
ie Enjayma a začnite vhodnú liečbu. Monitorujte pacienta minimálne dve hodiny po ukončení prvého podania infúzie z dôvodu výskytu prejavov alebo symptómov reakcie na infúziu a/alebo reakcie z precitlivenosti. Monitorujte pacienta jednu hodinu po ukončení nasledujúcich infúzií z dôvodu výskytu prejavov alebo symptómov reakcie na infúziu.
Podávanie infúzie v domácom prostredí
Podávanie infúzie v domácom prostredí má vykonať zdravotnícky pracovník.
Rozhodnutie zvážiť podávanie infúzie v domácom prostredí sa má zakladať na individuálnych klinických charakteristikách pacienta a individuálnych potrebách pacienta. Prechod z podávania
infúzie v klinickom zariadení na podávanie v domácom prostredí zahŕňa zabezpečenie adekvátnej
infraštruktúry a zdrojov, ktoré sú v súlade s pokynmi ošetrujúceho lekára. Podávanie infúzie Enjayma v domácom prostredí možno zvážiť u pacientov, ktorí dobre znášali podávanie infúzie v klinickom zariadení a nemali reakcie súvisiace s podávaním infúzie. Pri hodnotení vhodnosti pacienta na podávanie infúzie v domácom prostredí je potrebné zvážiť základné komorbidity pacienta a jeho schopnosť dodržiavať požiadavky podávania infúzie v domácom prostredí. Okrem toho sa majú zvážiť tieto kritériá:
• Pacient nesmie mať žiadne pretrvávajúce súbežné ochorenie, ktoré podľa názoru lekára môže pacienta vystaviť vyššiemu riziku pri podávaní infúzie v domácom prostredí ako pri podávaní infúzie v klinickom zariadení. Pred začatím podávania infúzie v domácom prostredí sa má vykonať komplexné zhodnotenie, aby sa zabezpečilo, že pacient je zdravotne stabilný.
• Pacient musel úspešne dostávať infúziu Enjayma v klinickom zariadení (nemocnica alebo ambulancia) minimálne tri mesiace pod dohľadom lekára alebo poskytovateľa zdravotnej starostlivosti, ktorý má skúsenosti s liečbou pacientov s CAD.
• Pacient musí byť ochotný a schopný dodržiavať postupy podávania infúzie v domácom prostredí a odporúčania ošetrujúceho lekára alebo poskytovateľa zdravotnej starostlivosti.
• Zdravotnícky pracovník, ktorý podáva infúziu v domácom prostredí, má byť k dispozícii celý čas počas podávania infúzie v domácom prostredí a minimálne 1 hodinu po podaní infúzie.
Ak sa u pacienta počas podávania infúzie v domácom prostredí vyskytnú nežiaduce reakcie, proces podávania infúzie sa má okamžite zastaviť, má sa začať vhodná medikamentózna liečba (pozri
časť 4.4) a má byť o tom informovaný ošetrujúci lekár. V takýchto prípadoch má ošetrujúci lekár rozhodnúť, či sa majú podať ďalšie infúzie, a ak áno, či sa majú infúzie podávať v nemocnici alebo
pod dohľadom v zariadení ambulantnej zdravotnej starostlivosti.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.4 Osobitné upozornenia a opatrenia pri používaní
Sledovateľnosť
Aby sa zlepšila (do)sledovateľnosť biologického lieku, má sa zrozumiteľne zaznamenať názov
a číslo šarže podaného lieku.
Infekcie
Enjaymo je zacielené na klasickú cestu komplementu (complement pathway, CP), a špecificky sa
viaže na proteínovú zložku komplementu 1, podzložku s (C1s), ktorá bráni štiepeniu proteínu
komplementu C4. Hoci lektínová a alternatívne cesty nie sú ovplyvnené, pacienti môžu mať zvýšenú náchylnosť na závažné infekcie, najmä infekcie spôsobené opuzdrenými baktériami, ako sú Neisseria meningitides, Streptococcus pneumoniae a Haemophilus influenza. Pred začatím liečby Enjaymom majú byť pacienti zaočkovaní proti opuzdreným baktériám, pozri nižšie „Očkovania“.
V klinických štúdiách s CAD sa u pacientov liečených Enjaymom hlásili závažné infekcie vrátane sepsy (pozri časť 4.8). Enjaymo sa nemá podávať pacientom s aktívnymi závažnými infekciami. Pacienti majú byť monitorovaní na skoré prejavy a symptómy infekcií a majú byť informovaní, aby v prípade výskytu takýchto symptómov okamžite vyhľadali lekársku pomoc.
Pacienti s vírusovou hepatitídou a HIV boli z klinických štúdií vylúčení. Pred liečbou a počas liečby musia pacienti informovať svojho lekára, ak im diagnostikovali hepatitídu B, hepatitídu C alebo infekciu HIV. Pri liečbe pacientov s hepatitídou B, hepatitídou C alebo infekciou HIV v anamnéze sa vyžaduje opatrnosť.
Očkovania
Pacientov očkujte podľa najaktuálnejších miestnych odporúčaní pre pacientov s pretrvávajúcimi deficitmi komplementu vrátane očkovacích látok proti meningokokom a streptokokom. Pacientov preočkujte v súlade s miestnymi odporúčaniami.
