie.
Pacienti s predchádzajúcou liečbou AR inhibítormi druhej generácie ako sú enzalutamid, apalutamid
a darolutamid alebo inhibítory enzýmu CYP17 ako je abiraterónacetát, ako aj pacienti, ktorí dostávali systémový kortikosteroid v dávke vyššej ako ekvivalent 10 mg prednizónu/deň v dobe počas 28 dní pred randomizáciou, boli vylúčení zo štúdie.
Celkom bolo randomizovaných 1 509 pacientov v pomere 2:1 na užívanie 600 mg darolutamidu perorálne dvakrát denne (n = 955) alebo zodpovedajúceho placeba (n = 554).
Všetci pacienti súbežne dostávali analóg hormónu uvoľňujúceho gonadotropín (GnRH) alebo mali bilaterálnu orchiektómiu. Do štúdie mohli byť zaradení pacienti s prítomnosťou panvových lymfatických uzlín < 2 cm v krátkej osi pod aortálnym rozdvojením. Neprítomnosť alebo prítomnosť metastáz bola hodnotená nezávislým centrálnym rádiologickým hodnotením. Do týchto analýz bolo zahrnutých 89 pacientov, ktorí boli retrospektívne identifikovaní s metastázami na začiatku štúdie. Randomizácia bola stratifikovaná podľa PSADT (≤ 6 mesiacov alebo > 6 mesiacov) a podľa liečby zameranej na osteoklasty pri vstupe do štúdie (áno alebo nie).
Nasledujúce demografické charakteristiky pacientov a charakteristiky ochorenia boli medzi liečebnými skupinami vyvážené. Medián veku bol 74 rokov (rozsah 48 - 95) a 9 % pacientov bolo vo veku 85 rokov alebo starších. Distribúcia podľa etnickej príslušnosti bola 79 % biela, 13 % ázijská
a 3 % čierna. Väčšina pacientov mala v čase diagnózy Gleasonovo skóre 7 alebo vyššie (73 %). Medián PSADT bol 4,5 mesiaca. 9 % pacientov podstúpilo orchiektómiu, 25 % pacientov podstúpilo prostatektómiu a 50 % pacientov podstúpilo aspoň jednu predchádzajúcu rádioterapiu. 76 % pacientov dostalo viac ako jednu predchádzajúcu hormonálnu liečbu. Pacienti mali pri vstupe do štúdie stav výkonnosti podľa Východnej kooperatívnej onkologickej skupiny (Eastern Cooperative Oncology Group Performance Status, ECOG PS) skóre 0 (69 %) alebo 1 (31 %).
Liečba darolutamidom pokračovala až do rádiografickej progresie ochorenia hodnotenej konvenčným zobrazovaním (CT, scintigrafia skeletu, NMR) zaslepeným centrálnym hodnotením, neprijateľnej toxicity alebo odstúpenia zo štúdie.
Primárnym koncovým ukazovateľom účinnosti bolo prežívanie bez metastáz (MFS). Sekundárnymi cieľovými ukazovateľmi boli: celkové prežívanie (OS), čas do progresie bolesti, čas do začatia prvej cytotoxickej chemoterapie karcinómu prostaty a čas do prvej symptomatickej kostnej príhody (definované ako výskyt niektorého z nasledujúcich stavov: externá rádioterapia na zmiernenie kostných príznakov, nová symptomatická patologická zlomenina kosti, kompresia miechy alebo ortopedický chirurgický zákrok súvisiaci s nádorom).
Liečba darolutamidom viedla k zlepšeniu MFS v porovnaní s placebom (pozri tabuľku 2 a obrázok 1). Výsledky MFS boli konzistentné v rámci všetkých podskupín pacientov bez ohľadu na PSADT, predchádzajúce použitie liekov cielených na kosti alebo lokoregionálne ochorenie. Ďalšie podskupiny s konzistentnými výsledkami MFS zahŕňali PSA na začiatku štúdie, Gleasonovo skóre pri diagnóze, vek, geografickú oblasť, ECOG PS na začiatku štúdie, etnikum a počet predchádzajúcich hormonálnych liečob.
Po primárnej analýze MFS po odslepení štúdie, bola pacientom, ktorí dostávali placebo, ponúknutá liečba darolutamidom v otvorenej fáze štúdie s možnosťou prekríženia. Spomedzi 554 pacientov
randomizovaných na placebo prešlo 170 (31 %) na liečbu darolutamidom. Analýza OS nebola upravená pre skresľujúce vplyvy prekríženia (cross-over).
V čase konečnej analýzy viedla liečba darolutamidom k štatisticky významnému zlepšeniu celkového prežívania v porovnaní s placebom (medián sa nedosiahol v žiadnej skupine, pozri tabuľku 2
a obrázok 2).
Liečba darolutamidom taktiež viedla k štatisticky významným oddialeniam času do progresie bolesti, času do začatia prvej cytotoxickej chemoterapie a času do prvej symptomatickej kostnej príhody v porovnaní s placebom (pozri tabuľku 2).
Všetky analýzy sa uskutočňovali v rámci kompletnej sady analýz.
Tabuľka 2: Výsledky účinnosti zo štúdie ARAMIS
Parameter
účinnosti
| Počet príhod (%)
| Medián (95 % IS)
| Pomer rizikab (95 % interval spoľahlivosti
[IS])
Hodnota p
(obojstranná)
|
Darolutamid
(n = 955)
|
Placeboa
(n = 554)
|
Darolutamid
(n = 955)
|
Placeboa
(n = 554)
|
Prežívanie bez metastáz
| 221 (23,1 %)
| 216 (39,0%)
| 40,4 mesiacov
(34,3; NR)
| 18,4 mesiaca
(15,5; 22,3)
| 0,413 (0,341; 0,500)
< 0,000001
|
Celkové prežívanie
| 148 (15,5 %)
| 106
(19,1 %)
| NR
(56,1; NR)
| NR
(46,9, NR)
| 0,685 (0,533; 0,881)
0,003048
|
Čas do progresie bolestic,d
| 251 (26,3 %)
| 178 (32,1%)
| 40,3 mesiaca
(33,2; 41,2)
| 25,4 mesiaca
(19,1; 29,6)
| 0,647 (0,533; 0,785)
0,000008b
|
Čas do začatia prvej cytotoxickej chemoterapie
| 127 (13,3 %)
| 98 (17,7%)
| NR
(NR, NR)
| NR
(NR, NR)
|
0,579 (0,444; 0,755)
< 0,000044
|
Čas do prvej symptomatickej kostnej príhody
| 29 (3,0%)
| 28 (5,1%)
| NR
(NR, NR)
| NR
(NR, NR)
| 0,484
(0,287; 0,815)
0,005294
|
a Vrátane 170 pacientov ktorí prešli v otvorenej fáze štúdie na liečbu darolutamidom.
b Pomer rizika < 1 v prospech darolutamidu.
c V prípade MFS a času do progresie bolesti sa za konečnú analýzu považuje analýza vykonaná v čase primárneho ukončenia.
d Pacient hlásil výsledok na základe hodnotenia dotazníka
Brief Pain Inventory-Short Form (Stručný'
dotazník intenzity bolesti) NR: Nedosiahnuté (
not reached).
Liečba darolutamidom tiež viedla k dlhšiemu prežívaniu bez progresie (PFS, medián 36,8 oproti
14,8 mesiaca, HR = 0,380, nominálna hodnota p < 0,000001) a dlhšiemu času do progresie PSA (medián 29,5 oproti 7,2 mesiaca, HR = 0,164, nominálna hodnota p < 0,000001). Konzistentnosť účinku sa pozorovala vo všetkých mierach prežívania (MFS, OS a PFS).
