
na vápnika v sére pred podaním dávky pod 1,87 mmol/l alebo nad 2,55 mmol/l, toto meranie sa má zopakovať
v nasledujúci deň.
5. Upravte dávku aktívneho vitamínu D alebo doplnku vápnika alebo oboch na základe hladiny vápnika v sére a klinického hodnotenia (t.j. prejavov a príznakov hypokalciémie a hyperkalciémie).
Navrhované úpravy dávkovania Natparu, aktívneho vitamínu D a doplnku vápnika na základe hladín
vápnika v sére sú uvedené nižšie:
H
ladina vápnika v sére pred podaním dávky
Nad
hornou hranicou normálnych hodnôt (upper
Upravte ako prvé Upravte ako druhé Upravte ako tretie
Natpar Aktívne formy
vitamínu D Doplnok vápnika
Zvážte zníženie alebo zastavenie liečby Natparom a
limit of normal, ULN) (2,55 mmol/l)*
Vyššia ako 2,25 mmol/l a
prehodnoťte ju
prostredníctvom merania hladiny vápnika v sére Zvážte zníženie
Znížte alebo prerušte** Znížte
Žiadna zmena alebo zníženie, ak podávanie
pod
hornou hranicou
normálnych hodnôt
(2,55 mmol/l)*
Menej ako alebo presne
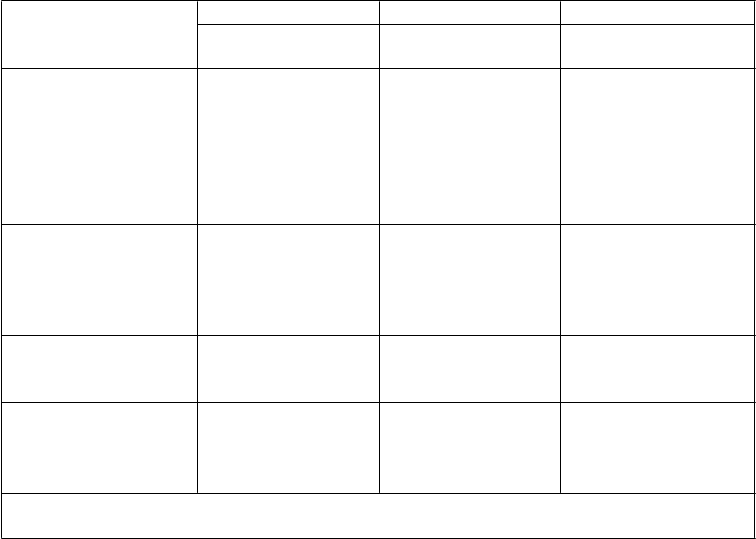
Žiadna zmena
Znížte alebo prerušte**
aktívneho vitamínu D bolo prerušené ešte pred touto titráciou
2,25 mmol/l a nad
2 mmol/l
Nižšie ako 2 mmol/l
Zvážte zvýšenie
po uplynutí aspoň 2 – 4
týždňov pri stabilnej dávke
Žiadna zmena Žiadna zmena
Zvýšte Zvýšte
*Hodnota ULN sa môže líšiť v závislosti od laboratória
**Prerušenie u pacientov dostávajúcich najnižšiu dávku, ktorá je k dispozícii
6. Opakujte 4. a 5. krok až kým sa nedosiahne cieľová koncentrácia vápnika v sére pred podaním dávky v rozsahu 2,0-2,25 mmol/l, až kým sa nepreruší podávanie aktívneho vitamínu D a až kým sa nedosiahne doplnenie vápnika na hladinu dostačujúcu na uspokojenie denných potrieb.
ÚpravadávkovaniaNatparupozačiatočnomobdobí
Počas titrácie sa musí monitorovať koncentrácia vápnika v sére (pozri časť 4.4).
Dávku Natparu možno zvyšovať o 25 mikrogramov približne každé 2 až 4 týždne, až po maximálnu dennú dávku 100 mikrogramov. Titráciu smerom nadol, až po minimum 25 mikrogramov možno urobiť kedykoľvek.
Odporúča sa zmerať hladinu vápnika v sére korigovanú na albumín 8 – 12 hodín po podaní Natparu. Ak hladina vápnika v sére po podaní dávky je > ULN, najprv znížte aktívny vitamín D a doplnky vápnika a monitorujte priebeh. Merania hladiny vápnika v sére pred podaním dávky a po nej sa majú opakovať a má sa potvrdiť, že sú v prijateľnom rozsahu predtým, ako sa zváži titrácia na vyššiu dávku Natparu. Ak hladina vápnika v sére po podaní dávky zostane > ULN, treba ešte väčšmi znížiť alebo prerušiť dávkovanie perorálnych doplnkov vápnika (tiež pozri tabuľku úprav v časti Začatie liečby Natparom).
Ak pri akéjkoľvek dávke Natparu hladina vápnika v sére po podaní dávky, korigovaná na albumín, prevýši ULN a nepodá sa žiadny aktívny vitamín D ani perorálny vápnik, alebo ak sú prítomné príznaky nasvedčujúce hyperkalciémii, dávka Natparu sa má znížiť (pozri časť 4.4).
