
í po užití poslednej dávky odporúča nedojčiť.
Fertilita
K dispozícii nie sú žiadne údaje týkajúce sa účinkov lieku Calquence na fertilitu u ľudí.
V predklinickej štúdii na samcoch a samiciach potkana s akalabrutinibom sa nepozorovali žiadne nežiaduce účinky na parametre fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Calquence nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Počas liečby akalabrutinibom sa však hlásila únava a závrat a pacientom, u ktorých sa vyskytnú tieto príznaky, sa má odporučiť, aby neviedli vozidlá ani neobsluhovali stroje, až kým príznaky neustúpia.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
U 1 040 pacientov liečených monoterapiou liekom Calquence boli najčastejšími (≥ 20 %) nežiaducimi
liekovými reakciami akéhokoľvek stupňa infekcia (66,7 %), bolesť hlavy (37,8 %), hnačka (36,7 %), tvorba podliatin (34,1 %), muskuloskeletálna bolesť (33,1 %), nevoľnosť (21,7 %), únava (21,3 %), kašeľ (21 %) a vyrážka (20,3 %). Najčastejšie (≥ 5 %) hlásené nežiaduce liekové reakcie ≥ 3. stupňa boli infekcia (17,6 %), leukopénia (14,3 %), neutropénia (14,2 %) a anémia (7,8 %).
U 223 pacientov liečených kombinovanou liečbou s liekom Calquence boli najčastejšími (≥ 20 %) nežiaducimi liekovými reakciami akéhokoľvek stupňa infekcia (74 %), muskuloskeletálna bolesť (44,8 %), hnačka (43,9 %), bolesť hlavy (43 %), leukopénia (31,8 %), neutropénia (31,8 %), kašeľ (30,5 %), únava (30,5 %), artralgia (26,9 %), nevoľnosť (26,9 %), závrat (23,8 %) a zápcha (20,2 %). Najčastejšie (≥ 5 %) hlásené nežiaduce liekové reakcie ≥ 3. stupňa boli leukopénia (30 %), neutropénia (30 %), infekcia (21,5 %), trombocytopénia (9 %) a anémia (5,8 %).
Tabuľkový zoznamnežiaducichreakciíV klinických štúdiách u pacientov užívajúcich Calquence ako liečbu hematologických malignít boli
identifikované nasledujúce nežiaduce liekové reakcie. Medián trvania liečby liekom Calquence hodnotený na základe sumárnych dát bol 26,2 mesiacov.
Nežiaduce liekové reakcie sú uvedené podľa triedy orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce liekové reakcie usporiadané podľa frekvencie výskytu, pričom najčastejšie sa vyskytujúce reakcie sú uvedené ako prvé. Navyše, príslušné kategórie frekvencie výskytu sú pre každú nežiaducu liekovú reakciu definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000); veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov). V rámci každej skupiny frekvencie výskytu sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 3: Nežiaduce liekové reakcie* u pacientov s hematologickými malignitami liečených monoterapiou akalabrutinibom (n = 1 040)
Trieda orgánových systémov MedDRA
|
MedDRA výraz
| Celková frekvencia výskytu
(všetky stupne CTCAE)
|
Frekvencia výskytu
CTCAE ≥ 3. stupňa†
|
Infekcie a nákazy
| Infekcia horných dýchacích ciest
| Veľmi časté (22 %)
| 0,8 %
|
Sinusitída
| Veľmi časté (10,7 %)
| 0,3 %
|
Pneumónia
| Časté (8,7 %)
| 5,1 %
|
Infekcia močových ciest
| Časté (8,5 %)
| 1,5 %
|
Nazofaryngitída
| Časté (7,4 %)
| 0 %
|
Bronchitída
| Časté (7,6 %)
| 0,3 %
|
Herpetické vírusové infekcie†
| Časté (5,9 %)
| 0,7 %
|
Aspergilové infekcie†
| Menej časté (0,5 %)
| 0,4 %
|
Reaktivácia hepatitídy B
| Menej časté (0,1 %)
| 0,1 %
|
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
| †
Druhá primárna malignita (SPM)
Nemelanómová kožná malignita†
SPM s výnimkou nemelanómového kožného karcinómu†
| Veľmi časté (12,2 %)
Časté (6,6 %) Časté (6,5 %)
| 4,1 %
0,5 %
3,8 %
|
Poruchy krvi
a lymfatického systému
| Neutropénia†
| Veľmi časté (15,7 %)
| 14,2 %
|
Anémia†
| Veľmi časté (13,8 %)
| 7,8 %
|
†
Trombocytopénia
| Časté (8,9 %)
| 4,8 %
|
Lymfocytóza
| Menej časté (0,3 %)
| 0,2 %
|
Poruchy metabolizmu a výživy
|
Syndróm nádorového rozpadu±
|
Menej časté (0,5 %)
|
0,4 %
|
Poruchy nervového systému
| Bolesť hlavy
| Veľmi časté (37,8 %)
| 1,1 %
|
Závrat
| Veľmi časté (13,4 %)
| 0,2 %
|
Poruchy srdca
a srdcovej činnosti
| †
Atriálna fibrilácia/flutter
|
Časté (4,4 %)
|
1,3 %
|
Poruchy ciev
| Tvorba podliatin†
Kontúzia Petechie Ekchymózy
| Veľmi časté (34,1 %)
Veľmi časté (21,7 %) Veľmi časté (10,7 %) Časté (6,3 %)
| 0 %
0 %
0 %
0 %
|
Hemorágia/hematóm†
Gastrointestinálna hemorágia
Intrakraniálna hemorágia
| Veľmi časté (12,6 %) Časté (2,3 %)
Časté (1 %)
| 1,8 %
0,6 %
0,5 %
|
Epistaxa
| Časté (7 %)
| 0,3 %
|
Poruchy gastrointestinálneho traktu
|
Hnačka
|
Veľmi časté (36,7 %)
|
2,6 %
|
Nevoľnosť
|
Veľmi časté (21,7 %)
|
1,2 %
|
Zápcha
|
Veľmi časté (14,5 %)
|
0,1 %
|
Vracanie
|
Veľmi časté (13,3 %)
|
0,9 %
|
Abdominálna bolesť†
|
Veľmi časté (12,5 %)
|
1 %
|
Poruch
y kože a podkožného tkaniva
|
Vyrážka†
|
Veľmi časté (20,3 %)
|
0,6 %
|
Poruch
y kostrovej a svalovej sústavy a spojivového tkaniva
|
Muskuloskeletálna bolesť†
|
Veľmi časté (33,1 %)
|
1,5 %
|
Artralgia
|
Veľmi časté (19,1 %)
|
0,7 %
|
Celkov
é poruchy a reakcie v mieste podania
|
Únava
|
Veľmi časté (21,3 %)
|
1,7 %
|
Asténia
|
Časté (5,3 %)
|
0,8 %
|
Laboratórne
a funkčné vyšetrenia
¶
(nález
y na základe
výsledkov vyšetrení)
|
Znížená hladina hemoglobínu§
|
Veľmi časté (42,6 %)
|
10,1 %
|
Znížený absolútny počet neutrofilov§
|
Veľmi časté (41,8 %)
|
20,7 %
|
Znížený počet trombocytov§
|
Veľmi časté (31,1 %)
|
6,9 %
|
*Podľa všeobecných terminologických kritérií pre nežiaduce udalosti podľa Národného inštitútu pre rakovinu (NCI
CTCAE) verzie 4.03.
†Zahŕňa viacnásobný výraz pre nežiaducu liekovú reakciu.
±Jeden prípad syndrómu nádorového rozpadu indukovaného liekom sa pozoroval v skupine s akalabrutinibom v štúdii
ASCEND.
§Predstavuje incidenciu nálezov laboratórnych a funkčných vyšetrení, nie hlásených nežiaducich udalostí.
¶Uvedené ako hodnoty stupňa CTCAE.
Tabuľka 4: Nežiaduce liekové reakcie* u pacientov s hematologickými malignitami liečenýchkombinovanou liečbou s akalabrutinibom (n = 223)
Trieda orgánových systémov MedDRA
|
MedDRA výraz
| Celková frekvencia výskytu
(všetky stupne CTCAE)
|
Frekvencia výskytu
CTCAE ≥ 3. stupňa†
|
Infekcie a nákazy
| Infekcia horných dýchacích ciest
| Veľmi časté (31,4 %)
| 1,8 %
|
Sinusitída
| Veľmi časté (15,2 %)
| 0,4 %
|
Nazofaryngitída
| Veľmi časté (13,5 %)
| 0,4 %
|
Infekcia močových ciest
| Veľmi časté (13 %)
| 0,9 %
|
Pneumónia
| Veľmi časté (10,8 %)
| 5,4 %
|
Bronchitída
| Časté (9,9 %)
| 0 %
|
Herpetické vírusové infekcie†
| Časté (6,7 %)
| 1,3 %
|
Progresívna multifokálna
leukoencefalopatia
|
Menej časté (0,4 %)
|
0,4 %
|
Reaktivácia hepatitídy B
| Menej časté (0,9 %)
| 0,1 %
|
Aspergilové infekcie†
| Veľmi zriedkavé (0 %)
| 0 %
|
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
| †
Druhá primárna malignita (SPM)
Nemelanómová kožná malignita†
SPM s výnimkou
nemelanómového kožného karcinómu†
| Veľmi časté (13 %)
Časté (7,6 %) Časté (6,3 %)
| 4,0 %
0,4 %
3,6 %
|
Tried
a orgánových systémov MedDRA
|
MedDRA výraz
|
Celkov
á frekvencia výskytu
(všetky stupne CTCAE)
|
Frekvenci
a výskytu
CTCAE ≥ 3. stupňa
†
|
Poruch
y krvi
a lymfatického systému
|
Neutropénia†
|
Veľmi časté (31,8 %)
|
30 %
|
Trombocytopénia†
|
Veľmi časté (13,9 %)
|
9 %
|
†
Anémia
|
Veľmi časté (11,7 %)
|
5,8 %
|
Lymfocytóza
|
Menej časté (0,4 %)
|
0,4 %
|
Poruchy metabolizmu
a výživy
|
Syndróm nádorového rozpadu±
|
Menej časté (1,8 %)
|
1,3 %
|
Poruch
y nervového systému
|
Bolesť hlavy
|
Veľmi časté (43 %)
|
0,9 %
|
Závrat
|
Veľmi časté (23,8 %)
|
0 %
|
Poruch
y srdca
a srdcovej činnosti
|
†
Atriálna fibrilácia/flutter
|
Časté (3,1 %)
|
0,9 %
|
Poruch
y ciev
|
Tvorba podliatin†
Kontúzia Petechie Ekchymózy
|
Veľmi časté (38,6 %)
Veľmi časté (27,4 %) Veľmi časté (11,2 %) Časté (3,1 %)
|
0 %
0 %
0 %
0 %
|
Hemorágia/hematóm†
Gastrointestinálna hemorágia
Intrakraniálna hemorágia
|
Veľmi časté (17,5 %)
Časté (3,6 %)
Menej časté (0,9 %)
|
1,3 %
0,9 %
0 %
|
Epistaxa
|
Časté (8,5 %)
|
0 %
|
Poruchy gastrointestinálneho traktu
|
Hnačka
|
Veľmi časté (43,9 %)
|
4,5 %
|
Nevoľnosť
|
Veľmi časté (26,9 %)
|
0 %
|
Zápcha
|
Veľmi časté (20,2 %)
|
0 %
|
Vracanie
|
Veľmi časté (19,3 %)
|
0,9 %
|
Abdominálna bolesť†
|
Veľmi časté (14,8 %)
|
1,3 %
|
Poruch
y kože
a podkožného tkaniva
|
Vyrážka†
|
Veľmi časté (30,9 %)
|
1,8 %
|
Poruch
y kostrovej a svalovej sústavy a spojivového tkaniva
|
Muskuloskeletálna bolesť†
|
Veľmi časté (44,8 %)
|
2,2 %
|
Artralgia
|
Veľmi časté (26,9 %)
|
1,3 %
|
Celkov
é poruchy a reakcie v mieste podania
|
Únava
|
Veľmi časté (30,5 %)
|
1,8 %
|
Asténia
|
Časté (7,6 %)
|
0,4 %
|
Laboratórne
a funkčné vyšetrenia¶
(nálezy na základe
výsledkov vyšetrení)
|
Znížený absolútny počet neutrofilov§
|
Veľmi časté (57,4 %)
|
35 %
|
Znížený počet trombocytov§
|
Veľmi časté (46,2 %)
|
10,8 %
|
Znížená hladina hemoglobínu§
|
Veľmi časté (43,9 %)
|
9 %
|
*Podľa všeobecných terminologických kritérií pre nežiaduce udalosti podľa Národného inštitútu pre rakovinu (NCI
CTCAE) verzie 4.03.
†Zahŕňa viacnásobný výraz pre nežiaducu liekovú reakciu.
±Jeden prípad syndrómu nádorového rozpadu indukovaného liekom sa pozoroval v skupine s akalabrutinibom v štúdii
ASCEND.
§Predstavuje incidenciu nálezov laboratórnych a funkčných vyšetrení, nie hlásených nežiaducich udalostí.
¶Uvedené ako hodnoty stupňa CTCAE.
Popis vybranýchnežiaducichreakciíUkončenie liečby a zníženie dávky v dôsledku nežiaducich reakciíSpomedzi 1 040 pacientov liečených monoterapiou liekom Calquence sa ukončenie liečby z dôvodu nežiaducich reakcií hlásilo u 9,3 % pacientov. Hlavné nežiaduce reakcie zahŕňali pneumóniu, trombocytopéniu a hnačku. Zníženia dávky z dôvodu nežiaducich reakcií sa hlásili u 4,2 % pacientov. Hlavné nežiaduce reakcie zahŕňali reaktiváciu hepatitídy B, sepsu a hnačku.
Spomedzi 223 pacientov liečených kombinovanou liečbou liekom Calquence sa ukončenie liečby z dôvodu nežiaducich reakcií hlásilo u 10,8 % pacientov. Hlavné nežiaduce reakcie zahŕňali pneumóniu, trombocytopéniu a hnačku. Zníženia dávky z dôvodu nežiaducich reakcií sa hlásili
u 6,7 % pacientov. Hlavné nežiaduce reakcie zahŕňali neutropéniu, hnačku a vracanie.
Staršie osobySpomedzi 1 040 pacientov v klinických štúdiách s monoterapiou liekom Calquence bolo 41 % vo veku viac ako 65 rokov a menej ako 75 rokov a 22 % bolo vo veku viac ako 75 rokov. Medzi pacientmi vo veku ≥ 65 rokov a mladšími pacientmi sa nepozorovali žiadne klinicky významné rozdiely
v bezpečnosti alebo účinnosti.
Spomedzi 223 pacientov v klinických štúdiách s liekom Calquence v kombinácii s obinutuzumabom bolo 47 % vo veku viac ako 65 rokov a menej ako 75 rokov a 26 % bolo vo veku viac ako 75 rokov. Medzi pacientmi vo veku ≥ 65 rokov a mladšími pacientmi sa nepozorovali žiadne klinicky významné rozdiely v bezpečnosti alebo účinnosti.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné

monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieK dispozícii nie je žiadna špecifická liečba predávkovania akalabrutinibom a doposiaľ neboli stanovené príznaky predávkovania. V prípade predávkovania je potrebné pacientov pozorne sledovať pre prejavy alebo príznaky nežiaducich reakcií a je potrebné podať vhodnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: cytostatiká, inhibítory proteínkinázy. ATC kód: L01EL02.
MechanizmusúčinkuAkalabrutinib je selektívny inhibítor Brutonovej tyrozínkinázy (BTK). BTK je signálna molekula dráh
receptora antigénu B-buniek (B-cell antigen receptor, BCR) a cytokínového receptora. V B-bunkách má signalizácia BTK za následok prežívanie a proliferáciu B-buniek a je potrebná pre bunkovú adhéziu, migráciu a chemotaxiu.
Akalabrutinib a jeho aktívny metabolit, ACP-5862, tvoria kovalentnú väzbu s cysteínovým zvyškom v aktívnom mieste BTK, čo vedie k ireverzibilnej inaktivácii BTK s minimálnymi necieľovými interakciami.
Far
m
akodynamické účinky
U pacientov s malignitami B-buniek, ktorí dostávali 100 mg akalabrutinibu dvakrát denne, sa medián
obsadenosti BTK v periférnej krvi v rovnovážnom stave v hodnote ≥ 95 % zachoval počas 12 hodín, čo viedlo k inaktivácii BTK počas odporúčaného dávkovacieho intervalu.
Elektrofyziológia srdca
Účinok akalabrutinibu na QTc interval sa hodnotil u 46 zdravých osôb mužského a ženského pohlavia
v randomizovanej, dvojito zaslepenej, detailnej QT štúdii s placebom a pozitívnymi kontrolnými
osobami. Pri supraterapeutickej dávke, 4-násobku maximálnej odporúčanej dávky, Calquence
v žiadnom klinicky významnom rozsahu nepredlžoval QT/QTc interval (napr. nie viac ako o 10 ms alebo o 10 ms) (pozri časti 4.4, 4.8 a 5.3).
Klinická účinnosťa bezpečnosť
Pacienti s CLL neliečenou v minulosti
Bezpečnosť a účinnosť lieku Calquence pri CLL, ktorá nebola v minulosti liečená, sa hodnotili v randomizovanej, multicentrickej, otvorenej štúdii fázy 3 (ELEVATE-TN) zahŕňajúcej 535 pacientov. Pacienti dostávali Calquence plus obinutuzumab, monoterapiu liekom Calquence alebo obinutuzumab plus chlorambucil. V štúdii ELEVATE-TN boli zahrnutí pacienti vo veku 65 rokov alebo starší alebo pacienti vo veku 18 až 65 rokov so súbežne existujúcimi zdravotnými stavmi, pričom 27,9 % pacientov malo CrCl < 60 ml/min. Spomedzi pacientov vo veku < 65 rokov malo
16,1 % medián skóre CIRS-G v hodnote 8. V štúdii mali pacienti povolené užívať antitrombotiká. Vylúčení boli pacienti, u ktorých sa vyžadovala antikoagulácia pomocou warfarínu alebo ekvivalentného antagonistu vitamínu K.
Pacienti boli randomizovaní v pomere 1:1:1 do 3 skupín na podávanie buď:
· Calquence plus obinutuzumab (Calquence + G): Calquence 100 mg sa podával dvakrát denne so začiatkom v 1. deň 1. cyklu až do progresie ochorenia alebo neakceptovateľnej toxicity. Obinutuzumab sa podával od 2. cyklu (maximálne počas 6 liečebných cyklov) a to 1. deň
100 mg, 2. deň 900 mg, 8. deň 1 000 mg, 15. deň 1 000 mg a následne počas 3.-7. cyklu sa podával vždy 1. deň cyklu v dávke 1 000 mg. Každý cyklus trval 28 dní.
· Monoterapiu liekom Calquence: Calquence 100 mg sa podával dvakrát denne až do progresie ochorenia alebo neakceptovateľnej toxicity.
· Obinutuzumab plus chlorambucil (GClb): Obinutuzumab a chlorambucil sa podávali počas maximálne 6 liečebných cyklov. Obinutuzumab 1 000 mg sa podával od prvého cyklu a to 1. deň 100 mg, 2. deň 900 mg, 8. deň 1 000 mg, 15. deň 1 000 mg a následne počas 2.-6. cyklu sa
podával vždy 1. deň cyklu v dávke 1 000 mg. Chlorambucil 0,5 mg/kg sa podával v 1. a 15. deň
1.-6. cyklu. Každý cyklus trval 28 dní.
Pacienti boli stratifikovaní podľa stavu mutácie delécie 17p (prítomná delécia vs. bez delécie), výkonnostného stavu podľa ECOG (0 alebo 1 oproti 2) a geografickej oblasti (Severná Amerika a Západná Európa oproti inej oblasti). Po potvrdení progresie ochorenia prešlo 45 pacientov randomizovaných v skupine GClb do skupiny s monoterapiou liekom Calquence. Východiskové
demografické charakteristiky a charakteristiky ochorenia populácie štúdie sú zhrnuté v tabuľke 5.
Tabuľka 5: Východiskové pacientske charakteristiky (štúdia ELEVATE-TN) u pacientov s CLL
neliečeno
u v minulosti
C
h
arakteristika
|
Calquenc
e plus obinutuzumab N = 179
|
Monoterapia
Calquence
N = 179
|
Obinutuzumab plus
chlorambucil N = 177
|
Vek, roky; medián (rozsah)
|
70 (41 – 88)
|
70 (44 – 87)
|
71 (46 – 91)
|
Mužské pohlavie; %
|
62
|
62
|
59,9
|
Biela rasa; %
|
91,6
|
95
|
93,2
|
Výkonnostný stav podľa ECOG 0 – 1; %
|
94,4
|
92,2
|
94,4
|
Medián času od diagnózy (mesiace)
|
30,5
|
24,4
|
30,7
|
Masívne ochorenie s uzlinami o veľkosti
≥ 5 cm; %
|
25,7
|
38
|
31,1
|
Cytogenetika/FISH kategória; %
Delécia 17p Delécia 11q Mutácia TP53
Nezmutovaný IGHV
Komplexný karyotyp (≥ 3 abnormality)
|
9,5
17,3
11,7
57,5
16,2
|
8,9
17,3
10,6
66,5
17,3
|
9
18,6
11,9
65,5
18,1
|
Rai štádium; %
0
I II III IV
|
1,7
30,2
20,1
26,8
21,2
|
0
26,8
24,6
27,9
20,7
|
0,6
28,2
27,1
22,6
21,5
|
Primárnym koncovým ukazovateľom bolo prežívanie bez progresie (progression-free survival, PFS)
v skupine s Calquence plus obinutuzumab (Calquence + G) oproti skupine s obinutuzumabom plus chlorambucil (GClb) na základe nezávislej hodnotiacej komisie (Independent Review Committee, IRC) podľa kritérií Medzinárodnej pracovnej skupiny pre chronickú lymfocytovú leukémiu (International Workshop on Chronic Lymphocytic Leukaemia, IWCLL) z roku 2008 so zapracovaním vysvetlenia lymfocytózy súvisiacej s liečbou (Cheson 2012). S mediánom sledovania 28,3 mesiacov PFS podľa IRC dokázalo 90% štatisticky významné zníženie rizika progresie ochorenia alebo úmrtia
z dôvodu CLL neliečenej v minulosti u pacientov v skupine s liekom Calquence + G v porovnaní so skupinou s GClb. Výsledky týkajúce sa účinnosti sú uvedené v tabuľke 6. Kaplanove-Meierove krivky PFS sú zobrazené na obrázku 1.
Tabuľka 6: Výsledky týkajúce sa účinnosti podľa hodnotenia IRC (štúdia ELEVATE-TN)u pacientov s CLL
| Calquence plus obinutuzumab N = 179
| Monoterapia
Calquence
N = 179
| Obinutuzumab plus chlorambucil N = 177
|
Prežívanie bez progresie*
|
Počet udalostí (%)
| 14 (7,8)
| 26 (14,5)
| 93 (52,5)
|
PD, n (%)
| 9 (5)
| 20 (11,2)
| 82 (46,3)
|
Úmrtia (%)
| 5 (2,8)
| 6 (3,4)
| 11 (6,2)
|
Medián (95 % IS), mesiace
| NR
| NR (34,2; NR)
| 22,6 (20,2; 27,6)
|
HR† (95 % IS)
| 0,10 (0,06; 0,17)
| 0,20 (0,13; 0,30)
| -
|
Hodnota p
| < 0,0001
| < 0,0001
| -
|
24-mesačný odhad, % (95 % IS)
| 92,7 (87,4; 95,8)
| 87,3 (80,9; 91,7)
| 46,7 (38,5; 54,6)
|
|
Calquenc
e plus obinutuzumab N = 179
|
Monoterapia
Calquence
N = 179
|
Obinutuzumab plus
chlorambucil N = 177
|
Celkov
é prežívanie
a
|
Úmrtia (%)
|
9 (5)
|
11 (6,1)
|
17 (9,6)
|
Pomer rizika (95 % IS)†
|
0,47 (0,21; 1,06)
|
0,60 (0,28; 1,27)
|
-
|
Miera najlepšej celkovej odpovede
*
(CR + CRi + nPR + PR)
|
ORR, n (%)
(95 % IS)
|
168 (93,9)
(89,3; 96,5)
|
153 (85,5)
(79,6; 89,9)
|
139 (78,5)
(71,9; 83,9)
|
Hodnota p
|
< 0,0001
|
0,0763
|
-
|
CR, n (%)
|
23 (12,8)
|
1 (0,6)
|
8 (4,5)
|
CRi, n (%)
|
1 (0,6)
|
0
|
0
|
nPR, n (%)
|
1 (0,6)
|
2 (1,1)
|
3 (1,7)
|
PR, n (%)
|
143 (79,9)
|
150 (83,8)
|
128 (72,3)
|
IS = interval spoľahlivosti; HR = pomer rizika; NR = nedosiahol sa; CR = úplná odpoveď; CRi = úplná odpoveď s neúplnou úpravou krvného obrazu; nPR = nodulárna čiastočná odpoveď; PR = čiastočná
odpoveď.
*Podľa hodnotenia IRC.
†Na základe stratifikovaného Coxovho modelu proporcionálnych rizík.
aMedián OS sa v oboch skupinách nedosiahol.
Obrázok 1: Kaplanova-Meierova krivka PFS podľa hodnotenia IRC (štúdia ELEVATE-TN)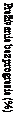 u pacientov s CLL (populácia podľa liečebného zámeru [intent-to-treat, ITT])
u pacientov s CLL (populácia podľa liečebného zámeru [intent-to-treat, ITT])Calquence+G
Calquence
GClb

Čas od randomizácie (mesiace)
Počet pacientov v riziku
|
Mesiac
| 0
| 3
| 6
| 9
| 12
| 15
| 18
| 21
| 24
| 27
| 30
| 33
| 36
| 39
|
Calquence
| 179
| 166
| 161
| 157
| 153
| 150
| 148
| 147
| 103
| 94
| 43
| 40
| 4
| 3
|
Calquence + G
| 179
| 176
| 170
| 168
| 163
| 160
| 159
| 155
| 109
| 104
| 46
| 41
| 4
| 2
|
GClb
| 177
| 162
| 157
| 151
| 136
| 113
| 102
| 86
| 46
| 41
| 13
| 13
| 3
| 2
|
Výsledky PFS pre Calquence s obinutuzumabom alebo bez neho boli konzistentné naprieč
podskupinami, vrátane vysokorizikových charakteristík. Vo vysokorizikovej populácii s CLL (delécia
17p, delécia 11q, mutácia TP53 alebo nezmutovaný IGHV) bol HR pre PFS pri Calquence
s obinutuzumabom alebo bez neho 0,08 [95 % IS (0,04; 0,15)] oproti obinutuzumabu plus chlorambucil 0,13 [95 % IS (0,08; 0,21)].
Tabuľka 7: Podskupinová analýza PFS (štúdia ELEVATE-TN)
|
Monoterapia Calquence
|
Calquence + G
|
N
|
P
o
mer
rizika
|
95 % IS
|
N
|
P
o
mer
rizika
|
95 % IS
|
Všetky osoby
|
179
|
0,20
|
(0,13; 0,30)
|
179
|
0,10
|
(0,06; 0,17)
|
Delécia 17p
Áno
Nie
|
19
160
|
0,20
0,20
|
(0,06; 0,64) (0,12; 0,31)
|
21
158
|
0,13
0,09
|
(0,04; 0,46) (0,05; 0,17)
|
Mutácia TP53
Áno
Nie
|
19
160
|
0,15
0,20
|
(0,05; 0,46) (0,12; 0,32)
|
21
158
|
0,04
0,11
|
(0,01; 0,22) (0,06; 0,20)
|
Delécia 17p
alebo/a mutácia TP53
Áno
Nie
|
23
156
|
0,23
0,19
|
(0,09; 0,61) (0,11; 0,31)
|
25
154
|
0,10
0,10
|
(0,03; 0,34) (0,05; 0,18)
|
Mutácia IGHV
Zmutovaný
Nezmutovaný
|
58
119
|
0,69
0,11
|
(0,31; 1,56) (0,07; 0,19)
|
74
103
|
0,15
0,08
|
(0,04; 0,52) (0,04; 0,16)
|
Delécia 11q
Áno
Nie
|
31
148
|
0,07
0,26
|
(0,02; 0,22) (0,16; 0,41)
|
31
148
|
0,09
0,10
|
(0,03; 0,26) (0,05; 0,20)
|
Komplexný
karyotyp
Áno
Nie
|
31
117
|
0,10
0,27
|
(0,03; 0,33) (0,16; 0,46)
|
29
126
|
0,09
0,11
|
(0,03; 0,29) (0,05; 0,21)
|
Pacienti s CLL, ktorí v minulosti dostávali aspoň jednu predchádzajúcu liečbu
Bezpečnosť a účinnosť lieku Calquence pri rekurentnej alebo refraktérnej CLL sa hodnotili
v randomizovanej, multicentrickej, otvorenej štúdii fázy 3 (ASCEND) zahŕňajúcej 310 pacientov, ktorí v minulosti dostávali aspoň jednu predchádzajúcu liečbu nezahŕňajúcu inhibítory BCL-2 alebo inhibítory receptora B-buniek. Pacienti dostávali monoterapiu liekom Calquence alebo liečbu podľa výberu skúšajúceho zahŕňajúcu buď idelalisib plus rituximab alebo bendamustín plus rituximab. Pacienti mali v štúdii povolené užívanie antitrombotík. Vylúčení boli pacienti, u ktorých sa vyžadovala antikoagulácia pomocou warfarínu alebo ekvivalentného antagonistu vitamínu K.
Pacienti boli randomizovaní v pomere 1:1 na podávanie buď:
· Calquence 100 mg dvakrát denne až do progresie ochorenia alebo neakceptovateľnej toxicity, alebo
· liečby podľa výberu skúšajúceho:
o idelalisib 150 mg dvakrát denne v kombinácii s rituximabom i.v. 375 mg/m2 v 1. deň prvého cyklu, po ktorom nasleduje 500 mg/m2 i.v. každé 2 týždne (4 dávky), potom
každé 4 týždne (3 dávky), t.j. celkovo 8 infúzií.
o bendamustín 70 mg/m2 (1. a 2. deň každého 28-dňového cyklu) v kombinácii
s rituximabom (375 mg/m2/500 mg/m2) v 1. deň každého 28-dňového cyklu počas až 6
cyklov.
Pacienti boli stratifikovaní podľa stavu mutácie delécie 17p (prítomná delécia vs. bez delécie), výkonnostného stavu podľa ECOG (0 alebo 1 oproti 2) a počtu predchádzajúcich terapií (1 až 3 oproti
≥ 4). Po potvrdení progresie ochorenia prešlo 35 pacientov randomizovaných v skupine s liečbou podľa výberu skúšajúceho (buď idelalisib plus rituximab alebo bendamustín plus rituximab) do skupiny s liekom Calquence. Východiskové demografické charakteristiky a charakteristiky ochorenia populácie štúdie sú zhrnuté v tabuľke 8.
Tabuľka 8: Východiskové pacientske charakteristiky (štúdia ASCEND) u pacientov s CLL
C
h
arakteristika
|
Monoterapia
Calquence
N = 155
|
Liečb
a podľa výberu
skúšajúceho: idelalisib + rituximab alebo bendamustín + rituximab N = 155
|
Vek, roky; medián (rozsah)
|
68 (32 – 89)
|
67 (34 – 90)
|
Mužské pohlavie; %
|
69,7
|
64,5
|
Biela rasa; %
|
93,5
|
91,0
|
Výkonnostný stav podľa ECOG; %
0
1
2
|
37,4
50,3
12,3
|
35,5
51,0
13,5
|
Medián času od diagnózy (mesiace)
|
85,3
|
79,0
|
Masívne ochorenie s uzlinami o veľkosti ≥ 5 cm; %
|
49,0
|
48,4
|
Medián počtu predchádzajúcich terapií pre CLL
(rozsah)
|
1 (1 – 8)
|
2 (1 – 10)
|
Počet predchádzajúcich terapií pre CLL; %
1
2
3
≥ 4
|
52,9
25,8
11,0
10,3
|
43,2
29,7
15,5
11,6
|
Cytogenetika/FISH kategória; %
Delécia 17p Delécia 11q Mutácia TP53
Nezmutovaný IGHV
Komplexný karyotyp (≥ 3 abnormality)
|
18,1
25,2
25,2
76,1
32,3
|
13,5
28,4
21,9
80,6
29,7
|
Rai štádium; %
0
I II III IV
|
1,3
25,2
31,6
13,5
28,4
|
2,6
20,6
34,8
11,6
29,7
|
Primárnym koncovým ukazovateľom bolo PFS na základe hodnotenia IRC podľa kritérií IWCLL
z roku 2008 so zapracovaním vysvetlenia lymfocytózy súvisiacej s liečbou (Cheson 2012).
S mediánom sledovania 16,1 mesiacov PFS dokázalo 69% štatisticky významné zníženie rizika úmrtia alebo progresie ochorenia u pacientov v skupine s liekom Calquence. Výsledky týkajúce sa účinnosti sú uvedené v tabuľke 9. Kaplanova-Meierova krivka PFS je zobrazená na obrázku 2.
Tabuľka 9: Výsledky týkajúce sa účinnosti podľa hodnotenia IRC (štúdia ASCEND)u pacientov s CLL
| Monoterapia Calquence
N = 155
| Liečba podľa výberu skúšajúceho: idelalisib + rituximab alebo bendamustín
+ rituximab
N = 155
|
Prežívanie bez progresie*
|
Počet udalostí (%)
| 27 (17,4)
| 68 (43,9)
|
PD, n (%)
| 19 (12,3)
| 59 (38,1)
|
Úmrtia (%)
| 8 (5,2)
| 9 (5,8)
|
Medián (95 % IS), mesiace
| NR
| 16,5 (14,0; 17,1)
|
HR† (95 % IS)
| 0,31 (0,20; 0,49)
|
Hodnota p
|
< 0,0001
|
15-mesačný odhad, % (95 % IS)
|
82,6 (75,0; 88,1)
|
54,9 (45,4; 63,5)
|
Celkov
é prežívanie
a
|
Úmrtia (%)
|
15 (9,7)
|
18 (11,6)
|
Pomer rizika (95 % IS)†
|
0,84 (0,42; 1,66)
|
-
|
Miera najlepšej celkovej odpovede
*
(CR + CRi + nPR + PR)**
|
ORR, n (%)
(95 % IS)
|
126 (81,3)
(74,4; 86,6)
|
117 (75,5)
(68,1; 81,6)
|
Hodnota p
|
0,2248
|
-
|
CR, n (%)
|
0
|
2 (1,3)
|
PR, n (%)
|
126 (81,3)
|
115 (74,2)
|
Dĺžka trvania odpovede (duration of response, DoR)
|
Medián (95 % IS), mesiace
|
NR
|
13,6 (11,9; NR)
|
IS = interval spoľahlivosti; HR = pomer rizika; NR = nedosiahol sa; CR = úplná odpoveď; CRi = úplná
odpoveď s neúplnou úpravou krvného obrazu; nPR = nodulárna čiastočná odpoveď; PR = čiastočná odpoveď; PD = progresia ochorenia.
*Podľa hodnotenia IRC.
aMedián OS sa v oboch skupinách nedosiahol. P < 0,6089 pre OS.
**CRi a nPR majú hodnotu 0.
†Na základe stratifikovaného Coxovho modelu proporcionálnych rizík.
Obrázok 2: Kaplanova-Meierova krivka PFS podľa hodnotenia IRC (ASCEND) u pacientov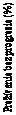 s CLL (populácia podľa liečebného zámeru [ITT])
s CLL (populácia podľa liečebného zámeru [ITT])Calquence
Liečba podľa výberu
skúšajúceho
Čas od randomizácie (mesiace)
Počet pacientov v riziku
|
Mesiac
| 0
| 1
| 2
| 3
| 4
| 5
| 6
| 7
| 8
| 9
| 10
| 11
| 12
| 13
| 14
| 15
| 16
| 17
| 18
| 19
| 20
| 21
| 22
| 23
|
Calquence
| 155
| 153
| 153
| 149
| 147
| 146
| 145
| 143
| 143
| 139
| 139
| 137
| 118
| 116
| 73
| 61
| 60
| 25
| 21
| 21
| 1
| 1
| 1
| 0
|
Liečba podľa
výberu skúšajúceho
| 155
| 150
| 150
| 146
| 144
| 142
| 136
| 130
| 129
| 112
| 105
| 101
| 82
| 77
| 56
| 44
| 39
| 18
| 10
| 8
| 0
|
|
|
|
Výsledky PFS pre Calquence boli konzistentné naprieč podskupinami, vrátane vysokorizikových
charakteristík. Vo vysokorizikovej populácii s CLL (delécia 17p, delécia 11q, mutácia TP53 alebo nezmutovaný IGHV) bol HR pre PFS 0,25 [95 % IS (0,16; 0,38)].
Tabuľka 10: Podskupinová analýza PFS podľa hodnotenia IRC (štúdia ASCEND)
|
Monoterapia Calquence
|
N
|
P
o
me
r rizika
|
95 % IS
|
Všetky osoby
|
155
|
0,27
|
(0,18; 0,40)
|
Delécia 17p
Áno
Nie
|
28
127
|
0,18
0,30
|
(0,07; 0,43) (0,19; 0,47)
|
Mutácia TP53
Áno
Nie
|
39
113
|
0,17
0,33
|
(0,08; 0,37) (0,21; 0,52)
|
Delécia 17p alebo mutácia TP53
Áno
Nie
|
45
108
|
0,16
0,34
|
(0,08; 0,34) (0,22; 0,55)
|
Mutácia IGHV
Zmutovaný
Nezmutovaný
|
33
118
|
0,30
0,28
|
(0,12; 0,76) (0,18; 0,43)
|
Delécia 11q
Áno
Nie
|
39
116
|
0,35
0,26
|
(0,16; 0,75) (0,16; 0,41)
|
Komplexný karyotyp
Áno
Nie
|
50
97
|
0,28
0,25
|
(0,15; 0,53) (0,15; 0,44)
|
Vo finálnej analýze, s mediánom sledovania 46,5 mesiacov pre Calquence a 45,3 mesiacov pre IR/BR,
sa pozorovalo 72 % zníženie rizika progresie ochorenia alebo úmrtia podľa hodnotenia skúšajúceho u pacientov v skupine s liekom Calquence. Medián PFS podľa hodnotenia skúšajúceho sa v skupine s liekom Calquence nedosiahol a v skupine s IR/BR bol 16,8 mesiacov. Výsledky týkajúce sa účinnosti podľa hodnotenia skúšajúceho sú uvedené v tabuľke 11. Kaplanova-Meierova krivka PFS podľa hodnotenia skúšajúceho je zobrazená na obrázku 3.
Tabuľka 11: Výsledky účinnosti vo finálnej analýze podľa hodnotenia skúšajúceho (štúdiaASCEND) u pacientov s CLL
| Monoterapia Calquence
N = 155
| Liečba podľa výberu skúšajúceho: idelalisib + rituximab alebo bendamustín
+ rituximab
N = 155
|
Prežívanie bez progresie*
|
Počet udalostí (%)
| 62 (40,0)
| 119 (76,8)
|
PD, n (%)
| 43 (27,7)
| 102 (65,8)
|
Úmrtia (%)
| 19 (12,3)
| 17 (11,0)
|
Medián (95 % IS), mesiace
| NR
| 16,8 (14,1; 22,5)
|
HR† (95 % IS)
| 0,28 (0,20; 0,38)
|
Celkové prežívaniea
|
Úmrtia (%)
| 41 (26,5)
| 54 (34,8 %)
|
Pomer rizika (95 % IS)†
| 0,69 (0,46; 1,04)
| -
|
IS = interval spoľahlivosti; HR = pomer rizika; NR = nedosiahol sa; PD = progresia ochorenia.
*Podľa hodnotenia skúšajúceho.
aMedián OS sa v oboch skupinách nedosiahol P = 0,0783 pre OS.
†Na základe stratifikovaného Coxovho modelu proporcionálnych rizík.
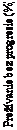 Obrázo
k 3: Kaplanova-Meierova krivka PFS podľa hodnotenia skúšajúceho vo finálnej analýze
(ASCEND) u pacientov s CLL
Calquence
Liečb
a podľa výberu skúšajúceho
Obrázo
k 3: Kaplanova-Meierova krivka PFS podľa hodnotenia skúšajúceho vo finálnej analýze
(ASCEND) u pacientov s CLL
Calquence
Liečb
a podľa výberu skúšajúceho
Čas od randomizácie (mesiace)
Mesiac
| 0
| 3
| 6
| 9
| 12
| 15
| 18
| 21
| 24
| 27
| 30
| 33
| 36
| 39
| 42
| 45
| 48
| 51
| 54
|
Calquence
| 155
| 151
| 143
| 139
| 133
| 128
| 121
| 117
| 111
| 110
| 100
| 94
| 85
| 80
| 79
| 52
| 21
| 4
| 0
|
Liečba podľa výberu
skúšajúceho
| 155
| 147
| 138
| 118
| 95
| 76
| 66
| 62
| 52
| 42
| 35
| 32
| 28
| 26
| 23
| 12
| 5
| 0
|
|
Vo finálnej analýze boli výsledky PFS podľa hodnotenia skúšajúceho pre Calquence konzistentné
naprieč podskupinami, vrátane vysokorizikových charakteristík a boli konzistentné s primárnou analýzou.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Calquence vo všetkých podskupinách pediatrickej populácie v liečbe CLL (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiFarmakokinetika (FK) akalabrutinibu a jeho aktívneho metabolitu, ACP-5862, sa skúmala u zdravých jedincov a u pacientov s malignitami B-buniek. Akalabrutinib vykazuje dávkovo úmernú FK
a akalabrutinib aj ACP-5862 vykazujú takmer lineárnu FK v rozsahu dávok 75 až 250 mg. Populačné FK modelovanie naznačuje, že FK akalabrutinibu a ACP-5862 je rovnaká u pacientov s rozdielnymi malignitami B-buniek. Pri odporúčanej dávke 100 mg dvakrát denne u pacientov s malignitami
B-buniek (vrátane CLL) bol geometrický priemer dennej plochy pod krivkou časového priebehu
plazmatickej koncentrácie v rovnovážnom stave (AUC24 hod) akalabrutinibu 1 679 ng•h/ml
a maximálna plazmatická koncentrácia (Cmax) akalabrutinibu 438 ng/ml, a pre ACP-5862 to boli hodnoty 4 166 ng•h/ml a 446 ng/ml, v uvedenom poradí.
AbsorpciaČas do dosiahnutia maximálnych plazmatických koncentrácií (Tmax) bol 0,5-1,5 hodiny pre akalabrutinib a 1,0 hodina pre ACP-5862. Absolútna biologická dostupnosť lieku Calquence bola
25 %.
Účino
k jedlanaakalabrutinib
U zdravých osôb neovplyvnilo podanie jednorazovej 75 mg dávky akalabrutinibu s vysokokalorickým
jedlom s vysokým obsahom tukov (približne 918 kalórií, 59 gramov sacharidov, 59 gramov tukov a 39 gramov proteínov) priemernú AUC v porovnaní s dávkovaním za podmienok nalačno. Výsledná Cmax sa znížila o 69 % a Tmax bol oneskorený o 1 – 2 hodiny.
Distribúcia
Rozsah reverzibilnej väzby na proteíny ľudskej plazmy bol pre akalabrutinib 99,4 % a pre ACP-5862
bol 98,8 %. Priemerný distribučný pomer krv-plazma in vitro bol pre akalabrutinib 0,8 a pre
ACP-5862 bol 0,7. Priemerný distribučný objem v rovnovážnom stave (Vss) bol pre akalabrutinib približne 34 l.
Biotransformácia/metabolizmus
Akalabrutinib sa in vitro metabolizuje prevažne prostredníctvom enzýmov CYP3A a v menšej miere
prostredníctvom konjugácie s glutatiónom a amidovou hydrolýzou. ACP-5862 bol identifikovaný ako hlavný metabolit v plazme, ktorý sa ďalej metabolizuje predovšetkým oxidáciou sprostredkovanou CYP3A, s geometrickým priemerom expozície (AUC), ktorý bol približne 2- až 3-násobne vyšší ako expozícia akalabrutinibu. ACP-5862 je pri inhibícii BTK približne o 50 % menej účinný ako akalabrutinib.
Štúdie in vitro naznačujú, že akalabrutinib pri klinicky relevantných koncentráciách neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, UGT1A1 ani UGT2B7 a je nepravdepodobné, že by ovplyvňoval klírens substrátov týchto CYP.
Štúdie in vitro naznačujú, že ACP-5862 pri klinicky relevantných koncentráciách neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4/5, UGT1A1 ani UGT2B7 a je nepravdepodobné, že by ovplyvňoval klírens substrátov týchto CYP.
Interakcie s transportnýmiproteínmi
Štúdie in vitro naznačujú, že akalabrutinib a ACP-5862 sú substrátmi P-gp a BCRP. Avšak je
nepravdepodobné, že by ich súbežné podávanie s inhibítormi BCRP malo za následok klinicky významné liekové interakcie. Súbežné podanie s inhibítorom OATP1B1/1B3 (600 mg rifampicínu, jednorazová dávka) viedlo k zvýšeniu Cmax akalabrutinibu 1,2-násobne a AUC 1,4-násobne (N = 24, zdravé osoby), čo nie je klinicky významné.
Akalabrutinib a ACP-5862 pri klinicky relevantných koncentráciách neinhibujú P-gp, OAT1, OAT3, OCT2, OATP1B1, OATP1B3 a MATE2-K. Akalabrutinib môže pri klinicky relevantných koncentráciách inhibovať intestinálny BCRP, zatiaľ čo ACP-5862 môže inhibovať MATE1 (pozri časť 4.5). Akalabrutinib pri klinicky relevantných koncentráciách neinhibuje MATE1, zatiaľ čo
ACP-5862 neinhibuje BCRP.
Eliminácia
Po podaní jednorazovej perorálnej dávky 100 mg akalabrutinibu bol terminálny eliminačný polčas
(t1/2) akalabrutinibu 1 až 2 hodiny. Hodnota t1/2 aktívneho metabolitu, ACP-5862, bola približne 7
hodín.
Priemerný zdanlivý perorálny klírens (CL/F) u pacientov s malignitami B-buniek bol pre akalabrutinib
134 l/hod a pre ACP-5862 bol 22 l/hod.
Po podaní jednorazovej 100 mg dávky rádioaktívne značeného [14C]-akalabrutinibu zdravým osobám sa 84 % dávky zachytilo v stolici a 12 % dávky sa zachytilo v moči, pričom menej ako 2 % dávky sa vylúčili ako nezmenený akalabrutinib.
Osobitné skupinypacientov
Na základe populačnej FK analýzy nemali vek (> 18 rokov), pohlavie, rasa (biela, afroamerická)
a telesná hmotnosť klinicky významné účinky na FK akalabrutinibu a jeho aktívneho metabolitu, ACP-5862.
Pediatrická populácia
U pacientov mladších ako 18 rokov sa neuskutočnili žiadne farmakokinetické štúdie s liekom
Calquence.
Porucha funkcie obličiek
Akalabrutinib podlieha minimálnej renálnej eliminácii. Farmakokinetická štúdia u pacientov s poruchou funkcie obličiek sa neuskutočnila.
Na základe populačnej FK analýzy sa u 408 jedincov s miernou poruchou funkcie obličiek (eGFR medzi 60 a 89 ml/min/1,73 m2 na základe odhadu podľa MDRD), 109 jedincov so stredne závažnou poruchou funkcie obličiek (eGFR medzi 30 a 59 ml/min/1,73 m2) v porovnaní so 192 jedincami
s normálnou funkciou obličiek (eGFR vyššia ako alebo rovnajúca sa 90 ml/min/1,73 m2) nepozoroval žiadny klinicky významný FK rozdiel. Farmakokinetika akalabrutinibu nebola charakterizovaná
u pacientov so závažnou poruchou funkcie obličiek (eGFR nižšia ako 29 ml/min/1,73 m2) ani
u pacientov s poruchou funkcie obličiek vyžadujúcou dialýzu. V klinických štúdiách neboli zahrnutí pacienti s hladinami kreatinínu vyššími ako 2,5-násobok ULN pre danú inštitúciu (pozri časť 4.2).
Porucha funkcie pečene
Akalabrutinib sa metabolizuje v pečeni. V špecializovaných štúdiách poruchy funkcie pečene bola expozícia (AUC) akalabrutinibu v porovnaní s osobami s normálnou funkciou pečene (n = 6) zvýšená
1,9-násobne, 1,5-násobne a 5,3-násobne u osôb s miernou (n = 6) (Childovo-Pughovo skóre A), stredne závažnou (n = 6) (Childovo-Pughovo skóre B) a závažnou (n = 8) (Childovo-Pughovo skóre C) poruchou funkcie pečene, v uvedenom poradí. Markery dôležité pre určenie eliminačnej kapacity
liečiv neboli významne ovplyvnené u osôb v skupine so stredne závažnou poruchou funkcie pečene, preto vplyv stredne závažnej poruchy funkcie pečene bol v tejto štúdii pravdepodobne podhodnotený. Na základe populačnej FK analýzy sa u osôb s miernou (n = 79) alebo stredne závažnou (n = 6) poruchou funkcie pečene (celkový bilirubín v hodnote medzi 1,5- až 3-násobkom ULN a akákoľvek hodnota AST) v porovnaní s osobami s normálnou (n = 613) funkciou pečene (celkový bilirubín
a AST v rámci ULN) nepozoroval žiadny klinicky významný rozdiel (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Karcinogenita
Štúdie karcinogénneho potenciálu s akalabrutinibom sa neuskutočnili.
Genotoxicita/mutagenita/fototoxicita
Akalabrutinib nebol mutagénny v teste bakteriálnej reverznej mutácie, v teste chromozómovej
aberácie in vitro ani v teste mikrojadierok v kostnej dreni u myší in vivo.
Na základe in vitro testov fototoxicity pomocou 3T3 bunkovej línie má akalabrutinib u ľudí nízke riziko fototoxicity.
Toxicita poopakovanompodávaní
U potkanov sa pri všetkých hladinách dávky v pankrease pozorovali mikroskopické nálezy
(hemorágia/pigment/zápal/fibróza v ostrovčekoch) minimálnej až miernej závažnosti. V štúdiách trvajúcich až 6 mesiacov s hladinou dávky bez pozorovaných nežiaducich účinkov (No Observed Adverse Effect Level, NOAEL), t.j. 30 mg/kg/deň u potkanov, sa v obličkách pozorovali nálezy minimálnej až miernej závažnosti (tubulárna bazofília, tubulárna regenerácia a zápal), ktoré neboli nežiaduce. Priemerné expozície (AUC) pri NOAEL u samcov a samíc potkana zodpovedajú
0,6-násobku a 1-násobku, v uvedenom poradí, klinickej expozície pri odporúčanej dávke 100 mg dvakrát denne. Najnižšia hladina dávky s pozorovanými nežiaducimi účinkami (Lowest Observed Adverse Effect Level, LOAEL), pri ktorej sa pozorovali reverzibilné nálezy týkajúce sa obličiek (stredne závažná tubulárna degenerácia) a pečene (nekróza individuálnych hepatocytov) v chronickej štúdii na potkanoch, bola 100 mg/kg/deň a poskytovala rozmedzie expozície 4,2-násobne väčšie ako je klinická expozícia pri odporúčanej dávke 100 mg dvakrát denne. V štúdiách trvajúcich 9 mesiacov
u psov bola hladina NOAEL 10 mg/kg/deň, čo zodpovedá expozícii 3-krát vyššej ako je klinická AUC pri odporúčanej klinickej dávke. U psov pri dávke 30 mg/kg/deň (9-násobok klinickej AUC) sa pozorovala minimálna degenerácia tubulov v obličkách, mierne zníženie hmotnosti sleziny
a prechodné minimálne až mierne zníženie množstva erytrocytov a zvýšenie ALT a ALP. Toxicity týkajúce sa srdca u potkanov (myokardiálna hemorágia, zápal, nekróza) a psov (perivaskulárny/ vaskulárny zápal) sa pozorovali len u zvierat, ktoré uhynuli počas štúdií a to pri dávkach vyšších ako je maximálna tolerovaná dávka (MTD). Expozície u potkanov a psov s nálezmi týkajúcimi sa srdca boli aspoň 6,8-násobkom a 25-násobkom klinickej AUC, v uvedenom poradí. Reverzibilitu nálezov týkajúcich sa srdca nebolo možné posúdiť, keďže tieto nálezy sa pozorovali iba pri dávkach vyšších ako MTD.
Reprodukčná toxikológia
U samcov alebo samíc potkana sa pri expozíciách v hodnote 10- alebo 9-násobku klinickej AUC pri
odporúčanej dávke, v uvedenom poradí, nepozorovali žiadne účinky na fertilitu.
U gravidných potkanov sa pri expozíciách v hodnote približne 9-násobku AUC pri odporúčanej dávke
100 mg dvakrát denne u pacientov nepozorovali žiadne účinky na embryonálno-fetálny vývin a prežívanie. V dvoch reprodukčných štúdiách na potkanoch sa pri expozíciách
v hodnote > 2,3-násobku klinickej expozície pri dávke 100 mg dvakrát denne pozorovala dystokia (zdĺhavý/náročný pôrod). V plazme plodu potkana bola potvrdená prítomnosť akalabrutinibu a jeho aktívneho metabolitu. Akalabrutinib a jeho aktívny metabolit boli prítomné v mlieku laktujúcich
potkanov.
V embryonálno-fetálnej štúdii na gravidných králikoch sa pri hladinách expozície, ktoré viedli k materskej toxicite a ktoré boli 2,4-násobne vyššie ako AUC pri odporúčanej dávke u ľudí, pozorovali znížená telesná hmotnosť plodu a oneskorená osifikácia.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsuly
celulóza, mikrokryštalická
oxid kremičitý, koloidný, bezvodý
kukuričný škrob, čiastočne predželatinovaný stearát horečnatý (E470b) karboxymetylškrob, sodná soľ
Obal kapsuly
želatína
oxid titaničitý (E171) žltý oxid železitý (E172) indigokarmín (E132)
Atrament potlače
šelak
čierny oxid železitý (E172) propylénglykol (E1 520) hydroxid amónny
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky.
6.4 Špeciálne upozornenia na uchovávanie
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Hliník/hliníkové blistre so symbolmi predstavujúcimi slnko/mesiac obsahujúce 6 alebo 8 tvrdých kapsúl. Škatuľky po 56 alebo 60 kapsúl.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
AstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)
EU/1/20/1479/001
EU/1/20/1479/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIE
Dátum prvej registrácie: 5. november 2020
10. DÁTUM REVÍZIE TEXTU
Podrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu
Tento liek je predmetom ďalšieho monitorovania. To umožní rýchle získanie nových informácií o bezpečnosti. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie. Informácie o tom, ako hlásiť nežiaduce reakcie, nájdete v časti 4.8.
1. NÁZOV LIEKUCalquence 100 mg filmom obalené tablety
2. KVALITATÍVNE A KVANTITATÍVNE ZLOŽENIEKaždá filmom obalená tableta obsahuje 100 mg akalabrutinibu (vo forme akalabrutinib-maleátu). Úplný zoznam pomocných látok, pozri časť 6.1.
3. LIEKOVÁ FORMAFilmom obalená tableta.
Oranžová, oválna, bikonvexná tableta s rozmerom 7,5 x 13 mm, s označením „ACA 100“ na jednej strane a bez označenia na druhej strane.
4. KLINICKÉ ÚDAJE4.1 Terapeutické indikácieCalquence v monoterapii alebo v kombinácii s obinutuzumabom je indikovaný dospelým pacientom na liečbu v minulosti neliečenej chronickej lymfocytovej leukémie (chronic lymphocytic leukaemia, CLL).
Calquence v monoterapii je indikovaný dospelým pacientom na liečbu chronickej lymfocytovej leukémie (CLL), ktorí dostávali aspoň jednu predchádzajúcu liečbu.
4.2 Dávkovanie a spôsob podávaniaLiečbu týmto liekom musí začať a viesť lekár, ktorý má skúsenosti s používaním protinádorových liekov.
DávkovanieOdporúčaná dávka je 100 mg akalabrutinibu dvakrát denne (čo zodpovedá celkovej dennej dávke
200 mg). Pre informácie týkajúce sa odporúčaného dávkovania obinutuzumabu si pozrite súhrn
charakteristických vlastností lieku s obsahom obinutuzumabu. Dávkovací interval je približne 12 hodín.
V liečbe liekom Calquence sa má pokračovať až do progresie ochorenia alebo neakceptovateľnej toxicity.
Úpravy dávkyNežiaduce reakcieOdporúčané úpravy dávky lieku Calquence pri nežiaducich reakciách ≥ 3. stupňa sú uvedené v tabuľke 1.
Tabuľka 1: Odporúčané úpravy dávky pri nežiaducich reakciách*Nežiaduca reakcia
| Výskyt
nežiaducej reakcie
| Úprava dávky
(Začiatočná dávka = 100 mg približne každých 12
hodín)
|
Trombocytopénia 3. stupňa s krvácaním,
Trombocytopénia 4. stupňa alebo
Neutropénia 4. stupňa
pretrvávajúca dlhšie ako 7 dní
Nehematologické toxicity 3. alebo vyššieho stupňa
| Prvýkrát
a druhýkrát
| Prerušte liečbu liekom Calquence.
Keď sa toxicita upraví na 1. stupeň alebo
východiskový stav, v liečbe liekom Calquence sa môže pokračovať v dávke 100 mg približne každých
12 hodín.
|
Tretíkrát
| Prerušte liečbu liekom Calquence.
Keď sa toxicita upraví na 1. stupeň alebo východiskový stav, v liečbe liekom Calquence sa môže pokračovať v zníženej frekvencii v dávke
100 mg jedenkrát denne.
|
Štvrtýkrát
| Ukončite liečbu liekom Calquence.
|
*Nežiaduce reakcie odstupňované podľa všeobecných terminologických kritérií pre nežiaduce udalosti
podľa Národného inštitútu pre rakovinu (National Cancer Institute Common Terminology Criteria for
Adverse Events, NCI CTCAE) verzie 4.03.
InterakcieOdporúčania týkajúce sa použitia lieku Calquence spolu s inhibítormi alebo induktormi CYP3A sú uvedené v tabuľke 2 (pozri časť 4.5).
Tabuľka 2: Použitie s inhibítormi alebo induktormi CYP3A
|
Súbežne podávaný liek
|
Odporúčané použitie lieku Calquence
|
Inhibítory
CYP3A
|
Silný inhibítor CYP3A
| Vyhnite sa súbežnému použitiu.
Ak sa tieto inhibítory budú podávať krátkodobo (ako napríklad antiinfektíva počas siedmich dní), liečbu liekom Calquence prerušte.
|
Stredne silný inhibítor
CYP3A
| Žiadna úprava dávky. U pacientov starostlivo sledujte výskyt nežiaducich reakcií, ak užívajú stredne silné
inhibítory CYP3A.
|
Slabý inhibítor CYP3A
| Žiadna úprava dávky.
|
Induktory
CYP3A
|
Silný induktor CYP3A
|
Vyhnite sa súbežnému použitiu.
|
Tablety akalabrutinibu sa môžu podávať súbežne s liečivami znižujúcimi žalúdočnú kyselinu
(inhibítory protónovej pumpy, antagonisty H2-receptorov, antacidá), na rozdiel od kapsúl akalabrutinibu, ktoré vykazujú zhoršenú absorpciu, keď sa podávajú s liečivami znižujúcimi žalúdočnú kyselinu (pozri časť 4.5).
Vynechaná dávka
Ak pacient vynechá dávku lieku Calquence a s užitím sa oneskorí o viac ako 3 hodiny, je potrebné ho
poučiť, aby ďalšiu dávku užil v bežnom naplánovanom čase. Na nahradenie vynechanej dávky sa nemá užívať dvojnásobná dávka lieku Calquence.
Osobitné skupinypacientov
Staršie osoby
U starších pacientov (vo veku ≥ 65 rokov) nie je potrebná žiadna úprava dávky (pozri časť 5.2).
Porucha funkcie obličiek
U pacientov s poruchou funkcie obličiek sa neuskutočnili žiadne špecifické klinické štúdie.
V klinických štúdiách s Calquence sa liečili pacienti s miernou alebo stredne závažnou poruchou funkcie obličiek. U pacientov s miernou alebo stredne závažnou poruchou funkcie obličiek (klírens kreatinínu vyšší ako 30 ml/min) nie je potrebná žiadna úprava dávky. Má sa udržiavať dostatočná hydratácia a pravidelne sa majú sledovať hladiny kreatinínu v sére. Pacientom so závažnou poruchou funkcie obličiek (klírens kreatinínu < 30 ml/min) sa má podávať Calquence iba v prípade, že prínos preváži riziko a u týchto pacientov sa majú starostlivo sledovať prejavy toxicity. U pacientov so závažnou poruchou funkcie obličiek a pacientov na dialýze nie sú k dispozícii žiadne údaje (pozri časť
5.2).
Porucha funkcie pečene
U pacientov s miernou alebo stredne závažnou poruchou funkcie pečene (Childovo-Pughovo skóre A, Childovo-Pughovo skóre B alebo celkový bilirubín medzi 1,5 – 3-násobkom hornej hranice normálu
[upper limit of normal, ULN] a akákoľvek hodnota AST) sa neodporúča žiadna úprava dávky. Avšak, u pacientov so stredne závažnou poruchou funkcie pečene sa majú dôkladne sledovať prejavy toxicity. Calquence sa neodporúča používať u pacientov so závažnou poruchou funkcie pečene (Childovo- Pughovo skóre C alebo celkový bilirubín > 3-násobok ULN a akákoľvek hodnota AST) (pozri časť
5.2).
Závažné srdcové ochorenie
Z klinických štúdií s Calquence boli vyradení pacienti so závažným kardiovaskulárnym ochorením.
Pediatrická populácia
Bezpečnosť a účinnosť Calquence u detí a dospievajúcich vo veku 0 až 18 rokov neboli stanovené. K dispozícii nie sú žiadne údaje.
Spôsob podávania
Calquence je určený na perorálne použitie. Tablety sa majú prehltnúť vcelku a zapiť vodou každý deň
približne v rovnakom čase, s jedlom alebo bez jedla (pozri časť 4.5). Tablety sa nemajú žuvať, drviť,
rozpúšťať ani deliť.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
4.
4 Osobitné upozornenia a opatrenia pri používaní
Hemorágia
U pacientov s hematologickými malignitami liečených monoterapiou liekom Calquence alebo
v kombinácii s obinutuzumabom sa vyskytli závažné hemoragické udalosti vrátane krvácania do
centrálneho nervového systému a gastrointestinálnej hemorágie, pričom niektoré mali fatálne následky. Tieto udalosti sa vyskytli u pacientov s trombocytopéniou aj u pacientov bez trombocytopénie. Celkovo boli krvácavé udalosti menej závažnými udalosťami vrátane tvorby podliatin a petechií (pozri časť 4.8).
Mechanizmus krvácavých udalostí nie je dostatočne objasnený.
Pacienti užívajúci antitrombotiká môžu mať zvýšené riziko hemorágie. Pri použití antitrombotík postupujte s opatrnosťou a zvážte dodatočné sledovanie prejavov krvácania, ak je súbežné použitie medicínsky nevyhnutné. Warfarín alebo iné antagonisty vitamínu K sa nemajú podávať súčasne
s liekom Calquence.
Zvážte pomer prínosu a rizika prerušenia liečby liekom Calquence počas aspoň 3 dní pred a po chirurgickom zákroku.
Infekcie
U pacientov s hematologickými malignitami liečených monoterapiou liekom Calquence
a v kombinácii s obinutuzumabom sa vyskytli závažné infekcie (bakteriálne, vírusové alebo mykotické) vrátane fatálnych udalostí. Tieto infekcie sa vyskytovali predovšetkým bez prítomnosti neutropénie 3. alebo 4. stupňa, pričom u 1,9 % všetkých pacientov bola hlásená neutropenická
infekcia. Vyskytli sa infekcie spôsobené reaktiváciou vírusu hepatitídy B (HBV) a vírusu herpes zoster
(HZV), aspergilóza a progresívna multifokálna leukoencefalopatia (PML) (pozri časť 4.8).
Vírusová reaktivácia
U pacientov užívajúcich Calquence sa hlásili prípady reaktivácie vírusu hepatitídy B. Pred začatím liečby liekom Calquence sa má stanoviť stav vírusu hepatitídy B (HBV). Ak majú pacienti pozitívnu sérológiu hepatitídy B, je potrebné sa pred začatím liečby poradiť s odborníkom na hepatálne ochorenia a pacienta je potrebné sledovať a manažovať podľa lokálnych medicínskych štandardov na prevenciu reaktivácie hepatitídy B.
V súvislosti s predchádzajúcou alebo súbežnou imunosupresívnou liečbou sa po použití Calquence hlásili prípady progresívnej multifokálnej leukoencefalopatie (PML) vrátane fatálnych prípadov. Pri diferenciálnej diagnóze u pacientov s novými alebo zhoršujúcimi sa neurologickými, kognitívnymi alebo behaviorálnymi prejavmi alebo príznakmi majú lekári zvážiť PML. Pri podozrení na PML sa majú vykonať vhodné diagnostické vyšetrenia a liečba liekom Calquence sa má prerušiť dovtedy, kým sa nevylúči PML. V prípade akýchkoľvek pochybností sa má zvážiť odporučenie pacienta
k neurológovi a vhodné diagnostické opatrenia na PML, vrátane vyšetrenia snímaním MRI prednostne s použitím kontrastnej látky, testovanie cerebrospinálnej tekutiny (cerebrospinal fluid, CSF) na JC vírusovú DNA a opakované neurologické vyšetrenia.
U pacientov so zvýšeným rizikom oportúnnych infekcií zvážte profylaxiu na základe štandardnej liečby. U pacientov sledujte prejavy a príznaky infekcie a liečte ich tak, ako je medicínsky vhodné.
Cytopénie
U pacientov s hematologickými malignitami liečených monoterapiou liekom Calquence
a v kombinácii s obinutuzumabom sa vyskytli cytopénie 3. alebo 4. stupňa súvisiace s liečbou vrátane neutropénie, anémie a trombocytopénie. Podľa zdravotnej indikácie sledujte úplný krvný obraz (pozri časť 4.8).
D
ruhé primárnemalignity
U pacientov s hematologickými malignitami liečených monoterapiou liekom Calquence
a v kombinácii s obinutuzumabom sa vyskytli druhé primárne malignity vrátane kožných a iných ako kožných karcinómov. Často sa hlásili kožné karcinómy. Sledujte u pacientov výskyt kožných karcinómov a odporučte im ochranu pred expozíciou slnečnému žiareniu (pozri časť 4.8).
Atriálna fibrilácia
U pacientov s hematologickými malignitami liečených monoterapiou liekom Calquence
a v kombinácii s obinutuzumabom sa vyskytla atriálna fibrilácia/flutter. U pacientov sledujte príznaky (napr. palpitácie, závrat, synkopa, bolesť v hrudníku, dyspnoe) atriálnej fibrilácie a atriálneho flutteru a podľa zdravotnej indikácie vykonajte EKG (pozri časti 4.5 a 4.2). U pacientov, u ktorých sa počas liečby liekom Calquence objaví atriálna fibrilácia, sa má vykonať dôkladné zhodnotenie rizika tromboembolického ochorenia. U pacientov s vysokým rizikom tromboembolického ochorenia sa má zvážiť striktne kontrolovaná liečba antikoagulanciami a alternatívne možnosti liečby voči lieku Calquence.
Iné lieky
Súbežné podávanie silných inhibítorov CYP3A spolu s liekom Calquence môže viesť k zvýšenej
expozícii akalabrutinibu a následne k vyššiemu riziku toxicity. Naopak, súbežné podávanie
s induktormi CYP3A môže viesť k zníženej expozícii akalabrutinibu a následne k riziku nedostatočnej účinnosti. Súbežnému používaniu so silnými inhibítormi CYP3A sa treba vyhýbať. Ak sa tieto inhibítory budú používať krátkodobo (ako sú antiinfektíva po dobu siedmich dní), liečba liekom Calquence sa má prerušiť. Ak sa podáva súčasne stredne silný inhibítor CYP3A, u pacientov je potrebné starostlivo sledovať prejavy toxicity (pozri časti 4.2 a 4.5). Súbežnému používaniu so silnými induktormi CYP3A4 sa treba vyhýbať z dôvodu rizika nedostatočnej účinnosti.
Calquence obsahujesodík
Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v dávke, t.j. v podstate zanedbateľné množstvo
sodíka.
4.5 Liekové a iné interakcie
Akalabrutinib a jeho aktívny metabolit sú primárne metabolizované prostredníctvom enzýmu 3A4 cytochrómu P450 (CYP3A4) a obidve chemické látky sú substrátmi P-gp a proteínu podmieňujúceho rezistenciu voči karcinómu prsníka (breast cancer resistance protein, BCRP).
Liečivá, ktorémôžuzvyšovaťplazmatickékoncentrácieakalabrutinibu
Inhibítory CYP3A/P-gp
Súbežné podávanie so silným inhibítorom CYP3A/P-gp (200 mg itrakonazolu jedenkrát denne počas 5
dní) zvýšilo u zdravých osôb (N = 17) Cmax akalabrutinibu 3,9-násobne a AUC 5,0-násobne.
Súbežnému používaniu so silnými inhibítormi CYP3A/P-gp sa treba vyhýbať. Ak sa silné inhibítory CYP3A/P-gp (napr. ketokonazol, konivaptán, klaritromycín, indinavir, itrakonazol, ritonavir, telaprevir, posakonazol, vorikonazol) budú podávať krátkodobo, liečba liekom Calquence sa má prerušiť (pozri časť 4.2).
Súbežné podávanie so stredne silnými inhibítormi CYP3A (400 mg flukonazolu ako jednorazová dávka alebo 200 mg isavukonazolu v opakovanej dávke počas 5 dní) zvýšilo u zdravých osôb Cmax
a AUC akalabrutinibu 1,4-násobne až 2-násobne, zatiaľ čo Cmax a AUC aktívneho metabolitu ACP-
5862 sa znížilo 0,65-násobne až 0,88-násobne v porovnaní s podávaním samotného akalabrutinibu. Pri
kombinovanom podávaní so stredne silnými inhibítormi CYP3A nie je potrebná žiadna úprava dávky. U pacientov je potrebné starostlivo sledovať výskyt nežiaducich reakcií (pozri časť 4.2).
Liečivá, ktorémôžuznižovaťplazmatickékoncentrácieakalabrutinibu
Induktory CYP3A
Súbežné podávanie so silným induktorom CYP3A (600 mg rifampicínu jedenkrát denne počas 9 dní)
znížilo u zdravých osôb (N = 24) Cmax akalabrutinibu o 68 % a AUC o 77 %.
Súbežnému používaniu so silnými induktormi aktivity CYP3A (napr. fenytoín, rifampicín, karbamazepín) sa treba vyhýbať. Súbežnému používaniu s ľubovníkom bodkovaným (Hypericum perforatum), ktorý môže nepredvídateľne znížiť plazmatické koncentrácie akalabrutinibu, sa treba vyhýbať.
Lieky znižujúce žalúdočnú kyselinu
Nepozorovali sa žiadne klinicky významné rozdiely vo farmakokinetike akalabrutinibu, keď sa
100 mg tableta akalabrutinibu používala súčasne s inhibítorom protónovej pumpy (20 mg rabeprazolu dvakrát denne počas 3 dní). Tablety akalabrutinibu sa môžu podávať súbežne s liečivami znižujúcimi žalúdočnú kyselinu (inhibítory protónovej pumpy, antagonisty H2-receptorov, antacidá), na rozdiel od kapsúl akalabrutinibu, ktoré vykazujú zhoršenú absorpciu, keď sa podávajú s liečivami znižujúcimi žalúdočnú kyselinu.
Liečivá, ktorýchplazmatickékoncentráciemôžubyťovplyvnenéliekomCalquence
Substráty CYP3A
Na základe údajov in vitro nie je možné vylúčiť, že akalabrutinib je inhibítorom CYP3A4 na črevnej úrovni a môže zvyšovať expozíciu substrátom CYP3A4 citlivým na črevný metabolizmus CYP3A. Pri súbežnom podávaní akalabrutinibu so substrátmi CYP3A4 s úzkym terapeutickým rozmedzím podávanými perorálne (napr. cyklosporín, ergotamín, pimozid) je potrebná opatrnosť.
Účinok akalabrutinibu na substráty CYP1A2
Štúdie in vitro naznačujú, že akalabrutinib indukuje CYP1A2. Súbežné podávanie akalabrutinibu so substrátmi CYP1A2 (napr. teofylín, kofeín) môže znížiť ich expozíciu.
Účinok akalabrutinibu a jeho aktívneho metabolitu, ACP-5862, na transportné systémy liekov
Akalabrutinib môže zvýšiť expozíciu súbežne podávaným substrátom BCRP (napr. metotrexát)
inhibíciou intestinálneho BCRP (pozri časť 5.2). Na minimalizáciu možnosti interakcie
v gastrointestinálnom trakte sa perorálne podávané substráty BCRP s úzkym terapeutickým rozmedzím, ako je napríklad metotrexát, majú užívať aspoň 6 hodín pred alebo po akalabrutinibe.
ACP-5862 môže zvýšiť expozíciu súbežne podávaným substrátom MATE1 (napr. metformín) inhibíciou MATE1 (pozri časť 5.2). Pacientov súbežne užívajúcich lieky s distribúciou závislou od MATE1 (napr. metformín) je potrebné sledovať pre prejavy zmenenej znášanlivosti v dôsledku zvýšenej expozície súbežne podávanému lieku počas užívania lieku Calquence.
4.6 Fertilita, gravidita a laktácia
Ženy vofertilnomveku
Ženám vo fertilnom veku je potrebné odporučiť, aby sa počas užívania lieku Calquence vyhli
gravidite.
Gravidita
Nie sú k dispozícii alebo je iba obmedzené množstvo údajov o použití akalabrutinibu u gravidných
žien. Na základe zistení zo štúdií na zvieratách môže počas gravidity expozícia akalabrutinibu predstavovať riziko pre plod. U potkanov sa pozorovala dystokia (náročný alebo zdĺhavý pôrod) a podávanie gravidným králikom bolo spojené so zníženým fetálnym rastom (pozri časť 5.3). Calquence sa nemá používať počas gravidity, pokiaľ klinický stav ženy nevyžaduje liečbu akalabrutinibom.
Dojčenie
Nie je známe, či sa akalabrutinib vylučuje do ľudského mlieka. K dispozícii nie sú žiadne údaje
o účinku akalabrutinibu na dojča alebo tvorbu mlieka. Prítomnosť akalabrutinibu a jeho aktívneho
metabolitu sa preukázala v mlieku laktujúcich potkanov. Riziko u dojčiat nemôže byť vylúčené. Dojčiacim matkám sa počas liečby liekom Calquence a počas 2 dní po užití poslednej dávky odporúča nedojčiť.
Fertilita
K dispozícii nie sú žiadne údaje týkajúce sa účinkov lieku Calquence na fertilitu u ľudí.
V predklinickej štúdii na samcoch a samiciach potkana s akalabrutinibom sa nepozorovali žiadne nežiaduce účinky na parametre fertility (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Calquence nemá žiadny alebo má zanedbateľný vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Počas liečby akalabrutinibom sa však hlásila únava a závrat a pacientom, u ktorých sa vyskytnú tieto príznaky, sa má odporučiť, aby neviedli vozidlá ani neobsluhovali stroje, až kým príznaky neustúpia.
4.8 Nežiaduce účinky
Súhrn bezpečnostnéhoprofilu
U 1 040 pacientov liečených monoterapiou liekom Calquence boli najčastejšími (≥ 20 %) nežiaducimi
liekovými reakciami akéhokoľvek stupňa infekcia (66,7 %), bolesť hlavy (37,8 %), hnačka (36,7 %), tvorba podliatin (34,1 %), muskuloskeletálna bolesť (33,1 %), nevoľnosť (21,7 %), únava (21,3 %), kašeľ (21 %) a vyrážka (20,3 %). Najčastejšie (≥ 5 %) hlásené nežiaduce liekové reakcie ≥ 3. stupňa boli infekcia (17,6 %), leukopénia (14,3 %), neutropénia (14,2 %) a anémia (7,8 %).
U 223 pacientov liečených kombinovanou liečbou s liekom Calquence boli najčastejšími (≥ 20 %) nežiaducimi liekovými reakciami akéhokoľvek stupňa infekcia (74 %), muskuloskeletálna bolesť (44,8 %), hnačka (43,9 %), bolesť hlavy (43 %), leukopénia (31,8 %), neutropénia (31,8 %), kašeľ (30,5 %), únava (30,5 %), artralgia (26,9 %), nevoľnosť (26,9 %), závrat (23,8 %) a zápcha (20,2 %). Najčastejšie (≥ 5 %) hlásené nežiaduce liekové reakcie ≥ 3. stupňa boli leukopénia (30 %), neutropénia (30 %), infekcia (21,5 %), trombocytopénia (9 %) a anémia (5,8 %).
Tabuľkový zoznamnežiaducichreakcií
V klinických štúdiách u pacientov užívajúcich Calquence ako liečbu hematologických malignít boli
identifikované nasledujúce nežiaduce liekové reakcie. Medián trvania liečby liekom Calquence
hodnotený na základe sumárnych dát bol 26,2 mesiacov.
Nežiaduce liekové reakcie sú uvedené podľa triedy orgánových systémov MedDRA. V rámci každej triedy orgánových systémov sú nežiaduce liekové reakcie usporiadané podľa frekvencie výskytu, pričom najčastejšie sa vyskytujúce reakcie sú uvedené ako prvé. Navyše, príslušné kategórie frekvencie výskytu sú pre každú nežiaducu liekovú reakciu definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000);
veľmi zriedkavé (< 1/10 000); neznáme (z dostupných údajov). V rámci každej skupiny frekvencie výskytu sú nežiaduce reakcie uvedené v poradí klesajúcej závažnosti.
Tabuľka 3: Nežiaduce liekové reakcie* u pacientov s hematologickými malignitami liečených monoterapiou akalabrutinibom (n = 1 040)
Trieda orgánových systémov MedDRA
|
MedDRA výraz
| Celková frekvencia výskytu
(všetky stupne CTCAE)
|
Frekvencia výskytu
CTCAE ≥ 3. stupňa†
|
Infekcie a nákazy
| Infekcia horných dýchacích ciest
| Veľmi časté (22 %)
| 0,8 %
|
Sinusitída
| Veľmi časté (10,7 %)
| 0,3 %
|
Pneumónia
| Časté (8,7 %)
| 5,1 %
|
Infekcia močových ciest
| Časté (8,5 %)
| 1,5 %
|
Nazofaryngitída
| Časté (7,4 %)
| 0 %
|
Bronchitída
| Časté (7,6 %)
| 0,3 %
|
Herpetické vírusové infekcie†
| Časté (5,9 %)
| 0,7 %
|
Aspergilové infekcie†
| Menej časté (0,5 %)
| 0,4 %
|
Reaktivácia hepatitídy B
| Menej časté (0,1 %)
| 0,1 %
|
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
| †
Druhá primárna malignita (SPM)
Nemelanómová kožná malignita†
SPM s výnimkou nemelanómového kožného karcinómu†
| Veľmi časté (12,2 %)
Časté (6,6 %) Časté (6,5 %)
| 4,1 %
0,5 %
3,8 %
|
Poruchy krvi
a lymfatického systému
| Neutropénia†
| Veľmi časté (15,7 %)
| 14,2 %
|
Anémia†
| Veľmi časté (13,8 %)
| 7,8 %
|
†
Trombocytopénia
| Časté (8,9 %)
| 4,8 %
|
Lymfocytóza
| Menej časté (0,3 %)
| 0,2 %
|
Poruchy metabolizmu a výživy
|
Syndróm nádorového rozpadu±
|
Menej časté (0,5 %)
|
0,4 %
|
Poruchy nervového systému
| Bolesť hlavy
| Veľmi časté (37,8 %)
| 1,1 %
|
Závrat
| Veľmi časté (13,4 %)
| 0,2 %
|
Poruchy srdca
a srdcovej činnosti
| †
Atriálna fibrilácia/flutter
|
Časté (4,4 %)
|
1,3 %
|
Poruchy ciev
| Tvorba podliatin†
Kontúzia Petechie Ekchymózy
| Veľmi časté (34,1 %)
Veľmi časté (21,7 %) Veľmi časté (10,7 %) Časté (6,3 %)
| 0 %
0 %
0 %
0 %
|
Hemorágia/hematóm†
Gastrointestinálna hemorágia
Intrakraniálna hemorágia
| Veľmi časté (12,6 %)
Časté (2,3 %) Časté (1 %)
| 1,8 %
0,6 %
0,5 %
|
Epistaxa
| Časté (7 %)
| 0,3 %
|
Poruchy gastrointestinálneho traktu
| Hnačka
| Veľmi časté (36,7 %)
| 2,6 %
|
Nevoľnosť
| Veľmi časté (21,7 %)
| 1,2 %
|
Zápcha
| Veľmi časté (14,5 %)
| 0,1 %
|
Vracanie
| Veľmi časté (13,3 %)
| 0,9 %
|
Abdominálna bolesť†
| Veľmi časté (12,5 %)
| 1 %
|
Poruchy kože a podkožného tkaniva
|
Vyrážka†
|
Veľmi časté (20,3 %)
|
0,6 %
|
Poruch
y kostrovej a svalovej sústavy a spojivového tkaniva
|
Muskuloskeletálna bolesť†
|
Veľmi časté (33,1 %)
|
1,5 %
|
Artralgia
|
Veľmi časté (19,1 %)
|
0,7 %
|
Celkov
é poruchy a reakcie v mieste podania
|
Únava
|
Veľmi časté (21,3 %)
|
1,7 %
|
Asténia
|
Časté (5,3 %)
|
0,8 %
|
Laboratórne
a funkčné vyšetrenia
¶
(nález
y na základe
výsledkov vyšetrení)
|
Znížená hladina hemoglobínu§
|
Veľmi časté (42,6 %)
|
10,1 %
|
Znížený absolútny počet neutrofilov§
|
Veľmi časté (41,8 %)
|
20,7 %
|
Znížený počet trombocytov§
|
Veľmi časté (31,1 %)
|
6,9 %
|
*Podľa všeobecných terminologických kritérií pre nežiaduce udalosti podľa Národného inštitútu pre rakovinu (NCI
CTCAE) verzie 4.03.
†Zahŕňa viacnásobný výraz pre nežiaducu liekovú reakciu.
±Jeden prípad syndrómu nádorového rozpadu indukovaného liekom sa pozoroval v skupine s akalabrutinibom v štúdii
ASCEND.
§Predstavuje incidenciu nálezov laboratórnych a funkčných vyšetrení, nie hlásených nežiaducich udalostí.
¶Uvedené ako hodnoty stupňa CTCAE.
Tabuľka 4: Nežiaduce liekové reakcie* u pacientov s hematologickými malignitami liečenýchkombinovanou liečbou s akalabrutinibom (n = 223)
Trieda orgánových systémov MedDRA
|
MedDRA výraz
| Celková frekvencia výskytu
(všetky stupne CTCAE)
|
Frekvencia výskytu
CTCAE ≥ 3. stupňa†
|
Infekcie a nákazy
| Infekcia horných dýchacích ciest
| Veľmi časté (31,4 %)
| 1,8 %
|
Sinusitída
| Veľmi časté (15,2 %)
| 0,4 %
|
Nazofaryngitída
| Veľmi časté (13,5 %)
| 0,4 %
|
Infekcia močových ciest
| Veľmi časté (13 %)
| 0,9 %
|
Pneumónia
| Veľmi časté (10,8 %)
| 5,4 %
|
Bronchitída
| Časté (9,9 %)
| 0 %
|
Herpetické vírusové infekcie†
| Časté (6,7 %)
| 1,3 %
|
Progresívna multifokálna
leukoencefalopatia
|
Menej časté (0,4 %)
|
0,4 %
|
Reaktivácia hepatitídy B
| Menej časté (0,9 %)
| 0,1 %
|
Aspergilové infekcie†
| Veľmi zriedkavé (0 %)
| 0 %
|
Benígne a malígne nádory, vrátane nešpecifikovaných novotvarov (cysty a polypy)
| †
Druhá primárna malignita (SPM)
Nemelanómová kožná malignita†
SPM s výnimkou nemelanómového kožného karcinómu†
| Veľmi časté (13 %)
Časté (7,6 %) Časté (6,3 %)
| 4,0 %
0,4 %
3,6 %
|
Poruchy krvi
a lymfatického systému
| Neutropénia†
| Veľmi časté (31,8 %)
| 30 %
|
Trombocytopénia†
| Veľmi časté (13,9 %)
| 9 %
|
†
Anémia
| Veľmi časté (11,7 %)
| 5,8 %
|
Lymfocytóza
| Menej časté (0,4 %)
| 0,4 %
|
Poruchy metabolizmu a výživy
|
Syndróm nádorového rozpadu±
|
Menej časté (1,8 %)
|
1,3 %
|
Poruchy nervového systému
| Bolesť hlavy
| Veľmi časté (43 %)
| 0,9 %
|
Závrat
| Veľmi časté (23,8 %)
| 0 %
|
Poruchy srdca
a srdcovej činnosti
| †
Atriálna fibrilácia/flutter
|
Časté (3,1 %)
|
0,9 %
|
Tried
a orgánových systémov MedDRA
|
MedDRA výraz
|
Celkov
á frekvencia výskytu
(všetky stupne CTCAE)
|
Frekvenci
a výskytu
CTCAE ≥ 3. stupňa
†
|
Poruch
y ciev
|
Tvorba podliatin†
Kontúzia Petechie Ekchymózy
|
Veľmi časté (38,6 %)
Veľmi časté (27,4 %) Veľmi časté (11,2 %) Časté (3,1 %)
|
0 %
0 %
0 %
0 %
|
Hemorágia/hematóm†
Gastrointestinálna hemorágia
Intrakraniálna hemorágia
|
Veľmi časté (17,5 %)
Časté (3,6 %)
Menej časté (0,9 %)
|
1,3 %
0,9 %
0 %
|
Epistaxa
|
Časté (8,5 %)
|
0 %
|
Poruchy gastrointestinálneho traktu
|
Hnačka
|
Veľmi časté (43,9 %)
|
4,5 %
|
Nevoľnosť
|
Veľmi časté (26,9 %)
|
0 %
|
Zápcha
|
Veľmi časté (20,2 %)
|
0 %
|
Vracanie
|
Veľmi časté (19,3 %)
|
0,9 %
|
Abdominálna bolesť†
|
Veľmi časté (14,8 %)
|
1,3 %
|
Poruch
y kože a podkožného tkaniva
|
Vyrážka†
|
Veľmi časté (30,9 %)
|
1,8 %
|
Poruch
y kostrovej a svalovej sústavy a spojivového tkaniva
|
Muskuloskeletálna bolesť†
|
Veľmi časté (44,8 %)
|
2,2 %
|
Artralgia
|
Veľmi časté (26,9 %)
|
1,3 %
|
Celkov
é poruchy a reakcie v mieste podania
'
|
Únava
|
Veľmi časté (30,5 %)
|
1,8 %
|
Asténia
|
Časté (7,6 %)
|
0,4 %
|
Laboratórne
a funkčné vyšetrenia¶
(nálezy na základe
výsledkov vyšetrení)
|
Znížený absolútny počet neutrofilov§
|
Veľmi časté (57,4 %)
|
35 %
|
Znížený počet trombocytov§
|
Veľmi časté (46,2 %)
|
10,8 %
|
Znížená hladina hemoglobínu§
|
Veľmi časté (43,9 %)
|
9 %
|
*Podľa všeobecných terminologických kritérií pre nežiaduce udalosti podľa Národného inštitútu pre rakovinu (NCI CTCAE) verzie 4.03.
†Zahŕňa viacnásobný výraz pre nežiaducu liekovú reakciu.
±Jeden prípad syndrómu nádorového rozpadu indukovaného liekom sa pozoroval v skupine s akalabrutinibom v štúdii
ASCEND.
§Predstavuje incidenciu nálezov laboratórnych a funkčných vyšetrení, nie hlásených nežiaducich udalostí.
¶Uvedené ako hodnoty stupňa CTCAE.
PopisvybranýchnežiaducichreakciíUkončenie liečby a zníženie dávky v dôsledku nežiaducich reakciíSpomedzi 1 040 pacientov liečených monoterapiou liekom Calquence sa ukončenie liečby z dôvodu nežiaducich reakcií hlásilo u 9,3 % pacientov. Hlavné nežiaduce reakcie zahŕňali pneumóniu, trombocytopéniu a hnačku. Zníženia dávky z dôvodu nežiaducich reakcií sa hlásili u 4,2 % pacientov. Hlavné nežiaduce reakcie zahŕňali reaktiváciu hepatitídy B, sepsu a hnačku.
Spomedzi 223 pacientov liečených kombinovanou liečbou liekom Calquence sa ukončenie liečby z dôvodu nežiaducich reakcií hlásilo u 10,8 % pacientov. Hlavné nežiaduce reakcie zahŕňali pneumóniu, trombocytopéniu a hnačku. Zníženia dávky z dôvodu nežiaducich reakcií sa hlásili
u 6,7 % pacientov. Hlavné nežiaduce reakcie zahŕňali neutropéniu, hnačku a vracanie.
Staršie osoby
Spomedzi 1 040 pacientov v klinických štúdiách s monoterapiou liekom Calquence bolo 41 % vo veku viac ako 65 rokov a menej ako 75 rokov a 22 % bolo vo veku viac ako 75 rokov. Medzi pacientmi vo veku ≥ 65 rokov a mladšími pacientmi sa nepozorovali žiadne klinicky významné rozdiely
v bezpečnosti alebo účinnosti.
Spomedzi 223 pacientov v klinických štúdiách s liekom Calquence v kombinácii s obinutuzumabom bolo 47 % vo veku viac ako 65 rokov a menej ako 75 rokov a 26 % bolo vo veku viac ako 75 rokov. Medzi pacientmi vo veku ≥ 65 rokov a mladšími pacientmi sa nepozorovali žiadne klinicky významné rozdiely v bezpečnosti alebo účinnosti.
Hlásenie podozrenínanežiaducereakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné
monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovanieK dispozícii nie je žiadna špecifická liečba predávkovania akalabrutinibom a doposiaľ neboli stanovené príznaky predávkovania. V prípade predávkovania je potrebné pacientov pozorne sledovať pre prejavy alebo príznaky nežiaducich reakcií a je potrebné podať vhodnú symptomatickú liečbu.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: cytostatiká, inhibítory proteínkinázy. ATC kód: L01EL02.
MechanizmusúčinkuAkalabrutinib je selektívny inhibítor Brutonovej tyrozínkinázy (BTK). BTK je signálna molekula dráh
receptora antigénu B-buniek (B-cell antigen receptor, BCR) a cytokínového receptora. V B-bunkách má signalizácia BTK za následok prežívanie a proliferáciu B-buniek a je potrebná pre bunkovú adhéziu, migráciu a chemotaxiu.
Akalabrutinib a jeho aktívny metabolit, ACP-5862, tvoria kovalentnú väzbu s cysteínovým zvyškom v aktívnom mieste BTK, čo vedie k ireverzibilnej inaktivácii BTK s minimálnymi necieľovými interakciami.
Farmakodynamické účinkyU pacientov s malignitami B-buniek, ktorí dostávali 100 mg akalabrutinibu dvakrát denne, sa medián
obsadenosti BTK v periférnej krvi v rovnovážnom stave v hodnote ≥ 95 % zachoval počas 12 hodín, čo viedlo k inaktivácii BTK počas odporúčaného dávkovacieho intervalu.
Elektrofyziológia srdcaÚčinok akalabrutinibu na QTc interval sa hodnotil u 46 zdravých osôb mužského a ženského pohlavia
v randomizovanej, dvojito zaslepenej, detailnej QT štúdii s placebom a pozitívnymi kontrolnými osobami. Pri supraterapeutickej dávke, 4-násobku maximálnej odporúčanej dávky, Calquence
v žiadnom klinicky významnom rozsahu nepredlžoval QT/QTc interval (napr. nie viac ako o 10 ms
alebo o 10 ms) (pozri časti 4.4, 4.8 a 5.3).
Klinická účinnosťa bezpečnosťPacienti s CLL neliečenou v minulostiBezpečnosť a účinnosť lieku Calquence pri CLL, ktorá nebola v minulosti liečená, sa hodnotili v randomizovanej, multicentrickej, otvorenej štúdii fázy 3 (ELEVATE-TN) zahŕňajúcej 535 pacientov. Pacienti dostávali Calquence plus obinutuzumab, monoterapiu liekom Calquence alebo obinutuzumab plus chlorambucil. V štúdii ELEVATE-TN boli zahrnutí pacienti vo veku 65 rokov alebo starší alebo pacienti vo veku 18 až 65 rokov so súbežne existujúcimi zdravotnými stavmi, pričom 27,9 % pacientov malo CrCl < 60 ml/min. Spomedzi pacientov vo veku < 65 rokov malo
16,1 % medián skóre CIRS-G v hodnote 8. V štúdii mali pacienti povolené užívať antitrombotiká. Vylúčení boli pacienti, u ktorých sa vyžadovala antikoagulácia pomocou warfarínu alebo ekvivalentného antagonistu vitamínu K.
Pacienti boli randomizovaní v pomere 1:1:1 do 3 skupín na podávanie buď:
· Calquence plus obinutuzumab (Calquence + G): Calquence 100 mg sa podával dvakrát denne so začiatkom v 1. deň 1. cyklu až do progresie ochorenia alebo neakceptovateľnej toxicity. Obinutuzumab sa podával od 2. cyklu (maximálne počas 6 liečebných cyklov) a to 1. deň
100 mg, 2. deň 900 mg, 8. deň 1 000 mg, 15. deň 1 000 mg a následne počas 3.-7. cyklu sa podával vždy 1. deň cyklu v dávke 1 000 mg. Každý cyklus trval 28 dní.
· Monoterapiu liekom Calquence: Calquence 100 mg sa podával dvakrát denne až do progresie ochorenia alebo neakceptovateľnej toxicity.
· Obinutuzumab plus chlorambucil (GClb): Obinutuzumab a chlorambucil sa podávali počas maximálne 6 liečebných cyklov. Obinutuzumab 1 000 mg sa podával od prvého cyklu a to 1. deň 100 mg, 2. deň 900 mg, 8. deň 1 000 mg, 15. deň 1 000 mg a následne počas 2.-6. cyklu sa
podával vždy 1. deň cyklu v dávke 1 000 mg. Chlorambucil 0,5 mg/kg sa podával v 1. a 15. deň
1.-6. cyklu. Každý cyklus trval 28 dní.
Pacienti boli stratifikovaní podľa stavu mutácie delécie 17p (prítomná delécia vs. bez delécie), výkonnostného stavu podľa ECOG (0 alebo 1 oproti 2) a geografickej oblasti (Severná Amerika a Západná Európa oproti inej oblasti). Po potvrdení progresie ochorenia prešlo 45 pacientov randomizovaných v skupine GClb do skupiny s monoterapiou liekom Calquence. Východiskové
demografické charakteristiky a charakteristiky ochorenia populácie štúdie sú zhrnuté v tabuľke 5.
Tabuľka 5: Východiskové pacientske charakteristiky (štúdia ELEVATE-TN) u pacientov s CLLneliečenou v minulostiCharakteristika
| Calquence plus obinutuzumab N = 179
| Monoterapia
Calquence
N = 179
| Obinutuzumab plus chlorambucil N = 177
|
Vek, roky; medián (rozsah)
| 70 (41 – 88)
| 70 (44 – 87)
| 71 (46 – 91)
|
Mužské pohlavie; %
| 62
| 62
| 59,9
|
Biela rasa; %
| 91,6
| 95
| 93,2
|
Výkonnostný stav podľa ECOG 0 – 1; %
| 94,4
| 92,2
| 94,4
|
Medián času od diagnózy (mesiace)
| 30,5
| 24,4
| 30,7
|
Masívne ochorenie s uzlinami o veľkosti
≥ 5 cm; %
| 25,7
| 38
| 31,1
|
Cytogenetika/FISH kategória; %
Delécia 17p Delécia 11q Mutácia TP53
Nezmutovaný IGHV
Komplexný karyotyp (≥ 3 abnormality)
|
9,5
17,3
11,7
57,5
16,2
|
8,9
17,3
10,6
66,5
17,3
|
9
18,6
11,9
65,5
18,1
|
C
h
arakteristika
|
Calquenc
e plus obinutuzumab N = 179
|
Monoterapia
Calquence
N = 179
|
Obinutuzumab
plus
chlorambucil N = 177
|
Rai štádium; %
0
I
II III IV
|
1,7
30,2
20,1
26,8
21,2
|
0
26,8
24,6
27,9
20,7
|
0,6
28,2
27,1
22,6
21,5
|
Primárnym koncovým ukazovateľom bolo prežívanie bez progresie (progression-free survival, PFS)
v skupine s Calquence plus obinutuzumab (Calquence + G) oproti skupine s obinutuzumabom plus chlorambucil (GClb) na základe nezávislej hodnotiacej komisie (Independent Review Committee, IRC) podľa kritérií Medzinárodnej pracovnej skupiny pre chronickú lymfocytovú leukémiu (International Workshop on Chronic Lymphocytic Leukaemia, IWCLL) z roku 2008 so zapracovaním vysvetlenia lymfocytózy súvisiacej s liečbou (Cheson 2012). S mediánom sledovania 28,3 mesiacov PFS podľa IRC dokázalo 90% štatisticky významné zníženie rizika progresie ochorenia alebo úmrtia
z dôvodu CLL neliečenej v minulosti u pacientov v skupine s liekom Calquence + G v porovnaní so skupinou s GClb. Výsledky týkajúce sa účinnosti sú uvedené v tabuľke 6. Kaplanove-Meierove krivky PFS sú zobrazené na obrázku 1.
Tabuľka 6: Výsledky týkajúce sa účinnosti podľa hodnotenia IRC (štúdia ELEVATE-TN)u pacientov s CLL
| Calquence plus obinutuzumab N = 179
| Monoterapia
Calquence
N = 179
| Obinutuzumab plus chlorambucil N = 177
|
Prežívanie bez progresie*
|
Počet udalostí (%)
| 14 (7,8)
| 26 (14,5)
| 93 (52,5)
|
PD, n (%)
| 9 (5)
| 20 (11,2)
| 82 (46,3)
|
Úmrtia (%)
| 5 (2,8)
| 6 (3,4)
| 11 (6,2)
|
Medián (95 % IS), mesiace
| NR
| NR (34,2; NR)
| 22,6 (20,2; 27,6)
|
HR† (95 % IS)
| 0,10 (0,06; 0,17)
| 0,20 (0,13; 0,30)
| -
|
Hodnota p
| < 0,0001
| < 0,0001
| -
|
24-mesačný odhad, %
(95 % IS)
| 92,7 (87,4; 95,8)
| 87,3 (80,9; 91,7)
| 46,7 (38,5; 54,6)
|
Celkové prežívaniea
|
Úmrtia (%)
| 9 (5)
| 11 (6,1)
| 17 (9,6)
|
Pomer rizika (95 % IS)†
| 0,47 (0,21; 1,06)
| 0,60 (0,28; 1,27)
| -
|
Miera najlepšej celkovej odpovede* (CR + CRi + nPR + PR)
|
ORR, n (%)
(95 % IS)
| 168 (93,9)
(89,3; 96,5)
| 153 (85,5)
(79,6; 89,9)
| 139 (78,5)
(71,9; 83,9)
|
Hodnota p
| < 0,0001
| 0,0763
| -
|
CR, n (%)
| 23 (12,8)
| 1 (0,6)
| 8 (4,5)
|
CRi, n (%)
| 1 (0,6)
| 0
| 0
|
nPR, n (%)
| 1 (0,6)
| 2 (1,1)
| 3 (1,7)
|
PR, n (%)
| 143 (79,9)
| 150 (83,8)
| 128 (72,3)
|
IS = interval spoľahlivosti; HR = pomer rizika; NR = nedosiahol sa; CR = úplná odpoveď; CRi = úplná
odpoveď s neúplnou úpravou krvného obrazu; nPR = nodulárna čiastočná odpoveď; PR = čiastočná odpoveď.
*Podľa hodnotenia IRC.
†Na základe stratifikovaného Coxovho modelu proporcionálnych rizík.
aMedián OS sa v oboch skupinách nedosiahol.
 Obrázok 1: Kaplanova-Meierova krivka PFS podľa hodnotenia IRC (štúdia ELEVATE-TN)
Obrázok 1: Kaplanova-Meierova krivka PFS podľa hodnotenia IRC (štúdia ELEVATE-TN)
 u pacientov s CLL (populácia podľa liečebného zámeru [intent-to-treat, ITT])
u pacientov s CLL (populácia podľa liečebného zámeru [intent-to-treat, ITT])
Calquence+G
Calquence
GClb
Čas od randomizácie (mesiace)
Počet pacientov v riziku
|
Mesiac
| 0
| 3
| 6
| 9
| 12
| 15
| 18
| 21
| 24
| 27
| 30
| 33
| 36
| 39
|
Calquence
| 179
| 166
| 161
| 157
| 153
| 150
| 148
| 147
| 103
| 94
| 43
| 40
| 4
| 3
|
Calquence + G
| 179
| 176
| 170
| 168
| 163
| 160
| 159
| 155
| 109
| 104
| 46
| 41
| 4
| 2
|
GClb
| 177
| 162
| 157
| 151
| 136
| 113
| 102
| 86
| 46
| 41
| 13
| 13
| 3
| 2
|
Výsledky PFS pre Calquence s obinutuzumabom alebo bez neho boli konzistentné naprieč
podskupinami, vrátane vysokorizikových charakteristík. Vo vysokorizikovej populácii s CLL (delécia
17p, delécia 11q, mutácia TP53 alebo nezmutovaný IGHV) bol HR pre PFS pri Calquence s obinutuzumabom alebo bez neho 0,08 [95 % IS (0,04; 0,15)] oproti obinutuzumabu plus chlorambucil 0,13 [95 % IS (0,08; 0,21)].
Tabuľka 7: Podskupinová analýza PFS (štúdia ELEVATE-TN)
| Monoterapia Calquence
| Calquence + G
|
N
| Pomer
rizika
| 95 % IS
| N
| Pomer
rizika
| 95 % IS
|
Všetky osoby
| 179
| 0,20
| (0,13; 0,30)
| 179
| 0,10
| (0,06; 0,17)
|
Delécia 17p
Áno
Nie
|
19
160
|
0,20
0,20
|
(0,06; 0,64) (0,12; 0,31)
|
21
158
|
0,13
0,09
|
(0,04; 0,46) (0,05; 0,17)
|
Mutácia TP53
Áno
Nie
|
19
160
|
0,15
0,20
|
(0,05; 0,46) (0,12; 0,32)
|
21
158
|
0,04
0,11
|
(0,01; 0,22) (0,06; 0,20)
|
Delécia 17p
alebo/a mutácia TP53
Áno
Nie
|
23
156
|
0,23
0,19
|
(0,09; 0,61) (0,11; 0,31)
|
25
154
|
0,10
0,10
|
(0,03; 0,34) (0,05; 0,18)
|
Mutácia IGHV Zmutovaný
Nezmutovaný
|
58
119
|
0,69
0,11
|
(0,31; 1,56) (0,07; 0,19)
|
74
103
|
0,15
0,08
|
(0,04; 0,52) (0,04; 0,16)
|
|
Monoterapia Calquence
|
Calquence + G
|
N
|
P
o
mer
rizika
|
95 % IS
|
N
|
P
o
mer
rizika
|
95 % IS
|
Delécia 11q
Áno
Nie
|
31
148
|
0,07
0,26
|
(0,02; 0,22) (0,16; 0,41)
|
31
148
|
0,09
0,10
|
(0,03; 0,26) (0,05; 0,20)
|
Komplexný
karyotyp
Áno
Nie
|
31
117
|
0,10
0,27
|
(0,03; 0,33) (0,16; 0,46)
|
29
126
|
0,09
0,11
|
(0,03; 0,29) (0,05; 0,21)
|
Pacienti s CLL, ktorí v minulosti dostávali aspoň jednu predchádzajúcu liečbu
Bezpečnosť a účinnosť lieku Calquence pri rekurentnej alebo refraktérnej CLL sa hodnotili
v randomizovanej, multicentrickej, otvorenej štúdii fázy 3 (ASCEND) zahŕňajúcej 310 pacientov, ktorí v minulosti dostávali aspoň jednu predchádzajúcu liečbu nezahŕňajúcu inhibítory BCL-2 alebo inhibítory receptora B-buniek. Pacienti dostávali monoterapiu liekom Calquence alebo liečbu podľa výberu skúšajúceho zahŕňajúcu buď idelalisib plus rituximab alebo bendamustín plus rituximab. Pacienti mali v štúdii povolené užívanie antitrombotík. Vylúčení boli pacienti, u ktorých sa vyžadovala antikoagulácia pomocou warfarínu alebo ekvivalentného antagonistu vitamínu K.
Pacienti boli randomizovaní v pomere 1:1 na podávanie buď:
· Calquence 100 mg dvakrát denne až do progresie ochorenia alebo neakceptovateľnej toxicity, alebo
· liečby podľa výberu skúšajúceho:
o idelalisib 150 mg dvakrát denne v kombinácii s rituximabom i.v. 375 mg/m2 v 1. deň prvého cyklu, po ktorom nasleduje 500 mg/m2 i.v. každé 2 týždne (4 dávky), potom
každé 4 týždne (3 dávky), t.j. celkovo 8 infúzií.
o bendamustín 70 mg/m2 (1. a 2. deň každého 28-dňového cyklu) v kombinácii
s rituximabom (375 mg/m2/500 mg/m2) v 1. deň každého 28-dňového cyklu počas až 6
cyklov.
Pacienti boli stratifikovaní podľa stavu mutácie delécie 17p (prítomná delécia vs. bez delécie), výkonnostného stavu podľa ECOG (0 alebo 1 oproti 2) a počtu predchádzajúcich terapií (1 až 3 oproti
≥ 4). Po potvrdení progresie ochorenia prešlo 35 pacientov randomizovaných v skupine s liečbou
podľa výberu skúšajúceho (buď idelalisib plus rituximab alebo bendamustín plus rituximab) do skupiny s liekom Calquence. Východiskové demografické charakteristiky a charakteristiky ochorenia populácie štúdie sú zhrnuté v tabuľke 8.
Tabuľka 8: Východiskové pacientske charakteristiky (štúdia ASCEND) u pacientov s CLLCharakteristika
| Monoterapia
Calquence
N = 155
| Liečba podľa výberu
skúšajúceho: idelalisib + rituximab alebo bendamustín + rituximab N = 155
|
Vek, roky; medián (rozsah)
| 68 (32 – 89)
| 67 (34 – 90)
|
Mužské pohlavie; %
| 69,7
| 64,5
|
Biela rasa; %
| 93,5
| 91,0
|
Výkonnostný stav podľa ECOG; %
0
1
2
|
37,4
50,3
12,3
|
35,5
51,0
13,5
|
Medián času od diagnózy (mesiace)
| 85,3
| 79,0
|
Masívne ochorenie s uzlinami o veľkosti ≥ 5 cm; %
| 49,0
| 48,4
|
C
h
arakteristika
|
Monoterapia Calquence
N = 155
|
Liečb
a podľa výberu skúšajúceho: idelalisib + rituximab alebo bendamustín + rituximab N = 155
|
Medián počtu predchádzajúcich terapií pre CLL (rozsah)
|
1 (1 – 8)
|
2 (1 – 10)
|
Počet predchádzajúcich terapií pre CLL; %
1
2
3
≥ 4
|
52,9
25,8
11,0
10,3
|
43,2
29,7
15,5
11,6
|
Cytogenetika/FISH kategória; % Delécia 17p
Delécia 11q
Mutácia TP53
Nezmutovaný IGHV
Komplexný karyotyp (≥ 3 abnormality)
|
18,1
25,2
25,2
76,1
32,3
|
13,5
28,4
21,9
80,6
29,7
|
Rai štádium; %
0
I
II III IV
|
1,3
25,2
31,6
13,5
28,4
|
2,6
20,6
34,8
11,6
29,7
|
Primárnym koncovým ukazovateľom bolo PFS na základe hodnotenia IRC podľa kritérií IWCLL
z roku 2008 so zapracovaním vysvetlenia lymfocytózy súvisiacej s liečbou (Cheson 2012).
S mediánom sledovania 16,1 mesiacov PFS dokázalo 69% štatisticky významné zníženie rizika úmrtia
alebo progresie ochorenia u pacientov v skupine s liekom Calquence. Výsledky týkajúce sa účinnosti sú uvedené v tabuľke 9. Kaplanova-Meierova krivka PFS je zobrazená na obrázku 2.
Tabuľka 9: Výsledky týkajúce sa účinnosti podľa hodnotenia IRC (štúdia ASCEND)u pacientov s CLL
| Monoterapia Calquence
N = 155
| Liečba podľa výberu
skúšajúceho: idelalisib +
rituximab alebo bendamustín
+ rituximab
N = 155
|
Prežívanie bez progresie*
|
Počet udalostí (%)
| 27 (17,4)
| 68 (43,9)
|
PD, n (%)
| 19 (12,3)
| 59 (38,1)
|
Úmrtia (%)
| 8 (5,2)
| 9 (5,8)
|
Medián (95 % IS), mesiace
| NR
| 16,5 (14,0; 17,1)
|
HR† (95 % IS)
| 0,31 (0,20; 0,49)
|
Hodnota p
| < 0,0001
|
15-mesačný odhad, % (95 % IS)
| 82,6 (75,0; 88,1)
| 54,9 (45,4; 63,5)
|
Celkové prežívaniea
|
Úmrtia (%)
| 15 (9,7)
| 18 (11,6)
|
Pomer rizika (95 % IS)†
| 0,84 (0,42; 1,66)
| -
|
Miera najlepšej celkovej odpovede* (CR + CRi + nPR + PR)**
|
ORR, n (%) (95 % IS)
| 126 (81,3) (74,4; 86,6)
| 117 (75,5) (68,1; 81,6)
|
Hodnota p
| 0,2248
| -
|
CR, n (%)
| 0
| 2 (1,3)
|
PR, n (%)
| 126 (81,3)
| 115 (74,2)
|
Dĺžka trvania odpovede (duration of response, DoR)
|
Medián (95 % IS), mesiace
|
NR
|
13,6 (11,9; NR)
|
IS = interval spoľahlivosti; HR = pomer rizika; NR = nedosiahol sa; CR = úplná odpoveď; CRi = úplná odpoveď s neúplnou úpravou krvného obrazu; nPR = nodulárna čiastočná odpoveď; PR = čiastočná odpoveď;
PD = progresia ochorenia.
*Podľa hodnotenia IRC.
aMedián OS sa v oboch skupinách nedosiahol. P < 0,6089 pre OS.
**CRi a nPR majú hodnotu 0.
†Na základe stratifikovaného Coxovho modelu proporcionálnych rizík.
Obrázok 2: Kaplanova-Meierova krivka PFS podľa hodnotenia IRC (ASCEND) u pacientov s CLL (populácia podľa liečebného zámeru [ITT])
s CLL (populácia podľa liečebného zámeru [ITT])Calquence
Liečba podľa výberu

skúšajúceho
Čas od randomizácie (mesiace)
Počet pacientov v riziku
|
Mesiac
| 0
| 1
| 2
| 3
| 4
| 5
| 6
| 7
| 8
| 9
| 10
| 11
| 12
| 13
| 14
| 15
| 16
| 17
| 18
| 19
| 20
| 21
| 22
| 23
|
Calquence
| 155
| 153
| 153
| 149
| 147
| 146
| 145
| 143
| 143
| 139
| 139
| 137
| 118
| 116
| 73
| 61
| 60
| 25
| 21
| 21
| 1
| 1
| 1
| 0
|
Liečba
podľa výberu skúšajúceho
| 155
| 150
| 150
| 146
| 144
| 142
| 136
| 130
| 129
| 112
| 105
| 101
| 82
| 77
| 56
| 44
| 39
| 18
| 10
| 8
| 0
|
|
|
|
Výsledky PFS pre Calquence boli konzistentné naprieč podskupinami, vrátane vysokorizikových
charakteristík. Vo vysokorizikovej populácii s CLL (delécia 17p, delécia 11q, mutácia TP53 alebo nezmutovaný IGHV) bol HR pre PFS 0,25 [95 % IS (0,16; 0,38)].
Tabuľka 10: Podskupinová analýza PFS podľa hodnotenia IRC (štúdia ASCEND)
|
Monoterapia Calquence
|
N
|
P
o
me
r rizika
|
95 % IS
|
Všetky osoby
|
155
|
0,27
|
(0,18; 0,40)
|
Delécia 17p
Áno
Nie
|
28
127
|
0,18
0,30
|
(0,07; 0,43) (0,19; 0,47)
|
Mutácia TP53
Áno
Nie
|
39
113
|
0,17
0,33
|
(0,08; 0,37) (0,21; 0,52)
|
Delécia 17p alebo mutácia TP53
Áno
Nie
|
45
108
|
0,16
0,34
|
(0,08; 0,34) (0,22; 0,55)
|
Mutácia IGHV
Zmutovaný
Nezmutovaný
|
33
118
|
0,30
0,28
|
(0,12; 0,76) (0,18; 0,43)
|
Delécia 11q
Áno
Nie
|
39
116
|
0,35
0,26
|
(0,16; 0,75) (0,16; 0,41)
|
Komplexný karyotyp
Áno
Nie
|
50
97
|
0,28
0,25
|
(0,15; 0,53) (0,15; 0,44)
|
Vo finálnej analýze, s mediánom sledovania 46,5 mesiacov pre Calquence a 45,3 mesiacov pre IR/BR,
sa pozorovalo 72 % zníženie rizika progresie ochorenia alebo úmrtia podľa hodnotenia skúšajúceho u pacientov v skupine s liekom Calquence. Medián PFS podľa hodnotenia skúšajúceho sa v skupine s liekom Calquence nedosiahol a v skupine s IR/BR bol 16,8 mesiacov. Výsledky týkajúce sa účinnosti podľa hodnotenia skúšajúceho sú uvedené v tabuľke 11. Kaplanova-Meierova krivka PFS podľa hodnotenia skúšajúceho je zobrazená na obrázku 3.
Tabuľka 11: Výsledky účinnosti vo finálnej analýze podľa hodnotenia skúšajúceho (štúdiaASCEND) u pacientov s CLL
| Monoterapia Calquence
N = 155
| Liečba podľa výberu skúšajúceho: idelalisib + rituximab alebo bendamustín
+ rituximab
N = 155
|
Prežívanie bez progresie*
|
Počet udalostí (%)
| 62 (40,0)
| 119 (76,8)
|
PD, n (%)
| 43 (27,7)
| 102 (65,8)
|
Úmrtia (%)
| 19 (12,3)
| 17 (11,0)
|
Medián (95 % IS), mesiace
| NR
| 16,8 (14,1; 22,5)
|
HR† (95 % IS)
| 0,28 (0,20; 0,38)
|
Celkové prežívaniea
|
Úmrtia (%)
| 41 (26,5)
| 54 (34,8 %)
|
Pomer rizika (95 % IS)†
| 0,69 (0,46; 1,04)
| -
|
IS = interval spoľahlivosti; HR = pomer rizika; NR = nedosiahol sa; PD = progresia ochorenia.
*Podľa hodnotenia skúšajúceho.
aMedián OS sa v oboch skupinách nedosiahol P = 0,0783 pre OS.
†Na základe stratifikovaného Coxovho modelu proporcionálnych rizík.
 Obrázo
k 3: Kaplanova-Meierova krivka PFS podľa hodnotenia skúšajúceho vo finálnej analýze
(ASCEND) u pacientov s CLL
Calquence
Liečb
a podľa výberu skúšajúceho
Obrázo
k 3: Kaplanova-Meierova krivka PFS podľa hodnotenia skúšajúceho vo finálnej analýze
(ASCEND) u pacientov s CLL
Calquence
Liečb
a podľa výberu skúšajúceho
Čas od randomizácie (mesiace)
Mesiac
| 0
| 3
| 6
| 9
| 12
| 15
| 18
| 21
| 24
| 27
| 30
| 33
| 36
| 39
| 42
| 45
| 48
| 51
| 54
|
Calquence
| 155
| 151
| 143
| 139
| 133
| 128
| 121
| 117
| 111
| 110
| 100
| 94
| 85
| 80
| 79
| 52
| 21
| 4
| 0
|
Liečba podľa výberu
skúšajúceho
| 155
| 147
| 138
| 118
| 95
| 76
| 66
| 62
| 52
| 42
| 35
| 32
| 28
| 26
| 23
| 12
| 5
| 0
|
|
Vo finálnej analýze boli výsledky PFS podľa hodnotenia skúšajúceho pre Calquence konzistentné
naprieč podskupinami, vrátane vysokorizikových charakteristík a boli konzistentné s primárnou analýzou.
Pediatrická populácia
Európska agentúra pre lieky udelila výnimku z povinnosti predložiť výsledky štúdií s liekom Calquence vo všetkých podskupinách pediatrickej populácie v liečbe CLL (informácie o použití v pediatrickej populácii, pozri časť 4.2).
5.2 Farmakokinetické vlastnostiFarmakokinetika (FK) akalabrutinibu a jeho aktívneho metabolitu, ACP-5862, sa skúmala u zdravých jedincov a u pacientov s malignitami B-buniek. Akalabrutinib vykazuje dávkovo úmernú FK
a akalabrutinib aj ACP-5862 vykazujú takmer lineárnu FK v rozsahu dávok 75 až 250 mg. Populačné FK modelovanie naznačuje, že FK akalabrutinibu a ACP-5862 je rovnaká u pacientov s rozdielnymi malignitami B-buniek. Pri odporúčanej dávke 100 mg dvakrát denne u pacientov s malignitami
B-buniek (vrátane CLL) bol geometrický priemer dennej plochy pod krivkou časového priebehu
plazmatickej koncentrácie v rovnovážnom stave (AUC24 hod) akalabrutinibu 1 679 ng•h/ml
a maximálna plazmatická koncentrácia (Cmax) akalabrutinibu 438 ng/ml, a pre ACP-5862 to boli hodnoty 4 166 ng•h/ml a 446 ng/ml, v uvedenom poradí.
Preukázalo sa, že tablety Calquence a kapsuly Calquence sú bioekvivalentné. Tablety Calquence obsahujú akalabrutinib-maleát, soľ akalabrutinibu, ktorá vykazuje vyššiu rozpustnosť pri vysokom pH ako báza akalabrutinibu, ktorá je liečivom kapsúl Calquence. Tablety Calquence majú preto lepšiu absorpciu v kombinácii s liečivami znižujúcimi žalúdočnú kyselinu.
Absorpcia
Čas do dosiahnutia maximálnych plazmatických koncentrácií (Tmax) bol 2-3,0 hodiny pre akalabrutinib a 0,5-4,0 hodín pre ACP-5862. Absolútna biologická dostupnosť lieku Calquence bola 25 %.
Účinok jedlanaakalabrutinib
U zdravých osôb neovplyvnilo podanie jednorazovej 100 mg dávky tabliet akalabrutinibu
s vysokokalorickým jedlom s vysokým obsahom tukov (približne 918 kalórií, 59 gramov sacharidov,
59 gramov tukov a 39 gramov proteínov) priemernú AUC v porovnaní s dávkovaním za podmienok
nalačno. Výsledná Cmax sa znížila o 54 % a Tmax bol oneskorený o 1 – 2 hodiny.
Distribúcia
Rozsah reverzibilnej väzby na proteíny ľudskej plazmy bol pre akalabrutinib 99,4 % a pre ACP-5862
bol 98,8 %. Priemerný distribučný pomer krv-plazma in vitro bol pre akalabrutinib 0,8 a pre
ACP-5862 bol 0,7. Priemerný distribučný objem v rovnovážnom stave (Vss) bol pre akalabrutinib približne 34 l.
Biotransformácia/metabolizmus
Akalabrutinib sa in vitro metabolizuje prevažne prostredníctvom enzýmov CYP3A a v menšej miere
prostredníctvom konjugácie s glutatiónom a amidovou hydrolýzou. ACP-5862 bol identifikovaný ako
hlavný metabolit v plazme, ktorý sa ďalej metabolizuje predovšetkým oxidáciou sprostredkovanou CYP3A, s geometrickým priemerom expozície (AUC), ktorý bol približne 2- až 3-násobne vyšší ako expozícia akalabrutinibu. ACP-5862 je pri inhibícii BTK približne o 50 % menej účinný ako akalabrutinib.
Štúdie in vitro naznačujú, že akalabrutinib pri klinicky relevantných koncentráciách neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, UGT1A1 ani UGT2B7 a je nepravdepodobné, že by ovplyvňoval klírens substrátov týchto CYP.
Štúdie in vitro naznačujú, že ACP-5862 pri klinicky relevantných koncentráciách neinhibuje CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4/5, UGT1A1 ani UGT2B7 a je nepravdepodobné, že by ovplyvňoval klírens substrátov týchto CYP.
Interakcie s transportnýmiproteínmi
Štúdie in vitro naznačujú, že akalabrutinib a ACP-5862 sú substrátmi P-gp a BCRP. Avšak je
nepravdepodobné, že by ich súbežné podávanie s inhibítormi BCRP malo za následok klinicky
významné liekové interakcie. Súbežné podanie s inhibítorom OATP1B1/1B3 (600 mg rifampicínu, jednorazová dávka) viedlo k zvýšeniu Cmax akalabrutinibu 1,2-násobne a AUC 1,4-násobne (N = 24, zdravé osoby), čo nie je klinicky významné.
Akalabrutinib a ACP-5862 pri klinicky relevantných koncentráciách neinhibujú P-gp, OAT1, OAT3, OCT2, OATP1B1, OATP1B3 a MATE2-K. Akalabrutinib môže pri klinicky relevantných koncentráciách inhibovať intestinálny BCRP, zatiaľ čo ACP-5862 môže inhibovať MATE1 (pozri časť 4.5). Akalabrutinib pri klinicky relevantných koncentráciách neinhibuje MATE1, zatiaľ čo
ACP-5862 neinhibuje BCRP.
Eliminácia
Po podaní jednorazovej perorálnej dávky 100 mg tabliet akalabrutinibu bol geometrický priemer
terminálneho eliminačného polčasu (t1/2) akalabrutinibu 1,4 hodín. Hodnota t1/2 aktívneho metabolitu, ACP-5862, bola 6,6 hodín.
Priemerný zdanlivý perorálny klírens (CL/F) u pacientov s malignitami B-buniek bol pre akalabrutinib
134 l/hod a pre ACP-5862 bol 22 l/hod.
Po podaní jednorazovej 100 mg dávky rádioaktívne značeného [14C]-akalabrutinibu zdravým osobám sa 84 % dávky zachytilo v stolici a 12 % dávky sa zachytilo v moči, pričom menej ako 2 % dávky sa vylúčili ako nezmenený akalabrutinib.
Osobitné skupinypacientov
Na základe populačnej FK analýzy nemali vek (> 18 rokov), pohlavie, rasa (biela, afroamerická)
a telesná hmotnosť klinicky významné účinky na FK akalabrutinibu a jeho aktívneho metabolitu, ACP-5862.
Pediatrická populácia
U pacientov mladších ako 18 rokov sa neuskutočnili žiadne farmakokinetické štúdie s liekom
Calquence.
Porucha funkcie obličiek
Akalabrutinib podlieha minimálnej renálnej eliminácii. Farmakokinetická štúdia u pacientov s poruchou funkcie obličiek sa neuskutočnila.
Na základe populačnej FK analýzy sa u 408 jedincov s miernou poruchou funkcie obličiek (eGFR medzi 60 a 89 ml/min/1,73 m2 na základe odhadu podľa MDRD), 109 jedincov so stredne závažnou poruchou funkcie obličiek (eGFR medzi 30 a 59 ml/min/1,73 m2) v porovnaní so 192 jedincami
s normálnou funkciou obličiek (eGFR vyššia ako alebo rovnajúca sa 90 ml/min/1,73 m2) nepozoroval
žiadny klinicky významný FK rozdiel. Farmakokinetika akalabrutinibu nebola charakterizovaná u pacientov so závažnou poruchou funkcie obličiek (eGFR nižšia ako 29 ml/min/1,73 m2) ani
u pacientov s poruchou funkcie obličiek vyžadujúcou dialýzu. V klinických štúdiách neboli zahrnutí
pacienti s hladinami kreatinínu vyššími ako 2,5-násobok ULN pre danú inštitúciu (pozri časť 4.2).
Porucha funkcie pečene
Akalabrutinib sa metabolizuje v pečeni. V špecializovaných štúdiách poruchy funkcie pečene bola expozícia (AUC) akalabrutinibu v porovnaní s osobami s normálnou funkciou pečene (n = 6) zvýšená
1,9-násobne, 1,5-násobne a 5,3-násobne u osôb s miernou (n = 6) (Childovo-Pughovo skóre A),
stredne závažnou (n = 6) (Childovo-Pughovo skóre B) a závažnou (n = 8) (Childovo-Pughovo skóre C) poruchou funkcie pečene, v uvedenom poradí. Markery dôležité pre určenie eliminačnej kapacity liečiv neboli významne ovplyvnené u osôb v skupine so stredne závažnou poruchou funkcie pečene, preto vplyv stredne závažnej poruchy funkcie pečene bol v tejto štúdii pravdepodobne podhodnotený. Na základe populačnej FK analýzy sa u osôb s miernou (n = 79) alebo stredne závažnou (n = 6) poruchou funkcie pečene (celkový bilirubín v hodnote medzi 1,5- až 3-násobkom ULN a akákoľvek hodnota AST) v porovnaní s osobami s normálnou (n = 613) funkciou pečene (celkový bilirubín
a AST v rámci ULN) nepozoroval žiadny klinicky významný rozdiel (pozri časť 4.2).
5.3 Predklinické údaje o bezpečnosti
Karcinogenita
Štúdie karcinogénneho potenciálu s akalabrutinibom sa neuskutočnili.
Genotoxicita/mutagenita/fototoxicita
Akalabrutinib nebol mutagénny v teste bakteriálnej reverznej mutácie, v teste chromozómovej
aberácie in vitro ani v teste mikrojadierok v kostnej dreni u myší in vivo.
Na základe in vitro testov fototoxicity pomocou 3T3 bunkovej línie má akalabrutinib u ľudí nízke riziko fototoxicity.
Toxicita poopakovanompodávaní
U potkanov sa pri všetkých hladinách dávky v pankrease pozorovali mikroskopické nálezy
(hemorágia/pigment/zápal/fibróza v ostrovčekoch) minimálnej až miernej závažnosti. V štúdiách trvajúcich až 6 mesiacov s hladinou dávky bez pozorovaných nežiaducich účinkov (No Observed Adverse Effect Level, NOAEL), t.j. 30 mg/kg/deň u potkanov, sa v obličkách pozorovali nálezy minimálnej až miernej závažnosti (tubulárna bazofília, tubulárna regenerácia a zápal), ktoré neboli nežiaduce. Priemerné expozície (AUC) pri NOAEL u samcov a samíc potkana zodpovedajú
0,6-násobku a 1-násobku, v uvedenom poradí, klinickej expozície pri odporúčanej dávke 100 mg dvakrát denne. Najnižšia hladina dávky s pozorovanými nežiaducimi účinkami (Lowest Observed Adverse Effect Level, LOAEL), pri ktorej sa pozorovali reverzibilné nálezy týkajúce sa obličiek (stredne závažná tubulárna degenerácia) a pečene (nekróza individuálnych hepatocytov) v chronickej štúdii na potkanoch, bola 100 mg/kg/deň a poskytovala rozmedzie expozície 4,2-násobne väčšie ako je klinická expozícia pri odporúčanej dávke 100 mg dvakrát denne. V štúdiách trvajúcich 9 mesiacov
u psov bola hladina NOAEL 10 mg/kg/deň, čo zodpovedá expozícii 3-krát vyššej ako je klinická AUC pri odporúčanej klinickej dávke. U psov pri dávke 30 mg/kg/deň (9-násobok klinickej AUC) sa pozorovala minimálna degenerácia tubulov v obličkách, mierne zníženie hmotnosti sleziny
a prechodné minimálne až mierne zníženie množstva erytrocytov a zvýšenie ALT a ALP. Toxicity týkajúce sa srdca u potkanov (myokardiálna hemorágia, zápal, nekróza) a psov (perivaskulárny/ vaskulárny zápal) sa pozorovali len u zvierat, ktoré uhynuli počas štúdií a to pri dávkach vyšších ako je maximálna tolerovaná dávka (MTD). Expozície u potkanov a psov s nálezmi týkajúcimi sa srdca boli aspoň 6,8-násobkom a 25-násobkom klinickej AUC, v uvedenom poradí. Reverzibilitu nálezov týkajúcich sa srdca nebolo možné posúdiť, keďže tieto nálezy sa pozorovali iba pri dávkach vyšších ako MTD.
Reprodukčná toxikológia
U samcov alebo samíc potkana sa pri expozíciách v hodnote 10- alebo 9-násobku klinickej AUC pri
odporúčanej dávke, v uvedenom poradí, nepozorovali žiadne účinky na fertilitu.
U gravidných potkanov sa pri expozíciách v hodnote približne 9-násobku AUC pri odporúčanej dávke
100 mg dvakrát denne u pacientov nepozorovali žiadne účinky na embryonálno-fetálny vývin a prežívanie. V dvoch reprodukčných štúdiách na potkanoch sa pri expozíciách
v hodnote > 2,3-násobku klinickej expozície pri dávke 100 mg dvakrát denne pozorovala dystokia
(zdĺhavý/náročný pôrod). V plazme plodu potkana bola potvrdená prítomnosť akalabrutinibu a jeho aktívneho metabolitu. Akalabrutinib a jeho aktívny metabolit boli prítomné v mlieku laktujúcich potkanov.
V embryonálno-fetálnej štúdii na gravidných králikoch sa pri hladinách expozície, ktoré viedli k materskej toxicite a ktoré boli 2,4-násobne vyššie ako AUC pri odporúčanej dávke u ľudí, pozorovali znížená telesná hmotnosť plodu a oneskorená osifikácia.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Jadro tablety
manitol (E421)
celulóza, mikrokryštalická (E460)
hydroxypropylcelulóza, čiastočne substituovaná (E463)
stearyl-fumarát sodný
Obal tabletyhypromelóza (E464)
kopovidón
oxid titaničitý (E171)
makrogol
triacylglyceroly, so stredne dlhým reťazcom žltý oxid železitý (E172)
červený oxid železitý (E172)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnosti2 roky.
6.4 Špeciálne upozornenia na uchovávanieTento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
6.5 Druh obalu a obsah baleniaHliník/hliníkové blistrové balenia so symbolmi predstavujúcimi slnko/mesiac obsahujúce 8 alebo 10
filmom obalených tabliet. Škatuľky po 56 alebo 60 tabliet. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciuVšetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIAstraZeneca AB
SE-151 85 Södertälje
Švédsko
8. REGISTRAČNÉ ČÍSLO (ČÍSLA)EU/1/20/1479/003
EU/1/20/1479/004
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 5. november 2020
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu