
br />
|
|
Obnovenie* ≤ 21 dní
|
Obnovenie* > 21 dní
|
15-50 %
|
100 %
|
50 %
|
< 15 %
|
100 %
|
33 %
|
*Obnovenie = počty krvných buniek ≥ počet nadir + (0,5 x [začiatočný počet – počet nadir])
Po úprave dávky sa má trvanie cyklu vrátiť späť na 28 dní.
Osobit né popul ácie Starší pacientiU starších pacientov sa neodporúčajú žiadne špecifické úpravy dávky. Keďže u starších pacientov je vyššia pravdepodobnosť zníženej funkcie obličiek, môže byť vhodné sledovať ich funkciu.
Pacienti s poruchou funkcie obličiekAzacitidín môže byť podávaný pacientom s poruchou funkcie obličiek bez úpravy úvodnej dávky (pozri časť 5.2). Ak sa vyskytne nevysvetliteľný pokles hydrogenuhličitanu v sére na menej ako 20 mmol/l, dávka sa má v nasledujúcom cykle znížiť o 50 %. Ak sa vyskytne nevysvetliteľný vzostup sérového kreatinínu alebo močovinového dusíka v krvi (
blood urea nitrogen, BUN) na úroveň ≥ 2- násobne vyššiu, ako sú východiskové hodnoty a nad horný limit normálnej hodnoty (
upper limit of normal, ULN), ďalší cyklus sa má odložiť dovtedy, kým sa hodnoty nevrátia na normálnu alebo východiskovú úroveň a v nasledujúcom liečebnom cykle sa má znížiť dávka o 50 % (pozri časť 4.4).
Pacienti s poruchou funkcie pečeneU pacientov s poruchou funkcie pečene neboli vykonané žiadne oficiálne štúdie (pozri časť 4.4). Pacienti so závažnou poruchou funkcie pečene sa majú pozorne sledovať kvôli výskytu nežiaducich
účinkov. U pacientov s poruchou funkcie pečene pred začatím liečby sa neodporúča žiadna špecifická
úprava úvodnej dávky a následné úpravy dávky majú vychádzať z hematologických laboratórnych hodnôt. Azacitidín je kontraindikovaný u pacientov s malígnymi pečeňovými nádormi v pokročilom štádiu (pozri časti 4.3 a 4.4).
PediatrickápopuláciaBezpečnosť a účinnosť azacitidínu u detí vo veku 0-17 rokov neboli doteraz stanovené. K dispozícii nie sú žiadne údaje.
SpôsobpodávaniaRekonštituovaný azacitidín sa má podávať subkutánnou injekciou do ramena, stehna alebo brucha. Miesta injekčnej aplikácie sa majú meniť. Nové injekcie sa majú aplikovať najmenej 2,5 cm od
predchádzajúceho miesta vpichu a nikdy nie do oblastí, v ktorých je miesto vpichu bolestivé, s
podliatinami, sčervenené alebo stvrdnuté.
Po rekonštitúcii sa suspenzia nemá filtrovať. Pokyny na rekonštitúciu lieku pred podaním, pozri časť
6.6.
4.3 KontraindikáciePrecitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Malígne pečeňové nádory v pokročilom štádiu (pozri časť 4.4).
Dojčenie (pozri časť 4.6).
4.4 Osobitné upozornenia a opatrenia pri používaníHematologickátoxicitaLiečba azacitidínom je spojená s anémiou, neutropéniou a trombocytopéniou, najmä počas prvých 2 cyklov (pozri časť 4.8). V prípade potreby (avšak aspoň pred každým liečebným cyklom) sa má stanoviť kompletný krvný obraz s cieľom sledovania odpovede a toxicity
. Po podaní odporúčanej
dávky v prvom cykle sa má dávka na základe hodnoty nadir a hematologickej odpovede pre následné cykly znížiť alebo jej podanie odložiť na neskôr (pozri časť 4.2). Pacienti majú byť poučení, aby ihneď hlásili prípady horúčky. Pacienti a lekári majú sledovať prejavy a príznaky krvácania.
Poruchafunkciepečene
U pacientov s poruchou funkcie pečene neboli vykonané žiadne oficiálne štúdie. U pacientov s
rozsiahlou nádorovou záťažou z dôvodu metastatického ochorenia bola hlásená progresívna pečeňová kóma a úmrtie počas liečby azacitidínom, a to najmä u pacientov s východiskovými hodnotami albumínu v sére < 30 g/l. Azacitidín je kontraindikovaný u pacientov s malígnymi pečeňovými nádormi v pokročilom štádiu (pozri časť 4.3).
Poruchafunkcieobličiek
Obličkové abnormality počínajúc od zvýšených hodnôt kreatinínu v sére až po zlyhanie obličiek a úmrtie boli pozorované u pacientov intravenózne liečených azacitidínom v kombinácii s inými
chemoterapeutikami. Okrem toho sa u 5 pacientov s chronickou myeloidnou leukémiou (CML), ktorí boli liečení azacitidínom a etopozidom, vyvinula renálna tubulárna acidóza, definovaná ako pokles
hladiny hydrogenuhličitanu v sére na < 20 mmol/l spojený s alkalickým močom a hypokaliémiou (draslík v sére < 3 mmol/l). Ak sa vyskytne nevysvetliteľný pokles hydrogenuhličitanu v sére (< 20 mmol/l) alebo vzostup sérového kreatinínu alebo BUN, dávka sa má znížiť alebo jej podanie odložiť
na neskôr (pozri časť 4.2).
Pacientom sa má odporučiť, aby okamžite hlásili oligúriu a anúriu poskytovateľovi zdravotnej
starostlivosti.
Hoci neboli zaznamenané žiadne klinicky významné rozdiely vo frekvencii nežiaducich reakcií medzi pacientmi s normálnou funkciou obličiek v porovnaní s pacientmi s poruchou funkcie obličiek, pacienti s poruchou funkcie obličiek sa majú dôkladne sledovať kvôli toxicite, pretože azacitidín a/alebo jeho metabolity sa primárne vylučujú obličkami (pozri časť 4.2).
Laboratórnetesty
Pred začatím liečby a pred každým liečebným cyklom je potrebné stanoviť hodnoty pečeňových testov, kreatinínu v sére a hydrogenuhličitanu v sére. Pred začatím liečby a v prípade potreby
(minimálne však pred každým liečebným cyklom) sa má stanoviť kompletný krvný obraz s cieľom
sledovania odpovede a toxicity, pozri tiež časť 4.8.
Ochoreniesrdcaapľúc
Pacienti s anamnézou závažného kongestívneho zlyhávania srdca, klinicky nestabilného ochorenia srdca alebo pľúc boli vylúčení z pivotných registračných štúdií (AZA PH GL 2003 CL 001 a AZA- AML-001), a preto nebola u týchto pacientov stanovená bezpečnosť a účinnosť azacitidínu. Posledné údaje z klinického skúšania u pacientov so známou anamnézou kardiovaskulárneho alebo pľúcneho ochorenia preukázali významné zvýšenie výskytu srdcových príhod v súvislosti s azacitidínom (pozri časť 4.8). Preto sa odporúča opatrnosť pri predpisovaní azacitidínu týmto pacientom. Pred liečbou a počas liečby je potrebné zvážiť kardiopulmonálne vyšetrenie.
Nekrotizujúcafasciitída
U pacientov liečených azacitidínom bola hlásená nekrotizujúca fasciitída, vrátane fatálnych prípadov. U pacientov, u ktorých sa rozvíja nekrotizujúca fasciitída, sa má liečba azacitidínom ukončiť a má sa
okamžite začať s vhodnou liečbou.
Syndrómzrozpadunádoru
Pacienti vykazujúci pred liečbou vysokú nádorovú záťaž sú ohrození syndrómom z rozpadu nádoru. Títo pacienti sa majú dôkladne sledovať a majú byť vykonané vhodné opatrenia.
4.5 Liekové a iné interakcie
Na základe údajov in vitro sa metabolizmus azacitidínu nezdá byť sprostredkovaný izoenzýmami cytochrómu P450 (CYP), UDP-glukuronosyltransferázami (UGT), sulfotransferázami (SULT) ani glutationtransferázami (GST). Interakcie súvisiace s týmito metabolickými enzýmami in vivo sú preto považované za nepravdepodobné.
Klinicky významné inhibičné alebo indukčné účinky azacitidínu na enzýmy cytochrómu P450 sú nepravdepodobné (pozri časť 5.2).
Neuskutočnili sa žiadne formálne klinické štúdie liekových interakcií s azacitidínom.
4.6 Fertilita, gravidita a laktácia
Ženyvo fertilnom veku/Antikoncepciaumužovažien
Ženy vo fertilnom veku a muži majú používať účinnú antikoncepciu počas liečby a 3 mesiace po liečbe.
Gravidita
Nie sú k dispozícii dostatočné údaje o použití azacitidínu u gravidných žien. Štúdie na myšiach preukázali reprodukčnú toxicitu (pozri časť 5.3). Nie je známe potenciálne riziko pre ľudí. Na základe
výsledkov zo štúdií na zvieratách a mechanizmu jeho účinku sa azacitidín nemá používať počas gravidity, najmä počas prvého trimestra, pokiaľ jeho použitie nie je jednoznačne nevyhnutné. Prínos
liečby sa má individuálne zvážiť vzhľadom na možné riziko pre plod.
Dojčenie
Nie je známe, či sa azacitidín/metabolity vylučujú do ľudského mlieka. Z dôvodu potenciálne závažných nežiaducich reakcií u dojčeného dieťaťa, je dojčenie počas liečby azacitidínom kontraindikované.
Fertilita
Nie sú k dispozícii žiadne údaje o vplyve azacitidínu na plodnosť u ľudí. U zvierat boli pri použití azacitidínu dokumentované nežiaduce reakcie na plodnosť samcov (pozri časť 5.3). Mužom sa má odporučiť, aby sa vyvarovali splodeniu dieťaťa počas liečby, pričom musia v priebehu liečby a ešte 3 mesiace po jej ukončení používať účinnú antikoncepciu. Pred začatím liečby sa má mužom odporučiť, aby navštívili poradňu ohľadne uchovania spermií.
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Azacitidín má malý alebo mierny vplyv na schopnosť viesť vozidlá a obsluhovať stroje. Pri používaní azacitidínu sa zaznamenala únava. Preto sa pri vedení vozidla alebo obsluhovaní strojov odporúča opatrnosť.
4.8 Nežiaduce účinky
Súhrn profilubezpečnosti
Dospelá populácia s MDS, CMML a AML (20-30 % blastov v kostnej dreni)
Nežiaduce reakcie považované za možno alebo pravdepodobne súvisiace s podávaním azacitidínu sa
vyskytli u 97 % pacientov.
Najčastejšie závažné nežiaduce reakcie pozorované v rámci pivotnej štúdie (AZA PH GL 2003 CL
001), ktoré boli tiež pozorované v podporných štúdiách (CALGB 9221 a CALGB 8921), zahŕňali febrilnú neutropéniu (8,0 %) a anémiu (2,3 %). Ďalšie závažné nežiaduce reakcie z týchto 3 štúdií
zahŕňali infekcie, ako je neutropenická sepsa (0,8 %) a zápal pľúc (2,5 %) (niektoré prípady s fatálnym
výsledkom), trombocytopénia (3,5 %), reakcie precitlivenosti (0,25 %) a prípady krvácania (napr. mozgové krvácanie [0,5 %], gastrointestinálne krvácanie [0,8 %] a intrakraniálne krvácanie [0,5 %]).
Najčastejšie hlásené nežiaduce reakcie počas liečby azacitidínom boli hematologické reakcie (71,4 %)
vrátane trombocytopénie, neutropénie a leukopénie (zvyčajne 3.-4. stupňa), gastrointestinálne reakcie (60,6 %) vrátane nevoľnosti, vracania (zvyčajne 1.-2. stupňa) alebo reakcie v mieste vpichu (77,1 %, zvyčajne 1.-2. stupňa).
Dospelá populácia vo veku 65 rokov alebo starší s AML s > 30 % blastov v kostnej dreni Najčastejšie závažné nežiaduce reakcie (≥ 10 %) pozorované z AZA-AML-001 v rámci skupiny s azacitidínom zahŕňali febrilnú neutropéniu (25,0 %), zápal pľúc (20,3 %) a pyrexiu (10,6 %). Iné menej často hlásené závažné nežiaduce reakcie v skupine s azacitidínom zahŕňali sepsu (5,1 %), anémiu (4,2 %), neutropenickú sepsu (3,0 %), infekciu močových ciest (3,0 %), trombocytopéniu (2,5
%), neutropéniu (2,1 %), celulitídu (2,1 %), závrat (2,1 %) a dyspnoe (2,1 %).
Najčastejšie hlásené nežiaduce reakcie (≥ 30 %) počas liečby azacitidínom boli gastrointestinálne príhody vrátane zápchy (41,9 %), nevoľnosti (39,8 %) a hnačky (36,9 %), (zvyčajne 1.-2. stupňa),
celkové poruchy a reakcie v mieste podania vrátane pyrexie (37,7 %; zvyčajne 1.-2.stupňa) a hematologické príhody vrátane febrilnej neutropénie (32,2 %) a neutropénie (30,1 %), (zvyčajne
3.-4. stupňa).
ZoznamnežiaducichreakciívtabuľkeNižšie uvedená tabuľka 1 obsahuje nežiaduce reakcie súvisiace s liečbou azacitidínom získané z hlavných klinických štúdií s MDS a AML a zo sledovania po uvedení lieku na trh.
Frekvencie výskytu sú definované nasledovne: veľmi časté (≥ 1/10), časté (≥ 1/100 až < 1/10), menej časté (≥ 1/1 000 až < 1/100), zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov). V rámci jednotlivých skupín frekvencií sú nežiaduce účinky
usporiadané v poradí klesajúcej závažnosti. Nežiaduce reakcie sú uvedené v tabuľke nižšie podľa najvyššej frekvencie pozorovanej v akejkoľvek z hlavných klinických štúdií.
Tabuľka 1: Nežiaduce reakcie hlásené u pacientov s MDS alebo AML liečených azacitidínom(klinické štúdie a obdobie po uvedení lieku na trh)Trieda orgánových
systémov
| Veľmi časté
| Časté
| Menej časté
| Zriedkavé
| Neznáme
|
Infekcie a nákazy
| zápal pľúc* (vrátane bakteriálneho, vírusového a plesňového), nazofaryngitída
| sepsa* (vrátane bakteriálnej, vírusovej a plesňovej), neutropenická sepsa*, infekcia dýchacích ciest (vrátane horných dýchacích ciest a
bronchitídy), infekcia močových ciest, celulitída, divertikulitída, orálna plesňová infekcia, sinusitída,
faryngitída, rinitída,
herpes simplex,
kožná infekcia
|
|
| nekrotizujúca fasciitída*
|
T
rieda orgánových systémov
|
V
eľmi časté
|
Č
asté
|
Menej časté
|
Z
riedkavé
|
N
eznáme
|
P
oruchy krvi a lymfatického systému
|
febrilná neutropénia*, neutropénia, leukopénia, trombocytopénia, anémia
|
pancytopénia*, zlyhanie kostnej drene
|
|
|
|
P
oruchy
im
unitného systému
|
|
|
reakcie z precitlivenosti
|
|
|
P
oruchy metabolizmu a výživy
|
anorexia, znížená chuť do jedla, hypokaliémia
|
dehydratácia
|
|
syndróm
z rozpadu
nádoru
|
|
P
sychické poruchy
|
nespavosť
|
zmätenosť, úzkosť
|
|
|
|
P
oruchy nervového systému
|
závrat, bolesť hlavy
|
intrakraniálne krvácanie*, synkopa, somnolencia, letargia
|
|
|
|
P
oruchy oka
|
|
krvácanie do oka, krvácanie do spojoviek
|
|
|
|
P
oruchy srdca
a srdcovej činnosti
|
|
perikardiálny výpotok
|
perikarditída
|
|
|
P
oruchy ciev
|
|
hypotenzia*, hypertenzia, ortostatická hypotenzia, hematóm
|
|
|
|
P
oruchy dýchacej sústavy, hrudníka a mediastína
|
dyspnoe, epistaxa
|
pleurálny výpotok, námahové dyspnoe, faryngolaryngeálna bolesť
|
|
intersticiálne ochorenie pľúc
|
|
P
oruchy gastrointestinálneho traktu
|
hnačka, vracanie, zápcha, nevoľnosť, bolesť brucha (vrátane bolesti hornej časti brucha a abdominálneho diskomfortu)
|
gastrointestinálne krvácanie* (vrátane krvácania z úst), krvácanie z hemoroidov, stomatitída, krvácanie ďasien, dyspepsia
|
|
|
|
P
oruchy pečene
a žlčových ciest
|
|
|
zlyhanie pečene*, progresívna hepatálna kóma
|
|
|
T
rieda orgánových systémov
|
V
eľmi časté
|
Č
asté
|
Menej časté
|
Z
riedkavé
|
N
eznáme
|
P
oruchy kože a podkožného tkaniva
|
petechie, svrbenie (vrátane generalizovaného), vyrážka, ekchymóza
|
purpura, alopécia, urtikária, erytém, makulárna vyrážka
|
akútna febrilná neutrofilná dermatóza, pyoderma gangrenosum
|
|
|
P
oruchy kostrovej a svalovej sústavy a spojivového tkaniva
|
artralgia, muskuloskeletálna bolesť (vrátane chrbta, kostí a bolesti končatín)
|
svalové spazmy,
myalgia
|
|
|
|
P
oruchy obličiek
a močových ciest
|
|
renálne zlyhanie*, hematúria, zvýšená hladina sérového kreatinínu
|
renálna tubulárna acidóza
|
|
|
C
elkové poruchy a reakcie v mieste podania
|
pyrexia*, únava, asténia, bolesť na hrudi, erytém v mieste vpichu injekcie, bolesť v mieste vpichu injekcie, reakcia v mieste vpichu injekcie (nešpecifikovaná)
|
podliatiny, hematóm, stvrdnutie, vyrážka, svrbenie, zápal, zmena sfarbenia
kože, uzlíky a krvácanie (v mieste
vpichu injekcie),
malátnosť, zimnica, krvácanie v mieste
zavedenia katétra
|
|
nekróza v mieste injekcie (v mieste vpichu)
|
|
L
aboratórne a funkčné vyšetrenia
|
pokles telesnej hmotnosti
|
|
|
|
|
*= zriedkavo sa zaznamenali fatálne prípady
Opis vybranýchnežiaducichreakciíHematologické nežiaduce reakcieNajčastejšie pozorované (≥ 10 %) hematologické nežiaduce reakcie súvisiace s liečbou azacitidínom zahŕňajú anémiu, trombocytopéniu, neutropéniu, febrilnú neutropéniu a leukopéniu, zvyčajne 3. alebo
4. stupňa. V priebehu prvých 2 cyklov existuje vyššie riziko výskytu týchto reakcií, potom sa u
pacientov tieto reakcie vyskytujú s menšou frekvenciou a obnovuje sa hematologická funkcia. Väčšina
hematologických nežiaducich reakcií bola kontrolovaná bežným sledovaním kompletného krvného obrazu a oneskorením podania azacitidínu v ďalšom cykle, profylaktickými antibiotikami a/alebo podľa potreby podpornou liečbou neutropénie rastovým faktorom (napr. G-CSF) a anémie alebo trombocytopénie transfúziami.
InfekcieMyelosupresia môže viesť k neutropénii a zvýšenému riziku infekcie. U pacientov dostávajúcich azacitidín boli hlásené závažné nežiaduce reakcie ako sú sepsa, vrátane neutropenickej sepsy, a zápal
pľúc, pričom niektoré mali fatálny výsledok. Infekcie možno kontrolovať použitím liekov proti
infekcii a podpornou liečbou neutropénie rastovým faktorom (napríklad G-CSF).
KrvácanieU pacientov dostávajúcich azacitidín sa môže vyskytnúť krvácanie. Boli hlásené závažné nežiaduce reakcie ako napríklad gastrointestinálne krvácanie a intrakraniálne krvácanie. U pacientov sa majú sledovať prejavy a príznaky krvácania, a to najmä u pacientov s existujúcou alebo s liečbou súvisiacou trombocytopéniou.
Precitlivenosť
U pacientov dostávajúcich azacitidín boli hlásené závažné reakcie z precitlivenosti. V prípade reakcie podobnej anafylaktickému šoku sa má liečba azacitidínom ihneď ukončiť a má sa začať vhodná symptomatická liečba.
Nežiaduce reakcie kože a podkožného tkanivaVäčšina nežiaducich reakcií kože a podkožného tkaniva súvisela s miestom vpichu injekcie. Žiadna z uvedených nežiaducich reakcií neviedla k prerušeniu podávania azacitidínu, ani k zníženiu dávky
azacitidínu počas pivotných štúdií. Väčšina nežiaducich reakcií sa vyskytla počas prvých 2 cyklov a v následných cykloch sa ich počet znižoval. Subkutánne nežiaduce reakcie, ako napríklad vyrážka, zápal
a svrbenie v mieste vpichu injekcie, vyrážka, erytém a kožné lézie, môžu vyžadovať súbežnú liečbu napríklad antihistaminikami, kortikosteroidmi a nesteroidovými protizápalovými liekmi (
non-steroidal anti-inflammatory drugs, NSAID). Tieto kožné reakcie sa musia odlíšiť od infekcií
mäkkých tkanív, ktoré sa niekedy objavujú v mieste vpichu injekcie. Po uvedení lieku na trh boli pri liečbe azacitidínom hlásené infekcie mäkkých tkanív, vrátane celulitídy a nekrotizujúcej fasciitídy, ktoré v zriedkavých prípadoch viedli k smrti. Pre postup pri liečbe infekčných nežiaducich reakcií pozri časť 4.8 Infekcie.
Gastrointestinálne nežiaduce reakcieNajčastejšie hlásené gastrointestinálne nežiaduce reakcie súvisiace s liečbou azacitidínom zahŕňali zápchu, hnačku, nevoľnosť a vracanie. Tieto nežiaduce reakcie boli liečené symptomaticky antiemetikami v prípade nevoľnosti a vracania, antidiaroikami v prípade hnačky a laxatívami a/alebo prípravkami na zmäkčenie stolice v prípade zápchy.
Renálne nežiaduce reakcieU pacientov liečených azacitidínom sa zaznamenali renálne abnormality v rozsahu od zvýšenej
hladiny kreatinínu v sére a hematúrie po renálnu tubulárnu acidózu, renálne zlyhanie a úmrtie (pozri časť 4.4).
Hepatálne nežiaduce reakcieU pacientov s výraznou nádorovou záťažou z dôvodu metastatického ochorenia sa počas liečby azacitidínom zaznamenalo zlyhanie pečene, progresívna hepatálna kóma a úmrtie (pozri časť 4.4).
Srdcové príhodyÚdaje z klinickej štúdie, ktoré povolilo zaradenie pacientov so známou anamnézou kardiovaskulárneho alebo pľúcneho ochorenia, preukázali štatisticky významné zvýšenie srdcových príhod u pacientov s novodiagnostikovanou AML liečenou azacitidínom (pozri časť 4.4).
Staršia populáciaK dispozícii je obmedzené množstvo informácií o bezpečnosti použitia azacitidínu u pacientov ≥ 85
rokov (so 14 [5,9 %] pacientmi ≥ 85 rokov v štúdii AZA-AML-001).
Hláseniepodozrenínanežiaducereakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili
akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.4.9 PredávkovaniePočas klinických štúdií sa vyskytol jeden prípad predávkovania azacitidínom. U tohto pacienta sa vyskytla hnačka, nevoľnosť a vracanie po podaní jednorazovej intravenóznej dávky približne 290 mg/m2, čo je takmer 4-násobok odporúčanej začiatočnej dávky.
V prípade predávkovania sa má u pacienta sledovať krvný obraz a podľa potreby sa mu má podať
podporná liečba. Nie je známe žiadne špecifické antidotum pri predávkovaní azacitidínom.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatiká, analógy pyrimidínu; ATC kód: L01BC07
Mechanizmus účinku
U azacitidínu sa predpokladá, že vykazuje svoje antineoplastické účinky pomocou viacerých
mechanizmov vrátane cytotoxicity pôsobiacej na abnormálne hematopoetické bunky v kostnej dreni a hypometylácie DNA. Cytotoxické účinky azacitidínu môžu byť výsledkom viacerých mechanizmov
vrátane inhibície syntézy DNA, RNA a proteínov, inkorporácie do RNA a DNA a aktivácie dráh
vedúcich k poškodeniu DNA. Neproliferujúce bunky sú relatívne necitlivé na azacitidín. Inkorporácia azacitidínu do DNA má za následok inaktiváciu DNA metyltransferáz, čo vedie k hypometylácii DNA. Hypometylácia DNA u aberantne metylovaných génov zapojených do regulácie normálneho bunkového cyklu, diferenciácie a dráh bunkovej smrti, môže spôsobiť opätovnú exprimáciu týchto génov a obnovenie funkcií potláčajúcich tvorbu nádorov v nádorových bunkách. Relatívny význam hypometylácie DNA v porovnaní s cytotoxicitou alebo inými účinkami azacitidínu na klinické výsledky nebol stanovený.
Klinická účinnosťabezpečnosť
Dospelá populácia (MDS, CMML a AML [20-30 % blastov v kostnej dreni])
Účinnosť a bezpečnosť azacitidínu boli sledované v medzinárodnej, multicentrickej, kontrolovanej, otvorenej, randomizovanej, komparatívnej štúdii fázy 3 s paralelnými skupinami (AZA PH GL 2003
CL 001) u dospelých pacientov so stredne-2 a vysokorizikovým MDS podľa medzinárodného prognostického hodnotiaceho systému (IPSS), u pacientov s refraktérnou anémiou s prevahou blastov
(refractory anaemia with excess blasts, RAEB), u pacientov s refraktérnou anémiou s prevahou
blastov v transformácii (refractory anaemia with excess blasts in transformation, RAEB-T) a u
pacientov s modifikovanou chronickou myelomonocytovou leukémiou (mCMML) podľa francúzsko- americko-britského (FAB) klasifikačného systému. Pacienti s RAEB-T (21-30 % blastov) sa podľa aktuálneho klasifikačného systému WHO považujú za pacientov s AML. Azacitidín spolu s najlepšou podpornou liečbou (best supportive care, BSC); (n = 179) sa porovnával s konvenčnými režimami liečby (conventional care regimens, CCR). CCR sa skladali zo samostatnej BSC (n = 105), nízkej dávky cytarabínu a BSC (n = 49) alebo zo štandardnej indukčnej chemoterapie a BSC (n = 25). Pacienti boli pred randomizáciou vyberaní svojím lekárom do 1 z 3 režimov CCR. Ak neboli pacienti randomizovaní na liečbu azacitidínom, boli liečení týmto vopred vybraným režimom. Ako súčasť kritérií pre zaradenie museli pacienti dosiahnuť výkonnostný stav podľa kritérií Eastern Cooperative Oncology Group (ECOG) na úrovni 0-2. Pacienti so sekundárnym MDS boli vylúčení zo štúdie. Primárnym koncovým ukazovateľom štúdie bola celkové prežívanie. Azacitidín sa podával vo forme subkutánnej dávky 75 mg/m2 denne po dobu 7 dní, po ktorých nasledovala 21-dňová prestávka (28- dňový liečebný cyklus), pričom medián počtu cyklov bol 9 (rozsah = 1-39) a priemerný počet cyklov bol 10,2. V rámci populácie intent to treat (ITT, populácia všetkých randomizovaných pacientov) bol stredný vek 69 rokov (rozsah bol 38 až 88 rokov).
V rámci ITT analýzy 358 pacientov (179 pacientov liečených azacitidínom a 179 liečených CCR), viedla liečba azacatidínom k mediánu prežívania 24,46 mesiacov oproti 15,02 mesiacom u pacientov liečených CCR, čo je rozdiel 9,4 mesiacov, so stratifikovanou log-rank p-hodnotou 0,0001. Miera rizika pre liečebný účinok bola 0,58 (95 % CI: 0,43; 0,77). Dvojročné miery prežívania boli 50,8 % u pacientov dostávajúcich azacitidín oproti 26,2 % u pacientov dostávajúcich CCR (p < 0,0001).

LEGENDA: AZA = azacitidín, CCR = konvenčné režimy liečby (
conventional care regimens), CI =
interval spoľahlivosti (
confidence interval), HR = miera rizika (
hazard ratio)
Prínosy liečby azacitidínom v dĺžke prežívania boli konzistentné oproti všetkým možnostiam liečby CCR (samostatne BSC, nízke dávky cytarabínu s BSC alebo štandardná indukčná chemoterapia s BSC) použitými v kontrolnej skupine.
Pri analýze cytogenetických podskupín IPSS bol zistený podobný medián celkového prežívania vo všetkých skupinách (priaznivá, intermediálna, nepriaznivá cytogenetika, vrátane monozómie 7).
V rámci analýz vekových podskupín bol vo všetkých skupinách (< 65 rokov, ≥ 65 rokov a ≥ 75 rokov)
zistený nárast mediánu celkového prežívania.
Liečba azacitidínom bola spojená s mediánom doby do úmrtia alebo do transformácie na AML 13,0 mesiaca oproti 7,6 mesiaca u pacientov podstupujúcich liečbu CCR, čo je zlepšenie o 5,4 mesiaca so stratifikovanou log-rank p-hodnotou 0,0025.
Liečba azacitidínom bola tiež spojená s poklesom cytopénií a s nimi súvisiacich príznakov. Liečba azacitidínom viedla k zníženej potrebe transfúzie erytrocytov a trombocytov. Z pacientov v skupine liečenej azacitidínom, ktorí boli na začiatku liečby závislí od transfúzie erytrocytov, sa 45,0 % počas liečby stalo nezávislými od transfúzie erytrocytov v porovnaní s 11,4 % pacientov v kombinácii skupín CCR (štatisticky významný (p < 0,0001) rozdiel bol 33,6 % (95 % CI: 22,4; 44,6)). U pacientov v skupine liečenej azacitidínom, ktorí boli na začiatku závislí od transfúzie erytrocytov a stali sa od nej nezávislými, bol medián doby nezávislosti od transfúzie erytrocytov 13 mesiacov.
Odpoveď bola vyhodnotená skúšajúcim alebo nezávislou komisiou (
Independent Review Committee,
IRC). Celková odpoveď (úplná remisia [CR] + čiastočná remisia [PR]) stanovená skúšajúcim bola 29
% v skupine liečenej azacitidínom a 12 % v kombinácii skupín CCR (p = 0,0001). Celková odpoveď
(CR + PR) stanovená IRC v štúdii AZA PH GL 2003 CL 001 bola 7 % (12/179) v skupine liečenej azacitidínom v porovnaní s 1 % (2/179) v kombinácii skupín CCR (p = 0,0113). Rozdiely medzi
hodnotením IRC a hodnotením skúšajúceho boli dôsledkom kritérií medzinárodnej pracovnej skupiny (International Working Group, IWG), vyžadujúcimi zlepšenie periférneho krvného obrazu a zachovanie tohto zlepšenia po dobu najmenej 56 dní. Prínos v prežívaní bol preukázaný aj u
pacientov, ktorí nedosiahli úplnú/čiastočnú odpoveď na liečbu azacitidínom. Hematologické zlepšenie
(veľké alebo malé) stanovené IRC sa dosiahlo u 49 % pacientov dostávajúcich azacitidín v porovnaní s 29 % pacientov v kombinácii skupín CCR (p < 0,0001).
U pacientov s jednou alebo viacerými cytogenetickými abnormalitami na začiatku liečby bolo percento pacientov s veľkou cytogenetickou odpoveďou podobné v skupine s azacitidínom a v kombinácii skupín CCR. Malá cytogenetická odpoveď bola štatisticky významne (p = 0,0015) vyššia v skupine s azacitidínom (34 %) v porovnaní s kombináciou skupín CCR (10 %).
Dospelá populácia vo veku 65 rokov alebo staršia s AML s > 30 % blastov v kostnej dreni Nižšie uvedené výsledky reprezentujú populáciu všetkých randomizovaných pacientov (ITT) študovanú v AZA-AML-001 (pozri časť 4.1 pre schválené indikácie).
Účinnosť a bezpečnosť azacitidínu boli sledované v medzinárodnej, multicentrickej, kontrolovanej, otvorenej štúdii fázy 3 s paralelnými skupinami u pacientov vo veku 65 rokov a starších s novodiagnostikovanou AML alebo sekundárnou AML s > 30 % blastov v kostnej dreni podľa klasifikácie WHO, ktorí neboli spôsobilí na HSCT. Azacitidín spolu s BSC (n=241) sa porovnával s CCR. CCR pozostával zo samotnej BSC (n=45), nízkej dávky cytarabínu spolu s BSC (n=158) alebo zo štandardnej intenzívnej chemoterapie s cytarabínom a antracyklínom spolu s BSC (n=44). Pred randomizáciou lekár vybral pacientom 1 z 3 režimov CCR. Ak pacienti neboli randomizovaní na liečbu azacitidínom, boli liečení vopred vybraným režimom. Ako súčasť kritérií pre zaradenie museli pacienti dosiahnuť výkonnostný stav podľa ECOG na úrovni 0-2 a vykazovať stredne alebo málo rizikové cytogenetické abnormality. Primárnym koncovým ukazovateľom štúdie bolo celkové prežívanie.
Azacitidín sa podával subkutánne v dávke 75 mg/m2/deň po dobu 7 dní, po ktorých nasledovala 21- dňová prestávka (28-dňový liečebný cyklus), pričom medián počtu cyklov bol 6 (rozsah: 1 až 28), u pacientov iba s BSC bol medián počtu cyklov 3 (rozsah: 1 až 20), u pacientov s nízkou dávkou cytarabínu bol medián počtu cyklov 4 (rozsah: 1 až 25) a u pacientov so štandardnou intenzívnou chemoterapiou bol medián počtu cyklov 2 (rozsah 1 až 3, indukčný cyklus plus 1 alebo 2 konsolidačné cykly).
Jednotlivé východiskové parametre boli porovnateľné medzi skupinami s azacitidínom a CCR. Medián veku pacientov bol 75,0 rokov (rozsah: 64 až 91 rokov), 75,2 % boli bielej rasy a 59,0 % boli muži. Na začiatku bolo 60,7 % pacientov klasifikovaných ako AML inak nešpecifikovaná, 32,4 % ako AML so zmenami podobnými myelodysplázii, 4,1% myeloidné neoplazmy súvisiace s terapiou a 2,9
% pacientov ako AML s rekurentnými genetickými abnormalitami podľa klasifikácie WHO.
V rámci ITT analýzy 488 pacientov (241 na azacitidíne a 247 na CCR) bola liečba azacitidínom spojená s mediánom prežívania 10,4 mesiacov verzus 6,5 mesiacov u pacientov liečených CCR, čo je rozdiel 3,8 mesiacov, so stratifikovanou log-rank p-hodnotou 0,1009 (obojstranná). Miera rizika pre liečebný účinok bola 0,85 (95 % CI=0,69; 1,03). Ročné miery prežívania boli 46,5 % u pacientov liečených azacitidínom verzus 34,3 % u pacientov na CCR.
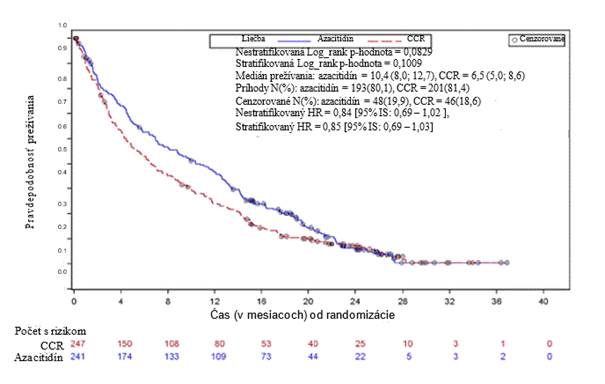
'
Coxov model proporcionálneho rizika upravený pre vopred špecifikované východiskové prognostické
faktory definovaný ako HR azacitidínu oproti CCR 0,80 (95% CI = 0,66; 0,99; p = 0,0355).
Okrem toho, hoci cieľom štúdie nebolo preukázanie štatisticky významného rozdielu pri porovnaní azacitidínu a vopred vybraných CCR terapeutických skupín, pri liečbe pacientov azacitidínom bola dĺžka prežívania väčšia v porovnaní s liečbou CCR - BSC samostatne a nízkou dávkou cytarabínu s BSC, a bola podobná v porovnaní so štandardnou intenzívnou chemoterapiou s BSC.
Vo všetkých vopred definovaných podskupinách veku (˂ 75 rokov a ≥ 75 rokov), pohlavia, rasy, výkonnostného stavu ECOG (0 alebo 1 a 2), východiskového cytogenetického rizika (stredné a malé), geografického regiónu, WHO klasifikácie AML (vrátane AML so zmenami podobnými myelodysplázii), východiskového počtu WBC (≤ 5 x109/l a > 5 x 109/l), východiskového počtu blastov v kostnej dreni (≤ 50 % a > 50 %) a predchádzajúcej MDS v anamnéze, bola tendencia k prínosu k celkovému prežívaniu (OS) v prospech azacitidínu. V niektorých vopred definovaných podskupinách pomer rizika celkového prežívania bol štatistického významu, vrátane pacientov s malým cytogenetickým rizikom, pacientov s AML so zmenami súvisiacimi s myelodyspláziou, pacientov <
75 rokov, žien a pacientov bielej rasy.
Hematologické a cytogenetické odpovede boli vyhodnotené skúšajúcim a IRC s podobnými výsledkami. Celková miera odpovede (úplná remisia [CR] + úplná remisia s neúplným obnovením krvného obrazu [CRi]), ktorú stanovila IRC, bola 27,8 % v skupine liečenej azacitidínom a 25,1 % v kombinácii skupín CCR (p = 0,5384). U pacientov, ktorí dosiahli CR alebo CRi, bola stredná doba trvania remisie u pacientov liečených azacitidínom 10,4 mesiacov (95% CI = 7,2; 15,2) a 12,3 mesiacov (95 % CI = 9,0; 17,0) u pacientov liečených CCR. Prínos v prežívaní bol taktiež preukázaný u pacientov, ktorí nedosiahli kompletnú odpoveď pri liečbe azacitidínom v porovnaní s CCR.
Liečba azacitidínom zlepšila periférny krvný obraz a viedla k zníženiu potreby transfúzií erytrocytov a trombocytov. Pacient bol považovaný za závislého na transfúzii erytrocytov alebo trombocytov na začiatku liečby, ak mal jednu alebo viac transfúzií erytrocytov alebo trombocytov počas 56 dní (8 týždňov) pri alebo pred randomizáciou, v tomto poradí. Pacient bol považovaný za nezávislého od transfúzie erytrocytov alebo trombocytov počas obdobia liečby, ak nemal žiadnu transfúziu
erytrocytov alebo trombocytov počas ktorýchkoľvek 56 po sebe idúcich dní počas sledovaného
obdobia.
Z pacientov v skupine s azacitidínom, ktorí boli na začiatku liečby závislí na transfúzii erytrocytov, sa počas obdobia liečby stalo 38,5 % (95 % CI = 31,1; 46,2) nezávislých na transfúzii erytrocytov, v porovnaní s 27,6 % (95 % CI = 20,9; 35,1) pacientov v kombinácii skupín CCR. U pacientov, ktorí boli závislí na transfúzii erytrocytov na začiatku liečby a počas liečby dosiahli nezávislosť na transfúzii, bola stredná doba trvania nezávislosti na transfúzii erytrocytov v skupine s azacitidínom
13,9 mesiacov a v skupine CCR sa nezávislosť nedosiahla.
Z pacientov v skupine s azacitidínom, ktorí boli na začiatku liečby závislí na transfúzii trombocytov, sa počas obdobia liečby stalo 40,6 % (95 % CI = 30,9; 50,8) nezávislých na transfúzii trombocytov, v porovnaní s 29,3 % (95 % CI = 19,7; 40,4) pacientov v kombinácii skupín CCR. U pacientov, ktorí boli závislí na transfúzii trombocytov na začiatku liečby a počas liečby dosiahli nezávislosť na transfúzii, bola stredná doba trvania nezávislosti na transfúzii trombocytov v skupine s azacitidínom
10,8 mesiacov a 19,2 mesiacov v skupine s CCR.
Kvalita života súvisiaca so zdravím (Health-Related Quality of Life, HRQoL) sa hodnotila pomocou dotazníka kvality života Európskej organizácie pre výskum a liečbu rakoviny (European Organization for Research and Treatment of Cancer Core Quality of Life Questionnaire, EORTC QLQ-C30). HRQoL údaje sa mohli analyzovať v podskupine celej populácie klinickej štúdie. Napriek tomu, že v analýze existujú obmedzenia, dostupné údaje naznačujú, že pacienti počas liečby azacitidínom nepociťujú významné zhoršenie kvality života.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po subkutánnom podaní jednorazovej dávky 75 mg/m2, sa azacitidín rýchlo absorboval s maximálnymi plazmatickými koncentráciami 750 ng/ml ± 403 ng/ml vyskytujúcimi sa po 0,5 h od
podania dávky (prvý bod odberu). Absolútna biologická dostupnosť azacitidínu po subkutánnom podaní v porovnaní s intravenóznym podaním (jednorazové dávky 75 mg/m2) bola na základe plochy
pod krivkou (AUC) približne 89 %.
Plocha pod krivkou a maximálna plazmatická koncentrácia (Cmax) subkutánneho podania azacitidínu boli približne priamo úmerné v medziach dávky 25 až 100 mg/m2.
Distribúcia
Po intravenóznom podaní bol priemerný distribučný objem 76 l ± 26 l a systémový klírens bol 147 l/h
± 47 l/h.
Biotransformácia
Na základe údajov in vitro sa metabolizmus azacitidínu nezdá byť sprostredkovaný izoenzýmami cytochrómu P450 (CYP), UDP-glukuronosyltransferázami (UGT), sulfotransferázami (SULT) ani glutationtransferázami (GST).
Azacitidín podlieha spontánnej hydrolýze a deaminácii sprostredkovanej cytidín-deaminázou. V S9 frakciách z ľudskej pečene bolo vytváranie metabolitov nezávislé od NADPH, čo naznačuje, že metabolizmus azacitidínu nebol sprostredkovaný izoenzýmami cytochrómu P450. In vitro štúdia azacitidínu s kultivovanými ľudskými hepatocytmi naznačuje, že koncentrácie od 1,0 μM až 100 μM
1A2, 2C19, 3A4 ani 3A5. V štúdiách zameraných na vyhodnotenie inhibície série izoenzýmov P450 (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 a 3A4) azacitidín v koncentrácii až do 100 μM nespôsoboval inhibíciu. Preto je indukcia alebo inhibícia CYP enzýmov azacitidínom pri klinicky dosiahnuteľných plazmatických koncentráciách nepravdepodobná.
Eliminácia
Azacitidín sa rýchlo vylučuje z plazmy so stredným polčasom eliminácie (t½) 41 ± 8 minút po subkutánnom podaní. Po subkutánnom podaní 75 mg/m2 azacitidínu jedenkrát denne počas 7 dní nedochádza k jeho akumulácii. Vylučovanie močom je primárnou cestou eliminácie azacitidínu a/alebo jeho metabolitov. Po intravenóznom a subkutánnom podaní 14C-azacitidínu sa 85 a 50 % podanej rádioaktivity vylúčilo močom a < 1 % sa vylúčilo stolicou.
Osobitnéskupinypacientov
Vplyv poruchy funkcie pečene (pozri časť 4.2), pohlavia, veku alebo rasy na farmakokinetické vlastnosti azacitidínu sa oficiálne neskúmal.
Poruchafunkcieobličiek
Porucha funkcie obličiek nemá významný vplyv na farmakokinetickú expozíciu azacitidínu po
jednorazovom a mnohonásobnom subkutánnom podaní. Po subkutánnom podaní jednorazovej dávky
75 mg/m2, sa priemerné hodnoty expozície (AUC a Cmax) u pacientov s ľahkou, stredne ťažkou alebo ťažkou poruchou funkcie obličiek zvýšili o 11-21 %, 15-27 %, a 41-66 %, v porovnaní s pacientmi s
normálnou funkciou obličiek. Avšak, expozícia bola v rámci rovnakého všeobecného rozmedzia expozícií pozorovaná u pacientov s normálnou funkciou obličiek. Azacitidín môže byť podávaný pacientom s poruchou funkcie obličiek bez úpravy úvodnej dávky za predpokladu, že títo pacienti sú sledovaní na toxicitu, pretože azacitidín a/alebo jeho metabolity sa primárne vylučujú obličkami.
Farmakogenomika
Účinok známeho polymorfizmu cytidín-deaminázy na metabolizmus azacitidínu sa oficiálne neskúmal.
5.3 Predklinické údaje o bezpečnosti
Azacitidín indukuje génové mutácie aj chromozómové aberácie v bunkových systémoch baktérií a cicavcov in vitro. Potenciálna karcinogenita azacitidínu bola hodnotená u myší a potkanov. Azacitidín vyvolával nádory hematopoetického systému u samíc myší pri intraperitoneálnom podávaní 3-krát týždenne po dobu 52 týždňov. Zvýšený výskyt nádorov v lymforetikulárnom systéme, pľúcach, mliečnych žľazách a koži bol pozorovaný u myší, ktorým bol podávaný azacitidín intraperitoneálne po dobu 50 týždňov. Štúdia tumorigenicity u potkanov odhalila zvýšený výskyt testikulárnych nádorov.
Štúdie zamerané na skorú embryotoxicitu u myší odhalili 44 % výskyt vnútromaternicových embryonálnych úmrtí (zvýšená resorpcia) po podaní jednorazovej intraperitoneálnej injekcie azacitidínu počas organogenézy. U myší, ktorým sa podával azacitidín v čase uzatvorenia tvrdého podnebia alebo pred ním, boli zistené vývojové abnormality mozgu. U potkanov azacitidín nespôsobil žiadne nežiaduce reakcie pri podávaní pred uhniezdením oplodneného vajíčka, bol však jednoznačne embryotoxický pri podávaní počas organogenézy. Abnormality plodu počas organogenézy u potkanov zahŕňajú anomálie centrálneho nervového systému (exencefália/encefalokéla), anomálie končatín (mikromélia, vbočená noha, syndaktýlia, oligodaktýlia) a iné (mikroftalmia, mikrognatia, gastroschíza, edém a abnormality rebier).
Podávanie azacitidínu samcom myší pred párením s neliečenými samicami myší viedlo k zníženiu plodnosti a strate potomstva počas následného embryonálneho a postnatálneho vývoja. Liečba samcov potkanov viedla k zníženiu hmotnosti semenníkov a nadsemenníkov, zníženiu počtu spermií, zníženiu frekvencie gravidity, zvýšeniu počtu abnormálnych embryí a k zvýšeným stratám embryí u oplodnených samíc (pozri časť 4.4).
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
manitol (E421)
6.2 Inkompatibility
Tento liek sa nesmie miešať s inými liekmi okrem tých, ktoré sú uvedené v časti 6.6.
6.3 Čas použiteľnosti
Neotvorená injekčná liekovka s práškom: 3 roky
Porekonštitúcii:
Keď sa azacitidín rekonštituuje pomocou vody na injekcie, ktorá sa neuchovávala v chladničke, chemická a fyzikálna stabilita rekonštituovaného lieku počas používania bola preukázaná pri teplote
25 °C počas 45 minút a pri teplote 2 °C až 8 °C počas 8 hodín.
Čas použiteľnosti rekonštituovaného lieku sa môže predĺžiť rekonštitúciou pomocou chladenej (2 °C až 8 °C) vody na injekcie. Keď sa azacitidín rekonštituuje pomocou chladenej (2 °C až 8 °C) vody na injekcie, chemická a fyzikálna stabilita rekonštituovaného lieku počas používania bola preukázaná pri teplote 2 °C až 8 °C počas 32 hodín.
Z mikrobiologického hľadiska sa má rekonštituovaný liek použiť okamžite. Ak sa nepoužije okamžite, za čas a podmienky uchovávania pred použitím zodpovedá používateľ, nesmie však byť dlhší ako 8 hodín pri teplote 2 °C až 8 °C, keď sa rekonštituoval pomocou vody na injekcie, ktorá sa
neuchovávala v chladničke a nie dlhší ako 32 hodín, keď sa rekonštituoval chladenou (2 °C až 8 °C)
vodou na injekcie.
6.4 Špeciálne upozornenia na uchovávanie
Neotvorenéinjekčnéliekovky
Tento liek nevyžaduje žiadne zvláštne podmienky na uchovávanie.
Rekonštituovanásuspenzia
Podmienky na uchovávanie po rekonštitúcii lieku, pozri časť 6.3.
6.5 Druh obalu a obsah balenia
Injekčná liekovka z bezfarebného skla uzatvorená butylkaučukovou zátkou potiahnutou ETFE a hliníkovým viečkom (biele pre 100 mg a oranžové pre 150 mg veľkosť balenia).
Veľkosti balenia:
1 injekčná liekovka obsahujúca 100 mg azacitidínu.
1 injekčná liekovka obsahujúca 150 mg azacitidínu. Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu a iné zaobchádzanie s liekom
Odporúčanianabezpečnézaobchádzaniesliekom
zaobchádzania a prípravy suspenzie azacitidínu musí postupovať opatrne. Je potrebné dodržiavať
postupy na bezpečné zaobchádzanie a likvidáciu protinádorových liekov.
Ak dôjde ku kontaktu rekonštituovaného azacitidínu s kožou, zasiahnuté miesto okamžite a dôkladne umyte mydlom a vodou. Ak sa dostane do kontaktu so sliznicami, zasiahnuté miesto dôkladne opláchnite vodou.
Tehotné zdravotnícke pracovníčky nemajú zaobchádzať s týmto liekom.
PostuprekonštitúcieAzacitidín sa má rekonštituovať vodou na injekcie. Čas použiteľnosti rekonštituovaného lieku sa môže
predĺžiť rekonštitúciou pomocou chladenej (2 °C až 8 °C) vody na injekcie. Podrobnosti o uchovávaní rekonštituovaného lieku sú uvedené v časti 6.3.
1. Pripravte si nasledovné pomôcky:
Injekčnú liekovku (injekčné liekovky) s azacitidínom, injekčnú liekovku (injekčné liekovky) s vodou na injekcie, nesterilné chirurgické rukavice, alkoholové tampóny, injekčnú striekačku
(injekčné striekačky) s ihlou (ihlami).
2. Odoberte príslušný objem vody na injekcie (pozri tabuľku nižšie) do injekčnej striekačky a dbajte na vytlačenie všetkého vzduchu z injekčnej striekačky.
Obsah injekčnej liekovky
| Objem vody na
injekcie
|
Výsledná koncentrácia
|
100 mg
| 4 ml
| 25 mg/ml
|
150 mg
| 6 ml
| 25 mg/ml
|
3. Zasuňte ihlu injekčnej striekačky obsahujúcej vodu na injekcie cez gumené viečko do
injekčnej liekovky s azacitidínom a vstreknite vodu na injekcie do injekčnej liekovky.
4. Vytiahnite injekčnú striekačku a ihlu a dôkladne pretrepte injekčnú liekovku, až kým sa nevytvorí
homogénna zakalená suspenzia. Po rekonštitúcii obsahuje každý ml suspenzie 25 mg azacitidínu. Rekonštituovaný liek je homogénna, zakalená suspenzia, bez aglomerátov.
Aksuspenziaobsahujeveľkéčasticealeboaglomeráty,trebajuzlikvidovať.Porekonštitúciisuspenziunefiltrujte,mohlobysatýmodstrániťliečivo.Musísazohľadniť,žefiltresanachádzajúvniektorýchadaptéroch,ihláchauzatvorenýchsystémoch;pretonapodanieliekuporekonštitúciisatakétosystémynemajúpoužívať.5. Očistite gumené viečko a zasuňte novú injekčnú striekačku s ihlou do injekčnej liekovky.
Prevráťte injekčnú liekovku hore dnom, pričom dbajte na to, aby bol hrot ihly pod hladinou tekutiny. Potiahnutím piesta dozadu natiahnite množstvo lieku požadované pre správnu dávku a dbajte na vytlačenie všetkého vzduchu z injekčnej striekačky. Vytiahnite injekčnú striekačku s ihlou z injekčnej liekovky a ihlu zlikvidujte.
6. Pevne nasaďte novú subkutánnu ihlu (odporúča sa kaliber 25) na injekčnú striekačku. Ihla sa nemá plniť pred injekčnou aplikáciou, aby sa znížil výskyt lokálnych reakcií v mieste vpichu.
7. Ak je potrebná viac ako 1 injekčná liekovka, zopakujte všetky vyššie uvedené kroky na
prípravu suspenzie. V dôsledku retencie v injekčnej liekovke a ihle nemusí byť možné natiahnuť všetku suspenziu z injekčnej liekovky.
8. Obsah dávkovacej injekčnej striekačky sa musí premiešať tesne pred podaním. Injekčná
striekačka naplnená rekonštituovanou suspenziou sa má pred podaním ponechať pri teplote približne 20-25 °C po dobu do 30 minút. Ak uplynie viac než 30 minút, suspenzia sa musí príslušným spôsobom zlikvidovať a musí sa pripraviť nová dávka. Na premiešanie rýchlo rolujte injekčnú striekačku medzi dlaňami dovtedy, kým sa nevytvorí homogénna, zakalená suspenzia.
Aksuspenziaobsahujeveľkéčasticealeboaglomeráty,trebajuzlikvidovať.Výpočetindividuálnejdávky
Celkovú dávku podľa plochy povrchu tela (
body surface area, BSA) možno vypočítať nasledovne: Celková dávka (mg) = dávka (mg/m2) x BSA (m2)
Nasledujúca tabuľka je uvedená iba ako príklad výpočtu individuálnych dávok azacitidínu na základe priemernej hodnoty BSA na úrovni 1,8 m2.
Dávka mg/m2
(% odporúčanej začiatočnej dávky)
| Celková dávka na základe hodnoty
BSA 1,8 m2
| Požadovaný počet injekčných
liekoviek
| Celkový potrebný
objem
rekonštituovanej
suspenzie
|
|
| 100 mg injekčná
liekovka
| 150 mg injekčná
liekovka
|
|
75 mg/m2 (100 %)
| 135 mg
| 2 injekčné
liekovky
| 1 injekčná
liekovka
| 5,4 ml
|
37,5 mg/m2 (50 %)
| 67,5 mg
| 1 injekčná
liekovka
| 1 injekčná
liekovka
| 2,7 ml
|
25 mg/m2 (33 %)
| 45 mg
| 1 injekčná
liekovka
| 1 injekčná
liekovka
| 1,8 ml
|
SpôsobpodávaniaRekonštituovaný azacitidín sa má podávať subkutánnou injekciou (ihlu zaveďte pod uhlom 45 – 90°)
do ramena, stehna alebo brucha použitím injekčnej ihly s kalibrom 25.
Dávky väčšie než 4 ml sa majú injekčne aplikovať do dvoch rôznych miest vpichu.Miesta injekčnej aplikácie sa majú meniť. Nové injekcie sa majú aplikovať najmenej 2,5 cm od predchádzajúceho miesta vpichu a nikdy nie do oblastí, v ktorých je miesto vpichu bolestivé,
s podliatinami, sčervenené alebo stvrdnuté.
Všetok nepoužitý liek alebo odpad vzniknutý z lieku sa má zlikvidovať v súlade s národnými požiadavkami.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIIEVER Valinject GmbH Oberburgau 3
4866 Unterach am Attersee
Rakúsko
8. REGISTRAČNÉ ČÍSLO44/0112/21-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie: 30. apríla 2021
10. DÁTUM REVÍZIE TEXTU
06/2021