 Obrázok 1: Kaplanove-Meierove krivky prežívania bez metastáz
D
arolutamid (n = 955)
Placebo (n = 554)
Obrázok 1: Kaplanove-Meierove krivky prežívania bez metastáz
D
arolutamid (n = 955)
Placebo (n = 554)
Pacienti s rizikom
Mesiace od randomizácie
Darolutamid
Placebo
 Obrázok 2: Kaplanove-Meierove krivky celkového prežívania
D
arolutamid (n = 955)
Placebo (n = 554)
Obrázok 2: Kaplanove-Meierove krivky celkového prežívania
D
arolutamid (n = 955)
Placebo (n = 554)
Pacienti s rizikom
Mesiace od randomizácie

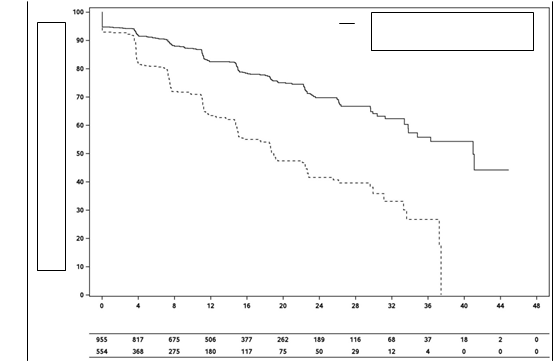 Darolutam
Placebo
Darolutam
Placebo
Pacienti, ktorí dostávali darolutamid v štúdii ARAMIS (dvojito zaslepená časť), preukázali významne
vyššiu potvrdenú mieru odpovede PSA (definovanú ako ≥ 50 % zníženie oproti východiskovým hodnotám) v porovnaní s pacientmi, ktorí dostávali placebo, 84,0 % oproti 7,9 % (rozdiel = 76,1 %, p < 0,000001 (nominálna hodnota p, len pre informáciu)).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s darolutamidom
vo všetkých podskupinách pediatrickej populácie pre malígne novotvary prostaty (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Všeobecný úvod
Darolutamid pozostáva z dvoch diastereomérov [(S, R)-darolutamid a (S, S)-darolutamid], ktoré sa
vzájomne premieňajú cez hlavný cirkulujúci metabolit nazývaný ketodarolutamid. In vitro vykazujú všetky tri látky podobnú farmakologickú aktivitu. Darolutamid je slabo rozpustný vo vodných rozpúšťadlách v širokom rozmedzí pH a je všeobecne rozpustnejší v organických rozpúšťadlách.
Absorpcia
Po perorálnom podaní 600 mg (2 tablety po 300 mg) sa maximálne plazmatické koncentrácie
darolutamidu 4,79 mg/l (variačný koeficient: 30,9 %) zvyčajne dosahujú približne 4 hodiny po podaní. Pomer dvoch diastereomérov, (S, R)-darolutamidu k (S, S)-darolutamidu sa zmenil z pomeru 1:1
v tablete na pomer približne 1:9 v plazme na základe údajov hodnôt AUC0 - 12 v rovnovážnom stave. Po perorálnom podaní spolu s jedlom sa rovnovážny stav dosiahne po 2 - 5 dňoch opakovaného dávkovania dvakrát denne.
Absolútna biologická dostupnosť v porovnaní s intravenóznou injekciou je približne 30 % po
perorálnom podaní tablety NUBEQA obsahujúcej 300 mg darolutamidu nalačno. Biologická dostupnosť darolutamidu sa pri podávaní s jedlom zvýšila 2,0- až 2,5-násobne. Podobné zvýšenie expozície sa pozorovalo pre hlavný metabolit ketodarolutamid.
Distribúcia
Zjavný distribučný objem darolutamidu po intravenóznom podaní je 119 l, čo naznačuje, že
darolutamid je široko distribuovaný v tele do intracelulárnych aj extracelulárnych tekutinových priestorov.
Darolutamid sa stredne (92 %) viaže na proteíny ľudskej plazmy bez rozdielu medzi dvoma diastereomérmi. Hlavný metabolit darolutamidu, ketodarolutamid, sa vo vysokej miere (99,8 %) viaže na plazmatické bielkoviny.