Vynechaná dávka
V prípade vynechania dávky sa Natpar musí podať akonáhle je to možné, a dodatočné exogénne zdroje
vápnika a/alebo aktívneho vitamínu D sa musia užiť v závislosti od príznakov hypokalciémie.
Prerušeniealeboukončenieliečby
Náhle prerušenie alebo ukončenie liečby Natparom môže viesť k závažnej hypokalciémii. Dočasné alebo trvalé prerušenie liečby Natparom musí byť sprevádzané monitorovaním hladín vápnika v sére a, podľa
potreby, úpravou príjmu exogénneho vápnika a/alebo aktívneho vitamínu D (pozri časť 4.4).
Osobitné populácie
Staršie osoby
Pozri časť 5.2.
Porucha funkcie obličiek
U pacientov s miernym až stredne závažným poškodením obličiek (klírens kreatinínu 30 až 80 ml/min) nie je potrebná žiadna úprava dávky. U pacientov so závažnou poruchou funkcie obličiek nie sú dostupné žiadne údaje (pozri časť 4.4).
Porucha funkcie pečene
U pacientov s miernym alebo stredne závažnej poruche funkcie pečene (celkové skóre 7 až 9 na stupnici podľa Childa-Pugha) nie je potrebná žiadna úprava dávky. U pacientov so závažnou poruchou funkcie pečene nie sú dostupné žiadne údaje (pozri časť 4.4).
Pediatrická populácia
Bezpečnosť a účinnosť Natparu u detí vo veku menej ako 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Natpar si môže pacient podať sám. Predpisujúci lekár alebo zdravotná sestra musia zaškoliť pacienta
v správnej injekčnej technike, najmä na začiatku používania.
Každá dávka musí byť podaná ako subkutánna injekcia raz denne, striedavo do jedného alebo druhého stehna.
Návod na rekonštitúciu lieku pred podaním a na použitie injekčného pera, pozri časť 6.6 a návod uvedený v písomnej informácii pre používateľa.
Natpar sa nesmie podávať intravenózne alebo intramuskulárne.
4.3 Kontraindikácie
Natpar je kontraindikovaný u pacientov:
- precitlivených na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1
- ktorí majú podstúpiť, alebo v minulosti podstúpili ožarovanie kostí
- s malignitami kostí alebo metastázami v kostiach
- ktorí sú vystavení zvýšenému základnému riziku osteosarkómu, ako sú pacienti s Pagetovou chorobou
kostí alebo dedičnými poruchami
- s neobjasnenými nárastmi hladiny alkalickej fosfatázy špecifickej pre kosti
- so pseudohypoparatyreoidizmom.
4.4 Osobitné upozornenia a opatrenia pri používaní
Vždy, keď sa Natpar podáva pacientovi, sa dôrazne odporúča zaznamenať názov a číslo série (lot) lieku, s cieľom zachovať spojitosť medzi pacientom a sériou (lot) lieku.
Cieľom liečby Natparom je dosiahnutie koncentrácie vápnika v sére pred podaním dávky vo výške
2,0 – 2,25 mmol/l a koncentrácie vápnika v sére < 2,55 mmol/l 8 – 12 hodín po podaní dávky.
Monitorovaniepacientovpočasliečby
Počas liečby Natparom sa musia monitorovať hladiny vápnika v sére pred podaním, a v niektorých prípadoch i po podaní dávky (pozri časť 4.2). V multicentrickom klinickom skúšaní boli hodnoty na albumín korigovaného vápnika v sére (albumin-corrected serum calcium, ACSC) 6 – 10 hodín po podaní dávky
v priemere o 0,25 mmol/l vyššie ako hodnoty pred podaním dávky, pričom maximálny pozorovaný nárast
bol 0,7 mmol/l. Dávky vápnika, vitamínu D alebo Natparu môže byť nutné znížiť, ak dôjde k hyperkalciémii
po podaní dávky, i v prípadoch keď sú koncentrácie vápnika pred podaním dávky prijateľné (pozri časť 4.2).
Hyperkalciémia
V klinických skúšaniach s Natparom bola hlásená hyperkalciémia. Hyperkalciémia sa bežne vyskytovala počas titračného obdobia, počas ktorého sa upravovali dávky perorálne podávaného vápnika, aktívneho vitamínu D a Natparu. Hyperkalciémiu možno minimalizovať pridržiavaním sa odporúčaného dávkovania, zohľadňovaním informácií získaných monitorovaním a pýtaním sa pacientov, či pociťujú akékoľvek príznaky hyperkalciémie. Ak sa rozvinie závažná hyperkalciémia (> 3,0 mmol/l alebo nad hornou hranicou
normálnych hodnôt a s príznakmi), treba zvážiť hydratáciu a dočasné zastavenie podávania Natparu, vápnika
a aktívneho vitamínu D, kým sa hladina vápnika v sére nevráti do normálneho rozsahu. Potom zvážte
pokračovanie podávania Natparu, vápnika a aktívneho vitamínu D v nižších dávkach (pozri časti 4.2 a 4.8).