Minimálne 2 týždne pred podaním prvej dávky Enjayma imunizujte pacientov bez očkovania proti opuzdreným baktériám v anamnéze. Ak je indikovaná urgentná liečba Enjaymom u neočkovaného pacienta, očkovaciu látku (očkovacie látky) mu podajte čo najskôr. Prínosy a riziká antibiotickej profylaxie na prevenciu infekcií u pacientov liečených Enjaymom neboli stanovené.
Reakcie z precitlivenosti
Tak ako pri iných liekoch s obsahom proteínov aj pri podávaní Enjayma sa môžu objaviť reakcie z
precitlivenosti vrátane anafylaxie. V klinických štúdiách sa po podaní Enjayma nepozorovali žiadne závažné reakcie z precitlivenosti. Ak sa vyskytnú reakcie z precitlivenosti, prerušte podávanie
Enjayma a začnite vhodnú liečbu.
Reakcie súvisiace spodávaníminfúzie
Podávanie Enjayma môže mať počas podávania infúzie alebo bezprostredne po podaní infúzie za
následok vznik reakcií súvisiacich s podávaním infúzie (pozri časť 4.8). U pacientov sa majú monitorovať reakcie súvisiace s podávaním infúzie, a ak sa reakcia objaví, infúzia sa má prerušiť a
má sa začať vhodná liečba.
Systémový lupuserythematosus(SLE)
Jedinci s dedičným deficitom klasickej cesty komplementu majú vyššie riziko vzniku SLE. Pacienti
so SLE boli z klinických štúdií s Enjaymom vylúčení. U pacientov liečených Enjaymom sa majú monitorovať prejavy a symptómy SLE a majú sa primerane vyhodnotiť. Enjaymo používajte
s opatrnosťou u pacientov so SLE aj u pacientov, u ktorých sa rozvinú prejavy a symptómy SLE.
Monitorovanie prejavov CADpoprerušeníliečbyEnjaymom
Účinky na hemolýzu po ukončení liečby slabnú. V prípade prerušenia liečby je preto potrebné u
pacientov monitorovať prejavy a symptómy hemolýzy.
Sodík
Tento liek obsahuje 3,5 mg na ml alebo 77 mg sodíka na injekčnú liekovku, čo zodpovedá 3,85 %
WHO odporúčaného maximálneho denného príjmu 2 g sodíka pre dospelú osobu.
4.5 Liekové a iné interakcie
Neuskutočnili sa žiadne interakčné štúdie. Nie je pravdepodobné, že by bolo Enjaymo kandidátom pre liekové interakcie sprostredkovaných cytochrómom P450, keďže ide o rekombinantný ľudský proteín. Interakcia sutimlimabu so substrátmi CYP sa neskúmala. Sutimlimab však u pacientov znižuje hladiny prozápalových cytokínov, ako je IL-6, o ktorom je známe, že potláča expresiu špecifických pečeňových enzýmov CYP450 (CYP1A2, CYP2C9, CYP2C19 a CYP3A4). Preto je potrebná opatrnosť pri začatí alebo ukončovaní liečby sutimlimabom u pacientov, ktorí dostávajú aj substráty CYP450 3A4, 1A2, 2C9 alebo 2C19, predovšetkým pri liekoch s úzkym terapeutickým indexom (ako je warfarín, karbamazepín, fenytoín a teofylín), a dávky sa majú podľa potreby upraviť.
4.6 Fertilita, gravidita a laktácia
Gravidita
Nie sú k dispozícii údaje o použití sutimlimabu u gravidných žien. Štúdie na zvieratách nepreukázali
priame alebo nepriame škodlivé účinky z hľadiska reprodukčnej toxicity (pozri časť 5.3).
Je známe, že ľudské protilátky IgG prechádzajú placentárnou bariérou; preto sa sutimlimab môže preniesť z matky na vyvíjajúci sa plod.
Ako preventívne opatrenie je vhodnejšie vyhnúť sa používaniu sutimlimabu počas gravidity. Sutimlimab sa má podávať počas gravidity len vtedy, ak je to jednoznačne indikované.
DojčenieJe známe, že ľudské IgG sa vylučujú do materského mlieka počas niekoľkých prvých dní po pôrode,
ktorých koncentrácie krátko na to klesajú na nízke. V dôsledku toho nemožno počas tohto krátkeho obdobia vylúčiť riziko pre dojčené dieťa. Nie je známe, či sa sutimlimab/metabolity vylučujú do
ľudského mlieka. Rozhodnutie, či ukončiť dojčenie alebo ukončiť/prerušiť liečbu sutimlimabom sa má urobiť po zvážení prínosu dojčenia pre dieťa a prínosu liečby pre ženu.
FertilitaÚčinky sutimlimabu na samčiu a samičiu fertilitu sa u zvierat neskúmali. V štúdiách s opakovaným
podávaním sutimlimabu s expozíciami až do približne 4-násobku odporúčanej dávky u ľudí sa u opíc cynomolgus nepozorovali žiadne účinky na reprodukčné orgány.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeEnjaymo nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje.
4.8 Nežiaduce účinkyZhrnutie bezpečnostného profiluNajčastejšie hlásené nežiaduce reakcie na Enjaymo v klinických štúdiách CADENZA a CARDINAL
boli bolesť hlavy, hypertenzia, infekcia močových ciest, infekcia horných dýchacích ciest, nazofaryngitída, nauzea, bolesť brucha, reakcie súvisiace s podávaním infúzie a cyanóza (hlásená ako
akrocyanóza).
Tabuľkový zoznamnežiaducichreakciíHodnotenie bezpečnosti Enjayma u pacientov s CAD bolo primárne založené na údajoch od
66 pacientov, ktorí sa zúčastnili randomizovanej, placebom kontrolovanej štúdie fázy 3 (CADENZA)
a otvorenej štúdie s jednou skupinou (CARDINAL).
V tabuľke 2 sú uvedené nežiaduce reakcie pozorované v štúdiách CADENZA a CARDINAL uvedené podľa triedy orgánových systémov a frekvencie s použitím nasledovných kategórií: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000
až < 1/1 000); veľmi zriedkavé (< 1/10 000). V rámci každej skupiny frekvencií sú nežiaduce reakcie usporiadané v poradí klesajúcej závažnosti.
Tabuľka 2 Zoznam nežiaducich reakcií v štúdiách CADENZA a CARDINAL MedDRA trieda orgánových systémov
| Veľmi časté
| Časté
|
Infekcie a nákazy
| Infekcie močových ciest
Cystitída
Infekcia horných dýchacích ciesta
Nazofaryngitídab
| Infekcie dolných dýchacích ciestc
Urosepsa
|
|
Gastroenteritída
Rinitída
|
Infekcia močových ciest spôsobená baktériou Escherichia
Bakteriálna infekcia močových ciest
Bakteriálna cystitída
Orálny herpes
Virémia Herpes simplex
Herpes zoster
Herpes simplex
|
Celkové poruchy a reakcie v mieste podania
|
|
Pyrexiaf
Pocit chladuf
Reakcie súvisiace s podávaním infúzief
|
Poruchy nervového systému
|
Bolesť hlavy
|
Auraf
Závratf*
|
Poruchy ciev
|
Hypertenziad
Cyanóza (hlásená ako akrocyanóza)
Raynaudov fenomén
|
Hypotenziaf*
Stresová kardiomyopatiaf
|
Poruchy gastrointestinálneho
traktu
|
Bolesť bruchae
Nevoľnosť
|
Hnačkaf
Dyspepsiaf
Aftózny vredf
|
Poruchy dýchacej sústavy, hrudníka a mediastína
|
|
Pocit nepohodlia v hrudníkuf*
|
Poruchy kože a podkožného tkaniva
|
|
Pruritusf*
|
a
Infekcie horných dýchacích ciest: infekcia horných dýchacích ciest, bronchitída a vírusová infekcia horných dýchacích ciest
bNazofaryngitída: nazofaryngitída, faryngitída
cInfekcie dolných dýchacích ciest: pneumónia spôsobená baktériou Klebsiella, pneumónia spôsobená ochorením COVID-19, infekcia dolných dýchacích ciest, vírusová infekcia dýchacích
ciest, infekcia dýchacích ciest, pneumónia
dHypertenzia: hypertenzia, zvýšený krvný tlak, esenciálna hypertenzia, hypertenzná kríza, hypertenzia bieleho plášťa
eBolesť brucha: bolesť brucha, bolesť v dolnej časti brucha, bolesť v hornej časti brucha, citlivosť
brucha
fReakcia súvisiaca s podávaním infúzie: všetky sa vyskytli do 24 hodín od začiatku podávania infúzie Enjayma. *Udalosti naznačujúce reakcie z precitlivenosti sú zahrnuté v tabuľke.
Závažné infekcieZo 66 pacientov, ktorí sa zúčastnili štúdií CADENZA a CARDINAL, sa závažné infekcie hlásili u 10
(15,2 %) pacientov. Medzi závažné infekcie uvedené v tabuľke nežiaducich reakcií patrí infekcia dýchacích ciest [pneumónia spôsobená baktériou Klebsiella (n=1), infekcia dýchacích ciest (n=1),
pneumónia spôsobená ochorením COVID-19 (n=1)], infekcia močových ciest [urosepsa (n=1) ,
infekcia močových ciest (n=1), bakteriálna infekcia močových ciest (n=1)], herpes zoster (n=1). Liečba sutimlimabom bola prerušená u jedného pacienta z dôvodu závažnej infekcie so smrteľným následkom, pneumónie spôsobenej baktériou Klebsiella. Neboli hlásené žiadne ďalšie smrteľné prípady infekcií. Pozri časť 4.4 ohľadom informácií o odporúčaniach očkovania proti závažným infekciám a na monitorovanie skorých prejavov a symptómov infekcií.
ImunogenitaImunogenita sutimlimabu sa hodnotila u pacientov s CAD v štúdiách CARDINAL a CADENZA na
začiatku liečby, počas obdobia liečby a na konci liečby (26. týždeň). U dvoch z 24 pacientov (8,3 %)
zaradených do štúdie CARDINAL, ktorí dostali aspoň jednu dávku sutimlimabu, sa vyvinuli protilátky proti lieku (
antidrug antibody, ADA) vyvolané liečbou. V štúdii CADENZA sa u 6 zo
42 pacientov liečených sutimlimabom (14,3 %) vyvinuli ADA vyvolané liečbou. ADA boli
prechodnej povahy s nízkym titrom a nesúviseli so zmenami farmakokinetického profilu, klinickej
odpovede alebo nežiaducimi udalosťami.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby
hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovanieU pacientov, u ktorých dôjde k predávkovaniu, sa odporúča okamžité prerušenie podávanie infúzie a dôkladné monitorovanie.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: Imunosupresíva, selektívne imunosupresíva, ATC kód: L04AA55
Mechanizmus účinkuSutimlimab je monoklonálna protilátka (mAb) IgG podtriedy 4 (IgG4), ktorá inhibuje klasickú cestu
(CP) a špecificky sa viaže na proteínovú zložku komplementu 1, podzložku s (C1s), serínovú proteázu, ktorá štiepi C4. Sutimlimab neinhibuje aktivity lektínovej a alternatívnej cesty komplementu. Inhibícia klasickej cesty komplementu na úrovni C1s zabraňuje ukladaniu opsonínov komplementu na povrchu červených krviniek, čo vedie k inhibícii hemolýzy u pacientov s CAD, zabraňuje tvorbe prozápalových anafylatoxínov C3a a C5a a „downstreamu“ terminálneho komplexu komplementu C5b-9.
Klinická účinnosť abezpečnosťU pacientov s CAD sa po prvom podaní infúzie Enjayma pozorovala viac ako 90 % inhibícia CP a
hladiny C4 sa vrátili na normálne úrovne (0,2 g/l) do jedného týždňa po prvej dávke Enjayma.
Bezpečnosť a účinnosť Enjayma u pacientov s chorobou chladových aglutinínov (CAD) sa hodnotili
v randomizovanej, dvojito zaslepenej, placebom kontrolovanej štúdii fázy 3 (CADENZA)
u 42 pacientov (n=22 na Enjayme a n=20 na placebe) a v otvorenej štúdii s jednou skupinou fázy 3 (CARDINAL) u 24 pacientov počas obdobia trvania 26 týždňov. Po ukončení šesťmesačných období
liečby (Časť A) pacienti v oboch štúdiách pokračovali v liečbe Enjaymom vo fáze predĺženia štúdie
hodnotiacej dlhodobú bezpečnosť a trvanie odpovede (Časť B) počas ďalších 12 mesiacov
(CADENZA) a 24 mesiacov (CARDINAL) po ukončení liečby posledného pacienta v Časti A. Obe štúdie zahŕňali 9-týždňové následné sledovanie po podaní poslednej dávky Enjayma. Kľúčovými kritériami vhodnosti boli východisková hladina hemoglobínu (Hb) ≤ 10 g/dl a aktívna hemolýza s hladinou bilirubínu nad normálnym referenčným rozsahom. Pacienti so syndrómom chladových aglutinínov (
cold agglutinin syndrome, CAS) boli vylúčení. Pacienti v štúdii CADENZA nemali v anamnéze transfúziu v priebehu 6 mesiacov ani viac ako jednu transfúziu krvi počas 12 mesiacov pred zaradením do štúdie, zatiaľ čo pacienti zaradení do štúdie CARDINAL mali v anamnéze aspoň jednu zdokumentovanú transfúziu krvi v priebehu 6 mesiacov pred zaradením do štúdie. Pacientom sa podávala intravenózna dávka 6 500 mg pri 39-< 75 kg alebo 7 500 mg Enjayma pri ≥ 75 kg počas približne 60 minút v 0. deň, 7. deň a potom každých 14 dní. Hlavné charakteristiky pri vstupe do štúdie skúmanej populácie sú zhrnuté v tabuľke 3 nižšie.
T
abuľka 3 Vstupné charakteristiky pacientov zaradených do klinických štúdií
Parameter
|
Štatistický parameter
|
CADENZ
A
|
CARD
INAL
|
|
|
Placebo
N=
20
|
E
njaymo
N=
22
|
E
njaymo
N=
24
|
Vek
|
Priemer
Min, Max
|
68,2
51, 83
|
65,3
46, 88
|
71,3
55, 85
|
Pohlavie
Muž
Žena
|
n (%)
|
4 (20,0)
16 (80,0)
|
5 (22,7)
17 (77,3)
|
9 (37,5)
15 (62,5)
|
Telesná hmotnosť
|
Priemer, kg
Min, Max
|
64,9
48, 95
|
66,8
39, 100
|
67,8
40, 112
|
Hemoglobín
|
Priemer, g/dl
|
9,33
|
9,15
|
8,59
|
Bilirubín (celkový)*
|
µmol/l
|
35,77
(1,75 x ULN)
|
41,17
(2 x ULN)
|
53,26
(2,6 × ULN†)
|
Transfúzia v
anamnéze
Za posledných
6 mesiacov
Za posledných
12 mesiacov
|
Priemerný
počet transfúzií (rozsah)
|
0
0
|
0
0,14 (0, 1)
|
3,2 (1, 19)
4,8 (1, 23)
|
FACIT† - Stupnica únavy
|
Priemer
|
32,99
|
31,67
|
32,5
|
*N=21 v štúdii CARDINAL; Placebo N=18 a Enjaymo N=20 v štúdii CADENZA, z údajov o
bilirubíne sa vylúčili pacienti s pozitívnym testom alebo bez dostupného výsledku testu na Gilbertov syndróm.
†ULN: Horná hranica normálu, FACIT: Funkčné hodnotenie liečby chronickej choroby (FACIT -
únava sa meria na stupnici od 0 (najhoršia únava) do 52 (bez únavy)
Štúdia CADENZAŠtyridsaťdva pacientov bolo randomizovaných na liečbu Enjaymom (n=22) alebo na podávanie
placeba (n=20) do 25. týždňa.
Účinnosť sa zakladala na podiele pacientov, ktorí splnili kritériá primárneho koncového ukazovateľa: zvýšenie hladiny Hb o ≥ 1,5 g/dl oproti východiskovej hodnote v časovom bode hodnotenia liečby (priemerná hodnota z 23., 25. a 26. týždňa), žiadna transfúzia krvi od 5. týždňa až do 26. týždňa a žiadna liečba CAD nad rámec toho, čo bolo povolené podľa protokolu od 5. týždňa až do 26. týždňa. Pacient dostal transfúziu krvi, ak dosiahol nasledovnú hranicu hemoglobínu: Hb < 7 g/dl alebo pri Hb
< 9 g/dl so symptómami. Zakázané terapie zahŕňali liečbu samotným rituximabom alebo kombináciu s cytotoxickými látkami.
Účinnosť sa ďalej hodnotila podľa nasledovných dvoch kľúčových sekundárnych koncových ukazovateľov: na základe účinku Enjayma na priemernú zmenu Hb oproti východiskovej hodnote a skóre stupnice únavy FACIT na posúdenie zmeny v kvalite života. Ďalšie sekundárne koncové ukazovatele boli: laboratórne merania hemolýzy vrátane priemernej zmeny celkového bilirubínu
oproti východiskovej hodnote. Zozbierané podporné údaje o účinnosti zahŕňali použitie transfúzie po piatich týždňoch liečby.
Výsledky účinnosti sú opísané v tabuľkách 4 a 5 nižšie.
Tabuľka 4 Výsledky účinnosti u pacientov s CAD v štúdii CADENZA – Časť A
Parameter
|
Štatistický parameter
|
Placebo
N=
20
|
E
njaymo
N=
22
|
Ú
činok liečby
|
R
eagujúci na liečbu
a
|
%
(95 % CI)
Pomer pravdepodobnosti (95 % CI)
p hodnota
|
3 (15,0) (3,2; 37,9)
|
16 (72,7) (49,8; 89,3)
|
15,94 (2,88; 88,04)
<0,001
|
H
e
m
oglobín
|
Priemerná zmena
oproti východiskovej hodnote (LS† priemer), g/dl
|
0,09
|
2,66
|
2,56
|
95 % CI LS
priemer
|
(-0,5; 0,68)
|
(2,09; 3,22)
|
(1,75; 3,38)
|
p hodnota
|
|
|
<0,001
|
P
riemerný počet transfúzií
(
5. týždeň až 26. týždeň)
|
n (SD)
|
0,5 (1,1)
|
0,05 (0,2)
|
NC
|
FACIT†- stupnica únavy
|
Priemer
|
33,66
|
43,15
|
|
Priemerná zmena oproti východiskovej hodnote (LS† priemer)
|
1,91
|
10,83
|
8,93
|
95 % CI LS
priemer
|
(-1,65;
5,46)
|
(7,45;
14,22)
|
(4; 13,85)
|
p hodnota
|
|
|
<0,001
|
C
elkový bilirubín*
|
Priemer, µmol/l
|
33,95
|
12,12
|
|
Priemerná zmena
oproti
východiskovej hodnote
|
-1,83
|
-22,13
|
NC
|
Počet normalizovaných
pacientov (%)
|
4 (22,2 %)
|
15 (88,2)
|
|
aReagujúci na liečbu bol definovaný ako pacient so zvýšenou hladinou Hb o ≥ 1,5 g/dl oproti
východiskovej hodnote v časovom bode hodnotenia liečby (priemerná hodnota z 23., 25. a
26. týždňa), bez transfúzie krvi od 5. týždňa do 26. týždňa a bez akejkoľvek liečby CAD nad rámec toho, čo bolo povolené podľa protokolu od 5. týždňa do 26. týždňa.
*N=18 pre placebo a N=17 pre Enjaymo, z údajov o bilirubíne boli vylúčení pacienti s pozitívnym testom alebo bez dostupného výsledku testu na Gilbertov syndróm
†LS: metóda najmenších štvorcov, FACIT: Funkčné hodnotenie liečby chronickej choroby, NC= Nepočítané
Priemerná zmena hemoglobínu (Hb) oproti východiskovej hodnote je znázornená na obrázku 1
nižšie.
O
brázok 1 Štúdia CADENZA Časť A: Graf priemernej zmeny hemoglobínu (g/dl) (+/- SE)
oproti východiskovej hodnote pri návšteve
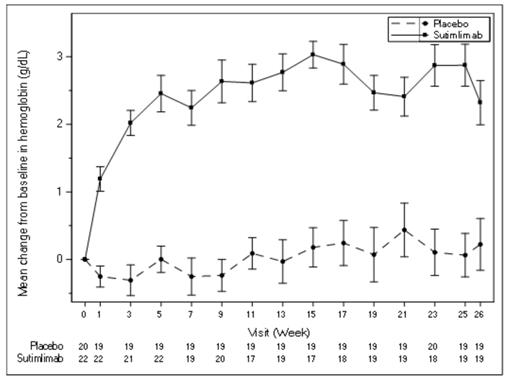
Priemerné hladiny bilirubínu pri návšteve sú znázornené na obrázku 2 nižšie.
