
cie alebo vyrážky môže byť nevyhnutné upraviť dávku alebo dočasne prerušiť liečbu (pozri časť 4.2).
A
ngioedém
V súvislosti s použitím Esbrietu po uvedení na trh boli hlásené prípady angioedému (v niektorých
prípadoch závažného) ako je opuch tváre, perí a/alebo jazyka, ktoré môžu súvisieť s ťažkosťami
s dýchaním alebo s pískavým dýchaním. Preto pacienti, u ktorých sa objavia prejavy alebo príznaky angioedému po podaní Esbrietu, musia okamžite prerušiť liečbu. Pacienti s angioedémom sa musia liečiť podľa zásad štandardnej zdravotnej starostlivosti. Esbriet sa nesmie používať u pacientov, ktorí majú v anamnéze angioedém spôsobený Esbrietom (pozri časť 4.3).
Závraty
U pacientov užívajúcich Esbriet boli hlásené závraty. Pacienti majú preto vedieť, ako reagujú na tento
liek, skôr než začnú vykonávať činnosti vyžadujúce duševnú bdelosť alebo koordináciu (pozri časť
4.7). V klinických štúdiách prebehla u väčšiny pacientov, u ktorých sa prejavili závraty, jedna udalosť
a väčšina udalostí ustúpila v priemere počas 22 dní. Ak závraty neustúpia alebo ak sa zhorší ich závažnosť, môže byť potrebné prispôsobiť dávku alebo dokonca prerušiť liečbu Esbrietom.
Únava
U pacientov užívajúcich Esbriet bola hlásená únava. Pacienti preto majú vedieť, ako reagujú na tento
liek, skôr než začnú vykonávať činnosti vyžadujúce duševnú bdelosť alebo koordináciu (pozri
časť 4.7).
Úbytok hmotnosti
U pacientov liečených Esbrietom bol hlásený úbytok hmotnosti (pozri časť 4.8). Lekári majú sledovať
hmotnosť pacienta a v prípade potreby podporiť zvýšenie príjmu kalórií, ak sa úbytok hmotnosti považuje za klinicky významný.
4.5 Liek ové a iné interakcie
Približne 70–80 % pirfenidónu sa metabolizuje prostredníctvom CYP1A2 s menším prispením iných
izoenzýmov CYP vrátane CYP2C9, 2C19, 2D6 a 2E1.
Konzumácia grapefruitového džúsu je spojená s inhibíciou CYP1A2 a počas liečby pirfenidónom sa
jej pacienti majú vyhýbať.
Fluvoxamín a inhibítory CYP1A2
V štúdii fázy 1 viedlo súbežné podávanie Esbrietu a fluvoxamínu (silný inhibítor CYP1A2
s inhibičnými účinkami na iné izoenzýmy CYP [CYP2C9, 2C19 a 2D6]) u nefajčiarov k 4-násobnému
zvýšeniu expozície pirfenidónu.
Esbriet je kontraindikovaný u pacientov, ktorí súbežne užívajú fluvoxamín (pozri časť 4.3). Fluvoxamín sa má pred začatím liečby Esbrietom vysadiť a počas liečby Esbrietom sa nemá užívať vzhľadom na znížený klírens pirfenidónu. Počas liečby pirfenidónom je potrebné vyhnúť sa ďalšej liečbe, ktorá inhibuje CYP1A2 a jeden alebo viac iných izoenzýmov CYP, ktoré sa podieľajú na metabolizme pirfenidónu (napr. CYP2C9, 2C19 a 2D6).
Extrapolácie podmienok in vitro a in vivo naznačujú, že silné a selektívne inhibítory CYP1A2 (napr. enoxacín) môžu zvýšiť expozíciu pirfenidónu približne 2 až 4-násobne. Ak sa nedá vyhnúť súbežnému použitiu Esbrietu so silným a selektívnym inhibítorom CYP1A2, dávka pirfenidónu sa má znížiť na
801 mg denne (jedna kapsula trikrát denne). Pacientov treba pozorne sledovať z hľadiska výskytu nežiaducich reakcií spojených s liečbou Esbrietu. Ak je to potrebné, vysaďte Esbriet (pozri časti 4.2 a
4.4).
Súčasné podávanie Esbrietu a 750 mg ciprofloxacínu (stredne silný inhibítor CYP1A2) zvyšovalo expozíciu pirfenidónu o 81 %. Ak je podávanie ciprofloxacínu v dávkach 750 mg dvakrát denne potrebné, dávku pirfenidónu je treba znížiť na 1602 mg denne (dve kapsule, trikrát denne). Esbriet je treba podávať s opatrnosťou, ak je ciprofloxacín podávaný v dávkach 250 mg alebo 500 mg raz alebo dvakrát denne.
Esbriet sa má užívať obozretne u pacientov liečených inými stredne silnými inhibítormi CYP1A2
(napr. amiodarón, propafenón).
Mimoriadna obozretnosť je tiež potrebná, keď sa inhibítory CYP1A2 používajú súbežne so silnými inhibítormi jedného alebo viacerých ďalších izoenzýmov CYP, ktoré sa podieľajú na metabolizme pirfenidónu, ako je CYP2C9 (napr. amiodarón, flukonazol), 2C19 (napr. chloramfenikol) a 2D6 (napr. fluoxetín, paroxetín).
Fajčenie cigariet a induktory CYP1A2
Interakčná štúdia fázy 1 hodnotila vplyv fajčenia cigariet (induktor CYP1A2) na farmakokinetiku
pirfenidónu. Expozícia pirfenidónu u fajčiarov bola 50 % v porovnaní s expozíciou u nefajčiarov. Fajčenie môže indukovať tvorbu pečeňových enzýmov, a teda zvyšovať klírens lieku a znižovať expozíciu. Počas liečby Esbrietom je potrebné vyhýbať sa súbežnému užívaniu silných induktorov CYP1A2 vrátane fajčenia na základe pozorovaného vzťahu medzi fajčením cigariet a potenciálom indukovať CYP1A2. Pacientom sa má odporučiť, aby prestali užívať silné induktory CYP1A2 a aby pred liečbou a počas liečby pirfenidónom nefajčili.
V prípade stredne silných induktorov CYP1A2 (napr. omeprazol) môže súbežné užitie teoreticky viesť
k zníženiu plazmatickej hladiny pirfenidónu.
Súbežné podávanie liekov, ktoré účinkujú ako silné induktory CYP1A2 a iných izoenzýmov CYP podieľajúcich sa na metabolizme pirfenidónu (napr. rifampicín), môže viesť k výraznému zníženiu plazmatickej hladiny pirfenidónu. Týmto liekom je potrebné vyhnúť sa vždy, keď je to možné.
4.6 Fertilita, gravidita a lak tácia
Gravidita
Nie sú k dispozícii údaje o použití Esbrietu u gravidných žien.
U zvierat dochádza k prieniku pirfenidónu a/alebo jeho metabolitov cez placentu s možnosťou
hromadenia pirfenidónu a/alebo jeho metabolitov v amniotickej tekutine.
Pri vysokých dávkach (≥1.000 mg/kg/deň) sa u potkanov pozorovalo predĺženie gestácie a zníženie životaschopnosti plodov.
Z dôvodu bezpečnosti sa neodporúča užívať Esbriet počas gravidity.
Dojčenie
Nie je známe, či sa pirfenidón alebo jeho metabolity vylučujú do ľudského mlieka. Dostupné
farmakokinetické údaje u zvierat preukázali vylučovanie pirfenidónu a/alebo jeho metabolitov do mlieka s možnosťou hromadenia pirfenidónu a/alebo jeho metabolitov v mlieku (pozri časť 5.3). Riziko pre dojčené dieťa sa nedá vylúčiť.
Po zvážení prínosu dojčenia pre dieťa a prínosu liečby Esbrietom pre matku sa musí rozhodnúť, či sa preruší dojčenie, alebo liečba Esbrietom.
Fertilita
V predklinických štúdiách sa nepozoroval nežiaduci vplyv na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Esbriet môže spôsobiť závraty a únavu, čo môže mať mierny vplyv na schopnosť viesť vozidlá alebo obsluhovať stroje. Pri výskyte týchto príznakov majú byť preto pacienti opatrní pri vedení vozidiel
a obsluhe strojov.
4.8 Nežiaduce účink y
Súhrnbezpečnostnéhoprofilu
K najčastejšie hláseným nežiaducim reakciám počas klinických štúdií skúmajúcich Esbriet v dávke
2.403 mg/deň v porovnaní s placebom patrila nauzea (32,4 % v porovnaní s 12,2 %), vyrážka (26,2 % v porovnaní so 7,7 %), hnačka (18,8 % v porovnaní so 14,4 %), únava (18,5 % v porovnaní s 10,4 %), dyspepsia (16,1 % v porovnaní s 5,0 %), anorexia (11,4 % v porovnaní s 3,5 %), bolesť hlavy (10,1 % v porovnaní so 7,7 %) a fotosenzitívna reakcia (9,3 % v porovnaní s 1,1 %).
Súhrnnežiaducichreakciíuvedenýchvtabuľke
Bezpečnosť Esbrietu sa hodnotila v klinických štúdiách zahŕňajúcich 1 650 dobrovoľníkov
a pacientov. Viac ako 170 pacientov bolo testovaných v nezaslepených štúdiách dlhšie ako päť rokov
a niektorí až do 10 rokov.
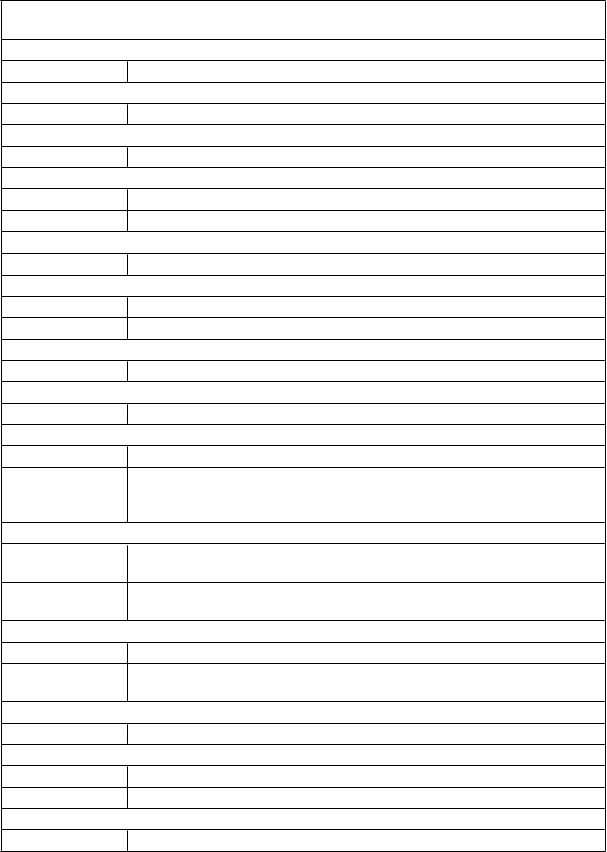
Tabuľka 1 uvádza nežiaduce reakcie hlásené s frekvenciou ≥2 % u 623 pacientov užívajúcich Esbriet v odporúčanej dávke 2.403 mg/deň v troch súhrnných pivotných štúdiách fázy 3. Nežiaduce reakcie hlásené v období po uvedení lieku na trh sú tiež uvedené v tabuľke 1. Nežiaduce reakcie sú uvedené podľa triedy orgánových systémov a v každej skupine frekvencií [veľmi časté (≥1/10), časté (≥1/100 až <1/10), menej časté (≥1/1 000 až <1/100), zriedkavé (≥1/10 000 až <1/1 000)] sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
 T
abuľk a 1 Nežiaduce reak cie podľa triedy orgánových systémov a frekvencie podľa
M
e
d
DRA
I
n
f
e
k
c
i
e a nákazy
T
abuľk a 1 Nežiaduce reak cie podľa triedy orgánových systémov a frekvencie podľa
M
e
d
DRA
I
n
f
e
k
c
i
e a nákazy
Časté infekcia horných dýchacích ciest; infekcia močových ciest
Poruchy k rvi a lymfatického systémuZriedkavé agranulocytóza1
Poruchy imunitného systémuMenej časté angioedém1
Poruchy metabolizmu a výživyVeľmi časté anorexia
Časté úbytok hmotnosti; znížená chuť do jedla
Psychick é poruchyČasté nespavosť
Poruchy nervového systému Veľmi časté bolesť hlavy
Časté závraty; somnolencia; dysgeúzia; letargia
Poruchy cievČasté návaly tepla
Poruchy dýchacej sústavy, hrudníka a mediastína Časté dyspnoe; kašeľ; produktívny kašeľ
Poruchy gastrointestinálneho traktuVeľmi časté dyspepsia; nauzea; hnačka
Časté gastroezofágová refluxová choroba; vracanie; abdominálna distenzia; abdominálny dyskomfort; abdominálna bolesť; bolesť v hornej časti brucha; žalúdočný dyskomfort; gastritída; zápcha; flatulencia
Poruchy pečene a žlčových ciestČasté zvýšená hladina ALT; zvýšená hladina AST; zvýšená hladina gama- glutamyltransferázy
Zriedkavé zvýšená hladina celkového bilirubínu v sére v kombinácii so zvýšenou
hladinou ALT a AST1
Poruchy k ože a podk ožného tkanivaVeľmi časté fotosenzitívna reakcia; vyrážka
Časté pruritus; erytém; suchá koža; erytematózna vyrážka; makulárna vyrážka;
svrbiaca vyrážka
Poruchy k ostrovej a svalovej sústavy a spojivového tkanivaČasté myalgia; artralgia
Celk ové poruchy a reakcie v mieste podaniaVeľmi časté únava
Časté asténia; bolesť na hrudníku iná ako srdcová
Úrazy, otravy a k omplik ácie liečebného postupuČasté Popálenina od slnka
1. Identifikované počas pozorovania po uvedení lieku na trh
Hlásenie podozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávk ovanie
Existuje málo klinických skúseností s predávkovaním. Zdravým dospelým dobrovoľníkom sa počas
12 dní opakovane podávali postupne zvyšované dávky pirfenidónu až do celkovej dávky 4 806 mg/deň vo forme šiestich 267 mg kapsúl trikrát denne. Nežiaduce reakcie boli mierne, prechodné a zhodovali sa s najčastejšie hlásenými nežiaducimi reakciami na pirfenidón.
V prípade podozrenia na predávkovanie sa má poskytnúť podporná lekárska starostlivosť vrátane
sledovania vitálnych známok a pozorného sledovania klinického stavu pacienta.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmak odynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresíva, iné imunosupresíva, kód ATC: L04AX05
Mechanizmus účinku pirfenidónu sa ešte celkom nestanovil. Z existujúcich údajov však vyplýva, že pirfenidón v rôznych systémoch in vitro a v živočíšnych modeloch pľúcnej fibrózy (fibróza vyvolaná bleomycínom a transplantáciou) prejavuje antifibrotické a protizápalové vlastnosti.
IPF je chronické fibrotické a zápalové ochorenie pľúc ovplyvnené syntézou a uvoľňovaním prozápalových cytokínov vrátane tumor nektrotizujúceho faktoru alfa (TNF-α) a interleukínu-1–beta (IL-1β) a zistilo sa, že pirfenidón znižuje hromadenie zápalových buniek ako reakciu na rôzne podnety.
Pirfenidón tlmí proliferáciu fibroblastov, tvorbu bielkovín a cytokínov spojených s fibrózou a zvýšenú biosyntézu a hromadenie mimobunkového matrix ako reakciu na cytokínové rastové faktory, ako je napríklad transformujúci rastový faktor beta (TGF-β) a rastový faktor odvodený z krvných doštičiek (PDGF).
Klinická účinnosť
Klinická účinnosť Esbrietu sa skúmala v štyroch multicentrických randomizovaných dvojito
zaslepených štúdiách fázy 3 kontrolovaných placebom u pacientov s IPF. Tri z týchto štúdií fázy 3
(PIPF-004, PIPF-006 a PIPF-016) boli mnohonárodné a jedna štúdia (SP3) sa uskutočnila v Japonsku.
Štúdie PIPF-004 a PIPF-006 porovnávali liečbu Esbrietom v dávke 2.403 mg/deň s placebom. Tieto štúdie boli takmer rovnaké, pokiaľ ide o dizajn, s niekoľkými výnimkami vrátane skupiny, v ktorej sa podávala stredne veľká dávka (1 197 mg/deň), v štúdii PIPF-004. V obidvoch štúdiách sa liečba podávala trikrát denne minimálne počas 72 týždňov. Primárnym cieľovým ukazovateľom v obidvoch štúdiách bola zmena hodnoty úsilnej vitálnej kapacity (Forced Vital Capacity, FVC), vyjadrenej
v percentách z referenčnej hodnoty, v 72. týždni v porovnaní s východiskovou hodnotou.
V štúdii PIPF-004 bol pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty,
v 72. týždni liečby v porovnaní s východiskovou hodnotou významne menší u pacientov užívajúcich
Esbriet (N = 174) v porovnaní s pacientmi užívajúcimi placebo (N = 174; p = 0,001, poradová (rank) analýza kovariancie - ANCOVA). Pri liečbe Esbrietom sa tiež dosiahol významne menší pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 24. týždni (p = 0,014), v 36. týždni
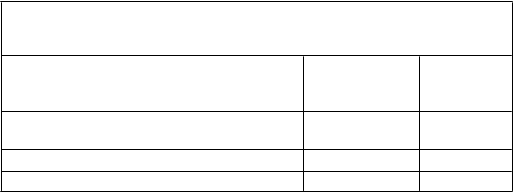
(p < 0,001), v 48. týždni (p < 0,001) a v 60. týždni (p < 0,001) v porovnaní s východiskovou hodnotou. V 72. týždni sa pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, o ≥ 10 % (čo je prahová hodnota poukazujúca na riziko úmrtia na IPF) v porovnaní s východiskovou hodnotou zaznamenal u 20 % pacientov užívajúcich Esbriet v porovnaní s 35 % pacientmi užívajúcimi placebo (tabuľka 2).
T
abuľk a 2 Vyhodnotenie zmeny hodnoty FVC, vyjadrenej v percentách
z referenčnej hodnoty, v 72. týždni v porovnaní s východiskovou hodnotou v štúdii PIPF-004, podľa k ategórií
P
i
r
f
e
n
i
d
ón
2.403 mg/deň
 (
N = 174)
P
l
acebo
(
N = 174)
(
N = 174)
P
l
acebo
(
N = 174)
Pokles o ≥10 % alebo úmrtie alebo
transplantácia pľúc
35 (20 %) 60 (34 %)
Pokles o menej ako 10 % 97 (56 %) 90 (52 %)
Žiadny pokles (zmena FVC >0 %) 42 (24 %) 24 (14 %)
Napriek tomu, že podľa vopred špecifikovanej poradovej (rank) ANCOVA nebol medzi pacientmi
užívajúcimi Esbriet a pacientmi užívajúcimi placebo žiadny rozdiel z hľadiska zmeny vzdialenosti prejdenej v šesťminútovom teste chôdzou (six minute w alk test, 6MWT) v 72. týždni v porovnaní s východiskovou hodnotou, v ad hoc analýze sa zistilo skrátenie vzdialenosti prejdenej v 6MWT
o ≥ 50 m u 37 % pacientov užívajúcich Esbriet v porovnaní so 47 % pacientmi užívajúcimi placebo
v štúdii PIPF-004.
V štúdii PIPF-006 sa pri liečbe Esbrietom (N = 171) v porovnaní s placebom (N = 173) nedosiahol
menší pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 72. týždni liečby
v porovnaní s východiskovou hodnotou (p = 0,501). Pri liečbe Esbrietom sa však dosiahol menší
pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 24. týždni (p < 0,001),
v 36. týždni (p = 0,011) a v 48. týždni (p = 0,005) v porovnaní s východiskovou hodnotou.
V 72. týždni sa pokles hodnoty FVC o ≥ 10 % zaznamenal u 23 % pacientov užívajúcich Esbriet
a u 27 % pacientov užívajúcich placebo (tabuľka 3).
Tabuľk a 3 Vyhodnotenie zmeny hodnoty FVC, vyjadrenej v percentách
z referenčnej hodnoty, v 72. týždni v porovnaní s východiskovou hodnotou v štúdii PIPF-006, podľa k ategórií
P
i
r
f
e
n
i
d
ón
2 403 mg/deň
 (
N = 171)
P
l
acebo
(
N = 173)
(
N = 171)
P
l
acebo
(
N = 173)
Pokles o ≥ 10 % alebo úmrtie alebo transplantácia pľúc
39 (23 %) 46 (27 %)
Pokles o menej ako 10 % 88 (52 %) 89 (51 %)
Žiadny pokles (zmena FVC > 0 %) 44 (26 %) 38 (22 %)
V štúdii PIPF-006 bolo skrátenie vzdialenosti prejdenej v 6MWT v 72. týždni v porovnaní
s východiskovou hodnotou významne menšie pri liečbe Esbrietom v porovnaní s placebom (p < 0,001,
poradová (rank) ANCOVA). V ad hoc analýze sa okrem toho zistilo skrátenie vzdialenosti prejdenej v 6MWT o ≥ 50 m u 33 % pacientov užívajúcich Esbriet v porovnaní so 47 % pacientov užívajúcimi placebo v štúdii PIPF-006.
V súhrnnej analýze prežívania v štúdiách PIPF-004 a PIPF-006 bola miera mortality v skupine
užívajúcej Esbriet v dávke 2 403 mg/deň 7,8 % v porovnaní s 9,8 % pri užívaní placeba (HR 0,77
[95 % IS, 0,47 - 1,28]).
Štúdia PIPF-016 porovnávala liečbu Esbrietom 2 403 mg/deň s placebom. Liek sa podával trikrát denne počas 52 týždňov. Primárnym cieľovým ukazovateľom bola zmena hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 52. týždni v porovnaní s východiskovou hodnotou. U celkovo
555 pacientov bol medián východiskovej hodnoty FVC na úrovni 68 % referenčnej hodnoty (rozpätie:
48 - 91 %) a medián východiskovej hodnoty DLCO na úrovni 42 % referenčnej hodnoty (rozpätie:
27 - 170 %). Dve percentá pacientov mali východiskovú hodnotu FVC pod 50 % referenčnej hodnoty
a 21 % pacientov malo východiskovú hodnotu DLCO pod 35 % referenčnej hodnoty.
V štúdii PIPF-016 bol pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty,
v 52. týždni liečby v porovnaní s východiskovou hodnotou významne menší u pacientov užívajúcich Esbriet (N = 278) v porovnaní s pacientmi užívajúcimi placebo (N = 277; p < 0,000001, poradová (rank) ANCOVA). Pri liečbe Esbrietom sa tiež dosiahol významne menší pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 13. týždni (p < 0,000001), v 26. týždni
(p < 0,000001) a v 39. týždni (p = 0,000002) v porovnaní s východiskovou hodnotou. V 52. týždni sa
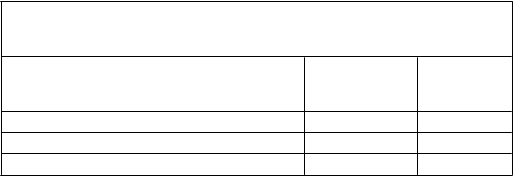
pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, o ≥ 10 % v porovnaní s východiskovou hodnotou alebo úmrtie zaznamenali u 17 % pacientov užívajúcich Esbriet v porovnaní s 32 % pacientmi užívajúcimi placebo (tabuľka 4).
Tabuľk a 4 Vyhodnotenie zmeny hodnoty FVC, vyjadrenej v percentách
z referenčnej hodnoty, v 52. týždni v porovnaní s východiskovou hodnotou v štúdii PIPF-016, podľa k ategórií
P
i
r
f
e
n
i
d
ón
2 403 mg/deň
 (
N = 278)
P
l
acebo
(
N = 277)
(
N = 278)
P
l
acebo
(
N = 277)
Pokles o ≥ 10 % alebo úmrtie 46 (17 %) 88 (32 %) Pokles o menej ako 10 % 169 (61 %) 162 (58 %)
Žiadny pokles (zmena FVC > 0 %) 63 (23 %) 27 (10 %)
V štúdii PIPF-016 bolo skrátenie vzdialenosti prejdenej v 6MWT v 52. týždni v porovnaní
s východiskovou hodnotou významne menšie u pacientov užívajúcich Esbriet v porovnaní s pacientmi
užívajúcimi placebo (p = 0,036, poradová (rank) ANCOVA); skrátenie vzdialenosti prejdenej
v 6MWT o ≥ 50 m sa zistilo u 26 % pacientov užívajúcich Esbriet v porovnaní s 36 % pacientov
užívajúcich placebo.
Vo vopred špecifikovanej, súhrnnej analýze štúdií PIPF-016, PIPF-004 a PIPF-006 bola celková
mortalita v 12. mesiaci v skupine užívajúcej Esbriet v dávke 2 403 mg/deň významne nižšia (3,5 %,
22 zo 623 pacientov) v porovnaní s placebom (6,7 %, 42 zo 624 pacientov), čo viedlo k zníženiu rizika celkovej mortality počas prvých 12 mesiacov o 48 % (HR 0,52 [95 % IS, 0,31 - 0,87], p = 0,0107,
log-rank test).
Štúdia (SP3) s japonskými pacientmi porovnávala pirfenidón v dávke 1 800 mg/deň (porovnateľná s dávkou 2 403 mg/deň podávanou v americkej a európskej populácii zo štúdie PIPF-004/006, a to v prepočte na telesnú hmotnosť) s placebom (pirfenidón: N = 110, placebo: N=109). Pri liečbe pirfenidónom sa dosiahol významne menší priemerný pokles vitálnej kapacity (VC) v 52. týždni (primárny cieľový ukazovateľ) v porovnaní s placebom (-0,09 ± 0,02 l pri pirfenidóne v porovnaní s -0,16 ±0,02 l pri placebe, p = 0,042).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Esbrietom vo
všetkých podskupinách pediatrickej populácie s IPF (informácie o použití v pediatrickej populácii,
pozri časť 4.2).
5.2 Farmak ok inetické vlastnosti
A
bsorpcia
Podávanie kapsúl Esbrietu s jedlom vedie k veľkému zníženiu Cmax (o 50 %) a k menšiemu vplyvu
na AUC v porovnaní s užívaním lieku bez jedla. Po perorálnom podaní jednorazovej dávky 801 mg
zdravým starším dospelým dobrovoľníkom (vo veku 50 - 66 rokov) s jedlom sa rýchlosť absorpcie pirfenidónu spomalila, kým AUC po podaní lieku s jedlom predstavovala približne 80 - 85 % hodnoty AUC pozorovanej po podaní lieku nalačno. Pri porovnávaní 801 mg tablety s tromi 267 mg kapsulami sa preukázala bioekvivalencia, keď sa podávali nalačno. Keď sa podávali s jedlom, 801 mg tableta splnila kritériá bioekvivalencie na základe meraní AUC v porovnaní s kapsulami, zatiaľ čo
90 % intervaly spoľahlivosti pre Cmax (108,26 % - 125,60 %) mierne prekročili hornú hranicu štandardných medzných hodnôt bioekvivalencie (90% IS: 80,00 % -125,00 %). Vplyv jedla na AUC pirfenidónu po perorálnom podaní bol medzi tabletou a kapsulami zhodný. V porovnaní s podaním nalačno, viedlo podanie ktorejkoľvek z uvedených liekových foriem s jedlom k zníženiu Cmax pirfenidónu, pričom pri tablete Esbrietu bolo zníženie Cmax mierne nižšie (o 40 %) ako pri kapsulách Esbrietu (o 50 %). U jedincov, ktorí užívali liek s jedlom, sa pozoroval znížený výskyt nežiaducich udalostí (nauzea a závraty) v porovnaní so skupinou, ktorá užívala liek nalačno. Odporúča sa preto podávať Esbriet s jedlom, aby sa znížil výskyt nauzey a závratov.
Absolútna biologická dostupnosť pirfenidónu u ľudí sa nestanovila. Distribúcia
Pirfenidón sa viaže na ľudské plazmatické bielkoviny, najmä na sérový albumín. Celková priemerná
väzba je od 50 % do 58 % v koncentráciách pozorovaných v klinických štúdiách (1 až 100 μg/ml). Priemerný zdanlivý distribučný objem v rovnovážnom stave po perorálnom podaní je približne 70 l, z čoho vyplýva, že distribúcia pirfenidónu do tkanív je nízka.
Biotransformácia
Približne 70–80 % pirfenidónu sa metabolizuje prostredníctvom CYP1A2 s menším prispením iných
izoenzýmov CYP vrátane CYP2C9, 2C19, 2D6 a 2E1. In vitro údaje naznačujú určitý farmakologicky relevantný účinok hlavného metabolitu (5-karboxy-pirfenidón) v koncentráciách vyšších ako maximálne plazmatické koncentrácie u pacientov s IPF. Môže to byť klinicky relevantné u pacientov
so stredne ťažkou poruchou obličiek, keď je zvýšená plazmatická expozícia 5-karboxy-pirfenidónu.
Eliminácia
Zdá sa, že perorálny klírens pirfenidónu je mierne saturovateľný. V štúdii s opakovanými dávkami skúmajúcej dávkové rozmedzie u zdravých starších dospelých, ktorým sa podávali dávky 267 mg do
1 335 mg trikrát denne, bol priemerný klírens znížený približne o 25 % pri dávke vyššej ako 801 mg
trikrát denne. Po podaní jednorazovej dávky pirfenidónu zdravým starším dospelým bol priemerný zdanlivý terminálny polčas eliminácie približne 2,4 hodiny. Približne 80 % perorálne podanej dávky pirfenidónu sa vylúči močom do 24 hodín po podaní dávky. Väčšina (>95 %) pirfenidónu sa vylúči vo forme metabolitu 5-karboxy-pirfenidónu a menej ako 1 % pirfenidónu sa vylúči močom v nezmenenej forme.
Osobitné skupiny pacientov
Poruchafunkciepečene
Farmakokinetika pirfenidónu a metabolitu 5-karboxy-pirfenidónu sa porovnávala u jedincov so
stredne ťažkou poruchou funkcie pečene (trieda B podľa Childa-Pugha) a u jedincov s normálnou funkciou pečene. Výsledky ukázali, že u pacientov so stredne ťažkou poruchou funkcie pečene bolo priemerné zvýšenie expozície pirfenidónu po jednorazovej dávke pirfenidónu 801 mg (3 x 267 mg kapsula) 60 %. Pirfenidón sa má používať obozretne u pacientov s ľahkou až stredne ťažkou poruchou funkcie pečene a pacienti musia byť pozorne sledovaní z hľadiska prejavov toxicity, najmä keď
súbežne užívajú známy inhibítor CYP1A2 (pozri časti 4.2 a 4.4). Esbriet je kontraindikovaný pri
ťažkej poruche pečene a pri ochorení pečene v terminálnom štádiu (pozri časti 4.2 a 4.3).
Poruchafunkcieobličiek
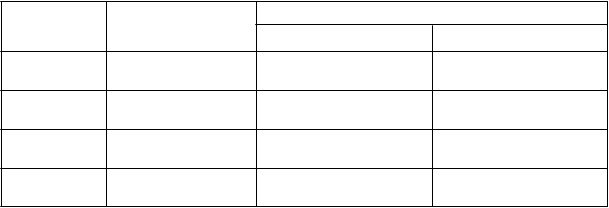
Vo farmakokinetike pirfenidónu sa u jedincov s ľahkou až ťažkou poruchou funkcie obličiek nepozorovali klinicky významné rozdiely v porovnaní s jedincami s normálnou funkciou obličiek. Materská látka sa predominantne metabolizuje najmä na 5-karboxy-pirfenidón. Priemer (SD) AUC0-∞
5-karboxy-pirfenidónu bol významne vyšší u pacientov v skupine so stredne ťažkou (p = 0,009) a ťažkou (p < 0,0001) poruchou funkcie obličiek v porovnaní so skupinou s normálnou funkciou obličiek; 100 (26,3) mg•h/l a 168 (67,4) mg•h/l v porovnaní s 28,7 (4,99) mg•h/l.
Sk
u
p
i
n
a s
p
o
r
u
c
h
o
u funkce
o
b
li
č
i
e
k
 Š
t
a
t
i
s tika
A
U
C
0-∞
(
m
g
• h/l)
P
i
r
f
e
n
i
d
ó
n 5-karboxy-pirfenidón
Š
t
a
t
i
s tika
A
U
C
0-∞
(
m
g
• h/l)
P
i
r
f
e
n
i
d
ó
n 5-karboxy-pirfenidón
normálna Priemer (SD) 42,6 (17,9) 28,7 (4,99)
n = 6 Medián (25.–75.) 42,0 (33,1–55,6) 30,8 (24,1–32,1)
a
ľahká Priemer (SD) 59,1 (21,5) 49,3
(14,6)
n = 6 Medián (25.–75.) 51,6 (43,7–80,3) 43,0 (38,8–56,8)
b
s tredne ťažká Priemer (SD) 63,5 (19,5) 100
(26,3)
n = 6 Medián (25.–75.) 66,7 (47,7–76,7) 96,3 (75,2–123)
c
ťažká Priemer (SD) 46,7 (10,9) 168
(67,4)
n = 6 Medián (25.–75.) 49,4 (40,7–55,8) 150 (123–248)
AUC0-∞ = plocha pod krivkou pre plazmatickú koncentráciu v rovnovážnom stave
ap-hodnota oproti normálu = 1,00 (párové porovnanie Bonferroniho testom) bp-hodnota oproti normálu = 0,009 (párové porovnanie Bonferroniho testom) cp-hodnota oproti normálu < 0,0001 (párové porovnanie Bonferroniho testom)
Expozícia 5-karboxy-pirfenidónu sa zvyšuje u pacientov so stredne ťažkou poruchou funkcie obličiek
3,5 krát alebo viackrát. Klinicky relevantná farmakodynamická aktivita metabolitu u pacientov so stredne ťažkou poruchou funkcie obličiek nemôže byť vylúčená. U pacientov s ľahkou poruchou funkcie obličiek, ktorí užívajú pirfenidón, nie je potrebná úprava dávkovania. Pirfenidón sa má používať s obozretnosťou u pacientov so stredne ťažkou poruchou funkcie obličiek. Použitie pirfenidónu je kontraindikované u pacientov s ťažkou poruchou funkcie obličiek (CrCl <30ml/min.) alebo pri ochorení obličiek v terminálnom štádiu vyžadujúcom dialýzu (pozri časti 4.2 a 4.3).
Populačné farmakokinetické analýzy zo 4 štúdií so zdravými dobrovoľníkmi alebo jedincami
s poruchou funkcie obličiek a jednej štúdie u pacientov s IPF nepreukázali klinicky významný vplyv veku, pohlavia alebo telesných proporcií na farmakokinetiku pirfenidónu.
5.3 Predk linick é údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko pre ľudí.
V štúdiách toxicity po opakovanom podávaní sa pozorovalo zvýšenie hmotnosti pečene u myší, potkanov a psov, ktoré bolo často sprevádzané centrilobulárnou hypertrofiou pečene. Po ukončení liečby sa stav vrátil do normálu. V štúdiách karcinogenicity na potkanoch a myšiach sa pozoroval zvýšený výskyt tumorov pečene. Tieto zistenia týkajúce sa pečene sú konzistentné s indukciou pečeňových mikrozomálnych enzýmov, čo je účinok, ktorý sa nepozoroval u pacientov užívajúcich Esbriet. Tieto zistenia sa nepovažujú za relevantné pre ľudí.
U potkaních samíc, ktorým sa podávala dávka 1 500 mg/kg/deň, čo bol 37-násobok dávky pre človeka,
teda 2 403 mg/deň, sa pozorovalo štatisticky významné zvýšenie výskytu tumorov maternice.
Z výsledkov mechanistických štúdií vyplýva, že výskyt tumorov maternice je pravdepodobne spojený
s dlhodobou nerovnováhou pohlavných hormónov sprostredkovaných dopamínom, čo u potkanov zahŕňa endokrinný mechanizmus špecifický pre druh, ktorý sa u ľudí nevyskytuje.
Reprodukčné toxikologické štúdie nepreukázali nežiaduce účinky na plodnosť samcov alebo samíc
potkanov, ani na postnatálny vývin potomstva a nezistil sa nijaký dôkaz teratogenity u potkanov (1.000 mg/kg/deň) alebo králikov (300 mg/kg/deň). U zvierat dochádza k prechodu pirfenidónu a/alebo jeho metabolitov cez placentu s možnosťou hromadenia pirfenidónu a/alebo jeho metabolitov v amniotickej tekutine. Pri vysokých dávkach (≥450 mg/kg/deň) mali potkany dlhší estrálny cyklus
a vysoký výskyt nepravidelných cyklov. Pri vysokých dávkach (≥1.000 mg/kg/deň) sa u potkanov
vyskytovala predĺžená gestácia a plody mali zníženú životaschopnosť. Štúdie na laktujúcich potkanoch naznačujú, že pirfenidón a/alebo jeho metabolity sa vylučujú do mlieka s potenciálnym hromadením pirfenidónu a/alebo jeho metabolitov v mlieku.
Na základe štandardných testov sa nezískal nijaký dôkaz o mutagénnom alebo genotoxickom účinku pirferidónu a pri testovaní expozície UV sa nezistil mutagénny účinok. Pri testovaní pod expozíciou UV bol pirferidón pozitívny vo fotoklastogénnom teste pľúcnych buniek čínskeho škrečka.
V prípade morčiat sa po perorálnom podaní pirferidónu a pri expozícii svetlu UVA/UVB pozorovala fototoxicita a podráždenie. Závažnosť fototoxických lézií sa minimalizovala použitím ochrany proti slnku.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsúl
mikrokryštalická celulóza,
sodná soľ kroskarmelózy,
povidón, magnéziumstearát
Obal kapsúl
oxid titaničitý (E171),
želatína
Farby nápisu
hnedá S-1-16530 alebo 03A2 farby obsahujúce:
šelak
čierny oxid železitý (E172) červený oxid železitý (E172) žltý oxid železitý (E172) propylénglykol
hydroxid amónny
6.2 Ink ompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
4 roky pre blistre.
3 roky pre fľaše.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte pri teplote do 30°C.
6.5 Druh obalu a obsah baleniaVeľkosti baleniaBalenienaúvodnú2-týždňovúliečbu7 x PVC/PE/PCTFE pretlačovacie pásy z hliníkovej fólie, z ktorých každý obsahuje 3 kapsuly (na dávkovanie v 1. týždni), zabalené spolu so 7x PVC/PE/PCTFE pretlačovacie pásy z hliníkovej fólie, z ktorých každý obsahuje 6 kapsúl (na dávkovanie v 2. týždni). Každé balenie obsahuje spolu
63 kapsúl.
Balenie na 4-týždňovúudržiavaciuliečbu14 x PVC/PE/PCTFE pretlačovacie pásy z hliníkovej fólie, z ktorých každý obsahuje 18 kapsúl
(zásoba na 2 dni). V PVC/PE/PCTFE perforovaných pretlačovacích pásoch z hliníkovej fólie je 14 x
18 kapsúl, spolu 252 kapsúl v balení.
250 ml biela fľaša z HDPE s detským bezpečnostným uzáverom obsahujúca 270 kapsúl. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuŽiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRoche Registration GmbH Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLO/ČÍSLAEU/1/11/667/001
EU/1/11/667/002
EU/1/11/667/003
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. februára 2011
Dátum posledného predĺženia registrácie: 8. septembra 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na w ebovej stránke Európskej agentúry pre lieky
http://w w w .ema.europa.eu.
1. NÁZOV LIEKU
Esbriet 267 mg filmom obalené tablety Esbriet 534 mg filmom obalené tablety Esbriet 801 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIE
Jedna filmom obalená tableta obsahuje 267 mg pirfenidónu. Jedna filmom obalená tableta obsahuje 534 mg pirfenidónu Jedna filmom obalená tableta obsahuje 801 mg pirfenidónu
Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMA
Filmom obalená tableta (tableta).
Esbriet 267 mg filmom obalené tablety sú žlté, oválne, približne 1,3 x 0,6 cm bikonvexné filmom obalené tablety s označením „PFD“.
Esbriet 534 mg filmom obalené tablety sú oranžové, oválne, približne 1,6 x 0,8 cm bikonvexné
filmom obalené tablety s označením „PFD“.
Esbriet 801 mg filmom obalené tablety sú hnedé, oválne, približne 2 x 0,9 cm bikonvexné filmom obalené tablety s označením „PFD“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutick é indik ácie
Esbriet je indikovaný dospelým na liečbu miernej až stredne závažnej idiopatickej pľúcnej fibrózy
(IPF).
4.2 Dávk ovanie a spôsob podávania
Liečbu Esbrietom majú začať a riadiť odborní lekári so skúsenosťami v diagnostike a liečbe IPF. Dávkovanie
Dospelí
Po začatí liečby sa dávka má titrovať až po odporúčanú dennú dávku 2 403 mg/deň počas 14 dní takto:
• 1. až 7. deň: 267 mg dávka podávaná trikrát denne (801 mg/deň),
• 8. až 14. deň: 534 mg dávka podávaná trikrát denne (1 602 mg/deň),
• od 15. dňa: 801 mg dávka podávaná trikrát denne (2 403 mg/deň).
Odporúčaná udržiavacia denná dávka Esbrietu je 801 mg trikrát denne s jedlom, celkovo
2 403 mg/deň.
Dávka vyššia ako 2 403 mg/deň sa neodporúča nijakému pacientovi (pozri časť 4.9).
Pacienti, ktorí 14 a viac po sebe nasledujúcich dní vynechajú liečbu Esbrietom, majú znova začať liečbu úvodným 2-týždňovým režimom titrácie až po odporúčanú dennú dávku.
Pri prerušení liečby na menej ako 14 po sebe nasledujúcich dní sa dávka môže vrátiť
k predchádzajúcej odporúčanej dennej dávke bez titrácie.
Úpravy dávky a inédôležitéčiniteletýkajúcesabezpečnéhoužívania
Gastrointestinálne udalosti: Pacientom, ktorí netolerujú liečbu v dôsledku gastrointestinálnych nežiaducich účinkov, je potrebné pripomenúť, aby liek užívali s jedlom. Ak symptómy pretrvávajú, dávka pirfenidónu sa môže znížiť na 267 mg - 534 mg, dva- až trikrát denne s jedlom, a dávka sa postupne môže znova zvyšovať až po odporúčanú dennú tolerovanú dávku. Ak symptómy pretrvávajú, pacienti môžu dostať inštrukcie na prerušenie liečby na jeden až dva týždne, aby symptómy ustúpili.
Fotosenzitívna reakcia alebo vyrážka: Pacientom, ktorí majú miernu až stredne závažnú
fotosenzitívnu reakciu alebo vyrážku, je potrebné pripomenúť, aby denne používali krém s ochranným faktorom a aby sa vyhýbali expozícii slnku (pozri časť 4.4). Dávka pirfenidónu sa môže znížiť na
801 mg denne (267 mg trikrát denne). Ak vyrážka pretrváva po 7 dňoch, liečba Esbrietom sa má
prerušiť na 15 dní. Dávka sa má potom znova postupne zvyšovať až po odporúčanú dennú dávku takým spôsobom, ako pri kumulatívnom stupňovaní dávky.
Pacientov, ktorí majú závažnú fotosenzitívnu reakciu alebo vyrážku, je potrebné informovať, aby prerušili liečbu a vyhľadali lekársku pomoc (pozri časť 4.4). Ak rozhodne lekár, Esbriet sa po odznení vyrážky môže začať znova užívať a dávka sa opäť môže postupne zvyšovať až po odporúčanú dennú dávku.
Funkcia pečene: V prípade výrazného zvýšenia hladiny alanínaminotransferázy a/alebo aspartátaminotransferázy (ALT/AST) so zvýšením bilirubínu alebo bez zvýšenia bilirubínu sa má dávka pirfenidónu prispôsobiť alebo liečba prerušiť podľa usmernení uvedených v časti 4.4.
Osobitné skupiny pacientov
Starší
U pacientov starších vo veku 65 rokov a viac nie je potrebné upraviť dávku (pozri časť 5.2).
Poruchafunkciepečene
U pacientov s ľahkou až stredne ťažkou poruchou funkcie pečene nie je potrebná úprava dávky (t. j. triedy A a B podľa Childa-Pugha). Keďže sa u niektorých jedincov s ľahkou až stredne ťažkou poruchou funkcie pečene môže zvýšiť plazmatická hladina pirfenidónu, je pri liečbe Esbrietom v tejto skupine pacientov potrebná obozretnosť. Liečba Esbrietom sa nemá používať u pacientov s ťažkou poruchou funkcie pečene alebo v terminálnom štádiu ochorenia pečene (pozri časti 4.3, 4.4 a 5.2).
Poruchafunkcieobličiek
U pacientov s ľahkou poruchou funkcie obličiek nie je potrebná úprava dávky. Esbriet sa má používať s obozretnosťou u pacientov so stredne ťažkou poruchou funkcie obličiek
(CrCl 30-50 ml/min). Liečba Esbrietom sa nemá používať u pacientov s ťažkou poruchou funkcie obličiek (CrCl <30 ml/min.) alebo pri ochorení obličiek v terminálnom štádiu vyžadujúcom dialýzu (pozri časti 4.3 a 5.2).
Pediatrickápopulácia
Použitie Esbrietu sa netýka pediatrickej populácie pre indikáciu IPF.
Spôsob podávania
Esbriet sa užíva perorálne. Tablety sa majú prehltnúť celé a zapiť vodou. Užíva sa s jedlom, aby sa
znížila možnosť výskytu nauzey a závratov (pozri časti 4.8 a 5.2).
4.3 Kontraindik ácie
• Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
• V anamnéze angioedém po liečbe pirfenidónom (pozri časť 4.4).
• Súbežné použitie fluvoxamínu (pozri časť 4.5).
• Závažná porucha funkcie pečene alebo ochorenie pečene v terminálnom štádiu (pozri časti 4.2
a 4.4).
• Závažná porucha funkcie obličiek (CrCl <30 ml/min.) alebo ochorenie obličiek v terminálnom
štádiu vyžadujúce dialýzu (pozri časti 4.2 a 5.2).
4.4 Osobitné upozornenia a opatrenia pri užívaní
Funkcia pečene
U pacientov liečených Esbrietom bolo hlásené viac ako trojnásobné zvýšenie hladín ALT a AST nad
hornú hranicu referenčného rozpätia (upper limit of normal, ULN). Tieto nálezy zriedkavo súviseli
so súbežnými zvýšeniami hladín celkového bilirubínu v sére. Testy funkcií pečene (ALT, AST
a bilirubín) sa majú vykonať pred začatím liečby Esbrietom, v mesačných intervaloch počas prvých
6 mesiacov a potom každé tri mesiace (pozri časť 4.8). Pri výraznom zvýšení pečeňových aminotransferáz sa má dávka lieku Esbriet upraviť alebo liečba prerušiť podľa usmernení, ktoré budú ďalej uvedené. U pacientov s potvrdeným zvýšením hladín ALT, AST alebo bilirubínu počas liečby môžu byť nevyhnutné nasledovné úpravy dávky:
OdporúčaniavprípadezvýšeniahladínALT/AST
U pacientov, ktorí majú po začatí liečby Esbrietom zvýšenú hladinu aminotransferázy
> 3 až ≤ 5 x ULN, sa majú vysadiť lieky sťažujúce interpretáciu nálezov, majú sa vylúčiť iné príčiny
a pacient má byť pozorne sledovaný. Ak je to klinicky vhodné, dávka Esbrietu sa má znížiť alebo liečba prerušiť. Keď sú výsledky testov na funkciu pečene v referenčnom rozpätí, dávka Esbrietu sa môže znova postupne zvyšovať až po odporúčanú dennú tolerovanú dávku.
Ak má pacient zvýšenú hladinu aminotransferázy ≤ 5 x ULN sprevádzanú symptómami alebo hyperbilirubinémiou, liečba Esbrietom sa má prerušiť a pacient nemá liek znova užívať.
Ak má pacient zvýšenú hladinu aminotransferázy > 5 x ULN, liečba Esbrietom sa má prerušiť
a pacient nemá liek znova užívať.
Poruchafunkciepečene
V prípade jedincov so stredne ťažkou poruchou funkcie pečene (t. j. trieda B podľa Childa-Pugha) sa expozícia pirfenidónu zvýšila o 60 %. Esbriet sa má používať obozretne u pacientov s existujúcou ľahkou až stredne ťažkou poruchou funkcie pečene (t. j. triedy A a B podľa Childa-Pugha) vzhľadom na možnú zvýšenú expozíciu pirfenidónu. Pacienti majú byť pozorne sledovaní z hľadiska prejavov toxicity, najmä ak súbežne užívajú známy inhibítor CYP1A2 (pozri časti 4.5 a 5.2). Esbriet sa neskúmal u jedincov s ťažkou poruchou funkcie pečene a nesmie sa používať u pacientov s ťažkou poruchou funkcie pečene (pozri časť 4.3).
Fotosenzitívna reakcia alebo vyrážka
Počas liečby Esbrietom sa pacienti majú vyhýbať vystaveniu priamemu slnečnému žiareniu (vrátane
horského slnka) alebo takéto vystavenie minimalizovať. Pacientov je potrebné informovať, aby denne používali krém s ochranným faktorom, nosili odev, ktorý chráni pred slnkom a aby sa vyhýbali iným liekom, o ktorých je známe, že spôsobujú fotosenzitivitu. Pacientov treba informovať, aby svojmu lekárovi nahlásili symptómy fotosenzitívnej reakcie alebo vyrážky. Závažné fotosenzitívne reakcie sú menej časté. V prípade miernej až závažnej fotosenzitívnej reakcie alebo vyrážky môže byť nevyhnutné upraviť dávku alebo dočasne prerušiť liečbu (pozri časť 4.2).
A
ngioedém
V súvislosti s použitím Esbrietu po uvedení na trh boli hlásené prípady angioedému (v niektorých
prípadoch závažného), ako je opuch tváre, pier a/alebo jazyka, ktoré môžu súvisieť s ťažkosťami
s dýchaním alebo s pískavým dýchaním. Preto pacienti, u ktorých sa objavia prejavy alebo príznaky angioedému po podaní Esbrietu, musia okamžite prerušiť liečbu. Pacienti s angioedémom sa musia liečiť podľa zásad štandardnej zdravotnej starostlivosti. Esbriet sa nesmie používať u pacientov, ktorí majú v anamnéze angioedém spôsobený Esbrietom (pozri časť 4.3).
Závraty
U pacientov užívajúcich Esbriet boli hlásené závraty. Pacienti majú preto vedieť, ako reagujú na tento
liek, skôr než začnú vykonávať činnosti vyžadujúce duševnú bdelosť alebo koordináciu (pozri
časť 4.7). V klinických štúdiách prebehla u väčšiny pacientov, u ktorých sa prejavili závraty, jedna
udalosť a väčšina udalostí ustúpila v priemere počas 22 dní. Ak závraty neustúpia alebo ak sa zhorší ich závažnosť, môže byť potrebné prispôsobiť dávku alebo dokonca prerušiť liečbu Esbrietom.
Únava
U pacientov užívajúcich Esbriet bola hlásená únava. Pacienti preto majú vedieť, ako reagujú na tento
liek, skôr než začnú vykonávať činnosti vyžadujúce duševnú bdelosť alebo koordináciu (pozri časť 4.7).
Úbytok hmotnosti
U pacientov liečených Esbrietom bol hlásený úbytok hmotnosti (pozri časť 4.8). Lekári majú sledovať
hmotnosť pacienta a v prípade potreby podporiť zvýšenie príjmu kalórií, ak sa úbytok hmotnosti považuje za klinicky významný.
4.5 Liek ové a iné interakcie
Približne 70 - 80 % pirfenidónu sa metabolizuje prostredníctvom CYP1A2 s menším prispením iných
izoenzýmov CYP vrátane CYP2C9, 2C19, 2D6 a 2E1.
Konzumácia grapefruitového džúsu je spojená s inhibíciou CYP1A2 a počas liečby pirfenidónom sa jej pacienti majú vyhýbať.
Fluvoxamín a inhibítory CYP1A2
V štúdii fázy 1 viedlo súbežné podávanie Esbrietu a fluvoxamínu (silný inhibítor CYP1A2
s inhibičnými účinkami na iné izoenzýmy CYP [CYP2C9, 2C19 a 2D6]) u nefajčiarov k 4-násobnému
zvýšeniu expozície pirfenidónu.
Esbriet je kontraindikovaný u pacientov, ktorí súbežne užívajú fluvoxamín (pozri časť 4.3). Fluvoxamín sa má pred začatím liečby Esbrietom vysadiť a počas liečby Esbrietom sa nemá užívať vzhľadom na znížený klírens pirfenidónu. Počas liečby pirfenidónom je potrebné vyhnúť sa ďalšej liečbe, ktorá inhibuje CYP1A2 a jeden alebo viac iných izoenzýmov CYP, ktoré sa podieľajú na metabolizme pirfenidónu (napr. CYP2C9, 2C19 a 2D6).
Extrapolácie podmienok in vitro a in vivo naznačujú, že silné a selektívne inhibítory CYP1A2 (napr. enoxacín) môžu zvýšiť expozíciu pirfenidónu približne 2- až 4-násobne. Ak sa nedá vyhnúť súbežnému použitiu Esbrietu so silným a selektívnym inhibítorom CYP1A2, dávka pirfenidónu sa má znížiť na 801 mg denne (267 mg trikrát denne). Pacientov treba pozorne sledovať z hľadiska výskytu nežiaducich reakcií spojených s liečbou Esbrietu. Ak je to potrebné, vysaďte Esbriet (pozri časti 4.2
a 4.4).
Súčasné podávanie Esbrietu a 750 mg ciprofloxacínu (stredne silný inhibítor CYP1A2) zvyšovalo expozíciu pirfenidónu o 81 %. Ak je podávanie ciprofloxacínu v dávkach 750 mg dvakrát denne potrebné, dávku pirfenidónu treba znížiť na 1 602 mg denne (534 mg trikrát denne). Esbriet treba podávať s opatrnosťou, ak je ciprofloxacín podávaný v dávkach 250 mg alebo 500 mg raz alebo dvakrát denne.
Esbriet sa má užívať obozretne u pacientov liečených inými stredne silnými inhibítormi CYP1A2
(napr. amiodarón, propafenón).
Mimoriadna obozretnosť je tiež potrebná, keď sa inhibítory CYP1A2 používajú súbežne so silnými inhibítormi jedného alebo viacerých ďalších izoenzýmov CYP, ktoré sa podieľajú na metabolizme pirfenidónu, ako je CYP2C9 (napr. amiodarón, flukonazol), 2C19 (napr. chloramfenikol) a 2D6 (napr. fluoxetín, paroxetín).
Fajčenie cigariet a induktory CYP1A2
Interakčná štúdia fázy 1 hodnotila vplyv fajčenia cigariet (induktor CYP1A2) na farmakokinetiku
pirfenidónu. Expozícia pirfenidónu u fajčiarov bola 50 % v porovnaní s expozíciou u nefajčiarov. Fajčenie môže indukovať tvorbu pečeňových enzýmov, a teda zvyšovať klírens lieku a znižovať expozíciu. Počas liečby Esbrietom je potrebné vyhýbať sa súbežnému užívaniu silných induktorov CYP1A2 vrátane fajčenia na základe pozorovaného vzťahu medzi fajčením cigariet a potenciálom indukovať CYP1A2. Pacientom sa má odporučiť, aby prestali užívať silné induktory CYP1A2 a aby pred liečbou a počas liečby pirfenidónom nefajčili.
V prípade stredne silných induktorov CYP1A2 (napr. omeprazol) môže súbežné užitie teoreticky viesť
k zníženiu plazmatickej hladiny pirfenidónu.
Súbežné podávanie liekov, ktoré účinkujú ako silné induktory CYP1A2 a iných izoenzýmov CYP podieľajúcich sa na metabolizme pirfenidónu (napr. rifampicín), môže viesť k výraznému zníženiu plazmatickej hladiny pirfenidónu. Týmto liekom je potrebné vyhnúť sa vždy, keď je to možné.
4.6 Fertilita, gravidita a lak tácia
Gravidita
Nie sú k dispozícii údaje o použití Esbrietu u gravidných žien.
U zvierat dochádza k prieniku pirfenidónu a/alebo jeho metabolitov cez placentu s možnosťou
hromadenia pirfenidónu a/alebo jeho metabolitov v amniotickej tekutine.
Pri vysokých dávkach (≥ 1 000 mg/kg/deň) sa u potkanov pozorovalo predĺženie gestácie a zníženie životaschopnosti plodov.
Ako preventívne opatrenie je vhodnejšie vyhnúť sa užívaniu Esbrietu počas gravidity.
Dojčenie
Nie je známe, či sa pirfenidón alebo jeho metabolity vylučujú do ľudského mlieka. Dostupné
farmakokinetické údaje u zvierat preukázali vylučovanie pirfenidónu a/alebo jeho metabolitov
do mlieka s možnosťou hromadenia pirfenidónu a/alebo jeho metabolitov v mlieku (pozri časť 5.3). Riziko pre dojčené dieťa sa nedá vylúčiť.
Rozhodnutie, či ukončiť dojčenie alebo či ukončiť liečbu Esbrietom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby Esbrietom pre matku.
Fertilita
V predklinických štúdiách sa nepozoroval nežiaduci vplyv na fertilitu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Esbriet môže spôsobiť závraty a únavu, čo môže mať mierny vplyv na schopnosť viesť vozidlá alebo obsluhovať stroje. Pri výskyte týchto príznakov majú byť preto pacienti opatrní pri vedení vozidiel
a obsluhe strojov.
4.8 Nežiaduce účink y
Súhrnbezpečnostnéhoprofilu
K najčastejšie hláseným nežiaducim reakciám počas klinických štúdií skúmajúcich Esbriet v dávke
2 403 mg/deň v porovnaní s placebom patrila nauzea (32,4 % v porovnaní s 12,2 %), vyrážka (26,2 % v porovnaní so 7,7 %), hnačka (18,8 % v porovnaní so 14,4 %), únava (18,5 % v porovnaní s 10,4 %), dyspepsia (16,1 % v porovnaní s 5,0 %), anorexia (11,4 % v porovnaní s 3,5 %), bolesť hlavy (10,1 % v porovnaní so 7,7 %) a fotosenzitívna reakcia (9,3 % v porovnaní s 1,1 %).
Súhrnnežiaducichreakciíuvedenýchvtabuľke
Bezpečnosť Esbrietu sa hodnotila v klinických štúdiách zahŕňajúcich 1 650 dobrovoľníkov
a pacientov. Viac ako 170 pacientov bolo testovaných v nezaslepených štúdiách dlhšie ako päť rokov
a niektorí až do 10 rokov.
Tabuľka 1 uvádza nežiaduce reakcie hlásené s frekvenciou ≥2 % u 623 pacientov užívajúcich Esbriet v odporúčanej dávke 2 403 mg/deň v troch súhrnných pivotných štúdiách fázy 3. Nežiaduce reakcie hlásené v období po uvedení lieku na trh sú tiež uvedené v tabuľke 1. Nežiaduce reakcie sú uvedené podľa triedy orgánových systémov a v každej skupine frekvencií [veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000)] sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
 T
abuľk a 1 Nežiaduce reak cie podľa triedy orgánových systémov a frekvencie podľa
M
e
d
DRA
I
n
f
e
k
c
i
e a nákazy
T
abuľk a 1 Nežiaduce reak cie podľa triedy orgánových systémov a frekvencie podľa
M
e
d
DRA
I
n
f
e
k
c
i
e a nákazy
Časté infekcia horných dýchacích ciest; infekcia močových ciest
Poruchy k rvi a lymfatického systémuZriedkavé agranulocytóza1
Poruchy imunitného systémuMenej časté angioedém1
Poruchy metabolizmu a výživyVeľmi časté anorexia
Časté úbytok hmotnosti; znížená chuť do jedla
Psychick é poruchyČasté nespavosť
Poruchy nervového systému Veľmi časté bolesť hlavy
Časté závraty; somnolencia; dysgeúzia; letargia
Poruchy cievČasté návaly tepla
Poruchy dýchacej sústavy, hrudníka a mediastína Časté dyspnoe; kašeľ; produktívny kašeľ
Poruchy gastrointestinálneho traktuVeľmi časté dyspepsia; nauzea; hnačka
Časté gastroezofágová refluxová choroba; vracanie; abdominálna distenzia; abdominálny dyskomfort; abdominálna bolesť; bolesť v hornej časti brucha; žalúdočný dyskomfort; gastritída; zápcha; flatulencia'
Poruchy pečene a žlčových ciestČasté zvýšená hladina ALT; zvýšená hladina AST; zvýšená hladina
gamaglutamyltransferázy
Zriedkavé zvýšená hladina celkového bilirubínu v sére v kombinácii so zvýšenou
hladinou ALT a AST1
Poruchy k ože a podk ožného tkanivaVeľmi časté fotosenzitívna reakcia; vyrážka
Časté pruritus; erytém; suchá koža; erytematózna vyrážka; makulárna vyrážka;
svrbiaca vyrážka
Poruchy k ostrovej a svalovej sústavy a spojivového tkanivaČasté myalgia; artralgia
Celk ové poruchy a reakcie v mieste podaniaVeľmi časté únava
Časté asténia; bolesť na hrudníku iná ako srdcová
Úrazy, otravy a k omplik ácie liečebného postupuČasté Popálenina od slnka
1. Identifikované počas pozorovania po uvedení lieku na trh
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 Predávk ovanie
Existuje málo klinických skúseností s predávkovaním. Zdravým dospelým dobrovoľníkom sa počas
12 dní opakovane podávali postupne zvyšované dávky pirfenidónu až do celkovej dávky 4 806 mg/deň vo forme šiestich 267 mg kapsúl trikrát denne. Nežiaduce reakcie boli mierne, prechodné a zhodovali sa s najčastejšie hlásenými nežiaducimi reakciami na pirfenidón.
V prípade podozrenia na predávkovanie sa má poskytnúť podporná lekárska starostlivosť vrátane
sledovania vitálnych známok a pozorného sledovania klinického stavu pacienta.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmak odynamické vlastnosti
Farmakoterapeutická skupina: Imunosupresíva, iné imunosupresíva, kód ATC: L04AX05
Mechanizmus účinku pirfenidónu sa ešte celkom nestanovil. Z existujúcich údajov však vyplýva, že pirfenidón v rôznych systémoch in vitro a v živočíšnych modeloch pľúcnej fibrózy (fibróza vyvolaná bleomycínom a transplantáciou) prejavuje antifibrotické a protizápalové vlastnosti.
IPF je chronické fibrotické a zápalové ochorenie pľúc ovplyvnené syntézou a uvoľňovaním prozápalových cytokínov vrátane tumor nektrotizujúceho faktora alfa (TNF-α) a interleukínu-1–beta (IL-1β) a zistilo sa, že pirfenidón znižuje hromadenie zápalových buniek ako reakciu na rôzne podnety.
Pirfenidón tlmí proliferáciu fibroblastov, tvorbu bielkovín a cytokínov spojených s fibrózou a zvýšenú biosyntézu a hromadenie mimobunkového matrix ako reakciu na cytokínové rastové faktory, ako je napríklad transformujúci rastový faktor beta (TGF-β) a rastový faktor odvodený z krvných doštičiek (PDGF).
Klinická účinnosť
Klinická účinnosť Esbrietu sa skúmala v štyroch multicentrických, randomizovaných, dvojito
zaslepených štúdiách fázy 3 kontrolovaných placebom u pacientov s IPF. Tri z týchto štúdií fázy 3
(PIPF-004, PIPF-006 a PIPF-016) boli mnohonárodné a jedna štúdia (SP3) sa uskutočnila v Japonsku.
Štúdie PIPF-004 a PIPF-006 porovnávali liečbu Esbrietom v dávke 2 403 mg/deň s placebom. Tieto štúdie boli takmer rovnaké, pokiaľ ide o dizajn, s niekoľkými výnimkami vrátane skupiny, v ktorej sa podávala stredne veľká dávka (1 197 mg/deň), v štúdii PIPF-004. V obidvoch štúdiách sa liečba podávala trikrát denne minimálne počas 72 týždňov. Primárnym cieľovým ukazovateľom v obidvoch štúdiách bola zmena hodnoty úsilnej vitálnej kapacity (Forced Vital Capacity, FVC), vyjadrenej
v percentách z referenčnej hodnoty, v 72. týždni v porovnaní s východiskovou hodnotou.
V štúdii PIPF-004 bol pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty,
v 72. týždni liečby v porovnaní s východiskovou hodnotou významne menší u pacientov užívajúcich
Esbriet (N = 174) v porovnaní s pacientmi užívajúcimi placebo (N = 174; p = 0,001, poradová (rank) analýza kovariancie - ANCOVA). Pri liečbe Esbrietom sa tiež dosiahol významne menší pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 24. týždni (p = 0,014), v 36. týždni
(p < 0,001), v 48. týždni (p < 0,001) a v 60. týždni (p < 0,001) v porovnaní s východiskovou hodnotou. V 72. týždni sa pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, o ≥ 10 % (čo je prahová hodnota poukazujúca na riziko úmrtia na IPF) v porovnaní s východiskovou hodnotou zaznamenal u 20 % pacientov užívajúcich Esbriet v porovnaní s 35 % pacientmi užívajúcimi placebo (tabuľka 2).
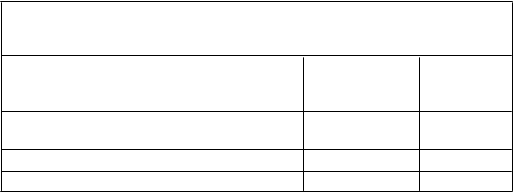
T
abuľk a 2 Vyhodnotenie zmeny hodnoty FVC, vyjadrenej v percentách
z referenčnej hodnoty, v 72. týždni v porovnaní s východiskovou hodnotou v štúdii PIPF-004, podľa k ategórií
P
i
r
f
e
n
i
d
ón
2 403 mg/deň
 (
N = 174)
P
l
acebo
(
N = 174)
(
N = 174)
P
l
acebo
(
N = 174)
Pokles o ≥ 10 % alebo úmrtie alebo transplantácia pľúc
35 (20 %) 60 (34 %)
Pokles o menej ako 10 % 97 (56 %) 90 (52 %)
Žiadny pokles (zmena FVC > 0 %) 42 (24 %) 24 (14 %)
Napriek tomu, že podľa vopred špecifikovanej poradovej (rank) ANCOVA nebol medzi pacientmi
užívajúcimi Esbriet a pacientmi užívajúcimi placebo žiadny rozdiel z hľadiska zmeny vzdialenosti prejdenej v šesťminútovom teste chôdzou (six minute w alk test, 6MWT) v 72. týždni v porovnaní s východiskovou hodnotou, v ad hoc analýze sa zistilo skrátenie vzdialenosti prejdenej v 6MWT
o ≥ 50 m u 37 % pacientov užívajúcich Esbriet v porovnaní so 47 % pacientmi užívajúcimi placebo
v štúdii PIPF-004.
V štúdii PIPF-006 sa pri liečbe Esbrietom (N = 171) v porovnaní s placebom (N = 173) nedosiahol
menší pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 72. týždni liečby
v porovnaní s východiskovou hodnotou (p = 0,501). Pri liečbe Esbrietom sa však dosiahol menší
pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 24. týždni (p < 0,001),
v 36. týždni (p = 0,011) a v 48. týždni (p = 0,005) v porovnaní s východiskovou hodnotou.
V 72. týždni sa pokles hodnoty FVC o ≥ 10 % zaznamenal u 23 % pacientov užívajúcich Esbriet
a u 27 % pacientov užívajúcich placebo (tabuľka 3).
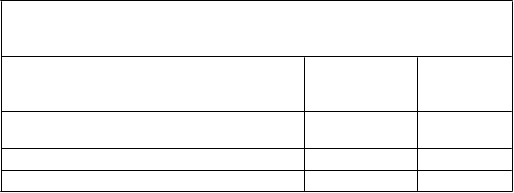
Tabuľk a 3 Vyhodnotenie zmeny hodnoty FVC, vyjadrenej v percentách
z referenčnej hodnoty, v 72. týždni v porovnaní s východiskovou hodnotou v štúdii PIPF-006, podľa k ategórií
P
i
r
f
e
n
i
d
ón
2 403 mg/deň
 (
N = 171)
P
l
acebo
(
N = 173)
(
N = 171)
P
l
acebo
(
N = 173)
Pokles o ≥ 10 % alebo úmrtie alebo transplantácia pľúc
39 (23 %) 46 (27 %)
Pokles o menej ako 10 % 88 (52 %) 89 (51 %)
Žiadny pokles (zmena FVC > 0 %) 44 (26 %) 38 (22 %)
V štúdii PIPF-006 bolo skrátenie vzdialenosti prejdenej v 6MWT v 72. týždni v porovnaní
s východiskovou hodnotou významne menšie pri liečbe Esbrietom v porovnaní s placebom (p < 0,001,
poradová (rank) ANCOVA). V ad hoc analýze sa okrem toho zistilo skrátenie vzdialenosti prejdenej v 6MWT o ≥ 50 m u 33 % pacientov užívajúcich Esbriet v porovnaní so 47 % pacientov užívajúcimi placebo v štúdii PIPF-006.
V súhrnnej analýze prežívania v štúdiách PIPF-004 a PIPF-006 bola miera mortality v skupine
užívajúcej Esbriet v dávke 2 403 mg/deň 7,8 % v porovnaní s 9,8 % pri užívaní placeba (HR 0,77
[95 % IS, 0,47 - 1,28]).
Štúdia PIPF-016 porovnávala liečbu Esbrietom 2 403 mg/deň s placebom. Liek sa podával trikrát denne počas 52 týždňov. Primárnym cieľovým ukazovateľom bola zmena hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 52. týždni v porovnaní s východiskovou hodnotou. U celkovo
555 pacientov bol medián východiskovej hodnoty FVC na úrovni 68 % referenčnej hodnoty (rozpätie:
48 - 91 %) a medián východiskovej hodnoty DLCO na úrovni 42 % referenčnej hodnoty (rozpätie:
27 - 170 %). Dve percentá pacientov mali východiskovú hodnotu FVC pod 50 % referenčnej hodnoty
a 21 % pacientov malo východiskovú hodnotu DLCO pod 35 % referenčnej hodnoty.
V štúdii PIPF-016 bol pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty,
v 52. týždni liečby v porovnaní s východiskovou hodnotou významne menší u pacientov užívajúcich Esbriet (N = 278) v porovnaní s pacientmi užívajúcimi placebo (N = 277; p < 0,000001, poradová (rank) ANCOVA). Pri liečbe Esbrietom sa tiež dosiahol významne menší pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, v 13. týždni (p < 0,000001), v 26. týždni
(p < 0,000001) a v 39. týždni (p = 0,000002) v porovnaní s východiskovou hodnotou. V 52. týždni sa
pokles hodnoty FVC, vyjadrenej v percentách z referenčnej hodnoty, o ≥ 10 % v porovnaní s východiskovou hodnotou alebo úmrtie zaznamenali u 17 % pacientov užívajúcich Esbriet v porovnaní s 32 % pacientmi užívajúcimi placebo (tabuľka 4).
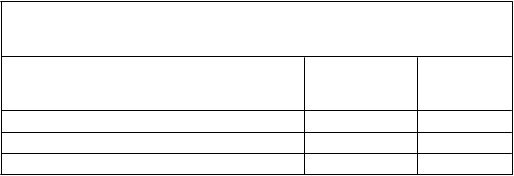
Tabuľk a 4 Vyhodnotenie zmeny hodnoty FVC, vyjadrenej v percentách
z referenčnej hodnoty, v 52. týždni v porovnaní s východiskovou hodnotou v štúdii PIPF-016, podľa k ategórií
P
i
r
f
e
n
i
d
ón
2 403 mg/deň
 (
N = 278)
P
l
acebo
(
N = 277)
(
N = 278)
P
l
acebo
(
N = 277)
Pokles o ≥ 10 % alebo úmrtie 46 (17 %) 88 (32 %) Pokles o menej ako 10 % 169 (61 %) 162 (58 %)
Žiadny pokles (zmena FVC > 0 %) 63 (23 %) 27 (10 %)
V štúdii PIPF-016 bolo skrátenie vzdialenosti prejdenej v 6MWT v 52. týždni v porovnaní
s východiskovou hodnotou významne menšie u pacientov užívajúcich Esbriet v porovnaní s pacientmi
užívajúcimi placebo (p = 0,036, poradová (rank) ANCOVA); skrátenie vzdialenosti prejdenej
v 6MWT o ≥ 50 m sa zistilo u 26 % pacientov užívajúcich Esbriet v porovnaní s 36 % pacientov
užívajúcich placebo.
Vo vopred špecifikovanej, súhrnnej analýze štúdií PIPF-016, PIPF-004 a PIPF-006 bola celková
mortalita v 12. mesiaci v skupine užívajúcej Esbriet v dávke 2 403 mg/deň významne nižšia (3,5 %,
22 zo 623 pacientov) v porovnaní s placebom (6,7 %, 42 zo 624 pacientov), čo viedlo k zníženiu rizika celkovej mortality počas prvých 12 mesiacov o 48 % (HR 0,52 [95 % IS, 0,31 - 0,87], p = 0,0107,
log-rank test).
Štúdia (SP3) s japonskými pacientmi porovnávala pirfenidón v dávke 1 800 mg/deň (porovnateľná s dávkou 2 403 mg/deň podávanou v americkej a európskej populácii zo štúdie PIPF-004/006, a to v prepočte na telesnú hmotnosť) s placebom (pirfenidón: N = 110, placebo: N=109). Pri liečbe pirfenidónom sa dosiahol významne menší priemerný pokles vitálnej kapacity (VC) v 52. týždni (primárny cieľový ukazovateľ) v porovnaní s placebom (-0,09 ± 0,02 l pri pirfenidóne v porovnaní s -0,16 ±0,02 l pri placebe, p = 0,042).
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Esbrietom
vo všetkých podskupinách pediatrickej populácie s IPF (informácie o použití v pediatrickej populácii,
pozri časť 4.2).
5.2 Farmak ok inetické vlastnosti
A
bsorpcia
Podávanie kapsúl Esbrietu s jedlom vedie k veľkému zníženiu Cmax (o 50 %) a k menšiemu vplyvu
na AUC v porovnaní s užívaním lieku bez jedla. Po perorálnom podaní jednorazovej dávky 801 mg
zdravým starším dospelým dobrovoľníkom (vo veku 50 - 66 rokov) s jedlom sa rýchlosť absorpcie pirfenidónu spomalila, kým AUC po podaní lieku s jedlom predstavovala približne 80 - 85 % hodnoty AUC pozorovanej po podaní lieku nalačno. Pri porovnávaní 801 mg tablety s tromi 267 mg kapsulami sa preukázala bioekvivalencia, keď sa podávali nalačno. Keď sa podávali s jedlom, 801 mg tableta splnila kritériá bioekvivalencie na základe meraní AUC v porovnaní s kapsulami, zatiaľ čo
90 % intervaly spoľahlivosti pre Cmax (108,26 % - 125,60 %) mierne prekročili hornú hranicu štandardných medzných hodnôt bioekvivalencie (90% IS: 80,00 % -125,00 %). Vplyv jedla na AUC pirfenidónu po perorálnom podaní bol medzi tabletou a kapsulami zhodný. V porovnaní s podaním nalačno, viedlo podanie ktorejkoľvek z uvedených liekových foriem s jedlom k zníženiu Cmax pirfenidónu, pričom pri tablete Esbrietu bolo zníženie Cmax mierne nižšie (o 40 %) ako pri kapsulách Esbrietu (o 50 %). U jedincov, ktorí užívali liek s jedlom, sa pozoroval znížený výskyt nežiaducich udalostí (nauzea a závraty) v porovnaní so skupinou, ktorá užívala liek nalačno. Odporúča sa preto podávať Esbriet s jedlom, aby sa znížil výskyt nauzey a závratov.
Absolútna biologická dostupnosť pirfenidónu u ľudí sa nestanovila. Distribúcia
Pirfenidón sa viaže na ľudské plazmatické bielkoviny, najmä na sérový albumín. Celková priemerná
väzba je od 50 % do 58 % v koncentráciách pozorovaných v klinických štúdiách (1 až 100 μg/ml). Priemerný zdanlivý distribučný objem v rovnovážnom stave po perorálnom podaní je približne 70 l, z čoho vyplýva, že distribúcia pirfenidónu do tkanív je nízka.
Biotransformácia
Približne 70–80 % pirfenidónu sa metabolizuje prostredníctvom CYP1A2 s menším prispením iných
izoenzýmov CYP vrátane CYP2C9, 2C19, 2D6 a 2E1. In vitro údaje naznačujú určitý farmakologicky relevantný účinok hlavného metabolitu (5-karboxy-pirfenidón) v koncentráciách vyšších ako maximálne plazmatické koncentrácie u pacientov s IPF. Môže to byť klinicky relevantné u pacientov
so stredne ťažkou poruchou obličiek, keď je zvýšená plazmatická expozícia 5-karboxy-pirfenidónu.
Eliminácia
Zdá sa, že perorálny klírens pirfenidónu je mierne saturovateľný. V štúdii s opakovanými dávkami
skúmajúcej dávkové rozmedzie u zdravých starších dospelých, ktorým sa podávali dávky od 267 mg
do 1 335 mg trikrát denne, bol priemerný klírens znížený približne o 25 % pri dávke vyššej ako
801 mg trikrát denne. Po podaní jednorazovej dávky pirfenidónu zdravým starším dospelým bol priemerný zdanlivý terminálny polčas eliminácie približne 2,4 hodiny. Približne 80 % perorálne podanej dávky pirfenidónu sa vylúči močom do 24 hodín po podaní dávky. Väčšina (> 95 %) pirfenidónu sa vylúči vo forme metabolitu 5-karboxy-pirfenidónu a menej ako 1 % pirfenidónu sa vylúči močom v nezmenenej forme.
Osobitné skupiny pacientov
Poruchafunkciepečene
Farmakokinetika pirfenidónu a metabolitu 5-karboxy-pirfenidónu sa porovnávala u jedincov
so stredne ťažkou poruchou funkcie pečene (trieda B podľa Childa-Pugha) a u jedincov s normálnou funkciou pečene. Výsledky ukázali, že u pacientov so stredne ťažkou poruchou funkcie pečene bolo priemerné zvýšenie expozície pirfenidónu po jednorazovej dávke pirfenidónu 801 mg (3 x 267 mg kapsula) 60 %. Pirfenidón sa má používať obozretne u pacientov s ľahkou až stredne ťažkou poruchou funkcie pečene a pacienti musia byť pozorne sledovaní z hľadiska prejavov toxicity, najmä keď
súbežne užívajú známy inhibítor CYP1A2 (pozri časti 4.2 a 4.4). Esbriet je kontraindikovaný pri ťažkej poruche pečene a pri ochorení pečene v terminálnom štádiu (pozri časti 4.2 a 4.3).
Porucha funkcieobličiek
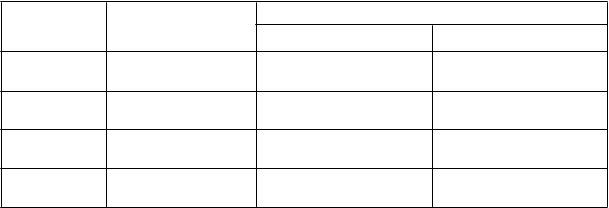
Vo farmakokinetike pirfenidónu sa u jedincov s ľahkou až ťažkou poruchou funkcie obličiek nepozorovali klinicky významné rozdiely v porovnaní s jedincami s normálnou funkciou obličiek. Materská látka sa predominantne metabolizuje najmä na 5-karboxy-pirfenidón. Priemer (SD) AUC0-∞
5-karboxy-pirfenidónu bol významne vyšší u pacientov v skupine so stredne ťažkou (p = 0,009) a ťažkou (p < 0,0001) poruchou funkcie obličiek v porovnaní so skupinou s normálnou funkciou obličiek; 100 (26,3) mg•h/l a 168 (67,4) mg•h/l v porovnaní s 28,7 (4,99) mg•h/l.
Sk
u
p
i
n
a s
p
o
r
u
c
h
o
u funkce
o
b
li
č
i
e
k
 Š
t
a
t
i
s tika
A
U
C
0-∞
(
m
g
• h/l)
P
i
r
f
e
n
i
d
ó
n 5-karboxy-pirfenidón
Š
t
a
t
i
s tika
A
U
C
0-∞
(
m
g
• h/l)
P
i
r
f
e
n
i
d
ó
n 5-karboxy-pirfenidón
normálna Priemer (SD) 42,6 (17,9) 28,7 (4,99)
n = 6 Medián (25.–75.) 42,0 (33,1–55,6) 30,8 (24,1–32,1)
a
ľahká Priemer (SD) 59,1 (21,5) 49,3
(14,6)
n = 6 Medián (25.–75.) 51,6 (43,7–80,3) 43,0 (38,8–56,8)
b
s tredne ťažká Priemer (SD) 63,5 (19,5) 100
(26,3)
n = 6 Medián (25.–75.) 66,7 (47,7–76,7) 96,3 (75,2–123)
c
ťažká Priemer (SD) 46,7 (10,9) 168
(67,4)
n = 6 Medián (25.–75.) 49,4 (40,7–55,8) 150 (123–248)
AUC0-∞ = plocha pod krivkou pre plazmatickú koncentráciu v rovnovážnom stave
ap-hodnota oproti normálu = 1,00 (párové porovnanie Bonferroniho testom) bp-hodnota oproti normálu = 0,009 (párové porovnanie Bonferroniho testom) cp-hodnota oproti normálu < 0,0001 (párové porovnanie Bonferroniho testom)
Expozícia 5-karboxy-pirfenidónu sa zvyšuje u pacientov so stredne ťažkou poruchou funkcie obličiek
3,5 krát alebo viackrát. Klinicky relevantná farmakodynamická aktivita metabolitu u pacientov so stredne ťažkou poruchou funkcie obličiek nemôže byť vylúčená. U pacientov s ľahkou poruchou funkcie obličiek, ktorí užívajú pirfenidón, nie je potrebná úprava dávkovania. Pirfenidón sa má používať s obozretnosťou u pacientov so stredne ťažkou poruchou funkcie obličiek. Použitie pirfenidónu je kontraindikované u pacientov s ťažkou poruchou funkcie obličiek (CrCl <30ml/min.) alebo pri ochorení obličiek v terminálnom štádiu vyžadujúcom dialýzu (pozri časti 4.2 a 4.3).
Populačné farmakokinetické analýzy zo 4 štúdií so zdravými dobrovoľníkmi alebo jedincami
s poruchou funkcie obličiek a jednej štúdie u pacientov s IPF nepreukázali klinicky významný vplyv
veku, pohlavia alebo telesných proporcií na farmakokinetiku pirfenidónu.
5.3 Predk linick é údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity
po opakovanom podávaní, genotoxicity a karcinogénneho potenciálu neodhalili žiadne osobitné riziko
pre ľudí.
V štúdiách toxicity po opakovanom podávaní sa pozorovalo zvýšenie hmotnosti pečene u myší, potkanov a psov, ktoré bolo často sprevádzané centrilobulárnou hypertrofiou pečene. Po ukončení liečby sa stav vrátil do normálu. V štúdiách karcinogenicity na potkanoch a myšiach sa pozoroval zvýšený výskyt tumorov pečene. Tieto zistenia týkajúce sa pečene sú konzistentné s indukciou pečeňových mikrozomálnych enzýmov, čo je účinok, ktorý sa nepozoroval u pacientov užívajúcich Esbriet. Tieto zistenia sa nepovažujú za relevantné pre ľudí.
U potkaních samíc, ktorým sa podávala dávka 1 500 mg/kg/deň, čo bol 37-násobok dávky pre človeka,
teda 2 403 mg/deň, sa pozorovalo štatisticky významné zvýšenie výskytu tumorov maternice.
Z výsledkov mechanistických štúdií vyplýva, že výskyt tumorov maternice je pravdepodobne spojený
s dlhodobou nerovnováhou pohlavných hormónov sprostredkovaných dopamínom, čo u potkanov zahŕňa endokrinný mechanizmus špecifický pre druh, ktorý sa u ľudí nevyskytuje.
Reprodukčné toxikologické štúdie nepreukázali nežiaduce účinky na plodnosť samcov alebo samíc
potkanov, ani na postnatálny vývin potomstva a nezistil sa nijaký dôkaz teratogenity u potkanov (1 000 mg/kg/deň) alebo králikov (300 mg/kg/deň). U zvierat dochádza k prechodu pirfenidónu a/alebo jeho metabolitov cez placentu s možnosťou hromadenia pirfenidónu a/alebo jeho metabolitov v amniotickej tekutine. Pri vysokých dávkach (≥ 450 mg/kg/deň) mali potkany dlhší estrálny cyklus
a vysoký výskyt nepravidelných cyklov. Pri vysokých dávkach (≥ 1 000 mg/kg/deň) sa u potkanov
vyskytovala predĺžená gestácia a plody mali zníženú životaschopnosť. Štúdie na laktujúcich potkanoch naznačujú, že pirfenidón a/alebo jeho metabolity sa vylučujú do mlieka s potenciálnym hromadením pirfenidónu a/alebo jeho metabolitov v mlieku.
Na základe štandardných testov sa nezískal nijaký dôkaz o mutagénnom alebo genotoxickom účinku pirferidónu a pri testovaní expozície UV sa nezistil mutagénny účinok. Pri testovaní pod expozíciou UV bol pirferidón pozitívny vo fotoklastogénnom teste pľúcnych buniek čínskeho škrečka.
V prípade morčiat sa po perorálnom podaní pirferidónu a pri expozícii svetlu UVA/UVB pozorovala fototoxicita a podráždenie. Závažnosť fototoxických lézií sa minimalizovala použitím ochrany proti slnku.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah tablety
mikrokryštalická celulóza,
sodná soľ kroskarmelózy,
povidón K30,
bezvodý koloidný oxid kremičitý,
magnéziumstearát
Filmový obal:
polyvinylalkohol,
oxid titaničitý (E171), makrogol 3350, mastenec,
267mgtableta
žltý oxid železitý (E172)
534 mgtableta
žltý oxid železitý (E172)
červený oxid železitý (E172)
801 mgtableta
červený oxid železitý (E172)
čierny oxid železitý (E172)
6.2 Ink ompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
2 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Fľaša z polyetylénu s vysokou hustotou (HDPE) s detským bezpečnostným a poistným uzáverom so závitom.
Veľkosti balenia
267mgfilmomobalenétablety
1 fľaša obsahujúca 21 filmom obalených tabliet
2 fľaše, každá s obsahom 21 filmom obalených tabliet (spolu 42 filmom obalených tabliet)
1 fľaša obsahujúca 42 filmom obalených tabliet
1 fľaša obsahujúca 90 filmom obalených tabliet
2 fľaše, každá s obsahom 90 filmom obalených tabliet (spolu 180 filmom obalených tabliet)
1 fľaša obsahujúca 180 filmom obalených tabliet
534 mg filmomobalenétablety
1 fľaša obsahujúca 21 filmom obalených tabliet
1 fľaša obsahujúca 90 filmom obalených tabliet
801 mg filmomobalenétablety
1 fľaša obsahujúca 90 filmom obalených tabliet
PVC/Aclar/PCTFE blister z hliníkovej fólie
Veľkosti balenia
267 mg filmom obalené tablety
1 blister s obsahom 21 filmom obalených tabliet (spolu 21)
2 blistre, každý s obsahom 21 filmom obalených tabliet (spolu 42)
4 blistre, každý s obsahom 21 filmom obalených tabliet (spolu 84)
8 blistrov, každý s obsahom 21 filmom obalených tabliet (spolu 168)
Balenie na úvodnú 2-týždňovú liečbu: multibalenie s obsahom 63 (1 balenie s obsahom 1 blister po 21
a 1 balenie s obsahom 2 blistre po 21) filmom obalených tabliet
Balenie na udržiavaciu liečbu: multibalenie s obsahom 252 (3 balenia, každý s obsahom 4 blistre po
21) filmom obalených tabliet
801 mg filmom obalené tablety
4 blistre, každý s obsahom 21 filmom obalených tabliet (spolu 84)
Balenie na udržiavaciu liečbu: multibalenie s obsahom 252 (3 balenia, každý s obsahom 4 blistre po
21) filmom obalených tabliet
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými
požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Roche Registration GmbH Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLAEU/1/11/667/005
EU/1/11/667/006
EU/1/11/667/007
EU/1/11/667/008
EU/1/11/667/009
EU/1/11/667/010
EU/1/11/667/011
EU/1/11/667/012
EU/1/11/667/013
EU/1/11/667/014
EU/1/11/667/015
EU/1/11/667/016
EU/1/11/667/017
EU/1/11/667/018
EU/1/11/667/019
EU/1/11/667/020
EU/1/11/667/021
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 28. februára 2011
Dátum posledného predĺženia registrácie: 08. septembra 2015
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na w ebovej stránke Európskej agentúry pre lieky
http://w w w .ema.europa.eu.