br />
Imunitne podmienená nefritída
> 1,5-3-násobok (ULN alebo východiskovej hodnoty)
3. stupeň s hladinou kreatinínu v sére > 3- násobok východiskovej hodnoty alebo > 3-6- násobok ULN; 4.
stupeň s hladinou kreatinínu v sére > 6- násobok ULN
2. stupeň počas > 1
Oddiaľte
podanie dávky
Natrvalo ukončite liečbu
Začnite liečbu prednizónom v dávke 1 až 2 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné znižovanie dávky
Imunitne podmienená vyrážka alebo dermatitída
týždňa Oddiaľte
podanie dávky
3. stupeň
Natrvalo
Začnite liečbu
prednizónom v dávke 1
až 2 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné
Imunitne podmienená myokarditída
Imunitne podmienená
4. stupeň
2. stupeň
3. alebo 4. stupeň alebo akýkoľvek stupeň
s pozitívnou biopsiou
ukončite liečbu
Oddiaľte podanie dávkyb
Natrvalo ukončite liečbu
Oddiaľte
znižovanie dávky
Začnite liečbu prednizónom v dávke 2 až 4 mg/kg/deň alebo ekvivalent, po ktorej má nasledovať postupné znižovanie dávky
Začnite liečbu prednizónom v dávke 1

myozitída/polymyozitída 2. alebo 3. stupeň
podanie dávky
až 4 mg/kg/deň alebo ekvivalent, po ktorej má
Nežiaduc
e reakcie
Závažnos
ťa Úprava liečby
IMFINZI
Natrvalo
Liečba kortikosteroidom, pokiaľ nie je uvedené inak
nasledovať postupné
Reakcie súvisiace s infúziou
4. stupeň
1. alebo 2. stupeň
3. alebo 4. stupeň
ukončite liečbuc Prerušte infúziu alebo
znížte rýchlosť infúzie Natrvalo ukončite
liečbu
Oddiaľte
znižovanie dávky
Môžete zvážiť premedikáciu na profylaxiu následných infúznych reakcií
Infekcia 3. alebo 4. stupeň
podanie dávky až do klinickej stabilizácie
Iné imunitne podmienené nežiaduce reakcie
3. stupeň Oddiaľte podanie dávky
Natrvalo
Zvážte úvodnú dávku
1 mg/kg/deň až
4 mg/kg/deň prednizónu alebo ekvivalent, po
4. stupeň
ukončite
liečbu
ktorej má nasledovať
postupné znižovanie dávky
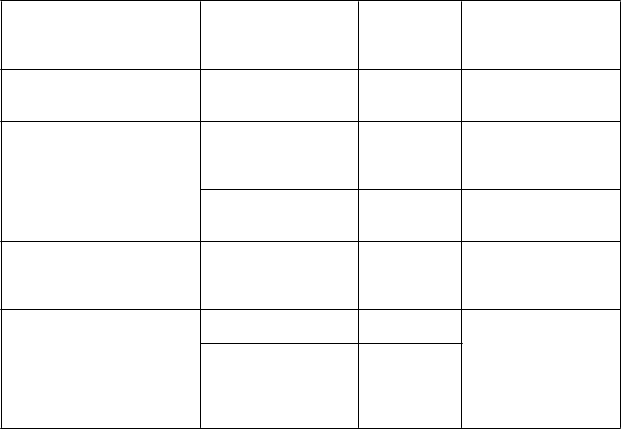
a bežné terminologické kritériá pre nežiaduce udalosti (Common Terminology Criteria for Adverse Events, CTCAE), verzia 4.03. ALT: alanínaminotransferáza; AST: aspartátaminotransferáza; ULN: horná hranica normálu.
b Ak sa odpoveď nedostaví do 3 až 5 dní napriek podaniu kortikosteroidov, okamžite začnite s doplnkovou imunosupresívnou liečbou. Po úprave (0. stupeň) sa má začať s postupným znižovaním dávky kortikosteroidu
a v jeho podávaní sa má pokračovať počas minimálne 1 mesiaca, po ktorom je možné opätovne podať
IMFINZI na základe klinického posúdenia.
c Natrvalo ukončite podávanie IMFINZI, ak nedôjde k úprave nežiaducej reakcie na ≤ 1. stupeň do 30 dní alebo ak sú prítomné prejavy respiračnej nedostatočnosti.
Pri suspektných imunitne podmienených nežiaducich reakciách sa má vykonať primerané zhodnotenie tak, aby sa potvrdila etiológia alebo vylúčili alternatívne etiológie. Ak dôjde k zhoršeniu alebo nedôjde k žiadnemu zlepšeniu, zvážte zvýšenie dávky kortikosteroidov a/alebo použitie doplnkových systémových imunosupresív. Po úprave na stupeň ≤ 1 sa má začať postupne znižovať dávka kortikosteroidu a so znižovaním sa má pokračovať počas minimálne 1 mesiaca. IMFINZI sa po oddialení podania dávky môže opätovne začať podávať do 12 týždňov, ak sa nežiaduce reakcie
upravili na stupeň ≤ 1 a ak sa dávka kortikosteroidu znížila na ≤ 10 mg prednizónu alebo ekvivalent denne. Pri opätovnom výskyte imunitne podmienených nežiaducich reakcií 3. alebo 4. stupňa (závažné alebo život ohrozujúce) sa má liečba IMFINZI natrvalo ukončiť.
Pri imunitne nepodmienených nežiaducich reakciách, zvážte oddialenie podania IMFINZI pri nežiaducich reakciách 2. a 3. stupňa až do úpravy na ≤ 1. stupeň alebo východiskový stav. Pri nežiaducich reakciách 4. stupňa (s výnimkou abnormalít laboratórnych hodnôt, pri ktorých sa má rozhodnutie o ukončení liečby vykonať na základe sprievodných klinických prejavov/príznakov a klinického posúdenia) sa má liečba IMFINZI natrvalo ukončiť.
Osobitné skupiny pacientovPediatrická populáciaBezpečnosť a účinnosť IMFINZI u detí a dospievajúcich mladších ako 18 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Staršie osoby
U starších pacientov (vo veku ≥ 65 rokov) nie je potrebná žiadna úprava dávky (pozri časť 5.1). Údaje u pacientov vo veku 75 rokov alebo starších sú obmedzené.
Porucha funkcie obličiek
U pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek sa neodporúča žiadna úprava dávky IMFINZI. Údaje u pacientov so závažnou poruchou funkcie obličiek sú príliš obmedzené na vyvodenie záverov pre túto populáciu(pozri časť 5.2).
Porucha funkcie pečene
Údaje u pacientov so stredne závažnou a závažnou poruchou funkcie pečene sú obmedzené. Z dôvodu
malého zapojenia procesov v pečeni do odbúravania durvalumabu sa u pacientov s poruchou funkcie pečene neodporúča žiadna úprava dávky IMFINZI, keďže sa neočakáva žiadny rozdiel v expozícii (pozri časť 5.2).
Spôsobpodávania
IMFINZI je určený na intravenózne použitie. Má sa podávať ako roztok na intravenóznu infúziu počas
60 minút (pozri časť 6.6).
Pokyny na riedenie lieku pred podaním, pozri časť 6.6.
4.3 Kontraindikácie
Precitlivenosť na liečivo (liečivá) alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Rozlíšiteľnosť
Na zlepšenie rozlíšiteľnosti biologických liekov sa má jasne zaznamenať obchodný názov a číslo šarže podávaného lieku.
Imunitnepodmienenápneumonitída
U pacientov dostávajúcich IMFINZI sa vyskytla imunitne podmienená pneumonitída alebo intersticiálna choroba pľúc, ktorá bola definovaná ako stav vyžadujúci použitie systémových kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu.