Obrázok 2 – Štúdia CADENZA Časť A: Graf priemerného bilirubínu (µmol/l) (+/- SE) pri návšteve (s vylúčením jedincov s pozitívnym testom alebo s neznámymi výsledkami testu na Gilbertov syndróm)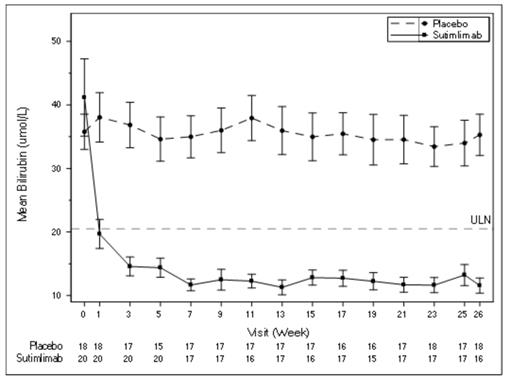
'
Kvalita života súvisiaca so zdravím
V Časti A sú zvýšenia priemerného skóre stupnice únavy FACIT uvedené na obrázku 3 nižšie.
Obrázok 3 – Štúdia CADENZA Časť A: Graf priemernej zmeny skóre stupnice únavy FACIT(SE) pri návšteve – pozorované – celý analyzovaný súbor
V Časti B sa priemerné hladiny hemoglobínu udržali na > 11 g/dl a pozorovala sa trvalá normalizácia priemerných hladín bilirubínu, čo poukazuje na trvalý pokles hemolýzy. Zlepšenia skóre stupnice únavy FACIT pozorované v Časti A sa zachovali.
Po poslednej dávke Enjayma v štúdii sa pozorovali prejavy a symptómy rekurentnej hemolýzy. Priemerný hemoglobín deväť týždňov po poslednej dávke v Časti B klesol o 2,41 g/dl (smerodajná odchýlka, SD: 2,21) a priemerný bilirubín sa zvýšil o 21,80 µmol/l (SD: 18,14) od posledných dostupných hodnôt počas liečby. Priemerné skóre stupnice únavy FACIT sa vrátilo na úrovne blízke východiskovým hodnotám na 31,29 s priemernou zmenou SD oproti východiskovej hodnote o -1,40 (11,48).
Štúdia CARDINALEnjaymo sa podávalo dvadsiatim štyrom pacientom do 25. týždňa.
Účinnosť sa zakladala na podiele pacientov, ktorí splnili kritériá primárneho koncového ukazovateľa: zvýšenie hladiny Hb o ≥ 2 g/dl oproti východiskovej hodnote alebo hladina Hb ≥ 12 g/dl v čase hodnotenia liečby (priemerná hodnota z 23., 25. a 26. týždňa), žiadna transfúzia krvi od 5. týždňa do
26. týždňa a žiadna liečba CAD nad rámec toho, čo bolo povolené podľa protokolu od 5. týždňa do
26. týždňa. Pacient dostal transfúziu krvi, ak dosiahol nasledovnú hranicu hemoglobínu: Hb < 7 g/dl alebo pri Hb < 9 g/dl so symptómami. Zakázané terapie zahŕňali liečbu samotným rituximabom alebo kombináciu s cytotoxickými látkami.
Účinnosť sa ďalej hodnotila podľa nasledovných sekundárnych koncových ukazovateľov: na základe účinku Enjayma na Hb a laboratórne merania hemolýzy vrátane priemernej zmeny celkového bilirubínu oproti východiskovej hodnote. Zmena v kvalite života sa hodnotila ako priemerná zmena oproti východiskovej hodnote skóre stupnice únavy FACIT ako sekundárny koncový ukazovateľ. Zozbierané podporné údaje o účinnosti zahŕňali použitie transfúzie po piatich týždňoch liečby.
Tabuľka 5 uvádza výsledky účinnosti u pacientov s CAD v štúdii CARDINAL.
Tabuľka 5 - Výsledky účinnosti u pacientov s CAD v štúdii CARDINAL – Časť A
Parameter
|
Štatistické parametre
| ENJAYMO N=24
|
Reagujúci na liečbua
| n (%)
| 13 (54)
|
Hemoglobín
| Priemerná zmena oproti východiskovej hodnote (LS† priemer), g/dl
95 % CI LS priemer
|
2,60 (0,74; 4,46)
|
Priemerný počet transfúzií
(5. týždeň až 26. týždeň)
|
n
|
0,9
|
Celkový bilirubín*
| Priemer, µmol/l
Priemerná zmena oproti východiskovej hodnote (LS† priemer)
Počet normalizovaných pacientov (%)
| 15,48 (0,76 × ULN†)
-38,18
13 (54,2)
|
FACIT†- stupnica únavy
| Priemer
Priemerná zmena oproti východiskovej hodnote (LS† priemer)
95 % CI LS priemer
| 44,26
10,85
(8,0; 13,7)
|
aReagujúci na liečbu bol definovaný ako pacient so zvýšenou hladinou Hb o ≥ 2 g/dl oproti
východiskovej hodnote alebo hladinou Hb ≥ 12 g/dl v čase hodnotenia liečby (priemerná hodnota z
23., 25. a 26. týždňa), bez transfúzie krvi od 5. týždňa do 26. týždňa a bez akejkoľvek liečby CAD
nad rámec toho, čo bolo povolené podľa protokolu od 5. týždňa do 26. týždňa.
*N=21 údaje o bilirubíne s vylúčením pacientov s Gilbertovým syndrómom
†LS: metóda najmenších štvorcov, ULN: Horná hranica normálu, FACIT: Funkčné hodnotenie liečby chronickej choroby
V Časti B sa priemerné hladiny hemoglobínu udržali na > 11 g/dl a pozorovala sa trvalá normalizácia priemerných hladín bilirubínu, čo poukazuje na trvalý pokles hemolýzy.
Po poslednej dávke Enjayma v štúdii sa pozorovali prejavy a symptómy rekurentnej hemolýzy. Priemerný hemoglobín deväť týždňov po poslednej dávke v Časti B klesol o 2,28 g/dl (SD: 1,80) a priemerný bilirubín sa zvýšil o 24,27 µmol/l (SD: 13,51) od posledných dostupných hodnôt počas liečby. Priemerné skóre stupnice únavy FACIT sa vrátilo k východiskovým hodnotám, s priemernou zmenou SD oproti východiskovým hodnotám pred liečbou o 1,05 (8,15).
Pediatrická populáciaEurópska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s Enjaymom vo
všetkých podskupinách pediatrickej populácie v liečbe primárnej choroby chladových aglutinínov
(informácie o použití v pediatrickej populácii, pozri časť 4.2).
Populácia starších pacientovVäčšina pacientov (43/66, 65 %) zaradených do klinických štúdií s Enjaymom na liečbu CAD bola
vo veku 65 rokov alebo starší. Hlásené klinické skúsenosti nezistili žiadne rozdiely v odpovediach na liečbu medzi pacientmi staršími ako 65 rokov a mladšími pacientmi.
5.2 Farmakokinetické vlastnostiFarmakokinetika (PK) sutimlimabu bola charakterizovaná u 24 pacientov (CARDINAL)
a 42 pacientov (CADENZA), čo zahŕňalo 51 pacientov liečených dávkou 6 500 mg a 15 pacientov
7 500 mg podľa odporúčaného dávkovania. Celkové expozície v rovnovážnom stave navrhovanej
dávkovacej schémy sú uvedené v tabuľke 6.
T
abuľka 6 - Priemerné (SD) parametre expozície v rovnovážnom stave
CARD
INAL a
CADENZ
A
|
D
ávka (mg)
|
C
m
in
(
µg/ml)*
|
AUC
S
S
(
µg·h/ml)*
|
Priemer (SD)
|
6 500 (n=51)
7 500 (n=15)
|
1 397 (721)
1 107 (661)
|
697 449 (256 234)
576 017 (253 776)
|
* Skratky: AUCss = plocha pod krivkou medzi 2 po sebe nasledujúcimi dávkami po dosiahnutí rovnovážneho stavu; Cmin = minimálna koncentrácia v rovnovážnom stave definovaná ako 1 hodina
pred podaním nasledujúcej dávky
Rovnovážny stav sa dosiahol v 7. týždni od začiatku liečby sutimlimabom s mierou kumulácie nižšou ako 2.
DistribúciaU pacientov s CAD bol distribučný objem v rovnovážnom stave v centrálnych a periférnych
kompartmentoch približne 5,8 l.
BiotransformáciaSutimlimab je proteín. Všeobecne je známe, že protilátky sa metabolizujú degradáciou na malé
peptidy a jednotlivé aminokyseliny.
ElimináciaPolčas eliminácie sutimlimabu závisí od plazmatickej koncentrácie. Terminálny polčas eliminácie
sutimlimabu v rovnovážnom stave založený na celkovom klírense (lineárny a nelineárny klírens) je
16 dní.
Linearita/nelinearitaPo jednorazových dávkach preukázal klírens sutimlimabu prudký začiatočný pokles pri dávkach
nižších ako 30 mg/kg (~ 2 g), medzi dávkami 60 a 100 mg/kg sutimlimabu sa zmenil na nezávislý od dávky.
Špeciálne populácieNa základe pohlavia, veku, poruchy funkcie pečene alebo poruchy funkcie obličiek sa nepozorovali
žiadne klinicky významné rozdiely vo farmakokinetike sutimlimabu. Hladiny expozície (Cmax, Cmin a
AUC) v rovnovážnom stave boli odhadnuté na základe dávky 6 500 mg (< 75 kg) a 7 500 mg
(> = 75 kg) podaných na 0. deň, 7. deň a potom každých 14 dní. Populačná farmakokinetická analýza
preukázala podobné parametre expozície medzi pohlaviami u 101 mužov a 95 žien zaradených do štúdie.
Populačná farmakokinetická analýza preukázala podobné parametre expozície u osôb rôznych rás
(94 belosi, 10 černosi, 42 aziati).
Populačná farmakokinetická analýza ukázala, že telesná hmotnosť a etnická príslušnosť (Japonci verzus iní ako Japonci) ovplyvnili farmakokinetiku sutimlimabu. Nižšia expozícia sa pozorovala u osôb s vyššou telesnou hmotnosťou. Na základe porovnania krížovej štúdie bola AUC0-168 sutimlimabu po dávke 30 až 100 mg/kg až o 38 % vyššia u osôb japonského pôvodu ako osôb iného pôvodu.
Farmakokinetický/farmakodynamickývzťah
Koncentrácia sutimlimabu vyššia ako 100 µg/ml viedla k maximálnej inhibícii CP. Navrhovaná schéma dávkovania viedla k dostatočnej expozícii sutimlimabu v rovnovážnom stave na dosiahnutie klinicky významných účinkov na Hb, bilirubín a celkové hladiny C4.
5.3 Predklinické údaje o bezpečnosti
V rozšírenej štúdii prenatálneho a postnatálneho vývoja (enhanced pre and postnatal development, ePPND) na opiciach cynomolgus sa neodhalili žiadne dôkazy nepriaznivých vývojových následkov po intravenóznom podávaní sutimlimabu od organogenézy až do pôrodu pri expozíciách približne 2-
3-násobku AUC u ľudí pri maximálnej odporúčanej dávke. V štúdiách s opakovaným podávaním sutimlimabu s expozíciami až do približne 4-násobku odporúčanej dávky u ľudí sa u opíc cynomolgus nepozorovali žiadne účinky na reprodukčné orgány.
Nevykonali sa žiadne štúdie na zvieratách na vyhodnotenie karcinogénneho potenciálu sutimlimabu. Predklinické údaje získané na základe predklinických štúdií na opiciach cynomolgus neodhalili
žiadne osobitné riziko pre ľudí.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
polysorbát 80 (E433)
chlorid sodný
dibázický fosforečnan sodný (E339)
monobázický fosforečnan sodný (E339)
voda na injekcie
6.2 Inkompatibility
Nevykonali sa žiadne štúdie kompatibility, preto sa tento liek nesmie miešať s inými liekmi.
6.3 Čas použiteľnosti
Zatvorená injekčná liekovka:
3 roky
Uchovávanie lieku pootvorení:
Chemická a fyzikálna stabilita pred použitím je preukázaná počas 16 hodín pri teplote 18°C až 25°C
alebo počas 72 hodín pri teplote 2°C až 8°C. Z mikrobiologického hľadiska sa má liek použiť okamžite.
Ak sa nepoužije okamžite, za čas a podmienky uchovávania pred použitím zodpovedá používateľ a za normálnych okolností nemajú presiahnuť 24 hodín pri 2°C až 8°C alebo 8 hodín pri izbovej teplote, pokiaľ sa otvorenie injekčnej liekovky a naplnenie infúzneho vaku nevykonalo za kontrolovaných a validovaných aseptických podmienok.
6.4 Špeciálne upozornenia na uchovávanie
Uchovávajte v chladničke (2°C–8°C).
Uchovávajte v pôvodnom obale na ochranu pred svetlom. Neuchovávajte v mrazničke.
Podmienky na uchovávanie po prvom otvorení injekčnej liekovky s liekom, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
22 ml roztoku v injekčnej liekovke (sklo typu I) so zátkou (butylová gumová), obrubou (hliník) a vyklápacím viečkom.
Každé balenie obsahuje 1 alebo 6 injekčných liekoviek. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
Enjaymo sa dodáva ako roztok v jednodávkovej injekčnej liekovke a má ho pripraviť zdravotnícky pracovník za použitia aseptickej techniky.
Príprava
1. Vyberte Enjaymo z chladničky. Na minimalizáciu penenia netraste.
2. Pred podaním injekčné liekovky vizuálne skontrolujte, či neobsahujú pevné častice a
či nezmenili farbu. Roztok je opalescenčná a bezfarebná až svetložltá tekutina. Nepodávajte, ak došlo k zmene farby alebo ak sú prítomné iné cudzie pevné častice.
3. Vypočítaný objem odoberte z príslušného počtu injekčných liekoviek na základe odporúčanej
dávky (pozri tabuľku 1) a dajte do prázdneho infúzneho vaku. Nepoužitú časť, ktorá zostala v injekčnej liekovke, zlikvidujte.
4. Pripravený roztok sa má podať okamžite. Podmienky uchovávania, pozri časť 6.3.
Podávanie
1. Pred podaním nechajte infúzny roztok dosiahnuť izbovú teplotu (18°C–25°C). Rýchlosť
podávanie infúzie nájdete v tabuľke 1, pozri časť 4.2. Infúzia sa má podávať počas 1-2 hodín v závislosti od telesnej hmotnosti pacienta. Infúziu podávajte iba pomocou polyétersulfónového (PES) membránového filtra s veľkosťou pórov 0,22 mikrónov. Môžu sa použiť ohrievače infúzií, neprekračujte teplotu 40°C.
2. Infúzny katéter a hadičky sa majú naplniť podávaným roztokom bezprostredne pred podaním infúzie a ihneď po ukončení podávania infúzie sa majú vypláchnuť dostatočným množstvom
(približne 20 ml) injekčného roztoku chloridu sodného s koncentráciou 9 mg/ml (0,9 %).
3. Nepozorovali sa žiadne inkompatibility medzi infúznym roztokom Enjayma a infúznymi vakmi vyrobenými z di-(2-etylhexyl)ftalátu (DEHP)-plastifikovaného polyvinylchloridu (PVC),
etylénvinylacetátu (EVA) a polyolefínu (PO); súpravami na podávanie liekov vyrobenými z
DEHP- plastifikovaného PVC, polypropylén (PP) bez obsahu DEHP a polyetylénu (PE) bez obsahu DEHP; a adaptérmi injekčných liekoviek vyrobenými z polykarbonátu (PC) a akrylonitril-butadién-styrénu (ABS).
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Sanofi B.V. Paasheuvelweg 25
1105 BP Amsterdam
Holandsko
8. REGISTRAČNÉ ČÍSLA
EU/1/22/1687/001
EU/1/22/1687/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 15. novembra 2022
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu