
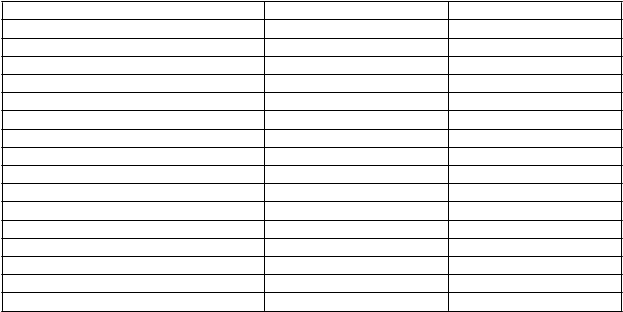
/>Poruchy nervového systému
Časté (> 1/100, < 1/10): bolesť hlavy
Poruchy kože a podkožného tkaniva
Časté (> 1/100, < 1/10): potenie
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté (> 1/100, < 1/10): artralgia a myalgia
Celkové ochorenia a reakcie v mieste podania
Časté (> 1/100, < 1/10): sčervenanie miesta vpichu, opuch miesta vpichu, stvrdnutie miesta vpichu, podliatina v mieste vpichu a bolesť miesta vpichu, horúčka, malátnosť, únava a chvenie
Väčšina týchto reakcií zvyčajne ustúpi do 1 až 2 dní bez liečby.
Nežiaduce
reakcie
z
klinických
skúšaní
u
d
etí
vo
veku
6
mesiacov
až
17
rokov
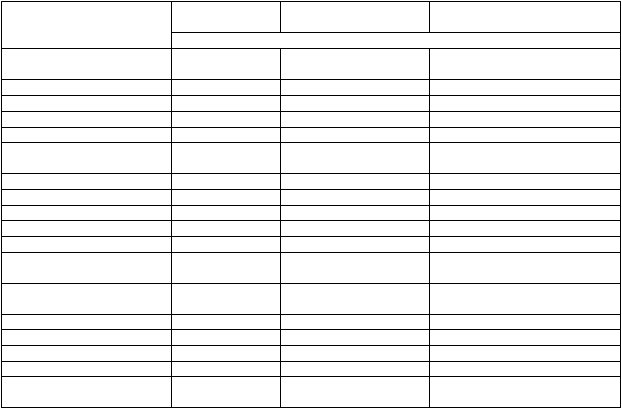
Klinické skúšanie (štúdia V87P6) bolo vykonané s vakcínou H5N1 kombinovanou s adjuvans
MF59C.1 (n=334) v porovnaní so sezónnou vakcínou proti chrípke (n=137).
Prvá dávka
Druhá dávka
(21 dní po prvej dávke)
Tretia dávka
(12 mesiacov po druhej dávke)
Batoľatá
(6 až < 36 mesiacov)
H5N1 vakcína s adjuvans
N
=145 N=138 N=124
Akékoľvek 76 % 68 % 80 % Lokálne 47 % 46 % 60 % Systémové 59 % 51 % 54 % Horúčka ≥ 38 °C (≥ 40 °C) 0 % 0 % 0 %
Akékoľvek iné nežiaduce účinky
54 % 49 % 35 %
D
eti (3 až < 9 rokov) N=96 N=93 N=85
Akékoľvek 72 % 68 % 79 % Lokálne 66 % 58 % 74 % Systémové 32 % 33 % 45 % Horúčka ≥ 38 °C (≥ 40 °C) 4 % 2 % 6 %
Akékoľvek iné nežiaduce účinky
36 % 31 % 19 %
A
d
o
l
escenti
(9 až < 18 rokov)
N
=93 N=91 N=83
Akékoľvek 91 % 82 % 89 % Lokálne 81 % 70 % 81 % Systémové 69 % 52 % 69 % Horúčka ≥ 38 °C (≥ 40 °C) 0 % 1 % 2 %
Akékoľvek iné nežiaduce účinky
30 % 27 % 22 %
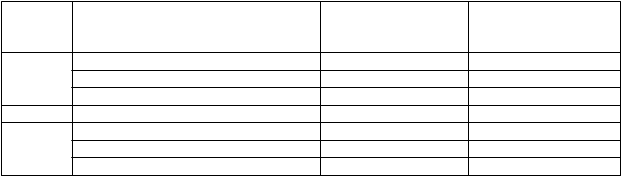
Focetria (H1N1v)
Nežiaduce reakcie pozorované týždeň po podaní vakcíny H1N1v Focetriau 77 detí vo veku
3 – 8 rokov a 80 detí a dospievajúcich vo veku 9 – 17 rokov, ktorým bola podaná vakcína s obsahom 7,5 µg, boli hlásené nasledovne:
1. injekcia 2. injekciaDeti (vo veku od 3 do 8 rokov) N = 77 N = 75Akákoľvek nežiaduca reakcia 74 % 69 % Lokálna 62 % 56 % Systémová 39 % 35 % Horúčka ≥ 38 °C až 38,9 °C 4 % 1 % Horúčka 39 °C až 39,9 °C 0 % 1 % Horúčka ≥ 40 °C 0 % 0 % Akákoľvek iná nežiaduca udalosť 14 % 17 % Dospievajúci (vo veku 9 až 17 rokov) N = 80 N = 79Akákoľvek nežiaduca reakcia 79 % 66 % Lokálna 70 % 58 % Systémová 45 % 30 % Horúčka ≥ 38 °C až 38,9 °C 3 % 1 % Horúčka 39 °C až 39,9 °C 0 % 0 % Horúčka ≥ 40 °C 0 % 0 % Akákoľvek iná nežiaduca udalosť 13 % 10 %Údaje u detí a dospievajúcich vo veku 3 – 17 rokov naznačujú mierne zníženie reaktogenity po druhej
dávke bez zvýšenia výskytu horúčky.

Veľmi časté reakcie hlásené u detí a dospievajúcich vo veku 3 až 17 rokov:
Bolesť, indurácia a erytém, malátnosť, myalgia, bolesť hlavy a únava.
Nežiaduce reakcie pozorované týždeň po podaní vakcíny H1N1v Focetria u 73 dojčiat vo veku 6 –
11 mesiacov a 73 batoliat vo veku 12 – 35 mesiacov, ktorým bola podaná vakcína s obsahom 7,5 µg, boli hlásené nasledovne:
1. injekcia 2. injekciaDojčatá (vo veku 6 až 11 mesiacov) N = 73 N = 68Akákoľvek nežiaduca reakcia 79 % 65 % Lokálna 44 % 26 % Systémová 70 % 56 % Horúčka ≥ 38 °C až 38,9 °C 11 % 9 % Horúčka 39 °C až 39,9 °C 3 % 4 % Horúčka ≥ 40 °C 0 % 0 % Akákoľvek iná nežiaduca udalosť 32 % 31 % Batoľatá (vo veku 12 až 35 mesiacov) N = 73 N = 71Akákoľvek nežiaduca reakcia 70 % 71 % Lokálna 51 % 49 % Systémová 60 % 49 % Horúčka ≥ 38 °C až 38,9 °C 10 % 11 % Horúčka 39 °C až 39,9 °C 4 % 1 % Horúčka ≥ 40 °C 1 % 0 % Akákoľvek iná nežiaduca udalosť 21 % 24 %Údaje u dojčiat a batoliat vo veku 6 – 35 mesiacov naznačujú mierne zníženie reaktogenity po druhej
dávke bez zvýšenia výskytu horúčky.
Veľmi časté reakcie hlásené u 146 dojčiat a batoliat vo veku 6 až 35 mesiacov:
Citlivosť, erytém, podráždenosť, nezvyčajný plač, spavosť, hnačka, vracanie a zmeny v stravovacích návykoch. Indurácia a ekchymóza boli veľmi častými reakciami u batoliat, ale nemej častými reakciami u dojčiat.
•
Postmarketingové sledovanieOkrem nežiaducich reakcií hlásených v klinických skúšaniach sa počas skúseností po uvedení vakcíny
H1N1v Focetria na trh hlásilo nasledovné:
Poruchy krvi a lymfatického systémuLymfadenopatia.
PoruchysrdcaasrdcovejčinnostiPalpitácia, tachykardia.
Celkové poruchy a reakcie v mieste podaniaAsténia.
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaSvalová slabosť, bolesť v končatinách.
Poruchy dýchacej sústavy, hrudníka a mediastínaKašeľ.
Poruchy kože a podkožného tkanivaGeneralizované kožné reakcie zahŕňajúce pruritus, urtikáriu alebo nešpecifickú vyrážku, angioedém.
Poruchy gastrointestinálneho traktu
Poruchy gastrointestinálneho traktu, ako je napr. nevoľnosť, bolesť brucha a hnačka.
Poruchy nervového systémuBolesť hlavy, závrat, somnolencia, synkopa. Neurologické poruchy, ako je napr. neuralgia, parestézia,
kŕče a neuritída.
Poruchy imunitného systémuAlergické reakcie, anafylaxia zahŕňajúca dyspnoe, bronchospazmus, opuch hrtana, ktorá v
zriedkavých prípadoch vedie k šoku.
Počas postmarketingových prieskumov s adjuvovanými interpandemickými trivalentnými vakcínami so zložením podobným vakcíne Foclivia (pandemická H1N1 vakcína adjuvovaná s MF59C.1) boli pozorované ďalšie nežiaduce reakcie:
Zriedkavé (> 1/10 000, < 1/1 000):
Trombocytopénia (niektoré veľmi zriedkavé prípady boli závažné s počtom trombocytov menej
ako 5 000 v mm3).
Veľmizriedkavé (< 1/10 000):
Zápal ciev s prechodným postihnutím obličiek a exsudačný multiformný erytém.
Neurologické poruchy, ako napríklad encefalomyelitída, neuritída a Guillainov-Barrého syndróm. Nežiaduce reakcie z postmarketingových prieskumov s pandemickou vakcínou: neaplikovateľné
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieNebol hlásený žiaden prípad predávkovania.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: chrípková vakcína, ATC kód: J07BB02
Táto časť popisuje klinické skúsenosti s pandemickými pohotovostnýmij H5N1 vakcínami po podaní dvoch dávok vakcíny s obsahom 7,5 mikrogramov.
Pandemické pohotovostné vakcíny obsahujú chrípkové antigény, ktoré sú odlišné od aktuálne sa vyskytujúcich vírusov chrípky. Tieto antigény sa môžu považovať za „nové“ antigény a simulujú situáciu, kedy je cieľová populácia pre vakcináciu imunologicky „neskúsená“. Údaje získané
s pandemickou pohotovostnou vakcínou podporia plán vakcinácie, ktorý sa pravdepodobne použije pre pandemickú vakcínu: údaje o klinickej účinnosti a bezpečnosti získané s pandemickými pohotovostnými vakcínami sú relevantné pre pandemické vakcíny.
Dospelí (18-60 rokov)
Bola vykonaná II. fáza klinického skúšania (štúdia V87P1) s vakcínou H5N1 kombinovanou
s adjuvans MF59C.1 u 312 zdravých dospelých osôb. Dve dávky vakcíny
H5N1 (A/Vietnam/1194/2004, adjuvantná dávka/ 7,5 µg hemaglutinínu (HA) boli podané v intervale
troch týždňov 156 osobám.
V ďalšom klinickom skúšaní (III. fáza) (štúdia V87P13) sa zúčastnilo 2693 dospelých osôb a boli im podané dve dávky vakcíny obsahujúcej H5N1 (A/Vietnam/1194/2004, adjuvantná dávka/ 7,5 µg HA) v intervale troch týždňov. Imunogenita bola hodnotená v podskupine (n=197) štúdie populácie.
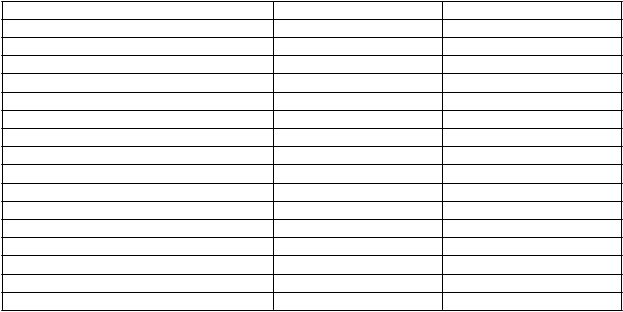
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči H5N1 A/Vietnam/1194/2004 u dospelých boli podľa analýzy (Single Radial Heamolysis, SRH) nasledujúce:
Anti-HA protilátka (SRH)
Štúdia V87P1
21 dní po 2. dávke
N=149
Štúdia V87P13
21 dní po 2. dávke
N=197
Miera séroprotekcie (95 % CI) 85 % (79-91) 91 % (87-95) Miera sérokonverzie (95 % CI) 85 % (78-90) 78 % (72-84) Faktor sérokonverzie (95 % CI) 7,74 (6,6-9,07) 4,03 (3,54-4,59)
Anti-HA protilátka (HI)
Štúdia V87P13
21 dní po 2. dávke
N=69
Štúdia V87P13
21 dní po 2. dávke
N=128
Počiatočný sérologický stav < 4 mm2 ≥ 4 mm2
Miera séroprotekcie (95 % CI)* 87 % (77-94) 94 % (88-97) Miera sérokonverzie (95 % CI)* 87 % (77-94) 73 % (65-81) Faktor sérokonverzie (95 % CI)** 8,87 (7,09-11) 2,71 (2,38-3,08)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
Výsledky mikroneutralizácie (MN) proti A/Vietnam/1194/2004 indikujú mieru séroprotekcie a mieru sérokonverzie v rozsahu od 67 % (60-74) do 85 % (78-90) a 65 % (58-72) až 83 % (77-89). Imunitná odpoveď na očkovanie hodnotená MN analýzou je v súlade s výsledkami získanými SRH analýzou.
Pretrvávanie protilátok po základnom očkovaní v tejto populácii bolo hodnotené hemaglutinačnou inhibíciou (HI), SRH a MN analýzami. V porovnaní s hladinami protilátok získanými v 43. deň
po ukončení základného očkovania boli hladiny protilátok v 202. deň znížené o 1/5 až 1/2 ich pôvodných hladín.
Staršie osoby (>60 rokov)
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči
H5N1 A/Vietnam/1194/2004 u osôb starších ako 60 rokov (obmedzený počet osôb starších
ako 70 rokov) merané SRH analýzou, hodnotené v dvoch klinických štúdiách boli nasledujúce:
Anti-HA protilátka (SRH)
Štúdia V87P1
21 dní po 2. dávke
N=84
Štúdia V87P13
21 dní po 2. dávke
N=210
Anti-HA protilátka (SRH)
Štúdia V87P13
21 dní po 2. dávke
N=66
Štúdia V87P13
21 dní po 2. dávke
N=143
Počiatočný sérologický stav < 4 mm2 ≥ 4 mm2
Miera séroprotekcie (95 % CI)* 82 % (70-90) 82 % (75-88) Miera sérokonverzie (95 % CI)* 82 % (70-90) 54 % (45-62) Faktor sérokonverzie (95 % CI)** 8,58 (6,57-11) 1,91 (1,72-2,12)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
Výsledky MN voči A/Vietnam/1194/2004 indikujú mieru séroprotekcie, mieru sérokonverzie
v rozsahu od 57 % (50-64) do 79 % (68-87) a 55 % (48-62) až 58 % (47-69). Imunitná odpoveď na očkovanie hodnotená analýzou MN je podobná s výsledkami získanými SRH analýzou a poukazuje na silnejšiu imunitnú odpoveď po úplnom základnom očkovaní u populácie starších osôb.
Pretrvávanie protilátok po základnom očkovaní v tejto populácii bolo hodnotené HI, SRH a MN analýzami. V porovnaní s hladinami protilátok získanými v 43. deň po ukončení základného očkovania boli hladiny protilátok v 202. deň znížené o 1/2 až 1/5 ich hladín po očkovaní. Až 50 % starších osôb očkovaných H5N1 vakcínou kombinovanou s MF59C získalo sérologickú ochranu na
6 mesiacov.
Posilňovacia dávka
Tretia (posilňovacia) dávka H5N1 vakcíny kombinovanej s MF59C bola podaná 6 mesiacov a viac
po základnom očkovaní. Výsledky sú uvedené podľa SRH analýzy.
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči
H5N1 A/Vietnam/1194/2004 merané SRH analýzou boli nasledujúce:
Štúdia V87P1 Dospelí,
posilňovacia dávka
(6 mesiacov po 2. dávke)
Štúdia V87P1 Staršie osoby,
posilňovacia dávka
(6 mesiacov po 2. dávke)


SRH N=71 N=38
Miera séroprotekcie (95 % CI) 89 % (79-95) 84 % (69-94) Miera sérokonverzie (95 % CI) 83 % (72-91) 63 % (46-78) Faktor sérokonverzie (95 % CI) 5,96 (4,72-7,53) 5,15 (3,46-7,66)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
Skúsenosti s použitím posilňovacej dávky u starších osôb sú obmedzené.
•
Podporné údaje u dospelých a starších osôbV dvoch štúdiách zameraných na zistenie účinných dávok dostávalo 78 dospelých adjuvovanú
pandemickú pohotovostnú vakcínu (H5N3 alebo H9N2). Dve dávky vakcíny s kmeňom
H5N3 (A/Duck/Singapur/97) v 3 rôznych dávkach (7,5; 15 a 30 µg HA/dávku) boli podané po troch
týždňoch od seba.
Vzorky séra sa testovali na pôvodný kmeň H5N3 a tiež na niekoľko izolovaných kmeňov H5N1.
Sérologické odpovede získané pomocou testu SRH ukázali, že 100 % pacientov dosiahlo sérologickú ochranu a 100 % dosiahlo sérologickú konverziu po dvoch injekčných podaniach dávky 7,5 µg.
V prípade adjuvovanej vakcíny sa tiež zistilo, že vyvolávala vznik protilátok proti kmeňom
H5N1 izolovaným v rokoch 2003 a 2004, ktoré vykazujú určitý antigénny posun v porovnaní s pôvodnými kmeňmi.
Dve dávky vakcíny obsahujúce kmeň H9N2 (A/Chicken/Hongkong/G9/97) v 4 rôznych dávkach (3,75; 7,5; 15 a 30 μg HA/dávku) boli podané po štyroch týždňoch od seba. Sérologické odpovede získané pomocou testu HI ukázali, že 92 % pacientov dosiahlo sérologickú ochranu a 75 % dosiahlo sérologickú konverziu po dvoch injekčných podaniach dávky 7,5 µg.
Skríženáreaktivita
Dospelí (18 – 60 rokov)
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátky voči
H5N1 A/turkey/Turkey/05 boli po podaní 2. dávky u dospelých vo veku 18-60 rokov merané SRH
a HI analýzami nasledujúce:
Anti-HA protilátka
Štúdia V87P1
21 dní po 2. dávke
N=70
Štúdia V87P13
21 dní po 2. dávke
N=197
SRH
HI
Miera séroprotekcie* (95 % CI) 70 % (58-80) 59 % (52-66) Miera sérokonverzie* (95 % CI) NA*** 49 % (42-56) Faktor sérokonverzie** (95 % CI) NA*** 2,37 (2,1-2,67)
N=69 N=197
Miera séroprotekcie (95 % CI) 36 % (25-49) 23 % (18-30) Miera sérokonverzie (95 % CI) NA*** 19 % (14-25) Faktor sérokonverzie (95 % CI) NA*** 1,92 (1,64-2,25)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
° merané HI analýzou ≥ 40
°° geometrický priemer pomerov HI
*** vo V87P1: počiatočný stav nebol testovaný
Výsledky MN z klinických štúdií v tabuľke vyššie preukázali mieru séroprotekcie proti A/turkey/Turkey/05 v rozsahu od 27 % (17-39) (V87P1) do 39 % (32-46) (V87P13) a mieru sérokonverzie 36 % (29-43) v štúdii V87P13. Výsledky MN v štúdii V87P13 preukázali GMR proti A/turkey/Turkey/05 2,77 (2,4-3,2).
Staršie osoby (>60 rokov)
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči
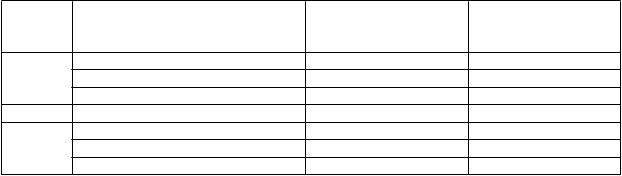
H5N1 A/turkey/Turkey/05 po druhej dávke u osôb starších ako 60 rokov merané SRH a HI analýzou, boli nasledujúce:
Anti-HA protilátka
Štúdia V87P1
21 dní po 2. dávke
N=37
Štúdia V87P13
21 dní po 2. dávke
N=207
SRH
HI
Miera séroprotekcie* (95 % CI) 57 % (39-73) 20 % (18-23) Miera sérokonverzie* (95 % CI) NA*** 48 % (41-55) Faktor sérokonverzie** (95 % CI) NA*** 1,74 (1,57-1,94)
N=36 N=208
Miera séroprotekcie (95 % CI) 36 % (21-54) 25 % (19-32) Miera sérokonverzie (95 % CI) NA*** 19 % (14-25) Faktor sérokonverzie (95 % CI) NA*** 1,79 (1,56-2,06)

* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
° merané HI analýzou ≥ 40
°° geometrický priemer pomerov HI
*** vo V87P1: počiatočný stav nebol testovaný
Výsledky MN z klinických štúdií v tabuľke vyššie preukázali mieru séroprotekcie proti A/turkey/Turkey/05 v rozsahu od 11 % (3-25) (štúdia V87P1) do 30 % (24-37) (štúdia V87P13) a mieru sérokonverzie 25 % (19-31) v štúdii V87P13. Výsledky MN v štúdii V87P13 preukázali GMR proti A/turkey/Turkey/05 2,01 (1,78-2,26).
• Údaje v pediatrickej populácii
Bolo vykonané klinické skúšanie (štúdia V87P6) vakcíny H5N1 kombinovanej s adjuvans
MF59C.1 u 471 detí vo veku od 6 mesiacov do 17 rokov. Dve dávky 7,5 mikrogramov boli podané v intervale troch týždňov a tretia dávka 12 mesiacov po prvej dávke. Po 3 týždňoch po 2. očkovaní (43. deň) všetky vekové skupiny (t.j. 6-35 mesiacov, 3-8 rokov a 9-17 rokov) dosiahli vysoké hladiny protilátok voči (A/Vietnam/1194/2004), ako bolo vyhodnotené SRH a HI analýzami a je uvedené
v tabuľke nižšie*. V tejto štúdii neboli pozorované žiadne závažné nežiaduce reakcie v súvislosti s vakcínou.
Batoľatá
(6- < 36 mesiacov)
Det
i
(3- < 9 rokov)
Dospievajúci
(9- < 18 rokov)
N=134 N=91 N=89
Miera séroprotekcie
(95 % CI)
43. deň
Faktor
HI sérokonverzie
(95 % CI)
43. deň až 1. deň Miera sérokonverzie (95 % CI)
43. Deň
97 % (92-99)
129 (109-151)
97 % (92-99)
97 % (91-99)
117 (97-142)
97 % (91-99)
89 % (80-94)
67 (51-88)
89 % (80-94)
N=133 N=91 N=90
SRH
Miera séroprotekcie
(95 % CI)
43. deň
Faktor sérokonverzie (95 % CI)
43. deň až 1. deň Miera sérokonverzie (95 % CI)
43. deň
100 % (97-100)
16 (14-18)
98 % (95-100)
100 % (96-100)
15 (13-17)
100 % (96-100)
100 % (96-100)
14 (12-16)
99 % (94-100)
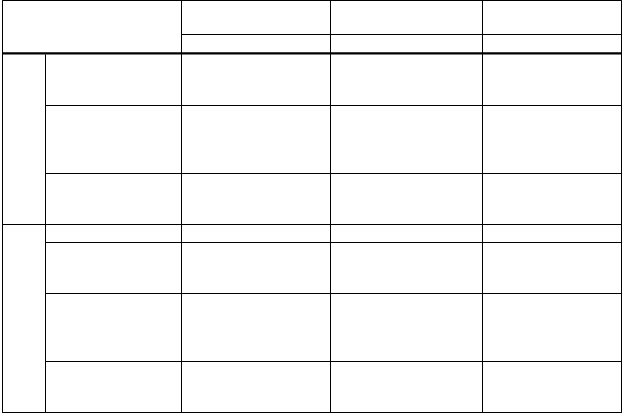
* Pretože chýbajú kritériá CHMP pre imunogenitu u detí, pre sérologické údaje získané po očkovaní detí sa použili kritériá CHMP pre imunogenitu použité na hodnotenie sezónnych chrípkových vakcín u dospelých. Význam klinickej ochrany je však neznámy.
Výsledky MN proti A/Vietnam/1194/2004 indikujú mieru séroprotekcie 99 % (95 % CI: 94-100), mieru sérokonverzie v rozsahu od 97 % (95 % CI: 91-99) do 99 % (95 % CI: 96-100) a GMR v rozsahu od 29 (95 % CI: 25-35) do 50 (95 % CI: 44-58).
Výsledky imunogenity vakcíny H1N1v Focetria (štúdia V111_03):
Miera séroprotekcie a miera sérokonverzie boli stanovené pomocou metódy HI a faktor
sérokonverzie bol vyjadrený ako geometrický priemer pomerov HI pre anti-HA protilátky k H1N1 po podaní jednej alebo dvoch 7,5 µg dávok vakcíny Focetria, hodnotený u 70 detí a dospievajúcich
(9 – 17 rokov), 60 detí (3 – 8 rokov), 58 detí (12 – 35 rokov) a 49 dojčiat (6 – 11 mesiacov). Vo všetkých vyššie uvedených vekových skupinách (v celkovej populácii a aj v podskupine séronegatívnych pacientov na začiatku) boli po 1. a aj po 2. dávke dosiahnuté kritériá imunogenity stanovené výborom CHMP pre dospelých (18 – 60 rokov).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s vakcínou Foclivia v jednej alebo vo viacerých podskupinách pediatrickej populácie na aktívnu imunizáciu proti vírusu chrípky A, podtyp H5N1. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
Liek Foclivia bol registrovaný za tzv. „mimoriadnych okolností“.
To znamená, že z vedeckých dôvodov nebolo možné získať všetky informácie o tomto lieku. Európska agentúra pre lieky (EMA) každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Neaplikovateľné.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané u vakcíny Foclivia a sezónnej vakcíny obsahujúcej adjuvans MF59C.1 neodhalili žiadne osobitné riziko pre ľudí na základe vykonaných konvenčných štúdií toxicity po opakovanej dávke, lokálnej znášanlivosti, fertility žien a reprodukčnej a vývinovej toxicity (do konca obdobia laktácie).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Chlorid sodný, chlorid draselný, dihydrogenfosforečnan draselný,
dihydrát hydrogenfosforečnanu sodného, hexahydrát chloridu horečnatého, dihydrát chloridu vápenatého,
voda na injekciu.
Ohľadom adjuvans, pozri časť 2.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
1 rok.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
0,5 ml v naplnenej injekčnej striekačke (sklenená, typ I) s piestovou zátkou (bróm-butylová guma). Balenia obsahujúce 1 a 10 kusov s ihlou alebo bez ihly. Injekčné striekačky bez ihly sú vybavené systémom Luer Lock.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6
.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Vakcína má pred použitím dosiahnuť izbovú teplotu. Pred použitím jemne potraste.
Pred podaním suspenziu vizuálne skontrolujte. V prípade akýchkoľvek častíc alebo nezvyčajného vzhľadu sa má vakcína zlikvidovať.
Pri používaní naplnenej injekčnej striekačky bez ihly dodávanej so systémom Luer Lock odstráňte kryt špičky odskrutkovaním proti smeru hodinových ručičiek. Po odobratí krytu pripojte ihlu na injekčnú striekačku naskrutkovaním v smere hodinových ručičiek až do zapadnutia. Po zapadnutí ihly na svoje miesto odstráňte ochranný kryt ihly a podajte vakcínu.
Všetka nepoužitá vakcína alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIISeqirus S.r.l.
Via del Pozzo 3/A, S. Martino
53035 Monteriggioni (SI) Taliansko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/09/577/001-002
EU/1/09/577/005-006
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. október 2009
Dátum posledného predĺženia registrácie: 19. október 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1
. NÁZOV LIEKU
Foclivia injekčná suspenzia
Pandemická chrípková vakcína (H5N1) (inaktivovaný adjuvovaný povrchový antigén)
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Povrchové antigény vírusu chrípky (hemaglutinín a neuraminidáza)* nasledujúcich kmeňov:
A/Vietnam/1194/2004 (H5N1) 7,5 mikrogramu**
na 0,5 ml dávku
* pomnožený na oplodnených slepačích vajciach zo zdravých kuracích kŕdľov
** vyjadrené v mikrogramoch hemaglutinínu.
Adjuvans MF59C.1 obsahuje:
Skvalén 9,75 miligramu Polysorbát 80 1,175 miligramu Sorbitantrioleát 1,175 miligramu Citrát sodný 0,66 miligramu Kyselina citrónová 0,04 miligramu
Táto vakcína spĺňa odporúčania Svetovej zdravotníckej organizácie (WHO) a rozhodnutie EÚ pre prípad pandémie.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčná suspenzia. Mliečnobiela tekutina.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Profylaxia chrípky pri oficiálne vyhlásenej pandemickej situácii.
Foclivia sa má používať v súlade s oficiálnym odporúčaním.
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Dospelí a staršie osoby: 0,5 ml vo zvolenom termíne.
Druhá dávka vakcíny sa má podať po uplynutí najmenej 3 týždňov.
Foclivia bola hodnotená u dospelých (vo veku 18 až 60 rokov) a starších osôb (vo veku nad 60 rokov)
po podaní po 1. a 22. dni podľa základnej očkovacej schémy.
K dispozícii sú obmedzené údaje o tretej dávke (posilňovacej) podanej 6 mesiacov po prvej dávke
(pozri časti 4.8 a 5.1).
Pediatrická populácia:
Bezpečnosť a účinnosť vakcíny Foclivia u osôb mladších ako 18 rokov neboli doteraz stanovené. V súčasnosti dostupné údaje o osobách vo veku 6 mesiacov až 18 rokov sú opísané v časti 5.1, ale neumožňujú uviesť odporúčania na dávkovanie.
Pre deti mladšie ako 6 mesiacov nie sú k dispozícii žiadne údaje.
Spôsob podávania
Očkovanie sa má vykonávať formou intramuskulárnej injekcie do deltového svalu alebo anterolaterálnej časti stehna (v závislosti od svalovej hmoty).
4.3 Kontraindikácie
Anamnéza anafylaktickej (t. j. život ohrozujúcej) reakcie na akékoľvek zložky alebo stopové zvyšky vajíčok, kuracích bielkovín, kanamycínu a neomycín sulfátu, formaldehydu
a cetyltrimetylamóniumbromidu (CTAB) tejto vakcíny. Pri pandemickej situácii však môže byť vhodné podávať vakcínu, pokiaľ sú pre prípad potreby okamžite dostupné prostriedky na resuscitáciu. Pozri časť 4.4.
4.4 Osobitné upozornenia a opatrenia pri používaní
Opatrne treba postupovať pri podávaní tejto vakcíny osobám so známou precitlivenosťou (inou než anafylaktická reakcia) na liečivo, na ktorúkoľvek z pomocných látok uvedených v časti 6.1, a na vajíčka, kuracie bielkoviny, kanamycín a neomycín, formaldehyd a cetyltrimetylamóniumbromid (CTAB).
Tak ako u všetkých injekčných vakcín, vždy má byť k dispozícii príslušná liečba a dohľad pre prípad
zriedkavej anafylaktickej reakcie po podaní vakcíny.
Ak to pandemická situácia umožňuje, u pacientov so závažným horúčkovitým ochorením alebo
s akútnou infekciou sa má očkovanie odložiť.
Foclivia sa nesmie v žiadnom prípade podávať intravaskulárne ani subkutánne. Preto je potrebné, aby lekári posúdili prínosy a možné riziká podávania vakcíny u jedincov s trombocytopéniou alebo
s akoukoľvek poruchou krvácania, ktoré by boli kontraindikáciou pre intramuskulárnu injekciu, pokiaľ
možný prínos nepreváži riziko krvácaní.
Odpoveď na protilátky u pacientov s endogénnou alebo iatrogénnou imunosupresiou môže byť nedostatočná.
Ochranná odpoveď nemusí byť vyvolaná u všetkých očkovaných osôb (pozri časť 5.1). Skrížená ochrana bola pozorovaná proti variantom vírusu súvisiacim s H5N1 počas klinického
skúšania (pozri časť 5.1).
V prípade podania druhej dávky je potrebné vziať do úvahy, že neexistujú žiadne údaje o bezpečnosti, imunogenite alebo o účinnosti, ktoré by podporili zameniteľnosť vakcíny Foclivia s inými monovalentnými H5N1 vakcínami.
Zatiaľ čo z používania vakcíny Foclivia nie sú k dispozícii žiadne údaje, boli hlásené prípady kŕčov
s horúčkou alebo bez nej u jedincov očkovaných vakcínou Focetria, ktorá je pandemickou
H1N1 vakcínou adjuvovanou s MF59.1, ktorá je podobná vakcíne Foclivia.
Väčšina febrilných kŕčov sa vyskytla u pediatrických jedincov. Niektoré prípady boli pozorované u jedincov s epilepsiou v anamnéze. Osobitná pozornosť sa má venovať jedincom, ktorí trpia epilepsiou a lekári majú informovať týchto jedincov (alebo ich rodičov) o možnosti objavenia sa kŕču (pozri časť 4.8).
Po podaní alebo dokonca pred podaním akejkoľvek vakcíny sa môže objaviť synkopa (mdloba), ako psychogénna odpoveď na pichnutie injekčnou ihlou. Môže byť sprevádzaná niekoľkými neurologickými prejavmi, ako je napr. prechodná porucha videnia, parestézia a tonicko-klonické kŕče končatín počas zotavovania sa. Je dôležité, aby boli zavedené postupy, ktoré predchádzajú poraneniu pri pádoch spôsobených mdlobou.
4.5 Liekové a iné interakcie
Foclivia sa nemá podávať súbežne s inými vakcínami. Ak je však súbežné podávanie s inými vakcínami indikované, očkovanie sa má vykonávať na odlišných končatinách. Treba poznamenať, že nežiaduce reakcie môžu byť intenzívnejšie.
Ak sa pacient lieči imunosupresívami, imunologická odpoveď môže byť znížená.
Po podaní vakcíny proti chrípke sa pri stanovovaní protilátok vírusu ľudskej imunitnej nedostatočnosti-1 (HIV-1), vírusu hepatitídy C a najmä HTLV-1 pomocou metódy ELISA môžu získať falošne pozitívne výsledky sérologického testu. V takýchto prípadoch je výsledok pri metóde Western blot negatívny. Tieto prechodné falošne pozitívne výsledky testu môžu byť spôsobené tvorbou protilátok IgM ako odpoveď na podanie vakcíny.
4.6 Fertilita, gravidita a laktácia
Gravidita
Neexistujú údaje o použití Foclivie u gravidných žien.
Údaje týkajúce sa bezpečnosti sú však k dispozícii u gravidných žien vystavených vakcíne Focetria (pandemická H1N1 vakcína podobná vakcíne Foclivia), ktorá obsahuje rovnaké množstvo MF59C.1 ako vakcína Foclivia. Nežiaduce udalosti, ktoré boli spontánne hlásené po jej uvedení na
trh a intervenčná štúdia nenaznačujú priame alebo nepriame škodlivé účinky na graviditu pri expozícii vakcíne Focetria. Okrem toho dve veľké observačné štúdie navrhnuté na posúdenie bezpečnosti pri expozícii vakcíne Focetria v gravidite nepreukázali žiadne zvýšenie výskytu gestačného diabetes mellitus, preeklampsie, potratov, pôrodov mŕtveho plodu, pôrodov s nízkou telesnou hmotnosťou, úmrtí novorodencov a vrodených malformácií medzi takmer 10 000 očkovanými gravidnými ženami
a ich potomkami v porovnaní s kontrolnou skupinou žien bez očkovania.
Je potrebné, aby lekári posúdili prínosy a možné riziká podávania vakcíny Foclivia gravidným ženám
pri zohľadnení oficiálnych odporúčaní.
Dojčenie
Vakcína sa môže používať počas laktácie.
Fertilita
Údaje týkajúce sa fertility nie sú k dispozícii.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Niektoré z účinkov uvedených v časti 4.8 „Nežiaduce účinky“ môžu ovplyvniť schopnosť viesť
vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn profilu bezpečnosti
Dospelí a staršie osoby (starší ako 18 rokov)
V klinických skúšaniach vykonaných s pandemickou pohotovostnou adjuvovanou H5N1 vakcínou u dospelých a starších osôb (viac informácií pozri časť 5.1) mala väčšina reakcií miernu povahu, krátku dĺžku trvania a bola kvalitatívne podobná reakciám, ktoré boli vyvolané štandardnými
sezónnymi vakcínami proti chrípke. Vo všeobecnosti je akceptované, že adjuvantný účinok, ktorý vedie k zvýšenej imunogenicite, je spojený s mierne vyššou frekvenciou výskytu lokálnych reakcií (najmä mierna bolesť) v porovnaní so štandardnými neadjuvovanými vakcínami proti chrípke. Po podaní druhej dávky sa objavilo menej reakcií v porovnaní s prvou dávkou.
Deti a dospievajúci vo veku 6 mesiacov až 17 rokov:
V klinickom skúšaní II fázy (Štúdia V87P6) sa u detí a dospievajúcich hodnotila bezpečnosť
pandemickej pohotovostnej adjuvovanej H5N1 vakcíny (viac informácií pozri v časti 5.1).
Bez ohľadu na vek bola reaktogenita vyššia po prvej dávke, ako po druhej dávke očkovania. Reaktogenita po tretej dávke podanej 12 mesiacov po druhej dávke bola vyššia, ako po prvej a aj druhej dávke. Percentuálny pomer jedincov, ktorí hlásili lokálne reakcie, bol vyšší ako v skupine s vyšším vekom, najmä z dôvodu vyššej miery hlásení bolesti. Najčastejšími vyžiadanými lokálnymi reakciami u batoliat boli erytém a citlivosť; podráždenosť a nezvyčajný plač boli najčastejšie hlásenými vyžiadanými systémovými reakciami. Najčastejšie hlásenou vyžiadanou lokálnou reakciou u detí a dospievajúcich bola bolesť a únava a bolesť hlavy boli najčastejšie hlásenými vyžiadanými systémovými reakciami. Medzi vekovými skupinami sa pri malom percentuálnom pomere hlásila horúčka. Údaje týkajúce sa bezpečnosti po prvej a druhej dávke u detí a dospievajúcich pri podobnej pandemickej vakcíne (Focetria H1N1v) naznačujú porovnateľný profil bezpečnosti ako je profil bezpečnosti, ktorý sa hlásil pri pandemickej pohotovostnej aH5N1 vakcíne (Foclivia).
Frekvencia výskytu nežiaducich reakcií v klinických skúšaniach a dohľade po uvedení vakcíny na trh
je uvedená nižšie.
• Klinické štúdie
Nežiaducereakciezklinickýchskúšaníudospelýchastaršíchosôb(staršíchako18rokov).
Výskyt nežiaducich reakcií bol hodnotený v štyroch klinických štúdiách s rôznymi kmeňmi chrípky a
s rozličným zložením (H5N3, H9N2 a H5N1), ktorých sa zúčastnilo približne 3696 dospelých
a starších osôb. Z týchto osôb bola 3618 osobám podaná pandemická pohotovostná vakcína Foclivia
(A/H5N1) (pozri časť 5.1).
Nežiaduce reakcie zistené počas klinických štúdií s pandemickou pohotovostnou vakcínou Foclivia sú uvedené nižšie.
Výskyt príznakov pozorovaných u pacientov vo veku nad 60 rokov bol nižší v porovnaní s populáciou vo veku 18 až 60 rokov.
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti:
Poruchy nervového systému
Časté (> 1/100, < 1/10): bolesť hlavy
Poruchy kože a podkožného tkaniva
Časté (> 1/100, < 1/10): potenie
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté (> 1/100, < 1/10): artralgia a myalgia
Celkové ochorenia a reakcie v mieste podania
Časté (> 1/100, < 1/10): sčervenanie miesta vpichu, opuch miesta vpichu, stvrdnutie miesta vpichu,
podliatina v mieste vpichu a bolesť miesta vpichu, horúčka, malátnosť, únava a chvenie
Väčšina týchto reakcií zvyčajne ustúpi do 1 až 2 dní bez liečby.
Nežiaduce
reakcie
z
klinických
skúšaní
u
d
etí
vo
veku
6
mesiacov
až
17
rokov
Klinické skúšanie (štúdia V87P6) bolo vykonané s vakcínou H5N1 kombinovanou s adjuvans
MF59C.1 (n=334) v porovnaní so sezónnou vakcínou proti chrípke (n=137).
1. injekcia 2. injekcia 3. injekcia
H5N1 vakcína s adjuvans
H5N1 vakcína s adjuvans
H5N1 vakcína s adjuvans
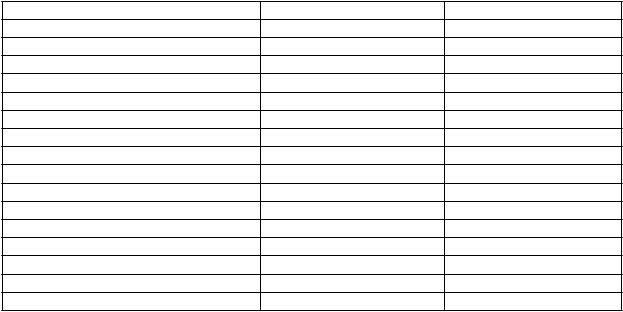
 Batoľatá (6 až < 36 mesiacov) N=145 N=138 N=124
Batoľatá (6 až < 36 mesiacov) N=145 N=138 N=124
Akékoľvek 76 % 68 % 80 % Lokálne 47 % 46 % 60 % Systémové 59 % 51 % 54 % Horúčka ≥ 38 °C (≥ 40 °C) 0 % 0 % 0 % Akékoľvek iné nežiaduce účinky 54 % 49 % 35 %
Deti (3 až < 9 rokov) N=96 N=93 N=85Akékoľvek 72 % 68 % 79 % Lokálne 66 % 58 % 74 % Systémové 32 % 33 % 45 % Horúčka ≥ 38 °C (≥ 40 °C) 4 % 2 % 6 % Akékoľvek iné nežiaduce účinky 36 % 31 % 19 %
Adolescenti (9 až < 18 rokov) N=93 N=91 N=83Akékoľvek 91 % 82 % 89 % Lokálne 81 % 70 % 81 % Systémové 69 % 52 % 69 % Horúčka ≥ 38 °C (≥ 40 °C) 0 % 1 % 2 % Akékoľvek iné nežiaduce účinky 30 % 27 % 22 %
Focetria (H1N1v)
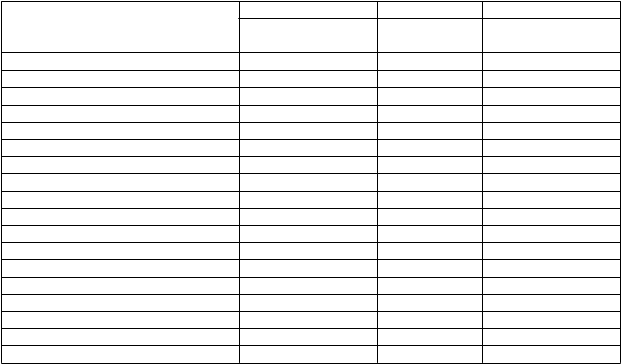
Nežiaduce reakcie pozorované týždeň po podaní vakcíny H1N1v Focetria u 77 detí vo veku 3 –
8 rokov a 80 detí a dospievajúcich vo veku 9 – 17 rokov, ktorým bola podaná vakcína s obsahom 7,5 µg, boli hlásené nasledovne:
1. injekcia 2. injekciaDeti (vo veku od 3 do 8 rokov) N = 77 N = 75Akákoľvek nežiaduca reakcia 74 % 69 % Lokálna 62 % 56 % Systémová 39 % 35 % Horúčka ≥ 38 °C až 38,9 °C 4 % 1 % Horúčka 39 °C až 39,9 °C 0 % 1 % Horúčka ≥ 40 °C 0 % 0 % Akákoľvek iná nežiaduca udalosť 14 % 17 % Dospievajúci (vo veku 9 až 17 rokov) N = 80 N = 79Akákoľvek nežiaduca reakcia 79 % 66 % Lokálna 70 % 58 % Systémová 45 % 30 % Horúčka ≥ 38 °C až 38,9 °C 3 % 1 % Horúčka 39 °C až 39,9 °C 0 % 0 % Horúčka ≥ 40 °C 0 % 0 % Akákoľvek iná nežiaduca udalosť 13 % 10 %Údaje u detí a dospievajúcich vo veku 3 – 17 rokov naznačujú mierne zníženie reaktogenity po druhej
dávke bez zvýšenia výskytu horúčky.
Veľmi časté reakcie hlásené u detí a dospievajúcich vo veku 3 až 17 rokov:
Bolesť, indurácia a erytém, malátnosť, myalgia, bolesť hlavy a únava.
Nežiaduce reakcie pozorované týždeň po podaní vakcíny H1N1v Focetria u 73 dojčiat vo veku 6 –
11 mesiacov a 73 batoliat vo veku 12 – 35 mesiacov, ktorým bola podaná vakcína s obsahom 7,5 µg, boli hlásené nasledovne:
1. injekcia 2. injekciaDojčatá (vo veku 6 až 11 mesiacov) N = 73 N = 68Akákoľvek nežiaduca reakcia 79 % 65 % Lokálna 44 % 26 % Systémová 70 % 56 % Horúčka ≥ 38 °C až 38,9 °C 11 % 9 % Horúčka 39 °C až 39,9 °C 3 % 4 % Horúčka ≥ 40 °C 0 % 0 % Akákoľvek iná nežiaduca udalosť 32 % 31 % Batoľatá (vo veku 12 až 35 mesiacov) N = 73 N = 71Akákoľvek nežiaduca reakcia 70 % 71 % Lokálna 51 % 49 % Systémová 60 % 49 % Horúčka ≥ 38 °C až 38,9 °C 10 % 11 % Horúčka 39 °C až 39,9 °C 4 % 1 % Horúčka ≥ 40 °C 1 % 0 % Akákoľvek iná nežiaduca udalosť 21 % 24 %Údaje u dojčiat a batoliat vo veku 6 – 35 mesiacov naznačujú mierne zníženie reaktogenity po druhej
dávke bez zvýšenia výskytu horúčky.
Veľmi časté reakcie hlásené u 146 dojčiat a batoliat vo veku 6 až 35 mesiacov:
Citlivosť, erytém, podráždenosť, nezvyčajný plač, spavosť, hnačka, vracanie a zmeny v stravovacích návykoch. Indurácia a ekchymóza boli veľmi častými reakciami u batoliat, ale nemej častými reakciami u dojčiat.
•
Postmarketingové sledovanieOkrem nežiaducich reakcií hlásených v klinických skúšaniach sa počas skúseností po uvedení vakcíny
H1N1v Focetria na trh hlásilo nasledovné:
Poruchy krvi a lymfatického systémuLymfadenopatia.
PoruchysrdcaasrdcovejčinnostiPalpitácia, tachykardia.
Celkové poruchy a reakcie v mieste podaniaAsténia.
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaSvalová slabosť, bolesť v končatinách.
Poruchy dýchacej sústavy, hrudníka a mediastínaKašeľ.
Poruchy kože a podkožného tkanivaGeneralizované kožné reakcie zahŕňajúce pruritus, urtikáriu alebo nešpecifickú vyrážku, angioedém.
Poruchy gastrointestinálneho traktuPoruchy gastrointestinálneho traktu, ako je napr. nevoľnosť, bolesť brucha a hnačka.
Poruchy nervového systému
Bolesť hlavy, závrat, somnolencia, synkopa. Neurologické poruchy, ako je napr. neuralgia, parestézia,
kŕče a neuritída.
Poruchy imunitného systémuAlergické reakcie, anafylaxia zahŕňajúca dyspnoe, bronchospazmus, opuch hrtana, ktorá v
zriedkavých prípadoch vedie k šoku.
Počas postmarketingových prieskumov s adjuvovanými interpandemickými trivalentnými vakcínami so zložením podobným vakcíne Foclivia (pandemická H1N1 vakcína adjuvovaná s MF59C.1) boli pozorované ďalšie nežiaduce reakcie:
Zriedkavé (> 1/10 000, < 1/1 000):
Trombocytopénia (niektoré veľmi zriedkavé prípady boli závažné s počtom trombocytov menej
ako 5 000 v mm3).
Veľmizriedkavé (< 1/10 000):
Zápal ciev s prechodným postihnutím obličiek a exsudačný multiformný erytém.
Neurologické poruchy, ako napríklad encefalomyelitída, neuritída a Guillainov-Barrého syndróm.
Nežiaduce reakcie z postmarketingových prieskumov s pandemickou vakcínou: neaplikovateľné
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieNebol hlásený žiaden prípad predávkovania.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: chrípková vakcína, ATC kód: J07BB02
Táto časť popisuje klinické skúsenosti s pandemickými pohotovostnýmij H5N1 vakcínami po podaní dvoch dávok vakcíny s obsahom 7,5 mikrogramov.
Pandemické pohotovostné vakcíny obsahujú chrípkové antigény, ktoré sú odlišné od aktuálne sa vyskytujúcich vírusov chrípky. Tieto antigény sa môžu považovať za „nové“ antigény a simulujú situáciu, kedy je cieľová populácia pre vakcináciu imunologicky „neskúsená“. Údaje získané
s pandemickou pohotovostnou vakcínou podporia plán vakcinácie, ktorý sa pravdepodobne použije pre pandemickú vakcínu: údaje o klinickej účinnosti a bezpečnosti získané s pandemickými pohotovostnými vakcínami sú relevantné pre pandemické vakcíny.
Dospelí (18-60 rokov)Bola vykonaná II. fáza klinického skúšania (štúdia V87P1) s vakcínou H5N1 kombinovanou
s adjuvans MF59C.1 u 312 zdravých dospelých osôb. Dve dávky vakcíny
H5N1 (A/Vietnam/1194/2004, adjuvantná dávka/ 7,5 µg hemaglutinínu (HA)) boli podané v intervale troch týždňov 156 osobám.
V ďalšom klinickom skúšaní (III. fáza) (štúdia V87P13) sa zúčastnilo 2693 dospelých osôb a boli im podané dve dávky vakcíny obsahujúcej H5N1 (A/Vietnam/1194/2004, adjuvantná dávka/ 7,5 µg HA) v intervale troch týždňov. Imunogenita bola hodnotená v podskupine (n=197) štúdie populácie.
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči H5N1 A/Vietnam/1194/2004 u dospelých boli podľa analýzy (Single Radial Heamolysis, SRH) nasledujúce:
Anti-HA protilátka (SRH)
Štúdia V87P1
21 dní po 2. dávke
N=149
Štúdia V87P13
21 dní po 2. dávke
N=197
Miera séroprotekcie (95 % CI) 85 % (79-91) 91 % (87-95) Miera sérokonverzie (95 % CI) 85 % (78-90) 78 % (72-84) Faktor sérokonverzie (95 % CI) 7,74 (6,6-9,07) 4,03 (3,54-4,59)
Anti-HA protilátka (HI)
Štúdia V87P13
21 dní po 2. dávke
N=69
Štúdia V87P13
21 dní po 2. dávke
N=128
Počiatočný sérologický stav < 4 mm2 ≥ 4 mm2
Miera séroprotekcie (95 % CI)* 87 % (77-94) 94 % (88-97) Miera sérokonverzie (95 % CI)* 87 % (77-94) 73 % (65-81) Faktor sérokonverzie (95 % CI)** 8,87 (7,09-11) 2,71 (2,38-3,08)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
Výsledky mikroneutralizácie (MN) proti A/Vietnam/1194/2004 indikujú mieru séroprotekcie a, mieru sérokonverzie v rozsahu od 67 % (60-74) do 85 % (78-90) a 65 % (58-72) až 83 % (77-89). Imunitná odpoveď na očkovanie hodnotená MN analýzou je v súlade s výsledkami získanými SRH analýzou.
Pretrvávanie protilátok po základnom očkovaní v tejto populácii bolo hodnotené hemaglutinačnou inhibíciou (HI), SRH a MN analýzami. V porovnaní s hladinami protilátok získanými v 43. deň po ukončení základného očkovania boli hladiny protilátok v 202. deň znížené o 1/5 až 1/2 ich pôvodných hladín.
Staršie osoby (>60 rokov)
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči
H5N1 A/Vietnam/1194/2004 u osôb starších ako 60 rokov (obmedzený počet osôb starších
ako 70 rokov) merané SRH analýzou, hodnotené v dvoch klinických štúdiách boli nasledujúce:
Anti-HA protilátka (SRH)
Štúdia V87P1
21 dní po 2. dávke
N=84
Štúdia V87P13
21 dní po 2. dávke
N=210
Miera séroprotekcie (95 % CI) 80 % (70-88) 82 % (76-87) Miera sérokonverzie (95 % CI) 70 % (59-80) 63 % (56-69) Faktor sérokonverzie (95 % CI) 4,96 (3,87-6,37) 2,9 (2,53-3,31)
Anti-HA protilátka (SRH)
Štúdia V87P13
21 dní po 2. dávke
N=66
Štúdia V87P13
21 dní po 2. dávke
N=143




Počiatočný sérologický stav < 4 mm2 ≥ 4 mm2
Miera séroprotekcie (95 % CI)* 82 % (70-90) 82 % (75-88) Miera sérokonverzie (95 % CI)* 82 % (70-90) 54 % (45-62) Faktor sérokonverzie (95 % CI)** 8,58 (6,57-11) 1,91 (1,72-2,12)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
Výsledky MN voči A/Vietnam/1194/2004 indikujú mieru séroprotekcie, mieru sérokonverzie
v rozsahu od 57 % (50-64) do 79 % (68-87) a 55 % (48-62) až 58 % (47-69). Imunitná odpoveď na očkovanie hodnotená analýzou MN je podobná s výsledkami získanými SRH analýzou a poukazuje na silnejšiu imunitnú odpoveď po úplnom základnom očkovaní u populácie starších osôb.
Pretrvávanie protilátok po základnom očkovaní v tejto populácii bolo hodnotené HI, SRH a MN analýzami. V porovnaní s hladinami protilátok získanými v 43. deň po ukončení základného očkovania boli hladiny protilátok v 202. deň znížené o 1/2 až 1/5 ich hladín po očkovaní. Až 50 % starších osôb očkovaných H5N1 vakcínou kombinovanou s MF59C získalo sérologickú ochranu na 6 mesiacov.
Posilňovacia dávka
Tretia (posilňovacia) dávka H5N1 vakcíny kombinovanej s MF59C bola podaná 6 mesiacov a viac
po základnom očkovaní. Výsledky sú uvedené podľa SRH analýzy.
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči
H5N1 A/Vietnam/1194/2004 merané SRH analýzou boli nasledujúce:
Štúdia V87P1 Dospelí,
posilňovacia dávka
(6 mesiacov po2. dávke)
Štúdia V87P1 Staršie osoby,
posilňovacia dávka
(6 mesiacov po2. dávke)

SRH N=71 N=38
Miera séroprotekcie (95 % CI) 89 % (79-95) 84 % (69-94) Miera sérokonverzie (95 % CI) 83 % (72-91) 63 % (46-78) Faktor sérokonverzie (95 % CI) 5,96 (4,72-7,53) 5,15 (3,46-7,66)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
Skúsenosti s použitím posilňovacej dávky u starších osôb sú obmedzené.
•
Podporné údaje u dospelých a starších osôbV dvoch štúdiách zameraných na zistenie účinných dávok dostávalo 78 dospelých adjuvovanú
pandemickú pohotovostnú vakcínu (H5N3 alebo H9N2). Dve dávky vakcíny s kmeňom
H5N3 (A/Duck/Singapur/97) v 3 rôznych dávkach (7,5; 15 a 30 µg HA/dávku) boli podané po troch
týždňoch od seba.
Vzorky séra sa testovali na pôvodný kmeň H5N3 a tiež na niekoľko izolovaných kmeňov H5N1.
Sérologické odpovede získané pomocou testu SRH ukázali, že 100 % pacientov dosiahlo sérologickú ochranu a 100 % dosiahlo sérologickú konverziu po dvoch injekčných podaniach dávky 7,5 µg.
V prípade adjuvovanej vakcíny sa tiež zistilo, že vyvolávala vznik protilátok proti kmeňom
H5N1 izolovaným v rokoch 2003 a 2004, ktoré vykazujú určitý antigénny posun v porovnaní s pôvodnými kmeňmi.
Dve dávky vakcíny obsahujúce kmeň H9N2 (A/chicken/Hongkong/G9/97) v 4 rôznych dávkach
(3,75; 7,5; 15 a 30 μg HA/dávku) boli podané po štyroch týždňoch od seba.
Sérologické odpovede získané pomocou testu HI ukázali, že 92 % pacientov dosiahlo sérologickú ochranu a 75 % dosiahlo sérologickú konverziu po dvoch injekčných podaniach dávky 7,5 µg.
S
krížená
reaktivita
Dospelí (18 – 60 rokov)
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátky voči
H5N1 A/turkey/Turkey/05 boli po podaní 2. dávky u dospelých vo veku 18-60 rokov merané SRH
a HI analýzami nasledujúce:
Anti-HA protilátka
Štúdia V87P1
21 dní po 2. dávke
N=70
Štúdia V87P13
21 dní po 2. dávke
N=197
SRH
HI
Miera séroprotekcie* (95 % CI) 70 % (58-80) 59 % (52-66) Miera sérokonverzie* (95 % CI) NA*** 49 % (42-56) Faktor sérokonverzie** (95 % CI) NA*** 2,37 (2,1-2,67)
N=69 N=197
Miera séroprotekcie (95 % CI) 36 % (25-49) 23 % (18-30) Miera sérokonverzie (95 % CI) NA*** 19 % (14-25) Faktor sérokonverzie (95 % CI) NA*** 1,92 (1,64-2,25)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
° merané HI analýzou ≥ 40
°° geometrický priemer pomerov HI
*** vo V87P1: počiatočný stav nebol testovaný
Výsledky MN z klinických štúdií v tabuľke vyššie preukázali mieru séroprotekcie proti A/turkey/Turkey/05 v rozsahu od 27 % (17-39) (V87P1) do 39 % (32-46) (V87P13) a mieru sérokonverzie 36 % (29-43) v štúdii V87P13. Výsledky MN v štúdii V87P13 preukázali GMR proti A/turkey/Turkey/05 2,77 (2,4-3,2).
Staršie osoby (>60 rokov)
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči
H5N1 A/turkey/Turkey/05 po druhej dávke u osôb starších ako 60 rokov merané SRH a HI analýzou, boli nasledujúce:
Anti-HA protilátka
Štúdia V87P1
21 dní po 2. dávke
N=37
Štúdia V87P13
21 dní po 2. dávke
N=207
SRH
HI
Miera séroprotekcie* (95 % CI) 57 % (39-73) 20 % (18-23) Miera sérokonverzie* (95 % CI) NA*** 48 % (41-55) Faktor sérokonverzie** (95 % CI) NA*** 1,74 (1,57-1,94)
N=36 N=208
Miera séroprotekcie (95 % CI) 36 % (21-54) 25 % (19-32) Miera sérokonverzie (95 % CI) NA*** 19 % (14-25) Faktor sérokonverzie (95 % CI) NA*** 1,79 (1,56-2,06)

* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
° merané HI analýzou ≥ 40
°° geometrický priemer pomerov HI
*** vo V87P1: počiatočný stav nebol testovaný
Výsledky MN z klinických štúdií v tabuľke vyššie preukázali mieru séroprotekcie proti A/turkey/Turkey/05 v rozsahu od 11 % (3-25) (štúdia V87P1) do 30 % (24-37) (štúdia V87P13) a mieru sérokonverzie 25 % (19-31) v štúdii V87P13. Výsledky MN v štúdii V87P13 preukázali GMR proti A/turkey/Turkey/05 2,01 (1,78-2,26).
• Údaje v pediatrickej populácii
Bolo vykonané klinické skúšanie (štúdia V87P6) vakcíny H5N1 kombinovanej s adjuvans
MF59C.1 u 471 detí vo veku od 6 mesiacov do 17 rokov. Dve dávky vakcíny 7,5 mikrogramov boli podané v intervale troch týždňov a tretia dávka 12 mesiacov po prvej dávke. Po 3 týždňoch po 2. očkovaní (43. deň) všetky vekové skupiny (t.j. 6-35 mesiacov, 3-8 rokov a 9-17 rokov) dosiahli vysoké hladiny protilátok voči (A/Vietnam/1194/2004), ako bolo vyhodnotené SRH a HI analýzami a je uvedené v tabuľke nižšie*. V tejto štúdii neboli pozorované žiadne závažné nežiaduce reakcie
v súvislosti s vakcínou.
Batoľatá
(6- < 36 mesiacov)
Det
i
(3- < 9 rokov)
Dospievajúci
(9- < 18 rokov)
N=134 N=91 N=89
Miera séroprotekcie
(95 % CI)
43. Deň
Faktor sérokonverzie
HI (95 % CI)
43. deň až 1. deň Miera sérokonverzie (95 % CI)
43. Deň
97 % (92-99)
129 (109-151)
97 % (92-99)
97 % (91-99)
117 (97-142)
97 % (91-99)
89 % (80-94)
67 (51-88)
89 % (80-94)
N=133 N=91 N=90
SRH
Miera séroprotekcie
(95 % CI)
43. deň
Faktor sérokonverzie
(95 % CI)
43. deň až 1. deň Miera sérokonverzie (95 % CI)
43. deň
100 % (97-100)
16 (14-18)
98 % (95-100)
100 % (96-100)
15 (13-17)
100 % (96-100)
100 % (96-100)
14 (12-16)
99 % (94-100)
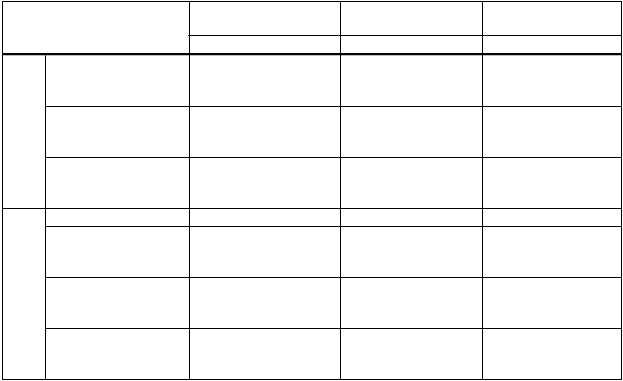
* Pretože chýbajú kritériá CHMP pre imunogenitu u detí, pre sérologické údaje získané po očkovaní detí sa použili kritériá CHMP pre imunogenitu použité na hodnotenie sezónnych chrípkových vakcín u dospelých. Význam klinickej ochrany je však neznámy.
Výsledky MN proti A/Vietnam/1194/2004 indikujú mieru séroprotekcie 99 % (95 % CI: 94-100), mieru sérokonverzie v rozsahu od 97 % (95 % CI: 91-99) do 99 % (95 % CI: 96-100) a GMR v rozsahu od 29 (95 % CI: 25-35) do 50 (95 % CI: 44-58).
Výsledky imunogenity vakcíny H1N1v Focetria (Štúdia V111_03):
Miera séroprotekcie a miera sérokonverzie stanovené pomocou metódy HI a faktor sérokonverzie vyjadrený ako pomer geometrických priemerov mier HI pre anti-HA protilátky k H1N1 po podaní jednej alebo dvoch 7,5 µg dávok vakcíny Focetria, vyhodnotený u 70 detí a dospievajúcich (9 –
17 rokov), 60 detí (3 – 8 rokov), 58 detí (12 – 35 rokov) a 49 dojčiat (6 – 11 mesiacov). Vo všetkých vyššie uvedených vekových skupinách (v celkovej populácii a aj v podskupine séronegatívnych
pacientov na začiatku) boli po 1. a 2. dávke dosiahnuté kritériá imunogenity stanovené výborom
CHMP pre dospelých (18 – 60 rokov).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s vakcínu Foclivia u jednej alebo vo viacerých podskupinách pediatrickej populácie na aktívnu imunizáciu proti vírusu chrípky A, podtyp H5N1. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
Liek Foclivia bol registrovaný za tzv. „mimoriadnych okolností“.
To znamená, že z vedeckých dôvodov nebolo možné získať všetky informácie tomto lieku.
Európska agentúra pre lieky (EMA) každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn charakteristických vlastností bude podľa potreby aktualizovať.
5
.2 Farmakokinetické vlastnosti
Neaplikovateľné.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané u vakcíny Foclivia a u sezónnej vakcíny obsahujúcej adjuvans MF59C.1 neodhalili žiadne osobitné riziko pre ľudí na základe vykonaných konvenčných štúdií toxicity po opakovanej dávke, lokálnej znášanlivosti, fertility žien a reprodukčnej a vývinovej toxicity (do konca obdobia laktácie).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Chlorid sodný, chlorid draselný, dihydrogenfosforečnan draselný,
dihydrát hydrogenfosforečnanu sodného, hexahydrát chloridu horečnatého, dihydrát chloridu vápenatého,
voda na injekciu.
Ohľadom adjuvans, pozri časť 2.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
1 rok.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
0,5 ml v injekčnej liekovke s jednou dávkou (sklenená, typ I) so zátkou (halo-butylová guma). Balenie obsahujúce 10 kusov.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Vakcína má pred použitím dosiahnuť izbovú teplotu. Pred použitím jemne potraste.
Pred podaním suspenziu vizuálne skontrolujte. V prípade akýchkoľvek častíc alebo nezvyčajného vzhľadu sa má vakcína zlikvidovať.
Všetka nepoužitá vakcína alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7
. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Seqirus S.r.l.
Via del Pozzo 3/A, S. Martino
53035 Monteriggioni (SI) Taliansko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/09/577/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. október 2009
Dátum posledného predĺženia registrácie: 19. október 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1
. NÁZOV LIEKU
Foclivia injekčná suspenzia vo viacdávkovom obale
Pandemická chrípková vakcína (H5N1) (inaktivovaný adjuvovaný povrchový antigén)
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Povrchové antigény vírusu chrípky (hemaglutinín a neuraminidáza)* nasledujúcich kmeňov:
A/Vietnam/1194/2004 (H5N1) 7,5 mikrogramu**
na 0,5 ml dávku
* pomnožený na oplodnených slepačích vajciach zo zdravých kuracích kŕdľov
** vyjadrené v mikrogramoch hemaglutinínu.
Adjuvans MF59C.1 obsahuje:
Skvalén 9,75 miligramu Polysorbát 80 1,175 miligramu Sorbitantrioleát 1,175 miligramu Citrát sodný 0,66 miligramu Kyselina citrónová 0,04 miligramu
Pomocné látky:
Tiomersal 0,05 miligramu
Toto je viacdávkový obal. Počty dávok na injekčnú liekovku, pozri časť 6.5.
Táto vakcína spĺňa odporúčania Svetovej zdravotníckej organizácie (WHO) a rozhodnutie EÚ pre prípad pandémie.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčná suspenzia. Mliečnobiela tekutina.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Profylaxia chrípky pri oficiálne vyhlásenej pandemickej situácii.
Foclivia sa má používať v súlade s oficiálnym odporúčaním.
4.2 Dávkovanie a spôsob podávania
Dávkovanie
Dospelí a staršie osoby: 0,5 ml vo zvolenom termíne.
Druhá dávka vakcíny sa má podať po uplynutí najmenej 3 týždňov.
Foclivia bola hodnotená u dospelých (vo veku 18 až 60 rokov) a starších osôb (vo veku nad 60 rokov)
po podaní po 1. a 22. dni podľa základnej očkovacej schémy.
K dispozícii sú obmedzené údaje o tretej dávke (posilňovacej) podanej 6 mesiacov po prvej dávke
(pozri časti 4.8 a 5.1).
Pediatrická populácia:
Bezpečnosť a účinnosť vakcíny Foclivia u osôb mladších ako 18 rokov neboli doteraz stanovené. V súčasnosti dostupné údaje o osobách vo veku 6 mesiacov až 18 rokov sú opísané v časti 5.1, ale neumožňujú uviesť odporúčania na dávkovanie.
Pre deti mladšie ako 6 mesiacov nie sú k dispozícii žiadne údaje.
Spôsob podávania
Očkovanie sa má vykonávať formou intramuskulárnej injekcie do deltového svalu alebo anterolaterálnej časti stehna (v závislosti od svalovej hmoty).
4.3 Kontraindikácie
Anamnéza anafylaktickej (t. j. život ohrozujúcej) reakcie na akékoľvek zložky alebo stopové zvyšky vajíčok, kuracích bielkovín, kanamycínu a neomycín sulfátu, formaldehydu
a cetyltrimetylamóniumbromidu (CTAB) tejto vakcíny. Pri pandemickej situácii však môže byť vhodné podávať vakcínu, pokiaľ sú pre prípad potreby okamžite dostupné prostriedky na resuscitáciu. Pozri časť 4.4.
4.4 Osobitné upozornenia a opatrenia pri používaní
Opatrne treba postupovať pri podávaní tejto vakcíny osobám so známou precitlivenosťou (inou než anafylaktická reakcia) na liečivo, na ktorúkoľvek z pomocných látok uvedených v časti 6.1, a na vajíčka, kuracie bielkoviny, kanamycín a neomycín, formaldehyd a cetyltrimetylamóniumbromid (CTAB).
Tak ako u všetkých injekčných vakcín, vždy má byť k dispozícii príslušná liečba a dohľad pre prípad
zriedkavej anafylaktickej reakcie po podaní vakcíny.
Ak to pandemická situácia umožňuje, u pacientov so závažným horúčkovitým ochorením alebo
s akútnou infekciou sa má očkovanie odložiť.
Foclivia sa nesmie v žiadnom prípade podávať intravaskulárne ani subkutánne.
Preto je potrebné, aby lekári posúdili prínosy a možné riziká podávania vakcíny u jedincov s trombocytopéniou alebo s akoukoľvek poruchou krvácania, ktoré by boli kontraindikáciou pre intramuskulárnu injekciu, pokiaľ možný prínos nepreváži riziko krvácaní.'
Odpoveď na protilátky u pacientov s endogénnou alebo iatrogénnou imunosupresiou môže byť nedostatočná.
Ochranná odpoveď nemusí byť vyvolaná u všetkých očkovaných osôb (pozri časť 5.1). Skrížená ochrana bola pozorovaná proti variantom vírusu súvisiacim s H5N1 počas klinického
skúšania (pozri časť 5.1).
V prípade podania druhej dávky je potrebné vziať do úvahy, že neexistujú žiadne údaje o bezpečnosti, imunogenite alebo o účinnosti, ktoré by podporili zameniteľnosť vakcíny Foclivia s inými monovalentnými H5N1 vakcínami.
Zatiaľ čo z používania vakcíny Foclivia nie sú k dispozícii žiadne údaje, boli hlásené prípady kŕčov
s horúčkou alebo bez nej u jedincov očkovaných vakcínou Focetria, ktorá je pandemickou
H1N1 vakcínou adjuvovanou s MF59.1, ktorá je podobná vakcíne Foclivia.
Väčšina febrilných kŕčov sa vyskytovala u pediatrických jedincov. Niektoré prípady boli pozorované u jedincov s epilepsiou v anamnéze. Osobitná pozornosť sa má venovať jedincom, ktorí trpia
epilepsiou a lekári majú informovať týchto jedincov (alebo ich rodičov) o možnosti objavenia sa kŕču
(pozri časť 4.8).
Po podaní alebo dokonca pred podaním akejkoľvek vakcíny sa môže objaviť synkopa (mdloba), ako psychogénna odpoveď na pichnutie injekčnou ihlou. Môže byť sprevádzaná niekoľkými neurologickými prejavmi, ako je napr. prechodná porucha videnia, parestézia a tonicko-klonické kŕče končatín počas zotavovania sa. Je dôležité, aby boli zavedené postupy, ktoré predchádzajú poraneniu pri pádoch spôsobených mdlobou.
4.5 Liekové a iné interakcie
Foclivia sa nemá podávať súbežne s inými vakcínami. Ak je však súbežné podávanie s inými vakcínami indikované, očkovanie sa má vykonávať na odlišných končatinách. Treba poznamenať, že nežiaduce reakcie môžu byť intenzívnejšie.
Ak sa pacient lieči imunosupresívami, imunologická odpoveď môže byť znížená.
Po podaní vakcíny proti chrípke sa pri stanovovaní protilátok vírusu ľudskej imunitnej nedostatočnosti-1 (HIV-1), vírusu hepatitídy C a najmä HTLV-1 pomocou metódy ELISA môžu získať falošne pozitívne výsledky sérologického testu. V takýchto prípadoch je výsledok pri metóde Western blot negatívny. Tieto prechodné falošne pozitívne výsledky testu môžu byť spôsobené tvorbou protilátok IgM ako odpoveď na podanie vakcíny.
4.6 Fertilita, gravidita a laktácia
Gravidita
Neexistujú údaje o použití Foclivie u gravidných žien.
Údaje týkajúce sa bezpečnosti sú však k dispozícii u gravidných žien vystavených vakcíne Focetria
(pandemická H1N1 vakcína podobná vakcíne Foclivia), ktorá obsahuje rovnaké množstvo
MF59C.1 ako vakcína Foclivia. Nežiaduce udalosti, ktoré boli spontánne hlásené po jej uvedení na trh a intervenčná štúdia nenaznačujú priame alebo nepriame škodlivé účinky na graviditu pri expozícii vakcíne Focetria. Okrem toho dve veľké observačné štúdie navrhnuté na posúdenie bezpečnosti pri expozícii vakcíne Focetria v gravidite nepreukázali žiadne zvýšenie výskytu gestačného diabetes mellitus, preeklampsie, potratov, pôrodov mŕtveho plodu, pôrodov s nízkou telesnou hmotnosťou, úmrtí novorodencov a vrodených malformácií medzi takmer 10 000 očkovanými gravidnými ženami
a ich potomkami v porovnaní s kontrolnou skupinou žien bez očkovania.
Je potrebné, aby lekári posúdili prínosy a možné riziká podávania vakcíny Foclivia gravidným ženám
pri zohľadnení oficiálnych odporúčaní.
Dojčenie
Vakcína sa môže používať počas laktácie.
Fertilita
Údaje týkajúce sa fertility nie sú k dispozícii.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Niektoré z účinkov uvedených v časti 4.8 „Nežiaduce účinky“ môžu ovplyvniť schopnosť viesť
vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinky
Súhrn profilu bezpečnosti
Dospelí a staršie osoby (starší ako 18 rokov)
V klinických skúšaniach vykonaných s pandemickou pohotovostnou adjuvovanou H5N1 vakcínou u dospelých a starších osôb (viac informácií pozri časť 5.1) mala väčšina reakcií miernu povahu, krátku dĺžku trvania a bola kvalitatívne podobná reakciám, ktoré boli vyvolané štandardnými sezónnymi vakcínami proti chrípke. Vo všeobecnosti je akceptované, že adjuvantný účinok, ktorý vedie k zvýšenej imunogenicite, je spojený s mierne vyššou frekvenciou výskytu lokálnych reakcií (najmä mierna bolesť) v porovnaní so štandardnými neadjuvovanými vakcínami proti chrípke. Po podaní druhej dávky sa objavilo menej reakcií v porovnaní s prvou dávkou.
Deti a dospievajúci vo veku 6 mesiacov až 17 rokov:
V klinickom skúšaní II fázy (Štúdia V87P6) sa u detí a dospievajúcich hodnotila bezpečnosť pandemickej pohotovostnej adjuvovanej H5N1 vakcíny (viac informácií pozri v časti 5.1). Bez ohľadu na vek bola reaktogenita vyššia po prvej dávke ako po druhej dávke očkovania.
Reaktogenita po tretej dávke podanej 12 mesiacov po druhej dávke bola vyššia ako po prvej a aj druhej dávke. Percentuálny pomer jedincov, ktorí hlásili lokálne reakcie, bol vyšší ako v skupine
s vyšším vekom, najmä z dôvodu vyššej miery hlásení bolesti. Najčastejšími vyžiadanými lokálnymi reakciami u batoliat boli erytém a citlivosť; podráždenosť a nezvyčajný plač boli najčastejšie hlásenými vyžiadanými systémovými reakciami. Najčastejšie hlásenou vyžiadanou lokálnou reakciou u detí a dospievajúcich bola bolesť a únava a bolesť hlavy boli najčastejšie hlásenými vyžiadanými systémovými reakciami. Medzi vekovými skupinami sa pri malom percentuálnom pomere hlásila horúčka. Údaje týkajúce sa bezpečnosti po prvej a druhej dávke u detí a dospievajúcich pri podobnej pandemickej vakcíne (Focetria H1N1v) naznačujú porovnateľný profil bezpečnosti ako je profil bezpečnosti, ktorý sa hlásil pri pandemickej pohotovostnej aH5N1 vakcíne (Foclivia).
Frekvencia výskytu nežiaducich reakcií v klinických skúšaniach a dohľade po uvedení vakcíny na trh
je uvedená nižšie.
• Klinické štúdie
Nežiaducereakciezklinickýchskúšaníudospelýchastaršíchosôb(staršíchako18rokov).
Výskyt nežiaducich reakcií bol hodnotený v štyroch klinických štúdiách s rôznymi kmeňmi chrípky
a s rozličným zložením (H5N3, H9N2 a H5N1), ktorých sa zúčastnilo približne 3696 dospelých
a starších osôb. Z týchto osôb bola 3618 osobám podaná pandemická pohotovostná vakcína Foclivia
(A/H5N1) (pozri časť 5.1).
Nežiaduce reakcie zistené počas klinických štúdií s pandemickou pohotovostnou vakcínou Foclivia sú uvedené nižšie.
Výskyt príznakov pozorovaných u pacientov vo veku nad 60 rokov bol nižší v porovnaní s populáciou vo veku 18 až 60 rokov.
V rámci jednotlivých skupín frekvencií sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti:
Poruchy nervového systému
Časté (> 1/100, < 1/10): bolesť hlavy
Poruchy kože a podkožného tkaniva
Časté (> 1/100, < 1/10): potenie
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
Časté (> 1/100, < 1/10): artralgia a myalgia
Celkové ochorenia a reakcie v mieste podania
Časté (> 1/100, < 1/10): sčervenanie miesta vpichu, opuch miesta vpichu, stvrdnutie miesta vpichu,
podliatina v mieste vpichu a bolesť miesta vpichu, horúčka, malátnosť, únava a chvenie
Väčšina týchto reakcií zvyčajne ustúpi do 1 až 2 dní bez liečby.
Nežiaducereakciezklinickýchskúšaníudetívoveku6mesiacovaž17rokov
Klinické skúšanie (štúdia V87P6) bolo vykonané s vakcínou H5N1 kombinovanou s adjuvans
MF59C.1 (n=334) v porovnaní so sezónnou vakcínou proti chrípke (n=137).
Prvá dávka Druhá dávka
(21 dní po prvej dávke)
H5N1 vakcína s adjuvans
Tretia dávka
(12 mesiacov po druhej dávke)

 Batoľatá (6 až < 36 mesiacov) N=145 N=138 N=124
Batoľatá (6 až < 36 mesiacov) N=145 N=138 N=124
Akékoľvek 76 % 68 % 80 % Lokálne 47 % 46 % 60 % Systémové 59 % 51 % 54 % Horúčka ≥ 38 °C (≥ 40 °C) 0 % 0 % 0 % Akékoľvek iné nežiaduce účinky 54 % 49 % 35 %
Deti (3 až < 9 rokov) N=96 N=93 N=85Akékoľvek 72 % 68 % 79 % Lokálne 66 % 58 % 74 % Systémové 32 % 33 % 45 % Horúčka ≥ 38 °C (≥ 40 °C) 4 % 2 % 6 % Akékoľvek iné nežiaduce účinky 36 % 31 % 19 %
Adolescenti (9 až < 18 rokov) N=93 N=91 N=83Akékoľvek 91 % 82 % 89 % Lokálne 81 % 70 % 81 % Systémové 69 % 52 % 69 % Horúčka ≥ 38 °C (≥ 40 °C) 0 % 1 % 2 % Akékoľvek iné nežiaduce účinky 30 % 27 % 22 %
Focetria (H1N1v)Nežiaduce reakcie pozorované týždeň po podaní vakcíny H1N1v Focetria u 77 detí vo veku 3 –
8 rokov a 80 detí a dospievajúcich vo veku 9 – 17 rokov, ktorým bola podaná vakcína s obsahom 7,5 µg, boli hlásené nasledovne:
1. injekcia 2. injekciaDeti (vo veku od 3 do 8 rokov) N = 77 N = 75Akákoľvek nežiaduca reakcia 74 % 69 % Lokálna 62 % 56 % Systémová 39 % 35 % Horúčka ≥ 38 °C až 38,9 °C 4 % 1 % Horúčka 39 °C až 39,9 °C 0 % 1 % Horúčka ≥ 40 °C 0 % 0 % Akákoľvek iná nežiaduca udalosť 14 % 17 % Dospievajúci (vo veku 9 až 17 rokov) N = 80 N = 79Akákoľvek nežiaduca reakcia 79 % 66 % Lokálna 70 % 58 % Systémová 45 % 30 % Horúčka ≥ 38 °C až 38,9 °C 3 % 1 % Horúčka 39 °C až 39,9 °C 0 % 0 % Horúčka ≥ 40 °C 0 % 0 % Akákoľvek iná nežiaduca udalosť 13 % 10 %Údaje u detí a dospievajúcich vo veku 3 – 17 rokov naznačujú mierne zníženie reaktogenity po druhej
dávke bez zvýšenia výskytu horúčky.
Veľmi časté reakcie hlásené u detí a dospievajúcich vo veku 3 až 17 rokov:
Bolesť, indurácia a erytém, malátnosť, myalgia, bolesť hlavy a únava.
Nežiaduce reakcie pozorované týždeň po podaní vakcíny H1N1v Focetria u 73 dojčiat vo veku 6 –
11 mesiacov a 73 batoliat vo veku 12 – 35 mesiacov, ktorým bola podaná vakcína s obsahom 7,5 µg, boli hlásené nasledovne:
1. injekcia 2. injekciaDojčatá (vo veku 6 až 11 mesiacov) N = 73 N = 68Akákoľvek nežiaduca reakcia 79 % 65 % Lokálna 44 % 26 % Systémová 70 % 56 % Horúčka ≥ 38 °C až 38,9 °C 11 % 9 % Horúčka 39 °C až 39,9 °C 3 % 4 % Horúčka ≥ 40 °C 0 % 0 % Akákoľvek iná nežiaduca udalosť 32 % 31 % Batoľatá (vo veku 12 až 35 mesiacov) N = 73 N = 71Akákoľvek nežiaduca reakcia 70 % 71 % Lokálna 51 % 49 % Systémová 60 % 49 % Horúčka ≥ 38 °C až 38,9 °C 10 % 11 % Horúčka 39 °C až 39,9 °C 4 % 1 % Horúčka ≥ 40 °C 1 % 0 % Akákoľvek iná nežiaduca udalosť 21 % 24 %Údaje u dojčiat a batoliat vo veku 6 – 35 mesiacov naznačujú mierne zníženie reaktogenity po druhej
dávke bez zvýšenia výskytu horúčky.
Veľmi časté reakcie hlásené u 146 dojčiat a batoliat vo veku 6 až 35 mesiacov:
Citlivosť, erytém, podráždenosť, nezvyčajný plač, spavosť, hnačka, vracanie a zmeny v stravovacích návykoch. Indurácia a ekchymóza boli veľmi častými reakciami u batoliat, ale nemej častými reakciami u dojčiat.
•
Postmarketingové sledovanieOkrem nežiaducich reakcií hlásených v klinických skúšaniach sa počas skúseností po uvedení vakcíny
H1N1v Focetria na trh hlásilo nasledovné:
Poruchy krvi a lymfatického systémuLymfadenopatia.
PoruchysrdcaasrdcovejčinnostiPalpitácia, tachykardia.
Celkové poruchy a reakcie v mieste podaniaAsténia.
Poruchy kostrovej a svalovej sústavy a spojivového tkanivaSvalová slabosť, bolesť v končatinách.
Poruchy dýchacej sústavy, hrudníka a mediastínaKašeľ.
Poruchy kože a podkožného tkanivaGeneralizované kožné reakcie zahŕňajúce pruritus, urtikáriu alebo nešpecifickú vyrážku, angioedém.
Poruchy gastrointestinálneho traktu
Poruchy gastrointestinálneho traktu, ako je napr. nevoľnosť, bolesť brucha a hnačka.
Poruchy nervového systémuBolesť hlavy, závrat, somnolencia, synkopa. Neurologické poruchy, ako je napr. neuralgia, parestézia,
kŕče a neuritída.
Poruchy imunitného systémuAlergické reakcie, anafylaxia zahŕňajúca dyspnoe, bronchospazmus, opuch hrtana, ktorá v
zriedkavých prípadoch vedie k šoku.
Počas postmarketingových prieskumov s adjuvovanými interpandemickými trivalentnými vakcínami so zložením podobným vakcíne Foclivia (pandemická H1N1 vakcína adjuvovaná s MF59C.1) boli pozorované ďalšie nežiaduce reakcie:
Zriedkavé (> 1/10 000, < 1/1 000):
Trombocytopénia (niektoré veľmi zriedkavé prípady boli závažné s počtom trombocytov menej ako 5 000 v mm3).
Veľmizriedkavé (< 1/10 000):
Zápal ciev s prechodným postihnutím obličiek a exsudačný multiformný erytém.
Neurologické poruchy, ako napríklad encefalomyelitída, neuritída a Guillainov-Barrého syndróm. Nežiaduce reakcie z postmarketingových prieskumov s pandemickou vakcínou: neaplikovateľné
Táto vakcína obsahuje tiomersal (organická zložka s obsahom ortuti) ako konzervačnú látku, a preto je
možné, že sa môže objaviť senzibilizačná reakcia (pozri časť 4.4).
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie prostredníctvom národného systému hlásenia uvedeného
v Prílohe V.
4.9 PredávkovanieNebol hlásený žiaden prípad predávkovania.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: chrípková vakcína, ATC kód: J07BB02
Táto časť popisuje klinické skúsenosti s pandemickými pohotovostnýmij H5N1 vakcínami po podaní dvoch dávok vakcíny s obsahom 7,5 mikrogramov.
Pandemické pohotovostné vakcíny obsahujú chrípkové antigény, ktoré sú odlišné od aktuálne sa vyskytujúcich vírusov chrípky. Tieto antigény sa môžu považovať za „nové“ antigény a simulujú situáciu, kedy je cieľová populácia pre vakcináciu imunologicky „neskúsená“. Údaje získané
s pandemickou pohotovostnou vakcínou podporia plán vakcinácie, ktorý sa pravdepodobne použije pre pandemickú vakcínu: údaje o klinickej účinnosti a bezpečnosti získané s pandemickými pohotovostnými vakcínami sú relevantné pre pandemické vakcíny.
Dospelí (18-60 rokov)
Bola vykonaná II. fáza klinického skúšania (štúdia V87P1) s vakcínou H5N1 kombinovanou
s adjuvans MF59C.1 u 312 zdravých dospelých osôb. Dve dávky vakcíny
H5N1 (A/Vietnam/1194/2004, adjuvantná dávka/ 7,5 µg hemagluitinínu (HA)) boli podané v intervale
troch týždňov 156 osobám.
V ďalšom klinickom skúšaní (III. fáza) (štúdia V87P13) sa zúčastnilo 2693 dospelých osôb a boli im podané dve dávky vakcíny obsahujúcej H5N1 (A/Vietnam/1194/2004, adjuvantná dávka/ 7,5 µg(HA) v intervale troch týždňov. Imunogenita bola hodnotená v podskupine (n=197) štúdie populácie.
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči H5N1 A/Vietnam/1194/2004 u dospelých boli podľa analýzy (Single Radial Heamolysis, SRH) nasledujúce:
Anti-HA protilátka (SRH)
Štúdia V87P1
21 dní po 2. dávke
N=149
Štúdia V87P13
21 dní po 2. dávke
N=197
Miera séroprotekcie (95 % CI) 85 % (79-91) 91 % (87-95) Miera sérokonverzie (95 % CI) 85 % (78-90) 78 % (72-84) Faktor sérokonverzie (95 % CI) 7,74 (6,6-9,07) 4,03 (3,54-4,59)
Anti-HA protilátka (HI)
Štúdia V87P13
21 dní po 2. dávke
N=69
Štúdia V87P13
21 dní po 2. dávke
N=128
Počiatočný sérologický stav < 4 mm2 ≥ 4 mm2
Miera séroprotekcie (95 % CI)* 87 % (77-94) 94 % (88-97) Miera sérokonverzie (95 % CI)* 87 % (77-94) 73 % (65-81) Faktor sérokonverzie (95 % CI)** 8,87 (7,09-11) 2,71 (2,38-3,08)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
Výsledky mikroneutralizácie (MN) proti A/Vietnam/1194/2004 indikujú mieru séroprotekcie a, mieru sérokonverzie v rozsahu od 67 % (60-74) do 85 % (78-90) a 65 % (58-72) až 83 % (77-89). Imunitná odpoveď na očkovanie hodnotená MN analýzou je v súlade s výsledkami získanými SRH analýzou.
Pretrvávanie protilátok po základnom očkovaní v tejto populácii bolo hodnotené hemaglutinačnou inhibíciou (HI), SRH a MN analýzami. V porovnaní s hladinami protilátok získanými v 43. deň po ukončení základného očkovania boli hladiny protilátok v 202. deň znížené o 1/5 až 1/2 ich pôvodných hladín.
Staršie osoby (>60 rokov)
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči
H5N1 A/Vietnam/1194/2004 u osôb starších ako 60 rokov (obmedzený počet osôb starších
ako 70 rokov) merané SRH analýzou, hodnotené v dvoch klinických štúdiách boli nasledujúce:
Anti-HA protilátka (SRH)
Štúdia V87P1
21 dní po 2. dávke
N=84
Štúdia V87P13
21 dní po 2. dávke
N=210
Anti-HA protilátka (SRH)
Štúdia V87P13
21 dní po 2. dávke
N=66
Štúdia V87P13
21 dní po 2. dávke
N=143
Počiatočný sérologický stav < 4 mm2 ≥ 4 mm2
Miera séroprotekcie (95 % CI)* 82 % (70-90) 82 % (75-88) Miera sérokonverzie (95 % CI)* 82 % (70-90) 54 % (45-62) Faktor sérokonverzie (95 % CI)** 8,58 (6,57-11) 1,91 (1,72-2,12)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
Výsledky MN voči A/Vietnam/1194/2004 indikujú mieru séroprotekcie, mieru sérokonverzie
v rozsahu od 57 % (50-64) do 79 % (68-87) a 55 % (48-62) až 58 % (47-69). Imunitná odpoveď na očkovanie hodnotená analýzou MN je podobná s výsledkami získanými SRH analýzou a poukazuje na silnejšiu imunitnú odpoveď po úplnom základnom očkovaní u populácie starších osôb.
Pretrvávanie protilátok po základnom očkovaní v tejto populácii bolo hodnotené HI, SRH a MN analýzami. V porovnaní s hladinami protilátok získanými v 43. deň po ukončení základného očkovania boli hladiny protilátok v 202. deň znížené o 1/2 až 1/5 ich hladín po očkovaní. Až 50 % starších osôb očkovaných H5N1 vakcínou kombinovanou s MF59C získalo sérologickú ochranu na
6 mesiacov.
Posilňovacia dávka
Tretia (posilňovacia) dávka H5N1 vakcíny kombinovanej s MF59C bola podaná 6 mesiacov a viac po
základnom očkovaní. Výsledky sú uvedené podľa SRH analýzy.
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči
H5N1 A/Vietnam/1194/2004 merané SRH analýzou boli nasledujúce:
Štúdia V87P1 Dospelí,
posilňovacia dávka
(6 mesiacov po 2. dávke)
Štúdia V87P1 Staršie osoby,
posilňovacia dávka
(6 mesiacov po2. dávke)
SRH N=71 N=38
Miera séroprotekcie (95 % CI) 89 % (79-95) 84 % (69-94) Miera sérokonverzie (95 % CI) 83 % (72-91) 63 % (46-78) Faktor sérokonverzie (95 % CI) 5,96 (4,72-7,53) 5,15 (3,46-7,66)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
Skúsenosti s použitím posilňovacej dávky u starších osôb sú obmedzené.
•
Podporné údaje u dospelých a starších osôbV dvoch štúdiách zameraných na zistenie účinných dávok dostávalo 78 dospelých adjuvovanú
pandemickú pohotovostnú vakcínu (H5N3 alebo H9N2). Dve dávky vakcíny s kmeňom
H5N3 (A/Duck/Singapur/97) v 3 rôznych dávkach (7,5; 15 a 30 µg HA/dávku) boli podané po troch
týždňoch od seba.
Vzorky séra sa testovali na pôvodný kmeň H5N3 a tiež na niekoľko izolovaných kmeňov H5N1.
Sérologické odpovede získané pomocou testu SRH ukázali, že 100 % pacientov dosiahlo sérologickú ochranu a 100 % dosiahlo sérologickú konverziu po dvoch injekčných podaniach dávky 7,5 µg.
V prípade adjuvovanej vakcíny sa tiež zistilo, že vyvolávala vznik protilátok proti kmeňom
H5N1 izolovaným v rokoch 2003 a 2004, ktoré vykazujú určitý antigénny posun v porovnaní s pôvodnými kmeňmi.
Dve dávky vakcíny obsahujúce kmeň H9N2 (A/chicken/Hongkong/G9/97) v 4 rôznych dávkach
(3,75; 7,5; 15 a 30 μg HA/dávku) boli podané po štyroch týždňoch od seba.
Sérologické odpovede získané pomocou testu HI ukázali, že 92 % pacientov dosiahlo sérologickú
ochranu a 75 % dosiahlo sérologickú konverziu po dvoch injekčných podaniach dávky 7,5 µg.
Skríženáreaktivita
Dospelí (18 – 60 rokov)
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátky voči
H5N1 A/turkey/Turkey/05 boli po podaní 2. dávky u dospelých vo veku 18-60 rokov merané SRH a
HI analýzami nasledujúce:
Anti-HA protilátka
Štúdia V87P1
21 dní po 2. dávke
N=70
Štúdia V87P13
21 dní po 2. dávke
N=197
SRH
HI
Miera séroprotekcie* (95 % CI) 70 % (58-80) 59 % (52-66) Miera sérokonverzie* (95 % CI) NA*** 49 % (42-56) Faktor sérokonverzie** (95 % CI) NA*** 2,37 (2,1-2,67)
N=69 N=197
Miera séroprotekcie (95 % CI) 36 % (25-49) 23 % (18-30) Miera sérokonverzie (95 % CI) NA*** 19 % (14-25) Faktor sérokonverzie (95 % CI) NA*** 1,92 (1,64-2,25)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
° merané HI analýzou ≥ 40
°° geometrický priemer pomerov HI
*** vo V87P1: počiatočný stav nebol testovaný
Výsledky MN z klinických štúdií v tabuľke vyššie preukázali mieru séroprotekcie proti A/turkey/Turkey/05 v rozsahu od 27 % (17-39) (V87P1) do 39 % (32-46) (V87P13) a mieru sérokonverzie 36 % (29-43) v štúdii V87P13. Výsledky MN v štúdii V87P13 preukázali GMR proti A/turkey/Turkey/05 2,77 (2,4-3,2).
Staršie osoby (>60 rokov)
Miera séroprotekcie*, miera sérokonverzie* a faktor sérokonverzie** pre anti-HA protilátku voči
H5N1 A/turkey/Turkey/05 po druhej dávke u osôb starších ako 60 rokov merané SRH a HI analýzou, boli nasledujúce:
Anti-HA protilátka
Štúdia V87P1
21 dní po 2. dávke
N=37
Štúdia V87P13
21 dní po 2. dávke
N=207
SRH
HI
Miera séroprotekcie* (95 % CI) 57 % (39-73) 20 % (18-23) Miera sérokonverzie* (95 % CI) NA*** 48 % (41-55) Faktor sérokonverzie** (95 % CI) NA*** 1,74 (1,57-1,94)
N=36 N=208
Miera séroprotekcie (95 % CI) 36 % (21-54) 25 % (19-32) Miera sérokonverzie (95 % CI) NA*** 19 % (14-25) Faktor sérokonverzie (95 % CI) NA*** 1,79 (1,56-2,06)
* merané SRH analýzou ≥ 25 mm2
** geometrický priemer pomerov SRH
° merané HI analýzou ≥ 40
°° geometrický priemer pomerov HI
*** vo V87P1: počiatočný stav nebol testovaný
Výsledky MN z klinických štúdií v tabuľke vyššie preukázali mieru séroprotekcie proti A/turkey/Turkey/05 v rozsahu od 11 % (3-25) (štúdia V87P1) do 30 % (24-37) (štúdia V87P13) a mieru sérokonverzie 25 % (19-31) v štúdii V87P13. Výsledky MN v štúdii V87P13 preukázali GMR proti A/turkey/Turkey/05 2,01 (1,78-2,26).
• Údaje v pediatrickej populácii
Bolo vykonané klinické skúšanie (štúdia V87P6) vakcíny H5N1 kombinovanej s adjuvans
MF59C.1 u 471 detí vo veku od 6 mesiacov do 17 rokov. Dve dávky vakcíny 7,5 mikrogramov boli podané v intervale troch týždňov a tretia dávka 12 mesiacov po prvej dávke. Po 3 týždňoch po 2. očkovaní (43. deň) všetky vekové skupiny (t.j. 6-35 mesiacov, 3-8 rokov a 9-17 rokov) dosiahli vysoké hladiny protilátok voči (A/Vietnam/1194/2004), ako bolo vyhodnotené SRH a HI analýzami a je uvedené v tabuľke nižšie*. V tejto štúdii neboli pozorované žiadne závažné nežiaduce reakcie v súvislosti s vakcínou.
Batoľatá
(6- < 36 mesiacov)
Det
i
(3- < 9 rokov)
Dospievajúci
(9- < 18 rokov)
Miera séroprotekcie
N=134 N=91 N=89
(95 % CI)
43. deň
Faktor sérokonverzie
HI (95 % CI)
43. deň až 1. deň Miera sérokonverzie (95 % CI)
43. deň
97 %
(92-99)
129 (109-151)
97 % (92-99)
97 %
(91-99)
117 (97-142)
97 % (91-99)
89 %
(80-94)
67 (51-88)
89 % (80-94)
N=133 N=91 N=90
SRH
Miera séroprotekcie
(95 % CI)
43. deň
Faktor sérokonverzie
(95 % CI)
43. deň až 1. deň Miera sérokonverzie (95 % CI)
43. deň
100 % (97-100)
16 (14-18)
98 % (95-100)
100 % (96-100)
15 (13-17)
100 % (96-100)
100 % (96-100)
14 (12-16)
99 % (94-100)

* Pretože chýbajú kritériá CHMP pre imunogenitu u detí, pre sérologické údaje získané po očkovaní detí sa použili kritériá CHMP pre imunogenitu použité na hodnotenie sezónnych chrípkových vakcín u dospelých. Význam klinickej ochrany je však neznámy.
Výsledky MN proti A/Vietnam/1194/2004 indikujú mieru séroprotekcie 99 % (95 % CI: 94-100), mieru sérokonverzie v rozsahu od 97 % (95 % CI: 91-99) do 99 % (95 % CI: 96-100) a GMR v rozsahu od 29 (95 % CI: 25-35) do 50 (95 % CI: 44-58).
Výsledky imunogenity vakcíny H1N1v Focetria (štúdia V111_03):
Miera séroprotekcie a miera sérokonverzie stanovené pomocou metódy HI a faktor
sérokonverzie vyjadrený ako geometrických priemer priemerov HI pre anti-HA protilátky k H1N1 po podaní jednej alebo dvoch 7,5 µg dávok vakcíny Focetria, hodnotený u 70 detí a dospievajúcich
(9 – 17 rokov), 60 detí (3 – 8 rokov), 58 detí (12 – 35 rokov) a 49 dojčiat (6 – 11 mesiacov). Vo všetkých vyššie uvedených vekových skupinách (v celkovej populácii a aj v podskupine séronegatívnych pacientov na začiatku) boli po 1. a aj po 2. dávke dosiahnuté kritériá imunogenity stanovené výborom CHMP pre dospelých (18 – 60 rokov).
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s vakcínou Foclivia v jednej alebo vo viacerých podskupinách pediatrickej populácie na aktívnu imunizáciu proti vírusu chrípky A, podtyp H5N1. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
Liek Foclivia bol registrovaný za tzv. „mimoriadnych okolností“.
To znamená, že z vedeckých dôvodov nebolo možné získať všetky informácie o tomto lieku. Európska agentúra pre lieky (EMA) každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn charakteristických vlastností bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Neaplikovateľné.
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané u vakcíny Foclivia a sezónnej vakcíny obsahujúcej adjuvans MF59C.1 neodhalili žiadne osobitné riziko pre ľudí na základe vykonaných konvenčných štúdií toxicity po opakovanej dávke, lokálnej znášanlivosti, fertility žien a reprodukčnej a vývinovej toxicity (do konca obdobia laktácie).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Chlorid sodný, chlorid draselný, dihydrogenfosforečnan draselný,
dihydrát hydrogenfosforečnanu sodného, hexahydrát chloridu horečnatého, dihydrát chloridu vápenatého,
tiomersal,
voda na injekciu.
Ohľadom adjuvans, pozri časť 2.
6.2 Inkompatibility
Nevykonali sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
1 rok.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C – 8°C). Neuchovávajte v mrazničke. Uchovávajte v pôvodnom obale na ochranu pred svetlom.
6.5 Druh obalu a obsah balenia
5,0 ml v injekčnej liekovke s 10 dávkami (sklenená, typ I) so zátkou (halo-butylová guma). Balenie obsahujúce 10 kusov.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6
.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Pred podaním suspenziu vizuálne skontrolujte. V prípade akýchkoľvek častíc alebo nezvyčajného vzhľadu sa má vakcína zlikvidovať.
Pred odobratím dávky (0,5 ml) vakcíny do injekčnej striekačky viacdávkovú injekčnú liekovku jemne pretrepte. Pred podaním má odobraté množstvo vakcíny dosiahnuť izbovú teplotu. Hoci viacdávkové injekčné liekovky vakcíny Foclivia obsahujú konzervačnú látku, ktorá zabraňuje rastu mikroorganizmov, minimalizácia rizika kontaminácie viacdávkovej injekčnej liekovky počas odberu každej dávky je zodpovednosťou používateľa.
Na štítok injekčnej liekovky si zaznamenajte dátum a čas odberu prvej dávky.
Medzi použitiami zabezpečte odporúčané podmienky na uchovávanie viacdávkovej injekčnej liekovky pri teplote 2°C až 8°C. Pokiaľ je to možné, viacdávková injekčná liekovka sa má použiť
počas 24 hodín po prvom odbere.
K dispozícii sú údaje, ktoré naznačujú, že viacdávkové injekčné liekovky sa môžu použiť až maximálne do 72 hodín po odobratí prvej dávky, avšak takého predĺženie obdobia na uchovávanie nemá byť uprednostňovanou voľbou.
Všetka nepoužitá vakcína alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIISeqirus S.r.l.
Via del Pozzo 3/A, S. Martino
53035 Monteriggioni (SI) Taliansko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/09/577/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 19. október 2009
Dátum posledného predĺženia registrácie: 19. október 2014
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.