
závažnosti týchto nežiaducich reakcií. Opatrnosť sa vyžaduje v liečbe
pacientov, ktorých aktuálny zdravotný stav môže byť zhoršený zvýšením tlaku krvi, hypokaliémiou (napr. u pacientov liečených srdcovými glykozidmi), alebo zadržiavaním tekutín (napr. u pacientov so zlyhaním srdca, závažnou alebo nestabilnou angínou pektoris, nedávnym infarktom myokardu alebo ventrikulárnou arytmiou a u pacientov s ťažkou poruchou funkcie obličiek).
Abiraterone Glenmark sa má používať opatrne u pacientov s kardiovaskulárnym ochorením v anamnéze. Zo štúdií fázy III s abiraterónom boli vylúčení pacienti s nekontrolovanou hypertenziou, klinicky významným ochorením srdca s dokumentovaným infarktom myokardu, alebo arteriálnymi trombotickými príhodami za posledných 6 mesiacov, závažnou alebo nestabilnou angínou alebo zlyhaním srdca triedy III alebo IV (štúdia 301) alebo zlyhávanie srdca triedy II až IV (štúdie 3011 a 302) podľa New York Heart Association (NYHA) alebo nameranou ejekčnou frakciou srdca < 50 %. V štúdiách 3011 a 302 boli vylúčení pacienti s atriálnou fibriláciou alebo inou srdcovou arytmiou, ktorá si vyžadovala liečbu. Bezpečnosť u pacientov s ejekčnou frakciou ľavej komory (LVEF) < 50 % alebo zlyhávaním srdca triedy III alebo IV podľa NYHA (v štúdii 301) alebo zlyhávaním srdca triedy II až IV podľa NYHA (v štúdiách 3011 a 302) nebola stanovená (pozri časti 4.8 a 5.1).
Pred začatím liečby u pacientov s významným rizikom kongestívneho zlyhania srdca (napr. zlyhávanie srdca, nekontrolovaná hypertenzia alebo srdcové príhody ako napríklad ischemická choroba srdca v anamnéze) je potrebné zvážiť posúdenie funkcie srdca (napr. echokardiogram). Pred začatím liečby abiraterónom sa má liečiť zlyhávanie srdca a majú sa optimalizovať funkcie srdca. Hypertenzia, hypokaliémia a zadržiavanie tekutín sa má upraviť a kontrolovať. Počas liečby sa má každé 2 týždne v prvých 3 mesiacoch, a potom na mesačnej báze sledovať tlak krvi, hladina draslíka v sére, zadržiavanie tekutín (nárast hmotnosti, periférny opuch) a iné prejavy a príznaky kongestívneho zlyhania srdca a abnormality sa majú upraviť. V súvislosti s liečbou s abiraterónom bolo u pacientov s hypokaliémiou pozorované predĺženie QT intervalu. Má sa posúdiť funkčnosť srdca ako je klinicky indikované, začať vhodná liečba a zvážiť prerušenie tejto liečby, ak sa vyskytne klinicky významný pokles vo funkčnosti srdca (pozri časť 4.2).
Hepatotoxicita a porucha funkcie pečene
V kontrolovaných klinických štúdiách sa vyskytlo výrazné zvýšenie hodnôt hepatálnych enzýmov, čo
viedlo k prerušeniu liečby alebo zmene dávkovania (pozri časť 4.8). Hladina sérových transamináz sa má merať pred začiatkom terapie, každé dva týždne počas prvých troch mesiacov liečby a potom raz mesačne. Ak sa objavia klinické príznaky alebo prejavy poukazujúce na hepatotoxicitu, je potrebné okamžite stanoviť sérové transaminázy. Ak sa kedykoľvek ALT alebo AST zvýši 5-násobne nad HHN, terapiu je
potrebné okamžite prerušiť a dôkladne monitorovať funkciu pečene. Obnovenie liečby je možné uskutočniť až po návrate hepatálnych testov pacienta na východiskové hodnoty, pričom sa liek podáva v zníženej
dávke (pozri časť 4.2).
Ak sa kedykoľvek počas terapie u pacientov objaví závažná hepatotoxicita (ALT alebo AST 20-násobne vyššia než HHN), liečba sa má ukončiť a u týchto pacientov sa už nemá obnoviť.
Pacienti s aktívnou alebo symptomatickou vírusovou hepatitídou boli vylúčení z klinických štúdií, z toho
dôvodu neexistujú žiadne údaje, ktoré by podporovali použitie lieku Abiraterone Glenmark v tejto populácii.
Neexistujú žiadne údaje o klinickej bezpečnosti a účinnosti viacnásobných dávok abiraterón-acetátu, keď sa podával pacientom so stredne ťažkou alebo ťažkou poruchou funkcie pečene (Childova-Pughova trieda B alebo C). Použitie lieku Abiraterone Glenmark sa má dôkladne posúdiť u pacientov so stredne ťažkou poruchou funkcie pečene, u ktorých má prínos zjavne prevážiť potenciálne riziko (pozri časti 4.2 a 5.2). Abiraterone Glenmark sa nemá používať u pacientov s ťažkou poruchou funkcie pečene (pozri časti 4.2, 4.3 a 5.2).
Po uvedení lieku na trh boli zriedkavo hlásené prípady akútneho zlyhania pečene a fulminantnej hepatitídy, niektoré so smrteľnými následkami (pozri časť 4.8).
Ukončenieliečbykortikosteroidmi a zvládaniestresovýchsituácií
V prípade, že sa u pacientov ukončí liečba prednizónom alebo prednizolónom, odporúča sa opatrnosť
a monitorovanie s ohľadom na adrenokortikálnu insuficienciu. Ak sa abiraterón naďalej podáva aj potom, ako bola ukončená liečba kortikosteroidmi, je potrebné pacientov sledovať z hľadiska príznakov nadbytku mineralokortikoidov (pozri informáciu vyššie).
U pacientov, ktorí dostávajú prednizón alebo prednizolón a sú vystavení neobvyklému stresu, je možné indikovať zvýšenú dávku kortikosteroidov pred stresujúcou situáciou, počas nej alebo po nej.
Hustotakostí
U mužov s metastatickým pokročilým karcinómom prostaty sa môže vyskytnúť zníženie kostnej hustoty.
Použitie lieku Abiraterone Glenmark v kombinácii s glukokortikoidmi môže tento účinok zosilniť.
Užívanieketokonazolu v minulosti
U pacientov, ktorí sa už predtým liečili na karcinóm prostaty ketokonazolom, možno očakávať nižší podiel
odpovedí.
Hyperglykémia
Užívanie glukokortikoidov môže zvýšiť hyperglykémiu, preto sa má u pacientov s diabetom často merať
hladina cukru v krvi.
Hypoglykémia
U pacientov s už existujúcim diabetom užívajúcich pioglitazón alebo repaglinid boli pri užívaní lieku Abiraterone Glenmark v kombinácii s prednizónom/prednizolónom hlásené prípady hypoglykémie (pozri časť 4.5); preto je u pacientov s diabetom potrebné sledovať hladinu cukru v krvi.
Použities chemoterapiou
Bezpečnosť a účinnosť súčasného užívania lieku Abiraterone Glenmark s cytotoxickou chemoterapiou nebola stanovená (pozri časť 5.1).
Potenciálneriziká
U mužov s metastatickým karcinómom prostaty, vrátane tých, ktorí sa liečia liekom Abiraterone Glenmark,
sa môže objaviť anémia a sexuálna dysfunkcia.
Účinkyna kostrovésvalstvoU pacientov liečených liekom Abiraterone Glenmark boli hlásené prípady myopatie a rabdomyolýzy. Väčšina prípadov sa vyskytla počas prvých 6 mesiacov liečby a vymizla po ukončení liečby abiraterónom.
U pacientov súbežne liečených liekmi, o ktorých je známe, že súvisia s myopatiou/rabdomyolýzou, sa
odporúča opatrnosť.
Interakcie s inýmiliekmiZ dôvodu rizika zníženej expozície abiraterónu sa počas liečby treba vyhnúť používaniu silných induktorov
CYP3A4 okrem prípadu, že by neexistovala žiadna alternatívna liečba (pozri časť 4.5).
Kombináciaabiraterónua prednizónu/prednizolónus Ra-223Liečba abiraterónom a prednizónom/prednizolónom v kombinácii s Ra-223 je kontraindikovaná (pozri časť
4.3) z dôvodu zvýšeného rizika zlomenín a tendencie k zvýšenej úmrtnosti medzi asymptomatickými a mierne symptomatickými pacientmi s karcinómom prostaty na základe pozorovania v klinických štúdiách.
Následnú liečbu s Ra-223 sa neodporúča začať skôr ako 5 dní po poslednom podaní lieku Abiraterone
Glenmark v kombinácii s prednizónom/prednizolónom.
PomocnélátkyTento liek obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej intolerancie,
celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať tento liek.
Abiraterone Glenmark 250 mg tabletyTento liek obsahuje menej ako 1 mmol sodíka (23 mg) v dávke štyroch tabliet s 250 mg, t. j. v podstate zanedbateľné množstvo sodíka.
Abiraterone Glenmark 500 mg filmom obalené tablety
|
|
Tento liek obsahuje 24 mg sodíka v dávke dvoch filmom obalených tabliet s 500 mg, čo zodpovedá 1 %
|
WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
|
|
| | |
4.5 Liekové a iné interakcieVplyv jedla na abiraterón-acetátPodávanie spolu s jedlom významne zvyšuje vstrebávanie abiraterón-acetátu. Účinnosť a bezpečnosť pri
podávaní spolu s jedlom nebola stanovená, preto sa tento liek nesmie užívať s jedlom (pozri časti 4.2 a 5.2)
.Interakcie s inýmiliekmiPotenciál iných liekov ovplyvniť expozície abiraterónuV klinickej interakčnej štúdii farmakokinetiky u zdravých jedincov predliečených rifampicínom, silným
induktorom CYP3A4, v dávke 600 mg denne počas 6 dní, po ktorej nasledovala jednorazová dávka abiraterón-acetátu 1 000 mg, sa priemerná plazmatická AUC∞ abiraterónu znížila o 55 %.
Používaniu silných induktorov CYP3A4 (napr., fenytoín, karbamazepín, rifampicín, rifabutín, rifapentín, fenobarbital, ľubovník bodkovaný [
Hypericum perforatum]) sa počas liečby treba vyhnúť okrem prípadu, že by neexistovala žiadna alternatívna liečba.
V samostatnej klinickej interakčnej štúdii farmakokinetiky u zdravých jedincov nemalo súbežné podávanie
ketokonazolu, silného inhibítora CYP3A4, klinicky významný vplyv na farmakokinetiku abiraterónu.
Potenciál ovplyvniť expozície iným liekomAbiraterón je inhibítor hepatálnych, lieky metabolizujúcich enzýmov CYP2D6 a CYP2C8.
V klinickej štúdii na stanovenie účinkov abiraterón-acetátu (spolu s prednizónom) na jednorazovú dávku dextrometorfánu, substrátu CYP2D6, sa systémová expozícia (AUC) dextrometorfánu zvýšila približne 2,9- násobne. AUC24 dextrorfánu, aktívneho metabolitu dextrometorfánu, sa zvýšila približne o 33 %.
Odporúča sa opatrnosť v prípade, že podávanie spolu s liekmi aktivovanými alebo metabolizovanými prostredníctvom CYP2D6, najmä s liekmi, ktoré majú úzky terapeutický index. Je potrebné zvážiť zníženie dávky liekov s úzkym terapeutickým indexom, ktoré sú metabolizované prostredníctvom CYP2D6.
Príklady liekov metabolizovaných prostredníctvom CYP2D6 zahŕňajú metoprolol, propranolol, dezipramín, venlafaxín, haloperidol, risperidón, propafenón, flekainid, kodeín, oxykodón a tramadol (posledné tri lieky potrebujú CYP2D6 na vytvorenie svojich aktívnych analgetických metabolitov).
V štúdii liekových interakcií s CYP2C8 u zdravých osôb sa AUC pioglitazónu zvýšila o 46 % a AUC M-III a M-IV, aktívnych metabolitov pioglitazónu, sa v oboch prípadoch znížili o 10 %, keď sa pioglitazón podával spolu s jednorazovou dávkou 1 000 mg abiraterón-acetátu.
U pacientov sa majú sledovať prejavy toxicity súvisiace so substrátom CYP2C8 s úzkym terapeutickým
indexom, ak sa užívajú súbežne. Príklady liekov metabolizovaných prostredníctvom CYP2C8 zahŕňajú pioglitazón a repaglinid (pozri časť 4.4).
Ukázalo sa, že hlavné metabolity abiraterón-sulfát a abiraterón-N-oxid-sulfát inhibujú in vitro vychytávanie hepatálnym transportérom OATP1B1, čo následne môže zvýšiť koncentráciu liekov vylučovaných prostredníctvom OATP1B1. K dispozícii nie sú žiadne klinické údaje, ktoré by potvrdili interakciu
súvisiacu s transportérom.
Užívanie s liekmi, o ktorých je známe, že predlžujú QT interval
Vzhľadom na to, že androgén-deprivačná liečba môže predlžovať QT interval, odporúča sa opatrnosť, keď sa abiraterón podáva s liekmi, o ktorých je známe, že predlžujú QT interval alebo s liekmi, ktoré môžu vyvolať torsades de pointes, ako sú antiarytmiká triedy IA (napr. chinidín, dizopyramid) alebo triedy III (napr. amiodarón, sotalol, dofetilid, ibutilid), metadón, moxifloxacín, antipsychotiká, atď.
Užívanie so spironolaktónom
Spironolaktón sa viaže na androgénový receptor a môže zvýšiť hladiny prostatického špecifického antigénu
(PSA). Užívanie s liekom Abiraterone Glenmark sa neodporúča (pozri časť 5.1).
4.6 Fertilita, gravidita a laktácia
Ženyv reprodukčnomveku
K dispozícii nie sú žiadne údaje o použití lieku Abiraterone Glenmark v gravidite u ľudí a tento liek nie je
určený na užívanie u žien v reprodukčnom veku.
Antikoncepcia u mužovažien
Nie je známe, či sú abiraterón alebo jeho metabolity prítomné v sperme. Ak pacient sexuálne žije s gravidnou partnerkou, je potrebné používať kondóm. Ak pacient sexuálne žije so ženou v reprodukčnom
veku, je potrebné používať kondóm spolu s ďalšou účinnou antikoncepčnou metódou. Štúdie na zvieratách preukázali reprodukčnú toxicitu (pozri časť 5.3).
Gravidita
Abiraterone Glenmark nie je určený na užívanie u žien a je kontraindikovaný u žien, ktoré sú alebo
potenciálne môžu byť gravidné (pozri časť 4.3 a 5.3).
Dojčenie
Abiraterone Glenmark nie je určený na užívanie u žien.
Fertilita
Abiraterón ovplyvnil fertilitu u samcov a samíc potkanov, ale tieto účinky boli úplne reverzibilné (pozri
časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Abiraterone Glenmark nemá žiadny alebo má len zanedbateľný vplyv na schopnosť viesť vozidlá a
obsluhovať stroje.
4.8 Nežiaduce účinkySúhrnbezpečnostnéhoprofiluV analýze nežiaducich reakcií zlúčených štúdií fázy III s abiraterónom, nežiaduce reakcie, ktoré boli
pozorované u ≥ 10 % pacientov, boli periférny edém, hypokaliémia, hypertenzia, infekcia močových ciest a
zvýšené hodnoty alanínaminotransferázy a/alebo zvýšené hodnoty aspartátaminotransferázy. Iné dôležité nežiaduce reakcie zahŕňajú poruchy srdca, hepatotoxicitu, zlomeniny a alergickú alveolitídu.
Abiraterone Glenmark môže vyvolávať hypertenziu, hypokaliémiu a retenciu tekutín ako farmakodynamický dôsledok svojho mechanizmu účinku. V štúdiách fázy III sa predpokladané mineralokortikoidné nežiaduce reakcie častejšie pozorovali u pacientov liečených abiraterón-acetátom než u pacientov, ktorí dostávali placebo: hypokaliémia 18 % oproti 8 %, hypertenzia 22 % oproti 16 % a retencia tekutín (periférny edém) 23 % oproti 17 %, v uvedenom poradí
. U pacientov liečených abiraterón- acetátom v porovnaní s pacientmi, ktorí dostávali placebo, sa pozoroval: 3. a 4. stupeň CTCAE (verzia 4.0) hypokaliémie u 6 % v porovnaní s 1 %; 3. a 4. stupeň CTCAE (verzia 4.0) hypertenzie u 7 % v porovnaní
s 5 % a retencia tekutín (periférny edém) 3. a 4. stupňa u 1 % v porovnaní s 1 % pacientov, v uvedenom
poradí.
Vo všeobecnosti sa mineralokortikoidné reakcie dali úspešne medicínsky zvládnuť. Súbežné podávanie
kortikosteroidu znižuje incidenciu a zmierňuje závažnosť uvedených nežiaducich reakcií (pozri časť 4.4).
TabuľkovýzoznamnežiaducichreakciíV klinických štúdiách u pacientov s metastatickým pokročilým karcinómom prostaty, ktorí užívali analóg
LHRH alebo ktorí boli v minulosti liečení orchiektómiou, sa abiraterón-acetát podával v dávke 1 000 mg denne v kombinácii s nízkou dávkou prednizónu alebo prednizolónu (5 mg alebo 10 mg denne v závislosti na indikácii).
Nežiaduce reakcie pozorované počas klinických štúdií a zo skúseností po uvedení lieku na trh sú uvedené nižšie podľa kategórie frekvencie. Kategórie frekvencie sú definované nasledovne: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1000); veľmi zriedkavé (< 1/10 000) a neznáme (z dostupných údajov).
V rámci jednotlivých skupín frekvencie sú nežiaduce účinky usporiadané v poradí klesajúcej závažnosti.