Prechod darolutamidu cez hematoencefalickú bariéru sa klinicky neskúmal. Expozícia mozgu darolutamidu v hodnotách AUC0 - 24 je však veľmi nízka, so 4,5 % plazmatickej expozície po jednorazovej dávke u potkanov a 1,9 - 3,9 % po opakovanej dávke u myší. To naznačuje nízky prechod darolutamidu cez neporušenú hematoencefalickú bariéru u potkanov a myší a nízku pravdepodobnosť, že darolutamid prechádza neporušenou hematoencefalickou bariérou u ľudí v klinicky významnom rozsahu.
Biotransformácia
Diastereoméry (S, R)-darolutamid a (S, S)-darolutamid sú schopné vzájomnej premeny cez metabolit
ketodarolutamid s preferenciou pre (S, S)-darolutamid.
Po jednorazovom perorálnom podaní 300 mg 14 C-darolutamidu podávaného vo forme perorálneho roztoku je ketodarolutamid jediný hlavný metabolit s približne 2-násobne vyššou celkovou expozíciou v plazme v porovnaní s darolutamidom. Darolutamid a ketodarolutamid spolu tvorili 87,4 % zo
14 C-rádioaktivity v plazme, čo naznačuje, že všetky ostatné metabolity majú malý význam. Darolutamid sa metabolizuje primárne oxidačným metabolizmom sprostredkovaným hlavne CYP3A4,
ako aj priamou glukuronidáciou sprostredkovanou prednostne UGT1A9 a UGT1A1. Okrem toho sa ukázalo, že hlavne izoformy AKR1C katalyzujú redukciu ketodarolutamidu na diastereoméry tejto látky.
Eliminácia
Účinný polčas darolutamidu a ketodarolutamidu v plazme pacientov je približne 20 hodín. Z dvoch
diastereomérov predstavujúcich darolutamid má (S, R)-darolutamid kratší účinný polčas 9 hodín v porovnaní s (S, S)-darolutamidom s účinným polčasom 22 hodín. Klírens darolutamidu po intravenóznom podaní bol 116 ml/min. (CV: 39,7 %). Celkovo 63,4 % materiálu súvisiaceho s látkou sa vylučuje močom (približne 7 % v nezmenenej podobe), 32,4 % sa vylučuje stolicou. Viac ako 95 % dávky sa vylúčilo do 7 dní po podaní.
Linearita/nelinearita
V rozsahu dávok 100 až 700 mg (po jednorazovej dávke a v ustálenom stave) sa expozícia dvom
diastereomérom a hlavnému metabolitu ketodarolutamidu lineárne zvyšuje takmer od dávky závislým spôsobom. Na základe nasýtenej absorpcie sa pri dávke 900 mg dvakrát denne nepozorovalo žiadne ďalšie zvýšenie expozície darolutamidu.
Špeciálne skupiny pacientov
Starší pacienti
Nepozorovali sa žiadne klinicky významné rozdiely vo farmakokinetických vlastnostiach darolutamidu (65 - 95 rokov).
Porucha funkcie obličiek
V klinickej farmakokinetickej štúdii boli hodnoty AUC a Cmax darolutamidu 2,5 a 1,6-krát vyššie
u pacientov s ťažkou poruchou funkcie obličiek (odhadovaná miera glomerulárnej filtrácie [eGFR] 15
až 29 ml/min./1,73 m2) v porovnaní so zdravými dobrovoľníkmi.
Populačná farmakokinetická analýza naznačuje 1,1-, 1,3- a približne 1,5-násobne vyššiu expozíciu (AUC) darolutamidu u pacientov s miernou, stredne ťažkou a ťažkou poruchou funkcie obličiek (eGFR 15 až 89 ml/min./1,73 m2) v porovnaní s pacientmi s normálnou funkciou obličiek.
Farmakokinetické vlastnosti darolutamidu sa neskúmali u pacientov s ochorením obličiek v konečnom štádiu, ktorí podstupovali dialýzu (eGFR < 15 ml/min./1,73 m2).
