
stane skôr alebo hladina bilirubínu
> 3-násobok ULN
Natrvalo ukončite liečbu
|
Imunitne podmienená kolitída alebo hnačka
|
2. stupeň
|
Oddiaľte podanie dávkyc
|
Začnite liečbu
prednizónom v dávke 1 až 2 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné znižovanie dávky
|
3. alebo 4. stupeň
|
Natrvalo ukončite liečbu
|
Intestinálna perforácia
AKÉHOKOĽVEK
stupňa
|
Natrvalo ukončite liečbu
|
V prípade podozrenia na intestinálnu
perforáciu sa ihneď poraďte s chirurgom
|
Imunitne podmienená hypertyreóza, tyreoiditída
|
2. – 4. stupeň
|
Oddiaľte podanie dávky až do
klinickej stabilizácie
|
Symptomatická liečba
|
Imunitne podmienená hypotyreóza
|
2. – 4. stupeň
|
Bez zmeny
|
Začnite substitučnú liečbu hormónom štítnej
žľazy podľa klinickej indikácie
|
Nežiaduc
e reakcie
|
Závažnos
ťa
|
Úprav
a liečby
|
Liečba
k
o
rtikosteroidom, pokiaľ nie je uvedené inakb
|
Imunitne podmienená insuficiencia nadobličiek, hypofyzitída/hypopituitarizmus
|
2. – 4. stupeň
|
Oddiaľte podanie dávky až do klinickej stabilizácie
|
Začnite liečbu
prednizónom v dávke 1 až 2 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné znižovanie dávky,
a hormonálnu substitučnú liečbu podľa klinickej indikácie
|
Imunitne podmienený diabetes mellitus 1. typu
|
2. – 4. stupeň
|
Bez zmeny
|
Začnite liečbu
inzulínom podľa klinickej indikácie
|
Imunitne podmienená nefritída
|
2. stupeň s hladinou
kreatinínu v sére
> 1,5 – 3-násobok (ULN alebo východiskovej hodnoty)
|
Oddiaľte podanie dávkyc
|
Začnite liečbu prednizónom v dávke 1 až 2 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné znižovanie dávky
|
3. stupeň s hladinou kreatinínu v sére
> 3-násobok východiskovej hodnoty alebo > 3 –
6-násobok ULN; 4. stupeň s hladinou kreatinínu v sére
> 6-násobok ULN
|
Natrvalo ukončite liečbu
|
Imunitne podmienená vyrážka alebo dermatitída (vrátane pemfigoidu)
|
2. stupeň počas > 1 týždňa alebo 3. stupeň
|
Oddiaľte podanie dávkyc
|
Začnite liečbu
prednizónom v dávke 1 až 2 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné znižovanie dávky
|
4. stupeň
|
Natrvalo ukončite liečbu
|
Imunitne podmienená myokarditída
|
2. – 4. stupeň
|
Natrvalo ukončite liečbu
|
Začnite liečbu prednizónom v dávke 2 až 4 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné znižovanie dávkyf
|
Imunitne podmienená myozitída/polymyozitída
|
2. alebo 3. stupeň
|
Oddiaľte podanie dávkyc,g
|
Začnite liečbu prednizónom v dávke 1 až 2 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné znižovanie dávky
|
4. stupeň
|
Natrvalo ukončite liečbu
|
Nežiaduc
e reakcie
|
Závažnos
ťa
|
Úprav
a liečby
|
Liečba
k
o
rtikosteroidom, pokiaľ nie je uvedené inakb
|
Reakcie súvisiace s infúziou
|
1. alebo 2. stupeň
|
Prerušte infúziu alebo znížte rýchlosť infúzie
|
Môžete zvážiť
premedikáciu na profylaxiu následných infúznych reakcií
|
3. alebo 4. stupeň
|
Natrvalo ukončite liečbu
|
Manažujte závažné
reakcie súvisiace s infúziou podľa ústavných štandardov, náležitých odporúčaní klinickej praxe a/alebo odporúčaní odborných spoločností
|
Imunitne podmienená myasténia gravis
|
2. – 4. stupeň
|
Natrvalo ukončite liečbu
|
Začnite liečbu prednizónom v dávke 1
až 2 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné
znižovanie dávky
|
Imunitne podmienená encefalitída
|
2. – 4. stupeň
|
Natrvalo ukončite liečbu
|
Začnite liečbu
prednizónom v dávke
1 až 2 mg/kg/deň alebo
ekvivalent, po ktorej má nasledovať postupné znižovanie dávky
|
Iné imunitne podmienené nežiaduce reakcieh
|
2. alebo 3. stupeň
|
Oddiaľte podanie dávkyc
|
Začnite liečbu
prednizónom v dávke
1 až 2 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné znižovanie dávky
|
4. stupeň
|
Natrvalo ukončite liečbu
|
Iné ako imunitne podmienené nežiaduce reakcie
|
2. a 3. stupeň
|
Oddiaľte podanie
dávky až kým sa stav neupraví na
≤ 1. stupeň alebo nevráti na východiskový stav
|
|
4. stupeň
|
Natrvalo ukončite liečbui
|
a Spoločné terminologické kritériá pre nežiaduce udalosti (Common Terminology Criteria for Adverse Events,
CTCAE), verzia 4.03. ALT: alanínaminotransferáza; AST: aspartátaminotransferáza; ULN: horná hranica normálu; BLV (baseline value): východisková hodnota.
b Po úprave na ≤ 1. stupeň sa má začať s postupným znižovaním dávky kortikosteroidu a v jeho podávaní sa má pokračovať počas minimálne 1 mesiaca. V prípade zhoršenia alebo absencie zlepšenia zvážte zvýšenie dávky
kortikosteroidu a/alebo použitie ďalších systémových imunosupresív.
c Po oddialení podania sa môže v liečbe IMJUDO a/alebo durvalumabom opätovne pokračovať v priebehu 12
týždňov, ak sa nežiaduce reakcie zmiernili na ≤ 1. stupeň a dávka kortikosteroidu bola znížená na ≤ 10 mg prednizónu alebo ekvivalent denne. Liečba IMJUDO a durvalumabom sa má v príslušných prípadoch natrvalo ukončiť pri rekurentných nežiaducich reakciách 3. stupňa.
d U pacientov s inou príčinou sa riaďte odporúčaniami pre zvýšené hladiny AST alebo ALT bez súbežných zvýšení hladiny bilirubínu.
e Ak sú východiskové hladiny AST a ALT menej ako ULN alebo sa rovnajú ULN u pacientov s postihnutím pečene, oddiaľte podanie alebo natrvalo ukončite liečbu durvalumabom na základe odporúčaní pre hepatitídu
bez postihnutia pečene.
f Ak sa odpoveď nedostaví do 2 až 3 dní napriek podaniu kortikosteroidov, okamžite začnite s doplnkovou imunosupresívnou liečbou. Po úprave (0. stupeň) sa má začať s postupným znižovaním dávky kortikosteroidu a v jeho podávaní sa má pokračovať počas minimálne 1 mesiaca.
g Natrvalo ukončite liečbu IMJUDO a durvalumabom, ak nedôjde k úprave nežiaducej reakcie na ≤ 1. stupeň do
30 dní alebo ak sú prítomné prejavy respiračnej insuficiencie.
h Zahŕňa imunitnú trombocytopéniu a pankreatitídu.
i S výnimkou abnormalít laboratórnych hodnôt 4. stupňa, pri ktorých má byť rozhodnutie o ukončení liečby
založené na sprievodných klinických prejavoch/príznakoch a klinickom posúdení.
Pri suspektných imunitne podmienených nežiaducich reakciách sa má vykonať primerané zhodnotenie tak, aby sa potvrdila etiológia alebo vylúčili alternatívne etiológie.
Osobitné skupinypacientov
Pediatrická populácia
Bezpečnosť a účinnosť IMJUDO u detí a dospievajúcich mladších ako 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Staršie osoby
U starších pacientov (vo veku ≥ 65 rokov) nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie obličiek
U pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek sa neodporúča žiadna úprava dávky IMJUDO. Údaje u pacientov so závažnou poruchou funkcie obličiek sú príliš obmedzené na vyvodenie záverov pre túto populáciu (pozri časť 5.2).
Porucha funkcie pečene
U pacientov s miernou alebo stredne závažnou poruchou funkcie pečene sa neodporúča žiadna úprava
dávky IMJUDO. IMJUDO sa neskúmal u pacientov so závažnou poruchou funkcie pečene (pozri časť
5.2).
Spôsob podávania
IMJUDO je určený na intravenózne použitie.
IMJUDO podajte pred podaním durvalumabu v rovnaký deň.
IMJUDO a durvalumab sa podávajú ako samostatné intravenózne infúzie. Informácie týkajúce sa podávania durvalumabu, pozri SPC durvalumabu.
Pokyny na riedenie lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Imunitne podmienenápneumonitída
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytla imunitne
podmienená pneumonitída alebo intersticiálna choroba pľúc, ktorá bola definovaná ako stav vyžadujúci použitie systémových kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu (pozri
časť 4.8). Pacientov je potrebné sledovať pre prejavy a príznaky pneumonitídy. Suspektnú pneumonitídu je potrebné potvrdiť rádiografickým zobrazovacím vyšetrením a vylúčiť iné infekčné etiológie a etiológie súvisiace s ochorením a má sa manažovať podľa odporúčaní v časti 4.2.
Imunitne podmienenáhepatitída
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytla imunitne
podmienená hepatitída, ktorá bola definovaná ako stav vyžadujúci použitie systémových kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu (pozri časť 4.8). Pred začatím liečby
a pred každou následnou infúziou sledujte hladinu alanínaminotransferázy, aspartátaminotransferázy,
celkového bilirubínu a alkalickej fosfatázy. Ďalšie sledovanie je potrebné zvážiť na základe klinického hodnotenia. Imunitne podmienená hepatitída sa má manažovať podľa odporúčaní v časti 4.2.
Imunitne podmienenákolitída
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytla imunitne
podmienená kolitída alebo hnačka, ktorá bola definovaná ako stav vyžadujúci použitie systémových
kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu (pozri časť 4.8). U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa hlásila intestinálna perforácia
a perforácia hrubého čreva. Pacientov je potrebné sledovať pre prejavy a príznaky kolitídy/hnačky
a intestinálnej perforácie a majú byť manažovaní podľa odporúčaní v časti 4.2.
Imunitnepodmienené endokrinopatie
Imunitnepodmienená hypotyreóza, hypertyreózaatyreoiditída
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytla imunitne
podmienená hypotyreóza, hypertyreóza a tyreoiditída, pričom hypotyreóza môže nasledovať po hypertyreóze (pozri časť 4.8). Pacientov je potrebné sledovať pre neobvyklé výsledky testov funkcie štítnej žľazy pred začatím liečby a pravidelne počas liečby a podľa indikácie na základe klinického vyšetrenia. Imunitne podmienená hypotyreóza, hypertyreóza a tyreoiditída sa má manažovať podľa odporúčaní v časti 4.2.
Imunitne podmienená insuficiencianadobličiek
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytla imunitne
podmienená insuficiencia nadobličiek (pozri časť 4.8). Pacientov je potrebné sledovať pre klinické
prejavy a príznaky insuficiencie nadobličiek. Pri symptomatickej insuficiencii nadobličiek majú byť pacienti manažovaní podľa odporúčaní v časti 4.2.
Imunitnepodmienený diabetes mellitus1.typu
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytol imunitne
podmienený diabetes mellitus 1. typu, ktorý sa môže najskôr prejavovať diabetickou ketoacidózou,
ktorá môže byť fatálna, pokiaľ sa nezachytí včas (pozri časť 4.8). Pacientov je potrebné sledovať pre klinické prejavy a príznaky diabetu mellitus 1. typu. Pri symptomatickom diabete mellitus 1. typu majú byť pacienti manažovaní podľa odporúčaní v časti 4.2.
Imunitne podmienená hypofyzitída/hypopituitarizmus
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytla imunitne
podmienená hypofyzitída alebo hypopituitarizmus (pozri časť 4.8). Pacientov je potrebné sledovať pre klinické prejavy a príznaky hypofyzitídy alebo hypopituitarizmu. Pri symptomatickej hypofyzitíde alebo hypopituitarizme majú byť pacienti manažovaní podľa odporúčaní v časti 4.2.
I
m
u
ni
t
ne podmienenánefritída
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytla imunitne
podmienená nefritída, ktorá bola definovaná ako stav vyžadujúci použitie systémových kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu (pozri časť 4.8). Pacientov je potrebné sledovať pre neobvyklé výsledky testov funkcie obličiek pred začatím liečby a pravidelne počas liečby a majú byť manažovaní podľa odporúčaní v časti 4.2.
Imunitne podmienenávyrážka
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytla imunitne
podmienená vyrážka alebo dermatitída (vrátane pemfigoidu), ktorá bola definovaná ako stav vyžadujúci použitie systémových kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu (pozri časť 4.8). U pacientov liečených inhibítormi PD-1 a CTLA-4 sa hlásili prípady
Stevensovho-Johnsonovho syndrómu alebo toxickej epidermálnej nekrolýzy. Pacientov je potrebné sledovať pre prejavy a príznaky vyrážky alebo dermatitídy a majú byť manažovaní podľa odporúčaní v časti 4.2.
Imunitne podmienená myokarditída
U pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom sa vyskytla imunitne
podmienená myokarditída, ktorá môže byť fatálna (pozri časť 4.8). Pacientov je potrebné sledovať pre prejavy a príznaky imunitne podmienenej myokarditídy a majú byť manažovaní podľa odporúčaní
v časti 4.2.
Iné imunitne podmienenénežiaduce reakcie
Vzhľadom na mechanizmus účinku tremelimumabu v kombinácii s durvalumabom sa môžu vyskytnúť
aj iné imunitne podmienené nežiaduce reakcie. U pacientov liečených tremelimumabom v kombinácii
s durvalumabom sa pozorovali nasledujúce imunitne podmienené nežiaduce reakcie: myasténia gravis,
myozitída, polymyozitída, meningitída, encefalitída, Guillainov-Barrého syndróm, imunitná trombocytopénia, neinfekčná cystitída a pankreatitída (pozri časť 4.8). Pacientov je potrebné sledovať pre prejavy a príznaky a majú byť manažovaní podľa odporúčaní v časti 4.2.