Tabuľka 1: Nežiaduce reakcie zistené v klinických štúdiách a po uvedení lieku na trhTrieda orgánových systémov
| Nežiaduca reakcia a frekvencia
|
Infekcie a nákazy
| veľmi časté: infekcia močových ciest
časté: sepsa
|
Poruchy endokrinného systému
| menej časté: adrenálna insuficiencia
|
Poruchy metabolizmu a výživy
| veľmi časté: hypokaliémia
časté: hypertriglyceridémia
|
Poruchy srdca a srdcovej činnosti
| časté: srdcové zlyhávanie*, angína pektoris, atriálna fibrilácia,
tachykardia
menej časté: iné arytmie
neznáme: infarkt myokardu, predĺženie QT intervalu
(pozri časti 4.4 a 4.5)
|
Poruchy ciev
| veľmi časté: hypertenzia
|
Poruchy dýchacej sústavy, hrudníka a
mediastína
| zriedkavé: alergická alveolitídaa
|
Poruchy gastrointestinálneho traktu
| veľmi časté: hnačka
časté: dyspepsia
|
Poruchy pečene a žlčových ciest
| veľmi časté: zvýšené hodnoty alanínaminotransferázy a/alebo
zvýšené hodnoty aspartátaminotransferázyb
zriedkavé: fulminantná hepatitída, akútne zlyhávanie pečene
|
Poruchy kože a podkožného tkaniva
| časté: vyrážka
|
Poruchy kostrovej a svalovej sústavy a
spojivového tkaniva
|
menej časté: myopatia, rabdomyolýza
|
Poruchy obličiek a močových ciest
|
časté: hematúria
|
C
e
l
k
ové poruchy a reakcie v mieste
podania
|
veľmi časté: periférny edém
|
Ú
r
azy, otravy a komplikácie liečebného
postupu
|
časté: zlomeniny**
|
|
|
* Pod srdcové zlyhávanie sa zaraďuje aj kongestívne srdcové zlyhávanie, dysfunkcia ľavej komory a zníženie
ejekčnej frakcie.
** Zlomeniny zahŕňajú osteoporózu a všetky zlomeniny okrem patologických zlomenín.
a Spontánne hlásenia zo skúseností po uvedení lieku na trh.
b Zvýšené hodnoty alanínaminotransferázy a/alebo zvýšené hodnoty aspartátaminotransferázy zahŕňajú zvýšenie ALT, zvýšenie AST a abnormálnu funkciu pečene.
U pacientov liečených abiraterón-acetátom sa vyskytli nasledujúce nežiaduce liekové reakcie 3. stupňa CTCAE (verzia 4.0): hypokaliémia 5 %; infekcia močových ciest 2 %; zvýšené hodnoty alanínaminotransferázy a/alebo zvýšené hodnoty aspartátaminotransferázy 4 %; hypertenzia 6 %;
zlomeniny 2 %; periférny edém, srdcové zlyhávanie a atriálna fibrilácia – každá reakcia s frekvenciou 1 %.
Hypertriglyceridémia a angína pektoris 3. stupňa CTCAE (verzia 4.0) sa vyskytli u < 1 % pacientov. Infekcia močových ciest, zvýšené hodnoty alanínaminotransferázy a/alebo zvýšené hodnoty
aspartátaminotransferázy, hypokaliémia, srdcové zlyhávanie, atriálna fibrilácia a zlomeniny 4. stupňa
CTCAE (verzia 4.0) sa vyskytli u < 1 % pacientov.
Vyššia incidencia hypertenzie a hypokaliémie bola pozorovaná v hormonálne citlivej populácii (štúdia
3011). Hypertenzia bola hlásená u 36,7 % pacientov v hormonálne citlivej populácii (štúdia 3011)
v porovnaní s 11,8 % v štúdii 301 a s 20,2 % v štúdii 302. Hypokaliémia bola pozorovaná u 20,4 %
pacientov v hormonálne citlivej populácii (štúdia 3011) v porovnaní s 19,2 % v štúdii 301 a s 14,9 % v
štúdii 302.
Incidencia a závažnosť nežiaducich udalostí bola vyššia v podskupine pacientov s východiskovou hodnotou
výkonnostného stavu ECOG 2 a tiež u starších pacientov (≥ 75 rokov).
Popis vybranýchnežiaducichreakciíKardiovaskulárne reakcieZ troch štúdií III. fázy boli vylúčení pacienti s nekontrolovanou hypertenziou, s klinicky významným ochorením srdca ako dokázaný infarkt myokardu alebo arteriálne trombotické príhody za posledných 6
mesiacov, závažnou alebo nestabilnou angínou pektoris, zlyhaním srdca triedy III alebo IV (štúdia
301) alebo zlyhaním srdca triedy II až IV (štúdie 3011 a 302) podľa Newyorskej kardiologickej spoločnosti
(NYHA, z angl. New York Heart Association) alebo nameranou ejekčnou frakciou srdca
< 50 %. Všetci zaradení pacienti (aktívne liečení aj liečení placebom) súbežne dostávali androgén-
deprivačnú terapiu, prevažne s použitím analógov LHRH, čo bolo spojené s cukrovkou, infarktom myokardu, cerebrovaskulárnou príhodou a náhlou kardiálnou smrťou. Incidencie kardiovaskulárnych
nežiaducich reakcií v štúdii III. fázy u pacientov užívajúcich abiraterón-acetát v porovnaní s pacientmi
užívajúcimi placebo, boli nasledovné: atriálna fibrilácia 2,6 % vs 2,0 %, tachykardia 1,9 % vs 1,0 %, angína
pektoris 1,7 % vs 0,8 %, zlyhávanie srdca 0,7 % vs 0,2 %, a arytmia 0,7 % vs 0,5 %.
HepatotoxicitaU pacientov liečených abiraterón-acetátom bola hlásená hepatotoxicita so zvýšenými hodnotami ALT, AST
a celkového bilirubínu. V rámci klinických štúdií fázy III bola hepatotoxicita 3. a 4. stupňa (napr. zvýšenie
ALT alebo AST na > 5-násobok hornej hranice normy (HHN) alebo zvýšenie bilirubínu na
> 1,5-násobok HHN) hlásená približne u 6 % pacientov, ktorí dostávali abiraterón-acetát, zvyčajne počas prvých 3 mesiacov od začiatku liečby. V štúdii 3011 bola hepatotoxicita 3. alebo 4. stupňa pozorovaná u 8,4
% pacientov liečených s abiraterónom. Desať pacientov dostávajúcich abiraterón ukončilo liečbu z dôvodu
hepatotoxicity; dvaja mali hepatotoxicitu 2. stupňa, šiesti mali hepatotoxicitu
3. stupňa a dvaja mali hepatotoxicitu 4. stupňa. V štúdii 3011 nezomrel z dôvodu hepatotoxicity ani jeden
pacient. V klinických štúdiách fázy III u pacientov, ktorí mali východiskové hodnoty ALT alebo AST zvýšené, bola väčšia pravdepodobnosť, že u nich dôjde k zvýšeným hodnotám hepatálnych testov v porovnaní s pacientmi, ktorí mali normálne východiskové hodnoty. Keď sa pozorovalo zvýšenie buď ALT alebo AST na > 5-násobok HHN, alebo zvýšenie bilirubínu na > 3-násobok HHN, podávanie abiraterón- acetátu sa prerušilo alebo sa ukončilo. Vo dvoch prípadoch došlo k výraznému zvýšeniu hodnôt hepatálnych testov (pozri časť 4.4). Títo dvaja pacienti s normálnou východiskovou funkciou pečene mali zvýšené ALT alebo AST na 15- až 40-násobok HHN a bilirubín na 2- až 6-násobok HHN. Po ukončení liečby sa u oboch pacientov hodnoty hepatálnych testov normalizovali a u jedného pacienta sa liečba obnovila bez toho, že by sa uvedené zvýšené hodnoty zopakovali. V štúdii 302 bolo pozorované zvýšenie hodnôt ALT alebo AST 3. alebo 4. stupňa u 35 (6,5 %) pacientov liečených abiraterón-acetátom. Zvýšené hodnoty aminotransferázy ustúpili u všetkých pacientov okrem 3 pacientov (2 s novými mnohonásobnými metastázami v pečeni a 1
so zvýšenou hodnotou AST približne 3 týždne po poslednej dávke abiraterón-acetátu). V klinických štúdiách fázy III bolo prerušenie liečby z dôvodu zvýšených hodnôt ALT a AST alebo abnormálnej funkcie
pečene hlásené u 1,1 % pacientov liečených abiraterón-acetátom a u 0,6 % pacientov liečených placebom;
žiadne úmrtie nebolo hlásené z dôvodu hepatotoxicity.
V klinických skúšaniach sa riziko hepatotoxicity zmiernilo vylúčením pacientov s východiskovou hepatitídou alebo významnými abnormalitami pečeňových testov. Zo štúdie 3011 boli vylúčení pacienti s východiskovou hodnotou ALT a AST > 2,5 x HHN, bilirubínom > 1,5 x HHN alebo
pacienti s aktívnou alebo symptomatickou vírusovou hepatitídou alebo chronickým ochorením pečene;
ascitom alebo poruchami krvácania kvôli dysfunkcii pečene. Zo štúdie 301 boli vylúčení pacienti
s východiskovými hodnotami ALT a AST ≥ 2,5-násobok HHN bez prítomnosti metastáz v pečeni a
> 5-násobku HHN, v prítomnosti metastáz v pečeni. Pacienti s metastázami v pečeni neboli vhodní a pacienti s východiskovou hladinou ALT a AST ≥ 2,5x HHN boli vylúčení zo štúdie 302.
Abnormálne hodnoty hepatálnych testov, ktoré sa vyskytli u pacientov zúčastňujúcich sa na klinických skúšaniach, boli rázne riešené požiadavkou na prerušenie liečby a povolením vrátiť sa k liečbe len po tom,
ak sa hodnoty hepatálnych testov u daného pacienta dostali späť na východiskové hodnoty (pozri časť 4.2). Pacienti so zvýšením ALT alebo AST na > 20-násobok HHN sa k liečbe nevrátili.
Bezpečnosť opakovanej liečby u týchto pacientov nie je známa. Mechanizmus hepatotoxicity nie je
objasnený.
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieSkúsenosti s predávkovaním abiraterón-acetátom u ľudí sú obmedzené.
Neexistuje žiadne špecifické antidotum. V prípade predávkovania je potrebné podávanie ukončiť a pristúpiť
k všeobecným podporným opatreniam, vrátane monitorovania arytmií, hypokaliémie a prejavov a
príznakov retencie tekutín. Zároveň je potrebné vyhodnotiť funkciu pečene.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: endokrinná liečba, iné antagonisty hormónov a príbuzné liečivá, ATC kód:
L02BX03
Mechanizmus účinkuAbiraterón-acetát sa konvertuje v podmienkach
in vivo na abiraterón, inhibítor biosyntézy androgénov. Konkrétne, abiraterón selektívne inhibuje enzým 17α-hydroxylázu/C17,20-lyázu (CYP17). Tento enzým sa nachádza a je potrebný na biosyntézu androgénov v tkanive semenníkov, nadobličiek a v nádorovom
tkanive prostaty. CYP17 katalyzuje premenu pregnenolónu a progesterónu na prekurzory testosterónu, DHEA a androsténdionu (v danom poradí) prostredníctvom 17α-hydroxylácie a štiepenia väzby C17,20. Inhibícia CYP17 má za následok taktiež zvýšenú tvorbu
mineralokortikoidov v nadobličkách (pozri časť 4.4).
Karcinóm prostaty citlivý na androgény odpovedá na liečbu, ktorá znižuje hladinu androgénov. Androgén- deprivačná terapia, ako je liečba analógmi LHRH alebo orchiektómia, znižuje tvorbu androgénov v semenníkoch, avšak nemá vplyv na tvorbu androgénov v nadobličkách alebo v nádore. Terapia abiraterónom znižuje sérový testosterón na nedetekovateľnú hladinu (pri použití komerčných testov) v prípade, že sa podáva spolu s analógmi LHRH (alebo orchiektómiou).
Farmakodynamickéúčinky
Abiraterón znižuje hladinu sérového testosterónu a iných androgénov na úroveň nižšiu než je hladina, ktorá
sa dosahuje použitím samotných analógov LHRH alebo orchiektómie. Je to dôsledok selektívnej inhibície enzýmu CYP17, ktorý je potrebný na biosyntézu androgénov. PSA slúži u pacientov
s karcinómom prostaty ako biomarker. V klinickej štúdii III. fázy s účasťou pacientov, u ktorých zlyhala
predchádzajúca chemoterapia s taxánmi, sa u 38 % pacientov liečených abiraterón-acetátom, oproti 10 %
pacientov, ktorí dostávali placebo, zaznamenal prinajmenšom 50 % pokles oproti východiskovej hladine
PSA.
Klinickáúčinnosťa bezpečnosť
Účinnosť bola stanovená v troch randomizovaných placebom kontrolovaných multicentrických klinických štúdiách III. fázy (štúdie 3011, 302 a 301) s pacientmi s mHSPC a mCRPC. Štúdia 3011 zahŕňala
pacientov, u ktorých bol novodiagnostikovaný (do 3 mesiacov od randomizácie) mHSPC a ktorí mali
vysokorizikové prognostické faktory. Prognóza s vysokým rizikom bola definovaná ako výskyt najmenej 2
z nasledujúcich 3 rizikových faktorov: (1) Gleasonovo skóre ≥ 8; (2) prítomnosť 3 alebo viacerých lézií na skene kosti; (3) prítomnosť merateľnej viscerálnej (s výnimkou ochorenia lymfatických uzlín) metastázy. V skupine s aktívnou liečbou sa abiraterón podával v dávke 1000 mg denne v kombinácii s nízkou dávkou prednizónu 5 mg jedenkrát denne a okrem toho ADT
(agonista LHRH alebo orchiektómia), čo predstavovalo štandardnú liečbu. Pacienti v kontrolnej skupine
dostávali ADT a placebá namiesto abiraterónu aj prednizónu. Do štúdie 302 boli zaradení pacienti, ktorí predtým nedostávali docetaxel; zatiaľ čo do štúdie 301 boli zaradení pacienti, ktorí dostávali predtým docetaxel. Pacienti užívali analóg LHRH alebo boli predtým liečení orchiektómiou. V skupine s aktívnou liečbou sa abiraterón podával v dávke 1000 mg denne v kombinácii s nízkou dávkou prednizónu alebo prednizolónu 5 mg dvakrát denne. Pacienti v kontrolnej skupine dostávali placebo a nízku dávku prednizónu alebo prednizolónu 5 mg dvakrát denne.
Zmeny v sérovej koncentrácii PSA samy osebe nie vždy predpovedajú klinický prínos. Z tohto dôvodu sa vo všetkých štúdiách odporúčalo, aby pacienti pokračovali v liečbe v štúdii až do dosiahnutia kritéria pre prerušenie liečby, tak ako je to opísané nižšie pre každú štúdiu.
Vo všetkých štúdiách nebolo povolené užívanie spironolaktónu, pretože spironolaktón sa viaže na
androgénový receptor a môže zvýšiť hladiny PSA.
Štúdia 3011 (pacienti s novodiagnostikovaným vysokorizikovým mHSPC)
V štúdii 3011 (n = 1199) bol medián veku zaradených pacientov 67 rokov. Počet pacientov liečených s
abiraterónom podľa rasy bol belosi 832 (69,4 %), aziati 246 (20,5 %), černosi alebo Afroameričania
25 (2,1 %), iní 80 (6,7 %), neznáma/nehlásená rasa 13 (1,1 %) a americkí Indiáni alebo pôvodní obyvatelia Aljašky 3 (0,3 %). Hodnota skóre výkonnostného stavu podľa ECOG bola 0 alebo 1 u 97 % pacientov. Pacienti so známou mozgovou metastázou, nekontrolovanou hypertenziou, významným ochorením srdca alebo zlyhaním srdca triedy II - IV podľa NYHA boli vylúčení. Pacienti, u ktorých bol metastatický karcinóm prostaty predtým liečený farmakoterapiou, rádioterapiou alebo chirurgicky, boli vylúčení, s výnimkou ADT do 3 mesiacov alebo 1 cyklu paliatívnej rádioterapie alebo chirurgického zákroku na liečbu príznakov následkom metastatického ochorenia. Združené primárne koncové ukazovatele účinnosti boli celkové prežívanie (OS, overall survival) a prežívanie bez rádiografickej progresie (rPFS, radiographic progression-free survival). Medián východiskového skóre bolesti podľa Brief Pain Inventory Short Form (BPI-SF) bol 2,0 v skupine s liekom aj v skupine s placebom. Okrem združených primárnych koncových
ukazovateľov bol prínos tiež hodnotený podľa času do udalosti súvisiacej so skeletom (SRE, skeletal- related event), času do ďalšej liečby karcinómu prostaty, času do začatia chemoterapie, času do progresie bolesti a času do progresie PSA. Liečba pokračovala až do progresie ochorenia, odvolania súhlasu s liečbou, výskytu neprijateľnej toxicity alebo úmrtia.
Prežívanie bez rádiografickej progresie bolo definované ako čas od randomizácie do výskytu rádiografickej progresie alebo úmrtia z akéhokoľvek dôvodu. Rádiografická progresia zahŕňala progresiu podľa skenu kostí (podľa modifikovaných kritérií PCWG2) alebo progresiu lézií mäkkých tkanív podľa CT alebo MRI (podľa RECIST 1.1).
Medzi liečenými skupinami bol pozorovaný významný rozdiel v rPFS (pozri Tabuľku 2 a Obrázok 1).
 Tabuľka 2: Prežívanie bez rádiografickej progresie – stratifikovaná analýza; populácia všetkých zaradených pacientov (Štúdia PCR3011)
Tabuľka 2: Prežívanie bez rádiografickej progresie – stratifikovaná analýza; populácia všetkých zaradených pacientov (Štúdia PCR3011)
Randomizovaní pacienti
AA-P
597
Placebo
602
Udalosť 239 (40,0 %) 354 (58,8 %)
Cenzurovaní 358 (60,0 %) 248 (41,2 %)
Čas do udalosti (mesiace)
Medián (95% IS) 33,02 (29,57; NO) 14,78 (14,69; 18,27)
Rozsah (0,0+; 41,0+) (0,0+; 40,6+)
Hodnota pa
< 0,0001
Pomer rizík
(
9
5 % IS)
b
0,466 (0,394; 0,550)
Poznámka: + = cenzurované pozorovanie, NO = nemožno odhadnúť. Pri definovaní udalosti rPFS sa berie do
úvahy rádiografická progresia a úmrtie. AA-P = osoby, ktoré dostali abiraterón-acetát a prednizón.
a Hodnota p je odvodená z log-rank testu stratifikovaného podľa skóre výkonnostného stavu ECOG (0/1 alebo 2)
a viscerálnej lézie (neprítomná alebo prítomná).
b Pomer rizík je odvodený zo stratifikovaného proporcionálneho modelu rizika. Pomer rizík < 1 v prospech AA-P.
