funkcie pečene nie je potrebná úprava dávky. U pacientov so stredne ťažkou poruchou funkcie pečene sú k dispozícii iba obmedzené údaje a u pacientov s ťažkou poruchou funkcie pečene nie sú dostupné žiadne údaje. Aprepitant sa má u týchto pacientov používať s opatrnosťou (pozri časti 4.4 a 5.2).
Spôsob podávaniaNa perorálne použitie.
Tvrdá kapsula sa má prehltnúť vcelku.
Aprepitant sa môže užiť s jedlom alebo bez jedla.
4.3 Kontraindikácie Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1.
Súbežné podanie s pimozidom, terfenadínom, astemizolom alebo cisapridom (pozri časť 4.5).
4.4 Osobitné upozornenia a opatrenia pri používaníPacienti so stredne ťažkou až ťažkou poruchou funkcie pečeneU pacientov so stredne ťažkou poruchou funkcie pečene sú k dispozícii iba obmedzené údaje a u pacientov s ťažkou poruchou funkcie pečene nie sú dostupné žiadne údaje. Aprepitant Stada sa má u týchto pacientov používať s opatrnosťou (pozri časť 5.2).
Interakcie s CYP3A4Aprepitant Stada sa má používať s opatrnosťou u pacientov, ktorí súbežne užívajú perorálne podávané liečivá primárne metabolizované CYP3A4 a s úzkym terapeutickým rozsahom, ako sú cyklosporín, takrolimus, sirolimus, everolimus, alfentanil, deriváty námeľových alkaloidov, fentanyl a chinidín (pozri časť 4.5). Ďalej je potrebné venovať zvýšenú pozornosť súbežnému podávaniu irinotekánu, pretože táto kombinácia môže vyústiť do zvýšenej toxicity.
Súbežné podávanie s warfarínom (substrát CYP2C9)U pacientov, ktorí sú dlhodobo liečení warfarínom, sa má pozorne monitorovať medzinárodný normalizovaný pomer (International Normalised Ratio, INR) počas liečby Aprepitantom Stada a počas 14 dní po každom trojdňovom cykle liečby Aprepitantom Stada (pozri časť 4.5).
Súbežné podávanie s hormonálnymi kontraceptívamiPočas podávania a 28 dní po ukončení podávania Aprepitantu Stada môže dôjsť k zníženiu účinnosti hormonálnych kontraceptív. Počas liečby Aprepitantom Stada a počas 2 mesiacov po užití poslednej dávky Aprepitantu Stada sa majú používať alternatívne nehormonálne zaisťovacie metódy antikoncepcie (pozri časť 4.5).
Pomocné látkyKapsuly Aprepitant Stada obsahujú sacharózu. Pacienti so zriedkavými dedičnými problémami intolerancie fruktózy, glukózo-galaktózovej malabsorpcie alebo deficitu sacharázy a izomaltázy nesmú užívať tento liek.
Kapsuly Aprepitant Stada obsahujú sodík. Tento liek obsahuje menej ako 1 mmol sodíka (23 mg) v kapsule, t.j. v podstate zanedbateľné množstvo sodíka.
4.5 Liekové a iné interakcieAprepitant (125 mg/80 mg) je substrát, stredne silný inhibítor a induktor CYP3A4. Aprepitant je tiež induktor CYP2C9. Počas liečby Aprepitantom Stada je CYP3A4 inhibovaný. Po ukončení liečby spôsobuje Aprepitant Stada prechodnú miernu indukciu CYP2C9, CYP3A4 a glukuronidácie. Nezdá sa, že aprepitant interaguje s P-glykoproteínovým transportérom, čo sa preukázalo neprítomnosťou interakcie aprepitantu s digoxínom.
Vplyv aprepitantu na farmakokinetiku iných liečivInhibícia CYP3A4Aprepitant (125 mg/80 mg) ako stredne silný inhibítor CYP3A4 môže zvýšiť plazmatické koncentrácie súbežne podávaných liečiv, ktoré sa metabolizujú cez CYP3A4. Počas 3-dňovej liečby Aprepitantom Stada sa môže celková expozícia perorálne podávaným substrátom CYP3A4 zvýšiť až približne trojnásobne. Na plazmatické koncentrácie intravenózne podávaných substrátov CYP3A4 sa očakáva nižší vplyv aprepitantu. Aprepitant Stada sa nesmie užívať súbežne s pimozidom, terfenadínom, astemizolom alebo cisapridom (pozri časť 4.3). Inhibícia CYP3A4 navodená aprepitantom by mohla viesť k zvýšeniu plazmatických koncentrácií týchto liečiv a potenciálne spôsobiť závažné alebo život ohrozujúce reakcie. Pri súbežnom podávaní Aprepitantu Stada a perorálne podávaných liečiv, ktoré sú primárne metabolizované CYP3A4 a s úzkym terapeutickým rozsahom, ako sú cyklosporín, takrolimus, sirolimus, everolimus, alfentanil, dihydroergotamín, ergotamín, fentanyl a chinidín, sa odporúča opatrnosť (pozri časť 4.4).
KortikosteroidyDexametazón: pri súbežnom užívaní s Aprepitantom Stada v režime 125 mg/80 mg sa má zvyčajná dávka perorálneho dexametazónu znížiť približne o 50 %. V klinických štúdiách chemoterapiou indukovanej nevoľnosti a vracania bola dávka dexametazónu zvolená s ohľadom na liekové interakcie (pozri časť 4.2). Aprepitant podávaný v režime 125 mg súbežne s perorálnym dexametazónom 20 mg v deň 1 a aprepitant 80 mg/deň podávaný súbežne s perorálnym dexametazónom 8 mg počas dní 2 až 5 zvýšil v dňoch 1 a 5 AUC dexametazónu, substrátu CYP3A4, 2,2-násobne.
Metylprednizolón: pri súbežnom užívaní s Aprepitantom Stada v režime 125 mg/80 mg sa má zvyčajná dávka intravenózne podávaného metylprednizolónu znížiť o približne 25 % a zvyčajná dávka perorálneho metylprednizolónu o približne 50 %. Aprepitant podávaný v režime 125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3 zvýšil AUC metylprednizolónu, substrátu CYP3A4, 1,3-násobne v deň 1 a 2,5-násobne v deň 3, keď sa v deň 1 súbežne podával metylprednizolón v dávke 125 mg intravenózne a v dňoch 2 a 3 v dávke 40 mg perorálne.
Počas kontinuálnej liečby metylprednizolónom sa môže AUC metylprednizolónu neskôr v priebehu 2 týždňov od úvodného podania dávky aprepitantu znížiť v dôsledku indukčného účinku aprepitantu na CYP3A4. Očakáva sa, že tento účinok môže byť výraznejší pri perorálne podávanom metylprednizolóne.
ChemoterapeutikáKeď bol aprepitant vo farmakokinetických štúdiách podávaný v režime 125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3, neovplyvnil farmakokinetiku docetaxelu podaného intravenózne v deň 1, ani vinorelbínu podaného intravenózne v deň 1 alebo deň 8. Keďže vplyv aprepitantu na farmakokinetiku perorálne podávaných substrátov CYP3A4 je väčší ako na farmakokinetiku intravenózne podávaných substrátov CYP3A4, nedá sa vylúčiť interakcia s perorálne podávanými chemoterapeutikami prevažne alebo čiastočne metabolizovanými CYP3A4 (napr. etopozid, vinorelbín). U pacientov, ktorí užívajú lieky prevažne alebo čiastočne metabolizované CYP3A4, sa odporúča opatrnosť a môže byť vhodné ďalšie sledovanie (pozri časť 4.4). Po súbežnom podaní aprepitantu a ifosfamidu boli pri používaní lieku po jeho uvedení na trh hlásené prípady neurotoxicity, potenciálnej nežiaducej reakcie ifosfamidu.
ImunosupresívaPočas 3-dňového režimu CINV sa v expozícii imunosupresívam metabolizovaným CYP3A4 (napr. cyklosporín, takrolimus, everolimus a sirolimus) predpokladá prechodný stredne silný vzostup nasledovaný miernym poklesom. Vzhľadom na krátke trvanie 3-dňového režimu a od času závislé obmedzené zmeny v expozícii sa počas 3 dní súbežného podávania s Aprepitantom Stada neodporúča zníženie dávky imunosupresíva.
MidazolamPri súbežnom užívaní Aprepitantu Stada (125 mg/80 mg) sa majú zohľadniť možné účinky zvýšených plazmatických koncentrácií midazolamu alebo iných benzodiazepínov, ktoré sa metabolizujú prostredníctvom CYP3A4 (alprazolam, triazolam).
Keď sa jednorazová perorálna dávka midazolamu 2 mg podala v dňoch 1 a 5 spolu s aprepitantom v režime 125 mg v deň 1 a 80 mg/deň v dňoch 2 až 5, zvýšil aprepitant AUC midazolamu, senzitívneho substrátu CYP3A4, 2,3-násobne v deň 1 a 3,3-násobne v deň 5.
V inej štúdii s intravenózne podávaným midazolamom sa aprepitant podal v dávke 125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3 a midazolam 2 mg bol podaný intravenózne pred podaním trojdňového režimu s aprepitantom a v dňoch 4, 8 a 15. Aprepitant zvýšil AUC midazolamu o 25 % v deň 4 a znížil AUC midazolamu o 19 % v deň 8 a o 4 % v deň 15. Tieto účinky sa nepovažovali za klinicky významné.
V tretej štúdii s intravenóznym a perorálnym podaním midazolamu sa aprepitant podával v dávke 125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3 spolu s ondansetrónom 32 mg v deň 1, dexametazónom 12 mg v deň 1 a 8 mg v dňoch 2 - 4. Táto kombinácia (t.j. aprepitant, ondansetrón a dexametazón) znížila AUC perorálneho midazolamu o 16 % v deň 6, o 9 % v deň 8, o 7 % v deň 15 a o 17 % v deň 22. Tieto účinky sa nepovažovali za klinicky významné.
S intravenóznym podaním midazolamu a aprepitantu bola ukončená ďalšia štúdia. 2 mg midazolamu boli intravenózne podané 1 hodinu po perorálnom podaní jednorazovej dávky aprepitantu 125 mg. Plazmatická AUC midazolamu sa zvýšila 1,5-násobne. Tento účinok sa nepovažoval za klinicky významný.
IndukciaAprepitant ako mierny induktor CYP2C9, CYP3A4 a glukuronidácie môže počas dvoch týždňov po začatí liečby znížiť plazmatické koncentrácie substrátov eliminovaných týmito cestami. Tento účinok sa môže prejaviť až po ukončení 3-dňovej liečby Aprepitantom Stada. Pre substráty CYP2C9 a CYP3A4 je indukcia prechodná, s maximálnym účinkom dosiahnutým 3-5 dní po ukončení 3-dňovej liečby Aprepitantom Stada. Účinok trvá počas niekoľkých dní, potom pomaly ustupuje a do dvoch týždňov od ukončenia liečby Aprepitantom Stada je klinicky nevýznamný. Mierna indukcia glukuronidácie sa tiež pozoruje pri podávaní 80 mg aprepitantu perorálne počas 7 dní. Údaje o účinkoch na CYP2C8 a CYP2C19 chýbajú. Ak sa počas tohto časového obdobia podáva warfarín, acenokumarol, tolbutamid, fenytoín alebo iné liečivá, o ktorých je známe, že sú metabolizované CYP2C9, odporúča sa opatrnosť.
WarfarínU pacientov dlhodobo liečených warfarínom sa má počas liečby Aprepitantom Stada a 2 týždne po každom 3-dňovom cykle liečby Aprepitantom Stada na nevoľnosť a vracanie vyvolané chemoterapiou dôsledne monitorovať protrombínový čas (INR) (pozri časť 4.4). Keď sa podal aprepitant v jednorazovej 125 mg dávke v deň 1 a 80 mg/deň v dňoch 2 a 3 zdravým jedincom stabilizovaným na dlhodobej liečbe warfarínom, aprepitant neovplyvnil plazmatickú AUC R(+) ani S(-) warfarínu v deň 3; 5 dní po ukončení liečby aprepitantom sa však minimálna koncentrácia S(-) warfarínu (substrátu CYP2C9) znížila o 34 % sprevádzaná 14 % poklesom INR.
TolbutamidAprepitant podávaný v dávke 125 mg v deň 1 a 80 mg/deň v dňoch 2 a 3 znížil AUC tolbutamidu (substrátu CYP2C9) o 23 % v deň 4, o 28 % v deň 8 a o 15 % v deň 15, keď sa tolbutamid podal v jednorazovej dávke 500 mg perorálne pred podaním aprepitantu v trojdňovom režime a v deň 4, 8 a 15.
Hormonálne kontraceptívaPočas podávania a 28 dní po ukončení podávania Aprepitantu Stada môže dôjsť k zníženiu účinnosti hormonálnych kontraceptív. Počas liečby Aprepitantom Stada a počas 2 mesiacov po poslednej dávke Aprepitantu Stada sa majú používať alternatívne nehormonálne zaisťovacie formy antikoncepcie.
V klinickej štúdii boli v dňoch 1 až 21 podávané jednorazové dávky perorálneho kontraceptíva obsahujúceho etinylestradiol a noretisterón s aprepitantom, ktorý sa podával v režime 125 mg v deň 8 a 80 mg/deň v dňoch 9 a 10 spolu s intravenóznym ondansetrónom 32 mg v deň 8 a perorálnym dexametazónom v dávke 12 mg v deň 8 a v dávke 8 mg/deň v dňoch 9, 10 a 11. V tejto štúdii sa počas dní 9 až 21 znížili minimálne (trough) koncentrácie etinylestradiolu až o 64 % a minimálne koncentrácie noretisterónu sa znížili až o 60 %.
Antagonisty 5-HT3V klinických interakčných štúdiách nemal aprepitant klinicky významný účinok na farmakokinetiku ondansetrónu, granisetrónu alebo hydrodolasetrónu (aktívny metabolit dolasetrónu).
Vplyv iných liekov na farmakokinetiku aprepitantuPri súbežnom podávaní Aprepitantu Stada s liečivami, ktoré inhibujú aktivitu CYP3A4 (napr. ketokonazol, itrakonazol, vorikonazol, posakonazol, klaritromycín, telitromycín, nefazodón a inhibítory proteázy), je potrebné postupovať opatrne, pretože sa predpokladá, že táto kombinácia bude mať za následok niekoľkonásobné zvýšenie plazmatickej koncentrácie aprepitantu (pozri časť 4.4).
Súbežnému podávaniu Aprepitantu Stada s liečivami, ktoré silne indukujú aktivitu CYP3A4 (napr. rifampicín, fenytoín, karbamazepín, fenobarbital), je potrebné sa vyhnúť, pretože táto kombinácia vedie k zníženým plazmatickým koncentráciám aprepitantu, čo môže spôsobiť zníženie účinnosti Aprepitantu Stada.
Súbežné podávanie Aprepitantu Stada s rastlinnými prípravkami obsahujúcimi ľubovník bodkovaný (
Hypericum perforatum) sa neodporúča.
KetokonazolPo podaní jednorazovej 125 mg dávky aprepitantu v deň 5 počas 10-dňového podávania ketokonazolu 400 mg/deň, silného inhibítora CYP3A4, došlo k približne 5-násobnému zvýšeniu AUC aprepitantu a k približne 3-násobnému zvýšeniu priemerného terminálneho polčasu aprepitantu.
RifampicínPo podaní jednorazovej 375 mg dávky aprepitantu v deň 9 počas 14-dňového podávania rifampicínu 600 mg/deň, silného induktora CYP3A4, došlo k zníženiu AUC aprepitantu o 91 % a k zníženiu priemerného terminálneho polčasu o 68 %.
Pediatrická populáciaInterakčné štúdie sa vykonali iba u dospelých.
4.6 Fertilita, gravidita a laktáciaAntikoncepcia u mužov a žienPočas podávania a 28 dní po ukončení podávania Aprepitantu Stada sa môže znížiť účinnosť hormonálnych kontraceptív. Počas liečby Aprepitantom Stada a počas 2 mesiacov po užití poslednej dávky Aprepitantu Stada sa majú používať alternatívne nehormonálne zaisťovacie metódy antikoncepcie (pozri časti 4.4 a 4.5).
GraviditaPre aprepitant nie sú k dispozícii žiadne klinické údaje o graviditách vystavených expozícii. Potenciál aprepitantu na reprodukčnú toxicitu nebol jasne stanovený, pretože v štúdiách na zvieratách sa nemohli dosiahnuť vyššie expozičné hladiny ako terapeutická expozícia u ľudí pri dávkach 125 mg/80 mg. Tieto štúdie nepreukázali priame alebo nepriame škodlivé účinky na graviditu, embryonálny/fetálny vývoj, pôrod alebo postnatálny vývoj (pozri časť 5.3). Možné účinky zmien neurokinínovej regulácie na reprodukciu nie sú známe. Aprepitant Stada sa nemá používať počas gravidity, pokiaľ to nie je úplne nevyhnutné.
DojčenieAprepitant sa vylučuje do mlieka laktujúcich potkanov. Nie je známe, či sa aprepitant vylučuje do ľudského mlieka, preto sa neodporúča dojčenie počas liečby Aprepitantom Stada.
FertilitaPotenciálne účinky aprepitantu na fertilitu neboli jasne stanovené, pretože v štúdiách na zvieratách sa nemohli dosiahnuť vyššie expozičné hladiny ako terapeutická expozícia u ľudí. Tieto štúdie fertility nepreukázali priame alebo nepriame škodlivé účinky na reprodukciu, fertilitu, embryonálny/fetálny vývoj alebo počet a pohyblivosť spermií (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať strojeAprepitant Stada môže mať malý vplyv na schopnosť viesť vozidlá, bicyklovať sa a obsluhovať stroje. Po podaní Aprepitantu Stada sa môže objaviť závrat a únava (pozri časť 4.8).
4.8 Nežiaduce účinkySúhrn bezpečnostného profiluBezpečnostný profil aprepitantu sa hodnotil u približne 6 500 dospelých vo viac ako 50 štúdiách a 184 detí a dospievajúcich v 2 pivotných pediatrických klinických skúšaniach.
U pacientov dostávajúcich vysoko emetogénnu chemoterapiu (HEC) boli najčastejšími nežiaducimi reakciami, pri ktorých bola hlásená vyššia incidencia u pacientov liečených režimom s aprepitantom ako u pacientov liečených štandardnou liečbou: štikútanie (4,6 % oproti 2,9 %), zvýšenie hladiny alanínaminotransferázy (ALT) (2,8 % oproti 1,1 %), dyspepsia (2,6 % oproti 2,0 %), zápcha (2,4 % oproti 2,0 %), bolesť hlavy (2,0 % oproti 1,8 %) a znížená chuť do jedla (2,0 % oproti 0,5 %). U pacientov dostávajúcich stredne emetogénnu chemoterapiu (MEC) najčastejšou nežiaducou reakciou, ktorá sa vyskytla s vyššou incidenciou u pacientov liečených režimom s aprepitantom než u pacientov liečených štandardnou liečbou, bola únava (1,4 % oproti 0,9 %).
Najčastejšími nežiaducimi reakciami hlásenými s väčšou incidenciou u pediatrických pacientov liečených režimom s aprepitantom ako s kontrolným režimom počas podávania emetogénnej protirakovinovej chemoterapie boli štikútanie (3,3 % oproti 0,0 %) a sčervenenie (1,1 % oproti 0,0 %).
Tabuľkový zoznam nežiaducich reakciíV združenej analýze štúdií HEC a MEC sa pozorovali nasledujúce nežiaduce reakcie s incidenciou vyššou pri aprepitante ako u dospelých alebo pediatrických pacientov liečených štandardnou liečbou alebo pri používaní lieku po jeho uvedení na trh. Kategórie frekvencie uvedené v tabuľke sú založené na štúdiách u dospelých; pozorované frekvencie v pediatrických štúdiách boli podobné alebo nižšie, pokiaľ nie sú uvedené v tabuľke. Niektoré menej časté nežiaduce reakcie v populácii dospelých sa nepozorovali v pediatrických štúdiách.
Frekvencie sú definované ako: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté (≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000) a veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov).
Trieda orgánových systémov
| Nežiaduca reakcia
| Frekvencia
|
Infekcie a nákazy
| kandidóza, stafylokoková infekcia
| zriedkavé
|
Poruchy krvi a lymfatického systému
| febrilná neutropénia, anémia
| menej časté
|
Poruchy imunitného systému
| reakcie z precitlivenosti vrátane anafylaktických reakcií
| neznáme
|
Poruchy metabolizmu a výživy
| znížená chuť do jedla
| časté
|
polydipsia
| zriedkavé
|
Psychické poruchy
| úzkosť
| menej časté
|
dezorientácia, euforická nálada
| zriedkavé
|
Poruchy nervového systému
| bolesť hlavy
| časté
|
závrat, somnolencia
| menej časté
|
kognitívna porucha, letargia, dysgeúzia
| zriedkavé
|
Poruchy oka
| konjunktivitída
| zriedkavé
|
Poruchy ucha a labyrintu
| tinitus
| zriedkavé
|
Poruchy srdca a srdcovej činnosti
| palpitácie
| menej časté
|
bradykardia, kardiovaskulárna porucha
| zriedkavé
|
Poruchy ciev
| nával tepla/sčervenenie
| menej časté
|
Poruchy dýchacej sústavy, hrudníka a mediastína
| štikútanie
| časté
|
orofaryngeálna bolesť, kýchanie, kašeľ, výtok hlienu z nosa do hltanu, podráždenie hrdla
| zriedkavé
|
Poruchy gastrointestinálneho traktu
| zápcha, dyspepsia
| časté
|
eruktácia, nauzea†, vracanie†, gastroezofágová refluxová choroba, bolesť brucha, sucho v ústach, flatulencia
| menej časté
|
perforácia vredu dvanástnika, stomatitída, abdominálna distenzia, tvrdá stolica, neutropenická kolitída
| zriedkavé
|
Poruchy kože a podkožného tkaniva
| vyrážka, akné
| menej časté
|
fotosenzitívna reakcia, hyperhidróza, seborea, kožná lézia, pruritická vyrážka, Stevensov-Johnsonov syndróm/toxická epidermálna nekrolýza
| zriedkavé
|
pruritus, urtikária
| neznáme
|
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
| svalová slabosť, svalové kŕče
| zriedkavé
|
Poruchy obličiek a močových ciest
| dyzúria
| menej časté
|
polakizúria
| zriedkavé
|
Celkové poruchy a reakcie v mieste podania
| únava
| časté
|
asténia, malátnosť
| menej časté
|
edém, diskomfort hrudníka, porucha chôdze
| zriedkavé
|
Laboratórne a funkčné vyšetrenia
| zvýšenie hladiny ALT
| časté
|
zvýšenie hladiny AST, zvýšenie hladiny alkalickej fosfatázy v krvi
| menej časté
|
potvrdená prítomnosť erytrocytov v moči, znížená hladina sodíka v krvi, pokles hmotnosti, zníženie počtu neutrofilov, prítomnosť glukózy v moči, zvýšené vylučovanie moču
| zriedkavé
|
† Nauzea a vracanie boli v prvých 5 dňoch liečby po chemoterapii parametrami účinnosti a až potom sa hlásili ako nežiaduce reakcie.
Opis vybraných nežiaducich reakciíProfily nežiaducich reakcií u dospelých boli v predĺžení štúdií HEC a MEC s viacnásobnými cyklami až na 6 ďalších cyklov chemoterapie vo všeobecnosti podobné tým, ktoré sa pozorovali v 1. cykle.
V ďalšej aktívne kontrolovanej klinickej štúdii u 1 169 dospelých pacientov dostávajúcich aprepitant a HEC bol profil nežiaducich reakcií vo všeobecnosti podobný profilu, ktorý sa pozoroval v iných štúdiách HEC s aprepitantom.
U dospelých pacientov liečených aprepitantom na pooperačnú nevoľnosť a vracanie (PONV) boli pozorované ďalšie nežiaduce reakcie a s vyššou incidenciou ako pri ondansetróne: bolesť v hornej časti brucha, abnormálne zvuky čriev, zápcha*, dyzartria, dyspnoe, znížená citlivosť, insomnia, mióza, nauzea, senzorická porucha, žalúdočné ťažkosti, subileus*, zníženie zrakovej ostrosti, sipot.
* Hlásené u pacientov užívajúcich vyššiu dávku aprepitantu.
Hlásenie podozrení na nežiaduce reakcieHlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v
Prílohe V.
4.9 PredávkovanieV prípade predávkovania je potrebné prerušiť liečbu Aprepitantom Stada a prejsť na celkovú podpornú liečbu a monitorovanie pacienta. Keďže aprepitant má antiemetický účinok, nemusí byť farmakologicky navodené zvracanie efektívne.
Aprepitant sa nedá odstrániť hemodialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI5.1 Farmakodynamické vlastnostiFarmakoterapeutická skupina: antiemetiká a lieky proti nevoľnosti, iné antiemetiká
ATC kód: A04AD12
Aprepitant je selektívny vysoko afinitný antagonista na neurokinínových 1 (NK
1) receptoroch pre ľudskú substanciu P.
3-dňový režim s aprepitantom u dospelýchV 2 randomizovaných, dvojito zaslepených štúdiách, ktoré celkovo zahŕňali 1 094 dospelých pacientov dostávajúcich chemoterapiu obsahujúcu cisplatinu ≥ 70 mg/m
2, sa porovnával režim s aprepitantom v kombinácii s ondansetrónom/dexametazónom (pozri časť 4.2) a štandardný režim (placebo plus ondansetrón 32 mg podaný intravenózne v deň 1 plus dexametazón 20 mg perorálne v deň 1 a 8 mg perorálne dvakrát denne počas dní 2 až 4). Aj keď sa v klinických skúšaniach použila intravenózna dávka 32 mg ondansetrónu, táto dávka už nie je odporúčanou dávkou. Príslušné informácie o dávkovaní pozri v informáciách vybraného lieku obsahujúceho antagonistu 5-HT
3.
Účinnosť vychádzala z hodnotenia nasledujúceho kompozitného ukazovateľa: úplná odpoveď (definovaná ako stav bez epizód vracania a bez použitia záchrannej liečby) hlavne počas 1. cyklu. Výsledky sa hodnotili pre každú individuálnu štúdiu a pre 2 štúdie v kombinácii.
V Tabuľke 1 je súhrn kľúčových výsledkov štúdií z kombinovanej analýzy.
Tabuľka 1
Percento odpovedajúcich dospelých pacientov, ktorí dostávali vysoko emetogénnu chemoterapiu, podľa liečebnej skupiny a fázy – 1. cyklus
| Režim s aprepitantom
| Štandardná liečba
| Rozdiely*
|
KOMPOZITNÉ UKAZOVATELE
| (N = 521)†
%
| (N = 524)†
%
|
%
|
(95 % IS)
|
|
|
|
|
|
Úplná odpoveď (žiadne vracanie a žiadna záchranná liečba)
|
Celkovo (0-120 hodín)
| 67,7
| 47,8
| 19,9
| (14,0, 25,8)
|
0-24 hodín
| 86,0
| 73,2
| 12,7
| (7,9, 17,6)
|
25-120 hodín
| 71,5
| 51,2
| 20,3
| (14,5, 26,1)
|
JEDNOTLIVÉ UKAZOVATELE
|
|
|
|
|
Bez vracania (žiadne epizódy vracania bez ohľadu na použitie záchrannej liečby)
|
Celkovo (0-120 hodín)
| 71,9
| 49,7
| 22,2
| (16,4, 28,0)
|
0-24 hodín
| 86,8
| 74,0
| 12,7
| (8,0, 17,5)
|
25-120 hodín
| 76,2
| 53,5
| 22,6
| (17,0, 28,2)
|
Bez signifikantnej nauzey (maximálna hodnota VAS < 25 mm na stupnici 0-100 mm)
|
Celkovo (0-120 hodín)
| 72,1
| 64,9
| 7,2
| (1,6, 12,8)
|
25-120 hodín
| 74,0
| 66,9
| 7,1
| (1,5, 12,6)
|
* Intervaly spoľahlivosti boli vypočítané bez úpravy podľa pohlavia a súbežnej chemoterapie, ktoré boli zahrnuté v primárnej analýze mier pravdepodobnosti a logistických modeloch.
† U jedného pacienta v režime s aprepitantom boli dostupné iba údaje z akútnej fázy a bol vylúčený z celkovej analýzy a analýzy oneskorenej fázy; u jedného pacienta so štandardnou liečbou boli dostupné iba údaje z oneskorenej fázy a bol vylúčený z celkovej analýzy a analýzy akútnej fázy.
Odhadovaný čas do prvého vracania v kombinovanej analýze je zobrazený na Kaplanovom-Meierovom grafe na obrázku 1.
Obrázok 1
Percento dospelých pacientov dostávajúcich vysoko emetogénnu chemoterapiu, ktorí boli bez vracania, v čase – 1. cyklus
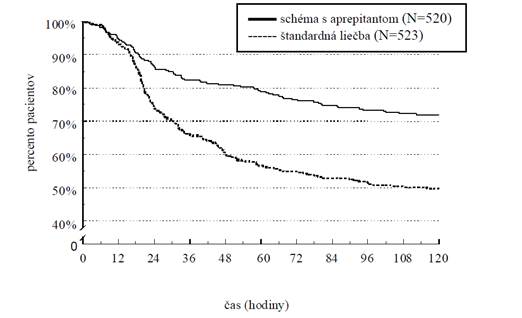
Štatisticky významné rozdiely v účinnosti sa pozorovali aj v každej z 2 individuálnych štúdií.
V tých istých 2 klinických štúdiách pokračovalo 851 dospelých pacientov v predĺžení s opakovanými cyklami až do 5 ďalších cyklov chemoterapie. Režim s aprepitantom si zjavne zachoval účinnosť počas všetkých cyklov.
V randomizovanej, dvojito zaslepenej štúdii u celkovo 866 dospelých pacientov (864 žien, 2 muži) dostávajúcich chemoterapiu, ktorá zahŕňala cyklofosfamid 750-1 500 mg/m
2 alebo cyklofosfamid 500-1 500 mg/m
2 a doxorubicín (< 60 mg/m
2) alebo epirubicín (< 100 mg/m
2), sa porovnával režim s aprepitantom v kombinácii s ondansetrónom/dexametazónom (pozri časť 4.2) so štandardnou liečbou (placebo plus ondansetrón 8 mg perorálne (dvakrát v deň 1 a každých 12 hodín v dňoch 2 a 3) plus dexametazón 20 mg perorálne v deň 1).
Účinnosť vychádzala z hodnotenia kompozitného ukazovateľa: úplná odpoveď (definovaná ako stav bez epizód vracania a bez použitia záchrannej liečby) hlavne počas 1. cyklu.
Súhrn kľúčových výsledkov štúdie je uvedený v Tabuľke 2.
Tabuľka 2
Percento dospelých pacientov odpovedajúcich na liečbu podľa liečebnej skupiny a fázy - 1. Cyklus stredne emetogénna chemoterapia
| Režim s aprepitantom
| Štandardná liečba
| Rozdiely*
|
KOMPOZITNÉ UKAZOVATELE
| (N = 433)†
%
| (N = 424)†
%
|
%
|
(95 % IS)
|
|
|
|
|
|
Úplná odpoveď (žiadne vracanie a žiadna záchranná liečba)
|
Celkovo (0-120 hodín)
| 50,8
| 42,5
| 8,3
| (1,6, 15,0)
|
0-24 hodín
| 75,7
| 69,0
| 6,7
| (0,7, 12,7)
|
25-120 hodín
| 55,4
| 49,1
| 6,3
| (-0,4, 13,0)
|
JEDNOTLIVÉ UKAZOVATELE
|
|
|
|
|
Bez vracania (žiadne epizódy vracania bez ohľadu na použitie záchrannej liečby)
|
Celkovo (0-120 hodín)
| 75,7
| 58,7
| 17,0
| (10,8, 23,2)
|
0-24 hodín
| 87,5
| 77,3
| 10,2
| (5,1, 15,3)
|
25-120 hodín
| 80,8
| 69,1
| 11,7
| (5,9, 17,5)
|
Bez signifikantnej nauzey (maximálna hodnota VAS < 25 mm na stupnici 0-100 mm)
|
Celkovo (0-120 hodín)
| 60,9
| 55,7
| 5,3
| (-1,3, 11,9)
|
0-24 hodín
| 79,5
| 78,3
| 1,3
| (-4,2, 6,8)
|
25-120 hodín
| 65,3
| 61,5
| 3,9
| (-2,6, 10,3)
|
* Intervaly spoľahlivosti boli vypočítané bez úpravy podľa vekovej kategórie (< 55 rokov, ≥ 55 rokov) a vyšetrovanej skupiny, ktoré boli zahrnuté v primárnej analýze mier pravdepodobnosti a logistických modeloch.
† U jedného pacienta v režime s aprepitantom boli dostupné iba údaje z akútnej fázy a bol vylúčený z celkovej analýzy a analýzy oneskorenej fázy.
V tej istej klinickej štúdii pokračovalo 744 dospelých pacientov v predĺžení s opakovanými cyklami až do 3 ďalších cyklov chemoterapie. Režim s aprepitantom si zjavne zachoval účinnosť počas všetkých cyklov.
V druhej multicentrickej, randomizovanej, dvojito zaslepenej klinickej štúdii s paralelnými skupinami sa režim s aprepitantom porovnával so štandardnou liečbou u 848 dospelých pacientov (652 žien, 196 mužov) dostávajúcich chemoterapiu, ktorá zahŕňala akúkoľvek intravenóznu dávku oxaliplatiny, karboplatiny, epirubicínu, idarubicínu, ifosfamidu, irinotekánu, daunorubicínu, doxorubicínu, intravenózneho cyklofosfamidu (< 1 500 mg/m
2) alebo intravenózneho cytarabínu (> 1 g/m
2). Pacienti dostávajúci režim s aprepitantom dostávali chemoterapiu na viaceré typy tumorov, ktoré zahŕňali 52 % karcinómov prsníka, 21 % gastrointestinálnych karcinómov vrátane kolorektálneho karcinómu, 13 % pľúcnych karcinómov a 6 % gynekologických karcinómov. Režim s aprepitantom v kombinácii s režimom s ondansetrónom/dexametazónom (pozri časť 4.2) sa porovnával so štandardnou liečbou (placebo v kombinácii s ondansetrónom 8 mg perorálne (dvakrát v deň 1 a každých 12 hodín v dňoch 2 a 3) plus dexametazón 20 mg perorálne v deň 1).
Účinnosť vychádzala z hodnotenia nasledujúcich primárnych a kľúčových sekundárnych cieľových ukazovateľov: bez vracania počas celého času (0 až 120 hodín po chemoterapii), hodnotenie bezpečnosti a tolerancie režimu s aprepitantom na chemoterapiou vyvolanú nevoľnosť a vracanie (CINV) a úplná odpoveď (definovaná ako stav bez vracania a bez použitia záchrannej liečby) počas celého času (0 až 120 hodín po chemoterapii). Ďalej sa hodnotilo percento bez signifikantnej nauzey počas celého času (0 až 120 hodín po chemoterapii) ako exploratívny cieľový ukazovateľ, a v akútnej a oneskorenej fáze ako post-hoc analýza.
Súhrn kľúčových výsledkov štúdie je uvedený v Tabuľke 3.
Tabuľka 3
Percento dospelých pacientov odpovedajúcich na liečbu podľa liečebnej skupiny a fázy v štúdii 2 - 1. Cyklus stredne emetogénna chemoterapia
| Režim s aprepitantom
| Štandardná liečba
| Rozdiely*
|
| (N = 425)
%
| (N = 406)
%
|
%
|
(95 % IS)
|
|
|
| '
|
|
Úplná odpoveď (žiadne vracanie a žiadna záchranná liečba)
|
Celkovo (0-120 hodín)
| 68,7
| 56,3
| 12,4
| (5,9, 18,9)
|
0-24 hodín
| 89,2
| 80,3
| 8,9
| (4,0, 13,8)
|
25-120 hodín
| 70,8
| 60,9
| 9,9
| (3,5, 16,3)
|
Bez vracania (žiadne epizódy vracania bez ohľadu na použitie záchrannej liečby)
|
Celkovo (0-120 hodín)
| 76,2
| 62,1
| 14,1
| (7,9, 20,3)
|
0-24 hodín
| 92,0
| 83,7
| 8,3
| (3,9, 12,7)
|
25-120 hodín
| 77,9
| 66,8
| 11,1
| (5,1, 17,1)
|
Bez signifikantnej nauzey (maximálna hodnota VAS < 25 mm na stupnici 0-100 mm)
|
Celkovo (0-120 hodín)
| 73,6
| 66,4
| 7,2
| (1,0, 13,4)
|
0-24 hodín
| 90,9
| 86,3
| 4,6
| (0,2, 9,0)
|
25-120 hodín
| 74,9
| 69,5
| 5,4
| (-0,7, 11,5)
|
* Intervaly spoľahlivosti boli vypočítané bez úpravy podľa pohlavia a regiónu, ktoré boli zahrnuté v primárnej analýze používajúcej logistické modely.
Prínos kombinovanej liečby s aprepitantom v celej populácii štúdie bol daný najmä výsledkami pozorovanými u pacientov so slabou kontrolou pri štandardnom režime, ako napr. u žien, i keď výsledky boli číselne lepšie bez ohľadu na vek, typ tumoru alebo pohlavie. Úplná odpoveď na režim s aprepitantom, resp. štandardnú liečbu sa dosiahla u 209/324 (65 %), resp. 161/320 (50 %) žien a 83/101 (82 %), resp. 68/87 (78 %) mužov.
Pediatrická populáciaV randomizovanej, dvojito zaslepenej, aktívnym komparátorom kontrolovanej klinickej štúdii, ktorá zahŕňala 302 detí a dospievajúcich (vo veku 6 mesiacov až 17 rokov) dostávajúcich stredne až vysoko emetogénnu chemoterapiu, bol režim s aprepitantom porovnávaný s kontrolným režimom na prevenciu CINV. Účinnosť režimu s aprepitantom bola vyhodnocovaná v jednorazovom cykle (1. cyklus). V nasledujúcich cykloch mali pacienti možnosť dostávať aprepitant v otvorenom usporiadaní štúdie (voliteľný 2. - 6. cyklus). V týchto voliteľných cykloch sa však účinnosť nehodnotila. Režim s aprepitantom pre dospievajúcich vo veku 12 až 17 rokov (n = 47) pozostával z kapsúl aprepitantu 125 mg perorálne v deň 1 a 80 mg/deň v deň 2 a 3 v kombinácii s ondansetrónom v deň 1. Režim s aprepitantom pre deti vo veku 6 mesiacov až menej ako 12 rokov (n = 105) pozostával z aprepitantu vo forme prášku na perorálnu suspenziu 3,0 mg/kg (až do 125 mg) perorálne v deň 1 a 2,0 mg/kg (až do 80 mg) perorálne v deň 2 a 3 v kombinácii s ondansetrónom v deň 1. Kontrolný režim u dospievajúcich vo veku 12 až 17 rokov (n = 48) a detí vo veku 6 mesiacov až menej ako 12 rokov (n = 102) pozostával z placeba pre aprepitant v deň 1, 2 a 3 v kombinácii s ondansetrónom v deň 1. Aprepitant alebo placebo sa podával 1 hodinu pred začiatkom chemoterapie a ondansetrón sa podával 30 minút pred začiatkom chemoterapie. Ako súčasť antiemetického režimu u pediatrických pacientov v obidvoch vekových skupinách bol povolený intravenózny dexametazón na základe rozhodnutia lekára. U pediatrických pacientov dostávajúcich aprepitant sa vyžadovalo zníženie dávky (50 %) dexametazónu. U pediatrických pacientov dostávajúcich kontrolný režim sa nevyžadovalo žiadne zníženie dávky. Z týchto pediatrických pacientov sa u 29 % v režime s aprepitantom a u 28 % v kontrolnom režime používal dexametazón ako súčasť režimu v 1. cykle.
Antiemetický účinok aprepitantu bol vyhodnocovaný počas 5-dňového (120 hodín) obdobia po začatí chemoterapie v deň 1. Primárnym cieľovým ukazovateľom bola úplná odpoveď v oneskorenej fáze (25 až 120 hodín po začatí chemoterapie) v 1. cykle. Súhrn kľúčových výsledkov štúdie je uvedený v Tabuľke 4.
Tabuľka 4
Počet (%) pediatrických pacientov s úplnou odpoveďou a bez vracania podľa liečebnej skupiny a fázy – 1. cyklus (populácia so zámerom liečiť)
| režim s aprepitantom n/m (%)
| kontrolný režim n/m (%)
|
PRIMÁRNY CIEĽOVÝ UKAZOVATEĽ
|
Úplná odpoveď* – oneskorená fáza
| 77/152 (50,7)†
| 39/150 (26,0)
|
ĎALŠIE VOPRED DEFINOVANÉ CIEĽOVÉ UKAZOVATELE
|
Úplná odpoveď* – akútna fáza
| 101/152 (66,4)‡
| 78/150 (52,0)
|
Úplná odpoveď* – celková fáza
| 61/152 (40,1)†
| 30/150 (20,0)
|
Bez vracania§ – celková fáza
| 71/152 (46,7)†
| 32/150 (21,3)
|
*Úplná odpoveď = bez vracania alebo pocitu na vracanie alebo napínania na vracanie bez použitia záchrannej liečby.
†p < 0,01 pri porovnaní s kontrolným režimom
‡p < 0,05 pri porovnaní s kontrolným režimom
§Bez vracania = bez vracania alebo pocitu na vracanie alebo napínania na vracanie
n/m = počet pacientov s požadovanou odpoveďou/počet pacientov zahrnutých v časovom bode.
Akútna fáza: 0 až 24 hodín po začatí chemoterapie.
Oneskorená fáza: 25 až 120 hodín po začatí chemoterapie.
Celková fáza: 0 až 120 hodín po začatí chemoterapie.
|
Odhadovaný čas do prvého vracania po začatí liečby chemoterapiou bol dlhší pri režime s aprepitantom (odhadovaný medián času do prvého vracania bol 94,5 hodín) v porovnaní so skupinou s kontrolným režimom (odhadovaný medián času do prvého vracania bol 26,0 hodín) ako je znázornené na Kaplanových-Meierových grafoch na obrázku 2.
Obrázok 2
Čas do prvej epizódy vracania od začiatku podávania chemoterapie – pediatrickí pacienti v celkovej fáze – 1. cyklus (populácia so zámerom liečiť)
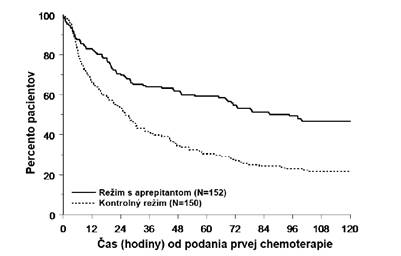
Analýza účinnosti v podskupinách v 1. cykle preukázala, že bez ohľadu na vekovú kategóriu, pohlavie, používanie dexametazónu pri antiemetickej profylaxii a emetogénnej chemoterapii, režim s aprepitantom poskytoval lepšiu kontrolu ako kontrolný režim s ohľadom na cieľové ukazovatele úplnej odpovede.
5.2 Farmakokinetické vlastnostiAprepitant vykazuje nelineárnu farmakokinetiku. Klírens aj absolútna biologická dostupnosť klesajú so stúpajúcou dávkou.
AbsorpciaPriemerná absolútna biologická dostupnosť perorálne podaného aprepitantu je 67 % pre 80 mg kapsulu a 59 % pre 125 mg kapsulu. Priemerná maximálna plazmatická koncentrácia (C
max) aprepitantu sa dosiahla približne po 4 hodinách (t
max). Perorálne podanie kapsuly aprepitantu spolu so štandardnými raňajkami, približne 800 kcal, malo za následok až 40 % zvýšenie AUC aprepitantu. Toto zvýšenie sa nepovažuje za klinicky významné.
Farmakokinetika aprepitantu je v rozsahu klinického dávkovania nelineárna. U zdravých mladých dospelých bolo zvýšenie AUC
0-∞ o 26 % väčšie, ako by zodpovedalo dávke po podaní jednorazových dávok v rozmedzí 80 mg a 125 mg po jedle.
Po jednorazovom perorálnom podaní 125 mg dávky aprepitantu v deň 1 a 80 mg raz denne v dňoch 2 a 3 bola AUC
0-24h (priemer ± SD) 19,6 ± 2,5 μg•h/ml v deň 1 a 21,2 ± 6,3 μg•h/ml v deň 3. C
max bola 1,6 ± 0,36 μg/ml v deň 1 a 1,4 ± 0,22 μg/ml v deň 3.
DistribúciaAprepitant sa silne viaže na proteíny, v priemere 97 %. Geometrický priemer zdanlivého distribučného objemu v rovnovážnom stave (Vd
ss) je u ľudí približne 66 l.
BiotransformáciaAprepitant je značne metabolizovaný. U zdravých mladých dospelých po podaní jednorazovej intravenóznej 100 mg dávky [
14C]-fosaprepitantu, proliečiva aprepitantu, tvorí aprepitant počas 72 hodín približne 19 % rádioaktivity v plazme, čo ukazuje na významnú prítomnosť metabolitov v plazme. V ľudskej plazme sa identifikovalo dvanásť metabolitov aprepitantu. Aprepitant sa do značnej miery metabolizuje oxidáciou na morfolínovom kruhu a jeho vedľajších reťazcoch a výsledné metabolity boli iba slabo aktívne.
In vitro štúdie s použitím ľudských hepatálnych mikrozómov naznačujú, že aprepitant je primárne metabolizovaný CYP3A4 a potenciálne s malým prispením CYP1A2 a CYP2C19.
ElimináciaAprepitant sa nevylučuje močom nezmenený. Metabolity sú vylučované v moči a prostredníctvom biliárnej exkrécie v stolici. Po podaní jednorazovej intravenóznej 100 mg dávky [
14C]-fosaprepitantu, proliečiva aprepitantu, zdravým jedincom sa 57 % rádioaktivity vylúčilo v moči a 45 % v stolici.
Plazmatický klírens aprepitantu je závislý od dávky, znižuje sa so zvyšujúcou dávkou a v rozsahu terapeutickej dávky sa pohybuje medzi približne 60 až 72 ml/min. Terminálny polčas sa pohybuje približne od 9 po 13 hodín.
Farmakokinetika v osobitných skupinách pacientovStarší pacienti: Po podaní jednorazovej perorálnej 125 mg dávky aprepitantu v deň 1 a 80 mg raz denne v dňoch 2 až 5 bola AUC
0-24h aprepitantu u starších pacientov (≥ 65 rokov) v deň 1 vyššia o 21 % a v deň 5 vyššia o 36 % v porovnaní s mladšími dospelými. C
max bola u starších v porovnaní s mladšími dospelými vyššia o 10 % v deň 1 a o 24 % v deň 5. Tieto rozdiely sa nepovažujú za klinicky relevantné.
U starších pacientov nie je nutná žiadna úprava dávky aprepitantu.
Pohlavie: Po perorálnom podaní jednorazovej 125 mg dávky aprepitantu je C
max aprepitantu o 16 % vyššia u žien ako u mužov. Polčas aprepitantu je u žien o 25 % nižší ako u mužov a jeho t
max sa dosahuje približne v rovnakom čase. Tieto rozdiely sa nepovažujú za klinicky relevantné. Podľa pohlavia nie je nutné upravovať dávku aprepitantu.
Porucha funkcie pečene: Mierna porucha funkcie pečene (trieda A podľa Childa-Pugha) neovplyvňuje klinicky relevantne farmakokinetiku aprepitantu. Pri miernej poruche funkcie pečene nie je potrebná úprava dávky. Z dostupných údajov nie je možné urobiť závery o vplyve stredne ťažkej poruchy funkcie pečene (trieda B podľa Childa-Pugha) na farmakokinetiku aprepitantu. U pacientov s ťažkou poruchou funkcie pečene (trieda C podľa Childa-Pugha) nie sú dostupné žiadne klinické alebo farmakokinetické údaje.
Porucha funkcie obličiek: Jednorazová 240 mg dávka aprepitantu sa podala pacientom s ťažkou poruchou funkcie obličiek (klírens kreatinínu < 30 ml/min) a pacientom v terminálnom štádiu renálneho ochorenia (ESRD) vyžadujúcom hemodialýzu.
U pacientov s ťažkou poruchou funkcie obličiek poklesla AUC
0-∞ celkového aprepitantu (neviazaného a viazaného na proteíny) v porovnaní so zdravými jedincami o 21 % a C
max poklesla o 32 %. U pacientov s ESRD na hemodialýze poklesla AUC
0-∞ celkového aprepitantu o 42 % a C
max poklesla o 32 %. Vzhľadom na mierne zníženie väzby aprepitantu na proteíny u pacientov s renálnym ochorením nebola AUC farmakologicky aktívneho neviazaného aprepitantu u pacientov s poruchou funkcie obličiek v porovnaní so zdravými jedincami signifikantne ovplyvnená. Hemodialýza, ktorá prebehla 4 až 48 hodín po podaní dávky, nemala na farmakokinetiku aprepitantu žiaden signifikantný vplyv, v dialyzáte sa vylúčilo menej ako 0,2 % dávky.
U pacientov s poruchou funkcie obličiek alebo u pacientov s ESRD na hemodialýze nie je potrebná úprava dávky Aprepitantu Stada.
Pediatrická populácia: Ako súčasť 3-dňového režimu sa pri podávaní kapsúl aprepitantu (125/80/80 mg) u dospievajúcich pacientov (vo veku 12 až 17 rokov) dosiahla hodnota AUC
0-24 hod nad 17 μg•h/ml v deň 1 s koncentráciami (C
min) na konci dňa 2 a 3 nad 0,4 μg/ml pri väčšine pacientov. Medián maximálnej plazmatickej koncentrácie (C
max) bol približne 1,3 μg/ml v deň 1 s výskytom približne za 4 hodiny. Ako súčasť 3-dňového režimu sa pri podávaní aprepitantu vo forme prášku na perorálnu suspenziu (3/2/2 mg/kg) u pacientov vo veku 6 mesiacov až menej ako 12 rokov dosiahla hodnota AUC
0-24 hod nad 17 μg•h/ml v deň 1 s koncentráciami (C
min) na konci dňa 2 a 3 nad 0,1 μg/ml pri väčšine pacientov. Medián maximálnej plazmatickej koncentrácie (C
max) bol približne 1,2 μg/ml v deň 1 s výskytom v rozmedzí 5 až 7 hodín.
Analýza populačnej farmakokinetiky aprepitantu u pediatrických pacientov (vo veku 6 mesiacov až 17 rokov) naznačuje, že pohlavie a rasa nemajú žiadny klinicky významný účinok na farmakokinetiku aprepitantu.
Vzťah medzi koncentráciou a účinkomZobrazovacie štúdie s pozitrónovou emisnou tomografiou (PET) s použitím vysoko špecifického značenia NK
1 receptora u zdravých mladých mužov preukázali, že aprepitant preniká do mozgu a obsadzuje NK
1 receptory v závislosti od dávky a plazmatickej koncentrácie. Predpokladá sa, že plazmatické koncentrácie aprepitantu, ktoré sa dosahujú po 3-dňovom režime podávania Aprepitantu Stada zabezpečujú obsadenie viac ako 95 % mozgových NK
1 receptorov.
5.3 Predklinické údaje o bezpečnostiPredklinické údaje získané na základe obvyklých štúdií toxicity po jednorazovom a opakovanom podávaní, genotoxicity, karcinogénneho potenciálu, reprodukčnej toxicity a vývinu neodhalili žiadne osobitné riziko pre ľudí. Treba však podotknúť, že systémová expozícia u hlodavcov bola podobná alebo dokonca nižšia ako terapeutická expozícia u ľudí pri dávke 125 mg/80 mg. Hoci sa v reprodukčných štúdiách pri ľudských expozičných hladinách nezistili žiadne nežiaduce účinky, expozície zvierat nie sú dostatočné na adekvátne zhodnotenie rizika u človeka.
V štúdiách juvenilnej toxicity na potkanoch liečených od 10. dňa po narodení do 63. dňa viedol aprepitant pri dávke 250 mg/kg dvakrát denne k skoršiemu otvoreniu vagíny u samíc a pri dávke10 mg/kg dvakrát denne k oneskorenému oddeleniu predkožky u samcov. Neobjavili sa žiadne limity klinicky významnej expozície. Neobjavili sa žiadne účinky na reprodukciu, fertilitu alebo prežívanie embrya/plodu a žiadne patologické zmeny v reprodukčných orgánoch. V štúdiách juvenilnej toxicity na psoch liečených od 14. dňa po narodení do 42. dňa sa pri dávke 6 mg/kg/deň pozoroval pokles hmotnosti semenníkov a veľkosti Leydigových buniek u samcov a pri dávke 4 mg/kg/deň zvýšenie hmotnosti maternice, hypertrofia maternice a cervixu a opuch vaginálnych tkanív u samíc. Neobjavili sa žiadne limity klinicky významnej expozície aprepitantu. Pri krátkodobej liečbe v súlade s odporúčaným dávkovacím režimom sa pri týchto nálezoch považuje za nepravdepodobné, že by boli klinicky významné.
6. FARMACEUTICKÉ INFORMÁCIE6.1 Zoznam pomocných látokObsah kapsulyhypromelóza
poloxamér
sacharóza
mikrokryštalická celulóza
Obal kapsuly (125 mg)želatína
laurylsíran sodný (E487)
oxid titaničitý (E 171)
červený oxid železitý (E 172)
Obal kapsuly (80 mg)želatína
laurylsíran sodný (E487)
oxid titaničitý (E 171)
Čierny atrament na potlačšelak
čierny oxid železitý (E 172)
propylénglykol (E1520)
6.2 InkompatibilityNeaplikovateľné.
6.3 Čas použiteľnosti30 mesiacov
6.4 Špeciálne upozornenia na uchovávanieUchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
6.5 Druh obalu a obsah balenia Aprepitant Stada je balený v papierovej škatuľke obsahujúcej zodpovedajúci počet blistrov s fóliou OPA/ALU/PVC-hliník s písomnou informáciou pre používateľa.
Aprepitant Stada 125 mg tvrdé kapsuly sa dodávajú v týchto veľkostiach balenia:
- Hliníkový blister obsahujúci jednu 125 mg kapsulu.
- 5 hliníkových blistrov, každý obsahuje jednu 125 mg kapsulu.
Aprepitant Stada 80 mg tvrdé kapsuly sa dodávajú v týchto veľkostiach balenia:
- Hliníkový blister obsahujúci jednu 80 mg kapsulu.
- Balenie na 2-dňovú liečbu obsahujúce dve 80 mg kapsuly.
- 5 hliníkových blistrov, každý obsahuje jednu 80 mg kapsulu.
Aprepitant Stada 80 mg a Aprepitant Stada 125 mg tvrdé kapsuly sa dodávajú v týchto veľkostiach balenia:
- Balenie na 3-dňovú liečbu obsahujúce jednu 125 mg kapsulu a dve 80 mg kapsuly.
Na trh nemusia byť uvedené všetky veľkosti balenia.
6.6 Špeciálne opatrenia na likvidáciu Žiadne zvláštne požiadavky na likvidáciu.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCIISTADA Arzneimittel AG
Stadastrasse 2-18
61118 Bad Vilbel
Nemecko
8. REGISTRAČNÉ ČÍSLA Aprepitant Stada 80 mg: 20/0031/19-S
Aprepitant Stada 125 mg: 20/0032/19-S
Aprepitant Stada 80 mg a Aprepitant Stada 125 mg: 20/0033/19-S
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTU02/2019