Hypokalciémia
Hypokalciémia, bežný klinický prejav hypoparatyreoidizmu, bola hlásená v klinických skúšaniach
s Natparom. Väčšina hypokalcemických udalostí, ku ktorým došlo v klinických skúšaniach, boli mierne až
stredné z hľadiská závažnosti. Riziko závažnej hypokalciémie bolo najväčšie po prerušení Natparu. Dočasné alebo trvalé prerušenie liečby Natparom musí byť sprevádzané monitorovaním hladín vápnika v sére a,
podľa potreby, zvýšením zdrojov exogénneho vápnika a/alebo aktívneho vitamínu D. Hypokalciémiu možno minimalizovať pridržiavaním sa odporúčaného dávkovania, zohľadňovaním informácií získaných monitorovaním a pýtaním sa pacientov, či pociťujú akékoľvek príznaky hypokalciémie (pozri časti 4.2
a 4.8).
Súbežné používanie s kardioglykozidmi
Hyperkalciémia akéhokoľvek pôvodu môže predisponovať na digitalisovú toxicitu. U pacientov
používajúcich Natpar súbežne s kardioglykozidmi (ako sú digoxín alebo digitoxín) monitorujte hladiny vápnika a kardioglykozidov v sére a pacientov, či sa u nich objavia prejavy a príznaky digitalisovej toxicity
(pozri časť 4.5).
Z
á
v
ažné
ochorenie
o
bličiek
alebo
pečene
Natpar sa má používať opatrne u pacientov so závažným ochorením obličiek alebo pečene, pretože takýto
pacienti neboli hodnotení v klinických skúšaniach.
Používanie u mladých dospelých
Natpar treba používať opatrne u mladých dospelých pacientov s otvorenými epifýzami, keďže títo pacienti môžu byť vystavení zvýšenému riziku osteosarkómu (pozri časť 4.3).
Používanie u starších pacientov
Klinické štúdie Natparu nezahrnovali dostatočné množstvo jedincov vo veku 65 rokov a starších na to, aby
sa stanovilo, či odpoveď u týchto jedincov sa líši od mladších jedincov.
Tachyfylaxia
U niektorých pacientov sa časom môže znížiť schopnosť Natparu zvyšovať hladinu vápnika. Aby sa to odhalilo, odpoveď koncentrácie vápnika v sére na podanie Natparu sa má pravidelne monitorovať, a v takom prípade treba zvážiť diagnózu tachyfylaxie.
Ak je koncentrácia 25-OH vitamínu D v sére nízka, primeraná suplementácia môže prinavrátiť odpoveď vápnika v sére na Natpar (pozri časť 4.2).
4.5 Liekové a iné interakcie
Hladiny vápnika v sére ovplyvňujú inotropické účinky kardioglykozidov. Kombinované použitie Natparu a kardioglykozidov (napr. digoxín alebo digitoxín) môže predisponovať pacientov na digitalisovú toxicitu, ak dôjde k vzniku hyperkalciémie. Nevykonali sa žiadne liekové interakčné štúdie s kardioglykozidmi a Natparom (pozri časť 4.4).
Treba sledovať pacientovu hladinu vápnika v sére v prípade akéhokoľvek lieku, ktorý mení hladinu vápnika
v sére (napr. lítium, tiazidy).
Súbežné podávanie kyseliny alendrónovej a Natparu môže viesť k zníženiu účinku šetriaceho vápnik (calcium sparing effect), čo môže rušiť normalizáciu hladiny vápnika v sére. Neodporúča sa súbežné používanie Natparu s bisfosfonátmi.
Natpar je bielkovina, ktorá neinhibuje pečeňové mikrozomálne enzýmy, ktoré metabolizujú liečivá (napr. izoenzýmy cytochrómu P450), ani nimi nie je metabolizovaná. Natpar sa neviaže na bielkoviny a má malý distribučný objem.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití Natparu u gravidných žien. Štúdie na zvieratách nepreukázali priame ani
nepriame účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Riziko u tehotných žien alebo vyvíjajúcich sa plodov nemôže byť vylúčené. Rozhodnutie, či začať alebo ukončiť liečbu Natparom počas gravidity sa má urobiť po zvážení známych rizík liečby v porovnaní
s prínosom pre ženu.
Dojčenie
Nie je známe, či sa Natpar vylučuje do materského mlieka.
Dostupné farmakologické údaje zo štúdií na zvieratách preukázali vylučovanie Natparu do mlieka (pozri časť 5.3).
Riziko u novorodencov/dojčiat nemôže byť vylúčené.. Rozhodnutie, či ukončiť dojčenie alebo ukončiť liečbu Natparom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú žiadne údaje o účinkoch Natparu na fertilitu u ľudí. Údaje na zvieratách nepoukazujú
na žiadne poškodenie fertility.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Natpar nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Keďže neurologické príznaky môžu byť prejavom nekontrolovaného hypoparatyreoidizmu, pacientov s poruchami kognície alebo pozornosti treba poučiť, aby sa zdržali vedenia vozidiel alebo obsluhy strojov, kým sa príznaky nepominú.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Najčastejšími nežiaducimi reakciami u pacientov liečených Natparom boli hyperkalciémia, hypokalciémia a s nimi spojené klinické prejavy zahrňujúce bolesť hlavy, hnačku, vracanie, parestéziu, hypoestéziu a
hyperkalciúriu. V klinických štúdiách boli tieto reakcie vo všeobecnosti z hľadiska závažnosti slabé až
mierne, prechodného trvania, a boli zvládnuté úpravami dávok Natparu, vápnika a/alebo aktívneho vitamínu D (pozri časti 4.4 a 5.1).
Tabuľkovýprehľadnežiaducichreakcií
Nežiaduce reakcie u pacientov liečených Natparom v štúdii kontrolovanej placebom sú uvedené nižšie podľa
triedy orgánových systémov databázy MedDRA a frekvencie. Frekvencie sú definované ako veľmi časté
(≥ 1/10) a časté (≥ 1/100 až < 1/10).
Trieda orgánových systémov Veľmi časté (≥ 1/10) Časté (≥ 1/100 až < 1/10) Poruchy metabolizmu a výživy hyperkalciémia, hypokalciémia hypomagneziémia†, tetánia†
Psychické poruchy úzkosť†, nespavosť*
Poruchy nervového systému bolesť hlavy*,†, hypoestézia†, parestézia†
Poruchy srdca a srdcovej
činnosti
ospalosť*
palpitácie*,†
Poruchy ciev hypertenzia*
Poruchy dýchacej sústavy, hrudníka a mediastína Poruchy gastrointestinálneho traktu
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Poruchy obličiek a močových
ciest
Celkové poruchy a reakcie v mieste podania
Laboratórne a funkčné
vyšetrenia
kašeľ†
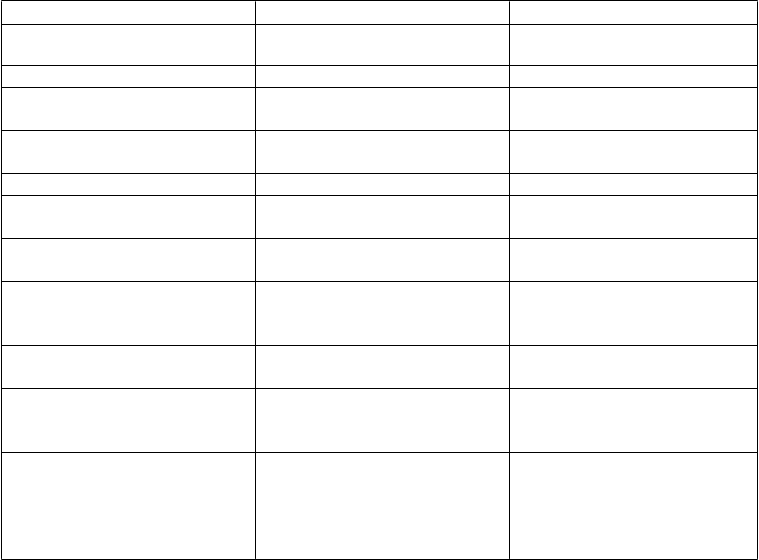
hnačka*,†, nauzea*, vracanie* bolesť v hornej časti brucha*
artralgia*, svalové kŕče† zášklby svalov†, bolesť kostí a svalov†, myalgia†, bolesť krku†, bolesť končatín hyperkalciúria†, polakizúria†
asténia*, bolesť v hrudníku†, únava, reakcie v mieste vpichu injekcie, smäd*
pozitívne anti-PTH protilátky, znížená hladina
25-hydroxycholekalciferolu v krvi†, znížená hladina vitamínu D
*Prejavy a príznaky potenciálne spojené s hyperkalciémiou, pozorované v klinických skúšaniach.
†Prejavy a príznaky potenciálne spojené s hypokalciémiou, pozorované v klinických skúšaniach..
Popis vybraných nežiaducich reakcií
Počas obdobia titrácie dávky boli bežne pozorované hyperkalciémia a hypokalciémia. Riziko závažnej hypokalciémie bolo najväčšie po prerušení Natparu (pozri časť 4.4).
R
eakcie v mieste vpichu injekcie
V štúdii kontrolovanej placebom sa u 9,5 % (8/84) pacientov liečených Natparom a u 15 % (6/40) pacientov,
ktorým bolo podané placebo, vyskytla reakcia v mieste vpichu injekcie, z ktorej všetky prípady boli miernej alebo strednej závažnosti.
ImunogenicitaV súlade s potenciálnymi imunogénnymi vlastnosťami liekov obsahujúcich peptidy, podanie Natparu môže spustiť tvorbu protilátok. V štúdii kontrolovanej placebom u dospelých s hypoparatyreoidizmom bola
incidencia protilátok proti hormónu prištítnych teliesok (PTH) 8,8 % (3/34) a 5,9 % (1/17) u pacientov,
ktorým bolo subkutánne podané 50 až 100 mikrogramov Natparu alebo placebo raz denne počas 24 týždňov.