Porucha funkcie pečene
V klinickej farmakokinetickej štúdii boli hodnoty Cmax a AUC darolutamidu 1,5- a 1,9-krát vyššie
u pacientov so stredne ťažkou poruchou funkcie pečene (Childovo-Pughovo skóre B) v porovnaní so zdravými dobrovoľníkmi. Nie sú údaje o pacientoch s ťažkou poruchou funkcie pečene (Childovo-
Pughovo skóre C).
Etnické rozdiely
Neboli pozorované žiadne klinicky významné rozdiely vo farmakokinetických vlastnostiach darolutamidu na základe etnicity (biela, japonská, ázijská iná než japonská, čierna alebo
africko-americká). Populačná farmakokinetická analýza ukázala 1,4-násobné zvýšenie expozície
(AUC) u japonských pacientov v porovnaní s pacientmi zo všetkých ostatných regiónov.
5
.3 Predklinické údaje o bezpečnosti
Systémová toxicita
V štúdiách toxicity po opakovanom podávaní u potkanov a psov boli hlavnými nálezmi zmeny
samčích reprodukčných orgánov (zníženie hmotnosti orgánov s atrofiou prostaty a nadsemenníkov). Tieto účinky sa vyskytli pri systémových expozíciách v rozmedzí predpokladanej expozície u ľudí alebo nižších (na základe porovnania hodnôt AUC). Medzi ďalšie zmeny reprodukčných tkanív patrilo minimálne zvýšenie vakuolizácie hypofýzy, atrofia a zníženie sekrécie semenných vezikúl
a mliečnych žliaz u potkanov, ako aj hypospermia semenníkov, dilatácia a degenerácia semenotvorných tubulov u psov. Zmeny v mužských reprodukčných orgánoch u oboch druhov boli
v súlade s farmakologickým účinkom darolutamidu a po 4- až 8-týždňovom zotavení sa zvrátili alebo čiastočne vymizli.
Embryotoxicita/teratogenita
Štúdie vývojovej toxicity sa nevykonali.
Reprodukčná toxicita
Štúdie reprodukčnej toxicity sa nevykonali. Avšak, na základe výsledkov štúdií toxicity po
opakovanom podávaní u potkanov a psov je pravdepodobné, že plodnosť u samcov je narušená, čo je v súlade s farmakologickým účinkom darolutamidu.
Genotoxicita a karcinogénnypotenciál
Darolutamid nevyvolával mutácie v teste mikrobiálnej mutagenézy (Ames). Pri vysokých
koncentráciách indukoval darolutamid in vitro štrukturálne chromozómové aberácie v kultivovaných ľudských lymfocytoch. V in vivo kombinovanom mikronukleovom teste kostnej drene a kométovom teste pečene a duodena potkanov sa však nepozorovala žiadna genotoxicita pri expozíciách presahujúcich maximálnu expozíciu u ľudí. Dlhodobé štúdie na zvieratách zamerané na vyhodnotenie karcinogénneho potenciálu darolutamidu sa nevykonali.
Farmakologická bezpečnosť
In vitro darolutamid slabo inhiboval draslíkový prúd hERG a vápnikový kanál typu L. In vivo, u psov
pod anestéziou darolutamid mierne skrátil trvanie intervalu QT, ale tento účinok sa nenašiel u psov pri vedomí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadrotablety
hydrogenfosforečnan vápenatý (E 341)
sodná soľ kroskarmelózy monohydrát laktózy
stearát horečnatý (E 470b)
povidón (E 1201)
Filmovýobal hypromelóza monohydrát laktózy makrogol (E 1521) oxid titaničitý (E 171)
6
.2 Inkompatibility
Neaplikovateľné.
6.3. Čas použiteľnosti3 roky
6.4. Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaBlistre z PVC/hliníkovej fólie obsahujúce 16 filmom obalených tabliet. Každé balenie obsahuje 96 alebo 112 filmom obalených tabliet.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBayer AG
51368 Leverkusen
Nemecko
8. REGISTRAČNÉ ČÍSLOEU/1/20/1432/001 112 filmom obalených tabliet

EU/1/20/1432/002 96 filmom obalených tabliet
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 27. marec 2020
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.