Reakcie súvisiaces infúziou
Pacientov je potrebné sledovať pre prejavy a príznaky reakcií súvisiacich s infúziou. U pacientov
dostávajúcich tremelimumab v kombinácii s durvalumabom sa hlásili závažné reakcie súvisiace
s infúziou (pozri časť 4.8). Reakcie súvisiace s infúziou majú byť manažované podľa odporúčaní v časti 4.2.
Pacienti vylúčenízklinických štúdií
Z klinických štúdií boli vylúčení pacienti s nasledujúcimi stavmi: Childovo-Pughovo skóre B alebo C,
trombóza hlavnej portálnej žily, transplantácia pečene, nekontrolovaná hypertenzia, mozgové metastázy v anamnéze alebo v súčasnosti, kompresia miechy, koinfekcia vírusom hepatitídy B a hepatitídy C, aktívne alebo v minulosti zdokumentované gastrointestinálne (GI) krvácanie
v priebehu 12 mesiacov, ascites vyžadujúci nefarmakologickú intervenciu v priebehu 6 mesiacov, hepatálna encefalopatia v priebehu 12 mesiacov pred začatím liečby, aktívne alebo v minulosti zdokumentované autoimunitné alebo zápalové ochorenia. Vzhľadom na absenciu údajov sa má tremelimumab v týchto populáciách používať s opatrnosťou po starostlivom zvážení možného prínosu/rizika na individuálnej báze.
Obsah sodíka
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v dávke, teda v podstate zanedbateľné
množstvo sodíka.
4.
5 Liekové a iné interakcie
Pred začatím podávania tremelimumabu sa neodporúča použitie systémových kortikosteroidov alebo imunosupresív, s výnimkou fyziologickej dávky systémových kortikosteroidov (≤ 10 mg/deň prednizónu alebo ekvivalent), z dôvodu ich možnej interferencie s farmakodynamickou aktivitou
a účinnosťou tremelimumabu. Systémové kortikosteroidy alebo iné imunosupresíva sa však môžu používať po začatí podávania tremelimumabu na liečbu imunitne podmienených nežiaducich reakcií (pozri časť 4.4).
Nevykonali sa žiadne formálne farmakokinetické (FK) liekové interakčné štúdie s tremelimumabom. Vzhľadom na to, že primárnymi eliminačnými dráhami durvalumabu sú proteínový katabolizmus sprostredkovaný retikuloendoteliálnym systémom alebo cieľovo sprostredkovaná dispozícia, neočakávajú sa žiadne metabolické liekové interakcie.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku
Ženy vo fertilnom veku majú počas liečby tremelimumabom a počas minimálne 3 mesiacov po
poslednej dávke tremelimumabu používať účinnú antikoncepciu.
Gravidita
K dispozícii nie sú žiadne údaje o použití tremelimumabu u gravidných žien. Na základe svojho
mechanizmu účinku má tremelimumab potenciál ovplyvniť udržanie gravidity a pri podávaní gravidným ženám môže spôsobiť poškodenie plodu. V reprodukčných štúdiách na zvieratách sa podávanie tremelimumabu gravidným makakom počas obdobia organogenézy nespájalo s materskou toxicitou ani žiadnymi účinkami na udržanie gravidity alebo embryonálno-fetálny vývin (pozri časť
5.3). O ľudskom IgG2 je známe, že prestupuje placentárnou bariérou. Tremelimumab sa neodporúča
používať počas gravidity a u žien vo fertilnom veku, ktoré počas liečby a počas minimálne 3 mesiacov po poslednej dávke nepoužívajú účinnú antikoncepciu.
Dojčenie
K dispozícii nie sú žiadne informácie týkajúce sa prítomnosti tremelimumabu v ľudskom mlieku,
absorpcie a účinkov na dojčené dieťa alebo účinkov na tvorbu mlieka. Ľudský IgG2 sa vylučuje do
ľudského mlieka. Vzhľadom na potenciálny výskyt nežiaducich reakcií na tremelimumab u dojčených detí sa dojčiacim ženám odporúča nedojčiť počas liečby a počas minimálne 3 mesiacov po poslednej dávke.
Fertilita
K dispozícii nie sú žiadne údaje týkajúce sa možných účinkov tremelimumabu na fertilitu u ľudí alebo zvierat. V štúdiách toxicity po opakovanom podávaní sa však pozorovala mononukleárna bunková infiltrácia do prostaty a maternice (pozri časť 5.3). Klinický význam týchto zistení pre fertilitu nie je známy.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Tremelimumab nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Bezpečnosť tremelimumabu 300 mg ako jednorazovej dávky v kombinácii s durvalumabom je založená na súhrnných údajoch od 462 pacientov s HCC (HCC skupina) zo štúdie HIMALAYA
a ďalšej štúdie u pacientov s HCC, štúdie 22. Najčastejšie (> 10 %) nežiaduce reakcie boli vyrážka (32,5 %), pruritus (25,5 %), hnačka (25,3 %), abdominálna bolesť (19,7 %), zvýšená hladina aspartátaminotransferázy/ zvýšená hladina alanínaminotransferázy (18,0 %), pyrexia (13,9 %), hypotyreóza (13,0 %), kašeľ/produktívny kašeľ (10,8 %), periférny edém (10,4 %) a zvýšená hladina lipázy (10,0 %) (pozri tabuľku 3).
Najčastejšie závažné nežiaduce reakcie (NCI CTCAE ≥ 3. stupňa) sú zvýšená hladina aspartátaminotransferázy/ zvýšená hladina alanínaminotransferázy (8,9 %), zvýšená hladina lipázy (7,1 %), zvýšená hladina amylázy (4,3 %) a hnačka (3,9 %).
Najčastejšie závažné nežiaduce reakcie sú kolitída (2,6 %), hnačka (2,4 %), pneumónia (2,2 %)
a hepatitída (1,7 %).
Frekvencia ukončenia liečby z dôvodu nežiaducich reakcií je 6,5 %. Najčastejšie nežiaduce reakcie vedúce k ukončeniu liečby sú hepatitída (1,5 %) a zvýšená hladina aspartátaminotransferázy/zvýšená hladina alanínaminotransferázy (1,3 %).
Závažnosť nežiaducich liekových reakcií sa hodnotila podľa kritérií CTCAE definujúcich 1. stupeň = mierne, 2. stupeň = stredne závažné, 3. stupeň = závažné, 4. stupeň = život ohrozujúce a 5. stupeň = smrteľné.