Súhrnne vo všetkých klinických štúdiách u pacientov s hypoparatyreoidizmom, po liečbe Natparom počas obdobia do 4 rokov bola miera incidencie imunogenicity 17/87 (19,5 %) a nezdalo sa, že by časom narastala. Týchto 17 pacientov malo nízky titer protilátok proti PTH a 3 z nich sa následne stali negatívnymi
na protilátky. Zjavná prechodná povaha protilátok proti PTH je pravdepodobne spôsobená ich nízkym titrom. Traja z týchto pacientov mali protilátky s neutralizujúcou aktivitou; títo pacienti si udržali klinickú odpoveď bez náznakov nežiaducich reakcií spojených s imunitou.
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V4.9 PredávkovaniePredávkovanie môže spôsobiť hyperkalciémiu, ktorej príznaky môžu zahrňovať búšenie srdca, zmeny na EKG, hypotenziu, nauzeu, vracanie, závrat a bolesť hlavy. Závažná hyperkalciémia môže byť život ohrozujúci stav, ktorý si vyžaduje súrnu lekársku starostlivosť a pozorné sledovanie (pozri časť 4.4).
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Homeostatiká vápnika, hormóny prištítnych teliesok a analógy, ATC kód: H05AA03
MechanizmusúčinkuEndogénny hormón prištítnych teliesok (PTH) je vylučovaný prištítnymi telieskami vo forme polypeptidu pozostávajúceho z 84 aminokyselín. PTH vykazuje svoj účinok prostredníctvom receptorov pre hormón
prištítnych teliesok na povrchu buniek, prítomných v kostiach, obličkách a nervovom tkanive. Receptory
pre hormón prištítnych teliesok patria do skupiny receptorov spriahnutých s G-proteínmi.
PTH má množstvo kľúčových fyziologických funkcií, ktoré zahrňujú jeho ústrednú úlohu v modulácii hladín vápnika a fosfátov v sére v úzkom rozmedzí, riadenie vylučovania vápnika a fosfátov obličkami, aktiváciu vitamínu D a udržiavanie normálneho kostného metabolizmu.
Natpar je vyrobený v
E. coli pomocou rekombinantnej DNA technológie a je identický so sekvenciou 84
aminokyselín endogénneho ľudského hormónu prištítnych teliesok.
FarmakodynamickéúčinkyPTH (1 – 84) je hlavným regulátorom homeostázy vápnika v plazme. V obličkách PTH (1 – 84) zvyšuje
renálnu tubulárnu reabsorpciu vápnika a podporuje vylučovanie fosfátov.
Celkovým účinkom PTH je zvýšenie koncentrácie vápnika v sére, zníženie vylučovania vápnika močom a
zníženie koncentrácie fosfátov v sére.
Natpar má rovnakú primárnu sekvenciu aminokyselín ako endogénny hormón prištítnych teliesok a možno
očakávať, že bude mať rovnaké fyziologické účinky.
Klinickáúčinnosťabezpečnosť
Bezpečnosť a klinická účinnosť Natparu u dospelých s hypoparatyreoidizmom sú odvodené z 1 randomizovanej štúdie kontrolovanej placebom a z otvorenej rozšírenej štúdie. V týchto štúdiách si Natpar podávali pacienti sami, pričom denné dávky boli v rozsahu od 25 do 100 mikrogramov v jednej subkutánnej injekcii.
Štúdia 1 - REPLACE
Cieľom tejto štúdie bolo udržať hladinu vápnika v sére pomocou Natparu, a zároveň znížiť dávku perorálne podávaného vápnika a aktívneho vitamínu D alebo ich vymeniť. Táto štúdia bola randomizovaná, dvojito
zaslepen, placebom kontrolovaná, multicentrická a trvala 24 týždňov. V rámci tejto štúdie boli pacienti s chronickým hypoparatyreoidizmom, ktorí užívali vápnik a aktívne formy vitamínu D (metabolit
vitamínu D alebo analógy) randomizovaní na Natpar (n = 84) alebo placebo (n = 40). Priemerný vek bol 47,3
rokov (rozsah 19 až 74 rokov); 79 % boli ženy. Pacienti mali hypoparatyreoidizmus v priemere 13,6 rokov.
Pri randomizácii boli dávky aktívnych foriem vitamínu D znížené o 50 % a pacienti boli zaradení do skupiny s Natparom 50 mikrogramov denne alebo do skupiny s placebom. Po randomizácii nasledovala 12-týždňová fáza titrácie Natparu a 12-týždňová fáza udržiavania dávky Natparu.
Deväťdesiat percent randomizovaných pacientov ukončilo 24 týždňov liečby.
Pre účinnosť analýzy boli jedinci, ktorí splnili tri zložky trojdielneho kritéria odpovede, považovaní
za pacientov odpovedajúcich na liečbu. Pacient odpovedajúci na liečbu bol definovaný pomocou zloženého koncového ukazovateľa primárnej účinnosti, čo bolo aspoň 50 %-né zníženie dávky aktívneho vitamínu D oproti východiskovej hodnote A aspoň 50 %-né zníženie dávky perorálne podávaného vápnika oproti východiskovej hodnote A koncentrácia celkového vápnika v sére korigovaná na albumín, ktorá je udržiavaná alebo normalizovaná v porovnaní s východiskovou hodnotou (≥ 1,875 mmol/l) a neprekročila hornú hranicu laboratórneho normálneho rozsahu.
Na konci liečby dosiahlo primárny koncový ukazovateľ 46/84 (54,8 %) pacientov liečených Natparom
v porovnaní s 1/40 (2,5 %) pacientov, ktorí dostávali placebo (p < 0,001).
V 24. týždni, v prípade pacientov, ktorí ukončili štúdiu, bolo 34/79 (43 %) pacientov liečených Natparom nezávislých od liečby aktívnym vitamínom D a neužívali viac ako 500 mg citrónanu vápenatého,
v porovnaní s 2/33 (6,1 %) pacientov, ktorí dostávali placebo (p < 0,001).
U šesťdesiatdeväť percent (58/84) jedincov randomizovaných na Natpar sa preukázalo zníženie dávky perorálne podávaného vápnika o ≥ 50 % v porovnaní so 7,5 % (3/40) jedincov randomizovaných na placebo. Priemerné percento zmeny z východiskovej hodnoty u perorálne podávaného vápnika bolo -51,8 %
(SD 44,6) u jedincov liečených Natparom v porovnaní so 6,5 % (SD 38,5) v placebovej skupine (p < 0,001). Okrem toho, u 87 % (73/84) pacientov liečených Natparom sa preukázalo ≥ 50 % zníženie dávky perorálne podávaného aktívneho vitamínu D v porovnaní so 45 % (18/40) v placebovej skupine.
Štúdia 2 - RACE
Štúdia 2 bola dlhotrvajúcou, otvorenou, rozšírenou štúdiou denných subkutánnych dávok Natparu pacientom s hypoparatyreoidizmom, ktorí ukončili predchádzajúce štúdie s Natparom.
Do štúdie bolo zaradených celkovo 49 pacientov. Pacienti dostávali denné dávky 25 mikrogramov, 50 mikrogramov, 75 mikrogramov alebo 100 mikrogramov po dobu približne 40 mesiacov (priemer 1 067 dní, rozsah 41 až 1 287 dní).
Výsledky preukazujú stálosť fyziologických účinkov Natparu počas 36 mesiacov, vrátane udržania priemerných, na albumín korigovaných hladín vápnika v sére (n = 36, 2,06 ± 0,17 mmol/l), zníženia vylučovania vápnika močom z východiskovej hodnoty (n = 36, -1,21 ± 5,5 mmol/24h), zníženia hladiny
fosfátov v sére (n=36, -0,22±0,29 mmol/l) a udržania normálnej hodnoty súčinu vápnika a fosforu (n=35,'
<4,4 mmol2/l2).
Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Natparom v jednej alebo vo viacerých podskupinách pediatrickej populácie s hypoparatyreoidizmom (informácie o použití
v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku.
Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Farmakokinetika Natparu po subkutánnom podaní do stehna jedincov s hypoparatyreoidizmom bola v súlade s farmakokinetikou pozorovanou u zdravých postmenopauzálnych žien, ktorým bol hormón prištítnych teliesok podávaný do stehna a brucha.
Absorpcia
Natpar podaný subkutánne mal absolútnu biologickú dostupnosť 53 %.
Distribúcia
Po intravenóznom podaní mal Natpar distribučný objem 5,35 l v rovnovážnom stave.
Biotransformácia
Štúdie in vitro a in vivo preukázali, že klírens Natparu je v prvom rade pečeňový proces, pričom obličky
zohrávajú menšiu úlohu.
Eliminácia
V pečeni je hormón prištítnych teliesok štiepený katepsínmi. V obličkách sa hormón prištítnych teliesok a
C-terminálne fragmenty vylučujú glomerulárnou filtráciou.
Farmakokinetický/farmakodynamickývzťah
Hormón prištítnych teliesok (rDNA) sa hodnotil v otvorenej farmakokinetickej/farmakodynamickej štúdii, v ktorej 7 pacienti s hypoparatyreoidizmom dostali jednorazové subkutánne dávky vo výške 50
a 100 mikrogramov, so 7-dňovým vymývacím intervalom medzi dávkami.
K maximálnym plazmatickým koncentráciám (priemerná hodnota Tmax) Natparu dochádza do 5 až 30 minút, a k druhému, obyčajne menšiemu maximu po 1 až 2 hodinách. Zdanlivý terminálny polčas (t1/2) bol 3,02 hodín pre dávku 50 mikrogramov a 2,83 hodín pre dávku 100 mikrogramov. Maximálne priemerné zvýšenia hladiny vápnika v sére, ku ktorým došlo v priebehu 12 hodín, boli približne 0,125 mmol/l pri dávke
50 mikrogramov a 0,175 mmol/l pri dávke 100 mikrogramov.
Účinok na metabolizmus minerálov
Liečba Natparom zvyšuje koncentráciu vápnika v sére u pacientov s hypoparatyreoidizmom, a k tomuto zvýšeniu dochádza v závislosti od dávky. Po jednorazovej injekcii hormónu prištítnych teliesok (rDNA) dosiahla priemerná hladina celkového vápnika v sére svoje maximum medzi 10 a 12 hodinami. Kalcemická odpoveď sa udržala dlhšie ako 24 hodín po podaní.
Vylučovanie vápnika močom
Liečba Natparom vedie k poklesu vylučovania vápnika močom o 13 % pri dávke 50 mikrogramov a o 23 %
pri dávke 100 mikrogramov až po minimum v priebehu 3 až 6 hodín; vylučovanie vápnika močom sa vráti
na hodnoty pred liečbou po 16 až 24 hodinách.
F
osfáty
Po injekcii Natparu, hladina fosfátov v sére klesá úmerne k hladine PTH (1 – 84) počas prvých 4 hodín a pretrváva počas 24 hodín po injekcii.
Aktívny vitamín D
Hladina 1,25-(OH)2D v sére sa zvyšuje po jednej dávke Natparu na maximálnu úroveň po približne 12 hodinách, pričom k návratu na hodnoty blízke začiatočným dochádza do 24 hodín. U dávky 50 mikrogramov bol pozorovaný väčší nárast hladín 1,25-(OH)2D v sére ako u dávky 100 mikrogramov, pravdepodobne kvôli priamej inhibícii obličkového enzýmu 25-hydroxyvitamín D-1-hydroxylázy sérovým vápnikom.
Osobitné populácie
Porucha funkcie pečene
Farmakokinetická štúdia u jedincov, ktorí netrpeli hypoparatyreoidizmom sa vykonala u 6 mužov a 6 žien
so stredne závažným poškodením pečene (skóre 7 – 9 na stupnici podľa Childa-Pugha [stupeň B]), ktorí boli
porovnávaní so zhodnou skupinou 12 jedincov s normálnou funkciou pečene. Po jednorazovej subkutánnej
dávke 100 mikrogramov boli priemerné hodnoty Cmax a Cmax korigovanej na začiatočný stav vyššie o 18 % až
20 % u jedincov so stredne závažným poškodením ako u jedincov s normálnou funkciou. Medzi dvomi
skupinami pečeňovej funkcie neboli žiadne zjavné rozdiely v časových profiloch celkovej koncentrácie vápnika v sére. U pacientov s miernym až stredne závažným poškodením pečene sa neodporúča úprava dávkovania Natparu. U pacientov so závažným poškodením pečene nie sú k dispozícii žiadne údaje.
Porucha funkcie obličiek
Farmakokinetika po jednorazovej subkutánnej dávke 100 mikrogramov Natparu bola hodnotená u 16 jedincov bez poškodenia (klírens kreatinínu (CLcr) > 80 ml/min) a u 16 jedincov s poškodením obličiek. Priemerná maximálna koncentrácia (Cmax) PTH po podaní 100 mikrogramov hormónu prištítnych teliesok (rDNA) jedincom s miernym až stredne závažným poškodením obličiek (CLcr 30 až 80 ml/min) bola približne o 23 % vyššia ako priemerná maximálna koncentrácia pozorovaná u jedincov s normálnou funkciou obličiek. Expozícia PTH, meraná pomocou hodnoty AUC0-posledná, bola približne o 3,9 % vyššia a hodnoty AUC0-posledná korigovanej na začiatočný stav, bola približne o 2,5 % vyššia ako expozícia PTH pozorovaná u jedincov s normálnou funkciou obličiek.
Na základe týchto výsledkov, u pacientov s miernym až stredne závažnou poruchou funkcie obličiek (CLcr
30 až 80 ml/min) nie je potrebná žiadna úprava dávky. U pacientov podstupujúcich obličkovú dialýzu neboli
vykonané žiadne štúdie. U pacientov so závažnou poruchou funkcie obličiek nie sú k dispozícii žiadne údaje.
Pediatrická populácia
Nie sú k dispozícii farmakokinetické údaje u pediatrických pacientov.
Staršie osoby
Klinické štúdie s Natparom nezahrňovali dostatočné množstvo jedincov vo veku 65 rokov a starších na to,
aby sa stanovilo, či odpoveď u týchto osôb sa líši od odpovede u mladších jedincov.
Pohlavie
V štúdii REPLACE sa nepozorovali žiadne klinicky významné rozdiely medzi pohlaviami.
Hmotnosť
Nie je potrebná žiadna úprava dávky na základe hmotnosti.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, mutagenity, toxicity
na fertilitu a všeobecnú reprodukciu, a miestnej znášanlivosti neodhalili žiadne osobitné riziko pre ľudí.
Potkany, ktorým boli podávané denné injekcie Natparu po dobu 2 rokov, mali nadmernú tvorbu kostnej hmoty závislú od dávky a zvýšený výskyt nádorov kostí, vrátane osteosarkómu, pravdepodobne spôsobený negenotoxickým mechanizmom. Klinický význam týchto zistení nie je známy, vzhľadom na rozdiely
vo fyziológii kosti medzi potkanmi a ľuďmi. V klinických skúšaniach neboli pozorované žiadne osteosarkómy.
Natpar nemal nepriaznivý vplyv na fertilitu alebo včasný vývoj embryí potkanov, embryofetálny vývoj
u potkanov a králikov, ani na pre- alebo postnatálny vývoj u potkanov. Minimálne množstvo Natparu sa
vylučuje do mlieka laktujúcich potkanov.
U opíc, ktoré po dobu 6 mesiacov denne dostávali subkutánne dávky, bol zvýšený výskyt obličkovej tubulárnej mineralizácie pri úrovniach expozície, ktoré boli 2,7-krát vyššie ako hladina klinickej expozície pri najvyššej dávke.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Prášok
Chlorid sodný
Manitol
Monohydrát kyseliny citrónovej
Hydroxid sodný (na úpravu pH)
Rozpúšťadlo
Metakrezol
Voda na injekcie
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
3 roky.
Rekonštituovaný roztok
Po rekonštitúcii bola chemická a fyzikálna stabilita počas používania roztoku preukázaná po dobu 14 dní pri skladovaní v chladničke (2 ºC – 8 ºC) a po dobu 3 dní pri skladovaní mimo chladničky pri teplote neprevyšujúcej 25 ºC počas 14-denného obdobia používania.
Pero s obsahom rekonštituovanej náplne udržiavajte dôkladne uzatvorené na ochranu pred svetlom.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C).
Neuchovávajte v mrazničke.
Náplň uchovávajte v príslušnom držiaku na náplne vo vonkajšom obale na ochranu pred svetlom.
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Dvojkomorová sklenená náplň v držiaku na náplne je vyhotovená zo skla typu I s 2 zátkami z brómbutylovej gumy a so zasúvacím krytom (hliník) s uzáverom z brómbutylovej gumy.
N
atpar 25 mikrogramov
Každá náplň v purpurovom držiaku na náplne obsahuje 350 mikrogramov hormónu prištítnych teliesok
(rDNA) vo forme prášku v prvej komôrke a 1 000 mikrolitrov rozpúšťadla v druhej komôrke (zodpovedajúce
14 dávkam).
Natpar 50 mikrogramov
Každá náplň v červenom držiaku na náplne obsahuje 700 mikrogramov hormónu prištítnych teliesok (rDNA)
vo forme prášku v prvej komôrke a 1 000 mikrolitrov rozpúšťadla v druhej komôrke (zodpovedajúce 14
dávkam).
Natpar 75 mikrogramov
Každá náplň v sivom držiaku na náplne obsahuje 1 050 mikrogramov hormónu prištítnych teliesok (rDNA)
vo forme prášku v prvej komôrke a 1 000 mikrolitrov rozpúšťadla v druhej komôrke (zodpovedajúce 14
dávkam).
Natpar 100 mikrogramov
Každá náplň v modrom držiaku na náplne obsahuje 1 400 mikrogramov hormónu prištítnych teliesok
(rDNA) vo forme prášku v prvej komôrke a 1 000 mikrolitrov rozpúšťadla v druhej komôrke (zodpovedajúce
14 dávkam).
Veľkosť balenia: Škatuľka obsahuje 2 náplne.
Na označenie rozličných síl sa používajú rozličné farby škatuliek/náplní:
25 mikrogramov - Purpurová
50 mikrogramov - Červená
75 mikrogramov - Sivá
100 mikrogramov - Modrá
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Hormón prištítnych teliesok (rDNA) sa podáva injekčne pomocou náplne s perom na opakované použitie. Jedno pero smie použiť iba jeden pacient. Pre každú injekciu sa musí použiť nová sterilná ihla. Používajte ihly na injekčné perá veľkosti 31G x 8 mm. Po rekonštitúcii musí byť tekutina bezfarebná a prakticky
bez obsahu cudzorodých častíc; hormón prištítnych teliesok (rDNA) sa nesmie použiť, ak je rekonštituovaný
roztok zakalený, sfarbený alebo obsahuje viditeľné častice.
NEPOTRIASAJTE počas rekonštitúcie alebo po nej; potriasanie môže spôsobiť denaturáciu liečiva. Predtým, ako použijete pero na opakované použitie, prečítajte si návod na použitie v písomnej informácii
pre používateľa.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Shire Pharmaceuticals Ireland Limited
5 Riverwalk
Citywest Business Campus
Dublin 24
Írsko
8. REGISTRAČNÉ ČÍSLO
EU/1/15/1078/001
EU/1/15/1078/002
EU/1/15/1078/003
EU/1/15/1078/004
9. DÁTUM PRVEJ REGISTRÁCIE/ PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.