
ť 5.2) nevyžadovali použitie rozdielneho začiatočného režimu dávkovania alebo rozdielnych krokov pre titráciu dávky na dosiahnutie dávkovania anagrelidu prispôsobeného individuálnemu pacientovi.
Počas klinického vývoja bolo približne 50 % pacientov liečených anagrelidom vo veku nad 60 rokov a nevyžadovali si žiadne vekovo špecifické úpravy dávok. Ako sa však očakávalo, u pacientov z tejto vekovej kategórie sa vyskytli závažné nežiaduce udalosti (najmä srdcové) v dvojnásobnej miere.
Porucha funkcie obličiek
Pre túto skupinu pacientov sú k dispozícii obmedzené farmakokinetické údaje. Pred začatím liečby sa majú najskôr stanoviť potenciálne riziká a prínosy liečby anagrelidom u pacientov s poruchou funkcie obličiek (pozri časť 4.3).
Porucha funkcie pečene
Pre túto skupinu pacientov sú k dispozícii obmedzené farmakokinetické údaje. Avšak pečeňový metabolizmus predstavuje hlavnú cestu vylučovania anagrelidu a preto sa môže predpokladať, že funkcia pečene ovplyvní tento proces. Preto sa neodporúča liečiť anagrelidom pacientov so stredne ťažkou alebo ťažkou poruchou funkcie pečene. Pred začatím liečby sa majú najskôr stanoviť potenciálne riziká a prínosy liečby anagrelidom u pacientov s miernou poruchou funkcie pečene (pozri
časti 4.3 a 4.4).
Pediatrická populácia
Bezpečnosť a účinnosť anagrelidu u detí neboli doteraz stanovené. Skúsenosti s použitím u detí a dospievajúcich sú veľmi obmedzené, anagrelid sa má u tejto skupiny pacientov používať
s opatrnosťou. V prípade chýbania osobitných pediatrických pokynov sú diagnostické kritéria WHO pre diagnózu ET u dospelých považované za relevantné pre pediatrickú populáciu. Diagnostické pokyny pre esenciálnu trombocytémiu majú byť dôsledne dodržiavané a v prípade neistoty sa má diagnóza pravidelne prehodnocovať, so snahou o jej odlíšenie od hereditárnej alebo sekundárnej trombocytózy, čo môže zahŕňať genetickú analýzu a biopsiu kostnej drene.
Cytoreduktívna terapia sa zvyčajne zvažuje u vysoko rizikových pediatrických pacientov.
Liečba anagrelidom sa má začať len pokiaľ pacient vykazuje prejavy progresie ochorenia alebo trpí trombózou. Ak sa liečba začne, prínosy a riziká liečby anagrelidom musia byť pravidelne sledované a potreba pokračovania liečby pravidelne vyhodnocovaná.
Cieľové hodnoty počtu doštičiek stanoví individuálne každému pacientovi ošetrujúci lekár.
Prerušenie liečby sa má zvážiť u pediatrických pacientov, ktorí nemajú uspokojivé odpovede na liečbu po približne 3 mesiacoch.
V súčasnosti sú dostupné údaje opísané v časti 4.4, 4.8, 5.1 a 5.2, ale neumožňujú uviesť odporúčania na dávkovanie.
Spôsob podávania
Na perorálne použitie. Kapsuly sa musia prehltnúť celé. Obsah kapsúl sa nemá drviť alebo riediť
v kvapaline.
4.3 Kontraindikácie
Precitlivenosť na liečivo alebo na ktorúkoľvek z pomocných látok uvedených v časti 6.1. Pacienti so stredne ťažkou alebo ťažkou poruchou funkcie pečene.
Pacienti so stredne ťažkou alebo ťažkou poruchou funkcie obličiek (klírens kreatinínu < 50 ml/min).
4.4 Osobitné upozornenia a opatrenia pri používaní
Porucha funkcie pečene
Pred začatím liečby sa majú najskôr stanoviť potenciálne riziká a prínosy liečby anagrelidom
u pacientov s miernou poruchou funkcie pečene. Neodporúča sa u pacientov so zvýšenou hladinou transamináz (viac ako 5-násobok hornej hranice normálnych hodnôt) (pozri časti 4.2 a 4.3).
Porucha funkcie obličiek
Pred začatím liečby sa majú najskôr stanoviť potenciálne riziká a prínosy liečby anagrelidom
u pacientov s poruchou funkcie obličiek (pozri časti 4.2 a 4.3).
Sledovanie
Liečba vyžaduje prísny klinický dozor nad pacientom, ktorý zahŕňa úplné vyšetrenie krvného obrazu
(hemoglobín a počet leukocytov a krvných doštičiek), zhodnotenie výsledkov vyšetrení funkcie pečene (ALT a AST), funkcie obličiek (sérový kreatinín a močovina) a elektrolytov (draslík, horčík a vápnik).
Krvné doštičky
Počet krvných doštičiek sa do 4 dní po ukončení liečby anagrelidom zvýši a vráti na úroveň pred
začatím liečby do 10 až 14 dní, pričom je možné, že dôjde k zvýšeniu nad hodnoty pred začatím liečby. Preto sa má počet krvných doštičiek často monitorovať.
Kardiovaskulárne
Zaznamenali sa závažné kardiovaskulárne nežiaduce reakcie vrátane prípadov torsade de pointes,
ventrikulárnej tachykardie, kardiomyopatie, kardiomegálie a kongestívneho zlyhania srdca (pozri
časť 4.8).
Anagrelid sa má používať s opatrnosťou u pacientov so známymi rizikovými faktormi, ktoré môžu predlžovať QT interval, ako napríklad kongenitálny syndróm dlhého QT intervalu, získané predĺženie QTc intervalu v anamnéze, lieky, ktoré môžu predĺžiť QTc interval a hypokaliémia.
Anagrelid sa má používať s opatrnosťou tiež u pacientov, ktorí môžu mať vyššiu maximálnu plazmatickú koncentráciu (Cmax) anagrelidu alebo jeho aktívneho metabolitu, 3-hydroxyanagrelidu, napr. pri poruche funkcie pečene alebo pri použití s CYP1A2 inhibítormi (pozri časť 4.5).
Odporúča sa dôkladné monitorovanie vplyvu na QTc interval.
Pred začiatkom liečby anagrelidom sa u všetkých pacientov odporúča vykonať predliečebné kardiovaskulárne vyšetrenie vrátane východiskového vyšetrenia EKG a echokardiografie. Všetci pacienti sa majú počas liečby pravidelne monitorovať (napr. pomocou EKG alebo echokardiografie) kvôli možným účinkom na kardiovaskulárny systém, ktoré môžu vyžadovať ďalšie kardiovaskulárne vyšetrenie. Hypokaliémia alebo hypomagneziémia sa musia pred podaním anagrelidu korigovať
a v priebehu terapie pravidelne monitorovať.
Anagrelid je inhibítorom cyklickej AMP-fosfodiesterázy III a pre jeho pozitívne inotropné
a chronotropné účinky sa anagrelid má používať s opatrnosťou u pacientov v akomkoľvek veku s diagnostikovaným ochorením srdca alebo s podozrením na toto ochorenie. Navyše sa vyskytli závažné kardiovaskulárne nežiaduce udalosti aj u pacientov bez podozrenia na ochorenie srdca
a s normálnymi výsledkami kardiovaskulárneho vyšetrenia pred liečbou.
Anagrelid sa má používať len ak potenciálne prínosy liečby prevýšia potenciálne riziká. Pulmonálna hypertenzia
U pacientov liečených anagrelidom boli hlásené prípady pulmonálnej hypertenzie. Pred začatím liečby
a počas liečby anagrelidom majú byť pacienti sledovaní z hľadiska prejavov a príznakov základného kardiopulmonálneho ochorenia.
Pediatrická populácia
Dostupné sú len veľmi obmedzené údaje o používaní anagrelidu u pediatrickej populácie a anagrelid
sa má používať u tejto skupiny pacientov s opatrnosťou (pozri časti 4.2, 4.8., 5.1 a 5.2).
Rovnako ako u dospelej populácie má byť pred liečbou a pravidelne počas liečby vykonané úplné vyšetrenie krvného obrazu a zhodnotenie výsledkov vyšetrení funkcie srdca, pečene a obličiek. Ochorenie môže progredovať do myelofibrózy alebo akútnej myeloblastovej leukémie (AML). Hoci miera takejto progresie nie je známa, u detí je priebeh ochorenia dlhší a preto je u nich zvýšené riziko malígnej transformácie v porovnaní s dospelými. Deti majú byť pravidelne monitorované kvôli progresii ochorenia v súlade so štandardnými klinickými postupmi ako sú fyzikálne vyšetrenie, zhodnotenie relevantných markerov ochorenia a biopsia kostnej drene.
Akékoľvek abnormality majú byť okamžite vyhodnotené a je potrebné urobiť vhodné opatrenia, ktoré môžu zahŕňať zníženie dávky, prerušenie alebo ukončenie.
Klinicky relevantné interakcie
Anagrelid je inhibítorom cyklickej AMP-fosfodiesterázy III (PDE III). Súbežné používanie anagrelidu
s inými inhibítormi PDE III ako napríklad s milrinónom, amrinónom, enoximónom, olprinónom a cilostazolom sa neodporúča.
Súbežné používanie anagrelidu a kyseliny acetylsalicylovej je spojené so závažnými hemoragickými príhodami (pozri časť 4.5).
Pomocné látky
Anagrelid Mylan obsahuje laktózu. Pacienti so zriedkavými dedičnými problémami galaktózovej
intolerancie, celkovým deficitom laktázy alebo glukózo-galaktózovou malabsorpciou nesmú užívať
tento liek.
Anagrelid Mylan obsahuje menej ako 1 mmol sodíka (23 mg) na kapsulu, to znamená, že je prakticky
„bez sodíka“.
4.5 Liekové a iné interakcie
Uskutočnili sa limitované farmakokinetické a/alebo farmakodynamické štúdie, skúmajúce možné interakcie medzi anagrelidom a inými liekmi.
Účinky iných liečiv na anagrelid
· Štúdie interakcií in vivo u ľudí preukázali, že digoxín a warfarín neovplyvňujú farmakokinetické
vlastnosti anagrelidu.
Inhibítory CYP1A2
· Anagrelid je primárne metabolizovaný prostredníctvom CYP1A2. Je známe, že CYP1A2 je inhibovaný niekoľkými liekmi, vrátane fluvoxamínu a enoxacínu, a tieto lieky by teoreticky mohli nepriaznivo ovplyvniť klírens anagrelidu.
Induktory CYP1A2
· Induktory CYP1A2 (ako napr. omeprazol) môžu znížiť expozíciu anagrelidu zvýšením hladiny jeho hlavného aktívneho metabolitu. Dopad na profily bezpečnosti a účinnosti anagrelidu nebol stanovený. Z tohto dôvodu sa pacientom užívajúcim súbežne induktory CYP1A2 odporúča klinické a biologické sledovanie. Pokiaľ je to potrebné, môže sa upraviť dávka anagrelidu.
Účinky anagrelidu na iné liečivá
· Anagrelid preukazuje limitovanú inhibičnú aktivitu voči CYP1A2, čo môže predstavovať
teoretickú možnosť interakcie s inými súbežne podávanými liekmi, ktoré zdieľajú rovnaký mechanizmus klírensu, napríklad teofylín.
· Anagrelid je inhibítorom PDE III. Účinky liekov s podobnými vlastnosťami, ako sú inotropné látky milrinón, enoximón, amrinón, olprinón a cilostazol, môže anagrelid zvýrazniť.
· Štúdie interakcií in vivo u ľudí preukázali, že anagrelid neovplyvňuje farmakokinetické vlastnosti digoxínu alebo warfarínu.
· Anagrelid podávaný v dávkach odporúčaných pri liečbe esenciálnej trombocytémie môže zosilniť účinky iných liekov, ktoré inhibujú alebo pozmeňujú funkciu krvných doštičiek, napr.
kyseliny acetylsalicylovej.
· V klinickej interakčnej štúdii so zdravými jedincami sa preukázalo, že súbežné podávanie opakovanej dávky anagrelidu 1 mg jedenkrát denne a kyseliny acetylsalicylovej 75 mg jedenkrát denne môže zvýšiť antiagregačné účinky každého liečiva na krvné doštičky
v porovnaní s podávaním samotnej kyseliny acetylsalicylovej. U niektorých pacientov s ET
súbežne liečených kyselinou acetylsalicylovou a anagrelidom sa vyskytli závažné hemorágie. Preto sa majú pred začatím liečby zhodnotiť potenciálne riziká súbežného používania anagrelidu s kyselinou acetylsalicylovou, predovšetkým u pacientov s vysoko rizikovým profilom pre krvácanie.
· Anagrelid môže u niektorých pacientov spôsobovať črevné poruchy a zhoršiť vstrebávanie hormonálnej perorálnej antikoncepcie.
Interakcie s jedlom
· Jedlo spomaľuje vstrebávanie anagrelidu, ale významne nemení systémovú expozíciu.
· Účinky jedla na biologickú dostupnosť sa nepovažujú za klinicky relevantné pri užívaní anagrelidu.
Pediatrická populácia
Interakčné štúdie sa uskutočnili len u dospelých.
4.6 Fertilita, gravidita a laktácia
Ženy vo fertilnom veku
Ženy vo fertilnom veku musia používať účinnú antikoncepciu počas liečby anagrelidom.
Gravidita
Nie sú k dispozícii dostatočné údaje o použití anagrelidu u gravidných žien. Štúdie na zvieratách
preukázali reprodukčnú toxicitu (pozri časť 5.3). Nie je známe potenciálne riziko u ľudí. Preto sa neodporúča užívať anagrelid počas gravidity.
Ak sa anagrelid užíva počas gravidity alebo pacientka počas užívania lieku otehotnie, má byť
upozornená na potenciálne riziko pre plod.
Dojčenie
Nie je známe, či sa anagrelid/metabolity vylučujú do ľudského mlieka. Dostupné údaje u zvierat
preukázali vylučovanie anagrelidu/metabolitov do mlieka. Riziko u novorodencov/dojčiat nemôže byť
vylúčené. Dojčenie má byť počas liečby anagrelidom ukončené.
Fertilita
Nie sú k dispozícii žiadne údaje o vplyve anagrelidu na fertilitu u ľudí. U samcov potkanov sa
nepozoroval žiadny vplyv anagrelidu na fertilitu alebo reprodukčné schopnosti. U samíc potkanov pri použití dávok prevyšujúcich terapeutické rozmedzie narušil anagrelid implantáciu (pozri časť 5.3).
4.7 Ovplyvnenie schopnosti viesť vozidlá a obsluhovať stroje
Počas klinického vývoja boli často hlásené závraty. Pacientom sa neodporúča sa viesť vozidlá alebo obsluhovať stroje, ak sa u nich počas liečby anagrelidom vyskytnú závraty.
4.8 Nežiaduce účinky
Súhrn bezpečnostného profilu
Bezpečnosť anagrelidu sa skúmala v 4 otvorených klinických štúdiách. V troch z týchto štúdií
u 942 pacientov, ktorí dostávali anagrelid v priemernej dávke približne 2 mg/deň, sa zhodnotila bezpečnosť.
V týchto štúdiách užívalo 22 pacientov anagrelid až po dobu 4 rokov.
V neskoršej štúdii sa hodnotila bezpečnosť u 3 660 pacientov, ktorí užívali anagrelid v priemernej dávke približne 2 mg/deň. V tejto štúdii dostávalo 34 pacientov anagrelid až po dobu 5 rokov.
Najčastejšie nežiaduce reakcie hlásené v súvislosti s anagrelidom boli bolesti hlavy, ktoré sa objavili
u približne 14 %, búšenie srdca u približne 9 %, retencia tekutín a nevoľnosť u približne 6 % a hnačky objavujúce sa u 5 % pacientov. Tieto nežiaduce reakcie na liek sa predpokladajú na základe farmakológie anagrelidu (inhibícia PDE III). Postupná titrácia dávkovania môže pomôcť zredukovať tieto účinky (pozri časť 4.2).
Tabuľkový súhrn nežiaducich reakcií
Nežiaduce reakcie, ktoré sa objavili v klinických skúšaniach, postregistračných bezpečnostných
skúšaniach a zo spontánnych hlásení, sú uvedené v tabuľke nižšie. V rámci tried orgánových systémov sú uvedené pod nasledovnými názvami: veľmi časté (≥ 1/10); časté (≥ 1/100 až < 1/10); menej časté
(≥ 1/1 000 až < 1/100); zriedkavé (≥ 1/10 000 až < 1/1 000), veľmi zriedkavé (< 1/10 000), neznáme (z dostupných údajov). V každej skupine frekvencií sú nežiaduce reakcie uvedené v poradí podľa klesajúcej závažnosti.
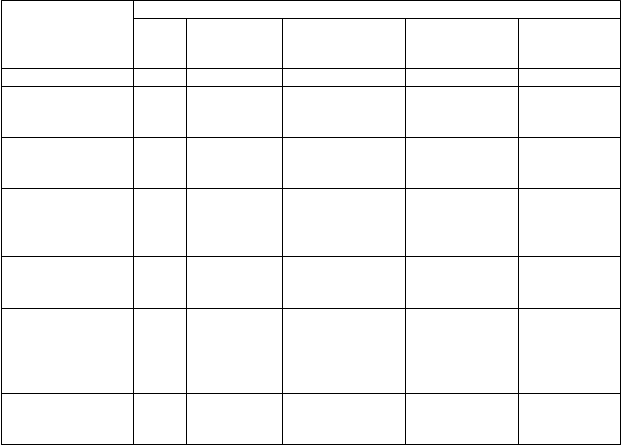
Trieda
orgánových systémov MedDRA
Poruchy krvi
a lymfatického
systému
Veľm
i časté
Frekvencia výskytu nežiaducich reakcií
Časté Menej časté Zriedkavé Neznáme
anémia pancytopénia trombocytopénia krvácanie ekchymóza
Poruchy metabolizmu a výživy
retencia tekutín
edém
strata hmotnosti
prírastok hmotnosti
Poruchy nervového systému
bolesť
hlavy
závraty depresia amnézia zmätenosť nespavosť parestézia znížená citlivosť nervozita
sucho v ústach
migréna dyzartria somnolencia poruchy koordinácie
Poruchy oka diplopia
poruchy videnia
Poruchy ucha a labyrintu Poruchy srdca a srdcovej činnosti
tachykardia palpitácie
komorová tachykardia kongestívne zlyhanie srdca fibrilácia predsiení supraventrikulárn a tachykardia arytmia hypertenzia synkopa
tinnitus
infarkt myokardu kardiomyopatia kardiomegália perikardiálny výpotok
angina pectoris posturálna hypotenzia vazodilatácia
torsade de pointes
Poruchy dýchacej sústavy, hrudníka a mediastína
pulmonálna hypertenzia pneumónia pleurálny výpotok dyspnoe
krvácanie z nosa
pľúcne infiltráty intersticiálna pľúcna choroba vrátane pneumonitídy a alergickej alveolitídy
 Poruchy gastrointestinálneh o traktu
Poruchy gastrointestinálneh o traktu
hnačka vracanie abdominálna bolesť nevoľnosť plynatosť
gastrointestinálne krvácanie pankreatitída anorexia
poruchy trávenia zápcha gastrointestinálne
kolitída gastritída krvácanie z ďasien
Trieda orgánových systémov MedDRA
Veľm
i časté
Frekvencia výskytu nežiaducich reakcií
Časté Menej časté Zriedkavé Neznáme
poruchy
Poruchy pečene a žlčových ciest
Poruchy kože a podkožného tkaniva
Poruchy kostrovej a svalovej sústavy a spojivového tkaniva
zvýšené hladiny pečeňových enzýmov
vyrážka alopécia pruritus sfarbenie kože artralgia myalgia
bolesť chrbta
suchá koža
hepatitída
Poruchy obličiek a močových ciest
impotencia zlyhanie obličiek noktúria
tubulointersti- ciálna
nefritída
Celkové poruchy
a reakcie v mieste podania
Laboratórne a funkčné vyšetrenia
únava bolesť hrudníka horúčka
triaška nevoľnosť slabosť
príznaky podobné chrípke bolesť
asténia
zvýšená hladina krvného kreatinínu
 Pediatrická populácia
Pediatrická populácia
48 pacientov vo veku 6-17 rokov (19 detí a 29 dospievajúcich) dostávalo anagrelid až 6,5 roka buď
v klinických štúdiách alebo ako boli súčasťou registra ochorenia (pozri časť 5.1).
Väčšina pozorovaných nežiaducich udalostí je medzi tými, ktoré sú uvedené v SPC. Avšak údaje o bezpečnosti sú obmedzené a neumožňujú vykonať zmysluplné porovnanie medzi dospelými
a pediatrickými pacientmi (pozri časť 4.4).
Hlásenie podozrení na nežiaduce reakcie
Hlásenie podozrení na nežiaduce reakcie po registrácii lieku je dôležité. Umožňuje priebežné monitorovanie pomeru prínosu a rizika lieku. Od zdravotníckych pracovníkov sa vyžaduje, aby hlásili akékoľvek podozrenia na nežiaduce reakcie na národné centrum hlásenia uvedené v Prílohe V.
4.9 PredávkovaniePo uvedení lieku na trh boli hlásené prípady úmyselného predávkovania anagrelidom. Medzi hlásené príznaky patrili sínusová tachykardia a vracanie. Príznaky odzneli po zavedení konzervatívnych opatrení.
Ukázalo sa, že anagrelid pri dávkach vyšších ako sa odporúča, spôsobuje zníženie krvného tlaku
s príležitostnými prípadmi hypotenzie. Jediná 5 mg dávka anagrelidu môže viesť k poklesu krvného tlaku, ktorý vo väčšine prípadov sprevádzajú závraty.
Špecifická protilátka pre anagrelid nie je známa. V prípade predávkovania je potrebné pozorné sledovanie klinického stavu pacienta, ktoré zahŕňa monitorovanie počtu krvných doštičiek pre možnú trombocytopéniu. Dávkovanie by sa malo znížiť, alebo ak je to potrebné aj prerušiť, až kým sa počet trombocytov nevráti do rozmedzia normálnych hodnôt.
5
. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Iné cytostatiká, ATC kód: L01XX35
Mechanizmus účinku
Presný mechanizmus účinku, ktorým anagrelid znižuje počet krvných doštičiek, nie je známy.
V štúdiách s bunkovými kultúrami anagrelid tlmil expresiu transkripčných faktorov, vrátane GATA-1 a FOG-1 potrebných pre megakaryocytopoézu, čo nakoniec viedlo k zníženiu produkcie krvných doštičiek.
Štúdie ľudskej megakaryocytopoézy in vitro potvrdili, že inhibičné pôsobenie anagrelidu na tvorbu krvných doštičiek u človeka je sprostredkované spomalením dozrievania megakaryocytov, zmenšovaním ich veľkosti a znížením ploidity. Dôkaz podobného pôsobenia in vivo sa pozoroval na bioptických vzorkách kostnej drene od liečených pacientov.
Anagrelid je inhibítorom cyklickej AMP-fosfodiesterázy III. Klinická účinnosťa bezpečnosť
Bezpečnosť a účinnosť anagrelidu ako látky znižujúcej počet krvných doštičiek sa hodnotili v štyroch
otvorených, nekontrolovaných klinických štúdiách (čísla štúdií: 700-012, 700-014, 700-999 a 13970-'
301) a zahŕňali viac ako 4 000 pacientov s myeloproliferatívnymi neoplazmami (MPN – myeloproliferative neoplasms). U pacientov s esenciálnou trombocytémiou bola úplná odpoveď definovaná ako zníženie počtu krvných doštičiek na hladinu pod 600 x 109/l alebo viac ako 50 % zníženie ich počtu v porovnaní s počiatočným stavom a udržanie tohto zníženia po dobu najmenej
4 týždňov. V štúdiách 700-012, 700-014, 700-999 a 13970-301 sa doba úplnej odpovede pohybovala od 4 do 12 týždňov. Klinický prospech z hľadiska trombo-hemoragických príhod nebol presvedčivo dokázaný.
Vplyv na srdcovú frekvenciu a QTc interval
Vplyv dvoch rôznych dávok anagrelidu (jednorazové dávky 0,5 mg a 2,5 mg) na srdcovú frekvenciu a QTc interval bol hodnotený v dvojito zaslepenej, randomizovanej, placebom a aktívnym komparátorom kontrolovanej, skríženej štúdií u zdravých dospelých mužov a žien.
K vzostupu srdcovej frekvencie v závislosti od dávky došlo v priebehu prvých 12 hodín, pričom maximálny vzostup sa objavil v čase maximálnej koncentrácie. Maximálna zmena priemernej tepovej frekvencie nastala za 2 hodiny po podaní lieku a bola +7,8 úderov za minútu pre dávku 0,5 mg
a +29,1 úderov za minútu pre dávku 2,5 mg.
Prechodné predĺženie priemerného QTc bolo pozorované u oboch dávok v čase zrýchlenia tepovej frekvencie a maximálne zmeny priemerného QTcF (QT interval korigovaný metódou podľa Fridericia) boli +5,0 ms po 2 hodinách pre dávku 0,5 mg a +10,0 ms po 1 hodine pre dávku 2,5 mg.
Pediatrická populácia
V otvorenej klinickej štúdii u 8 detí a 10 dospievajúcich (vrátane pacientov, ktorí predtým neboli
liečení anagrelidom alebo boli pred štúdiou liečení anagrelidom po dobu až 5 rokov) sa medián počtu krvných doštičiek znížil na kontrolované hladiny po 12 týždňoch liečby. Priemerná denná dávka mala tendenciu byť vyššia u dospievajúcich.
V štúdii pediatrického registra bol medián počtu krvných doštičiek v porovnaní s hodnotou zistenou v čase diagnózy znížený a bol udržiavaný až do 18 mesiacov u 14 pediatrických pacientov s ET liečených anagrelidom (4 deti, 10 dospievajúcich). V skorších otvorených štúdiách bolo pozorované zníženie mediánu počtu krvných doštičiek u 7 detí a 9 dospievajúcich liečených od 3 mesiacov do
6,5 roka.
Priemerná celková denná dávka anagrelidu zo všetkých štúdií u pediatrických pacientov s ET bola značne variabilná, ale celkové údaje naznačujú, že dospievajúcim by sa mohli podávať podobné začiatočné a udržiavacie dávky ako dospelým a nižšie začiatočné dávky 0,5 mg/deň by boli vhodnejšie pre deti vo veku viac ako 6 rokov (pozri časti 4.2., 4.4., 4.8 a 5.2). U všetkých pediatrických pacientov je potrebná opatrná titrácia dennej špecifickej dávky pacienta.
Referenčný liek obsahujúci anagrelid bol registrovaný za tzv. mimoriadnych okolností.
To znamená, že pre zriedkavosť výskytu ochorenia nebolo možné získať všetky informácie o tomto lieku.
Európska agentúra pre lieky každý rok posúdi nové dostupné informácie o tomto lieku a tento súhrn charakteristických vlastností lieku bude podľa potreby aktualizovať.
5.2 Farmakokinetické vlastnosti
Absorpcia
Po perorálnom podaní anagrelidu u človeka sa minimálne 70 % vstrebáva z gastrointestinálneho
traktu. U jedincov, ktorým sa podá nalačno sa maximálna plazmatická koncentrácia dosiahne približne po 1 hodine od podania. Farmakokinetické údaje od zdravých jedincov ukazujú, že jedlo znižuje Cmax anagrelidu o 14 %, ale zvyšuje hodnoty AUC o 20 %. Jedlo tiež znížilo Cmax aktívneho metabolitu 3- hydroxyanagrelidu o 29 %, hoci nemalo žiadny účinok na hodnoty AUC.
Biotransformácia
Anagrelid je primárne metabolizovaný prostredníctvom CYP1A2 za tvorby 3-hydroxyanagrelidu,
ktorý je ďalej metabolizovaný prostredníctvom CYP1A2 na neaktívny metabolit 2-amino-5,6-dichlór-
3,4-dihydrochinazolín.
Eliminácia
Plazmatický polčas anagrelidu je krátky, približne 1,3 hodiny a ako sa predpokladalo na základe tohto
polčasu, akumulácia anagrelidu v plazme sa nepreukázala. Menej ako 1 % sa vyskytuje v moči vo forme anagrelidu. Priemerné množstvo 2-amino-5,6-dichlór-3,4-dihydrochinazolínu vylúčené močom zodpovedá približne 18 – 35 % podanej dávky.
Navyše tieto výsledky nevykazujú známky samoindukcie klírensu anagrelidu. Linearita
Proporcionalita dávky sa prejavila v rozmedzí dávok 0,5 mg až 2 mg.
Pediatrická populácia
Farmakokinetické údaje u detí a dospievajúcich, ktorým sa podal anagrelid nalačno (vekový rozsah 7 -
16 rokov) s esenciálnou trombocytémiou ukazujú, že expozícia (Cmax a AUC) anagrelidu normalizovaná na dávku, mala tendenciu byť vyššia u detí/dospievajúcich v porovnaní s dospelými. Taktiež sa pozorovala tendencia k zvýšenej expozícii normalizovanej dávky aktívneho metabolitu.
Starší
Farmakokinetické údaje u starších pacientov nalačno s ET (vekový rozsah 65 - 75 rokov) v porovnaní s dospelými pacientami nalačno (vekový rozsah 22 – 50 rokov) ukazujú, že Cmax a AUC anagrelidu boli o 36 % a 61 % vyššie u starších pacientov v uvedenom poradí, ale Cmax a AUC aktívneho metabolitu 3-hydoxyanagrelidu boli o 42 % a 37 % nižšie u starších pacientov v uvedenom poradí. Tieto rozdiely pravdepodobne spôsobil nižší presystémový metabolizmus anagrelidu na 3- hydroxyanagrelid u starších pacientov.
5.3 Predklinické údaje o bezpečnosti
Toxicita po opakovanom podaní
Po opakovanom perorálnom podaní anagrelidu psom, sa pri dávke 1 mg/kg/deň alebo vyššej
pozorovali subendokardiálne krvácanie a fokálna nekróza myokardu u samcov a samíc, pričom samce boli senzitívnejšie. Dávka, pri ktorej nebol pozorovaný účinok (NOEL - no observed effect level)
u samcov psov (0,3 mg/kg/deň) zodpovedá 0,1; 0,1 a 1,6-násobku AUC u ľudí pre dávku anagrelidu
2 mg/deň, a metabolity BCH24426 a RL603, v uvedenom poradí.
Reprodukčná toxikológia
Fertilita
U samcov potkanov bolo zistené, že anagrelid pri perorálnych dávkach až do 240 mg/kg/deň (˃1 000 - násobok dávky 2 mg/deň, na základe plochy povrchu tela), nemá žiadny vplyv na fertilitu
a reprodukčné schopnosti. U samíc potkanov bolo pri dávke 30 mg/kg/deň pozorované zvýšenie pred a poimplantačných strát a pokles priemerného počtu živých embryí. NOEL (10 mg/kg/deň) pre tento účinok bola 143, 12 a 11-násobne vyššia ako AUC u ľudí po podaní dávky anagrelidu 2 mg/deň
a metabolitov BCH24426 a RL603, v uvedenom poradí.
Štúdie embryofetálneho vývoja
U potkanov a králikov boli toxické dávky anagrelidu pre matku spojené so zvýšenou resorpciou embrya a úmrtnosťou plodu.
V štúdii prenatálneho a postnatálneho vývoja u samíc potkanov vyvolával anagrelid pri perorálnych dávkach ≥10 mg/kg iný ako nežiaduci účinok v podobe predĺženia doby gravidity. Pri dávke NOEL (3 mg/kg/deň), AUC pre anagrelid a metabolity BCH24426 a RL603 bola 14-, 2- a 2-násobne vyššia ako AUC u ľudí po podaní perorálnej dávky anagrelidu 2 mg/deň.
Anagrelid pri dávke ≥ 60 mg/kg zvýšil dobu trvania pôrodov u chovných samíc a mortalitu plodov. Pri dávke NOEL (30 mg/kg/deň), AUC pre anagrelid a metabolity BCH24426 a RL603 bola 425-, 31-
a 13-násobne vyššia ako AUC u ľudí po podaní perorálnej dávky anagrelidu 2 mg/deň, v uvedenom poradí.
Mutagénny a karcinogénny potenciál
Štúdie genotoxického potenciálu anagrelidu nezistili žiadne mutagénne alebo klastogénne účinky.
Počas dvojročnej štúdie karcinogenity na potkanoch boli pozorované neneoplastické a neoplastické nálezy, ktoré súviseli alebo sa pripisovali prehnanému zvýšenému farmakologickému účinku. V rámci nich sa výskyt adrenálnych (nadobličkových) feochromocytómov zvýšil v porovnaní s kontrolnými zvieratami u samcov pri všetkých úrovniach dávok (≥ 3 mg/kg/deň) a u samíc pri dávke 10 mg/kg/deň a vyššej. Najnižšia dávka u samcov (3 mg/kg/deň) zodpovedá 37-násobku expozície (AUC) u človeka po podaní dávky1 mg dvakrát denne. Adenokarcinómy maternice epigenetického pôvodu mohli súvisieť s enzýmovou indukciou rodiny enzýmov CYP1. Boli pozorované u samíc prijímajúcich
30 mg/kg/deň, čo zodpovedá 572-násobku expozície (AUC) u človeka po podaní dávky1 mg dvakrát denne.
6. FARMACEUTICKÉ INFORMÁCIE
6.1 Zoznam pomocných látok
Obsah kapsuly
laktóza
monohydrát laktózy
sodná soľ kroskarmelózy povidón (K29/32) mikrokryštalická celulóza stearan horečnatý
Obal kapsuly
Anagrelid Mylan 0,5 mg tvrdé kapsuly
želatína
oxid titaničitý (E171)
Anagrelid Mylan 1 mg tvrdé kapsuly želatína
oxid titaničitý (E171)
čierny oxid železitý (E172)
6.2 Inkompatibility
Neaplikovateľné.
6.3 Čas použiteľnosti
3 roky
6.4 Špeciálne upozornenia na uchovávanie
Anagrelid Mylan 0,5 mg tvrdé kapsuly
Uchovávajte v pôvodnom obale na ochranu pred svetlom a vlhkosťou. Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie.
Anagrelid Mylan 1 mg tvrdé kapsuly
Uchovávajte v pôvodnom obale na ochranu pred vlhkosťou.
Tento liek nevyžaduje žiadne zvláštne teplotné podmienky na uchovávanie.
6.5 Druh obalu a obsah balenia
Fľaše z polyetylénu s vysokou hustotou (HDPE) s objemom 30 ml alebo 75 ml a s poistným patentným polypropylénovým (PP) detským bezpečnostným uzáverom a s vysušovadlom.
Veľkosť balenia: 100 tvrdých kapsúl.
6.6 Špeciálne opatrenia na likvidáciu
Žiadne zvláštne požiadavky.
7. DRŽITEĽ ROZHODNUTIA O REGISTRÁCII
Mylan S.A.S
117 Allée des Parcs, Saint-Priest,
69800, Francúzsko
8. REGISTRAČNÉ ČÍSLA
EU/1/17/1256/001
EU/1/17/1256/002
9. DÁTUM PRVEJ REGISTRÁCIE/PREDĹŽENIA REGISTRÁCIEDátum prvej registrácie:
10. DÁTUM REVÍZIE TEXTUPodrobné informácie o tomto lieku sú dostupné na internetovej stránke Európskej agentúry pre lieky
http://www.ema.europa.eu.