
rubínu, liečba sa musí ukončiť a neodporúča sa jej opätovné začatie.
Ak dôjde k vzostupu hladín ALT alebo AST na > 5-násobok ULN, ktorý nie je spojený s akýmkoľvek vzostupom hladiny bilirubínu, liečba sa má prerušiť. Liečba sa môže opätovne začať dávkou 120 mg podávanou s.c. injekciou raz za štyri týždne, keď sa hladiny ALT a AST vrátia do referenčného rozpätia a posúdi sa pomer prínosu a rizika liečby pre pacienta. Ak sa rozhodne o opätovnom začatí liečby, pečeňové parametre sa musia pozorne sledovať a ak sa zaznamená akékoľvek následné zvýšenie hladín ALT/AST a/alebo bilirubínu, liečba sa musí ukončiť a neodporúča sa jej opätovné začatie (pozri časti 4.4 a 4.8).
Tabuľka 2: Odporúčaná dávka pre opätovné začatie liečby po vzostupe hladín pečeňovýchtransaminázPosledná podaná dávka
| Odporúčaná dávka pre opätovné začatie liečby
|
Pred menej ako
12 týždňami
| Liečba sa má opätovne začať odporúčanou dávkou podávanou raz za
4 týždne.
|
Pred 12 a viac týždňami
| Liečba sa má opätovne začať odporúčanou dávkou podanou
v 0.*, 2. a 4. týždni a potom raz za 4 týždne.
|
* „0. týždeň“ znamená čas prvého podania po opätovnom začatí liečby.
Odporúčanie na úpravu dávky pri neutropéniiAk je počet neutrofilov nižší ako 1,0 x109/l a potvrdený opakovane vykonaným testom, liečba sa má
prerušiť, až kým počet neutrofilov nebude > 1,0 x109/l.
Odporúčanie na úpravu dávky pri nízkom počte krvných doštičiekAk je počet krvných doštičiek nižší ako 75 x109/l a potvrdený opakovane vykonaným testom, liečba sa má prerušiť, až kým počet krvných doštičiek nebude ≥ 75 x109/l.
Osobitné skupiny pacientovPediatrická populáciaDávkovanie u dospievajúcich pacientov vo veku ≥ 12 rokov s telesnou hmotnosťou ≥ 40 kg
a dospelých pacientov je rovnaké (pozri časti 5.1 a 5.2). Bezpečnosť a účinnosť satralizumabu u detí s telesnou hmotnosťou < 40 kg neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
Staršie osobyU pacientov vo veku ≥ 65 rokov nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie obličiekBezpečnosť a účinnosť satralizumabu neboli formálne skúmané u pacientov s poruchou funkcie
obličiek. U pacientov s miernou poruchou funkcie obličiek sa neodporúča žiadna úprava dávky (pozri
časť 5.2).
Porucha funkcie pečeneBezpečnosť a účinnosť satralizumabu neboli skúmané u pacientov s poruchou funkcie pečene.
K dispozícii nie sú žiadne údaje (pozri časť 5.2).
Počas liečby satralizumabom boli pozorované vzostupy hladín pečeňových enzýmov (pozri časti 4.4 a 4.8). Úprava dávky je uvedená vyššie v odseku Odporúčanie na úpravu dávky pri abnormálnych hladinách pečeňových enzýmov.
Spôsob podávania
Satralizumab 120 mg sa podáva s.c. injekciou pomocou jednodávkovej PFS. Má sa podať celý obsah
(1 ml) PFS.
Odporúčané miesta vpichu sú brucho a stehno. Miesta vpichu sa majú striedať a injekcie sa nikdy nemajú podať do materských znamienok, jaziev, ani do miest, kde je koža citlivá, pomliaždená, červená, stvrdnutá alebo porušená.
Podrobné pokyny na podávanie satralizumabu sú uvedené na konci písomnej informácie pre používateľa.
Podanie samotným pacientom a/alebo opatrovateľom
Prvá injekcia musí byť podaná pod dohľadom kvalifikovaného zdravotníckeho pracovníka (healthcare
professional, HCP).
Po náležitej inštruktáži o príprave a podaní injekcie môže dospelý pacient/opatrovateľ podávať všetky ďalšie dávky v domácom prostredí, ak ošetrujúci lekár rozhodne, že je to vhodné a dospelý pacient/opatrovateľ ovláda techniku podávania injekcie.
Pacienti/opatrovatelia majú ihneď vyhľadať lekársku pomoc, ak sa u pacienta objavia príznaky závažných alergických reakcií, a majú si u svojho HCP overiť, či sa v liečbe môže, alebo nemôže pokračovať.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov a číslo
šarže podaného lieku.
Infekcie
U pacientov s aktívnou infekciou má byť podanie satralizumabu odložené, kým sa infekcia dostane
pod kontrolu (pozri časť 4.2).
U pacientov, ktorí dostávajú liečbu satralizumabom, sa odporúča dbať na včasné odhalenie
a diagnostiku infekcie. U pacienta, u ktorého sa rozvinie akákoľvek závažná alebo oportúnna infekcia, má byť liečba odložená a má sa začať vhodná liečba za pozorného sledovania pacienta. Pacienti majú byť poučení, aby v prípade prejavov a príznakov infekcií čo najskôr vyhľadali lekársku pomoc, aby bola možná včasná diagnostika infekcií. Pacientom má byť poskytnutá karta upozornení pre pacienta.