U pacientov podstupujúcich rádioterapiu pľúc sa často pozoruje radiačná pneumonitída a klinické prejavy pneumonitídy a radiačnej pneumonitídy sú veľmi podobné. V štúdii PACIFIC sa u pacientov, ktorí v priebehu 1 až 42 dní pred vstupom do skúšania absolvovali minimálne 2 cykly súbežnej chemoradiačnej liečby, vyskytla pneumonitída alebo radiačná pneumonitída u 161 (33,9 %) pacientov v liečebnej skupine s IMFINZI a 58 (24,8 %) v skupine s placebom, vrátane udalostí 3. stupňa (3,4 % oproti 3,0 %) a 5. stupňa (1,1 % oproti 1,7 %) (pozri časť 4.8).
Pacientov je potrebné sledovať pre prejavy a príznaky pneumonitídy alebo radiačnej pneumonitídy. Pacientov so suspektnou pneumonitídou je potrebné vyšetriť rádiografickým snímaním a majú byť manažovaní podľa odporúčaní v časti 4.2.
Imunitnepodmienenáhepatitída
U pacientov dostávajúcich IMFINZI sa vyskytla imunitne podmienená hepatitída, ktorá bola definovaná ako stav vyžadujúci použitie systémových kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu (pozri časť 4.8). Pacientov je potrebné sledovať pre neobvyklé výsledky testov funkcie pečene pred začatím liečby a pravidelne počas liečby IMFINZI a podľa indikácie na základe klinického vyšetrenia. Imunitne podmienená hepatitída sa má manažovať podľa odporúčaní v časti
4.2.
I
m
unitne
podmienená
kolitída
U pacientov dostávajúcich IMFINZI sa vyskytla imunitne podmienená kolitída alebo hnačka, ktorá bola definovaná ako stav vyžadujúci použitie systémových kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu (pozri časť 4.8). Pacientov je potrebné sledovať pre prejavy a príznaky kolitídy alebo hnačky a majú byť manažovaní podľa odporúčaní v časti 4.2.
Imunitnepodmienenéendokrinopatie
Hypotyreóza a hypertyreóza
U pacientov dostávajúcich IMFINZI sa vyskytla imunitne podmienená hypotyreóza a hypertyreóza (vrátane tyreoiditídy), pričom hypotyreóza môže nasledovať po hypertyreóze (pozri časť 4.8). Pacientov je potrebné sledovať pre neobvyklé výsledky testov funkcie štítnej žľazy pred začatím liečby a pravidelne počas liečby a podľa indikácie na základe klinického vyšetrenia. Imunitne podmienená hypotyreóza a hypertyreóza (vrátane tyreoiditídy) sa má manažovať podľa odporúčaní v časti 4.2.
Insuficiencia nadobličiek
U pacientov dostávajúcich IMFINZI sa vyskytla imunitne podmienená insuficiencia nadobličiek (pozri časť 4.8). Pacientov je potrebné sledovať pre klinické prejavy a príznaky insuficiencie nadobličiek. Pri symptomatickej insuficiencii nadobličiek majú byť pacienti manažovaní podľa odporúčaní v časti 4.2.
Diabetes mellitus 1. typu
U pacientov dostávajúcich IMFINZI sa vyskytol imunitne podmienený diabetes mellitus 1. typu (pozri časť 4.8). Pacientov je potrebné sledovať pre klinické prejavy a príznaky diabetu mellitus 1. typu. Pri symptomatickom diabete mellitus 1. typu majú byť pacienti manažovaní podľa odporúčaní v časti 4.2.
Hypofyzitída/hypopituitarizmus
U pacientov dostávajúcich IMFINZI sa vyskytla imunitne podmienená hypofyzitída alebo hypopituitarizmus (pozri časť 4.8). Pacientov je potrebné sledovať pre klinické prejavy a príznaky hypofyzitídy alebo hypopituitarizmu. Pri symptomatickej hypofyzitíde alebo hypopituitarizme majú byť pacienti manažovaní podľa odporúčaní v časti 4.2.
Imunitnepodmienenánefritída
U pacientov dostávajúcich IMFINZI sa vyskytla imunitne podmienená nefritída, ktorá bola definovaná ako stav vyžadujúci použitie systémových kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu (pozri časť 4.8). Pacientov je potrebné sledovať pre neobvyklé výsledky testov funkcie obličiek pred začatím liečby a pravidelne počas liečby IMFINZI a majú byť manažovaní podľa odporúčaní v časti 4.2.
Imunitnepodmienenávyrážka
U pacientov dostávajúcich IMFINZI sa vyskytla imunitne podmienená vyrážka alebo dermatitída, ktorá bola definovaná ako stav vyžadujúci použitie systémových kortikosteroidov a nemala žiadnu jasnú alternatívnu etiológiu (pozri časť 4.8). U pacientov liečených inhibítormi PD-1 sa hlásili prípady Stevensovho-Johnsonovho syndrómu alebo toxickej epidermálnej nekrolýzy. Pacientov je potrebné sledovať pre prejavy a príznaky vyrážky alebo dermatitídy a majú byť manažovaní podľa odporúčaní
v časti 4.2.
Inéimunitnepodmienenénežiaducereakcie
Vzhľadom na mechanizmus účinku IMFINZI sa môžu vyskytnúť aj iné imunitne podmienené
nežiaduce reakcie. Nasledujúce imunitne podmienené nežiaduce reakcie sa hlásili u menej ako 1 % pacientov liečených monoterapiou IMFINZI v klinických skúšaniach (n = 1 889): myokarditída, myozitída, polymyozitída. Pacientov je potrebné sledovať pre prejavy a príznaky a majú byť manažovaní podľa odporúčaní v časti 4.2. U pacientov v programe klinických štúdií sa hlásili prípady pankreatitídy. Pacientov je potrebné sledovať pre prejavy a príznaky a majú byť manažovaní podľa odporúčaní v časti 4.2 pre iné imunitne podmienené nežiaduce reakcie.
Reakcie
súvisiace
s infúziou
Pacientov je potrebné sledovať pre prejavy a príznaky reakcií súvisiacich s infúziou. U pacientov dostávajúcich IMFINZI sa hlásili závažné reakcie súvisiace s infúziou (pozri časť 4.8). Reakcie súvisiace s infúziou majú byť manažované podľa odporúčaní v časti 4.2.
Pacientivylúčenízklinickýchskúšaní
Zo štúdie PACIFIC boli vylúčení pacienti s nasledujúcimi stavmi: východiskové výkonnostné skóre
podľa ECOG ≥ 2; aktívne alebo v minulosti zdokumentované autoimunitné ochorenie v priebehu 2 rokov pred vstupom do štúdie; imunodeficiencia v anamnéze; závažné imunitne podmienené nežiaduce reakcie v anamnéze; zdravotné stavy vyžadujúce systémovú imunosupresiu, s výnimkou fyziologickej dávky systémových kortikosteroidov (≤ 10 mg/deň prednizónu alebo ekvivalent); aktívna tuberkulóza alebo hepatitída typu B alebo C alebo infekcia HIV alebo pacienti, ktorí dostali živú atenuovanú očkovaciu látku v priebehu 30 dní pred alebo po začiatku liečby IMFINZI. Vzhľadom na absenciu údajov sa má durvalumab v týchto populáciách používať s opatrnosťou po starostlivom zvážení možného prínosu/rizika na individuálnej báze.
4.5 Liekové a iné interakcie
Pred začatím podávania durvalumabu sa neodporúča použitie systémových kortikosteroidov alebo imunosupresív, s výnimkou fyziologickej dávky systémových kortikosteroidov (≤ 10 mg/deň prednizónu alebo ekvivalent), z dôvodu ich možnej interferencie s farmakodynamickou aktivitou
a účinnosťou durvalumabu. Systémové kortikosteroidy alebo iné imunosupresíva sa však môžu používať po začatí podávania durvalumabu na liečbu imunitne podmienených nežiaducich reakcií (pozri časť 4.4).
Nevykonali sa žiadne formálne farmakokinetické (FK) liekové interakčné štúdie s durvalumabom. Vzhľadom na to, že primárnymi eliminačnými dráhami durvalumabu sú proteínový katabolizmus sprostredkovaný retikuloendoteliálnym systémom alebo cieľovo sprostredkovaná dispozícia, neočakávajú sa žiadne metabolické liekové interakcie.
4.6 Fertilita, gravidita a laktácia
Ženyvofertilnomveku
Ženy vo fertilnom veku majú počas liečby durvalumabom a počas minimálne 3 mesiacov po poslednej
dávke durvalumabu používať účinnú antikoncepciu.
Gravidita
K dispozícii nie sú žiadne údaje o použití durvalumabu u gravidných žien. Na základe svojho
mechanizmu účinku má durvalumab potenciál ovplyvniť udržanie gravidity a v alogénnych modeloch gravidity u myší sa preukázalo, že narušenie signalizácie PD-L1 vedie k zvýšenej miere potratu plodu. Štúdie s durvalumabom na zvieratách nenaznačujú reprodukčnú toxicitu (pozri časť 5.3). O ľudskom IgG1 je známe, že prestupuje placentárnou bariérou a v štúdiách na zvieratách sa potvrdil prestup durvalumabu placentou. Durvalumab môže pri podaní gravidnej žene spôsobiť poškodenie plodu
a neodporúča sa používať počas gravidity a u žien vo fertilnom veku, ktoré počas liečby a počas minimálne 3 mesiacov po poslednej dávke nepoužívajú účinnú antikoncepciu.
Dojčenie
Nie je známe, či sa durvalumab vylučuje do ľudského materského mlieka. Dostupné toxikologické
údaje u opíc Cynomolgus preukázali nízke hladiny durvalumabu v materskom mlieku v 28. deň po pôrode (pozri časť 5.3). Protilátky môžu u ľudí prechádzať do materského mlieka, potenciál pre absorpciu a poškodenie novorodenca však nie je známy. Možné riziko u dojčeného dieťaťa však nemôže byť vylúčené. Rozhodnutie, či ukončiť dojčenie alebo ukončiť alebo prerušiť liečbu durvalumabom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
Fertilita
K dispozícii nie sú žiadne údaje týkajúce sa možných účinkov durvalumabu na fertilitu u ľudí alebo zvierat.
4.
7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Durvalumab nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrnbezpečnostnéhoprofilu
Bezpečnosť IMFINZI (10 mg/kg) bola hodnotená v štúdii PACIFIC (n = 475) u pacientov s lokálne pokročilým, neresekovateľným NSCLC, ktorí dokončili liečbu minimálne 2 cyklov súbežnej chemorádioterapie počas 1 až 42 dní pred začatím štúdie. Najčastejšími nežiaducimi reakciami v tejto populácii pacientov boli kašeľ (40,2 % oproti 30,3 % v skupine s placebom), infekcie horných dýchacích ciest (26,1 % oproti 11,5 % v skupine placebom) a vyrážka (21,7 % oproti 12,0 % v skupine s placebom). Najčastejšou nežiaducou reakciou 3.-4. stupňa bola pneumónia (6,5 % oproti 5,6 %
v skupine s placebom). Celkový výskyt nežiaducich reakcií 3. alebo 4. stupňa bol 12,8 % v skupine
IMFINZI oproti 9,8 % v skupine s placebom.
Tabuľkovýzoznamnežiaducich reakcií
V tabuľke 2 je uvedený výskyt nežiaducich reakcií u pacientov s lokálne pokročilým, neresekovateľným NSCLC v štúdii PACIFIC, na základe frekvencie výskytu daného typu nežiaducej reakcie bez ohľadu na kauzalitu hodnotenú skúšajúcim. Nežiaduce liekové reakcie sú uvedené podľa triedy orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce liekové reakcie uvedené v poradí klesajúcej frekvencie výskytu. Príslušné kategórie frekvencie výskytu sú pre každú nežiaducu liekovú reakciu definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov). V rámci každej skupiny frekvencie výskytu sú nežiaduce liekové reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 2: Nežiaduce liekové reakcie u pacientov s lokálne pokročilým, neresekovateľným
NSCLC liečených IMFINZI v dávke 10 mg/kg
akýkoľvek stupeň (%) 3.-4. stupeň (%)
Infekcie a nákazy
infekcie horných dýchacích ciesta Veľmi časté 26,1 0,4
pneumóniab,c Veľmi časté 17,1 6,5
infekcie zubov a mäkkých tkanív ústd
Časté 3,6 0
orálna kandidóza Časté 3,2 0
chrípka Časté 2,5 0
Poruchy endokrinného systému
hypotyreózae Veľmi časté 11,6 0,2
hypertyreózaf Časté 8,2 0 insuficiencia nadobličiek Menej časté 0,2 0 diabetes mellitus 1. typu Menej časté 0,2 0,2
hypofyzitída/
hypopituitarizmus
Zriedkavég < 0,1 < 0,1
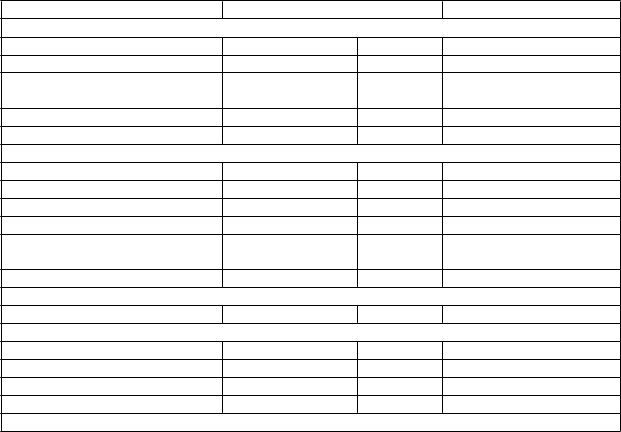
diabetes insipidus Zriedkavég < 0,1 < 0,1
Poruchy srdca a srdcovej činnostimyokarditída Zriedkavég < 0,1 < 0,1
Poruchy dýchacej sústavy, hrudníka a mediastínakašeľ/produktívny kašeľh Veľmi časté 40,2 0,6
pneumonitídab Veľmi časté 12,6 1,7
dysfónia Časté 3,8 0
intersticiálna choroba pľúc Menej časté 0,6 0
Poruchy gastrointestinálneho traktu
akýkoľve
k stupeň (%) 3.-4. stupeň (%)
hnačka Veľmi časté 18,3 0,6 abdominálna bolesťi Veľmi časté 10,1 0,4 kolitídaj Časté 1,1 0,2
Poruchy pečene a žlčových ciest
zvýšená hladina aspartátaminotransferázy alebo zvýšená hladina alanínaminotransferázyk
Časté 6,1 1,9
hepatitídac,l Menej časté 0,6 0
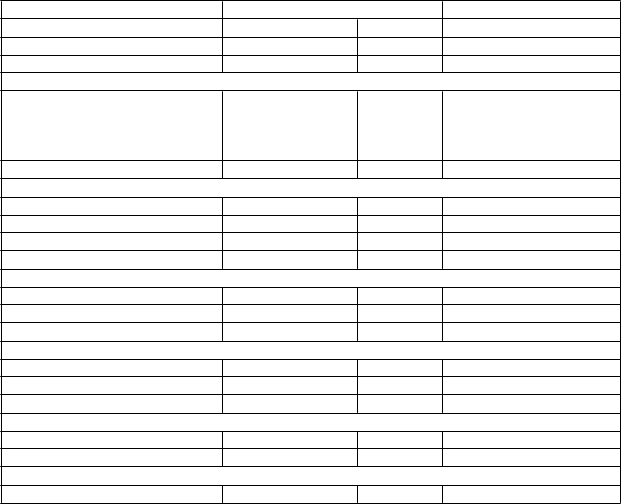
Poruchy kože a podkožného tkanivavyrážkam Veľmi časté 21,7 0,6 pruritusn Veľmi časté 12,4 0 dermatitída Časté 1,5 0 nočné potenie Časté 2,3 0
Poruchy kostrovej a svalovej sústavy a spojivového tkanivamyalgia Časté 8,0 0,2
myozitída Menej časté 0,4 0
polymyozitídac Zriedkavég < 0,1 < 0,1
Poruchy obličiek a močových ciestzvýšená hladina kreatinínu v krvi Časté 4,6 0,2 dyzúria Časté 2,3 0 nefritídao Menej časté 0,4 0
Celkové poruchy a reakcie v mieste podaniapyrexia Veľmi časté 14,7 0,2
periférny edém Časté 7,8 0
Úrazy, otravy a komplikácie liečebného postupureakcia súvisiaca s infúzioup Časté 1,9 0
a zahŕňa laryngitídu, nazofaryngitídu, peritonzilárny absces, faryngitídu, rinitídu, sinusitídu, tonzilitídu, tracheobronchitídu a infekciu horných dýchacích ciest.
b zahŕňa infekciu pľúc, pneumóniu vyvolanú
Pneumocystis jirovecii, pneumóniu, adenovírusovú pneumóniu, bakteriálnu pneumóniu, cytomegalovírusovú pneumóniu, hemofilovú pneumóniu, pneumóniu vyvolanú druhom
Klebsiella, nekrotizujúcu pneumóniu, pneumokokovú pneumóniu a streptokokovú pneumóniu.
c fatálna pneumonitída a fatálna pneumónia boli medzi liečebnou skupinou IMFINZI a skupinou s placebom
v štúdii PACIFIC hlásené v podobnej miere; fatálna hepatitída a fatálna polymyozitída boli hlásené v ostatných klinických skúšaniach.
d zahŕňa gingivitídu, infekciu úst, periodontitídu, dentálnu pulpitídu, zubný absces a zubnú infekciu.
e zahŕňa autoimunitnú hypotyreózu a hypotyreózu.
f zahŕňa hypertyreózu, autoimunitnú tyreoiditídu, tyreoiditídu, subakútnu tyreoiditídu a Basedowovu chorobu.
g frekvencia vychádza z prípadov, ktoré sa nepozorovali v štúdii PACIFIC, ale pozorovali v ostatných klinických skúšaniach (n = 1 889).
h zahŕňa kašeľ a produktívny kašeľ.
i zahŕňa abdominálnu bolesť, bolesť v spodnej časti brucha, bolesť v hornej časti brucha a bolesť v bokoch.
j zahŕňa kolitídu, enteritídu, enterokolitídu a proktitídu.
k zahŕňa zvýšenú hladinu alanínaminotransferázy, zvýšenú hladinu aspartátaminotransferázy, zvýšenú hladinu pečeňového enzýmu a zvýšenú hladinu aminotransferáz.
l zahŕňa hepatitídu, autoimunitnú hepatitídu, toxickú hepatitídu, hepatocelulárne poškodenie, akútnu hepatitídu a hepatotoxicitu.
m zahŕňa erytematóznu vyrážku, generalizovanú vyrážku, makulárnu vyrážku, makulopapulárnu vyrážku, papulárnu vyrážku, pruritickú vyrážku, pustulárnu vyrážku, erytém, ekzém a vyrážku.
n zahŕňa generalizovaný pruritus a pruritus.
o zahŕňa autoimunitnú nefritídu, tubulointersticiálnu nefritídu, nefritídu, glomerulonefritídu a membranóznu glomerulonefritídu.
p zahŕňa reakciu súvisiacu s infúziou a urtikáriu s nástupom v deň podania dávky alebo 1 deň po podaní dávky.
PopisvybranýchnežiaducichreakciíIMFINZI sa najčastejšie spája s imunitne podmienenými nežiaducimi reakciami. Väčšina z nich,
liečby IMFINZI. Údaje k nasledujúcim imunitne podmieneným nežiaducim reakciám odrážajú údaje zo spojenej bezpečnostnej databázy 1 889 pacientov, ktoré zahŕňajú štúdiu PACIFIC a dve ďalšie štúdie (otvorené klinické skúšanie s viacerými liečebnými skupinami u pacientov s pokročilými solídnymi nádormi a otvorená štúdia u pacientov s lokálne pokročilým alebo metastatickým NSCLC). V rámci všetkých štúdií sa IMFINZI podával v dávke 10 mg/kg každé dva týždne. Usmernenia
k manažmentu týchto nežiaducich reakcií sú uvedené v časti 4.4.
Imunitne podmienená pneumonitída
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI (n = 1 889 s viacerými typmi nádorov)
sa imunitne podmienená pneumonitída vyskytla u 79 (4,2 %) pacientov vrátane pneumonitídy
3. stupňa u 12 (0,6 %) pacientov, pneumonitídy 4. stupňa u 1 (< 0,1 %) pacienta a pneumonitídy 5. stupňa u 5 (0,3 %) pacientov. Medián času do výskytu pneumonitídy bol 53 dní (rozsah: 1 – 341 dní). Štyridsaťpäť zo 79 pacientov dostávalo liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne) a 2 pacienti dostávali tiež liečbu infliximabom. Liečba IMFINZI sa ukončila u 26 pacientov. Pneumonitída sa upravila u 42 pacientov.
V štúdii PACIFIC sa imunitne podmienená pneumonitída vyskytla častejšie u pacientov, ktorí absolvovali súbežnú chemoradiačnú liečbu v priebehu 1 až 42 dní pred vstupom do štúdie (10,7 %) ako u iných pacientov v spojenej bezpečnostnej databáze (2,0 %).
V štúdii PACIFIC (n = 475 v skupine s IMFINZI a n = 234 v skupine s placebom) sa imunitne podmienená pneumonitída vyskytla u 51 (10,7 %) pacientov v liečebnej skupine s IMFINZI a u 16 (6,8 %) pacientov v skupine s placebom, vrátane pneumonitídy 3. stupňa u 8 (1,7 %) pacientov
liečených IMFINZI oproti 6 (2,6 %) pacientom užívajúcim placebo a pneumonitídy 5. stupňa (fatálna) u 4 (0,8 %) pacientov liečených IMFINZI oproti 3 (1,3 %) pacientom užívajúcim placebo. Medián času do výskytu pneumonitídy bol v liečebnej skupine s IMFINZI 53 dní (rozsah: 1 – 341 dní) oproti
55,5 dní (rozsah: 0 – 231 dní) v skupine s placebom. V liečebnej skupine s IMFINZI dostávalo 44 z 51 pacientov systémové kortikosteroidy, vrátane 28 pacientov, ktorí dostávali liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne) a 2 pacienti dostávali tiež liečbu infliximabom. V skupine s placebom dostávalo 11 zo 16 pacientov systémové kortikosteroidy, vrátane 9 pacientov, ktorí dostávali liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Pneumonitída sa upravila u 27 pacientov v liečebnej skupine
s IMFINZI oproti 6 pacientom v skupine s placebom.
Imunitne podmienená hepatitída
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa imunitne podmienená hepatitída
vyskytla u 19 (1,0 %) pacientov, vrátane hepatitídy 3. stupňa u 11 (0,6 %) pacientov a hepatitídy 5. stupňa (fatálna) u 1 (< 0,1 %) pacienta. Medián času do výskytu hepatitídy bol 70 dní (rozsah: 15 –
312 dní). Trinásť z 19 pacientov dostávalo liečbu vysokými dávkami kortikosteroidov (minimálne
40 mg prednizónu alebo ekvivalent denne). Jeden pacient dostával tiež liečbu mykofenolátom. Liečba
IMFINZI sa ukončila u 4 pacientov. Hepatitída sa upravila u 13 pacientov.
Imunitne podmienená kolitída
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa imunitne podmienená kolitída alebo
hnačka vyskytla u 31 (1,6 %) pacientov, vrátane kolitídy alebo hnačky 3. stupňa u 6 (0,3 %) pacientov a kolitídy alebo hnačky 4. stupňa u 1 (< 0,1 %) pacienta. Medián času do výskytu kolitídy alebo hnačky bol 74 dní (rozsah: 1 – 365 dní). Šestnásť z 31 pacientov dostávalo liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Jeden pacient dostával tiež liečbu infliximabom. Liečba IMFINZI sa ukončila u 8 pacientov. Kolitída alebo hnačka sa upravila
u 23 pacientov.
Imunitne podmienené endokrinopatie
Hypotyreóza
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa imunitne podmienená hypotyreóza
vyskytla u 137 (7,3 %) pacientov, vrátane hypotyreózy 3. stupňa u 1 (< 0,1 %) pacienta. Medián času do výskytu hypotyreózy bol 85 dní (rozsah: 9 – 378 dní). Spomedzi 137 pacientov, 134 pacientov dostávalo hormonálnu substitučnú liečbu a dvaja pacienti dostávali na liečbu hypotyreózy vysoké
dávky kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne) nasledované hormonálnou substitúciou. Pre hypotyreózu sa liečba IMFINZI neukončila u žiadneho pacienta.
Hypertyreóza
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa imunitne podmienená hypertyreóza vyskytla u 34 (1,8 %) pacientov, pričom sa nevyskytli žiadne prípady hypotyreózy 3. alebo 4. stupňa.
Medián času do výskytu hypertyreózy bol 41 dní (rozsah: 14 – 195 dní). Dvadsaťšesť z 34 pacientov dostávalo medikamentóznu liečbu (tiamazol, karbimazol, propyltiouracil alebo betablokátor), 12 pacientov dostávalo liečbu tyroxínom, keď hypertyreóza prešla do hypotyreózy, 12 pacientov
dostávalo systémové kortikosteroidy a 3 z 12 pacientov dostávali liečbu vysokými dávkami systémových kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Pre hypertyreózu sa liečba IMFINZI neukončila u žiadneho pacienta. U ôsmich pacientov sa po hypertyreóze vyskytla hypotyreóza.
Insuficiencia nadobličiek
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa imunitne podmienená insuficiencia
nadobličiek vyskytla u 7 (0,4 %) pacientov, vrátane insuficiencie nadobličiek 3. stupňa u 1 (< 0,1 %) pacienta. Medián času do výskytu insuficiencie nadobličiek bol 141 dní (rozsah: 70 – 265 dní). Všetkých 7 pacientov dostávalo systémové kortikosteroidy; 2 zo 7 pacientov dostávali liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Pre insuficienciu nadobličiek sa liečba IMFINZI neukončila u žiadneho pacienta. Insuficiencia nadobličiek sa upravila u 1 pacienta.
Diabetes mellitus 1. typu
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa imunitne podmienený diabetes mellitus 1. typu vyskytol u 1 (< 0,1 %) pacienta (3. stupeň). Liečba IMFINZI sa pre diabetes mellitus
1. typu v tomto prípade ukončila. Tento 1 pacient dostával liečbu inzulínom.
Hypofyzitída/hypopituitarizmus
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa imunitne podmienený hypopituitarizmus vyskytol u 1 (< 0,1 %) pacienta (3. stupeň). Tento 1 pacient dostával liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne) a liečba IMFINZI sa v tomto prípade neukončila.
Imunitne podmienená nefritída
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa imunitne podmienená nefritída
vyskytla u 3 (0,2 %) pacientov, vrátane nefritídy 3. stupňa u 1 (< 0,1 %) pacienta. Medián času do výskytu nefritídy bol 95 dní (rozsah: 28 – 239 dní). Dvaja (0,1 %) pacienti dostávali liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Liečba IMFINZI sa ukončila u všetkých 3 pacientov. Nefritída sa upravila u 2 pacientov.
Imunitne podmienená vyrážka
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa imunitne podmienená vyrážka alebo
dermatitída vyskytla u 30 (1,6 %) pacientov, vrátane vyrážky alebo dermatitídy 3. stupňa u 7 (0,4 %) pacientov. Medián času do výskytu vyrážky alebo dermatitídy bol 74 dní (rozsah: 1 – 365 dní). Jedenásť z 30 pacientov dostávalo liečbu vysokými dávkami kortikosteroidov (minimálne 40 mg prednizónu alebo ekvivalent denne). Liečba IMFINZI sa ukončila u 2 pacientov. Vyrážka alebo dermatitída sa upravila u 18 pacientov.
Reakcie súvisiace s infúziou
V spojenej bezpečnostnej databáze s monoterapiou IMFINZI sa reakcie súvisiace s infúziou vyskytli
u 35 (1,9 %) pacientov, vrátane reakcií 3. stupňa u 5 (0,3 %) pacientov.
Abnormality laboratórnych hodnôt
V rámci pacientov liečených durvalumabom v štúdii PACIFIC bol podiel pacientov, u ktorých sa vyskytlo zhoršenie abnormalít laboratórnych hodnôt oproti východiskovým hodnotám, nasledovný:
38,5 % (všetky stupne), 2,3 % (3. – 4. stupeň) pre zvýšenú hladinu alanínaminotransferázy, 36,0 %
(všetky stupne), 2,8 % (3. – 4. stupeň) pre zvýšenú hladinu aspartátaminotransferázy, 16,3 % (všetky stupne) pre zvýšenú hladinu kreatinínu, 26,5 % (všetky stupne) pre zvýšenú hladinu TSH > ULN
a viac ako východisková hladina, 31,9 % (všetky stupne) pre zníženú hladinu TSH < LLN a menej ako východisková hladina.
ImunogenitaSpomedzi 1 570 pacientov, ktorí boli liečení IMFINZI v dávke 10 mg/kg každé 2 týždne a boli
hodnotiteľní na prítomnosť protilátok proti lieku (anti-drug antibodies, ADA), bol výsledok na ADA vyvolané liečbou pozitívny u 2,9 % (45/1 570) pacientov. Neutralizujúce protilátky (neutralizing antibodies, nAb) proti durvalumabu sa detegovali u 0,5 % (8/1 570) pacientov. Prítomnosť ADA nemala klinicky významný vplyv na bezpečnosť. K dispozícii nie je dostatočný počet pacientov na stanovenie vplyvu ADA na účinnosť. Na základe populačnej FK analýzy sa u pacientov pozitívnych na prítomnosť ADA očakáva mierne nižšia expozícia, zníženie FK expozície je však menej ako 30 % v porovnaní s typickým pacientom a nepovažuje sa za klinicky významné.
StaršieosobyMedzi staršími (≥ 65 rokov) a mladšími pacientmi sa nehlásili žiadne celkové rozdiely v bezpečnosti. Údaje u pacientov s NSCLC vo veku 75 rokov alebo starších sú obmedzené.
Hláseniepodozrenínanežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieK dispozícii nie sú žiadne údaje o predávkovaní durvalumabom. V prípade predávkovania je potrebné pacientov pozorne sledovať pre prejavy alebo príznaky nežiaducich reakcií a je potrebné okamžite začať vhodnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: cytostatiká, monoklonálne protilátky. ATC kód: L01XC28.
Mechanizmus účinkuExpresia proteínu predstavujúceho ligand receptora programovanej bunkovej smrti 1 (programmed
cell death ligand-1, PD-L1) je adaptívnou imunitnou odpoveďou, ktorá nádorom pomáha uniknúť pred odhalením a elimináciou imunitným systémom. PD-L1 môže byť indukovaný zápalovými signálmi (napr. interferón gama) a môže byť exprimovaný na nádorových bunkách aj na imunitných bunkách spojených s nádorom v mikroprostredí nádoru. PD-L1 blokuje funkciu a aktiváciu T-buniek prostredníctvom interakcie s PD-1 a CD80 (B7.1). Väzbou na svoje receptory PD-L1 znižuje aktivitu, proliferáciu a produkciu cytokínov cytoxických T-buniek.
Durvalumab je plne ľudská monoklonálna protilátka typu imunoglobulínu G1 kapa (IgG1κ), ktorá selektívne blokuje interakciu PD-L1 s PD-1 a CD80 (B7.1). Durvalumab neindukuje od protilátok závislú bunkovú cytotoxicitu (antibody dependent cell-mediated cytotoxicity, ADCC). Selektívna blokáda interakcií PD-L1/PD-1 a PD-L1/CD80 posilňuje protinádorové imunitné odpovede a zvyšuje aktiváciu T-buniek.
Klinickáúčinnosťa bezpečnosťÚčinnosť IMFINZI sa skúmala v štúdii PACIFIC, randomizovanej, dvojito zaslepenej, placebom kontrolovanej multicentrickej štúdii u 713 pacientov s lokálne pokročilým, neresekovateľným NSCLC. Pacienti absolvovali minimálne 2 cykly zvolenej chemoterapie na báze platiny spolu
s rádioterapiou v priebehu 1 až 42 dní pred vstupom do štúdie a mali výkonnostný stav podľa ECOG 0
alebo 1. Deväťdesiatdva percent pacientov dostalo celkovú dávku ožiarenia 54 až 66 Gy. Zo štúdie boli vylúčení pacienti, u ktorých došlo k progresii po chemoradiačnej liečbe, pacienti
s predchádzajúcou expozíciou akejkoľvek anti-PD-1 alebo anti-PD-L1 protilátke, pacienti s aktívnym alebo v minulosti zdokumentovaným autoimunitným ochorením v priebehu 2 rokov pred vstupom do štúdie; s imunodeficienciou v anamnéze; so závažnými imunitne podmienenými nežiaducimi reakciami v anamnéze; so zdravotnými stavmi vyžadujúcimi systémovú imunosupresiu, s výnimkou fyziologickej dávky systémových kortikosteroidov; s aktívnou tuberkulózou alebo hepatitídou typuB alebo C alebo infekciou HIV alebo pacienti, ktorí dostali živú atenuovanú očkovaciu látku v priebehu
30 dní pred alebo po začiatku liečby IMFINZI. Pacienti boli randomizovaní v pomere 2:1 na podávanie IMFINZI v dávke 10 mg/kg (n = 476) alebo placeba v dávke 10 mg/kg (n = 237) vo forme intravenóznej infúzie každé 2 týždne až počas 12 mesiacov alebo do neakceptovateľnej toxicity, či potvrdenej progresie ochorenia. Randomizácia bola stratifikovaná podľa pohlavia, veku (< 65 rokov oproti ≥ 65 rokov) a fajčenia (fajčiar oproti nefajčiarovi). Pacienti s kontrolovaným ochorením po 12 mesiacoch mali možnosť byť po progresii ochorenia opätovne liečení. Zhodnotenie stavu nádoru sa vykonávalo každých 8 týždňov počas prvých 12 mesiacov a následne každých 12 týždňov.
Pacienti boli zaradení bez ohľadu na úroveň expresie nádorovej PD-L1. Ak boli k dispozícii archívne vzorky nádorového tkaniva odobratého pred chemoradiačnou liečbou, retrospektívne sa analyzovali na expresiu PD-L1 na nádorových bunkách použitím metódy VENTANA PD-L1 (SP263) IHC. Zo 713 randomizovaných pacientov poskytlo 63 % pacientov vzorku tkaniva, ktorá mala dostatočná kvalitu a kvantitu na stanovenie expresie PD-L1 a u 37 % bola expresia neznáma.
Demografické charakteristiky a východiskové charakteristiky ochorenia boli medzi skupinami štúdie rovnomerne vyvážené. Východiskové demografické charakteristiky celkovej populácie štúdie boli nasledovné: muži (70 %), vek ≥ 65 rokov (45 %), vek ≥ 75 rokov (8 %), biela rasa (69 %), ázijská rasa (27 %), iná rasa (4 %), fajčiar v súčasnosti (16 %), bývalý fajčiar (75 %), nefajčiar (9 %), výkonnostný stav podľa ECOG 0 (49 %), výkonnostný stav podľa ECOG 1 (51 %). Charakteristiky ochorenia boli nasledovné: štádium IIIA (53 %), štádium IIIB (45 %), histologický podtyp skvamózny (46 %), neskvamózny (54 %). U 451 pacientov s dostupnou expresiou PD-L1 bolo 67 % s TC ≥ 1 % [PD-L1'
TC 1-24 % (32 %), PD-L1 TC ≥ 25 % (35 %)] a 33 % bolo TC < 1 %.
Dvomi primárnymi ukazovateľmi štúdie boli prežívanie bez progresie (progression-free survival, PFS) a celkové prežívanie (overall survival, OS) pri IMFINZI oproti placebu. Sekundárne ukazovatele účinnosti zahŕňali PFS po 12 mesiacoch (PFS 12) a 18 mesiacoch (PFS 18) od randomizácie a čas od randomizácie do druhej progresie (time from randomisation to second progression, PFS2). PFS sa posudzovalo zaslepeným nezávislým centrálnym hodnotením (blinded independent central review, BICR) podľa RECIST verzie 1.1.
Štúdia preukázala štatisticky významné zlepšenie PFS v liečebnej skupine s IMFINZI v porovnaní so skupinou s placebom [pomer rizika (hazard ratio, HR) = 0,52 (95% IS: 0,42; 0,65), p < 0,0001]. Štúdia preukázala štatisticky významné zlepšenie OS v liečebnej skupine s IMFINZI v porovnaní so
skupinou s placebom [HR = 0,68 (95% IS: 0,53; 0,87), p = 0,00251]. Pozri tabuľku 3 a obrázky 1 a 2.
Tabuľka 3: Výsledky účinnostivštúdiiPACIFICa
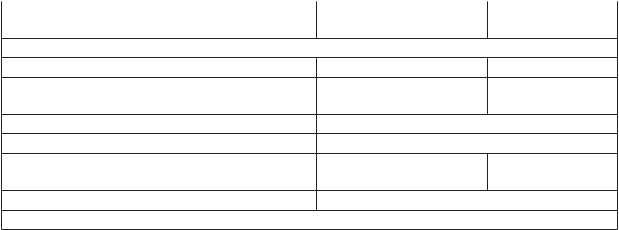 IMFINZI
(
n = 476)
OS
Placebo
(
n = 237)
IMFINZI
(
n = 476)
OS
Placebo
(
n = 237)
Počet úmrtí (%) 183 (38,4 %) 116 (48,9 %)
Medián (mesiace) (95% IS)
NR (34,7; NR)
28,7 (22,9; NR)
HR (95% IS) 0,68 (0,53; 0,87)
2-stranná p-hodnota 0,00251
OS po 24 mesiacoch (%) (95% IS)
66,3 %
(61,7 %; 70,4 %)
55,6 %
(48,9 %; 61,3 %)
p-hodnota 0,005
PFS
IMFINZI
(
n = 476)
Placebo
(
n = 237)
Počet prípadov (%) 214 (45,0 %) 157 (66,2 %)
Medián PFS (mesiace) (95% IS)
16,8 (13,0; 18,1)
5,6 (4,6; 7,8)
HR (95% IS) 0,52 (0,42; 0,65)
p-hodnota p < 0,0001
PF
S po 12 mesiacoch (%) (95% IS)
PF
S po 18 mesiacoch (%) (95% IS)
PFS2
Mediá
n PFS2
b
(mesiace)
(95% IS)
55,9 %
(51,0 %; 60,4 %)
44,2 %
(37,7 %; 50,5 %)
28,3 (25,1; 34,7)
35,3 %
(29,0 %; 41,7 %)
27,0 %
(19,9 %; 34,5 %)
17,1 (14,5; 20,7)
HR (95% IS) 0,58 (0,46; 0,73)
p-hodnota p < 0,0001
a Analýza OS bola vykonaná približne 13 mesiacov po primárnej analýze PFS.
b PFS2 je definované ako čas od dátumu randomizácie až do dátumu druhej progresie (definovanej podľa miestnej štandardnej klinickej praxe) alebo úmrtia.
NR: nedosiahol sa
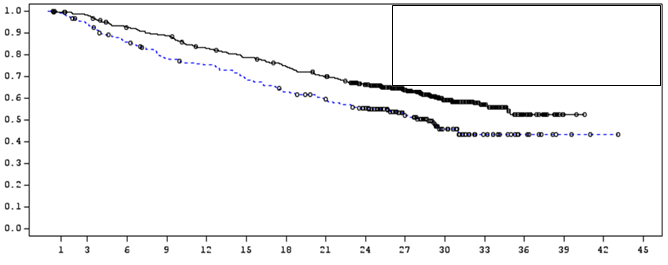
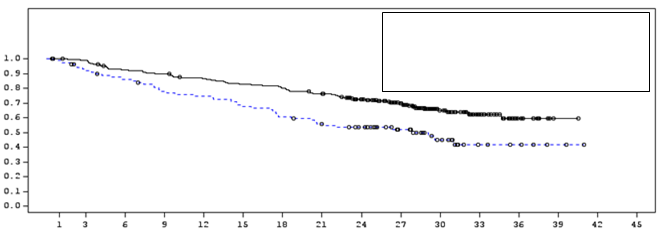
Obrázok 1: Kaplanova-Meierova krivka OSMedián OS (95 % IS)
IMFINZI NR (34,7; NR) Placebo 28,7 (22,9; NR)
Pomer rizika (95 % IS): 0,68 (0,53; 0,87)
IMFINZI
Placebo
Čas od randomizácie
Počet pacientov v riziku
Mesiac 0 3 6 9 12 15 18 21 24 27 30 33 36 39 42 45
IMFINZI 476 464 431 415 385 364 343 319 274 210 115 57 23 2 0 0
Placebo 237 220 198 178 170 155 141 130 117 78 42 21 9 3 1 0
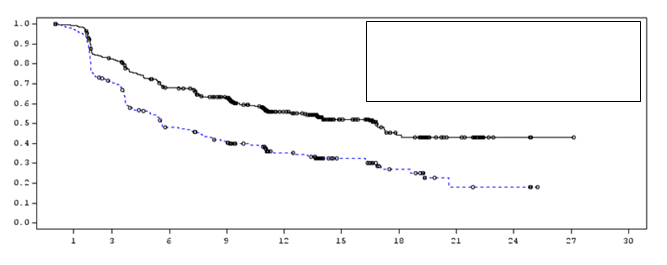
 Obrázok 2: Kaplanova-Meierova krivka PFS
Obrázok 2: Kaplanova-Meierova krivka PFS
Medián PFS (95 % IS)
IMFINZI 16,8 (13,0; 18,1) Placebo 5,6 (4,6; 7,8)
Pomer rizika (95 % IS): 0,52 (0,42; 0,65)
IMFINZI Placebo
Počet pacientov v riziku
Čas od randomizácie (mesiace)

Mesiac 0 3 6 9 12 15 18 21 24 27 30
IMFINZI 476 377 301 264 159 86 44 21 4 1 0
Placebo 237 163 106 87 52 28 15 4 3 0 0
Zlepšenia PFS a OS v prospech pacientov dostávajúcich IMFINZI v porovnaní s pacientmi dostávajúcimi placebo boli pozorované konzistentne vo všetkých vopred definovaných analyzovaných podskupinách, vrátane etnickej príslušnosti, veku, pohlavia, fajčenia v anamnéze, stavu mutácie EGFR a histológie.
Post-hoc analýzy podskupiny na základe expresie PD-L1Na vyhodnotenie účinnosti na základe expresie PD-L1 nádorovými bunkami (≥ 25 %, 1-24 %, ≥ 1 %,
< 1 %) a u pacientov, u ktorých nebolo možné stanoviť stav PD-L1 (PD-L1 neznáme) boli vykonané doplňujúce analýzy podskupiny. Výsledky PFS a OS sú zhrnuté na obrázkoch č. 3, 4, 5 a 6.
Obrázok 3: Kaplanova-Meierova krivka OS pre PD-L1 TC ≥ 1 %Medián OS (95 % IS)
IMFINZI NR (NR, NR) Placebo 29,1 (17,7; NR)
Pomer rizika (95 % IS): 0,53 (0,36; 0,77)
IMFINZI
Placebo

Počet pacientov v riziku
Čas od randomizácie (mesiace)

Mesiac 0 3 6 9 12 15 18 21 24 27 30 33 36 39 42 45
IMFINZI 212 208 193 187 178 171 165 156 134 105 62 34 12 1 0 0
Placebo 91 81 75 67 64 58 52 46 41 29 17 7 5 2 0 0
Obrázok 4: Kaplanova-Meierova krivka PFS pre PD-L1 TC ≥ 1 %
Medián PFS (95 % IS)
IMFINZI 17,8 (16,9; NR) Placebo 5,6 (3,6; 11,0)

Pomer rizika (95 % IS): 0,46 (0,33; 0,64)
IMFINZI Placebo
Čas od randomizácie (mesiace)
Počet pacientov v riziku
Mesiac 0 3 6 9 12 15 18 21 24 27
IMFINZI 212 174 143 127 82 52 30 14 1 0
Placebo 91 59 39 34 20 13 8 4 3 0
Obrázok 5. Stromový graf (forest plot) OS podľa expresie PD-L1
Všetci pacienti PD-L1 TC ≥ 1 % PD-L1 TC ≥ 25 % PD-L1 TC 1-24 % PD-L1 TC < 1 % PD-L1 neznáme
Prípady/n (%)
IMFINZI Placebo HR (95 % IS)
183/476 (38,4 %) 116/237 (48,9 %) 0,68 (0,53; 0,87)
70/212 (33,0 %) 45/91 (49,5 %) 0,53 (0,36; 0,77)
37/115 (32,2 %) 23/44 (52,3 %) 0,46 (0,27; 0,78)
33/97 (34,0 %) 22/47 (46,8 %) 0,60 (0,35; 1,03)
41/90 (45,6 %) 19/58 (32,8 %) 1,36 (0,79; 2,34)
72/174 (41,4 %) 52/88 (59,1 %) 0,62 (0,43; 0,89)
 Obrázok 6: Stromový graf (forest plot) PFS podľa expresie PD-L1
Obrázok 6: Stromový graf (forest plot) PFS podľa expresie PD-L1
Všetci pacienti PD-L1 TC ≥ 1% PD-L1 TC ≥ 25% PD-L1 TC 1-24% PD-L1 TC < 1%
PD-L1 neznáme
Prípady/n (%)IMFINZI Placebo HR (95%, IS)214/476 (45,0 %) 157/237 (66,2 %) 0,52 (0,42; 0,65)
84/212 (39,6 %) 59/91 (64,8 %) 0,46 (0,33; 0,64)
48/115 (41,7 %) 31/44 (70,5 %) 0,41 (0,26; 0,65)
36/97 (37,1 %) 28/47 (59,6 %) 0,49 (0,30; 0,80)
49/90 (54,4 %) 40/58 (69,0 %) 0,73 (0,48; 1,11)
81/174 (46,6 %) 58/88 (65,9 %) 0,59 (0,42; 0,83)
Celkovo bol profil bezpečnosti durvalumabu v podskupine PD-L1 TC ≥ 1 % konzistentný s
populáciou so zámerom liečiť, rovnako ako v podskupine PD-L1 TC < 1 %.
Výsledky hlásené pacientom
Príznaky hlásené pacientom, aktivita a kvalita života súvisiaca so zdravím (HRQoL) boli zozbierané s použitím dotazníka EORTC QLQ-C30 a jeho modulu pre karcinóm pľúc (EORTC QLQ-LC13). Dotazníky LC13 a C30 sa hodnotili na začiatku, každé 4 týždne počas prvých 8 týždňov, následne každých 8 týždňov až do uplynutia obdobia liečby alebo do ukončenia liečby IMFINZI kvôli toxicite alebo progresii ochorenia. Dodržiavanie (compliance) bolo podobné medzi liečebnými skupinami
s IMFINZI a placebom (83 % oproti 85,1 % vyplnených hodnotiteľných dotazníkov celkovo).
Medzi skupinami s IMFINZI a placebom sa východiskovo nepozorovali žiadne rozdiely v príznakoch hlásených pacientom, aktivite a HRQoL. Počas trvania štúdie až do 48. týždňa sa medzi skupinami
s IMFINZI a placebom nepozorovali žiadne klinicky významné rozdiely v príznakoch, aktivite
a HRQoL (na základe hodnotenia rozdielu ≥ 10 bodov).
Pediatrickápopulácia
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s durvalumabom vo všetkých podskupinách pediatrickej populácie v liečbe malígnych nádorov (s výnimkou nádorov centrálneho nervového systému, nádorov hematopoetického a lymfoidného tkaniva) (informácie
o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
FK durvalumabu sa skúmala u 1 902 pacientov so solídnymi nádormi pri dávkach siahajúcich od 0,1 do 20 mg/kg podávaných intravenózne jedenkrát každé dva, tri alebo štyri týždne. FK expozícia sa pri dávkach < 3 mg/kg zvýšila viac ako úmerne dávke (nelineárna FK) a pri dávkach ≥ 3 mg/kg sa zvýšila úmerne dávke (lineárna FK). Rovnovážny stav sa dosiahol približne v 16. týždni. Na základe populačnej FK analýzy, ktorá zahŕňala 1 878 pacientov s rozsahom dávky ≥ 10 mg/kg každé 2 týždne, bol geometrický priemer distribučného objemu v rovnovážnom stave (Vss) 5,64 l. Klírens (clearance, CL) durvalumabu sa v priebehu času znížil, čo viedlo ku geometrickému priemeru klírensu
v rovnovážnom stave (CLss) v hodnote 8,16 ml/h v 365. deň; zníženie CLss nebolo považované za
klinicky významné. Terminálny biologický polčas (t1/2) bol na základe východiskového CL približne
18 dní. Primárnymi eliminačnými dráhami durvalumabu sú proteínový katabolizmus sprostredkovaný retikuloendoteliálnym systémom alebo cieľovo sprostredkovaná dispozícia.
Osobitnéskupinypacientov
Vek (19 – 96 rokov), telesná hmotnosť (34 – 149 kg), pohlavie, pozitívny stav protilátok proti liečivu (ADA), hladiny albumínu, hladiny LDH, hladiny kreatinínu, solubilný PD-L1, typ nádoru, rasa alebo výkonnostný stav podľa ECOG nemali žiadny klinicky významný vplyv na FK durvalumabu.
Pacientis poruchoufunkcieobličiek
Mierna (klírens kreatinínu (creatinine clearance, CrCL) 60 až 89 ml/min) a stredne závažná porucha
funkcie obličiek (klírens kreatinínu (CrCL) 30 až 59 ml/min) nemali žiadny klinicky významný vplyv na FK durvalumabu. Vplyv závažnej poruchy funkcie obličiek (CrCL 15 až 29 ml/min) na FK durvalumabu nie je známy.
Pacientis poruchoufunkciepečene
Mierna porucha funkcie pečene (hladina bilirubínu ≤ ULN a hladina AST > ULN alebo hladina bilirubínu > 1,0- až 1,5-násobok ULN a akákoľvek hladina AST) nemala žiadny klinicky významný vplyv na FK durvalumabu. Vplyv stredne závažnej poruchy funkcie pečene (hladina bilirubínu > 1,5- až 3-násobok ULN a akákoľvek hladina AST) alebo závažnej poruchy funkcie pečene (hladina bilirubínu > 3-násobok ULN a akákoľvek hladina AST) na farmakokinetiku durvalumabu nie je známy. Avšak, keďže monoklonálne protilátky IgG nie sú eliminované primárne prostredníctvom hepatálnych dráh, neočakáva sa, že zmena funkcie pečene ovplyvní expozíciu durvalumabu.
5.3 Predklinické údaje o bezpečnosti
Karcinogenitaa mutagenita
Karcinogénny a genotoxický potenciál durvalumabu sa neskúmal.
Reprodukčnátoxikológia
Podľa hlásení v literatúre zohrávajú dráhy PD-1/PD-L1 ústrednú úlohu v udržaní gravidity tým, že
zachovávajú imunitnú znášanlivosť matky voči plodu a v alogénnych modeloch gravidity u myší sa preukázalo, že narušenie signalizácie PD-L1 vedie k zvýšenej miere potratu plodu. V reprodukčných štúdiách na zvieratách sa podávanie durvalumabu gravidným opiciam Cynomolgus od potvrdenia gravidity až do pôrodu pri hladinách expozície približne 18-násobne vyšších ako sú hladiny pozorované pri klinickej dávke durvalumabu 10 mg/kg (na základe AUC) spájalo s prestupom cez placentu, ale nie s maternálnou toxicitou alebo s účinkami na embryonálno-fetálny vývin, výsledok gravidity alebo postnatálny vývin. V 28. deň po pôrode sa v materskom mlieku opíc Cynomolgus zistili zanedbateľné hladiny durvalumabu.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
histidín
monohydrát histidíniumchloridu dihydrát trehalózy
polysorbát 80
voda na injekcie
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.
3 Čas použiteľnosti
Neotvoren
á
injekčná
liekovka
3 roky.
Zriedenýroztok
Ak sa nepoužije okamžite, chemická a fyzikálna stabilita IMFINZI počas používania sa preukázala
počas nie viac ako 24 hodín pri teplote 2 °C až 8 °C alebo počas 4 hodín pri izbovej teplote až do
25 °C od času prepichnutia zátky injekčnej liekovky do začiatku podávania.
6.4 Špeciálne upozornenia na uchovávanie Uchovávajte v chladničke (2 °C – 8 °C). Neuchovávajte v mrazničke.
Uchovávajte v pôvodnom obale na ochranu pred svetlom. Podmienky na uchovávanie po riedení lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
2,4 ml koncentrátu v injekčnej liekovke zo skla typu 1 so zátkou z elastoméru a sivým odklápacím hliníkovým tesnením obsahujúcich 120 mg durvalumabu. Veľkosť balenia s 1 injekčnou liekovkou.
10 ml koncentrátu v injekčnej liekovke zo skla typu 1 so zátkou z elastoméru a bielym odklápacím hliníkovým tesnením obsahujúcich 500 mg durvalumabu. Veľkosť balenia s 1 injekčnou liekovkou.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Prípravaroztoku
IMFINZI sa dodáva v jednodávkovej injekčnej liekovke a neobsahuje žiadne konzervačné látky, musí
sa preto dodržiavať aseptický postup.
· Liek vizuálne skontrolujte pre prítomnosť tuhých častíc a zmenu sfarbenia. IMFINZI je číry až opalescenčný, bezfarebný až svetložltý roztok. Injekčnú liekovku vyraďte, ak je roztok zakalený, má zmenenú farbu alebo spozorujete viditeľné častice. Injekčnú liekovku nepretrepávajte.
· Z injekčnej liekovky (injekčných liekoviek) IMFINZI odoberte požadovaný objem a preneste ho do intravenózneho (i.v.) vaku obsahujúceho injekčný roztok chloridu sodného 9 mg/ml
(0,9 %) alebo injekčný roztok glukózy 50 mg/ml (5 %). Zriedený roztok premiešajte opatrným prevrátením. Konečná koncentrácia zriedeného roztoku má byť v rozsahu 1 mg/ml a 15 mg/ml. Roztok neuchovávajte v mrazničke ani nepretrepávajte.
· Zlikvidujte akýkoľvek nepoužitý podiel, ktorý zostal v injekčnej liekovke. Podávanie
· Infúzny roztok podajte intravenózne počas 60 minút cez intravenóznu súpravu obsahujúcu sterilný, proteíny málo viažuci, zaradený filter s veľkosťou pórov 0,2 alebo 0,22 mikrometrov.
· Nepodávajte súbežne s inými liekmi cez rovnakú infúznu súpravu.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/18/1322/002 120 mg injekčná liekovka
EU/1/18/1322/001 500 mg injekčná liekovka
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.