Obrázok 1: Kaplanova-Meierova krivka prežívania bez rádiografickej progresie; populácia všetkých zaradených pacientov (štúdia PCR3011)
Štatisticky významné zlepšenie celkového prežívania v prospech AA-P plus ADT bolo pozorované s 34 % znížením rizika úmrtia v porovnaní s placebom plus ADT (HR = 0,66; 95 % IS: 0,56; 0,78; p < 0,0001) (pozri Tabuľku 3 a Obrázok 2).
Tabuľka 3: Celkové prežívanie pacientov liečených s abiraterón-acetátom alebo placebom v štúdiiPCR3011 (analýza všetkých zaradených pacientov)
C
e
l
k
ové prežívanie (mesiace) Abiraterón-acetát s prednizónom
(
N = 597)
Placebo
(
N = 602)
Úmrtia (%) 275 (46 %) 343 (57 %)
Medián prežívania (mesiace) (95 % IS) 53,3
(48,2; NO)
Pomer rizík (95% IS)1 0,66 (0,56; 0,78)

NO = nemožno odhadnúť
36,5
(33,5; 40,0)
1 Pomer rizík je odvodený zo stratifikovaného proporcionálneho modelu rizika. Pomer rizík < 1 v prospech abiraterón-
acetátu s prednizónom.
Obrázok 2: Kaplanova-Meierova krivka celkového prežívania; populácia všetkých zaradenýchpacientov (štúdia PCR3011)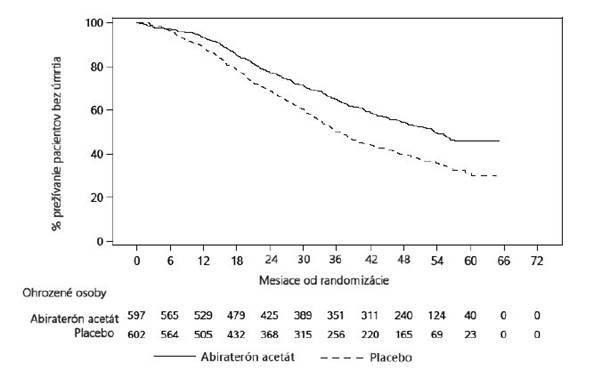
Analýzy podskupín sú konzistentne v prospech liečby s abiraterón-acetátom. Účinok liečby AA-P na rPFS a OS vo vopred stanovených podskupinách bol priaznivý a zhodný s celkovou skúšanou populáciou, s výnimkou podskupiny s hodnotou skóre ECOG 2, kde sa nepozorovala žiadna tendencia k prínosu liečby; avšak malá veľkosť vzorky (n = 40) obmedzuje prijatie akéhokoľvek významného záveru.
Okrem pozorovaných zlepšení celkového prežívania a rPFS, sa prínos liečby s abiraterónom v porovnaní s placebom preukázal vo všetkých perspektívne definovaných meraniach sekundárnych koncových ukazovateľov.
Štúdia 302 (pacienti, ktorí predtým nedostávali chemoterapiu)Do tejto štúdie boli zaradení pacienti, ktorí boli asymptomatickí alebo mierne symptomatickí a ktorým doposiaľ nebola klinicky indikovaná chemoterapia. Skóre 0-1 najhoršej bolesti (BPI-SF) počas posledných
24 hodín bolo považované za asymptomatické a skóre 2-3 bolo považované za mierne symptomatické.
V štúdii 302 (n = 1088) bol medián veku zaradených pacientov 71 rokov u pacientov liečených abiraterónom plus prednizón alebo prednizolón a 70 rokov u pacientov liečených placebom plus prednizón alebo prednizolón. Počet pacientov liečených abiraterónom podľa rasy bol kaukazská rasa 520 (95,4 %), čierna rasa 15 (2,8 %), ázijská rasa 4 (0,7 %) a iné 6 (1,1 %). Hodnota skóre výkonnostného stavu podľa Eastern Cooperative Oncology Group (ECOG) bola 0 u 76 % pacientov a 1 u 24 % pacientov v oboch ramenách. Päťdesiat percent pacientov malo len metastázy kostí, ďalších 31 % pacientov malo metastázy kostí a mäkkých tkanív alebo lymfatických uzlín a 19 % pacientov malo len metastázy mäkkých tkanív alebo lymfatických uzlín. Pacienti s viscerálnymi metastázami boli vylúčení. Združenými primárnymi koncovými ukazovateľmi účinnosti boli celkové prežívanie a prežívanie bez rádiografickej progresie (rPFS). Okrem združených primárnych koncových ukazovateľov účinnosti bol prínos tiež hodnotený podľa času do použitia opiátov na bolesť spôsobenú rakovinou, času do začatia cytotoxickej chemoterapie, času do zhoršenia skóre výkonnostného stavu ECOG o ≥ 1 stupeň a času do progresie PSA na základe kritérií PCWG2 (Prostate Cancer Working Group-2). Liečba v štúdii bola prerušená v čase jednoznačnej klinickej progresie. Liečba mohla byť tiež prerušená v čase potvrdenia rádiografickej progresie podľa uváženia skúšajúceho.
Prežívanie bez rádiografickej progresie (rPFS) bolo posúdené použitím štúdií sekvenčného snímkovania, ako je to definované kritériami PCWG2 (pre lézie kostí) a modifikovanými kritériami RECIST (Response Evaluation Criteria In Solid Tumors) (pre lézie mäkkých tkanív). Analýza rPFS použila centrálne posúdenie vyhodnotenia rádiografickej progresie.
Pri plánovanej analýze rPFS bolo k dispozícii 401 udalostí, 150 (28 %) pacientov liečených abiraterónom a 251 (46 %) pacientov liečených placebom malo rádiografický dôkaz progresie alebo zomrelo. Medzi liečenými skupinami bol pozorovaný výrazný rozdiel v rPFS (pozri Tabuľku 4 a Obrázok 3).
Tabuľka 4: Štúdia 302: Prežívanie bez rádiografickej progresie u pacientov liečených buď abiraterón-acetátom alebo placebom v kombinácii s prednizónom alebo prednizolónom sanalógmi LHRH alebo predchádzajúcou orchiektómiou
abiraterón-acetátom alebo placebom v kombinácii s prednizónom alebo prednizolónom sanalógmi LHRH alebo predchádzajúcou orchiektómiou
A
b
i
rat
erón-acetát
Placebo
(N = 546) (N = 542)
Prežívanie bez rádiografickej
progresie (rPFS)
Progresia alebo úmrtie 150 (28 %) 251 (46 %)
Medián rPFS v mesiacoch
(95 % IS)
nebolo dosiahnuté
(11,66; NO)
8,3 (8,12; 8,54)
hodnota p* < 0,0001
Pomer rizík**
(95 % IS)

NO = nemožno odhadnúť
0,425 (0,347; 0,522)
* p-hodnota je odvodená z log-rank testu stratifikovaného podľa východiskového skóre ECOG (0 alebo
1)
** pomer rizík < 1 v prospech abiraterón-acetátu
Obrázok 3: Kaplanove-Meierove krivky prežívania bez rádiografickej progresie u pacientov liečených buď abiraterón-acetátu alebo placebom v kombinácii s prednizónom alebo prednizolónom s analógmi LHRH alebo predchádzajúcou orchiektómiou

AA = abiraterón-acetátu
Údaje o pacientoch boli naďalej zbierané až do termínu druhej predbežnej analýzy celkového prežívania (OS, z angl. overall survival). Rádiografické posúdenie rPFS vykonané skúšajúcim následne po analýze citlivosti sa uvádza v Tabuľke 5 a na Obrázku 4.
Šesťstosedem (607) pacientov malo rádiografickú progresiu alebo zomrelo: 271 (50 %) v skupine s abiraterón-acetátom a 336 (62 %) v skupine s placebom. Liečba abiraterón-acetátom znížila riziko rádiografickej progresie alebo úmrtia o 47 % v porovnaní s placebom (HR = 0,530; 95 % IS: [0,451; 0,623], p < 0,0001). Medián rPFS bol 16,5 mesiaca v skupine s abiraterón-acetátom a 8,3 mesiaca v skupine s placebom.
Tabuľka 5: Štúdia 302: Prežívanie bez rádiografickej progresie u pacientov liečených buď abiraterón-acetátom alebo placebom v kombinácii s prednizónom alebo prednizolónom s analógmi LHRH alebo predchádzajúcou orchiektómiou (druhá predbežná analýza celkového prežívania podľa posúdenia skúšajúcim)
A
b
i
rat
erón-acetát
Placebo
(N = 546) (N = 542)
Prežívanie bez rádiografickej
progresie (rPFS)
Progresia alebo úmrtie 271 (50 %) 336 (62 %)

Medián rPFS v mesiacoch
(95 % IS)
16,5 (13,80; 16,79)
8,3 (8,05; 9,43)
hodnota p* < 0,0001
Pomer rizík** 0,530 (0,451; 0,623)
(95 % IS)
* p-hodnota je odvodená z log-rank testu stratifikovaného podľa východiskového skóre ECOG (0 alebo
1)
** pomer rizík < 1 v prospech abiraterón-acetátu
Obrázok 4: Kaplanove-Meierove krivky prežívania bez rádiografickej progresie u pacientov liečených buď abiraterón-acetátom alebo placebom v kombinácii s prednizónom alebo prednizolónom s analógmi LHRH alebo predchádzajúcou orchiektómiou (druhá predbežná analýza celkového prežívania podľa posúdenia skúšajúcim)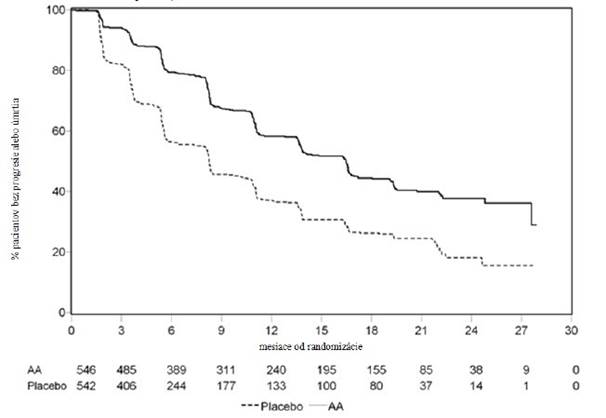
AA = abiraterón-acetát
Po zaznamenaní 333 úmrtí bola vykonaná plánovaná priebežná analýza OS. Štúdia bola odslepená na základe pozorovaného významného klinického prínosu a pacientom v skupine s placebom bola ponúknutá liečba s abiraterónom. Celkové prežívanie bolo dlhšie s abiraterón-acetátom ako s placebom s 25 % znížením rizika úmrtia (HR = 0,752; 95 % IS: [0,606; 0,934], p=0,0097), ale údaje o OS neboli vyhodnotiteľné a priebežné výsledky nedosiahli vopred stanovenú hranicu pre ukončenie pre štatistickú významnosť (pozri Tabuľku 6). Prežívanie sa sledovalo aj po tejto priebežnej analýze.
Po zaznamenaní 741 úmrtí bola vykonaná plánovaná záverečná analýza OS (medián sledovania
49 mesiacov). Zomrelo 65 % (354 z 546) pacientov liečených s abiraterónom, v porovnaní so 71 % (387 z
542) pacientov dostávajúcich placebo. Štatisticky významný prínos pre OS v prospech skupiny liečenej abiraterón-acetátom sa preukázal 19,4 % znížením rizika úmrtia (HR=0,806; 95 % IS: [0,697; 0,931],
p=0,0033) a zlepšením mediánu OS o 4,4 mesiaca (abiraterón 34,7 mesiacov, placebo 30,3 mesiacov)
(pozri Tabuľku 6 a Obrázok 5). Toto zlepšenie sa preukázalo, aj keď 44 % pacientov v skupine s placebom
dostávalo abiraterón-acetát ako následnú liečbu.
Tabuľka 6: Štúdia 302: Celkové prežívanie u pacientov liečených buď abiraterón-acetátom alebo placebom v kombinácii s prednizónom alebo prednizolónom s analógmi LHRH alebo predchádzajúcou orchiektómiou
Priebežná analýza prežívania
|
A
b
i
raterón
(
N = 546)
|
Placebo
(
N = 542)
|
Úmrtia (%)
|
147 (27 %)
|
186 (34 %)
|
Medián prežívania (mesiace)
|
nebolo dosiahnuté
|
27,2
|
(95 % IS)
|
(NO; NO)
|
(25,95; NO)
|

p-hodnota* 0,0097
Pomer rizík**(95% IS) 0,752 (0,606; 0,934)
Záverečná analýza prežívania Úmrtia'
| 354 (65 %)
| 387 (71 %)
|
Medián celkového prežívania
| 34,7 (32,7; 36,8)
| 30,3 (28,7; 33,3)
|
v mesiacoch (95 % IS)
|
|
|
p-hodnota* 0,0033
Pomer rizík** (95 % IS) 0,806 (0,697; 0,931)
NO = nemožno odhadnúť
* p-hodnota je odvodená z log-rank testu stratifikovaného podľa východiskového skóre ECOG (0 alebo
1)
** pomer rizík < 1 v prospech abiraterón-acetátu
Obrázok 5: Kaplanove-Meierove krivky prežívania pacientov liečených buď abiraterónom alebo placebom v kombinácii s prednizónom alebo prednizolónom s analógmi LHRH alebo predchádzajúcou orchiektómiou, záverečná analýza AA = abiraterón-acetát
Okrem pozorovaných zlepšení celkového prežívania a rPFS, sa prínos liečby abiraterón-acetátu
v porovnaní s placebom preukázal vo všetkých meraniach sekundárnych koncových ukazovateľov
nasledovne:
Čas do progresie PSA na základe kritérií PCWG2: Medián času do progresie PSA bol 11,1 mesiaca
u pacientov dostávajúcich abiraterón-acetát a 5,6 mesiaca u pacientov dostávajúcich placebo (HR = 0,488;
95 % IS: [0,420; 0,568], p < 0,0001). Čas do progresie PSA bol pri liečbe abiraterón-acetátom približne dvojnásobný (HR = 0,488). Podiel pacientov s potvrdenou odpoveďou PSA bol vyšší v skupine
s abiraterón-acetátom ako v skupine s placebom (62 % oproti 24 %; p < 0,0001). U pacientov s merateľným ochorením mäkkých tkanív bol pri liečbe abiraterón-acetátom pozorovaný výrazne väčší počet úplných a čiastočných odpovedí nádorov.
Čas do použitia opiátov na bolesť spôsobenú rakovinou: Medián času do použitia opiátov na bolesť súvisiacu s karcinómom prostaty bol v čase záverečnej analýzy 33,4 mesiaca u pacientov dostávajúcich abiraterón-acetát a 23,4 mesiaca u pacientov dostávajúcich placebo (HR = 0,721; 95 % IS: [0,614; 0,846], p < 0,0001).
Čas do začatia cytotoxickej chemoterapie: Medián času do začatia cytotoxickej chemoterapie bol
25,2 mesiaca u pacientov dostávajúcich abiraterón-acetát a 16,8 mesiaca u pacientov dostávajúcich placebo
(HR = 0,580; 95 % IS: [0,487; 0,691], p < 0,0001).
Čas do zhoršenia skóre výkonnostného stavu ECOG o ≥ 1 stupeň: Medián času do zhoršenia výkonnostného stavu ECOG o ≥ 1 stupeň bol 12,3 mesiaca u pacientov dostávajúcich abiraterón-acetát a
10,9 mesiaca u pacientov dostávajúcich placebo (HR = 0,821; 95 % IS: [0,714; 0,943], p = 0,0053).
Nasledujúce koncové ukazovatele štúdie preukázali štatisticky významnú výhodu v prospech liečby abiraterón-acetátom:
Objektívna odpoveď: Objektívna odpoveď bola definovaná ako podiel pacientov s merateľným Ochorením, ktorí dosiahli úplnú alebo čiastočnú odpoveď podľa kritérií RECIST (za cieľovú léziu sa považovala lymfatická uzlina s východiskovou veľkosťou ≥ 2 cm). Podiel pacientov s merateľným ochorením na začiatku liečby, ktorí dosiahli objektívnu odpoveď, bol 36 % v skupine s abiraterón-acetátom a 16 % v skupine s placebom (p < 0,0001).
Bolesť: Liečba abiraterón-acetátom výrazne znížila riziko progresie priemernej intenzity bolesti o 18 % v porovnaní s placebom (p = 0,0490). Medián času do progresie bol 26,7 mesiaca v skupine s abiraterón- acetátom a 18,4 mesiaca v skupine s placebom.
Čas do zhoršenia vo FACT-P (celkové skóre): Liečba abiraterón-acetátom znížila riziko zhoršenia FACTP (celkové skóre) o 22 % v porovnaní s placebom (p = 0,0028). Medián času do zhoršenia vo FACT-P (celkové skóre) bol 12,7 mesiaca v skupine s abiraterón-acetátom a 8,3 mesiaca v skupine s placebom.
Štúdia 301 (pacienti, ktorí predtým dostávali chemoterapiu)
Do štúdie 301 boli zaradení pacienti, ktorí predtým dostávali docetaxel. U pacientov sa nevyžadovala
progresia ochorenia pri docetaxeli, pretože toxicita vyplývajúca z tejto chemoterapie mohla viesť k
prerušeniu liečby. Pacienti pokračovali v užívaní liečby v štúdii až do progresie PSA (potvrdená 25 % zvýšením nad pacientovu východiskovú/najnižšiu hodnotu) spolu s rádiografickou progresiou definovanou v protokole a symptomatickou alebo klinickou progresiou. Pacienti, ktorí boli predtým liečení na karcinóm prostaty ketokonazolom, boli vylúčení z tejto štúdie. Primárnym koncovým ukazovateľom účinnosti bolo celkové prežívanie.
Medián veku zaradených pacientov bol 69 rokov (rozsah 39 - 95). Počet pacientov liečených abiraterón-
acetátom podľa rasy bol kaukazská rasa 737 (93,2 %), čierna rasa 28 (3,5 %), ázijská rasa 11 (1,4 %) a iné
14 (1,8 %). Jedenásť percent zaradených pacientov malo hodnotu skóre výkonnostného stavu ECOG 2;
70 % malo rádiografický dôkaz progresie ochorenia s progresiou PSA alebo bez nej; 70 % v minulosti
absolvovalo jednu cytotoxickú chemoterapiu a 30 % absolvovalo dve takéto chemoterapie. Metastázy v
pečeni boli prítomné u 11 % pacientov liečených abiraterón-acetátom.
V plánovanej analýze uskutočnenej po tom, ako sa zaznamenalo 552 úmrtí, sa zistilo, že zomrelo 42 % (333 zo 797) pacientov liečených abiraterónom v porovnaní s 55 % (219 z 398) pacientov, ktorí dostávali placebo. Bolo pozorované štatisticky významné zlepšenie mediánu celkového prežívania u pacientov liečených abiraterón-acetátom (pozri Tabuľku 7).
 Tabuľka 7: Celkové prežívanie pacientov liečených buď abiraterón-acetátom alebo placebom v kombinácii s prednizónom alebo prednizolónom s analógmi LHRH alebo predchádzajúcou orchiektómiouAbiraterón-acetát Placebo(N = 797) (N = 398)
Tabuľka 7: Celkové prežívanie pacientov liečených buď abiraterón-acetátom alebo placebom v kombinácii s prednizónom alebo prednizolónom s analógmi LHRH alebo predchádzajúcou orchiektómiouAbiraterón-acetát Placebo(N = 797) (N = 398) Primárna analýza prežívania
Primárna analýza prežívania Úmrtia (%)
Medián prežívania (mesiace) (95 % IS)
333 (42 %)
14,8 (14,1; 15,4)
219 (55 %)
10,9 (10,2; 12,0)
p-hodnotaa
Pomer rizík (95% IS)b
< 0,0001
0,646 (0,543; 0,768)
A
k
t
u
alizovaná analýza prežívania
Úmrtia (%) 501 (63 %) 274 (69 %)

Medián prežívania (mesiace) 15,8 (14,8; 17,0) 11,2 (10,4; 13,0) (95 % IS)
Pomer rizík (95 % IS)b 0,740 (0,638; 0,859)
a p-hodnota je odvodená z log-rank testu stratifikovaného podľa skóre výkonnostného stavu ECOG (0-1 vs.
2), skóre bolesti (absentujúca vs. prítomná), počtu predtým absolvovaných režimov chemoterapií (1 vs. 2)
a typu progresie ochorenia (iba PSA vs. rádiografická).
b Pomer rizík je odvodený zo stratifikovaného proporcionálneho modelu rizika. Pomer rizík < 1 v prospech
abiraterón-acetátu.
Vo všetkých časových bodoch hodnotenia po niekoľkých úvodných mesiacoch liečby zostal nažive vyšší podiel pacientov liečených abiraterón-acetátom v porovnaní s podielom pacientov liečených placebom (pozri Obrázok
6).
O
brázok 6: Kaplanove-Meierove krivky prežívania pacientov liečených buď abiraterón-acetátom alebo placebom v kombinácii s prednizónom alebo prednizolónom s analógmi LHRH alebo predchádzajúcou orchiektómiou
AA = abiraterón-acetát
Analýza prežívania u podskupín poukázala na konzistentný prínos v prežívaní pri liečbe abiraterón-
acetátom (pozri Obrázok 7).
Obrázok 7: Celkové prežívanie podľa podskupín: pomer rizík a 95 % interval spoľahlivosti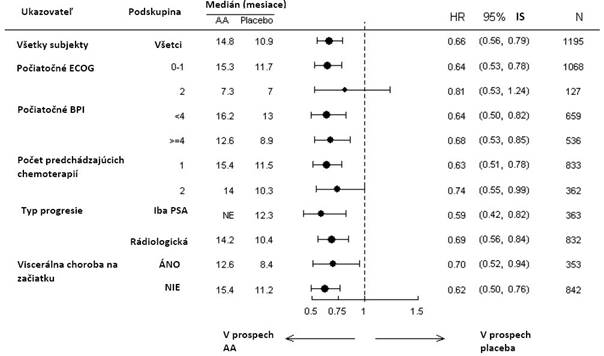
AA = abiraterón-acetát; BPI = dotazníková metóda hodnotenia bolesti (z angl. Brief Pain Inventory); IS = interval spoľahlivosti; ECOG = skóre výkonnostného stavu podľa Východnej spolupracujúcej onkologickej skupiny (z angl. Eastern Cooperative Oncology Group performance score); HR = pomer rizík; NE = nehodnotiteľné (z angl. not evaluable)
Navyše k pozorovanému zlepšeniu v celkovom prežívaní, všetky sekundárne koncové ukazovatele štúdie poukazovali v prospech abiraterón-acetátu a boli štatisticky významné po upravení pre viacnásobné testovanie, a to:
U pacientov, liečených abiraterón-acetátom sa potvrdila významne vyššia celková miera odpovede PSA (definované ako ≥ 50 % pokles oproti východiskovému stavu) v porovnaní s pacientmi, ktorí dostávali placebo, 38 % oproti 10 %, p < 0,0001.
Medián času do progresie PSA bol 10,2 mesiaca u pacientov liečených abiraterón-acetátom a 6,6 mesiaca u pacientov, ktorí dostávali placebo (HR = 0,580; 95 % IS: [0,462; 0,728], p < 0,0001).
Medián prežívania bez rádiografickej progresie ochorenia bol 5,6 mesiaca u pacientov liečených abiraterón-acetátom a 3,6 mesiaca u pacientov, ktorí dostávali placebo (HR = 0,673; 95 % IS: [0,585;
0,776], p < 0,0001).
Bolesť
Podiel pacientov so zmiernením bolesti bol štatisticky významne vyšší v skupine s abiraterónom než
v skupine s placebom (44 % oproti 27 %, p = 0,0002). Pacient so zmiernením bolesti bol definovaný ako ten, ktorý pocítil najmenej 30 % pokles oproti východiskovému stavu v skóre intenzity najhoršej bolesti
BPI-SF počas posledných 24 hodín bez akéhokoľvek zvýšenia skóre použitia analgetík pozorovaného pri dvoch po sebe idúcich hodnoteniach v intervale štyroch týždňov. Jedine pacienti s východiskovým skóre
bolesti ≥ 4 a najmenej jedným skóre bolesti po vstupe do štúdie podliehali analýze (N = 512) z hľadiska
zmiernenia bolesti.
Progresia bolesti sa zaznamenala u nižšieho podielu pacientov liečených abiraterónom v porovnaní
s pacientmi, ktorí dostávali placebo, a to v 6. (22 % oproti 28 %), 12. (30 % oproti 38 %) a 18. mesiaci
(35 % oproti 46 %). Progresia bolesti bola definovaná ako zvýšenie oproti východiskovému stavu ≥ 30 % v skóre intenzity najhoršej bolesti BPI-SF počas predchádzajúcich 24 hodín bez poklesu skóre použitia analgetík pozorovaného pri dvoch po sebe nasledujúcich návštevách, alebo ako ≥ 30 % zvýšenie skóre použitia analgetík pozorované pri dvoch po sebe nasledujúcich návštevách. Čas do progresie bolesti pri
25. percentile bol 7,4 mesiaca v skupine s abiraterónom oproti 4,7 mesiaca v skupine s placebom.
Udalosti súvisiaceso skeletom
Udalosti súvisiace so skeletom boli zaznamenané u nižšieho podielu pacientov v skupine s abiraterónom
v porovnaní so skupinou s placebom v 6. mesiaci (18 % oproti 28 %), 12. mesiaci (30 % oproti 40 %) a
18. mesiaci (35 % oproti 40 %). Čas do prvej udalosti súvisiacej so skeletom pri 25. percentile v skupine s abiraterónom bol dvojnásobný oproti času v kontrolnej skupine – 9,9 mesiaca oproti 4,9 mesiaca.
Udalosť súvisiaca so skeletom bola definovaná ako patologická zlomenina, kompresia miechy, paliatívna
rádioterapia kosti alebo chirurgický zákrok na kosti.
Pediatrickápopulácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s referenčným liekom obsahujúcim abiraterón-acetát vo všetkých podskupinách pediatrickej populácie pre pokročilú rakovinu prostaty. Informácie o použití v pediatrickej populácii, pozri časť 4.2.
5.2 Farmakokinetické vlastnosti
Po podaní abiraterón-acetátu sa farmakokinetika abiraterónu a abiraterón-acetátu skúmala u zdravých jedincov, pacientov s metastatickým pokročilým karcinómom prostaty a u jedincov bez karcinómu s poruchou funkcie pečene alebo obličiek. Abiraterón-acetát sa rýchlo konvertuje v podmienkach in vivo na abiraterón, inhibítor biosyntézy androgénov (pozri časť 5.1).
A
b
sorpcia
Po perorálnom podaní abiraterón-acetátu nalačno je čas do dosiahnutia maximálnej koncentrácie abiraterónu v plazme približne 2 hodiny.
Podanie abiraterón-acetátu s jedlom, v porovnaní s podaním nalačno, vedie až k 10-násobnému (AUC) a až k 17-násobnému (Cmax) zvýšeniu priemernej systémovej expozície abiraterónu, v závislosti od obsahu tuku v jedle. Ak vezmeme do úvahy bežnú rozmanitosť obsahu a zloženia jedla, užívanie abiraterónu spolu s jedlom má potenciál viesť k vysoko variabilným expozíciám. Z tohto dôvodu sa Abiraterone Glenmark nesmie užívať spolu s jedlom. Má sa užívať minimálne jednu hodinu pred jedlom alebo minimálne dve hodiny po jedle. Tablety treba prehĺtať celé a zapíjať vodou (pozri časť 4.2).
Distribúcia
Väzba na plazmatické bielkoviny 14C-abiraterónu v ľudskej plazme je 99,8 %. Zdanlivý distribučný
objem je približne 5 630 l, čo poukazuje na to, že abiraterón sa extenzívne distribuuje do periférnych tkanív.
Biotransformácia
Po perorálnom podaní 14C-abiraterón-acetatátu vo forme kapsúl sa abiraterón-acetát hydrolyzuje na
abiraterón, ktorý sa následne metabolizuje, vrátane sulfatácie, hydroxylácie a oxidácie prevažne v pečeni. Väčšina cirkulujúcej rádioaktivity (približne 92 %) sa zistila vo forme metabolitov abiraterónu. Spomedzi
15 detekovateľných metabolitov sú 2 hlavné metabolity – abiraterón-sulfát a abiraterón-N-oxid-sulfát;
každý z nich predstavuje približne 43 % celkovej rádioaktivity.
Eliminácia
Priemerný biologický polčas abiraterónu v plazme je približne 15 hodín podľa údajov získaných od zdravých jedincov. Po perorálnom podaní 14C-abiraterón-acetátu v dávke 1 000 mg sa približne 88 % rádioaktívnej dávky zistilo v stolici a približne 5 % v moči. Hlavné zložky prítomné v stolici sú nezmenený abiraterón-acetát a abiraterón (približne 55 % a 22 % podanej dávky, v uvedenom poradí).
Porucha funkcie pečene
Farmakokinetika abiraterón-acetátu sa skúšala u jedincov s už existujúcou ľahkou alebo stredne ťažkou
poruchou funkcie pečene (Childova-Pughova trieda A a B, v uvedenom poradí) ako aj
u zdravých jedincov v kontrolnej skupine. Systémová expozícia abiraterónu po jednorazovej perorálnej dávke 1 000 mg sa zvýšila približne o 11 % u jedincov s ľahkou o 260 % u jedincov so stredne ťažkou, už
existujúcou poruchou funkcie pečene. Priemerný polčas abiraterónu sa predlžuje na približne 18 hodín
u jedincov s ľahkou poruchou funkcie pečene a na približne 19 hodín u jedincov so stredne ťažkou
poruchou funkcie pečene.
V ďalšej štúdii sa farmakokinetika abiraterónu skúmala u jedincov s už existujúcou ťažkou (n = 8) poruchou funkcie pečene (Childova-Pughova trieda C) a u 8 zdravých kontrolných jedincov s normálnou funkciou pečene. AUC abiraterónu sa zvýšila o približne 600 % a frakcia voľného lieku sa zvýšila o 80 % u jedincov s ťažkou poruchou funkcie pečene v porovnaní s jedincami s normálnou funkciou pečene.
Nie je potrebná úprava dávkovania u pacientov s už existujúcou ľahkou poruchou funkcie pečene. Použitie abiraterón-acetátu sa má dôkladne posúdiť u pacientov so stredne ťažkou poruchou funkcie pečene, u ktorých by mal prínos jasne prevážiť potenciálne riziko (pozri časti 4.2 a 4.4). Abiraterón-acetát sa nemá používať u pacientov s ťažkou poruchou funkcie pečene (pozri časti 4.2, 4.3 a 4.4).
U pacientov, u ktorých sa rozvinie počas liečby hepatotoxicita, môže byť potrebné pozastaviť liečbu alebo
upraviť dávkovanie (pozri časť 4.2 a 4.4).
Porucha funkcie obličiek
Farmakokinetika abiraterón-acetátu sa porovnávala u pacientov v terminálnom štádiu ochorenia obličiek
so stabilným rozvrhom hemodialýzy oproti zodpovedajúcej kontrolnej skupine jedincov
s normálnou funkciou obličiek. Systémová expozícia abiraterónu sa po jednorazovej perorálnej dávke
1 000 mg u jedincov v terminálnom štádiu ochorenia obličiek liečených dialýzou nezvýšila. Podávanie
(pozri časť 4.2). Nie sú však klinické skúsenosti u pacientov s karcinómom prostaty a ťažkou poruchou funkcie obličiek. U týchto pacientov sa odporúča opatrnosť.
5.3 Predklinické údaje o bezpečnostiVo všetkých štúdiách toxicity na zvieratách boli výrazne znížené hladiny cirkulujúceho testosterónu.
V dôsledku toho boli zaznamenané znížené hmotnosti orgánov a morfologické a/alebo histopatologické
zmeny reprodukčných orgánov a adrenálnych, podmozgových a prsných žliaz. Všetky zmeny vykazovali úplnú alebo čiastočnú reverzibilitu. Zmeny reprodukčných orgánov a orgánov citlivých na androgén sú konzistentné s farmakológiou abiraterónu. Všetky hormonálne zmeny súvisiace s liečbou zanikli alebo sa javili ako ustupujúce po 4-týždňovom období rekonvalescencie.
V štúdiách fertility u samcov i samíc potkanov abiraterón-acetát znižoval fertilitu, čo bolo úplne reverzibilné 4 až 16 týždňov po ukončení podávania abiraterón-acetátu.
V štúdii vývojovej toxicity u potkanov abiraterón-acetát ovplyvňoval graviditu, vrátane zníženej fetálnej hmotnosti a prežívania. Boli pozorované vplyvy na vonkajšie pohlavné orgány, aj keď abiraterón-acetát nebol teratogénny.
V týchto štúdiách fertility a vývojovej toxicity vykonaných na potkanoch súviseli všetky účinky
s farmakologickou aktivitou abiraterónu.
Okrem zmien na reprodukčných orgánoch, pozorovaných vo všetkých štúdiách toxicity na zvieratách, predklinické údaje založené na konvenčných farmakologických štúdiách bezpečnosti, toxicity po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí. V 6-mesačnej štúdii s transgénnymi (Tg.rasH2) myšami nebol abiraterón-acetát karcinogénny.
V 24-mesačnej štúdii karcinogenity na potkanoch zvyšoval abiraterón-acetát incidenciu novotvarov
z intersticiálnych buniek semenníkov. Tento nález sa považuje za súvisiaci s farmakologickou aktivitou
abiraterónu a je špecifický pre potkany. Abiraterón-acetát nebol karcinogénny u samíc potkanov.
Liečivo abiraterón predstavuje environmentálne riziko pre vodné prostredie, a to najmä pre ryby.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokAbiraterone Glenmark 250 mg tablety:laktóza, monohydrát celulóza, mikrokryštalická
kroskarmelóza, sodná soľ
povidón laurylsíran sodný
koloidný bezvodý oxid kremičitý stearát horečnatý
Abiraterone Glenmark 500 mg filmom obalené tablety:
Jadro tabletylaktóza, monohydrát celulóza, mikrokryštalická kroskarmelóza, sodná soľ hypromelóza
laurylsíran sodný
koloidný bezvodý oxid kremičitý stearát horečnatý
Obal tablety polyvinylalkohol
oxid titaničitý (E 171)

makrogol mastenec
červený oxid železitý (E 172)
čierny oxid železitý (E 172)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnosti2 roky.
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaAbiraterone Glenmark 250 mg tabletyOkrúhle biele HDPE fľaše s detským bezpečnostným polypropylénovým skrutkovacím uzáverom.
Každé balenie obsahuje jednu fľašu so 120 tabletami.
Abiraterone Glenmark 500 mg filmom obalené tablety
|
|
PVC/PVDC/hliníkové blistre.
|
|
Filmom obalené tablety môžu byť zabalené do priehľadných alebo nepriehľadných (bielych) blistrov.
|
Každé blistrové balenie obsahuje 56, 56 x 1, 60, 60 x 1, 90 alebo 90 x 1 filmom obalených tabliet.
|
|
| | | |

Okrúhle, biele HDPE fľaše s detským bezpečnostným polypropylénovým skrutkovacím uzáverom.
Každé balenie obsahuje jednu fľašu so 60 filmom obalenými tabletami.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomNa základe mechanizmu účinku môže tento liek poškodiť vyvíjajúci sa plod, preto tehotné ženy alebo ženy, ktoré môžu byť tehotné nesmú zaobchádzať s týmto liekom bez ochrany, napr. rukavíc.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami. Tento liek môže predstavovať riziko pre vodné prostredie (pozri časť 5.3).
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIGlenmark Pharmaceuticals s.r.o., Hvězdova 1716/2b, 140 78 Praha 4, Česká republika
8. REGISTRAČNÉ ČÍSLAAbiraterone Glenmark 250 mg tablety: 44/0226/21-S
Abiraterone Glenmark 500 mg filmom obalené tablety: 44/0227/21-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE Dátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTU
08/2021