Očkovania
Živé a živé atenuované (oslabené) očkovacie látky sa nemajú podávať súbežne so satralizumabom,
pretože klinická bezpečnosť nebola stanovená. Interval medzi podaním živých očkovacích látok
a začiatkom liečby satralizumabom má byť v súlade s aktuálnymi odporúčaniami na očkovanie
v súvislosti s imunomodulačnými alebo imunosupresívnymi liečivami.
K dispozícii nie sú žiadne údaje o vplyve očkovania u pacientov liečených satralizumabom. Odporúča sa, aby boli všetci pacienti podrobení aktuálne platným imunizáciám v súlade s aktuálnymi odporúčaniami imunizácie ešte pred začatím liečby satralizumabom.
Pečeňové enzýmy
Počas liečby satralizumabom boli pozorované mierne až stredne závažné vzostupy hladín pečeňových
transamináz, tieto vzostupy boli väčšinou pod 5-násobkom ULN (pozri časť 4.8).
Hladiny ALT a AST sa majú kontrolovať raz za štyri týždne počas prvých troch mesiacov liečby, potom raz za tri mesiace počas jedného roka a následne podľa klinickej indikácie.
U pacientov s hladinami ALT alebo AST > 5-násobok ULN sa má liečba satralizumabom ukončiť
(pozri časť 4.2).
Počet neutrofilov
Po liečbe satralizumabom sa vyskytli poklesy počtu neutrofilov (pozri časť 4.8). Počet neutrofilov sa
má skontrolovať po 4 až 8 týždňoch od začiatku liečby a následne podľa klinickej indikácie.
Odporúčania na prerušenie podávania dávky sú uvedené v časti 4.2.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie.
V populačných farmakokinetických (pharmacokinetic, PK) analýzach sa nezistil žiadny vplyv azatioprínu (AZA), perorálnych kortikosteroidov (OC) alebo mofetil-mykofenolátu (MMF) na klírens satralizumabu.
Štúdie vykonané v podmienkach in vitro aj in vivo ukázali, že cytokíny, ako napríklad IL-6, potláčajú
expresiu špecifických pečeňových enzýmov CYP450 (CYP1A2, CYP2C9, CYP2C19 a CYP3A4).
Preto je potrebná opatrnosť, keď sa liečba satralizumabom začína alebo ukončuje u pacientov, ktorí dostávajú aj substráty CYP450 3A4, 1A2, 2C9 alebo 2C19, najmä substráty s úzkym terapeutickým indexom (napríklad warfarín, karbamazepín, fenytoín a teofylín), a v prípade potreby sa majú upraviť ich dávky.
Vzhľadom na predĺžený terminálny polčas satralizumabu môže účinok satralizumabu pretrvávať niekoľko týždňov po ukončení liečby.
4.6 Fertilita, gravidita a laktácia
Gravidita
K dispozícii nie sú žiadne údaje o použití satralizumabu u gravidných žien. Štúdie na opiciach
nepreukázali škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu Enspryngu počas gravidity.
Dojčenie
Nie je známe, či sa satralizumab vylučuje do ľudského materského mlieka. Je známe, že ľudský IgG sa
vylučuje do materského mlieka počas prvých dní po pôrode a následne v krátkom čase klesá na nízke koncentrácie; riziko pre dojčené deti preto nemôže byť vylúčené počas tohto krátkeho obdobia. Po jeho uplynutí sa použitie Enspryngu počas dojčenia môže zvážiť, iba ak je to klinicky potrebné.
Fertilita
K dispozícii nie sú žiadne klinické údaje o vplyve satralizumabu na fertilitu ľudí. Štúdie na zvieratách
nepreukázali žiadnu poruchu samčej alebo samičej fertility (pozri časť 5.3).
4
.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Enspryng nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkySúhrn bezpečnostného profiluNajčastejšie hlásené nežiaduce reakcie boli: bolesť hlavy (19,2 %), artralgia (13,5 %), znížený počet
bielych krviniek (13,5 %), hyperlipidémia (13,5 %) a reakcie súvisiace s podávaním injekcie (12,5 %).
Tabuľkový zoznam nežiaducich reakciíV tabuľke 3 sú zhrnuté nežiaduce reakcie, ktoré boli hlásené v súvislosti s použitím satralizumabu
v monoterapii alebo v kombinácii s IST v klinických skúšaniach.
Nežiaduce reakcie z klinických skúšaní (tabuľka 3) sú uvedené podľa triedy orgánových systémov
MedDRA. Nežiaduce reakcie sú prezentované prostredníctvom počtu nežiaducich udalostí
na 100 pacientorokov a pomocou údajov o frekvencii. Zodpovedajúca kategória frekvencie pre každú nežiaducu reakciu je založená na údajoch o frekvencii a na nasledujúcej konvencii: veľmi časté
(≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000
až < 1/1 000), veľmi zriedkavé (< 1/10 000).
Tabuľka 3: Nežiaduce reakcieTrieda orgánových systémov
| Frekvencia
|
| Veľmi časté
| Časté
|
Poruchy krvi a lymfatického systému
|
| Hypofibrinogenémia
|
Poruchy metabolizmu a výživy
| Hyperlipidémia
|
|
Psychické poruchy
|
| Insomnia
|
Poruchy nervového systému
| Bolesť hlavy
| Migréna
|
Poruchy srdca a srdcovej činnosti
|
| Bradykardia
|
Poruchy ciev
|
| Hypertenzia
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
| Alergická rinitída
|
Poruchy gastrointestinálneho traktu
|
| Gastritída
|
Poruchy kože a podkožného tkaniva
|
| Vyrážka, pruritus
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| Artralgia
| Muskuloskeletálna
stuhnutosť
|
Celkové poruchy a reakcie v mieste podania
| Reakcie súvisiace
s podávaním injekcie
| Periférny edém
|
Laboratórne a funkčné vyšetrenia
| Znížený počet bielych
krviniek
| Znížený počet neutrofilov, znížený počet krvných
doštičiek, zvýšené hladiny
transamináz, zvýšená hladina bilirubínu v krvi, zvýšená telesná hmotnosť
|
Opis vybraných nežiaducich reakcií
Reakcie súvisiace s podávaním injekcie (injection-related reactions, IRR)
IRR hlásené u pacientov liečených satralizumabom boli prevažne mierne až stredne závažné a väčšina
z nich sa vyskytla do 24 hodín po podaní injekcie. Najčastejšie hlásené systémové príznaky boli
hnačka a bolesť hlavy. Najčastejšie hlásené lokálne reakcie v mieste vpichu boli začervenanie, erytém,
pruritus, vyrážka a bolesť.
Telesná hmotnosť
V období dvojito zaslepenej liečby bolo zvýšenie telesnej hmotnosti o ≥ 15 % v porovnaní s telesnou
hmotnosťou na začiatku štúdie pozorované u 3,8 % pacientov liečených satralizumabom
(v monoterapii alebo v kombinácii s IST) v porovnaní s 2,7 % pacientov, ktorí dostávali placebo
(alebo placebo plus IST).
Laboratórne abnormality
Neutrofily
V období dvojito zaslepenej liečby boli poklesy počtu neutrofilov pozorované u 31,7 % pacientov liečených satralizumabom (v monoterapii alebo v kombinácii s IST) v porovnaní s 21,6 % pacientov, ktorí dostávali placebo (alebo placebo plus IST). Poklesy počtu neutrofilov boli väčšinou prechodné alebo občasné.
9,6 % pacientov, ktorí dostávali satralizumab, malo počet neutrofilov pod 1 x 109/l v porovnaní s 5,4 % pacientov, ktorí dostávali placebo (alebo placebo plus IST).
Krvné doštičky
V období dvojito zaslepenej liečby sa poklesy počtu krvných doštičiek (pod 150 x 109/l) vyskytli
u 24,0 % pacientov liečených satralizumabom (v monoterapii alebo v kombinácii s IST) v porovnaní s 9,5 % pacientov, ktorí dostávali placebo alebo placebo plus IST. Znížený počet krvných doštičiek nesúvisel s krvácavými príhodami.
Poklesy počtu krvných doštičiek boli väčšinou prechodné a neboli pod 75 x 109/l.
Pečeňové enzýmy
V období dvojito zaslepenej liečby sa vzostupy hladín ALT alebo AST v uvedenom poradí vyskytli u 27,9 % a 18,3 % pacientov liečených satralizumabom (v monoterapii alebo v kombinácii s IST)
v porovnaní s 12,2 % a 13,5 % pacientov, ktorí dostávali placebo alebo placebo plus IST. Tieto vzostupy boli väčšinou pod 3-násobkom ULN, boli prechodné a upravili sa bez prerušenia liečby satralizumabom.
Vzostupy hladín ALT alebo AST na > 3-násobok ULN sa v uvedenom poradí vyskytli u 2,9 %
a 1,9 % pacientov liečených satralizumabom (v monoterapii alebo v kombinácii s IST). Tieto vzostupy neboli spojené so zvýšeniami hladiny celkového bilirubínu.
U jedného (1 %) pacienta, ktorý dostával satralizumab v kombinácii s IST, sa pozorovali vzostupy hladiny ALT nad 5-násobok ULN po 4 týždňoch od začatia liečby; po ukončení liečby sa hladina ALT normalizovala a u tohto pacienta nebola liečba satralizumabom opätovne začatá (pozri časti 4.2 a 4.4).
Lipidové parametre
V období dvojito zaslepenej liečby došlo u 10,6 % pacientov, ktorí dostávali satralizumab
(v monoterapii alebo v kombinácii s IST), k vzostupom hladiny celkového cholesterolu nad
7,75 mmol/l v porovnaní s 1,4 % pacientov, ktorí dostávali placebo (alebo placebo plus IST);
u 20,2 % pacientov, ktorí dostávali satralizumab, došlo k vzostupom hladín triacylglycerolov nad
3,42 mmol/l v porovnaní s 10,8 % pacientov, ktorí dostávali placebo.
Pediatrická populácia
Bezpečnosť a účinnosť satralizumabu boli skúmané u 9 detí vo veku ≥ 12 rokov. Predpokladá sa, že
frekvencia, typ a závažnosť nežiaducich reakcií u detí vo veku od 12 rokov sú rovnaké ako u dospelých.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieV prípade predávkovania má byť pacient pozorne sledovaný, symptomaticky liečený a ak je to
potrebné, majú sa začať podporné opatrenia.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: imunosupresíva, inhibítory interleukínu, ATC kód: L04AC19
Mechanizmus účinkuSatralizumab je rekombinantná humanizovaná monoklonálna protilátka (
monoclonal antibody, mAb)
podtriedy imunoglobulínu G2 (IgG2), ktorá sa viaže na rozpustný aj na membránovo-viazaný ľudský receptor pre interleukín-6 (IL-6R), a tým zabraňuje downstreamovej signalizácii IL-6 sprostredkovanej týmito receptormi.
Hladiny IL-6 sú zvýšené v likvore (mozgovomiechovom moku) a v sére pacientov s NMO
a s NMOSD počas fáz aktivity ochorenia. Funkcie IL-6 sa podieľajú na patogenéze NMO a NMOSD vrátane aktivácie B-lymfocytov, diferenciácie B-lymfocytov na plazmoblasty a produkcie patologických autoprotilátok, napr. proti AQP4, čo je proteín, ktorý vytvára vodný kanál v CNS
a ktorý je exprimovaný hlavne astrocytmi, aktivácie a diferenciácie Th17 lymfocytov, inhibície
regulačných T-lymfocytov a zmien priepustnosti hematoencefalickej bariéry.
Farmakodynamické účinkyV klinických štúdiách so satralizumabom u pacientov s NMO a s NMOSD boli pozorované poklesy
hladín C-reaktívneho proteínu (CRP), fibrinogénu a komplementu (zložiek C3, C4 a hemolytickej aktivity komplementu - CH50).
Klinická účinnosť a bezpečnosťÚčinnosť a bezpečnosť satralizumabu boli hodnotené v dvoch pivotných klinických skúšaniach
fázy III u pacientov s NMOSD (diagnostikovanou ako AQP4-IgG séropozitívna alebo séronegatívna
NMO, t. j. neuromyelitis optica s pozitivitou alebo negativitou protilátok proti akvaporínu-4 (AQP4-IgG) v sére [Wingerchuckove kritériá z roku 2006], alebo ako AQP4-IgG séropozitívne NMOSD, t. j. ochorenia zo spektra neuromyelitis optica s pozitivitou protilátok proti akvaporínu-4 (AQP4-IgG) v sére [Wingerchuckove kritériá z roku 2007]).
Štúdia BN40898 zahŕňala dospelých a dospievajúcich pacientov s NMOSD vo veku 12 - 74 rokov, ktorí boli liečení stabilnou IST, mali aspoň 2 relapsy v posledných 2 rokoch pred skríningom (aspoň jeden relaps v priebehu 12 mesiacov pred skríningom) a skóre EDSS (Expanded Disability Status Scale – rozšírená škála funkčnej nespôsobilosti) 0 až 6,5; zatiaľ čo štúdia BN40900 zahŕňala dospelých pacientov vo veku 18 – 74 rokov, ktorí nedostávali IST ako základnú liečbu, mali aspoň
1 relaps alebo prvý atak v priebehu posledných 12 mesiacov pred skríningom a skóre EDSS 0 až 6,5.
Obidve štúdie zahŕňali približne 30 % AQP4-IgG séronegatívnych pacientov s NMO.
Účinnosť sa v obidvoch štúdiách hodnotila na základe času do výskytu prvého relapsu potvrdeného nezávislým výborom pre posudzovanie klinických cieľových ukazovateľov (Clinical Endpoint Committee, CEC), pričom relaps bol definovaný vopred špecifikovaným zhoršením skóre EDSS
a skóre funkčných systémov (Functional System Score, FSS), hodnotených do 7 dní po tom, ako pacient nahlásil príznaky (potvrdený [adjudicated] relaps).
Štúdia BN40898 (známa aj ako SA-307JG alebo SAkuraSky)
Štúdia BN40898 bola randomizované, multicentrické, dvojito zaslepené, placebom kontrolované
klinické skúšanie hodnotiace účinok satralizumabu v kombinácii so stabilnou IST (perorálne kortikosteroidy [oral corticosteroids, OC] v dávke až do 15 mg/deň [ekvivalent prednizolónu], azatioprín [AZA] v dávke až do 3 mg/kg/deň alebo mofetil-mykofenolát [MMF] v dávke
až do 3 000 mg/deň; dospievajúci dostávali kombináciu AZA a OC alebo MMF a OC). Dvojito zaslepená fáza štúdie zahŕňala 83 AQP4-IgG séropozitívnych a séronegatívnych pacientov
(76 dospelých a 7 dospievajúcich). Pacientom boli prvé 3 jednotlivé dávky satralizumabu 120 mg alebo zodpovedajúceho placeba podané subkutánnou (s.c.) injekciou do oblasti brucha alebo stehna raz za 2 týždne počas prvých 4 týždňov a potom im bol skúšaný liek podávaný raz za 4 týždne. Dizajn štúdie a základné charakteristiky skúmanej populácie sú uvedené v tabuľke 4.
Tabuľka 4: Dizajn štúdie a základné charakteristiky AQP4-IgG séropozitívnych pacientov v štúdii BN40898
Názov štúdie
|
Štúdia BN40898
(AQP4-IgG séropozitívni: N = 55; ITT*: N = 83)
|
Dizajn štúdie
|
Skúmaná populácia
|
Dospievajúci a dospelí pacienti s NMO alebo s NMOSD,
liečení stabilnou IST
Vek 12 – 74 rokov, ≥ 2 relapsy v posledných 2 rokoch pred skríningom (s aspoň jedným relapsom v priebehu
12 mesiacov pred skríningom), skóre EDSS 0 až 6,5
|
Dĺžka trvania štúdie
na zhodnotenie účinnosti
|
„Event-driven“** (26 potvrdených relapsov) Medián obdobia sledovania: satralizumab 139,4 týždňa, placebo 40,2 týždňa (v ITT: 115,1 týždňa a 42,5 týždňa
v uvedenom poradí)
|
Liečebné skupiny, randomizácia v pomere 1:1
|
Skupina A: satralizumab 120 mg s.c.
Skupina B: placebo
|
Z
á
kladné charakteristiky AQP4-IgG séropozitívnych pacientov
|
Satralizumab + IST (n = 27)
|
Placebo + IST (n = 28)
|
Diagnóza, n (%): NMO
NMOSD
|
19 (70,4)
8 (29,6)
|
14 (50,0)
14 (50,0)
|
Priemerný vek v rokoch (SD) (min. – max.)
|
44,4 (15,7) (13 – 73)
|
43,4 (12,9) (14 – 65)
|
Staršie osoby (≥ 65 rokov), n (%)
|
3 (11,1)
|
1 (3,6)
|
Dospievajúci (≥ 12 až < 18 rokov), n (%)
|
1 (3,7)
|
2 (7,1)
|
Distribúcia podľa pohlavia, n (%) muži / n (%) ženy
|
0 / 27 (100)
|
0 / 28 (100)
|
Imunosupresívna terapia (IST), n (%):
perorálne kortikosteroidy (OC)
azatioprín (AZA)
mofetil-mykofenolát (MMF) AZA + OC***
MMF + OC***
|
14 (51,9)
11 (40,7)
1 (3,7)
0
1 (3,7)
|
13 (46,4)
11 (39,3)
3 (10,7)
0
1 (3,6)
|
* Populácia všetkých randomizovaných pacientov (
Intention-To-Treat, ITT)
** („Event-driven“ – dĺžka trvania štúdie bola závislá od dosiahnutia vopred určeného počtu sledovanej udalosti, v tomto prípade potvrdeného relapsu.) Pacienti, ktorí boli liečení záchrannou liečbou a u ktorých sa nevyskytol žiadny potvrdený relaps, mohli byť zaradení do otvorenej predĺženej (
open label extension, OLE) fázy štúdie
a boli cenzurovaní v primárnej analýze účinnosti.
*** Kombinácia povolená u dospievajúcich pacientov.
Š
túdia BN40900 (známa aj ako SA-309JG alebo SAkuraStar)
Štúdia BN40900 bola randomizované, multicentrické, dvojito zaslepené, placebom kontrolované
klinické skúšanie hodnotiace účinok satralizumabu v monoterapii v porovnaní s placebom. Štúdia
zahŕňala 95 AQP4-IgG séropozitívnych a séronegatívnych dospelých pacientov. Pacientom boli prvé
3 jednotlivé dávky satralizumabu 120 mg alebo zodpovedajúceho placeba podané subkutánnou (s.c.) injekciou do oblasti brucha alebo stehna raz za 2 týždne počas prvých 4 týždňov a potom im bol skúšaný liek podávaný raz za 4 týždne.
Dizajn štúdie a základné charakteristiky skúmanej populácie sú uvedené v tabuľke 5.
Tabuľka 5: Dizajn štúdie a základné charakteristiky AQP4-IgG séropozitívnych pacientov v štúdii BN40900
Názov štúdie
| Štúdia BN40900
(AQP4-IgG séropozitívni: N = 64; ITT*: N = 95)
|
Dizajn štúdie
|
Skúmaná populácia
| Dospelí pacienti s NMO alebo s NMOSD
Vek 18 – 74 rokov, ≥ 1 relaps alebo prvý atak v priebehu posledných 12 mesiacov pred skríningom, skóre EDSS 0 až 6,5. Pacienti buď dostali predchádzajúcu liečbu zameranú na prevenciu relapsu NMOSD, alebo nedostali žiadnu predchádzajúcu liečbu.
|
Dĺžka trvania štúdie
na zhodnotenie účinnosti
| „Event-driven“ (44 potvrdených relapsov, alebo 1,5 roka
po dátume randomizácie posledného zaradeného pacienta, podľa
toho, k čomu došlo skôr)
Medián obdobia sledovania: satralizumab 96,7 týždňa, placebo 60,1 týždňa (v ITT: 95,4 týždňa a 60,5 týždňa v uvedenom poradí)
|
Liečebné skupiny,
randomizácia v pomere 2:1
| Monoterapia:
Skupina A: satralizumab 120 mg s.c.
Skupina B: placebo
|
Základné charakteristiky AQP4-IgG séropozitívnych pacientov
| Satralizumab (n = 41)
| Placebo (n = 23)
|
Diagnóza, n (%): NMO
NMOSD
|
26 (63,4)
15 (36,6)
|
15 (65,2)
8 (34,8)
|
Priemerný vek v rokoch
(SD)
(min. – max.)
|
46,0 (12,0) (22 – 70)
|
40,1 (11,5) (20 – 56)
|
Staršie osoby (≥ 65 rokov), n (%)
|
1 (2,4)
|
0
|
Distribúcia podľa pohlavia,
n (%) muži / n (%) ženy
|
10 (24,4) / 31 (75,6)
|
1 (4,3) / 22 (95,7)
|
* Populácia všetkých randomizovaných pacientov (
Intention-To-Treat, ITT)'
Primárna
ú
činnosť
U AQP4-IgG séropozitívnych pacientov sa relatívne riziko výskytu potvrdeného relapsu znížilo
o 79 % (pomer rizík (
Hazard Ratio, HR) [95 % IS]: 0,21 [0,06 – 0,75]) v štúdii BN40898 a o 74 % (HR [95 % IS]: 0,26 [0,11 – 0,63]) v štúdii BN40900 (pozri grafy 1 a 2). Keď boli údaje zo štúdií BN40898 a BN40900 združené, zistilo sa, že liečba satralizumabom s IST alebo bez IST viedla
k zníženiu celkového rizika o 75 % (HR [95 % IS]; 0,25 (0,12 – 0,50]) u AQP4-IgG séropozitívnych pacientov. V 48. týždni bolo 85,7 % AQP4-IgG séropozitívnych pacientov liečených satralizumabom ešte stále bez potvrdeného relapsu, keď sa používal v kombinácii s IST alebo v monoterapii,
v porovnaní s 58,7 % pacientov v skupine s placebom. V 96. týždni bolo 81,4 % AQP4-IgG séropozitívnych pacientov liečených satralizumabom ešte stále bez potvrdeného relapsu, keď sa používal v kombinácii s IST alebo v monoterapii, v porovnaní so 47,2 % pacientov v skupine
s placebom. Účinnosť nebola významná u AQP4-IgG séronegatívnych pacientov.
Graf 1: Štúdia BN40898 – čas do výskytu prvého potvrdeného relapsu počas dvojitozaslepenej fázy u AQP4-IgG séropozitívnych pacientov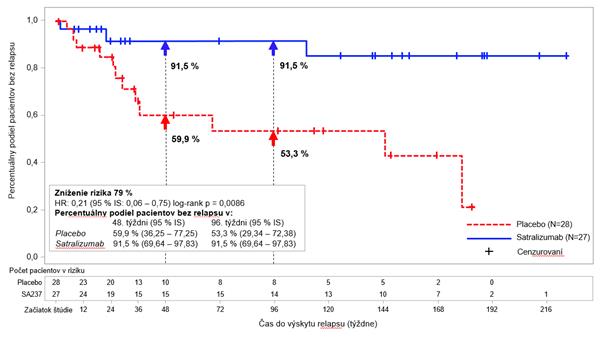
Graf 2: Štúdia BN40900 – čas do výskytu prvého potvrdeného relapsu počas dvojito
z
a
slepenej fázy u AQP4-IgG séropozitívnych pacientov
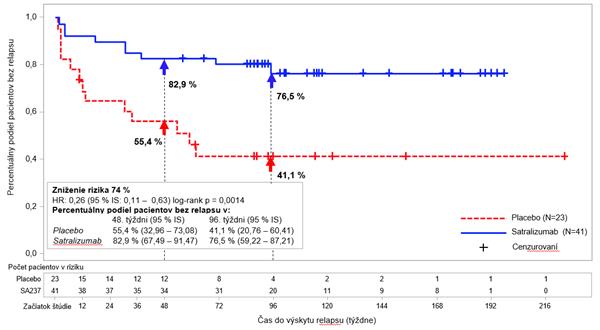
Liečba satralizumabom u AQP4-IgG séropozitívnych pacientov znížila ročný výskyt potvrdených
relapsov (
annualized rate of adjudicated relapses, ARR) o 88 % (pomer výskytu [
rate ratio, RR] = 0,122, 95 % IS: 0,027 – 0,546; p = 0,0039) v štúdii BN40898 a o 90 % (RR = 0,096,
95 % IS: 0,020 – 0,473; p = 0,0086) v štúdii BN40900 v porovnaní s liečbou placebom.
U AQP4-IgG séropozitívnych pacientov liečených satralizumabom bola v porovnaní s pacientmi liečenými placebom potreba záchrannej liečby (napr. kortikosteroidy, intravenózny imunoglobulín a/alebo aferéza [vrátane plazmaferézy alebo výmeny plazmy]) znížená o 61 % (pomer šancí [
odds ratio, OR] = 0,3930, 95 IS: 0,1343 – 1,1502; p = 0,0883) v štúdii BN40898 a o 74 % (OR = 0,2617,
95 % IS: 0,0862 – 0,7943; p = 0,0180) v štúdii BN40900.
Liečba satralizumabom u AQP4-IgG séropozitívnych pacientov znížila riziko výskytu závažného relapsu definovaného zvýšením skóre EDSS o ≥ 2 body oproti skóre EDSS pri predchádzajúcom hodnotení o 85 % (čas do výskytu prvého závažného potvrdeného relapsu počas dvojito zaslepenej fázy, HR = 0,15, 95 % IS: 0,02 – 1,25; p = 0,0441) v štúdii BN40898 a o 79 % (HR = 0,21,
95 % IS: 0,05 – 0,91; p = 0,0231) v štúdii BN40900 v porovnaní s liečbou placebom.
Kľúčové sekundárne cieľové ukazovateleZmena v bolesti a únave v 24. týždni v porovnaní s východiskovým stavom nebola dosiahnutá
v štúdiách BN40898 a BN40900.
Otvorená predĺžená fázaAnalýzy dlhodobejších údajov zahŕňajúcich OLE fázu (na základe relapsu liečeného záchrannou
liečbou) ukázali, že po 120 týždňoch liečby bolo 58 % a 73 % AQP4-IgG séropozitívnych pacientov liečených satralizumabom ešte stále bez relapsu, keď sa satralizumab podával ako prídavná liečba alebo v monoterapii, v uvedenom poradí.
I
m
unog
enita
V štúdii fázy III, BN40898 (kombinácia s IST), a v štúdii fázy III, BN40900 (v monoterapii) sa
protilátky proti lieku (anti-drug antibodies, ADA) v uvedenom poradí zaznamenali
u 41 % a 71 % pacientov, ktorí dostávali satralizumab v dvojito zaslepenej fáze. Schopnosť ADA
neutralizovať väzbu satralizumabu nie je známa.
Expozícia bola nižšia u ADA pozitívnych pacientov, avšak nezistil sa žiadny vplyv ADA
na bezpečnosť a žiadny zreteľný vplyv na účinnosť či na farmakodynamické markery svedčiace
o interakcii medzi liečivom a cieľovou štruktúrou (target engagement).
Liečba satralizumabom viedla k podobnému zníženiu rizika výskytu potvrdeného relapsu u pacientov v štúdiách fázy III aj napriek rozdielnemu výskytu ADA medzi týmito štúdiami.
Pediatrická populácia
V štúdii BN40898 bolo zaradených 7 dospievajúcich pacientov počas dvojito zaslepenej fázy.
Ich priemerný vek bol 15,4 roka a medián telesnej hmotnosti bol 79,6 kg. Väčšina pacientov bola ženského pohlavia (n = 6). Štyria pacienti boli belosi, 2 boli černosi/Afroameričania a 1 bol Ázijec. Traja (42,9 %) dospievajúci pacienti boli AQP4-IgG séropozitívni pri skríningu (2 v skupine
s placebom a 1 v skupine so satralizumabom). Počas dvojito zaslepenej fázy sa potvrdený relaps
vyskytol u 1 z 3 dospievajúcich v skupine s placebom a u 1 zo 4 dospievajúcich v skupine
so satralizumabom. Vzhľadom na malú veľkosť vzorky sa pomer rizík pre primárny cieľový ukazovateľ, ktorým bol čas do výskytu prvého potvrdeného relapsu, v tejto podskupine nevypočítal. Dvaja ďalší dospievajúci pacienti boli zaradení do otvorenej (open-label) fázy štúdie.
Európska agentúra pre lieky udelila odklad z povinnosti predložiť výsledky štúdií s Enspryngom v jednej alebo vo viacerých podskupinách pediatrickej populácie pri liečbe NMOSD (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika satralizumabu bola charakterizovaná u zdravých dobrovoľníkov japonského
a belošského (kaukazského) pôvodu, ako aj u pacientov s NMO a s NMOSD. Farmakokinetika
u pacientov s NMO a s NMOSD, ktorým bola podávaná odporúčaná dávka, bola charakterizovaná pomocou metód populačných PK analýz založených na databáze 154 pacientov.
Priebeh krivky závislosti koncentrácie satralizumabu od času u pacientov s NMO alebo s NMOSD bol
presne opísaný pomocou dvojkompartmentového populačného PK modelu s paralelnou lineárnou
a cieľovou štruktúrou sprostredkovanou (Michaelisovou-Mentenovej) elimináciou a s.c. absorpciou prvého poriadku. Parametre klírensu a distribučného objemu satralizumabu boli alometricky škálované podľa telesnej hmotnosti (prostredníctvom funkcie výkonu [power function] s fixným koeficientom výkonu rovným 0,75 pre parametre klírensu a 1 pre parametre distribučného objemu). Preukázalo sa,
že telesná hmotnosť je významným kovariantom, pričom u pacientov vážiacich 123 kg (97,5. percentil
distribúcie podľa telesnej hmotnosti) v porovnaní so 60 kg pacientom bol klírens zvýšený o 71,3 %
a Vc o 105 %.
Rovnovážna farmakokinetika sa dosiahla po období podávania nasycovacej dávky (8 týždňov)
a hodnoty Cmin, Cmax a AUC v rovnovážnom stave boli takéto (priemer (±SD)): Cmin: 19,7 (12,2) µg/ml, Cmax: 31,5 (14,9) µg/ml a AUC: 737 (386) µg.ml/deň.
Absorpcia
Rýchlostná konštanta absorpcie satralizumabu bola 0,0104/h a zodpovedala polčasu absorpcie
približne 3 dni (66 hodín) pri podávaní odporúčanej dávky (pozri časť 4.2). Biologická dostupnosť
bola vysoká (85,4 %).
Distribúcia
Satralizumab podlieha dvojfázovej distribúcii. Distribučný objem v centrálnom kompartmente bol
3,46 l a distribučný objem v periférnom kompartmente bol 2,07 l. Klírens medzi kompartmentami bol
14 ml/h.
Biotransformácia
Metabolizmus satralizumabu sa priamo neskúmal, pretože protilátky podliehajú výlučne katabolizmu.
Eliminácia
Celkový klírens satralizumabu je závislý od koncentrácie. Lineárny klírens (predstavujúci približne
polovicu z celkového klírensu v rovnovážnom stave pri podávaní odporúčanej dávky pacientom
s NMO a s NMOSD) je odhadnutý na 2,50 ml/h. Súvisiaci terminálny polčas t1/2 je približne 30 dní
(rozmedzie 22 – 37 dní) na základe údajov združených zo štúdií fázy 3.
Osobitné skupiny pacientov
Populačné farmakokinetické analýzy u dospelých pacientov s NMO alebo s NMOSD ukázali, že vek,
pohlavie a rasa významne neovplyvnili farmakokinetiku satralizumabu. I keď telesná hmotnosť ovplyvnila farmakokinetiku satralizumabu, neodporúčajú sa žiadne úpravy dávky v súvislosti s týmito demografickými charakteristikami.
Pediatrická populácia
Údaje získané u 8 dospievajúcich pacientov (vo veku 13 – 17 rokov), ktorí dostávali dávkovaciu
schému pre dospelých, ukázali, že populačné PK parametre satralizumabu sa významne nelíšia od tých, ktoré sa pozorovali v populácii dospelých. Preto nie je potrebná žiadna úprava dávky.
Staršie osoby
Neuskutočnili sa žiadne štúdie zamerané na preskúmanie PK satralizumabu u pacientov
vo veku ≥ 65 rokov, avšak pacienti s NMO alebo s NMOSD vo veku medzi 65 a 74 rokmi boli zahrnutí do klinických štúdií BN40898 a BN40900.
Porucha funkcie obličiek
Neuskutočnila sa žiadna formálna štúdia vplyvu poruchy funkcie obličiek na PK satralizumabu. Avšak
pacienti s miernou poruchou funkcie obličiek (klírens kreatinínu ≥ 50 ml/min a < 80 ml/min) boli zahrnutí do štúdií fázy III. Na základe populačnej PK analýzy nemá porucha funkcie obličiek žiadny vplyv na PK satralizumabu, čo je v súlade so známymi mechanizmami klírensu satralizumabu. Preto nie je potrebná žiadna úprava dávky.
Porucha funkcie pečene
Neuskutočnila sa žiadna formálna štúdia vplyvu poruchy funkcie pečene na PK satralizumabu (pozri
časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Predklinické údaje získané na základe obvyklých farmakologických štúdií bezpečnosti, toxicity
po opakovanom podávaní a reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí.
Karcinogenita
Neuskutočnili sa žiadne štúdie karcinogenity na hlodavcoch stanovujúce karcinogénny potenciál
satralizumabu. V 6-mesačnej štúdii chronickej toxicity na opiciach rodu Cynomolgus sa nepozorovali žiadne proliferujúce lézie.
Genotoxicita
Neuskutočnili sa žiadne štúdie stanovujúce mutagénny potenciál satralizumabu. Nepredpokladá sa, že
protilátky spôsobujú účinky na DNA.
Reprodukčná toxicita
Podávanie satralizumabu v prenatálnom období a postnatálna expozícia satralizumabu u gravidných
opíc a ich potomkov nespôsobili žiadne nežiaduce účinky na zvieracie matky, vývin plodu, výsledok gravidity ani na prežívanie a vývin mláďat vrátane schopnosti učenia sa.
Koncentrácie satralizumabu v materskom mlieku boli veľmi nízke (< 0,9 % zodpovedajúcich plazmatických hladín u matky).
Fertilita
U opíc sa počas dlhodobej liečby satralizumabom nepozorovali žiadne účinky na reprodukčné orgány
samcov alebo samíc.
Syndróm uvoľňovania cytokínov
Na základe in vitro štúdií s ľudskou krvou sa riziko uvoľňovania prozápalových cytokínov počas
liečby satralizumabom považuje za nízke v zmysle výskytu a zvýšenia hladín cytokínov.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
histidín
kyselina asparágová arginín
poloxamér 188
voda na injekcie
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
2 roky
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2 °C – 8 °C).
Neuchovávajte v mrazničke. Nepoužite injekčnú striekačku, ak zmrzla.
Injekčnú striekačku vždy uchovávajte v suchu.
PFS uchovávajte vo vonkajšej škatuľke na ochranu pred svetlom a vlhkosťou.
Ak injekčná striekačka ešte nebola otvorená a uchováva sa vonkajšej škatuľke, môže byť ponechaná mimo chladničky pri teplote do 30 °C počas jedného obdobia trvajúceho najviac 8 dní. Po uchovávaní pri izbovej teplote sa liek nemá vrátiť späť do chladničky a má sa buď použiť, alebo zlikvidovať.
6.5 Druh obalu a obsah balenia1 ml roztoku v PFS (polymér) s integrovanou ihlou z nehrdzavejúcej ocele, ktorá je vybavená pevným krytom ihly z chlórovaného butylkaučuk-polyprolylénu a uzatvorená chlórovaným butylkaučukovým vrchnákom. PFS je označená a pozostáva z automatického ochranného krytu ihly, piesta a predĺžených úchytiek na prsty (
extended finger flanges, EFF).
Veľkosť balenia po 1 PFS a multibalenie 3 (3 balenia po 1) PFS. Na trh nemusia byť uvedené všetky
veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekomPo vybratí škatuľky z chladničky sa má zapečatená škatuľka otvoriť a PFS sa má uchopiť za valec a opatrne vybrať zo škatuľky. Je dôležité nechať PFS dosiahnuť izbovú teplotu tak, že sa počká
30 minút predtým, ako sa liek začne podávať.
Liek sa nemá použiť, ak je tekutina zakalená, má zmenenú farbu, obsahuje viditeľné častice alebo
ak ktorákoľvek časť PFS javí známky poškodenia.
Injekcia sa musí podať ihneď po odstránení vrchnáka a najneskôr do 5 minút, aby sa zabránilo vysušeniu lieku a upchatiu ihly. Ak sa naplnená injekčná striekačka nepoužije do 5 minút
po odstránení vrchnáka, musíte ju vyhodiť do nádoby odolnej proti prepichnutiu a použiť novú naplnenú injekčnú striekačku.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIRoche Registration GmbH Emil-Barell-Strasse 1
79639 Grenzach-Wyhlen
Nemecko
8. REGISTRAČNÉ ČÍSLOEU/1/21/1559/001
EU/1/21/1559/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.