Tabuľkový zoznamnežiaducich reakciíPokiaľ nie je uvedené inak, v tabuľke 3 je uvedený výskyt nežiaducich reakcií (adverse reaction,
ADR) u pacientov liečených tremelimumabom 300 mg v kombinácii s durvalumabom v HCC skupine
zahŕňajúcej 462 pacientov. Nežiaduce reakcie sú uvedené podľa triedy orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú ADR uvedené v poradí klesajúcej frekvencie výskytu. Príslušné kategórie frekvencie výskytu sú pre každú ADR definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až
< 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov). V rámci každej skupiny sú frekvencie výskytu ADR uvedené v poradí klesajúcej závažnosti.
| Tremelimumab 300 mg v kombinácii s durvalumabom
(n = 462)
| Nežiaduca reakcia
| Frekvencia výskytu akékoľvek stupňa
| Frekvencia výskytu 3. – 4. stupňa
| Infekcie a nákazy
| infekcie horných dýchacích ciesta
| Časté
| 39 (8,4 %)
|
|
| pneumóniab
| Časté
| 20 (4,3 %)
| Časté
| 6 (1,3 %)
| chrípka
| Časté
| 10 (2,2 %)
|
|
| infekcie zubov a mäkkých tkanív ústc
| Časté
| 6 (1,3 %)
|
|
| orálna kandidóza
| Menej časté
| 3 (0,6 %)
|
|
| Poruchy krvi a lymfatického systému
| imunitná trombocytopéniad
| Neznáme
|
|
|
| Poruchy endokrinného systému
| hypotyreózae
| Veľmi časté
| 60 (13,0 %)
|
|
| hypertyreózaf
| Časté
| 44 (9,5 %)
| Menej časté
| 1 (0,2 %)
| tyreoiditídag
| Časté
| 8 (1,7 %)
|
|
| insuficiencia nadobličiek
| Časté
| 6 (1,3 %)
| Menej časté
| 1 (0,2 %)
| hypopituitarizmus/hypofyzitída
| Menej časté
| 4 (0,9 %)
|
|
|
|
|
Tabuľka 3: Nežiaduce reakcie u pacientov s HCC liečených tremelimumabom 300 mg v kombinácii s durvalumabom
|
T
remelimuma
b 300 mg v kombinácii s durvalumabom
(
n = 462)
|
N
e
žiaduc
a reakcia
|
Frekvenci
a výskytu akékoľvek stupňa
|
Frekvencia výskytu 3. – 4. stupňa
|
diabetes insipidusd
|
Neznáme
|
|
|
|
diabetes mellitus 1. typud
|
Neznáme
|
|
|
|
Poruch
y nervového systému
|
myasténia gravis
|
Menej časté
|
2 (0,4 %)
|
|
|
meningitída
|
Menej časté
|
1 (0,2 %)
|
Menej časté
|
1 (0,2 %)
|
Guillainov-Barrého syndrómd
|
Neznáme
|
|
|
|
encefalitídad
|
Neznáme
|
|
|
|
Poruchy srdca a srdcovej činnosti
|
myokarditída
|
Menej časté
|
2 (0,4 %)
|
|
|
Po
r
uchy dýchacej sústavy, hrudníka a mediastína
|
kašeľ/produktívny kašeľ
|
Veľmi časté
|
50 (10,8 %)
|
Menej časté
|
1 (0,2 %)
|
Pneumonitídah
|
Časté
|
11 (2,4 %)
|
Menej časté
|
1 (0,2 %)
|
dysfónia
|
Menej časté
|
4 (0,9 %)
|
|
|
intersticiálna choroba pľúc
|
Menej časté
|
1 (0,2 %)
|
|
|
Poruch
y gastrointestinálneho traktu
|
hnačka
|
Veľmi časté
|
117 (25,3 %)
|
Časté
|
18 (3,9 %)
|
abdominálna bolesťi
|
Veľmi časté
|
91 (19,7 %)
|
Časté
|
10 (2,2 %)
|
zvýšená hladina lipázy
|
Časté
|
46 (10,0 %)
|
Časté
|
33 (7,1 %)
|
zvýšená hladina amylázy
|
Časté
|
41 (8,9 %)
|
Časté
|
20 (4,3 %)
|
kolitídaj
|
Časté
|
16 (3,5 %)
|
Časté
|
12 (2,6 %)
|
pankreatitídak
|
Časté
|
6 (1,3 %)
|
Menej časté
|
3 (0,6 %)
|
intestinálna perforáciad
|
Neznáme
|
|
|
|
perforácia hrubého črevad
|
Neznáme
|
|
|
|
Poruchy pečene a žlčových ciest
|
zvýšená hladina aspartátaminotransferázy/ zvýšená hladina alanínaminotransferázyl
|
Veľmi časté
|
83 (18,0 %)
|
Časté
|
41 (8,9 %)
|
hepatitídam
|
Časté
|
23 (5,0 %)
|
Časté
|
8 (1,7 %)
|
Poruchy kože a podkožného tkaniva
|
vyrážkan
|
Veľmi časté
|
150 (32,5 %)
|
Časté
|
14 (3,0 %)
|
pruritus
|
Veľmi časté
|
118 (25,5 %)
|
|
|
dermatitídao
|
Časté
|
6 (1,3 %)
|
|
|
nočné potenie
|
Časté
|
6 (1,3 %)
|
|
|
pemfigoid
|
Menej časté
|
1 (0,2 %)
|
|
|
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
myalgia
|
Časté
|
16 (3,5 %)
|
Menej časté
|
1 (0,2 %)
|
myozitída
|
Menej časté
|
3 (0,6 %)
|
Menej časté
|
1 (0,2 %)
|
polymyozitída
|
Menej časté
|
1 (0,2 %)
|
Menej časté
|
1 (0,2 %)
|
Poruchy obličiek a močových ciest
|
zvýšená hladina kreatinínu v krvi
|
Časté
|
21 (4,5 %)
|
Menej časté
|
2 (0,4 %)
|
dyzúria
|
Časté
|
7 (1,5 %)
|
|
|
nefritídap
|
Menej časté
|
3 (0,6 %)
|
Menej časté
|
2 (0,4 %)
|
neinfekčná cystitídad
|
Neznáme
|
|
|
|
Celkové poruchy a reakcie v mieste podania
|
pyrexia
|
Veľmi časté
|
64 (13,9 %)
|
Menej časté
|
1 (0,2 %)
|
periférny edémq
|
Veľmi časté
|
48 (10,4 %)
|
Menej časté
|
2 (0,4 %)
|
Úrazy, otravy a komplikácie liečebného postupu
|
reakcia súvisiaca s infúziour
|
Časté
|
6 (1,3 %)
|
|
|
a Zahŕňa nazofaryngitídu, faryngitídu, rinitídu, tracheobronchitídu a infekciu horných dýchacích ciest.
b Zahŕňa pneumóniu vyvolanú Pneumocystis jirovecii a pneumóniu.
c Zahŕňa periodontitídu, dentálnu pulpitídu, zubný absces a zubnú infekciu.
d Nežiaduca reakcia sa nepozorovala v HCC skupine, hlásila sa však u pacientov liečených durvalumabom alebo durvalumab + tremelimumab v klinických štúdiách sponzorovaných spoločnosťou AstraZeneca.
e Zahŕňa zvýšenú hladinu hormónu stimulujúceho štítnu žľazu v krvi, hypotyreózu a imunitne podmienenú
hypotyreózu.
f Zahŕňa zníženú hladinu hormónu stimulujúceho štítnu žľazu v krvi a hypertyreózu.
g Zahŕňa autoimunitnú tyreoiditídu, imunitne podmienenú tyreoiditídu, tyreoiditídu a subakútnu tyreoiditídu.
h Zahŕňa imunitne podmienenú pneumonitídu a pneumonitídu.
i Zahŕňa abdominálnu bolesť, bolesť v spodnej časti brucha, bolesť v hornej časti brucha a bolesť v bokoch.
j Zahŕňa kolitídu, enteritídu a enterokolitídu.
k Zahŕňa pankreatitídu a akútnu pankreatitídu.
l Zahŕňa zvýšenú hladinu alanínaminotransferázy, zvýšenú hladinu aspartátaminotransferázy, zvýšenú hladinu
pečeňových enzýmov a zvýšenú hladinu aminotransferáz.
m Zahŕňa autoimunitnú hepatitídu, hepatitídu, hepatocelulárne poškodenie, hepatotoxicitu a imunitne podmienenú hepatitídu.
n Zahŕňa ekzém, erytém, vyrážku, makulárnu vyrážku, makulopapulóznu vyrážku, papulóznu vyrážku a pruritickú vyrážku.
o Zahŕňa dermatitídu a imunitne podmienenú dermatitídu.
p Zahŕňa autoimunitnú nefritídu a imunitne podmienenú nefritídu.
q Zahŕňa periférny edém a periférny opuch.
r Zahŕňa reakciu súvisiacu s infúziou a urtikáriu.
Popis vybranýchnežiaducichreakcií
Údaje nižšie vyjadrujú informácie týkajúce sa významných nežiaducich reakcií pre tremelimumab
300 mg v kombinácii s durvalumabom v HCC skupine (n = 462).
Imunitne podmienená pneumonitída
V HCC skupine (n = 462) sa imunitne podmienená pneumonitída vyskytla u 6 (1,3 %) pacientov,
z toho 3. stupňa u 1 (0,2 %) pacienta a 5. stupňa (fatálna) u 1 (0,2 %) pacienta. Medián času do výskytu pneumonitídy bol 29 dní (rozsah: 5 – 774 dní). Šesť pacientov dostávalo systémové kortikosteroidy a 5 z týchto 6 pacientov dostávalo liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Jeden pacient dostával tiež iné imunosupresíva. Liečba sa ukončila u 2 pacientov. Pneumonitída sa upravila u 3 pacientov.
Imunitne podmienená hepatitída
V HCC skupine (n = 462) sa imunitne podmienená hepatitída vyskytla u 34 (7,4 %) pacientov, z toho
3. stupňa u 20 (4,3 %) pacientov, 4. stupňa u 1 (0,2 %) pacienta a 5. stupňa (fatálna) u 3 (0,6 %) pacientov. Medián času do výskytu hepatitídy bol 29 dní (rozsah: 13 – 313 dní). Všetci pacienti dostávali systémové kortikosteroidy a 32 z týchto 34 pacientov dostávalo liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Deväť pacientov dostávalo tiež iné imunosupresíva. Liečba sa ukončila u 10 pacientov. Hepatitída sa upravila u 13 pacientov.
Imunitne podmienená kolitída
V HCC skupine (n = 462) sa imunitne podmienená kolitída alebo hnačka vyskytla u 31 (6,7 %)
pacientov, z toho 3. stupňa u 17 (3,7 %) pacientov. Medián času do výskytu kolitídy alebo hnačky bol
23 dní (rozsah: 2 – 479 dní). Všetci pacienti dostávali systémové kortikosteroidy a 28 z týchto 31 pacientov dostávalo liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Štyria pacienti dostávali tiež iné imunosupresíva. Liečba sa ukončila u 5 pacientov. Kolitída alebo hnačka sa upravila u 29 pacientov.
V štúdiách nad rámec HCC skupiny sa u pacientov dostávajúcich tremelimumab v kombinácii s durvalumabom pozorovala intestinálna perforácia (zriedkavé).
Imunitne
podmienenéendokrinopatie
I
munitne podmienená hypotyreóza
V HCC skupine (n = 462) sa imunitne podmienená hypotyreóza vyskytla u 46 (10,0 %) pacientov.
Medián času do výskytu hypotyreózy bol 85 dní (rozsah: 26 – 763 dní). Jeden pacient dostával liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne).
U všetkých pacientov sa vyžadovala ďalšia liečba vrátane hormonálnej substitučnej liečby. Hypotyreóza sa upravila u 6 pacientov. U 4 pacientov imunitne podmienenej hypotyreóze predchádzala imunitne podmienená hypertyreóza.
Imunitne podmienená hypertyreóza
V HCC skupine (n = 462) sa imunitne podmienená hypertyreóza vyskytla u 21 (4,5 %) pacientov,
z toho 3. stupňa u 1 (0,2 %) pacienta. Medián času do výskytu hypertyreózy bol 30 dní (rozsah: 13 –
60 dní). Štyria pacienti dostávali systémové kortikosteroidy a všetci z týchto štyroch pacientov dostávali liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). U dvadsiatich pacientov sa vyžadovala ďalšia liečba (tiamazol, karbimazol, propyltiouracil, chloristan, blokátor kalciového kanála alebo betablokátor). Pre hypertyreózu sa liečba ukončila'
u jedného pacienta. Hypertyreóza sa upravila u 17 pacientov.
Imunitne podmienená tyreoiditída
V HCC skupine (n = 462) sa imunitne podmienená tyreoiditída vyskytla u 6 (1,3 %) pacientov. Medián času do výskytu tyreoiditídy bol 56 dní (rozsah: 7 – 84 dní). Dvaja pacienti dostávali systémové kortikosteroidy a 1 z týchto 2 pacientov dostával liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). U všetkých pacientov sa vyžadovala ďalšia liečba vrátane hormonálnej substitučnej liečby. Tyreoiditída sa upravila u 2 pacientov.
Imunitne podmienená insuficiencia nadobličiek
V HCC skupine (n = 462) sa imunitne podmienená insuficiencia nadobličiek vyskytla u 6 (1,3 %)
pacientov, z toho 3. stupňa u 1 (0,2 %) pacienta. Medián času do výskytu insuficiencie nadobličiek bol
64 dní (rozsah: 43 – 504 dní). Všetci pacienti dostávali systémové kortikosteroidy a 1 z týchto 6 pacientov dostával liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Insuficiencia nadobličiek sa upravila u 2 pacientov.
Imunitne podmienený diabetes mellitus 1. typu
V štúdiách nad rámec HCC skupiny sa u pacientov dostávajúcich tremelimumab v kombinácii
s durvalumabom pozoroval imunitne podmienený diabetes mellitus 1. typu (menej časté).
Imunitne podmienená hypofyzitída/hypopituitarizmus
V HCC skupine (n = 462) sa imunitne podmienená hypofyzitída/hypopituitarizmus vyskytli u 5
(1,1 %) pacientov. Medián času do výskytu týchto udalostí bol 149 dní (rozsah: 27 – 242 dní). Štyria pacienti dostávali systémové kortikosteroidy a 1 z týchto 4 pacientov dostával liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). U troch pacientov sa vyžadovala tiež endokrinná liečba. Hypofyzitída/hypopituitarizmus sa upravili u 2 pacientov.
Imunitne podmienená nefritída
V HCC skupine (n = 462) sa imunitne podmienená nefritída vyskytla u 4 (0,9 %) pacientov, z toho
3. stupňa u 2 (0,4 %) pacientov. Medián času do výskytu nefritídy bol 53 dní (rozsah: 26 – 242 dní). Všetci pacienti dostávali systémové kortikosteroidy a 3 z týchto 4 pacientov dostávali liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Liečba sa ukončila
u 2 pacientov. Nefritída sa upravila u 3 pacientov.
Imunitnepodmienená vyrážka
V HCC skupine (n = 462) sa imunitne podmienená vyrážka alebo dermatitída (vrátane pemfigoidu)
vyskytla u 26 (5,6 %) pacientov, z toho 3. stupňa u 9 (1,9 %) pacientov a 4. stupňa u 1 (0,2 %)
pacienta. Medián času do výskytu vyrážky alebo dermatitídy bol 25 dní (rozsah: 2 – 933 dní). Všetci pacienti dostávali systémové kortikosteroidy a 14 z týchto 26 pacientov dostávalo liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Jeden pacient dostával iné imunosupresíva. Liečba sa ukončila u 3 pacientov. Vyrážka alebo dermatitída sa upravila u 19 pacientov.
ImunogenitaRovnako, ako pri všetkých proteínoch na terapeutické účely, pri tremelimumabe existuje potenciál
imunogenity. Imunogenita tremelimumabu je založená na súhrnných údajoch u 2 075 pacientov, ktorí
boli liečení tremelimumabom v dávke 75 mg alebo 1 mg/kg a boli hodnotiteľní na prítomnosť protilátok proti liečivu (anti-drug antibodies, ADA). Dvestopäťdesiatdva pacientov (12,1 %) malo pozitívny výsledok testu na ADA vyvolané liečbou. Neutralizujúce protilátky proti tremelimumabu sa detegovali u 10,0 % (208/2 075) pacientov. Prítomnosť ADA nemala vplyv na farmakokinetiku tremelimumabu a nepozoroval sa žiadny zjavný účinok na účinnosť a bezpečnosť.
V štúdii HIMALAYA spomedzi 182 pacientov, ktorí boli liečení tremelimumabom 300 mg ako jednorazovej dávky v kombinácii s durvalumabom a boli hodnotiteľní na prítomnosť ADA proti tremelimumabu, malo 20 (11,0 %) pacientov pozitívny výsledok testu na ADA vyvolané liečbou. Neutralizujúce protilátky proti tremelimumabu sa detegovali u 4,4 % (8/182) pacientov. Prítomnosť ADA nemala zjavný účinok na farmakokinetiku alebo bezpečnosť.
Staršie osobyÚdaje u HCC pacientov vo veku 75 rokov alebo starších sú obmedzené.
Hlásenie podozrenínanežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieK dispozícii nie sú žiadne údaje o predávkovaní tremelimumabom. V prípade predávkovania je potrebné pacientov pozorne sledovať pre prejavy alebo príznaky nežiaducich reakcií a je potrebné okamžite začať vhodnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Iné monoklonálne protilátky a konjugáty protilátky s liečivom. ATC
kód: L01FX20
Mechanizmus účinkuAntigén spojený s cytotoxickými T-lymfocytmi (cytotoxic T lymphocyte-associated antigen, CTLA-4)
je primárne exprimovaný na povrchu T-lymfocytov. Interakcia CTLA-4 s jeho ligandmi, CD80
a CD86, obmedzuje aktiváciu efektorových T-buniek prostredníctvom mnohých možných mechanizmov, predovšetkým však cez obmedzenie kostimulačnej signalizácie prostredníctvom CD28.
Tremelimumab je selektívna, plne ľudská protilátka typu IgG2, ktorá blokuje interakciu CTLA-4
s CD80 a CD86, čím posilňuje aktiváciu a proliferáciu T-buniek, čo vedie k zvýšenej diverzite T- buniek a posilnenej protinádorovej aktivite.
Kombinácia tremelimumabu, inhibítora CTLA-4, a durvalumabu, inhibítora PD-L1, má za následok zlepšenie protinádorových odpovedí pri metastatickom nemalobunkovom karcinóme pľúc. Na syngénnych nádorových modeloch myší viedla duálna blokáda PD-L1 a CTLA-4 k zvýšenej protinádorovej aktivite.
Klinická účinnosť
HCC –štúdia HIMALAYA
Účinnosť IMJUDA 300 mg ako jednorazovej dávky v kombinácii s durvalumabom sa hodnotila
v štúdii HIMALAYA, randomizovanej, otvorenej, multicentrickej štúdii u pacientov s potvrdeným neresekovateľným HCC (uHCC), ktorí v minulosti nedostávali systémovú liečbu pre HCC. Do štúdie boli zahrnutí pacienti s ochorením v štádiu C alebo B podľa BCLC (Barcelona Clinic Liver Cancer) (nespôsobilí na lokoregionálnu liečbu) a Childovým-Pughovým skóre A.
Zo štúdie boli vylúčení pacienti s mozgovými metastázami alebo anamnézou mozgových metastáz, koinfekciou vírusom hepatitídy B a hepatitídy C; aktívnym alebo v minulosti zdokumentovaným gastrointestinálnym (GI) krvácaním v priebehu 12 mesiacov; ascitom vyžadujúcim nefarmakologickú intervenciu v priebehu 6 mesiacov; hepatálnou encefalopatiou v priebehu 12 mesiacov pred začatím liečby; aktívnymi alebo v minulosti zdokumentovanými autoimunitnými alebo zápalovými ochoreniami.
Zahrnutí boli aj pacienti s ezofagovými varixami s výnimkou pacientov s aktívnym alebo v minulosti zdokumentovaným GI krvácaním v priebehu 12 mesiacov pred zaradením do štúdie.
Randomizácia bola stratifikovaná podľa makrovaskulárnej invázie (MVI) (áno oproti nie), etiológie ochorenia pečene (potvrdený vírus hepatitídy B oproti potvrdený vírus hepatitídy C oproti iné)
a výkonnostného stavu podľa ECOG (0 oproti 1). V štúdii HIMALAYA bolo randomizovaných 1 171
pacientov v pomere 1:1:1 na podávanie:
· D: durvalumab 1 500 mg každé 4 týždne
· IMJUDO 300 mg ako jednorazová dávka + durvalumab 1 500 mg; následne durvalumab
1 500 mg každé 4 týždne
· S: sorafenib 400 mg dvakrát denne
Zhodnotenie stavu nádoru sa vykonávalo každých 8 týždňov počas prvých 12 mesiacov a následne každých 12 týždňov. Hodnotenie prežívania sa vykonávalo každý mesiac počas prvých 3 mesiacov po ukončení liečby a následne každé 2 mesiace.
Primárnym ukazovateľom bolo celkové prežívanie (overall survival, OS). Sekundárne ukazovatele zahŕňali prežívanie bez progresie (progression-free survival, PFS), mieru objektívnej odpovede (objective response rate, ORR) a dĺžku trvania odpovede (duration of response, DoR) na základe hodnotenia skúšajúceho podľa RECIST verzie 1.1.
Demografické charakteristiky a východiskové charakteristiky ochorenia boli medzi skupinami štúdie rovnomerne vyvážené. Východiskové demografické charakteristiky celkovej populácie štúdie boli nasledovné: muži (83,7 %), vek < 65 rokov (50,4 %), biela rasa (44,6 %), ázijská rasa (50,7 %), čierna alebo afroamerická rasa (1,7 %), iná rasa (2,3 %), výkonnostný stav podľa ECOG 0 (62,6 %); Childovo-Pughovo skóre A (99,5 %), makrovaskulárna invázia (25,2 %), extrahepatálne šírenie
(53,4 %), východisková hodnota AFP < 400 ng/ml (63,7 %), východiskové AFP ≥ 400 ng/ml
(34,5 %), vírusová etiológia: hepatitída B (30,6 %), hepatitída C (27,2 %), neinfikovaní (42,2 %), hodnotiteľné údaje PD-L1 (86,3 %), pozitivita TAP (Tumour area positivity) ≥ 1 % (38,9 %), PD-L1
TAP < 1 % (48,3 %) [analýza Ventana PD-L1 (SP263)].
Výsledky sú uvedené v tabuľke 4 a na obrázku 1.
Tabuľk
a 4: Výsledky účinnosti IMJUDA 300 mg v kombinácii s durvalumabom oproti režimu S
v štúdii HIMALAYA
|
IMJUD
O 300 mg v kombinácii
s durvalumabom
(
n = 393)
|
S
(
n = 389)
|
Dĺžka trvania sledovania
|
Medián dĺžky sledovania (mesiace)
|
33,2
|
32,2
|
OS
|
Počet úmrtí (%)
|
262 (66,7)
|
293 (75,3)
|
Med
i
án OS (mesiace) (95 % IS)
|
16,4 (14,2; 19,6)
|
13,8 (12,3; 16,1)
|
HR (95 % IS)
|
0,78 (0,66; 0,92)
|
p-hodnotab
|
0,0035
|
PFS
|
Počet udalostí (%)
|
335 (85,2)
|
327 (84,1)
|
Med
i
án PFS (mesiace)
(9
5 % IS)
|
3,78
(
3,68; 5,32)
|
4,07
(
3,75; 5,49)
|
HR (95 % IS)
|
0,90 (0,77; 1,05)
|
ORR
|
OR
R n (%)
c
|
79 (20,1)
|
20 (5,1)
|
Úplná odpoveď n (%)
|
12 (3,1)
|
0
|
Čiastočná odpoveď n (%)
|
67 (17,0)
|
20 (5,1)
|
DoR
|
Med
i
án DoR (mesiace)
|
22,3
|
18,4
|
a Vypočítané pomocou reverznej Kaplanovej-Meierovej metódy (s reverzným cenzorovacím indikátorom).
b Na základe alfa spotrebnej funkcie podľa Lana-DeMetsa (Lan-DeMets alpha spending function) s hraničným testom podľa O’Briena-Fleminga (O'Brien Fleming type boundary) a skutočným počtom pozorovaných udalostí
bola hranica pre dosiahnutie štatistickej významnosti pre IMJUDO 300 mg + durvalumab oproti režimu S 0,0398 (
Lan a DeMets, 1983).
c Potvrdená úplná odpoveď.
NR = nedosiahlo sa, IS = interval spoľahlivosti
 Obrázok 1: Kaplanova-Meierova krivka OS
Obrázok 1: Kaplanova-Meierova krivka OSMedián OS (95 % IS)
IMJUDO 300 mg
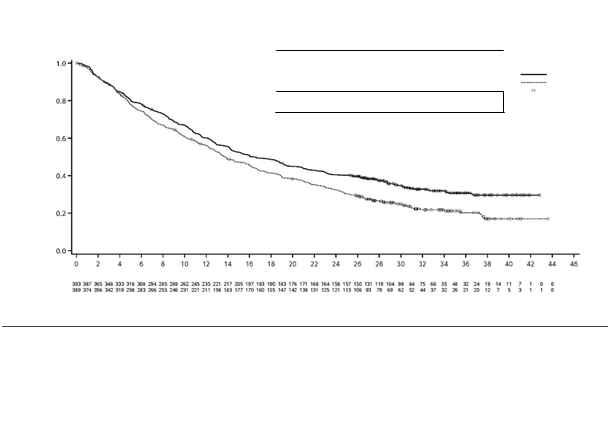
+ durvalumab
16,4 (14,2 – 19,6)
S 13,8 (12,3 – 16,1)
Pomer rizika (95 % IS) 0,78 (0,66; 0,92)
IMJUDO 300 mg + d
S Cenzorované
IMJUDO 300 mg + d
S
Čas od randomizácie (mesiace)

Pediatrická populácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s tremelimumabom vo všetkých podskupinách pediatrickej populácie v liečbe malígnych nádorov (s výnimkou nádorov
centrálneho nervového systému, nádorov hematopoetického a lymfoidného tkaniva) (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika (FK) tremelimumabu sa hodnotila pre tremelimumab v monoterapii a aj v kombinácii s durvalumabom.
FK tremelimumabu sa skúmala u pacientov pri dávkach v rozsahu od 75 mg do 750 mg alebo
10 mg/kg podávaných intravenózne jedenkrát každé 4 alebo 12 týždňov ako monoterapia alebo pri
jednorazovej dávke 300 mg. FK expozícia sa pri dávkach ≥ 75 mg zvýšila úmerne dávke (lineárna FK). Rovnovážny stav sa dosiahol približne v 12. týždni. Na základe populačnej FK analýzy, ktorá zahŕňala pacientov liečených tremelimumabom v monoterapii alebo v kombinácii s inými liekmi
s rozsahom dávky ≥ 75 mg (alebo 1 mg/kg) každé 3 alebo 4 týždne, bol odhadovaný klírens (CL)
tremelimumabu 0,309 l/deň a distribučný objem (Vd) 6,33 l. Terminálny polčas bol približne 14,2 dní.
Osobitné skupinypacientov
Vek (18 – 87 rokov), telesná hmotnosť (34 – 149 kg), pohlavie, pozitívny stav protilátok proti liečivu
(ADA), hladiny albumínu, hladiny LDH, hladiny kreatinínu, typ nádoru, rasa alebo výkonnostný stav
podľa ECOG/WHO nemali žiadny klinicky významný vplyv na FK tremelimumabu.
Pacienti s poruchou funkcieobličiek
Mierna (klírens kreatinínu (creatinine clearance, CrCL) 60 až 89 ml/min) a stredne závažná porucha
funkcie obličiek (klírens kreatinínu (CrCL) 30 až 59 ml/min) nemala žiadny klinicky významný vplyv na FK tremelimumabu. Vplyv závažnej poruchy funkcie obličiek (CrCL 15 až 29 ml/min) na FK tremelimumabu nie je známy.
Pacienti s poruchoufunkciepečene
Mierna porucha funkcie pečene (hladina bilirubínu ≤ ULN a hladina AST > ULN alebo hladina
bilirubínu > 1,0- až 1,5-násobok ULN a akákoľvek hladina AST) a stredne závažná porucha funkcie
pečene (hladina bilirubínu > 1,5- až 3-násobok ULN a akákoľvek hladina AST) nemala žiadny klinicky významný vplyv na FK tremelimumabu. Vplyv závažnej poruchy funkcie pečene (hladina bilirubínu > 3-násobok ULN a akákoľvek hladina AST) na FK tremelimumabu nie je známy; avšak, keďže monoklonálne protilátky IgG nie sú primárne eliminované prostredníctvom hepatálnych dráh, neočakáva sa, že zmena funkcie pečene ovplyvní expozíciu tremelimumabu.
5.3 Predklinické údaje o bezpečnosti
Toxikológia u zvierat
V chronickej 6-mesačnej štúdii na makakoch sa liečba tremelimumabom spájala s dávkou súvisiacim
výskytom pretrvávajúcej hnačky a kožnej vyrážky, chrást a otvorených rán, ktoré limitovali dávku. Tieto klinické prejavy sa spájali tiež so zníženou chuťou do jedla a telesnou hmotnosťou
a opuchnutými periférnymi lymfatickými uzlinami. Histopatologické nálezy korelujúce
s pozorovanými klinickými prejavmi zahŕňali reverzibilný chronický zápal slepého čreva a hrubého čreva, mononukleárnu bunkovú infiltráciu do kože a hyperpláziu v lymfoidných tkanivách.
V slinnej žľaze, pankrease (acinárny), štítnej žľaze, prištítnych telieskach, nadobličkách, srdci, pažeráku, jazyku, periportálnej oblasti pečene, kostrovom svale, prostate, maternici, hypofýze, oku (spojovka, extraokulárne svaly) a choroidálneho plexu mozgu sa pozorovalo od dávky závislé zvýšenie výskytu a závažnosti mononukleárnej bunkovej infiltrácie so zápalom mononukleárnych buniek alebo bez zápalu. V tejto štúdii sa nezistila žiadna hladina NOAEL u zvierat liečených najnižšou dávkou 5 mg/kg/týždeň; stredná dávka 15 mg/kg/týždeň sa však považovala za najvyššiu
možnú dávku, ktorá nespôsobuje závažnú toxicitu (highest non-severely toxic dose, HNSTD). Táto dávka poskytla na expozícii založenú bezpečnostnú hranicu 1,77 pre klinicky relevantnú expozíciu.
Karcinogenita a mutagenita
Karcinogénny a genotoxický potenciál tremelimumabu sa neskúmal.
Reprodukčná toxikológia
Štúdie fertility s tremelimumabom na zvieratách sa nevykonali. V štúdiách toxicity po opakovanom
podávaní sa pozorovala mononukleárna bunková infiltrácia do prostaty a maternice. Keďže štúdie fertility u zvierat sa s tremelimumabom neuskutočnili, klinický význam týchto zistení pre fertilitu nie je známy. V reprodukčných štúdiách sa podávanie tremelimumabu gravidným makakom počas obdobia organogenézy nespájalo s materskou toxicitou alebo účinkami na spontánne potraty, hmotnosť plodov alebo externé, viscerálne, skeletálne abnormality alebo hmotnosť vybraných fetálnych orgánov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
histidín
monohydrát histidínium-chloridu dihydrát trehalózy
dihydrát edetanu disodného polysorbát 80
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Neotvorená injekčnáliekovka
4 roky pri 2 °C – 8 °C.
Zriedený roztok
Chemická a fyzikálna stabilita v rámci používania sa preukázala počas 28 dní pri teplote 2 °C až 8 °C
a počas 48 hodín pri izbovej teplote (až do 25 °C) od času prípravy.
Z mikrobiologického hľadiska sa má pripravený infúzny roztok použiť okamžite. Ak sa nepoužije okamžite, za čas uchovávania v rámci používania a podmienky uchovávania pred použitím zodpovedá používateľ a zvyčajne by nemali presiahnuť 24 hodín pri teplote 2 °C až 8 °C alebo 12 hodín pri izbovej teplote (až do 25 °C), pokiaľ riedenie neprebehlo v kontrolovaných a validovaných aseptických podmienkach.
Absencia rastu mikroorganizmov v pripravenom infúznom roztoku sa preukázala počas 28 dní pri teplote 2 °C až 8 °C a počas 48 hodín pri izbovej teplote (až do 25 °C) od času prípravy.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C).
Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom. Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Dostupné sú dve balenia lieku IMJUDO:
· 1,25 ml (celkovo 25 mg tremelimumabu) koncentrátu v injekčnej liekovke zo skla typu I so zátkou z elastoméru a fialovým odklápacím hliníkovým tesnením. Veľkosť balenia s 1 jednodávkovou injekčnou liekovkou.
· 15 ml (celkovo 300 mg tremelimumabu) koncentrátu v injekčnej liekovke zo skla typu I so zátkou z elastoméru a tmavomodrým odklápacím hliníkovým tesnením. Veľkosť balenia s 1 jednodávkovou injekčnou liekovkou.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Príprava roztoku
IMJUDO sa dodáva v jednodávkovej injekčnej liekovke a neobsahuje žiadne konzervačné látky, musí
sa preto dodržiavať aseptický postup.
· Liek vizuálne skontrolujte na prítomnosť tuhých častíc a zmenu sfarbenia. IMJUDO je číry až slabo opalescenčný, bezfarebný až svetložltý roztok. Injekčnú liekovku vyraďte, ak je roztok zakalený, má zmenenú farbu alebo spozorujete viditeľné častice. Injekčnú liekovku nepretrepávajte.
· Z injekčnej liekovky (injekčných liekoviek) IMJUDO odoberte požadovaný objem a preneste ho do intravenózneho vaku obsahujúceho injekčný roztok chloridu sodného 9 mg/ml (0,9 %) alebo injekčný roztok glukózy 50 mg/ml (5 %). Zriedený roztok premiešajte opatrným prevrátením. Konečná koncentrácia zriedeného roztoku má byť v rozsahu 0,1 mg/ml
a 10 mg/ml. Roztok neuchovávajte v mrazničke ani nepretrepávajte.
· Je nevyhnutná opatrnosť na zabezpečenie sterility pripraveného roztoku.
· Po odobratí lieku z injekčnej liekovky opätovne neprepichujte injekčnú liekovku.
· Zlikvidujte akýkoľvek nepoužitý podiel, ktorý zostal v injekčnej liekovke. Podávanie
· Infúzny roztok podajte intravenózne počas 60 minút cez intravenóznu súpravu obsahujúcu
sterilný, proteíny málo viažuci, in-line filter s veľkosťou pórov 0,2 alebo 0,22 mikrometrov.
· Nepodávajte súbežne s inými liekmi cez rovnakú infúznu súpravu.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/22/1713/001 25 mg injekčná liekovka
EU/1/22/1713/002 300 mg injekčná liekovka
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu