
šie korigovanej zrakovej ostrosti (BCVA) o ≥30 písmen v porovnaní s posledným stanovením zrakovej ostrosti;
o subretinálneho krvácania postihujúceho stred fovey, alebo ak rozsah krvácania je
≥50% celkovej plochy lézie;
• Dávka sa nemá podať, ak bol uskutočnený alebo plánovaný intraokulárny chirurgický zákrok počas uplynulých alebo nasledujúcich 28 dní.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné klinické skúšania.
Prídavné použitie fotodynamickej liečby (PDT) verteporfínom a Eylei sa neskúmalo, preto sa bezpečnostný profil nestanovil.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú dispozícii žiadne údaje o použití afliberceptu u gravidných žien.
Štúdie na zvieratách preukázali embryo-fetálnu toxicitu po vysokej systémovej expozícii (pozri časť
5.3).
Aj keď je systémová expozícia po podaní do oka veľmi nízka, Eylea sa neodporúča počas gravidity, pokiaľ potenciálny prínos nepreváži potenciálne riziko pre plod.
Laktácia
Nie je známe či sa aflibercept vylučuje do ľudského mlieka. Riziko pre dojčené dieťa nemožno vylúčiť.
Eylea sa neodporúča počas laktácie. Po zohľadnení prínosu dojčenia pre dieťa a prínosu liečby pre ženu je treba rozhodnúť, či prerušiť dojčenie alebo liečbu Eyleou..
Fertilita
Výsledky zo štúdií na zvieratách s vysokou systémovou expozíciou naznačujú, že aflibercept môže narušiť fertilitu samcov a samíc (pozri časť 5.3). Takéto účinky sa neočakávajú po očnom podaní
s veľmi nízkou systémovou expozíciou.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Injekcia Eylea má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje, vzhľadom k možným dočasným poruchám videnia súvisiacich s aplikáciou injekcie alebo očným vyšetrením. Pacienti
nesmú viesť vozidlá alebo obsluhovať stroje, kým sa ich zrakové funkcie dostatočne nezlepšia.
4.8 Nežiaduce účinky
Súhrnprofilubezpečnosti
Bezpečnosť liečby Eyleou sa sledovala celkovo v dvoch klinických skúšaniach vo fáze 3 s 1
824 pacientmi počas 96 týždňov a z toho 1 223 pacientov bolo liečených dávkou 2 mg.
Závažné nežiaduce reakcie súvisiace s podaním injekcie sa vyskytli pri menej ako 1 z 1 000 intravitreálnych injekčných podaní Eylei a zahŕňali endoftalmitídu, traumatickú kataraktu a prechodne zvýšený vnútroočný tlak (pozri časť 4.4).
Najčastejšie nežiaduce reakcie (u minimálne 5 % pacientov liečených Eyleou) boli spojovkové krvácanie (26,7 %), bolesť oka (10,3 %), odlúčenie sklovca (8,4 %), katarakta (7,9 %), zákaly sklovca (7,6 %) a zvýšený vnútroočný tlak (7,2 %).
Tabuľkovýzoznamnežiaducichreakcií
Nižšie popísané údaje bezpečnosti zahŕňajú všetky nežiaduce reakcie z klinických skúšaní vo fáze 3 s vlhkou vekom podmienenou degeneráciou makuly pravdepodobne súvisiacimi s podaním injekcie alebo liekom samotným počas 96 týždňov trvania klinického skúšania.
Nežiaduce reakcie sú zoradené podľa triedy orgánového systému a frekvencie pomocou nasledovnej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10,000 to < 1/1,000 pacientov).
Trieda orgánového systému
|
Veľmi časté
|
Časté
|
Menej časté
|
Zriedkavé
|
Poruchy imunitného
systému
|
|
| Precitlivenosť *)
|
|
Poruchy oka
| Spojovkové
krvácanie, bolesť oka
| Odlúpenie sietnice,
trhlina v pigmentovom epiteli sietnice,
odlúpenie pigmentového epitelu sietnice, degenerácia sietnice,
katarakta,
nukleárna katarakta, subkapsulárna katarakta,
erózia rohovky,
zvýšený vnútroočný tlak, rozmazané videnie, zákaly sklovca, opuch
rohovky,
odlúčenie sklovca, bolesť v mieste podania injekcie,
pocit cudzieho telesa v očiach, zvýšené slzenie, opuch očného viečka,
krvácanie v mieste podania injekcie,
hyperémia spojoviek okulárna hyperémia
| Endoftalmitída**),
trhlina v sietnici, krvácanie do sklovca,
kortikálna katarakta, lentikulárne opacity,
porušenie epitelu rohovky,
erózia rohovky, podráždenie
v mieste podania,
abnormálne pocity v oku,
podráždenie viečka, vitritída,
uveitída,
irititída, iridocyklitída,
zápal prednej očnej komory
| Hypopyon
|
*) zahŕňa reakcie precitlivenosti
**) pozitívna a negatívna kultúra endoftalmitídy
Popis vybraných nežiaducich reakciíV klinických skúšaniach fázy III pri vlhkej AMD sa u pacientov užívajúcich antitrombotické látky vyskytla zvýšená incidencia spojovkového krvácania. Tento zvýšený výskyt sa porovnal u pacientov liečených ranibizumabom a Eyleou.
Arteriálne trombembolické príhody (ATEs) sú nežiaduce príhody potenciálne súvisiace so systémovou inhibíciou VEGF. Po intravitreálnom použití inhibítorov VEGF existuje teoretické riziko arteriálnych
trombembolických príhod.
Arteriálne trombembolické príhody, podľa definície kritérií „Antiplatelet Trialists’ Collaboration“ (APTC) zahŕňajú nefatálny infarkt myokardu, nefatálnu cievnu mozgovú príhodu alebo vaskulárnu smrť (vrátane úmrtí z neznámej príčiny). Incidencia v klinických skúšaniach (VIEW1 a VIEW2) vo fáze 3 s vlhkou vekom podmienenou degeneráciou makuly počas 96 týždňov trvania klinického skúšania bola 3,3 % (60 z 1 824) v kombinovanej skupine pacientov liečených Eyleou v porovnaní s
3,2 % (19 z 595) pacientov liečených ranibizumabom (pozri časť 5.1).
Tak ako pri všetkých terapeutických proteínoch, aj pri Eyle existuje možnosť imunogenity.
4.9 Predávkovanie
V klinických skúšaniach sa použili dávky až do 4 mg v mesačných intervaloch a vyskytli sa ojedinelé prípady predávkovaní s 8 mg.
Predávkovanie zvýšeným objemom injekcie môže zvýšiť vnútroočný tlak. Preto sa má v prípade predávkovania monitorovať vnútroočný tlak a ak to považuje ošetrujúci lekár za nevyhnutné, má sa začať primeraná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologiká /antineovaskularizačné liečiva
ATC kód: S01LA05
Aflibercept je rekombinantný fúzny proteín, ktorý obsahuje fragmenty extracelulárnych domén ľudských VEGF receptorov 1 a 2 naviazaných na Fc fragment ľudského IgG1.
Aflibercept je vytvorený rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka
(CHO) K1.
Aflibercept pôsobí ako solubilný náhradný receptor, ktorý viaže VEGF-A a PlGF s vyššou afinitou
ako ich prirodzené receptory, a tým môže inhibovať väzbovosť a aktiváciu týchto analogických VEGF
receptorov.
Spôsobúčinku
Vaskulárny endoteliálny rastový faktor-A (VEGF-A) a placentárny rastový faktor (PlGF) sú členmi rodiny angiogénnych faktorov VEGF, ktoré môžu pôsobiť ako silné mitogénne, chemotaktické faktory a faktory vaskulárnej permeability endotelových buniek. VEGF pôsobí prostredníctvom dvoch receptorov tyrozínkináz; VEGFR-1 a VEGFR-2 prítomných na povrchu endotelových buniek. PlGF
sa viaže iba na VEGFR-1, ktorý je prítomný aj na povrchu leukocytov. Nadmerná aktivácia týchto receptorov sprostredkovaná VEGF-A môže mať za následok patologickú neovaskularizáciu
a nadmernú vaskulárnu permeabilitu. PlGF môže spolupôsobiť s VEGF-A v týchto procesoch, a tiež
je známe, že podporuje infiltráciu leukocytmi a vaskulárny zápal.
Farmakodynamickéúčinky
Vlhká vekom podmienená degenerácia makuly sa prejavuje patologickou choroidálnou neovaskularizáciou (CNV). Presakovanie krvi a tekutiny z CNV môže spôsobiť zhrubnutie sietnice alebo opuch a/alebo krvácanie pod sietnicu/do sietnice, čo vedie k strate zrakovej ostrosti.
U pacientov liečených Eyleou (jednou injekciou mesačne tri po sebe nasledujúce mesiace, po ktorých sa pokračuje jednou injekciou každé 2 mesiace) sa krátko po začatí liečby znížilo zhrubnutie sietnice
a znížila sa priemerná veľkosť ložiska CNV, čo je v súlade s výsledkami pozorovanými po liečbe
0,5 mg ranibizumabom mesačne.
V klinickom skúšaní VIEW1 bolo priemerné zníženie zhrubnutia sietnice pri optickej koherenčnej tomografii (OCT) (-130 respektíve -129 mikrónov v 52. týždni v skupinách klinického skúšania s 2 mg Eylei každé dva mesiace a ranibizumabom 0,5 mg mesačne (v uvedenom poradí)). V klinickom skúšaní VIEW2 tiež v 52. týždni bolo priemerné zníženie zhrubnutia sietnice pri OCT (-149 respektíve -139 mikrónov v skupinách klinického skúšania s 2 mg Eylei každé dva mesiace a ranibizumabom 0,5 mg mesačne (v uvedenom poradí)).
Redukcia veľkosti CNV a zníženie zhrubnutia sietnice sa vo všeobecnosti zachovali v druhom roku klinických skúšaní.
K
l
i
n
i
cká
účinnosť
a
bezpečnosť
Bezpečnosť a účinnosť Eylei sa hodnotili v dvoch randomizovaných, multicentrických, dvojito zaslepených, aktívne kontrolovaných klinických skúšaniach u pacientov s vlhkou vekom podmienenou
degeneráciou makuly. Celkovo sa liečilo 2 412 pacientov a účinnosť sa hodnotila (1 817 s Eyleou)
v dvoch klinických skúšaniach (VIEW1 a VIEW2). V každom klinickom skúšaní boli pacienti náhodne pridelení v pomere 1:1:1:1 do 1 zo 4 dávkovacích schém:
1) 2 mg Eylei sa podávali každých 8 týždňov po 3 začiatočných mesačných dávkach (Eylea 2Q8);
2) 2 mg Eylei sa podávali každé 4 týždne (Eylea 2Q4);
3) 0,5 mg Eylei sa podávalo každé 4 týždne (Eylea 0.5Q4); a
4) 0,5 mg ranibizumabu sa podávalo každé 4 týždne (ranibizumab 0.5Q4). Vekové rozpätie pacientov od 49 do 99 rokov s mediánom 76 rokov.
V druhom roku klinických skúšaní pokračovali pacienti v používaní dávky v sile, na ktorú boli v úvode randomizovaní, no modifikovaná dávkovacia schéma sa nastavila podľa vyhodnotenia zrakových a anatomických výsledkov protokolom definovaným maximálnym dávkovacím intervalom
12 týždňov.
V obidvoch klinických skúšaniach bol primárnym koncovým ukazovateľom účinnosti podiel pacientov liečených podľa protokolu, u ktorých zostal zachovaný vízus, definovaný stratou menej ako
15 písmen zrakovej ostrosti v 52. týždni v porovnaní s východiskovým stavom.
V klinickom skúšaní VIEW1 v 52. týždni malo 95,1 % pacientov v liečebnej skupine Eylea
2Q8 zachovaný vízus , v porovnaní s 94,4 % pacientmi v skupine ranibizumab 0.5Q4. Liečba Eyleou ukázala, že je non- inferiórna a klinicky ekvivalentná so skupinou ranibizumab 0.5Q4.
V klinickom skúšaní VIEW2 v 52. týždni malo 95,6 % pacientov v liečebnej skupine Eylea
2Q8 zachovaný vízus ,v porovnaní s 94,4 % pacientmi v skupine ranibizumab 0.5Q4. Liečba Eyleou ukázala, že je non- inferiórna a klinicky ekvivalentná so skupinou ranimizumab 0.5Q4.
Detailné výsledky zo súhrnnej analýzy oboch klinických skúšaní sú uvedené nižšie v Tabuľke a na
Obrázku.
T
abuľka:
V
ý
sledky
účinnosti
v
52.
týždni
(
primárna
analýza)
a
96.
týždni;
súhrnné údaje
z klinických
skúšaníVIEW1aVIEW2B)
Výsledok účinnosti
| Eylea 2Q8 E)
(2 mg Eylea každých 8 týždňov po 3 začiatočných mesačných dávkach)
(n = 607)
| Ranibizumab 0.5Q4
(ranibizumab 0,5 mg každé
4 týždne)
(n = 595)
|
| 52. týždeň
| 96. týždeň G)
| 52. týždeň
| 96. týždeň G)
|
Priemerný počet injekcií
| 7,6
| 11,2
| 12,3
| 16,5
|
Priemerný počet injekcií počas 2.roka (52. až 96.
týždeň)
|
|
4.1
|
|
4.6
|
Podiel pacientov so zachovanou zrakovou ostrosťou
(strata < 15 písmen BCVAA)) (v skupine podľa protokolu)
|
95,33%B)
|
92,42 %
|
94,42 % B)
|
91,60 %
|
RozdielC)
(95 % CI)D)
|
0,9 %
(-1,7, 3,5)F)
|
0,8 %
(- 2,3, 3,8)F)
|
|
|
Medián zmeny v BCVA podľa meraní ETDRSA) skóre písmen oproti východiskovému stavu
|
8,40
|
7,62
|
8,74
|
7,89
|
Rozdiel v LS A) medián
(ETDRS písmená)C)
(95 % CI)D)
|
-0,32
(-1,87, 1,23)
|
-0,25
(-1,98, 1,49)
|
|
|
Podiel pacientov, ktorí získali minimálne 15 písmen zrakovej ostrosti oproti východiskovému stavu
|
30,97 %
|
33,44 %
|
32,44 %
|
31,60 %
|
RozdielC)
(95 % CI)D)
|
-1,5 %
(-6,8, 3,8)
|
1,8 %
(-3,5, 7,1)
|
|
|
A) BCVA: Best Corrected Visual Acuity (Najlepšie korigovaná zraková ostrosť)
ETDRS: Early Treatment Diabetic Retinopaty Study (Klinické skúšanie včasnej liečby diabetickej retinopatie)
LS: Least square means derived from ANCOVA (Priemery najmenších štvorcov vypočítané na základe modelu ANCOVA)
B) Celkový analyzovaný súbor (Full Analysis Set, FAS), Výsledky z posledného dokumentovaného vyšetrenia
(Last Observation Carried Forward, LOCF) pre všetky analýzy okrem podielu pacientov so zachovanou zrakovou ostrosťou v 52. týždni, čo je skupina podľa protokolu (Per Protocol Set, PPS)
C) Rozdiel je hodnota skupiny s Eyleou mínus hodnota skupiny s ranibizumabom. Pozitívna hodnota zvýhodňuje
Eyleu.
D) Interval spoľahlivosti (Confidence interval, CI) vypočítaný pomocou normálneho rozdelenia
E) Po začatí liečby tromi mesačnými dávkami
F) Interval spoľahlivosti ležiaci úplne nad -10 %,ktorý poukazuje na non-inferioritu Eylei voči ranibizumabu
G) Na začiatku 52. týždňa, u všetkých skupín pacientov liečených pomocou modifikovanej formy štvrťročnej liečby sa aplikovala dávka každé 4 týždne, ale nie menej ako jedenkrát počas každých 12 týždňov na základe
vopred špecifikovaných kritérií opakovanej liečby
Obrázok 1. Priemerná zmena zrakovej ostrosti oproti východiskovému stavu do 96. týždňa pre súhrnné údaje z klinických skúšaní View1 a View2

*) Od východiskového stavu do 52. týždňa sa Eylea podávala každých 8 týždňov po 3 začiatočných mesačných dávkach. . Od východiskového stavu do 52. týždňa sa ranibizumab 0,5mg podával každé 4 týždne. Na začiatku
52. týždňa, u všetkých skupín pacientov liečených pomocou modifikovanej formy štvrťročnej liečby sa aplikovala dávka každé 4 týždne, ale nie menej ako jedenkrát počas každých 12 týždňov na základe vopred špecifikovaných kritérií opakovanej liečby.
Podiel pacientov, ktorí v 96.týždni získali minimálne 15 písmen zrakovej ostrosti v porovnaní s východiskovým stavom bol 33,44 % v skupine Eylea 2Q8 a 31,60 % v skupine ranibizumab 0.5Q4.
Súhrnné údaje z klinických skúšaní VIEW1 a VIEW2 s Eyleou preukázali klinicky významné zmeny voči východiskovému stavu pri vopred špecifikovanom sekundárnom ukazovateli účinnosti stanoveným National Eye Institute Visual Function Questionnaire (NEI VFQ-25). Intenzita týchto zmien bola podobná tým, ktoré boli publikované v klinických skúšaniach a zodpovedali získaniu 15 písmen s najlepšie korigovanou zrakovou ostrosťou (Best Corrected Visual Acuity, BCVA).
Žiadne klinicky významné rozdiely sa nezistili medzi Eyleou a referenčným liekom ranibizumabom
v zmenách celkového skóre NEI VFQ-25 a podškálach (aktivity vyžadujúce videnie do blízka, aktivity vyžadujúce videnie do diaľky a aktivity závislé špecificky na víze ) v 52. týždni od východiskového stavu.
Zníženia priemernej plochy CNV boli preukázané vo všetkých dávkovacích skupinách v obidvoch klinických skúšaniach.
Výsledky účinnosti vo všetkých hodnotených podskupinách (napr. vek, pohlavie, rasa, východisková zraková ostrosť, typ lézie, veľkosť lézie) v každom klinickom skúšaní a v súhrnnej analýze a boli zhodné s výsledkami v celkovej populácii.
V druhom roku klinických skúšaní bola vo všeobecnosti účinnosť zachovaná počas posledného hodnotenia v 96. týždni. V druhom roku klinických skúšaní sa u 2-4% pacientov požadovala aplikácia všetkých dávok na mesačnej báze a u tretiny pacientov sa požadovala aspoň jedna dávka v intervale jedného mesiaca počas liečby.
Starší
p
acienti
V klinických skúšaniach bolo približne 89 % (1 616/1 817) pacientov randomizovaných na liečbu Eyleou vo veku 65 rokov alebo starších a približne 63 % (1 139/1 817) bolo vo veku 75 rokov alebo starších.
Detiadospievajúci
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky klinických skúšaní pre Eyleu vo všetkých vekových podskupinách detí a dospievajúcich s vlhkou vekom podmienenou degeneráciou makuly (pre informácie o použití u detí a dospievajúcich, pozri časť 4.2 ).
5.2 Farmakokinetické vlastnosti
Na dosiahnutie lokálnych účinkov v oku sa Eylea podáva priamo do sklovca. Absorpcia/Distribúcia
Aflibercept je po intravitreálnom podaní pomaly absorbovaný z oka do systémového obehu
a v systémovom obehu je prevažne pozorovaný ako inaktívny, stabilný komplex s VEGF. Iba „voľný aflibercept“ je však schopný viazať endogénny VEGF.
Vo farmakokinetickej podštúdiiso 6 pacientmi s častým odberom vzoriek boli v priebehu 1 až 3 dní po intravitreálnom podaní injekcie 2 mg maximálne plazmatické koncentrácie voľného afliberceptu (systémová C max ) nízke, so strednou hodnotou približne 0,02 mikrogramu/ml (rozsah od 0 do 0,054)
a takmer u všetkých pacientov boli nedetekovateľné dva týždne po podaní. Aflibercept sa nekumuluje v plazme pri podávaní intravitreálne každé 4 týždne.
Stredná maximálna plazmatická koncentrácia voľného afliberceptu je približne 50 až 500-násobne nižšia ako koncentrácia afliberceptu potrebná na inhibíciu biologickej aktivity systémového VEGF
o 50 % na modeloch zvierat, u ktorých sa pozorovali zmeny krvného tlaku po cirkulujúcich hladinách voľného afliberceptu dosahujúceho približne 10 mikrogramov/ml a vrátili sa na východiskovú hodnotu
po poklese hladín pod približne 1 mikrogram/ml. Odhaduje sa, že po intravitreálnom podaní 2 mg pacientom je stredná maximálna plazmatická koncentrácia voľného afliberceptu viac ako 100- násobne nižšia ako koncentrácia afliberceptu potrebná na naviazanie maximálne polovice
systémového VEGF (2,91 mikrogramov/ml) v klinickom skúšaní so zdravými dobrovoľníkmi. Preto nie sú pravdepodobné systémové farmakodynamické účinky, ako sú zmeny tlaku krvi.
Eliminácia
Keďže Eylea je liek proteínového charakteru, nevykonali sa žiadne metabolické klinické skúšania.
Voľný aflibercept viaže VEGF za vzniku stabilného, inertného komplexu. Predpokladá sa, že tak ako iné veľké proteíny aj voľný i viazaný aflibercept sa budú rozkladať proteolytickým katabolizmom.
Poškodeniefunkcieobličiek
U pacientov s poškodením funkcie obličiek sa s Eyleou nevykonali žiadne špeciálne klinické skúšania.
Farmakokinetická analýza pacientov v klinickom skúšaní VIEW2, v ktorej malo 40 % poškodenú funkciu obličiek (24 % mierne, 15 % stredne ťažko a 1 % ťažko) neodhalila žiadne rozdiely
v súvislosti s plazmatickými koncentráciami liečiva po intravitreálnom podávaní každé 4 alebo
8 týždňov.
5.3 Predklinické údaje o bezpečnosti
V predklinických štúdiách sa toxické účinky pri opakovanom podávaní pozorovali iba pri podstatne vyšších systémových expozíciách, ako je maximálna expozícia u ľudí po intravitreálnom podaní stanovenej klinickej dávky, čo poukazuje na malý význam pre klinické použitie.
Erózie a ulcerácie dýchacieho epitelu v nosových mušliach opíc liečených afliberceptom intravitreálne sa pozorovali pri vyšších systémových expozíciách, ako je maximálna expozícia u ľudí. Systémová
expozícia na základe Cmax a AUC voľného afliberceptu bola približne 200- respektíve 700-násobne vyššia (v uvedenom poradí) v porovnaní so zodpovedajúcimi hodnotami pozorovanými u ľudí po intravitreálnej dávke 2 mg. Pri hladine bez pozorovaného nepriaznivého účinku (No Observed Adverse Effect Level, NOAEL) 0,5 mg/oko u opíc bola systémová expozícia 42- respektíve 56- násobne vyššia (v uvedenom poradí) na základe Cmax a AUC.
Na mutagénny alebo karcinogénny potenciál afliberceptu sa nerobili žiadne štúdie.
V štúdii embryofetálneho vývinu spôsobil aflibercept na gravidných králikoch po intravenóznom podávaní (3 až 60 mg/kg) embryofetálnu toxicitu (teratogenita pri všetkých testovaných dávkach). Pri najnižšej testovanej dávke v tejto štúdii (3 mg/kg) boli systémové expozície na základe Cmax a AUC voľného afliberceptu približne 2 900- respektíve 600-násobne vyššie (v uvedenom poradí) v porovnaní so zodpovedajúcimi hodnotami pozorovanými u ľudí po intravitreálnej dávke 2 mg.
Účinky na fertilitu samcov a samíc sa hodnotili ako časť 6-mesačnej štúdie na opiciach
s intravenóznym podávaním afliberceptu v rozsahu dávok od 3 do 30 mg/kg. Pri všetkých hladinách dávok sa pozorovali chýbajúce alebo nepravidelné menštruácie, ktoré súviseli so zmenami hladín samičích pohlavných hormónov a zmeny morfológie a pohyblivosti spermií. Na základe Cmax a AUC voľného afliberceptu pozorovaných pri intravenóznej dávke 3 mg/kg boli systémové expozície približne 4 900-násobne respektíve 1 500-násobne vyššie (v uvedenom poradí), než expozícia pozorovaná u ľudí po intravitreálnej dávke 2 mg. Všetky zmeny boli reverzibilné.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Polysorbát 20
Monohydrát dihydrogénfosforečnanu sodného (na úpravu pH) Heptahydrát monohydrogénfosforečnanu sodného (na úpravu pH) Chlorid sodný
Sacharóza
Voda na injekciu
6.2 Inkompatibility
Nerobili sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (pri teplote 2°C až 8°C). Neuchovávajte v mrazničke.
Naplnenú injekčnú striekačku uchovávajte vo jej blistri a vo vonkajšom obale na ochranu pred svetlom.
Pred použitím sa môže neotvorené blistrové balenie Eylei uchovávať pri izbovej teplote (25 °C) počas
24 hodín. Po otvorení blistra dodržiavajte aseptické podmienky.
6.5 Druh obalu a obsah balenia
90 mikrolitrov roztoku v naplnenej injekčnej striekačke (sklo typu I), ktorá je označená čiernou ryskou
dávkovania, s piestovou zátkou (z elastomérovej gumy) a s nadstavcom so závitom („Luer lock“)
s koncovým uzáverom (z elastomérovej gumy). Veľkosť balenia 1.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomNaplnená injekčná striekačka je len na jednorazové použitie.
Neotvárajte blister s naplneným injekčným roztokom mimo čistej sály na podávanie.
Vzhľadom k tomu, že naplnená injekčná striekačka obsahuje väčší objem (90 mikrolitrov) ako odporúčaná dávka (50 mikrolitrov), časť objemu obsiahnutého v injekčnej striekačke, musí byť odstránený pred podaním.
Injekčný roztok pred podaním vizuálne skontrolujte na prítomnosť cudzorodých častíc a/alebo zmenu zafarbenia alebo zmenu fyzikálneho vzhľadu. Naplnenú injekčnú striekačku nepoužívajte ak sú viditeľné častice, zakalenie alebo zmena farby. V prípade, že niečo také zistíte, liek zlikvidujte.
Na intravitreálne podanie injekcie sa má použiť injekčná ihla 30 G x ½ palca.
Pokyny na používanie naplnenej injekčnej striekačky:1. Keď ste pripravený na podanie Eylei, otvorte škatuľku a vyberte sterilné blistrové balenie.
Opatrne otvorte blistrové balenie, tak aby ste zabezpečili sterilitu jeho obsahu. Injekčnú striekačku položte na sterilný podnos, pokým sa pripravíte na kompletizáciu.
2. Použitím aseptického postupu vyberte injekčnú striekačku zo sterilného blistrového balenia.

3. Pri odstraňovaní uzáveru injekčnej striekačky držte injekčnú striekačku v jednej ruke, zatiaľ čo palcom a ukazovákom druhej ruky uchopíte uzáver injekčnej striekačky. Upozornenie: Uzáver injekčnej striekačky vytiahnite (neotáčajte ani nekrúťte uzáverom).
4. Nevyťahujte piest, aby sa zamedzilo strate sterility lieku.
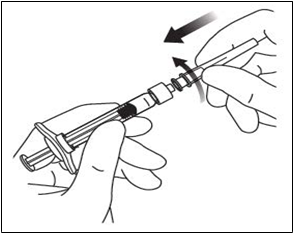
5. Použitím aseptického postupu pootočením pevne nasaďte injekčnú ihlu na nadstavec so závitom („Luer lock“) na injekčnú striekačku.
6. Z injekčnej ihly odstráňte plastový kryt.
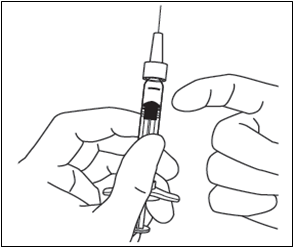
7. Injekčnú striekačku s ihlou držte smerom nahor, skontrolujte v nej prítomnosť bubliniek. V prípade prítomnosti bubliniek injekčnú striekačku jemne poklepte prstom, kým bublinky nevystúpia na povrch.
8. Na odstránenie všetkých bubliniek a nadbytočného lieku pomaly zatlačte na piest, aby ste
zarovnali cylindrickú spodnú časť kužeľovitého okraja piestu s čiernou ryskou dávky na injekčnej striekačke (čo zodpovedá 50 mikrolitrom).
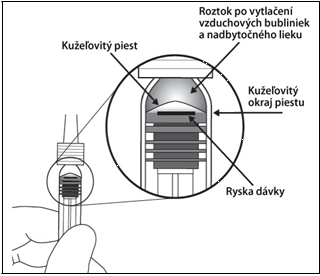
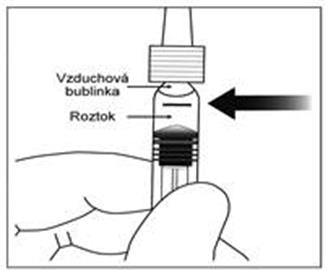
9. Naplnená injekčná striekačka je len na jednorazové použitie.
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBayer Pharma AG
13342 Berlin
Nemecko
8. REGISTRAČNÉ ČÍSLO9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.
1. NÁZOV LIEKU
Eylea 40 mg/ml injekčný roztok v injekčnej liekovke
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
1 ml injekčného roztoku obsahuje 40 mg afliberceptu*.
Každá injekčná liekovka obsahuje 100 mikrolitrov, čo zodpovedá 4 mg afliberceptu. To poskytuje použiteľné množstvo na podanie jednorazovej dávky 50 mikrolitrov obsahujúcich 2 mg afliberceptu.
*Aflibercept, je fúzny proteín, ktorý obsahuje fragmenty extracelulárnych domén ľudských VEGF (vaskulárny endoteliálny rastový faktor) receptorov 1 a 2 naviazaných na Fc fragment ľudského IgG1 a vytvorený rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka (CHO) K1.
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Injekčný roztok (roztok)
Číry, bezfarebný až bledožltý, izo-osmotický roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikácie
Eylea je indikovaná na liečbu neovaskulárnej (vlhkej) vekom podmienenej degenerácie makuly
(VPDM ) u dospelých (pozri časť 5.1).
4.2 Dávkovanie a spôsob podávania
Eylea je len na intravitreálne podanie.
Eyleu musí podávať iba kvalifikovaný lekár so skúsenosťami s podávaním intravitreálnych injekcií. Dávkovanie
Odporúčaná dávka Eylei sú 2 mg afliberceptu, čo zodpovedá 50 mikrolitrom.
Liečba Eyleou sa začína jednou injekciou mesačne tromi po sebe nasledujúcimi dávkami, po ktorých sa pokračuje jednou injekciou každé 2 mesiace. Medzi injekciami nie je potrebné monitorovanie.
Po prvých 12 mesiacoch liečby Eyleou, možno na základe zrakových a anatomických výsledkov liečebný interval predĺžiť.
Vtomto prípade má monitorovanie určovať lekár a môže byť častejšie ako samotné plánované podávanie injekcií.
O
sobitné skupiny pacientov
Pacienti s poškodením funkcie pečene a/alebo obličiek
Žiadne špecifické klinické skúšania s Eyleou sa nevykonali u pacientov s poškodením funkcie pečene a/alebo obličiek.
Dostupné údaje nenaznačujú potrebu úpravy dávky Eylei u týchto pacientov (pozri časť 5.2).
Starší pacienti
Nie sú potrebné žiadne zvláštne opatrenia.
Deti a dospievajúci
Bezpečnosť a účinnosť sa u detí a dospievajúcich neskúmala. U detí a dospievajúcich neexistuje žiadne použitie Eylei v indikácií na vlhkú vekom podmienenú degeneráciu makuly.
Spôsob podania
Intravitreálne podanie injekcií musí vykonať, v súlade s lekárskymi štandardmi a platnými postupmi,
kvalifikovaný lekár so skúsenosťami s podávaním intravitreálnych injekcií. Vo všeobecnosti sa musí zabezpečiť adekvátna anestézia a asepsa vrátane podania širokospektrálneho lokálneho antibiotika
(napr. jódovaný povidón sa nanáša na kožuokolooka,očnéviečkoapovrchoka). Odporúča sa chirurgická dezinfekcia rúk, použitie sterilných rukavíc, sterilného rúška a sterilného spekula očného
viečka (alebo náhrady).
Injekčná ihla sa zavádza 3,5-4,0 mm za limbom do dutiny sklovca, vyhýba sa horizontálnemu poludníku a smeruje do centra očnej gule. Potom sa aplikuje objem injekcie 0,05 ml; pri následných injekciách sa má použiť iné miesto na sklére.
Okamžite po intravitreálnom podaní injekcie by mali byť pacienti monitorovaní kvôli zvýšeniu vnútroočného tlaku. Primeraný monitoring má pozostávať z kontroly perfúzie terča zrakového nervu alebo tonometrie. Ak je to potrebné, majú byť k dispozícii sterilné nástroje na paracentézu.
Po intravitreálnom podaní injekcie majú byť pacienti poučení, aby bez meškania hlásili akékoľvek symptómy navodzujúce endoftalmitídu (napr. bolesť oka, sčervenanie oka, fotofóbia, rozmazané videnie).
Každá injekčná liekovka sa má použiť len na liečbu jedného oka.
Injekčná liekovka obsahuje viac ako je odporúčaná dávka 2 mg. Extrahovateľný objem injekčnej liekovky (100 mikrolitrov) sa nesmie celý použiť. Prebytočné množstvo sa má pred aplikáciou odstrániť.
Pri aplikácií celkového množstva môže dôjsť k predávkovaniu. Na odstránenie vzduchových bubliniek a nadbytočného lieku pomaly zatlačte na piest aby ste zarovnali cylindrickú spodnú časť kužeľovitého okraja piestu s čiernou ryskou dávky na injekčnej liekovke (čo zodpovedá 50 mikrolitrom, t.j. 2 mg afliberceptu).
Po podaní injekcie sa musí všetok nepoužitý liek zlikvidovať. Pokyny na zaobchádzanie s liekom, pozri časť 6.6.
4.3 Kontraindikácie
- precitlivenosť na liečivo aflibercept alebo na ktorúkoľvek z pomocných látok uvedených v časti
6.1.
- Aktívna alebo suspektná očná alebo periokulárna infekcia.
- Aktívny závažný vnútroočný zápal.
4.4 Osobitné upozornenia a opatrenia pri používaní
Endoftalmitída
Podanie intravitreálnych injekcií vrátane injekcií afliberceptu sa spájalo s endoftalmitídou (pozri časť
4.8). Pri podávaní Eylei sa musia vždy používať náležité aseptické injekčné postupy. Pacienti majú byť poučení, aby bez meškania hlásili akékoľvek symptómy nasvedčujúce endoftalmitíde a tieto symptómy sa majú primerane manažovať.
Zvýšenývnútroočnýtlak
V priebehu 60 minút po podaní intravitreálnej injekcie, vrátane injekcií Eylei, sa pozorovali zvýšenia vnútroočného tlaku (pozri časť 4.8). Osobitná opatrnosť je potrebná u pacientov s nedostatočne
kontrolovaným glaukómom (nepodávajte Eyleu pokým je vnútroočný tlak ≥ 30 mmHg). Vo všetkých prípadoch sa preto musí monitorovať a primerane manažovať vnútroočný tlak aj perfúzia terča
zrakového nervu.
Imunogenita
Eylea je terapeutický proteín, preto je možná imunogenicita s Eyleaou (pozri časť 4.8).
Pacienti majú byť poučení, aby hlásili akékoľvek znaky alebo symptómy nasvedčujúce vnútroočnému zápalu, napríklad bolesť, fotofóbia alebo začervenanie, ktoré môžu byť klinickým príznakom
hypersenzitivity.
Systémovéúčinky
Systémové nežiaduce účinky, vrátane mimoočného krvácania a arteriálnych tromboembolických príhod, sa hlásili po intravitreálnom použití inhibítorov VEGF, že tieto účinky sa môžu týkať inhibície VEGF.
Iné
Tak ako pri iných intravitreálnych liečbach s anti-VEGF sa na liečbu vekom podmienenej degenerácie makuly vzťahuje tiež nasledovné:
• Bezpečnosť a účinnosť liečby Eyleou pri súčasnom podaní do oboch očí sa systematicky neskúmali.
• Rizikové faktory spojené so vznikom trhliny v pigmentovom epiteli sietnice po liečbe anti- VEGF pri vlhkej VPDM zahŕňajú rozsiahle a/alebo vysoké odlúpenie pigmentového epitelu sietnice. Pri začatí liečby Eyleou je potrebná opatrnosť u pacientov s týmito rizikovými faktormi trhlín v pigmentovom epiteli sietnice.
• Liečba sa má ukončiť u pacientov s regmatogénnym odlúpením sietnice alebo makulárnymi dierami 3. alebo 4. stupňa.
• Existuje teoretické riziko arteriálnych tromboemolických príhodpo intravitreálnom použití inhibítorov VEGF (vaskulárny endoteliálny rastový faktor), pozri časť 4.8
• V prípade poškodenia sietnice sa má dávkovanie prerušiť a liečba sa nemá opäť začať, kým sa dostatočne obnoví.
• Dávka sa nemá podať a v liečbe sa nemá pokračovať skôr ako počas najbližšej plánovanej návštevy v prípade:
o poklesu najlepšie korigovanej zrakovej ostrosti (BCVA) o ≥30 písmen v porovnaní s posledným stanovením zrakovej ostrosti;
o subretinálneho krvácania postihujúceho stred fovey, alebo ak rozsah krvácania je ≥50%
celkovej plochy lézie;
• Dávka sa nemá podať, ak bol uskutočnený alebo plánovaný intraokulárny chirurgický zákrok počas uplynulých alebo nasledujúcich 28 dní.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné klinické skúšania.
Prídavné použitie fotodynamickej liečby (PDT) verteporfínom a Eylei sa neskúmalo, preto sa bezpečnostný profil nestanovil.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú dispozícii žiadne údaje o použití afliberceptu u gravidných žien.
Štúdie na zvieratách preukázali embryo-fetálnu toxicitu po vysokej systémovej expozícii (pozri časť
5.3).
Aj keď je systémová expozícia po podaní do oka veľmi nízka, Eylea sa neodporúča počas gravidity, pokiaľ potenciálny prínos nepreváži potenciálne riziko pre plod.
Laktácia
Nie je známe či sa aflibercept vylučuje do ľudského mlieka. Riziko pre dojčené dieťa nemožno vylúčiť.
Eylea sa neodporúča počas laktácie. Po zohľadnení prínosu dojčenia pre dieťa a prínosu liečby pre ženu je treba rozhodnúť, či prerušiť dojčenie alebo liečbu Eyleou.
Fertilita
Výsledky zo štúdií na zvieratách s vysokou systémovou expozíciou naznačujú, že aflibercept môže narušiť fertilitu samcov a samíc (pozri časť 5.3). Takéto účinky sa neočakávajú po očnom podaní
s veľmi nízkou systémovou expozíciou.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Injekcia Eylea má mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje, vzhľadom k možným dočasným poruchám videnia súvisiacich s aplikáciou injekcie alebo očným vyšetrením. Pacienti
nesmú viesť vozidlá alebo obsluhovať stroje, kým sa ich zrakové funkcie dostatočne nezlepšia.
4.8 Nežiaduce účinky
Súhrnprofilubezpečnosti
Bezpečnosť liečby Eyleou sa sledovala celkovo v dvoch klinických skúšaniach vo fáze 3 s 1
824 pacientmi počas 96 týždňov a z toho 1 223 pacientov bolo liečených dávkou 2 mg.
Závažné nežiaduce reakcie súvisiace s podaním injekcie sa vyskytli pri menej ako 1 z 1 000 intravitreálnych injekčných podaní Eylei a zahŕňali endoftalmitídu, traumatickú kataraktu a prechodne zvýšený vnútroočný tlak (pozri časť 4.4).
Najčastejšie nežiaduce reakcie (u minimálne 5 % pacientov liečených Eyleou) boli spojovkové krvácanie (26,7 %), bolesť oka (10,3 %), odlúčenie sklovca (8,4 %), katarakta (7,9 %), zákaly sklovca (7,6 %) a zvýšený vnútroočný tlak (7,2 %).
T
abuľkový
zoznam
nežiaducich
reakcií
Nižšie popísané údaje bezpečnosti zahŕňajú všetky nežiaduce reakcie (závažné a nezávažné) z klinických skúšaní vo fáze 3 s vlhkou vekom podmienenou degeneráciou makuly pravdepodobne súvisiacimi s podaním injekcie alebo liekom samotným počas 96 týždňov trvania klinického skúšania.
Nežiaduce reakcie sú zoradené podľa triedy orgánového systému a frekvencie pomocou nasledovnej konvencie: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100) , zriedkavé (≥ 1/10,000 to < 1/1,000 pacientov).
Trieda
orgánového systému
|
Veľmi časté
|
Časté
|
Menej časté
|
Zriedkavé
|
Poruchy imunitného systému
|
|
| Precitlivenosť *)
|
|
Poruchy oka
| Spojovkové
krvácanie, bolesť oka
| Odlúpenie sietnice,
trhlina v pigmentovom epiteli
sietnice,
odlúpenie pigmentového epitelu sietnice,degenerácia sietnice,
katarakta,
nukleárna katarakta, subkapsulárna katarakta,
erózia rohovky, zvýšený vnútroočný
tlak,
rozmazané videnie, zákaly sklovca, opuch rohovky,
odlúčenie sklovca, bolesť v mieste podania injekcie,
pocit cudzieho telesa v očiach,
zvýšené slzenie, opuch očného viečka,
krvácanie v mieste podania injekcie, hyperémia spojoviek
okulárna hyperémia
| Endoftalmitída**),
trhlina v sietnici, krvácanie do sklovca,
kortikálna katarakta,
lentikulárne opacity, porušenie epitelu rohovky,
erózia rohovky, podráždenie v mieste
podania, abnormálne pocity v oku,
podráždenie očného viečka,
vitritída, uveitída,
irititída, iridocyklitída,
zápal prednej očnej
komory
| Hypopyon
|
*) zahŕňa reakcie precitlivenosti
**) pozitívna a negatívna kultúra endoftalmitídy
Popis vybraných nežiaducich reakciíV klinických skúšaniach fázy III pri vlhkej AMD sa u pacientov užívajúcich antitrombotické látky vyskytla zvýšená incidencia spojovkového krvácania. Tento zvýšený výskyt sa porovnal u pacientov liečených ranibizumabom a Eyleou.
Arteriálne trombembolické príhody (ATEs) sú nežiaduce príhody potenciálne súvisiace so systémovou inhibíciou VEGF. Po intravitreálnom použití inhibítorov VEGF existuje teoretické riziko arteriálnych trombembolických príhod.
Arteriálne trombembolické príhody, podľa definície kritérií „Antiplatelet Trialists’ Collaboration“ (APTC) zahŕňajú nefatálny infarkt myokardu, nefatálnu cievnu mozgovú príhodu alebo vaskulárnu smrť (vrátane úmrtí z neznámej príčiny). Incidencia v klinických skúšaniach (VIEW1 a VIEW2) vo fáze 3 s vlhkou vekom podmienenou degeneráciou makuly počas 96 týždňov trvania klinického skúšania bola 3,3 % (60 z 1 824) v kombinovanej skupine pacientov liečených Eyleou v porovnaní s
3,2 % (19 z 595) pacientov liečených ranibizumabom (pozri časť 5.1).
Tak ako pri všetkých terapeutických proteínoch, aj pri Eylei existuje možnosť imunogenity.
4.9 Predávkovanie
V klinických skúšaniach sa použili dávky až do 4 mg v mesačných intervaloch a vyskytli sa ojedinelé prípady predávkovaní s 8 mg.
Predávkovanie zvýšeným objemom injekcie môže zvýšiť vnútroočný tlak. Preto sa má v prípade predávkovania monitorovať vnútroočný tlak a ak to považuje ošetrujúci lekár za nevyhnutné, má sa začať primeraná liečba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Oftalmologiká / antineovaskularizačné liečivá
ATC kód: S01LA05
Aflibercept je rekombinantný fúzny proteín, ktorý obsahuje fragmenty extracelulárnych domén ľudských VEGF receptorov 1 a 2 naviazaných na Fc fragment ľudského IgG1.
Aflibercept je vytvorený rekombinantnou DNA technológiou v bunkách ovárií čínskeho škrečka
(CHO) K1.
Aflibercept pôsobí ako solubilný náhradný receptor, ktorý viaže VEGF-A a PlGF s vyššou afinitou'
ako ich prirodzené receptory, a tým môže inhibovať väzbovosť a aktiváciu týchto analogických VEGF
receptorov.
Spôsobúčinku
Vaskulárny endoteliálny rastový faktor-A (VEGF-A) a placentárny rastový faktor (PlGF) sú členmi rodiny angiogénnych faktorov VEGF, ktoré môžu pôsobiť ako silné mitogénne, chemotaktické faktory
a faktory vaskulárnej permeability endotelových buniek. VEGF pôsobí prostredníctvom dvoch
receptorov tyrozínkináz; VEGFR-1 a VEGFR-2 prítomných na povrchu endotelových buniek. PlGF sa viaže iba na VEGFR-1, ktorý je prítomný aj na povrchu leukocytov. Nadmerná aktivácia týchto receptorov sprostredkovaná VEGF-A môže mať za následok patologickú neovaskularizáciu
a nadmernú vaskulárnu permeabilitu. PlGF môže spolupôsobiť s VEGF-A v týchto procesoch, a tiež je známe, že podporuje infiltráciu leukocytmi a vaskulárny zápal..
Farmakodynamickéúčinky
Vlhká vekom podmienená degenerácia makuly sa prejavuje patologickou choroidálnou neovaskularizáciou (CNV). Presakovanie krvi a tekutiny z CNV môže spôsobiť zhrubnutie sietnice
alebo opuch a/alebo krvácanie pod sietnicu/do sietnice, čo vedie k strate zrakovej ostrosti.
U pacientov liečených Eyleou (jednou injekciou mesačne tri po sebe nasledujúce mesiace, po ktorých sa pokračuje jednou injekciou každé 2 mesiace) sa krátko po začatí liečby znížilo zhrubnutie sietnice
a znížila sa priemerná veľkosť ložiska CNV, čo je v súlade s výsledkami pozorovanými po liečbe
0,5 mg ranibizumabom mesačne.
V klinickom skúšaní VIEW1 bolo priemerné zníženie zhrubnutia sietnice pri optickej koherenčnej tomografii (OCT) (-130 respektíve -129 mikrónov v 52. týždni v skupinách klinického skúšania s 2
mg Eylei každé dva mesiace a ranibizumabom 0,5 mg mesačne (v uvedenom poradí)). V klinickom
skúšaní VIEW2 tiež v 52. týždni bolo priemerné zníženie zhrubnutia sietnice pri OCT (-149 respektíve -139 mikrónov v skupinách klinického skúšania s 2 mg Eylei každé dva mesiace a ranibizumabom 0,5 mg mesačne (v uvedenom poradí)).
Redukcia veľkosti CNV a zníženie zhrubnutia sietnice sa vo všeobecnosti zachovali v druhom roku klinických skúšaní.
Klinickáúčinnosťabezpečnosť
Bezpečnosť a účinnosť Eylei sa hodnotili v dvoch randomizovaných, multicentrických, dvojito zaslepených, aktívne kontrolovaných klinických skúšaniach u pacientov s vlhkou vekom podmienenou degeneráciou makuly. Celkovo sa liečilo 2 412 pacientov a účinnosť sa hodnotila (1 817 s Eyleou)
v dvoch klinických skúšaniach (VIEW1 a VIEW2). V každom klinickom skúšaní boli pacienti náhodne pridelení v pomere 1:1:1:1 do 1 zo 4 dávkovacích schém:
1) 2 mg Eylei sa podávali každých 8 týždňov po 3 začiatočných mesačných dávkach (Eylea 2Q8);
2) 2 mg Eylei sa podávali každé 4 týždne (Eylea 2Q4);
3) 0,5 mg Eylei sa podávalo každé 4 týždne (Eylea 0.5Q4); a
4) 0,5 mg ranibizumabu sa podávalo každé 4 týždne (ranibizumab 0.5Q4).
Vekové rozpätie pacientov od 49 do 99 rokov s mediánom 76 rokov.
V druhom roku klinických skúšaní pokračovali pacienti v používaní dávky v sile, na ktorú boli v úvode randomizovaní, no modifikovaná dávkovacia schéma sa nastavila podľa vyhodnotenia zrakových a anatomických výsledkov protokolom definovaným maximálnym dávkovacím intervalom
12 týždňov.
V obidvoch klinických skúšaniach bol primárnym koncovým ukazovateľom účinnosti podiel pacientov liečených podľa protokolu, u ktorých zostal zachovaný vízus, definovaný stratou menej ako
15 písmen zrakovej ostrosti v 52. týždni v porovnaní s východiskovým stavom.
V klinickom skúšaní VIEW1 v 52. týždni malo 95,1 % pacientov v liečebnej skupine Eylea
2Q8 zachovaný vízus , v porovnaní s 94,4 % pacientmi v skupine ranibizumab 0.5Q4. Liečba Eyleou ukázala, že je non- inferiórna a klinicky ekvivalentná so skupinou ranibizumab 0.5Q4.
V klinickom skúšaní VIEW2 v 52. týždni malo 95,6 % pacientov v liečebnej skupine Eylea
2Q8 zachovaný vízus ,v porovnaní s 94,4 % pacientmi v skupine ranibizumab 0.5Q4. Liečba Eyleou ukázala, že je non- inferiórna a klinicky ekvivalentná so skupinou ranibizumab 0.5Q4.
Detailné výsledky zo súhrnnej analýzy oboch klinických skúšaní sú uvedené nižšie v Tabuľke a na
Obrázku.
T
abuľka:
V
ý
sledky
účinnosti
v
52.
týždni
(
primárna
analýza)
a
96.
týždni;
súhrnné údaje
z klinických
skúšaníVIEW1aVIEW2B)
Výsledok účinnosti
| Eylea 2Q8 E)
(2 mg Eylea každých 8 týždňov po 3 začiatočných mesačných dávkach)
(n = 607)
| Ranibizumab 0.5Q4
(ranibizumab 0,5 mg každé
4 týždne)
(n = 595)
|
| 52. týždeň
| 96. týždeň G)
| 52. týždeň
| 96. týždeň G)
|
Priemerný počet injekcií
| 7,6
| 11,2
| 12,3
| 16,5
|
Priemerný počet injekcií počas 2.roka (52. až 96. týždeň)
|
|
4,1
|
|
4,6
|
Podiel pacientov so zachovanou zrakovou ostrosťou
(strata < 15 písmen BCVAA)) (v skupine podľa protokolu)
|
95,33%B)
|
92,42 %
|
94,42 % B)
|
91,60 %
|
RozdielC)
(95 % CI)D)
|
0,9 %
(-1,7, 3,5)F)
|
0,8 %
(- 2,3, 3,8)F)
|
|
|
Medián zmeny v BCVA podľa meraní ETDRSA) skóre písmen oproti východiskovému stavu
|
8,40
|
7,62
|
8,74
|
7,89
|
Rozdiel v LS A) medián
(ETDRS písmená)C)
(95 % CI)D)
|
-0,32
(-1,87, 1,23)
|
-0,25
(-1,98, 1,49)
|
|
|
Podiel pacientov, ktorí získali minimálne 15 písmen zrakovej ostrosti oproti východiskovému stavu
|
30,97 %
|
33,44 %
|
32,44 %
|
31,60 %
|
RozdielC)
(95 % CI)D)
|
-1,5 %
(-6,8, 3,8)
|
1,8 %
(-3,5, 7,1)
|
|
|
A) BCVA: Best Corrected Visual Acuity (Najlepšie korigovaná zraková ostrosť)
ETDRS: Early Treatment Diabetic Retinopaty Study (Klinické skúšanie včasnej liečby diabetickej retinopatie)
LS: Least square means derived from ANCOVA (Priemery najmenších štvorcov vypočítané na základe modelu ANCOVA)
B) Celkový analyzovaný súbor (Full Analysis Set, FAS), Výsledky z posledného dokumentovaného vyšetrenia
(Last Observation Carried Forward, LOCF) pre všetky analýzy okrem podielu pacientov so zachovanou zrakovou ostrosťou v 52. týždni, čo je skupina podľa protokolu (Per Protocol Set, PPS)
C) Rozdiel je hodnota skupiny s Eyleou mínus hodnota skupiny s ranibizumabom. Pozitívna hodnota zvýhodňuje
Eyleu.
D) Interval spoľahlivosti (Confidence interval, CI) vypočítaný pomocou normálneho rozdelenia
E) Po začatí liečby tromi mesačnými dávkami
F) Interval spoľahlivosti ležiaci úplne nad -10 %,ktorý poukazuje na non-inferioritu Eylei voči ranibizumabu
G) Na začiatku 52. týždňa, u všetkých skupín pacientov liečených pomocou modifikovanej formy štvrťročnej liečby sa aplikovala dávka každé 4 týždne, ale nie menej ako jedenkrát počas každých 12 týždňov na základe
vopred špecifikovaných kritérií opakovanej liečby
Obrázok 1. Priemerná zmena zrakovej ostrosti oproti východiskovému stavu do 96. týždňa pre súhrnné údaje z klinickýchskúšaní View1 a View2
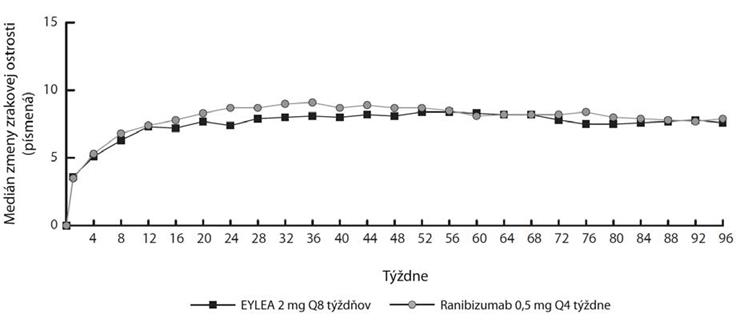
*) Od východiskového stavu do 52.týždňa sa Eylea podávala každých 8 týždňov po 3 začiatočných mesiačných dávkach. Od východiskového stavu do 52. týždňa sa ranibizumab 0,5 mg podával každé 4 týždne.Na začiatku
52. týždňa, u všetkých skupín pacientov liečených pomocou modifikovanej formy štvrťročnej liečby sa aplikovala dávka každé 4 týždne, ale nie menej ako jedenkrát počas každých 12 týždňov na základe vopred špecifikovaných kritérií opakovanej liečby
Podiel pacientov, ktorí v 96. týždni získali minimálne 15 písmen zrakovej ostrosti v porovnaní s východiskovým stavom bol 33,44 % v skupine Eylea 2Q8 a 31,60 % v skupine ranibizumab 0.5Q4.
Súhrnné údaje z klinických skúšaní VIEW1 a VIEW2 s Eyleou preukázali klinicky významné zmeny voči východiskovému stavu pri vopred špecifikovanom sekundárnom ukazovateli účinnosti stanoveným National Eye Institute Visual Function Questionnaire (NEI VFQ-25). Intenzita týchto zmien bola podobná tým, ktoré boli publikované v klinických skúšaniach a zodpovedali získaniu 15 písmen s najlepšie korigovanou zrakovou ostrosťou (Best Corrected Visual Acuity, BCVA).
Žiadne klinicky významné rozdiely sa nezistili medzi Eyleou a referenčným liekom ranibizumabom v zmenách celkového skóre NEI VFQ-25 a podškálach (aktivity vyžadujúce si videnie do blízka, aktivity vyžadujúce videnie do diaľky a aktivity závislé špecificky na víze ) v 52. týždni od východiskového stavu.
Zníženia priemernej plochy CNV boli preukázané vo všetkých dávkovacích skupinách v obidvoch klinických skúšaniach.
Výsledky účinnosti vo všetkých hodnotených podskupinách (napr. vek, pohlavie, rasa, východisková zraková ostrosť, typ lézie, veľkosť lézie) v každom klinickom skúšaní a v súhrnnej analýze a boli zhodné s výsledkami v celkovej populácii.
V druhom roku klinických skúšaní bola vo všeobecnosti účinnosť zachovaná počas posledného hodnotenia v 96. týždni. V druhom roku klinických skúšaní sa u 2-4% pacientov sa požadovala aplikácia všetkých dávok na mesačnej báze a u tretiny pacientov sa požadovala aspoň jedna dávka v intervale jedného mesiaca počas liečby.
StaršípacientiV klinických skúšaniach bolo približne 89 % (1 616/1 817) pacientov randomizovaných na liečbu Eyleou vo veku 65 rokov alebo starších a približne 63 % (1 139/1 817) bolo vo veku 75 rokov alebo starších.
D
eti
a
dospievajúci
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky klinických skúšaní pre Eyleu vo všetkých vekových podskupinách detí a dospievajúcich s vlhkou vekom podmienenou degeneráciou makuly (pre informácie o použití u detí a dospievajúcich, pozri časť 4.2 ).
5.2 Farmakokinetické vlastnosti
Na dosiahnutie lokálnych účinkov v oku sa Eylea podáva priamo do sklovca. Absorpcia/Distribúcia
Aflibercept je po intravitreálnom podaní pomaly absorbovaný z oka do systémového obehu
a v systémovom obehu je prevažne pozorovaný ako inaktívny, stabilný komplex s VEGF. Iba „voľný aflibercept“ je však schopný viazať endogénny VEGF.
Vo farmakokinetickej pod-štúdii so 6 pacientmi s častým odoberom vzoriek boli v priebehu 1 až 3 dní po intravitreálnom podaní injekcie 2 mg maximálne plazmatické koncentrácie voľného afliberceptu (systémová Cmax ) nízke, so strednou hodnotou približne 0,02 mikrogramu/ml (rozsah od 0 do 0,054)
a takmer u všetkých pacientov boli nedetekovateľné dva týždne po podaní. Aflibercept sa nekumuluje
v plazme pri podávaní intravitreálne každé 4 týždne.
Stredná maximálna plazmatická koncentrácia voľného afliberceptu je približne 50 až 500-násobne nižšia ako koncentrácia afliberceptu potrebná na inhibíciu biologickej aktivity systémového VEGF
o 50 % na modeloch zvierat, u ktorých sa pozorovali zmeny krvného tlaku po cirkulujúcich hladinách voľného afliberceptu dosahujúceho približne 10 mikrogramov/ml a vrátili sa na východiskovú hodnotu po poklese hladín pod približne 1 mikrogram/ml. Odhaduje sa, že po intravitreálnom podaní 2 mg
pacientom je stredná maximálna plazmatická koncentrácia voľného afliberceptu viac ako 100- násobne nižšia ako koncentrácia afliberceptu potrebná na naviazanie maximálne polovice
systémového VEGF (2,91 mikrogramov/ml) v klinickom skúšaní so zdravými dobrovoľníkmi. Preto nie sú pravdepodobné systémové farmakodynamické účinky, ako sú zmeny tlaku krvi.
Eliminácia
Keďže Eylea je liek proteínového charakteru, nevykonali sa žiadne metabolické klinické skúšania.
Voľný aflibercept viaže VEGF za vzniku stabilného, inertného komplexu. Predpokladá sa, že tak ako iné veľké proteíny aj voľný i viazaný aflibercept sa budú rozkladať proteolytickým katabolizmom.
Poškodeniefunkcieobličiek
U pacientov s poškodením funkcie obličiek sa s Eyleou nevykonali žiadne špeciálne klinické skúšania.
Farmakokinetická analýza pacientov v klinickom skúšaní VIEW2, v ktorej malo 40 % poškodenú funkciu obličiek (24 % mierne, 15 % stredne ťažko a 1 % ťažko) neodhalila žiadne rozdiely
v súvislosti s plazmatickými koncentráciami liečiva po intravitreálnom podávaní každé 4 alebo
8 týždňov.
5.3 Predklinické údaje o bezpečnosti
V predklinických štúdiách sa toxické účinky pri opakovanom podávaní pozorovali iba pri podstatne vyšších systémových expozíciách, ako je maximálna expozícia u ľudí po intravitreálnom podaní stanovenej klinickej dávky, čo poukazuje na malý význam pre klinické použitie.
Erózie a ulcerácie dýchacieho epitelu v nosových mušliach opíc liečených afliberceptom intravitreálne sa pozorovali pri vyšších systémových expozíciách, ako je maximálna expozícia u ľudí. Systémová expozícia na základe Cmax a AUC voľného afliberceptu bola približne 200- respektíve 700-násobne vyššia (v uvedenom poradí) v porovnaní so zodpovedajúcimi hodnotami pozorovanými u ľudí po intravitreálnej dávke 2 mg. Pri hladine bez pozorovaného nepriaznivého účinku (No Observed
Adverse Effect Level, NOAEL) 0,5 mg/oko u opíc bola systémová expozícia 42- respektíve 56- násobne vyššia (v uvedenom poradí) na základe Cmax a AUC.
Na mutagénny alebo karcinogénny potenciál afliberceptu sa nerobili žiadne štúdie.
V štúdii embryofetálneho vývinu spôsobil aflibercept na gravidných králikoch po intravenóznom podávaní (3 až 60 mg/kg) embryofetálnu toxicitu (teratogenita pri všetkých testovaných dávkach). Pri najnižšej testovanej dávke v tejto štúdii (3 mg/kg) boli systémové expozície na základe Cmax a AUC voľného afliberceptu približne 2 900- respektíve 600-násobne vyššie (v uvedenom poradí) v porovnaní so zodpovedajúcimi hodnotami pozorovanými u ľudí po intravitreálnej dávke 2 mg.
Účinky na fertilitu samcov a samíc sa hodnotili ako časť 6-mesačnej štúdie na opiciach
s intravenóznym podávaním afliberceptu v rozsahu dávok od 3 do 30 mg/kg. Pri všetkých hladinách dávok sa pozorovali chýbajúce alebo nepravidelné menštruácie, ktoré súviseli so zmenami hladín
samičích pohlavných hormónov a zmeny morfológie a pohyblivosti spermií. Na základe Cmax a AUC
voľného afliberceptu pozorovaných pri intravenóznej dávke 3 mg/kg boli systémové expozície približne 4 900-násobne respektíve 1 500-násobne vyššie (v uvedenom poradí), než expozícia pozorovaná u ľudí po intravitreálnej dávke 2 mg. Všetky zmeny boli reverzibilné.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Polysorbát 20
Monohydrát dihydrogénfosforečnanu sodného (na úpravu pH) Heptahydrát monohydrogénfosforečnanu sodného (na úpravu pH)
Chlorid sodný
Sacharóza
Voda na injekciu
6.2 Inkompatibility
Nerobili sa štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (pri teplote 2°C až 8°C). Neuchovávajte v mrazničke.
Injekčnú liekovku uchovávajte vo vonkajšom obale na ochranu pred svetlom.
Pred použitím sa môže neotvorená liekovka Eylei uchovávať pri izbovej teplote (25 °C) počas
24 hodín. Po otvorení dodržiavajte aseptické podmienky.
6.5 Druh obalu a obsah balenia
100 mikrolitrov roztoku v injekčnej liekovke (sklo typu I) so zátkou (z elastomérovej gumy)
a injekčná ihla s filtrom 18 G. Veľkosť balenia 1.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Injekčná liekovka je len na jednorazové použitie.
Vzhľadom k tomu, že injekčná liekovka obsahuje väčší objem (100 mikrolitrov) ako odporúčaná dávka (50 mikrolitrov), časť objemu v injekčnej liekovke, musí byť odstránený pred podaním.
Injekčný roztok pred podaním vizuálne skontrolujte na prítomnosť cudzorodých častíc a/alebo zemnu zafarbenia alebo fyzikálneho vzhľadu. Injekčnú liekovku nepoužívajte ak sú viditeľné častice, zakalenie alebo zmena farby. V prípade, že niečo také zistíte, liek zlikvidujte.
Na intravitreálne podanie injekcie sa má použiť injekčná ihla 30 G x ½ palca.
Pokyny na používanie injekčnej liekovky:
1. Odstráňte plastové viečko a vydezinfikujte vonkajšiu časť gumenej zátky injekčnej liekovky.
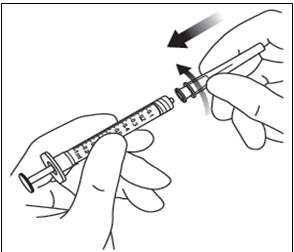
2. Nasaďte 18 G injekčnú ihlu s 5-mikrónovým filtrom
dodanú v škatuľke k 1-ml sterilnej injekčnej striekačke s nadstavcom so závitom („Luer lock“).
3. Vtlačte injekčnú ihlu s filtrom do stredu zátky injekčnej liekovky až kým sa injekčná ihla nedotkne
dna injekčnej liekovky.
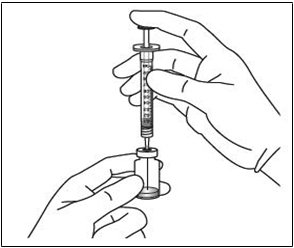
4. Použitím aseptického postupu odoberte celý obsah injekčnej liekovky s Eyleou do injekčnej striekačky
držaním injekčnej liekovky vo zvislej polohe, mierne naklonenej, tak aby sa uľahčilo úplné odobratie
obsahu. Na ochranu proti vzduchu, sa uistite, aby sa ihla úplne ponorila do roztoku. Pokračujte s naklonením liekovky .
5. Dbajte na to, aby ste pri vyprázdnení injekčnej liekovky dostatočne potiahli piest, aby sa injekčná ihla
s filtrom úplne vyprázdnila.
6. Odstráňte injekčnú ihlu s filtrom a náležite ju znehodnoťte.
Poznámka: Injekčná ihla s filtrom sa nesmie použiť na intravitreálne podanie injekcie.
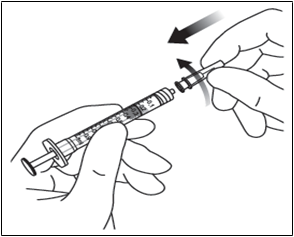
7. Použitím aseptického postupu pootočením pevne nasaďte injekčnú ihlu 30 G x ½ palca na nadstavec so závitom („Luer lock“) na injekčnú striekačku.
8. Keď ste pripravený na podanie Eylei, z injekčnej ihly odstráňte plastový kryt.

9. Injekčnú striekačku s ihlou držte smerom nahor, skontrolujte v nej prítomnosť bubliniek. V prípade prítomnosti bubliniek injekčnú striekačku jemne poklepte prstom, kým bublinky nevystúpia na povrch..
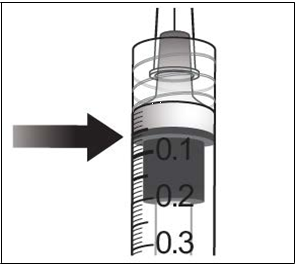

10. Na odstránenie všetkých bubliniek a nadbytočného lieku pomaly zatlačte na piest, aby ste zarovnali
hrot piestu s ryskou, ktorá označuje na injekčnej striekačke 0, 05 ml.
11. Injekčné liekovky sú len na jednorazové použitie.
Nepoužitý liek alebo odpad vzniknutý z lieku má byť zlikvidovaný v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIBayer Pharma AG
13342 Berlin
Nemecko
8. REGISTRAČNÉ ČÍSLO
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.