
70" valign="top"> · Okamžite prerušte infúziu liekom
MINJUVI a liečte prejavy a príznaky.
· Keď prejavy a príznaky ustúpia alebo sa znížia na 1. stupeň, pokračujte v infúzii lieku MINJUVI maximálne
pri 50 % rýchlosti, pri ktorej sa reakcia vyskytla. Ak sa u pacienta do 1 hodiny nevyskytne ďalšia reakcia a životné
funkcie sú stabilné, rýchlosť infúzie sa
môže zvýšiť každých 30 minút podľa znášanlivosti maximálne na rýchlosť, pri ktorej sa reakcia vyskytla.
|
3. stupeň (závažný)
| · Okamžite prerušte infúziu liekom
MINJUVI a liečte prejavy a príznaky.
· Keď prejavy a príznaky ustúpia alebo sa znížia na 1. stupeň, pokračujte v infúzii lieku MINJUVI maximálne
pri 25 % rýchlosti, pri ktorej sa reakcia
vyskytla. Ak sa u pacienta do 1 hodiny nevyskytne ďalšia reakcia a životné funkcie sú stabilné, rýchlosť infúzie sa môže zvýšiť každých 30 minút podľa znášanlivosti maximálne
na 50 % rýchlosti, pri ktorej sa reakcia vyskytla.
· Ak sa po opätovnom pokuse reakcia vráti, okamžite zastavte infúziu.
|
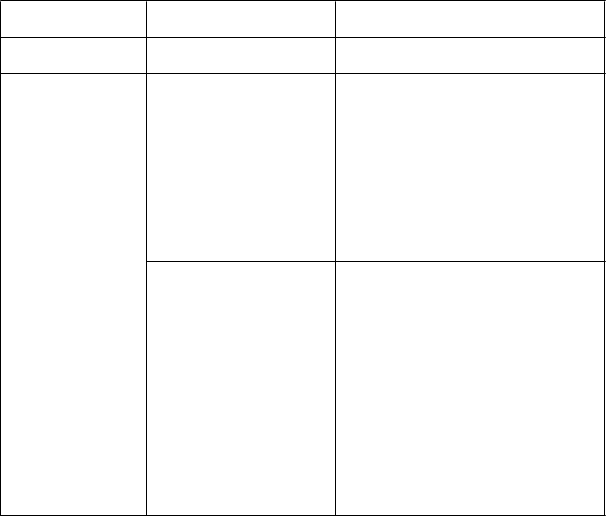
Nežiaduca reakcia
|
Z
ávažnosť
|
Úprava dávky
|
|
4. stupeň (život ohrozujúci)
|
· Okamžite zastavte infúziu a natrvalo ukončite podávanie lieku MINJUVI.
|
Myelosupresia
|
Počet krvných doštičiek nižší ako 50 000/μl
|
· Vysaďte liek MINJUVI a lenalidomid a každý týždeň monitorujte celkový krvný obraz, kým počet krvných
doštičiek nebude 50 000/μl alebo vyšší.
· Pokračujte v užívaní lieku MINJUVI
v rovnakej dávke a lenalidomidu
v zníženej dávke, ak sa krvné doštičky vrátia na ≥ 50 000/μl. Úpravy dávkovania sú uvedené v súhrne charakteristických vlastností lieku pre lenalidomid.
|
|
Počet neutrofilov nižší ako 1 000/μl po dobu
najmenej 7 dní
alebo
počet neutrofilov nižší ako 1 000/μl so zvýšením telesnej teploty na 38 °C alebo viac
alebo
počet neutrofilov nižší ako 500/μl
|
· Vysaďte liek MINJUVI a lenalidomid a každý týždeň monitorujte celkový
krvný obraz, kým počet neutrofilov
nebude 1 000/μl alebo vyšší.
· Pokračujte v užívaní lieku MINJUVI
v rovnakej dávke a lenalidomidu
v zníženej dávke, ak sa neutrofily vrátia na ≥ 1 000/μl. Úpravy dávkovania sú uvedené v súhrne charakteristických vlastností lieku pre lenalidomid.
|
Osobitné
skupiny
pacientov
Pediatrická populácia
Bezpečnosť a účinnosť lieku MINJUVI u detí mladších ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Starší pacientiU starších pacientov (≥ 65 rokov) nie je potrebná úprava dávky.
Porucha funkcie obličiekU pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek nie je potrebná úprava dávky (pozri časť 5.2). Nie sú k dispozícii žiadne údaje o odporúčaných dávkach u pacientov so závažnou poruchou funkcie obličiek.
Porucha funkcie pečeneU pacientov s miernou poruchou funkcie pečene nie je potrebná úprava dávky (pozri časť 5.2). Nie sú k dispozícii žiadne údaje o odporúčaných dávkach u pacientov so stredne závažnou alebo závažnou poruchou funkcie pečene.
SpôsobpodávaniaLiek MINJUVI je určený na intravenózne použitie po rekonštitúcii a zriedení.
· Pri prvej infúzii 1. cyklu má byť rýchlosť intravenóznej infúzie 70 ml/h počas prvých 30 minút.
Potom sa má rýchlosť infúzie zvýšiť, aby sa dokončila prvá infúzia v priebehu 2,5 hodiny.
· Všetky následné infúzie sa majú podať v priebehu 1,5 až 2 hodín.
· V prípade nežiaducich reakcií zvážte odporúčané úpravy dávky uvedené v tabuľke č. 1.
· Liek MINJUVI sa nesmie podávať súbežne s inými liekmi prostredníctvom tej istej infúznej súpravy.
· Liek MINJUVI sa nesmie podávať ako intravenózna rýchla injekcia ani bolus.
Pokyny na rekonštitúciu a riedenie lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo šarže podaného lieku.
Reakciesúvisiacesinfúziou
Môžu sa vyskytnúť reakcie súvisiace s infúziou, ktoré boli hlásené častejšie počas prvej infúzie (pozri časť 4.8). Pacienti majú byť počas celej infúzie pozorne sledovaní. Pacientov je potrebné poučiť, aby kontaktovali svojich zdravotníckych pracovníkov, ak sa u nich vyskytnú prejavy a príznaky reakcií súvisiacich s infúziou vrátane horúčky, zimnice, vyrážky alebo problémov s dýchaním do 24 hodín od podania infúzie. Pred začatím infúzie tafasitamabu sa má pacientom podať premedikácia. Na základe závažnosti reakcie súvisiacej s infúziou sa má podávanie infúzie tafasitamabu prerušiť alebo ukončiť
a má sa začať príslušná liečba (pozri časť 4.2).
Myelosupresia
Liečba tafasitamabom môže spôsobiť závažnú a/alebo ťažkú myelosupresiu vrátane neutropénie, trombocytopénie a anémie (pozri časť 4.8). Kompletný krvný obraz sa má monitorovať počas celej
liečby a pred podaním každého liečebného cyklu. Na základe závažnosti nežiaducej reakcie sa má infúzia tafasitamabu vysadiť (pozri tabuľku č. 1). Úpravy dávkovania sú uvedené v súhrne charakteristických vlastností lieku pre lenalidomid.
Neutropénia
Počas liečby tafasitamabom bola hlásená neutropénia vrátane febrilnej neutropénie. Je potrebné zvážiť podanie faktorov stimulujúcich kolónie granulocytov (granulocyte colony-stimulating factors,
G-CSF), najmä u pacientov s neutropéniou 3. alebo 4. stupňa. Všetky príznaky alebo prejavy vzniku infekcie je potrebné predvídať, vyhodnotiť a liečiť.
Trombocytopénia
Počas liečby tafasitamabom bola hlásená trombocytopénia. Je potrebné zvážiť vysadenie súbežných liekov, ktoré môžu zvýšiť riziko krvácania (napr. inhibítory krvných doštičiek, antikoagulanciá). Pacientov treba poučiť, aby okamžite hlásili prejavy alebo príznaky modrín alebo krvácania.
Infekcie
Smrteľné a závažné infekcie vrátane oportúnnych infekcií sa vyskytli u pacientov počas liečby tafasitamabom. Tafasitamab sa má podávať pacientom s aktívnou infekciou iba vtedy, ak je infekcia primerane liečená a dobre kontrolovaná. Pacienti s anamnézou opakujúcich sa alebo chronických infekcií môžu byť vystavení zvýšenému riziku infekcie a majú byť primerane monitorovaní. Pacientov je potrebné poučiť, aby kontaktovali svojich zdravotníckych pracovníkov, ak sa vyskytne horúčka alebo iný dôkaz o možnej infekcii, ako je zimnica, kašeľ alebo bolesť pri močení.
Syndrómrozpadunádoru
U pacientov s vysokou nádorovou záťažou a rýchlo proliferujúcim nádorom môže byť zvýšené riziko syndrómu rozpadu nádoru. U pacientov s DLBCL bol počas liečby tafasitamabom pozorovaný syndróm rozpadu nádoru. Pred liečbou tafasitamabom sa majú prijať vhodné opatrenia/profylaxia
v súlade s miestnymi odporúčaniami. Počas liečby tafasitamabom sa má u pacientov dôkladne monitorovať výskyt syndrómu rozpadu nádoru.
Očkovanie
Bezpečnosť očkovania živými vakcínami po liečbe tafasitamabom nebola skúmaná a očkovanie živými vakcínami sa neodporúča súbežne s liečbou tafasitamabom.
Pomocnélátky
Tento liek obsahuje 37,0 mg sodíka na 5 injekčných liekoviek (dávka pre pacienta s telesnou hmotnosťou 83 kg), čo zodpovedá 1,85 % WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
4.6 Fertilita, gravidita a laktácia
Liečba tafasitamabom v kombinácii s lenalidomidom sa nemá začať u pacientok, pokiaľ nie je vylúčené tehotenstvo. Pozrite si aj súhrn charakteristických vlastností lieku pre lenalidomid.
Ženyvplodnomveku/antikoncepciaužien
Ženy v plodnom veku majú byť poučené, že majú používať účinnú antikoncepciu počas liečby tafasitamabom a najmenej 3 týždne po skončení liečby.
Gravidita
Neuskutočnili sa štúdie reprodukčnej toxicity a vývinu tafasitamabu.
Nie sú k dispozícii údaje o použití tafasitamabu u gravidných žien. Je však známe, že IgG prechádza placentou a tafasitamab môže spôsobiť depléciu B-buniek plodu na základe farmakologických vlastností (pozri časť 5.1). V prípade expozície počas gravidity sa má u novorodencov monitorovať deplécia B-buniek a vakcinácie živými vakcínami proti vírusom sa majú odložiť, až kým sa počet
B-buniek dieťaťa nevráti na normálnu hodnotu (pozri časť 4.4).
Tafasitamab sa neodporúča užívať počas gravidity a u žien vo fertilnom veku nepoužívajúcich antikoncepciu.
Lenalidomid môže spôsobiť embryofetálne poškodenie a je kontraindikovaný počas gravidity a u žien vo fertilnom veku, pokiaľ nie sú splnené všetky podmienky programu prevencie gravidity počas užívania lenalidomidu.
Dojčenie
Nie je známe, či sa tafasitamab vylučuje do ľudského mlieka. Je však známe, že materský IgG sa vylučuje do materského mlieka. Nie sú k dispozícii žiadne údaje o použití tafasitamabu u dojčiacich žien a riziko u dojčiat nemôže byť vylúčené. Ženy majú byť poučené, aby počas liečby tafasitamabom a aspoň 3 mesiace po poslednej dávke nedojčili.
Fertilita
Neuskutočnili sa žiadne špecifické štúdie na vyhodnotenie účinku tafasitamabu na fertilitu. V štúdii toxicity po opakovanom podávaní u zvierat neboli pozorované žiadne nežiaduce účinky na reprodukčné orgány samcov a samíc (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Liek MINJUVI nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. U pacientov používajúcich tafasitamab však bola hlásená únava, čo je potrebné vziať do úvahy v prípade vedenia vozidla alebo obsluhy strojov.
4.8 Nežiaduce účinky
Súhrn
profilu
bezpečnosti
Najčastejšie nežiaduce reakcie sú: infekcie (73 %), neutropénia (51 %), asténia (38 %), anémia (36 %), hnačka (36 %), trombocytopénia (31 %), kašeľ (26 %), periférny edém (24 %), pyrexia (24 %), znížená chuť do jedla (22 %).
Najčastejšie závažné nežiaduce reakcie boli infekcia (26 %) vrátane pneumónie (7 %) a febrilná neutropénia (6 %).
Trvalé ukončenie liečby tafasitamabom z dôvodu nežiaducej reakcie sa vyskytlo u 15 % pacientov. Najčastejšie nežiaduce reakcie vedúce k trvalému ukončeniu liečby tafasitamabom boli infekcie
a nákazy (5 %), poruchy nervového systému (2,5 %) a poruchy dýchacej sústavy, hrudníka a mediastína (2,5 %).
Frekvencia úpravy dávky alebo prerušenia v dôsledku nežiaducich reakcií bola 65 %. Najčastejšie nežiaduce reakcie vedúce k prerušeniu liečby tafasitamabom boli poruchy krvi a lymfatického systému
(41 %).
SúhrnnýzoznamnežiaducichreakciíNežiaduce reakcie hlásené v klinických skúšaniach sú zoradené podľa tried orgánových systémov podľa databázy MedDRA a podľa frekvencie. Frekvencie nežiaducich reakcií vychádzajú z hlavného skúšania fázy 2 MOR208C203 (L-MIND) s 81 pacientmi. Pacienti boli vystavení tafasitamabu
v priemere 7,7 mesiaca. Frekvencie nežiaducich reakcií z klinických skúšaní sú založené na frekvenciách nežiaducich udalostí všetkých príčin, kde časť udalostí nežiaducich reakcií môže mať iné príčiny ako liek, napr. ochorenie, iné lieky alebo nesúvisiace príčiny.
Frekvencie sú definované ako: veľmi časté (≥1/10); časté (≥1/100 až <1/10); menej časté
(≥1/1 000 až <1/100); zriedkavé (≥1/10 000 až <1/1000); veľmi zriedkavé (<1/10 000) a neznáme
(z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Trieda orgánových systémov
| Frekvencia
| Nežiaduce reakcie
| Infekcie a nákazy
| Veľmi časté
| Bakteriálne, vírusové a hubové infekcie+
vrátane oportúnnych infekcií so smrteľnými následkami (napr. bronchopulmonálna aspergilóza, bronchitída, zápal pľúc a infekcia močových ciest)
| Časté
| Sepsa (vrátane neutropenickej sepsy)
| Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
| Časté
| Karcinóm bazálnych buniek
| Poruchy krvi a lymfatického systému
| Veľmi časté
| Febrilná neutropénia+, neutropénia+, trombocytopénia+, anémia, leukopénia+
| Časté
| Lymfopénia
| Poruchy imunitného systému
| Časté
| Hypogamaglobulinémia
| Poruchy metabolizmu a výživy
| Veľmi časté
| Hypokaliémia, znížená chuť do jedla
| Časté
| Hypokalciémia, hypomagneziémia
| Poruchy nervového systému
| Časté
| Bolesť hlavy, parestézia, dysgeúzia
| Poruchy dýchacej sústavy, hrudníka a mediastína
| Veľmi časté
| Dyspnoe, kašeľ
| Časté
| Exacerbácia chronickej obštrukčnej choroby pľúc, upchatie nosa
| Poruchy gastrointestinálneho traktu
| Veľmi časté
| Hnačka, zápcha, vracanie, nevoľnosť, bolesť brucha
| Poruchy pečene a žlčových ciest
| Časté
| Hyperbilirubinémia, zvýšené transaminázy
(vrátane zvýšenia ALT a/alebo AST), zvýšená gama-glutamyltransferáza
|
|
|
Tabuľka č. 2: Nežiaduce reakcie u pacientov s recidivujúcim alebo refraktérnym DLBCL, ktorí užívali tafasitamab v klinickom skúšaní MOR208C203 (L-MIND)
Trieda orgánových systémov
|
Frekvencia
|
Nežiaduce reakcie
|
Poruchy kože a podkožného tkaniva
|
Veľmi časté
|
Vyrážky (zahŕňa rôzne typy vyrážok, napr. vyrážky, makulopapulárne vyrážky, svrbivé vyrážky, erytematózne vyrážky)
|
Časté
|
Svrbenie, alopécia, erytém, hyperhidróza
|
Poruchy kostrovej a svalovej sústavy
a spojivového tkaniva
|
Veľmi časté
|
Bolesť chrbta, svalové kŕče
|
Časté
|
Artralgia, bolesť končatín, bolesť kostrovej a svalovej sústavy
|
Poruchy obličiek a močových ciest
|
Časté
|
Zvýšená hladina kreatinínu v krvi
|
Celkové poruchy a reakcie v mieste podania
|
Veľmi časté
|
Asténia (vrátane malátnosti), únava, periférny edém, pyrexia
|
Časté
|
Zápal sliznice
|
Laboratórne a funkčné vyšetrenia
|
Časté
|
Zníženie hmotnosti, zvýšenie C-reaktívneho proteínu
|
Úrazy, otravy a komplikácie liečebného postupu
|
Časté
|
Reakcie súvisiace s infúziou
|
+ Ďalšie informácie o tejto nežiaducej reakcii sú uvedené v nasledujúcom texte.
V porovnaní s výskytom pri kombinovanej liečbe s lenalidomidom sa výskyt nehematologických nežiaducich reakcií pri monoterapii tafasitamabom znížil najmenej o 10 % pre zníženú chuť do jedla, asténiu, hypokaliémiu, zápchu, nevoľnosť, svalové kŕče, dyspnoe a zvýšenie C-reaktívneho proteínu.
Popis vybraných nežiaducich reakciíMyelosupresiaLiečba tafasitamabom môže spôsobiť závažnú alebo ťažkú myelosupresiu vrátane neutropénie, trombocytopénie a anémie (pozri časti 4.2 a 4.4).
V štúdii L-MIND sa myelosupresia (t. j. neutropénia, febrilná neutropénia, trombocytopénia,
leukopénia, lymfopénia alebo anémia) vyskytla u 65,4 % pacientov liečených tafasitamabom. Myelosupresia bola liečená redukciou alebo prerušením lenalidomidu, prerušením tafasitamabu a/alebo podaním G-CSF (pozri časti 4.2 a 4.4). Myelosupresia viedla k prerušeniu liečby tafasitamabom u 41 % a k ukončeniu liečby tafasitamabom u 1,2 %.
Neutropénia/febrilná neutropéniaVýskyt neutropénie bol 51 %. Výskyt neutropénie 3. alebo 4. stupňa bol 49 % a febrilnej neutropénie 3. alebo 4. stupňa 12 %. Medián trvania akejkoľvek nežiaducej reakcie neutropénie bol 8 dní
(rozsah 1 – 222 dní); medián času do nástupu prvého výskytu neutropénie bol 49 dní
(rozsah 1 – 994 dní).
TrombocytopéniaVýskyt trombocytopénie bol 31 %. Výskyt trombocytopénie 3. alebo 4. stupňa bol 17 %. Medián trvania akejkoľvek nežiaducej reakcie trombocytopénie bol 11 dní (rozsah 1 – 470 dní); medián času
do nástupu prvého výskytu trombocytopénie bol 71 dní (rozsah 1 – 358 dní).
AnémiaVýskyt anémie bol 36 %. Výskyt anémie 3. alebo 4. stupňa bol 7 %. Medián trvania akejkoľvek nežiaducej anemickej reakcie bol 15 dní (rozmedzie 1 – 535 dní); medián času do nástupu prvej
anémie bol 49 dní (rozsah 1 – 1 129 dní).
Keď pacienti v štúdii L-MIND prešli z tafasitamabu a lenalidomidu v kombinovanej liečebnej fáze na tafasitamab samotný v rozšírenej fáze monoterapie, výskyt hematologických udalostí sa znížil najmenej o 20 % pri neutropénii, trombocytopénii a anémii; pri monoterapii tafasitamabom nebol hlásený žiadny výskyt febrilnej neutropénie (pozri časti 4.2 a 4.4).
Infekcie
V štúdii L-MIND sa infekcie vyskytli u 73 % pacientov. Výskyt infekcií 3. alebo 4. stupňa bol 28 %. Najčastejšie hlásenými infekciami 3. alebo vyššieho stupňa boli pneumónia (7 %), infekcie dýchacích
ciest (4,9 %), infekcie močových ciest (4,9 %) a sepsa (4,9 %). Infekcia bola smrteľná u < 1 % pacientov (pneumónia) do 30 dní od poslednej liečby.
Medián času do prvého nástupu infekcie 3. alebo 4. stupňa bol 62,5 dňa (4 – 1 014 dní). Medián
trvania akejkoľvek infekcie bol 11 dní (1 – 392 dní). Odporúčania týkajúce sa liečby infekcií sú uvedené v časti 4.4.
Infekcia viedla k prerušeniu podávania tafasitamabu u 27 % a k ukončeniu podávania tafasitamabu u 4,9 % pacientov.
ReakciesúvisiacesinfúziouV štúdii L-MIND sa reakcie súvisiace s infúziou vyskytli u 6 % pacientov. Všetky reakcie súvisiace s infúziou boli 1. stupňa a ustúpili v deň výskytu. Osemdesiat percent týchto reakcií sa vyskytlo
počas 1. alebo 2. cyklu. Medzi príznaky patrili triaška, návaly tepla, dyspnoe a hypertenzia (pozri časti 4.2 a 4.4).
ImunogenicitaU 245 pacientov liečených tafasitamabom neboli pozorované žiadne protilátky proti tafasitamabu, ktoré by sa vyskytli počas liečby, alebo ktorých hladiny by boli zvýšené počas liečby. Už existujúce protilátky proti tafasitamabu boli zistené u 17/245 pacientov (6,9 %) bez vplyvu na farmakokinetiku, účinnosť alebo bezpečnosť tafasitamabu.
Osobitné skupiny pacientovStaršípacientiZ 81 pacientov liečených v štúdii L-MIND bolo 56 (69 %) pacientov vo veku > 65 rokov. Pacienti vo veku > 65 rokov mali numericky vyšší výskyt závažných nežiaducich udalostí vzniknutých počas
liečby (
treatment emergent adverse events, TEAE) (55 %) ako pacienti vo veku ≤ 65 rokov (44 %).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieV prípade predávkovania sa majú pacienti pozorne sledovať, či sa u nich nevyskytujú prejavy alebo príznaky nežiaducich reakcií, a podľa potreby sa má poskytnúť podporná starostlivosť.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Cytostatiká, monoklonálne protilátky, ATC kód: L01XC35.
MechanizmusúčinkuTafasitamab je monoklonálna protilátka s posilneným Fc fragmentom, ktorá sa zameriava na antigén
CD19 exprimovaný na povrchu nezrelých B a zrelých B-lymfocytov.
Po naviazaní sa na CD19 tafasitamab sprostredkúva lýzu B-buniek prostredníctvom:
· zapojenia imunitných efektorových buniek, ako sú prirodzené zabíjače, γδ T-bunky a fagocyty,
· priamej indukcie bunkovej smrti (apoptóza).
Modifikácia Fc vedie k zvýšenej bunkovej cytotoxicite závislej od protilátok a k bunkovej fagocytóze závislej od protilátok.
Farmakodynamické
účinky
U pacientov s recidivujúcim alebo refraktérnym DLBCL tafasitamab viedol k zníženiu počtu B-buniek v periférnej krvi. Zníženie v porovnaní s východiskovým počtom B-buniek dosiahlo 97 % po ôsmich dňoch liečby v štúdii L-MIND. Maximálne zníženie B-buniek približne o 100 % (medián) bolo dosiahnuté do 16 týždňov liečby.
Aj keď deplécia B-buniek v periférnej krvi je merateľný farmakodynamický účinok, priamo nekoreluje s depléciou B-buniek v pevných orgánoch ani v malígnych depozitoch.
Klinickáúčinnosť
Tafasitamab v kombinácii s lenalidomidom, po ktorom nasledovala monoterapia tafasitamabom, bol skúmaný v štúdii L-MIND, nezaslepenej multicentrickej štúdii s jedným ramenom. Táto štúdia bola vykonaná u dospelých pacientov s relapsujúcim alebo refraktérnym DLBCL
po 1 až 3 predchádzajúcich systémových liečbach DLBCL, ktorí v čase skúšania neboli kandidátmi na chemoterapiu s vysokými dávkami, po ktorej nasledovala ASCT alebo ktorí odmietli ASCT. Jedna
z predchádzajúcich systémových liečob musela zahŕňať cielenú liečbu CD20. Zo štúdie boli vylúčení pacienti so závažnou poruchou funkcie pečene (celkový bilirubín v sére > 3 mg/dl) a pacienti
s poruchou funkcie obličiek (CrCL< 60 ml/min.), ako aj pacienti s anamnézou alebo dôkazom klinicky významného kardiovaskulárneho ochorenia, ochorenia CNS a/alebo iného systémového ochorenia.
Pacienti so známou anamnézou dvojitej/trojitej prestavby génu DLBCL (double/triple-hit DLBCL)
boli tiež vylúčení pri zaradení do štúdie.
Počas prvých troch cyklov pacienti dostávali 12 mg/kg tafasitamabu infúziou v 1., 8., 15. a 22. deň každého 28-dňového cyklu plus počiatočnú dávku vo 4. deň 1. cyklu. Potom bol tafasitamab podávaný v 1. a 15. deň každého cyklu až do progresie ochorenia. Premedikácia vrátane antipyretík, blokátorov receptorov histamínu H1 a H2 a glukokortikosteroidov bola podaná 30 až 120 minút pred prvými
tromi infúziami tafasitamabu.
Pacienti samostatne užívali 25 mg lenalidomidu denne v 1. až 21. deň každého 28-dňového cyklu, maximálne 12 cyklov.
Do štúdie L-MIND bolo zaradených celkom 81 pacientov. Medián veku bol 72 rokov (rozmedzie 41 až 86 rokov), 89 % pacientov boli belosi a 54 % bolo mužov. Z 81 pacientov 74 (91,4 %) malo skóre výkonnosti ECOG 0 alebo 1, a 7 (8,6 %) malo skóre ECOG 2. Medián počtu predchádzajúcich liečob bol dva (rozsah: 1 až 4), pričom 40 pacientov (49,4 %) dostávalo jednu
predchádzajúcu liečbu a 35 pacientov (43,2 %) dostávalo 2 predchádzajúce línie liečby. Piati pacienti (6,2 %) mali 3 predchádzajúce línie liečby a 1 (1,2 %) mal 4 predchádzajúce línie liečby. Všetci pacienti užívali predchádzajúcu liečbu, ktorá obsahovala CD20. Osem pacientov malo diagnózu DLBCL premenenej z lymfómu nízkeho stupňa. Pätnásť pacientov (18,5 %) malo primárne refraktérne ochorenie, 36 (44,4 %) bolo refraktérnych voči svojej poslednej predchádzajúcej liečbe
a 34 (42,0 %) bolo refraktérnych voči rituximabu. Deväť pacientov (11,1 %) dostalo predchádzajúcu
ASCT. Primárnymi dôvodmi pre pacientov, ktorí nie sú kandidátmi na ASCT, boli vek (45,7 %), refraktérnosť na záchrannú chemoterapiu (23,5 %), komorbidity (13,6 %) a odmietnutie chemoterapie s vysokými dávkami/ASCT (16,0 %).'
Jeden pacient užíval tafasitamab, ale nie lenalidomid. Zostávajúcich 80 pacientov dostalo aspoň jednu dávku tafasitamabu a lenalidomidu. Všetci pacienti zaradení do štúdie L-MIND mali diagnózu DLBCL na základe lokálnej patológie. Podľa centrálneho patologického preverenia však u 10 pacientov nemohol byť klasifikovaný DLBCL.
Medián trvania expozície liečbe bol 9,2 mesiaca (rozmedzie: 0,23; 54,67 mesiacov). Tridsaťdva
(39,5 %) pacientov dokončilo 12 cyklov tafasitamabu. Tridsať (37,0 %) pacientov dokončilo 12 cyklov lenalidomidu.
Primárnym ukazovateľom účinnosti bola najlepšia miera objektívnej odpovede (objective response rate, ORR), definovaná ako podiel účastníkov s úplnou a čiastočnou odpoveďou hodnotený nezávislou kontrolnou komisiou (independent review committee, IRC). Ďalšie ukazovatele účinnosti zahŕňali trvanie odpovede (duration of response, DoR), prežívanie bez progresie ochorenia (progression-free
survival, PFS) a celkové prežívanie (
overall survival, OS). Výsledky účinnosti sú zhrnuté v tabuľke č. 3.
Parameter účinnosti
| Tafasitamab + lenalidomid
(N = 81 [ITT]*)
| 30-NOV-2019 cut-off
(analýza 24 mesiacov)
| 30-OCT-2020 cut-off
(analýza 35 mesiacov)
| Primárny ukazovateľ
| Najlepšia miera objektívnej odpovede (podľa IRC)
| Celková miera odpovede, n (%) (95 % IS)
| 46 (56,8)
[45,3; 67,8]
| 46 (56,8)
[45,3; 67,8]
| Miera úplnej odpovede, n (%) (95 % IS)
| 32 (39,5)
[28,8; 51,0]
| 32 (39,5)
[28,8; 51,0]
| Miera čiastočnej odpovede, n (%) (95 % IS)
| 14 (17,3)
[9,8; 27,3]
| 14 (17,3)
[9,8; 27,3]
| Sekundárne ukazovatele
| Celkové trvanie odpovede (úplná + čiastočná odpoveď)a
| Medián, mesiace
(95 % IS)
| 34,6
[26,1, NR]
| 43,9
[26,1; NR]
|
|
|
Tabuľka č. 3: Výsledky účinnosti u pacientov s recidivujúcim alebo refraktérnym difúznym veľkobunkovým B-lymfómom v štúdii MOR208C203 (L-MIND)ITT (
intention to treat) = úmysel liečiť sa; NR (
not reached) = nedosiahnuté
*Jeden pacient užíval len tafasitamab
IS: binomický presný interval spoľahlivosti pomocou Clopperovej Pearsonovej metódy
a Odhady podľa Kaplana-Meiera
Celkové prežívanie (OS) bolo v štúdii sekundárnym ukazovateľom. Po mediáne času ďalšieho sledovania 42,7 mesiaca (95 % IS: 38,0; 47,2) bol medián OS 31,6 mesiaca (95 % IS: 18,3; nedosiahnuté).
Spomedzi ôsmich pacientov, ktorí mali DLBCL transformovaný z predchádzajúceho indolentného lymfómu, malo sedem pacientov objektívnu odpoveď (traja pacienti CR, štyria pacienti PR) a jeden pacient mal stabilné ochorenie ako najlepšiu odpoveď na liečbu tafasitamabom + lenalidomidom.
StaršípacientiV súbore ITT malo 36 z 81 pacientov ≤ 70 rokov a 45 z 81 pacientov > 70 rokov. Neboli pozorované žiadne celkové rozdiely v účinnosti u pacientov ≤ 70 rokov v porovnaní s pacientmi > 70 rokov.
PediatrickápopuláciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky skúšaní s liekom MINJUVI vo všetkých podskupinách pediatrickej populácie pri difúznom veľkobunkovom B- lymfóme (informácie o použití v pediatrickej populácii, pozri časť 4.2).
Tento liek bol registrovaný s tzv. podmienkou. To znamená, že sa očakávajú ďalšie údaje o tomto lieku.
Európska agentúra pre lieky najmenej raz ročne posúdi nové informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnostiAbsorpcia, distribúcia, biotransformácia a eliminácia boli zdokumentované na základe populačnej farmakokinetickej analýzy.
AbsorpciaNa základe analýzy tafasitamabu v kombinácii s lenalidomidom boli priemerné minimálne koncentrácie tafasitamabu v sére (± smerodajná odchýlka) 179 (± 53) μg/ml počas týždenného (plus dodatočná dávka na 4. deň 1. cyklu) intravenózneho podávania 12 mg/kg. Počas podávania
každých 14 dní od 4. cyklu boli priemerné minimálne koncentrácie v sére 153 (± 68) μg/ml. Celkové maximálne sérové koncentrácie tafasitamabu boli 483 (± 109) μg/ml.
Distribúcia
Celkový distribučný objem tafasitamabu bol 9,3 l.
Biotransformácia
Presná cesta, ktorou sa tafasitamab metabolizuje, nebola charakterizovaná. Tafasitamab je humánna IgG monoklonálna protilátka a očakáva sa, že sa rozloží na malé peptidy a aminokyseliny prostredníctvom katabolických dráh rovnakým spôsobom ako endogénne IgG.
Eliminácia
Klírens tafasitamabu bol 0,41 l/deň a terminálny polčas eliminácie bol 16,9 dňa. Po dlhodobých pozorovaniach sa zistilo, že klírens tafasitamabu sa časom znížil na 0,19 l/deň po dvoch rokoch.
Osobitnéskupinypacientov
Vek, telesná hmotnosť, pohlavie, veľkosť nádoru, typ ochorenia, počet B-buniek alebo absolútny počet lymfocytov, protilátky proti lieku, hladiny laktátdehydrogenázy a sérového albumínu nemali relevantný vplyv na farmakokinetiku tafasitamabu. Vplyv rasy a etnického pôvodu na farmakokinetiku tafasitamabu nie je známy.
Porucha funkcie obličiek
Vplyv poruchy funkcie obličiek sa v špecializovaných klinických skúšaniach formálne netestoval, pri miernej až stredne závažnej poruche funkcie obličiek (klírens kreatinínu (CrCL) ≥ 30 a < 90 ml/min
odhadnutý podľa Cockcroftovej-Gaultovej rovnice) sa však nepozorovali žiadne klinicky významné
rozdiely vo farmakokinetike tafasitamabu. Vplyv ťažkej poruchy funkcie obličiek až konečného štádia ochorenia obličiek (CrCL < 30 ml/min) nie je známy.
Porucha funkcie pečene
Vplyv poruchy funkcie pečene sa v špecializovaných klinických skúšaniach formálne netestoval, pri miernej poruche funkcie pečene (celkový bilirubín ≤ horná hranica normálnej hodnoty (upper limit of normal, ULN) a aspartátaminotransferáza (AST) > ULN alebo celkový bilirubín 1 až 1,5-násobok ULN a akákoľvek AST) však neboli pozorované žiadne klinicky významné rozdiely vo farmakokinetike tafasitamabu. Vplyv stredne závažnej až ťažkej poruchy funkcie pečene (celkový bilirubín > 1,5-násobok ULN a akýkoľvek AST) nie je známy.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje neodhalili žiadne osobitné riziko pre ľudí. Štúdietoxicitypoopakovanompodávaní
Ukázalo sa, že tafasitamab je vysoko špecifický pre antigén CD19 na B-bunkách. Štúdie toxicity po intravenóznom podávaní opiciam cynomolgus nepreukázali žiadny iný účinok ako očakávanú farmakologickú depléciu B-buniek v periférnej krvi a v lymfoidných tkanivách. Tieto zmeny po ukončení liečby ustúpili.
Mutagenita/karcinogenita
Keďže tafasitamab je monoklonálna protilátka, štúdie genotoxicity a karcinogenity sa nevykonali, pretože takéto testy nie sú pre túto molekulu v navrhovanej indikácii relevantné.
Reprodukčnátoxicita
Štúdie reprodukčnej toxicitya vývinu , ako aj špecifické štúdie na hodnotenie účinkov na fertilitu sa s tafasitamabom nevykonali. V 13-týždňovej štúdii toxicity po opakovanom podávaní na opiciach cynomolgus sa však nepozorovali žiadne nežiaduce účinky na reprodukčné orgány u samcov a samíc a žiadne účinky na dĺžku menštruačného cyklu u samíc.
6
. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
dihydrát citrónanu sodného monohydrát kyseliny citrónovej dihydrát trehalózy
polysorbát 20
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6. Neboli pozorované žiadne inkompatibility so štandardnými infúznymi materiálmi.
6.3 Čas použiteľnosti
Neotvorenáinjekčnáliekovka
3 roky
Rekonštituovanýroztok(predzriedením)
Bolo preukázané, že liek má počas používania chemickú a fyzikálnu stabilitu až 24 hodín pri teplote 2 °C – 25 ºC.
Z mikrobiologického hľadiska sa má rekonštituovaný roztok ihneď spotrebovať, pokiaľ spôsob rekonštitúcie nevylučuje riziko vzniku mikrobiálnej kontaminácie. Ak sa nepoužije ihneď, za uchovávanie počas používania a za podmienky uchovávania zodpovedá používateľ. Neuchovávajte v mrazničke ani nepretrepávajte.
Zriedenýroztok(nainfúziu)
Bolo preukázané, že liek má počas používania chemickú a fyzikálnu stabilitu maximálne 36 hodín pri teplote 2 °C – 8 °C a potom až 24 hodín pri teplote do 25 ºC.
Z mikrobiologického hľadiska sa má zriedený roztok použiť okamžite. Ak sa nepoužije ihneď, za čas uchovávania počas používania a za podmienky pred použitím zodpovedá používateľ a zvyčajne nemá byť dlhší ako 24 hodín pri teplote 2 - 8 °C, pokiaľ sa riedenie nevykonalo za kontrolovaných
a validovaných aseptických podmienok. Neuchovávajte v mrazničke ani nepretrepávajte.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C).
Injekčnú liekovku uchovávajte vo vonkajšom obale na ochranu pred svetlom. Podmienky na uchovávanie po rekonštitúcii a zriedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Injekčná liekovka z číreho skla typu I s butylovou gumovou zátkou, hliníkovým tesnením a plastovým výklopným uzáverom obsahujúca 200 mg tafasitamabu. Veľkosť balenia po 1 injekčnej liekovke.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Liek MINJUVI sa dodáva v sterilných jednorazových injekčných liekovkách bez konzervačných látok. Liek MINJUVI sa má pred intravenóznou infúziou rekonštituovať a zriediť.
Pri rekonštitúcii a riedení používajte vhodné aseptické postupy.
Pokynytýkajúcesarekonštitúcie
· Dávku tafasitamabu stanovte na základe telesnej hmotnosti pacienta vynásobením 12 mg telesnou hmotnosťou pacienta (kg). Potom vypočítajte počet potrebných injekčných liekoviek tafasitamabu (každá injekčná liekovka obsahuje 200 mg tafasitamabu) (pozri časť 4.2).
· Pomocou sterilnej injekčnej striekačky jemne pridajte 5,0 ml sterilnej vody na injekcie do každej injekčnej liekovky lieku MINJUVI. Prúd nasmerujte na steny každej injekčnej liekovky a nie priamo na lyofilizovaný prášok.
· Rekonštituovanú injekčnú liekovku (rekonštituované injekčné liekovky) jemne otáčajte, aby ste podporili rozpustenie lyofilizovaného prášku. Netrepte ani nemiešajte prudko. Nevyberajte
obsah, kým sa všetky pevné látky úplne nerozpustia. Lyofilizovaný prášok sa má rozpustiť do 5 minút.
· Rekonštituovaný roztok má vyzerať ako bezfarebný až mierne žltý roztok. Pred pokračovaním sa vizuálnou kontrolou presvedčte, či nie sú prítomné pevné častice alebo zmena farby. Ak je roztok zakalený, zmenil farbu alebo obsahuje viditeľné častice, injekčnú liekovku (injekčné
liekovky) zlikvidujte.
Pokynynariedenie· Má sa použiť infúzny vak obsahujúci 250 ml injekčného roztoku chloridu sodného 9 mg/ml
(0,9 %).
· Vypočítajte celkový objem potrebného rekonštituovaného roztoku tafasitamabu 40 mg/ml. Z
infúzneho vaku natiahnite objem rovnajúci sa tomuto objemu a odobratý objem zlikvidujte.
· Odoberte celkový vypočítaný objem (ml) rekonštituovaného roztoku tafasitamabu z injekčnej liekovky (injekčných liekoviek) a pomaly ho pridajte do infúzneho vaku s chloridom
sodným 9 mg/ml (0,9 %). Zlikvidujte všetku nepoužitú časť tafasitamabu, ktorá zostala v injekčnej liekovke.
· Konečná koncentrácia zriedeného roztoku má byť od 2 mg/ml do 8 mg/ml tafasitamabu.
· Intravenózny vak jemne premiešajte pomalým prevracaním vaku. Nepretrepávajte.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIIncyte Biosciences Distribution B.V. Paasheuvelweg 25
1105 BP Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/21/1570/001
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 26. august 2021
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.